
Submitted:
24 December 2024
Posted:
25 December 2024
You are already at the latest version
Abstract
Pancreatic cancer has the lowest 5-year survival rate (13%) among major cancers and is the third leading cause of cancer-related deaths in the United States. The high lethality of this cancer is attributed to its insidious onset, late-stage diagnosis, rapid progression, and limited treatment options. Addressing these challenges requires a deeper understanding of the complex tumor microenvironment to identify novel therapeutic targets. Newer approaches like adoptive cell therapy have shown remarkable success in treating hematological malignancies but their application in solid tumors particularly pancreatic cancer is still in the early stages of development. ACT broadly involves isolating immune cells (T lymphocytes, Natural Killer cells, and macrophages) from the patient followed by genetic engineering to enhance and mount a specific anti-tumor response. Various ACT modalities are under investigation for pancreatic cancer, including chimeric antigen receptor T cells (CAR-T), chimeric antigen receptor NK cells (CAR-NK), tumor-infiltrating lymphocytes (TIL), T cell receptor (TCR) engineered T cells, cytokine-induced killer cells (CIK). Major hurdles have been identifying actionable tumor antigens and delivering focused cellular therapies to overcome the immunosuppressive and dense fibrotic stroma surrounding the pancreatic cancer. Further studies are needed to explore the limitations faced by cellular therapy in pancreatic cancer and identify novel combination treatment approaches in order to improve clinical outcomes.
Keywords:
pancreatic ductal adenocarcinoma
; adoptive cell therapy
; chimeric antigen receptor T cells (CAR-T)
; chimeric antigen receptor NK cells (CAR-NK)
; tumor-infiltrating lymphocytes (TIL)
; T cell receptor (TCR) engineered T cells
; cytokine-induced killer cells (CIK)
; oncolytic virus
1. Introduction
Pancreatic cancer is among the most lethal malignancies, representing a significant global health challenge. In 2024, it is projected to cause approximately 51,000 deaths out of 66,000 newly diagnosed cases in the United States alone [1]. With a 5-year survival rate of just 13%, pancreatic cancer has the lowest survival rate among major cancers. Its incidence is similar among African-American and Caucasian populations. Despite accounting for only 3% of new cancer diagnoses in the United States, pancreatic cancer is currently the third leading cause of cancer-related deaths and is expected to become the second in the near future [2]. Globally, the European region exhibits the highest age-standardized incidence and mortality rates, while the Southeast Asia region reports the lowest [3]. The high lethality of pancreatic cancer is attributed to its insidious onset, late-stage diagnosis, aggressive progression, and limited treatment options. Addressing these challenges requires a deeper understanding of the tumor microenvironment (TME) to identify novel therapeutic targets and expand treatment options, ultimately improving patient outcomes in the long term.
Pancreatic cancer risk factors are broadly categorized into modifiable and non-modifiable factors [4]. Key modifiable risk factors include smoking, excessive alcohol consumption, diets high in red or processed meats, obesity, chronic pancreatitis, and infections such as Helicobacter pylori. These factors contribute to the higher incidence observed in developed countries. Non-modifiable risk factors include advanced age, male gender, ethnicity, specific blood groups, microbiota composition, genetic predisposition, and diabetes mellitus. A comprehensive understanding of these risk factors is critical for developing effective prevention and intervention strategies.
2. Current Management of Pancreatic Ductal Adenocarcinoma
Pancreatic ductal adenocarcinomas (PDAC) account for approximately 90% of primary pancreatic cancers, with the remainder comprising less common types such as squamous, acinar, signet-ring (exocrine), ampullary, neuroendocrine, and undifferentiated carcinomas [5]. PDAC is typically stratified for risk and management using the tumor-node-metastasis (TNM) system outlined in the 8th edition of the American Joint Committee on Cancer (AJCC) staging manual [6]. While TNM staging informs treatment and prognosis, it does not include resection status, which is critical for surgical planning. Another widely used classification system focuses on tumor resectability and the presence of distant metastatic disease at diagnosis. Based on this approach, PDAC is categorized as resectable (R), borderline resectable (BR), locally advanced (LA), or metastatic [7,8,9,10]. Resectability is determined by the degree of tumor involvement with surrounding arteries and veins, typically assessed in a multidisciplinary setting. In R-PDAC, there is no tumor contact with adjacent blood vessels. BR-PDAC involves some tumor contact with blood vessels, with the expectation that systemic chemotherapy (CT) or radiation therapy (RT) can convert these cases to R-PDAC. LA-PDAC, a less clearly defined category, includes tumors with significant involvement of major arteries (e.g., celiac trunk or superior mesenteric artery interface >180°) or veins, rendering both resection and vascular reconstruction infeasible.
Following stratification, treatment plans are tailored to the disease stage. For R-PDAC and BR-PDAC, neoadjuvant chemotherapy (NAT), often combined with RT, is now preferred prior to surgical resection. In LA-PDAC, NAT helps identify patients who may benefit from subsequent surgery. For metastatic PDAC and certain LA cases, clinical trial enrollment is recommended. Systemic chemotherapy, using regimens such as FOLFIRINOX or gemcitabine/nab-paclitaxel (G/NP), remains the cornerstone of treatment for advanced PDAC. However, outcomes for metastatic PDAC remain poor, with a 5-year survival rate of only 3% [11,12].
Immunotherapy in PDAC focuses on leveraging the tumor microenvironment (TME) and the host immune system [13]. Immune checkpoint inhibitors (ICIs), targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), have shown efficacy in mismatch repair-deficient (MMR-D) tumors, but this subgroup represents only 2% of PDAC patients [14,15,16]. While ICIs have revolutionized outcomes for esophageal, liver, and biliary tract cancers, their success in mismatch repair-proficient PDAC remains limited [17,18,19,20,21,22,23,24,25,26]. Emerging immunotherapy approaches, including oncolytic virus therapy (OVT), adoptive cell transfer therapy (ACT), and cancer vaccines, hold promise for all tumor types, including PDAC [20]. Among these, chimeric antigen receptor (CAR) T-cell therapy is being actively investigated as a novel therapeutic strategy in PDAC. The urgent need for new targets and treatment modalities highlights the potential of immunotherapy as a critical avenue for improving outcomes in this challenging and aggressive disease.
3. Adoptive Cell Therapy in PDAC
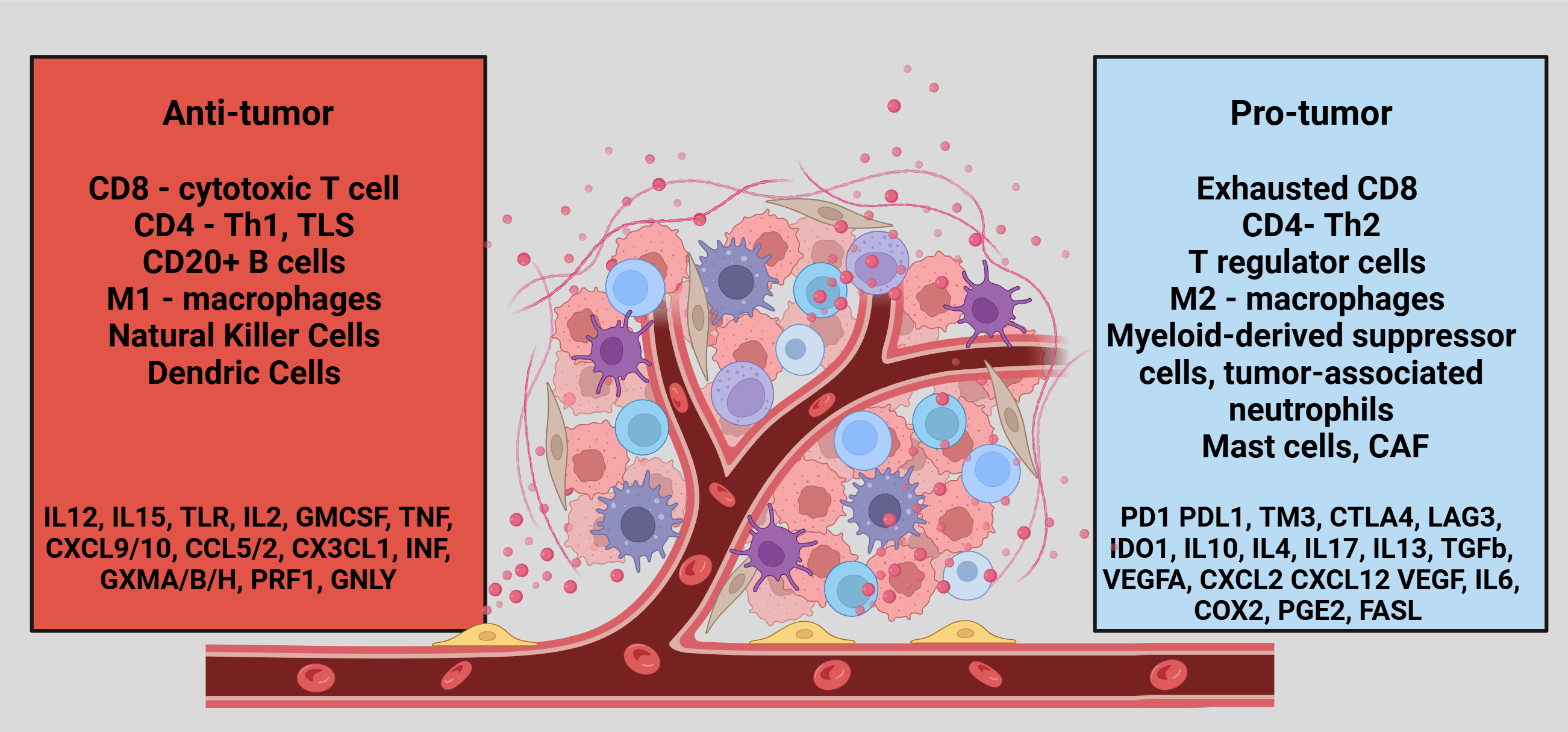
TME is a complex ecosystem that surrounds tumor cells, comprising various immune cell populations that play critical roles in maintaining its pro-tumorigenic nature [18]. These immune cells include lymphocytes (T and B cells), macrophages, natural killer (NK) cells, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), neutrophils, and mast cells, as illustrated in Figure 1. Each cell type contributes uniquely to the dynamic interplay within the TME, promoting tumor growth, immune evasion, and resistance to therapy. Immune-related TME is important not only for the effectiveness of ICI or other cellular therapy modalities but also for the outcomes, affecting both prognosis and treatment response. High infiltration of anti-tumor immune cells significantly improved the outcomes of PDA irrespective of the ICI use [27,28,29,30,31,32,33,34].
There is a growing interest in targeting TME to treat PDAC, and ACT is emerging as a key strategy in this effort [20,35]. While ACT has shown remarkable success in treating hematological malignancies, its application in solid tumors, including PDAC, remains in the early stages of development [36,37]. ACT broadly involves isolating immune cells—such as T lymphocytes, NK cells, and macrophages—from the patient, followed by their re-engineering and genetic modification to enhance their anti-tumor activity [38]. Various ACT modalities are under investigation for PDAC, including chimeric antigen receptor T cells (CAR-T), chimeric antigen receptor NK cells (CAR-NK), tumor-infiltrating lymphocytes (TILs), T cell receptor (TCR)-engineered T cells, and cytokine-induced killer cells (CIK-cells). These approaches are currently being evaluated in clinical trials to improve outcomes for this challenging malignancy. The following sections will delve deeper into these ACT modalities and their potential impact on PDAC treatment.
4. CAR- T in PDAC
CAR-T cell is a form of ACT that redirects a patient’s T cells to specifically target cancer cells through genetic engineering [39]. CARs are synthetic receptors designed with four main components: an extracellular antigen-binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains [37]. Since the development of first-generation CARs in 1989, subsequent generations have undergone significant advancements to enhance clinical efficacy [40]. To date, five generations of CAR-T cells have been developed, each featuring modifications to the domain structure and the inclusion of additional co-stimulatory molecules. Newer generations of CAR-T cells demonstrate improved T cell activation, enhanced efficacy, and greater persistence, with the ability to rapidly expand and survive long after infusion. However, CAR-T cell therapy's success heavily depends on identifying actionable tumor-specific antigens. This remains a significant challenge for PDAC despite the therapy’s transformative impact on hematological malignancies. We summarized the results of completed early-phase clinical trials (Phase I or I/II) in Table 1 and ongoing trials in Table 2.
The principal toxicities associated with CAR-T cell therapy are cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [61]. CRS presents clinically with a spectrum of symptoms, ranging from mild flu-like manifestations to severe vasodilatory shock and end-organ dysfunction, potentially leading to life-threatening complications. Management of CRS involves supportive care, including symptomatic treatment, and the use of tocilizumab, an interleukin-6 (IL-6) receptor antagonist, with or without corticosteroids, depending on the severity of the condition. ICANS typically develops after the onset of CRS and exhibits a range of neurological symptoms, from temporary cognitive deficits to fatal cerebral edema. The management of ICANS is stratified by severity: mild cases are treated with supportive measures, severe cases with corticosteroids, and anti-IL-6 therapy is employed only if ICANS occurs alongside CRS. The underlying factors contributing to these toxicities include antigen overlap between cancerous and normal tissues, leading to off-target effects and an exaggerated immune response triggered by CAR-T cell activation.
5. Tumor-Infiltrating Lymphocyte Therapy (TIL) in PDAC
TIL-based ACT involves isolating TILs from tumor tissues, expanding them in vitro, and reinfusing them into patients to identify and destroy tumor cells [62]. TIL therapy, which has shown promising results in solid tumors such as melanoma, breast, and ovarian cancers, is now being investigated for PDAC [63,64,65,66,67,68,69,70,71,72,73,74,75,76]. In a meta-analysis that examined PDAC-TME, a higher CD8+ T cell subgroup was associated with significant survival benefits, highlighting the potential of TIL-therapy in PDAC [77]. TILs therapy has unique advantages, including its ability to target tumor-specific neoantigens due to the presence of multiple T-cell receptor clones, its ease of extraction from tumor tissue owing to the high number of effector memory T cells, and its low toxicity profile since it utilizes autologous cells without genetic modification [78,79]
In a study involving 17 patients, including 5 with PDAC, the best response was observed in a PDAC patient with stable disease for 17 months [79]. However, this patient, who had liver and peritoneal metastases, exhibited no response at the primary tumor site. Overall, no objective responses (OR) were recorded among the PDAC cohort, with 3 achieving stable disease (SD) and 2 progressing (PD). Progression-free survival (PFS) and overall survival (OS) for PDAC patients were 2.43 months and 14.49 months, respectively, which were worse compared to the overall study population (PFS: 2.53 months, OS: 18.86 months) [70]. Bone marrow suppression emerged as a concerning high-grade adverse event across the entire study cohort. TIL therapy for PDAC remains in its early stages, and ongoing clinical trials are summarized in Table 3. Continued research is essential to optimize this approach and improve patient outcomes with PDAC.
6. Oncolytic Virus
The development of oncolytic virus (OV) therapy began in the 1990s, but its integration into clinical practice has accelerated only within the last decade [80]. OVs are genetically engineered viruses designed to selectively infect, replicate within, and ultimately destroy cancer cells. The first OV therapy to receive regulatory approval was talimogene laherparepvec (T-VEC or OncoVEXGM-CSF), approved in 2015 for the treatment of refractory melanoma [81]. Since then, several other OVs—including H-1 parvovirus, VCN-01 adenovirus, LOAd703 adenovirus, and pelareorep reovirus—have shown promising preclinical efficacy in pancreatic cancer cell lines [82,83,84,85]. In an early clinical study reported in 2018, the HF10 virus was injected directly into the primary tumor of patients with locally advanced pancreatic ductal adenocarcinoma (PDAC), alongside systemic erlotinib and gemcitabine therapy [86]. Among the nine patients who completed the study, three achieved PR, four had SD, and two experienced disease progression (PD). Severe adverse events such as bone marrow suppression, gastrointestinal perforation, and liver dysfunction were reported but were unrelated to OV therapy.
Subsequent investigations have demonstrated the safety and potential efficacy of other OVs, such as VCN-01 and LOAd703, when combined with chemotherapy (e.g., gemcitabine/nab-paclitaxel) or immune checkpoint inhibitors (ICIs) like pembrolizumab [85,87,88,89]. These studies highlight the potential of OVs to act synergistically with existing therapies. OVs also enhance the effectiveness of CAR-T cell therapy in solid tumors by improving tumor-associated antigen presentation, increasing T cell infiltration and tumor penetration, and mitigating immune suppression within the tumor microenvironment. This combination generates a more robust and durable anti-tumor response, making OVs a promising adjunctive therapy in PDAC treatment [90]. We discussed ongoing OV therapy trials in Table 3.
7. Genetically Modified T Cell Therapy
NK cells play a pivotal role as part of the body’s first line of defense against cancer. Genetically engineered NK cells have demonstrated the ability to mount specific and targeted anti-tumor responses, offering a promising avenue for cancer immunotherapy [91]. Preclinical studies involving chimeric antigen receptor NK (CAR-NK) cells targeting prostate stem cell antigen (PSCA) and mesothelin in pancreatic ductal adenocarcinoma (PDAC) have shown encouraging results, raising hope for their clinical application in select patient populations [92,93]. Compared to CAR-T cells, CAR-NK cells offer potential advantages, including reduced toxicity due to their shorter half-life and distinct cytokine profile, as well as a lower likelihood of inducing alloreactivity, making them suitable for "off-the-shelf" therapeutic products [20,94]. However, several limitations hinder their clinical implementation. These include technical challenges in manufacturing, poor tumor infiltration, and the short half-life of NK cells, which necessitates repeated administrations to sustain therapeutic effects. [95,96]. We discussed ongoing CAR-NK trials in Table 3.
In addition to NK cells, T cells can be genetically modified to express tumor-specific T-cell receptors (TCRs), enabling robust and precise anti-tumor responses [97]. Commonly targeted antigens in TCR therapy include mesothelin (MSLN), epidermal growth factor receptor (EGFR), claudin 18.2 (CLDN), CD133, and human epidermal growth factor receptor 2 (HER2). Notably, a study by Leidner et al. demonstrated that TCRs targeting mutant KRAS (KRAS12D) elicited responses in patients with metastatic PDAC, highlighting the potential of TCR-based therapies in this challenging cancer type [98]. We discussed ongoing TCR trials in Table 3.
8. Cytokine-Induced Killer (CIK) Cells
CIK cells are a heterogeneous group of CD8+ T cells that exhibit a hybrid phenotype, combining features of both T cells and natural killer (NK) cells. These cells are generated by incubating human-derived peripheral lymphocytes with anti-CD3 antibodies and cytokines [99,100,101]. CIK cells have shown potential to enhance the efficacy of other anti-cancer therapies, such as immune checkpoint inhibitors (ICIs) and chemotherapy, by amplifying anti-tumor responses [102].
Preclinical and clinical studies have demonstrated synergistic effects when CIK therapy is combined with chemotherapy in pancreatic cancer [100,101]. n a randomized study evaluating the addition of the chemotherapy agent S-1 to CIK therapy, a slight improvement in progression-free survival (PFS) was observed (2.5 months vs. 2.9 months, p = 0.03), although overall survival (OS) was comparable between the groups (6.1 months vs. 6.6 months, p = 0.09) [101]. Hematological toxicity was similar across both groups, but the incidence of non-infectious fever was significantly higher in the CIK group (32% vs. 3.3%, p = 0.004). In another study involving 47 patients with advanced pancreatic ductal adenocarcinoma (PDAC), median OS and PFS were notably higher in the group treated with dendritic cell-CIK (DC-CIK) therapy combined with S-1 (212 and 136 days, respectively) compared to those receiving DC-CIK therapy alone (128 and 85 days), chemotherapy alone (141 and 92 days), or supportive care only (52 and 43 days) [103]. These findings suggest that CIK-based therapies, particularly when combined with other modalities, hold promise for improving outcomes in advanced PDAC patients. We discussed ongoing CIK cells therapy in PDA in Table 3.
9. Conclusions
PDAC is a highly aggressive and challenging cancer that requires a multimodal approach to identify new therapeutic targets and develop innovative treatments. A comprehensive understanding of the TME in PDAC is critical to overcoming the limitations of current therapies and improving clinical outcomes. Cancer immunotherapy, particularly CAR-T therapy, has shown remarkable success in hematological malignancies and is now expanding its scope to solid tumors, including PDAC. However, the effectiveness of CAR-T therapy in PDAC is limited by factors such as tumor heterogeneity, T-cell exhaustion, and the suppressive influence of tumor-associated immune cells within the TME. Advances in next-generation CAR-T therapies, coupled with combination strategies integrating other treatment modalities, hold promise for addressing these challenges and unlocking the potential of CAR-T cell therapy in PDAC.
Author Contributions
Conceptualization, A.M.; resources, D.S.; writing—original draft preparation, D.S.; writing—review and editing, A.M.; visualization, A.M.; supervision, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
None.
Institutional Review Board Statement
Not applicable. Institutional data was not used for this project.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
biorender.com was used to make figures.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology. 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Ilic, I.; Ilic, M. International patterns in incidence and mortality trends of pancreatic cancer in the last three decades: A joinpoint regression analysis. World J Gastroenterol. 2022, 28, 4698–4715. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; et al. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Roalsø, M.; Aunan, J.R.; Søreide, K. Refined TNM-staging for pancreatic adenocarcinoma – Real progress or much ado about nothing? European Journal of Surgical Oncology. 2020, 46, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B. What Makes a Pancreatic Cancer Resectable? American Society of Clinical Oncology Educational Book. 2018, 300–305. [Google Scholar] [CrossRef]
- Vernuccio, F.; Messina, C.; Merz, V.; Cannella, R.; Midiri, M. Resectable and Borderline Resectable Pancreatic Ductal Adenocarcinoma: Role of the Radiologist and Oncologist in the Era of Precision Medicine. Diagnostics (Basel). 2021, 11. [Google Scholar] [CrossRef]
- Isaji, S.; Mizuno, S.; Windsor, J.A.; Bassi, C.; Fernández-Del Castillo, C.; Hackert, T.; et al. International consensus on definition and criteria of borderline resectable pancreatic ductal adenocarcinoma 2017. Pancreatology. 2018, 18, 2–11. [Google Scholar] [CrossRef]
- Barcellini, A.; Peloso, A.; Pugliese, L.; Vitolo, V.; Cobianchi, L. Locally Advanced Pancreatic Ductal Adenocarcinoma: Challenges and Progress. Onco Targets Ther. 2020, 13, 12705–12720. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. New England Journal of Medicine. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O. Cancer immunotherapy. Cancer Biother Radiopharm. 2011, 26, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Nasar, N.; Eikenboom, E.; Seier, K.; Gonen, M.; Wagner, A.; Jarnagin, W.R.; et al. Survival of patients with microsatellite instability-high and Lynch syndrome-associated pancreatic ductal adenocarcinomas. J Clin Oncol. 2024, 42, 640. [Google Scholar] [CrossRef]
- Velcheti, V.; Schalper, K. Basic Overview of Current Immunotherapy Approaches in Cancer. Am Soc Clin Oncol Educ Book. 2016, 35, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020, 27, S87–s97. [Google Scholar] [CrossRef]
- Sangro, B.; Chan, S.L.; Kelley, R.K.; Lau, G.; Kudo, M.; Sukeepaisarnjaroen, W.; et al. Four-year overall survival update from the phase III HIMALAYA study of tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. Ann Oncol. 2024, 35, 448–457. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J Clin Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Ruth He, A.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evidence. 2022. [CrossRef]
- Farhangnia, P.; Khorramdelazad, H.; Nickho, H.; Delbandi, A.-A. Current and future immunotherapeutic approaches in pancreatic cancer treatment. Journal of Hematology & Oncology. 2024, 17, 40. [Google Scholar]
- Callahan, M.; Amin, A.; Kaye, F.J.; Morse, M.A.; Taylor, M.H.; Peltola, K.J.; et al. Nivolumab monotherapy or combination with ipilimumab with or without cobimetinib in previously treated patients with pancreatic adenocarcinoma (CheckMate 032). J Immunother Cancer. 2024, 12. [Google Scholar] [CrossRef]
- Renouf, D.J.; Loree, J.M.; Knox, J.J.; Topham, J.T.; Kavan, P.; Jonker, D.; et al. he CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nature Communications. 2022, 13, 5020. [Google Scholar] [CrossRef]
- Padrón, L.J.; Maurer, D.M.; O'Hara, M.H.; O'Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med. 2022, 28, 1167–1177. [Google Scholar] [CrossRef]
- Bockorny, B.; Macarulla, T.; Semenisty, V.; Borazanci, E.; Feliu, J.; Ponz-Sarvise, M.; et al. Motixafortide and Pembrolizumab Combined to Nanoliposomal Irinotecan, Fluorouracil, and Folinic Acid in Metastatic Pancreatic Cancer: The COMBAT/KEYNOTE-202 Trial. Clin Cancer Res. 2021, 27, 5020–5027. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncology. 2019, 5, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Miksch, R.C.; Schoenberg, M.B.; Weniger, M.; Bösch, F.; Ormanns, S.; Mayer, B.; et al. Prognostic Impact of Tumor-Infiltrating Lymphocytes and Neutrophils on Survival of Patients with Upfront Resection of Pancreatic Cancer. Cancers (Basel). 2019, 11. [Google Scholar] [CrossRef]
- Panahi, M.; Rezagholizadeh, F.; Mollazadehghomi, S.; Farhangnia, P.; Niya, M.H.K.; Ajdarkosh, H.; et al. The association between CD3+ and CD8+tumor-infiltrating lymphocytes (TILs) and prognosis in patients with pancreatic adenocarcinoma. Cancer Treatment and Research Communications. 2023, 35, 100699. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; et al. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: a systematic review and meta-analysis. Eur J Cancer. 2020, 132, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wu, M.; Guo, L.; Zuo, Q. Pretreatment blood neutrophil/lymphocyte ratio is associated with metastasis and predicts survival in patients with pancreatic cancer. Bulletin du Cancer. 2018, 105, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. British Journal of Cancer. 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, S.; Gao, Q. An integrated overview of the immunosuppression features in the tumor microenvironment of pancreatic cancer. Frontiers in immunology. 2023, 14, 1258538. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; et al. The Immune Microenvironment in Pancreatic Cancer. Int J Mol Sci. 2020, 21. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity. 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Hartupee, C.; Nagalo, B.M.; Chabu, C.Y.; Tesfay, M.Z.; Coleman-Barnett, J.; West, J.T.; et al. Pancreatic cancer tumor microenvironment is a major therapeutic barrier and target. Frontiers in immunology. 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer. 2021, 9. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer Journal 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Tano, Z.E.; Varghese, A.M.; Adusumilli, P.S. CAR T-cell therapy for pancreatic cancer. J Surg Oncol. 2017, 116, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw Dejenie, T.; Tiruneh G/Medhin, M.; Dessie Terefe, G.; Tadele Admasu, F.; Wale Tesega, W.; Chekol Abebe, E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Human Vaccines & Immunotherapeutics. 2022, 18, 2114254. [Google Scholar]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Miyazaki, Y.; Tsukasa, K.; Matsubara, S.; Yoshimitsu, M.; Takao, S. CD133 facilitates epithelial-mesenchymal transition through interaction with the ERK pathway in pancreatic cancer metastasis. Molecular Cancer. 2014, 13, 15. [Google Scholar] [CrossRef]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; et al. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br J Cancer. 2008, 98, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Moody, A.E.; Guha, P.; Hardaway, J.C.; Prince, E.; LaPorte, J.; et al. HITM-SURE: Hepatic immunotherapy for metastases phase Ib anti-CEA CAR-T study utilizing pressure enabled drug delivery. J Immunother Cancer. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Xu, W.; Ji, S.; et al. Diagnostic and prognostic value of carcinoembryonic antigen in pancreatic cancer: a systematic review and meta-analysis. OncoTargets and Therapy 2017, 10, 4591–4598. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Clarke, L.; Pal, A.; Buchwald, P.; Eglinton, T.; Wakeman, C.; et al. A Review of the Role of Carcinoembryonic Antigen in Clinical Practice. Ann Coloproctol. 2019, 35, 294–305. [Google Scholar] [CrossRef] [PubMed]
- van Manen, L.; Groen, J.V.; Putter, H.; Vahrmeijer, A.L.; Swijnenburg, R.-J.; Bonsing, B.A.; et al. Elevated CEA and CA19-9 serum levels independently predict advanced pancreatic cancer at diagnosis. Biomarkers. 2020, 25, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Kishiwada, M.; Hayasaki, A.; Chipaila, J.; Maeda, K.; Noguchi, D.; et al. Role of Serum Carcinoma Embryonic Antigen (CEA) Level in Localized Pancreatic Adenocarcinoma: CEA Level Before Operation is a Significant Prognostic Indicator in Patients With Locally Advanced Pancreatic Cancer Treated With Neoadjuvant Therapy Followed by Surgical Resection: A Retrospective Analysis. Annals of Surgery. 2022, 275, e698–e707. [Google Scholar]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018, 3. [Google Scholar] [CrossRef]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J Clin Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opinion on Biological Therapy. 2021, 21, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Johnston, F.M.; Tan, M.C.B.; Tan, B.R.; Jr Porembka, M.R.; Brunt, E.M.; Linehan, D.C.; et al. Circulating Mesothelin Protein and Cellular Antimesothelin Immunity in Patients with Pancreatic Cancer. Clinical Cancer Research. 2009, 15, 6511–6518. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Molecular Therapy. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- QIC, *!!! REPLACE !!!*; Liu, C.; Gong, J.; Li, J.; Liu, D.; Wang, X.; et al. Claudin18.2-targeted chimeric antigen receptor T cell-therapy for patients with gastrointestinal cancers: Final results of CT041-CG4006 phase 1 trial. J Clin Oncol. 2024, 42, 2501. [Google Scholar]
- Qin, S.; Tian, W.; Li, M.; Wei, H.; Sun, L.; Xie, Q.; et al. 1054P A phase Ia study to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy of a modular CLDN18.2-targeting PG CAR-T therapy (IBI345) in patients with CLDN18.2+ solid tumors. Ann Oncol. 2023, 34, S638. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy. 2020, 22, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.L.; Lertpiriyapong, K.; Cocco, L.; Martelli, A.M.; Libra, M.; Candido, S.; et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Advances in Biological Regulation. 2015, 59, 65–81. [Google Scholar] [CrossRef]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal Growth Factor Receptor in Pancreatic Cancer. Cancers. 2011, 3, 1513–1526. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Yarmand-Bagheri, R. The role of HER2 in angiogenesis. Semin Oncol. 2001, 28, 27–32. [Google Scholar] [CrossRef]
- Schmidts, A.; Wehrli, M.; Maus, M.V. Toward Better Understanding and Management of CAR-T Cell–Associated Toxicity. Annual review of medicine 2021, 72, 365–82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers. 2022, 14, 4160. [Google Scholar] [CrossRef]
- Betof Warner, A.; Corrie, P.G.; Hamid, O. Tumor-Infiltrating Lymphocyte Therapy in Melanoma: Facts to the Future. Clinical Cancer Research. 2023, 29, 1835–1854. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Sadri, M.; Najafi, A.; Rahimi, A.; Baghernejadan, Z.; Khorramdelazad, H.; et al. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front Immunol. 2022, 13, 1018962. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; et al. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. JNCI: Journal of the National Cancer Institute. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Ann Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Ellebaek, E.; Iversen, T.Z.; Junker, N.; Donia, M.; Engell-Noerregaard, L.; Met, Ö.; et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. Journal of Translational Medicine. 2012, 10, 169. [Google Scholar] [CrossRef]
- Ben-Avi, R.; Farhi, R.; Ben-Nun, A.; Gorodner, M.; Greenberg, E.; Markel, G.; et al. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol Immunother. 2018, 67, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Kradin, R.L.; Boyle, L.A.; Preffer, F.I.; Callahan, R.J.; Barlai-Kovach, M.; Strauss, H.W.; et al. Tumor-derived interleukin-2-dependent lymphocytes in adoptive immunotherapy of lung cancer. Cancer Immunol Immunother. 1987, 24, 76–85. [Google Scholar] [CrossRef]
- Freedman, R.S.; Kudelka, A.P.; Kavanagh, J.J.; Verschraegen, C.; Edwards, C.L.; Nash, M.; et al. Clinical and biological effects of intraperitoneal injections of recombinant interferon-gamma and recombinant interleukin 2 with or without tumor-infiltrating lymphocytes in patients with ovarian or peritoneal carcinoma. Clin Cancer Res. 2000, 6, 2268–2278. [Google Scholar]
- Fujita, K.; Ikarashi, H.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T.; et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995, 1, 501–507. [Google Scholar] [PubMed]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: a pilot study. Oncoimmunology. 2018, 7, e1502905. [Google Scholar] [CrossRef] [PubMed]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget. 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- O’Malley, D.; Lee, S.; Psyrri, A.; Sukari, A.; Thomas, S.; Wenham, R.; et al. 492 Phase 2 efficacy and safety of autologous tumor-infiltrating lymphocyte (TIL) cell therapy in combination with pembrolizumab in immune checkpoint inhibitor-naïve patients with advanced cancers. Journal for ImmunoTherapy of Cancer. 2021, 9, A523–A524. [Google Scholar]
- Li, J.; Chen, Q.Y.; He, J.; Li, Z.L.; Tang, X.F.; Chen, S.P.; et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. Oncoimmunology. 2015, 4, e976507. [Google Scholar] [CrossRef]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; et al. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: a systematic review and meta-analysis. European Journal of Cancer. 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers (Basel). 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.; Knisely, A.; Vining, D.; Kopetz, S.; Overman, M.J.; Javle, M.; et al. Efficacy and safety of autologous tumor-infiltrating lymphocytes in recurrent or refractory ovarian cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. J Immunother Cancer. 2024, 12. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Prospects for combined use of oncolytic viruses and CAR T-cells. Journal for ImmunoTherapy of Cancer. 2017, 5, 90. [Google Scholar] [CrossRef]
- Johnson, D.B.; Puzanov, I.; Kelley, M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015, 7, 611–619. [Google Scholar] [CrossRef] [PubMed]
- McAuliffe, P.F.; Jarnagin, W.R.; Johnson, P.; Delman, K.A.; Federoff, H.; Fong, Y. Effective treatment of pancreatic tumors with two multimutated herpes simplex oncolytic viruses. J Gastrointest Surg. 2000, 4, 580–588. [Google Scholar] [CrossRef]
- Toda, M.; Martuza, R.L.; Rabkin, S.D. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol Ther. 2000, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.-R.; Kaowinn, S.; Moon, J.; Soh, J.; Kang, H.Y.; Jung, C.-R.; et al. Oncotropic H-1 parvovirus infection degrades HIF-1α protein in human pancreatic cancer cells independently of VHL and RACK1. Int J Oncol. 2015, 46, 2076–2082. [Google Scholar] [CrossRef]
- Musher, B.L.; Smaglo, B.G.; Abidi, W.; Othman, M.; Patel, K.; Jawaid, S.; et al. A phase I/II study of LOAd703, a TMZ-CD40L/4-1BBL-armed oncolytic adenovirus, combined with nab-paclitaxel and gemcitabine in advanced pancreatic cancer. J Clin Oncol. 2022, 40, 4138. [Google Scholar] [CrossRef]
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer. 2018, 18, 596. [Google Scholar] [CrossRef] [PubMed]
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Álvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J Immunother Cancer. 2021, 9. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Bazan-Peregrino, M.; Gil-Martín, M.; Álvarez, R.; Macarulla, T.; Riesco-Martinez, M.C.; et al. Phase I, multicenter, open-label study of intravenous VCN-01 oncolytic adenovirus with or without nab-paclitaxel plus gemcitabine in patients with advanced solid tumors. J Immunother Cancer. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Chen, S.; Xie, P.; Loghmani, H.; Heineman, T.; Kalyan, A.; et al. Combination of pembrolizumab and pelareorep promotes anti-tumour immunity in advanced pancreatic adenocarcinoma (PDAC). Br J Cancer. 2023, 129, 782–790. [Google Scholar] [CrossRef]
- Ponterio, E.; Haas, T.L.; De Maria, R. Oncolytic virus and CAR-T cell therapy in solid tumors. Frontiers in immunology. 2024, 15, 1455163. [Google Scholar] [CrossRef] [PubMed]
- Froelich, W. CAR NK Cell Therapy Directed Against Pancreatic Cancer. Oncology Times. 2021, 43, 46. [Google Scholar] [CrossRef]
- Teng, K.Y.; Mansour, A.G.; Zhu, Z.; Li, Z.; Tian, L.; Ma, S.; et al. Off-the-Shelf Prostate Stem Cell Antigen-Directed Chimeric Antigen Receptor Natural Killer Cell Therapy to Treat Pancreatic Cancer. Gastroenterology. 2022, 162, 1319–1333. [Google Scholar] [CrossRef]
- Da, Y.; Liu, Y.; Hu, Y.; Liu, W.; Ma, J.; Lu, N.; et al. STING agonist cGAMP enhances anti-tumor activity of CAR-NK cells against pancreatic cancer. Oncoimmunology. 2022, 11, 2054105. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Farrukh, H.; Chittepu, V.; Xu, H.; Pan, C.X.; Zhu, Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef]
- Wang, K.; Wang, L.; Wang, Y.; Xiao, L.; Wei, J.; Hu, Y.; et al. Reprogramming natural killer cells for cancer therapy. Molecular Therapy. 2024, 32, 2835–2855. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.-F.; Rivas, J.M.; et al. T-cell receptor-based therapy: an innovative therapeutic approach for solid tumors. Journal of Hematology & Oncology. 2021, 14, 102. [Google Scholar]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N Engl J Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Zhang, Y.; Schmidt-Wolf, I.G.H. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. Journal of Cellular Physiology. 2020, 235, 9291–9303. [Google Scholar] [CrossRef]
- Choi, J.H.; Nam, G.H.; Hong, J.-m.; Cho, I.R.; Paik, W.H.; Ryu, J.K.; et al. Cytokine-Induced Killer Cell Immunotherapy Combined With Gemcitabine Reduces Systemic Metastasis in Pancreatic Cancer: An Analysis Using Preclinical Adjuvant Therapy-Mimicking Pancreatic Cancer Xenograft Model. Pancreas. 2022, 51, 1251–1257. [Google Scholar] [CrossRef]
- Wang, M.; Shi, S.B.; Qi, J.L.; Tang, X.Y.; Tian, J. S-1 plus CIK as second-line treatment for advanced pancreatic cancer. Med Oncol. 2013, 30, 747. [Google Scholar] [CrossRef]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; et al. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Frontiers in immunology. 2017, 8, 774. [Google Scholar] [CrossRef]
- Jiang, N.; Qiao, G.; Wang, X.; Morse, M.A.; Gwin, W.R.; Zhou, L.; et al. Dendritic Cell/Cytokine-Induced Killer Cell Immunotherapy Combined with S-1 in Patients with Advanced Pancreatic Cancer: A Prospective Study. Clin Cancer Res. 2017, 23, 5066–5073. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Summarizing the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma.
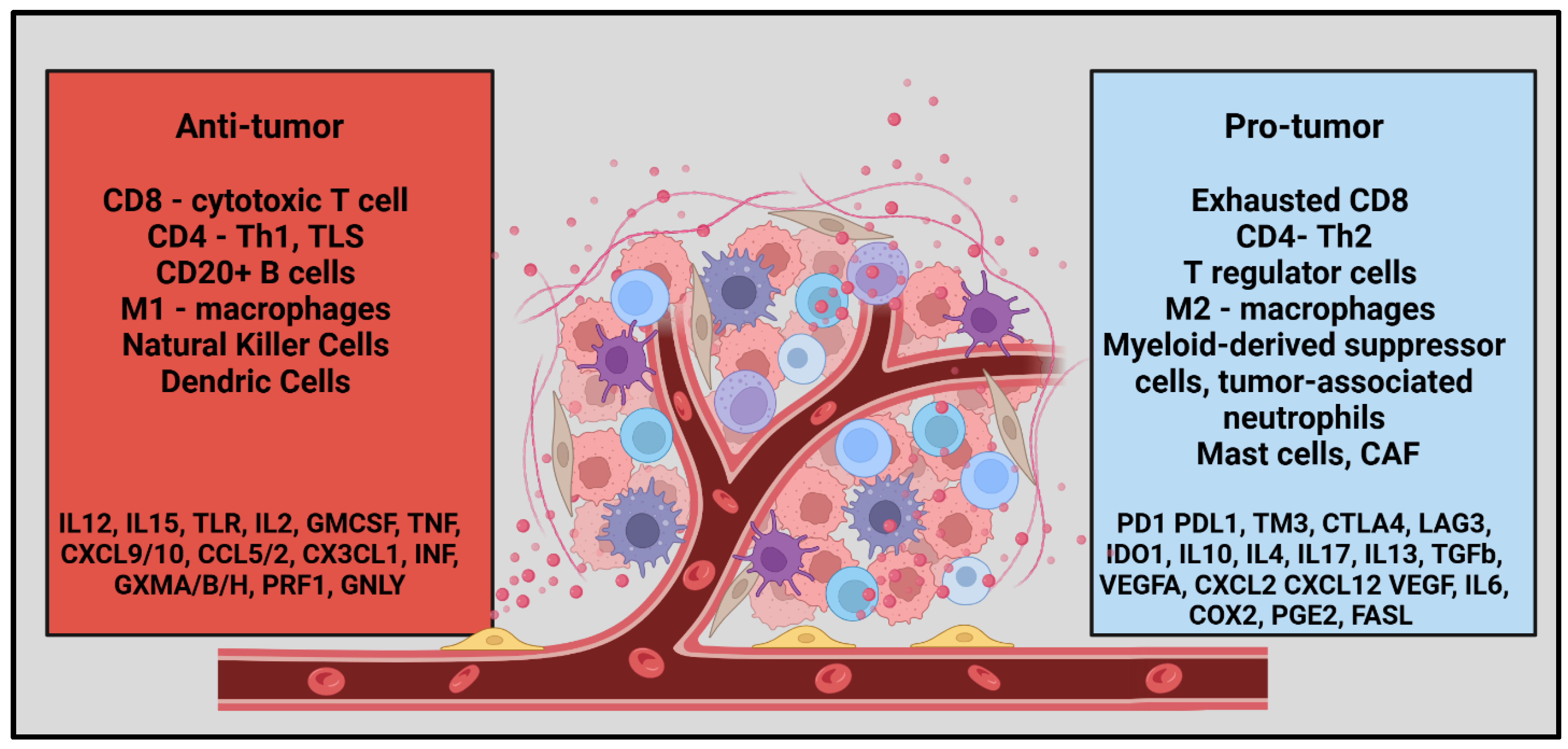

Table 1.
Current evidence of CAR-T cell therapies in pancreatic ductal adenocarcinoma.
| Trial | Target | Outcomes | Adverse effects | Notes on the target |
| NCT02541370* [41] (n=23) |
CD-133 (B) PDAC – 7/23 |
PR – 2 SD – 3 PD - 2 |
Hyperbilirubinemia, Anemia, Leucopenia, Thrombocytopenia, Anorexia, and Mucosal hyperemia | It is a transmembrane protein and the most commonly expressed cancer stem cell marker in several cancer types [42]. Correlates with histologic type, lymphatic invasion, and metastasis in pancreatic cancer[43]. |
| NCT02850536 [44] (n=5) |
CEA | OS – 23.2m DOR – 13m |
Fever, Electrolyte abnormalities, Hypertension | It can be elevated in PDAC, and a level > 7.2 ng/ml in LA PDAC is often associated with systemic disease [45,46,47,48]. |
| NCT01897415 [49] (n=6) |
Mesothelin | SD – 2 PD – 4 |
Abdominal pain, Back pain Dysgeusia, Gastritis |
It is an important factor in pancreatic growth by promoting proliferation and inhibiting apoptosis through p53-dependent and p53-independent pathways [50,51]. Mesothelin-specific T cells were generated in 50% of pancreatic cancer patients in a study [52]. |
| NCT02159716 [53] (n=15) |
Mesothelin (B) PDAC – 5/15 |
PD – 3 SD - 2 |
Anemia, Lymphopenia, Fatigue, Dysgeusia, DIC | |
| NCT03874897 [54] (n=37) |
Claudin 18.2 (B) PDAC – 5/37 |
PD – 1 SD – 3 PR – 1 |
Lymphopenia, Neutropenia, Anemia, Thrombocytopenia, Elevated conjugated bilirubin, Elevated aminotransferase, Hypokalemia, Pyrexia | It is a transmembrane protein that controls the paracellular space through which molecules pass in the epithelial and endothelial tissues and is essential for normal membrane barrier function [46]. It is overexpressed in various cancers and plays an important role in the progression of pancreatic neoplasms. Claudin types could be tumor-specific. |
| NCT05199519 [55] (n=7) |
Claudin 18.2 (B) PDAC – 2/5 |
PR – 1 SD – 1 |
Neutropenia, Anorexia | |
| NCT01869166 [56] (n=14) |
EGFR | PR – 4 SD – 8 PD - 2 |
Lymphocytopenia, Pleural effusion, Pulmonary interstitial exudation, Dermatitis Herpetiformis, Gastrointestinal hemorrhage |
It plays a crucial role in normal cellular growth, prevention of apoptosis and development of metastasis in many types of cancer [57]. There are 4 receptors in the EGF family HER1, HER2, HER3, HER4 [58]. |
| NCT01935843 [59] (n=11) |
HER2 (B) PDAC – 2/11 |
SD - 2 | Anemia, Lymphopenia Fever, Fatigue Transaminase elevation Gastrointestinal hemorrhage |
It is a cell-membrane protein involved in promoting cell division & differentiation and contributes to tumor progression by triggering angiogenesis [60] |
* Phase I/II, B – basket trials PDAC – pancreatic ductal, adenocarcinoma, PR – partial response, SD – stable disease, PD – progressive disease, OS – overall survival, DOR – duration of response, CEA – carcinoembryonic antigen, EGFR - epidermal growth factor receptor, HER2 - human epidermal growth factor receptor 2, DIC - disseminated intravascular coagulation.
Table 2.
Ongoing CAR-T Trials in Pancreatic Ductal Adenocarcinoma.
| Trial | Phase | Size | Target | Primary outcome | Secondary outcomes |
| NCT06464965 | I | 30 |
Claudin 18.2 |
MTD, DLT | ORR, DCR, OS, PFS, DOR |
| NCT05472857 | I | 30 | AE, MTD | ORR, DOR, DCR PFS, | |
| NCT04404595 | Ib | 110 | AE, MTD, DLT, ORR | ORR, DOR, DCR, PFS, OS, HRQoL | |
| NCT04581473 | I/II | 192 | AE, MTD, PFS | ORR, DCR, DOR, OS, PFS | |
| NCT05393986 | I | 63 | DLT, MTD | AE, PK, ORR, DOR, DCR, OS, PFS | |
| NCT05275062 | I | 6 | AE | ORR, DCR, OS, PFS, CAR -T %, Tumor marker, RR, IM92 Ab | |
| NCT06126406 | I | 60 |
CEA |
AE, DLT | DCR, AUCS, CMAX, TMAX, CEA content |
| NCT06043466 | I | 30 | Dose range, , DLT, MTD | DCR, AUCS, CMAX, TMAX, CEA content | |
| NCT06010862 | I | 36 | AE, MTD | DCR, ORR, DOR, OS PFS, AUCS, CMAX, TMAX, CEA content | |
| NCT05736731 | I/II | 160 | DLT, RP2D, ORR | A2B530%, Cytokine analysis | |
| NCT04660929 | I | 48 |
HER 2 |
AE, Feasibility of manufacturing, CT - 0508 | ORR, PFS |
| NCT03740256 | I | 45 | DLT | ORR, DCR, OS, PFS, AEs grade 3 |
|
| NCT06051695 | I/ II | 230 | Mesothelin | DLT, RP2D, ORR | A2B694 persistence, Cytokine analysis |
| NCT05239143 | I | 180 | MUC1 - C | MTD, R2PD, ORR | - |
| NCT06158139 | I | 27 | B7-H3 | AE, CRS, Neurotoxicity | DLT, OS, PFS, DCR, ORR, B7-H3 expression |
| NCT02830724 | I/II | 124 | CD 70 | AE within 2 weeks, RR | AE within 6 weeks) |
MTD – Maximum tolerated dose, DLT – Dose-dependent toxicity, AE – adverse events, CRS – cytokine release syndrome, RX – treatment, R2PD – recommended phase 2 dose, QOL – quality of life, ORR – Objective response rate, CR – complete response, PR – partial response, DOR – duration of overall response, DOCR – duration of overall complete response, DCR – disease control rate, RRR – radiographic response rate, OS – overall survival, PFS – progression-free survival, , RR – response rate (PR + CR). HRQoL – Health related Quality of Life, PK – pharmacokinetics, CEA – carcinoembryonic antigen, ACUS – area under the curve, CMAX – highest concentration of CEA CAR-T cells expanded, TMAX - time to reach the highest concentration.
Table 3.
Ongoing Adoptive Cell Therapy Trials in Pancreatic Ductal Adenocarcinoma.
| Trial | Phase | Size | Target | Outcomes | |
| TIL-therapy | NCT05098197 | I | 50 | - | TRAE, ORR, DCR, DOR, PFS, OS |
| NCT03935893 | II | 240 | - | ORR, CRR, DOR, DCR, PFS, OS | |
| NCT04426669 | I/II | 20 | - | MTD, PE, AE PFS, OS |
|
| NCT01174121 | II | 332 | - | RR, AE Efficacy |
|
| NCT05098197 | I | 50 | - | TRAE, ORR, DCR, DOR, PFS, OS | |
| Oncolytic virus | NCT03740256 (adenovirus) |
I | 45 | HER2 | DLT ORR, DCR, PFS, OS, >grade 3 AE |
| NCT02705196 (adenovirus) |
I/II | 55 | DLT ORR, OS |
||
| NCT05860374 (herpes virus) |
I | 20 | TRAE, SIR, LA DCR, DOR, QoLA |
||
| NCT05886075 (herpes virus) |
I | 24 | AE, SIR, LA DCR, DOR, QoLA |
||
| NCT06508307 | I | 21 | MTD, DLT, TEAE, LA ORR, DOR, PFS Viral distribution, lymphocyte ratio, cytokine levels, immunogenicity |
||
| NCT05076760 | I | 61 | MTD, AE, ORR DCR, DOR, PFS, OS Exploratory biomarker analysis |
||
| CAR-NK | NCT03941457 | I/II | 9 | ROBO1 | TRAE |
| NCT02839954 | I/II | 10 | MUC1 | TRAE ORR |
|
| NCT03841110 | I | 64 | NK cell + ICI | DLT ORR and DOR |
|
| NCT06464965 | I | 30 | Claudin18.2 | MTD and DLT ORR, DCR, PFS, OS, and DOR |
|
| NCT05922930 | I/II | TROP2 | DLT, ORR and PFS | ||
| Cytokine-induced killer (CIK) cells | NCT03509298 | II | 90 | CIK with anti-CD3-MUC1 bispecific antibody | ORR, PFS, TTP, DCR, OS, SSR |
| NCT05955157 | II/III randomized |
52 | DC-CIK _ S-1 vs. S-1 | TRAE, Hematological CBR Efficacy |
|
| T-cell receptor-engineered T-cells | NCT04809766 | I | 15 | Mesothelin | TRAE ORR, PFS, OS |
| NCT05438667 | I | 18 | KRAS | OS, PFS, TTP, EFS, DFS, DoE AE, CMAX, TMAX, AUC, TCR-T cell number, peak value of cytokines |
|
| NCT06487377 | I | 12 | KRAS | DLT, TRAE, SAE ORR, DCR, DOR, TTR, OS, PFS, TCR-T cell counts, TCR gene copies, anti-drug antibodies, changes in tumor markers |
|
| NCT04146298 | I/II | 30 | KRAS | TRAE, ORR OS, TCR transduced T cell % |
|
| NCT06054984 | I | 18 | RAS/TP 53 | TRAE, CMAX, TMAX, AUC ORR, DCR, PFS, OS Change in tumor size, biomarker |
|
| NCT06043713 | I | 24 | KRAS | AE, DLT, MTD CBR, ORR, SD, ORR, PFS, OS, changes in TME |
|
| NCT05877599 | I | 162 | TP53 | DLT, AE, SAE, TRAE, ORR, BOR, DOR, CBR, TTR, PFS | |
| NCT06218914 | I | 24 | KRAS | DLT, AE, SAE ORR, BOR, DOR, CBR, TTR, PFS, OS |
|
| NCT06105021 | I/II | 100 | KRAS | OBD, DLT, SAE, TEAE ORR, DOR, PFS, TTR, CBR, OS |
|
| NCT04622423 | Observational | 475 | Tumor mutational burden, Gene expression profile, Antigenic landscape, T cell repertoire, OS, PFS, Change in tumor marker | ||
| NCT05964361 | I/II | 10 | WT-1 | Leukapheresis %, SAE, BOR, DOR, ORR, DCR, PFS, OS, QoLA | |
| NCT03190941 | I/II | 110 | KRAS | RR, TRAE | |
| NCT03745326 | I/II | 70 | KRAS | RR, TRAE | |
| NCT04810910 | I | 20 | Personalized Neo-antigen vaccine |
TRAE, RFS, OS, CD4/CD8 | |
TIL – tumor infiltrating lymphocytes, CAR – chimeric antigen receptor, NK – natural killer cells, TRAE – treatment related adverse events, ORR – Objective response rate, CR – complete response, PR – partial response, DOR – duration of response, OS – overall survival, PFS – progression-free survival, MTD – Maximum tolerated dose, PE - preliminary efficacy, LA – lab abnormalities, RR – response rate, DCR – disease control rate, QoLA – Quality of life assessment, SIR - Systemic Immune Response, DLT – Dose-dependent toxicity, TTP – time to progression, SSR – symptom remission rate, CBR – clinical benefit rate, Cmax – peak plasma concentration, Tmax – peak time, AUC – area under concentration, BOR - best overall response.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



