Submitted:
04 January 2025
Posted:
06 January 2025
You are already at the latest version
Abstract
Acute Myeloid Leukemia (AML) is a heterogeneous hematological malignancy that poses a sig-nificant therapeutic challenge due to its high recurrence rate and demanding treatment regimens. Increasing evidence suggests that endoplasmic reticulum (ER) stress and downstream activation of the unfolded protein response (UPR) pathway plays a key role in the pathogenesis of AML. ER stress is triggered by the accumulation of misfolded or unfolded proteins within the ER. This causes activation of the UPR to restore cellular homeostasis. However, the UPR can shift from promoting survival to inducing apoptosis under prolonged or excessive stress conditions. AML cells can manipulate the UPR pathway to evade apoptosis, promoting tumor progression and re-sistance against various therapeutic strategies. This review provides the current knowledge on ER stress in AML and its prognostic and therapeutic implications.
Keywords:
acute myeloid leukemia
; endoplasmic reticulum stress
; unfolded protein response
; hematopoietic stem cells
; IRE1α
; PERK
; ATF6
1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy characterized by abnormal clonal proliferation and differentiation of hematopoietic stem cells (HSCs). The 2022 WHO classification based on key mutations and genetic alterations distinguishes 32 subtypes of AML [1]. AML represents nearly 80% of cases of leukemia in individuals over 60 years old [2]. Despite advancements in AML treatments, managing this aggressive malignancy remains challenging. Overall survival rates are significantly affected by the persistence of measurable residual disease (MRD), which often leads to relapse [3]. These challenges underscore the need for novel therapeutic targets and reliable biomarkers to enhance treatment efficacy and to improve patient prognosis.
The pathogenesis of AML involves chromosomal rearrangements, gene mutations, inflammation, epigenetic reprogramming, and deregulated cellular metabolism. The most common risk factors for developing AML include previous exposure to chemotherapy or radiotherapy, toxic substances, and a history of hematologic malignancies. Secondary AML represents 10% to 30% of all AML cases. The likelihood of developing AML rises with age, with a median onset of 68 years. It is commonly observed among men, Caucasians, and Pacific Islanders/Alaskan natives [4,5]. One of the most frequent mutations in newly diagnosed AML patients is the internal tandem duplication (ITD) of the FLT3 gene. Almost 30% of AML cases present this alteration and are linked to a rapidly progressing disease with a poor prognosis [6,7]. Common genetic mutations of interest include alterations in the NPM1, DNMT3A, CCAAT/enhancer-binding protein (CEBPA), IDH1, and IDH2 genes, along with mutations in RUNX1 and TP53. Additionally, notable chromosomal translocations encompass t(8;21), inv(16), and t(15;17) [8,9,10].
Recent studies have drawn attention to the critical role of endoplasmic reticulum (ER) stress in AML’s development and progression [11,12]. The ER is an essential organelle in eukaryotic cells. When proteins misfold or accumulate within the ER, a condition known as ER stress occurs. To restore balance, cells activate an adaptive mechanism known as the unfolded protein response (UPR). However, the UPR may lead to apoptotic cell death if the stress is too severe. The activation of the UPR and ER stress has been associated with the development of several cancers, including colorectal cancer, lung cancer, multiple myeloma, chronic lymphocytic leukemia, and AML [13,14,15,16,17]. Chronic ER stress and prolonged UPR activation are also associated with diseases such as neurodegenerative disorders, diabetes, autoimmune diseases, and cardiovascular diseases [18,19]. In the context of AML, UPR, and ER stress act as both a double-edged sword and a critical modulator of the disease, playing a significant role in leukemogenesis [20]. Moreover, targeting ER stress in AML has emerged as a promising strategy for overcoming resistance and improving treatment outcomes. AML cells often exhibit ER stress reactions that may hinder treatments' success [21,22]. What's more, ER stress can be a prognostic biomarker in AML [23]. Thus, this review presents the current knowledge we have gathered on the multiple roles of ER stress in AML.
2. Pathogenesis of ER Stress and the UPR
The ER plays many essential roles in the cell, including synthesizing, folding, and structurally maturing proteins, storing intracellular calcium ions, synthesizing and storing lipids, and metabolizing glucose. Disruptions in protein folding and modification can accumulate misfolded or unfolded proteins in the ER lumen, resulting in ER stress. This stress triggers the activation of the unfolded protein response (UPR) signaling pathway. The UPR is an adaptive response that promotes cell survival or activates apoptosis when homeostasis is not maintained [24]. The UPR consists of three primary signaling branches: inositol-requiring enzyme-1 (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor-6 (ATF6) [25,26]. The IRE1α- X-box-binding protein 1 (XBP1) pathway is the most conserved branch of the UPR. IRE1 is the oldest UPR sensor, first identified in yeast Saccharomyces cerevisiae [27]. The spliced version of XBP1, known as XBP1s, regulates the upregulation of a broad range of UPR-related genes. These genes are involved in various processes of ER-associated degradation (ERAD) [28]. ER stress causes caspase-mediated cleavage of IRE1. This process generates a stable fragment of IRE1 that includes an ER-lumenal domain and a transmembrane segment. The cleavage separates the stress-sensing and signaling portions of IRE1, leading to a reduction in its activity [29]. PERK is an ER transmembrane receptor that remains inactive while associated with BiP chaperones. When unfolded or misfolded proteins accumulate in the ER lumen due to stress, BiP dissociates from PERK, leading to its activation through oligomerization and trans-autophosphorylation. This active kinase phosphorylates eIF2α, inhibiting global protein translation and causing a G1 phase cell cycle arrest. It also preferentially translates ATF4 to help restore cell homeostasis. If ER stress persists and the pro-adaptive UPR fails, PERK may activate pro-apoptotic signals via CHOP, promoting apoptosis [30]. ATF4 regulates genes essential for stress adaptation, but during prolonged ER stress, it can also activate CCAAT-enhancer-binding protein homologous protein (CHOP) genes, triggering apoptosis [31,32]. The UPR also becomes activated during hypoxia, infection, altered calcium homeostasis, abnormalities in lipid metabolism, and oxidative stress [33,34,35].
2.1. ER Stress and the UPR in HSCs
HSCs are the foundation of the hematopoietic system, playing a crucial role in the long-term maintenance and production of all mature blood cell lineages throughout an organism's lifespan [36,37]. HSCs are exposed to harsh conditions in the bone marrow. The bone marrow niche has low oxygen concentrations, making hematopoietic stem and progenitor cells (HSPCs) susceptible to hypoxia, which can further increase ER stress.
In low-oxygen environments, hypoxia-inducible factors (HIFs) activate, mediating a significant adaptive response to hypoxia that involves multiple pathways necessary for maintaining cellular homeostasis. It has been reported that HIF-2α-deficient HSPCs show higher levels of reactive oxygen species (ROS) generation (Figure 1). Elevated ROS can lead to ER stress and trigger apoptosis by activating the UPR. Additionally, ROS can cause genomic instability by damaging DNA, which may contribute to chemotherapy resistance [38,39]. Specific adaptive pathways of UPR, such as the IRE1α-XBP1 pathway, help protect HSCs from apoptosis caused by ER stress, whereas an IRE1α knockout results in decreased reconstitution of HSCs [40]. Conversely, increased activation of PERK in HSCs makes them more sensitive to ER stress [41].
In vitro studies show that IL-4 induces apoptosis in HSCs and downregulates transcription factors involved in megakaryocyte development. IL-4Rαhigh HSCs are particularly sensitive to ER stress-induced apoptosis [42].
Moreover, research indicates that a deficiency of UFBP1, a vital component of the Ufm1 conjugation system essential for hematopoiesis and erythroid differentiation, leads to increased ER stress and activation of the UPR [43]. Depletion of Uba5 also leads to increased ER stress and decreased levels of transcription factors. In contrast, the knockdown of ASC1 affects the expression of transcription factors without directly impacting ER stress. ASC1 interacts with the promoters of GATA-1 and KLF1 in a UFBP1-dependent manner, emphasizing the interconnected roles of UFBP1, ASC1, and other Ufm1-related components in the survival and differentiation of hematopoietic cells by stabilizing ER function and supporting erythroid gene expression [43]. RCAD/Ufl1, an E3 ligase in the Ufm1 system, regulates DDRGK1, ASC1, ER stress, and autophagy. Its loss disrupts hematopoiesis, causing severe anemia, heightened ER stress, DNA damage, p53 activation, and, ultimately, the death of HSCs [44].
3. Pathogenesis of ER Stress and the UPR in AML
Recent studies indicate that AML cells exploit UPR pathways to evade apoptosis, facilitating tumor progression and contributing to their resistance to various therapeutic approaches [20,45,46,47]. The microenvironment in AML influences ER stress levels, affecting disease progression and response to treatment [38,48]. Leukemic cells interact with their surroundings in ways that can exacerbate ER stress, such as by releasing cytokines and competing for metabolic resources. Additionally, the activation of genes like MYC in AML increases ER tension, which affects various signaling pathways and supports cellular proliferation [48,49].
In AML cases with FLT3-ITD mutations, ER stress, and UPR signaling are critical (Table 1). Cells with these mutations rely on UPR signaling to manage the instability of the mutant FLT3 protein, leading to ER stress caused by improper protein maturation [45,50]. AML cells without ITD mutation exhibiting higher levels of ER stress predominantly rely on the mitochondrial pathway for glucose metabolism. In contrast, AML cells with ITD mutation and lower ER stress favor the glycolytic pathway [51]. Many new therapies are also being developed that target these mutations (Table 1).
The CEBPA gene plays a vital role in developing myeloid cells and is frequently disrupted in AML. The chaperone calreticulin (CALR) involved in the UPR can bind to a stem-loop structure in the five prime regions of CEBPA mRNA, consequently blocking its translation. This mechanism has been primarily observed in specific AML subtypes characterized by gene rearrangements such as t(3;21) or inv(16). Additionally, the XBP1s were identified in 17.4% of AML patients, and this group exhibited significantly higher expression levels of UPR target genes, including CALR, GRP78, and CHOP [52]. The sustained activation of XBP1s expression in AML cells induces apoptosis both in vitro and in vivo. Moreover, moderate XBP1s expression sensitizes these cells to chemotherapeutic treatments [13]. Furthermore, in vitro studies revealed that calreticulin expression may increase through the ATF6 pathway in myeloid leukemia cells, inhibiting CEBPA protein expression both in vitro and in leukemic cells from patients with an activated UPR [52,53].
In myeloid leukemic cells, the overexpression of protein disulfide isomerase (PDI) inhibited the translation of CEBPA without affecting its transcription. Conversely, when PDI was inhibited, the levels of CEBPA protein were restored. It was also discovered that PDI interacts directly with calreticulin. The expression of PDI and a subsequent decrease in CEBPA protein levels were observed when ER stress was induced in these leukemic cells. Notably, 25.4% of AML patients showed signs of ER stress along with elevated levels of PDI. These findings suggest that PDI, as part of the ER stress-associated complex, inhibits the translation of CEBPA and disrupts myeloid differentiation in AML patients experiencing an activated state UPR [54].
Chromatin immunoprecipitation sequencing (ChIP-seq) identified specific target genes of XBP1s, including the long non-coding RNA (lncRNA) MIR22HG, which serves as the precursor transcript of microRNA-22-3p (miR-22-3p). Upregulation of miR-22-3p by XBP1s, or enforced expression of miR-22, significantly decreases cell viability and enhances the sensitivity of leukemic cells to chemotherapy. The intracellular effects of miR-22-3p are mediated, at least in part, by targeting the mRNA encoding the deacetylase sirtuin-1 (SIRT1), a well-established pro-survival factor. Consequently, the novel XBP1s/miR-22/SIRT1 regulatory axis could be pivotal in leukemic cells' proliferation and chemotherapeutic response [13].
O-GlcNAcylation may aid AML cell survival by modulating the UPR, mainly by inhibiting its pro-apoptotic pathways. Increased levels of O-GlcNAc transferase (OGT) and O-GlcNAc hydrolase (OGA) are associated with heightened expression of UPR-related genes. Moreover, UPR-associated transcription factors may boost the hexosamine biosynthetic pathway activity, promoting O-GlcNAcylation. This process disrupts the PERK-CHOP pathway, reducing apoptosis and supporting AML cell survival under stress conditions, such as hypoxia and proteotoxicity in the bone marrow [55].
4. Prognostic Implications of ER Stress Markers in AML
The prognostic significance of ER stress markers in AML has gained increasing attention due to their correlation with treatment outcomes and disease progression. Studies indicate that high levels of the UPR regulator XBP1, a marker of ER stress, correlate with a poor prognosis in AML with FLT3-ITD mutation. Furthermore, XBP1 and IRE1α levels are elevated in FLT3-ITD AML cells compared to those in healthy individuals [45].
In AML cells, lower levels of CEBPA lead to higher levels of DDIT3. This increase in DDIT3 can boost cell death due to stress in the ER and interfere with the process of forming leukemia cells. The balance between C/EBPα forms, C/EBPα-p30 and C/EBPα-p42, also affects how cells react to chemotherapy. A low ratio of C/EBPα-p42 to C/EBPα-p30 in AML cells is linked to resistance against the BCL2 inhibitor venetoclax [60].
A recent study using RNA-seq data from TCGA-LAML focused on genes related to ER stress and identified 42 genes linked to the prognosis of AML. From this group, a 13-gene risk score model was developed and validated. It includes genes such as MBTPS1, SESN2, BCAP31, CHAC1, CALR, UBE2G2, EIF2AK4, CREB3, AIFM1, WFS1, CTH, PTPN1, TMTC4. This model effectively classified patients into high- and low-risk groups, with lower risk scores associated with better survival outcomes [23]. Analysis of 483 pediatric AML patients from a Children's Oncology Group trial showed that low valosin-containing protein (VCP) expression was significantly linked to better five-year survival rates—81% in low VCP patients versus 63% in those with higher levels. VCP is a critical component of ERAD. This association is held regardless of the chemotherapy regimen, with or without bortezomib (Btz). VCP was also an independent predictor of outcomes, with lower VCP levels correlating with higher levels of UPR proteins IRE1 and GRP78. Furthermore, five-year overall survival in patients with low VCP, moderately high IRE1, and high GRP78 improved after treatment with daunorubicin and etoposide combined with Btz compared to daunorubicin and etoposide alone [61].
5. Direct Targeting of ER Stress in AML
The potential of IRE1α inhibition, a component of the UPR, by agents such as MKC-3946, 2-hydroxy-1-naphthaldehyde (HNA), STF-083010, and toyocamycin, to disrupt XBP1 mRNA splicing and show cytotoxic effects against AML cells is a promising research area [62,63,64,65,66]. This inhibition triggers caspase-dependent apoptosis and leads to G1 cell cycle arrest, partly regulated by Bcl-2 family proteins, G1 phase control proteins (such as p21cip1, p27kip1, and cyclin D1), and chaperone proteins [67]. Notably, murine bone marrow cells lacking XBP1 demonstrate resistance to the growth-inhibitory effects of IRE1α inhibitors. Furthermore, they combine HNA with either Btz or arsenic trioxide (AS2O3) to produce synergistic cytotoxicity in AML cells. This synergy is characterized by increased phosphorylation of JNK and decreased phosphorylation of PI3K and MAPK. Additionally, inhibiting IRE1α RNase activity leads to the upregulation of several microRNAs, including miR-34a, in AML cells. In addition, the suppression of miR-34a provides resistance against HNA treatment [53,68].
Moreover, another study indicates that STF-083010 effectively reduced cell survival, inhibited cell growth, and induced apoptosis in AML cell lines and patient samples, all while preserving healthy cells. This selective impact remained consistent even with XBP1 depletion in a mouse model or STF treatment of healthy human cells. Additionally, combining STF-083010 with the FLT3 TKI quizartinib (AC220) significantly increased malignant cell death compared to either treatment alone, indicating a promising dual-target strategy for AML therapy [45].
Further research reveals that combining an IRE1α RNase inhibitor MKC8866 with proteasome inhibitors (Btz and carfilzomib) significantly reduces XBP1s levels. This combination therapy enhances cell death in AML cells, including the resistant CD34+CD38− subpopulation, and impairs their capacity for clonogenic growth [69].
6. Effect of New Therapy on ER Stress in AML
6.1. JUN Inhibition in AML
Inhibiting JUN through short hairpin RNA (shRNA) significantly reduces the viability of AML cells and hampers disease progression in vivo. RNA sequencing analyses indicate that JUN inhibition diminishes the UPR transcriptional activity. Specifically, JUN is activated via MEK signaling in response to ER stress. It binds to the promoters of essential UPR effectors, such as XBP1 and ATF4, stimulating their transcription and assisting AML cells' edition to ER pressure. Furthermore, shRNA-mediated suppression of XBP1 or ATF4 triggers apoptosis in AML cells. It extends disease latency in vivo, implying that the decreased survival associated with JUN inhibition is linked to the loss of pro-survival UPR signaling [70] (Table 2).
6.2. NMT Inhibition in AML
N-myristoylation, a protein modification essential for survival signaling and metabolism, is catalyzed by the enzymes NMT1 and NMT2, which show variable expression in AML cell lines and patient samples. Low NMT2 expression correlates with poor patient outcomes. Zelenirstat, a pioneering pan-NMT inhibitor, has successfully and significantly inhibited myristoylation in AML cells, causing the degradation of Src family kinases, ER stress, apoptosis, and cell death. This successful inhibition underscores the potential of Zelenirstat as a treatment for AML (Table 2). It was tolerated in vivo and reduced leukemic burden in various AML models. LSC-enriched fractions, especially in the OCI-AML22 model, were notably sensitive to myristoylation inhibition. Zelenirstat also disrupted mitochondrial complex I function and oxidative phosphorylation, which is crucial for LSC survival [71].
6.3. Proteasome Inhibition in AML
Proteotoxic stress, induced by proteasome inhibition, is emerging as a potential AML treatment strategy. Btz is a selective proteasome inhibitor approved by the FDA for treating multiple myeloma. It shows efficacy partly by triggering ER stress-dependent apoptosis [72]. In in vitro study, treatment with proteasome inhibitors such as Btz and carfilzomib increased XBP1s in KG1a and U937 cell lines [69].
AML cells treated with Btz showed down-regulation of the m6A regulator WTAP due to increased oxidative stress, as indicated by elevated ROS levels and HMOX-1 activation. In addition, Btz has been linked to ER stress, which further contributes to proteotoxicity. This down-regulation can be reversed by the reducing agent N-acetylcysteine. The study also identified modified m6A circRNAs affected by Btz, including circHIPK3, which is involved in protein folding and regulation of oxidative stress [73].
A combination of low doses of the differentiating agent retinoic acid (R), the proteasome inhibitor Btz (B), and the oxidative stress inducer arsenic trioxide (A) shows potent cytotoxic effects on FLT3-ITD+ AML cells by inducing ER stress and oxidative stress (Table 2). However, AML cells gain resistance to RBA in co-culture with bone marrow stromal cells. High-dose ascorbic acid overcomes this resistance, and the RBA-ascorbic acid combination significantly extends survival in a murine FLT3-ITD+ AML model without toxicity. Notably, the treatment disrupts AML-BMSC interactions, causing actin cytoskeleton alterations, increased nuclear thickness, and cytosolic relocalization of YAP in BMSCs [74].
6.4. Malachite Green-Mediated Photodynamic Therapy (PDT) in AML
PDT is a therapy that utilizes the cytotoxic properties of visible light in the presence of a photosensitizer and oxygen. A recent study investigated the relationship between PDT's anti-leukemic effects and ER stress induction in a subtype of AML-acute promyelocytic leukemia cells. The cells were incubated with various concentrations of malachite green and then exposed to specific light conditions. Cell viability was assessed using the MTT assay, and the expression of ER stress markers PERK and GRP78 was evaluated through immunocytochemical staining. The results showed that the combination of malachite green and light significantly decreased cell viability compared to controls treated with malachite green alone, light alone, or neither. Additionally, the treatment group significantly upregulated the expression of PERK and GRP78 [75].
6.5. Staphylococcal Enterotoxins in AML
Staphylococcus aureus (S. aureus) is a common pathogen in patients undergoing therapy for AML. It produces various virulence factors, including Staphylococcal enterotoxins A (SEA) and B (SEB). SEA and SEB significantly alter AML cell behavior. The treatment with SEA and SEB increased AML cell proliferation and resistance to cytarabine while promoting cell migration and invasion, affecting approximately 50% of the cells. Transcriptomic analysis and gene set enrichment analyses, validated by PCR, revealed dysregulation of immune-related genes and pathways. This dysregulation may enable AML cells to evade immune responses and survive in the hostile environment created by these toxins, potentially through the ER stress signaling pathway [76].
6.6. Camalexin in AML
Camalexin is a phytoalexin, a naturally occurring antimicrobial compound produced by plants in response to stress, such as pathogen attacks. It inhibits the growth of various cancer cell lines [77]. In AML cells, camalexin induces apoptosis through a caspase-dependent mitochondrial pathway, with ER stress as an upstream regulator. Treatment with camalexin significantly increases the levels of ROS and enhances the activity of superoxide dismutase (SOD) and catalase. Additionally, it raises oxidized glutathione (GSSG) levels while lowering the reduced glutathione (GSH) levels. These changes suggest that ROS generation is crucial for the ER stress and apoptosis triggered by camalexin. In a xenograft mouse model, camalexin effectively suppressed tumor growth without causing noticeable toxicity, indicating that its antitumor effects are mediated through ROS-induced ER stress, leading to mitochondrial apoptosis [78].
6.7. PLK4 Inhibition in AML
Polo-like kinase 4 (PLK4) is a crucial regulator of centriole duplication. Abnormal PLK expression is linked to cancer development and progression [79,80]. PLK4 is overexpressed in AML cells. The knockdown of PLK4, or its specific inhibition using centrinone, leads to G2/M phase cell cycle arrest (Table 2). The suppression of cyclin B1 and Cdc2 expression and the promotion of pro-apoptotic protein levels cause this arrest. Furthermore, targeting PLK4 enhances the expression of proteins associated with ER stress, such as GRP78, ATF4, ATF6, and CHOP [81].
6.8. Venetoclax in AML
Venetoclax, a drug that targets and inhibits BCL-2, is used to treat AML, often in combination with various agents to enhance therapeutic effects. However, resistance and recurrence remain significant challenges. In vitro and in vivo experiments using AML cell lines, particularly Molm13 and THP-1, have shown that metformin and venetoclax work synergistically to inhibit cell growth and promote apoptosis. The combination of these drugs significantly increases the expression of CHOP, a marker of ER stress. Moreover, reducing CHOP expression through knockdown techniques decreases the apoptosis induced by this drug combination. Additionally, the combination of metformin and venetoclax has demonstrated strong anti-leukemia effects in xenograft models and bone marrow samples from AML patients. These findings suggest that pairing metformin and venetoclax could provide a robust and safe treatment option for AML, warranting further clinical studies [21] (Table 2). A low C/EBPα p42 to p30 ratio in AML cells is linked to resistance against the BCL-2 inhibitor venetoclax, as BCL-2 is a primary target of DDIT3. However, combining venetoclax with ER stress inducers such as tunicamycin and sorafenib can overcome this resistance. These findings indicate that AML patients with a low C/EBPα p42/p30 ratio may not respond well to venetoclax monotherapy but could benefit from combined treatment approaches that include ER stress-inducing agents [60].
6.9. PAD Inhibition in AML
Peptidylarginine deiminases (PADs) are a group of enzymes that facilitate post-translational deimination or citrullination of target proteins. This process alters their structure and function and modulates gene regulation [82,83]. Elevated levels of PAD2 and PAD4 have been linked to the progression of acute myeloid leukemia (AML). In vitro studies indicate that the pan-PAD inhibitor BB-Cl-Amidine (BB-Cl-A) induces apoptosis in AML cells, a response not elicited by the PAD4-specific inhibitor. Treatment with BB-Cl-A resulted in the activation of the ER stress response, as evidenced by increased levels of p-PERK and p-eIF2α, ultimately promoting apoptosis in AML cells. The findings advise that PAD2 is essential for maintaining ER homeostasis and mobile survival, indicating that inhibiting PAD2 with BB-Cl-A may also provide a promising therapeutic approach for AML [84] (Table 2).
6.10. G protein–Coupled Estrogen Receptor-1 (GPER) in AML
The GPER has different roles from estrogen receptors ERα and ERβ, and its activation by specific agonists suppresses tumors in various cancers [85]. The proteins S100A8 and S100A9 are stored in neutrophils as a stable heterodimer known as calprotectin. Calprotectin is released in response to infection and activates the innate immune response [86]. S100A9 is extensively overexpressed in AML, and therapeutic strategies aimed toward concentrating on this protein were proven to reduce the viability of AML cells, promoting apoptosis efficiently. This reduction in cell survival is mainly attributed to the downregulation of mTOR and ER pathways.
When S100A9 is silenced, it significantly alters the cellular environment, affecting extracellular acidification and mitochondrial metabolism, essential for energy production and normal cellular function. Furthermore, the S100A9 inhibitor tasquinimod has been shown to primarily disrupt mitochondrial characteristics, highlighting its potential as a targeted therapy.
Significantly, strategies focusing on S100A9 impair the increase of AML cells and enhance their susceptibility to the anti-most cancer agent venetoclax. A high-quality decrease in the anti-apoptotic proteins BCL-2 and c-MYC stages, crucial for most cancer cell survival, characterizes this more advantageous sensitivity. This twin action of S100A9-targeting approaches underscores their capacity as a promising avenue for improving remedy outcomes in AML [87] (Table 2).
Recent research shows that LNS8801, a synthetic agonist of the G protein-coupled estrogen receptor (GPER), selectively induces apoptosis in human AML cells. Studies on AML cell lines and patient samples reveal that LNS8801 inhibits AML cell growth without impacting normal mononuclear cells. Although GPER is present in healthy and malignant myeloid cells, LNS8801’s anti-AML effects appear independent of GPER signaling. Instead, LNS8801 triggers AML cell death mainly through a caspase-dependent apoptosis pathway driven by ROS production and activation of the ER stress response, primarily via IRE1α [88] (Table 2).
6.11. N-acetyltransferase 10 (NAT10) Inhibition in AML
NAT10 is an enzyme with acetyltransferase and RNA binding activities. It serves as a writer enzyme for the N4-acetylcytidine modification of mRNAs. NAT10 is overexpressed in multiple malignancies, including AML, and is linked to poor prognosis, making it a potential therapeutic target [89]. In an in vitro study, NAT10 inhibition—achieved through shRNA knockdown and pharmacological inhibitors—reduced cell proliferation, caused G1 cell cycle arrest, and increased apoptosis in AML cells (Table 2). Mechanistically, NAT10 inhibition reduced CDK2, CDK4, Cyclin D1, and Cyclin E levels while increasing p16 and p21, which are critical for cell cycle regulation. Additionally, NAT10 inhibition induced ER stress by upregulating GRP78 and promoting caspase 12 cleavage, activating the UPR via increased IRE1, CHOP, and PERK levels. This response activated apoptosis by upregulating pro-apoptotic Bax and Bak and downregulating anti-apoptotic Bcl-2 [90].
6.12. p53-MDM2 Axis Inhibition in AML
The MDM2 proto-oncogene (MDM2) is a negative regulator of the p53 protein [91]. Dysregulation of MDM2 is associated with poorer outcomes in AML. Studies are ongoing to determine the efficacy of blocking the p53-MDM2 pathway in AML. MDM2 antagonists, such as Nutlin-3a, show limited efficacy alone, prompting the exploration of combination treatments. Nutlin-3a with Triptolid shows a synergistic effect; it inhibits cell proliferation and induces mitochondrial-mediated apoptosis in p53 wild-type (wt) AML cells, in vivo and ex vivo. This combination also reduces tumor growth and leukemia burden in an AML xenograft model (Table 2). Moreover, the combination shows partial effectiveness in some p53-deficient cases. Nutlin-3a enhanced the expression of p53 target genes PUMA and p21, while Triptolide decreased levels of anti-apoptotic factors XIAP and Mcl-1. The combined treatment amplified these effects. Triptolide also operates through p53-independent mechanisms by disrupting the MYC-ATF4 axis, leading to ER stress induction [92].
6.13. Lysine Demethylase 6A (KDM6A) in AML
KDM6A is a member of the Jumonji-C (JmjC) domain-containing family of histone demethylases and plays an essential role during embryonic development. Mutations of KDM6A are found in various neoplasms, including multiple myeloma, acute lymphoid leukemia, and pancreatic adenocarcinoma [93].
KDM6A regulates immunogenic cell death (ICD) in AML through epigenetic control of genes involved in ER stress and immune signaling pathways. Loss of KDM6A enhances the immunogenicity of AML cells and sensitizes them to immunomodulatory therapies. Targeting KDM6A-deficient AML subtypes with agents that induce ICD or combine epigenetic modulation with immunotherapy could be a promising therapeutic strategy. Moreover, KDM6A deficiency imparts features similar to "BRCAness," making AML cells more sensitive to PARP inhibitors like olaparib. Treatment with olaparib further induced ICD and increased the expression of NKG2D ligands (ULBP1, ULBP5) on AML cells. Combining olaparib with the KDM6A inhibitor, GSK-J4 showed additive effects in promoting ICD and immune activation [94].
7. Conclusions
Overtime, the ER stress and UPR pathway have become established in the pathogenesis of many diseases. However, the role of UPR and ER stress in AML is still poorly understood. Recent research shows that ER stress has a dual role in AML, from promoting leukemic cell survival and resistance to chemotherapy to the possibility of using this pathway in treatment, prognosis, and disease stratification. AML is characterized by high heterogeneity and a large number of genetic variations, which is why treatment is so difficult. The hope for an improved prognosis comes from new therapies that target specific mutations found in AML. Another area of improvement in prognosis is targeting ER stress therapy in AML. Currently, the only therapy targeting ER stress directly is the inhibition of IRE1α. IRE1α inhibitors have demonstrated effectiveness in inducing apoptosis in AML cells. Nevertheless, these studies have only been conducted in vitro. There is no research with these inhibitors in vivo in animal models or clinical trials with patients. However, there are many therapies where ER stress is involved indirectly. The use of targeted therapy for ER stress in AML appears to be more effective when combined with other drugs rather than as a monotherapeutic treatment. Intriguingly, ER stress biomarkers may also be helpful as prognostic and stratification factors. ER stress biomarker levels in samples from AML patients are associated with five-year survival rates. Coming research should focus on the results of IRE1α inhibition in animal models and clinical trials. Additional studies confirming the role of ER stress biomarkers, such as prognostic and stratification factors, are also necessary.
Author Contributions
Conceptualization, W.W., and A.S.; methodology, W.W., and G.G.; formal analysis, A.S. and I.M.; investigation, W.W., A.S., G.G., and N.S.; resources, W.R.-K. and I.M.; writing—original draft preparation W.W., A.S., and N.S.; writing—review and editing, A.S., and I.M.; visualization, W.W., and N.S.; supervision, A.S, and I.M.; project administration, A.S., and I.M.; funding acquisition, I.M. All authors have read and agreed to the published version of the manuscript.
Funding
The study was financed from the funds of the Medical University of Lodz, Poland, no. 503/1-156-07/503-11-001.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated in the present study may be requested from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. R I Med J (2013) 2020, 103, 38–40. [Google Scholar] [PubMed]
- Short, N.J.; Zhou, S.; Fu, C.; Berry, D.A.; Walter, R.B.; Freeman, S.D.; Hourigan, C.S.; Huang, X.; Nogueras Gonzalez, G.; Hwang, H.; et al. Association of Measurable Residual Disease With Survival Outcomes in Patients With Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. JAMA Oncology 2020, 6, 1890–1899. [Google Scholar] [CrossRef]
- Wachter, F.; Pikman, Y. Pathophysiology of Acute Myeloid Leukemia. Acta Haematol 2024, 147, 229–246. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of Acute Myeloid Leukemia: Recent Progress and Enduring Challenges. Blood Reviews 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Hebestreit, K.; Gröttrup, S.; Emden, D.; Veerkamp, J.; Ruckert, C.; Klein, H.-U.; Müller-Tidow, C.; Dugas, M. Leukemia Gene Atlas – A Public Platform for Integrative Exploration of Genome-Wide Molecular Data. PLOS ONE 2012, 7, e39148. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Kiyoi, H.; Nakao, M.; Iwai, T.; Misawa, S.; Okuda, T.; Sonoda, Y.; Abe, T.; Kahsima, K.; Matsuo, Y.; et al. Internal Tandem Duplication of the FLT3 Gene Is Preferentially Seen in Acute Myeloid Leukemia and Myelodysplastic Syndrome among Various Hematological Malignancies. A Study on a Large Series of Patients and Cell Lines. Leukemia 1997, 11, 1605–1609. [Google Scholar] [CrossRef]
- Estey, E.; Hasserjian, R.P.; Döhner, H. Distinguishing AML from MDS: A Fixed Blast Percentage May No Longer Be Optimal. Blood 2022, 139, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, O.K.; Porwit, A.; Orazi, A.; Hasserjian, R.P.; Foucar, K.; Duncavage, E.J.; Arber, D.A. The International Consensus Classification of Acute Myeloid Leukemia. Virchows Arch 2023, 482, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic Mutations Precede Acute Myeloid Leukemia Years before Diagnosis. Nat Med 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Hawkins, E.; Parker, R.E.; Ross, M.; Kmieciak, M.; Reshko, L.B.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A.; et al. Co-Administration of the mTORC1/TORC2 Inhibitor INK128 and the Bcl-2/Bcl-xL Antagonist ABT-737 Kills Human Myeloid Leukemia Cells through Mcl-1 down-Regulation and AKT Inactivation. Haematologica 2015, 100, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, H.; Miao, Z.; Yu, Y.; Gai, D.; Zhang, G.; Ge, L.; Shen, X. Endoplasmic Reticulum Stress-Related Signature Predicts Prognosis and Immune Infiltration Analysis in Acute Myeloid Leukemia. Hematology 2023, 28, 2246268. [Google Scholar] [CrossRef]
- Philippe, C.; Jaud, M.; Féral, K.; Gay, A.; Van Den Berghe, L.; Farce, M.; Bousquet, M.; Pyronnet, S.; Mazzolini, L.; Rouault-Pierre, K.; et al. Pivotal Role of the Endoplasmic Reticulum Stress-Related XBP1s/miR-22/SIRT1 Axis in Acute Myeloid Leukemia Apoptosis and Response to Chemotherapy. Leukemia 2024, 38, 1764–1776. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Beilankouhi, E.A.V.; Sajadi, M.A.; Alipourfard, I.; Hassani, P.; Valilo, M.; Safaralizadeh, R. Role of the ER-Induced UPR Pathway, Apoptosis, and Autophagy in Colorectal Cancer. Pathology - Research and Practice 2023, 248, 154706. [Google Scholar] [CrossRef]
- Féral, K.; Jaud, M.; Philippe, C.; Di Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules 2021, 11, 199. [Google Scholar] [CrossRef]
- Śniegocka, M.; Liccardo, F.; Fazi, F.; Masciarelli, S. Understanding ER Homeostasis and the UPR to Enhance Treatment Efficacy of Acute Myeloid Leukemia. Drug Resist Updat 2022, 64, 100853. [Google Scholar] [CrossRef] [PubMed]
- Panahi, M.; Hase, Y.; Gallart-Palau, X.; Mitra, S.; Watanabe, A.; Low, R.C.; Yamamoto, Y.; Sepulveda-Falla, D.; Hainsworth, A.H.; Ihara, M.; et al. ER Stress Induced Immunopathology Involving Complement in CADASIL: Implications for Therapeutics. Acta Neuropathol Commun 2023, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Vijayalalitha, R.; Archita, T.; Juanitaa, G.R.; Jayasuriya, R.; Amin, K.N.; Ramkumar, K.M. Role of Long Non-Coding RNA in Regulating ER Stress Response to the Progression of Diabetic Complications. Curr Gene Ther 2023, 23, 96–110. [Google Scholar] [CrossRef]
- Doron, B.; Abdelhamed, S.; Butler, J.T.; Hashmi, S.K.; Horton, T.M.; Kurre, P. Transmissible ER Stress Reconfigures the AML Bone Marrow Compartment. Leukemia 2019, 33, 918–930. [Google Scholar] [CrossRef]
- Hua, L.; Yang, N.; Li, Y.; Huang, K.; Jiang, X.; Liu, F.; Yu, Z.; Chen, J.; Lai, J.; Du, J.; et al. Metformin Sensitizes AML Cells to Venetoclax through Endoplasmic Reticulum Stress—CHOP Pathway. British Journal of Haematology 2023, 202, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Qu, J.; Lin, D.; Feng, K.; Tan, M.; Liao, H.; Zeng, L.; Xiong, Q.; Huang, J.; Chen, W. Reduced Proteolipid Protein 2 Promotes Endoplasmic Reticulum Stress-Related Apoptosis and Increases Drug Sensitivity in Acute Myeloid Leukemia. Mol Biol Rep 2023, 51, 10. [Google Scholar] [CrossRef]
- Pan, M.; Zhou, J.; Jiao, C.; Ge, J. Bioinformatics Analysis of the Endoplasmic Reticulum Stress-Related Prognostic Model and Immune Cell Infiltration in Acute Myeloid Leukemia. Hematology 2023, 28, 2221101. [Google Scholar] [CrossRef] [PubMed]
- Bolland, H.; Ma, T.S.; Ramlee, S.; Ramadan, K.; Hammond, E.M. Links between the Unfolded Protein Response and the DNA Damage Response in Hypoxia: A Systematic Review. Biochemical Society Transactions 2021, 49, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 Signaling Affects Cell Fate during the Unfolded Protein Response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. ER Stress and Distinct Outputs of the IRE1α RNase Control Proliferation and Senescence in Response to Oncogenic Ras. Proceedings of the National Academy of Sciences 2017, 114, 9900–9905. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional Induction of Genes Encoding Endoplasmic Reticulum Resident Proteins Requires a Transmembrane Protein Kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-Mediated Unconventional mRNA Splicing and S2P-Mediated ATF6 Cleavage Merge to Regulate XBP1 in Signaling the Unfolded Protein Response. Genes Dev 2002, 16, 452–466. [Google Scholar] [CrossRef]
- Shemorry, A.; Harnoss, J.M.; Guttman, O.; Marsters, S.A.; Kőműves, L.G.; Lawrence, D.A.; Ashkenazi, A. Caspase-Mediated Cleavage of IRE1 Controls Apoptotic Cell Commitment during Endoplasmic Reticulum Stress. eLife 2019, 8, e47084. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Current Molecular Medicine 16, 533–544. [CrossRef]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating Transcription Factor 4 Is Translationally Regulated by Hypoxic Stress. Molecular and Cellular Biology 2004, 24, 7469–7482. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H. CHOP Is a Multifunctional Transcription Factor in the ER Stress Response. The Journal of Biochemistry 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Agellon, L.B.; Michalak, M. Chapter One - Calcium Signaling and Endoplasmic Reticulum Stress. In International Review of Cell and Molecular Biology; Marchi, S., Galluzzi, L., Eds.; Inter-Organellar Ca Signaling in Health and Disease - Part B; Academic Press, 2021; Vol. 363, pp. 1–20.
- Banerjee, T.; Grabon, A.; Taylor, M.; Teter, K. cAMP-Independent Activation of the Unfolded Protein Response by Cholera Toxin. Infection and Immunity 2021, 89. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-Induced Unfolded Protein Response Feedback Attenuates Ferroptosis via PERK/ATF4/HSPA5 Pathway in Glioma Cells. Journal of Experimental & Clinical Cancer Research 2019, 38, 402. [Google Scholar] [CrossRef]
- Becker, A.J.; McCulloch, E.A.; Till, J.E. Cytological Demonstration of the Clonal Nature of Spleen Colonies Derived from Transplanted Mouse Marrow Cells. 1963.
- Calvanese, V.; Nguyen, A.T.; Bolan, T.J.; Vavilina, A.; Su, T.; Lee, L.K.; Wang, Y.; Lay, F.D.; Magnusson, M.; Crooks, G.M.; et al. MLLT3 Governs Human Haematopoietic Stem-Cell Self-Renewal and Engraftment. Nature 2019, 576, 281–286. [Google Scholar] [CrossRef]
- Rouault-Pierre, K.; Lopez-Onieva, L.; Foster, K.; Anjos-Afonso, F.; Lamrissi-Garcia, I.; Serrano-Sanchez, M.; Mitter, R.; Ivanovic, Z.; de Verneuil, H.; Gribben, J.; et al. HIF-2α Protects Human Hematopoietic Stem/Progenitors and Acute Myeloid Leukemic Cells from Apoptosis Induced by Endoplasmic Reticulum Stress. Cell Stem Cell 2013, 13, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Carroll, K.S. Redox Regulation of Epidermal Growth Factor Receptor Signaling through Cysteine Oxidation. Biochemistry 2012, 51, 9954–9965. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, M.; Jin, X.; Ney, G.; Yang, K.B.; Peng, F.; Cao, J.; Iwawaki, T.; Del Valle, J.; Chen, X.; et al. Adaptive Endoplasmic Reticulum Stress Signalling via IRE1α–XBP1 Preserves Self-Renewal of Haematopoietic and Pre-Leukaemic Stem Cells. Nat Cell Biol 2019, 21, 328–337. [Google Scholar] [CrossRef]
- van Galen, P.; Kreso, A.; Mbong, N.; Kent, D.G.; Fitzmaurice, T.; Chambers, J.E.; Xie, S.; Laurenti, E.; Hermans, K.; Eppert, K.; et al. The Unfolded Protein Response Governs Integrity of the Haematopoietic Stem-Cell Pool during Stress. Nature 2014, 510, 268–272. [Google Scholar] [CrossRef]
- Gao, A.; Xu, S.; Li, Q.; Zhu, C.; Wang, F.; Wang, Y.; Hao, S.; Dong, F.; Cheng, H.; Cheng, T.; et al. Interlukin-4 Weakens Resistance to Stress Injury and Megakaryocytic Differentiation of Hematopoietic Stem Cells by Inhibiting Psmd13 Expression. Sci Rep 2023, 13, 14253. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Pi, W.; Sivaprakasam, S.; Zhu, X.; Zhang, M.; Chen, J.; Makala, L.; Lu, C.; Wu, J.; Teng, Y.; et al. UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLOS Genetics 2015, 11, e1005643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhu, X.; Zhang, Y.; Cai, Y.; Chen, J.; Sivaprakasam, S.; Gurav, A.; Pi, W.; Makala, L.; Wu, J.; et al. RCAD/Ufl1, a Ufm1 E3 Ligase, Is Essential for Hematopoietic Stem Cell Function and Murine Hematopoiesis. Cell Death Differ 2015, 22, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Jaquet, T.; Preisinger, C.; Bütow, M.; Tillmann, S.; Chatain, N.; Huber, M.; Koschmieder, S.; Kirschner, M.; Jost, E.; Brümmendorf, T.H.; et al. The Unfolded Protein Response Mediates Resistance in AML and Its Therapeutic Targeting Enhances TKI Induced Cell Death in FLT3-ITD + AML. Blood 2021, 138, 2246. [Google Scholar] [CrossRef]
- Zhou, C.; Li, J.; Du, J.; Jiang, X.; Xu, X.; Liu, Y.; He, Q.; Liang, H.; Fang, P.; Zhan, H.; et al. HMGCS1 Drives Drug-Resistance in Acute Myeloid Leukemia through Endoplasmic Reticulum-UPR-Mitochondria Axis. Biomedicine & Pharmacotherapy 2021, 137, 111378. [Google Scholar] [CrossRef]
- Benito, J.; Zeng, Z.; Konopleva, M.; Wilson, W.R. Targeting Hypoxia in the Leukemia Microenvironment. International Journal of Hematologic Oncology 2013, 2, 279–288. [Google Scholar] [CrossRef]
- Huiting, L.N.; Samaha, Y.; Zhang, G.L.; Roderick, J.E.; Li, B.; Anderson, N.M.; Wang, Y.W.; Wang, L.; Laroche, F.J.F.; Choi, J.W.; et al. UFD1 Contributes to MYC-Mediated Leukemia Aggressiveness through Suppression of the Proapoptotic Unfolded Protein Response. Leukemia 2018, 32, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, B.; Iosue, I.; Djodji Damas, N.; Mangiavacchi, A.; Chiaretti, S.; Messina, M.; Padula, F.; Guarini, A.; Bozzoni, I.; Fazi, F.; et al. Critical Role of C-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes & Cancer 2011, 2, 585–592. [Google Scholar] [CrossRef]
- Chan, P.; Stolz, J.; Kohl, S.; Chiang, W.-C.; Lin, J.H. Endoplasmic Reticulum Stress in Human Photoreceptor Diseases. Brain Research 2016, 1648, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Turos-Cabal, M.; Sánchez-Sánchez, A.M.; Puente-Moncada, N.; Herrera, F.; Antolin, I.; Rodríguez, C.; Martín, V. FLT3-ITD Regulation of the Endoplasmic Reticulum Functions in Acute Myeloid Leukemia. Hematol Oncol 2024, 42, e3281. [Google Scholar] [CrossRef]
- Schardt, J.A.; Mueller, B.U.; Pabst, T. Chapter Thirteen - Activation of the Unfolded Protein Response in Human Acute Myeloid Leukemia. In Methods in Enzymology; Conn, P.M., Ed.; The Unfolded Protein Response and Cellular Stress, Part A; Academic Press, 2011; Vol. 489, pp. 227–243.
- Sun, H.; Lin, D.-C.; Guo, X.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-Driven pro-Survival Pathways Is a Promising Therapeutic Application in Acute Myeloid Leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef]
- Haefliger, S.; Klebig, C.; Schaubitzer, K.; Schardt, J.; Timchenko, N.; Mueller, B.U.; Pabst, T. Protein Disulfide Isomerase Blocks CEBPA Translation and Is Up-Regulated during the Unfolded Protein Response in AML. Blood 2011, 117, 5931–5940. [Google Scholar] [CrossRef] [PubMed]
- Schauner, R.; Cress, J.; Hong, C.; Wald, D.; Ramakrishnan, P. Single Cell and Bulk RNA Expression Analyses Identify Enhanced Hexosamine Biosynthetic Pathway and O-GlcNAcylation in Acute Myeloid Leukemia Blasts and Stem Cells. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Follow-up of Patients with R/R FLT3-Mutation-Positive AML Treated with Gilteritinib in the Phase 3 ADMIRAL Trial. Blood 2022, 139, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Erba, H.P.; Montesinos, P.; Kim, H.-J.; Patkowska, E.; Vrhovac, R.; Žák, P.; Wang, P.-N.; Mitov, T.; Hanyok, J.; Kamel, Y.M.; et al. Quizartinib plus Chemotherapy in Newly Diagnosed Patients with FLT3-Internal-Tandem-Duplication-Positive Acute Myeloid Leukaemia (QuANTUM-First): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Frankfurt, O.; Schuh, A.C.; Döhner, H.; Martinelli, G.; Patel, P.A.; Raffoux, E.; et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination With Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J Clin Oncol 2021, 39, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Kim, H.T.; Soiffer, R.J.; Levis, M.J.; Li, S.; Kim, A.S.; Mims, A.S.; DeFilipp, Z.; El-Jawahri, A.; McAfee, S.L.; et al. Enasidenib as Maintenance Following Allogeneic Hematopoietic Cell Transplantation for IDH2-Mutated Myeloid Malignancies. Blood Advances 2022, 6, 5857–5865. [Google Scholar] [CrossRef]
- Du, M.; Wang, M.; Liu, M.; Fu, S.; Lin, Y.; Huo, Y.; Yu, J.; Yu, X.; Wang, C.; Xiao, H.; et al. C/EBPα-P30 Confers AML Cell Susceptibility to the Terminal Unfolded Protein Response and Resistance to Venetoclax by Activating DDIT3 Transcription. Journal of Experimental & Clinical Cancer Research 2024, 43, 79. [Google Scholar] [CrossRef]
- Hoff, F.W.; Qiu, Y.; Brown, B.D.; Gerbing, R.B.; Leonti, A.R.; Ries, R.E.; Gamis, A.S.; Aplenc, R.; Kolb, E.A.; Alonzo, T.A.; et al. Valosin-Containing Protein (VCP/P97) Is Prognostically Unfavorable in Pediatric AML, and Negatively Correlates with Unfolded Protein Response Proteins IRE1 and GRP78: A Report from the Children’s Oncology Group. PROTEOMICS – Clinical Applications 2023, 17, 2200109. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.-T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 Splicing by Inhibition of IRE1α Is a Promising Therapeutic Option in Multiple Myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Khan, M.H.; Cai, M.; Deng, J.; Yu, P.; Liang, H.; Yang, F. Anticancer Function and ROS-Mediated Multi-Targeting Anticancer Mechanisms of Copper (II) 2-Hydroxy-1-Naphthaldehyde Complexes. Molecules 2019, 24, 2544. [Google Scholar] [CrossRef]
- Hatokova, Z.; Evinova, A.; Racay, P. STF-083010 an Inhibitor of IRE1α Endonuclease Activity Affects Mitochondrial Respiration and Generation of Mitochondrial Membrane Potential. Toxicol In Vitro 2023, 92, 105652. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Djibo, R.; Darracq, A.; Calendo, G.; Zhang, H.; Henry, R.A.; Andrews, A.J.; Baylin, S.B.; Madzo, J.; Najmanovich, R.; et al. Selective CDK9 Inhibition by Natural Compound Toyocamycin in Cancer Cells. Cancers (Basel) 2022, 14, 3340. [Google Scholar] [CrossRef]
- Yang, Z.; Huo, Y.; Zhou, S.; Guo, J.; Ma, X.; Li, T.; Fan, C.; Wang, L. Cancer Cell-Intrinsic XBP1 Drives Immunosuppressive Reprogramming of Intratumoral Myeloid Cells by Promoting Cholesterol Production. Cell Metab 2022, 34, 2018–2035.e8. [Google Scholar] [CrossRef] [PubMed]
- Gebert, M.; Bartoszewska, S.; Opalinski, L.; Collawn, J.F.; Bartoszewski, R. IRE1-Mediated Degradation of Pre-miR-301a Promotes Apoptosis through Upregulation of GADD45A. Cell Commun Signal 2023, 21, 322. [Google Scholar] [CrossRef]
- Sun, H.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; Chien, W.; Müller-Tidow, C.; et al. Abstract 510: Inhibition of IRE1α-Driven pro-Survival Pathways Is a Promising Therapeutic Application in Acute Myeloid Leukemia. Cancer Research 2014, 74, 510. [Google Scholar] [CrossRef]
- Creedican, S.; Robinson, C.M.; Mnich, K.; Islam, M.N.; Szegezdi, E.; Clifford, R.; Krawczyk, J.; Patterson, J.B.; FitzGerald, S.P.; Summers, M.; et al. Inhibition of IRE1α RNase Activity Sensitizes Patient-Derived Acute Myeloid Leukaemia Cells to Proteasome Inhibitors. Journal of Cellular and Molecular Medicine 2022, 26, 4629–4633. [Google Scholar] [CrossRef]
- Zhou, C.; Martinez, E.; Di Marcantonio, D.; Solanki-Patel, N.; Aghayev, T.; Peri, S.; Ferraro, F.; Skorski, T.; Scholl, C.; Fröhling, S.; et al. JUN Is a Key Transcriptional Regulator of the Unfolded Protein Response in Acute Myeloid Leukemia. Leukemia 2017, 31, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Gamma, J.M.; Liu, Q.; Beauchamp, E.; Iyer, A.; Yap, M.C.; Zak, Z.; Ekstrom, C.; Pain, R.; Kostiuk, M.A.; Mackey, J.R.; et al. Zelenirstat Inhibits N-Myristoyltransferases to Disrupt Src Family Kinase Signaling and Oxidative Phosphorylation, Killing Acute Myeloid Leukemia Cells. Molecular Cancer Therapeutics 2024, OF1–OF12. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, Y.; Hu, K.; Lu, J. GRP78 Inhibitor HA15 Increases the Effect of Bortezomib on Eradicating Multiple Myeloma Cells through Triggering Endoplasmic Reticulum Stress. Heliyon 2023, 9, e19806. [Google Scholar] [CrossRef] [PubMed]
- Iaiza, A.; Mazzanti, G.; Goeman, F.; Cesaro, B.; Cortile, C.; Corleone, G.; Tito, C.; Liccardo, F.; De Angelis, L.; Petrozza, V.; et al. WTAP and m6A-Modified circRNAs Modulation during Stress Response in Acute Myeloid Leukemia Progenitor Cells. Cell. Mol. Life Sci. 2024, 81, 276. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, F.; Śniegocka, M.; Tito, C.; Iaiza, A.; Ottone, T.; Divona, M.; Travaglini, S.; Mattei, M.; Cicconi, R.; Miglietta, S.; et al. Retinoic Acid and Proteotoxic Stress Induce AML Cell Death Overcoming Stromal Cell Protection. Journal of Experimental & Clinical Cancer Research 2023, 42, 223. [Google Scholar] [CrossRef]
- Çalişkan, M.; Bayrak, G.; Özlem Çalişkan, S. Anti-Leukemic Effect of Malachite Green-Mediated Photodynamic Therapy by Inducing ER Stress in HL-60 Cells. Medical Records 2024, 6, 8–13. [Google Scholar] [CrossRef]
- Turk, S.; Yanpar, H.; Baesmat, A.S.; Canli, S.D.; Cinar, O.E.; Malkan, U.Y.; Turk, C.; Haznedaroglu, I.C.; Ucar, G. Enterotoxins A and B Produced by Staphylococcus Aureus Increase Cell Proliferation, Invasion and Cytarabine Resistance in Acute Myeloid Leukemia Cell Lines. Heliyon 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Pilatova, M.; Ivanova, L.; Kutschy, P.; Varinska, L.; Saxunova, L.; Repovska, M.; Sarissky, M.; Seliga, R.; Mirossay, L.; Mojzis, J. In Vitro Toxicity of Camalexin Derivatives in Human Cancer and Non-Cancer Cells. Toxicol In Vitro 2013, 27, 939–944. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, G.; Wu, W.; Yao, S.; Han, X.; He, D.; He, J.; Zheng, G.; Zhao, Y.; Cai, Z.; et al. Camalexin Induces Apoptosis via the ROS-ER Stress-Mitochondrial Apoptosis Pathway in AML Cells. Oxid Med Cell Longev 2018, 2018, 7426950. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Yu, Q.; Yang, N.; Xiao, Z.; Song, C.; Zhang, R.; Yang, S.; Liu, Z.; Deng, H. Therapeutic Potential of Targeting Polo-like Kinase 4. Eur J Med Chem 2024, 265, 116115. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell Biology. Reversible Centriole Depletion with an Inhibitor of Polo-like Kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhong, L.; Chu, X.; Wan, P.; Liu, Z.; Lu, Y.; Zhang, Z.; Wang, X.; Zhou, Z.; Shao, X.; et al. Downregulation of Polo-like Kinase 4 Induces Cell Apoptosis and G2/M Arrest in Acute Myeloid Leukemia. Pathol Res Pract 2023, 243, 154376. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylarginine Deiminases in Citrullination, Gene Regulation, Health and Pathogenesis. Biochim Biophys Acta 2013, 1829, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Uysal-Onganer, P.; D’Alessio, S.; Mortoglou, M.; Kraev, I.; Lange, S. Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. Int J Mol Sci 2021, 22, 1396. [Google Scholar] [CrossRef]
- Sun, Y.-N.; Ma, Y.-N.; Jia, X.-Q.; Yao, Q.; Chen, J.-P.; Li, H. Inducement of ER Stress by PAD Inhibitor BB-Cl-Amidine to Effectively Kill AML Cells. Curr Med Sci 2022, 42, 958–965. [Google Scholar] [CrossRef]
- Ambrosini, G.; Natale, C.A.; Musi, E.; Garyantes, T.; Schwartz, G.K. The GPER Agonist LNS8801 Induces Mitotic Arrest and Apoptosis in Uveal Melanoma Cells. Cancer Res Commun 2023, 3, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Marinković, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ Res 2020, 127, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Satilmis, H.; Vandewalle, N.; Verheye, E.; De Bruyne, E.; Menu, E.; De Beule, N.; De Becker, A.; Ates, G.; Massie, A.; et al. Targeting S100A9 Protein Affects mTOR-ER Stress Signaling and Increases Venetoclax Sensitivity in Acute Myeloid Leukemia. Blood Cancer J 2023, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Doepner, M.; Weissenrieder, J.; Majer, A.D.; Mercado, S.; Estell, A.; Natale, C.A.; Sung, P.J.; Foskett, J.K.; Carroll, M.P.; et al. LNS8801 Inhibits Acute Myeloid Leukemia by Inducing the Production of Reactive Oxygen Species and Activating the Endoplasmic Reticulum Stress Pathway. Cancer Res Commun 2023, 3, 1594–1606. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Lu, Y.; Cao, J.; Lin, L.; Chen, X.; Zhou, Z.; Pu, J.; Chen, G.; Ma, X.; Deng, Q.; et al. N-Acetyltransferase NAT10 Controls Cell Fates via Connecting mRNA Cytidine Acetylation to Chromatin Signaling. Sci Adv 2024, 10, eadh9871. [Google Scholar] [CrossRef]
- Zi, J.; Han, Q.; Gu, S.; McGrath, M.; Kane, S.; Song, C.; Ge, Z. Targeting NAT10 Induces Apoptosis Associated With Enhancing Endoplasmic Reticulum Stress in Acute Myeloid Leukemia Cells. Front Oncol 2020, 10, 598107. [Google Scholar] [CrossRef]
- Abdullazade, S.; Behrens, H.-M.; Krüger, S.; Haag, J.; Röcken, C. MDM2 Amplification Is Rare in Gastric Cancer. Virchows Arch 2023, 483, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Deng, S.; Deng, M.; Shi, Y.; Zhong, M.; Ding, L.; Jiang, Y.; Zhou, Y.; Carter, B.Z.; Xu, B. Therapeutic Synergy of Triptolide and MDM2 Inhibitor against Acute Myeloid Leukemia through Modulation of P53-Dependent and -Independent Pathways. Exp Hematol Oncol 2022, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cui, J.; Zhao, Y.; Liu, X.; Chen, L.; Xia, Y.; Wang, Y.; Chen, S.; Sun, S.; Shi, B.; et al. KDM6A-ARHGDIB Axis Blocks Metastasis of Bladder Cancer by Inhibiting Rac1. Mol Cancer 2021, 20, 77. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.K.; Bose, A.; Chattopadhyay, A.; Ghosh, S.; Bhowmik, S.; Samanta, S.K.; Bhattacharyya, M.; Sengupta, A. KDM6A Modulates Anti-Tumor Immune Response By Integrating Immunogenic Cell Death in Human Acute Myeloid Leukemia. Blood 2023, 142, 2753. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the main molecular consequences of the conditions that prevail in the bone marrow niche for HSC cells. A low-oxygen environment increases endoplasmic reticulum (ER) stress levels. Increased ER stress causes an increase in Unfolded Protein Response (UPR) activity. The production of Reactive Oxygen Species (ROS) also increases. Consequently, the activity of hypoxia-inducible factors (HIFs) likewise increases.
Figure 1.
Schematic representation of the main molecular consequences of the conditions that prevail in the bone marrow niche for HSC cells. A low-oxygen environment increases endoplasmic reticulum (ER) stress levels. Increased ER stress causes an increase in Unfolded Protein Response (UPR) activity. The production of Reactive Oxygen Species (ROS) also increases. Consequently, the activity of hypoxia-inducible factors (HIFs) likewise increases.
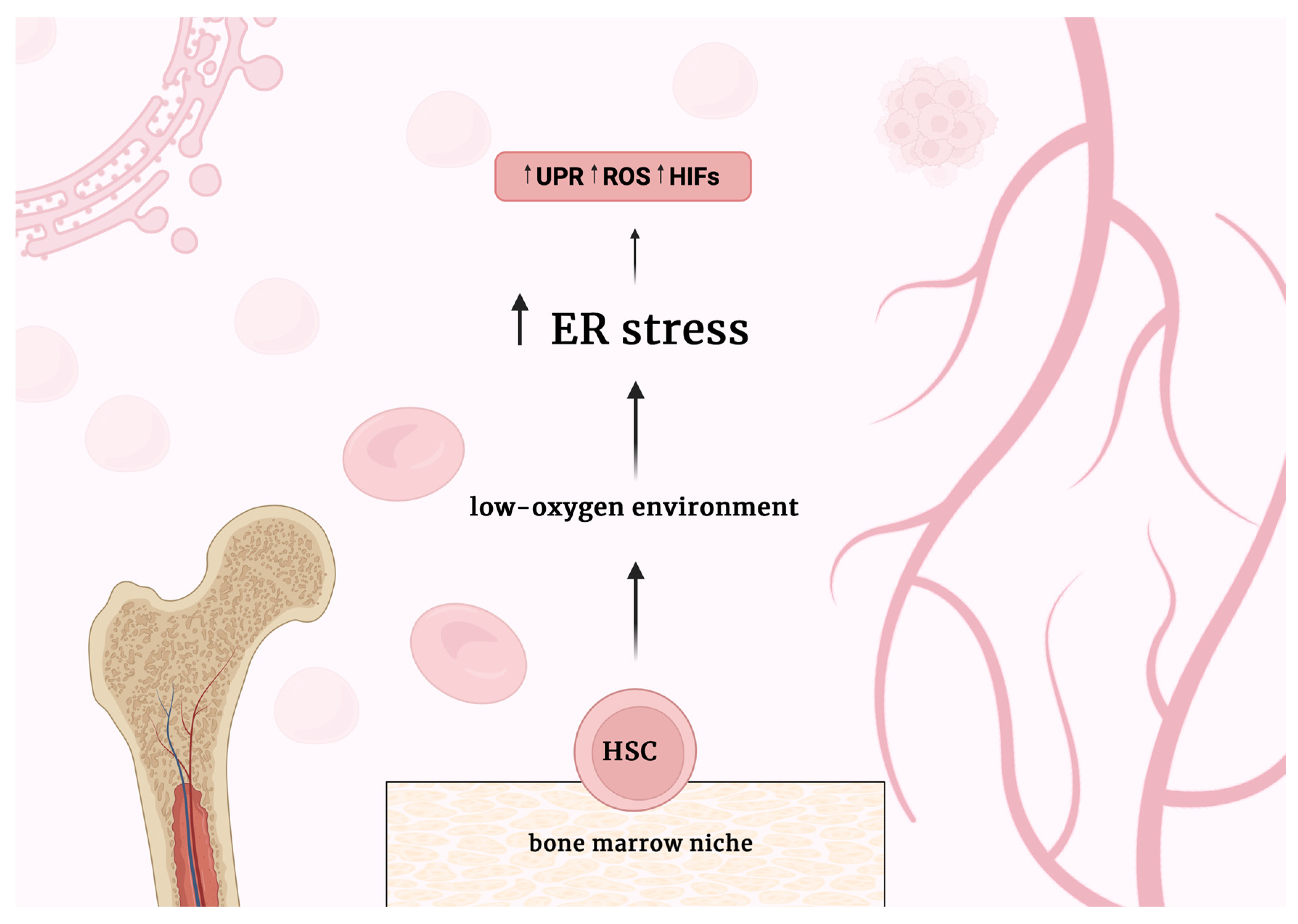

Table 1.
Targeted therapies for the most common genetic mutations in AML.
| Target | Compound Name | In combination | Current Status | Results |
| FTL3 | Gilteritinib | - | FDA-approved | FDA-approved Gilteritinib for treatment of adult patients who have relapsed or refractory AML with a FLT3 mutation based on ADMIRAL trial (NCT02421939) [56]. |
| Quizartinib | Cytarabine + anthracycline | FDA-approved | FDA-approved Quizartinib for treatment of adult patients with newly AML that is FLT3 ITD-positive baseod on QuANTUM-First trial (NCT02668653) [57]. | |
| Crenolanib | - | Phase II | Complete Response (CR)/CRi 14,3%, Partial Response (PR) 16,1%, Overall Response Ratio (ORR) 30.4%, safe, well tolerated. [NCT01657682]. | |
| IDH1 | Ivosidenib | Gilteritinib | Phase I | Data unpublished [NCT05756777]. |
| Ivosidenib | Azacitidine | Phase 1b/2 | ORR 78,3%, CR 60,9%, safe, well tolerated [58]. | |
|
IDH2 |
Enasidenib |
- |
Phase I | Two-year progression-free 69%, overall survival 74%, safe, well tolerate [59]. |
| Phase II | Data unpublished [NCT04203316] | |||
| Enasidenib | Gilteritinib | Phase I | Data unpublished [NCT05756777] | |
|
NPM1 |
Selinexor |
Homoharringtonine + daunorubicin + cytarabine or Granulocyte Colony-Stimulating Factor + aclacinomycin + cytarabine | Phase III | Data unpublished [NCT05726110] |
| Revumenib | Azacitidine + venetoclax | Phase III | Data unpublished [NCT06652438] | |
| Eltanexor | Venetoclax | Phase I | Data unpublished [NCT06399640] | |
| Ziftomenib | - | Phase I/II | Data unpublished [NCT04067336] | |
| DS-1594b | Azacitidine + venetoclax or Cyclophosphamide + Dexamethasone + Vincristine + Rituximab | Phase I/II | Data unpublished [NCT04752163] | |
| BMF-219 | - | Phase I | Data unpublished [NCT05153330] |
Table 2.
New AML therapy affecting ER stress.
| Target | Compound Name | In combination | Current Status | Results |
| JUN | shJUN-1 and shJUN-2 knockdown | - | Preclinical phase | Inhibition of XBP1 or ATF4 via shRNA leads to apoptosis in AML cells and significantly prolongs disease latency in vivo, linking reduced survival from JUN inhibition to decreased pro-survival UPR signaling [70]. |
| NMT | Zelenirstat | - | Preclinical phase | Zelenirstat was well tolerated in vivo and caused the inhibition of AML cell lines [71]. |
| Proteasome | MKC8866 | Btz or carfilzomib | Preclinical phase | MKC8866, in combination with proteasome inhibitors, significantly reduces XBP1s levels and increases cell death in AML cell lines and patient-derived AML cells [69]. |
| Btz | - | Phase I | Data unpublished [NCT00077467]. | |
| Retinoic acid | Btz and arsenic trioxide | Preclinical phase | The combination exhibits strong cytotoxic effects on FLT3-ITD+ AML cell lines and primary blasts from patients due to disrupted ER homeostasis and increased oxidative stress [74]. | |
| PLK4 | Centrinone | - | Preclinical phase | Inhibition of PLK4 induces apoptosis, G2/M, and ER stress in AML cells [81] |
| BCL-2 | Venetoclax |
Metformin | Preclinical phase | The combination of metformin and venetoclax showed enhanced anti-leukemic activity with acceptable safety in patients with AML [21]. |
| PAD | BB-Cl-Amidine | - | Preclinical phase | BB-Cl-A activates the ER stress response and, due to this, effectively induces apoptosis in the AML cells [84]. |
| GPER | S100A9 siRNA knockdown | Venetoclax |
Preclinical phase | S100A9 knockdown significantly increases venetoclax sensitivity in AML cells [87]. |
| LNS8801 | - | Preclinical phase | LNS8801 induced AML cell death mainly through a caspase-dependent apoptosis pathway, inducing levels of ROS and ER stress response pathways, including IRE1α [88]. | |
| NAT10 | NAT10 shRNA knockdown | - | Preclinical phase | Targeting NAT10 promotes ER stress, triggers the UPR pathway, and activates the Bax/Bcl-2 axis in AML cells [90]. |
| MDM2 | Nutlin-3a | Triptolide | Preclinical phase | The combination exhibits a significant antileukemia effect through both p53-dependent and independent mechanisms, the latter involving the disruption of the MYC-ATF4 axis-mediated ER stress [92]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



