
Submitted:
25 December 2024
Posted:
26 December 2024
You are already at the latest version
Abstract
Dermatitis is commonly treated using Western medicine, systemic medication, or phototherapy. However, long-term monitoring and side effects remain a concern. Chinese herbal medicine has gained attention as an alternative. Although studies have explored the antioxidant and anti-inflammatory effects of quercetin (Q) and its derivative rutin (RT) on the skin, quercitrin (QI) and hyperoside (HP) have not yet been thoroughly investigated. This research aims to compare these four Chinese herbal medicines for their potential to improve dermatitis and explore their mechanisms. CM-H2DCFDA staining, Western blotting and ELISA analysis showed that Q exhibited the best antioxidant and anti-inflammatory effects, mainly inhibiting free radical production and affecting downstream pathways, including MEK1/2-Erk1/2-c-Fos, JNK1/2-c-Jun, and traditional IκB-NF-κB pathway. We also confirmed the anti-inflammatory effect of Q in a mouse model of atopic dermatitis (AD). In conclusion, though less potent than Q, its derivatives still possess anti-inflammatory and antioxidant abilities.
Keywords:
quercetin
; quercetin derivatives
; anti-oxidation
; anti-inflammation
; NF-kB
; reactive oxygen species
; mitogen-activated protein kinase
1. Introduction
The keratinocytes (KCs) are the skin’s epidermis’s main structural components and barrier function. Through mechanical and innate immune antibacterial mechanisms, KCs protect the body from external factors [1]. They interact with the immune system by altering the microbiome through lipid production [1]. These immune cells release various cytokines, chemokines, danger-associated molecular patterns (DAMPs), and lipid mediators [2,3]. KCs express Toll-like receptors (TLRs), including TLR1, -2, -3, -5, -6, and -10, which are receptors for pathogen antigen recognition. When triggered, they lead to the production of inflammatory cytokines and the initiation of immune responses [4,5,6]. Therefore, KCs in the epidermis play an important role in barrier functions and initiating immune responses.
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease, characterized by complex etiology, including genetic and environmental factors leading to epidermal and immune system abnormalities. It is characterized by itching, eczema, and skin barrier defects. Tumor necrosis factor-α (TNF-α) signaling in keratinocytes releases various pro-inflammatory cytokines and chemokines, such as interleukin (IL)-1β, IL-6, and IL-8 [7]. Additionally, TNF-α induces skin barrier dysfunction, thereby increasing skin permeability and promoting the infiltration of immune cells (including T cells and macrophages) into the skin [8]. This process makes the skin more susceptible to allergens and irritants, further exacerbating the inflammatory response in AD. Targeting TNF-α with anti-TNF-α therapies has shown promise in reducing inflammation and improving symptoms in AD [7,8].
When the skin is exposed to ultraviolet and visible light, ROS are also generated. They act as secondary signaling molecules within cells, exacerbating skin aging, inflammation, photosensitivity, and carcinogenesis. However, they can also regulate homeostasis in the human body. Both KCs and fibroblasts in the skin possess NADPH oxidase (NOX), which produces small amounts of ROS that can promote the growth and differentiation of both cell types. Additionally, both KCs and endothelial cells in the skin contain xanthine oxidase. Studies have indicated that ROS or lipid radicals are produced via XO when LPS binds to TLR, participating in the early inflammatory response of the skin. After 24 hours, free radicals are mainly produced through NOX activity. Therefore, the source of ROS seems to depend on the inflammatory state of the skin damage. ROS can also trigger various biological responses by activating transcription factors or kinases, such as AP-1, MAPK, and nuclear factor kappa B (NF-κB).
Quercetin (Q) is a plant flavonoid compound with antioxidative, anti-inflammatory, and photoprotective properties, making it a potential therapeutic application in dermatopathology. In 2020, HaCaT cells were stimulated with a mixture of 50 ng/mL IL-4, 50 ng/mL IL-13, and 10 ng/mL TNF-α to simulate AD conditions. Pre-treatment with 1.5 μM Q reduced the expression of IL-1β, IL-6, IL-8, Thymic stromal lymphopoietin (TSLP), matrix metalloprotease (MMP)-1, MMP-2, and MMP-9, but increased the expression of Superoxide dismutase-1 (SOD1), SOD2, Catalase (CAT), Glutathione peroxidase (GPx), and IL-10. Additionally, Q promotes wound healing through ERK1/2 and NF-κB but does not affect P-STAT6. Therefore, Q alleviates AD symptoms through its antioxidant and anti-inflammatory effects [9]. Q 3-O-glycosides (Q3Gs) are a major class of Q glycosides, with structures formed by glycosidic bonds at the C3 position of the Q carbon ring. Various glycosyl groups bind to the C3 position of the Q carbon ring to create different 3-O-glycosides. Quercitrin (QI), also known as 3,5,7,3’,4’-OH, 3-rhamnosyl Q, Q 3-O-R-L-rhamnoside, or 3-rhamnosyl Q, also has extensive biological activities and is thus considered a classic derivative of 3-O-glycosides [10]. Due to its glycoside form, QI has physical and chemical properties different from Q [11]. The Jegal et al., demonstrated that QI has the efficacy to treat allergies and allergy-related contact dermatitis [12]. Hyperoside (HP) is a type of Q3Gs, with a β-D-galactopyranoside residue (known as Q-3-O-β-D-galactopyranoside) attached at the C3 position of the Q carbon ring. HP possesses antioxidant properties, with the position of the -OH groups being one of the most critical factors. The 3’,4’-o-diphenol hydroxyl structure on the B ring confers strong antioxidant activity. The 5-OH and 7-OH groups also contribute to the strong antioxidant effect of HP, making it a good antioxidant. Its pharmacological effects include cancer prevention and protection of the brain, neurons, heart, kidneys, lungs, blood vessels, bones, joints, and liver. The Charachit et al., revealed in 2020 that QI and HP alleviates UVB-induced human keratinocyte damage and oxidative stress by regulating the MAPK and Akt pathways [13]. Rutin (RT) is also a type of Q3Gs, a non-toxic polyphenolic natural flavonoid compound known as Q-3-O-rutinoside and vitamin P. In addition to its bioactive properties of antioxidation, anti-inflammation, and anti-tumor activity, many studies are currently on its structure and activity in skin applications [14,15,16,17,18,19,20]. It has shown good therapeutic effects on skin aging [21,22] and treating inflammatory skin diseases such as psoriasis [23], atopic and allergic dermatitis [24]. RT has already been developed into a clinical drug, indicating that Q3Gs have become an important source of innovative medicines. Although many studies have investigated the effects of Q and RT on the skin, the comparative activities of Q3Gs are still largely unknown. This research aims to investigate and compare the antioxidant and anti-inflammatory properties of Q3Gs in an in vitro model using TNF-α to induce inflammation in KCs. Q and RT will act as positive controls. The anti-inflammatory effects of the most effective Chinese herb will also be validated in an in vivo mouse model of AD.
2. Results
2.1. Cell Toxicity of Q And Its Derivatives in HaCaT Cells
Using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to analyze the cytotoxicity of the Q and its derivatives on HaCaT cells, the results in Figure 1A show that after 24 hours, Q and its derivatives at concentrations below 5 µM do not cause significant cytotoxicity, with cell viability remaining above 90%. At concentrations below 50 µM, cell viability is still above 80%, possibly due to dilution with 0.1% DMSO (results not shown). For subsequent 24-hour experiments on Q and its derivatives, we might select concentrations of 50 µM or below for analysis. In addition, the results in Figure 1B show that after 24 hours, using 10 or 40 ng/mL TNF-α does not cause cell apoptosis. Therefore, subsequent studies will choose the appropriate drug concentration based on research needs, such as conditions that generate oxidative stress or inflammatory responses [25].
2.2. The Antioxidant Capacity of Q And Its Derivatives in HaCaT Cells
The antioxidant capacity of Q and its derivatives was assessed using the DPPH radical scavenging assay and the CM-H2DCFDA fluorescence labeling of intracellular radical production. As shown in Figure 2A, treating with 25 µM Q and its derivatives resulted in less than 50% DPPH radical scavenging ability. However, at 50 µM, the scavenging rate exceeded 50%, though the effect was still limited and contrary to other literature. Increasing the concentration of all drugs to 100 µM resulted in approximately 80% scavenging rate, but this concentration caused cytotoxicity in cells (except for RT). To avoid comparing the antioxidant activity of these drugs at the same concentration, subsequent experiments can use the highest safe concentration for each drug on HaCaT cells to observe their intracellular antioxidant capacity, potentially reflecting similar antioxidant effects as reported in other literature and indirectly comparing their DPPH radical scavenging rates. Further we investigated the intracellular antioxidant capacity of these drugs. HaCaT cells were pre-treated with 25 µM or 50 µM Q and its derivatives for 2 hours, followed by stimulation with 40 ng/mL TNF-α for 24 hours, and analyzed using CM-H2DCFDA fluorescence labeling. As shown in Figure 2B, TNF-α alone significantly increased intracellular ROS production, showing a twofold increase compared to the control group. Pre-treatment with Q and its derivatives significantly inhibited TNF-α-induced intracellular ROS production. Thus, these four drugs demonstrated excellent intracellular radical scavenging ability, with Q achieving the antioxidant effect of other derivatives at a lower dose (25 µM) compared to 50 µM [25].
2.3. The Anti-Inflammatory Ability of Q And Its Derivatives in HaCaT Cells [25]
We utilized TNF-α to induce an inflammatory response in HaCaT cells and observed the expression of different inflammatory mediators. TNF-α induced cyclooxygenase 2 (COX-2) expression increased over time. Further investigation of the anti-inflammatory effects of Q and its derivatives showed that pre-treatment with 25 µM Q and 50 µM QI, HP, or RT effectively inhibited TNF-α-induced COX-2 protein expression. However, it was not possible to determine which drug was most effective (results not shown). Therefore, we pre-treated HaCaT cells with a lower dose of 10 µM. The results showed that at 24 and 48 hours, COX-2 expression induced by TNF-α was still inhibited, with QI and HP showing slightly better inhibitory effects (Figure 3A). We also observed whether TNF-α induced intercellular adhesion molecule (ICAM)-1 expression, an important adhesion protein in monocyte adhesion to skin cells during inflammatory responses. As shown in Figure 3C, TNF-α induced ICAM-1 protein expression at 24 hours, and only Q at 10 µM significantly inhibited TNF-α-induced ICAM-1 expression, followed by HP, although the effect was insignificant. QI and RT had no significant impact on ICAM-1 expression. Additionally, whether Q and its derivatives affect TNF-α induced the expression of pro-inflammatory cytokines IL-6 and IL-8, as shown in Figure 3B, TNF-α significantly induced the production of IL-6 and IL-8. However, at the same concentration, only Q significantly inhibited TNF-α-induced IL-6 and IL-8 production, while the Q derivatives only slightly inhibited IL-8 expression. In conclusion, Q and its derivatives had the best anti-inflammatory effects, with Q showing the most significant impact on inflammatory mediators.
2.4. The Effect of Q on DNCB-Induced AD Mice Skin Damage and Inflammation [26]
Based on the results of cellular experiments, we used Q, the best-performing herbal extract among the four tested, in a DNCB-induced AD mice model to verify its anti-inflammatory effects in vivo. The dorsal skin of the female mice was shaved and sensitized cutaneously (skin smear) with DNCB. Mice were then given Q (40 μM) cutaneously from days 14 to 28 (Figure 4A). Compared to normal mice, DNCB-sensitized mice exhibited significant AD-like symptoms on the shoulder and ears, including edema, erythema, scarring, and epidermal peeling (Figure 4B). Since DNCB causes ear swelling in this experimental model, we measured ear thickness on day 28. The AD (DNCB-sensitized mice) group showed significant ear swelling compared to the AD + Q (40 μM) group, which significantly inhibited ear swelling in mice (Figure 4B, right panel). To study the effect of Q on AD-like skin lesions, ear and shoulder back skin sections were stained with H/E to examine epidermal thickness and eosinophil infiltration. As shown in Figure 4C, compared to the Normal group, the AD group exhibited thickened epidermal layers and increased eosinophil infiltration (Figure 4C, middle panel). The AD + Q (40 μM) group significantly reduced epidermal thickness and eosinophil infiltration compared to the AD group (Figure 4C, right panel). Additionally, mast cell infiltration in the ears and shoulder back skin was also inhibited (Figure 4D). Next, we examined antibody and cytokine levels to assess whether quercetin can modulate allergic or inflammatory responses in the serum. Compared to the AD group, the AD + Q (40 μM) group significantly reduced the levels of COX-2, ICAM-1, IL-4, and IFN-γ (Figure 4E). Furthermore, the AD + Q (40 μM) group also significantly inhibited the production of IgE (results not shown). These results indicate that topical application of Q alleviates symptoms and inflammation in AD-like mice [26].
2.5. Effect of Q and Its Derivatives on TNF-α Induced Activation of MAPK Pathways [25]
Further, we investigate whether Q and its derivatives affect the activity of Erk1/2, JNK1/2, and c-Jun, thereby influencing TNF-α-induced ICAM-1 expression. As shown in Figures 5A,B, pre-treatment of cells with 50 µM Q and its derivatives significantly inhibited the phosphorylation of Erk2 and JNK1, both expressed the prominent phenotype in HaCaT cells, after 30 minutes of TNF-α stimulation, with Q showing the most potent inhibitory effect, while QI significantly inhibited JNK1 phosphorylation at 5 minutes. Since the transcription factors of ICAM-1 include AP-1 (such as c-Jun and c-Fos), we further observed whether Q and its derivatives affect AP-1 phosphorylation. Figure 5C shows that only Q significantly inhibited c-Jun phosphorylation at different time points. Based on the results from Figure 3C, Figure 4, and Figure 5B,C, we can infer that Q has the best anti-inflammatory ability among Q and its derivatives. It mainly inhibits the activation of the JNK1/c-Jun or MEK1/2-Erk1/2-c-Fos pathways, thereby reducing TNF-α-induced ICAM-1 expression or the production of other inflammatory mediators, such as IL-6 and IL-8, thus reducing the inflammatory response [25].
2.6. Effect of Q and Its Derivatives on TNF-α Induced NF-κB (p65) Pathway [25]
NF-κB is involved in many inflammatory responses and mediators expression. Once IκBα and NF-κB (p65) are phosphorylated, the phosphorylated IκBα separates from NF-κB (p65) in the cytoplasm and follows a degradation pathway. The phosphorylated NF-κB (p65) gains kinase activity after separating from IκBα and translocates to the nucleus to initiate gene transcription. Therefore, we further investigated whether Q and its derivatives affect the activation of the NF-κB (p65) pathway downstream of TNF-α-TNFR signaling. Figure 6A shows that TNF-α induced strong IκBα phosphorylation at 5 minutes, followed by degradation at 10-15 minutes. However, Q and its derivatives had little effect on IκBα phosphorylation. At 30-60 minutes, newly synthesized IκBα was observed, and Q and its derivatives influenced IκBα phosphorylation at 30 minutes. Among these drugs, only Q significantly inhibited TNF-α induced IκBα phosphorylation at 30 minutes [25].
Furthermore, as shown in Figure 6B, Q and its derivatives inhibited NF-κB (p65) phosphorylation between 15-30 minutes. We further observed whether Q and its derivatives affect the translocation of phosphorylated NF-κB (p65) (with kinase activity) to the nucleus. TNF-α stimulation for 30 minutes was used as the condition for NF-κB (p65) nuclear translocation. As shown in Figure 6C, the separation of nuclear/cytoplasmic proteins indicated that TNF-α stimulation for 30 minutes significantly reduced the amount of NF-κB (p65) in the cytoplasm and significantly increased its amount in the nucleus. Q and its derivatives slightly reduced the translocation of NF-κB (p65) from the cytoplasm to the nucleus. The fluorescence staining results in Figure 6D also confirmed that phosphorylated NF-κB (p65) was concentrated in the nucleus under TNF-α stimulation. However, pre-treatment with Q and its derivatives caused phosphorylated NF-κB (p65) to disperse in both the cytoplasm and nucleus, but the inhibition of NF-κB (p65) nuclear translocation was not very significant. Based on these results, we speculate that Q and its derivatives mainly affect TNF-α induced phosphorylation of IκBα and NF-κB (p65), thereby influencing the expression of inflammatory proteins such as COX-2 and ICAM-1 [25].
2.7. Intracellular Signaling Pathways Involved in TNF-α Induced Expression of COX-2 and ICAM-1
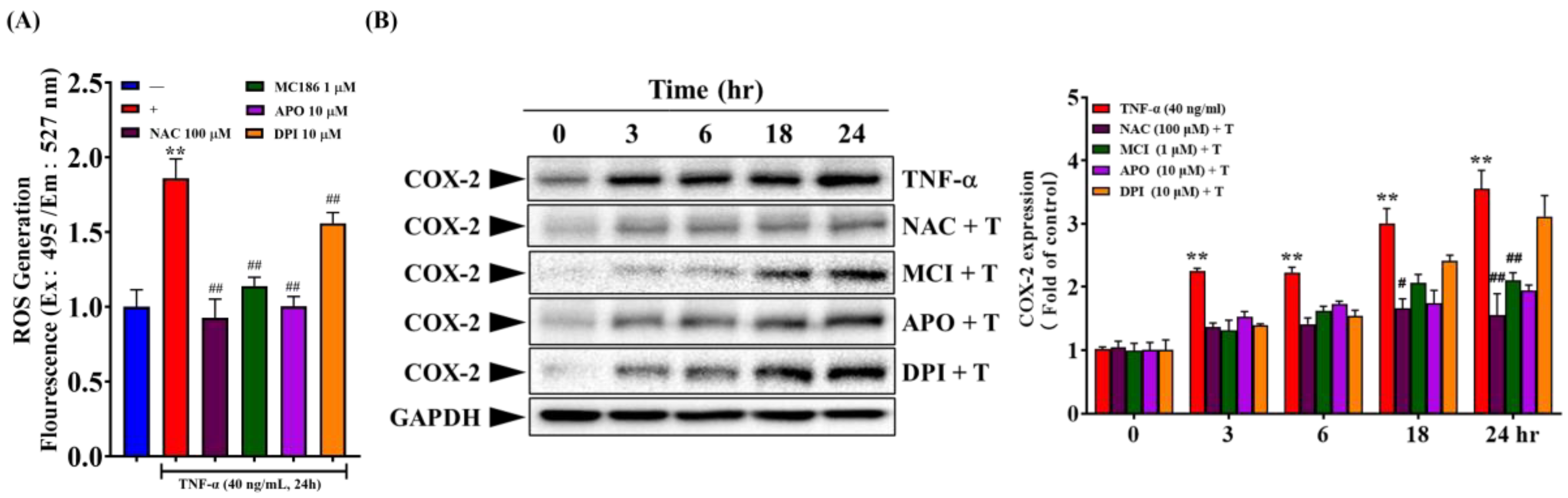
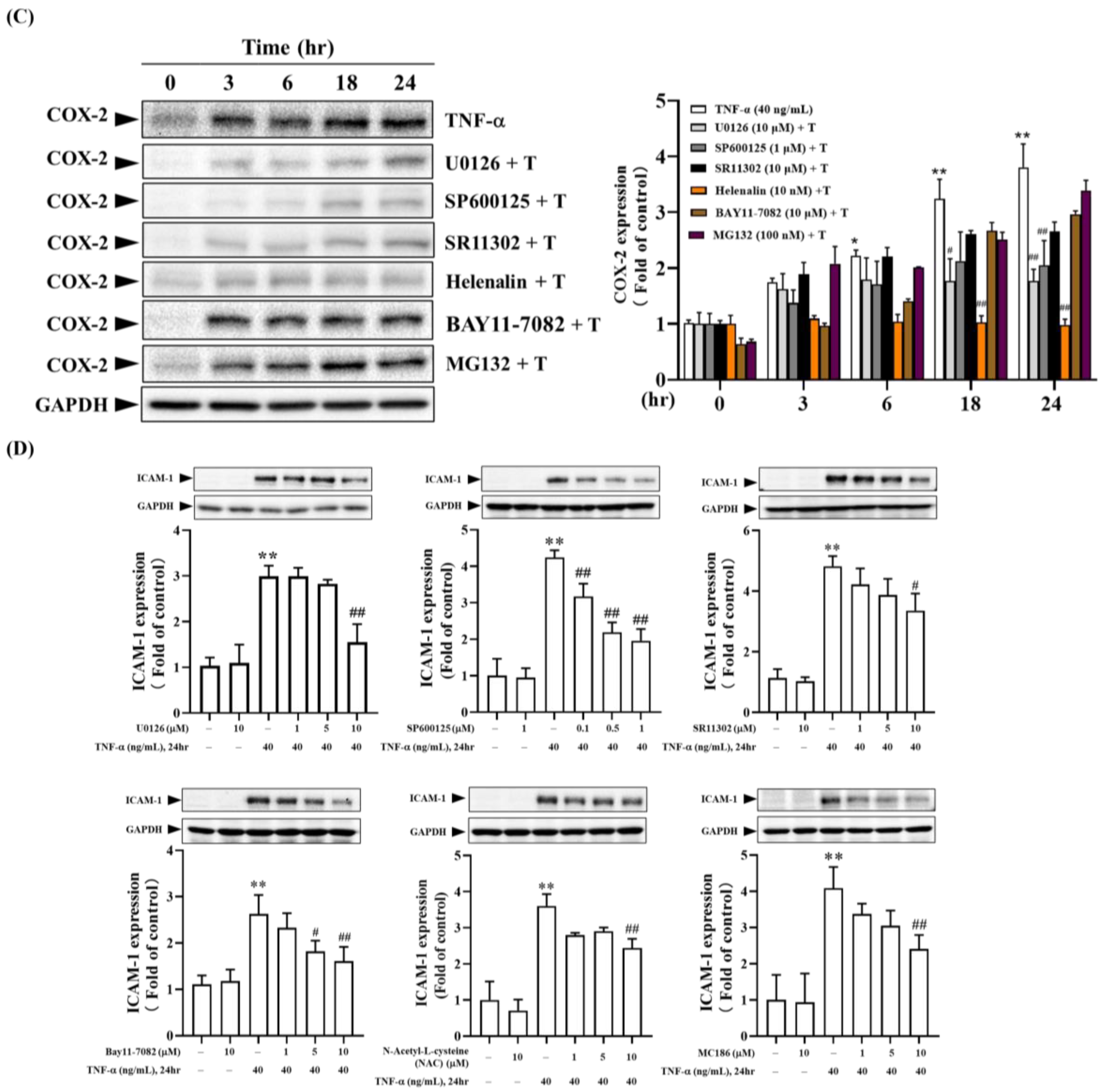
The downstream signaling pathways of TNF-α-TNFR are involved in many skin inflammation and the expression of inflammatory mediators. As indicated in Figure 2B, the binding of TNF-α to TNFR results in the overproduction of intracellular free radicals. Therefore, we analyzed whether intracellular free radicals participate in TNF-α induced expression of COX-2 and ICAM-1. As shown in Figure 7A, pre-treatment of cells with broad-spectrum free radical scavengers (NAC and MCI186) and NOX inhibitors (APO and DPI) effectively inhibited the production of intracellular free radicals. Additionally, these free radical inhibitors affected the expression of COX-2 and ICAM-1, showing time and dose-dependent inhibition of TNF-α-induced expression of COX-2 and ICAM-1 (Figure 7B,D). Further, we investigate whether the Mitogen-Activated Protein Kinases (MAPKs) pathway participates in TNF-α induced expression of COX-2 and ICAM-1. As shown in Figure 7C,D, pre-treatment of HaCaT cells with U0126 (MEK1/2 inhibitor), SP600125 (JNK1/2 inhibitor), and SR11302 (AP-1 inhibitor) demonstrated dose-dependent inhibition of TNF-α-induced expression of COX-2 and ICAM-1. Notably, 10 µM U0126 and SR11302 and 0.1~1 µM SP600125 significantly inhibited TNF-α induced ICAM-1 expression. Additionally, high concentrations of U0126 and SP600125 significantly inhibited TNF-α-induced COX-2 expression after 24 hours.
Based on Figure 6, we know that the binding of TNF-α to TNFR activates the NF-κB pathway. We further investigated whether the NF-κB pathway participates in TNF-α induced expression of COX-2 and ICAM-1. Figure 7 shows that BAY11-7082 (IκBα pharmacological inhibitor) exhibited dose-dependent significant inhibition of TNF-α-induced ICAM-1 expression. However, it had a less substantial effect on COX-2 expression (Figure 7B). MG132 (proteasome inhibitor) had little impact on TNF-α-induced ICAM-1 expression, indicating that the degradation of phosphorylated IκBα does not play a critical role in regulating the expression of inflammatory mediators. On the other hand, Helenalin (NF-κB inhibitor) significantly inhibited TNF-α induced COX-2 expression, suggesting that NF-κB activation plays a vital role in TNF-α induced COX-2 expression (Figure 7B). In summary, the binding of TNF-α to TNFR induces the production of intracellular free radicals, further activating downstream MEK1/2-Erk1/2-c-Fos, JNK1/2-c-Jun, and the conventional IκBα-NF-κB pathways, which regulate the expression of ICAM-1 and COX-2 [25,27].
3. Discussion
This research compares four Chinese herbal medicines regarding their effectiveness in improving dermatitis. Utilizing CM-H2DCFDA staining, Western blotting, and ELISA analysis, it was found that herbal medicine Q exhibited the best antioxidant and anti-inflammatory effects. Q primarily inhibits free radical production and affects downstream pathways, including MEK1/2-Erk1/2-c-Fos, JNK1/2-c-Jun, and the traditional IκBα-NF-κB pathway. The anti-inflammatory effects of Q were further validated using a mouse model of AD. Although Q’s derivatives are less potent, they still possess notable anti-inflammatory and antioxidant properties.
Mitochondrial production of reactive oxygen species (ROS) is typically regarded as a key contributor to oxidative stress. During skin infections and inflammation, neutrophils and macrophages exhibit vigorous NOX activity, leading to the production of large amounts of O2•- in the skin. Additionally, ROS-induced lipid oxidation and iron overload contribute to ferroptosis, which plays a role in the development of skin conditions such as psoriasis, skin cancer, and collagen diseases [28]. Therefore, if Chinese herbal medicines can reduce ROS production within cells, they may slow or prevent the progression or worsening of skin diseases.
Results from Figure 2 demonstrated that Q and its derivatives at 25 µM exhibited less than 50% DPPH radical scavenging ability, indicating moderate antioxidant capacity. At 50 µM, the scavenging rate exceeded 50%, showing a more potent effect, although still limited compared to other studies. At 100 µM, the scavenging rate reached approximately 80%, indicating high antioxidant capacity, but this concentration caused cytotoxicity in cells (except for RT). Q achieved significant intracellular radical scavenging at a lower dose (25 µM), while its derivatives required 50 µM to achieve similar effects. Therefore, in terms of antioxidant action, Q is more effective at lower concentrations compared to its derivatives.
In the study illustrated in Figure 3, TNF-α was used to induce an inflammatory response in HaCaT cells, and various inflammatory mediators were measured. Pre-treatment with 25 µM of Q and 50 µM of QI, HP, or RT effectively inhibited TNF-α-induced COX-2 expression, with QI and HP demonstrating slightly better effects at a lower dose of 10 µM. For ICAM-1 expression, only 10 µM of Q significantly inhibited TNF-α induction, followed by HP. Q also significantly suppressed the production of pro-inflammatory cytokines IL-6 and IL-8, while the derivatives had lesser effects. In conclusion, Q demonstrated the most pronounced anti-inflammatory effects among the compounds tested.
AD is a chronic inflammatory skin condition closely associated with inflammation and oxidative stress. Oxidative stress can damage keratinocytes (skin cells), increase dermal inflammation, and compromise skin barrier function [29], exacerbating AD symptoms such as itching, redness, and skin lesions [30]. To verify its anti-inflammatory effects in vivo, the study utilized Q, the top-performing herbal extract, in a DNCB-induced AD mouse model. Results from Figure 4 indicated that DNCB-sensitized mice showed significant AD-like symptoms, including edema, erythema, scarring, and epidermal peeling. Treatment with Q (40 μM) significantly reduced ear swelling and diminished epidermal thickness, as well as eosinophil and mast cell infiltration in the skin. Additionally, Q significantly lowered levels of COX-2, ICAM-1, IL-4, IFN-γ, and IgE in the serum. Overall, topical application of Q effectively alleviated symptoms and inflammation in AD-like mice, demonstrating its strong anti-inflammatory efficacy.
The effects of Q and its derivatives on several signaling pathways related to the expression of TNF-α-induced inflammatory mediators were analyzed. Pre-treatment with 50 µM of Q significantly inhibited the phosphorylation of Erk2, JNK1, and c-Jun, showing the most potent effect among the tested compounds. This inhibition decreases the activation of the JNK1/c-Jun and MEK1/2-Erk1/2-c-Fos pathways, resulting in reduced ICAM-1 expression and lower production of inflammatory mediators such as IL-6 and IL-8 (Figure 5).
Additionally, Q and its derivatives affected the NF-κB pathway. Although their effects on IκBα phosphorylation were limited, Q was notably more effective at inhibiting the phosphorylation of both IκBα and NF-κB (p65) at the 30-minute mark, compared to all other tested compounds. This inhibition also impacted the translocation of NF-κB (p65) to the nucleus, thereby reducing its activity (Figure 6). In summary, Q demonstrated significant inhibitory effects on inflammatory signaling pathways, particularly JNK1/c-Jun, MEK1/2-Erk1/2-c-Fos, and NF-κB, resulting in decreased expression of COX-2, ICAM-1, and other inflammatory mediators.
The study investigated how the downstream signaling pathways of TNF-α-TNFR are involved in skin inflammation and the expression of inflammatory mediators. When TNF-α binds to its receptor TNFR, it leads to the overproduction of intracellular free radicals. Pre-treatment with broad-spectrum free radical scavengers and NOX inhibitors effectively decreased free radical production and reduced the expression of COX-2 and ICAM-1. Additionally, the MAPK pathway plays a role in TNF-α-induced expression of COX-2 and ICAM-1, with specific inhibitors U0126, SP600125, and SR11302 demonstrating dose-dependent inhibition (see Figure 7A,B). The NF-κB pathway, activated by the binding of TNF-α to TNFR, shows that inhibitors such as BAY11-7082 significantly decrease ICAM-1 expression, though the effect on COX-2 expression is less pronounced. Furthermore, Helenalin was found to significantly inhibit TNF-α-induced COX-2 expression, underscoring the critical role of NF-κB in COX-2 regulation (refer to Figure 7C,D). In summary, the binding of TNF-α to TNFR triggers the production of intracellular free radicals, activating the MEK1/2-Erk1/2-c-Fos, JNK1/2-c-Jun, and IκBα-NF-κB pathways, which together regulate the expression of ICAM-1 and COX-2.
The compounds Q and its derivatives (Q3Gs) examined in this study are classified as flavonoids. The structural variations among these compounds primarily stem from the number and positioning of hydroxyl groups in the flavone structure, which in turn influence their biological activities [31,32]. For instance, an increased number of hydroxyl groups can enhance their antioxidant and anti-inflammatory properties. HP shares a structural similarity with RT and Q, but it contains two additional hydroxyl groups in the flavone part. These hydroxyl groups, along with a glucose moiety, boost its antioxidant and anti-inflammatory effects by scavenging free radicals and inhibiting inflammation. Furthermore, the presence of the 3’,4’-o-diphenol hydroxyl structure on the B ring, as well as the 5-OH and 7-OH groups, significantly contribute to its potent antioxidant effects [33,34]. Additionally, the double bond between C-2 and C-3 has also been linked to increased antioxidant activity [35]. However, the results from Figure 2 and Figure 3A,B indicate that HP does not significantly outperform Q and other Q3Gs in terms of antioxidant and anti-inflammatory effects. Research has demonstrated that certain Q3Gs are absorbed more efficiently than the Q aglycone when taken orally. Glycosylation improves the water solubility of Q, thereby enhancing its bioavailability [32]. QI, on the other hand, is characterized by low lipophilicity, poor membrane permeability, and limited oral bioavailability, which restrict its applications in the food and pharmaceutical sectors. Modifying the hydroxyl groups on the core or the glycoside side chain of QI by adding different acyl groups can enhance its lipid solubility and bioavailability [36]. The glycoside component of QI, when combined with the aglycone, significantly improves its solubility in polar solvents and facilitates absorption via glucose transporters located in the intestinal mucosa [37]. Consequently, QI—which includes both glycosides and aglycone—is absorbed more readily than Q or other Q derivatives [38]. Due to its unique chemical structure and the two hydroxyl groups on the catechol B ring, QI can participate in ROS scavenging both indirectly (by chelating or binding to Fe2+) and directly, displaying excellent antioxidant capabilities. Nonetheless, results from Figure 2 reveal that QI still does not exhibit superior antioxidant effects.
Q is notable for its significant anti-inflammatory and antioxidant properties compared to the other compounds studied, consistent with findings from previous studies [31]. In plants, Q predominantly exists as water-soluble glycosides, which are converted into their aglycone form by β-glucosidases in the small intestine. This conversion facilitates passive diffusion through the intestinal lining, potentially aided by the sodium/glucose cotransporter-1 [39,40]. Once in the body, Q can be oxidized to form derivatives such as quercetin-quinone and quercetin-quinone methides [41,42]. The unabsorbed portion of Q is degraded by colonic microbiota, producing phenolic acids that are then absorbed and transported to the liver for further conjugation [43,44]. Therefore, Q can enter cells either through passive diffusion or by utilizing transport proteins [45]. However, Q has poor water solubility and undergoes significant first-pass metabolism, resulting in low bioavailability, which limits its clinical applications. Recent studies have investigated nanoformulations and enzymatically modified forms of Q to enhance its uptake and delivery [31]. Inside the cell, Q interacts with various cellular components and binds to cellular receptors and signaling molecules [45]. For instance, Q can interfere with the binding of the viral Spike protein to cellular ACE2, as indicated by in-silico docking studies [46,47]. Molecular docking has also demonstrated Q’s capacity to form stable complexes with proteins such as mTOR, Parkin, AKT, Beclin-1, P62, LC3A, Pink-1, and PI3K, which are involved in cell growth, proliferation, and survival. By modulating these pathways, Q can influence cellular processes such as autophagy and apoptosis [48].
Although both Q and its derivatives (Q3Gs) provide significant antioxidant and anti-inflammatory benefits for the skin, Q may have a slight advantage in overall effectiveness, particularly for soothing and protecting sensitive skin. However, glycosides typically have better bioavailability due to their enhanced solubility and absorption [32], potentially making them more effective for therapeutic purposes. Therefore, the optimal choice depends on individual skin needs and responses.
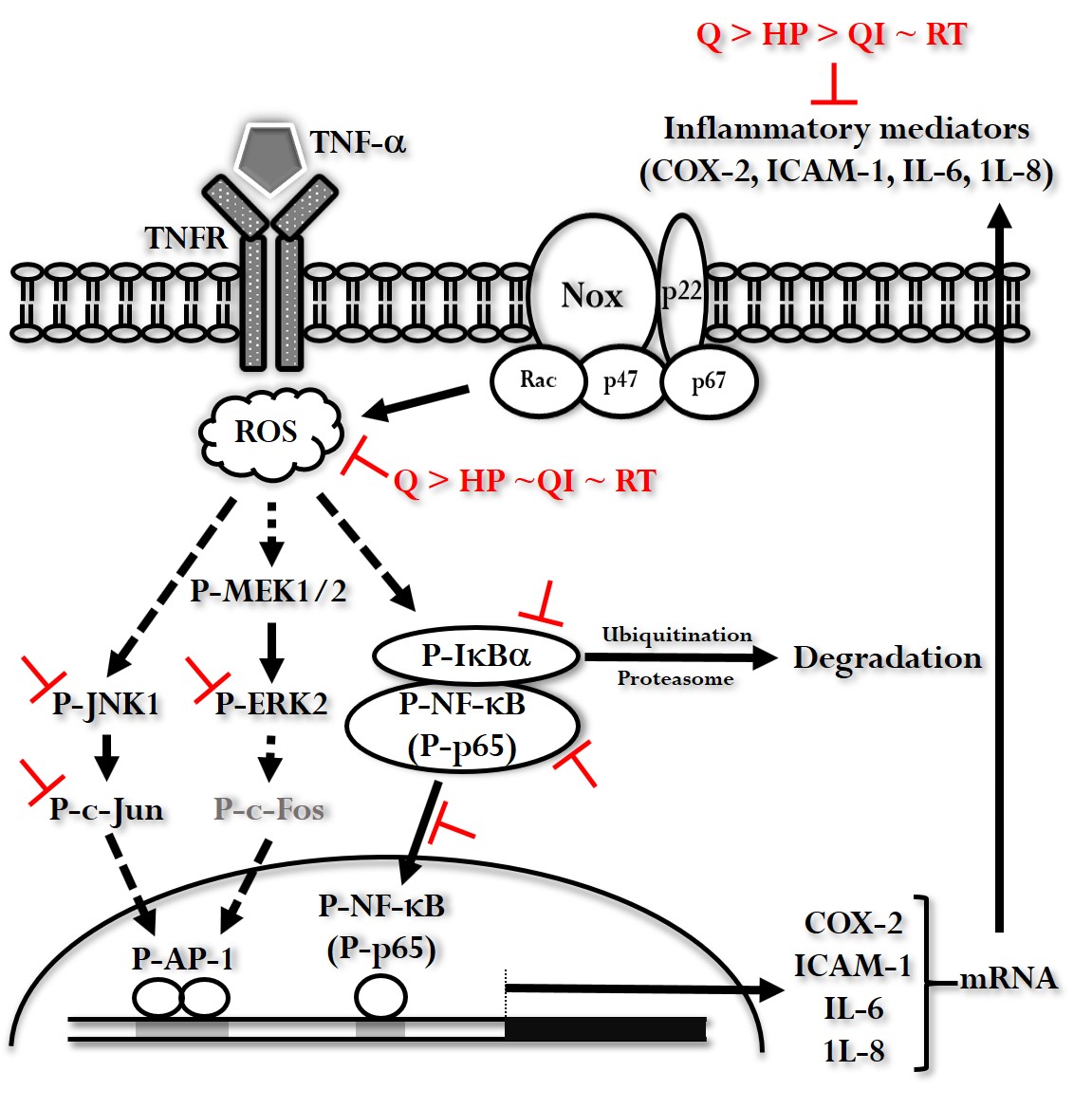
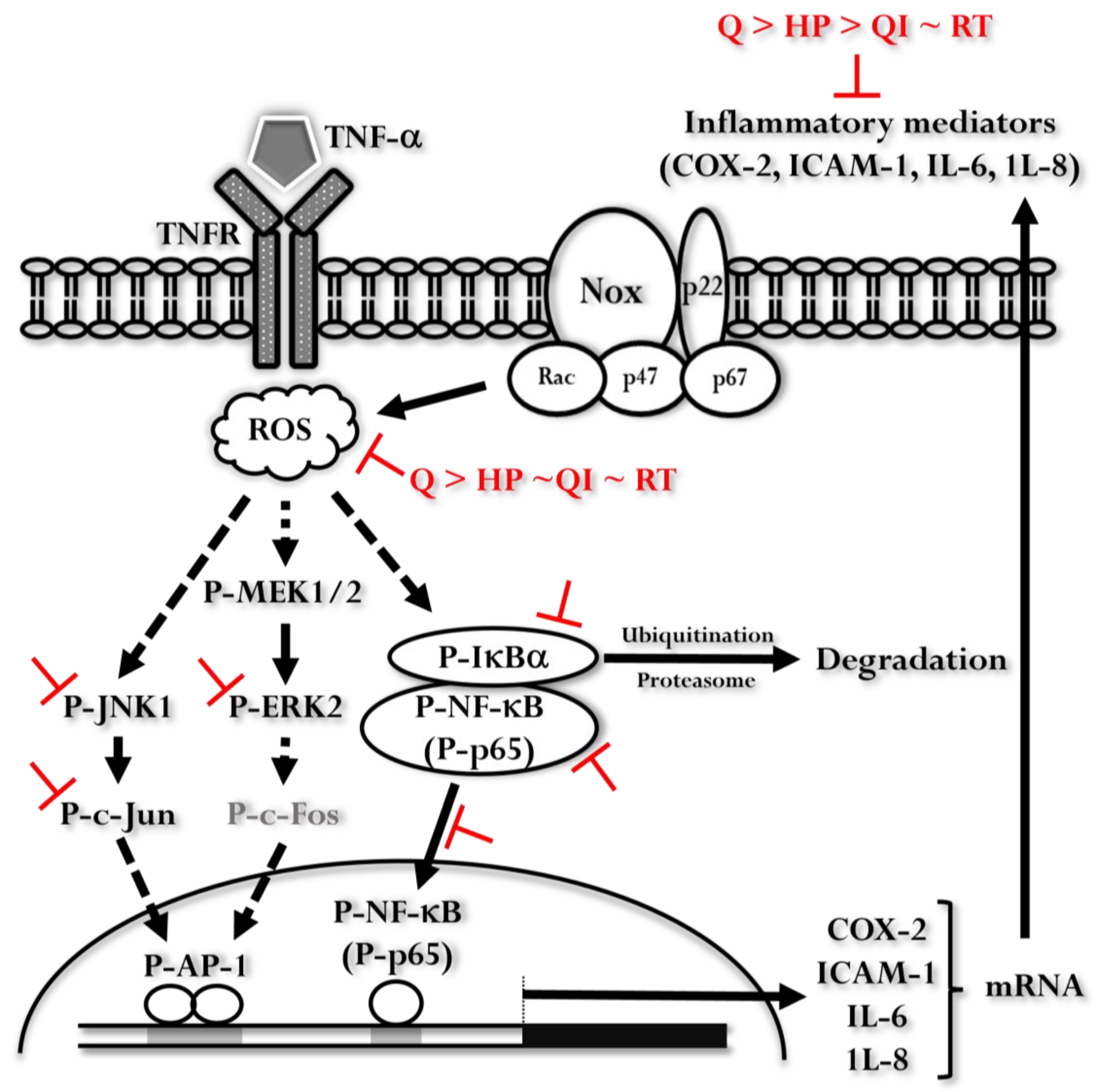
Figure 8.
The illustration represents the signaling pathways influenced by Q and its derivatives in the context of TNF-α-induced inflammatory responses. When TNF-α binds to its receptor (TNFR), it leads to an increase in reactive oxygen species (ROS) and the activation of the MEK1/2-Erk2, JNK1-c-Jun, and IκBα-NF-κB pathways. These activations ultimately result in the expression of inflammatory mediators such as COX-2, ICAM-1, IL-6, and IL-8. Q and its derivatives inhibit various steps in these pathways. Among the tested compounds, Q exhibits the most significant inhibitory effect.
Figure 8.
The illustration represents the signaling pathways influenced by Q and its derivatives in the context of TNF-α-induced inflammatory responses. When TNF-α binds to its receptor (TNFR), it leads to an increase in reactive oxygen species (ROS) and the activation of the MEK1/2-Erk2, JNK1-c-Jun, and IκBα-NF-κB pathways. These activations ultimately result in the expression of inflammatory mediators such as COX-2, ICAM-1, IL-6, and IL-8. Q and its derivatives inhibit various steps in these pathways. Among the tested compounds, Q exhibits the most significant inhibitory effect.
4. Materials and Methods
4.1. Cell Viability Analysis
The 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess the cytotoxicity of drugs in cells. Human keratinocyte cells (HaCaT cells, from the Cell Line Services) were seeded in a 96-well plate with Dulbecco’s modified medium (DMEM, Gibco BRL) containing 10% fetal bovine serum (FBS, HyClone), penicillin-resistant streptomycin (Thermo Fisher Scientific Inc., MA, USA) and 0.25 µg/mL amphotericin B solution (Merk Sigma-Aldrich, Darmstadt, Germany). The cells were cultured at 37 ° C in a 5% CO2 incubator. When cell confluence reached approximately 80%, the medium was replaced with serum-free 0% FBS DMEM for 24 hours, followed by treatment with different drug concentrations for specified times. After treatment, the medium was replaced with 0.5 mg/mL of MTT reagent and cells were incubated at 37 °C with 5% CO2 for 1 hour. Purple crystals formed and the supernatant was carefully aspirated from the wells. Next, 100 µL of dimethyl sulfoxide (DMSO) was added to dissolve the purple crystals, and the plate was incubated in the dark for 30 minutes. The optical density (OD570) was measured. Cell viability was calculated using the formula: Cell viability= (Absorbance of the test sample - Absorbance of the blank group / Absorbance of the untreated control group) × 100%.
4.2. Antioxidant Activity Detection
4.2.1. DPPH Radical Scavenging Assay
Prepare a 200 µM DPPH (Merk Sigma-Aldrich) solution in ethanol. Prepare 25 or 50 µM concentrations of Chinese herbal medicines and vitamin C (used as a positive control) in 0.1 M phosphate buffer. Add 10 µL of each test sample to a transparent 96-well plate, followed by 100 µL of DPPH reagent. Incubate for 12 minutes. Analyze absorbance at 517 nm using a SpectraMax® i3x multifunctional microplate reader (Molecular Devices, San Jose, CA, USA). Calculate the radical scavenging rate using the formula: Radical clearance rate (%) = 1 - [(ODsample- ODblank)/(ODcontrol - ODblank)] × 100.
4.2.2. Intracellular Free Radical Measurement
CM-H2DCFDA (Invitrogen™) fluorescence staining assessed intracellular free radical levels—culture HaCaT cells in a 96-well black plate with transparent bottom. When cell confluence reaches approximately 80%, replace the medium with serum-free DMEM for 24 hours. Pretreat cells with different concentrations of Q and its derivatives or free radical scavengers (N-Acetyl-L-cysteine (NAC), MCI186, Apocynin (APO), Diphenyleneiodonium (DPI) (Merk Sigma-Aldrich) for 2 hours, followed by stimulation with 40 ng/mL TNF-α (R&D Systems, Minneapolis, MN, USA) for 24 hours, replace the medium with 8 µM CM-H2DCFDA fluorescent dye (prepared in DMSO and diluted in 0% FBS DMEM) and incubate in the dark at 37 ° C with 5% CO2 for 30 minutes. Wash with PBS and add 100 µL of PBS. Analyze fluorescence using a multifunctional SpectraMax® i3x microplate reader (Ex: 495/9 nm; Em: 527/15 nm).
4.3. Anti-Inflammatory Activity Detection
4.3.1. Detection of COX-2 and ICAM-1 Inflammatory Proteins
Western Blotting was used to detect COX-2 and ICAM-1 expression. After treating HaCaT cells with Q (from Merk Sigma-Aldrich) and its derivatives (QI from ChemFaces, Wuhan, China; HP from Nature Standard, Shanghai, China; RT from Merk Sigma-Aldrich), use RIPA lysis buffer to lyse them and collect total protein. Quantify protein concentration using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific Inc.). 30% Acrylamide/Bis Solution, 29:1 (Bio-Rad Laboratories, CA, USA) is used to prepare 10% separation gel (lower gel) and 6% stacking gel (upper gel). Loading each sample into the wells of the stacking gel. Start electrophoresis at 60 volts until the dye front reaches the lower gel, then increase the voltage to 80-100 volts until the dye front reaches the gel bottom. After electrophoresis, proteins are transferred from the gel to a PVDF membrane (Merck Millipore, MA, USA) using a Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). Wash the membrane with 1X pH 7.4 TBST (25 mM Tris, 150 mM NaCl, 0.05% Tween-20). Block the membrane with a blocking buffer (Visual Protein Biotech Corporation, Taipei, Taiwan) at room temperature for 1 hour. Incubate with primary antibodies against COX-2 (Cell Signaling Technology, MA, USA) or ICAM-1 (Cell Signaling Technology) at 4°C overnight. The next day, incubate with secondary antibodies at room temperature for 1 hour. Exposing the membrane to enhanced chemiluminescence (ECL) reagent (Energenesis Biomedical, Taipei, TW). Use a ChemiDox XRS+ imaging system (Bio-Rad Laboratories) to capture images and Image Lab™ 5.0 Software (Bio-Rad Laboratories) for quantification.
4.3.2. Detection of IL-6 and IL-8 Inflammatory Cytokines
ELISA was used to detect IL-6 and IL-8 levels (Bioss Inc., MA, USA) in TNF-α-treated HaCaT cells. After treating HaCaT cells with various drugs, collect the supernatant as samples, add 100 µL of standard (diluted series), and sample to each well of a 96-well transparent plate. Incubate at 37°C in the dark for 90 minutes. After washing and then adding 100 µL of IL-6 or IL-8 primary antibody for incubation at 37°C in the dark for 60 minutes. After washing and then adding 100 µL of substrate for incubation at 37°C in the dark for 15 minutes. Add 100 µL of stop solution to each well. Measure the absorbance at 450 nm using a SpectraMax® i3x Multi-Mode Microplate Reader and calculate the concentration using Arigo’s official free software GainData® (https://www.arigobio.com/elisa-calculator).
4.4. Detection of Intracellular Signaling Pathways in TNF-α Induced COX-2 and ICAM-1 Expression
When HaCaT cells cultured in a 12-well plate reach 80% confluency, replace the medium with 0% FBS DMEM and incubate for 24 hours. Pretreat cells with free radical scavengers (NAC, MCI186, APO, or DPI), MAPKs inhibitors (MEK1/2 inhibitor: U0126, JNK1/2 inhibitor: SP600125), transcription factor AP-1 inhibitor (SR11302), IκBα inhibitor (BAY11-7082), proteasome inhibitor (MG132), or NF-κB inhibitor (Helenalin) (all inhibitors from Merk Sigma-Aldrich) for 2 hours and then Stimulate cells with 40 ng/ml TNF-α (prepared in 0.1% BSA) for 24 hours. Inhibitors are prepared in DMSO and diluted with 0% FBS DMEM to the required concentration. After the incubation, wash cells with PBS and lyse using RIPA lysis buffer to collect protein lysate. Measure the protein concentration of each sample using a Pierce™ BCA Protein Assay Kit and adjust concentrations to be uniform. Prepare samples for Western blotting to analyze changes in COX-2 and ICAM-1 inflammatory protein levels.
4.5. Detection of Effects of Q and Its Derivatives on TNF-α Induced NF-κB and MAPKs Pathways
4.5.1. NF-κB And MAPKs Pathways Activation Analysis
HaCaT cells cultured in a 12-well plate grew to reach 80% confluency. The medium was replaced with 0% FBS DMEM and incubated for 24 hours. Treat cells with Q and its derivatives (prepared in DMSO and diluted with 0% FBS DMEM to the required concentration) for 2 hours and then stimulate the cells with 40 ng/mL TNF-α for 0-60 minutes or 30 minutes. Wash cells with PBS and use RIPA lysis buffer to collect the protein lysate. Quantify protein concentration using a Pierce™ BCA Protein Assay Kit and adjust concentrations accordingly. Analyze the NF-κB pathway: P-NF-κB (p65) and P-IκBα; the MAPKs pathway: P-Erk1/2, P-JNK1/2, and P-c-Jun (Cell Signaling Technology) using Western Blotting.
4.5.2. Analysis of Cytoplasmic and Nuclear NF-κB (p65) Distribution
Culture HaCaT cells in a 10 cm dish. After treatment with Q and its derivatives and TNF-α, wash with PBS and scrape cells into a centrifuge tube using 0.05% Trypsin. Terminate Trypsin activity with an equal volume of 10% FBS DMEM. Centrifuge at 1,500 rpm for 5 minutes, resuspend the cell pellet in 1 mL PBS, and transfer to a microcentrifuge tube. Centrifuge at 500 g, 4°C for 5 minutes, discard the supernatant and retain the cell pellet. Sequentially add Cytoplasmic Extraction Buffer (CE buffer) and NP40, vortex vigorously, and let sit for 5 minutes. Centrifuge at 1,000 g, 4°C for 5 minutes, and collect the supernatant (cytoplasmic sample). Repeat the procedure with CE buffer for the nuclear fraction. Add Nuclear Extraction Buffer, vortex vigorously, and centrifuge at 15,000 g, 4°C for 10 minutes to obtain the nuclear sample. Prepare samples for Western Blotting to analyze the distribution of NF-κB (p65) in the cytoplasm and nucleus.
In addition, culture HaCaT cells in a 6-well plate. After treatment with drugs and TNF-α, wash cells with PBS. Fix cells with 2% paraformaldehyde in cell culture medium for 2 minutes, followed by 2% paraformaldehyde in PBS for 10 minutes. Wash with PBS and incubate with 0.3% Triton-X100 in PBS for 15 minutes to increase cell membrane permeability. Wash with PBS and incubate with Blocking Buffer (0.3 M Glycine and 3% BSA in PBS) for 1 hour to reduce non-specific antibody binding. Incubate cells with NF-κB (p65) antibody (diluted 1:200 in Blocking Buffer) at 4°C for 24 hours. Wash with TPBS (PBS also contains 0.05% Tween-20) and dry, and incubate cells with FITC-conjugated secondary antibody (diluted 1:1000 in Blocking Buffer) at room temperature for 1 hour. Wash with TPBS and dry. Incubate cells with Hoechst 33342 (diluted 1:1000 in PBS, Invitrogen™) at room temperature for 5 minutes. Wash with PBS. Place the plate under a fluorescence microscope and capture images. Use ImageJ software for image overlay and analysis.
4.6. DNCB-Induced AD-like Mouse Model
All animal experimental protocols were approved by the Animal Care Committee of Chang Gung University of Science and Technology and Chang Gung University (IACUC approval number: CGU110-157). The female BALB/c mice aged 8 weeks were procured from the National Laboratory Animal Center (Taiwan) and housed at a consistent temperature (23±2oC) in the air-controlled conventional animal room at the Animal Center of Chang Gung University. After 1 week of acclimation, 1-chloro-2,4-dinitrobenzene (DNCB; Sigma–Aldrich, MA, USA) was applied to both the shoulder back of skin and ears to induce AD-like symptoms. One day after complete dorsal hair removal, 100 μL 0.5% DNCB in an acetone: olive oil mixture (3: 1 vol/vol) was smeared to shaved dorsal skin and 20 μL was applied to ears on days 1–3, 8-10. On days 14, 18, 21, 25, and 28, mice were challenged with 100 mL 1% DNCB on the shoulder back of skin and 20 mL 1% DNCB on each ear. Mice were divided into three groups: (1) Normal control-mice were sensitized and challenged using a mixture of acetone and olive oil in a 3:1 volume ratio.; (2) AD group-mice were sensitized and challenged with DNCB; (3) AD+Q group-mice similarly sensitized and challenged with DNCB but received an additional topical treatment of 40 μM Q. Each group has six mice. All described treatments were applied during days 14-28, as shown in Figure 1A.
4.6.1. Measurement of Ear Thickness
On the 29th day, ear swelling was measured using a dial thickness gauge (Mitutoyt, Tokyo, Japan) to minimize human error, with the same operator performing all measurements.
4.6.2. Detection of IgE, BUN, CRE, GOT, and GPT
Blood was collected from the ocular venous plexus, centrifuged at 6000 rpm for 5 minutes to obtain serum, which was then stored at -80°C. High-sensitivity kits (BD Biosciences) were used to measure IgE, IgG, GOT, and GPT levels in the serum.
4.6.3. Histopathology and Inflammation Detection
Skin samples from different treatment groups were fixed in 10% formaldehyde, dehydrated through graded alcohols, and embedded in paraffin. Sections of 5 μm thickness were stained with H/E for tissue structure and eosinophil distribution, and with toluidine blue for monocyte infiltration, and examined under a light microscope.
4.6.4. mRNA Expression of Inflammatory Mediators
After measuring ear thickness, skin samples were collected and ground for analysis using Real-time PCR. Total RNA was extracted using Trizol reagent (Invitrogen™) and converted to cDNA. The expression of COX-2, ICAM-1, IL-4, and INF-γ mRNA was quantified with SoFast EvaGreen Supermix and primers on a CFX Connect Real-time PCR Detection System (Bio-Rad Laboratories), using the 2-ΔΔCt method.
4.6.5. COX-2 Protein Expression
Skin samples from the treated areas were ground and analyzed for COX-2 protein expression using Western blotting. The preparation of protein samples, experimental conditions, and procedures are as described above.
5. Conclusions
This study highlights Q’s superior antioxidant and anti-inflammatory effects compared to three its derivatives. Q effectively inhibits free radical production and influences key pathways such as MEK1/2-Erk1/2-c-Fos, JNK1/2-c-Jun, and the IκBα-NF-κB pathway. Its anti-inflammatory benefits were also confirmed in a mice model of AD. Although Q’s derivatives are less potent, they still possess notable anti-inflammatory and antioxidant properties.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, C.Y.C.; methodology, H.T.W.; T.C.K.; C.C.Y.; formal analysis, H.T.W.; T.C.K.; C.C.Y.; investigation, C.Y.C., G.H.Y.; resources, C.Y.C., G.H.Y., H.C.L.; data curation, C.Y.C., G.H.Y.; writing—original draft preparation, C.Y.C., G.H.Y.; writing—review and editing, C.Y.C.; funding acquisition, C.Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Science and Technology, Taiwan, grant number MOST 111-2320-B-255-002 and NSTC 113-2314-B-182-010; Chang Gung Medical Research Foundation, Taiwan under grant number CMRPF3M0041, CMRPF3M0031, CMRPF3M0032, and, CORPF3P0011, CORPF3P0012; Chang Gung University of Science and Technology, Taiwan under grant number ZRRPF3M0091-9, ZRRPF3N0101-6, and ZRRPF3P0091-6.
Acknowledgments
Cheng C.Y. acknowledges the equipment and technical assistance provided by the Research Center for Chinese Herbal Medicine at Chang Gung University of Science and Technology.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AD: Atopic dermatitis |
| FBS: Fetal bovine serum |
| HP: Hyperoside |
| ICAM: Intercellular adhesion molecule |
| IL: Interleukin |
| KCs: Keratinocyte Cells |
| NOX: NADPH oxidase |
| Q: Quercetin |
| Q3Gs: Q 3-O-glycosides |
| QI: Quercitrin |
| ROS: Reactive oxygen molecules |
| RT: Rutin |
| TNF-α: Tumor necrosis factor-α |
References
- Evtushenko, N. A.; Beilin, A. K.; Kosykh, A. V.; Vorotelyak, E. A.; Gurskaya, N. G., Keratins as an Inflammation Trigger Point in Epidermolysis Bullosa Simplex. International journal of molecular sciences 2021, 22, (22). [CrossRef]
- Elias, P. M.; Williams, M. L.; Holleran, W. M.; Jiang, Y. J.; Schmuth, M., Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. Journal of lipid research 2008, 49, (4), 697-714. [CrossRef]
- Chambers, E. S.; Vukmanovic-Stejic, M., Skin barrier immunity and ageing. Immunology 2020, 160, (2), 116-125. [CrossRef]
- Medzhitov, R., Toll-like receptors and innate immunity. Nature Reviews Immunology 2001, 1, (2), 135-145.
- Baker, B. S.; Ovigne, J. M.; Powles, A. V.; Corcoran, S.; Fry, L., Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: modulation of TLR expression in chronic plaque psoriasis. British Journal of Dermatology 2003, 148, (4), 670-679. [CrossRef]
- Köllisch, G.; Kalali, B. N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M., Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005, 114, (4), 531-541. [CrossRef]
- Udommethaporn, S.; Tencomnao, T.; McGowan, E. M.; Boonyaratanakornkit, V., Assessment of Anti-TNF-α Activities in Keratinocytes Expressing Inducible TNF- α: A Novel Tool for Anti-TNF-α Drug Screening. PLOS ONE 2016, 11, (7), e0159151.
- Humeau, M.; Boniface, K.; Bodet, C., Cytokine-Mediated Crosstalk Between Keratinocytes and T Cells in Atopic Dermatitis. Front Immunol 2022, 13, 801579. [CrossRef]
- Beken, B.; Serttas, R.; Yazicioglu, M.; Turkekul, K.; Erdogan, S., Quercetin Improves Inflammation, Oxidative Stress, and Impaired Wound Healing in Atopic Dermatitis Model of Human Keratinocytes. Pediatr Allergy Immunol Pulmonol 2020, 33, (2), 69-79. [CrossRef]
- Chen, J.; Li, G.; Sun, C.; Peng, F.; Yu, L.; Chen, Y.; Tan, Y.; Cao, X.; Tang, Y.; Xie, X. J. P. R., Chemistry, pharmacokinetics, pharmacological activities, and toxicity of Quercitrin. 2022, 36, (4), 1545-1575.
- Chen, J.; Li, G.; Sun, C.; Peng, F.; Yu, L.; Chen, Y.; Tan, Y.; Cao, X.; Tang, Y.; Xie, X.; Peng, C., Chemistry, pharmacokinetics, pharmacological activities, and toxicity of Quercitrin. 2022, 36, (4), 1545-1575.
- Jegal, J.; Park, N.-J.; Lee, S.-Y.; Jo, B.-G.; Bong, S.-K.; Kim, S.-N.; Yang, M. H., Quercitrin, the Main Compound in Wikstroemia indica, Mitigates Skin Lesions in a Mouse Model of 2,4-Dinitrochlorobenzene-Induced Contact Hypersensitivity. 2020, 2020, (1), 4307161. [CrossRef]
- Charachit, N.; Sukhamwang, A.; Dejkriengkraikul, P.; Yodkeeree, S., Hyperoside and Quercitrin in Houttuynia cordata Extract Attenuate UVB-Induced Human Keratinocyte Cell Damage and Oxidative Stress via Modulation of MAPKs and Akt Signaling Pathway. 2022, 11, (2), 221. [CrossRef]
- Pinzaru, I.; Tanase, A.; Enatescu, V.; Coricovac, D.; Bociort, F.; Marcovici, I.; Watz, C.; Vlaia, L.; Soica, C.; Dehelean, C., Proniosomal Gel for Topical Delivery of Rutin: Preparation, Physicochemical Characterization and In Vitro Toxicological Profile Using 3D Reconstructed Human Epidermis Tissue and 2D Cells. Antioxidants 2021, 10, (1), 85. [CrossRef]
- Chaudhary, M.; Khan, A.; Gupta, M., Skin ageing: Pathophysiology and current market treatment approaches. Current aging science 2020, 13, (1), 22-30. [CrossRef]
- Baldisserotto, A.; Vertuani, S.; Bino, A.; De Lucia, D.; Lampronti, I.; Milani, R.; Gambari, R.; Manfredini, S., Design, synthesis and biological activity of a novel Rutin analogue with improved lipid soluble properties. Bioorganic & medicinal chemistry 2015, 23, (1), 264-271. [CrossRef]
- Cosco, D.; Failla, P.; Costa, N.; Pullano, S.; Fiorillo, A.; Mollace, V.; Fresta, M.; Paolino, D., Rutin-loaded chitosan microspheres: characterization and evaluation of the anti-inflammatory activity. Carbohydrate polymers 2016, 152, 583-591. [CrossRef]
- ben Sghaier, M.; Pagano, A.; Mousslim, M.; Ammari, Y.; Kovacic, H.; Luis, J., Rutin inhibits proliferation, attenuates superoxide production and decreases adhesion and migration of human cancerous cells. Biomedicine & Pharmacotherapy 2016, 84, 1972-1978. [CrossRef]
- Peres, D.; De Oliveira, C.; Da Costa, M.; Tokunaga, V.; Mota, J.; Rosado, C.; Consiglieri, V.; Kaneko, T.; Velasco, M.; Baby, A., Rutin increases critical wavelength of systems containing a single UV filter and with good skin compatibility. Skin Research and Technology 2016, 22, (3), 325-333. [CrossRef]
- Pivec, T.; Kargl, R.; Maver, U.; Bračič, M.; Elschner, T.; Žagar, E.; Gradišnik, L.; Stana Kleinschek, K., Chemical structure–antioxidant activity relationship of water–based enzymatic polymerized Rutin and its wound healing potential. Polymers 2019, 11, (10), 1566. [CrossRef]
- Choi, S. J.; Lee, S.-N.; Kim, K.; Joo, D. H.; Shin, S.; Lee, J.; Lee, H. K.; Kim, J.; Kwon, S. B.; Kim, M. J., Biological effects of rutin on skin aging. International journal of molecular medicine 2016, 38, (1), 357-363.
- Kostyuk, V.; Potapovich, A.; Albuhaydar, A. R.; Mayer, W.; De Luca, C.; Korkina, L., Natural Substances for Prevention of Skin Photoaging: Screening Systems in the Development of Sunscreen and Rejuvenation Cosmetics. Rejuvenation Res 2018, 21, (2), 91-101. [CrossRef]
- Kumar, P.; Vaidya, V.; Sakpal, G., Formulation and development of rutin and gallic acid loaded herbal gel for the treatment of psoriasis and skin disease. J. Sci. Technol 2020, 5, 192-203.
- Choi, J. K.; Kim, S.-H., Rutin suppresses atopic dermatitis and allergic contact dermatitis. Experimental Biology and Medicine 2013, 238, (4), 410-417. [CrossRef]
- 王湘婷. 評估槲皮素衍生物在人體角質形成細胞上的生物活性. 長庚科技大學, 桃園縣, 2023.
- 郭庭君. 探討槲皮素對皮膚角質上皮細胞抗氧化、抗發炎的影響. 長庚科技大學, 桃園縣, 2017.
- 楊靖婕. 探討不同金銀花初萃物與其活性分子對皮膚細胞生物活性效果評估. 長庚科技大學, 桃園縣, 2022.
- Nakai, K.; Tsuruta, D., What Are Reactive Oxygen Species, Free Radicals, and Oxidative Stress in Skin Diseases? International journal of molecular sciences 2021, 22, (19).
- Raimondo, A.; Serio, B.; Lembo, S., Oxidative Stress in Atopic Dermatitis and Possible Biomarkers: Present and Future. Indian Journal of Dermatology 2023, 68, (6), 657-660. [CrossRef]
- Alessandrello, C.; Sanfilippo, S.; Minciullo, P. L.; Gangemi, S., An Overview on Atopic Dermatitis, Oxidative Stress, and Psychological Stress: Possible Role of Nutraceuticals as an Additional Therapeutic Strategy. International Journal of Molecular Sciences 2024, 25, (9), 5020. [CrossRef]
- Mirza, M. A.; Mahmood, S.; Hilles, A. R.; Ali, A.; Khan, M. Z.; Zaidi, S. A. A.; Iqbal, Z.; Ge, Y., Quercetin as a Therapeutic Product: Evaluation of Its Pharmacological Action and Clinical Applications—A Review. Pharmaceuticals 2023, 16, (11), 1631. [CrossRef]
- Makino, T.; Shimizu, R.; Kanemaru, M.; Suzuki, Y.; Moriwaki, M.; Mizukami, H., Enzymatically Modified Isoquercitrin, α-Oligoglucosyl Quercetin 3-<i>O</i>-Glucoside, Is Absorbed More Easily than Other Quercetin Glycosides or Aglycone after Oral Administration in Rats. Biological and Pharmaceutical Bulletin 2009, 32, (12), 2034-2040. [CrossRef]
- Kang Na, R.; Son Yun, G.; Kim Jeong, Y., 천연물질 퀘르세틴 유도체의 다양한 구조 및 효소 저해 활성. 생명과학회지 2024, 34, (9), 656-665.
- Zhao, J.; Li, Y.; Wang, F.; Jia, B.; Liu, C.; Wang, R., Study of 6 flavonoids compounds for the scavenging superoxide anion free radical ability and the structure-activity relationships. China Med Herald 2014, 11, 7-10.
- Cai, Y. Z.; Mei, S.; Jie, X.; Luo, Q.; Corke, H., Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci 2006, 78, (25), 2872-88.
- Wang, X.; Kong, J. J. S. w. G. C. x. b. C. J. o. B., Enzymatic synthesis of acylated quercetin 3-O-glycosides: a review. 2021, 37, (6), 1900-1918. [CrossRef]
- Gee, J. M.; DuPont, M. S.; Rhodes, M. J.; Johnson, I. T. J. F. R. B.; Medicine, Quercetin glucosides interact with the intestinal glucose transport pathway. 1998, 25, (1), 19-25.
- Morand, C.; Manach, C.; Crespy, V.; Remesy, C. J. F. R. R., Quercetin 3-O-β-glucoside is better absorbed than other quercetin forms and is not present in rat plasma. 2000, 33, (5), 667-676. [CrossRef]
- Ader, P.; Wessmann, A.; Wolffram, S., Bioavailability and metabolism of the flavonol quercetin in the pig. Free Radical Biology and Medicine 2000, 28, (7), 1056-1067.
- Murota, K.; Terao, J., Antioxidative flavonoid quercetin: implication of its intestinal absorption and metabolism. Archives of biochemistry and biophysics 2003, 417, (1), 12-17. [CrossRef]
- Rietjens, I. M.; Boersma, M. G.; van der Woude, H.; Jeurissen, S. M.; Schutte, M. E.; Alink, G. M., Flavonoids and alkenylbenzenes: mechanisms of mutagenic action and carcinogenic risk. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2005, 574, (1-2), 124-138.
- van der Woude, H.; Alink, G. M.; van Rossum, B. E.; Walle, K.; van Steeg, H.; Walle, T.; Rietjens, I. M., Formation of transient covalent protein and DNA adducts by quercetin in cells with and without oxidative enzyme activity. Chemical research in toxicology 2005, 18, (12), 1907-1916.
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C., Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. The American journal of clinical nutrition 2005, 81, (1), 230S-242S. [CrossRef]
- Thilakarathna, S. H.; Rupasinghe, H. V., Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, (9), 3367-3387. [CrossRef]
- Xiong, F.; Zhang, Y.; Li, T.; Tang, Y.; Song, S.-Y.; Zhou, Q.; Wang, Y., A detailed overview of quercetin: implications for cell death and liver fibrosis mechanisms. Frontiers in Pharmacology 2024, 15.
- Pan, B.; Fang, S.; Zhang, J.; Pan, Y.; Liu, H.; Wang, Y.; Li, M.; Liu, L., Chinese herbal compounds against SARS-CoV-2: puerarin and quercetin impair the binding of viral S-protein to ACE2 receptor. Computational and structural biotechnology journal 2020, 18, 3518-3527.
- Roy, A. V.; Chan, M.; Banadyga, L.; He, S.; Zhu, W.; Chrétien, M.; Mbikay, M., Quercetin inhibits SARS-CoV-2 infection and prevents syncytium formation by cells co-expressing the viral spike protein and human ACE2. Virology Journal 2024, 21, (1), 29.
- Guo, Z.; Zhang, J.; Li, M.; Xing, Z.; Li, X.; Qing, J.; Zhang, Y.; Zhu, L.; Qi, M.; Zou, X., Mechanism of action of quercetin in regulating cellular autophagy in multiple organs of Goto-Kakizaki rats through the PI3K/Akt/mTOR pathway. Frontiers in Medicine 2024, 11. [CrossRef]
Figure 1.
Analysis of the toxicity of Q and its derivatives in HaCaT cells. HaCaT cells were treated with (A) Q, QI, HP and RT; (B) 10 and 40 ng/mL TNF-α for 24 hours, and cell viability was evaluated using the MTT assay by measuring OD570 nm. Each experimental group was repeated thrice, and results were presented as Mean ± SD. One-way analysis of variance (ANOVA) statistical analysis with Dunnett’s multiple comparison test was used to evaluate significant differences between groups. * p < 0.05, **p < 0.01 indicate significance compared to the untreated group [25].
Figure 1.
Analysis of the toxicity of Q and its derivatives in HaCaT cells. HaCaT cells were treated with (A) Q, QI, HP and RT; (B) 10 and 40 ng/mL TNF-α for 24 hours, and cell viability was evaluated using the MTT assay by measuring OD570 nm. Each experimental group was repeated thrice, and results were presented as Mean ± SD. One-way analysis of variance (ANOVA) statistical analysis with Dunnett’s multiple comparison test was used to evaluate significant differences between groups. * p < 0.05, **p < 0.01 indicate significance compared to the untreated group [25].
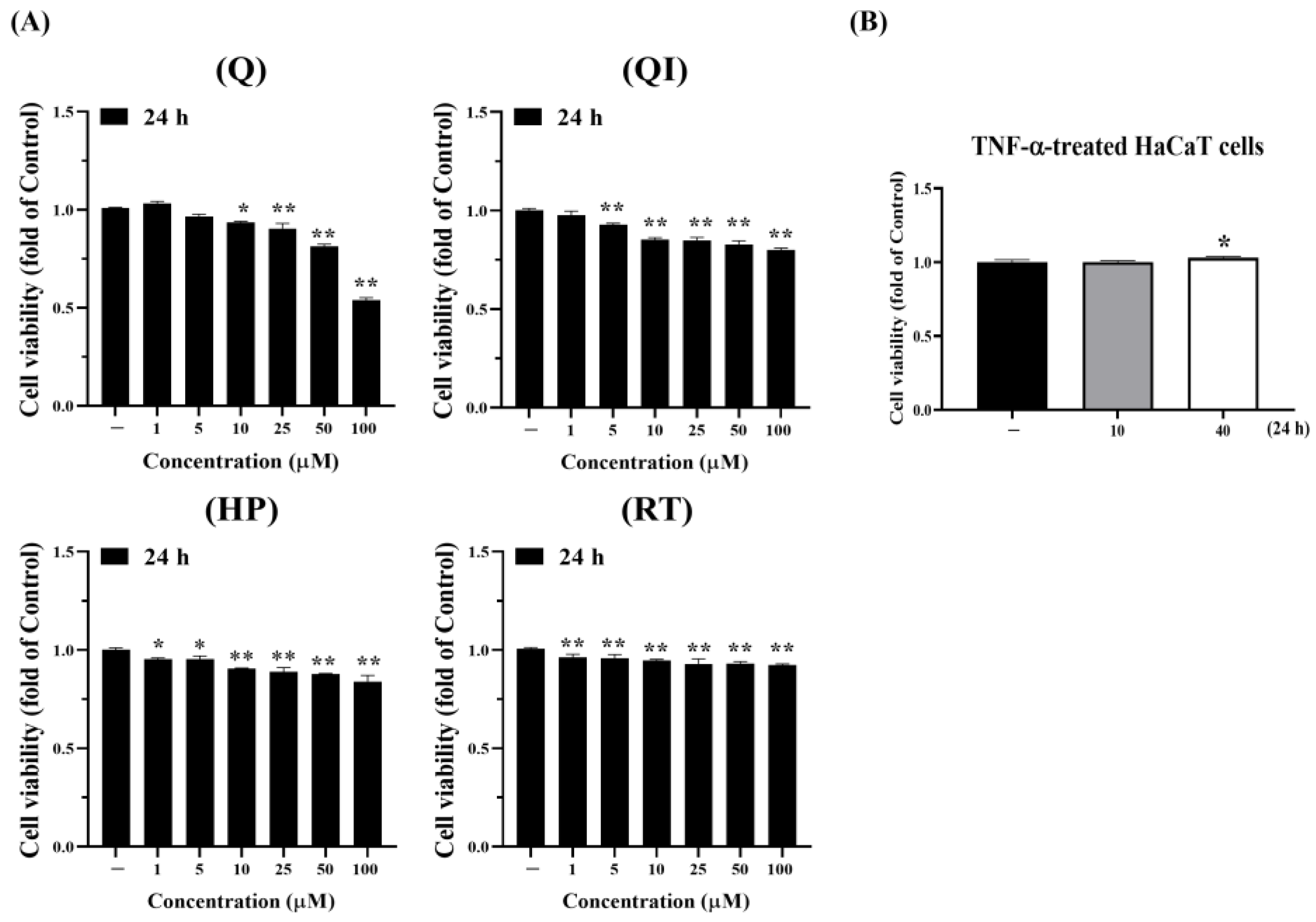
Figure 2.
Comparison of the antioxidant effect of Q and its derivatives. (A) At 25 or 50 µM, Q, QI, HP and RT were incubated separately with DPPH radicals for 12 minutes. Detect the absorbance value 517. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. Two-way ANOVA statistical analysis was used, followed by Tukey’s multiple comparison test to assess significant differences between groups. *p < 0.05, **p < 0.01 indicate significance compared to 25 µM Vit C (positive control); #p < 0.05, ##p < 0.01 indicate significance compared to 50 µM Vit C; $$p < 0.01 indicates significance compared to the same drug at 25 µM. (B) HaCaT cells were pretreated with the highest safe concentration of Q, QI, HP, and RT for 2 hours, followed by stimulation with 40 ng/ml TNF-α for 24 hours. CM-H2DCFDA fluorescence staining was used to assess intracellular free radicals after 30 minutes. Detection Ex: 495/9 nm; Em: 527/15 nm fluorescence value. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. One-way ANOVA statistical analysis with Dunnett’s multiple comparison tests was used to evaluate significant differences between groups. **p < 0.01 indicates significance compared to the untreated group; ##p < 0.01 indicates significance compared to the TNF-α-stimulated group [25].
Figure 2.
Comparison of the antioxidant effect of Q and its derivatives. (A) At 25 or 50 µM, Q, QI, HP and RT were incubated separately with DPPH radicals for 12 minutes. Detect the absorbance value 517. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. Two-way ANOVA statistical analysis was used, followed by Tukey’s multiple comparison test to assess significant differences between groups. *p < 0.05, **p < 0.01 indicate significance compared to 25 µM Vit C (positive control); #p < 0.05, ##p < 0.01 indicate significance compared to 50 µM Vit C; $$p < 0.01 indicates significance compared to the same drug at 25 µM. (B) HaCaT cells were pretreated with the highest safe concentration of Q, QI, HP, and RT for 2 hours, followed by stimulation with 40 ng/ml TNF-α for 24 hours. CM-H2DCFDA fluorescence staining was used to assess intracellular free radicals after 30 minutes. Detection Ex: 495/9 nm; Em: 527/15 nm fluorescence value. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. One-way ANOVA statistical analysis with Dunnett’s multiple comparison tests was used to evaluate significant differences between groups. **p < 0.01 indicates significance compared to the untreated group; ##p < 0.01 indicates significance compared to the TNF-α-stimulated group [25].
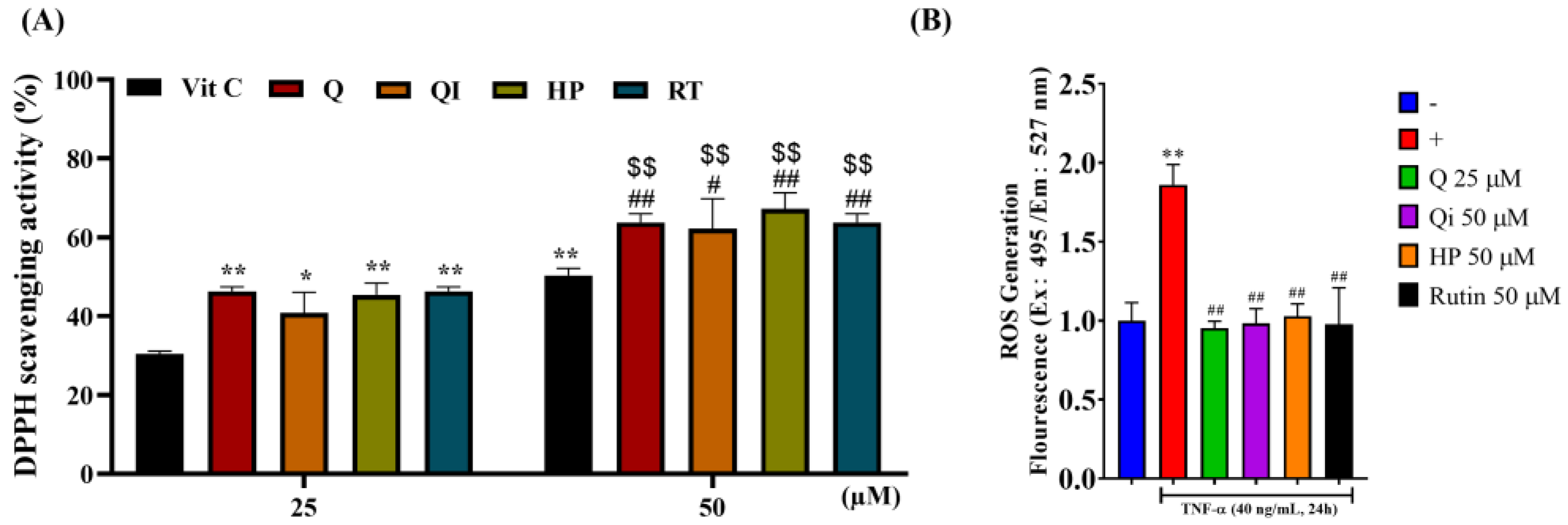
Figure 3.
Comparison of the anti-inflammatory abilities of Q and its derivatives. (A) HaCaT cells were pretreated with 10 µM Q and its derivatives for 2 hours, followed by stimulation with 40 ng/ml TNF-α for 24 and 48 hours to induce an inflammatory response. Western blot was used to analyze the COX-2 protein. (B) The highest dose (50 µM) of each Chinese herbal medicine was pretreated for 2 hours; (C) Chinese herbal medicines were pretreated with 10, 25, and 50 µM, (B, C) followed by treatment with 40 ng/mL TNF-α treatment for 24 hours. (B) ELISA was used to measure IL-6 and IL-8 levels. (C) Western blot was used to analyze the ICAM-1 protein. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. A one-way ANOVA statistical analysis with Dunnett’s multiple comparison test assessed significant differences between groups. *p < 0.05, **p < 0.01 indicate significance compared to the untreated group; #p < 0.05, ##p < 0.01 indicate significance compared to the TNF-α-stimulated group [25].
Figure 3.
Comparison of the anti-inflammatory abilities of Q and its derivatives. (A) HaCaT cells were pretreated with 10 µM Q and its derivatives for 2 hours, followed by stimulation with 40 ng/ml TNF-α for 24 and 48 hours to induce an inflammatory response. Western blot was used to analyze the COX-2 protein. (B) The highest dose (50 µM) of each Chinese herbal medicine was pretreated for 2 hours; (C) Chinese herbal medicines were pretreated with 10, 25, and 50 µM, (B, C) followed by treatment with 40 ng/mL TNF-α treatment for 24 hours. (B) ELISA was used to measure IL-6 and IL-8 levels. (C) Western blot was used to analyze the ICAM-1 protein. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. A one-way ANOVA statistical analysis with Dunnett’s multiple comparison test assessed significant differences between groups. *p < 0.05, **p < 0.01 indicate significance compared to the untreated group; #p < 0.05, ##p < 0.01 indicate significance compared to the TNF-α-stimulated group [25].
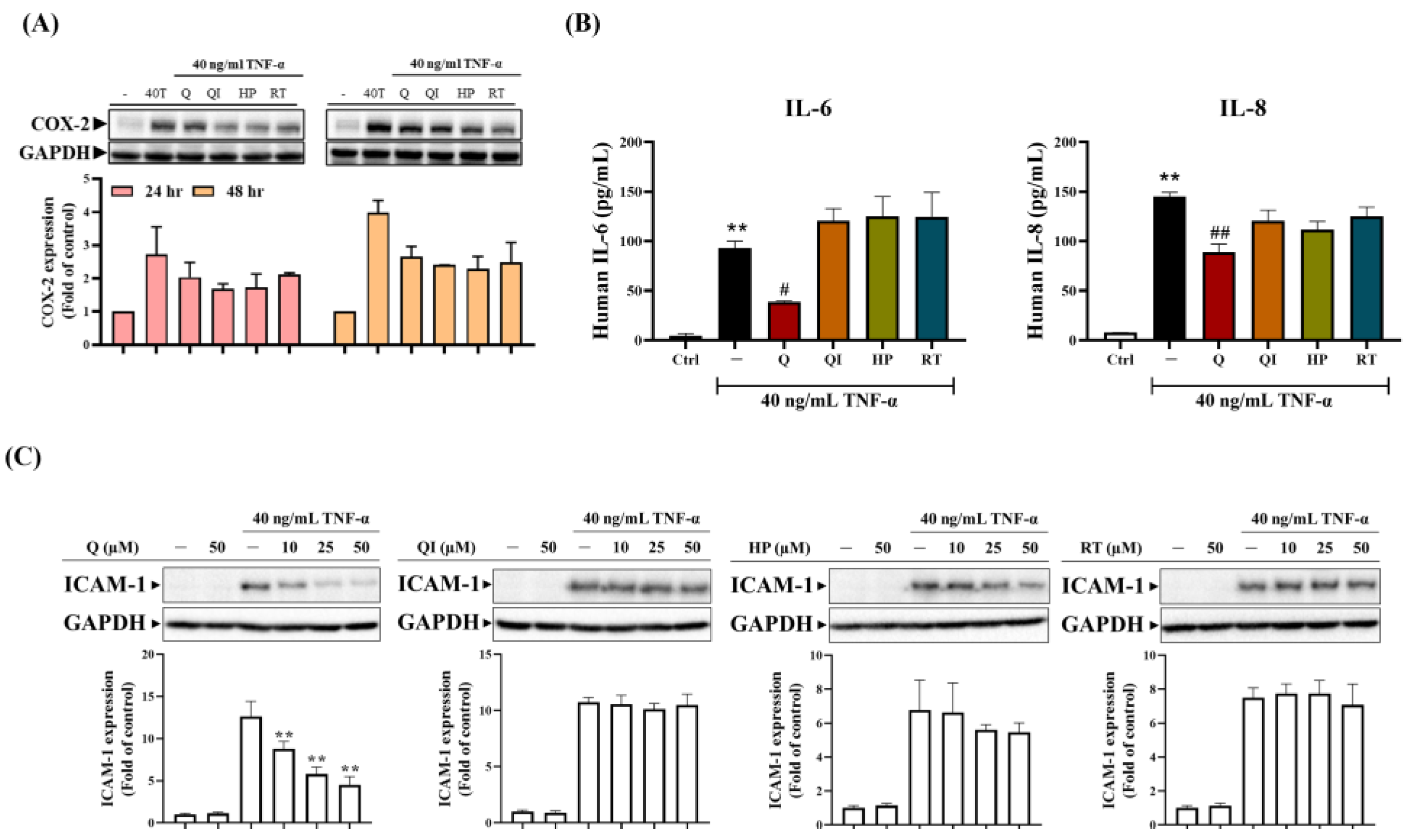
Figure 4.
The effects of Q on inflammation and skin features on DNCB-induced AD mice. The animal experiment design is shown in (A) and divided into three groups: N group (Normal control mice), AD group (sensitized control mice), and AD+Q group (sensitized mice topically treated with 40 mM Q). Each group has six mice. (B) Images of the ear and skin of the shoulder back were taken with a digital camera, and ear thickness was calculated. (C, D) The ear and skin of shoulder back sections were stained with (C) H&E (for eosinophil) and (D) toluidine blue staining (for mast cell infiltration) in the N group, AD group, and AD+Q group (200 x magnification). Red lines indicate skin thickness; arrows indicate mast cells. (E) The skin of the shoulder back was homogenized and analyzed by Western blotting using an anti-COX-2 and anti-GAPDH antibody (as a loading control); The mRNA expression of ICAM-1, IL-4, or IFN-γ was determined by Real-Time PCR. Data expressed as means ± SD. of at least six mice per group for all groups. Using One-way ANOVA statistical analysis and Dunnett’s multiple comparisons test for interpreting significant differences between groups. **p < 0.01 indicates a significant difference compared to the Normal group; ##p < 0.01 indicates a significant difference compared to the AD group [26].
Figure 4.
The effects of Q on inflammation and skin features on DNCB-induced AD mice. The animal experiment design is shown in (A) and divided into three groups: N group (Normal control mice), AD group (sensitized control mice), and AD+Q group (sensitized mice topically treated with 40 mM Q). Each group has six mice. (B) Images of the ear and skin of the shoulder back were taken with a digital camera, and ear thickness was calculated. (C, D) The ear and skin of shoulder back sections were stained with (C) H&E (for eosinophil) and (D) toluidine blue staining (for mast cell infiltration) in the N group, AD group, and AD+Q group (200 x magnification). Red lines indicate skin thickness; arrows indicate mast cells. (E) The skin of the shoulder back was homogenized and analyzed by Western blotting using an anti-COX-2 and anti-GAPDH antibody (as a loading control); The mRNA expression of ICAM-1, IL-4, or IFN-γ was determined by Real-Time PCR. Data expressed as means ± SD. of at least six mice per group for all groups. Using One-way ANOVA statistical analysis and Dunnett’s multiple comparisons test for interpreting significant differences between groups. **p < 0.01 indicates a significant difference compared to the Normal group; ##p < 0.01 indicates a significant difference compared to the AD group [26].
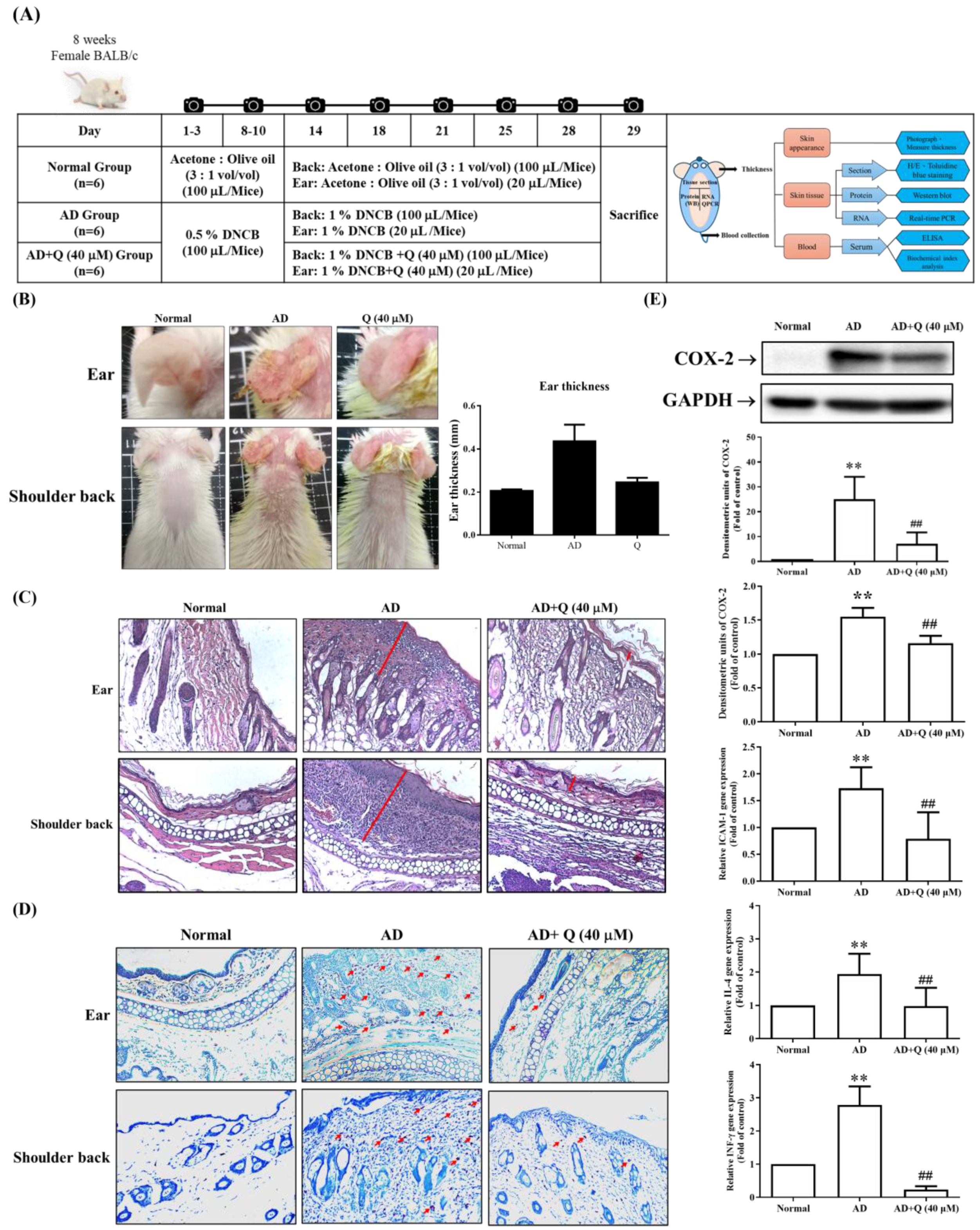
Figure 5.
Impact of Q and its derivatives on TNF-α-Induced MAPK pathway activation. Q and its derivatives were pretreated with the same concentration (50 µM) in HaCaT cells for 2 hours. Subsequently, cells were stimulated with 40 ng/mL TNF-α at different time points (0, 5, 10, 15, 30, and 60 minutes). Western blot was used to analyze the expression of (A) P-Erk2, (B) P-JNK1, (C) P-c-Jun, and (D) GAPDH. GAPDH served as the loading control for the cytoplasmic protein. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. The two-way ANOVA statistical analysis with Tukey’s multiple comparison test evaluated significant differences between groups. *p< 0.05 indicates significance compared to the untreated group; #p < 0.05 indicates significance compared to the TNF-α-stimulated group at the same time point [25].
Figure 5.
Impact of Q and its derivatives on TNF-α-Induced MAPK pathway activation. Q and its derivatives were pretreated with the same concentration (50 µM) in HaCaT cells for 2 hours. Subsequently, cells were stimulated with 40 ng/mL TNF-α at different time points (0, 5, 10, 15, 30, and 60 minutes). Western blot was used to analyze the expression of (A) P-Erk2, (B) P-JNK1, (C) P-c-Jun, and (D) GAPDH. GAPDH served as the loading control for the cytoplasmic protein. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. The two-way ANOVA statistical analysis with Tukey’s multiple comparison test evaluated significant differences between groups. *p< 0.05 indicates significance compared to the untreated group; #p < 0.05 indicates significance compared to the TNF-α-stimulated group at the same time point [25].
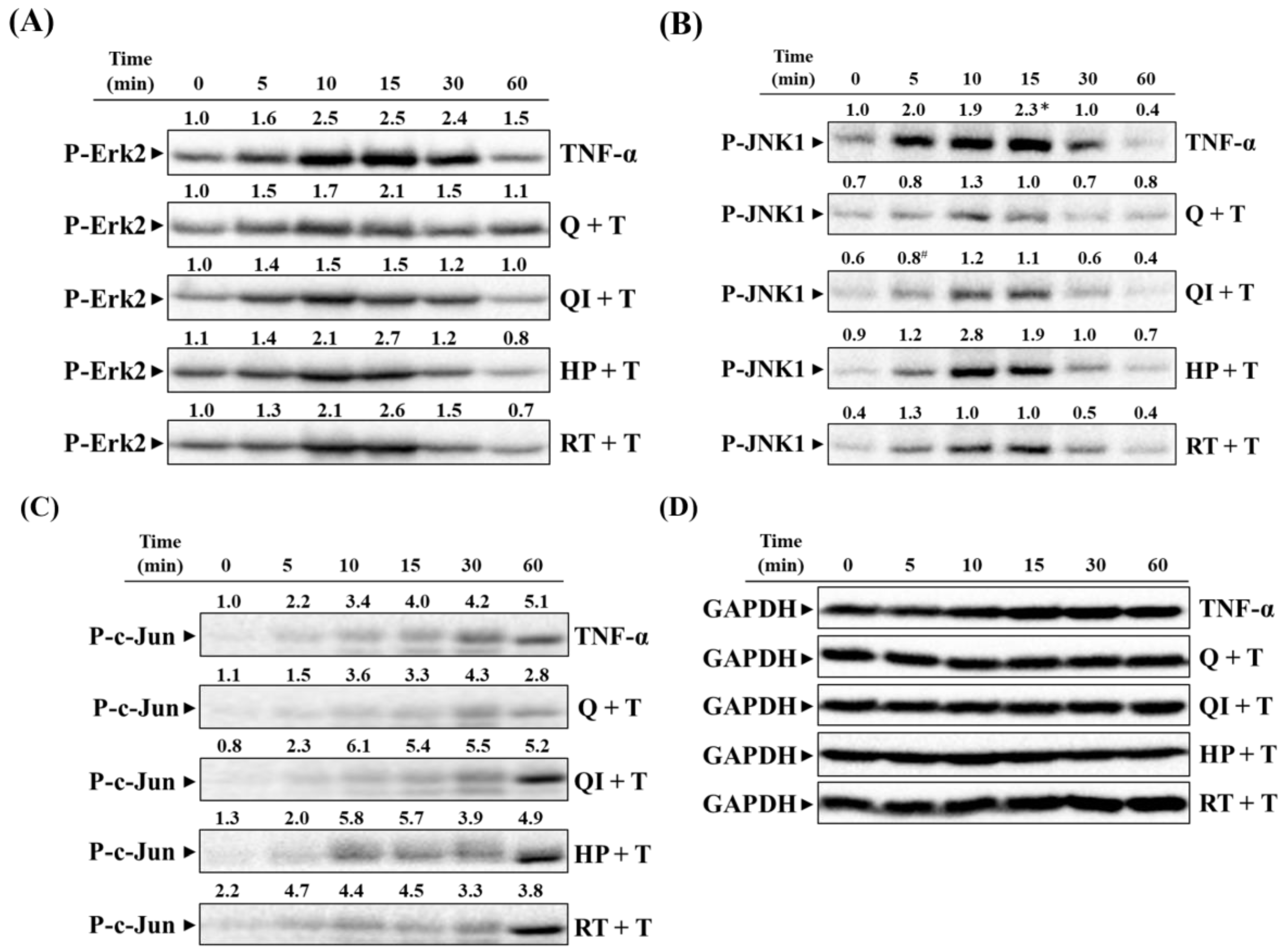
Figure 6.
Impact of Q and its derivatives on TNF-α-Induced NF-κB Pathway. (A, B) Q and its derivatives were pretreated with the same concentration (50 µM) in HaCaT cells for 2 hours, followed by stimulation with 40 ng/mL TNF-α at different time points (0, 5, 10, 15, 30, 60 minutes). (A, B) Western blot was used to analyze the trends of (A) P-IκBα and (B) P-NF-κB (p65) expression. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. Two-way ANOVA statistical analysis with Tukey’s multiple comparison test evaluated significant differences between groups. ##p < 0.01 indicates significance compared to the TNF-α-stimulated group at the same time point. (C) After drug treatment, cells were collected and prepared as nuclear and cytoplasmic protein samples. Western blot was used to analyze P-NF-κB (p65) expression in the nucleus/cytoplasm. Lamin B1 was the nuclear protein loading control, and GAPDH was the cytoplasmic protein loading control. (D) After drug treatment, cells were fixed, permeabilized, and stained with FITC-labeled antibodies against P-NF-κB (p65) (green fluorescence). Hoechst 33342 was used to stain cell nuclei (blue fluorescence). Fluorescence microscopy was used for observation in random fields [25].
Figure 6.
Impact of Q and its derivatives on TNF-α-Induced NF-κB Pathway. (A, B) Q and its derivatives were pretreated with the same concentration (50 µM) in HaCaT cells for 2 hours, followed by stimulation with 40 ng/mL TNF-α at different time points (0, 5, 10, 15, 30, 60 minutes). (A, B) Western blot was used to analyze the trends of (A) P-IκBα and (B) P-NF-κB (p65) expression. Each experimental group was repeated at least three times, and the results are presented as Mean ± SD. Two-way ANOVA statistical analysis with Tukey’s multiple comparison test evaluated significant differences between groups. ##p < 0.01 indicates significance compared to the TNF-α-stimulated group at the same time point. (C) After drug treatment, cells were collected and prepared as nuclear and cytoplasmic protein samples. Western blot was used to analyze P-NF-κB (p65) expression in the nucleus/cytoplasm. Lamin B1 was the nuclear protein loading control, and GAPDH was the cytoplasmic protein loading control. (D) After drug treatment, cells were fixed, permeabilized, and stained with FITC-labeled antibodies against P-NF-κB (p65) (green fluorescence). Hoechst 33342 was used to stain cell nuclei (blue fluorescence). Fluorescence microscopy was used for observation in random fields [25].
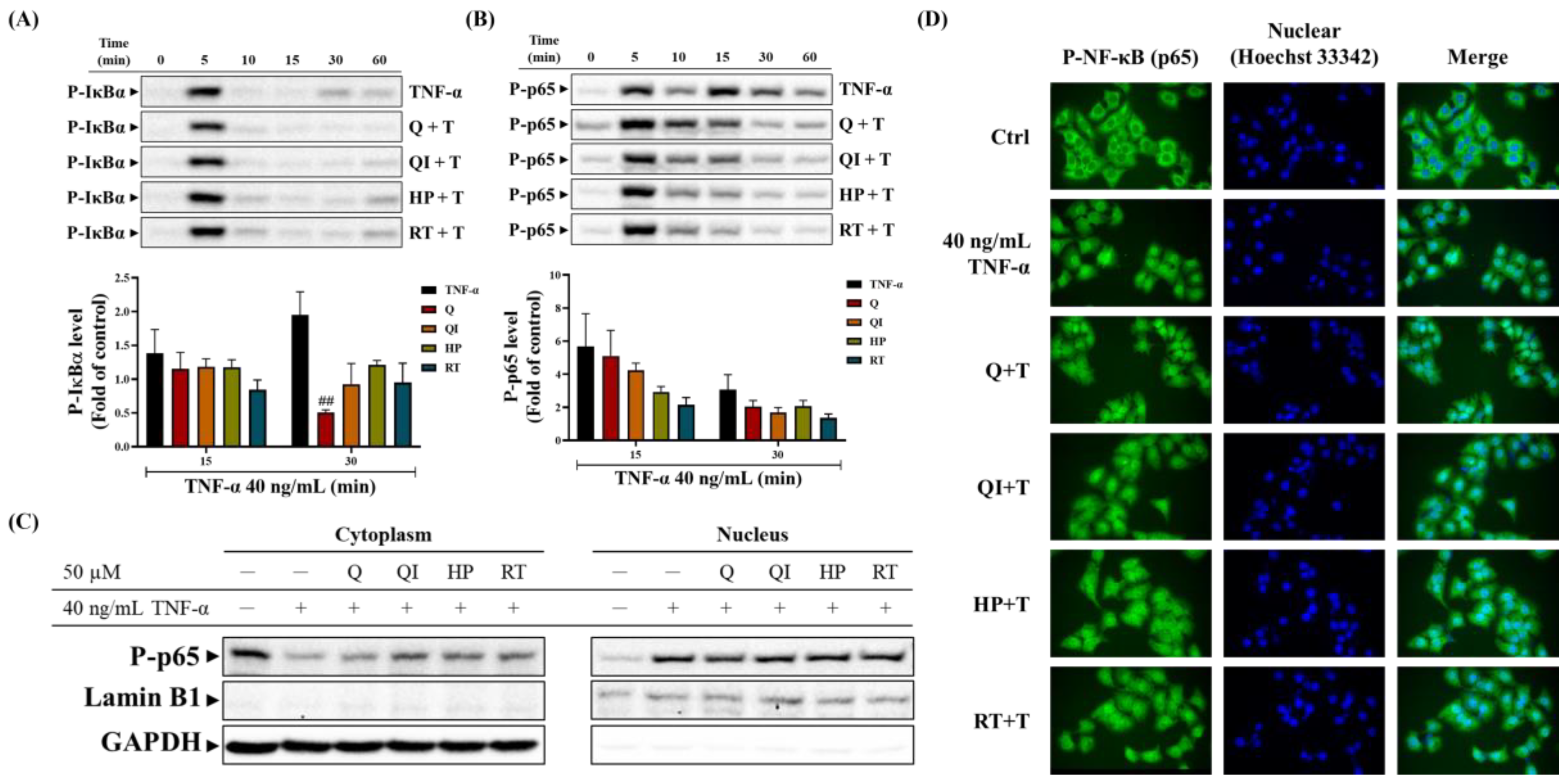
Figure 7.
Involvement of intracellular signaling pathways in TNF-α-induced COX-2 and ICAM-1 expression. (A, B) HaCaT cells were pretreated with 100 µM NAC, 1 µM MCI186, 10 µM APO, 10 µM DPI, 10 µM U0126, 1 µM SP600125, 10 µM SR11302, 10 nM Helenalin, 10 µM BAY11-7082, or 100 nM MG132 for 2 hours, followed by stimulation with 40 ng/mL TNF-α for (A, D) 24 hours; (B, C) at 0, 3, 6, 18, and 24 hours. (A) CM-H2DCFDA fluorescence staining was used to observe intracellular free radical levels after 30 minutes. (B~D) Western blot was used to analyze COX-2 or ICAM-1 protein expression, with GAPDH as the cytoplasmic protein loading control. Each experimental group was repeated thrice, and results were presented as Mean ± SD. One-way ANOVA statistical analysis was used, with Dunnett’s multiple comparison test for (A, D) and Tukey’s multiple comparison test for (B, C). **p < 0.01 indicates significance compared to the untreated group; #p < 0.05, ##p < 0.01 indicate significance compared to the TNF-α-stimulated group at the same time point [25,27].
Figure 7.
Involvement of intracellular signaling pathways in TNF-α-induced COX-2 and ICAM-1 expression. (A, B) HaCaT cells were pretreated with 100 µM NAC, 1 µM MCI186, 10 µM APO, 10 µM DPI, 10 µM U0126, 1 µM SP600125, 10 µM SR11302, 10 nM Helenalin, 10 µM BAY11-7082, or 100 nM MG132 for 2 hours, followed by stimulation with 40 ng/mL TNF-α for (A, D) 24 hours; (B, C) at 0, 3, 6, 18, and 24 hours. (A) CM-H2DCFDA fluorescence staining was used to observe intracellular free radical levels after 30 minutes. (B~D) Western blot was used to analyze COX-2 or ICAM-1 protein expression, with GAPDH as the cytoplasmic protein loading control. Each experimental group was repeated thrice, and results were presented as Mean ± SD. One-way ANOVA statistical analysis was used, with Dunnett’s multiple comparison test for (A, D) and Tukey’s multiple comparison test for (B, C). **p < 0.01 indicates significance compared to the untreated group; #p < 0.05, ##p < 0.01 indicate significance compared to the TNF-α-stimulated group at the same time point [25,27].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



