Submitted:
24 December 2024
Posted:
25 December 2024
You are already at the latest version
Abstract
The calcium-sensing receptor (CaSR) is a validated therapeutic target in the treatment of hyperparathyroidism and related diseases. The CaSR ago-positive allosteric modulator (PAM), AC265347 (1), exhibits a chemically and pharmacologically unique profile compared to current approved CaSR PAM therapeutics. Herein we report a series of ‘next-generation’ analogues of AC265347 investigating the impact of structural modifications at the stereogenic centre on CaSR PAM activity. Compounds 5 and 7b featuring the alcohol functional group showed ago-PAM profiles comparable to (1), whilst compounds 6, 7, 8 & 9 devoid of this functionality were ‘pure’ PAMs with no intrinsic agonism. These novel chemical tools provide an opportunity to explore the therapeutic potential of AC265367-like PAMs as a function of affinity, cooperativity and intrinsic agonism.
Keywords:
positive allosteric modulator
; PAM
; calcium-sensing receptor
; AC265347
; stereogenic centre
1. Introduction
AC265347 (1) (Figure 1), identified by ACADIA Pharmaceuticals from a screening library, is composed of a benzothiazole scaffold, a stereogenic centre bearing a tertiary alcohol and a dimethyl-substituted phenyl motif. This molecule is a positive allosteric modulator of the calcium-sensing receptor (CaSR) with intrinsic agonism (ago-PAM) and structurally distinct from the typical naphthylalkylamine class of CaSR modulators (e.g. cinacalcet (2), Figure 1). The initial in vivo study used the racemic mixture of AC265347 and demonstrated potent suppression of PTH release in rodents, consistent with a CaSR PAM profile [1]. Of note, the S-enantiomer had 10-fold greater potency than the R-enantiomer [1,2]. Further analysis confirmed AC265347’s binding selectivity for the CaSR with no off-target activation at the GABAb receptors and PTH1 receptors, thus validating its therapeutic potential [1]. The evaluation of AC265347 as a CaSR therapeutic was discontinued for undisclosed reasons with the last publication in 2011, however, this unique molecule warrants further investigation based on its ago-PAM activity accompanied by an improved side-effect profile.
The work described herein is an extension of our previously published research [3] and primarily focussed on implementing structural modifications at the stereogenic centre of AC265347 (1) and elucidating the ensuing pharmacology. We sought to provide further understanding of the key attributes of the active motif to guide new rational drug design towards generating lead drug candidates at this important therapeutic target for treating CaSR-related disorders. Our efforts utilised the recently published cryo-EM CaSR structures to inform and rationalise medicinal chemistry efforts towards the development of compounds with improved CaSR activity, specifically: (i) affinity of the allosteric ligand for the free receptor (KB); cooperativity factors defining the magnitude and direction of the allosteric ligand’s effect on the orthosteric ligand’s affinity (α) and/or downstream efficacy (β); and the intrinsic agonist efficacy of the allosteric ligand (τB) [4].
The work of Gustaffson et al. [2] examined the importance of the hydroxy group at the stereogenic centre by completely removing the H-bonding functional group as depicted in analogues 3 and 4 (Figure 2). Upon investigation, the sp3-hybridised analogue 3 was reported to be approximately 100-fold less potent than AC265347, while the sp2-hybridised olefinic analogue 4 lost all activity at the CaSR. Conclusively, the hydroxy group was deemed crucial for activity, and suspected to be involved in weak H-bond interactions [2]. However, previous AC265347 hydroxy group exploration was limited to complete removal of the H-bonding functional group [2]. To extend the previous findings we sought to diversify the current SAR library by making structural modifications targeting the hydroxy group.
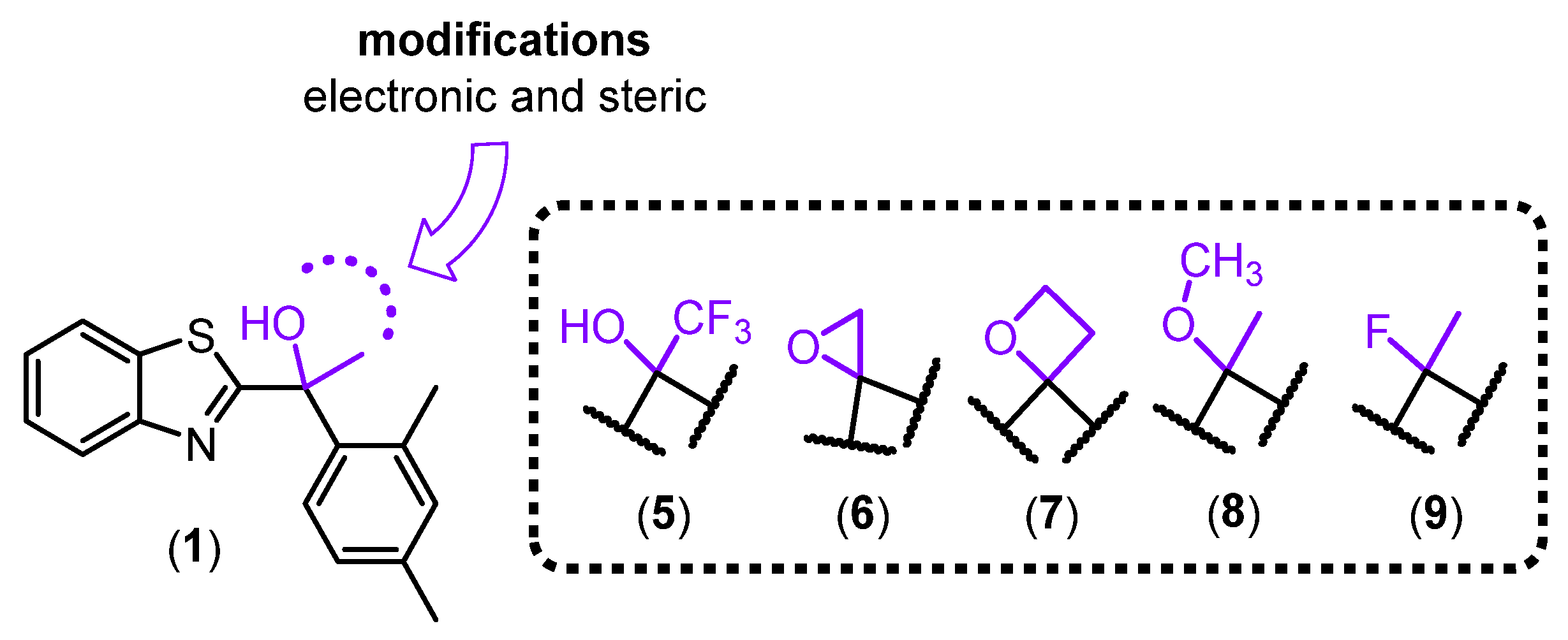
Herein, we aimed to delineate the role of H-bond acceptor and donor predicted to partake in binding to the CaSR. The suite of analogues (Figure 3) includes subtle structural modification to the OH group, for example, by substitution to a methoxy group (8). This modification removes of the H-bond donor property, thus allowing examination of the H-bond acceptor ability alone. Bioisosteric replacement of the OH group for a fluorine (9) is commonly used in medicinal chemistry because of its similarity in size, polarity and electronegativity between oxygen and fluorine atoms [5]. The substitution strategy is thought to provide metabolic stability, mainly due to the high C-F bond energy and conformational disturbance to avoid recognition by enzymes [6]. The replacement of OH for F is restricted to the H-bond acceptor ability, thus allows examination of the H-bond donor property provided by the hydroxy hydrogen atom. Additionally, the epoxide (6) and oxetane (7) were synthesised to challenge the importance of spatial orientation and rotational freedom on PAM activity at the CaSR. While the oxetane derivative angles the bonds almost perpendicularly at ~90o, the epoxide derivative is more constrained with a bond angle at 60o (Figure 3), thereby bringing the O and C atoms in significantly closer proximity compared to the parent compound (~105o) and increases the relative distance between the aromatic motifs. We also included a previously published analogue (5) [3] whereby the methyl substituent at the stereocentre was replaced with trifluoromethyl to enable further comparisons. We anticipate changes in allosteric effect with these structural changes, thus revealing the importance in maintaining the rotational freedom at the stereogenic centre and allowing us to dissect the role of H-bond donor/acceptor properties.
By generating a focused library of analogues with rational structural modifications on lead compound 1, we sought to delineate the structural motif responsible for affinity, cooperativity or agonism at the CaSR. Analogues were subjected to Ca2+i mobilisation assays and the activity quantified using the operational model of agonism and allosteric modulation (refer to section 3.3). Affinity, cooperativity and agonism were extracted and compared to the reference compounds 1 and 5. Herein, we describe the synthetic strategies, characterisation and pharmacological evaluations of the designed analogues.
2. Results and Discussion
2.1. Chemistry
Our SAR study focused on investigating the importance of the H-bond donor and acceptor properties of parent compound 1. To further address the limitation in our understanding, we removed the H-bond donor component by replacing the hydroxy group for methoxy, fluorine, epoxide and oxetane as H-bond acceptor moieties. The employment of epoxide (6) and oxetane (7) motifs would challenge the spatial importance of the H-bond acceptor and the methyl group at the stereogenic centre with their intrinsic rigidity. The methoxy substitution (8) simply provided greater flexibility compared to 6 and 7, however structurally bulkier than a hydroxy group. Replacing the hydroxy group for a fluorine (9) though retained H-bond acceptor ability, satisfied steric bulk and electronegativity, the differences between oxygen and fluorine atoms could result in a different pharmacological profile [5]. The included literature compound, (5), surveyed the biochemical effect of isoteric replacement of the methyl substituent for trifluoromethyl at the stereogenic centre. Herein, we describe the synthetic pathways and challenges encountered in the generation of analogues that examine the importance of the hydroxy group in 1.
The O-cyclised series synthesis contained two analogues, epoxide 6 and oxetane 7 (Scheme 1), to examine the effect of removing the H-bond donor property and the importance of spatial orientation by introducing rigidity to the compound structure. The reaction proceeded with the lithiated benzothiazole species generated in situ as a nucleophile attacking the carbonyl carbon of selected electrophiles [7]. The intramolecular cyclisation resulted in epoxide 6 and oxetane 7 analogues.
Characteristic proton splitting of epoxide hydrogens of 6 at δ 3.4 and 3.7 ppm as doublets, each integrating to one proton were observed as anticipated. The small coupling constant of 5.9 Hz for geminal protons supported the formation of the epoxide product 6. HRMS analysis found m/z 282.0959 which reinforces the molecular formula C17H15NOS and identity of 6.
While nucleophilic addition occurred between the lithiated benzothiazole and the selected carbonyl carbon followed by O-cyclisation with bromide ion as the leaving group to form oxetane 7 (as illustrated in Figure 9), a significant by-product corresponding to the terminal alkene 7b was also detected. Besides acting as an intended nucleophile, the lithiated species of benzothiazole can also act as a base, thereby deprotonating the α-hydrogen. This would subsequently allow the formation of a terminal alkene via elimination resulting in the allylic alcohol 7b product, thereby preventing the desired intramolecular cyclisation.
Our initial column purification attempt collected both oxetane 7 and allylic alcohol 7b due to their small difference in Rf values. This prompted a change in purification strategy to which preparative Thin Layer Chromatography (TLC) was employed followed by characterisation of oxetane 7 and allylic alcohol 7b. Distinctive signals corresponding to oxetane methylene hydrogen atoms H3 and H4 in the 1H NMR spectrum were observed whereby all protons on the oxetane ring are non-equivalent because of the stereogenic centre. In addition, DEPT 13C NMR analysis found C3 and C4 at 34 and 67 ppm, respectively, as secondary carbon signals thus reinforcing the successful formation of oxetane 7. Characterisation of allylic alcohol 7b found terminal alkene 1H NMR resonances at δ 6.6, 5.5 and 5.4 ppm as doublet-of-doublets integrating to one proton each. The highly deshielded signals observed in the spectrum are characteristic of alkene protons; convincing evidence to support the formation of by-product 7b [8]. The generation of epoxide 6 and oxetane 7 concluded the series of O-cyclised analogues to examine the effect of installing rigid groups at the stereogenic centre and the importance of the H-bond donor property. Additionally, we address the importance of H-bond interactions by simple substitution of the hydroxy (1) to methoxy group (8) and fluorine substituent (9). While the simple modification of methoxy 8 removed the H-bond donor effect, fluorine 9 maintains H-bond acceptor ability, however, carries a different electronegativity compared to the oxygen atom.
The synthesis of O-alkylated 8 began with AC265347 generated in house utilising commercially available acetophenone and benzothiazole. The O-alkylation reaction employed sodium hydride (NaH) as a base to deprotonate the hydroxy group to allow for nucleophilic attack of the methyl iodide (MeI) to take place (Scheme 3) [9]. This reaction resulted in a low conversion of 25-30% with the addition 1 and 2 equivalents of NaH and MeI, respectively. Thus, 2 and 2.5 equivalents were used to optimise the conversion of product to 50% by HPLC. This transformation was supported by 1H NMR with the absence of the hydroxy proton signal previously observed at δ 3.7 ppm (1H) for alcohol 1 and the presence of a downfield singlet resonance at δ 3.2 ppm (3H) for the methoxy group of 8.
The synthesis of 9 utilised safer XtalFluor-M over the commonly used but hazardous DAST and Deoxo-Fluor as deoxofluorinating agents (Scheme 3). DBU was employed as a preferred promoter to minimise formation of dimerised ether and sulfinate by-products [10]. The resulting crude reaction product suggested a minor conversion of product evident on LCMS over the course of 18 h. Gradient column of straight toluene followed by 20:1 toluene: EtOAc furnished the desired product 9 in a low yield of 12%. To confirm the identity of the newly formed product, we performed 19F NMR alongside 1H NMR, 13C NMR and HRMS experiments. The 19F NMR spectrum reassured the presence of fluorine atom within the compound structure at δ -96 ppm as a singlet. Additionally, the absence of the characteristic hydroxy signal at δ 3.7 ppm on the 1H NMR spectrum pointed to successful deoxofluorination of alcohol 1 to produce fluorinated compound 9. A large coupling constant is typically reported for C-F bond in the 13C NMR spectrum which is consistent with our observation for alkyl fluoride 9 at δ 98 ppm as a doublet (J = 172 Hz). Carbon atoms two-bonds away from fluorine expectedly displayed smaller coupling constants (21-30 Hz). Smaller coupling constants for carbon atoms three or more bonds away were also observed on the 13C NMR spectrum of alkyl fluoride 9 (see experimental section for more detail).
2.2. Pharmacology
Each of the synthesised compounds were assessed for modulation of Ca2+o-stimulated Ca2+i mobilisation in recombinant HEK293 cells stably expressing inducible wild-type human CaSR (FlpIn-Trex-HEK293-CaSR) to determine their affinity (KB) for the allosteric site, cooperativity effect on the orthosteric ligand’s affinity (α) and/or efficacy (β), and potential intrinsic agonism (τB) (Table 1) [4]. Figure 4A depicts the concentration-response curve for the reference compound AC265347 (1) demonstrating (i) the characteristic leftward shift with increasing concentrations of 1 which is indicative of positive allosteric modulation (cooperativity) of the endogenous orthosteric ligand Ca2+, and (ii) the characteristic upward shift with increasing concentrations of 1 in the absence of Ca2+ (vehicle) indicative of allosteric agonism; hence the classification of 1 as an ago-PAM.
Removal of the H-bond donor ability and constraining the rotational freedom of oxygen as an epoxide to generate compound 6, a modest reduction in cooperativity and loss of intrinsic agonism was observed compared to 1. The prominent reduction in agonism was evident by the lack of baseline increase in the Ca2+o concentration-response curve (Figure 4B). A similar trend was observed for oxetane-containing derivative, 7. The data collected from both epoxide 6 and oxetane 7 (Table 1, Figure 4B & 4C) suggests low tolerability of a more constrained spatial orientation. Disruption of the potential intramolecular interaction between the alcohol O and benzothiazole S atoms may also contribute to the reduced activity. Interestingly, novel compound 7b, discovered as a byproduct during the synthesis of 7, retained affinity, cooperativity and agonism comparable to 1 (Figure 4D). Substituting the methyl for an ethenyl group in 7b did not hinder hydroxy group rotational freedom, which potentially retained CaSR allosteric activity. This feature was also observed with our previously published derivative (5) containing the CF3 as a substitute for CH3 whilst maintaining the OH group, and exhibiting an ago-PAM profile (Table 1) comparable to 1.
The minor modification resulting from replacing the hydroxy for methoxy group, 8, had no effect on affinity, while cooperativity was reduced and agonism eliminated compared to 1 (Table 1, Figure 4E). The decreasing trend in cooperativity devoid of agonism may be a consequence of removing the H-bond donor properties. Previous SAR study performed by Gustaffson et al. [2], suggested the importance of the hydroxy group pertaining to H-bond interactions. The methoxy analogue 8 revealed hydroxy oxygen H-bond acceptor properties may play a crucial role in maintaining affinity, while H-bond donor properties have little impact on affinity.
Replacement of the hydroxy group for the fluoro substituent was supported by the ability of fluorine to make H-bond interactions that mimic those of the hydroxy group. Fluorine is known as a bioisostere of OH with similarity in polarity and size [11]. Also, fluorine is hydrophobic compared to oxygen, therefore its inclusion can enhance oral bioavailability and display a more druglike profile [12,13]. Despite the similarities shared between a hydroxy group and fluorine, fluorinated compound 9 saw a reduction in cooperativity with no detectable agonism compared to 1 (Table 1, Figure 4F). Non-covalent interaction between the hydroxy O and sulfur atoms may form a pseudo 5-membered ring, to which the removal of the hydrogen atom will disrupt the desired orientation thus potentially resulting in reduced activity. Overall, compounds 6, 7, 8 & 9, although displaying reduced affinity and cooperativity, did exhibit a relatively ‘pure’ PAM profile devoid of intrinsic agonism.
2.3. Molecular Docking Study
Currently, all known small molecule modulators of the CaSR bind to an allosteric pocket within the 7-transmembrane domain. The amino acid E837 is of particular interest, as it is a critical residue for the activity of the arylalkylamine class of CaSR modulators that includes cinacalcet. Previous work by Gustaffson et al. [2], in addition to in-house modelling, suggested AC265347 forms a weak hydrogen bond interaction with E837. Molecular docking was undertaken to identify whether the structural changes in the prepared AC265347 analogues could be linked to a difference in binding conformation that would relate to the pharmacological characteristics observed. Our modelling predicts AC265347 (Figure 5, panel A) binds towards the top of the 7-transmembrane domain, with the benzothiazole core oriented towards the bottom of the domain and the dimethyl-substituted phenyl motif extending towards extracellular space. The docking indicates the alcohol functional group of the more active S-enantiomer (eutomer) of AC265347 is oriented towards and predicted to interact with E837 via a hydrogen bond, while the alcohol of the less active R-enantiomer (distomer) faces away from E837. The difference in conformation and associated molecular interactions may account for the 10-fold increased activity of the AC265347 eutomer. The binding poses of the trifluoromethyl (5), allylic alcohol (7b, figure 5, panel C) and methoxy analogues (8) match the orientation of AC265347 eutomer, however, their positions are slightly displaced due to increased steric hinderance and/or electronic effects at the stereocentre (e.g. there was a distance of 1.9 Å between the stereogenic carbon centres of compounds 1 and 7b; panel C). The epoxide (6, figure 5, panel D) and oxetane (7) variants are both positioned so that the stereocentre is towards the left side of the binding pocket near L776 and F684 and therefore facing away from E837. The binding pose of compound 9, with the tertiary alcohol substituted for a fluorine (figure 5, panel B), matches that of AC265347 due to their similar molecular sizes. Our findings show that compounds (6, 7, 8 and 9) that are incapable of forming the predicted hydrogen bond with E837 results in loss of intrinsic agonism. This is consistent with the previously published mutagenesis data highlighting the importance of this residue [14]. This docking study suggests that the hydrogen bond capability of the tertiary alcohol at the stereocentre is important for the intrinsic agonism of this chemotype.
3. Materials and Methods
3.1. Materials
3.1.1. Chemistry
All solvents and chemicals were purchased from standard suppliers and were used without any further purification. 1H NMR and 13C NMR spectra were acquired at 400.13 and 100.62 MHz, respectively, on a Bruker Avance III Nanobay 400 MHz NMR spectrometer coupled to the BACS 60 automatic sample changer and equipped with a 5 mm PABBO BB-1H/ D Z-GRD probe. All spectra obtained were processed using MestReNova software (v.6.0). Chemical shifts (δ) for all 1H NMR spectra are reported in parts per million (ppm) using deuterated solvents as reference. The data for all spectra are reported in the following format: chemical shift (δ), (multiplicity, coupling constants J (Hz), integral), where the multiplicity is defined as s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, and m = multiplet. 13C NMR were routinely carried out as J-modulated spin-echo (JMOD) or Distortionless Enhancement by Polarisation Transfer (DEPT) experiments, all 13C NMR δ are reported in ppm. Thin layer chromatography (TLC) was carried out routinely on silica gel 60F254 precoated plates (0.25 mm, Merck). Flash column chromatography was carried out using Davisil LC60A silica gel, 40-63 μm.
Liquid chromatography mass spectrometry (LCMS) was performed on one of two instruments; either an Agilent 6100 Series Single Quad LC/MS or an Agilent 1200 Series HPLC (equipped with a 1200 Series G13111A Quaternary Pump, G1329A Thermostatted Autosampler, and a G1314B Variable Wavelength Detector) and the data was processed using LC/MSD Chemstation Rev.B.04.01 SP1 coupled with Easy Access Software. Both systems were equipped with a Reverse Phase Luna C8(2) (5 μm, 50 × 4.6 mm, 100 Å) column maintained at 30 oC. An MeCN gradient (5-100%) was used to obtain optimal separation, where 4 min were required for the gradient to reach 100% MeCN and maintained for a further 3 min before requiring 3 min to return to the initial gradient of 5% MeCN (total run time = 10 min). Solvent A = 0.1% aqueous formic acid; Solvent B = MeCN/ 0.1% formic acid.
The purity and retention time of final products were determined using analytical HPLC and high-resolution mass spectrometry (HRMS). Analytical HPLC was carried out using an Agilent 1260 Infinity Analytical HPLC fitted with a Zorbax Eclipse Plus C18 Rapid Resolution column (100 mm × 4.60 mm, 3.5 μm) using a binary solvent system: solvent A of 0.1% aqueous TFA; solvent B of 0.1% TFA in MeCN. Gradient elution was achieved over 10 min using 95% A + 5% B to 100% B over 9 min, and 100% B maintained for 1 min at a flow rate of 1 mL/min monitored at both 214 and 254 nm. HRMS were conducted on an Agilent 6224 TOP LC/MS Mass Spectrometer coupled to an Agilent 1290 Infinity. All data was acquired and reference mas corrected via dual-spray electrospray ionisation (ESI) source. Each scan or data point on the total ion chromatogram (TIC) is average of 13700 transients, producing one spectrum per second. Mass spectra were created by averaging the scans across each peak and background subtracted against the first 10 seconds of the TIC. Data acquisition was carried out using the Agilent Mass Hunter Data Acquisition software version B.05.00 Build 5.0.5042 and analysis was performed using Mass Hunter Qualitative Analysis version B.05.00 Build 5.0.519.13.
3.1.2. Pharmacology
Flp-InTM TREXTM Human Embryonic Kidney (HEK) 293 cells were purchased from Invitrogen (Carlsbad, USA). Fluo-8-AM (acetoxymethyl ester) was purchased from Abcam (Cambridge, USA). Dulbecco’s Modified Eagle’s Medium (high glucose), poly-D-lysine, hygromycin B, blasticidin HCl, tetracycline and all other reagents were purchased from Sigma Aldrich (St Louis, USA). Ionomycin was purchased from Cayman Chemicals (Michigan, USA).
FlpIn HEK293 TRex cells stably expressing cmyc-tagged WT CaSR were grown in DMEM supplemented with 5% FBS and antibiotic selection (200 μg/mL hygromycin; 5 μg/mL blasticidin). All cells were maintained at 37 °C in a 5% CO2 humidified incubator, grown to confluence, and then seeded in 96-well culture plates. The day before assays, cells were seeded onto poly-D-lysine–coated, clear bottom 96-well plates in assay medium (DMEM supplemented with 5% dialysed FBS) at a density of 80,000 cells/well.
3.2. Chemical Synthesis of Compounds
General Procedure A [16] (lithiation)
To a solution of benzothiazole (1.1 equiv.) in dry THF (3 mL) at -78 oC was added nBuLi solution (2.5 M, 1.3 equiv.) in a dropwise fashion and stirred for 1 h. Then a pre-cooled substituted acetophenone (1 equiv.) in dry THF solution was subsequently added dropwise to the reaction mixture then allowed to reach r.t and stirred for a further 24 h. The resulting mixture was quenched by saturated NH4Cl (5 mL) and extracted with EtOAc (3 × 10 mL) and then washed with saturated brine. The combined organic layer was dried over anhydrous Na2SO4 then concentrated under reduced pressure.
General Procedure B [7] (O-cyclisation)
Preparation of lithiobenzothiazole was carried out in accordance with general procedure A. To a solution of lithiobenzothiazole was added halo-substituted ketone (0.7 equiv to benzothiazole) in dry THF (5 mL) at -78 oC. The reaction mixture was allowed to warm to r.t after an hour and stirred for another 5 h. The resulting mixture was quenched with sat. NH4Cl (5 mL), extracted with EtOAc, washed with brine, dried over Na2SO4 and evaporated under vacuum.
Synthesis of 1-(Benzo[d]thiazol-2-yl)-1-(2,4-dimethylphenyl)ethan-1-ol (1) [3]
Synthesis as described in general procedure A. Purification using silica gel column chromatography (5% acetone in toluene) was performed to afford product as white crystal (133 mg, 70%). 1H NMR (401 MHz, CDCl3) δ 8.01 (d, J = 8.2 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.51 – 7.45 (m, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.98 (s, 1H), 3.73 (s, 1H), 2.33 (s, 3H), 2.14 (s, 3H), 2.11 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 179.95 (C), 152.26 (C), 138.76 (C), 138.53 (C), 137.66 (C), 135.93 (C), 133.47 (CH), 126.45 (CH), 126.32 (CH), 126.25 (CH), 125.30 (CH), 123.16 (CH), 121.88 (CH), 31.33 (CH3), 21.35 (CH3), 21.06 (CH3). HRMS (m/z): C17H17NOS requires [M+H]+ 284.1104; found 284.1115.
Synthesis of 2-(2-(2,4-Dimethylphenyl)oxiran-2-yl)benzo[d]thiazole (6)
Synthesis as described in general procedure B. The resulting crude was purified via silica gel column chromatography (8:1 petroleum ether: EtOAc) to afford the product as a light-yellow paste (89 mg, 40%). 1H NMR (401 MHz, CDCl3) δ 8.00 (ddd, J = 8.2, 1.2, 0.7 Hz, 1H), 7.83 (ddd, J = 7.9, 1.3, 0.6 Hz, 1H), 7.49 – 7.42 (m, 2H), 7.37 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.14 – 7.10 (m, 1H), 7.09 – 7.05 (m, 1H), 3.71 (d, J = 5.9 Hz, 1H), 3.44 (d, J = 5.9 Hz, 1H), 2.38 (s, 3H), 2.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.89 (C), 153.77 (C), 139.22 (C), 137.86 (C), 135.50 (C), 132.17 (C), 131.43 (CH), 128.76 (CH), 126.65 (CH), 126.17 (CH), 125.34 (CH), 123.58 (CH), 121.69 (CH), 60.25 (C), 57.60 (CH2), 21.38 (CH3), 19.59 (CH3). HPLC: tR 6.027 min, >95% purity (214 & 254 nm). HRMS (m/z): C17H15NOS requires [M+H]+ 282.0947; found 282.0959.
Synthesis of 2-(2-(2,4-Dimethylphenyl)oxetan-2-yl)benzo[d]thiazole (7)
Synthesis as described in general procedure B. Purification using silica gel column chromatography (10:1 petroleum ether: EtOAc) afforded product mixture 7 and 7b. Subsequently, preparative TLC purification method (40:1 toluene: EtOAc) separated 37 (14 mg, 9%) as a clear oil and 7b as a white solid (9 mg, 6%). 1H NMR (401 MHz, CDCl3) δ 8.07 (ddd, J = 8.2, 1.2, 0.6 Hz, 1H), 7.81 (ddd, J = 8.0, 1.3, 0.7 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.47 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 7.35 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.15 (d, J = 7.9 Hz, 1H), 6.98 (s, 1H), 4.94 (ddd, J = 8.4, 7.3, 5.7 Hz, 1H), 4.66 (dt, J = 9.0, 5.9 Hz, 1H), 3.87 (ddd, J = 11.0, 8.4, 6.0 Hz, 1H), 3.23 (ddd, J = 11.0, 9.0, 7.3 Hz, 1H), 2.35 (s, 3H), 2.11 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 175.91 (C), 153.52 (C), 139.02 (C), 138.23 (C), 134.80 (C), 132.18 (CH), 126.54 (CH), 126.03 (CH), 125.27 (CH), 124.93 (CH), 123.71 (CH), 121.88 (CH), 121.23(C), 87.80 (C), 66.71 (CH2), 33.60 (CH2), 21.20 (CH3), 19.85 (CH3). HPLC: tR 8.921 min, >95% purity (214 & 254 nm). HRMS (m/z): C18H17NOS requires [M+H]+ 296.1104; found 296.1105.
1-(Benzo[d]thiazol-2-yl)-1-(2,4-dimethylphenyl)prop-2-en-1-ol (7b):1H NMR (401 MHz, CDCl3) δ 8.02 (ddd, J = 8.2, 1.2, 0.6 Hz, 1H), 7.83 (ddd, J = 7.9, 1.3, 0.6 Hz, 1H), 7.48 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 7.43 – 7.34 (m, 2H), 7.01 (d, J = 8.5 Hz, 2H), 6.64 (dd, J = 17.1, 10.5 Hz, 1H), 5.47 (dd, J = 17.1, 0.9 Hz, 1H), 5.41 (dd, J = 10.5, 0.9 Hz, 1H), 3.81 (s, 1H), 2.32 (s, 3H), 2.16 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 177.71 (C), 152.44 (C), 141.66 (CH), 138.72 (C), 137.97 (C), 137.92 (C), 136.01 (C), 133.54 (CH), 128.04 (CH), 126.29 (2C), 125.39 (CH), 123.34 (CH), 121.89 (CH), 115.57 (CH2), 79.89 (C), 21.32 (CH3), 21.08 (CH3). HRMS (m/z): C18H17NOS requires [M+H]+ 296.1104; found 296.1111.
Synthesis of 2-(1-(2,4-Dimethylphenyl)-1-methoxyethyl)benzo[d]thiazole (8)
Synthesis was modified from literature procedure [9]. To a solution of 2 (80.2 mg, 0.28 mmol) in THF (5 mL) was added dropwise to a suspension of NaH (from 55% dispersion in oil, 2 equiv.) in THF. When hydrogen evolution had ceased, methyl iodide (2.5 equiv.) was added to the solution and stirred for 5 h. The crude mixture was quenched with NH4Cl (15 mL), extracted with ether (3 x 10 mL), washed with brine, dried over Na2SO4 and evaporated under vacuum. The resulting crude was purified via preparative TLC (20:1 toluene: acetone) to afford desired product as a white solid (40%). 1H NMR (401 MHz, CDCl3) δ 7.97 (ddd, J = 8.2, 1.3, 0.7 Hz, 1H), 7.86 (ddd, J = 7.9, 1.3, 0.7 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.42 (ddd, J = 8.2, 7.2, 1.3 Hz, 1H), 7.34 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.07 (d, J = 8.0 Hz, 1H), 6.96 (s, 1H), 3.21 (s, 3H), 2.33 (s, 3H), 2.08 (s, 3H), 2.04 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 179.01 (C), 153.08 (C), 138.28 (C), 137.73 (C), 136.60 (C), 135.55 (C), 133.26 (CH), 128.00 (CH), 126.44 (CH), 125.77 (CH), 124.94 (CH), 123.46 (CH), 121.69 (CH), 82.31 (C), 77.16 (CH3), 50.53 (CH3), 25.32 (CH3), 21.10 (CH3). HPLC: tR 6.388 min, >95% purity (214 & 254 nm). HRMS (m/z): C18H19NOS requires [M+H]+ 298.1260; found 298.1273.
Synthesis of 2-(1-(2,4-Dimethylphenyl)-1-fluoroethyl)benzo[d]thiazole (9)
Synthesis was modified from literature procedure [10]. To a solution of 2 (53 mg, 0.187 mmol) in anhydrous DCM was cooled to -78 oC followed by the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.5 equiv.) and XtalFluor-M (1.5 equiv.) under nitrogen. The reaction mixture was allowed to warm to room temperature and stirred for 24 h. The resulting mixture was quenched with a 5% aqueous sodium bicarbonate (5 mL) solution and extracted with DCM (3 × 5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting crude was purified via silica gel column chromatography (straight toluene then 20:1 toluene: EtOAc) to afford the product as a clear paste (11.5%). 1H NMR (401 MHz, CDCl3) δ 8.05 (ddd, J = 8.2, 1.2, 0.7 Hz, 1H), 7.87 (ddd, J = 8.0, 1.3, 0.6 Hz, 1H), 7.51 – 7.46 (m, 1H), 7.46 – 7.43 (m, 1H), 7.40 (ddd, J = 7.9, 7.2, 1.2 Hz, 1H), 7.06 (d, J = 8.0 Hz, 1H), 7.00 (s, 1H), 2.33 (s, 3H), 2.27 (d, J = 23.0 Hz, 3H), 2.21 (d, J = 2.6 Hz, 3H). 19F NMR (376 MHz, CDCl3) δ -96.78. 13C NMR (101 MHz, CDCl3) δ 174.21 (d, J = 29.6 Hz, C), 152.91 (C), 138.94 (d, J = 1.4 Hz, C), 136.55 (d, J = 21.1 Hz, C), 136.29 (d, J = 1.9 Hz, C), 135.82 (C), 133.26 (CH), 126.60 (CH), 126.21 (CH), 126.08 (d, J = 9.7 Hz, CH), 125.60 (CH), 123.88 (CH), 121.85 (CH), 97.80 (d, J = 172.2 Hz, C), 27.39 (d, J = 25.5 Hz, CH3), 21.12 (d, J = 5.2 Hz, CH3), 21.08 (CH3). HPLC: tR 6.400 min, >95% purity (214 & 254 nm). HRMS (m/z): C17H16FNS requires [M+H]+ 286.1060; found 286.1073.
3.3. Pharmacological Evaluation of Compounds
3.3.1. Functional Assay - Ca2+i Mobilisation
Ca2+i mobilisation assays were performed as previously described [14,17]. Specifically, FlpIn-TRex-HEK293-CaSR stable cell lines were seeded at 80 000 cells/well in poly-D-lysine (50 μg/mL) coated clear 96 well plates and incubated overnight at 37 °C, 5% CO2, in the presence of 100 ng/mL tetracycline to induce CaSR expression. Cells were washed in assay buffer (150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2, 10 mM D-glucose, 10 mM HEPES, 0.1 mM CaCl2, 0.5% BSA, 4 mM probenecid at pH 7.4) and loaded with 1 μM Fluo-8® AM in assay buffer for 1 hour. Agonist and PAMs (0, 0.03, 0.1, 0.3, 1, 3 or 10 μM) were co-added and measurements of Ca2+i mobilisation were performed in duplicate at 37 °C using a Flexstation 1 or 3 (Molecular Devices; Sunnyvale, CA, USA). Fluorescence was detected for 20 s to establish a baseline response and then a further 60 s at 490 nm excitation and 520 nm emission. Data were normalised to the % of ionomycin response. Ionomycin (1 μM) was used as an internal assay control.
3.3.2. Data Statistical Analysis
Data describing the interaction between Ca2+o and the PAMs were fitted to the following operational model of agonism and allosteric modulation [18,19,20], which was modified to accommodate the presence of ambient agonist [21]:
where EC50 is the agonist concentration (M) that produces a half maximal response; KB is the affinity of the allosteric ligand; τB is the efficacies of the orthosteric agonist and allosteric ligand, respectively; α and β describe the allosteric effects on orthosteric agonist affinity and efficacy, respectively; [A] and [B] are the orthosteric agonist and allosteric ligand concentrations (μM), respectively; Em is the maximal system response; nT is the slope of the transducer function linking agonist receptor occupancy to response and was constrained to unity; nB is the slope of agonist binding linking agonist concentration to occupancy; and [C] is the ambient Ca2+o concentration present in the assay buffer.
All non-linear regression analysis was performed in GraphPad Prism 7 (GraphPad Software,
San Diego, CA). Affinity, cooperativity and efficacy parameters were estimated as logarithms [22]. A one-way ANOVA with Dunnett’s multiple comparisons post-test was used to determine statistical differences between PAM pKB, Logαβ and LogτB, where significance was defined as p<0.05.
3.4. Computational Modelling of Compounds
Structural information obtained from the Cryo-EM PDBID:7M3G [15] was used as the basis for docking. Docking was performed using ICM, where the candidate was placed in the 7TM binding pocket that contained evocalcet. PAMs were initially placed in the centre of the presumed binding site before being extensively sampled by biased probability Monte Carlo sampling in internal coordinates. Therefore, the hydrogen bonding, van der Waals, and hydrophobic and electrostatic potential of the CaSR 7TM binding cavity were calculated to create a “grid potential map,” which was used to score the binding potential of randomised conformations of the PAM. Binding scores were calculated as described previously [23]. Ten poses with the lowest calculated binding scores were retained for each PAM. These poses were further re-fined using a “flexible receptor” approach, by undertaking biased probability Monte Carlo optimisation of receptor residue sidechains [24], while simultaneously sampling PAM shape and position by Monte Carlo randomisation. Final results were assessed using a combination of ICM docking scores and calculated binding energies [25,26], coupled with manual inspection for agreement with mutagenesis data for AC265347 (1) and published SAR data [4,14].
4. Conclusions
A series of novel analogues generated from AC265347 (1) were synthesised in this SAR study. The library of analogues provided insight into the importance of the tertiary alcohol feature and concomitant H-bonding profile at the stereogenic centre to the ago-PAM profile of 1. Structural modifications to the hydroxy motif afforded compounds exhibiting a ‘pure’ PAM profile (epoxide 6, oxetane 7, methoxy 8 and fluorinated compound 9) with partially reduced affinity and cooperativity and devoid of intrinsic agonism. This abolition of agonism demonstrated a close correlation between intrinsic agonism and the hydroxy group, perhaps suggesting the role of the hydroxy group in allosteric CaSR agonism. Moreover, alterations in spatial orientation (epoxide 6 and oxetane 7) and replacement of hydroxy (fluoro analogue 9) that can interrupt the potential non-covalent intramolecular interaction between the sulfur and oxygen atoms led to a diminution of agonist activity at the CaSR. The study extends the previous work by Gustaffson et al. exploring the significance of the hydroxy group on CaSR allosteric activity. Furthermore, the SAR investigation herein has enriched the key structural features for activity at the CaSR, and identified novel chemical tools to ascertain the therapeutic potential of AC265367-like PAMs devoid of intrinsic agonism.
Supplementary Materials
Not applicable.
Author Contributions
Conceptualization, A.D., K.J.G., K.L. and B.C.; methodology, L.V.D., A.D., T.M.J., K.J.G., K.L. and B.C.; software, L.V.D., J.D., A.N.K., T.M.J., K.J.G. and K.L.; validation, L.V.D., J.D., A.D., K.J.G., K.L. and B.C.; formal analysis, L.V.D., J.D., A.D., A.N.K., T.M.J., K.J.G., K.L. and B.C.; investigation, L.D.V., J.D. A.D., K.J.G., K.L. and B.C.; resources, A.D., K.J.G., K.L. and B.C.; data curation, L.D.V., J.D., K.J.G. and K.L.; writing—original draft preparation, L.D.V., J.D., K.J.G., K.L. and B.C.; writing—review and editing, L.V.D., J.D., A.D., A.N.K., T.M.J., K.J.G., K.L. and B.C.; visualization, L.V.D., J.D., K.J.G., K.L.; supervision, A.D., K.J.G., K.L. and B.C.; project administration, K.L., K.J.G. and B.C.; funding acquisition, K.L., K.J.G. and B.C.
Funding
This research was funded in part by the Australian Research Council (Discovery project grant DP170104228) and the National Health and Medical Research Council of Australia (Project grant 1138891). KL and KJG are Australian Research Council Future Fellows (fellowships FT160100075 and FT170100392, respectively). TMJ is a National Health and Medical Research Council Emerging Leader Fellow (fellowship 2008341).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
The authors wish to thank Dr Jason Dang and Dr William O’Malley for spectroscopic support, and Dr Shane Hellyer and Dr Jiayin Diao for providing scientific input on the in vitro pharmacology.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ma, J.-N.; Owens, M.; Gustafsson, M.; Jensen, J.; Tabatabaei, A.; Schmelzer, K.; Olsson, R.; Burstein, E.S. Characterization of highly efficacious allosteric agonists of the human calcium-sensing receptor. J Pharmacol Exp Ther 2011, 337, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.; Jensen, J.; Bertozzi, S.M.; Currier, E.A.; Ma, J.-N.; Burstein, E.S.; Olsson, R. Discovery of a class of calcium sensing receptor positive allosteric modulators 1-(benzothiazol-2-yl)-1-phenylethanols. Bioorg Med Chem Lett 2010, 20, 5918–5921. [Google Scholar] [CrossRef] [PubMed]
- Dinh, L.V.; DeBono, A.; Keller, A.N.; Josephs, T.M.; Gregory, K.J.; Leach, K.; Capuano, B. Development of AC265347-Inspired Calcium-Sensing Receptor Ago-Positive Allosteric Modulators. ChemMedChem 2021, 16, 3451–3462. [Google Scholar] [CrossRef]
- Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 2007, 28, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L. Selective Fluorination in Drug Design and Development: An Overview of Biochemical Rationales. CTMC 2006, 6, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Welch, J. T. Advances in the preparation of biologically active organofluorine compounds. Tetrahedron 1987, 43, 3123–3197. [Google Scholar] [CrossRef]
- Chikashita, H.; Ishibaba, M.; Ori, K.; Itoh, K. General Reactivity of 2-Lithiobenzothiazole to Various Electrophiles and the Use as a Formyl Anion Equivalent in the Synthesis of α-Hydroxy Carbonyl Compounds. Bull Chem Soc Jpn 1988, 61, 3637–3648. [Google Scholar] [CrossRef]
- Morrill, C.; Morrill, C.; Grubbs, R.H. Highly Selective 1,3-Isomerization of Allylic Alcohols via Rhenium Oxo Catalysis. J Am Chem Soc 2005, 127, 2842–2843. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.G.C.; Bruneau, P.; Crawley, G.C.; Edwards, M.P.; Foster, S.J.; Girodeau, J.M.; Kingston, J.F.; McMillan, R.M. (Methoxyalkyl)thiazoles: a new series of potent, selective, and orally active 5-lipoxygenase inhibitors displaying high enantioselectivity. J Med Chem 1991, 34, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- L’Heureux, A.; Beaulieu, F.; Bennett, C.; Bill, D.R.; Clayton, S.; LaFlamme, F.; Mirmehrabi, M.; Tadayon, S.; Tovell, D.; Couturier, M. Aminodifluorosulfinium Salts: Selective Fluorination Reagents with Enhanced Thermal Stability and Ease of Handling. J Org Chem 2010, 75, 3401–3411. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J Phys Chem 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Hevey, R. Bioisosteres of carbohydrate functional groups in glycomimetic design. Biomimetics 2019, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Rychlewski, J.; Rychlewska, U. Effects of Substitution of OH Group by F Atom for Conformational Preferences of Fluorine-Substituted Analogues of (R,R)-Tartaric Acid, Its Dimethyl Diester, Diamide, and N,N,N‘,N‘-Tetramethyl Diamide. Ab Initio Conformational Analysis. J Am Chem Soc 1999, 121, 1912–1921. [Google Scholar] [CrossRef]
- Leach, K.; Gregory, K.J.; Kufareva, I.; Khajehali, E.; Cook, A.E.; Abagyan, R.; Conigrave, A.D.; Sexton, P.M.; Christopoulos, A. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res 2016, 26, 574–592. [Google Scholar] [CrossRef]
- Gao, Y.; Robertson, M.J.; Rahman, S.N.; et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature 2021, 595, 455–459. [Google Scholar] [CrossRef]
- Anthony, N.J.; Andresen, B.M.; Northrup, A.B.; Childers, K.K.; Donofrio, A.; Miller, T.A.; Liu, Y.; Machacek, M.R.; Woo, H.C.; Spencer, K.B.; Ellis, J. M.; Altman, M. D.; Romeo, E.T.; Guay, D.; Grimm, J.; Lebrun, M.; Robichaud, J.S.; Wang, L.; Dubois, B.; Deng, Q. Thiazole-substituted Aminoheteroaryls as spleen Tyrosine Kinase Inhibitors, in WO 2014/176210 Al, 2014.
- Keller, A.N.; Kufareva, I.; Josephs, T.M.; Diao, J.; Mai, V.T.; Conigrave, A.D.; Christopoulos, A.; Gregory, K.J.; Leach, K. Identification of global and ligand-specific calcium sensing receptor activation mechanisms. Mol Endocrinol 2018, 93, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, S.D.; Albold, S.; Wang, T.; Chen, A.; May, L.; Leach, K.; Gregory, K.J. Selective" Class C G Protein-Coupled Receptor Modulators Are Neutral or Biased mGlu(5) Allosteric Ligands. Mol Pharmacol 2018, 93, 504–514. [Google Scholar] [CrossRef]
- Leach, K.; Loiacono, R.E.; Felder, C.C.; McKinzie, D.L.; Mogg, A.; Shaw, D.B.; Sexton, P.M.; Christopoulos, A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacol 2010, 35, 855–869. [Google Scholar] [CrossRef]
- Gregory, K.J.; Giraldo, J.; Diao, J.; Christopoulos, A.; Leach, K. Evaluation of operational models of agonism and allosterism at receptors with multiple orthosteric binding sites. Mol Pharmacol 2020, 97, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Davey, A.E.; Leach, K.; Valant, C.; Conigrave, A.D.; Sexton, P.M.; Christopoulos, A. Positive and Negative Allosteric Modulators Promote Biased Signaling at the Calcium-Sensing Receptor. Endocrinology 2012, 153, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Assessing the distribution of parameters in models of ligand–receptor interaction: to log or not to log. Trends Pharmacol Sci 1998, 19, 351–357. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. (1999). Derivation of sensitive discrimination potential for virtual ligand screening. RECOMB99 3rd International Conference on Computational Molecular Biology (p. 312). Lyon, France.
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Totrov, M.; Abagyan, R. Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit 1999, 12, 177–190. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of two chemical classes of CaSR positive allosteric modulators (PAMs): benzothiaziole (AC265347, 1) and naphthylalkylamine (cinacalcet, 2).
Figure 1.
Chemical structures of two chemical classes of CaSR positive allosteric modulators (PAMs): benzothiaziole (AC265347, 1) and naphthylalkylamine (cinacalcet, 2).
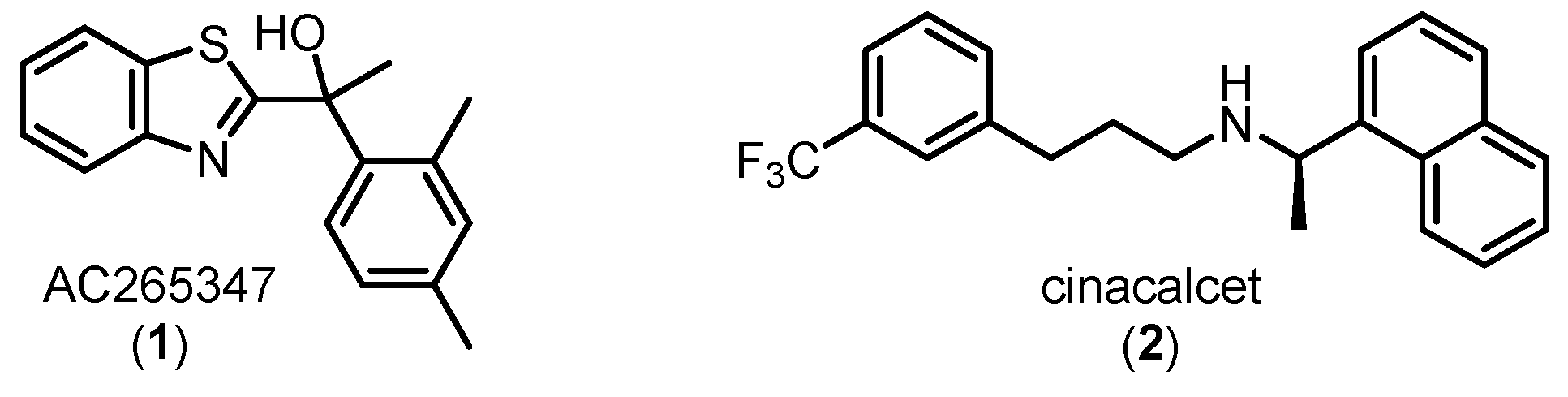
Figure 2.
Previous structural modifications at the stereogenic centre (*) of AC265347 investigating the importance of H-bonding interactions.
Figure 2.
Previous structural modifications at the stereogenic centre (*) of AC265347 investigating the importance of H-bonding interactions.
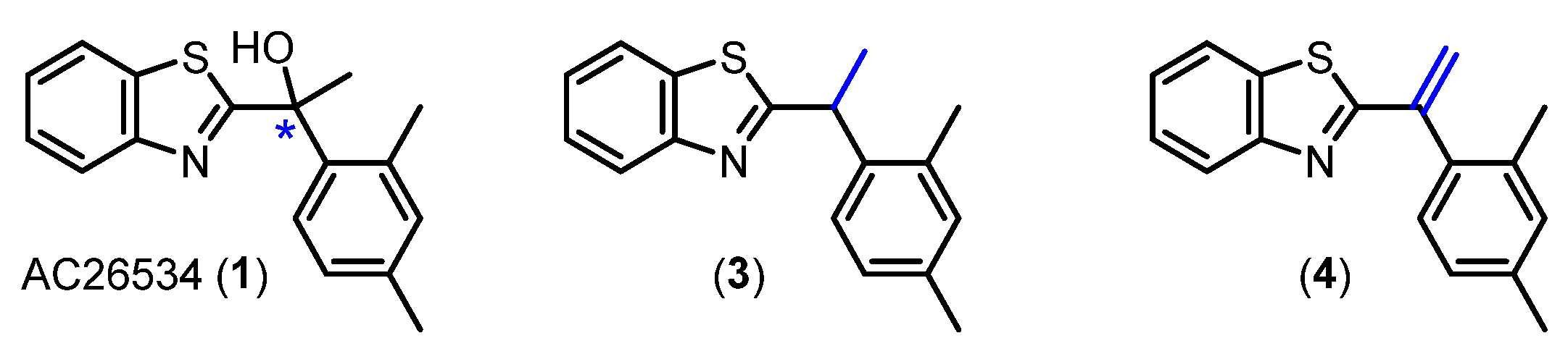
Figure 3.
Proposed structural modifications to the hydroxy group at the stereogenic centre of AC265347 (1).
Figure 3.
Proposed structural modifications to the hydroxy group at the stereogenic centre of AC265347 (1).
Scheme 1.
Synthesis of O-cyclised analogues epoxide 6 and oxetane 7a. aReagents and conditions: i) nBuLi, THF, N2, -78 oC (6 (40%), 7 (9%) and 7b (6%)).
Scheme 1.
Synthesis of O-cyclised analogues epoxide 6 and oxetane 7a. aReagents and conditions: i) nBuLi, THF, N2, -78 oC (6 (40%), 7 (9%) and 7b (6%)).
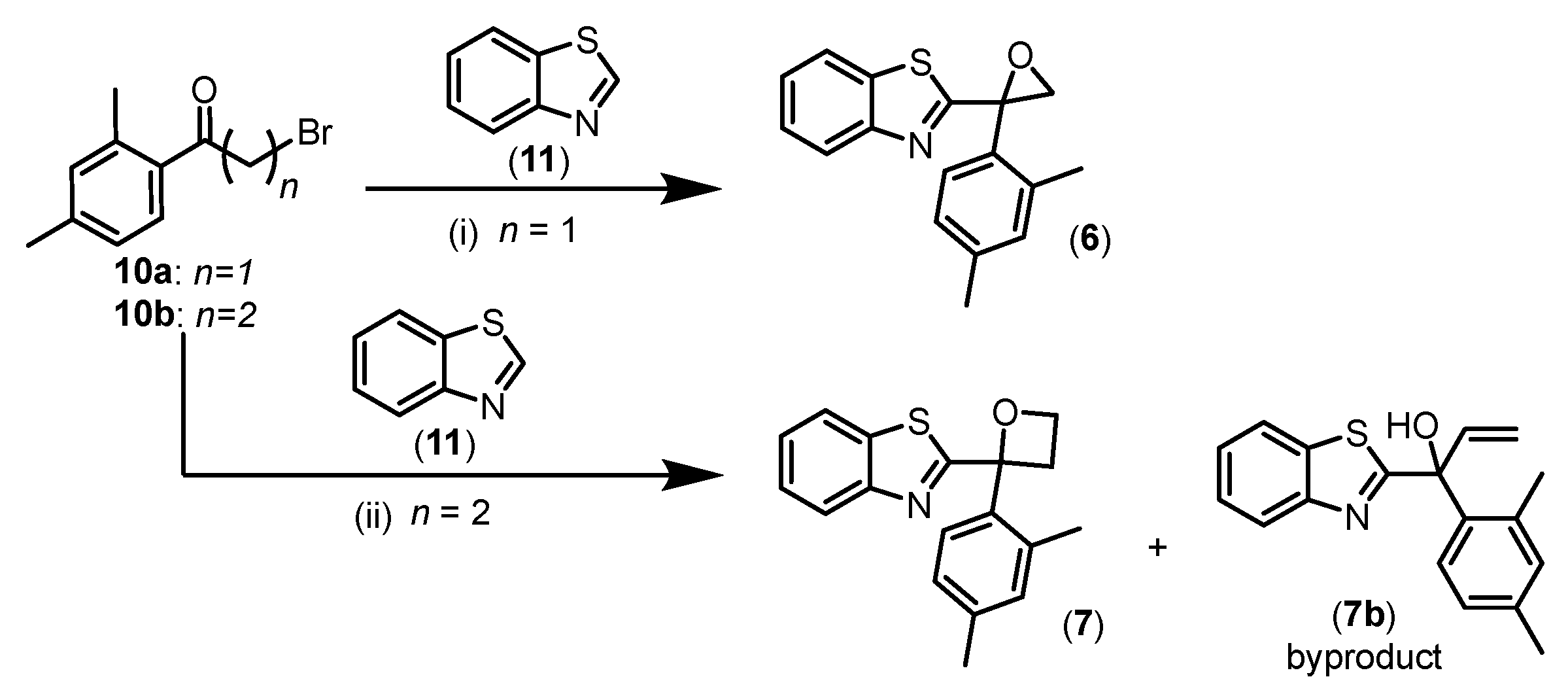
Figure 3.
Proposed mechanism for the cyclisation of oxetane 7 and allylic alcohol by-product 7b.
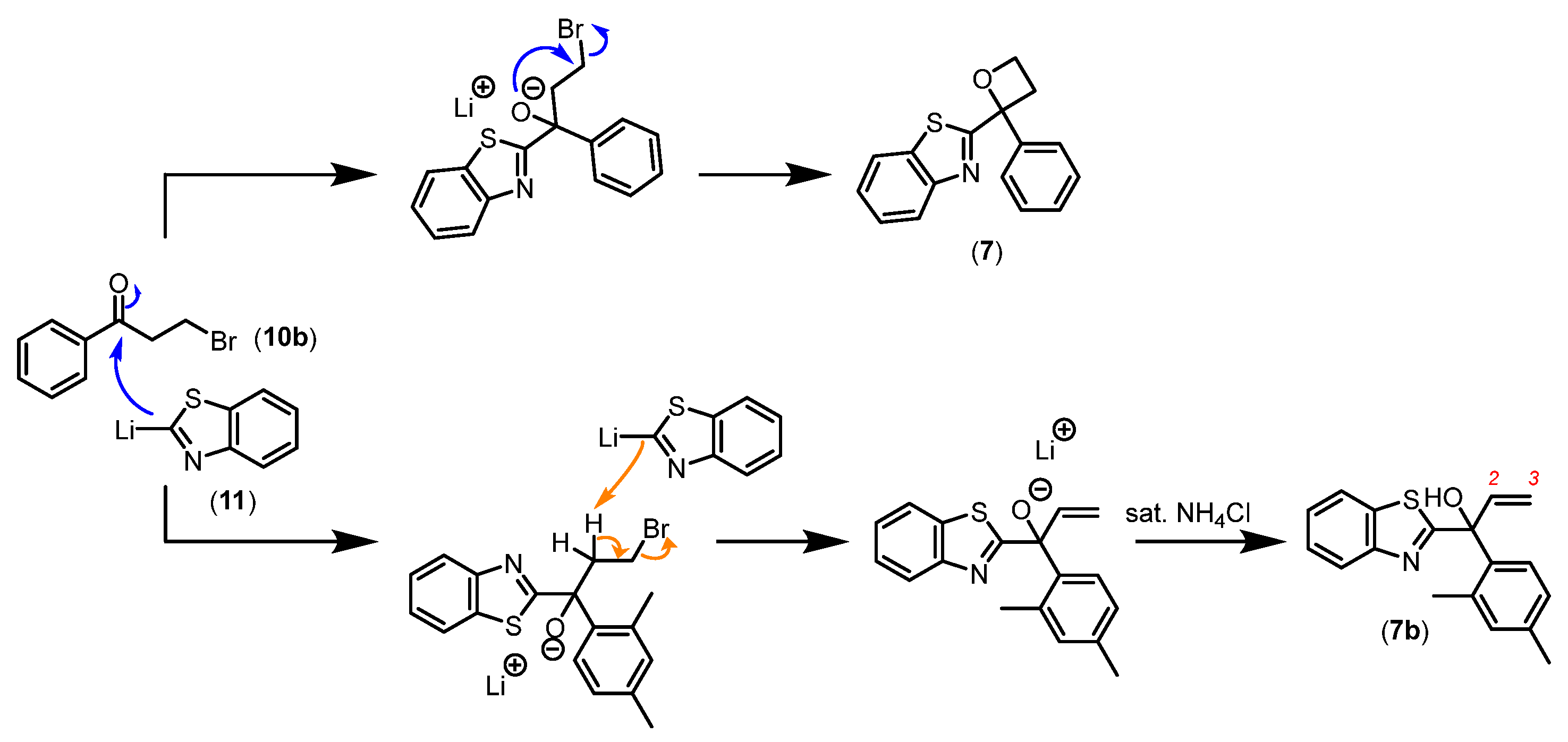
Scheme 3.
Synthesis of methoxy 8 and fluoro 9 analogues to investigate the biochemical effect of compounds without H-bond donor propertiesa. aReagents and conditions: i) nBuLi, THF, N2(g), -78 oC, 70%; ii) NaH, MeI, THF, 50%; iii) XtalFluor-M, DBU, dry DCM, -78 oC to rt, 12%.
Scheme 3.
Synthesis of methoxy 8 and fluoro 9 analogues to investigate the biochemical effect of compounds without H-bond donor propertiesa. aReagents and conditions: i) nBuLi, THF, N2(g), -78 oC, 70%; ii) NaH, MeI, THF, 50%; iii) XtalFluor-M, DBU, dry DCM, -78 oC to rt, 12%.
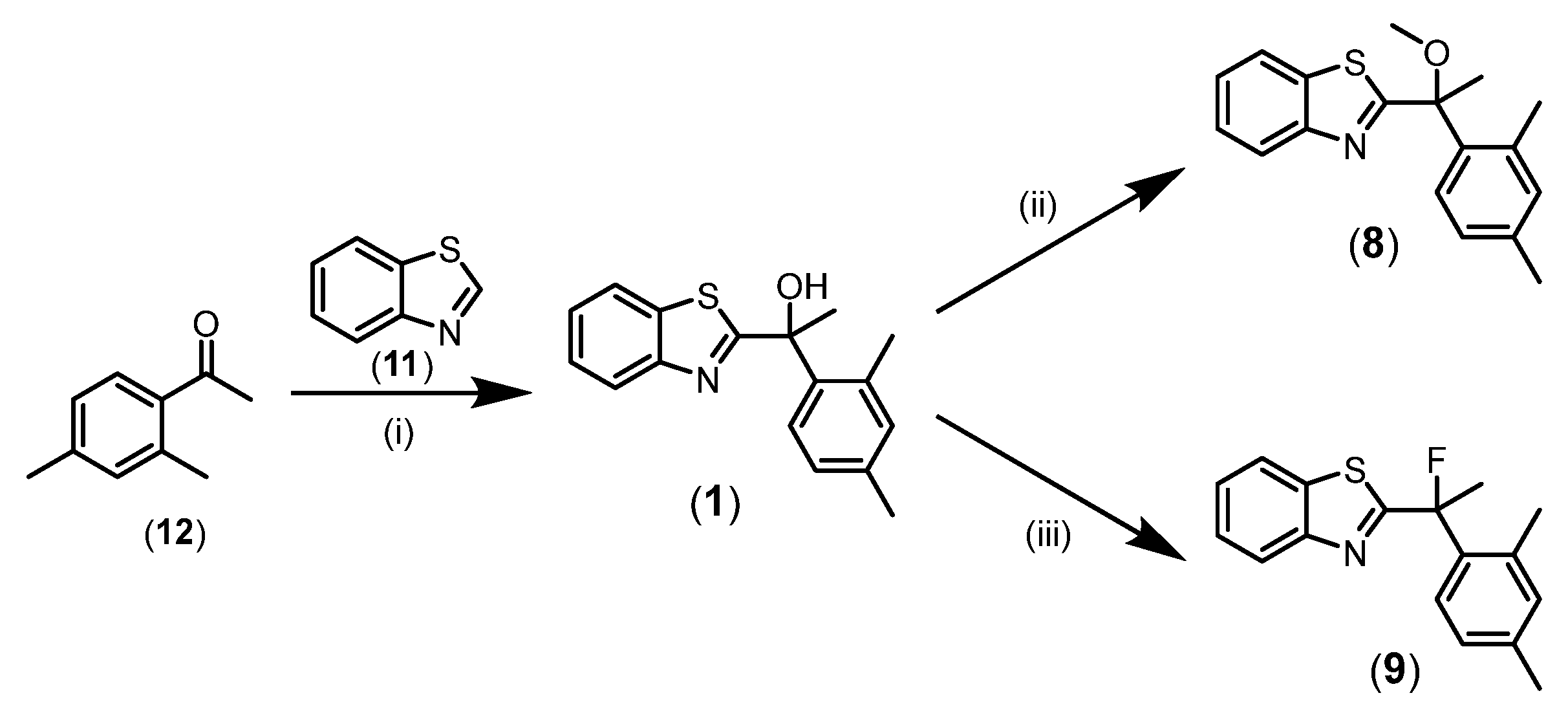
Figure 4.
Pharmacological evaluation of AC265347 (1) and derivatives for PAM activity using Ca2+i mobilisation assays. Concentration-response curves are shown for Ca2+o in the absence or presence of various concentrations of epoxide 6 (panel B), oxetane 7 (panel C), allylic alcohol by-product 7b (panel D), methoxy 8 (panel E) and fluoro 9 (panel F) compared to parent compound AC265347 (1) (panel A). Data are mean ± SD pooled from n≥3 independent experiments performed in duplicate. Curves through the data points are the best fit of Eq.1 to the data.
Figure 4.
Pharmacological evaluation of AC265347 (1) and derivatives for PAM activity using Ca2+i mobilisation assays. Concentration-response curves are shown for Ca2+o in the absence or presence of various concentrations of epoxide 6 (panel B), oxetane 7 (panel C), allylic alcohol by-product 7b (panel D), methoxy 8 (panel E) and fluoro 9 (panel F) compared to parent compound AC265347 (1) (panel A). Data are mean ± SD pooled from n≥3 independent experiments performed in duplicate. Curves through the data points are the best fit of Eq.1 to the data.
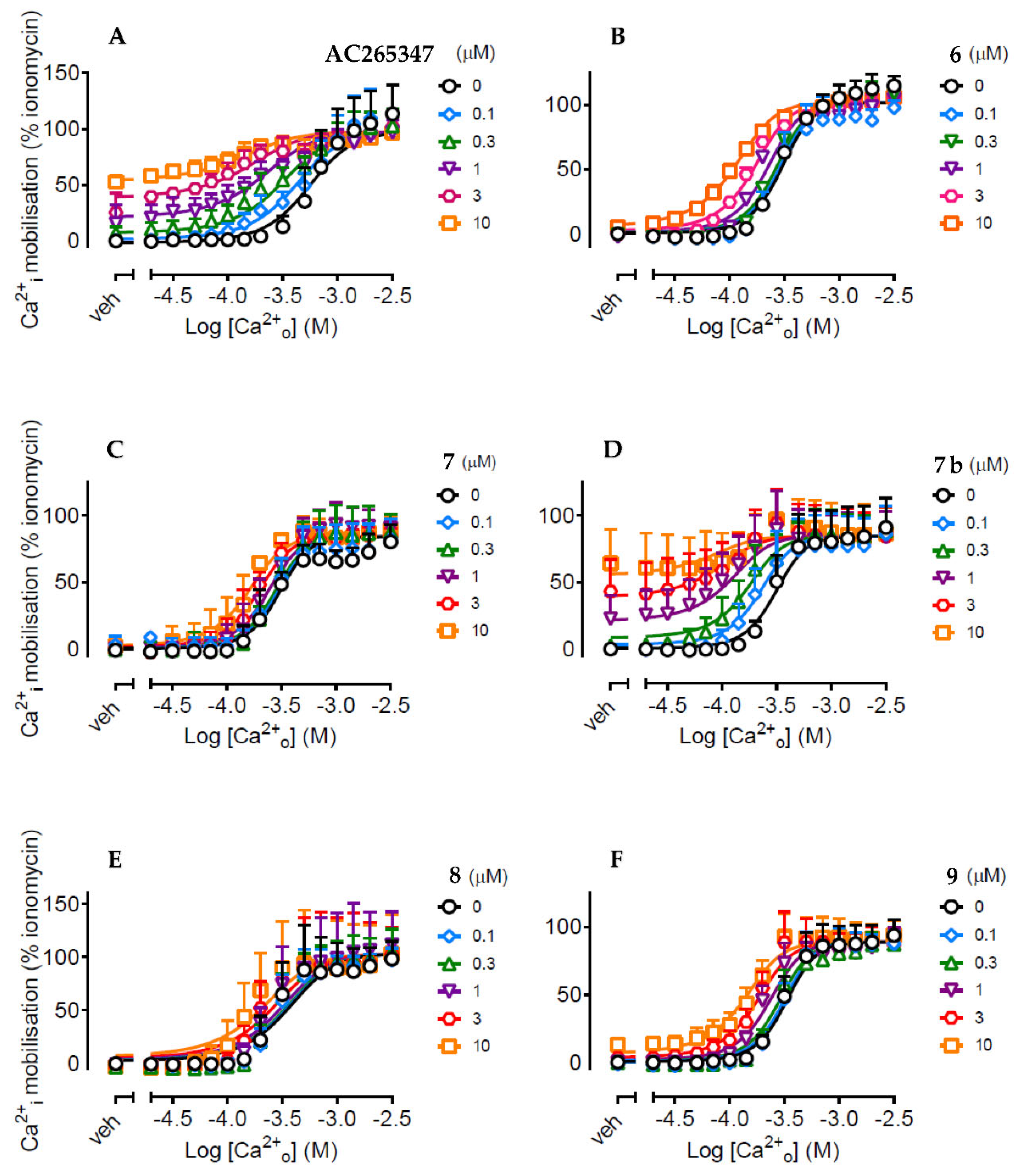
Figure 5.
Proposed binding mode of representative compounds: (A) AC265347 (1), (B) compound 9, (C) overlay of AC265347 (1) and compound 7b with a distance of 1.9 Å between the stereogenic carbon centres and (D) compound 6 in the CaSR 7TM in an active model derived from the Cryo-EM structure PDBID:7M3G [15].
Figure 5.
Proposed binding mode of representative compounds: (A) AC265347 (1), (B) compound 9, (C) overlay of AC265347 (1) and compound 7b with a distance of 1.9 Å between the stereogenic carbon centres and (D) compound 6 in the CaSR 7TM in an active model derived from the Cryo-EM structure PDBID:7M3G [15].
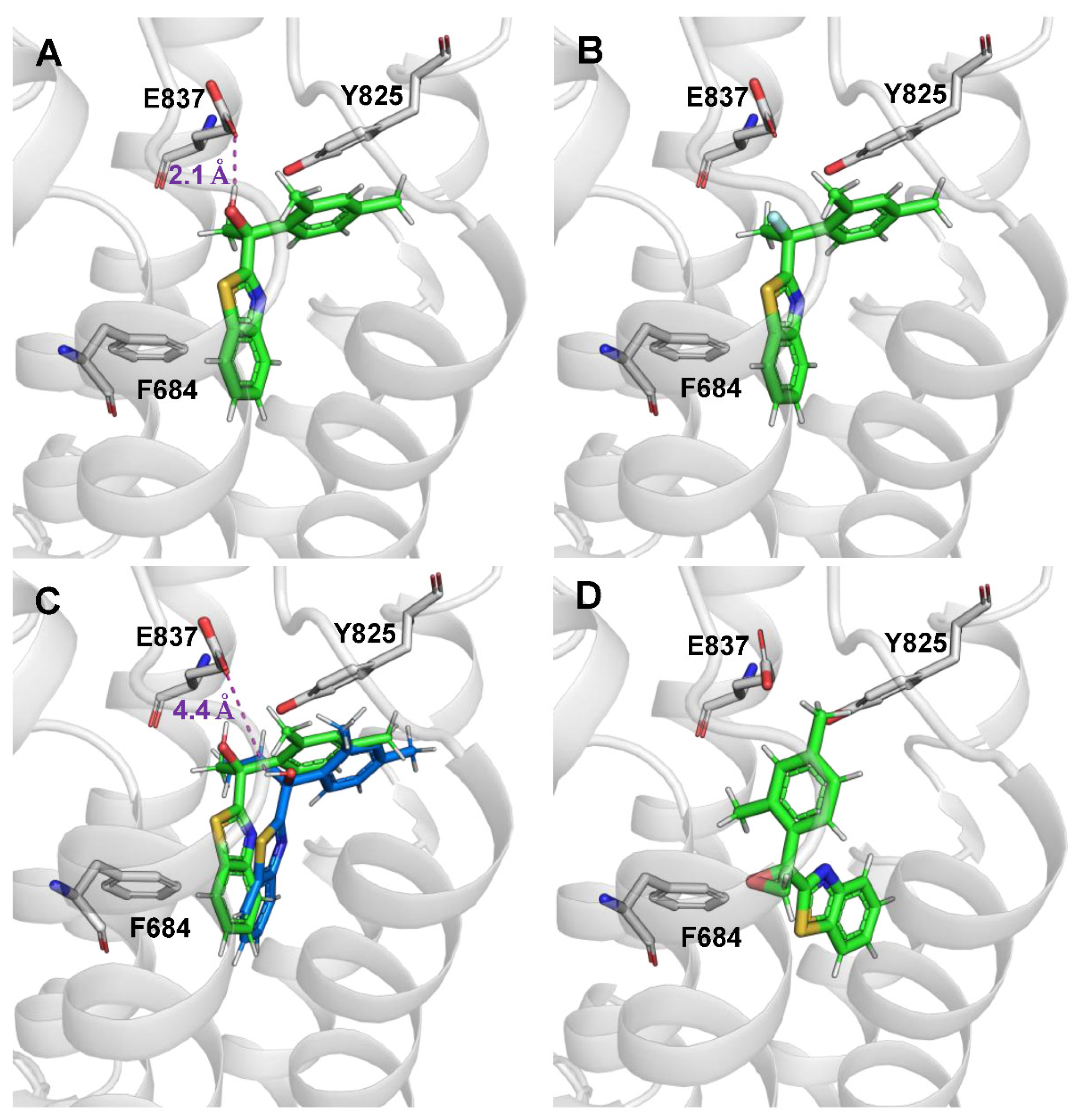

Table 1.
SAR surrounding the AC265347 hydroxy group and pharmacological profiles of compounds. Functional affinity (pKB), positive allosteric cooperativity (αβ) and intrinsic (allosteric) agonism (τB) were determined using Ca2+i mobilisation assays in FlpIn-Trex-HEK293-CaSR cells. Data are mean ± SD estimated from at least three independent experiments (n) performed in duplicate.
Table 1.
SAR surrounding the AC265347 hydroxy group and pharmacological profiles of compounds. Functional affinity (pKB), positive allosteric cooperativity (αβ) and intrinsic (allosteric) agonism (τB) were determined using Ca2+i mobilisation assays in FlpIn-Trex-HEK293-CaSR cells. Data are mean ± SD estimated from at least three independent experiments (n) performed in duplicate.
| Entry | Structure | pKB ± SD KB, μM) |
Logαβ ± SD (αβ) |
LogτB ± SD (τB) |
n |
|---|---|---|---|---|---|
| AC265347 (1)a | 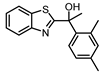 |
5.1 ± 0.09 (7.9) |
2.5 ± 0.9 (316) |
0.5 ± 0.2 (3.2) |
5 |
| 5a | 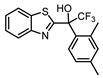 |
5.2 ± 0.2 (6.3) |
1.6 ± 0.9 (40) |
0.7 ± 0.2 (5.0) |
3 |
| 6 | 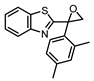 |
4.7 ± 0.4 (20) |
1.5 ± 0.4 (32) |
ND | 3 |
| 7 | 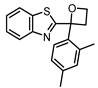 |
4.6 ± 0.2 (25) |
0.9 ± 0.1 (7.9) |
ND | 3 |
| 7b | 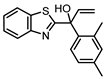 |
4.7 ± 0.4 (20) |
2.5 ± 0.4 (316) |
0.6 ± 0.4 (4.0) |
4 |
| 8 | 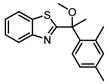 |
4.8 ± 0.6 (16) |
1.1 ± 0.5 (16) |
ND | 3 |
| 9 | 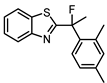 |
4.8 ± 0.5 (16) |
1.6 ± 0.3 (40) |
ND | 4 |
aPreviously published data [3]; ND - negligible or no response detected.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



