Submitted:
18 December 2024
Posted:
26 December 2024
Read the latest preprint version here
Preprints on COVID-19 and SARS-CoV-2
Abstract
Mitochondrial flux, the processes by which the mitochondrial compartment of the cell fuses, fissions and is degraded, underlies a complex spectrum of diseases. Pathological functioning of the innate immune system is underpinned by signalling pathways previously studied for their role reconfiguring cellular metabolic requirements. This review attempts to establish the physiologic state of mitochondrial flux across the cell, explain the process and regulation of mitochondrial biogenesis and autophagic degradation, before examining how disruption of these processes is intrinsic to a number of disease states. Particular attention is paid to how bioenergetic demands of the tissues are modulated in viral SARS-CoV, hepatitis B & C infection and asthma.
Keywords:
Mitochondrial flux
; mitophagy
; autophagy
; innate immunity
; OXPHOS
; metabolism
; asthma
; SARS-CoV-2
1. Introduction
Mitochondria are the primary bioenergetic organelles of cellular metabolism and their function underlies much of the metabolic activity throughout the organism. While their roles in oxidative phosphorylation (OXPHOS), the tricarboxylic acid (TCA) cycle, electron transport chain (ETC), production of reactive oxygen species (ROS) and oxidative or age-related damage are most widely understood, they also hold diverse roles as constituent components of the innate immune system. This review first approaches the role of mitochondrial flux as a central feature of the physiologic mitochondrial compartment – the continuously fusing and fissioning mitochondrial component of the cell – before explaining mechanisms of mitochondrial biogenesis and autophagy. After detailing the regulatory mechanisms that control this process, these topics are explored in the context of specific diseases.
2. The physiologic Mitochondrion
2.1. Mitochondrial Flux is an Essential Feature of the Mitochondrial Compartment
The physiological state of the mitochondrial compartment of the cell is one of flux. Mitochondrial morphology is heterogeneous and depends on the phenotype and function of the tissue, ranging from dense reticular networks in cells with high OXPHOS requirements to more discrete, small, separated organelles in tissues dependent on anaerobic glycolysis (1,2). It is a dynamic process whereby mitochondria continuously undergo fusion and fission, the “right” configuration being controlled by cellular and extracellular factors, and it is essential to the normal function of the cell.
Fission protein 1 (Fis1) acting in conjunction with dynamin related peptide 1 (Drp1) are the primary fission regulating peptides of the outer membrane. While Fis1 is an outer membrane protein, Drp1 is a cytosolic peptide recruited to the outer mitochondrial membrane (3,4). Excessive Drp1 activation has been implicated in autosomal recessive juvenile Parkinsonism, which results from failure of mitochondrial fission processes and impaired mitophagy (5,6). Fusion is controlled mainly by GTPases mitofusin 1 and 2 (Mfn1, Mfn2) in the outer membrane and optic atrophy protein 1 (Opa1, also known as Mgm1) in the inner membrane. Their dysfunction has been implicated in diseases such as dominant optic atrophy (7) and Charcot-Marie-Tooth 2A (8).
Morphologically, failure of fusion proteins results in fragmentation of the mitochondrial compartment and disorder of the cellular OXPHOS system (9–11). Cells requiring a highly energy-dense profile display a rich, closely spaced reticular mitochondrial network that provides a key highway for metabolite and waste transport while allowing continued traffic of mitochondrial proteins that are encoded by nuclear genomes (12). Mitochondrial flux is controlled by two distinct and opposing processes – mitochondrial biogenesis (mitogenesis) and autophagy of defective mitochondria (mitophagy).
2.2. Mitogenesis
Mitochondrial biogenesis is a complex process requiring extensive crosstalk between nuclear and mitochondrial apparatus. The mitochondrial genome consists of around 16.5 thousand base pairs. 13 peptides are transcribed from mitochondrial DNA (mtDNA) and produced within the mitochondria: 7 of the 45 ETC complex I components, 1 component of the 11 comprising ETC complex III, 3 of the 13 comprising ETC complex IV and 2 of the 18 required for ETC complex V. A further 22 tRNA and 2 mitochondrial ribosomal RNA genes are also present, without which mitochondrial protein transcription and translation could not take place (13). The several thousand other components required to produce functioning mitochondria are derived from nuclear-encoded proteins which must be transcribed, translated, processed, and transported into the mitochondria.
mtDNA is housed within protein nucleoids consisting of high mobility group (HMG) proteins, particularly mitochondrial transcription factor A (TFAM). These proteins assist in structural stability, replication and transcription and are essential to mitochondrial biogenesis (14,15). This is conducted by a mtDNA replication apparatus which, while described in the literature, is nevertheless poorly understood. mtDNA polymerase (Polγ) is a nuclear encoded protein composed of two subunits, encoded by genes POLG (held at locus 15q25) and POLG2 (held at locus 17q24.1). The process of mtDNA transcription is highly conserved in eukaryotes and is upregulated when cellular OXPHOS demands are increased. Pol γ acts with Twinkle (the mtDNA helicase) and mitochondrial single stranded DNA binding protein (mtSSB) to initiate mtDNA replication (16,17). Failure of Pol γ results in a variety of monogenic hereditary and sporadic disorders including microcerebrohepatopathy, Alpers-Huttenlocher syndrome (a triad of neurodevelopmental regression, seizures and liver failure) and progressive external ophthalmoplegia. Characteristic molecular features of these diseases include multiple mtDNA deletions, cytochrome c deficiency within muscle fibres and gross depletion of mtDNA throughout the body (18,19). Germline failure of mtSSB is lethal in utero while localised inactivation in myocardial tissue results in cardiomyopathy in murine models (17).
2.3. Mitophagy
Mitophagy is the autophagic process by which mitochondria are degraded and recycled into their component parts. When mitochondria malfunction, damage associated molecular patterns (DAMPs) such as mtDNA and ROS are produced. Recognised by a host of pattern recognition receptors (PRRs), defective mitochondria are then selectively sequestered for disposal via lysosomal degradation (20).
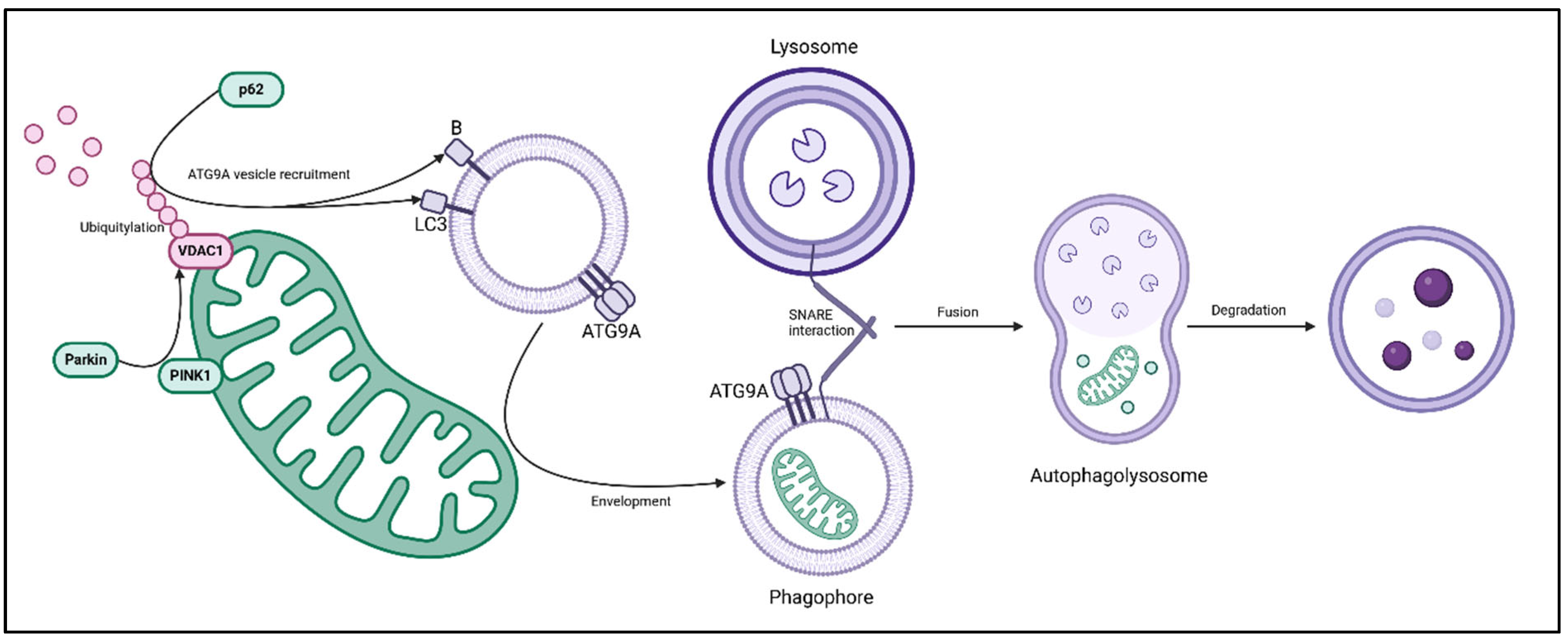
Phosphatase and tensin homolog (PTEN) induced kinase 1 (PINK1) – Parkin interaction constitutes the classic mitophagy pathway. PINK1 is a serine/threonine kinase containing mitochondrial targeting sequence, bearing mitochondrial membrane side and a cytoplasmic facing kinase (21), and it is activated by depolarisation of the mitochondrial membrane (22,23). Parkin (so named for its aetiologic role in the pathogenesis of several variants of autosomal recessive hereditary Parkinson disease (5,6)) is a cytosolic E3 ubiquitin ligase which locates to the mitochondrial membrane when activated by PINK1. Activated Parkin chain-ubiquitylates N-terminally fused lysine residues on voltage dependent anion channel 1 (VDAC1) (21,22), along with Mfn1 and Mfn2 (21,24,25), resulting in the recruitment of autophagic adaptor protein p62 (also referred to as sequestrome-1 or SQSTM1). p62 is the link between the ubiquitylation pathway proteins PINK1-Parkin and the autophagy related (ATG) proteins subsequently responsible for vesicle interactions and lysosomal envelopment of the organelle. p62 interacts with ATG8 homologues LC3 and B of the phagophore vesicle complex (26), allowing the defective organelle to be enveloped by the expanding phagophore membrane. At this point, activation of ATG9a allows fusion of the phagophore with the lysosome via SNARE-SNARE protein interaction (27), resulting in the introduction of proteolytic enzymes and organelle degradation within the autolysosome (28), as described in Figure 1.
Parkin independent mitophagy is less well explored and appears to function via proteins involved in physiologic mitochondrial flux. Parkin knockout cells with defective mitochondria produce mitophagy by PINK1 interaction with Drp1, resulting in fracture of the outer mitochondrial membrane and chain-ubiquitylation of the inner membrane (29). In Drp1 and Opa1 knockout (“mitochondrial stasis”) hepatocytes, p62 recruits cullin-RING scaffold protein associated ubiquitin ligases Keap1 and Rbx1, allowing an alternative PINK1-Parkin independent method of ubiquitylation (30). An alternative method has been demonstrated in hypoxic cells. BNip3 and Nix (members of the Bcl-2 pro-apoptotic protein family (31)) prevent hypoxia inducible factor 1α (HIF-1α) stabilisation, resulting in the degradation of Mfn1, Mfn2 and translocase of outer membrane protein 20 (TOM20). This prevents fusion, continued use of damaged organelle segments and failure of mitochondrial membrane integrity (32). It remains to be seen whether Parkin independent mitophagy pathways occur concurrently to classic PINK1-Parkin autophagy or are truly independent of it and only occur when components are defective.
Phosphase and tensin homolog induced kinase 1 (PINK1) is a mitochondrial membrane bound protein. On depolarisation of the mitochondrial membrane, PINK1 recruits cytoplasmic Parkin protein, an E3 ubiquitin ligase. When activated, Parkin chain-ubiquitylates lysine residues on voltage dependent anion channel 1 (VDAC1). This signals p62, which interacts with autophagy related protein 8 (ATG8) homologues LC3 and B, resulting in envelopment of the defective organelle within an ATG9A phagophore. SNARE-SNARE interaction with lysosomes causes phagophore-lysosomal fusion and results in the formation of the autophagolysosome, the contents of which are subsequently degraded. (Created in BioRender.com)
3. Regulation of Flux and Metabolism
3.1. The PPAR-PGC-1 Axis
Nuclear transcription factors peroxisome proliferator-activated receptors (PPAR) hold a central role in determining the metabolic profile of cells and tissues. PPAR isoforms have heterogenous expression throughout the organism, their different localisation allowing fine control of the metabolic needs of the tissue (33). Activated PPARγ and PPARβ stimulate nuclear transcription of OXPHOS proteins and ETC subunits, along with stimulating mitochondrial transcription of mtDNA encoded genes (34). Nuclear stimulators of PPARγ include thyroid hormone T3 (35–37), prostaglandin J2 (33), long chain fatty acids (38) and thiazolidinedione pharmaceuticals such as rosiglitazone (39). Within the nucleus PPARs exist as a heterodimer bound to retinoid X receptor (RXR) which, on ligand activation, binds PPAR-responsive regulatory elements within the genome activating transcription (40). With regards to PPARγ (and to a certain extent PPARα (41)), this includes fatty acid binding protein 2 (aP2), acyl-CoA binding protein (ACBP), lipoprotein lipase (LPL), fatty acid translocase/transport protein (FAT/FATP also known as cluster of differentiation 36 or CD36), acyl-CoA synthetase (ACS), glycerol kinase (GyK), insulin receptor substates 1 and 2 (IRS1 and IRS2) and glucose transporter 4 (Glut4). The net result of this is increased mobilisation of fats and fatty acids, their import to the cell, processing into usable metabolites and eventually, oxidative phosphorylation (33). Acting via a similar mechanism, PPARδ acts in skeletal muscle to upregulate cytochrome c, uncoupling proteins 2 and 3 (UCP2 and UCP3) (42) along with TCA cycle proteins succinate dehydrogenase and citrate synthase, resulting in a shift towards muscle fibres with higher OXPHOS potential (43).
PPAR isoforms act with PPARγ coactivator 1 (PGC-1) to provide upregulation of diverse mitochondrial functions. In brown fat tissue, this is most prominently the upregulation of uncoupling protein 1 (UCP1), essential for thermogenesis (44). While PPARs act mainly to induce transcription of proteins responsible for lipid metabolism, the TCA cycle and OXPHOS components, PGC-1 is the primary nuclear stimulator of mitogenesis, ultimately marshalling over 1100 nuclear genes via several downstream transcription factors (45–47). Well studied targets of the PPARγ-PGC1 axis include nuclear respiratory factor 2 (NRF2), which activates TFAM to upregulate mtDNA replication and transcription (48), and UCP2 and paraoxonase-2 (PON2), which allow enhanced action to mitigate the production of reactive oxygen species produced within mitochondria (47).
3.2. The Role of AMPK, Sirtuins and mTOR
The major link element between lipid metabolism, TCA proteins, mitogenesis and mitophagy are NAD-dependent deacylase sirtuin proteins, themselves activated by AMP-protein kinase (AMPK) and mammalian target of rapamycin (mTOR). These pathways are highly conserved from single celled eukaryotes to mammals and have a complex role in the integration of nutrient sensing, metabolic demands and cell stress or damage, and their outcomes converge on mitophagy, mitogenesis and cell proliferative processes.
AMPK is activated when the AMP/ATP ratio increases within in the cell, indicating low balance of ready-use energy. AMPK is a major stimulator of mitochondrial flux, mitophagy and mitogenesis and it is through this mechanism that cells with low energy balance, increased ROS and stress act to repair and replace defective components of the mitochondrial compartment (49).
Sirtuins are responsible for deacylation of histone proteins and are heavily linked to ageing processes. Of the seven sirtuin homologues known in mammalian cells, Sirt2 has been best studied for its role in longevity and is primarily localised within the nucleolus where it promotes genome stability in mitotically active cells (50,51). Genome sequencing of Sirt3, 4 and 5 appear to show mitochondrial targeting signals and confocal laser scanning microscopy of proteins tagged with MitoTracker red show localisation within the mitochondrial compartment (52,53). Sirt3 translocates to the mitochondrial compartment from the nucleus at times of cellular stress, such as when DNA damage occurs (54). Deacylation targets of Sirt1, Sirt3 and Sirt5 include PGC-1α in the nucleus (55) and BNip3 (56), Opa1(57) and Parkin (58) in the mitochondria, while AMPK also activates PINK1 and Fis1 (59). PINK1 and the PINK1-Parkin pathway appear to play an important role in mediating mitochondrial flux – overexpression of PINK1 results in increased Drp1 activation and fragmentation of mitochondrial compartment while underexpression of PINK1 results in excessive fusion (60).
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase complex which in its active state stimulates ribosome biogenesis, protein synthesis and metabolism via the IRS-PI3K pathway, while inhibiting mitophagy. It is a major regulator of cellular growth and anabolism and, via interaction with a number of signalling pathways, responds to insulin stimulation and cellular energy levels (61). mTOR signalling has been well documented owing to interaction with apoptotic apparatus via the PI3K/Akt/mTOR signalling cascade (62). With regards to mitophagy, mTOR acta via the phosphorylation of tuberous sclerosis complex 2 (TSC2), maintained within endoplasmic reticular linkages to mitochondria, to negatively interact with FK506 binding protein 8 (FKBP8), which when active initiates LC3 vesicle-dependent autophagy (63). In nutrient starvation, AMPK inactivates mTOR, allowing unopposed FKBP8 action, upregulating mitophagy (64). Previously assumed to be separate processes, significant crosstalk exists between the PI3k/Akt/mTOR cascade and the unfolded protein response (UPR), which mediates metabolic and apoptotic response to endoplasmic reticular stress. Collectively, mTOR and the UPR provide the integration pathways to balance organismal metabolic requirements with nutrient availability. Unfavourable growth conditions or hypoxia produce UPR activation and mTOR inactivation, upregulating mitophagy, reducing the OXPHOS requirements of the cell, and balancing the metabolic requirements of the tissue with the nutrients available (65).
3.3. Direct Action Via Mitochondrial Receptors
Direct regulatory action on mitochondria is established in the literature, although poorly understood, and there remains much scope for future research. In macrophages, glucocorticoid action causes increased polarisation of the mitochondrial membrane, import of TCA cycle metabolites and upregulation of mitochondrial biogenesis (66). Steroid and thyroid hormone receptors are found within the mitochondrial matrix, all nuclear encoded yet allowing for direct endocrine action within the mitochondria. Two major mitochondrial isoforms of the glucocorticoid receptor (GR), GRα and GRβ have been detected, two isoforms of the oestrogen receptor (ER), ERα and ERβ along with two isoforms of the thyroid receptor c-erbAα and c-erbAβ (67).
New studies indicate that on ligand binding, cytoplasmic glucocorticoid receptors interact with pyruvate dehydrogenase complex proteins and translocate to within the mitochondrial compartment of the cell, where they upregulate TCA cycle activity (66). It was noted in 1977 that isolated rat mitochondria respond to thyroid hormone T3 stimulation with increased OXPHOS activity (68) and the presence of matrix receptors for these hormones explains this finding. In addition to acting on nuclear transcription factors, T3 and steroid hormone administration results in increased mitochondrial genome expression by acting on c-erbA receptors to activate TFAM, increasing transcription of mitochondrial protein subunits (35).
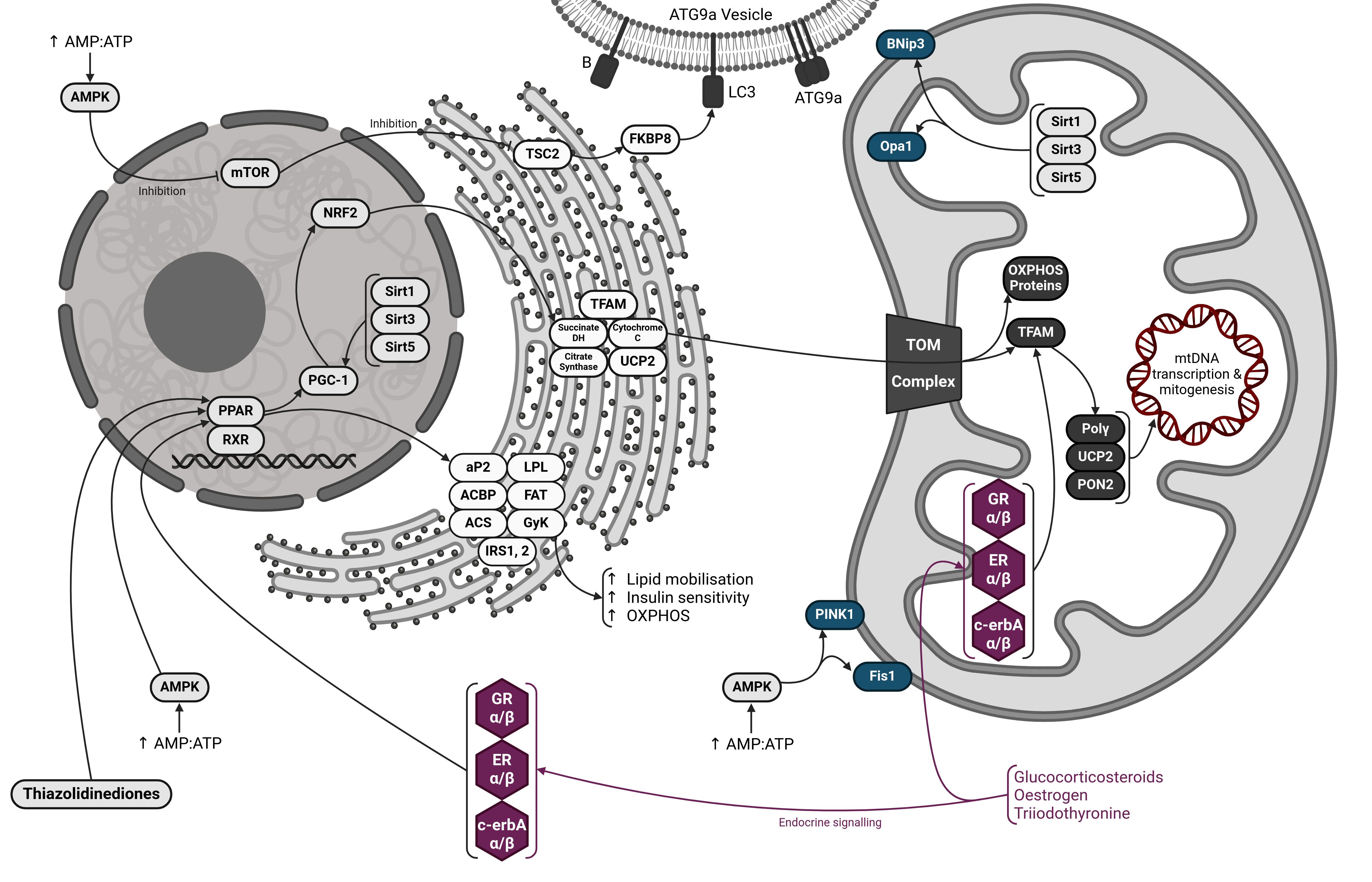
Figure 2.
Control of Flux, Mitophagy, Mitogenesis & Metabolism.
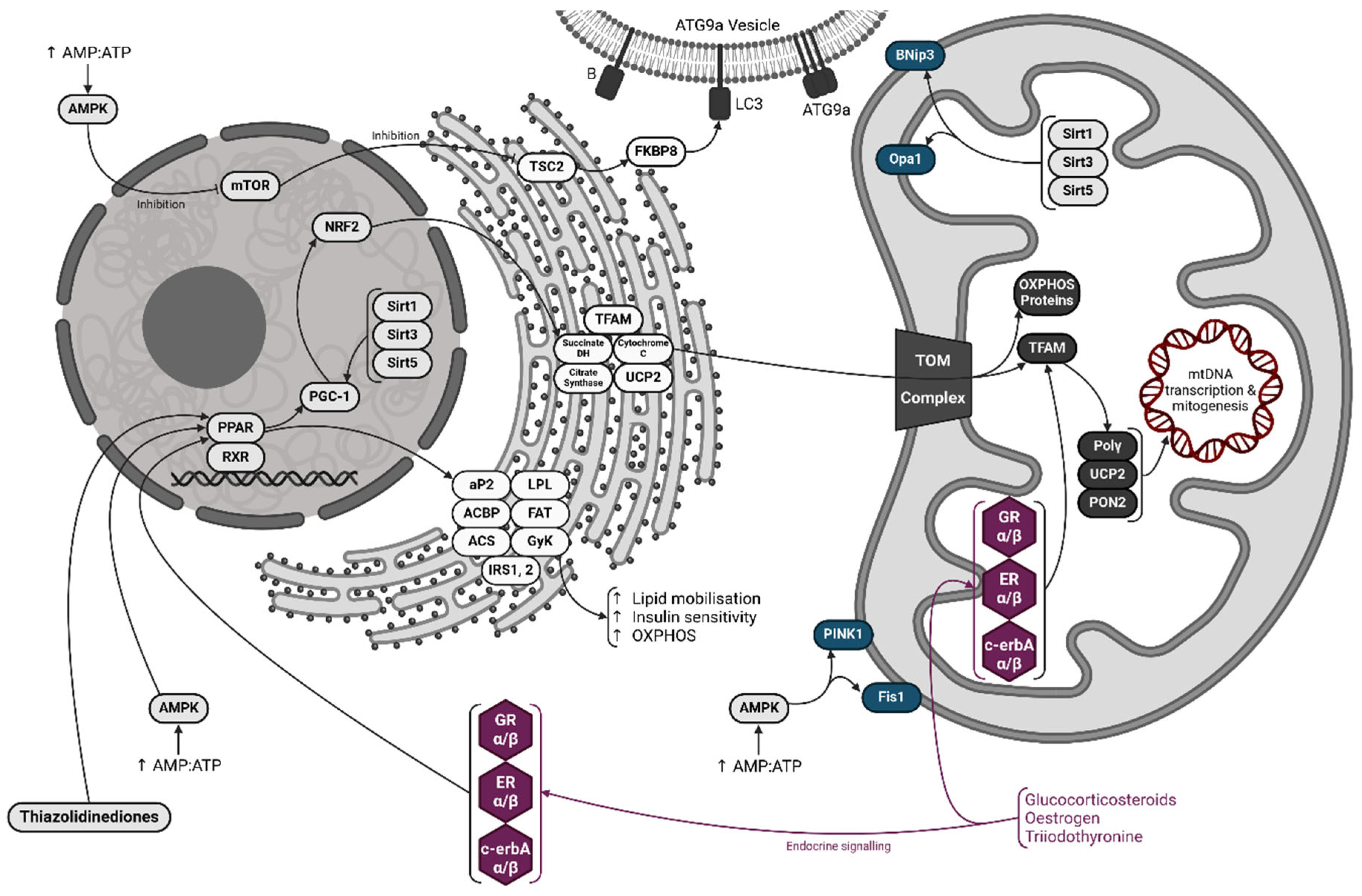
Peroxisome proliferator-activated receptors (PPARs) are nuclear transcription factors responsible for control of lipid metabolism, oxidative phosphorylation (OXPHOS) and mitochondrial biogenesis. On ligand binding, PPARs associate with the retinoic acid X receptor (RXR) and bind PPAR-associated regulatory elements of the genome. This stimulates the production of proteins such as fatty acid binding protein 2 (aP2), acyl-CoA binding protein (ACBP), lipoprotein lipase (LPL), fatty acid translocase/transport protein (FAT/FATP also known as cluster of differentiation 36 or CD36), acyl-CoA synthetase (ACS), glycerol kinase (GyK), insulin receptor substates 1 and 2 (IRS1 and IRS2). The net result of this is increased lipid mobilisation, insulin sensitivity and OXPHOS.
PPARs also control PPAR coactivator 1 (PGC-1) homologues, which are the master regulators of mitochondrial biogenesis. Acting via nuclear respiratory factor 2 (NRF2), they upregulate production of key OXPHOS proteins including succinate dehydrogenase (succinate DH), cytochrome c, citrate synthase and uncoupling protein 2 (UCP2). These proteins are conveyed to the mitochondrial compartment through dense networks of endoplasmic reticulum before entering the organelle through translocase of outer membrane (TOM) complexes. PGC-1 homologues also stimulate production of mitochondrial transcription factor A (TFAM), which upregulates mtDNA polymerase (Polγ). Along with UCP2 and paraoxonase-2 (PON2), Polγ increases replication and transcription of mitochondrial DNA (mtDNA), production of mtDNA encoded OXPHOS proteins and mitogenesis.
Adenosine monophosphate kinase (AMPK) is activated when the AMP:ATP ratio within the cell increases – a key indicator of low energy balance. AMPK is a modulator of lipid metabolism, mitochondrial flux and mitogenesis. Acting on PPARs, it increases lipid mobilisation, insulin sensitivity and OXPHOS; while downstream it also increases mtDNA replication and transcription via the PPAR-PGC-1 axis. Active AMPK also inhibits mammalian target of rapamycin (mTOR). In its resting state, mTOR constitutively inhibits tuberous sclerosis protein 2 (TSC2). When mTOR is inactivated by AMPK, TSC2 is free to interact with FK506 binding protein 8 (FKBP8), which is an activator of microtubule-associated protein 1A/1B-light Chain 3 (LC3), a membrane bound protein present in autophagy related protein 9a (ATG9a) vesicles, required for phagophore formation. Active AMPK also stimulates phosphase and tensin homolog induced kinase 1 (PINK1), a key component of PINK1-Parkin mitophagy, and fission protein 1 (Fis1), required for proper fission during mitochondrial flux. Increased flux and mitophagy augments increased mitogenesis, lipid mobilisation and OXPHOS – the mitochondrial component of the cell is renewed, balancing the metabolic capabilities with organismal requirements.
Sirtuin (Sirt) proteins are linked to ageing and longevity and in their core role deacylate histone proteins, allowing genes to be transcribed. In the nucleus Sirt1, 3 and 5 activate PGC-1 homologues, leading to increased mitogenesis. Within the mitochondria, their secondary role includes upregulating flux proteins optic atrophy 1 protein (Opa1) and Bcl-2 Interacting Protein 3 (BNip3).
Other than via AMPK or Sirt proteins, the systems can be activated by endocrine signalling by glucocorticosteroids, oestrogen or triiodothyronine (T3). Cytosolic glucocorticoid receptors (GRα and β), oestrogen receptors (ERα and β) and T3 receptors (c-erbAα and β) converge on PPAR signalling. Within the mitochondrion, they activate TFAM, allowing dual nuclear-mitochondrial activation and signalling for metabolic requirements. Thiazolidinediones, which are novel anti-diabetic therapies, also target the PPAR-PGC-1 axis, explaining their efficacy in weight loss and altering metabolic function. (Created in BioRender.com)
4. Mitochondria – A Component of the Innate Immune System
4.1. Inflammatory Cytokine Production
Mitochondrial flux and mitophagy play key roles in regulating inflammatory processes. Failure of the mitochondrial fusion-fission cycle results in depolarisation of the mitochondrial membrane, organelle swelling and the formation of “megamitochondria” (69–72). ROS induced damage and mtDNA are recognised as DAMPs, and their resultant leak into the cytoplasm and extracellular environment causes activation of inflammatory processes. The most important PRRs in mitochondrial contexts are Toll-like receptors (TLRs), particularly TLR9. TLR9 is responsive to mtDNA and expressed both intracellularly in membranous vesicles and extracellularly on macrophages (73). Of further note are retinoic acid inducible gene I (RIG-I), which binds non-cell double stranded DNA (dsDNA) (74), and melanoma differentiation associated protein 5 (MDA5), sensing non-cell RNA (75).
Secretion of pro-inflammatory IL-1β and IL-18 by macrophages is dependent on activation of the “inflammasome”, a cytoplasmic protein-complex responsible for cleavage and activation of caspase-1. “Canonical” inflammasomes consist of pro-caspase 1, an aspartate specific cysteine protease (ASC) adaptor protein and a PRR – in this context, nucleotide-binding oligomerisation domain-like receptors (NOD-like receptors or NLRs) (76). When the NLR is activated, pro-caspase 1 is converted to its active form, allowing enzymatic cleavage of pro-IL-1β and other cytokine progenitors to their active form (77). “Non-canonical” inflammasomes do not depend on NLRs and result in gasdermin D cleavage by caspase-11, allowing IL-1β secretion and pyroptosis (78,79).
Mitochondrial antiviral signalling protein (MAVS) plays a key role as an intermediate between nuclear transcription factors NFκB and interferon regulatory factors (IRFs) and the production of interferons during infection of the cell (74,75). In the resting state MAVS is bound to Mfn2 on the mitochondrial outer membrane. When the RIG-I and MDA5 complex responds to non-cell genetic material, they activate MAVS, which binds to TANK-binding kinase 1 (TBK1) and inhibitor of NFκB kinase ϵ (IKKϵ), translocating to the nucleus and activating IRFs to upregulate transcription of interferons. The cyclic GMP-AMP synthase – stimulator of interferon genes (cGAS-STING) pathway is a similar mechanism that responds to the presence of dsDNA converging directly on TBK1 and IKKϵ (80,81). TLR9 activation by mtDNA acts via adaptor protein MyD88 and a series of IKK adaptors to converge on NFκB, which when translocated to the nucleus upregulates transcription of pro-inflammatory cytokines TNFα, IL-1 and IL-6 (74).
While mitochondrial ROS have long been recognised as a potential trigger of NLRP inflammasomes, a constitutive level of ROS is also an essential component of the function of the innate immune system (73). Transcription of MAVS is negatively regulated by ROS levels, and constitutive production of ROS by NADPH oxidase enzymes appears to be required for RIG-I mediated activation of IRF3 and production of proinflammatory cytokines (82).
4.2. SARS-CoV-2 Infection and Post-Acute Sequelae
The role of canonical and non-canonical inflammasome activation under viral infection is now well understood, and its core role in SARS-Cov-2 inflammation provides greater understanding of the pathogenesis of COVID-19 while also suggesting novel treatment options. SARS-CoV-2 open reading frames (ORFs) are translated from positive-strand RNA and interfere with autophagic processes, disrupting Parkin ubiquitylation and autophagolysosome fusion. This resultant metabolic derangement, along with suppression of the interferon production RIG-I/MAVS and cGAS-STING pathways, causes failure of innate immune antiviral processes (83–86). Of deeper interest are complex interactions between viral entities and mitochondrial structural and transport proteins. RNA-GPS studies early in the COVID-19 pandemic predicted interactions between viral polypeptides and subunits of ETC complexes I, III and IV (87,88). Fluorescence microscopy located SARS-CoV-2 genetic material within the mitochondrial matrix while indicating a consequent failure of mitophagy (89). Viral accessory protein ORF3c has been demonstrated to utilise TOM20 to locate itself within the mitochondrial compartment of the cell where it too disrupts mitochondrial flux (85). Pathological studies have also found that viral genetic material and polypeptide components of SARS-CoV-2 infection persist within post-mortem specimens (90,91), at times for years (92). The implication is that elements of the SARS-CoV-2 virus can sequester themselves within the mitochondrial component of the organism to evade immune surveillance and response. The inflammatory response to viral invasion, accumulation of defective components of the mitochondrial compartment and failure of tissue OXPHOS systems forms a putative theory for the pathogenesis of post-acute sequelae in SARS-CoV-2 infection (83).
4.3. Disruption of Mitochondrial Innate Immunity in Hepatitis B & C Infection
Hepatitis B (HBV) is an encapsulated DNA virus, while hepatitis C (HCV) is an RNA virus, yet both result in persistent and progressive liver damage, the outcome of which is hepatic fibrosis, cirrhosis and death. HBV and HCV gain access to the liver via an organ-specific multi-receptor complex, after which the viral particles are unencapsulated, the nucleocapsid is released into the cytoplasm and the viral DNA (HBV) or positive sense viral RNA (HCV) can act as material for translation of viral peptides (93).
HBV enhances its replicative ability by hijacking mitophagic processes, initially by stimulating excessive organelle fission by promoting Drp1, and ultimately by the upregulation of the PINK1-Parkin axis and degradation of Mfn2. The outcome of this process is persistence of the infected cell beyond usual viability, reducing ROS production and maintaining limited OXPHOS activity while using glycolysis metabolites to provide useful substrates for virion production (94,95). In HCV, viral RNA is recognised by RIG-I and MDA5 PRRs, which trigger MAVS assisted IRF and NFκB based interferon response (96). Viral non-structural (NS) proteins NS3 and NS4 are serine proteases that render MAVS ineffective by cleaving it from Mfn2, thus preventing RIG-I/MDA5 signalling (97), while also interfering with essential cofactors of TLR signalling (98). Spread of the virus between hepatocytes results in a state of persistent inflammation in newly infected areas as pro-inflammatory pathways are activated, while chronically infected areas have interferon production suppressed by PRR interference (99).
HCV infection appears to increase mitophagy via an indirect route by suppression of mTOR, which when active suppresses mitophagy (100) or unfolded protein response (UPR) (101) pathways, although whether this occurs in HCV infection to the same extent as in HBV is controversial within the literature (102,103). The converse is true in alcoholic hepatitis (AH), where hepatocytes from AH patients display reduced Drp1 and inactive mitochondrial fission. Electron micrograph studies show the accumulation of megamitochondria, while metabolomic studies show increased ROS, cell stress, cytosolic mtDNA and activation of cGAS-STING interferon signalling (104).
4.4. Nascent Connections Between Mitophagy, Asthma and COPD
Asthma as a disease is characterised by chronic airway inflammation, respiratory symptoms such as coughing and wheezing, varying in intensity over time (105). Inappropriate mitophagy plays a role in the development of asthma in response to particulate matter. Genetic studies indicate that single nucleotide polymorphisms of ATG5 and SQSTM1 genes highly predispose to the development of asthma, while electron micrographs of bronchial biopsy material show increased autophagosome activity in patients with reduced lung function (106). The mTOR pathway appears to play a role in the development of asthma, as evidenced by murine models where mTOR blockade through rapamycin alleviates symptoms of inflammation. Asthmatic mice produced by allergic sensitisation using cigarette smoke extract showed significantly decreased neutrophil count and markers of inflammation when treated with intraperitoneal infusions of rapamycin (107). A similar study showed increased LC3 autophagosome formation in sensitised mice, with autophagy-related double-membrane vesicles more prominent in eosinophils from sensitised mice than in controls (108). Histopathological examination of human large airway epithelial tissue demonstrates increased expression of ATG5 protein in severe cases of asthma with airway remodelling, the changes in correspondence to the severity of the remodelling process (109). As the disease progresses, transforming growth factor β (TGF-β) signalling produces fibrosis while ultimately downregulating PINK1-Parkin autophagic processes, mitophagy and mitogenesis (110).
Similar effects have been demonstrated in COPD, and rapamycin and mTOR blockade remain targets of interest in development of pharmaceutical treatments for pulmonary inflammatory disease (111). The role of the PINK1-Parkin mitophagy pathway in pathogenesis was demonstrated by showing that PINK1 deficiency was protective against cell death and necroptosis in response to cigarette smoke in mouse models (112). A similar study demonstrated the LC3 deficient mice had reduced airspace enlargement in response to cigarette smoke extract than control animals (113).
5. Conclusions
The mitochondrial compartment plays a central role in our understanding of innate immunity. Rather than considering the metabolic aspects of mitochondria separate from their now well-defined role in the innate immune system, both roles are complementary and essential to the other’s proper function. The metabolic phenotype of a tissue is the outward expression of a configuration requirement demanded by systemic and local homeostasis, which is to say that the metabolic role of mitochondria underpin their function within the immune system. Pathological change in tissue is accompanied by change in the metabolic phenotype of component cells, underpinned by changes in the configuration of cellular mitochondria. To this end, we refer not to singular mitochondrion but the mitochondrial compartment of a tissue. This terminology encapsulates the dynamic nature of mitochondrial flux – a highly regulated continuum of fusion-fission events throughout which the bioenergetic and immune components of the cell can remain in balance with the demands of the organism.
Via a complex, cascading network of extra- and intracellular signals the OXPHOS metabolic component of the cell is modulated in response to stress, starvation, pathogenic invasion, and other modalities of disease. Certain invaders such as SARS-CoV-2, HBV and HCV have evolved to take advantage of this apparatus. Our innate antiviral response to both acute and chronic SARS-CoV-2 infection can be characterised as a battle of control over E3 ubiquitin ligases, as viral material attempts to sequester itself within defective organelles that are resistant to proper patterns of mitochondrial flux.
In asthma, autophagic processes are upregulated and correspond to disease severity. Previous treatment methods have focused on targeting inflammatory pathways in the hopes that this would ameliorate the symptoms. While in mild to moderate cases this may prove effective, severe asthma has both a marked deleterious effect on the individual and high mortality. Further studies elucidating the role of mTOR signalling, PINK1-Parkin autophagy and downstream LC3 and ATG5 mediated autophagosome activity are warranted. Greater understanding of the pathophysiology of asthma enables the development of novel therapeutic interventions that should revolutionise the treatment of a widespread and severely debilitating illness.
Author Contributions
CGW: conceptualisation, writing – original draft, writing – review & editing
Funding
Not applicable
Ethical approval
Not applicable
Consent to participate / publication
Not applicable
Availability of data and materials
Not applicable
Conflicts of Interest
The authors declare that there are no conflicts of interest
Abbreviations
- ACBP - Acyl-CoA Binding Protein
- AH - Alcoholic Hepatitis
- AMPK - AMP-Protein Kinase
- ATG - Autophagy-Related
- BNip3 - Bcl-2 Interacting Protein 3
- CD36 - Cluster of Differentiation 36 (also known as FAT/FATP – Fatty Acid Translocase/Transport Protein)
- cGAS - Cyclic GMP-AMP Synthase
- COPD - Chronic Obstructive Pulmonary Disease
- DAMPs - Damage Associated Molecular Patterns
- Drp1 - Dynamin-Related Protein 1
- ETC - Electron Transport Chain
- FAT/FATP - Fatty Acid Translocase/Transport Protein (also known as CD36)
- Fis1 - Fission Protein 1
- FKBP8 - FK506 Binding Protein 8
- Glut4 - Glucose Transporter 4
- GR - Glucocorticoid Receptor
- HCV - Hepatitis C Virus
- HIF-1α - Hypoxia Inducible Factor 1 Alpha
- HMG - High Mobility Group
- HBV - Hepatitis B Virus
- IKKϵ - Inhibitor of NFκB Kinase Epsilon
- IL - Interleukin
- IRFs - Interferon Regulatory Factors
- IRS - Insulin Receptor Substrate
- Keap1 - Kelch-like ECH-associated Protein 1
- LC3 - Microtubule-associated Protein 1A/1B-light Chain 3
- LPL - Lipoprotein Lipase
- MAVS - Mitochondrial Antiviral Signalling Protein
- MDA5 - Melanoma Differentiation Associated Protein 5
- Mfn1/Mfn2 - Mitofusin 1 / Mitofusin 2
- mtDNA - Mitochondrial DNA
- mtSSB - Mitochondrial Single-Stranded DNA Binding Protein
- mTOR - Mammalian Target of Rapamycin
- NFκB - Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells
- NLRs - Nucleotide-Binding Oligomerisation Domain-like Receptors
- NRF2 - Nuclear Respiratory Factor 2
- OXPHOS - Oxidative Phosphorylation
- PINK1 - PTEN Induced Kinase 1
- PON2 - Paraoxonase-2
- PPAR - Peroxisome Proliferator-Activated Receptor
- PRRs - Pattern Recognition Receptors
- RIG-I - Retinoic Acid Inducible Gene I
- ROS - Reactive Oxygen Species
- RXR - Retinoid X Receptor
- SARS-CoV - Severe Acute Respiratory Syndrome Coronavirus
- SNARE - Soluble NSF Attachment Protein Receptor
- SQSTM1 - Sequestrome-1 (also known as p62)
- TBK1 - TANK-binding Kinase 1
- TCA - Tricarboxylic Acid
- TFAM - Mitochondrial Transcription Factor A
- TLR - Toll-like Receptor
- TOM - Translocase of Outer Membrane
- TSC1/TSC2 - Tuberous Sclerosis Complex 1 / Tuberous Sclerosis Complex 2
- UCP - Uncoupling Protein
- UPR - Unfolded Protein Response
- VDAC1 - Voltage Dependent Anion Channel 1
References
- Bakeeva LE, Chentsov YuS null, Skulachev VP. Mitochondrial framework (reticulum mitochondriale) in rat diaphragm muscle. Biochim Biophys Acta. 1978 Mar 13;501(3):349–69.
- Kayar SR, Claassen H, Hoppeler H, Weibel ER. Mitochondrial distribution in relation to changes in muscle metabolism in rat soleus. Respir Physiol. 1986 Apr;64(1):1–11.
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003 Sep 19;278(38):36373–9.
- van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb Perspect Biol. 2013 Jun;5(6):a011072.
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998 Apr 9;392(6676):605–8.
- Valente EM, Abou-Sleiman PM, Caputo V; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Alexander C, Votruba M, Pesch UEA; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004 May;36(5):449–51.
- Meeusen SL, Nunnari J. How mitochondria fuse. Current Opinion in Cell Biology. 2005 Aug 1;17(4):389–94.
- Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, et al. Mitochondrial Inner-Membrane Fusion and Crista Maintenance Requires the Dynamin-Related GTPase Mgm1. Cell. 2006 Oct 20;127(2):383–95.
- Chan, DC. Dissecting Mitochondrial Fusion. Developmental Cell. 2006 Nov 1;11(5):592–4.
- Iqbal S, Ostojic O, Singh K, Joseph AM, Hood DA. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle & Nerve. 2013;48(6):963–70.
- Wallace, DC. Mitochondrial genetic medicine. Nat Genet. 2018 Dec;50(12):1642–9.
- MacAlpine DM, Perlman PS, Butow RA. The high mobility group protein Abf2p influences the level of yeast mitochondrial DNA recombination intermediates in vivo. Proc Natl Acad Sci U S A. 1998 Jun 9;95(12):6739–43.
- Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. 2011 Aug 16;108(33):13534–9.
- Copeland WC, Longley MJ. Mitochondrial genome maintenance in health and disease. DNA Repair (Amst). 2014 Jul;19:190–8.
- Jiang M, Xie X, Zhu X, Jiang S, Milenkovic D, Misic J, et al. The mitochondrial single-stranded DNA binding protein is essential for initiation of mtDNA replication. Sci Adv. 2021 Jul;7(27):eabf8631.
- Longley MJ, Clark S, Man CYW, Hudson G, Durham SE, Taylor RW, et al. Mutant POLG2 Disrupts DNA Polymerase γ Subunits and Causes Progressive External Ophthalmoplegia. The American Journal of Human Genetics. 2006 Jun 1;78(6):1026–34.
- Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019 Jan;15(1):40–52.
- Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2019 Apr 21;16(1):3–17.
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010 Feb;12(2):119–31.
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. Journal of Cell Biology. 2010 Apr 19;189(2):211–21.
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010 Nov 29;191(5):933–42.
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008 Dec 1;183(5):795–803.
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AHV, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010 Dec 15;19(24):4861–70.
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007 Aug 17;282(33):24131–45.
- Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 17(10):2680–8.
- Nishimura T, Tooze SA. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020 ;6(1):32.
- Oshima Y, Cartier E, Boyman L, Verhoeven N, Polster BM, Huang W, et al. Parkin-independent mitophagy via Drp1-mediated outer membrane severing and inner membrane ubiquitination. J Cell Biol. 2021 Apr 14;220(6):e202006043.
- Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, et al. Mitochondrial Stasis Reveals p62-mediated Ubiquitination in Parkin-independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018 Oct 2;28(4):588-604.e5.
- Dorn, GW. Mitochondrial Pruning by Nix and BNip3: An Essential Function for Cardiac-Expressed Death Factors. J Cardiovasc Transl Res. 2010 Aug;3(4):374–83.
- Sulkshane P, Ram J, Thakur A, Reis N, Kleifeld O, Glickman MH. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021 Jun 17;45:102047.
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013 May;19(5):557–66.
- Christofides A, Konstantinidou E, Jani C, Boussiotis VA. The role of Peroxisome Proliferator-Activated Receptors (PPAR) in immune responses. Metabolism. 2021 Jan;114:154338.
- Wrutniak-Cabello C, Casas F, Cabello G. Thyroid hormone action in mitochondria. J Mol Endocrinol. 2001 Feb;26(1):67–77.
- Marín-García, J. Thyroid hormone and myocardial mitochondrial biogenesis. Vascul Pharmacol. 2010;52(3–4):120–30.
- Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. 2018 Jan;24(1):39–49.
- Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. 2014 Jan;53:124–44.
- Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am J Pathol. 2009 Feb;174(2):519–33.
- Barish GD, Narkar VA, Evans RM. PPARδ: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006 Mar 1;116(3):590–7.
- Vega RB, Huss JM, Kelly DP. The Coactivator PGC-1 Cooperates with Peroxisome Proliferator-Activated Receptor α in Transcriptional Control of Nuclear Genes Encoding Mitochondrial Fatty Acid Oxidation Enzymes. Mol Cell Biol. 2000 Mar;20(5):1868–76.
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003 Dec 23;100(26):15924–9.
- Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, et al. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003 Dec;17(15):2299–301.
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998 Mar 20;92(6):829–39.
- Andersson U, Scarpulla RC. PGC-1-Related Coactivator, a Novel, Serum-Inducible Coactivator of Nuclear Respiratory Factor 1-Dependent Transcription in Mammalian Cells. Mol Cell Biol. 2001 Jun;21(11):3738–49.
- Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:10.1042/bse0470069.
- Jamwal S, Blackburn JK, Elsworth JD. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol Ther. 2021 Mar;219:107705.
- Gureev AP, Shaforostova EA, Popov VN. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front Genet. 2019 ;10:435.
- Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018 Feb;19(2):121–35.
- Longo VD, Kennedy BK. Sirtuins in Aging and Age-Related Disease. Cell. 2006 Jul 28;126(2):257–68.
- Haigis MC, Guarente LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006 Jan 11;20(21):2913–21.
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide–dependent deacetylase. J Cell Biol. 2002 Aug 19;158(4):647–57.
- Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD- dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002 Oct 15;99(21):13653–8.
- Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007 Apr 15;21(8):920–8.
- Wang S, Wan T, Ye M, Qiu Y, Pei L, Jiang R, et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1α/mitochondrial biosynthesis pathway. Redox Biol. 2018 Apr 5;17:89–98.
- Yao J, Wang J, Xu Y, Guo Q, Sun Y, Liu J, et al. CDK9 inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy. 18(8):1879–97.
- Wang R, Xu H, Tan B, Yi Q, Sun Y, Xiang H, et al. SIRT3 promotes metabolic maturation of human iPSC-derived cardiomyocytes via OPA1-controlled mitochondrial dynamics. Free Radic Biol Med. 2023 Feb 1;195:270–82.
- Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. 2015 Feb 20;11(2):253–70.
- Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018 Jul 5;23(1):86-100.e6.
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008 ;105(19):7070–5.
- Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell. 2006 Feb 10;124(3):471–84.
- Fulda, S. Synthetic lethality by co-targeting mitochondrial apoptosis and PI3K/Akt/mTOR signaling. Mitochondrion. 2014 Nov 1;19:85–7.
- Shirane-Kitsuji M, Nakayama KI. Mitochondria: FKBP38 and mitochondrial degradation. Int J Biochem Cell Biol. 2014 Jun;51:19–22.
- Inoki K, Zhu T, Guan KL. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell. 2003 Nov 26;115(5):577–90.
- Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012 May;22(5):274–82.
- Auger JP, Zimmermann M, Faas M, Stifel U, Chambers D, Krishnacoumar B, et al. Metabolic rewiring promotes anti-inflammatory effects of glucocorticoids. Nature. 2024 May;629(8010):184–92.
- Psarra AMG, Sekeris CE. Steroid and thyroid hormone receptors in mitochondria. IUBMB Life. 2008 Apr;60(4):210–23.
- Goffart S, Wiesner RJ. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp Physiol. 2003 Jan;88(1):33–40.
- Wakabayashi, T. Structural changes of mitochondria related to apoptosis: swelling and megamitochondria formation. Acta Biochim Pol. 1999;46(2):223–37.
- Karbowski M, Kurono C, Wozniak M, Ostrowski M, Teranishi M, Nishizawa Y, et al. Free radical-induced megamitochondria formation and apoptosis. Free Radic Biol Med. 1999 Feb;26(3–4):396–409.
- Wakabayashi, T. Megamitochondria formation - physiology and pathology. J Cell Mol Med. 2002;6(4):497–538.
- Shang Y, Li Z, Cai P, Li W, Xu Y, Zhao Y, et al. Megamitochondria plasticity: Function transition from adaption to disease. Mitochondrion. 2023 Jul 1;71:64–75.
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune response by inhibiting NALP3 inflammasome-mediated mitochondrial DNA release. Nat Immunol. 2011 Mar;12(3):222–30.
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006 May;24(5):633–42.
- Vazquez C, Horner SM. MAVS Coordination of Antiviral Innate Immunity. J Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Molecular Cell. 2002 Aug 1;10(2):417–26.
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011 Oct 16;479(7371):117–21.
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015 Oct;526(7575):666–71.
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proceedings of the National Academy of Sciences. 2017 Jun 6;114(23):E4612–20.
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe. 2015 Jun 10;17(6):811–9.
- Soucy-Faulkner A, Mukawera E, Fink K, Martel A, Jouan L, Nzengue Y, et al. Requirement of NOX2 and Reactive Oxygen Species for Efficient RIG-I-Mediated Antiviral Response through Regulation of MAVS Expression. PLOS Pathogens. 2010 Jun 3;6(6):e1000930.
- Ward C, Schlichtholz B. Post-Acute Sequelae and Mitochondrial Aberration in SARS-CoV-2 Infection. International Journal of Molecular Sciences. 2024 Jan;25(16):9050.
- Hou P, Wang X, Wang H, Wang T, Yu Z, Xu C, et al. The ORF7a protein of SARS-CoV-2 initiates autophagy and limits autophagosome-lysosome fusion via degradation of SNAP29 to promote virus replication. Autophagy. 19(2):551–69.
- Mozzi A, Oldani M, Forcella ME, Vantaggiato C, Cappelletti G, Pontremoli C, et al. SARS-CoV-2 ORF3c impairs mitochondrial respiratory metabolism, oxidative stress, and autophagic flux. iScience. 2023 Jul 21;26(7):107118.
- Li X, Hou P, Ma W, Wang X, Wang H, Yu Z, et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol. 2022 Jan;19(1):67–78.
- Wu KE, Fazal FM, Parker KR, Zou J, Chang HY. RNA-GPS Predicts SARS-CoV-2 RNA Residency to Host Mitochondria and Nucleolus. Cell Syst. 2020 Jul 22;11(1):102-108.e3.
- Stukalov A, Girault V, Grass V, Karayel O, Bergant V, Urban C, et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature. 2021 Jun;594(7862):246–52.
- Shang C, Liu Z, Zhu Y, Lu J, Ge C, Zhang C, et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front Microbiol. 2021;12:780768.
- Roden AC, Boland JM, Johnson TF, Aubry MC, Lo YC, Butt YM, et al. Late Complications of COVID-19A Morphologic, Imaging, and Droplet Digital Polymerase Chain Reaction Study of Lung Tissue. Arch Pathol Lab Med. 2022 Jul 1;146(7):791–804.
- Zollner A, Koch R, Jukic A, Pfister A, Meyer M, Rössler A, et al. Postacute COVID-19 is Characterized by Gut Viral Antigen Persistence in Inflammatory Bowel Diseases. Gastroenterology. 2022 Aug 1;163(2):495-506.e8.
- Goh D, Lim JCT, Fernaíndez SB, Joseph CR, Edwards SG, Neo ZW, et al. Case report: Persistence of residual antigen and RNA of the SARS-CoV-2 virus in tissues of two patients with long COVID. Frontiers in Immunology [Internet]. 2022 [cited 2024 Jan 24];13. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2022.939989.
- Manns MP, Buti M, Gane E, Pawlotsky JM, Razavi H, Terrault N, et al. Hepatitis C virus infection. Nat Rev Dis Primers. 2017 Mar 2;3(1):1–19.
- Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PLoS Pathog. 2013 Dec 5;9(12):e1003722.
- Huang XY, Li D, Chen ZX, Huang YH, Gao WY, Zheng BY, et al. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp Cell Res. 2018 Jul 1;368(1):75–83.
- Schwerk J, Negash A, Savan R, Gale M. Innate Immunity in Hepatitis C Virus Infection. Cold Spring Harb Perspect Med. 2021 Feb;11(2):a036988.
- Wong MT, Chen SSL. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell Mol Immunol. 2016 Jan;13(1):11–35.
- Ferreon JC, Ferreon ACM, Li K. Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J Biol Chem. 2005, 280, 20483–20492. [Google Scholar] [CrossRef] [PubMed]
- Wilkins C, Woodward J, Lau DT -Y., Barnes A, Joyce M, McFarlane N, et al. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry,. Hepatology. 2013 Feb;57(2):461–9.
- Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy. 2013 Feb 1;9(2):175–95.
- Sir D, Chen W ling, Choi J, Wakita T, Yen TSB, Ou J hsiung J. Induction of Incomplete Autophagic Response by Hepatitis C Virus via the Unfolded Protein Response. Hepatology. 2008 Oct;48(4):1054–61.
- Wang L, Ou J hsiung J. Hepatitis C Virus and Autophagy. Biol Chem. 2015 Nov;396(11):1215–22.
- Ma X, McKeen T, Zhang J, Ding WX. Role and Mechanisms of Mitophagy in Liver Diseases. Cells. 2020 Mar 31;9(4):837.
- Ma X, Chen A, Melo L, Clemente-Sanchez A, Chao X, Ahmadi AR, et al. Loss of hepatic DRP1 exacerbates alcoholic hepatitis by inducing megamitochondria and mitochondrial maladaptation. Hepatology. 2023 Jan 1;77(1):159–75.
- Levy ML, Bacharier LB, Bateman E, Boulet LP, Brightling C, Buhl R, et al. Key recommendations for primary care from the 2022 Global Initiative for Asthma (GINA) update. NPJ Prim Care Respir Med. 2023 Feb 8;33(1):7.
- Poon AudreyH, Chouiali F, Tse SM, Litonjua AugustoA, Hussain SNA, Baglole CJ, et al. Genetic and histological evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol. 2012 Feb;129(2):569–71.
- Lee HS, Park HW. Role of mTOR in the Development of Asthma in Mice With Cigarette Smoke-Induced Cellular Senescence. J Gerontol A Biol Sci Med Sci. 2021 Nov 1;77(3):433–42.
- Liu JN, Suh DH, Trinh HKT, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. 2016 Jul;48(7):e243.
- McAlinden KD, Deshpande DA, Ghavami S, Xenaki D, Sohal SS, Oliver BG, et al. Autophagy Activation in Asthma Airways Remodeling. Am J Respir Cell Mol Biol. 2019 May;60(5):541–53.
- Wrana JL, Attisano L. The Smad pathway. Cytokine & Growth Factor Reviews. 2000 Apr 1;11(1):5–13.
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014 Sep 2;124(9):3987–4003.
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014 Sep 2;124(9):3987–4003.
- Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, et al. Egr-1 Regulates Autophagy in Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. PLoS One. 2008 Oct 2;3(10):e3316.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



