Submitted:
31 December 2024
Posted:
02 January 2025
You are already at the latest version
Abstract
It has long been clear that mitochondria play important roles in metastatic disease, including regulatory contributions. Nonetheless, our knowledge in this area was too fragmentary to be of practical clinical use. However, over the last decade a robust community of investigators has extended our knowledge such that it is practical to begin to build testable theories. Moreover, these theories can plausibly give clinicians actionable opportunities to effectively attack advanced, currently treatment-resistant carcinomas.
Our goal here is to contextualize and review important features of this recent new data base. We will also illustrate the kind of testable speculative theories that might be built on this foundation.
Specifically, we will explore the evidence that many or most advanced, treatment-resistant tumors converge on a uniform state in which mitochondria play a universal, indispensable regulatory role (Figure 1 [1]). Speculative hypotheses of this form are robustly testable/falsifiable. Moreover, such theories suggest a specific novel approach to clinical attack on advanced, currently treatment-resistant carcinomas.
Keywords:
cancer
; mitochondria
; MIPS
; resistance
; lipid
; CPI-613
; ferroptosis
; sulfasalazine
I. Cybrids and Nuclear Transplantation: Direct Evidence for a Causal Role for Mitochondria in Cancer
A seminal body of early work directly and strongly suggests that a cytoplasmic component is essential for the properties of a large fraction of advanced malignancies, in addition to nuclear genetic/epigenetic components. These essential, replicating cytosolic regulatory components largely or entirely consist of mitochondria [2–4] (also see text below).
Though vast amounts of work remain to be done, currently available data in this area are strongly consistent with the following picture. Advanced cancer is a cell state, maintained by pathological manipulation/redeployment of specific regulatory circuitry. This same circuity is deployed for cytodetermination/differentiation in normal developmental processes including, most importantly, wound healing, as discussed in detail in section V. below. This malignant state can be specified by machinery that has a level of internal redundancy such that it can specify this state or set of closely related states in somewhat variable ways that, nonetheless, all converge on universal properties in advanced malignancies (see sections V. and VI. for details).
As noted, this state-determining decision making/implementation machinery includes both well-established nuclear genetic/epigenetic components and less well characterized mitochondrial circuit elements. The relative contributions of proximate cancer-producing genetic/epigenetic changes in the nucleus and mitochondrial regulatory elements apparently varies significantly between individual tumors (op cit.; immediately below), in spite of the apparently universal outcome (V. and VI.).
Nuclear transplantation and cybrid experiments provide unique analytical opportunities in this context as follows.
First, in nuclear transplantation, a nucleus removed from a malignant cell largely devoid of cytoplasmic elements is injected into an enucleated egg cell. These studies have shown that a malignant nucleus is able to direct remarkable levels of normal development having been separated from its malignant cytoplasm [5,6]. These results are consistent with the hypothesis that stably replicable altered mitochondrial regulatory circuits can be essential to malignancy.
Second, cybrid experiments are of the following form. Using centrifugation, nuclei can be driven through the cell membrane yielding a cytoplast (nucleus-free cytoplasm) and a karyoplast (nucleus surrounded by a small plasma membrane envelope largely free of cytoplasm). Using fusion techniques a karyoplast from one cell line can be introduced into the cytoplast of another (technique described in [7]). Deployment of this technique and its use to specifically implicate mitochondria is well illustrated in [8].
Using cybrid experiments, diverse observations indicate that altered mitochondrial regulatory circuits have dramatically different levels of importance in different tumors. [We will argue below that these differences do not necessarily indicate significant difference in ultimate, fundamental regulatory circuit logic.]
Several examples of the range of results from cybrid experiments are illuminating as follows.
At one extreme, xenograft tumorigenicity of rho-minus (mitochondrial DNA-free) HeLa cells was fully recovered when their mitochondria were replaced with normal human fibroblast mitochondria [9]. This result argues that genetic or epigenetic mitochondrial alterations are unimportant to Hela tumor formation. At the opposite extreme, Israel and Schaeffer [10] showed that in a malignant NIH 3T3 derivative line the cytoplast from this derivative was sufficient to produce tumorigenicity when fused with a nucleus from a non-malignant NIH 3T3 line. This particular malignancy appears to be entirely dependent on cytoplasmic, presumably mitochondrial, regulatory information.
A relatively large fraction of cybrid case studies produce results intermediate between these two extremes, where the full aggressiveness of tumor forming ability depends both on genetic/epigenetic changes in karyoplasts (nuclei) and in cytoplasts (clearly or presumably mitochondria)(see, for example, [8,11,12]).
Thus, these experiments indicate that a common pattern is genetic/epigenetic alteration of both nuclear and mitochondrial regulatory circuitry cooperatively producing full blown malignancy. Specifically, a common outcome is that advanced, treatment-resistant cancer includes indispensable mitochondrial and nuclear contributions.
As we will discuss in more detail below, it is common that advanced cancers depend on redox signaling circuits, including central mitochondrial elements (see, for example, [8,12])(Figure 1).
The following examples are illustrative of this point. Electron transport complex 1 (ETC-CI) mutations are commonly associated with increased metastasis and reactive oxygen species (ROS) production (herein ROS will refer to what is ultimately hydrogen peroxide, H2O2; reviewed in [13,14]). Notably, it has been proposed that ROS production interferes with proper CI assembly/stability, creating a positive feedback for ROS production [15]. Likewise, interfering with antioxidant NADPH production pathway mimics pro-advanced-malignancy effects of ETC mitochondrial genome mutations [16]. As expected from mitochondrial ROS control of HIF1a, mitochondrial mutations supporting advanced malignancy enhance HIFa expression, contributing to the malignant state [17,18]; see further discussion in VIa. Also notable in this context, resistance to cell death from anoikis following matrix detachment is one feature of malignancy. One way in which this survival is achieved is through enhancement of antioxidant-feeding NADPH driven by AMPK activation, implicating redox regulation [19].
Though mitochondrial mutations are common in advanced, treatment-resistant tumors (above), we currently have no clear evidence that such mutations are universal in advanced disease. The crucial issue is what this phenomenology tells us about advanced carcinoma in the general case. Given multiple mitochondrial genotypes in the same cell, sometimes including rare minor variants, (heteroplasmy [20–22]), it is conceivable that causally essential mitochondrial mutations are more common in advanced cancer than we currently recognize. Independently of the resolution of this issue, these data make it clear that additional elevation of mitochondrial ROS through potentially diverse mechanisms is nearly universal in driving advanced, treatment-resistant cancer [23–25].
Note that the differences in detail of the cases above do not necessarily imply differences in fundamental mechanism (Figure 1). We will return to this issue in section VI where we will argue all these cases, and potentially all advanced cancers, result from the same underlying regulatory circuitry set point.
Another especially important feature of the mitochondrial regulatory component of cancer is its role in advanced treatment resistance [26,27].
The idea that cancer is a mitochondrial disease, rather than a purely nuclear disease, is long-standing, but widely under-appreciated (reviewed in [2–4]). Our goal in this review will be to explore the evidence that this view of cancer is likely necessary to build a clinically useful, universal theory of advanced, treatment-resistant cancer (VI.).
In summary, what these data tell us is that mitochondria are, most commonly, replicating regulatory controllers providing crucial regulatory elements engendering advanced, treatment-resistant cancer. Moreover, mitochondrial ROS regulatory signals play a common, possibly universal role, in this regulatory status. In the following sections we will build a more complete picture of mitochondrial control of advanced malignancy, ultimately discussing the clinical implications of the research community’s improving understanding.
II. Introduction to the Mitochondrial Information Processing System (MIPS) as a Central Causal Agent of Malignancy
Oxygen levels in the earth’s atmosphere have increased in stages over the last 2.5 billion years. The most recent increase occurred around 600 million years ago, correlating with the emergence of large metazoan organisms, dependent on high oxygen levels [28]. These ancestral animals evolved to be dependent on oxygen-based metabolism in mitochondria, as has long been recognized. More recently, it has emerged that the oxygen-dependent production of reactive oxygen species for signaling evolved in parallel in the original common ancestor of all surviving metazoans. These pervasive ROS-dependent regulatory systems are, thus, ancient and highly conserved [30–33]. Among them are the decision-making circuitry hijacked and modified in the emergence and progression of cancer (V. and VI.). Because proteins readily evolve to sense redox signals, primarily through oxidation-sensitive cysteine residues, ROS is uniquely suited to coordinate the global state of a cell or a tissue [29,33–35]. We begin here with a brief review of the oxygen-dependent signaling functions of mitochondria.
Due to the historical sequence of biological discoveries, mitochondria are still commonly thought of as primarily metabolic organelles, burning reduced carbon nutrients with oxygen to generate ATP and drive metabolic cycles, primarily the TCA cycle, to inter-transform metabolites for biosynthetic purposes.
While these metabolic functions are indispensable, a robustly growing body of insight over the last 40 years has shown that mitochondria also have radically different functions that are equally vital to oxygen-dependent metazoan. These include ROS signaling, commonly also generated by mitochondria [1,36–43].
Most importantly, mitochondria represent nodes in cellular circuits that execute vital regulatory decision-making tasks. Mitochondria absorb diverse signals in communication with one another and other organelles [1,38–40] (Figure 1). These circuits are then capable of changing set points in diverse ways and thereby generating any of a variety of signals that control/change the state of the resident cell (op cit.; below). As we will discuss below, these set point changes appear to depend, at least in part, on positive feedback loops between mitochondrial ROS and normally functioning genes that have come to be called oncogenes/tumor suppressors in view of the pathological roles of their malfunctions.
Figure 1.
Illustrated are selected signaling processes forming elements of feedback loops involved in mitochondrial information processing system (MIPS) decision making (reviewed in [1]). This machinery consists of elements (organelles and molecules) that interact to create networks capable of making decisions based on incoming and circulating information. [lyso = lysozomes, LD = lipid droplets, ER = endoplasmic reticulum, ROS = reactive oxygen species, especially H2O2.]. In this context, mitochondria are initially targets of diverse incoming external information, including hormones, proteins/peptides, Ca++, and ROS. We will explore the hypothesis that the MIPS machinery regulates all advanced, treatment-resistant cancers in a specific, universal fashion. Our focus will be on the ROS-based elements of MIPS function. This MIPS hardware is central to the control of normal developmental events. Especially important is decision making/execution in the wound healing machinery, in turn, apparently universally hijacked and modified to produce all carcinomas. Key to MIPS regulatory decision-making circuitry is that it has set points that can change, in turn, directing changes in functional cell state. In cancer, this machinery is genetically/epigenetically modified so that a significant subset of tumor cells is pushed toward permanent progenitor-like status (text). The key to the lethality of cancer is the significant subpopulation locked in this progenitor-like, perpetually replicating cell state (Figure 2). The details of the working hypothesis we review lead naturally to a potentially effective, universal clinical approach to dealing with otherwise treatment-resistant advanced cancers.
Figure 1.
Illustrated are selected signaling processes forming elements of feedback loops involved in mitochondrial information processing system (MIPS) decision making (reviewed in [1]). This machinery consists of elements (organelles and molecules) that interact to create networks capable of making decisions based on incoming and circulating information. [lyso = lysozomes, LD = lipid droplets, ER = endoplasmic reticulum, ROS = reactive oxygen species, especially H2O2.]. In this context, mitochondria are initially targets of diverse incoming external information, including hormones, proteins/peptides, Ca++, and ROS. We will explore the hypothesis that the MIPS machinery regulates all advanced, treatment-resistant cancers in a specific, universal fashion. Our focus will be on the ROS-based elements of MIPS function. This MIPS hardware is central to the control of normal developmental events. Especially important is decision making/execution in the wound healing machinery, in turn, apparently universally hijacked and modified to produce all carcinomas. Key to MIPS regulatory decision-making circuitry is that it has set points that can change, in turn, directing changes in functional cell state. In cancer, this machinery is genetically/epigenetically modified so that a significant subset of tumor cells is pushed toward permanent progenitor-like status (text). The key to the lethality of cancer is the significant subpopulation locked in this progenitor-like, perpetually replicating cell state (Figure 2). The details of the working hypothesis we review lead naturally to a potentially effective, universal clinical approach to dealing with otherwise treatment-resistant advanced cancers.
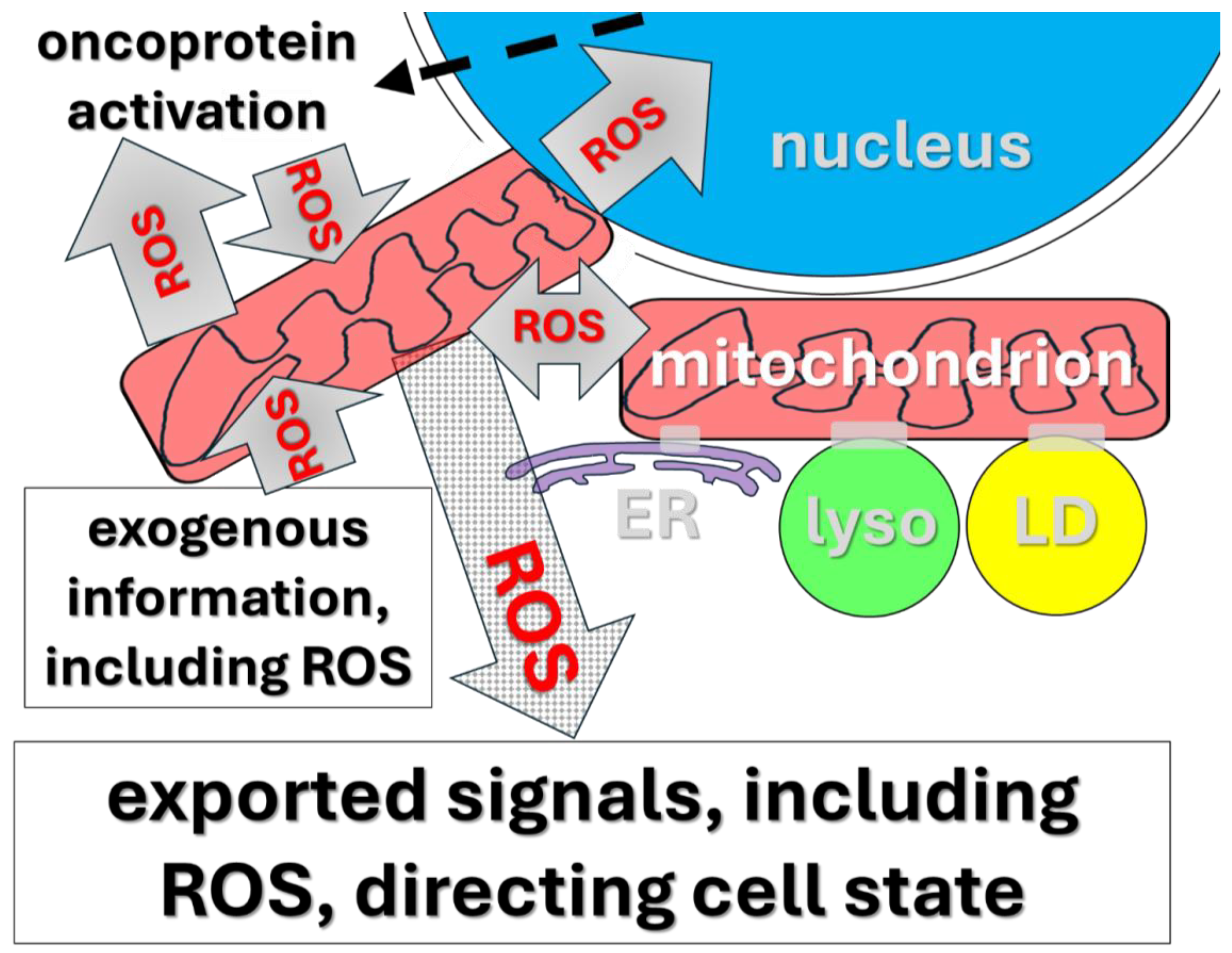
Metazoans face the vastly demanding challenge of undergoing precisely controlled cellular state change on rather dramatic scales. Such state changes include regulated cell growth as well as diverse local cases of determination and differentiation. The adaptive goals of these state changes include not only exquisitely regulated developmental and physiological processes, they also entail wound healing. As noted, malfunction of these processes is also crucial to cancer as we will see in more detail in section V. A key regulatory feature of these diverse processes now appears clearly to include a class of decision-making circuits in which mitochondria are central nodes. This class of circuits has recently come to be called the “mitochondrial information processing system” (MIPS; [1,39,43]). Mitochondria function in MIPS with the same formal logic as electronic circuit elements, though with greater sophistication and dimensionality.
The totality of the MIPS story is well beyond the scope of this review (see [1] for a detailed review). The important detail here is that mitochondria form networks with one another and with diverse additional organelles to create circuits designed to integrate information (Figure 1), thereby making and implementing decisions analogously to electronic or neurological circuits [1,43–45].
In this review, we will focus on one specific component of the MIPS, redox signaling, both for information processing/decision making and for decision implementation/execution [1,38,44–52] .
More specifically, we will argue that evolved redox-dependent cellular decision-making serves to organize and execute wound healing. It has long been recognized that carcinomas are “wounds that do not heal” (reviewed [53–55]). Elements of the same MIPS activities that control normal cytodevelopment are also implicated in wound healing. As discussed in detail in section V, there is extensive, diverse evidence that this wound healing MIPS function is illicitly mis- and over-activated to produce cancer. Moreover, it is highly plausible that elements of this pathological MIPS function evolve to be further hyper-activated to produce most or all cases of advanced, treatment-resistant carcinoma.
Among useful data supporting this general picture are the following (see additional examples in IV. and V.). For example, mitochondrial formation of extensive networks that lead to substantial changes in ROS signaling (hyperfusion) is associated with the activation of the integrated stress response (ISR; [56,57]). The cause/effect relationships between hyperfusion and other known ISR regulatory elements, including GCN2 and PERK, remain ambiguous. For example, PERK activation and hyperfusion could represent elements of a positive feedback loop. Nonetheless, further clarification of the relationship between MIPS decision making and ISR activation would be fruitful.
An additional example of the role of MIPS in cancer is illuminating in this regard. The TSC/mTORC1 pathway makes an apparently binary decision between grow and do-not-grow and is directly implicated in diverse cancers [58,59]. Moreover, this machinery senses levels of amino acids to make these decisions with the grow decision driven by elevated amino acid levels (op cit.). This process occurs in the context of mTORC1 association with lysosomes and lysosomes form informational contacts with mitochondria [60,61](Figure 1). This is a property expected of a system one of whose main goals is contributing to the wound scab (die and contribute matrix for wound healing) versus cellular scar (live, divide, develop, heal wound) decision (see VIa. for more detail). The well characterized behavior/role of the TSC/mTORC1 in malignancy is consistent with mis-setting of the system to grow, thus contributing to the decision to be malignant [58,59].
The participation of the lysosomal system in apparently feeding information to mitochondria illustrates the general principles of how MIPS works. Specifically, the mitochondria/lysosome contact sites allow interorganellar signaling, including transmission from lysosome to mitochondrion down the strong interorganellar Ca++ and H+ concentration gradients [62,63]. Further, mTORC1-mediated transfer of cholesterol signals to synapsed mitochondria contributes to aflatoxin-induced cell death [64].
Thus, the mitochondrial/lysosomal synapse behaves as expected of an element of the MIPS circuitry, in this case controlling cell growth and death. Given that mitochondria directly detect oxygen levels, including in healing wounds, these phenomena look like an element of mitochondrial integration of information to determine circuit set/change, in turn, to control cell state. As noted, malfunction of this system is implicated in cancer (V. and VI.).
Our understanding of MIPS is now substantial and continues to grow robustly [1]. Cells have an extensive, diverse array of processes for generating reactive oxygen species and a complete discussion of these processes is beyond our scope here (reviewed in detail in [43]). We will primarily focus here on mitochondrial generation of and response to H2O2. Additional general insight into decision-making based on redox signals can be found in [65,66].
III. Crucial Details of the Mitochondrial ROS Production Response
In following sections we will repeatedly encounter ROS production under conditions where it exerts (or appears to) a causal regulatory role. In some cases, the intracellular source of that ROS will be unknown or unreported. In other cases, extramitochondrial sources will be implicated as (potentially) causal, including cytosolic or plasma membrane NADPH oxidases (NOXs) [66–69]; see especially Fukai and Ushio-Fukai [70]. It is crucial to recognize that mitochondria are likely to also be involved in all these cases. Several details are germane.
First, mitochondria themselves behave as a ROS-based communication circuits, including amplifying received ROS signals. For example, this process can be seen in cases referred to as ROS-induced ROS release (RIRR)[49–52,71].
Secondly, extra-mitochondrial H2O2 can readily enter the mitochondrial matrix, presumably by diffusion [72]. However, strikingly, H2O2 produced in the matrix apparently cannot readily escape the mitochondrion by simple diffusion (op cit.). This unexpected observation suggests that ROS transfer from the mitochondrial matrix to external targets may require specialized connections between the mitochondria and the target. These cases include inter-organelle contacts such as between mitochondria and nuclei [73,74]. Moreover, there is evidence for specialized mitochondrial interaction with individual target molecules, including the PTEN regulator [74a-c.] and, plausibly, other oncogenes and tumor suppressors we will discuss below (Vc.). Thus, mitochondria are efficient detectors of external ROS signals and, apparently selective transmitters/amplifiers of ROS signals, including during RIRR-like processes.
An illustrative initial example of this relationship is cell layer delamination, a central wound-healing-like process that occurs at various points during development. In one case of this process mitochondrial ROS is causally implicated, though NOX is not excluded [75]. In a second analysis of the same case NOX ROS is causally implicated, though mitochondrial ROS is not excluded [76].
Again, mitochondrial ROS generation in response to exogenous ROS appears to be universal; thus, MIPS is apparently inevitably engaged by any cellular ROS signaling process. Even ROS signaling cascades initiated outside mitochondria, apparently generally rapidly engage the MIPS machinery. MIPS is commonly ultimately responsible for making and implementing decisions (IV. and V.). Thus, when ROS regulated cellular processes are implicated, we will generally operate on the working hypothesis that MIPS is causally engaged.
Finally, there is a consistent pattern in diverse studies indicating that elevated mitochondrial ROS is responsible for the transition from stem cells (quiescent) to progenitor cells (actively growing). This results in extensive cell division before differentiation in normal tissues or indefinite replication without extensive differentiation in malignant cells (Figure 2; text below). This is observed in mammals, zebra fish, and Drosophila (flies), indicating an ancient conserved system. This pattern is observed in both normal development and wound healing [43,67,75,77–81].
These processes include diverse feedback loops between ROS and oncogene activation, a logic expected of decision-making systems (op. cit.; Figure 1 and section V.).
IV. Evidence for MIPS ROS-Dependent Decision Making: Animal Development
As we discuss in detail in section Vb., the presumptively MIPS-regulated processes most central in considering the cancer problem are those involving stem and progenitor cells in wound healing. However, superficially more macroscopic developmental decision making presumptively controlled by the MIPS helps us develop a clearer, more confident grasp of the behavior of this control circuitry. Examples of currently understood redox control of animal development will give us this view (reviewed in detail in [82]). We will pay particular attention to the powerful Drosophila genetic system with emphasis on the evidence that these regulatory systems are ancient, conserved and shared with mammals (V.).
As we consider these cases, recall that robust ROS signals are always likely to engage MIPS (III.).
Petrova, et al., [31] show that the transition from quiescent egg to developmentally active embryo in Drosophila correlates with and is dependent on a massive wave of oxidation of protein cysteines. This indicates that initiation of development is managed by systemic redox signaling. These authors also review evidence that similar processes are involved in early mammalian development.
Owusu-Ansah [88] provide compelling evidence that mitochondrial ROS controls the G1-S cell cycle checkpoint throughout Drosophila development.
Sing, et al. [84] show that specialized cadherins, mediating cell-cell interactions and adhesion in Drosophila, undergo proteolytic cleavage to release a peptide fragment that enters the mitochondrion to interact with and control electron transport system complex 1 (ETC-CI). This control process, in turn, manages mitochondrial functions, presumably including ROS generation. Mutational inactivation of this cadherin engenders systemic anatomical abnormalities.
As reviewed in Oswald, et al. [85] and Percio, et al. [32] there is an extensive body of evidence in mammals, arthropods, and nematodes, that major developmental events shaping the properties of neurons are regulated by ROS, generated by both NADPH oxidases (NOX) and mitochondrial sources.
Coffman, et al. [86] show that the establishment of the oral/anal axis in the sea urchin embryo is determined by differential local distribution of mitochondria and the redox gradient thereby produced. Similar phenomenology is seen in diverse additional embryonic developmental cases [87–92] .
Amblard, et al.[93] show that local ROS values pattern the developing zebra fish brain, at least in part, by controlling Engrailed protein expression.
Kiss et al. [95] and Paffenholz. et al. [94] show that development of the mouse inner ear balance organ requires ROS signaling.
Chartier, et al. [96] show that the Crumbs regulatory gene manages epithelial polarity in both mammals and arthropods through control of ROS generation.
In summary, redox signaling, frequently or always ultimately involving mitochondrial ROS, is remarkably pervasive throughout metazoan development. In the following section we will focus particularly on analogous developmental processes directly germane to understanding cancer emergence and progression. These processes will be those involved in individual stem and progenitor cells executing the specialized use of developmental machinery for wound healing. We will find there, again, that ROS regulation of these processes is pervasive. In section VI. Below, we will turn to how these insights might allow us to begin to build a clear, universal theory of the origin and behavior of advanced, treatment-resistant cancers.
V. Evidence for MIPS ROS-Dependent Decision-Making When Stem/Progenitor Cells and Wound Healing Machinery Are Modified Specifically to Produce Advanced, Treatment-Resistant Carcinoma
a). Overview of Important General Features
While the cases in the preceding section contribute to our confidence in the central regulatory role of the MIPS machinery, there is a particular feature of animal development that is more directly relevant to the cancer problem. There is a large body of histological evidence that carcinomas are complex tissues reminiscent of the granulation stage of normal wound healing. However, in carcinomas this stage does not progress efficiently to maturation, differentiation, and resolution phases [53–55]. Thus, carcinomas are “wounds that do not heal.” Tumors appear to reflect permanent arrest in a state that is only transient in normal wound healing.
Before deeper examination of details, several broader issues will prove valuable. Relatively robust redox (ROS) signaling is a feature of cancers and its strength generally increases with progression, including mitochondrial ROS (reviewed in [23,25]). This can be assessed directly or through expression of mitochondrial ROS-regulated processes (reviewed in [97]), including HIF [23], Akt [98], and Nrf2 [23,99]. These ROS increases can also be scored by corresponding, compensatory levels of anti-oxidant defense responses, also elevated in advanced cancers [19,97,100]. Again, recall that robust cellular ROS signals are always likely to engage or derive directly from MIPS (III.).
Studying ROS signaling during normal solid tissue terminal differentiation is relatively difficult. Thus, our best knowledge in this domain is in lymphocyte development, especially B cell maturation into differentiated plasma cells [101]. While changes in ROS generation as differentiation progresses suggest somewhat elevated production, coordinated upregulation of antioxidant systems (including the Nrf2 machinery) suggests that onset of differentiation may be signaled by reduced net ROS levels [101,102]. Though our ignorance in this area remains rather pervasive, a viable hypothesis is that progenitor-like carcinoma cells are prevented from entering differentiation as a byproduct of permanently elevated and/or reconfigured ROS signaling in these cells.
A recent discovery supports the hypothesis that mitochondrial ROS signals driving the onset of progenitor cell differentiation require ROS release from mitochondria complex III (CIII) of the electron transport system [103]. These experiments were done in Drosophila, but it is likely that this behavior is universal in all animals (ibid.). ROS released from CIII can emerge directly to the exterior of the inner mitochondrial membrane [104,105]; whereas most mitochondrial ROS is formed within the matrix [106], requiring different modes/spatial targets of export [72]. Thus, it is plausible that terminal differentiation requires a transition in the details of mitochondria ROS signaling which could be forestalled at the set point of the MIPS signaling sustaining tumor progenitor cell growth.
b) Fundamentals of Cytological Structure of Carcinomas
As this section unfolds, we will explore additional details of the cytodevelopmental machinery that is deployed for wound healing. Specifically, cytological histogenesis hardware is central to wound healing. Universally, in every tissue, this wound healing machinery consists of largely quiescent stem cells that can be mobilized to divide to spin off progenitor cells in response to wounding. Progenitor cells are capable of extensive rounds of replication producing a large population of cells that can then make the decision to undergo cytodifferentiation replacing damaged tissue in wound healing [107].
In view of this, we can be more specific about advanced cancers. Their bulk is made up substantially or primarily of tumor versions of progenitor cells which progress to differentiation inefficiently and defectively (see discussion of [101] immediately above; [108–110]). One strong piece of evidence for the majority of cancer bulk consisting of cells locked in the dividing progenitor state is the common tumor expression of high levels of Myc. In normal adult tissues Myc is always expressed in progenitor cells and virtually exclusively in these cells [111–114].
Tumors also contain small populations of stem-like cells, indicating that tumor cells may occasionally oscillate between stem-like and progenitor-like cells [27,115]. Cancers appear to be able to arise from either stem or progenitor populations and to retain the capacity to reciprocally change state between stem and progenitor status [116–120].
It will also be important to the discussion below to be aware of an extensive literature indicating that the behavior of stem and progenitor cells is regulated by ROS signaling, with increased ROS commonly driving emergence of the proliferative progenitor state [121–128]. This apparently universal picture is consistent with involvement of MIPS signaling in specifying stem and progenitor-like cancer cell states.
Tumor stem cells grow slowly and are hard to kill therapeutically, possibly because of their developmental commitment to maintain low ROS levels (immediately above and scab/scar decision making in VIa.). Thus, targeting stem cells therapeutically is challenging. However, because these cells are such small populations and grow so slowly, if clinicians could permanently, recurrently kill replicating progenitor-like cells it should be possible to achieve stable clinical management.
This targeting of progenitor-like cells is the implicit goal of essentially all current standards of care. The practical problem is that these progenitor-like tumor cells rapidly evolve resistance (VIa.) so that perennial treatment quickly becomes futile. In the last section of this review, we will discuss how this technical barrier might be overcome. To define the context and background to this goal we first need to understand more about the MIPS control of stem and progenitor cell behavior. Wound healing is a uniquely opportune way to achieve this. Mitochondrial ROS (presumptive MIPS involvement) and mitochondrial morphological changes are features of wound healing [80].
c) Oncogenes and ROS Signaling Engage MIPS Decision Making in Wound Healing and Carcinoma
Studying the molecular details of wound healing in mammals is cumbersome and our knowledge is relatively fragmentary. However, there is now extensive evidence that the wound healing machinery/process is ancient and highly conserved throughout metazoans so that we are free to choose more suitable experimental systems.
Studies with diverse metazoan experimental systems identify genes involved in wound healing in vertebrates, worms, and flies. Strikingly and importantly, these genes commonly turn out to be what are traditionally referred to as oncogenes and tumor suppressors as a result of their earlier identified roles in mammalian cancer. These observations constitute strong direct molecular evidence for the wounds-that-do-not-heal hypothesis for carcinomas. Equivalently, these data strongly suggest the hypothesis that cancer arises from malfunction of wound healing hardware. Further consistent with this hypothesis is the long-recognized causal connection between recurrent wounding and cancer origins, for example, chronic asbestos micro-wounding of lung epithelium correlates with mesotheliomas [128a-c]. The following are examples of additional empirical evidence supporting this view.
First, src homologs in Drosophila are essential for wound healing processes, including healing of wounded embryonic epithelium [129], controlling cell polarity in embryonic wound healing [79], and transduction of wounding signals [130,131].
Second, ras homologs in Drosophila contribute to wound healing in imaginal discs [132].
Both src and ras are generalized information transducers, of course; however, one of the central processes in which they participate is control of cell growth decisions (see, for example, [129,132]).
The next crucial step here is to explore the role of ROS signaling in oncogene participation in wound healing, in preparation for understanding ROS signaling in tumorigenesis. It is universally observed in all tested metazoan systems that a strong ROS signal is locally mounted at wound sites and is essential for successful healing. Examples of data supporting this insight are as follows.
First, Xenopus tail amputation is immediately followed by a robust, sustained ROS signal at the point of transection. Moreover, interfering with this ROS signal chemically or genetically interferes with tail regeneration (wound healing). Wnt and Notch, also having oncogene activity, are essential to this wound healing process. Moreover, the Wnt signal in this process is ROS induced [133].
Second, several studies of wound healing in Drosphila show ROS production at the wound site [75,134,135].
Third, robust ROS signals are also induced at wound sites in Caenorhabditis (worms) [77,136].
It is also important here that oncogenes involved in wound healing can act as ROS receptors/responders as follows.
First, src signaling can be activated by ROS in Drosophila [79,130] and in mammals [137,138].
Second, all ras family members require activation by ROS oxidation of highly conserved sulfhydryl groups on these proteins [139–141].
Third, rhoG oncogene protein signaling molecule is ROS activated in Drosophila wound healing [142,143]. The rhoA molecule is ROS activated in mammals [144]
Fourth, there is evidence that oncogenic FoxO transcription factor both responds to ROS and controls ROS levels, consistent with this being a feedback loop [145,146]. We will see other oncogene/ROS feedback loops immediately below.
Fifth, in mammals p53 can be activated by ROS and, reciprocally, control ROS levels [147], another likely oncogene/ROS feedback loop.
Sixth, oncogene EGFR receptor tyrosine kinase can be activated by ROS [148].
As noted, there is substantial evidence that oncogenes are not only activated by ROS but also drive ROS production, constructing potential positive feedback loops (Figure 1)[149]. Such loops are expected to be essential components of MIPS decision making, including their cooptation for emergence and progression of cancer to which we will return below (Figure 1). The following are examples of oncogene driving ROS production. Also see Raimondi, et al. [150] for a general review of these phenomena. Note that oncogene-driven ROS elevation in cancer commonly results from upregulation of oncogene expression levels and/or mutational activation of oncogenes. Such mutations are expected to initiate circuit resetting through MIPS feedback elements (Figure 1; references below).
First, ras-driven ROS enables malignant proliferation in mammalian systems and plays a similar role in normal zebra fish development [151]. In view of ROS activation of ras (above) this likely represents a positive feedback loop suitable for circuit setting, including through MIPS. Similarly, Kras activation drives a ROS signal in human tumor cells [139,152–154].
Second, the PI3K/Akt pathway is very frequently mutationally activated in cancer. This activation is commonly driven by ROS inactivation of PTEN, functioning normally as a tumor suppressor [155,156]. PI3K/Akt activation, in turn, drives ROS production [157,158]) Again, this is a likely feedback loop.
Third, Myc expression can drive elevated ROS levels possibly through its manipulation of mitochondrial metabolic control [159,160].
It is also notable in this context that some tumor suppressor genes are, likewise, implicated in wound healing. This aspect of tumor suppressors is less well studied. Cases that have been examined support the larger view here that cancer production/suppression machinery actually evolved to be wound healing machinery, including the following.
First, as noted above, p53 (which sometimes acts as a tumor suppressor) is implicated in management of wound healing and ROS signaling [78,81,161].
Second, grainyhead-like transcription factors (GRHLs) behave as tumor suppressors and control cytodifferentiation processes, including wound healing (see [162] and references therein). Notably, ROS levels are influenced by GRHL expression [163].
Third, gastrokines act as tumor suppressors and control wound healing in GI tract epithelium [164]. Gastrokines regulate ROS levels [165].
Fourth, Tenascin-X (TNX) is implicated in both wound healing and as a tumor suppressor [166].
Fifth, the PTEN gene acts as a tumor suppressor and controls wound healing [167]. As noted above, PTEN both controls ROS production (through Akt) and responds to ROS signals.
Sixth, FOXO1 is implicated both in wound healing and as a tumor suppressor [168]. FOXO1 is both activated by ROS and implicated in ROS control [169,170], once again a likely feedback loop.
Seventh, Kruppel-like factor 6 (KLF6) behaves as a tumor suppressor and regulates wound healing [171,172]. KLF6 is both activated by ROS and implicated in control of ROS levels [173,174].
Eighth, SIRT1 can act as a tumor suppressor and is implicated in ROS control [175–177]. SIRT1 is well known to participate in wound healing [178].
Nineth and more generally, the epithelial-mesenchymal transition (EMT) is central to tumorigenesis and to wound healing. Genetic analysis of EMT control strongly supports the hypothesis that genes controlling this process are also commonly oncogene-like and/or tumor suppressor-like [179].
An additional attractive hypothesis for how mitochondrial ROS output might be modulated is as follows. The alpha-ketoglutarate dehydrogenase (KGDH or OGDH) enzyme complex is a key, rate-limiting, regulatory step in the TCA cycle. KGDH also acts as a context-dependent tumor suppressor (reviewed in [180]). Notably, there is significant circumstantial evidence that electrons produced by KGDH are differentially partitioned into ATP synthesis (with ancillary ROS production) or NADPH (supporting ROS elimination, antioxidation) (op cit. and [181]). This property allows KGDH to control net mitochondrial ROS levels. Such control of net ROS could, for example, be determined by differential KGDH association in metabolons also containing ETC-CI (ROS) or the mitochondrial transhydrogenase (NNT; [182,183]), perhaps in response to posttranslational modification of the enzyme [184,185]. Moreover, KGDH both produces ROS and responds to ROS in an autoregulatory feedback circuit [186,187]. In overview, KGDH is likely to be a central hub controlling net mitochondrial ROS output and future investigation of details of this process, especially in healing wounds and cancer cells, will be of especially high importance. Further consistent with this view, KGDH behavior is implicated in malignancy, though in incompletely understood ways which could include antioxidant/NADPH production [188–196].
d) Tying Threads Together
Our knowledge of oncogenes and tumor suppressors is not exhaustive; nor is our understanding of the MIPS decision making system. Nonetheless, the cases we have reviewed immediately above support a simple, clear overall working hypothesis. These data illustrate the pervasiveness of empirical support for this updated form of the wounds-that-do-not-heal hypothesis for carcinomas. Very strikingly, most or all oncogenes and tumor suppressors normally function to control wound healing at the cytological level. This machinery is modified and/or redeployed to produce the malignant state, apparently universally. Moreover, the nuclear genetic machinery components apparently universally consist of proteins that normally control stem and progenitor cell behavior exploiting circuits that usually (or always) involve ROS signaling feedback loops. These ROS signals commonly involve mitochondrial ROS, though are sometimes apparently initially triggered by extramitochondrial sources (III.). The role of mitochondrial genome mutations engendering oncogene-produced ROS and acting as casual agents in cancer (above; see, for example, [8,11,12]) also strongly supports this hypothesis.
Thus, it seems plausible to define the wounds-that-do-not-heal hypothesis for cancer more precisely as emerging from mis-deployment of wound healing machinery in a very specific sequence of events. It is fruitful to begin with normal wound healing. Wounding activates oncogenes (src, for example, [13,197]), thereby initiating oncogene/ROS feedback loops. Such loops will also feed ROS into mitochondria. If this wounding signal is robust enough, stable ROS-based feedback loops involving active oncogenes and MIPS will initiate the wound healing process. Initiating this stable loop system represents a developmental decision-making event of the sort MIPS is designed for.
Alternatively, carcinomas can result from the mis-activation of the normal MIPS circuit setting-dependent wound healing feedback loops. More specifically, such pathological, tumorigenic redox signals initially arise from a variety of nuclear genetic/epigenetic triggering of oncogene/ROS feedback loops and/or initial mutational elevation of mitochondrial ROS production. ROS from these sources/loops activates elevates sustained mitochondrial ROS production. Persistence of this signal includes self-activation through RIRR-like processes [48–50]. See Figure 1 and Figure 2.
The implication of this view is that all carcinomas will share and depend on the same fundamental underlying processes. Indeed, as noted, remarkably, oncogenes/tumor suppressors and their ROS feedback loops are substantially, perhaps entirely, coextensive with genes controlling wound healing. As we come to understand these processes more thoroughly, we can expect to gain clinical efficacy. We will propose a more detailed version of this picture immediately below and explore how these refinements might lead to improved clinical approaches.
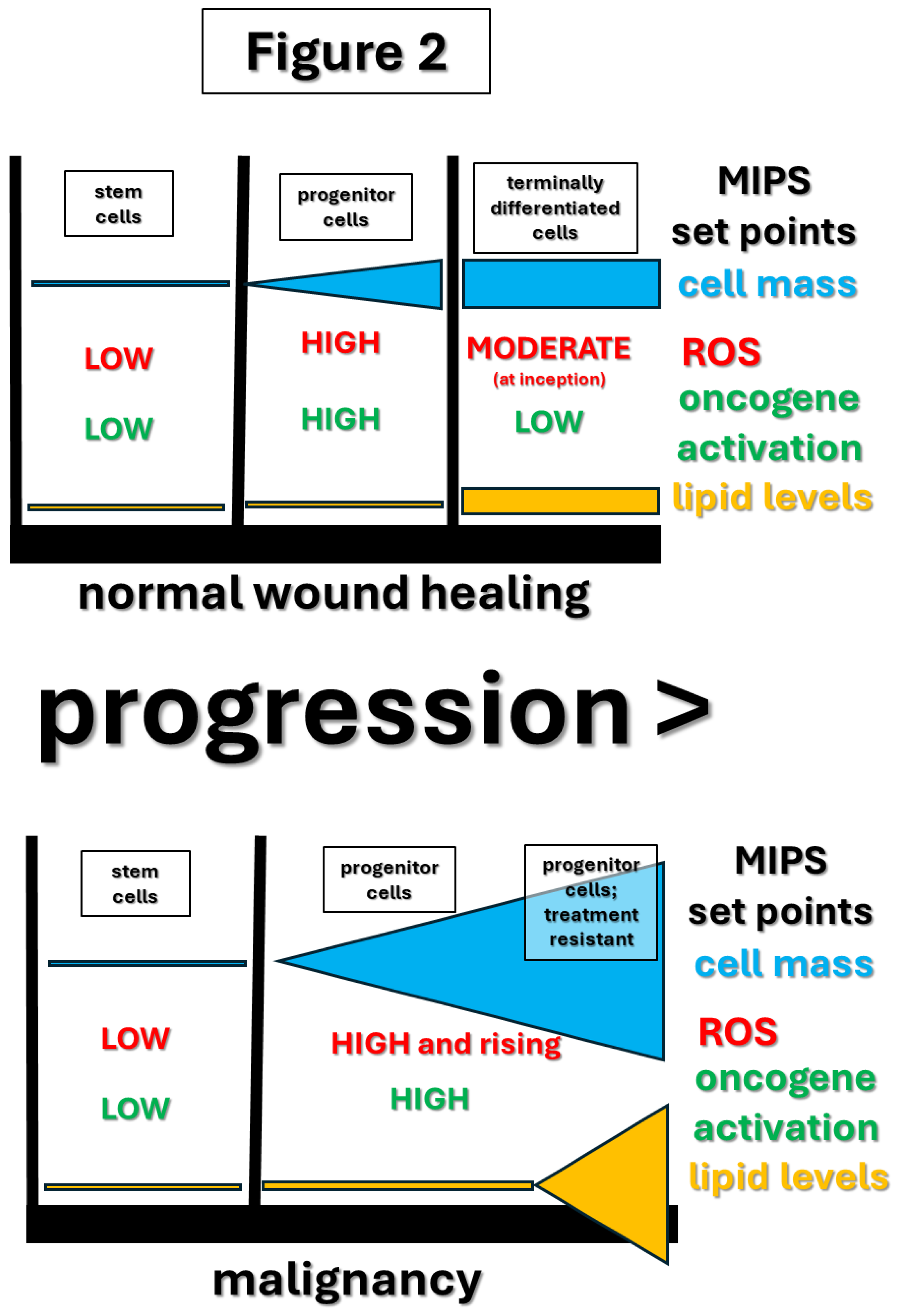
Figure 2.
Diagrammed is the development of normal wound healing cells (top) - stem cells, to progenitor cells, to ultimately mature, terminally differentiated cells. The progenitor-like subpopulation of tumor cells (bottom), which retain their progenitor-like, perpetual growth status, based on current evidence, is crucial to clinical lethality (text). Progenitor-like MIPS circuit set point (Figure 1; text) drives the tumor cell subpopulation permanently locked in the progenitor-like state [108,109,110]. The differences in capacity for differentiation result from genetic/epigenetic changes in MIPS-centered cell state decision making. The tumor progenitor state lock is plausibly hypothesized to be produced by genetic/epigenetic changes in oncogene/ROS and mitochondrial ROS feedback loops (Figure 1 and text). While most differentiated normal cells contain modest numbers of lipid droplets [228], advanced, treatment-resistant tumor cells commonly have much larger numbers of lipid droplets ([215]; reviewed in [204]). There is strong evidence that elevated lipid levels are responsible for treatment resistance [204,215], most likely through its capacity to drive powerful antioxidant synthesis as discussed in detail in the text.
Figure 2.
Diagrammed is the development of normal wound healing cells (top) - stem cells, to progenitor cells, to ultimately mature, terminally differentiated cells. The progenitor-like subpopulation of tumor cells (bottom), which retain their progenitor-like, perpetual growth status, based on current evidence, is crucial to clinical lethality (text). Progenitor-like MIPS circuit set point (Figure 1; text) drives the tumor cell subpopulation permanently locked in the progenitor-like state [108,109,110]. The differences in capacity for differentiation result from genetic/epigenetic changes in MIPS-centered cell state decision making. The tumor progenitor state lock is plausibly hypothesized to be produced by genetic/epigenetic changes in oncogene/ROS and mitochondrial ROS feedback loops (Figure 1 and text). While most differentiated normal cells contain modest numbers of lipid droplets [228], advanced, treatment-resistant tumor cells commonly have much larger numbers of lipid droplets ([215]; reviewed in [204]). There is strong evidence that elevated lipid levels are responsible for treatment resistance [204,215], most likely through its capacity to drive powerful antioxidant synthesis as discussed in detail in the text.
VI. Dealing with Advanced, Treatment-Resistant Cancer: Exemplar of a Working Theory for the Role of ROS in MIPS Signaling in the Presence of the Elevated Lipid Levels Characteristic/Causal of Treatment Resistance
a) Context and Fundamentals of a Working Theory
We reviewed above the extensive, diverse body of evidence that carcinomas result from mal-deployment of the cytodevelopmental machinery evolved for normal wound healing. Moreover, key features of these cytodevelopmental processes are likely controlled by the MIPS-centered redox regulatory machinery (Figure 1). It remains unclear why the progenitor-like carcinoma cells generally advance poorly through differentiation. However, incompletely understood elements of redox signaling are likely to be implicated in blocking the MIPS regulatory circuit setting necessary to permit differentiation (see [198] and references therein). See immediately below for further discussion.
The emerging new level of insight generated by the community, in turn, provides a foundation to begin building more specific and general theories of advanced, metastatic, treatment-resistant malignancy. Traditionally, treatment resistance was thought to arise from changes in drug target sites and/or increased ability to export xenobiotic drugs [199,200]. However, there is emerging evidence that common or universal changes in lipid metabolism are also essential for advanced resistance. Before discussing what the form of such a theory might be, we must clarify additional functional features of advanced disease (again, apparently controlled by/emerging from MIPS regulation).
Available evidence strongly supports the hypothesis that all clinical standards of care resulting in debulking of advanced tumors do so by triggering progenitor-like cell death through driving a large increase in mitochondrial ROS and triggering the scab cell death response. This feature includes chemotherapy [201], radiotherapy [115] and immunotherapy [202]. Consistent with this view, endogenous reducing potential is required for treatment resistance [203]. Moreover, the evidence that strong additional elevation in ROS levels is causal for therapy-induced cell death is diverse [115,201,202].
These data predict two important things. First is that treatment-resistant tumors are expected to have elevated capacity for antioxidant generation, especially through NADPH-driven systems. In fact, there is a large body of evidence that resistant tumors usually or almost always have strikingly elevated levels of lipid stores (recently reviewed in [204]; also see [205]). Lipids can provide robust flows of NADPH through NNT use of mitochondrial electron flux from lipid oxidative metabolism (reviewed in [182,183]). As expected, lipids are well known to act as in vivo antioxidants (reviewed in [206]).
Primary tumors differ dramatically in the morphology of their mitochondria, including fragmented versus elongated [207,208], reviewed in [209]. This is consistent with altered regulation of mitochondrial ROS production. In general, the dependence of treatment-resistant tumor cells on expanded lipid catabolism will drive mitochondrial specialization for elevated oxphos, as observed (reviewed in [209]). It is especially notable here that mitochondria in advanced tumor cells commonly form direct contacts with lipid droplets, facilitating efficient lipid catabolism [208,210].
Second, as noted, it is likely the ROS-induced tumor cell death is produced by a conserved decision-making system designed to allow normal cells in a healing wound to survive and actively participate in wound healing (scar) or undergo cell death to contribute to the matrix for wound healing (scab) [211].
The scab/scar decision by individual stem/progenitor cells is expected to be made through the MIPS machinery in view of its properties discussed above. Diverse cell death decisions are well known to be controlled by mitochondria, consistent with this view [212]. Mitochondrial control of cell death by ferroptosis [213] will prove to be particularly important to us below. It is reasonable to hypothesize that the logic of this machinery is as follows.
Cells in wounds will generally produce ROS in response to oxygen-deprivation; it appears that at limiting oxygen levels oxygen is preferentially diverted to ROS production [38,214]. Survival of cells in wounds (scar decision) is expected to depend on sufficient nutrient access to produce enough anti-oxidant capacity to forestall the scab decision driven by elevated ROS [211]. Thus, it is reasonable to predict that nutrient-dense lipid stores would be a primary feature supporting wound/tumor progenitor cell survival (make scar decision). [Lipid accumulation in healing wounds is poorly studied and this is an urgent future research priority.] This picture predicts a rationale for the necessity of the observed pattern of strong lipid accumulation in treatment-resistant tumors [204,215].
In a related speculation, at low lipid levels characteristic of early tumors there is expected to be insufficient mitochondrial antioxidant formation [182,183] to prevent redox inhibition of the highly redox-sensitive aconitase TCA cycle enzyme [206,216]. Aconitase inhibition drives glucose and glutamine carbon into citrate exported from mitochondria for lipogenesis. This, in turn, drives fatty acid synthesis, including high production of the intermediate malonyl-CoA. Malonyl-CoA also serves as an inhibitor of CPT1 and, thus, limits mitochondrial fatty acid uptake [217]. Collectively, these processes drive accelerating steady state lipid accumulation.
As this process drives increasing levels of fat deposits in lipid droplets (LDs), mass action is expected to ultimately drive sufficient antioxidant production to relieve aconitase inhibition. This, in turn, will engender reduced malonyl-CoA and derepression of CPI1, establishing a higher lipid consumption regime.
Thus, the steady state levels of lipids maintained by these processes will be determined by currently uncharacterized regulatory set points. Nonetheless, it is plausible to hypothesize that these set points in advanced, treatment-resistant cancers drive accumulation of the observed high levels of lipid stores in these tumors [215] (reviewed in [204]). This state also allows lipid catabolism in support of NADPH production. Thus, these enhanced lipid stores are expected to support sufficiently robust antioxidant generation to manage the robust mitochondrial redox response to most therapeutic agents [206], thereby engendering treatment resistance.
In view of this relatively clear, plausible mechanistic picture, it follows that lipid accumulation is likely to be MIPS-controlled. While our direct knowledge of this issue remains poor, we do know that elevated mitochondrial ROS drives pathways that, in turn, induce enhanced lipogenesis and lipid import, including HIF-1 and, possibly, Nrf2 [218–220]. We also know that Akt activation drives lipogenesis [221,222]. Moreover, inhibitors of tyrosine kinase upstream of Akt inhibit the lipid catabolism necessary for resistance [215], indicating multifarious control of lipid accumulation/metabolism. As noted, much more work remains to be done to understand the mechanistic details underlying lipid accumulation and catabolism, presumably including for antioxidant generation.
Thus, the doubly elevated ROS levels in advanced cancer likely arise from oxygen deprivation, from intrinsic ROS elevation in progenitor-like tumor cells, and from activated oncogene-driven ROS generation (discussed in Vc. above). Collectively, these effects are plausibly expected to drive the observed exceptionally elevated lipid levels in advanced, treatment-resistant tumors (reviewed in [204,223]). We are now positioned to discuss a plausible framework for a general theory of advanced, treatment-resistant carcinoma with the following elements.
A MIPS set point with elevated mitochondrial ROS arises in one of two general ways, under active selection for survival and growth in the competitive tumor environment (reviewed in [224]) as follows. On the one hand, mitochondrial mutations resulting in elevated ROS production through mutant oxphos machinery, is often seen in advanced cancers (see, for example, [8,13,14,15]). This mitochondrial ROS elevation can be produced by and drive oncogene activation through any of the various ROS/oncogene feedback loops discussed above (Figure 1) [35,74,145,147,157,169,170,173–177]. On the other hand, genetic/epigenetic oncogene activation or tumor suppressor inactivation (ibid.) can equally well activate any of various oncogene/ROS positive feedback loops (ibid.). It follows that activation of the MIPS lipid control process discussed immediately above is, thereby, expected to be further intensified in advanced, treatment-resistant tumors.
Thus, these processes may begin at relatively mild degrees with tumor initiation. With knock-on genetic/epigenetic changes enhancing the aggressiveness of this MIPS circuit setting, in turn, drives changes in tumor cell state. We will address immediately below how this picture helps us think about general clinical approaches with likely improved and, plausibly, universal potency. But first, it is useful to note that there is more direct evidence for this hypothesis as follows.
A compelling experimental study by the Bonini group [74] supports and further enriches this picture. Their work showed that elevated src expression drove substantially enhanced mitochondrial ROS generation, producing a robustly drug-resistant malignant phenotype. Remarkably, these investigators also showed that this mitochondrial ROS elevation directly drove epigenetic reprogramming of wound healing/malignancy genetic programs (through redox effects on chromatin structure), including EMT-driving gene products. As src can be activated by ROS [138] it is quite conceivable that a feedback loop driven by a ROS-increasing mitochondrial mutations [79] or mutational activation of src would initiate a stable clone of MIPS-controlled treatment-resistant disease through an extension of this experimentally validated event sequence.
b) Using This Theoretical Picture to Design Potentially Powerful, General Attack Strategies Against Advanced, Treatment-Resistant Carcinomas
The large lipid accumulation in treatment-resistant cancers has relevant features in addition to driving antioxidant/NADPH synthesis. Specifically, just as oxygen-deprived tumors (and presumably healing wounds) dump excess metabolic NADH reducing potential through reduction of pyruvate to lactate, they also use an exotic lipid desaturase enzyme class to accomplish the same effect [225]. Therefore, the large lipid stores in treatment-resistant, hypoxic cancers have a relatively high level of polyunsaturated fatty acids (PUFAs). This is important because PUFAs are the targets of cascade peroxidation that, in turn, triggers cell death by ferroptosis. This cascade is commonly initiated by mitochondrial ROS [205,213,223,226].
In view of these features, the therapeutic goal in attacking treatment-resistant tumors is to generate enough mitochondrial ROS to breach the antioxidant defense threshold and trigger the runaway lipid peroxidation initiating ferroptotic death (ibid.). This can be achieved by some combination of further elevating tumor mitochondrial ROS and interfering with tumor antioxidant defenses. One element of the antioxidant response that can protect from ferroptotic death includes glutathione system elements whose synthesis, in turn, can be targeted with small molecule drugs, including sulfasalazine which is FDA approved for other purposes [226]. However, sulfasalazine is not tumor specific in its effects and, thus, tolerable doses are limited, in turn, constraining clinical efficacy. Analogously, current standards of care agents for cancer are, likewise, not very tumor specific and, thus, have side-effect limitations on their ability to induce mitochondrial ROS.
Therefore, we need agents that will attenuate antioxidant defenses and enhance mitochondrial ROS tumor specifically. We developed a drug family that is able to induce robust tumor-specific mitochondrial ROS signals, CPI-613, thus having limited side effects [187,227]. Resistance to this drug is dependent on very high levels of cellular lipid accumulation and this resistance can be substantially reduced by inhibition of fatty acid catabolism [215], though these inhibitors also face the lack of tumor selectivity. This is consistent with the picture that fatty acid driven NADPH (antioxidant) production [182,183] is essential to resistance. We and others are looking for tumor-specific inhibitors of anti-oxidant capability to couple with tumor specific ROS effects of the form of CPI-613. As far as we are aware, there is no approach, other than combinations of agents of this form, that will allow successful attack on the main group of treatment-resistant cancers. If such a combination were to become available, it is expected to be pan-carcinomic in clinical effectiveness for the reasons discussed immediately above.
In summary, the vast body of insight from the research community has allowed progress toward a plausible theory of a general approach to targeting treatment-resistant cancers. The challenges now are to further test and refine such theories and, in turn, to practically implement the approaches they allow.
Author Contributions
Both authors contributed equally to this manuscript and to the experimental work from our group referred to herein.
Funding
Our group’s published reviewed herein was funded by a FUSION award from Stony Brook University Renaissance School of Medicine and by royalty income from the CPI-613 agent.
Data Availability Statement
No original data are described within this review.
Acknowledgments
We are grateful to our colleagues at Stony Brook University in the Biochemistry and Cell Biology Department and the Lipid Signaling and Metabolism in Cancer (LSMC) group for diverse helpful discussions.
Conflicts of Interest
Bingham and Zachar invented and patented the CPI-613 drug family, through Stony Brook University. CPI-613 patents were licensed to Cornerstone Pharmaceuticals for clinical development.
References
- Picard, M.; Shirihai, O.S. Mitochondrial signal transduction. Cell Metabolism, 2022, 34(11), 1620-1653. [CrossRef] [PubMed Central]
- Reinhard, T.N. Cancer as a mitochondrial metabolic disease. Frontiers in Cell and Developmental Biology, 2015, 3. [CrossRef] [PubMed Central]
- Seyfried, T.N.; Chinopoulos, C. Can the mitochondrial metabolic theory explain better the origin and management of cancer than can the somatic mutation theory? Metabolites, 2021, 11(9). [CrossRef] [PubMed Central]
- Krotofil, M.; Tota, M.; Siednienko, J.; Donizy, P. Emerging paradigms in cancer metastasis: ghost mitochondria, vasculogenic mimicry, and polyploid giant cancer cells. Cancers, 2024,16(20). [CrossRef] [PubMed Central]
- McKinnell, R.G.; Deggins, B.A.; Labat, D.D. Transplantation of pluripotential nuclei from triploid frog tumors. Science, 1969, 165(3891), 394-+. [CrossRef] [PubMed]
- Hochedlinger, K.; Blelloch, R.; Brennan, C.; Yamada, Y.; Kim, M.J.; Chin, L.; Jaenisch, R. Reprogramming of a melanoma genome by nuclear transplantation. Genes & Development, 2004, 18(15), 1875-1885. [CrossRef] [PubMed Central]
- Clark, M.A; Shay, J.W.; Goldstein, L. Techniques for purifying L-cell karyoplasts with minimal amounts of cytoplasm. Somatic Cell Genetics, 1980, 6(4), 455-464. [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma,Y.; Hayashi, J<.-I.; ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science, 2008, 320(5876), 661-664. [CrossRef] [PubMed]
- Hayashi, J.I., Takemitsu, M.; Nonaka, I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa-cells by introduction of mitochondrial-DNA from normal human cells. Somatic Cell and Molecular Genetics, 1992, 18(2), 123-129. [CrossRef] [PubMed]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic mediation of malignancy. In Vitro Cell Dev Biol, 1988, 24(5), 487-490. [CrossRef] [PubMed]
- Shidara, Y.; Yamagata, K.; Kanamori, T.; Nakano, K.; Kwong, J.Q.; Manfredi, G.; Oda, H.; Ohta, S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Research, 2005, 65(5), 1655-1663. [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun; C.Q.; Hall, J.; Lim; SD; Issa, M.M.; Flanders. W.D.; Hosseini, S.H. ; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America, 2005, 102(3), 719-724. [CrossRef] [PubMed Central]
- Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Méndez, R; Enríquez, J.A.; Fernández-Silva, P.; Martínez-Lostao, L.; Martín Villalba, M.; et al. (2019). Mutations in the ND2 subunit of mitochondrial complex I are sufficient to confer increased tumorigenic and metastatic potential to cancer cells. Cancers, 11(7). [CrossRef] [PubMed Central]
- Moreno-Loshuertos, R.; Acín-Pérez, R.; Fernández-Silva, P.; Movilla, N.; Pérez-Martos, A.; de Cordoba, S.R.; Gallardo, M.E.; Enriquez, J.A. (2006). Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nature Genetics, 38(11), 1261-1268. [CrossRef] [PubMed]
- Gasparre, G.; Porcelli, A.M.; Lenaz, G.; Romeo, G. Relevance of mitochondrial genetics and metabolism in cancer development. Cold Spring Harbor Perspectives in Biology, 2013, 5(2). [CrossRef] [PubMed Central]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L. J.; Yagi T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. Journal of Clinical Investigation, 2013, 123(3), 1068-1081. [CrossRef] [PubMed Central]
- Pouysségur, J.; Mechta-Grigoriou, F. Redox regulation of the hypoxia-inducible factor. Biological Chemistry, 2006, 387(10-11), 1337-1346. [CrossRef] [PubMed]
- Sun, Q.M.; Zhou, H.; Binmadi, N.O.; Basile, J.R. Hypoxia-inducible factor-1-mediated regulation of semaphorin 4D affects tumor growth and vascularity. Journal of Biological Chemistry, 2009, 284(46), 32066-32074. [CrossRef] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature, 2012, 485(7400), 661-+. [CrossRef] [PubMed Central]
- Almeida, J., Alves, J.M., Valecha, M., Prado-Lopez, S., Alvarino, P., Fonseca, M.M.; Cameselle-Teijeiro, J.M.; Chantada, D.; Fonseca, M.M.; Posada, D. Single-cell mtDNA heteroplasmy in colorectal cancer. Genomics, 2022, 114(2). [CrossRef] [PubMed]
- Guo, Y.; Li, C.I.; Sheng, Q.H.; Winther, J.F.; Cai, Q.Y.; Boice, J.D.; Shyr, Y. Very low-level heteroplasmy mtDNA variations are inherited in humans. Journal of Genetics and Genomics, 2013, 40(12), 607-615. [CrossRef] [PubMed Central]
- Pérez-Amado, C.J.; Bazan-Cordoba, A.; Hidalgo-Miranda, A.; Jiménez-Morales, S. Mitochondrial heteroplasmy shifting as a potential biomarker of cancer progression. International Journal of Molecular Sciences, 2021, 22(14). [CrossRef] [PubMed Central]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery, 2013, 12(12), 931-947. [CrossRef] [PubMed]
- Wong, H.S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radical Biology and Medicine, 2019,130, 140-150. [CrossRef] [PubMed]
- Shah, M. A.; Rogoff, H. A. Implications of reactive oxygen species on cancer formation and its treatment. Seminars in Oncology, 2021, 48(3), 238-245. [CrossRef] [PubMed]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.,; Lin, T.T.; Chang, H.J.; Kuo, S.-J.; Chen, S.-T.; Liu, C.S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. Journal of Experimental & Clinical Cancer Research, 2019, 38. [CrossRef] [PubMed Central]
- Garimella, S.V.; Gampa, S.C.; Chaturvedi, P. Mitochondria in cancer stem cells: From an innocent bystander to a central player in therapy resistance. Stem Cells and Cloning-Advances and Applications, 2023, 6, 19-41. [CrossRef] [PubMed Central]
- Reinhard, C. T., & Planavsky, N. J. The history of ocean oxygenation. Annual Review of Marine Science, 2022, 14, 331-353. [CrossRef] [PubMed]
- Poole, L. B. The basics of thiols and cysteines in redox biology and chemistry. Free Radical Biology and Medicine, 2015, 80, 148-157. [CrossRef] [PubMed Central]
- Kuksal, N.; Chalker, J.; Mailloux, R.J. Progress in understanding the molecular oxygen paradox - Function of mitochondrial reactive oxygen species in cell signaling. Biological Chemistry, 2017, 398(11), 1209-1227. [CrossRef]
- Petrova, B.; Liu, K.K.; Tian, C.P.; Kitaoka, M.; Freinkman, E.; Yang, J.; Orr-Weaver, T.L. Dynamic redox balance directs the oocyte-to-embryo transition via developmentally controlled reactive cysteine changes. Proceedings of the National Academy of Sciences of the United States of America, 2018, 115(34), E7978-E7986. [CrossRef] [PubMed Central]
- Percio, A.; Cicchinelli, M.; Masci, D.; Summo, M.; Urbani, A.; Greco, V. Oxidative cysteine post translational modifications drive the redox code underlying neurodegeneration and amyotrophic lateral sclerosis. Antioxidants, 2024, 13(8). [CrossRef] [PubMed Central]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radical Biology and Medicine, 2019, 140, 14-27. [CrossRef] [PubMed Central]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Molecular Cell, 2021, 81(18), 3691-3707. [CrossRef] [PubMed]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: function or dysfunction? Oncogene, 2024b, 43(5), 295-303. [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P. T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia -: A mechanism of O2 sensing. Journal of Biological Chemistry, 2000, 275(33), 25130-25138. [CrossRef] [PubMed]
- Jones, D.P.; Sies, H. The redox code. Antioxidants & Redox Signaling, 2015, 23(9), 734-746. [CrossRef] [PubMed Central]
- Chandel, N.S. Evolution of mitochondria as signaling organelles. Cell Metabolism, 2015, 22(2), 204-206. [CrossRef] [PubMed]
- Picard, M. Mitochondrial synapses: intracellular communication and signal integration. Trends in Neurosciences, 2015, 38(8), 468-474. [CrossRef] [PubMed]
- Picard, M.; McManus, M.J.; Csordás, G.; Várnai, P., Dorn, G. W.; Williams, D.; Hajnóczky, G.; Wallace, D. C. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nature Communications, 2015, 6. [CrossRef] [PubMed Central]
- Reczek, C.R., & Chandel, N.S. The two faces of reactive oxygen species in cancer. In T. Jacks & C. L. Sawyers (Eds.), Annual Review of Cancer Biology, 2017, (Vol. 1, pp. 79-98). [CrossRef]
- Picard, M.; Sandi, C. The social nature of mitochondria: Implications for human health. Neuroscience and Biobehavioral Reviews, 2021, 120, 595-610. [CrossRef] [PubMed Central]
- Sinenko, S.A.; Starkova, T Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological signaling functions of reactive oxygen species in stem cells: From flies to man. Frontiers in Cell and Developmental Biology, 2021, 9. [CrossRef]
- Mukherjee, R.; Sinha, S.; Luker, G.D.; Ghosh, P. Interlinked switch circuits of biological intelligence. Trends in Biochemical Sciences, 2024, 49(4), 286-289. [CrossRef] [PubMed]
- Haran, T.K.; Keren, L. From genes to modules, from cells to ecosystems. Molecular Systems Biology, 2022, 18(5). [CrossRef] [PubMed Central]
- Humphries, M.D.; Gurney, K. Making decisions in the dark basement of the brain: A look back at the GPR model of action selection and the basal ganglia. Biological Cybernetics, 2021, 115(4), 323-329. [CrossRef] [PubMed]
- Idelchik, M.D.S.; Begley, U.; Begley, T.J.; Melendez, J.A. (2017). Mitochondrial ROS control of cancer. Seminars in Cancer Biology, 2017, 47, 57-66. [CrossRef] [PubMed Central]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell, 2015, 163(3), 560-569. [CrossRef] [PubMed Central]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta, 2006, 1757(5-6), 509-517. [CrossRef] [PubMed]
- Zhou, L.F.; Aon, M.A.; Almas, T.; Cortassa, S. Winslow, R.L., & O'Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. Plos Computational Biology, 2010, 6(1). [CrossRef] [PubMed Central]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological Reviews, 2014, 94(3), 909-950. [CrossRef] [PubMed]
- Zandalinas, S.I.; Mittler, R. ROS-induced ROS release in plant and animal cells. Free Radical Biology and Medicine, 2018, 122, 21-27. [CrossRef] [PubMed]
- Millare, B.; O'Rourke, B; Trayanova, N. Hydrogen peroxide diffusion and scavenging shapes mitochondrial network instability and failure by sensitizing ROS-induced ROS release. Scientific Reports, 2020, 10(1). [CrossRef] [PubMed Central]
- Dvorak, H.F.; Flier, J.; Frank, H. Tumors - wounds that do not heal - similarities between tumor stroma generation and wound healing. New England Journal of Medicine, 1986, 315(26), 1650-1659. Retrieved from <Go to ISI>://WOS:A1986F365700006. [PubMed]
- Ribatti, D. The contribution of Harold F. Dvorak to the study of tumor angiogenesis and stroma generation mechanisms. Endothelium-Journal of Endothelial Cell Research, 2007, 14(3), 131-135. [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal-a historical perspective with a focus on the fundamental roles of increased vascular permeability and clotting. Semin Thromb Hemost, 2019, 45(6), 576-592. [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Reports, 2018, 22(11), 2827-2836. [CrossRef] [PubMed Central]
- Abdullah, M. O.; Zeng, R. X.; Margerum, C. L.; Papadopoli, D.; Monnin, C.; Punter, K. B.; Chu, C.; Al-Rofaidi, M.; Al-Tannak, N.F.; Berardi Y. W.; et al. Mitochondrial hyperfusion via metabolic sensing of regulatory amino acids. Cell Reports, 2022, 40(7) 111198. [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends in Cell Biology, 2014, 24(7), 400-406. [CrossRef] [PubMed Central]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nature Cell Biology, 2019, 21(2), 133-142. [CrossRef] [PubMed]
- Aston, D.; Capel, R.A.; Ford, K L.; Christian, H.C.; Mirams, G.R.; Rog-Zielinska, E.A.; Kohl, P.; Galione, A.; Burton, R.A.B.; Derek A Terrar, D.A. High resolution structural evidence suggests the sarcoplasmic reticulum forms microdomains with acidic stores (lysosomes) in the heart. Scientific Reports, 2017, 7. [CrossRef] [PubMed Central]
- Cisneros, J.; Belton, T.B.B.; Shum, G.C.C.; Molakal, C.G.G.; Wong, Y.C.C. Mitochondria-lysosome contact site dynamics and misregulation in neurodegenerative diseases. Trends in Neurosciences, 2022, 45(4), 312-322. [CrossRef] [PubMed Central]
- Jain, A.; Zoncu, R. Organelle transporters and inter-organelle communication as drivers of metabolic regulation and cellular homeostasis. Molecular Metabolism, 2022, 60. [CrossRef] [PubMed] [PubMed Central]
- Sun, T.; Wang, H.; Zhang, X.F.; Ling, P.X.; Xiao, Y.; Chen, Q.X.; Chen, X.Y. Ion monitoring at nanoscale sites of interorganelle membrane contact in living cells. Small Structures, 2024. 5(4). [CrossRef]
- Lin, J.X.; Xu, C.Y.; Wu, X.M.; Che, L.; Li, T.Y.; Mo, S.M.; Guo, D-B.; Lin, Z.N. ; Lin, Y.C. Rab7a-mTORC1 signaling-mediated cholesterol trafficking from the lysosome to mitochondria ameliorates hepatic lipotoxicity induced by aflatoxin B1 exposure. Chemosphere, 2023, 320. [CrossRef] [PubMed]
- Mailloux, R.J. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxidative Medicine and Cellular Longevity, 2018, 2018. [CrossRef] [PubMed Central]
- Nam, H.J.; Park, Y.Y.; Yoon, G.; Cho, H.; Lee, J.H. Co-treatment with hepatocyte growth factor and TGF-β1 enhances migration of HaCaT cells through NADPH oxidase-dependent ROS generation. Experimental and Molecular Medicine, 2010, 42(4), 270-279. [CrossRef] [PubMed Central]
- André-Lévigne, D.; Modarressi, A.; Pepper, M.S.; Pittet-Cuénod, B. Reactive oxygen species and NOX enzymes are emerging as key players in cutaneous wound repair. International Journal of Molecular Sciences, 2017, 18(10). [CrossRef] [PubMed Central]
- Boudreau, H.E.; Leto, T.L. Model systems to investigate NOX-dependent cell migration and invasiveness. In Knaus, U.G.; Leto, T.L. (Eds.), NADPH Oxidases: Methods and Protocols, 2019, (Vol. 1982, pp. 473-485).
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nature Reviews Molecular Cell Biology, 2024, 25(9), 701-719. [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells, 2020,9(8). [CrossRef] [PubMed Central]
- Kuznetsov, A. V., Javadov, S., Saks, V., Margreiter, R., & Grimm, M. (2017). Synchronism in mitochondrial ROS flashes, membrane depolarization and calcium sparks in human carcinoma cells. Biochim Biophys Acta Bioenerg, 1858(6), 418-431. [CrossRef]
- Pak, V.V.; Ezerina, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N. M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metabolism, 2020, 31(3), 642-+. [CrossRef] [PubMed Central]
- Desai, R.; East, D.A.; Hardy, L.; Faccenda, D.; Rigon, M.; Crosby, J; Alvarez, J.M.S.; Singh, A.; Mainenti, M.; Hussey, L.K.; et al. Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Science Advances, 2020, 6(51). [CrossRef] [PubMed Central]
- Palma, F.R.; Coelho, D.R.; Pulakanti, K.; Sakiyama, M.J.; Huang, Y.P.; Ogata, F.T.; Danes, J.M.; Meyer, A.; Furdui, C.M.; Spitz, D.R.; et al. Histone H3.1 is a chromatin-embedded redox sensor triggered by tumor cells developing adaptive phenotypic plasticity and multidrug resistance. Cell Reports, 2024a, 43(3). [CrossRef] [PubMed Central]
- Zhu, Y.; Hoell, P.; Ahlemeyer, B.; Krieglstein, J. PTEN: A crucial mediator of mitochondria-dependent apoptosis. Apoptosis, 2006, 11(2), 197-207. [CrossRef] [PubMed]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death and Differentiation, 2013, 20(12), 1631-1643. [CrossRef] [PubMed Central]
- Kreis, P.; Leondaritis, G.; Lieberam, I.; Eickholt, B. J. Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Frontiers in Molecular Neuroscience, 2014. 7. [CrossRef] [PubMed Central]
- Patel, S.; Narasimha, M. Mitochondrial ROS regulates cytoskeletal and mitochondrial remodeling to tune cell and tissue dynamics in a model for wound healing. Developmental Cell, 2014, 28(3), 239-252. [CrossRef] [PubMed]
- Fujisawa, Y.; Shinoda, N.; Chihara, T.; Miura, M. ROS regulate caspase-dependent cell delamination without apoptosis in the Drosophila pupal notum. IScience, 2020, 23(8), 101413. [CrossRef] [PubMed Central]
- Xu, S.H.; Chisholm, A.D. C elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. 2014, 31(1), 48-60. [CrossRef] [PubMed Central]
- Sener, A.; Çevik, Ö.; Dogan, Ö.; Altindis, N.G.; Aksoy, H.; Okuyan, B. The effects of topical melatonin on oxidative stress, apoptosis signals, and p53 protein expression during cutaneous wound healing. Turkish Journal of Biology, 2015, 39(6), 888-895. [CrossRef]
- Hunter, M.V.; Willoughby, P.M.; Bruce, A.E.E.; Fernandez-Gonzalez, R. Oxidative stress orchestrates cell polarity to promote embryonic wound healing. Developmental Cell, 2018, 47(3), 377-+. [CrossRef] [PubMed]
- Xu, S.Q.; Li,S. Y.; Bjorklund, M.; Xu, S.H. Mitochondrial fragmentation and ROS signaling in wound response and repair. Cell Regeneration, 2022, 11(1). [CrossRef] [PubMed]
- Chen, X.N.; Ran, X.Y.; Wei, XB.; Zhu, L. F.; Chen, S.D.; Liao, Z.Y.; Xu, K.; Xia, W.D. Bioactive glass 1393 promotes angiogenesis and accelerates wound healing through ROS/P53/MMP9 signaling pathway. Regenerative Therapy, 2024, 26, 132-144. [CrossRef] [PubMed Central]
- Hebbar, S.; Knust, E. Reactive oxygen species (ROS) constitute an additional player in regulating epithelial development. Bioessays, 2021, 43(8). [CrossRef] [PubMed]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nature Genetics, 2008, 40(3), 356-361. [CrossRef] [PubMed]
- Sing, A.; Tsatskis, Y.; Fabian, L.; Hester, I.; Rosenfeld, R.; Serricchio, M.; Yau, N.; Bietenhader, M.; Shanbhag, R.; Jurisicova , A.; et al. The atypical cadherin fat directly regulates mitochondrial function and metabolic state. Cell, 2014, 158(6), 1293-1308. [CrossRef] [PubMed]
- Oswald, M.C.W.; Garnham, N.; Sweeney, S.T; Landgraf, M. Regulation of neuronal development and function by ROS. FEBS Letters, 2018, 592(5), 679-691. [CrossRef] [PubMed Central]
- Coffman, J.A.; McCarthy, J.J.; Dickey-Sims, C.; Robertson, A.J. Oral-aboral axis specification in the sea urchin embryo II. Mitochondrial distribution and redox state contribute to establishing polarity in Strongylocentrotus purpuratus. Developmental Biology, 2004, 273(1), 160-171. [CrossRef] [PubMed]
- Hernández-García, D.; Wood, C.D.; Castro-Obregón, S.; Covarrubias, L. Reactive oxygen species: A radical role in development? Free Radic Biol Med, 2010, 49(2), 130-143. [CrossRef] [PubMed]
- Coffman, J.A.; Su, Y.H. Redox regulation of development and regeneration. Current Opinion in Genetics & Development, 2019, 57, 9-15. [CrossRef] [PubMed]
- Breus, O.; Dickmeis, T. Genetically encoded thiol redox-sensors in the zebrafish model: lessons for embryonic development and regeneration. Biological Chemistry, 2021. 402(3), 363-378. [CrossRef] [PubMed]
- Salas-Vidal, E.; Mendez-Cruz, F.J.; Ramírez-Corona, A.; Reza-Medina, B. Oxygen, reactive oxygen species and developmental redox networks: Evo-devo evil-devils? International Journal of Developmental Biology, 2021, 65(4-6), 345-356. [CrossRef] [PubMed]
- Madan, S.; Uttekar, B.; Chowdhary, S.; Rikhy, R. Mitochondria lead the way: Mitochondrial dynamics and function in cellular movements in development and disease. Frontiers in Cell and Developmental Biology, 2022, 9. [CrossRef] [PubMed Central]
- Garfinkel, A.M., Mnatsakanyan, N., Patel, J.H., Wills, A.E., Shteyman, A., Smith, P.J. S.; Alavian, K.N.; Jonas, E.A.; Khokha, M.K. Mitochondrial leak metabolism induces the Spemann-Mangold Organizer via Hif-1a in Xenopus. Developmental Cell, 2023, 58(22), 2597-+. [CrossRef] [PubMed Central]
- Amblard, I.; Thauvin, M.; Rampon, C.; Queguiner, I.; Pak, V.V., Belousov, V; Alain Prochiantz, A.; Volovitch M.; Joliot, A. ; Vriz, S. H2O2 and Engrailed 2 paracrine activity synergize to shape the zebrafish optic tectum. Communications Biology, 2020, 3(1). [CrossRef] [PubMed Central]
- Paffenholz, R.; Bergstrom, R. A.; Pasutto, F.; Wabnitz, P.; Munroe, R. J.; Jagla, W.; Heinzmann, U.; Marquardt, A.; Bareiss, A.; Laufs, J. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev, 2004, 18(5), 486-491. [CrossRef] [PubMed Central]
- Kiss, P.J., Knisz, J., Zhang, Y.Z., Baltrusaitis, J., Sigmund, C. D., Thalmann, R.; Smith R.J.H.; Verpy E.; Bánfi, B. Inactivation of NADPH oxidase organizer results in severe imbalance. Current Biology, 2006, 16(2), 208-213. [CrossRef] [PubMed]
- Chartier, F.J.M.; Hardy, É.; Laprise, P. Crumbs limits oxidase-dependent signaling to maintain epithelial integrity and prevent photoreceptor cell death. Journal of Cell Biology, 2012, 198(6), 991-998. [CrossRef] [PubMed Central]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radical Research, 2010, 44(5), 479-496. [CrossRef] [PubMed Central]
- Yousefi, B.; Samadi, N.; Ahmadi, Y. Akt and p53R2, partners that dictate the progression and invasiveness of cancer. DNA Repair, 2014, 22, 24-29. [CrossRef] [PubMed]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental activation of Nrf2 restricts the progression of Nrf2-activated malignant tumors. Cancer Research, 2020, 80(16), 3331-3344. [CrossRef] [PubMed]
- Jeon, S.M., Chandel, N.S., & Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature, 2012, 485(7400), 661-+. [CrossRef] [PubMed Central]
- Bertolotti, M.; Sitia, R.; Rubartelli, A. On the redox control of b lymphocyte differentiation and function. Antioxidants & Redox Signaling, 2012, 16(10), 1139-1149. [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nature Reviews Cancer, 2022, 22(5), 280-297. [CrossRef] [PubMed]
- Kahlon, U., Ricca, F.D., Pillai, S.J., Olivetta, M., Tharp, K.M., Jao, L.E.; Dudin, O.; McDonald, K.; Aydogan, M.G. A mitochondrial redox switch licenses the onset of morphogenesis in animals. bioRxiv., 2024,. [CrossRef] [PubMed Central]
- Muller, F.L.; Liu, Y.H.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. Journal of Biological Chemistry, 2004, 279(47), 49064-49073. [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Biology and Medicine, 2016, 100, 14-31. [CrossRef] [PubMed]
- Sies, H.; Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nature Reviews Molecular Cell Biology, 2020, 21(7), 363-383. [CrossRef] [PubMed]
- Witte, M.B.; Barbul, A. General principles of wound healing. Surgical Clinics of North America, 1997, 77(3), 509-+. [CrossRef] [PubMed]
- Suetsugi, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133 hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochemical and Biophysical Research Communications, 2006, 351(4), 820-824. [CrossRef] [PubMed]
- Donnenberg, V.S.; Landreneau, R.J.; Donnenberg, A.D. Tumorigenic stem and progenitor cells: Implications for the therapeutic index of anti-cancer agents. Journal of Controlled Release, 2007, 122(3), 385-391. [CrossRef] [PubMed Central]
- de Souza, V.B.; Schenka, A A. Cancer stem and progenitor-like cells as pharmacological targets in breast cancer treatment. Breast Cancer (Auckl), 2015, 9(Suppl 2), 45-55. [CrossRef] [PubMed Central]
- Kwan, K.Y., Shen, J., & Corey, D.P. C-MYC Transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports, 2015, 4(1), 47-60. [CrossRef] [PubMed Central]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. Journal of Experimental & Clinical Cancer Research, 2018, 37. [CrossRef] [PubMed]
- Wang, X.L.; Ma, Y.X.; Xu, R.J.; Ma, J.J.; Zhang, H.C.; Qi, S. B.; Xu, J-H.; Qin, X-Z; Zhang, H-N.; Liu, C-M.; et al. c-Myc controls the fate of neural progenitor cells during cerebral cortex development. Journal of Cellular Physiology, 2020, 235(4), 4011-4021. [CrossRef] [PubMed]
- Cai, C.H.; Hu, X.Y.; Dai, P.B.; Zhang, T.R.; Jiang, M.; Wang, L.F.; Hua, W.; Fan, Y.; Han, X.-X.; Gao, Z.L. c-Myc regulates neural stem cell quiescence and activation by coordinating the cell cycle and mitochondrial remodeling. Signal Transduction and Targeted Therapy, 2021, 6(1). [CrossRef] [PubMed Central]
- Diehn, M., Cho, R W., Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature, 2009, 458(7239), 780-U123. [CrossRef] [PubMed Central]
- Eberhart, C.G. Even cancers want commitment: Lineage identity and medulloblastoma formation. Cancer Cell, 2008,14(2), 105-107. [CrossRef] [PubMed Central]
- Persson, A.I.; Petritsch, C.; Swartling, F.J.; Itsara, M.; Sim, F.J.; Auvergne, R; Goldenberg, D.D.; Vandenberg, S.R.; Nguyen K.N.; Yakovenko, S; et al. Non-stem cell origin for oligodendroglioma. Cancer Cell, 2010, 18(6), 669-682. [CrossRef] [PubMed Central]
- Tu, S.M. Cancer: a "stem-cell" disease? Cancer Cell International, 2013, 13. [CrossRef] [PubMed Central]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone. H.; Freeman-Mills, L.; et al. Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell, 2016, 30(4), 578-594. [CrossRef] [PubMed Central]
- Haag, D., Mack, N., da Silva, P. B. G., Statz, B., Clark, J., Tanabe, K.; Sharma, T.; Jäger, J.; Jones, D.T.W.;et al. H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell, 2021,39(3), 407-422.e13. [CrossRef] [PubMed]
- Piccoli, C.; D'Aprile, A.; Ripoli, M.; Scrima, R.; Lecce, L.; Boffoli, D.; Tabilio, A.; Capitanio, N. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constitutively reactive oxygen species. Biochemical and Biophysical Research Communications, 2007, 353(4), 965-972. [CrossRef] [PubMed]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature, 2009, 461(7263), 537-U109. [CrossRef] [PubMed Central]
- Motohashi, H.; Kimura, M.; Fujita, R.; Inoue, A.; Pan, X.Q.; Takayama, M.; Katsuoka, F.; Aburatani, H.; Bresnick,E.H.; Yamamoto, M. NF-E2 domination over Nrf2 promotes ROS accumulation and megakaryocytic maturation. Blood, 2010, 115(3), 677-686. [CrossRef] [PubMed Central]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development, 2014, 141(22), 4206-4218. [CrossRef] [PubMed Central]
- Urao, N.; Ushio-Fukai, M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radical Biology and Medicine, 2013, 54, 26-39. [CrossRef] [PubMed Central]
- di Bello, G.; Vendemiale, G.;Bellanti, F. Redox cell signaling and hepatic progenitor cells. European Journal of Cell Biology, 2018, 97(8), 546-556. [CrossRef] [PubMed]
- Maraldi,T.; Angeloni, C.; Prata, C.; Hrelia, S. NADPH oxidases: Redox regulators of stem cell fate and function. Antioxidants, 2021,10(6). [CrossRef] [PubMed Central]
- Sommers, O.; Tomsine, R.A.; Khacho, M. Mitochondrial dynamics drive muscle stem cell progression from quiescence to myogenic differentiation. Cells, 2024, 13(21). [CrossRef] [PubMed Central]
- Khandia, R., & Munjal, A. Interplay between inflammation and cancer. In R. Donev (Ed.), Inflammatory Disorders, Pt A, 2020, (Vol. 119, pp. 199-245). 0.1016/bs.apcsb.2019.09.004.
- 15th Report on Carcinogens. Rep Carcinog, (2021). 15. [CrossRef] [PubMed]
- Nigam, M.; Mishra, A.P., Deb; V. K., Dimri, D.B.; Tiwari, V.; Bungau, S.G.; Bungau, A.F. ; Radu, A.F. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomedicine & Pharmacotherapy, 2023, 164. [CrossRef] [PubMed]
- Tsarouhas, V.; Yao, L.Q.; Samakovlis, C. Src kinases and ERK activate distinct responses to Stitcher receptor tyrosine kinase signaling during wound healing in Drosophila. Journal of Cell Science, 2014, 127(8), 1829-1839. [CrossRef] [PubMed]
- Evans, I.R.; Rodrigues, F.; Armitage, E.L.; Wood, W. Draper/CED-1 mediates an ancient damage response to control inflammatory blood cell migration in vivo. Current Biology, 2015, 25(12), 1606-1612. [CrossRef] [PubMed Central]
- Srinivasan, N., Gordon, O., Ahrens, S., Franz, A., Deddouche, S., Chakravarty, P.; David Phillips, D,; Yunus, A.A.; Rosen , M.R.; Valente, R.S. ; et al. Actin is an evolutionarily-conserved damage-associated molecular pattern that signals tissue injury in Drosophila melanogaster. Elife, 2016, 5. [CrossRef] [PubMed Central]
- Floc'hlay, S.; Balaji, R.; Stanković, D.; Christiaens, V.M.; González-Blas, B., De Winter, S.; Hulselmans, G.J.; De Waegeneer, M.; Quan, X.; Koldere, D.; et al. Shared enhancer gene regulatory networks between wound and oncogenic programs. Elife, 2023, 12. [CrossRef] [PubMed Central]
- Love, N.R.; Chen, Y.Y.; Ishibashi, S.; Kritsiligkou, P.; Lea, R.; Koh, Y.; Gallop, J.L.; Dorey, K.; Amaya, E. Amputation-induced reactive oxygen species are required for successful Xenopus tadpole tail regeneration. Nature Cell Biology, 2013, 15(2), 222-228. [CrossRef] [PubMed Central]
- Patel, P.H.; Penalva, C.; Kardorff, M.; Roca, M.; Pavlovic, B.; Thiel, A.; Aurelio A Teleman, A.A.; Edgar, B.A. Damage sensing by a Nox-Ask1-MKK3-p38 signaling pathway mediates regeneration in the adult Drosophila midgut. Nature Communications, 2019,10. [CrossRef] [PubMed Central]
- Serras, F. The sooner, the better: ROS, kinases and nutrients at the onset of the damage Response in Drosophila. Frontiers in Cell and Developmental Biology, 2022, 10. [CrossRef] [PubMed Central]
- Pooranachithra, M.; Suruthi, K.; Prabhanand, B. J.; Deepa, M.; Sekhar, D.; Venkateswaran, K.; Gajbhiye Rahul, G.; Ravichandiran. V.; Balamurugan, K. Survival upon Staphylococcus aureus mediated wound infection in Caenorhabditis elegans and the mechanism entailed. Microbial Pathogenesis, 2021, 157. [CrossRef] [PubMed]
- MacKay, C.E.; Knock, G.A. Control of vascular smooth muscle function by Src-family kinases and reactive oxygen species in health and disease. Journal of Physiology-London, 2015, 593(17), 3815-3828. [CrossRef] [PubMed Central]
- Inumaru, J.; Nagano, O.; Takahashi, E.; Ishimoto, T.; Nakamura, S.; Suzuki, Y.; Niwa S.-I.; Umezawa, K.; Tanihara, H.; Saya, H. Molecular mechanisms regulating dissociation of cell-cell junction of epithelial cells by oxidative stress. Genes to Cells, 2009, 14(6), 703-716. [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman B.; Mutlu G.M.; Budinger, R.A.S.; Chandel, N. S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(19), 8788-8793. [CrossRef]
- Hobbs, G.A.; Bonini, M.G.; Gunawardena, H.P.; Chen, X.; Campbell, S.L. Glutathiolated ras: Characterization and implications for ras activation. Free Radical Biology and Medicine, 2013, 57, 221-229. [CrossRef] [PubMed Central]
- Messina, S.; De Simone, G.; Ascenzi, P. Cysteine-based regulation of redox-sensitive Ras small GTPases. Redox Biology, 2019, 26. [CrossRef] [PubMed Central]
- Muliyil, S.; Narasimha, M. Mitochondrial ROS regulates cytoskeletal and mitochondrial remodeling to tune cell and tissue dynamics in a model for wound healing. Dev Cell, 2014, 28(3), 239-252. [CrossRef] [PubMed]
- Fujisawa, Y.; Shinoda, N.; Chihara, T.; Miura, M. ROS regulate caspase-dependent cell delamination without apoptosis in the Drosophila pupal notum. Iscience, 2020, 23(8). [CrossRef] [PubMed Central]
- Aghajanian, A.; Wittchen, E. S.; Campbell, S. L.; Burridge, K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. Plos One, 2009, 4(11), e8045. [CrossRef] [PubMed Central]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.O.P.; Lazo-Kallanian, S.; Williams, I.R; et al. FoxOs are critical mediators of hematopoietic.
- stem cell resistance to physiologic oxidative stress. Cell, 2007, 128(2), 325-339. [CrossRef] [PubMed]
- de Keizer, P.L.J.; Burgering, B.M.T.; Dansen, T.B. Forkhead Box O as a sensor, mediator, and regulator of redox signaling. Antioxidants & Redox Signaling, 2011,14(6), 1093-1106. [CrossRef] [PubMed]
- Liu, B., Chen, Y.M., & Clair, D.K.S. ROS and p53: A versatile partnership. Free Radical Biology and Medicine, 2008, 44(8), 1529-1535. [CrossRef] [PubMed Central]
- Truong, T.H.; Ung, P.M.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular basis for redox activation of epidermal growth factor receptor kinase. Cell Chemical Biology, 2016, 23(7), 837-848. [CrossRef]
- Hocaoglu, H.; Sieber, M. Mitochondrial respiratory quiescence: A new model for examining the role of mitochondrial metabolism in development. Seminars in Cell & Developmental Biology, 2023,138, 94-103. [CrossRef] [PubMed Central]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: a dangeROS liaison. British Journal of Cancer, 2020, 122(2), 168-181. [CrossRef] [PubMed Central]
- Ogrunc, M., Di Micco, R., Liontos, M., Bombardelli, L., Mione, M., Fumagalli, M.; Gorgoulis, V.G.; d'Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ, 2014, 21(6), 998-1012. [CrossRef]
- Iskandar, K.; Foo, J.; Liew, A.Q.X.; Zhu, H.Y.X.; Raman, D.; Hirpara, J.L.; Leong, Y.Y.; Babak, M.V.; Kirsanova, A.A.; Armand, A.-S.; et al. A novel mTORC2-AKT-ROS axis triggers mitofission and mitophagy-associated execution of colorectal cancer cells upon drug-induced activation of mutant KRAS. Autophagy, 2024, 20(6), 1418-1441. [CrossRef] [PubMed Central]
- Hu, Y.M; Lu, W.Q.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras G12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Research, 2012, 22(2), 399-412. [CrossRef] [PubMed Central]
- Liou, G.Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.Z.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. ;Mutant Kras-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Reports, 2016, 14(10), 2325-2336. [CrossRef] [PubMed Central]
- Kitagishi, Y.; Matsuda, S. Redox regulation of tumor suppressor PTEN in cancer and aging (Review). Int J Mol Med, 2013, 31(3), 511-515. [CrossRef] [PubMed]
- Nakanishi, A.; Wada, Y.; Kitagishi, Y.; Matsuda, S. Link between PI3K/AKT/PTEN pathway and NOX protein in diseases. Aging and Disease, 2014, 5(3), 203-211. [CrossRef] [PubMed Central]
- Brookes, P.S.; Yoon, Y.S.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology-Cell Physiology, 2004, 287(4), C817-C833. [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in cancer. Front Oncol, 2018, 8, 160. [CrossRef] [PubMed Central]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Molecular Cell, 2002, 9(5), 1031-1044. [CrossRef] [PubMed]
- Lee, K.M., Giltnane, J.M., Balko, J.M., Schwarz, L.J., Guerrero-Zotano, A.L., Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders M.E.; et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metabolism, 2017, 26(4), 633-+. [CrossRef] [PubMed Central]
- Yang, B.W., Zhu, W.Z., Zheng, Z.D., Chai, R.F., Ji, S.H., Ren, G H.; Liu, T.; Liu, Z.; Song, T.; Li, F. ; et al. Fluctuation of ROS regulates proliferation and mediates inhibition of migration by reducing the interaction between DLC1 and CAV-1 in breast cancer cells. In Vitro Cellular & Developmental Biology-Animal, 2017, 53(4), 354-362. [CrossRef] [PubMed]
- Farris, J.C., Pifer, P.M., Zheng, L., Gottlieb, E., Denvir, J.; Frisch, S. M. Grainyhead-like 2 reverses the metabolic changes induced by the oncogenic epithelial-mesenchymal transition: Effects on anoikis. Molecular Cancer Research, 2016,14(6), 528-538. [CrossRef] [PubMed Central]
- Frisch, S.M.; Farris, J.C.; Pifer, P.M. Roles of Grainyhead-like transcription factors in cancer. Oncogene, 2017, 36(44), 6067-6073. [CrossRef] [PubMed]
- Menheniott, T.R.; Kurklu, B.; Giraud, A.S. Gastrokines: stomach-specific proteins with putative homeostatic and tumor suppressor roles. American Journal of Physiology-Gastrointestinal and Liver Physiology, 2013, 304(2), G109-G121. [CrossRef] [PubMed]
- Yoon, J.H., Kang, Y.H., Choi, Y.J., Park, I.S., Nam, S.W., Lee, J.Y.; Lee, Y.S.; Park, W.S. Gastrokine 1 functions as a tumor suppressor by inhibition of epithelial-mesenchymal transition in gastric cancers. Journal of Cancer Research and Clinical Oncology, 2011, 137(11), 1697-1704. [CrossRef] [PubMed]
- Okuda-Ashitaka, E.; Matsumoto, K. Tenascin-X as a causal gene for classical-like Ehlers-Danlos syndrome. Frontiers in Genetics, 2023, 14. [CrossRef] [PubMed Central]
- Pulido, R. PTEN inhibition in human disease therapy. Molecules, 2018, 23(2). [CrossRef] [PubMed Central]
- Zeng, Y.B.; Liang, X.H.; Zhang, G.X.; Jiang, N.; Zhang, T.; Huang, J.Y.; Zhang, L.; Zeng, X.C. miRNA-135a promotes hepatocellular carcinoma cell migration and invasion by targeting forkhead box O1. Cancer Cell International, 2016, 16. [CrossRef] [PubMed Central]
- ter Horst, E.N.; Hahn, N.E.; Geerts, D.; Musters, R.J.P.; Paulus, W.J., van Rossum, A. C.; Meischl, C.; Piek, J.J.; Niessen, H.W.M.; et al. p47phox-dependent reactive oxygen species stimulate nuclear translocation of the FoxO1 transcription factor during metabolic inhibition in cardiomyoblasts. Cell Biochemistry and Biophysics, 2018, 76(3), 401-410. [CrossRef] [PubMed Central]
- Liang, X.Y.; Su, Y.J.; Huo, Y.B. Forkhead box protein O1 (FoxO1) /SERPINB1 ameliorates ROS production in diabetic nephropathy. Food Science & Nutrition, 2021, 9(1), 44-51. [CrossRef] [PubMed Central]
- Li, Y.Y.; Zheng, Q.M.; Zhang, N.; Zhang, M. The Kruppel-like factor 6 represses proliferation and invasion of cutaneous malignant melanoma. Polish Journal of Pathology, 2023, 74(3), 194-202. [CrossRef] [PubMed]
- Syafruddin, S.E., Mohtar, M.A., Nazarie, W.; Low, T. Y. Two sides of the same coin: The roles of KLF6 in physiology and pathophysiology. Biomolecules, 2020, 10(10). [CrossRef]
- Urtasun, R.; Cubero, F.J.; Nieto, N. Oxidative stress modulates KLF6Full and its splice variants. Alcoholism-Clinical and Experimental Research, 2012, 36(11), 1851-1862. [CrossRef] [PubMed Central]
- Kourdova, L.T., Miranda, A.L., Racca, A.C., Rojas, M.L., Del Puerto, M.C., Castro, C; Susana Genti-Raimond, S. ; Panzetta-Dutari, G.M. Downregulation of kruppel-like factor 6 expression modulates extravillous trophoblast cell behavior by increasing reactive oxygen species. Placenta, 2022, 127, 62-72. [CrossRef] [PubMed]
- Han, M.K.; Song, E.K.; Guo, Y.; Ou, X.; Mantel, C.; Broxmeyer, H.E. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell, 2008, 2(3), 241-251. [CrossRef] [PubMed Central]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. Plos One, 2013, 8(9). [CrossRef] [PubMed Central]
- Di Sante, G.; Pestell, T.G.; Casimiro, M.C.; Bisetto, S.; Powell, M.J.; Lisanti, M.; Cordon-Cardo, P.C.; Castillo-Martin, M.; Bonal, D.M.; Debattisti, V.; et al. Loss of Sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays Park2 translocation to mitochondria. American Journal of Pathology, 2015, 185(1), 266-279. [CrossRef] [PubMed Central]
- Qiang, L.; Sample, A.; Liu, H.; Wu, X.Y.; He, Y.Y. Epidermal SIRT1 regulates inflammation, cell migration, and wound healing. Scientific Reports, 2017, 7. [CrossRef] [PubMed Central]
- Gasparics, A.; Sebe, A. Forward genetic screens as tools to investigate role and mechanisms of EMT in cancer. Cancers, 2022, 14(23). [CrossRef] [PubMed Central]
- Hansen, G.E.; Gibson, G.E. The α-ketoglutarate dehydrogenase complex as a hub of plasticity in neurodegeneration and regeneration. International Journal of Molecular Sciences, 2022, 23(20). [CrossRef] [PubMed Central]
- Wagner, M.; Bertero, E.; Nickel, A.; Kohlhaas, M.; Gibson, G.E.; Heggermont, W.; Stephane Heymans, S.; Maack, C. (2020). Selective NADH communication from α-ketoglutarate dehydrogenase to mitochondrial transhydrogenase prevents reactive oxygen species formation under reducing conditions in the heart. Basic Research in Cardiology, 115(5). [CrossRef]
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature, 2019, 573(7773), 291-+. [CrossRef] [PubMed Central]
- Nesci, S.; Trombetti, F.; Pagliarani, A. Nicotinamide nucleotide transhydrogenase as a sensor of mitochondrial biology. Trends in Cell Biology, 2020, 30(1), 1-3. [CrossRef] [PubMed]
- Lagranha, C.J.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circulation Research, 2010, 106(11), 1681-U1646. [CrossRef] [PubMed Central]
- Green, S.R.; Storey, K.B. Regulation of the α-ketoglutarate dehydrogenase complex during hibernation in a small mammal, the Richardson's ground squirrel (Urocitellus richardsonii). Biochimica Et Biophysica Acta-Proteins and Proteomics, 2020, 1868(9). [CrossRef] [PubMed]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radical Research, 2011, 45(1), 29-36. [CrossRef] [PubMed Central]
- Stuart, S D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab, 2014, 2(1), 4. [CrossRef] [PubMed Central]
- Sen, T.; Sen, N.; Noordhuis, M.G.; Ravi, R.; Wu, T.C.; Ha, P. K.; Sidransky, D. ; Hoque, M. O. OGDHL is a modifier of AKT-dependent signaling and NF-κB Function. Plos One, 2012, 7(11). [CrossRef] [PubMed Central]
- Anderson, N.M.; Li,D.; Peng, H L.; Laroche, F.J.F.,; Mansour, M.R.; Gjini, E.; Aioub, M.; Helman, D.J.; J E Roderick, J.E.; Cheng, T.; et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia, 2016, 30(6), 1365-1374. [CrossRef] [PubMed Central]
- Ilic, N.; Birsoy, K.; Aguirre, A.J.; Kory, N.; Pacold, M.E.; Singh, S.; Susan E Moody, S.E.; DeAngelo, J.D.; Spardy, N.A.; Freinkman, E. ; et al. . PIK3CAb mutant tumors depend on oxoglutarate dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America, 2017,114(17), E3434-E3443. [CrossRef] [PubMed Central]
- Lu, X.; Wu, N.; Yang, W.L.; Sun, J.; Yan; K.M.; Wu, J. OGDH promotes the progression of gastric cancer by regulating mitochondrial bioenergetics and Wnt/β-catenin signal pathway. Oncotargets and Therapy, 2019, 12, 7489-7500. [CrossRef] [PubMed Central]
- Lu, X.; Yang, P.F.; Zhao, X.R.; Jiang, M.Z.; Hu, S.J.; Ouyang, Y.N.; Zeng, L,; Wu, J. OGDH mediates the inhibition of SIRT5 on cell proliferation and migration of gastric cancer. Experimental Cell Research, 2019, 382(2-3). [CrossRef] [PubMed]
- Khalaj-Kondori, M.; Hosseinnejad, M.; Hosseinzadeh, A.; Sharif, S. B.; Hashemzadeh, S. Aberrant hypermethylation of OGDH gene promoter in sporadic colorectal cancer. Current Problems in Cancer, 2020. 44(1). [CrossRef] [PubMed]
- Li, M.M.; Wu, P.D.; Yang, Z.C.; Deng, S.W;, Ni, L.Y.; Zhang, Y. F.; Jin, L.; Pan, Y. miR-193a-5p promotes pancreatic cancer cell metastasis through SRSF6-mediated alternative splicing of OGDHL and ECM1. American Journal of Cancer Research, 2020, 10(1), 38-+. Retrieved from <Go to ISI>://WOS:000508940800003. [PubMed]
- Anderson, N.M.; Qin, X.D.; Finan, J.M.; Lam, A.; Athoe, J., Missiaen, R.;Skuli, N.; Kennedy, A.; Saini, A.S.; Tao, T.; et al. Metabolic enzyme DLST promotes tumor aggression and reveals a vulnerability to OXPHOS inhibition in high-risk neuroblastoma. Cancer Research, 2021, 81(17), 4417-4430. [CrossRef] [PubMed Central]
- Shen, N.; Korm, S.; Karantanos, T.; Li, D., Zhang, X.Y., Ritou, E.; Xu, H.; Lam, A.; English, J.; Zong, W.-X,; et al. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Communications Biology, 2021, 4(1). [CrossRef] [PubMed Central]
- Ponte, S.; Carvalho, L.; Gagliardi, M.; Campos, I.; Oliveira, P.J.; Jacinto, A. Drp1-mediated mitochondrial fission regulates calcium and F-actin dynamics during wound healing. Biology Open, 2020, 9(5). [CrossRef] [PubMed Central]
- Oka, S.; Tsuzuki, T.; Hidaka, M.; Ohno, M.; Nakatsu, Y.; Sekiguchi, M. Endogenous ROS production in early differentiation state suppresses endoderm differentiation via transient FOXC1 expression. Cell Death Discovery, 2022, 8(1). [CrossRef] [PubMed Central]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey S.; et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell International, 2015, 15. [CrossRef] [PubMed Central]
- Wang, X.; Zhang, H.; Chen, X.; Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019 ,2(2):141-160. Epub 2019 Jun 19. [CrossRef] [PubMed Central]
- Yang, G.G.; Zhang, H.; Zhang, D.Y.; Cao, Q.; Yang, J.; Ji, L.N.; Mao, Z.W. Cancer-specific chemotherapeutic strategy based on the vitamin K3 mediated ROS regenerative feedback and visualized drug release in vivo. Biomaterials, 2018,185, 73-85. [CrossRef]
- Liu, R.L.; Peng, L.Y.; Zhou, L.; Huang, Z.; Zhou, C.W.; Huang, C.H. Oxidative stress in cancer immunotherapy: Molecular mechanisms and potential applications. Antioxidants, 2022, 11(5). [CrossRef] [PubMed Central]
- Gu, X.H.; Mu, C.Y.; Zheng, R.J., Zhang; Z., Zhang, Q.; Liang, T.B. The cancer antioxidant regulation system in therapeutic resistance. Antioxidants, 2024, 13(7). [CrossRef] [PubMed Central]
- Bingham, P. M.; Zachar, Z. Toward a unifying hypothesis for redesigned lipid catabolism as a clinical target in advanced, treatment-resistant carcinomas. Int J Mol Sci, 2023, 24(18). [CrossRef] [PubMed Central]
- Jiang, X.J.; Stockwell, B.R.; Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nature Reviews Molecular Cell Biology, 2021, 22(4), 266-282. [CrossRef] [PubMed Central]
- Bartolacci, C.; Andreani, C.; El-Gammal, Y.; Scaglioni, P.P. Lipid metabolism regulates oxidative stress and ferroptosis in ras-driven cancers: a perspective on cancer progression and therapy. Frontiers in Molecular Biosciences, 2021, 8. [CrossRef] [PubMed Central]
- Benard, G.; Bellance, N.; Jose, C., Melser, S.; Nouette-Gaulain, K.; Rossignol, R. Multi-site control and regulation of mitochondrial energy production. Biochimica Et Biophysica Acta-Bioenergetics, 2010, 1797(6-7), 698-709. [CrossRef] [PubMed]
- Han, M.Q.; Bushong, E.A.; Segawa, M.; Tiard, A.; Wong, A.L.; Brady, M.R.; Momcilovic, M.; Wolf, D.M.; Zhang, R.; Petcherski. A.; et al. Spatial mapping of mitochondrial networks and bioenergetics in lung cancer. Nature, 2023, 615(7953), 712-719. [CrossRef] [PubMed Central]
- Giedt, R.J., Fumene Feruglio, P., Pathania, D., Yang, K.S., Kilcoyne, A., Vinegoni, C.;
- Mitchison, T.J.; Weissleder, R. Computational imaging reveals mitochondrial morphology as a biomarker of cancer phenotype and drug response. Scientific Reports, 2016, 6. [CrossRef] [PubMed Central]
- Talari, N.K.; Mattam, U.; Meher, N.K.; Paripati, A K.; Mahadev, K.; Krishnamoorthy, T.; Sepuri, N.B. Lipid-droplet associated mitochondria promote fatty-acid oxidation through a distinct bioenergetic pattern in male Wistar rats. Nature Communications, 2023, 14(1). [CrossRef] [PubMed Central]
- Zhou, S.X.; Xie, M.B.; Su, J.J.; Cai, B.J.; Li, J.A.; Zhang, K. New insights into balancing wound healing and scarless skin repair. Journal of Tissue Engineering, 2023, 14. [CrossRef]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and cell death. Nat Cell Biol, 2024, 26(9), 1434-1446. [CrossRef] [PubMed]
- Gao, M.H., Yi, J.M., Zhu, J.J., Minikes, A.M., Monian, P., Thompson, C.B., & Jiang, X.J. Role of mitochondria in ferroptosis. Molecular Cell, 2019, 73(2), 354-363.e3. [CrossRef] [PubMed Central]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.P.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Paul T Schumacker, P.T.Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metabolism, 2005, 1(6), 401-408. [CrossRef] [PubMed]
- Guardado Rivas, M. O.; Stuart, S.D.; Thach, D.; Dahan, M.; Shorr, R.; Zachar, Z.; Bingham, P.M. Evidence for a novel, effective approach to targeting carcinoma catabolism exploiting the first-in-class, anti-cancer mitochondrial drug, CPI-613. Plos One, 2022,. 17(6). [CrossRef] [PubMed Central]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Report, 2014, 19(1), 8-15. [CrossRef] [PubMed Central]
- Ngo, J., Choi, D.W., Stanley, I.A., Stiles, L., Molina, A.J.A., Chen, P.H.; Lako, A.; Sung, I.C.H.; Goswami, R.; Kim, M-Y.; et al. Mitochondrial morphology controls fatty acid utilization by changing CPT1 sensitivity to malonyl-CoA. EMBO J, 2023, 42(11), e111901. [CrossRef] [PubMed Central]
- Huang, J.S.; Tabbi-Anneni, I.; Gunda, V.; Wang, L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. American Journal of Physiology-Gastrointestinal and Liver Physiology, 2010, 299(6), G1211-G1221. [CrossRef] [PubMed Central]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. Journal of Cell Science, 2012, 125(14), 3485-3493. [CrossRef] [PubMed Central]
- Tong, Z.K., Du, X.K., Zhou, Y., Jing, F.X., Ma, J.P., Feng, Y.Y.; Lou, S.; Wang, Q.; Dong, Z.X.; et al. ROS Drp1-mediated mitochondrial fission promotes pulmonary fibrosis progression through the regulation of lipid metabolic reprogramming by ROS/HIF-1α. Cellular Signalling, 2024, 117. [CrossRef]
- Calvisi, D.F; Wang, C.M.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology, 2011, 140(3), 1071-U1542. [CrossRef] [PubMed Central]
- Li, L.; Wang, C.M.; Calvisi, D.F.; Evert, M.; Pilo, M.G.; Jiang, L.J.; Yuneva, M.; Chen, X. SCD1 expression is dispensable for hepatocarcinogenesis induced by AKT and Ras oncogenes in mice. Plos One, 2013, 8(9). [CrossRef] [PubMed Central]
- Jiang, X.J.; Stockwell, B.R.; Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nature Reviews Molecular Cell Biology, 2021, 22(4), 266-282. [CrossRef] [PubMed Central]
- Tasdogan, A.; McFadden, D.G.; Mishra, P. Mitochondrial DNA haplotypes as genetic modifiers of cancer. Trends in Cancer, 2020, 6(12), 1044-1058. [CrossRef] [PubMed]
- Kim, W.; Deik, A.; Gonzalez, C.; Gonzalez, M.E.; Fu, F.F.; Ferrari, M.; Churchhouse, C.L.; Florez, J.C.; Jacobs, S.B.R.; Rhee, E. P. Polyunsaturated fatty acid desaturation is a mechanism for glycolytic NAD recycling. Cell Metabolism, 2019, 29(4), 856-+. [CrossRef] [PubMed Central]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. L. Broadening horizons: the role of ferroptosis in cancer. Nature Reviews Clinical Oncology, 2021, 18(5), 280-296. [CrossRef] [PubMed]
- Alistar, A.; Morris, B.M.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; Zachar, Z,; Bingham, P,M.; et al. (2017). Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncology, 2017, 18(6), 770-778. [CrossRef] [PubMed Central]
- Walther, T.C.; Farese, R.V. Lipid droplets and cellular lipid metabolism. In R. D. Kornberg (Ed.), Annual Review of Biochemistry,2012, Vol 81 (Vol. 81, pp. 687-714). [CrossRef] [PubMed Central]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



