Submitted:
19 December 2024
Posted:
20 December 2024
You are already at the latest version
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer, able to thrive in a challenging tumor microenvironment. Current standard therapies, including surgery, radiation, chemotherapy, and chemoradiation, have shown a dismal survival prognosis, resulting in less than a year of life in the metastatic setting. The pressing need to find better therapeutic methods brought about the discovery of new targeted therapies against the infamous KRAS mutations, the major oncological drivers of PDAC. The most common KRAS mutation is KRASG12D, which causes a conformational change in the protein that constitutively activates downstream signaling pathways driving cancer hallmarks. Novel anti-KRASG12D therapies have been developed for solid-organ tumors, including small compounds, pan-RAS inhibitors, protease inhibitors, chimeric T-cell receptors, and therapeutic vaccines. This comprehensive review summarizes the current knowledge on the biology of KRAS-driven PDAC, the latest therapeutic options that have been experimentally validated, and developments in ongoing clinical trials.
Keywords:
PDAC
; KRASG12D
; Targeted Therapies
; Tumor Microenvironment
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common pancreatic cancer, accounting for more than 90% of cases. It is notorious for its poor prognosis, with a 5-year survival rate of less than 10%, largely due to late diagnosis, rapid disease progression, and limited therapeutic options [1,2]. Despite advancements in oncology, PDAC remains resistant to most conventional treatments such as surgery, chemotherapy, and radiation, highlighting the urgent need for more effective, targeted therapeutic strategies.
A pivotal factor in PDAC’s aggressiveness is the high prevalence of mutations in the KRAS gene, with over 90% of patients exhibiting activating mutations in this oncogene [3]. Among these mutations, KRASG12D is the most frequently observed, accounting for approximately 40% of all KRAS mutations in PDAC [4]. These mutations lead to the constitutive activation of the KRAS protein, which is integral to cell proliferation, survival, and metabolism, as it persistently drives several key downstream signaling pathways including RAF-MEK-ERK and PI3K-AKT-mTOR [1,5]. This constant signaling promotes uncontrolled cell division, inhibits apoptosis, and contributes to the hallmark features of cancer, including angiogenesis and immune evasion.
For decades, the KRAS protein was considered “undruggable,” primarily because of its lack of a deep binding pocket and its high affinity for guanosine triphosphate (GTP) [6]. While efforts to target other members of the RAS family, such as HRAS and NRAS, have seen some success, KRAS-targeted therapies lagged significantly behind. The breakthrough came with the discovery of KRASG12C inhibitors, such as sotorasib and adagrasib, which have shown remarkable efficacy in cancers harboring this specific mutation, particularly non–small cell lung cancer [7,8]. These inhibitors covalently bind to the cysteine residue in the KRASG12C mutant, stabilizing the protein in its inactive Guanosine Diphosphate (GDP)-bound state. Unfortunately, this strategy does not apply to KRASG12D, which lacks the cysteine residue necessary for covalent binding, necessitating the development of alternative therapeutic approaches [9,10].
The development of selective KRASG12D inhibitors has been a significant focus of recent research, culminating in the identification of MRTX1133. Unlike KRASG12C inhibitors, MRTX1133 is a non-covalent inhibitor that binds to both the active (GTP-bound) and inactive (GDP-bound) forms of KRASG12D with high specificity and potency [11,12]. Preclinical models have shown that MRTX1133 can inhibit KRASG12D-driven tumors in vitro and in vivo, particularly in PDAC, where the mutation is prevalent. Early studies demonstrated its ability to shrink tumors and inhibit downstream signaling with minimal off-target effects on wild-type KRAS, making it a highly promising candidate for clinical development [12]. Since then, more small compounds against KRAS mutations have been developed, such as pan-KRAS inhibitor BI-2493, protease inhibitor HRS-4642 targeting KRASG12D, and immunotherapies designed for KRASG12D PDAC.
While KRASG12D inhibitors represent a leap forward, PDAC poses additional challenges due to its dense stromal environment and high level of tumor heterogeneity. PDAC is characterized by a fibrotic stroma that impairs drug delivery and fosters an immune-suppressive microenvironment, which reduces the effectiveness of targeted therapies and immunotherapies alike [13,14]. Moreover, the high degree of genetic variability within PDAC tumors means that even successful inhibition of KRASG12D often leads to adaptive resistance through alternative signaling pathways, such as the activation of PI3K-AKT-mTOR or the induction of epithelial-mesenchymal transition [15,16]. These compensatory mechanisms allow tumors to bypass KRAS blockade, making combination therapies an important area of focus.
Immunotherapy is also emerging as a potential strategy to overcome some of the limitations of KRAS-targeted treatments. CAR T-cell therapy, which involves engineering a patient’s T cells to recognize and attack tumor-specific antigens, has shown early promise in preclinical models of KRASG12D-driven cancers. Additionally, therapeutic vaccines that target mutant KRAS proteins, such as Moderna’s mRNA-5671, are being investigated to elicit an immune response capable of recognizing and eliminating KRAS-mutant tumor cells [7]. While these approaches are still in early stages, they offer a complementary strategy to directly target KRAS and may help overcome some of the challenges posed by the tumor microenvironment and resistance mechanisms.
Despite significant progress in developing KRASG12D-targeted therapies, several obstacles remain. The complexity of the tumor microenvironment, genetic heterogeneity, and the rapid emergence of resistance pathways mean that combination therapies, involving multiple inhibitors or the addition of immune-modulating agents, are likely to be required to achieve long-term therapeutic success [12]. As new inhibitors like MRTX1133 move toward clinical trials, understanding these challenges will be crucial for optimizing treatment regimens and improving outcomes for PDAC patients. This review focuses on KRASG12D as a strong driver of PDAC, details the different therapeutic approaches targeting this mutation, and summarizes ongoing clinical trials.
2. KRAS Mutations Drive Progressive Neoplastic Differentiation of the Pancreas
PDAC is the most common and lethal form of pancreatic cancer. There are 3 PanIN grades, with increasing levels of disorganization and nuclear abnormalities, finally transitioning into PDAC. During this transition, tumor suppressor genes CDKN2A, p53, and SMAD4 are inactivated [17].
a. KRAS Is Pivotal in Cancer Progression
The worst mutation in terms of prognosis is KRASG12D [18]. KRAS encodes 21-kDa small GTPases, which cycle between a GTP-bound state (“on”) to a GDP-bound state (“off”). This cycle is mediated by guanine nucleotide exchange factors which activate RAS by aiding the exchange of GDP into GTP. On the other hand, GTPase-activating proteins drive RAS-mediated GTP hydrolysis and hence inactivation. The constitutive activation of Ras results in the persistent stimulation of downstream signaling factors, resulting in the activation of the major hallmarks of cancer [19].
To proliferate, cancer cells must have sufficient energy and biosynthetic building blocks. KRAS promotes glucose uptake by increasing glucose transporter GLUT1, which increases the speed by which glycolytic activity occurs and lactate is produced [20,21]. Such alteration confers a distinct survival advantage evidenced in oncogenic KRAS-bearing cell lines [20].
Mutant KRAS increases GLUT1 expression and other rate-limiting glycolytic enzyme genes, such as hexokinase 1 and 2, phosphofructokinase-1 (Pfk1), and lactate dehydrogenase A, responsible for converting pyruvate to lactate [16]. KRAS upregulates rate-limiting enzymes for hexosamine, and enzymes involved in the pentose phosphate pathway [16]. These pathways have been deeply involved in cancer progression. Additionally, PDAC has a low vascular density and dense stromal components. These aspects are a challenge to the penetration of PDAC and delivery of therapies, such as antibodies [13].
b. KRAS Plays an Important Role in PDAC
In PDAC, oncogenic KRAS manages glutamine expression through a non-canonical pathway [22]. The first step leading to PDAC is the progressive differentiation of normal pancreatic ductal epithelium into precancerous lesions, known as pancreatic intraepithelial neoplasia (PanIN). The KRAS gene mutation is an early event initiating over 90% of low-grade PanIN lesions [23]. PDAC cells prefer low intracellular oxygen species (ROS) levels [24]. Intriguingly, KRASG12D increases transcription of nuclear factor erythroid 2-related factor (NRF2), which activates the ROS detoxification program, reducing the intracellular environment [25]. Unlike what was initially thought, KRAS-driven PDAC requires low levels of ROS for optimal growth [26,27]. This requirement could be explained by the opposite roles of ROS: while at low levels ROS may be promoting cell growth, at high levels these species are cytotoxic [28].
Autophagy is required for KRAS-driven growth. The process of autophagy is a highly conserved mechanism that degrades the intracellular components and promotes reprogrammed cell survival when there are metabolic stresses, by providing ATP and building blocks like amino acids, sugars, lipids, and nucleosides [29]. Autophagy generally is controlled by multiple autophagy-related genes (ATGs), as well as multiple signaling components and growth factors [30]. Autophagy can either cause or inhibit tumor formation [31,32]. There is strong evidence suggesting that in RAS-dependent PDAC, autophagy sustains tumor growth [33]. Indeed, high autophagy levels were detected in pancreatic primary tumors and cell lines. Deleting ATG5 and inhibiting autophagy with chloroquine was able to suppress PDAC growth both in cell lines and in animals [34]. On the other hand, the survival of KRASG12D-driven animals with tumors was increased using chloroquine. Suppressing autophagy was associated with increased ROS, less mitochondrial oxidative phosphorylation, and higher DNA damage [34]. On the same note, in cell lines including PANC-1, high basal autophagy was needed to sustain growth and survival [35]. These studies concluded that autophagy and mitophagy are required [36] for PDAC to produce bioenergetic intermediates for the TCA cycle and remove damaged mitochondria. Autophagy-driven cell death or senescence was further promoted by oncogenic KRASV12 overexpression, further showing that there is a crosstalk between RAS and autophagy based on different cell types and genetics [37,38,39]. Many studies have shown that KRAS mutation in pancreatic cancer is significantly correlated with shorter overall survival, whereas only a few studies have shown an absence of a correlation [40,41,42,43,44,45,46,47]. Recently, a study of 803 patients with PDAC showed that those with KRASG12R had similar overall survival compared to those with wild-type KRAS (38 vs. 34 months, respectively), but those with KRASG12D and KRASQ61 had shorter overall survival (22 months and 20 months, respectively) [48,49].
c. The Distribution of KRAS Mutations in PDAC
KRAS is the most mutated gene in PDAC (95%), according to several exome sequencing experiments [3,50]. The most common mutation in KRAS occurs at the second exon at codon 12 (G12), on the first or second nucleotide, resulting in a conformational change at the site where GTP binds, hence changing the GTP hydrolysis rate [51]. KRAS mutations are cancer type specific, with 98% of PDAC KRAS mutations occurring at codon 12, codon 13 (G13), or codon 61 (Q61) [4,5]. These alterations hinder the intrinsic activity of KRAS GTPase activity, blocking the interaction between KRAS and GAPs. Consequently, KRAS becomes constitutively activated and persistently stimulates downstream signaling pathways driving cancer hallmarks [19]. Of the codon 12 mutations, the most frequent are G12D (GAT, 40% of all KRAS mutations), G12V (GTT, 33%), and G12R (CGT, 15%), as well as in G12C (TGT), G12A (GCT), and G12S (AGT) [48]. Other common mutations at the second exon occur on codon 13, accounting for 7% of KRAS mutations (G13D, G13C, G13S, and G13R); additional mutations are seen at codon 61 of the third exon (Q61H, Q61R, Q61K, and Q61L) and codons 117 and 146 of the fourth exon (K117 and A146) [52]. Further, the sera of patients with KRAS-mutated PDAC contained point mutations in codons 12 and 13 [53]. Moreover, other highly common KRAS mutations in PDAC are KRASG12V, which was mutated in 32.5% of cases, and G12R in 17.1%; less common are G12C (1.7%), G12A/S/L/I (1.4%), G13C/D/P/H/R (1.2%), Q61H (4.8%), Q61K (0.5%), and others (0.5%) [54]. In our analysis of 2188 PDAC patients using cBioPortal data, 93.7% of patients had KRAS mutations, with the following mutations: G12D (seen in 40.4%), G12V (32.1%), G12R (15.9%), Q61H (4.9%), Q61R (1.7%), G12C (1.2%), other (3.2%) (cBioPortal accessed on September 4, 2024; Figure 1).
3. The KRASG12D Mutation in Solid-Organ Tumors Including Pancreatic Cancer
The RAS protein is frequently mutationally activated (HRAS, KRAS, and NRAS) in human cancer. This gene is frequently mutated in the top 3 cancers associated with mortality in the US (lung, colorectal, and pancreatic). KRAS is the most commonly mutated RAS isoform, and nearly all PDACs are KRAS dependent [55].
According to the Catalogue of Somatic Mutations in Cancer (COSMIC), a useful tool providing access to comprehensive human tumor mutation databases. KRAS mutations were expressed in about 22% of analyzed tumors, versus 8% for NRAS and 3% for HRAS [55]. The RAS isoform mutations are codon-specific. About 80% of KRAS mutations occur at codon 12, whereas only a few occur at codon 61. On the contrary, around 60% of NRAS mutations happen at codon 61, in contrast to the 35% encountered at codon 12. HRAS has 40% of its mutations on codon 12 and 40% on codon 61. At the DNA level, KRAS and NRAS have identical sequences encoding Gly12 and Glu61. Additionally, these oncogenic mutations of KRAS, NRAS, and HRAS are in the same amino-acid regions, producing equal effects on the encoded protein and its activity [55].
The KRASG12D mutation appears in 40% of PDAC patients, encoding a GAT sequence, which produces aspartic acid instead of guanine at amino acid position 12 [52]. Among the various KRAS mutations, KRASG12D is the most frequently correlated with worse survival [40,41,42,43,44,45,46,47,49,56,57,58].
The KRASG21D mutation alone has been shown to result in PDAC formation and its protracted onset [59,60]. Additionally, when coupled with a Tp53-inactivating mutation (Tp53R157H [61]), Smad4 mutation [62], or Cdkn2a mutation [60,63], an acceleration in the formation of PanIN can be observed, often rapidly progressing into metastatic PDAC.
In a mouse model of KRASG12D-driven PDAC, the loss of the wild-type allele was associated with metastatic disease. Such wild-type allele loss is also observed in human PDAC [64,65]. Interestingly, upon KRAS inactivation in PDAC that is KRASG12D-driven, rapid regression of primary and metastatic tumor growth can be observed in the presence of Tp53 deficiency [16,66]. This shows how KRAS inhibition in p53-deficient PDAC mice could be a useful therapeutic strategy, as will be discussed in the next chapter. Additionally, KRASG12D-driven pancreatic tumor formation was significantly impaired in Rac1-deficient mice [67], Pak1-deficient mice or those treated with anti-Pak1 [68], Rala/Ralb-deficient mice [69], Pik3ca variant–bearing mice [70,71], in mice deficient in both Mek1 and Mek2 or mice deficient in both Erk1 and Erk2 [72], and Raf1/Craf deficient mice [69,72,73].
Ras mutation can be caused by many genotoxic agents. One of the most frequent carcinogenic chemicals is methyl nitrosourea, which targets the second base on the 12th codon of HRAS and KRAS, generating a G12D mutation in many cancer types [74,75]. In comparison, UV radiation is known to target pyrimidine dimers, mutating RASQ61 [76]. In lung cancer, a distinctive couple of G12C mutations in KRAS occur, namely GC into TA. These G12C mutations are associated with in vitro bulky DNA formation driven by tobacco smoke products [77]. In smokers, this mutation is very common [78]. In pancreatic and colorectal cancers, this mutation is far less abundant [79,80,81]. In colorectal cancer, codon 13 mutation of KRAS is very common. In advanced colorectal cancer, patients with G13D mutation did not show a response to cetuximab therapy, an anti–epidermal growth factor receptor [82].
In thyroid carcinoma, significant numbers of mutations of all RAS isoforms have been observed, linked to ionizing radiation and other chemical carcinogens [83]. There is a pattern across RAS isoforms. In this type of cancer, 95% of NRAS mutations happen at codon 61 and 66% of KRAS mutations at codon 12. HRAS has 40% of its mutations on codon 12 and 50% on codon 61. Each codon has distinctive mutation patterns, with KRAS mutation at codon 12 often showing G12D, while HRAS favors G21V [82].
DNA damage and repair following exposure to carcinogens (e.g., benzo[a]pyrene diol epoxide, or BPDE) has also been associated with RAS isoforms. The preferred binding site for BPDE on KRAS is on codon 12 [84]. While the amino acid sequence encoded by each isoform is close to identical across many species, the DNA shows significant variation. The exon 1 variation produces different secondary structures, such as hairpin loops and G-quadruplex structures [85]. In AML, the expression of NRAS is relatively higher [86]. Additionally, some evidence shows that BPDE-driven DNA damage repair at codon 12 of KRAS was less efficient than that in HRAS and NRAS [87]. In short, the 12th codon is both the most targeted by BPDE and is the least repaired across the isoforms. These reasons could potentially explain why KRAS mutations occur so frequently in cancers [55].
4. Latest Anti-KRASG12D Therapies
Recently, small compounds have been developed that can target KRASG12D in PDAC. Unlike KRASG12C inhibitors, selective inhibitors of KRASG12D should bind with high affinity, without a requirement to covalently bind to the mutant KRAS protein. MRTX1133 is the first noncovalent KRASG12D small molecule with high affinity (Kd as low as 0.8nM) and is a powerful inhibitor, with nanomolar IC50 values (5 nM), shown to have efficacy in vivo in xenografted mice [88]. Among the advantages of small inhibitors compared to antibodies are oral delivery, more stability at room temperature, lower cost to manufacture, a great tumor distribution because of their relatively smaller size, and the variety of mechanisms they work through.
Another inhibitor of G12D came from the recent search for pan-RAS inhibitors, which led to the discovery of BI-2865. The IC50 of BI-2865 was 140 nM for BaF3 pro B cell line overexpressing KRASG12C, KRASG12D, and KRASA146V. A structural analog of this compound is BI-2493, which was optimized for in vivo administration. The antitumor effect of BI-2493 was associated with inhibition of phospho-ERK, and DUSP6 expression, in addition to favorable pharmacokinetics [89]. However, additional studies are required to further demonstrate the efficacy of BI-2493 and BI-2865 in different animal models.
The anti-tumor efficacy of the anti-KRASG12D drug HRS-4642 with proteasome inhibitor carfilzomib has been demonstrated against KRASG12D-mutant of various solid tumor cancer types both in vitro and in vivo. Either as a single agent or in combination with carfilzomib, a selective proteasome inhibitor, the drug was able to reshape the tumor microenvironment, making it more immune-permissive and eliciting an immune response against cancer [90].
ERAS-4693 and ERAS-5024 are two oral anti-KRASG12D drugs recently generated for solid tumors. In PDAC xenografts, these drugs were given intermittently and showed strong anti-tumor activity. However, there was a strong dose-limiting toxicity, limiting the feasibility of using this drug. More toxicological studies showed that MRGPRX2, linked to pseudo-allergic reactions, was agonized by both ERAS-4693 and ERAS-5024 [91].
The first oral covalent KRASG12D-selective inhibitor is RMC-9805. This drug forms a stable, high-affinity novel tri-complex with KRASG12D [92]. It exploits the intracellular chaperone cyclophilin A, forming a non-covalent complex. Overall, the agent forms a binary complex with S-IIP of KRASG12D-GTP bound KRASG12D, forming a tri-complex of KRAS, cyclophilin-A, and RMC-9805, leading to a covalent G12D cross-linkage, blocking the irreversible downstream binding of KRAS effectors. The interaction causes a selective and persistent modification of KRASG12D by disrupting the downstream KRASG12D signaling effectors (e.g., RAF kinases), thus inducing apoptosis and inhibiting cell proliferation. The authors observed that RMC-9805 was more active in PDAC and non–small cell lung carcinoma than in colorectal cancer models [93].
ASP3082 is the first KRASG12D degrader, which remarkably inhibits KRASG12D-mutated cancer models. It is a targeted protein degrader that uses proteolysis-targeting chimera technology to achieve its purpose. The drug entails an E3 ubiquitin ligase-binding moiety conjugated to a KRASG12D-binding moiety, via a linker. Once administered, the KRASG12D degrader targets and binds with the KRASG12D moiety of the KRASG12D-mutated protein and E3 ligase-binding moiety forming a ternary complex. This binding induces an E3 ligase-mediated ubiquitination and proteosome-mediated degradation of the KRASG12D-mutated protein. Subsequently, the KRASG12D-mediated signaling and activation of the downstream survival pathway is prevented, causing KRASG12D-driven tumor repression [94]. ASP3082 showed growth-inhibitory activity in KRASG12D-mutated PDAC. Additionally, in vitro, ASP3082 was able to inhibit mutated KRASG12D compared to 9000 other proteins, but this was not seen in the wild-type cancer cells. The inhibitory activity of this drug has also been shown in vivo in mice that received an intravenous administration once a week. The effect was dose-dependent and statistically significant, resulting in tumor inhibition with without body weight loss, indicating that it could potentially work without toxicity [95].
Chimeric T cell receptors (CAR-T) are an evolving type of therapy customized for each patient. They are generated by collecting T cells from patients and engineering them in the laboratory to recognize a specific patient’s antigen to be then re-injected in the patients. Autologous KRASG12D HLA-C*08:02-restricted T-cell receptor (TCR) gene-engineered T lymphocytes, known as NT-112, target and inhibit KRASG12D with potential antineoplastic activity [96]. The treatment of a PDAC patient with NT-112 showed successful regression of visceral metastases (overall partial response of 72%, according to the Response Evaluation Criteria in Solid Tumors) for 6 months. Moreover, the study inferred that more than 2% of all the patient’s circulating peripheral-blood T cells had the engineered T cells 6 months after the cell transfer. The overall response rate in the PDAC was mediated in this patient with the help of this TCR gene therapy [97]. Another TCR anti-KRASG12D has been tested in phase I/II clinical trials (NCT03745326). Recently, a study has shown that NT-112 was able to induce tumor clearance in two independent models in vivo. NT-112 T cells were associated with low-frequency chromosomal translocation events (<0.1%) between on-target and off-target Cas9 cleavage sites [98].
A major concern with KRASG12D-targeting drugs has been that they can inhibit wild-type KRAS, causing toxicities, even though in preclinical data, such concern has not been observed yet. This makes it crucial for these drugs to be validated in clinical settings, with a small number of patients first, in a dose-escalation manner. Figure 2 summarizes current anti-KRASG12D therapeutic interventional strategies.
5. The Role of an Immune-Permissive Tumor Microenvironment
A recent study using spatial genomics of 20 patients and bulk RNA sequencing of 100 tumors has brought to light enhanced epithelial-mesenchymal transition in KRASG12D and increased NKFβ in patients with KRASG12R pancreatic cancer [99]. RMC-9805 in combination with anti-PD-1 therapy showed synergetic activity by shaping a favorable immune microenvironment through cytokines. This reiterates the importance of the tumor immune microenvironment in anti-KRASG12D therapy [93]. The role of the tumor microenvironment in mediating the efficacy of KRASG12D inhibition by MRTX1133 and subsequent resistance mechanisms has been recently investigated using spatial transcriptomics, proteomics, and single-cell RNA sequencing. The drug was associated with higher levels of antigen-presenting cells, T cells, and tumor-restraining fibroblasts close to the cancer cells. These first shreds of evidence are suggestive of a remodeling of the local tumor microenvironment to facilitate the response to the KRASG12D inhibition. The single cell-sequencing data discovered CDK8 as an intrinsic resistance mediator, whereas CXCL2 was an extrinsic resistance mediator to the MRTX1133 drug [100]. With this knowledge in mind, multiple immune and cell-cycle markers could be pivotal candidates to be targeted at the same time for optimal KRASG12D inhibition in the PDAC context.
6. Ongoing Clinical Developments
Multiple clinical trials are testing the safety and efficacy of KRASG12D therapy in solid tumors including PDAC. A phase I trial of HRS-4642 has demonstrated the drug’s safety in 18 patients with solid tumors [101]. RMC-9805 has been tested in early-phase I/IIb clinical trials of KRASG12D solid tumors (NCT06040541). The study is testing the safety and tolerability of the drug in 290 patients with KRAS-driven solid tumors. ASP3082 has been tested in a dose-escalation phase I study to test its safety and tolerability in a small group of patients with metastatic solid tumors (3-12 patients). The next expansion study may enroll less than 20 patients. The safety and tolerability will be evaluated (NCT05382559) [102].
Vaccines have been tested against KRASG12D (Figure 2). The GI-4000 vaccine exhibited a striking effect in a mouse model, reducing the tumor burden by >80% when compared to the adjuvant alone. The vaccines elicited a strong immune response in these animals, resulting in the secretion of Th1 cytokines. However, the release of Th2-related cytokines was minimal [103]. A phase II clinical trial testing GI-4000 against KRASG12C/D/V in lung carcinoma showed that 50% of patients showed an immune response to mutant KRASG12C/D/V, and overall survival showed a positive trend [104]. Another KRAS vaccine is mRNA-5671 (Moderna). This tetravalent vaccine is formulated in a lipid nanoparticle that targets four of the most commonly occurring KRAS mutations, G12D, G12V, G13D, and G12C. Once administered, mRNA-5671 is taken up and translated by APCs. After translation, the epitopes are presented by major histocompatibility complex molecules expressed on the APCs. Consequently, both cytotoxic T-lymphocytes and memory T cells are activated against tumor cells harboring these specific KRAS mutations [103,105]. Another KRASG12D vaccine, for PDAC and colorectal cancer, is ELI-002 2P. It enhances lymph node delivery and immune response through the use of amphiphile modifications of G12D and G12R KRAS peptides when given together with CpG oligonucleotide adjuvant. Encouraging immunogenicity and relapse-free survival was observed in 25 patients (20 with PDAC and 5 with colorectal cancer) in a phase I dose-escalation study (NCT04853017). Ongoing clinical trials in solid tumors, including PDAC, are summarized in Table 1.
7. Discussion
Among the most lethal malignancies, PDAC is one of the most difficult to treat, and the current standard methods have been so far proven inefficient. Moreover, PDAC is usually diagnosed at late stages, when the tumor has already metastasized to other parts of the body, further limiting the available therapeutic tools.
Given this need, the efficacy of novel gene therapies or targeted therapies is currently being tested in laboratory and clinical settings. Among the advantages of covalent drugs is that they can bind strongly to their targets even at low concentrations because of their picomolar IC50 concentrations; and they are more selective, lowering the risk of adverse effects; moreover, their duration of action is longer. Among the disadvantages are off-target toxicity and the risk of having excess immunogenicity [106]. Noncovalent drugs could address some of these important issues, providing that their dose of administration is fine-tuned to obtain reasonable therapeutic indexes. New noncovalent drugs have been tested. Besides the MRTX1133 KRASG12D inhibitor, BI-2865 is a pan-KRAS inhibitor that also binds to GDP-associated wild-type and mutant KRAS with high affinity. The latter drug was later optimized into BI-2493 for in vivo use [107]. However, their therapeutic efficacy should be reiterated in more genetically modified animal models.and further explored in both animal and clinical trial. Additionally, in cell lines and organoids, resistance to the anti-KRASG12D MRTX1133 was shown to be mediated by the PI3K-AKT-mTOR pathway and epithelial-mesenchymal-transition proteins [108]. It appears a co-evolution of resistance to the anti-KRASG12D exists in the human body. Therefore, multiple combinational therapies, including the modulation of a favorable immune microenvironment, could be the more appropriate approach to PDAC.
Despite significant progress in therapies targeting KRASG12D, several limitations persist. A key challenge is the intrinsic heterogeneity of KRAS mutations and the complex genetic landscape of PDAC beyond KRAS itself. Various mutations and emerging variants complicate the development of universal and effective therapies. Additionally, concomitant mutations in genes such as p53, SMAD4, and CDKN2A can influence therapeutic responses, necessitating combinatorial and personalized approaches (9, 62, 63).
Although KRASG12D is the predominant mutation, other variants, such as KRASG12C and KRASG13D, further complicate the development of inhibitors with broad efficacy. KRASG12C inhibitors have shown promise in certain cancers, but their efficacy in PDAC remains unclear due to genetic variability and the complex tumor microenvironment. Several studies underscore the challenges of targeting KRAS mutations. KRASG12C inhibitors like sotorasib and adagrasib have demonstrated potential, particularly in non–small cell lung cancer, but their success in PDAC is less definitive due to the tumor’s genetic heterogeneity and challenging microenvironment. KRASG12C is also known to promote a pro-inflammatory tumor microenvironment, which may undermine the long-term efficacy of these inhibitors in solid tumors like PDAC. Furthermore, KRAS variants—including KRASG12D, KRASG12C, and KRASG13D—exhibit different biochemical properties, such as varying rates of GTP hydrolysis and interactions with downstream effectors. This variability complicates the development of inhibitors that can effectively target multiple KRAS variants across diverse tumor types [108,109,110]. Beyond targeting specific KRAS subtypes, there is an urgent need for clinical trials to validate the efficacy and safety of these novel drugs in larger populations and more complex preclinical models [88,89]. Alarmingly, resistance models are emerging, with some studies highlighting the activation of alternative pathways, such as PI3K-AKT-mTOR and epithelial-mesenchymal transition–related proteins, which may counteract the inhibitory effects of anti-KRASG12D therapies [111].
As technology advances, faster ways of making new compounds are evolving that could best target KRAS. First and later generations of translational targeted anti-KRAS therapies hold the promise of improving PDAC.
Conclusions: Currently, there are multiple experimental analyses towards KRAS-derived targets, but they have not been incorporated in the clinical setting as much as perhaps they should be. For a long time, treating KRAS was considered to be impossible, mainly because it was very difficult to find a drug that could bind to it with high sensitivity to the complicated chemical structure of this protein. The high affinity for GTP/GDP made this task much harder. However, with recent advancements of new small molecules that can reversibly or irreversibly bind to KRAS with high affinity, we have a possible impossibility for the treatment of cancers highly reliant on KRAS, such as PDAC.
Author Contributions
Navid Sobhani: Conceptualization, Supervision, Data curation, Visualization, Writing – original draft, Reviewing and editing. Matteo Pittacolo: Conceptualization, Data curation, Conceptualization, Software, Visualization, Writing - original draft. Alberto D’Angelo: Conceptualization, Investigation, Writing and editing. Giovanni Marchegiani: Conceptualization, Formal analysis, Supervision, Writing – original draft, Reviewing and editing.
Acknowledgments
We would like also to acknowledge Ms. Sara J Bronson for her professional editorial service.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014, 74, 2913-2921. [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin 2023, 73, 17-48. [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801-1806. [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res 2020, 80, 2969-2974. [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 2014, 39, 91-100. [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548-551. [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020, 383, 1207-1217. [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021, 384, 2371-2381. [CrossRef]
- McGregor, L.M.; Jenkins, M.L.; Kerwin, C.; Burke, J.E.; Shokat, K.M. Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes. Biochemistry 2017, 56, 3178-3183. [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry 2020, 63, 6679-6693. [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J Med Chem 2022, 65, 3123-3133. [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat. Med. 2022, 28, 2171-2182. [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861-868. [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin Cancer Res 2012, 18, 4266-4276. [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101-105. [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656-670. [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006, 20, 1218-1249. [CrossRef]
- Valsangkar, N.P.; Ingkakul, T.; Correa-Gallego, C.; Mino-Kenudson, M.; Masia, R.; Lillemoe, K.D.; Fernández-del Castillo, C.; Warshaw, A.L.; Liss, A.S.; Thayer, S.P. Survival in ampullary cancer: potential role of different KRAS mutations. Surgery 2015, 157, 260-268. [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011, 11, 761-774. [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555-1559. [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 2011, 7, 523. [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101-105. [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730-733.e739. [CrossRef]
- Kong, B.; Qia, C.; Erkan, M.; Kleeff, J.; Michalski, C.W. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol 2013, 4, 246. [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106-109. [CrossRef]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Oxidative stress in apoptosis and cancer: an update. Arch Toxicol 2012, 86, 1649-1665. [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010, 107, 8788-8793. [CrossRef]
- Matés, J.M.; Segura, J.A.; Campos-Sandoval, J.A.; Lobo, C.; Alonso, L.; Alonso, F.J.; Márquez, J. Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol 2009, 41, 2051-2061. [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344-1348. [CrossRef]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy - unanswered questions. J Cell Sci 2011, 124, 161-170. [CrossRef]
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in cancer: good, bad, or both? Cancer Res 2006, 66, 9349-9351. [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev 2011, 25, 1999-2010. [CrossRef]
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 2008, 99, 1813-1819. [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011, 25, 717-729. [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011, 25, 460-470. [CrossRef]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol 2009, 29, 4495-4507. [CrossRef]
- Wang, Y.; Wang, X.D.; Lapi, E.; Sullivan, A.; Jia, W.; He, Y.W.; Ratnayaka, I.; Zhong, S.; Goldin, R.D.; Goemans, C.G.; et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A 2012, 109, 13325-13330. [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009, 23, 798-803. [CrossRef]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011, 42, 23-35. [CrossRef]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol 2016, 7, e157. [CrossRef]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Ormanns, S.; Haas, M.; Modest, D.P.; Kirchner, T.; et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol 2013, 48, 544-548. [CrossRef]
- Gu, Y.; Ji, Y.; Jiang, H.; Qiu, G. Clinical Effect of Driver Mutations of KRAS, CDKN2A/P16, TP53, and SMAD4 in Pancreatic Cancer: A Meta-Analysis. Genet Test Mol Biomarkers 2020, 24, 777-788. [CrossRef]
- Ogura, T.; Yamao, K.; Hara, K.; Mizuno, N.; Hijioka, S.; Imaoka, H.; Sawaki, A.; Niwa, Y.; Tajika, M.; Kondo, S.; et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol 2013, 48, 640-646. [CrossRef]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol 2018, 4, e173420. [CrossRef]
- Rachakonda, P.S.; Bauer, A.S.; Xie, H.; Campa, D.; Rizzato, C.; Canzian, F.; Beghelli, S.; Greenhalf, W.; Costello, E.; Schanne, M.; et al. Somatic mutations in exocrine pancreatic tumors: association with patient survival. PLoS ONE 2013, 8, e60870. [CrossRef]
- Shin, S.H.; Kim, S.C.; Hong, S.M.; Kim, Y.H.; Song, K.B.; Park, K.M.; Lee, Y.J. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas 2013, 42, 216-222. [CrossRef]
- Kawesha, A.; Ghaneh, P.; Andrén-Sandberg, A.; Ograed, D.; Skar, R.; Dawiskiba, S.; Evans, J.D.; Campbell, F.; Lemoine, N.; Neoptolemos, J.P. K-ras oncogene subtype mutations are associated with survival but not expression of p53, p16(INK4A), p21(WAF-1), cyclin D1, erbB-2 and erbB-3 in resected pancreatic ductal adenocarcinoma. Int J Cancer 2000, 89, 469-474. [CrossRef]
- Zhang, Z.; Zhang, H.; Liao, X.; Tsai, H.I. KRAS mutation: The booster of pancreatic ductal adenocarcinoma transformation and progression. Front Cell Dev Biol 2023, 11, 1147676. [CrossRef]
- Yousef, A.; Yousef, M.; Chowdhury, S.; Abdilleh, K.; Knafl, M.; Edelkamp, P.; Alfaro-Munoz, K.; Chacko, R.; Peterson, J.; Smaglo, B.G.; et al. Impact of KRAS mutations and co-mutations on clinical outcomes in pancreatic ductal adenocarcinoma. NPJ Precis Oncol 2024, 8, 27. [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399-405. [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. MEK in cancer and cancer therapy. Pharmacol Ther 2014, 141, 160-171. [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2020, 17, 153-168. [CrossRef]
- Nakano, Y.; Kitago, M.; Matsuda, S.; Nakamura, Y.; Fujita, Y.; Imai, S.; Shinoda, M.; Yagi, H.; Abe, Y.; Hibi, T.; et al. KRAS mutations in cell-free DNA from preoperative and postoperative sera as a pancreatic cancer marker: a retrospective study. Br J Cancer 2018, 118, 662-669. [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin Oncol 2021, 48, 10-18. [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012, 72, 2457-2467. [CrossRef]
- Herrera, M.; García, E.; Diaz, D.C.; Ruiz-Garcia, E.; Miyagui Adame, S.; Zilli Hernandez, S.; Lopez, M.; Carbajal, B.; Calderillo Ruiz, G. Real-world impact on overall survival of oxaliplatin-based adjuvant chemotherapy in older patients with colorectal cancer in Mexico. J. Clin. Oncol. 2024, 42, 61-61. [CrossRef]
- Philip, P.A.; Azar, I.; Xiu, J.; Hall, M.J.; Hendifar, A.E.; Lou, E.; Hwang, J.J.; Gong, J.; Feldman, R.; Ellis, M.; et al. Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin Cancer Res 2022, 28, 2704-2714. [CrossRef]
- Nusrat, F.; Khanna, A.; Jain, A.; Jiang, W.; Lavu, H.; Yeo, C.J.; Bowne, W.; Nevler, A. The Clinical Implications of KRAS Mutations and Variant Allele Frequencies in Pancreatic Ductal Adenocarcinoma. J Clin Med 2024, 13, 2103. [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437-450. [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003, 17, 3112-3126. [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469-483. [CrossRef]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res 2007, 67, 8121-8130. [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A 2006, 103, 5947-5952. [CrossRef]
- Grabocka, E.; Pylayeva-Gupta, Y.; Jones, M.J.; Lubkov, V.; Yemanaberhan, E.; Taylor, L.; Jeng, H.H.; Bar-Sagi, D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell 2014, 25, 243-256. [CrossRef]
- Lim, K.H.; Ancrile, B.B.; Kashatus, D.F.; Counter, C.M. Tumour maintenance is mediated by eNOS. Nature 2008, 452, 646-649. [CrossRef]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012, 122, 639-653. [CrossRef]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719-730, 730.e711-717. [CrossRef]
- Chow, H.Y.; Jubb, A.M.; Koch, J.N.; Jaffer, Z.M.; Stepanova, D.; Campbell, D.A.; Duron, S.G.; O’Farrell, M.; Cai, K.Q.; Klein-Szanto, A.J.; et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res 2012, 72, 5966-5975. [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 2018, 8. [CrossRef]
- Castellano, E.; Sheridan, C.; Thin, M.Z.; Nye, E.; Spencer-Dene, B.; Diefenbacher, M.E.; Moore, C.; Kumar, M.S.; Murillo, M.M.; Grönroos, E.; et al. Requirement for interaction of PI3-kinase p110α with RAS in lung tumor maintenance. Cancer Cell 2013, 24, 617-630. [CrossRef]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957-968. [CrossRef]
- Blasco, R.B.; Francoz, S.; Santamaría, D.; Cañamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 2011, 19, 652-663. [CrossRef]
- Karreth, F.A.; Frese, K.K.; DeNicola, G.M.; Baccarini, M.; Tuveson, D.A. C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov 2011, 1, 128-136. [CrossRef]
- Barbacid, M. ras oncogenes: their role in neoplasia. Eur J Clin Invest 1990, 20, 225-235. [CrossRef]
- Zarbl, H.; Sukumar, S.; Arthur, A.V.; Martin-Zanca, D.; Barbacid, M. Direct mutagenesis of Ha-ras-1 oncogenes by N-nitroso-N-methylurea during initiation of mammary carcinogenesis in rats. Nature 1985, 315, 382-385. [CrossRef]
- Törmänen, V.T.; Pfeifer, G.P. Mapping of UV photoproducts within ras proto-oncogenes in UV-irradiated cells: correlation with mutations in human skin cancer. Oncogene 1992, 7, 1729-1736.
- Seo, K.Y.; Jelinsky, S.A.; Loechler, E.L. Factors that influence the mutagenic patterns of DNA adducts from chemical carcinogens. Mutat Res 2000, 463, 215-246. [CrossRef]
- Le Calvez, F.; Mukeria, A.; Hunt, J.D.; Kelm, O.; Hung, R.J.; Tanière, P.; Brennan, P.; Boffetta, P.; Zaridze, D.G.; Hainaut, P. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res 2005, 65, 5076-5083. [CrossRef]
- Capella, G.; Cronauer-Mitra, S.; Pienado, M.A.; Perucho, M. Frequency and spectrum of mutations at codons 12 and 13 of the c-K-ras gene in human tumors. Environ Health Perspect 1991, 93, 125-131. [CrossRef]
- Porta, M.; Crous-Bou, M.; Wark, P.A.; Vineis, P.; Real, F.X.; Malats, N.; Kampman, E. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: etiopathogenic similarities, differences and paradoxes. Mutat Res 2009, 682, 83-93. [CrossRef]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer 2003, 3, 733-744. [CrossRef]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. Jama 2010, 304, 1812-1820. [CrossRef]
- Sipos, J.A.; Mazzaferri, E.L. Thyroid cancer epidemiology and prognostic variables. Clin Oncol (R Coll Radiol) 2010, 22, 395-404. [CrossRef]
- Hu, W.; Feng, Z.; Tang, M.S. Preferential carcinogen-DNA adduct formation at codons 12 and 14 in the human K-ras gene and their possible mechanisms. Biochemistry 2003, 42, 10012-10023. [CrossRef]
- Wright, B.E.; Reimers, J.M.; Schmidt, K.H.; Reschke, D.K. Hypermutable bases in the p53 cancer gene are at vulnerable positions in DNA secondary structures. Cancer Res 2002, 62, 5641-5644.
- Gougopoulou, D.M.; Kiaris, H.; Ergazaki, M.; Anagnostopoulos, N.I.; Grigoraki, V.; Spandidos, D.A. Mutations and expression of the ras family genes in leukemias. Stem Cells 1996, 14, 725-729. [CrossRef]
- Denissenko, M.F.; Pao, A.; Pfeifer, G.P.; Tang, M. Slow repair of bulky DNA adducts along the nontranscribed strand of the human p53 gene may explain the strand bias of transversion mutations in cancers. Oncogene 1998, 16, 1241-1247. [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. Journal of Medicinal Chemistry 2022, 65, 3123-3133. [CrossRef]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023, 619, 160-166. [CrossRef]
- Flores-Gómez, A.A.; Drosten, M. HRS-4642: The next piece of the puzzle to keep KRAS in check. Cancer Cell 2024, 42, 1157-1159. [CrossRef]
- Brooun, A.; Zhang, J.; Li, C.; Lam, R.; Cheng, H.; Shoemaker, R.; Daly, J.; Olaharski, A. The pharmacologic and toxicologic characterization of the potent and selective KRAS G12D inhibitors ERAS-4693 and ERAS-5024. Toxicol Appl Pharmacol 2023, 474, 116601. [CrossRef]
- Knox, J.E.; Jiang, J.; Burnett, G.L.; Liu, Y.; Weller, C.E.; Wang, Z.; McDowell, L.; Steele, S.L.; Chin, S.; Chou, K.J.; et al. Abstract 3596: RM-036, a first-in-class, orally-bioavailable, Tri-Complex covalent KRASG12D(ON) inhibitor, drives profound anti-tumor activity in KRASG12D mutant tumor models. Cancer Research 2022, 82, 3596-3596. [CrossRef]
- Jiang, L.; Menard, M.; Weller, C.; Wang, Z.; Burnett, L.; Aronchik, I.; Steele, S.; Flagella, M.; Zhao, R.; Evans, J.W.W.; et al. Abstract 526: RMC-9805, a first-in-class, mutant-selective, covalent and oral KRASG12D(ON) inhibitor that induces apoptosis and drives tumor regression in preclinical models of KRASG12D cancers. Cancer Research 2023, 83, 526-526. [CrossRef]
- KRAS G12D degrader ASP3082. NIH Website.
- Nagashima, T.; Inamura, K.; Nishizono, Y.; Suzuki, A.; Tanaka, H.; Yoshinari, T.; Yamanaka, Y. ASP3082, a First-in-class novel KRAS G12D degrader, exhibits remarkable anti-tumor activity in KRAS G12D mutated cancer models. Eur. J. Cancer 2022, 174, S30. [CrossRef]
- autologous KRAS G12D-specific HLA-C*08:02-restricted TCR gene engineered T lymphocytes NT-112. NIH Website.
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N Engl J Med 2022, 386, 2112-2119. [CrossRef]
- Ma, J.; Tubb, V.; Mojadidi, M.; Meade, K.; Kong, X.; Kroon, P.; Sareen, A.; Stringer, B.; Krijgsman, O.; Claasen, Y.; et al. Non-clinical evaluation of NT-112, an autologous T cell product engineered to express an HLA-C*08:02-restricted TCR targeting KRAS G12D and resistant to TGF-b inhibition. J. Clin. Oncol. 2024, 42, e14533-e14533. [CrossRef]
- McIntyre, C.A.; Grimont, A.; Park, J.; Meng, Y.; Sisso, W.J.; Seier, K.; Jang, G.H.; Walch, H.; Aveson, V.G.; Falvo, D.J.; et al. Distinct clinical outcomes and biological features of specific <em>KRAS</em> mutants in human pancreatic cancer. Cancer Cell 2024, 42, 1614-1629. [CrossRef]
- McAndrews, K.M.; Mahadevan, K.K.; Li, B.; Sockwell, A.M.; Morse, S.J.; Kelly, P.J.; Kirtley, M.L.; Conner, M.R.; Patel, S.I.; Khumbar, S.V.; et al. Abstract B092: CDK8 and CXCL2 remodel the tumor microenvironment to contribute to KRASG12D small molecule inhibition resistance in pancreatic ductal adenocarcinoma. Cancer Research 2024, 84, B092-B092. [CrossRef]
- Zhou, C.; Li, W.; Song, Z.; Zhang, Y.; Zhang, Y.; Huang, D.; Yang, Z.; Zhou, M.; Mao, R.; Huang, C.; et al. LBA33 A first-in-human phase I study of a novel KRAS G12D inhibitor HRS-4642 in patients with advanced solid tumors harboring KRAS G12D mutation. Ann. Oncol. 2023, 34, S1273. [CrossRef]
- Tolcher, A.W.; Park, W.; Wang, J.S.; Spira, A.I.; Janne, P.A.; Lee, H.-J.; Gill, S.; LoRusso, P.; Herzberg, B.; Goldman, J.W.; et al. Trial in progress: A phase 1, first-in-human, open-label, multicenter, dose-escalation and dose-expansion study of ASP3082 in patients with previously treated advanced solid tumors and KRAS G12D mutations. J. Clin. Oncol. 2023, 41, TPS764-TPS764. [CrossRef]
- Pan, J.; Zhang, Q.; Sei, S.; Shoemaker, R.H.; Lubet, R.A.; Wang, Y.; You, M. Immunoprevention of KRAS-driven lung adenocarcinoma by a multipeptide vaccine. Oncotarget 2017, 8, 82689-82699. [CrossRef]
- Chaft, J.E.; Litvak, A.; Arcila, M.E.; Patel, P.; D’Angelo, S.P.; Krug, L.M.; Rusch, V.; Mattson, A.; Coeshott, C.; Park, B.; et al. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer 2014, 15, 405-410. [CrossRef]
- mRNA-derived KRAS-targeted vaccine V941. NIH Website.
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: a rational approach to drug discovery. RSC Med Chem 2020, 11, 876-884. [CrossRef]
- Kingwell, K. A non-covalent inhibitor with pan-KRAS potential. Nat Rev Drug Discov 2023, 22, 622. [CrossRef]
- Dilly, J.; Hoffman, M.T.; Abbassi, L.; Li, Z.; Paradiso, F.; Parent, B.D.; Hennessey, C.J.; Jordan, A.C.; Morgado, M.; Dasgupta, S.; et al. Mechanisms of resistance to oncogenic KRAS inhibition in pancreatic cancer. Cancer Discov 2024, 7, 1137-1357. [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J Exp Clin Cancer Res 2022, 41, 27. [CrossRef]
- Batrash, F.; Kutmah, M.; Zhang, J. The current landscape of using direct inhibitors to target KRAS(G12C)-mutated NSCLC. Exp Hematol Oncol 2023, 12, 93. [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol 2018, 15, 709-720. [CrossRef]
Figure 1.
G12 mutational rates in PDAC. cBioPortal analysis from 2188 pancreatic ductal adenocarcinoma patients. The mutational distribution of the G12 is shown in the pie chart at left with the percentage of patients. Microsatellite instability (MSI) pie chart at the lower left (stable MSI in blue and NA in gray). Overall survival (OS) pie chart at the lower right (deceased in orange and living in green).
Figure 1.
G12 mutational rates in PDAC. cBioPortal analysis from 2188 pancreatic ductal adenocarcinoma patients. The mutational distribution of the G12 is shown in the pie chart at left with the percentage of patients. Microsatellite instability (MSI) pie chart at the lower left (stable MSI in blue and NA in gray). Overall survival (OS) pie chart at the lower right (deceased in orange and living in green).
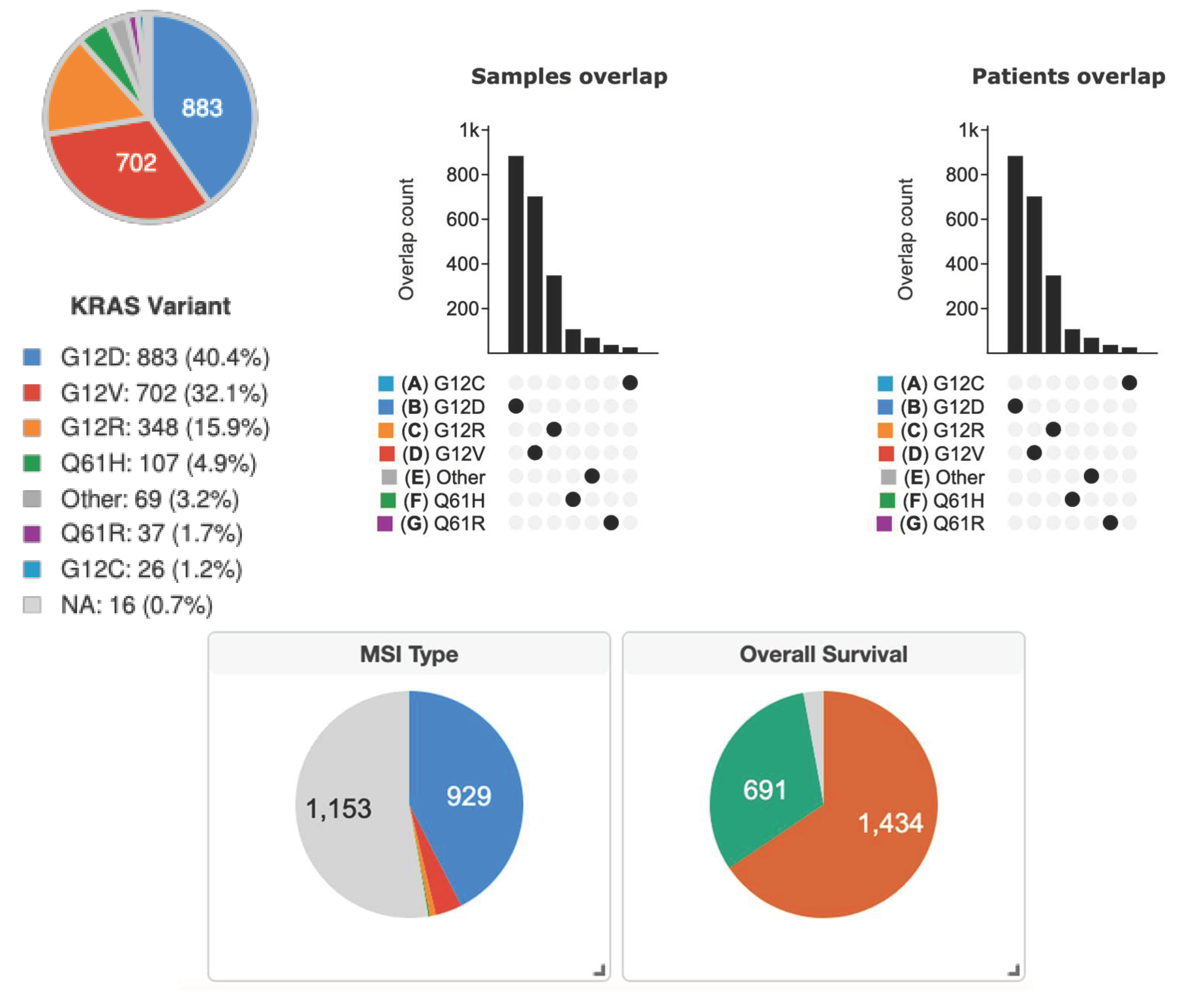
Figure 2.
KRASG12D therapies. The KRAS pathway and therapeutic anti-KRASG12D, vaccination, and CAR-T therapies are considered at the forefront of pancreatic ductal adenocarcinoma research.
Figure 2.
KRASG12D therapies. The KRAS pathway and therapeutic anti-KRASG12D, vaccination, and CAR-T therapies are considered at the forefront of pancreatic ductal adenocarcinoma research.
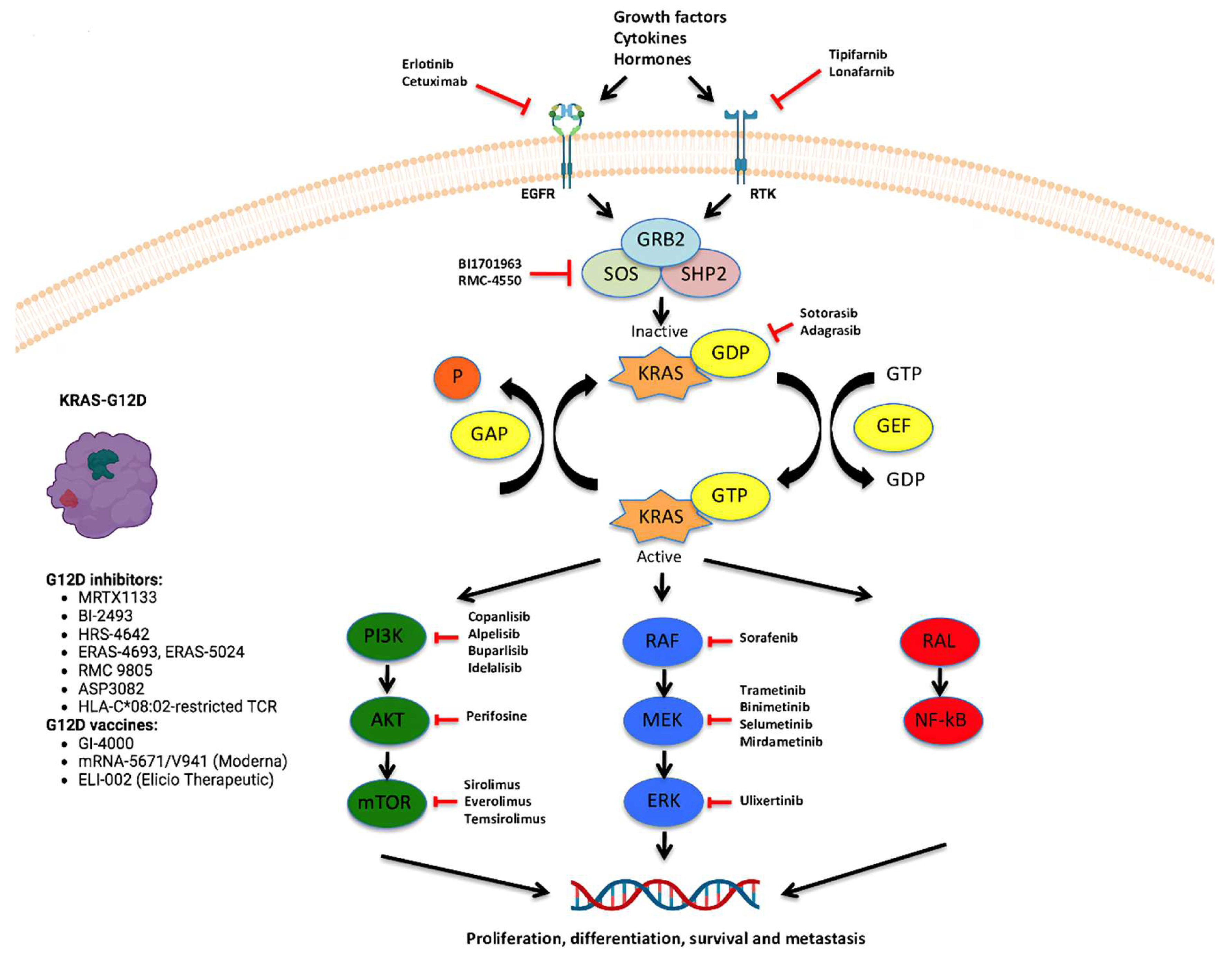

Table 1.
Ongoing clinical trials testing anti-KRASG12D therapies in solid tumors.
| Trial Identifier | Investigation Plan | Viral Vaccine/ Drug |
Clinical Setting | Primary Endpoint | Phase | Trial Status |
|---|---|---|---|---|---|---|
| NCT06385925 | Non-randomized, sequential assignment, open label | TSN1611 | First line | DLT | I/II | Recruiting |
| NCT06385678 | Non-randomized, single group assignment, open label | HRS-4642; adebrelimab; SHR-A1921; chemotherapy: pemetrexed, cisplatin, carboplatin |
First line | DLT, RP2D, ORR | Ib/II | Recruiting |
| NCT06500676 | Single group assignment, open label | GFH375 | First line | DLT, ORR | I/II | Recruiting |
| NCT05737706 | Non-randomized, sequential assignment, open label | MRTX1133 | Second or later line | DLT, ORR, DOR, PFS, OS | I/II | Recruiting |
| NCT06478251 | Non-randomized, parallel assignment, open label | NW-301 TCR-T, NW-301D TCR-T | First line | DLT | I | Recruiting |
| NCT06403735 | Non-randomized, single group assignment, open label | QLC1101 | First line | DLT | I | Recruiting |
| NCT03745326 | Non-randomized, sequential assignment, open label | Drug: cyclophosphamide Drug: fludarabine Drug: aldesleukin Biological: anti-KRAS G12D mTCR peripheral blood lymphocytes |
First line | DLT; PR+CR | I/II | Recruiting |
| NCT06218914 | Sequential assignment, open label | NT-112 | First line | DLT | I | Recruiting |
| NCT05533463 | Non-randomized, single group assignment | HRS-4642 | First line | DLT | I | Recruiting |
| NCT06364696 | Non-randomized, sequential assignment, open label | ASP4396 | First or later line | DLT, ORR, DOR, DCR, PFS, OS | I | Recruiting |
| NCT06040541 | Non-randomized, parallel assignment, open label | RMC-9805 RMC-6236 |
Second line | AEs, DLT | I/Ib | Recruiting |
| NCT05382559 | Non-randomized, sequential assignment, open label | ASP3082; cetuximab; chemotherapies |
First line | AEs, DLT | I | Recruiting |
| NCT06546150 | Single assignment, open label | RE002 T cell | First line | AEs | I | Not yet recruiting |
| NCT05254184 | Single assignment, open label | Mutant KRAS-targeted long peptide vaccine | First line | AEs | I | Recruiting |
| NCT06484790 | Non-randomized, parallel assignment, open label | NW-301V; NW-301D |
First line | DLT | I | Recruiting |
| NCT06484556 | Non-randomized, parallel assignment, open label | NW-301V; NW-301D |
First line | DLT | I | Recruiting |
| NCT06487377 | Single assignment, 3+3 dose escalation, open label | IX001 TCR-T | First or Later line | AEs, DLT | I | Recruiting |
| NCT06428500 | Non-randomized, sequential assignment, open label | QTX3046 | First or Later line | TEAEs, DLTs | I | Recruiting |
| NCT06520488 | Single group assignment, open label | HRS-4642 | First or Later line | MTD | Ib/II | Not-Yet Recruiting |
| NCT06227377 | Non-randomized, single group assignment, open label | QTX3034 | First line | DLT, TEAEs | I | Recruiting |
| NCT05726864 | Randomized, sequential assignment, open label | ELI-002 7P | First or Later line | AEs | Ia, Ib, II | Recruiting |
| NCT05786924 | Non-randomized, sequential assignment, open label | BDTX-4933 | First line | MTD | I | Recruiting |
| NCT06447662 | Non-randomized, sequential assignment, open label | PF-07934040 | First or later line | AEs, DLT | I/IIa and IIb | Recruiting |
| NCT06179160 | Non-randomized, sequential assignment, open label | INCB161734; cetuximab; retifanlimab | First line | DLTs, TEAEs | I | Recruiting |
| NCT05846516 | Non-randomized, sequential assignment, open label | VSV-GP154; ATP150; ATP152; ezabenlimab |
First line | DLT, DFS |
Ib | Recruiting |
| NCT05983159 | Non-randomized, parallel assignment, open label | Alpelisib; mirdametinib |
First line | VM-PSOM | II | Not-Yet Recruiting |
| NCT06208124 | Treatment, single group assignment, open label | IMM-6-415 | First or later lines | DLT | I/IIa | Recruiting |
| NCT05585320 | Non-randomized, parallel assignment, open label | IMM-1-104 monotherapy (treatment group A); IMM-1-104 + modified gemcitabine/nab-paclitaxel (treatment group B); IMM-1-104 + modified FOLFIRINOX (treatment group C) |
First line or later line | AEs, DLTs, RP2D, OS |
I/IIa | Recruiting |
AE, adverse event; DCR, disease control rate; DFS, disease-free survival; DLT, dose-limiting toxicity; DOR, duration of response; MTD, maximum tolerated dose; ORR, overall response rate; OS, overall survival; PR+CR, partial response and complete response; RP2D, recommended phase II dose; TEAE, treatment-emergent adverse event.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



