
Submitted:
27 December 2024
Posted:
30 December 2024
You are already at the latest version
Abstract
Background/Objectives: Predicting the effects of mutations in proteins and DNA on the binding free energy of protein-DNA complexes is crucial for understanding how DNA variants impact wild-type cellular function. As many cellular interactions involve protein-DNA binding, accurately predicting changes in binding free energy (ΔΔG) is valuable for distinguishing pathogenic mutations from benign ones. Methods: The study describes the further development of the SAMPDI-3D machine learning method, which is trained on an expanded database of experimentally measured ΔΔGs. This enhanced model incorporates new features, including the 3D structure of the mutant protein, structural features of the mutant structure, and a position-specific scoring matrix (PSSM). Benchmarking was conducted using 5-fold cross-validation. Results: The updated SAMPDI-3D model achieves a Pearson Correlation Coefficient (PCC) of 0.68 for mutations in proteins and a PCC of 0.80 for mutations in DNA. These results represent significant improvements over existing tools. Additionally, the method’s rapid execution time enables genome-scale predictions. Conclusions: The advancements in SAMPDI-3D enhance its predictive performance and utility for analyzing mutations in protein-DNA complexes. By leveraging structural information and an expanded training dataset, SAMPDI-3D provides researchers with a more accurate and efficient tool for mutation analysis, contributing to the identification of pathogenic variants and improving our understanding of cellular function.
Keywords:
1. Introduction
2. Materials and Methods
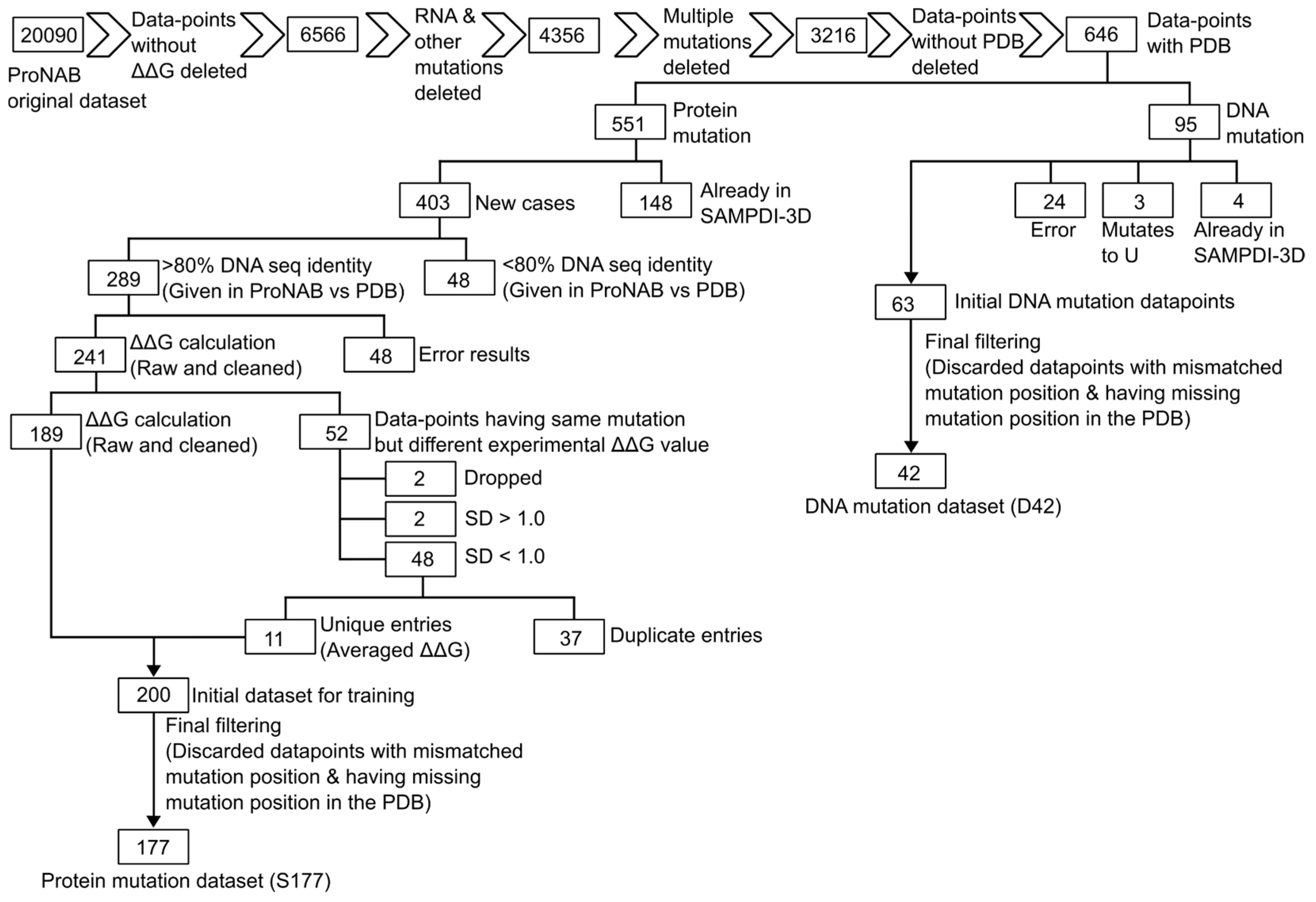
2.1. Data Cleaning (ProNAB)
2.2. Training Dataset for Protein Mutations
2.3. Training Dataset for DNA Mutations
2.4. Key Features in SAMPDI-3D v2 Machine Learning Model
2.4.1. Protein Mutation
- (a)
- Nucleotide Amino Acid contacts: Total number of interactions between the protein and the DNA.
- (b)
- Base amino acid hydrogen-bonds: Total number of hydrogen-bonds nucleotide bases in the DNA and protein residues.
- (c)
- Phosphate amino acid hydrogen bonds: Total number of hydrogen bonds between nucleotide phosphate and protein residues.
- (d)
- Base amino acid stacks: Total number of stacks identified in the protein-DNA complex structure between the nucleotide bases and protein residues.
2.4.2. DNA Mutation
2.5. Machine Learning Model Training
2.6. Selecting the Optimal Machine Learning Approach Using PyCaret
2.7. Hyperparameter Tuning and Advanced Model Training
3. Results
3.1. Dataset (Protein Mutations) Comparison of SAMPDI-3D and Newly Curated Dataset from ProNAB
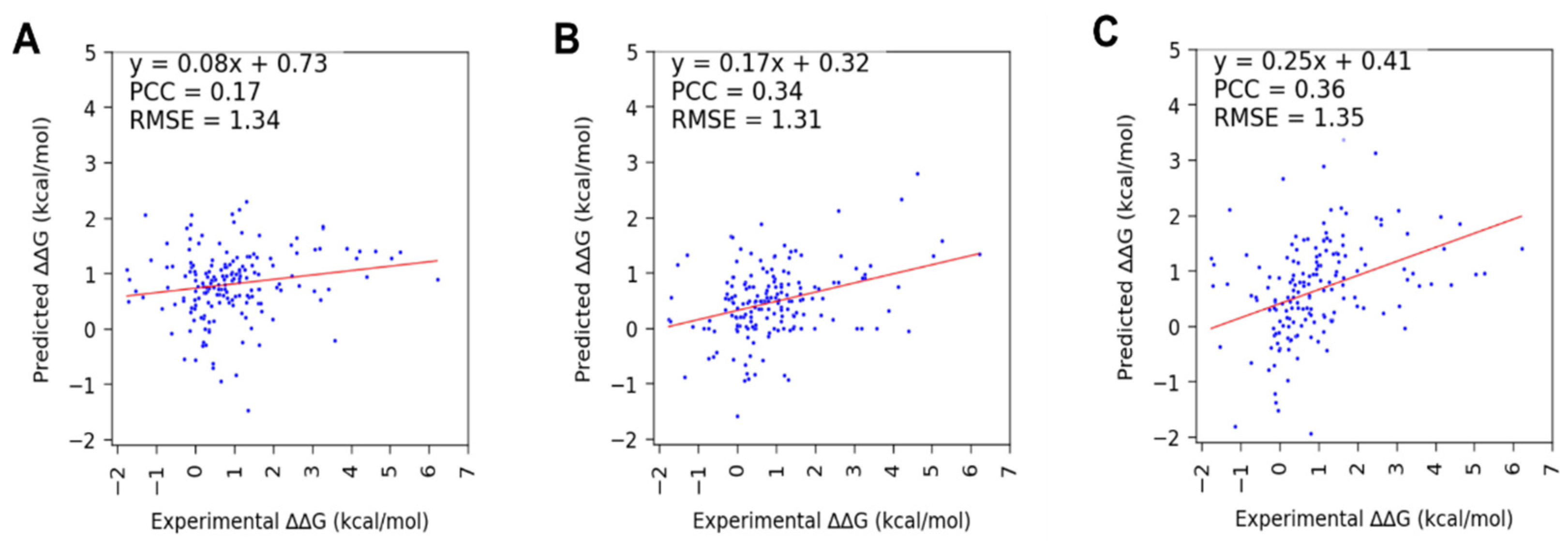
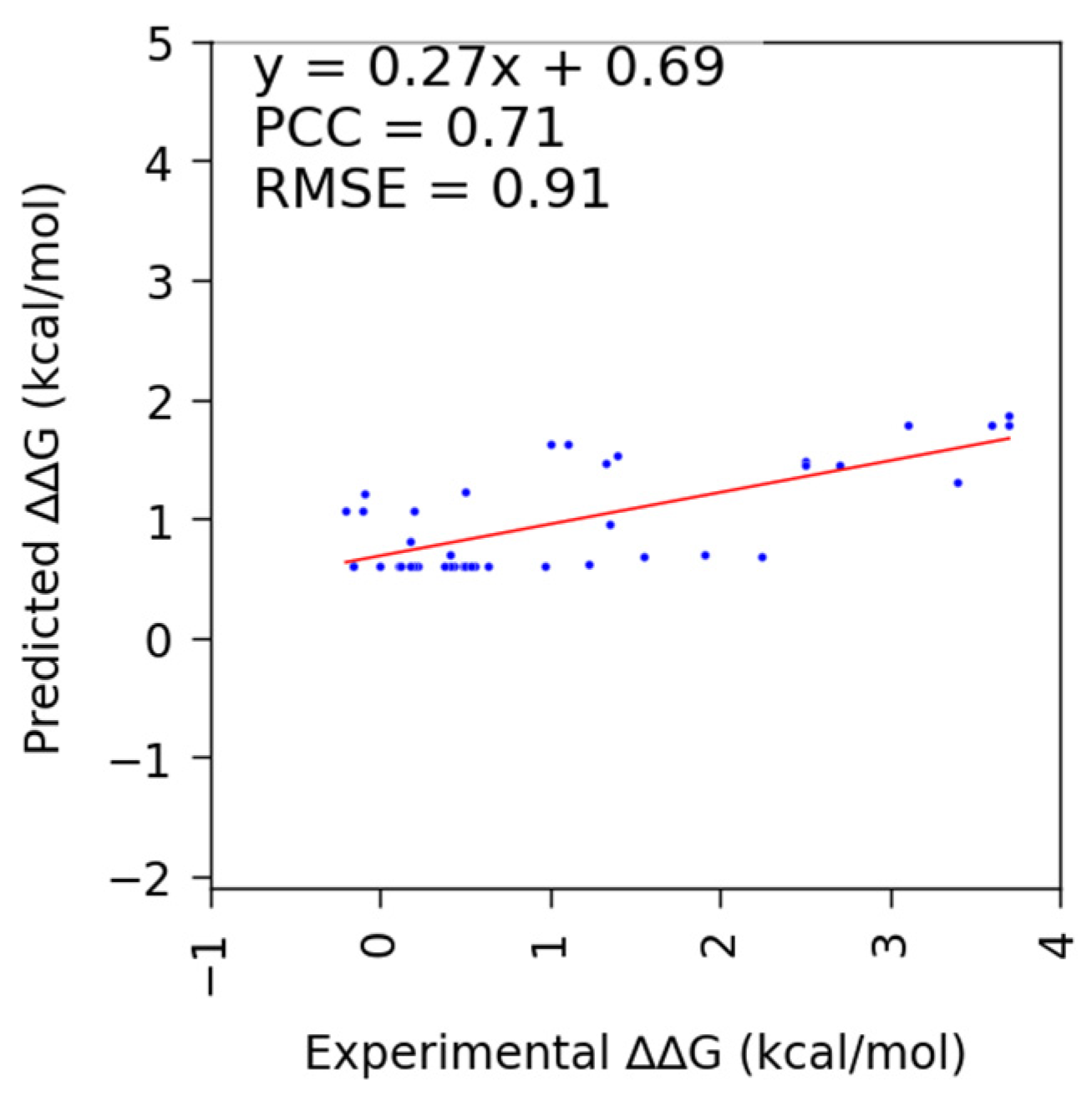
3.2. Performance of SAMPDI-3D and Other Available Methods on S177 and D42 (Newly Curated from ProNAB) Datasets
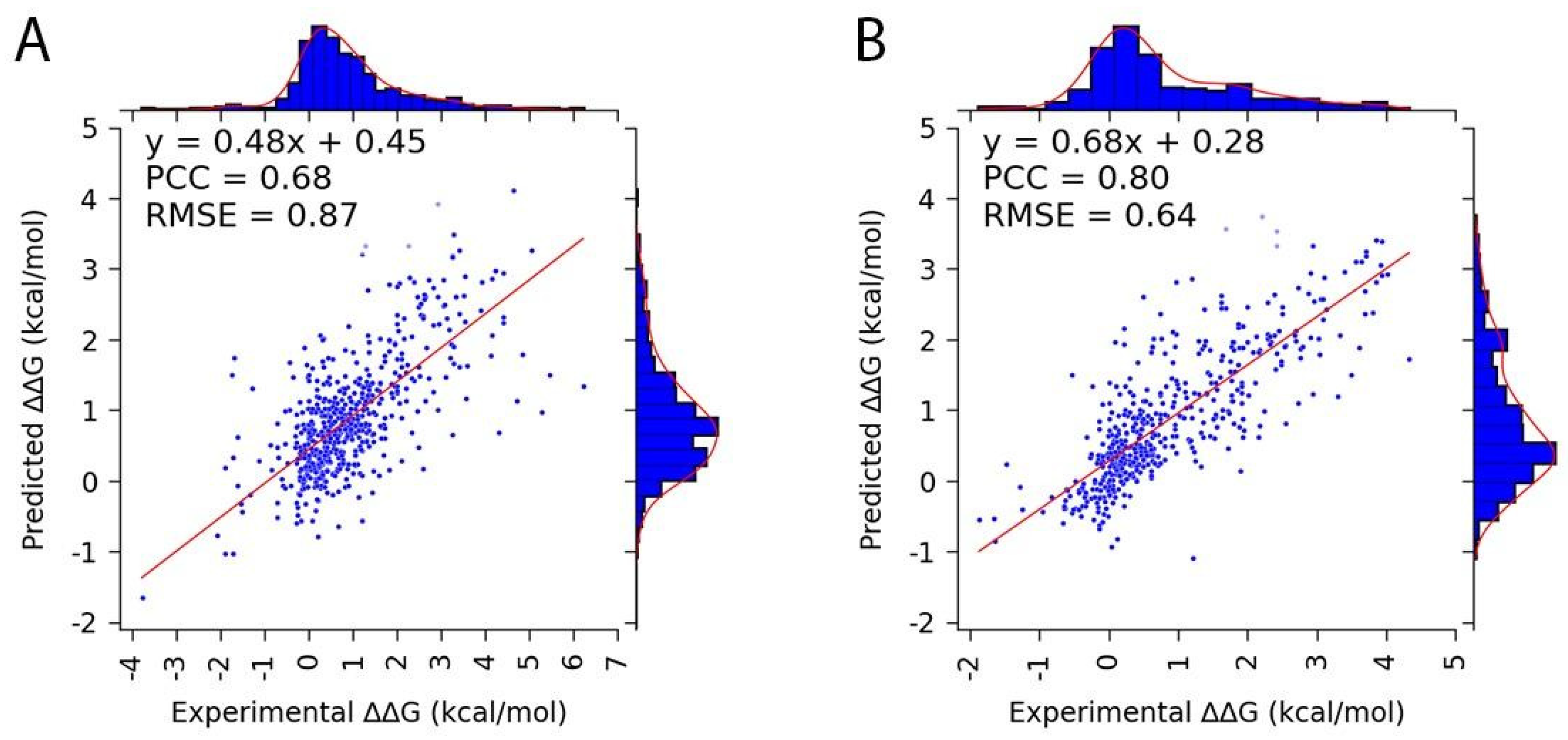
3.3. Performance of SAMPDI-3D v2 on Protein and DNA Mutation Databases
3.4. Web Server Implementation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCC | Pearson correlation coefficient |
| MSE | Mean squared error |
| RMSE | Root mean squared error |
References
- Bendel, A.M.; Faure, A.J.; Klein, D.; Shimada, K.; Lyautey, R.; Schiffelholz, N.; Kempf, G.; Cavadini, S.; Lehner, B.; Diss, G. The Genetic Architecture of Protein Interaction Affinity and Specificity. Nat. Commun. 2024, 15, 8868. [CrossRef]
- Vigneault, F.; Guérin, S.L. Regulation of Gene Expression: Probing DNA-Protein Interactions in Vivo and in Vitro. Expert Rev. Proteomics 2005, 2, 705–718. [CrossRef]
- Göös, H.; Kinnunen, M.; Salokas, K.; Tan, Z.; Liu, X.; Yadav, L.; Zhang, Q.; Wei, G.-H.; Varjosalo, M. Human Transcription Factor Protein Interaction Networks. Nat. Commun. 2022, 13, 766. [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [CrossRef]
- Aggarwal, B.D.; Calvi, B.R. Chromatin Regulates Origin Activity in Drosophila Follicle Cells. Nature 2004, 430, 372–376. [CrossRef]
- Wang, D.; Qian, X.; Sanchez-Solana, B.; Tripathi, B.K.; Durkin, M.E.; Lowy, D.R. Cancer-Associated Point Mutations in the DLC1 Tumor Suppressor and Other Rho-GAPs Occur Frequently and Are Associated with Decreased Function. Cancer Res. 2020, 80, 3568–3579. [CrossRef]
- Pifer, P.M.; Yates, E.A.; Legleiter, J. Point Mutations in Aβ Result in the Formation of Distinct Polymorphic Aggregates in the Presence of Lipid Bilayers. PloS One 2011, 6, e16248. [CrossRef]
- Kramers, C.; Danilov, S.M.; Deinum, J.; Balyasnikova, I.V.; Scharenborg, N.; Looman, M.; Boomsma, F.; de Keijzer, M.H.; van Duijn, C.; Martin, S.; et al. Point Mutation in the Stalk of Angiotensin-Converting Enzyme Causes a Dramatic Increase in Serum Angiotensin-Converting Enzyme but No Cardiovascular Disease. Circulation 2001, 104, 1236–1240. [CrossRef]
- Zeviani, M.; DiDonato, S. Neurological Disorders Due to Mutations of the Mitochondrial Genome. Neuromuscul. Disord. NMD 1991, 1, 165–172. [CrossRef]
- Calianese, D.C.; Noji, T.; Sullivan, J.A.; Schoch, K.; Shashi, V.; McNiven, V.; Ramos, L.L.P.; Jordanova, A.; Kárteszi, J.; Ishikita, H.; et al. Substrate Specificity Controlled by the Exit Site of Human P4-ATPases, Revealed by de Novo Point Mutations in Neurological Disorders. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2415755121. [CrossRef]
- Bi, M.; Su, W.; Li, J.; Mo, X. Insights into the Inhibition of Protospacer Integration via Direct Interaction between Cas2 and AcrVA5. Nat. Commun. 2024, 15, 3256. [CrossRef]
- Hellman, L.M.; Fried, M.G. Electrophoretic Mobility Shift Assay (EMSA) for Detecting Protein–Nucleic Acid Interactions. Nat. Protoc. 2007, 2, 1849–1861.
- Garner, M.M.; Revzin, A. A Gel Electrophoresis Method for Quantifying the Binding of Proteins to Specific DNA Regions: Application to Components of the Escherichia Coli Lactose Operon Regulatory System. Nucleic Acids Res. 1981, 9, 3047–3060. [CrossRef]
- Fried, M.; Crothers, D.M. Equilibria and Kinetics of Lac Repressor-Operator Interactions by Polyacrylamide Gel Electrophoresis. Nucleic Acids Res. 1981, 9, 6505–6525. [CrossRef]
- Freire, E.; Mayorga, O.L.; Straume, M. Isothermal Titration Calorimetry. Anal. Chem. 1990, 62, 950A-959A. [CrossRef]
- Velázquez-Campoy, A.; Ohtaka, H.; Nezami, A.; Muzammil, S.; Freire, E. Isothermal Titration Calorimetry. Curr. Protoc. Cell Biol. 2004, Chapter 17, Unit 17.8. [CrossRef]
- Bastos, M.; Abian, O.; Johnson, C.M.; Ferreira-da-Silva, F.; Vega, S.; Jimenez-Alesanco, A.; Ortega-Alarcon, D.; Velazquez-Campoy, A. Isothermal Titration Calorimetry. Nat. Rev. Methods Primer 2023, 3, 17. [CrossRef]
- Capelli, D.; Scognamiglio, V.; Montanari, R. Surface Plasmon Resonance Technology: Recent Advances, Applications and Experimental Cases. TrAC Trends Anal. Chem. 2023, 163, 117079, doi:. [CrossRef]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface Plasmon Resonance: A Versatile Technique for Biosensor Applications. Sensors 2015, 15, 10481–10510. [CrossRef]
- Lameirinhas, R.A.M.; Torres, J.P.N.; Baptista, A.; Martins, M.J.M. A New Method to Analyse the Role of Surface Plasmon Polaritons on Dielectric-Metal Interfaces. IEEE Photonics J. 2022, 14, 1–9.
- Berger, M.F.; Bulyk, M.L. Universal Protein-Binding Microarrays for the Comprehensive Characterization of the DNA-Binding Specificities of Transcription Factors. Nat. Protoc. 2009, 4, 393–411. [CrossRef]
- Berger, M.F.; Bulyk, M.L. Protein Binding Microarrays (PBMs) for Rapid, High-Throughput Characterization of the Sequence Specificities of DNA Binding Proteins. Methods Mol. Biol. Clifton NJ 2006, 338, 245–260. [CrossRef]
- Pantier, R.; Chhatbar, K.; Alston, G.; Lee, H.Y.; Bird, A. High-Throughput Sequencing SELEX for the Determination of DNA-Binding Protein Specificities in Vitro. STAR Protoc. 2022, 3, 101490. [CrossRef]
- Biedner, B.; Yassur, Y. Effect of Resection of Lateral Rectus Muscle in Undercorrected Esotropia. Ophthalmol. J. Int. Ophtalmol. Int. J. Ophthalmol. Z. Augenheilkd. 1987, 195, 45–48. [CrossRef]
- Rastogi, C.; Rube, H.T.; Kribelbauer, J.F.; Crocker, J.; Loker, R.E.; Martini, G.D.; Laptenko, O.; Freed-Pastor, W.A.; Prives, C.; Stern, D.L.; et al. Accurate and Sensitive Quantification of Protein-DNA Binding Affinity. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E3692–E3701. [CrossRef]
- Dantas Machado, A.C.; Cooper, B.H.; Lei, X.; Di Felice, R.; Chen, L.; Rohs, R. Landscape of DNA Binding Signatures of Myocyte Enhancer Factor-2B Reveals a Unique Interplay of Base and Shape Readout. Nucleic Acids Res. 2020, 48, 8529–8544. [CrossRef]
- Zhao, Y.; Ruan, S.; Pandey, M.; Stormo, G.D. Improved Models for Transcription Factor Binding Site Identification Using Nonindependent Interactions. Genetics 2012, 191, 781–790. [CrossRef]
- Li, G.; Panday, S.K.; Peng, Y.; Alexov, E. SAMPDI-3D: Predicting the Effects of Protein and DNA Mutations on Protein–DNA Interactions. Bioinformatics 2021, 37, 3760–3765. [CrossRef]
- Peng, Y.; Sun, L.; Jia, Z.; Li, L.; Alexov, E. Predicting Protein-DNA Binding Free Energy Change upon Missense Mutations Using Modified MM/PBSA Approach: SAMPDI Webserver. Bioinforma. Oxf. Engl. 2018, 34, 779–786. [CrossRef]
- Pires, D.E.V.; Ascher, D.B. mCSM-NA: Predicting the Effects of Mutations on Protein-Nucleic Acids Interactions. Nucleic Acids Res. 2017, 45, W241–W246. [CrossRef]
- Nguyen, T.B.; Myung, Y.; de Sá, A.G.C.; Pires, D.E.V.; Ascher, D.B. mmCSM-NA: Accurately Predicting Effects of Single and Multiple Mutations on Protein-Nucleic Acid Binding Affinity. NAR Genomics Bioinforma. 2021, 3, lqab109–lqab109. [CrossRef]
- Zhang, N.; Chen, Y.; Zhao, F.; Yang, Q.; Simonetti, F.L.; Li, M. PremPDI Estimates and Interprets the Effects of Missense Mutations on Protein-DNA Interactions. PLOS Comput. Biol. 2018, 14, e1006615. [CrossRef]
- Si-Rui Xiao; Yao-Kun Zhang; Kai-Yu Liu; Yu-Xiang Huang; Rong Liu PNBACE: An Ensemble Algorithm to Predict the Effects of Mutations on Protein-Nucleic Acid Binding Affinity. BMC Biol. 2024. [CrossRef]
- Harini, K.; Srivastava, A.; Kulandaisamy, A.; Gromiha, M.M. ProNAB: Database for Binding Affinities of Protein–Nucleic Acid Complexes and Their Mutants. Nucleic Acids Res. 2022, 50, D1528–D1534. [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [CrossRef]
- Prabakaran, P.; An, J.; Gromiha, M.M.; Selvaraj, S.; Uedaira, H.; Kono, H.; Sarai, A. Thermodynamic Database for Protein-Nucleic Acid Interactions (ProNIT). Bioinforma. Oxf. Engl. 2001, 17, 1027–1034. [CrossRef]
- Liu, L.; Xiong, Y.; Gao, H.; Wei, D.-Q.; Mitchell, J.C.; Zhu, X. dbAMEPNI: A Database of Alanine Mutagenic Effects for Protein–Nucleic Acid Interactions. Database 2018, 2018. [CrossRef]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and Non-Redundant UniProt Reference Clusters. Bioinformatics 2007, 23, 1282–1288. [CrossRef]
- Schäffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the Accuracy of PSI-BLAST Protein Database Searches with Composition-Based Statistics and Other Refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [CrossRef]
- Moon, C.P.; Fleming, K.G. Side-Chain Hydrophobicity Scale Derived from Transmembrane Protein Folding into Lipid Bilayers. Proc. Natl. Acad. Sci. 2011, 108, 10174–10177. [CrossRef]
- Shapovalov, M.V.; Dunbrack Jr., R.L. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure 2011, 19, 844–858. [CrossRef]
- Pommié, C.; Levadoux, S.; Sabatier, R.; Lefranc, G.; Lefranc, M.-P. IMGT Standardized Criteria for Statistical Analysis of Immunoglobulin V-REGION Amino Acid Properties. J. Mol. Recognit. JMR 2004, 17, 17–32. [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A Series of PDB-Related Databanks for Everyday Needs. Nucleic Acids Res. 2015, 43, D364–D368. [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637, doi:. [CrossRef]
- Lu, X.-J.; Bussemaker, H.J.; Olson, W.K. DSSR: An Integrated Software Tool for Dissecting the Spatial Structure of RNA. Nucleic Acids Res. 2015, 43, e142–e142. [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack Jr., R.L. Improved Prediction of Protein Side-Chain Conformations with SCWRL4. Proteins Struct. Funct. Bioinforma. 2009, 77, 778–795, doi:. [CrossRef]
- Ali, M. PyCaret: An Open Source, Low-Code Machine Learning Library in Python 2020.
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794.

| Model | PCC | RMSE (kcal/mol) |
|---|---|---|
| CatBoost Regressor | 0.65 | 1.32 |
| Extra Trees Regressor | 0.65 | 1.28 |
| Gradient Boosting Regressor | 0.64 | 1.16 |
| Light Gradient Boosting Machine | 0.64 | 1.24 |
| Random Forest Regressor | 0.63 | 1.28 |
| Extreme Gradient Boosting | 0.63 | 1.28 |
| AdaBoost Regressor | 0.61 | 1.30 |
| Linear Regression | 0.46 | 1.46 |
| Ridge Regression | 0.43 | 1.49 |
| Huber Regressor | 0.32 | 0.56 |
| Model | PCC | RMSE (kcal/mol) |
|---|---|---|
| CatBoost Regressor | 0.71 | 0.75 |
| Random Forest Regressor | 0.69 | 0.77 |
| Extra Trees Regressor | 0.68 | 0.79 |
| Gradient Boosting Regressor | 0.67 | 0.79 |
| Extreme Gradient Boosting | 0.67 | 0.79 |
| Light Gradient Boosting Machine | 0.65 | 0.81 |
| K Neighbors Regressor | 0.62 | 0.84 |
| AdaBoost Regressor | 0.59 | 0.86 |
| Decision Tree Regressor | 0.45 | 0.94 |
| Decision Tree Regressor | 0.45 | 0.98 |
| Model | PCC (Best Iteration) |
RMSE (kcal/mol) (Best Iteration) |
|---|---|---|
| Extreme Gradient Boosting | 0.67 | 0.89 |
| CatBoost Regressor | 0.66 | 0.91 |
| Gradient Boosting Regressor | 0.66 | 0.89 |
| Extra Trees Regressor | 0.65 | 0.91 |
| Light Gradient Boosting Machine | 0.65 | 0.9 |
| AdaBoost Regressor | 0.64 | 0.92 |
| Random Forest Regressor | 0.63 | 0.91 |
| Models | PCC (Best Iteration) |
RMSE (kcal/mol) (Best Iteration) |
|---|---|---|
| Extreme Gradient Boosting | 0.78 | 0.69 |
| CatBoost Regressor | 0.77 | 0.69 |
| Light Gradient Boosting Machine | 0.77 | 0.7 |
| Random Forest Regressor | 0.76 | 0.7 |
| Extra Trees Regressor | 0.76 | 0.7 |
| Gradient Boosting Regressor | 0.76 | 0.7 |
| K Neighbors Regressor | 0.74 | 0.73 |
| Mutated Residue | S419 (Count) | S419 (%) | S177 (Count) | S177 (%) |
|---|---|---|---|---|
| Alanine (A) | 296 | 70.64 | 108 | 61.02 |
| Cysteine (C) | 6 | 1.43 | 0 | 0 |
| Aspartic acid (D) | 5 | 1.19 | 4 | 2.26 |
| Glutamic acid (E) | 6 | 1.43 | 13 | 7.34 |
| Phenylalanine (F) | 9 | 2.15 | 4 | 2.26 |
| Glycine (G) | 9 | 2.15 | 7 | 3.95 |
| Histidine (H) | 6 | 1.43 | 1 | 0.56 |
| Isoleucine (I) | 2 | 0.48 | 0 | 0 |
| Lysine (K) | 13 | 3.1 | 5 | 2.82 |
| Leucine (L) | 12 | 2.86 | 4 | 2.26 |
| Methionine (M) | 6 | 1.43 | 7 | 3.95 |
| Asparagine (N) | 5 | 1.19 | 3 | 1.69 |
| Proline (P) | 2 | 0.48 | 2 | 1.13 |
| Glutamine (Q) | 7 | 1.67 | 5 | 2.82 |
| Arginine (R) | 9 | 2.15 | 3 | 1.69 |
| Serine (S) | 9 | 2.15 | 5 | 2.82 |
| Threonine (T) | 6 | 1.43 | 3 | 1.69 |
| Valine (V) | 7 | 1.67 | 3 | 1.69 |
| Tryptophan (W) | 1 | 0.24 | 0 | 0 |
| Tyrosine (Y) | 3 | 0.72 | 0 | 0 |
| Mutation | Method | PCC | RMSE |
| Protein | SAMPDI-3D | 0.17 | 1.34 |
| mCSM-NA | 0.34 | 1.31 | |
| PremPDI | 0.36 | 1.35 | |
| DNA | SAMPDI-3D | 0.71 | 0.91 |
| Mutation | PCC (Best Iteration) |
Average PCC (50 Iterations) |
Number of Features |
| Protein | 0.68 | 0.65 ± 0.05 | 49 |
| DNA | 0.80 | 0.77 ± 0.06 | 35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



