Submitted:
23 December 2024
Posted:
24 December 2024
You are already at the latest version
Abstract
Background: Triple-negative breast cancer (TNBC) is a highly aggressive subtype with limited effective treatments available, including targeted therapies, often leading to poor prognosis. Mitotic checkpoint kinase BUB1 is frequently overexpressed in TNBC and correlates with poor survival outcomes suggesting its potential as a therapeutic target. This study explores the cytotoxicity of TNBC cells to BUB1 inhibition, alone or in combination with radiation and demonstrates that ferroptosis, an iron-dependent form of programmed cell death, has a role.
Methods: TNBC cell lines (SUM159, MDA-MB-231, and BT-549) were treated with a BUB1 inhibitor BAY1816032 (BUB1i) alone or in combination with the ferroptosis activator RSL3 with or without 4 Gy irradiation. Cell viability assays were conducted to assess treatment effects, and qPCR analyses measured expression of key ferroptosis markers including ACSL4 and PTGS2, and GPX4 and SLC7A11 expression. Ferroptosis specificity was confirmed through co-treatment with the ferroptosis inhibitor Ferrostatin-1 (F-1).
Results: In all TNBC cell lines studied, BUB1 inhibition significantly induced ferroptosis, marked by increased expression of ACSL4 and PTGS2 and decreased expression of GPX4 and SLC7A11. The combination of BUB1i with RSL3 further amplified these ferroptosis markers, suggesting at least an additive effect, which was not present with the combination of BUB1i and radiation. Co-treatment with Ferrostatin-1 reversed the expression of ferroptosis markers, suggesting that BUB1i mediated cell death may involve ferroptosis signaling in TNBC cell lines.
Conclusions: This study demonstrates that BUB1 inhibition may independently induce ferroptosis in TNBC cell lines which is enhanced when combined with a ferroptosis activator. Further research is warranted to delineate molecular mechanism of BUB1 mediated ferroptosis in TNBC.
Keywords:
BAY1816032
; DNA damage
; Ferroptosis
; radiation
; TNBC
1. Introduction
Triple-negative breast cancer (TNBC) is a subtype of breast cancer characterized by the absence of estrogen, progesterone, and HER2 receptors, rendering it unresponsive to conventional hormone therapies and HER2-targeted treatments [1]. Additionally, chemotherapy options for TNBC although standard of care, are not always effective [2]. While the FDA has approved several newer therapies for TNBC, cytotoxic chemotherapies such as carboplatin, docetaxel, and doxorubicin remain the mainstay of treatment [3-5]. Novel therapeutic approaches, including immunotherapies like atezolizumab and pembrolizumab, and antibody-drug conjugates such as sacituzumab govitecan and trastuzumab deruxtecan (for low/ultra-low HER2 expression) provide targeted treatments for different TNBC subtypes [5-7]. Furthermore, PARP inhibitors like olaparib and talazoparib have proven effective in BRCA-mutated TNBC cases [5,8,9]. Despite these advancements, TNBC continues to pose a significant clinical challenge due to its aggressive nature, high recurrence rates, and poor prognosis [10]. Consequently, the development of better therapeutic strategies that specifically target TNBC cells is needed.
One potential new therapeutic strategy against TNBC is to exploit ferroptosis [11-14]. Ferroptosis is a unique form of cell death, distinct from apoptosis, necrosis, and autophagy. It is specifically characterized by the accumulation of lipid peroxides within cellular membranes, driven by intracellular iron and the generation of reactive oxygen species (ROS) leading to cell-death [15,16]. Ferroptosis exploits the unique vulnerabilities of certain cancer types including TNBC [17-19]. Several agents, including erastin and sulfasalazine have been shown to induce ferroptosis in TNBC cells by targeting key metabolic pathways related to oxidative stress and iron metabolism [20,21]. A combination of siramesine and lapatinib was shown to trigger ferroptosis in MDA-MB-231 and SKBR3 cells [22] while DMOCPTL, a derivative of parthenolide, was identified as a potent inducer of ferroptosis and apoptosis, offering a novel approach for targeting TNBC [23]. DMOCPTL induces ferroptosis by promoting the ubiquitination and degradation of GPX4, a critical enzyme that protects cells from lipid peroxidation [23]. These findings underscore the therapeutic promise of ferroptosis induction in TNBC.
A recent study has identified a role for mitotic checkpoint serine/threonine kinase BUB1 in regulating ferroptosis in cancer cells [24]. BUB1 plays a critical role in ensuring proper chromosomal segregation during mitosis [25]. We identified that pharmacological inhibition or CRISPR ablation of BUB1 reduced tumor growth and improved the efficacy of radiotherapy, chemotherapy, and chemoradiotherapy in pre-clinical models of TNBC [26,27] and lung cancer [28]. While BUB1 has been traditionally studied in the context of its role in mitotic regulation, new evidence linking BUB1 to ferroptosis in pancreatic cancer cells [24] prompted us to hypothesize that BUB1 regulates ferroptosis in TNBC cells. In the present study, we sought to test whether BUB1 inhibition induces ferroptosis in TNBC cell lines and if these effects are altered when combined with irradiation. Previous research has demonstrated that ferroptosis can be triggered by a variety of mechanisms, including iron overload and oxidative stress, both of which are exacerbated by the disruption of cellular division [29]. Given that TNBC cells exhibit high rates of proliferation and are often dependent on intact mitotic checkpoints to maintain genomic stability [30], we hypothesized that BUB1 inhibition would create conditions vulnerable to ferroptosis.
Recently, we demonstrated that BUB1 is overexpressed in TNBC, and that this overexpression correlates with poorer patient outcomes [27]. Additionally, we confirmed that both pharmacological inhibition and genomic ablation of BUB1 were cytotoxic [27] supporting a new therapeutic strategy. Here, we employed a pharmacological BUB1 inhibitor BAY1816032 in multiple TNBC cell lines and assessed cell viability in the presence of the ferroptosis activator RSL3 and the inhibitor Ferrostatin-1. We also investigated the combined effects of BUB1 inhibition and irradiation to determine whether there is any potential therapeutic synergy between these treatments. Additionally, we evaluated how BUB1 inhibition influences the expression of key ferroptosis-related genes. Our data provides a foundation for developing approaches that combine BUB1 inhibition with ferroptosis induction to improve treatments for TNBC.
2. Materials and methods
2.1. Chemicals
BUB1 kinase inhibitor BAY1816032 (Catalog No. HY-103020), ferroptosis inducer RSL3 (Catalog No. HY-100218A), and ferroptosis inhibitor Ferrostatin-1 (Catalog No. HY-100579) were obtained from MedChemExpress (Monmouth Junction, NJ, USA).
2.2. Cell lines and culture
The SUM159 cell line was originally established by Stephen P. Ethier and was sourced from Sofia Merajver at the University of Michigan. SUM159 cells were cultured in HAM’s F-12 media (Catalog No. 31765305, Thermo Fisher Scientific; Waltham, MA, USA) supplemented with 5% FBS, 10 mM HEPES, 1 μg/mL Hydrocortisone, 6 μg/mL Insulin, and 1% Penicillin-Streptomycin. The MDA-MB-231 cell line, obtained from the American Type Culture Collection (ATCC), was grown in DMEM media (Catalog No. 30-2002, ATCC) supplemented with 10% FBS and 1% Penicillin-Streptomycin. BT-549 cells were maintained in RPMI-1640 media (Catalog No. 30-2001, ATCC) supplemented with 10% FBS, 0.023 U/mL Insulin, and 1% Penicillin-Streptomycin. All cell lines were maintained at 37°C in a 5% CO2 incubator and passaged when they reached 70% confluence, with passage numbers for SUM159, MDA-MB-231, and BT-549 ranging from P20 to P25. Mycoplasma contamination was regularly tested using the MycoAlert PLUS kit (Lonza, Cat. No. LT07-705).
2.3. Drug treatment and radiation
The stock solutions of BAY1816032 (20 mM), RSL3 (10 mM), and Ferrostatin-1 (10 mM) were made in DMSO and stored at -80°C. For each experiment, a new vial was thawed, and any unused stock solution was discarded. Working concentrations were prepared in media containing serum and supplements. Irradiation was conducted 1 hour after drug treatment using a CIX-3 orthovoltage unit (Xstrahl Inc, Suwanee, GA), operating at 320 kV and 10 mA with a 1 mm Cu filter.
2.4. Cell viability
SUM159, MDA-MB-231, and BT-549 cells were seeded at a density of 2,000 cells per well in 96-well plates. After 24 hours, the cells were treated with BAY1816032 (1 μM), RSL3 (62.5 nM), Ferrostatin-1 (10 µM), or a combination of BAY1816032 with RSL3 and Ferrostatin-1 for 72 hours. Cytotoxicity was evaluated using the alamarBlue cell viability kit (Thermo Fisher Scientific, Cat. No. DAL1100) following the manufacturer’s instructions. Absorbance was measured at 570 nm using a Synergy H1 Hybrid Reader (BioTek Instruments, Winooski, VT, USA), and the results were compared to those of vehicle/mock-treated cells. All the experiments were performed in triplicates and were repeated at least three times. The data was analyzed and plotted on GraphPad Prism (V9).
2.5. Quantitative PCR
Cells (1.5 x 105) were seeded in 6-well plates 24 hours before treatment with BUB1i (1 μM), RSL3 (0.25 µM), or Ferrostatin-1 (10 µM) along with irradiation (4 Gy). After 24 hours, the cells were harvested and stored at -80°C. Total RNA was extracted using TRIzol (Catalog No. 15596026, Thermo Fisher Scientific), and RNA concentration was determined on Nanodrop 2000c (Thermo Fisher Scientific). RNA was reverse transcribed into cDNA with the Super Script III Reverse Transcriptase kit (Catalog No. 18080044, Thermo Fisher Scientific), dNTPs (Catalog No. R0191, Thermo Fisher Scientific), and Random Primers (Catalog No. 48190011, Thermo Fisher Scientific). Quantitative PCR (qPCR) was conducted using Takyon Low ROX SYBR 2X MasterMix (Catalog No. UF-LSMT-B0701, Eurogentec) and KiCqStart pre-designed SYBR green gene-specific primers in a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). The expression levels of each gene were normalized to GAPDH for each experiment. Primer sequences are provided in Table 1. All qRT-PCR reactions were performed in triplicate, and all experiments were repeated at least three times.

3. Results
3.1. BUB1 inhibition increases cell death induced by ferroptosis activator RSL3
The effect of BUB1 inhibitor BAY1816032 with ferroptosis inhibitor RSL3 on cell viability was evaluated in SUM159, MDA-MB-231 and BT-549 cells (Figure 1). A dose-response experiment was conducted in SUM159 cells to determine the effective concentrations of BUB1i, RSL3, and Ferrostatin-1 (Figure S1). Single agent IC70-80 (BAY1816032 1 μM, RSL3 62.5 nM and Ferrostatin-1 10 μM) were selected for drug-combination experiments.
BUB1 inhibitor (BUB1i) alone reduced cell viability in TNBC cell lines SUM159, MDA-MB-231, and BT-549 by 20-30% which significantly increased when BUB1i was combined with the ferroptosis activator RSL3 (Figure 1A-1C). There was a slight increase in cell death when cells were irradiated (4 Gy; Fig 1D-1F). BUB1i and RSL3 combination treatment had the most significant effect on SUM159 cells (Figure 1A and 1D).
3.2. Ferroptosis inhibitor Ferrostatin-1 reverses BUB1 inhibition-induced cell death
Cells were treated with BUB1i in combination with Ferrostatin-1 (F-1), a well-established ferroptosis inhibitor (Figure 2). Co-treatment with F-1 mitigated the cell death caused by BUB1 inhibition, with cell viability reducing the BUB1 inhibition-induced cytotoxicity toward levels comparable to untreated controls (Figure 2A-2C). F-1 mediated reversal in cell viability was also observed in irradiated cells (Figure 2D-2F). Interestingly, F-1 did not completely abrogate the effect of BUB1i in all cell lines and whether radiation was included or not. Nonetheless, the observation consistent across SUM159, MDA-MB-231, and BT-549 cell lines that a reduction in cytotoxicity occurred when ferroptosis was reduced further supports that BUB1i-induced cell death was at least partly due to ferroptosis.
3.3. BUB1 inhibition alters expression of key ferroptosis genes
qRT-PCR assays were performed to measure the effect of BUB1 inhibition on ferroptosis markers in TNBC cell lines. Cells were treated with either a mock control or 1 μM BUB1 inhibitor (BUB1i) for 24 hours and total RNA was isolated to assess the expression of ACSL4 (Acyl-CoA Synthetase Long Chain Family Member 4), PTGS2 (Prostaglandin-Endoperoxide Synthase 2), GPX4 (Glutathione Peroxidase 4), and SLC7A11 (Solute Carrier Family 7 Member 11) in SUM159, MDA-MB-231, and BT-549 cells. We observed that BUB1 inhibition significantly upregulated the expression of key ferroptosis markers ACSL4 and PTGS2 (Figure 3A and 3C) and downregulated GPX4 and SLC7A11 (Figure 3B and 3D) compared to the mock treatment.
TNBC cells were treated with sham or 4 Gy radiation for 24 hours and monitored for the expression of ACSL4, PTGS2, GPX4 and SLC7A11 genes (Figure S2) in an attempt to confirm published reports that radiation causes ferroptotic cell death [31]. As expected, ACSL4 and PTGS2 expression increased in irradiation cells compared to sham-irradiated cells while GPX4 and SLC7A11 expression decreased. We next evaluated if BUB1 played any role in radiation-mediated ferroptosis in TNBC cells. The cell lines were treated with 1 μM BUB1 inhibitor (BUB1i) and 4 Gy irradiation for 24 hours and the expressions of the same four genes were evaluated. BUB1i combined with irradiation significantly upregulated ACSL4 expression in all three cell lines compared to radiation-only treated cells (Figure 4A), suggesting enhanced ferroptotic activity. BUB1i significantly reduced the expression of GPX4 compared to radiation-only treated cells (Figure 4B). The downregulation of GPX4 in irradiated cells treated with BUB1i underscores the role of this treatment in overcoming cellular resistance to ferroptosis. Furthermore, BUB1i caused a marked increase in PTGS2 and decreases in SLC7A11 expression compared to radiation-only (Fig 4C-D), confirming the activation of ferroptotic signaling. This response was notably higher in irradiated cells than in non-irradiated cells, indicating at least an additive effect of BUB1 inhibition and radiation in amplifying ferroptosis-related oxidative stress (Figure S3). The collective upregulation of ACSL4 and PTGS2, coupled with the downregulation of GPX4 and SLC7A11, strongly indicates a pro-ferroptotic shift in TNBC cells treated with BUB1i and irradiation (Figure S3). These changes suggest that the combination treatment disrupts cellular defenses against lipid peroxidation, promoting ferroptosis as a potential mechanism for radiosensitization in TNBC.
We next assessed if BUB1i affected the expression of ferroptosis genes long term, TNBC cell lines were treated for 72 hours with BUB1i, and 4 Gy radiation and gene expression was quantitated. ACSL4 expression remained elevated in irradiated BUB1i-treated cells across all cell lines (Figure S4A), indicating sustained lipid peroxidation and prolonged ferroptotic response. GPX4 expression continued to be suppressed at 72 hours (Figure S4B), suggesting that antioxidant defenses remained compromised well after initial treatment, thus perpetuating the pro-ferroptotic state. PTGS2 expression also stayed elevated (Figure S4C), highlighting ongoing oxidative stress, while SLC7A11 remained downregulated (Figure S4D), reinforcing the limited availability of glutathione. This extended suppression of ferroptosis defenses further supports a role for BUB1 in potentially mediating radiation-induced ferroptotic cell death. A role for BUB1 in radiation-mediated ferroptotic cell death was further confirmed by using BUB1 depleted cell lines [27]. BUB1 CRISPR knockout (KO) TNBC cells were sham- or 4 Gy irradiated, and gene expression was evaluated after 72 hours. In SUM159 and MDA-MB-231 BUB1 KO cells, ACSL4, GPX4, PTGS2, and SLC7A11 expression were comparable to those of BUB1i-treated cells. A pro-ferroptotic effect of radiation and BUB1 KO was evident in the expression of ACSL4 and PTGS2 (Figure S5-A and S5-C), with no pro-ferroptotic effect evident in the other markers in BUB1 KO cells.
BUB1i and RSL3 regulate ferroptosis markers in TNBC cell lines
The expression of ferroptosis markers ACSL4, GPX4, PTGS2 and SLC7A11 was assessed in SUM159, MDA-MB-231, and BT-549 cell lines that were treated with BUB1i and ferroptosis activator RSL3 alone or in combination for 24 hours. The combined treatment of BUB1i and RSL3 significantly upregulated ACSL4 expression across all cell lines compared to individual treatments or mock controls (Figure 5A). Expression of GPX4 was notably downregulated in cells treated with BUB1i and RSL3 (Figure 5B) while PTGS2 expression was significantly elevated and SLC7A11 expression was significantly reduced in BUB1i and RSL3-treated cells (Figure 5D).
3.4. Ferrostatin-1 reverses BUB1i induced ferroptosis in TNBC cell lines
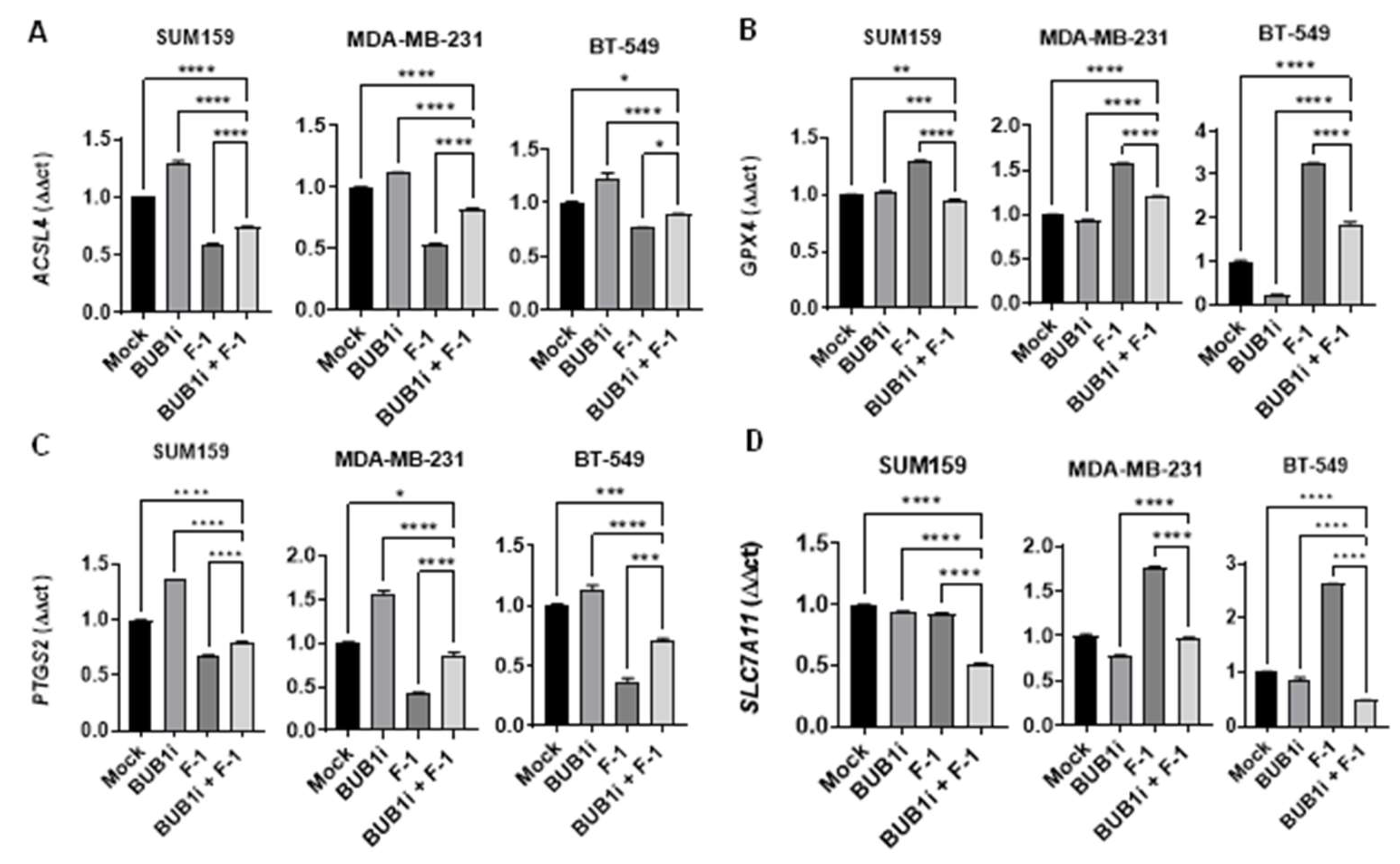
TNBC cell lines were treated with BUB1 inhibitor and Ferrostatin-1 and ferroptosis marker expression was evaluated to further demonstrate the specificity of ferroptosis induced by BUB1 inhibition. Co-treatment with F-1 effectively reduced the expression of ACSL4 and PTGS2 in BUB1i-treated cells (Figure 6A and 6C), indicating that F-1 suppresses the ferroptotic response initiated by BUB1 inhibition. In the presence of F-1, GPX4 expression increased minimally (Figure 6B) while SLC7A11 expression remained stable (Figure 6D) compared to the BUB1i treatment without F-1.
4. Discussion
Our recent studies demonstrated that BUB1 was significantly overexpressed in several breast cancers including TNBC compared to normal tissues and its overexpression was associated with poorer patient survival [27]. Additionally, BUB1 was expressed higher in TNBC cell lines (SUM159, MDA-MB-231, and BT-549) compared to non-cancerous cells. Together, these observations provided a compelling rationale for investigating BUB1 inhibition as a radiosensitization strategy in TNBC [27]. Building on our findings and recent studies that radiation may induce ferroptosis [32], we wanted to test the hypothesis that BUB1, either alone or in combination with radiation, could induce ferroptosis and potentially serve as a novel therapeutic approach.
Ferroptosis, an iron-dependent form of cell death driven by lipid peroxidation, has emerged as a promising therapeutic target, particularly in apoptosis-resistant cancers such as TNBC [33]. This study reveals that BUB1 inhibition significantly promotes ferroptosis in TNBC cells, especially when combined with the ferroptosis activator RSL3 but is likely independent of irradiation (Figure 1). The ability of F-1 to reverse BUB1i-induced ferroptosis suggests exploiting BUB1 as a therapeutic target to induce ferroptosis in TNBC (Figure 2). These findings underscore the therapeutic potential of BUB1 as a target in TNBC, where treatment options are limited due to a lack of hormone receptors and poor responsiveness to conventional therapies. BUB1 inhibition significantly alters the expression of ferroptotic genes, indicating its potential to induce a ferroptotic response in TNBC cell lines. This is demonstrated by the upregulation of ACSL4 and PTGS2 (Figures 3A and 3C) alongside the downregulation of GPX4 and SLC7A11 (Figures 3B and 3D). These changes suggest that BUB1 inhibition alone is sufficient to trigger ferroptosis. Furthermore, the pronounced upregulation of ACSL4 and PTGS2, combined with the suppression of GPX4 and SLC7A11, highlights an at-least-additive effect of BUB1 inhibition and RSL3 in driving ferroptosis in TNBC cells. ACSL4 is crucial for incorporating polyunsaturated fatty acids into membrane phospholipids, enhancing lipid peroxidation—a hallmark of ferroptosis [34]. This aligns with studies by Guo et al., who reported that ACSL4 upregulation is essential for sensitizing cancer cells to ferroptosis, particularly in contexts where traditional apoptotic pathways are inactive [35]. In TNBC, a subtype that often resists apoptosis, targeting ferroptosis through BUB1 inhibition could be a viable approach for inducing cell death. GPX4 is essential for detoxifying lipid peroxides, and its inhibition is a common feature in ferroptosis [36]. Downregulating GPX4 through BUB1 inhibition reduces the cellular defense against ferroptotic cell death, a finding consistent with studies showing that GPX4 is a critical regulator of ferroptosis and that its suppression enhances cancer cell susceptibility to lipid peroxidation-induced cell death [36]. The observed changes in ferroptotic gene expression highlight BUB1's potential as a novel therapeutic target to induce cell death in TNBC through ferroptotic mechanisms. These findings align with growing evidence that targeting oxidative stress and lipid peroxidation pathways could provide innovative treatment strategies for aggressive cancers such as TNBC [37].
To confirm the specificity of ferroptosis in BUB1i-treated TNBC cells, we used F-1. Our results show that F-1 successfully reversed cell viability loss caused by BUB1 inhibition (Figure 2), indicating that the cell death observed was ferroptosis-specific. This finding complements the work of Skouta et al., who demonstrated that Ferrostatin-1 inhibits lipid peroxidation, thereby preventing ferroptotic cell death in cells undergoing oxidative stress-induced lipid damage [38]. In the context of our study, the reversal of ferroptosis markers by F-1 provides a confirmation that the cell death induced by BUB1 inhibition is mediated through ferroptotic mechanisms, validating BUB1’s role in ferroptosis regulation in TNBC. This suggests that F-1 protects against ferroptosis by sustaining antioxidant defenses and that BUB1 inhibition may induce ferroptotic cell death. Moreover, BUB1 KO experiments demonstrated increased ferroptotic marker expression and increased sensitivity to irradiation (Figure S5), indicating that BUB1 itself is crucial for maintaining ferroptotic activity under radiotherapy. This is consistent with Wang et al., who reported that BUB1 is integral to various cancer-related cell death pathways including ferroptosis and it influences oxidative stress and lipid metabolism [39].
Our findings indicate that targeting BUB1, especially in combination with ferroptosis activators like RSL3 may be a promising therapeutic approach for TNBC. This approach leverages ferroptosis to overcome the resistance typically seen with apoptosis-based therapies. Given that ferroptosis bypasses apoptotic mechanisms, which are often inactive in TNBC, therapies that induce ferroptosis could be particularly effective in this subtype. Recent studies suggest that targeting ferroptosis can improve therapeutic outcomes in various cancers, including pancreatic [24] and colorectal cancers [40], by complementing traditional treatments and reducing tumor resistance. Our study adds to this growing body of research, suggesting that BUB1-targeted therapy could similarly benefit TNBC patients, particularly those who may not respond well to existing therapies. Artemisinin derivatives, such as artesunate, disrupt iron homeostasis, increasing free iron levels and lipid peroxidation [41], and enhance ferroptosis by modulating the p38 and ERK signaling pathways, as observed in glioblastoma cells [42]. Similarly, sorafenib, a multikinase inhibitor, promotes ferroptosis by inhibiting the cystine/glutamate antiporter (System Xc−), leading to glutathione depletion and lipid peroxidation [43]. Clinical trials, including the ARTIC M33/2 study on oral artesunate in metastatic breast cancer [44] and the Phase II trial (N0336) evaluating sorafenib [45], underscore the potential of ferroptosis-targeting agents despite some challenges in clinical efficacy. These findings emphasize the importance of regulatory targets like GPX4 and System Xc− in ferroptosis, highlighting the promise of novel therapeutic strategies to overcome cancer resistance mechanisms. Our study of BUB1 inhibition aligns with these approaches, supporting this pathway to leverage ferroptosis in aggressive cancers like TNBC [46,47].
5. Conclusion
In conclusion, this study highlights the potential of BUB1 inhibition as a therapeutic strategy to induce ferroptosis in TNBC. Further, in vivo studies and clinical trials will be essential to validate the efficacy of BUB1-targeted treatments, possibly combined with ferroptosis activators. These insights contribute to the ongoing development of innovative therapies aimed at exploiting ferroptosis for cancer treatment and suggest that BUB1 inhibition should be further investigated as a potential strategy to induce ferroptosis and enhance therapeutic efficacy in aggressive cancers like TNBC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Financial support
This work was supported by NCI R21 (1R21CA252010-01A1), the HFHS Research Administration Start up, the HFHS Proposal Development Award, the HFHS-Rad Onc Start Up, and the Game on Cancer award to Shyam Nyati. We also thank HFCI for providing a Translational Oncology Postdoctoral Fellowship to Sushmitha Sriramulu.
Author Contributions
S.S. performed all the experiments with help from S.T. S.S. also drafted the manuscript with input from all other authors. S.S, S.T, S.L.B., F.S., B.M., and S.N. analyzed the data. S.N. conceptualized and directed the study.
Conflicts of Interest
S.S., S.T., S.L.B., S.N.: no COI, F.S.: Varian Medical Systems Inc.—Honorarium and travel reimbursement for lectures and talks, Varian Noona—Member of Medical Advisory Board—Honorarium (no direct conflict), B.M.: Research support from Varian, ViewRay, and Philips (no direct conflict).
References
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-c. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Research 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Li Y, Zhang H, Merkher Y, Chen L, Liu N, Leonov S, Chen Y. Recent advances in therapeutic strategies for triple-negative breast cancer. Journal of Hematology & Oncology. 2022;15(1):121.
- Obidiro O, Battogtokh G, Akala EO. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics. 2023;15(7).
- Zhu S, Wu Y, Song B, Yi M, Yan Y, Mei Q, Wu K. Recent advances in targeted strategies for triple-negative breast cancer. Journal of Hematology & Oncology. 2023;16(1):100.
- Zagami P, Carey LA. Triple negative breast cancer: Pitfalls and progress. npj Breast Cancer. 9: 2022;8(1), 2022.
- Bou Zerdan, M.; Ghorayeb, T.; Saliba, F.; Allam, S.; Bou Zerdan, M.; Yaghi, M.; Bilani, N.; Jaafar, R.; Nahleh, Z. Triple Negative Breast Cancer: Updates on Classification and Treatment in 2021. Cancers 2022, 14, 1253. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Jove, M.; Gombos, A.; Awada, A. Cytotoxic chemotherapy: Still the mainstay of clinical practice for all subtypes metastatic breast cancer. Critical Reviews in Oncology/Hematology 2016, 100, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Speers, C.; Nyati, S. Present and Future of Immunotherapy for Triple-Negative Breast Cancer. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Alaluf, E.; Shalamov, M.M.; Sonnenblick, A. Update on current and new potential immunotherapies in breast cancer, from bench to bedside. Frontiers in Immunology 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Turati, A.; Rosato, A.; Carpanese, D. Sacituzumab govitecan in triple-negative breast cancer: from bench to bedside, and back. Frontiers in Immunology 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol 2021, 16, 255–282. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer☆. Annals of Oncology 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Xiong, N.; Wu, H.; Yu, Z. Advancements and challenges in triple-negative breast cancer: a comprehensive review of therapeutic and diagnostic strategies. Front Oncol 2024, 14, 1405491. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, Y.; Jiang, Y.; Bu, J.; Gu, X. Targeting ferroptosis, the achilles' heel of breast cancer: A review. Frontiers in pharmacology, 2022, 13, 1036140. [Google Scholar] [CrossRef]
- https://www.frontiersin.org/articles/10.3389/fphar.2022. 1036.
- . [CrossRef]
- https://europepmc. 9709.
- https://europepmc.org/articles/PMC9709426?pdf=render.
- Qi, X.; Wan, Z.; Jiang, B.; Ouyang, Y.; Feng, W.; Zhu, H.; Tan, Y.; He, R.; Xie, L.; Li, Y. Inducing ferroptosis has the potential to overcome therapy resistance in breast cancer. Front Immunol 2022, 13, 1038225. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, Y.; Yu, Q.; Song, J.; Jin, Y.; Gao, X. Compounds targeting ferroptosis in breast cancer: progress and their therapeutic potential. Front Pharmacol 2023, 14, 1243286. [Google Scholar] [CrossRef] [PubMed]
- Ge, A.; He, Q.; Zhao, D.; Li, Y.; Chen, J.; Deng, Y.; Xiang, W.; Fan, H.; Wu, S.; Li, Y.; et al. Mechanism of ferroptosis in breast cancer and research progress of natural compounds regulating ferroptosis. J Cell Mol Med 2024, 28, e18044. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-l.; Huang, Z.-j.; Lin, Z.-t.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: past, present and future. Cell Death & Disease 2020, 11, 88. [Google Scholar] [CrossRef]
- Yan, H.-f.; Zou, T.; Tuo, Q.-z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: mechanisms and links with diseases. Signal Transduction and Targeted Therapy 2021, 6, 49. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell 2024, 42, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Jia, Y.; Dai, E.; Liu, J.; Kang, R.; Tang, D.; Han, L.; Zhong, Y.; Meng, L. Ferroptotic therapy in cancer: benefits, side effects, and risks. Molecular Cancer 2024, 23, 89. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Ge, A.; He, Q.; Zhao, D.; Li, Y.; Chen, J.; Deng, Y.; Xiang, W.; Fan, H.; Wu, S.; Li, Y.; et al. Mechanism of ferroptosis in breast cancer and research progress of natural compounds regulating ferroptosis. Journal of Cellular and Molecular Medicine 2024, 28, e18044. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front Pharmacol 2020, 11, 239. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis 2016, 7, e2307. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, X.; Liu, C.; Ge, W.; Wang, Q.; Hao, X.; Wang, M.; Chen, Y.; Zhang, Q. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol 2021, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, X.; Kong, L.; Pan, Z.; Chen, G. BUB1 Promotes Gemcitabine Resistance in Pancreatic Cancer Cells by Inhibiting Ferroptosis. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Klebig, C.; Korinth, D.; Meraldi, P. Bub1 regulates chromosome segregation in a kinetochore-independent manner. J Cell Biol 2009, 185, 841–858. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Walker, E.; Nyati, S. BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy. Biomolecules 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Chen, W.-M.; Hassan, O.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Green, M.D.; Davis, A.J.; Speers, C.; et al. BUB1 regulates non-homologous end joining pathway to mediate radioresistance in triple-negative breast cancer. Journal of Experimental & Clinical Cancer Research 2024, 43, 163. [Google Scholar] [CrossRef]
- Thoidingjam, S.; Sriramulu, S.; Hassan, O.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Gadgeel, S.; Nyati, S. BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer. Cancers 2024, 16, 3291. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, Y.e.; Chen, X.; Zhong, H.; Wang, Y. Ferroptosis in life: To be or not to be. Biomedicine & Pharmacotherapy 2023, 159, 114241. [Google Scholar] [CrossRef]
- Thu, K.L.; Soria-Bretones, I.; Mak, T.W.; Cescon, D.W. Targeting the cell cycle in breast cancer: towards the next phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.E.; Kendig, C.B.; An, N.; Fazilat, A.Z.; Churukian, A.A.; Griffin, M.; Pan, P.M.; Longaker, M.T.; Dixon, S.J.; Wan, D.C. Role of ferroptosis in radiation-induced soft tissue injury. Cell Death Discovery 2024, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Research 2020, 30, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, D.; Li, S.; Xiao, J.; Zhu, Z. Ferroptosis: the emerging player in remodeling triple-negative breast cancer. Front Immunol 2023, 14, 1284057. [Google Scholar] [CrossRef]
- Jia, B.; Li, J.; Song, Y.; Luo, C. ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Guo, N. Identification of ACSL4 as a biomarker and contributor of ferroptosis in clear cell renal cell carcinoma. Transl Cancer Res 2022, 11, 2688–2699. [Google Scholar] [CrossRef]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in cell death, autophagy, and disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef]
- Nandi, I.; Ji, L.; Smith, H.W.; Avizonis, D.; Papavasiliou, V.; Lavoie, C.; Pacis, A.; Attalla, S.; Sanguin-Gendreau, V.; Muller, W.J. Targeting fatty acid oxidation enhances response to HER2-targeted therapy. Nature Communications 2024, 15, 6587. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. Journal of the American Chemical Society 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, X.; Kong, L.; Pan, Z.; Chen, G. BUB1 Promotes Gemcitabine Resistance in Pancreatic Cancer Cells by Inhibiting Ferroptosis. Cancers 2024, 16, 1540. [Google Scholar] [CrossRef]
- Yan, H.; Talty, R.; Aladelokun, O.; Bosenberg, M.; Johnson, C.H. Ferroptosis in colorectal cancer: a future target? British Journal of Cancer 2023, 128, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.-X.; Jiang, X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death & Differentiation 2020, 27, 242–254. [Google Scholar] [CrossRef]
- Song, Q.; Peng, S.; Che, F.; Zhu, X. Artesunate induces ferroptosis via modulation of p38 and ERK signaling pathway in glioblastoma cells. J Pharmacol Sci 2022, 148, 300–306. [Google Scholar] [CrossRef]
- Li, Q.; Chen, K.; Zhang, T.; Jiang, D.; Chen, L.; Jiang, J.; Zhang, C.; Li, S. Understanding sorafenib-induced ferroptosis and resistance mechanisms: Implications for cancer therapy. European Journal of Pharmacology 2023, 955, 175913. [Google Scholar] [CrossRef] [PubMed]
- von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Osburg, J.; Storch-Hagenlocher, B.; Sertel, S.; Elsässer, M.; Remppis, B.A.; Edler, L.; Munzinger, J.; et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res Treat 2017, 164, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Aspitia, A.; Morton, R.F.; Hillman, D.W.; Lingle, W.L.; Rowland, K.M., Jr.; Wiesenfeld, M.; Flynn, P.J.; Fitch, T.R.; Perez, E.A. Phase II trial of sorafenib in patients with metastatic breast cancer previously exposed to anthracyclines or taxanes: North Central Cancer Treatment Group and Mayo Clinic Trial N0336. J Clin Oncol 2009, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, Z. Recent progress in ferroptosis: inducers and inhibitors. Cell Death Discovery 2022, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduction and Targeted Therapy 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Effects of BUB1 inhibition and ferroptosis activation on TNBC cell viability. (A-C) Viability assays in TNBC cell lines (SUM159, MDA-MB-231, BT-549) show BUB1 inhibitor (BUB1i, 1 µM) alone reduces viability, while co-treatment with ferroptosis activator RSL3 (62.5 nM) further decreases survival. (D-F) Adding irradiation (4 Gy) to BUB1i and RSL3 slightly increases cell death. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Figure 1.
Effects of BUB1 inhibition and ferroptosis activation on TNBC cell viability. (A-C) Viability assays in TNBC cell lines (SUM159, MDA-MB-231, BT-549) show BUB1 inhibitor (BUB1i, 1 µM) alone reduces viability, while co-treatment with ferroptosis activator RSL3 (62.5 nM) further decreases survival. (D-F) Adding irradiation (4 Gy) to BUB1i and RSL3 slightly increases cell death. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
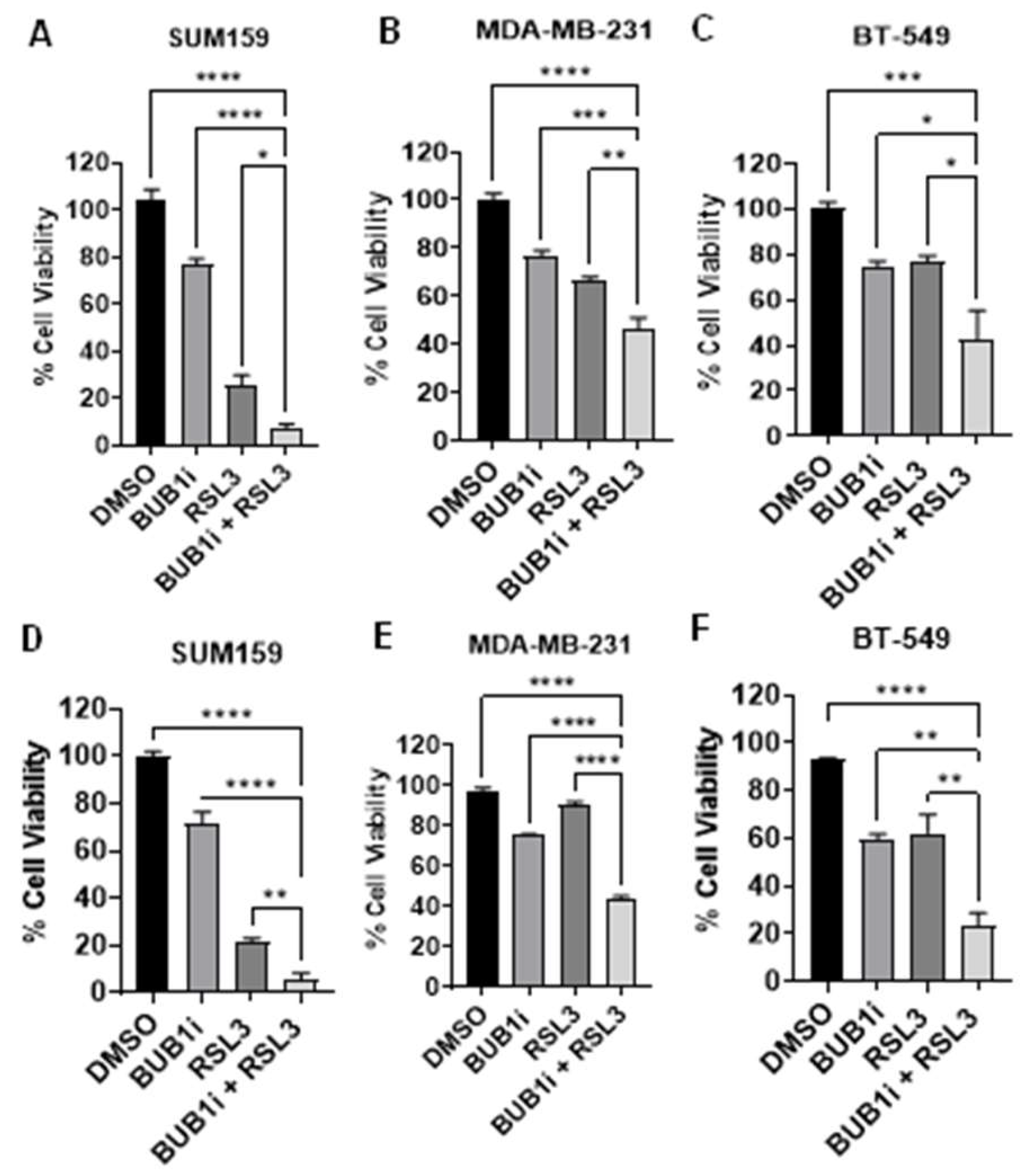
Figure 2.
Ferrostatin-1 confirms ferroptosis-specific cell death induced by BUB1 inhibition in TNBC cell lines. TNBC cell lines (SUM159, MDA-MB-231, BT-549) were treated with BUB1 inhibitor (BUB1i, 1 µM) alone or with Ferrostatin-1 (F-1, 10 µM). (A-C) In non-irradiated cells, F-1 reversed BUB1i-induced cell death, restoring viability to control levels. (D-F) F-1 mediated reversal in cell viability even in irradiated cells (4 Gy). P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Figure 2.
Ferrostatin-1 confirms ferroptosis-specific cell death induced by BUB1 inhibition in TNBC cell lines. TNBC cell lines (SUM159, MDA-MB-231, BT-549) were treated with BUB1 inhibitor (BUB1i, 1 µM) alone or with Ferrostatin-1 (F-1, 10 µM). (A-C) In non-irradiated cells, F-1 reversed BUB1i-induced cell death, restoring viability to control levels. (D-F) F-1 mediated reversal in cell viability even in irradiated cells (4 Gy). P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
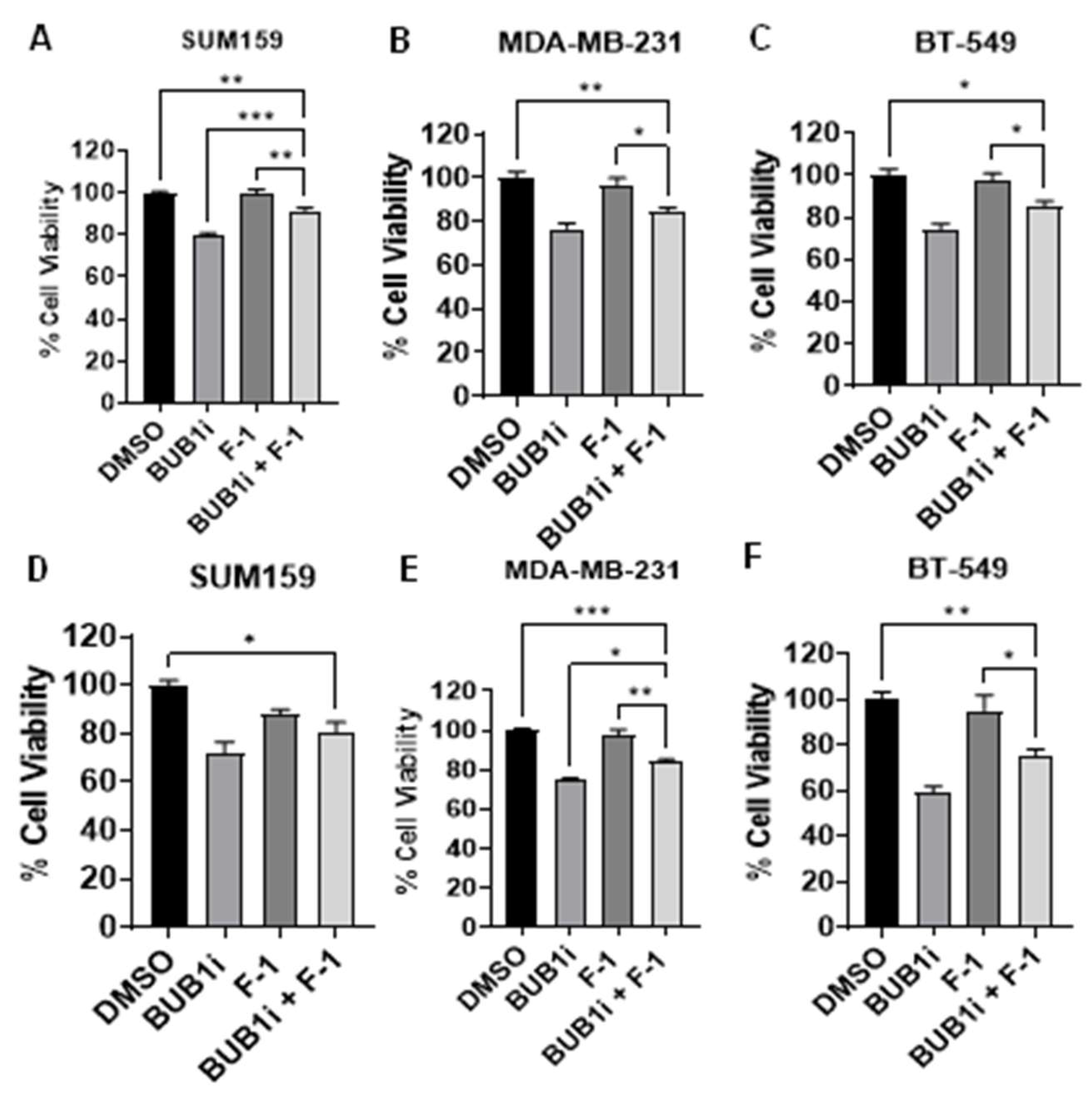
Figure 3.
BUB1 inhibition alters ferroptosis marker expression in TNBC cell lines. TNBC cell lines (SUM159, MDA-MB-231, BT-549) were treated with BUB1 inhibitor (1 µM) or mock control for 24 hours, and ferroptosis markers were assessed via qPCR. (A) ACSL4 was significantly upregulated (B) GPX4 was significantly downregulated (C) PTGS2 was significantly upregulated, and (D) SLC7A11 was significantly downregulated suggesting that BUB1 inhibition may trigger a ferroptotic response in TNBC cell lines. P-values were defined as ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Figure 3.
BUB1 inhibition alters ferroptosis marker expression in TNBC cell lines. TNBC cell lines (SUM159, MDA-MB-231, BT-549) were treated with BUB1 inhibitor (1 µM) or mock control for 24 hours, and ferroptosis markers were assessed via qPCR. (A) ACSL4 was significantly upregulated (B) GPX4 was significantly downregulated (C) PTGS2 was significantly upregulated, and (D) SLC7A11 was significantly downregulated suggesting that BUB1 inhibition may trigger a ferroptotic response in TNBC cell lines. P-values were defined as ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
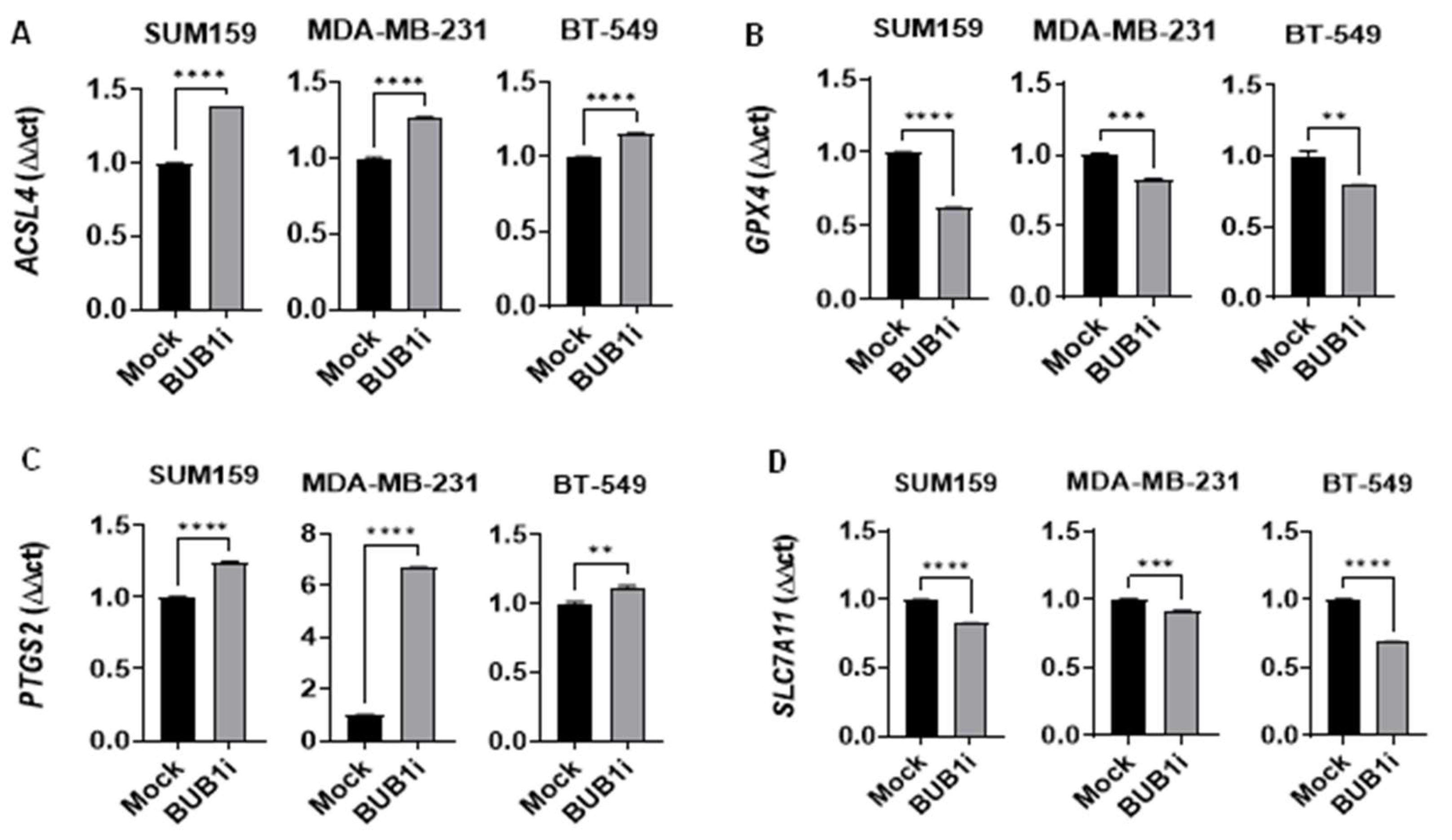
Figure 4.
Upregulation of ferroptosis markers in TNBC cell lines following BUB1 inhibition and irradiation. TNBC cell lines (SUM159, MDA-MB-231, BT-549) were treated with BUB1 inhibitor (1 µM) and irradiation (4 Gy) for 24 hours, and ferroptosis marker expression was analyzed by qPCR. (A) ACSL4 levels increased, indicating enhanced lipid peroxidation. (B) GPX4 was significantly downregulated, reducing antioxidant defenses. (C) PTGS2 expression was elevated, reflecting increased oxidative stress. (D) SLC7A11 was downregulated, impairing cystine import and sensitizing cells to ferroptosis. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Figure 4.
Upregulation of ferroptosis markers in TNBC cell lines following BUB1 inhibition and irradiation. TNBC cell lines (SUM159, MDA-MB-231, BT-549) were treated with BUB1 inhibitor (1 µM) and irradiation (4 Gy) for 24 hours, and ferroptosis marker expression was analyzed by qPCR. (A) ACSL4 levels increased, indicating enhanced lipid peroxidation. (B) GPX4 was significantly downregulated, reducing antioxidant defenses. (C) PTGS2 expression was elevated, reflecting increased oxidative stress. (D) SLC7A11 was downregulated, impairing cystine import and sensitizing cells to ferroptosis. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
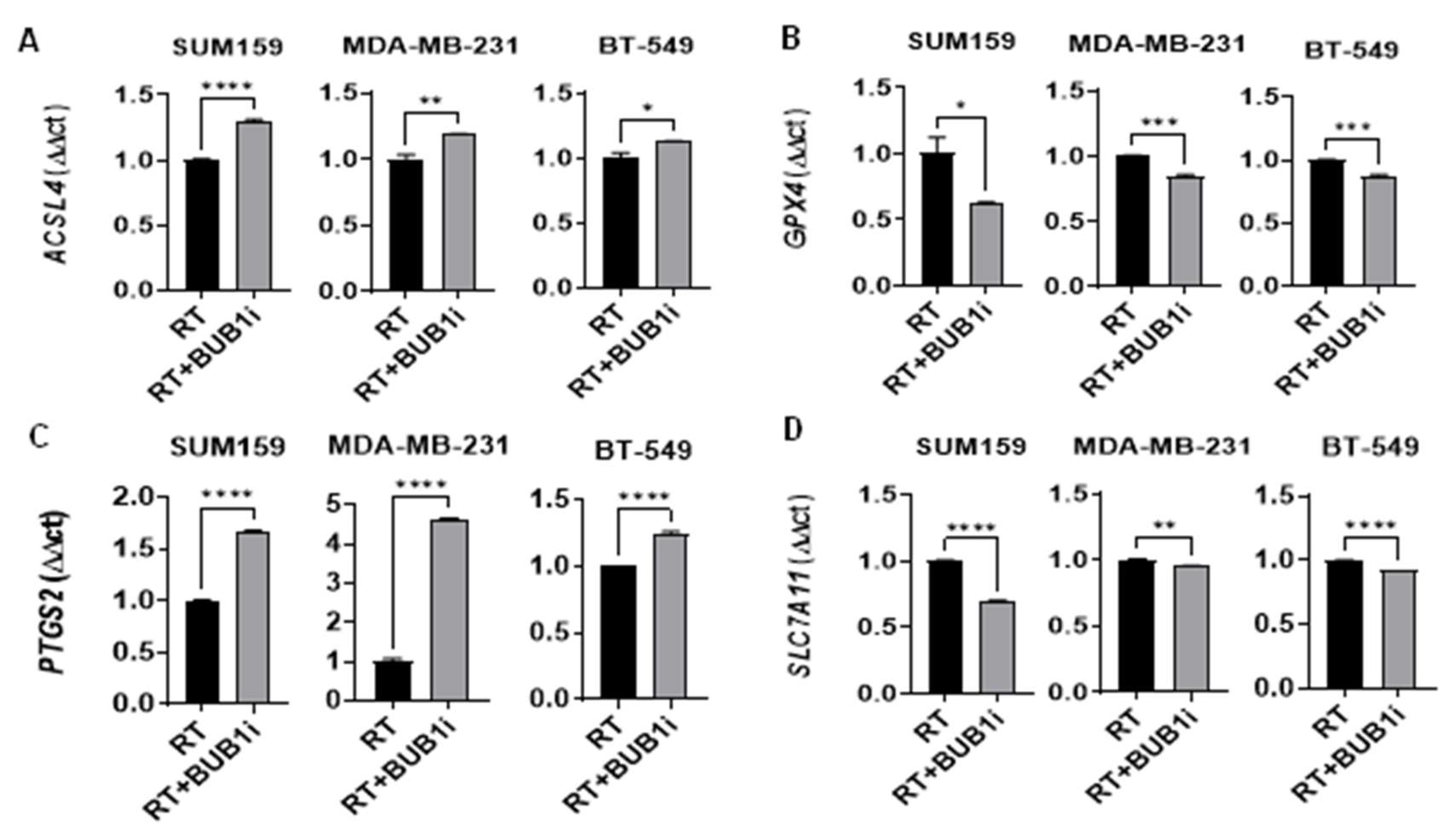
Figure 5.
Regulation of ferroptosis markers by BUB1i and RSL3 in TNBC cell lines. Ferroptosis markers ACSL4, GPX4, PTGS2, and SLC7A11 were analyzed in SUM159, MDA-MB-231, and BT-549 cells treated for 24 hours with BUB1i (1 µM), RSL3 (0.25 µM), or their combination. (A) Combined treatment significantly increased ACSL4 expression and (C) elevated PTGS2 levels, indicating enhanced lipid peroxidation and oxidative stress. (B) GPX4 was markedly downregulated, reducing antioxidant defenses, while (D) SLC7A11 was significantly decreased, impairing cystine import and glutathione synthesis. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Figure 5.
Regulation of ferroptosis markers by BUB1i and RSL3 in TNBC cell lines. Ferroptosis markers ACSL4, GPX4, PTGS2, and SLC7A11 were analyzed in SUM159, MDA-MB-231, and BT-549 cells treated for 24 hours with BUB1i (1 µM), RSL3 (0.25 µM), or their combination. (A) Combined treatment significantly increased ACSL4 expression and (C) elevated PTGS2 levels, indicating enhanced lipid peroxidation and oxidative stress. (B) GPX4 was markedly downregulated, reducing antioxidant defenses, while (D) SLC7A11 was significantly decreased, impairing cystine import and glutathione synthesis. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
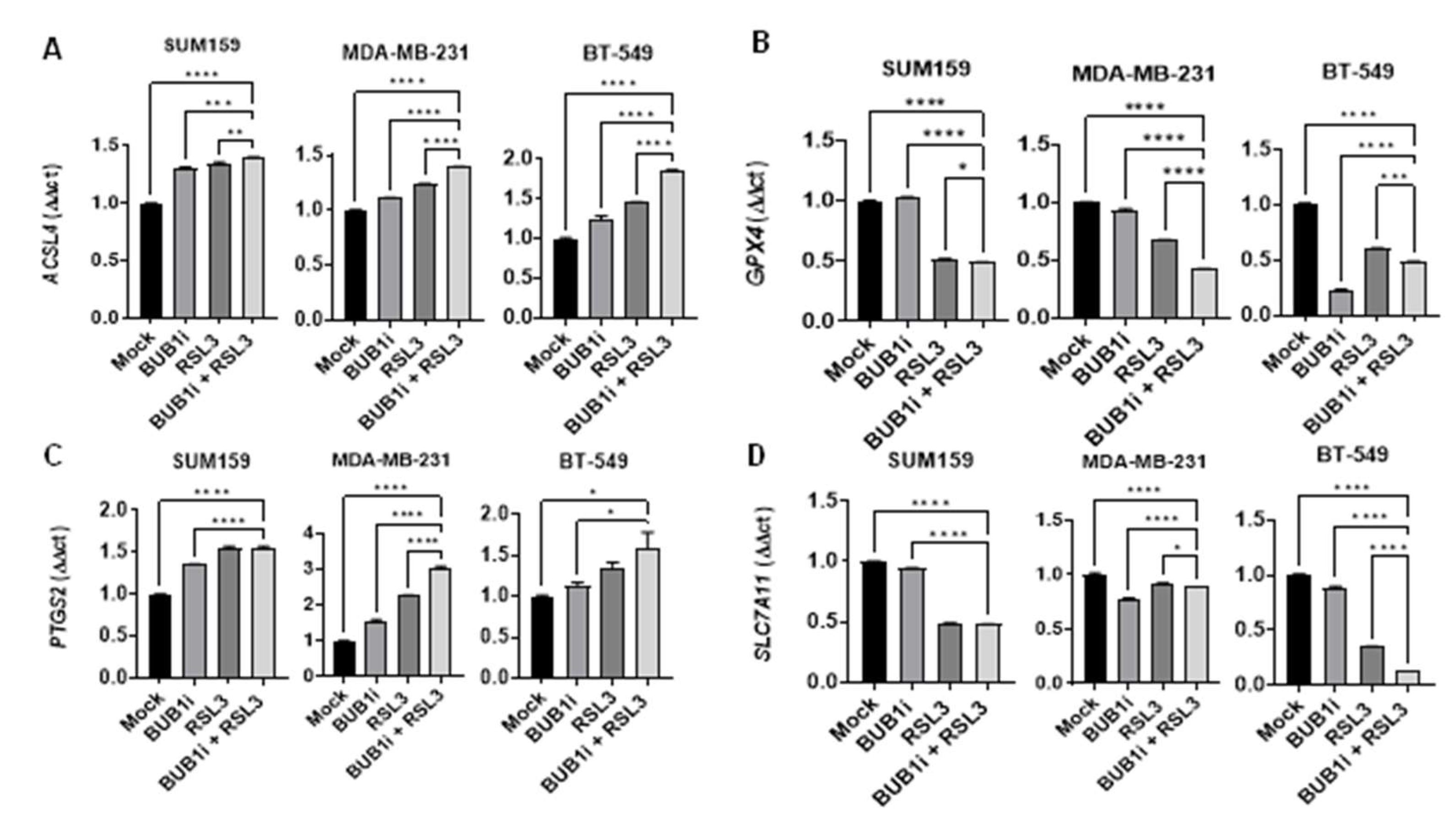
Figure 6.
Ferrostatin-1 reverses BUB1i induced ferroptosis in TNBC cell lines. TNBC cell lines were treated with BUB1 inhibitor (1 µM) alone or combined with Ferrostatin-1 (F-1, 10 µM) for 24 hours. (A) F-1 co-treatment significantly reduced ACSL4 and (C) PTGS2 expression (B) GPX4 showed a slight increase with F-1, while (D) SLC7A11 levels remained unchanged compared to BUB1i-only treatment. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Figure 6.
Ferrostatin-1 reverses BUB1i induced ferroptosis in TNBC cell lines. TNBC cell lines were treated with BUB1 inhibitor (1 µM) alone or combined with Ferrostatin-1 (F-1, 10 µM) for 24 hours. (A) F-1 co-treatment significantly reduced ACSL4 and (C) PTGS2 expression (B) GPX4 showed a slight increase with F-1, while (D) SLC7A11 levels remained unchanged compared to BUB1i-only treatment. P-values were defined as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



