Submitted:
18 December 2024
Posted:
19 December 2024
Read the latest preprint version here
Abstract
Background/Objectives: As neurodegenerative diseases become increasingly prevalent, there is a growing interest in natural therapeutic agents to address neuroinflammation, a critical factor in these disorders. With their historical use in traditional medicine, medicinal plants offer the potential for mitigating neuroinflammation. This review aimed to evaluate the effects of medicinal plants on neuroinflammation by conducting a thorough search across reputable databases. Summary: We reviewed studies that investigated the impact of medicinal plants on neuroinflammation, focusing on effective dosages, mechanisms of action, and clinical implications. We revealed the anti-inflammatory potential of plants such as Cleistocalyx nervosum var. paniala, Curcuma longa, Cannabis sativa, and Dioscorea nipponica. Although these studies highlight significant anti-inflammatory effects, variability in dosages and phytochemical compositions indicates the need for further research. Conclusions: Future studies should emphasize animal models, standardized protocols, and comprehensive safety assessments to facilitate the translation of findings into clinical trials. Additionally, integrating advanced methodologies such as genetic studies and nanomedicine could further enhance these plants' therapeutic potential.
Keywords:
Medicinal Plants
; Neuroinflammation
; Molecular Mechanisms
; NF-κB
; HO-1
; Nrf2
1. Introduction
The increase in life expectancy has been accompanied by a rise in the prevalence of age-related neurodegenerative diseases (NDs). Maintaining neuronal function and cognitive ability depends on the brain's high energy demands, as it is one of the most energetically active organs despite not performing mechanical work like the skeletal muscles or heart [1,2,3,4]. Typically, the human brain consumes approximately 3.5 ml of oxygen per 100 grams of brain tissue per minute [5,6]. This high oxygen consumption makes neurons susceptible to reactive oxygen species (ROS) damage and subsequent inflammation [7-13].
Neuroinflammation is characterized by activating the brain's innate immune system in response to inflammatory stimuli. This response involves the activation of microglia and astrocytes, the release of pro-inflammatory cytokines and chemokines, ROS production, and sometimes the infiltration of peripheral leukocytes into the central nervous system (CNS). While neuroinflammation initially acts as a protective mechanism, sustained or maladaptive inflammation has been identified as a critical factor in the progression of various neurological diseases, including NDs, psychiatric disorders, pain syndromes, stroke, and traumatic brain injury [4,14,15,16,17,18,19].
Addressing neuroinflammation to reduce disease severity and improve patient outcomes is a promising strategy against neurodegeneration. From a molecular perspective, there are several conventional drug targets for neuroinflammation, such as enzymes, receptors, and ion channels [19,20,21,22,23,24]. However, the high cost of synthetic drugs presents a challenge, emphasizing the need for alternative approaches [25,26]. This has heightened interest in naturally occurring medicinal plants known for their antioxidant, anti-inflammatory, and neuroprotective properties. These plants are often more cost-effective and have been safely utilized in treatments for thousands of years [27,28].
A substantial body of literature underscores the beneficial effects of dietary phytochemicals and medicinal plants on overall health and longevity [29,30,31,32]. Medicinal-plant-based drug discovery remains a vital and underexplored area of research with substantial potential [33]. Despite pharmaceutical advances, plants have historically been a rich source of therapeutic compounds, yet many of their potential benefits are still untapped [34]. By comprehensively investigating these plants, researchers can uncover novel compounds that could offer significant breakthroughs in treating various conditions. Specifically, this approach could lead to the discovery of new treatments for complex issues such as neuroinflammation and neurodegeneration, which are often challenging to address with current therapies [12,35].
Despite previous efforts, existing reviews have yet to fully explore the potential of medicinal plants in regulating neuroinflammation. Tyler et al. [36] offered valuable insights into the therapeutic potential of plants for neurodegenerative diseases. However, their review needed a systematic examination of the literature addressing medicinal plants targeting neuroinflammation. Their focus was broader, with a greater emphasis on ethnobotanical aspects rather than exclusively discussing neuroinflammation. In contrast, Suk et al. [37] explored the regulation of neuroinflammation by herbal medicine and its implications for neurodegenerative diseases. Their review centered predominantly on traditional medicines and their flavonoid derivatives. Additionally, this review, published in 2005, needs to be updated and would benefit from an update incorporating recent advancements in the field. This study aims to fill these gaps by thoroughly reviewing the current literature. The manuscript includes a table (Table 1) that compiles research on medicinal plants' role as potential neuroinflammation modulators. Figure 1 illustrates the microglia's distinct phenotypes, M1 (pro-inflammatory) and M2 (anti-inflammatory).
2. Nuclear Factor Kappa B (NF-κB): A Central Player in the Development of Neuroinflammation and Neurodegenerative Conditions
In 1986, Sen and Baltimore discovered that, in B lymphocytes, a protein binds to a specific DNA sequence, called NF-kB, that will be acting in case of extensive inflammation [50]. From the moment B cells' maturation induces NF-kB, it is possible to note that this element has fundamental biological importance. As a starting point, it is essential to know that NF-kB is in latency in the cell. With a stimulus that can be inflammatory, a series of ordered responses begin to occur, and if the problem is resolved, the path of latency returns. In this scenario, the cells of the innate immune system are activated so that they are sufficient to fight minor infections or, in the case of smaller organisms, be their only line of defense. In the case of extensive inflammation, the adaptive immune system cells involve T and B cells. The T cell receptor (TCR) allows T lymphocytes to recognize antigens processed by antigen-presenting cells (APCs), which require the activation of NF-kB [51,52].
Initially, the most important thing to know is that nuclear factor-κB (NF-κB) exists as hetero or homodimers in a family of protein complexes [53]. These dimers make up a family: RelA, RelB, c-Rel, NF-kB1 (p50), and NF-kB2 (p52) -transcription factors intrinsically involved with inflammatory processes [54]. The NF- κB transcription factor has a fundamental role in regulating several aspects through the action of genes, ranging from cell multiplication to multicellular immune and inflammatory responses. For the functions to occur without complications, the NF-κB system must be adequately regulated, as well as the formation of protein monomers that will form its dimeric family responsible for linking the DNA. [55]. NF-kB activity is regulated at the mRNA level, so transcriptional signals control this transcription factor [53].
It is essential to know that the cytoplasm is predominantly where NF-kB is located, so it is inactive as it is in the form of a complex associated with IκB proteins [56,57]. NF-kB is responsible for mediating survival factors and an antioxidant cellular response fundamental in microcirculatory disturbance, as it acts on their development [58,59]. However, when oxidative stress happens, NF-kB translocates to the nucleus from the NF-κB–IκBα complex. It is in the nucleus where the initiation of transcription of proinflammatory enzymes - such as COX-2, NADPH oxidase, and sPLA2 - as well as proinflammatory cytokines, like TNF-α, IL-1β, and IL-6, occurs. [56]
NF-κB has the property of transcribing genes, which may be proinflammatory, triggering a series of events related to neuroinflammation. [54]. The two catalytic subunits, IKKα and IKKβ, and the regulatory subunit NEMO comprise the IkB kinase complex (IKK) [60]. Two different pathways activate NF-κB, so the canonical pathway is primarily responsible for inflammation and is stimulated by Tool Like Receptors (TLRs). First, IκBα captures the p65/p50 dimers in the cytoplasm. Consequently, certain pro-inflammatory stimuli, such as pathogens or cytokines, release p65/p50 dimers through the phosphorylation cascade so that IκBα is degraded. Soon after, the cognatekB motif is linked to the p65/p50 translocated in the nucleus, activating the NF-κB gene. [54]. The regulatory protein MyD88 (myeloid differentiation primary response 88) mediates the canonical process [60]. In this sense, there is a negative feedback mechanism since the temporary activation of NF-kB is related to the induction of negative regulators, such as IκBα, A20, and p105 [61].
The non-canonical NF-κB pathway has the RelB/p52 as the transcription factor and maintains an intimate relationship with IKKα homodimers, considering that it depends on them to occur. [61]. Interestingly, in certain types of cells, some members of NF-kB, such as RELA, associate with p100 and are activated through the induction of p100 processing [62]. In this context, regulating this pathway is only possible through the NF-kB-inducing kinase (NIK) expression. Thus, NIK recruitment to TRAF2 at a steady state is mediated through the TNF receptor-associated factor (TRAF3), resulting in the ubiquitination of NIF and its consequent degradation. Therefore, the NF-kB complex remains inactive in the cytoplasm due to low levels of endogenous NIF. Upon activation of the non-canonical NF-κB pathway, TRAF3 proteolysis is induced by TRAF2 – this degradation of TRAF3 releases and accumulates NIF. After this, phosphorylation of p100 by IKKα heterodimers is induced by NIK, promoting an incomplete deterioration that makes p52 available. Ultimately, p52-RelB heterodimers translocate to the nucleus, allowing target genes to be transcribed. [63].
Since the host may be exposed to pathogens that cause tissue damage and infections, inflammation is a protective response of the body capable of promoting the degradation of pathogens and wound healing [62]. In the case of neuroinflammation, the central nervous system promotes an immune response, which is sometimes related to the etiology and course of several psychiatric and neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and multiple sclerosis (MS). In this sense, NF-kB in the glial cells has a central role in the regulation of chronic neuroinflammation, that is neurodegenerative [64,65], bearing in mind that various diseases can be exacerbated or intensified in cases of uncontrolled inflammation [66,67].
3. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2): A Novel Approach to Address Oxidative Stress and Neuroinflammation in Neurodegenerative Disorders
The term oxidative stress was introduced into the literature in 1985 with the publication of a book called "Oxidative Stress", which began a series of studies related to the topic, such as Redox Biology. Several updates, including the action of redox signaling, promoted a redefinition of the initial concept [68]. Firstly, it is essential to understand that oxidative stress is essential for maintaining the organism even at a cellular level. This process is characterized by an imbalance between the production and consumption of oxidative agents, altering homeostasis: on several occasions, the body's ability to neutralize these reactive species decreases, or there is an increase in Reactive Oxygen Species (ROS) production [69,70]. ROS have well-known kinetic, thermodynamic, and physicochemical properties and are produced in a reaction in the mitochondrial chain, encompassing the I and III Complexes, after 4 steps of reduction of 1 electron from an oxygen molecule. In this reaction, the species that are called reactive oxygen species are the 3 primary ones, namely: superoxide anion (O2•‒), hydrogen peroxide (H2O2), and the hydroxyl radical (HO•) [71,72].
It is important to understand that Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2), which was discovered in 1994 [73], remains low until the moment when it is induced by an oxidative increase inside the cell, resulting in degradation of this protein and its movement to the nucleus. In principle, Nrf2 is a basic leucine zipper protein (bZIP) that belongs to the cap'n'collar (CNC) family of transcription factors, which also comprises NRF1, NRF3, and p45 NF-E2 and includes 605 amino acids [74-76]. It is interesting to note that Nrf2 exerts protective action against inflammation by inhibiting NF-kB [77]. In an oxidative stress scenario, Nrf2 is the most important regulator of phase II genes and binds to antioxidant response elements (ARE). Establishing a Keap1/Nrf2 complex in the cytoplasm makes it possible to affirm that the Nrf2/ARE pathway is organized by the negative regulator Kelch-like epichlorohydrin-related proteins (Keap1). Nrf2 phosphorylation is induced through Keap1 so that it forms heterodimers (small musculoaponeurotic fibrosarcoma oncogene homolog, Nrf2-Maf) after being translocated to the nucleus [78-80]. Some pathways indicate that phosphorylation is occurring, and they are the ones that activate Nfr2: PI3K (phosphatidylinositol 3-kinase), CK2 (casein kinase 2), protein kinase C (PKC) and MAP-Ks (protein kinases activated by mitogen) [81]. The control of many antioxidant pathways is carried out through Nfr2 target genes. One can mention, as an antioxidant pathway, the synthesis and regeneration of glutathione (GSH) and the synthesis and recovery of thioredoxin [82].
In its structure, Nrf2 has 7 regions called Nrf2-ECH homology domain (Neh), from Neh1 to Neh7. It is the CNC-bZIP region, surrounded by the Neh1 domain, that makes it possible to bind Nrf2 to DNA, through the dimerization mechanism with small musculoaponeurotic fibrosarcoma (Maf) proteins. The Neh2 domain is responsible for allowing the interaction of Nrf2 with the Keap1 protein, in addition to containing two dragons, DLG and ETGE, of high and low affinity, respectively. Furthermore, for the deterioration of Nrf2 to occur through the action of Keap1-dependent ubiquitin, the role of Neh2 becomes fundamental since the lysine residues present in it participate in the degradation process. The Neh3 domain, positioned at the carboxyl terminus of Nrf2, is responsible for modulating the transcriptional activation of the DNA-binding protein chromo-ATPase helicase (CHD6), which is involved in the transactivation of ARE-dependent genes. In which cAMP (CBP) interacts with the binding protein, transactivation activity involves the Neh4 and Neh5 domains. The Neh6 domain has DSGIS and DSAPGS, which are involved in the β-transducin repeat (β-TrCP), which is a component of proteins that are associated with the negative control of Nrf2 through SCF/ β-TrCP. Finally, the Neh7 domain is related to the retinoic acid receptor α (RARα), which inhibits the expression of the Nrf2 target gene [83-88].
As previously mentioned, ARE are the sequences present in cytoprotective proteins that are expressed through the induction of compounds that cause oxidative stress linked to Nfr2, so that they represent a fundamental antioxidant in the body. ARE are also called Electrophilic Response Element (EpRE), represented by the following sequence: 5´-gagTcACaGTgAGtCggCAaaatt-3´ (capital letters mean essential nucleotides) [87,88]. The ARE pathway has some downstream effectors that are worth highlighting: the detoxification system that includes Phase 1 (oxidize/reduce), Phase II (conjugation enzymes), and Phase III (drug efflux carriers) [89]. Given the relevance of the ARE, it can be stated that its activation triggers the manifestation of enzymes, such as heme oxygenase 1 (HO-1) and NAD(P)H: quinone oxidoreductase 1 (NQO1) [90]. HO-1, in its role as an antioxidant enzyme, is the one that Nrf2 induces most, a fact that guarantees an antioxidant/oxidant balance in the cellular environment in the event of a harmful process [91]. Not only Keap1, but certain other factors, such as histone acetylation, microRNA (miRNA) regulation, and DNA methylation, control the Nrf2/ARE pathway [92]
Given the fact that the Nfr2 pathway is activated mainly as a result of oxidative stress involving ROS and ARE inducers, we can state that these inducers are endogenous (such as NO and H2O2), exogenous (such as quercetin and oleanolic acid), therapeutic (such as nimesulide), phytochemicals (such as genistein and curcumin) and environmental agents (e.g. paraquat and some metals) [93,94]
It is also essential to highlight the relevance of Keap1, which has the following functional domains: Kelch or DGR, NTR, CTR, bric-a-brac (BTB), and intervening region (IVR). Through two Kelch domains, Keap1 is established as a homodimer in which each dimer binds to an Nrf2 molecule, with one binding site having high affinity (ETGF motif) and the other having weak affinity (DLG motif). These two motifs are found in the Neh2 domain of Nrf2. It is through the DRG domain that Keap 1 and Nfr2 are linked. Furthermore, the E3 ubiquitin ligase complex (Rbx-1) binds to Keap1 through the action of cullin-3, which is an adapter. Because of this process, the 26S proteasome directs Nfr2 to ubiquitination and consequent destruction [74,78,84,95]. It is interesting to note that the manifestation of the target gene occurs as a result of Nrf2 nuclear translocation, and three cysteines residues present in Keap1 (Cys273, Cys151, and Cys288) play a central role in this process [88,96]. Given that Nrf2 is degraded, it is necessary to point out the two ways: Keap1-independent degradation (which occurs in situations of oxidative stress) and homeostatic Keap1-dependent degradation [97]. For the Keap1-independent Nrf2/ARE pathway to be activated, some factors also play a fundamental role, such as AMP-activated protein kinase (AMPK) and protein kinase C (PKC) [98].
When analyzing the relationship between Keap1 and Nrf2, it is possible to state that the most recognized model is the “hinge and latch.” In this scenario, there are two reasons at work: ETGE, which, to attach Nrf2 to Keap1, functions as a "hinge," and DLG, which acts as a type of "latch." Combining these two motifs stimulates oxidation, reducing the amount of low-affinity DEG and preventing the ubiquitination of Nrf2 and its consequent degradation. As a result, there is an accumulation of Nrf2, which inserts into the nucleus [98-101]. Throughout the process in which the stress-inducible protein p62 activates Nrf2, the DLG latch is disjointed by inhibitors of the Keap1-Nrf2 relationship [102].
Under conditions of oxidative stress, Nrf2 is neither ubiquitinated nor proteasomal degraded, which results in an abundance of this compound through de novo protein synthesis and consequent deactivation of the E3 ligase adapter activity, promoting the translocation of Nrf2 to the nucleus from the moment its quantity exceeds that of Keap1. Musculoaponeurotic fibrosarcomas (sMaf), such as MafG, MafF, and MafK [103], proteins assemble with Nrf2 to create transcriptionally active heterodimers. In this context, in the promoters of many target genes (involving detoxification enzymes and antioxidants), ARE are identified by the NRF2-sMaf heterodimer [104-106].
Given the relevance of Nrf2 for numerous processes in the body, it is important to highlight its relationship with neuroinflammation and neurodegeneration. Firstly, it is essential to point out that Nrf2 is expressed in the central nervous system (CNS), which involves cells such as microglia and neurons [107]. Microglial cells play a central role in neuroinflammation, and through two distinct activation pathways, they change from the M0 state (resting state) to the M1 or M2 state (activated state). The M1 state is pro-inflammatory and provides pro-inflammatory cytokines (including tumor necrosis factor-α (TNF-α) and IL-6), increasing the amount of tissue damage through the generation of ROS [66,88,108]. Given this, increased levels of oxidative markers and signaling, in addition to constituent parts of damaged cells, could be seen in patients with diseases such as Parkinson's disease (PD), Multiple sclerosis (MS), and Alzheimer's disease (DA) [76,109]. Therefore, activation of the Nrf2-ARE pathway induces the action of antioxidant proteins and detoxifying enzymes, which defend the body from inflammatory damage [110]. In this scenario, Nrf2 has activators that can neutralize the injuries caused by ROS that promote neurodegeneration [111].
4. Impact of the NLR (Nucleotide-binding Domain and Leucine-rich Repeat Containing) Family Pyrin Domain Containing 3 (NLRP3) Inflammasome on Neuroinflammation: Exploring a Promising Therapeutic Target for Neuroinflammation
Human beings have two forms of immune system defense against pathogens: innate and acquired. In this scenario, some molecules play a central role: pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP). PAMPs are nonspecific microbial molecules that pattern recognition receptors localize and detect, in addition to being fundamental for maintaining the pathogenicity of a group of pathogens. DAMPs can be a molecule, no matter which one, that is exposed during any cell injury or damage process [112-114]. It is interesting to note that mitochondria are responsible for producing energy in the form of ATP by the cell, so that, consequently, they generate reactive oxygen species (ROS). Given that ROS releases cells from damaged mitochondria and this process releases organelles with some damage, it can be assumed that some mitochondrial DAMPs are released extracellularly. In this context, immune system cells are activated by inflammatory activation, releasing more mitochondria and creating a cyclical process that increases inflammation [115].
Furthermore, it is essential to highlight the role of pattern recognition receptors (PRRs), which are responsible for recognizing PAMPs and are distributed both on the cell membrane and in the cytoplasm. Activation of PRRs promotes the generation of pro-inflammatory cytokines and type I interferon (interferon-α and interferon-β), which were stimulated by downstream signaling cascades. The PRRs found in the membrane are mainly formed by Toll-Like receptors (TLRs) and c-type lectin (CLRs). The PRRs, which are in the cytoplasm, are made up, in particular, of the NOD-Like receptors (NLRs), which play a key role in inflammation and are constituted by C-terminal leucine-rich repeats (LRR) and nucleotide-binding domain (NBD/NOD/NACHT) [116-119].
Firstly, it is interesting to understand that, in 2002, Dr. Jurg Tschopp accomplished the brilliant feat of detecting and identifying Pycard/ASC, caspase-1, caspase-5, and NALP1, which are characterized as a complex capable of activating caspases, in addition to the pyrin domain containing 1), which has the function of maturating IL-1β, creating the concept of inflammasome [120]. In view of this, it is essential to highlight that inflammasomes are an oligomeric and cytosolic multiprotein complex and have the function of acting as mediators in immune responses activated by pathogens or tissue injuries, being activated after the identification of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), in addition to homeostasis-altering molecular processes (HAMPs). In this scenario, the activation of the cysteine protease caspase-1 is coordinated by inflammasomes, so the secretion of the pro-inflammatory cytokines IL-18 and interleukin-1β (IL-1β) occurs after proteolytic processing [121-123].
It is necessary to highlight that the best-determined and analyzed inflammasomes are NLRP3 (which contains 3 PYD and is part of the NLR family), NLPR1 (which is also a member of the NLR family and has a leucine-rich repeat of the nucleotide-binding domain), NAIP-NLRC4, pyrin, and AIM2 (which is Absent in Melanoma 2) [124-126].
In this scenario, there are two types of inflammasome: canonical, such as those from the NLR family (NLRP1, NLRP3, NLRC4) and AIM2; and non-canonical ones, which are composed of Malt1, CARD9, Bcl-10, caspase-8, and ASC, and it´s commonly dependent on caspase-4 or caspase-5 [122,123,127]. The NLR family has 3 partitions: pyrin domains (PYDs) or an N-terminal caspase recruitment domain (CARD) and an NOD that is centrally located and surrounded by C-terminal leucine-rich repeats (LRRs) [128].
Following this line of reasoning, the canonical pathway occurs when LRRs are related to the NACHT domain and prevent the generation of inflammasomes when there is no presence of DAMPs and PAMPs stimulation. When PAMPs or DAMPs are identified by LRR, the adapter protein ASC relates to the Pyrin-Pyrin structural domain and is attracted through the NLR protein. After this, the CARD-CARD domain allows the pro-caspase-1 effector domain to join the ASC and overlap into an inflammasome, causing oligomerization and consequent formation of an inflammasome complex that contains a heptamer. Of equal importance, it is necessary to point out the non-canonical inflammasome pathway, which depends on caspase-4/5/11. The transcription of pro-IL-1β and pro-IL-18 occurs when lipopolysaccharide (LPS) from Gram-negative bacilli is recognized by TLR4, which promotes the activation of NF-kB. The secretion of pro-caspase-11 is related to this process through the expression of type 1 IFNs caused by Trif/IRF3 [116].
With the advancement of studies on NLRP3, it is essential to highlight that its discovery is linked to its expressions in the immune system, such as in TCD4+ and TCD8+ cells, and a mild expression in other tissues of the immune system, in the thymus and the lymph nodes, in addition to neurons, myeloid cells, and muscle cells [129,130]. The NLRP3 inflammasome has three components, namely: caspase-1, which is characterized as an effector that promotes the maturation of IL-1β and IL-18, which are inflammatory cytokines; the NLRP3, which is the intracellular sensor; and the ASC (apoptosis-associated speck-like protein containing a CARD), which is an adapter [131-133]. NLRP3 has three domains, namely: NACHT, the central oligomerization domain, and NOD, the binding domain, which is related to ATPase activity; the amino-terminal pyrin domain (PYD), which is related to protein-protein interaction; and a carboxy-terminal leucine-rich repeat (LRR) domain [134,135].
There are three mechanisms of NLRP3 activation: canonical, non-canonical, and alternative pathways. However, before canonical activation, the NLRP3 inflammasome must be prepared to control the expression of components present in the inflammasome, such as NLRP3 itself, pro-IL-1β, and procaspase-1 [136]. In the first stage, which is called the priming stage or signal one and requires activation of myeloid differentiation primary response protein (MyD88) [137], certain PAMPs and DAMPs relate to pattern-recognition receptors (PRRs), such as NLRs, NOD, or TLRs. It promotes an intense induction and transcription of NF-kB, which causes an increase in the expression of the NLRP3 protein. After this, the deubiquitination of NLRP3 and the ubiquitination and phosphorylation of the ASC protein occur, which allows the assembly of the inflammasome. Furthermore, this step is also responsible for inducing post-transductional modifications (PTMs) of NLRP3, which make NLRP3 stable in a state without activity and self-suppressed, but with an appropriate signal [117,131,138-140]. Following the activation step, the NLRP3 oligomer, pro-caspase 1, and ASC are prepared to structure a complex during oligomerization [141]. This phase is known as inflammasome assembly or signal 2, and it is activated by certain molecular and cellular stimuli such as PAMPs, DAMPs, ATP, and ion influx. In this scenario, the pro-inflammatory cytokines IL-18 and IL-1B and the pyroptosis effector protein, Gasdermin-D (GSDMD), are cleaved by the pro-caspase-1 enzyme so that IL-18 and IL-1B are released extracellularly after the N-terminal fragments of GSDMD create pores in the plasma membrane. It is also important to point out that ionic and osmotic flow occurs through these pores, and the influx of calcium (Ca2+) and efflux of potassium (K+) promote cell turgidity so that the influx of Ca2+ releases cellular vesicles and the efflux of K+ promotes an intensification of inflammasome activation [142-147]. Furthermore, the adaptive immune response is induced by IL-18 and IL-1B [148]. In humans, caspase-4 triggers the non-canonical response, which occurs as a reaction after Gram-negative bacteria infect the organism [149]. Unlike the previous pathways, in the alternative path, LPS stimulation induces the maturation and consequent secretion of IL-1, in addition to the activation of caspase-1. Furthermore, this pathway requires Syk activity, caspase 4/5, and Ca2+ flux influenced by LPS and mediated by CD14/TLR4 [138].
Given the working mechanism of NLRP3, it is possible to state that it has an intrinsic relationship with neuroinflammation and neurodegenerative diseases. The activation of NLRP3 inflammasomes in the central nervous system is related to misfolded proteins and amyloids where, in pathological situations, these proteins have the role of being a signaling molecule that initiates the transcription and expression of NLRP3 and IL-1β, producing a cascade inflammatory character and positive feedback [150]. Studies have shown that patients with Parkinson's disease (PD) have, in their cerebrospinal fluid (CSF), high concentrations of IL-1β and IL-18, in addition to upregulated gene expression of caspase-1, ACS, and NLRP3, as well as increased protein expression of IL -1β, caspase-1 and NLRP3 in cells with only one nucleus in peripheral blood [151,152]. In this sense, the activation of microglial inflammasomes stimulated by α-synuclein and TLR is related to the progression of Parkinson's disease [153,154]. Alzheimer's disease (AD) is also related to the high production of caspase-1, IL-18, and IL-1β for the activation of NLRP3 inflammasome induced by Aβ in microglia during the positive feedback loop, which is produced during the systemic neuroinflammation [155-157]. It is interesting to note that a decrease in Aβ level and an improvement in memory impairment of APP/PS1/NLRP3 -/- mice is related to a knockout of NLRP3 [158]. Therefore, the NLRP3 inflammasome has gained space since its relevance in neurodegenerative and neuroinflammatory diseases gained importance in new research [159-161].
5. JAK/STAT: An Evergreen and Unconventional Pathway in Neuroinflammation and Neurological Dysfunctions
Firstly, it is essential to understand that Janus Kinase or “just another kinase” (JAK) is constituted as a family of transmembrane tyrosine kinases present inside the cell, involving tyrosine kinase 2 (TYK2), JAK1, JAK2 and JAK 3 and, through the action of the Signal Transducers and Activators of Transcription (STAT) pathway, transduce information mediated by, mainly, type I/II cytokines, also phosphorylating signaling molecules carrying Src-Homology 2 (SH2) domains. STATs, which have seven constituents in their family (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6), are characterized as cytoplasmic proteins that, at the end of activation, are recruited and phosphorylated at the C-terminal area in the cytoplasm [162,163]. In this scenario, JAK/STAT has the role of being the pathway responsible for transducing extracellular signals into transcriptional information and adjusting physiological processes [164]. The STAT protein consisting of an N-terminal domain is STAT1, the coiled-coil domain makes up STAT3 and STAT1, the DNA-binding domain forms STAT1, STAT2, and STAT3, Linker is the constituent domain of STAT1 and STAT3, and Src-homology 2 (SH2) that makes up STAT1, STAT3, STAT4, and STAT5a/b, in addition to the C-terminal transcriptional activation domain (TAD), responsible for forming STAT1, STAT2, STAT3, STAT4, STAT5a/b and STAT6 [164].
The fundamental issue to understand about the JAK/STAT pathway is that its regulatory mechanism can be positive and negative. If positive, the following cytokines are involved: growth factors (GFs), interferons (INFs), interleukins, colony-stimulating factors (CSFs), chemokines (CFs), and tumor necrosis factors (TNFs). If the regulation is negative, the protein inhibitors of activated statistics (PIASs), suppressors of cytokine signaling (SOCSs), and protein tyrosine phosphatases (PTPs) are the regulators involved [165,166]. Interestingly, the development of autoimmune diseases is closely related to the suspension of the JAK/STAT signaling pathway, in which the SOCS protein plays an essential role [167,168]. SOCS1 impacts the induction of HLA-DR and CD86 expressions in dendritic cells, so this variant has been characterized as a genetic sign of Multiple Sclerosis [169]. Furthermore, STAT1 and STAT4 act in Th1-type immune reactions in a crucial way, as STAT3 and STAT6 are closely related to Th2 and Th17-type immune responses [170].
More than 50 different cytokines carry out the canonical activation of the JAK/STAT pathway. At its beginning, an extracellular ligand, usually a cytokine, interferon, or growth factor (GF), binds to its receptor, promoting its dimerization. After this, the receptor subunits are targeted and undergo oligomerization, and through the Box1 and Box2 cytokine domains, a homologous area close to the cell membrane joins the region actively functioning in JAK. It is known that numerous activation receptors can bind to a type of JAK, as can be seen in the case of JAK3, which IL-2 and IL-4 activate. JAK phosphorylation is induced by catalyzed tyrosine residues, which also allow the creation of anchoring areas for wandering amino acids. Finally, STAT form homo- and hetero-dimers and translocate into the nucleus, binding sites that are originated by the activation of JAK, using the SH2 structural domain, resulting in the transcription of different other target genes [171-175]. Then, the tyrosine residues of STAT elements are phosphorylated by JAKs, providing STAT activation [176].
A possible relationship between STAT and inflammation in NK and CD8+ T cells is between the potential response to cytokines and their correspondence with the variable amounts of STAT1 and STAT4, which are determining factors. However, changes in the amount of STAT5 affect the functioning of T cells [177].
The proliferation and activity of cells present in the brain, such as neurons and astrocytes, are regulated by the JAK/STAT pathway. The growth of neural stem cells (NSCs) does not stop during brain development and can be observed in neurogenic areas, including the olfactory bulb's subventricular zone (SVZ). The JAK/STAT pathway regulates NSC growth so that cytokines, especially IL-15, are expressed by adult NSCs in the SVZ, stimulating the action of STAT1, STAT3, and STAT5. Interestingly, blocking JAK prevents NSC multiplication [178].
In this scenario, neurodegenerative diseases are also strictly linked to the JAK/STAT pathway. Alzheimer's disease is related to a dysfunction in the JAK2/STAT3 pathway, in which nicotinic acetylcholine receptors activate this pathway and lower the Aβ neurotoxicity [179-182]. The pathogenesis of AD was also related to the inactivation of STAT3, in which its activated/phosphorylated configuration (p-STAT3) had a considerable decline in hippocampal neurons of patients with the pathology compared to the control group [183]. Studies have shown that, in mice, high amounts of Aβ intensify tyrosine phosphorylation and the transcriptional functioning of TYK2, which is related to a high phosphorylation of STAT3 in brains with Alzheimer's disease. Furthermore, when inhibited, JAK/STAT3 signaling blocks microglia and astrocytes' activation in animal neurodegeneration prototypes [184]. The results of Porro et al. demonstrated that inactivation of the JAK/STAT pathway interrupted the amount of secretion of pro-inflammatory cytokines, promoting a rise in the level of IL-4 and IL-10, causing a change in the protective phenotype, attenuating the inflammation present in neurodegeneration [185].
Through the activation of the JAK/STAT pathway, the polarization of pro-inflammatory macrophages becomes possible, having an intrinsic relationship with IFN-γ, so that a deficiency in this interferon, presented by mice, demonstrated a reduction in the amount of neuron loss present in the substantia nigra through the induction of de-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), that is also related to the Parkinson’s Disease [186]. In their study, Qin et al. [187] demonstrated the relationship between the JAK/STAT pathway in microglial activation, its vital importance in the recruitment of cytokines and chemokines, and its function in Parkinson's disease. In research, possible therapy using a JAK/STAT pathway blockade through the JAK1 and JAK2 inhibitor, AZD1480. The action of α-SYN activated the JAK/STAT pathway in macrophages and microglia, and the role of AZD1480 was to block the major histocompatibility complex Class II, which is induced by α-SYN and, consequently, decreases the activation of STAT1 and STAT3 due to inflammatory gene inhibition in macrophages and microglia. The results showed that therapy with AZD1480 promoted an interruption of neuroinflammation stimulated by α-SYN, preventing microglia activation, the passage of CD4+ T cells and macrophages, and the formation of pro-inflammatory cytokines and chemokines. Notably, in vivo, the JAK/STAT pathway disruption prevented the deterioration of dopaminergic neurons. Therefore, Qin et al. demonstrated the relationship between neuroinflammation and neurodegeneration and the JAK/STAT pathway suppression. In this context, inhibition of the already activated JAK/STAT pathway may be a positive alternative for treating Parkinson's disease [188].
6. Neuroinflammation and Microglial Activation: Charting the Path Forward Alzheimer's Disease, Parkinson's Disease, and Multiple Sclerosis
At the beginning of the 20th century, the Spanish neuroscientist Pío del Río Hortega identified what he named "microglia," which, at the time, he believed to be a new species of cell present in the brain capable of carrying out phagocytosis. Currently, studies have identified that microglia come from erythro-myeloid progenitors (EMPs), also known as C-KIT+/ CD41+, present in the yolk sac and multiply from embryonic day 8 (E8) [189]. Microglial cells are constituted as macrophages found in the brain to control neural activity and homeostasis and communicate with other cells of the central nervous system (CNS), such as astrocytes, neurons, and oligodendrocytes [190-192].
It is crucial to understand that between 5 and 10% of cells in the central nervous system (CNS) are microglia, and these have different morphologies that are closely related to their function and the local environment, such as amoeboid, medusa, rod, and branched [193]. It is essential to understand that, in a scenario of damage or injury, microglia begin to have an amoeboid morphology with larger cell bodies and smaller branches [194,195]. Furthermore, microglial cells work with the blood-brain barrier (BBB) to defend the brain against damage and attack pathogens [196].
When installed in the brain, self-renewal is how microglia maintain themselves, requiring the action of transcription factors, such as interferon regulatory factor 8 and PU.1, and cytokines, including colony-stimulating factor (CSF)-1 and interleukin (IL)-34 [197]. For microglia to develop and be preserved, some components are essential, namely extracellular signal-regulated kinases (ERK), which allow the multiplication and maintenance of microglia, and the colony-stimulating factor 1 receptor (CSF1R), which configures as a tyrosine kinase receptor capable of propagating intracellular signals that have a central role in microglial multiplication and CSF-1 activates it and (IL)-34 [198,199]. It is essential to point out that an accelerated depletion of microglia occurs after CSF-1R blockade by pharmacological means [194]. What differentiates microglial cells from macrophages in the adult brain is the fact that microglia are capable of expressing P2RY12/P2RY13, TMEM119, and CD11B, having a low expression of CD45, while circulating macrophages reveal a high expression of CD45 and CD11B [200].
By expressing more than 100 sensory genes, microglia become capable of establishing a link with the microenvironment in which they are located. Among these genes, we can mention complement system receptors, which help them interpret these signals to act as necessary, in addition to Tlr2, P2yr12, and Siglech [201].
There are some mechanisms for regulating microglial activity that play a fundamental role and deserve to be highlighted. Pattern Recognition Receptors (PRRs) are related to regulating innate immunity, recognizing PAMPs and DAMPs. Cytokine receptors are associated with the production of several cytokines, such as transforming growth factor-β (TGFβ), tumor necrosis factor-α (TNFα), and interleukins (ILs), since, in the brain, microglia are the largest producer of cytokines. Chemokine receptors are linked to G proteins (GPCRs), called CCL1 receptors (CCR1), CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CXCR1, CXCR2, CXCR3, CXCR4 and CXCR5. Neurotransmitter receptors participate in the regulation of interactivity between neurons and microglia through the recognition of adenosine triphosphate (ATP) and its metabolites, and it includes cholinergic receptors, glutamate receptors, adrenergic receptors, purinoceptors, and cannabinoid receptors. There is also expression in microglia of myeloid cell receptor-2 (TREM2), which is a receptor present on the surface of cells that is part of the extensive family of Immunoglobulins (Ig) and is triggered through the recognition of pathogens by Toll-like receptors (TLRs), so that TREM2 establishes a relationship with the adapter DNAX-protein- 12 activation (DAP12), present in the internal part of the cell. In addition to these receptors, others participate in this regulation, such as phosphatidylserine receptors, scavenger receptors, and Fc receptors [202-205].
Microglial cells are classified by a macrophage-derived system that can sometimes be simplistic: M1 (classical) and M2 (alternative). First, M1, which is activated by interferon-gamma (IFN-γ) and lipopolysaccharide (LPS) and has a pro-inflammatory and neurodegenerative profile, is related to the formation of nitric acid, proteases and pro-inflammatory cytokines ([IL-6], interleukin [IL]-1β, TNF-α and IL-12) through the expression of NADPH oxidase, that produces inducible nitric oxide synthase (iNOS), and also through the action of the transcription factor signal transducer and activator of transcription 1 (STAT1), which acts via Janus kinase (JAK) 1/JAK2 signaling. In addition, there is also expression of the major histocompatibility complex II (MHC II), the costimulatory molecules CD45 and CD47, integrins such as CD11b and CD11c, and Fc receptors, which can intensify neural injuries and have harmful impacts on neurodegenerative diseases. In the case of M2, with an anti-inflammatory and healing profile, it has the expression of anti-inflammatory cytokines (TGF-β, IL-4, IL-10, and IL-13), Th2 recruiting factor CC chemokine ligand 2 (CCL2), arginase 1 (ARG1), CD206 and others. M2 microglia are responsible for generating fibroblast growth factor (FGF), colony-stimulating factor-1 (CSF-1), insulin-like growth factor-1 (IGF-1), in addition to neurotrophic growth factors such as brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF1). In this context, it is interesting to highlight that the "classically activated" M1 phenotype can have its polarization blocked in microglia after a spinal cord injury caused by the deletion of the TMEM16F protein, which acts as an ion channel that depends on Ca2+ and phospholipid scramblase [192,206-212]. Interestingly, some microglia, as they synthesize pro-inflammatory and anti-inflammatory markers, have an intermediate phenotype. In this context, IL-4 and IL-13 stimulate M2a-type microglia, which incites CD206, arginase, and chitinase 3-like 3. On the other hand, the M2b phenotype is characterized as an intermediate state of microglia due to the secretion of restorative and inflammatory markers. Furthermore, once the inflammatory issue is resolved, M2c encourages tissue remodeling and repair, forming CD206, transforming growth factor beta (TGF-β) and CD163 [213,214].
Also noteworthy is the ability of microglia to express CD11b, which is also known as a marker “priming”, such as MHC-II [215], Transmembrane Protein 119 (TMEM119), Sal-like protein 1 (SALL1), Purinergic Receptor P2Y12 (P2Y12) and Purinergic Receptor P2Y13 (P2Y13), involved in the immune response of the adult brain. Furthermore, it is interesting to note that microglia constantly investigate the CNS to have a relationship with processes such as protein clustering, phagocytosis, secretion of soluble factors, and refinement of the neural circuit. In a scenario of CNS damage, microglia are activated in stages and a complex manner, responding through the action of signaling molecules, such as chemokines and cytokines, to repair the tissue [203,204,216].
It is essential to understand that, to protect the vigilant phenotype of microglia and regulate their activity, the CD200-CD200R and CX3CL1-CX3CR1 axes are vital. However, as a result of aging, there is a reduction in communicability through these axes, a fact that is related to the decrease in the expression of CX3CR1 and the neural ligands CX3CL1 and CD200, creating a pro-inflammatory scenario [201,217,218]. Following this line of reasoning, in the brain of an elderly person, chronic inflammation promotes immunosuppression that damages the distribution pattern of microglia and causes a severe reduction in branching [218]. Interestingly, only in the white matter are the number of microglia positive for MAC-2, which is an indicator of subpopulations of microglia that perform phagocytosis, positively regulated, which does not occur in the gray matter. Therefore, certain elderly people, called "Superagers," had fewer microglia in the cortical white matter and, consequently, had better cognitive function [219].
Alzheimer's disease (AD) is the multifactorial neurodegenerative cause of dementia disease with the highest incidence, and this fact can be observed in the prediction that in the year 2050, in the world, the number of patients with AD will be 139 million [220]. The enzymes beta (β) and gamma (γ)-secretases are responsible for cleaving the amyloid precursor protein (APP), causing the aggregation of amyloid β-peptide (Aβ) that will not only form Aβ plaques, but also compacts them and influence their growth, and, from the moment these plaques form in the brain, their strictly close contact with microglia begins, making them reactive [221,222]. In this context, in AD, there is an aggregation of β-amyloid plaques (Aβ) on the outside of the cell, as well as an accumulation and intraneural clustering of hyperphosphorylated tau protein (pTau), which is also called neurofibrillary tangles (NFTs), being that these facts are related to the decrease in the number of neurons and, consequently, in synapses in the cortical and subcortical areas of the brain [223]. In this scenario, through the action of Aβ, microglia secrete pro-inflammatory cytokines and neurotoxic elements, such as IFN-γ, TNF-α, IL-1, and IL-6, in addition to ROS, so that TNF-α is configured as a of the most relevant cytokines in AD [224]. Moreover, microglial activation in AD has two distinct consequences. The first is related to the clearance and phagocytosis of Aβ. In contrast, the second, through the kB pathway, promotes the release of many inflammatory mediators, causing neuroinflammatory responses [225]. It is essential to understand that, with genetic studies that recognized more than 25 genetic loci strictly related to the possibility of developing AD, most of the ordinary genetic variants (Sp1l, ApoE) or even the uncommon ones (Cd33, Trem2) act by encoding proteins which are mainly or solely expressed in microglia, so that the transition to disease-associated microglia (DAM) has an intrinsic relationship with these genes and stimulating the receptor expressed on myeloid cells 2 (TREM2) and TYRO protein tyrosine kinase binding protein (TiroBP) [226-228]. According to a study by Rangaraju et al., the DAM has divisions, namely pro-inflammatory, anti-inflammatory, and homeostatic phenotypes. The genes Cst2, CD44, and Nampt, belonging to the pro-inflammatory DAM, were discovered in the human brain proteome, revealing a correlation with AD [229]. It is essential to understand that the secretion of tau protein is carried out by microglia, which intensifies neuronal deterioration, in addition to contributing to the senescence of neighboring microglia, causing an indefinite repetition of these events, which increases the risk of developing AD [230].
Microglia have an intrinsic relationship with AD, as can be analyzed in numerous studies, including that by Sun et al. [231], who observed 1542 genes that were differentially expressed in AD, covering changes characteristic of the state of microglia and AD disease phase, which relate to 12 microglial transcriptional states that have been observed in 443 individuals. By investigating post-mortem brain samples at an advanced age from 217 patients with AD and 226 individuals considered as controls, the transcriptome of 152,459 unique microglial nuclei was analyzed in 6 brain areas. The first step was to collect 174,420 immune cells to perform in silico brain screening of single-nucleus RNA-seq (snRNA-seq) datasets using previously known marker genes (STAR Methods), which formed 16 clusters, Among these, 12 clusters are microglia (0-8 and 10012) and are marked by CD74, CSF1R, and C3 so that they were determined as different microglial states based on their molecular signatures and functionalities. The homeostatic microglia (MG0) and its intense expression of homeostatic markers CX3CR1 and P2RY12 were subject to annotation and observation, as well as the neuron surveillance microglia (MG1), which has a fierce expression of neurotransmitter receptors, and MG3, which demonstrated great improvement in ribosome biogenesis. Furthermore, three inflammatory states were described (MG2, MG8, and MG10), with an intense expression of both cytokines and cytokine receptors (IL1B, IL4R, IL17R, IL15, IL10RA, and CCL3), which are closely related to immune responses, highlighting signaling through the action of cytokines and the structuring of the inflammatory reaction. From this, the study analyzed 423 individuals considered controls, with early and late AD in the prefrontal cortex, evaluating the statistical significance of the divergences in cellular fractions. It was demonstrated that in AD, MG4, and MG8 presented fractions with a considerable increase, unlike MG1, which presented a decrease in the fraction. Furthermore, using IBA1 pan-macrophage markers used in immunohistochemistry, it was possible to observe that individuals with AD have nuclei that express P2RY12 with comparatively smaller fractions. With the analysis of the increased expression of PPARG evidenced in snRNA-seq data, an increased amount was observed in the fraction of PPARG+P2RY12+ cells in AD patients. Linked to this is an enrichment of FOXP1+LRRK2+ microglia in this group, demonstrating an MG8 inflammatory state. The discoveries made through in situ hybridization confirm that inflammatory states, together with the state of lipid processing, establish a correlation with diseases.
In the study by Crapser et al. [232], there is a focus on the condensed extracellular matrix (ECM), which is called the perineuronal network, which is linked to the regularization of plasticity in conjunction with astrocytes and synaptic terminals, establishing the modern “tetrapartite synapse.” The formation of PNNs occurs mainly around parvalbumin (PV) + GABAergic interneurons present in all brain areas during the process of closing the crucial plasticity interval, allowing a "locking" of proximal synapses and ensuring harmony and preservation synaptic, which happens from the moment in which microglial development is responsible for completing synaptic pruning. After PNN ablation, the reconsolidation and regeneration of remote memories are impaired since memory fragments that were recently formed have the potential to affect the integrity and regeneration of previously assimilated information. In addition, the abnormal functioning of interneurons related to cognitive impairments in AD explains the alteration of the structure of PNNs, which modifies not only the transmission of synapses but also the constitution of the ion channel or neurotransmitter receptor of synapses. In the study, an aggressive sample of the disease was used in mice of the 5xFAD type, and the results were confirmed by a long immunohistochemical (IHC) investigation of human cortical tissue, and, in both, there was essentially a loss of PNNs in the brain. It was also discovered that 5xFAD brain areas with a high incidence of the disease present an accelerated PNN deficit, and the decrease in the number of PV + interneurons occurs following the impairment of PNN regions and the integrity of the structure. It is essential to highlight that networks that present abnormal morphology in the AD brain are related to microglial activation so that PNN staining demonstrates colocalization in mouse and human microglia. Through the action of the selective colony-stimulating factor 1 receptor (CSF1R) inhibitor PLX5622, the study reveals that the chronic reduction of microglia in the periods following the evolution of the Aβ plaque in 5xFAD mice, as well as during its progress, prevents, efficiently, the PNN loss. Therefore, it is concluded that the change in microglia due to the disease helps the occurrence of plaque-dependent depletion of PNNs in the human brain and in mice with AD.
A study by Cheng et al. [226] aimed to present the relationship between microglial activation and the expression of protein 2 of the calcium homeostasis modulating family (Calhm2), which is elevated in 5 × FAD mice, carrying 5 mutations in the AD gene. Firstly, it is essential to point out that intracellular calcium levels are increased by the action of Aβ, resulting in the activation of the NLRP3 inflammasome in microglia. In this sense, microglial activation is instigated by interferon-γ (IFN-γ) and lipopolysaccharide (LPS), so nicardipine, a calcium channel blocker, inhibits such activation. Furthermore, proteins from the calcium homeostasis modulating family (Calhm, Calhm1, Calhm2 and Calhm3) have gained space in current science. For example, we can observe Calhm1, which has an essential influence on forming Aβ, calcium homeostasis, and the fragility of neuronal cells that suffer the toxic effects driven by Aβ. Note also the correspondence between the incidence of AD and the P86L mutation of Calhm1. Linked to this, the study showed increased Calhm2 levels in mice with AD. This protein acts to regulate microglial inflammatory activation and calcium influx, and a decrease in Aβ deposition, dysfunctions in cognition, and neuroinflammation associated with AD result from both the conventional Calhm2 KO and the microglia-specific KO of Calhm2.
Parhizkar et al. [233] demonstrated that the single ε4 allele of Apolipoprotein E (APOE), as well as the sequential variants in the gene responsible for encoding the stimulatory receptor expressed in myeloid cells 2 (TREM2), have an intrinsic relationship with the probability of developing AD. The importance of Trem2 is widely discussed, including preserving the energetic mechanism, phagocytosis of Aβ fibrils, uptake of mortar cells, and chemotaxis. This fact can be observed in microglia that react to damage in the brain, in which ApoE and Trem2 are intensely upregulated. ApoE, a prominent component of amyloid plaques, can ensure their clustering and deposition. In the study, APPPS1 transgenic mice were used, with intra-hippocampal injections of brain extracts carrying Aβ being administered, to compare the results of Trem2 in more mature and Aβ plaques not experimentally seeded. Small animal positron tomography (μPET) was also used to produce longitudinal images to verify the process of microglial activation and amyloid depletion. The last step was to observe the loss of Trem2 activity and its relationship with the deposition of ApoE in amyloid plaques in mice and the gathering of microglia, as well as cases of AD in which there is a change in Trem2 coding. In AD situations without TREM2 variants, excessive concentrations of microglia were detected around amyloid plaques. At the same time, a much lower frequency was observed in those AD patients in which TREM2 variants present loss of function, and the microglia that cluster around amyloid plaques promote an increase in ApoE expression. It was recently discovered that those mice that exhibit either a paucity of ApoE or express the p.R47H TREM2 variant, which is related to AD, show a surprising decrease in plaque compaction. Therefore, Parhizkar et al., by demonstrating the action of microglia on amyloid plaque fibrillary, which varies with its activation state, allowed the study to have a high degree of clinical relevance, considering that longitudinal amyloid μPET catheterizes Itself as a readout for amyloid-based therapies.
Other studies have also linked the influence of the ApoE protein with microglial activation and activity, as can be seen in the research by Chernyaeva et al. [234], which demonstrated the interaction between the ApoE protein and the complement regulatory factor H (FH). In this scenario, neurotoxicity and clearance mediated by Aβ1-42 are influenced by the specific binding of FH and ApoE, a fact related to the complement activator C1q, which is located in Aβ brain plaques and related to the pathogenesis of AD.
The study by Meilandt et al. [235], together with the findings of Parhizkar et al. [233], indicates that the functioning of microglia that are dependent on Trem2 can limit plaque formation at an early stage of AD so that, in soluble Aβ models, this occurs through sequestration and degradation, in addition to an increased occurrence of Aβ absorption in plaque systems present during the course of the disease. This research investigated the function of Trem2 during microglial activation, neuronal dystrophy in the PS2APP species of β-amyloidosis, and plaque aggregation. Using Campbell-Switzer silver staining, age- and sex-related impacts of Trem2 deletion in excess plaque analyzed were observed in both female PS2APP;Trem2ko and PS2APP;Trem2ko mice. This research investigated the function of Trem2 during microglial activation, neuronal dystrophy in the PS2APP species of β-amyloidosis, and plaque aggregation. Using Campbell-Switzer silver staining, age- and sex-related impacts of Trem2 deletion in excess plaque analyzed were observed in both female PS2APP;Trem2ko mice and female PS2APP;Trem2ko mice. In PS2APP;Trem2koA mice, of all ages and of both sexes, microglial accumulation in the vicinity of the plaque and other forms of activation had an extreme reduction and, as a consequence, the plaques in the brains of these animals presented a more diffuse structure compared to those in PS2APP brains;Trem2wt. This research investigated the function of Trem2 during microglial activation, neuronal dystrophy in the PS2APP species of β-amyloidosis, and plaque aggregation. Using Campbell-Switzer silver staining, age- and sex-related impacts of Trem2 deletion in excess plaque analyzed were observed in both female PS2APP;Trem2ko mice and female PS2APP;Trem2ko mice. In PS2APP;Trem2koA mice, of all ages and both sexes, microglial accumulation in the vicinity of the plaque and other forms of activation had an extreme reduction and, as a consequence, the plaques in the brains of these animals presented a more diffuse structure compared to those in PS2APP;Trem2wt brains. In PS2APP;Trem2ko mice, the depletion of the proximal dendritic column of the plaque was accentuated, a fact that highlighted the relationship between the activation of Trem2-dependent microglia in the surroundings of the plaque and its defending capacity for neurons, differentiating itself from functions interceded by complement and by microglia that cooperate to deplete β-amyloid model synapses. In the study, superior cognitive functionalities in mice with β-amyloid pathology and slowed AD development in humans were related to increased PET signal for TSPO ligands, a "marker of neuroinflammation." The probes used in the clinic (18F-Florbetapir, 11C-PiB) demonstrate stronger labeling than molecular probes (X-34, Thioflavin S and methoxy-X04), which relates to mice with scarce Trem2 and their plaques that cause damage to proximal neurites, a fact that helps clarify the reasons why cognitive reduction correlates more efficiently with the reduction of synapses and tau pathology than with the identification of cerebral amyloid.
Studies by Wang et al. [236] corroborate the influence of TREM2 on AD through the protein tyrosine kinase SYK, demonstrating that the activation of microglia via SYK is not possible using the TREM2R47H variant, present in humans and related to the high probability and development of AD. Furthermore, microglia that exhibit an insufficiency of SYC were found unable to engulf Aβ plaques, intensifying disease development and behavioral-related damage. A path to immunotherapy was shown in the study, in which, in mice capable of expressing the TREM2R47H allele, an antibody against a receptor that activates SYK, called CLEC7A, was inserted systemically, resulting in possible new options for the treatment of AD.
In their discovery, Qiao et al. [237] highlighted the relationship between a greater probability of developing AD and the p.H157Y variant of TREM2, which is in the cleavage region of it is an extracellular domain and is also considered rare. By generating a new Trem2 H157Y knock-in mouse, using CRISPR/Cas9 technology, the study demonstrated that an improvement in synaptic plasticity and an increase in TREM2 elimination were caused by Trem2 H157Y and, in the existence of amyloid pathology, it was even responsible for increasing the speed of Aβ clearance, in addition to reducing not only the amyloid load but also dystrophic neurites, the toxic Aβ oligomer and gliosis in the subsequent phases of amyloid pathology. Therefore, the study demonstrated that Trem2 H157Y may be advantageous for reducing amyloid pathology and its toxicity, in addition to benefiting brain activities, and can negatively control neuroinflammatory pathways related to neurodegeneration.
In the research by Prakash et al. [238], the relationship between lipid droplets (LDs) produced by microglia, which occurs after exposure to Aβ, and their increase upon the approach of amyloid plaques were studied using the brain of a mouse model with AD 5xFAD and humans with AD. Microglia full of LD showed a deficiency in Aβ phagocytosis, with the generation of LD being subject to age and disease progression. Through unbiased lipidomic assessment, a considerable reduction in free fatty acids (FFA) was discovered, while long-chain triacylglycerols (TAGs) rose. In this sense, for the conversion of FFAs into TAGs to occur, the enzyme DGAT2 becomes necessary and is also responsible for promoting the generation of LD in microglia. The abundance of the DGAT2 enzyme in the cerebral microglia of patients and brains with 5xFAD is evident in the research, with microglial uptake of Aβ benefiting after pharmacological inhibition of DGAT2. Considering that lipids, during chronic inflammation as occurs in AD, induce cytotoxicity, it was discovered that such toxic molecules can be metabolized in TAGs by microglia, a type of defense not only for the microglia but also for neurons. Nevertheless, the structure and functioning of microglia are affected by this process. Therefore, the study generates evidence that could lead to a path in which DGAT2 inhibition is characterized as a means capable of boosting the protective characteristic of microglia in AD and other neurodegenerative disorders with abundant protein accumulation and LD deposition.
In the study by Kellogg et al. [239], the major histocompatibility complex I (MHC-I) is related to the regulation of the eradication of synapses linked to the development and pathology of tau in AD, presenting an increased expression during brain aging in mice and humans so that microglia are the origin of both classical MHC-I and non-classical MHC-I. In research, two antigen-independent MHC-I receptors gained prominence: paired immunoglobulin-like receptors type 2 (Pilrs) and receptors from the leukocyte immunoglobulin-like receptor subfamily (Lilrs). These receptors are induced in microglia in aging and AD processes and have ITIMs (immunoreceptor tyrosine-based inhibition motifs) and ITAMs (immunoreceptor tyrosine-based activation motifs), and, in the recognition cell, both are capable of inciting activation signals. Syc activity, which acts as a regulator of microglial phenotype, is activated by ITAMs and inhibited by ITIMs. The function of Syc, depending on the receptor, can be modulated through the union of Pilr and Lilr receptors with MHC-1, which may include ITIM or ITAM cascades. Through the MHC-I receptors and Pilr and Lilr family receptors found in microglia, MHC-I can control the microglial phenotype together with ITAM, ITIM, and Syc. Kellogg et al. also described a possible activity of microglial MHC-I, that would be related to p16INK4A, a canonical marker of senescence induced in microglia was discovered.
A recent study demonstrated a possible relationship between diet and the risk of developing AD. Velazquez et al. [240] investigated the vitamin nutrient choline, similar to vitamin B, in several foods in the human diet, such as chicken liver, eggs, and milk, and its involvement in the pathogenesis of AD. Choline has the function of donating methyl and preceding the formation of cell membranes, in addition to acting as a precursor of the neurotransmitter acetylcholine, which is reduced in the brain of AD patients and is responsible for putting the alpha7 nicotinic acetylcholine receptor (α7nAchR) into operation, also having the function of an agonist of Sigma- 1R (σ1R). Both receptors act in the CNS, controlling its immune response and, if uncontrolled, favor AD development. Over a while, female APP/PS1 and NonTg mice were fed a choline supplementation (Ch+) (5.0 g/kg choline chloride) or exposed to a controlled amount of choline (1.1 g/kg choline) and evaluated in the Morris water maze using neuropathological and spatial memory tests. Among the results, both a considerable decrease in the β-amyloid plaque burden and an improvement in spatial memory could be observed in APP/PS1 mice. The effects of the research demonstrated reduced amyloidogenic processing of APP, a negative structuring of α7nAch and σ1 receptors, and decreases in microglial activation linked to pathology. Therefore, the data found on Ch+ throughout life can bring premises to science, considering its relationship with a possible attenuation of the risk of AD.
Studies on Parkinson's Disease (PD) gained visibility in the scientific community, considering that, in 2020, in the world, more than 9 million individuals lived with the pathology, ranking second as the most common neurodegenerative disease [241,242]. Among the factors that are related to a greater risk of developing PD are advanced age, being male, having a family history of the disease, and even living in a rural area and drinking untreated water [243]. The main symptoms associated with the disease are apathy [244], pain [245], bradykinesia [246], cognitive impairment, and psychiatric changes [247], in addition to motor impairment, so tremor affects 75% of patients [248,249]. It is essential to understand that some genes are linked to the pathogenesis of PD, such as KAT8 and KANSL1, which are related to mitophagy, and PARKIN and Phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1), in which, if presented with mutations, they can be causes of autosomal recessive PD of early origin [250,251]. Other genes, such as leucine-rich repeat kinase 2 (LRRK2) and DJ-1 deglucase protein, in addition to the PINK1 gene already mentioned, control microglial activity in healthy physiological circumstances, which can involve inflammatory processes up to phagocytosis. Mutations in these genes, as occurs in PD, cause an increase in the number of inflammatory markers and, consequently, ROS increase, promoting, on the one hand, an impairment in the function of PINK1 and DJ-1, and, on the other hand, an improvement in the function of LRRK2 and PARKIN [252].
In pathology, dopaminergic neurons, present in the substantia nigra pars compacta (SNpc), degenerate gradually and for a long time, and the filamentous protein α-synuclein (α-Syn) makes up the protein inclusions that are called Lewy bodies [253]. It is essential to understand that α-synuclein can be presented as β-sheet-containing fibrils and plays the secondary role of a seed element for the aggregation of α-synuclein, with self-aggregation being a pathological function. In PD, a decrease in α-synuclein clearance is related to chaperone-mediated macroautophagy and autophagy, favoring the formation of α-synuclein inclusions. In cases where neurons overexpress α-synuclein, exosomes are released, which can provoke a premature reaction from microglia [254,255]. Furthermore, via the activation of inflammasomes, especially NLRP3, the secretion of pro-inflammatory cytokines, such as IL18 and IL1β, are stimulated by α-Syn aggregates, a fact that establishes a negative correlation with the development of PD [256,257]. α-Syn, in physiological situations, is also responsible for regulating the storage of dopamine in synaptic vesicles and, consequently, establishes a relationship with the expression and functioning of the vesicular monoamine transporter 2 (VMAT2) in neurons in the substantia nigra, corroborating its relationship with PD. In the interaction between α-Syn aggregates and VMAT2, a decrease in dopamine vesicular storage space would lead to a deficiency in synaptic transmission and, as an effect, the death of neurons [257,258].
The importance of α-synuclein (α-syn) and the cell-to-cell transfer of α-syn can be seen in the study by George et al. [259], in which microglia activated by IL-4 or LPS are capable of stimulating aggregation of α-syn, with an increase in the amount of pro-inflammatory cytokines, a fact that is related to the pathogenesis of PD.
It is necessary to point out that an increase in the functioning of the NLRP3 inflammasome causes a deficit in microglial autophagy in PD, leading to a change to a neurotoxic condition in microglia [260]. The study by Yan et al. [261] explored the relationship between the microglial NLRP3 inflammasome, the PARKIN gene, and PD. Through the neuronal expression of NLRP3, PARKIK polyubiquitination occurs and triggers neurodegeneration, so the objective of the research was to understand the function of PARKIK in the activation of NLRP3 using the overexpression of PARKIK in microglia following treatment with lipopolysaccharide (LPS). Three tests were performed on PARKIN knockout mice and wild-type mice to qualify motor activity: open field test, rotarod test, and pole test. It was then discovered that, in LPS-induced mice, the decrease in PARKIN function caused an abundant assembly of microglial NLRP3, contributing to motor deficiency and a reduction in the number of dopaminergic neurons present in the substantia nigra. Therefore, evidence suggests that regulating microglial NLRP3 activation by PARKIN via polyubiquitination ameliorates neurodegeneration in PD.
As previously discussed, Calhm, a membrane protein, played an important role in AD and, for Bo et al. [262], PD also has a relationship with this modulator, which is 1-Methyl-4-phenyl-1,2,3, 6-tetrahydropyridine (MPTP)-induced. The research demonstrated that microglial Calhm2 is responsible for, in microglia, modulating the EFhd2-STAT3 pathway, in which EFhd2 is configured as a calcium-binding protein and STAT3 as a protein that, if overactivated, causes the release of pro-inflammatory factors in microglia, resulting in neuronal reduction.
Basurco et al. [263] demonstrated in his study the relationship between innate immune reaction and PD. After the insertion of serotype 9 of the adeno-associated virus, capable of expressing the α-synuclein and mCherry genes, into the substantia nigra, purification of astrocytes (ACSA2+) and myeloid cells (CD11b+) from both the striatum and midbrain was carried out, to sequence RNA in large volume. CD11b+ cells, in models with PD in the midbrain, demonstrated a distinct M2-like anti-inflammatory transcriptomic profile. At the same time, those in the striatum exhibited a pro-inflammatory state comparable to microglia linked to the pathology. The phagocytosis of dopaminergic neuronal bodies in the midbrain was carried out by microglia and infiltrated macrophages, which had increased their quantity. It is interesting to note that astrocytes and microglia presented activation states inherent to the situation throughout the process of α-synuclein-dependent degeneration, with divergent contributions to the inflammatory response and, as a result, highlighted new paths for the treatment of PD through inflammatory response and microglia.
Multiple Sclerosis (MS) is a complex disease of the central nervous system (CNS) that is chronic, neurodegenerative, inflammatory, and autoimmune, which affects more than 2 million people worldwide [264,265]. Although the etiology of the pathology still seems uncertain, some conditions are related to a greater risk of developing MS, including biological sex, genetic constitution, and geographic factors [266]. With advancing age, the degeneration of oligodendrocytes and oligodendrocyte precursor cells increases, which promotes more significant degradation of myelin, a fact that is related to the ongoing neurodegeneration of the pathology, in addition to the fact that the capacity for remyelination is reduced as the years go by years [267]. A large number of receptors are expressed in both macrophages and microglia, such as scavenger receptors (SR), Fc receptors, and complement receptors (CR) so they are related to myelin uptake and act in the pathogenesis of MS [268]. Even in the early stages of the generation of damage in MS, it is possible to observe the activation of microglia, showing a continuous increase in the density of macrophages/microglia as the pathology progresses [269].
Ren et al. [270] demonstrated the relationship between Quaking (Qki), an RNA-binding protein, and the control of phagocytosis carried out by microglia in the process of demyelination and remyelination. The results point to an intensification in the neuroprotective activity of microglia in cases of CNS injuries and neurodegenerative pathologies, such as MS, a fact that is related to more significant phagocytosis by Qki. Therefore, the essential role of microglia in remyelination was confirmed [271]. Moreover, disruption of the blood-brain barrier (BBB) depends on microglial activation. This injury to the BBB is characterized as a recurrent and fundamental aspect of the progression of MS, demonstrated by the study by Yu et al. [272], further highlighting a possible manipulation of microglia to protect the BBB and ensure an improvement in inflammatory demyelination.
7. Exploring Medicinal Plants in Neuroinflammation: Comprehensive Insights on Effects, Dosage, Mechanisms, and Clinical Applications
The studies on various medicinal plants and their phytochemicals present a compelling overview of potential therapeutic agents against neuroinflammation. For instance, the research on Cleistocalyx nervosum var. paniala indicates that the extract significantly reduces pro-inflammatory markers such as Cyclooxygenase-2 (COX-2) and inducible Nitric Oxide Synthase (iNOS) while inhibiting the activation of Nuclear Factor kappa B (NF-κB) and its downstream effects. The effective doses, ranging from 5 to 100 μg/mL, reveal its capability to modulate the inflammatory response in Tumor Necrosis Factor-alpha (TNF-α)- and Lipopolysaccharide (LPS)-stimulated BV-2 cells [38] [39]. These findings underscore the potential of this plant as a source of new anti-inflammatory agents, particularly for neurodegenerative diseases. However, future research should focus on in vivo models to corroborate these results and rigorous safety assessments to establish a comprehensive profile in clinical settings.
The efficacy of Curcuma longa is well-documented in the context of neuroinflammation, mainly through the action of curcumin and its derivatives. The included studies demonstrated a marked decrease in inflammatory cytokines and a reduction in NF-κB activation in LPS-stimulated BV-2 cells. The doses tested ranged from 12.5 to 200 μg/mL, highlighting the potential for Curcuma longa as a natural alternative for treating neuroinflammation and oxidative stress-related disorders [40] [41]. Despite its promise, the variability in effective dosing observed across studies indicates a need for more standardized protocols and clinical trials encompassing diverse populations to evaluate its efficacy comprehensively.
Cannabis sativa has garnered attention due to its diverse compounds, including cannabinoids that exhibit anti-inflammatory properties in LPS-stimulated BV-2 cells. The included study indicated significant reductions in pro-inflammatory cytokines while also increasing levels of endogenous cannabinoids, suggesting a dual role in inflammation modulation and pain management [42]. While these findings open avenues for novel treatments targeting neuroinflammation and chronic pain, the study's limitations, such as sparse clinical evidence and the challenges of standardizing cannabis strains, necessitate further research that focuses on specific cannabinoid profiles and their therapeutic implications in clinical settings.
The study on Dioscorea nipponica also reveals a promising mechanism for regulating neuroinflammation through the modulation of iNOS and COX-2 expression in both in vitro and in vivo models. The effective doses were 10 to 100 μg/mL for in vitro studies and 60 mg/kg for in vivo applications [43]. Moreover, increased expression of Brain-Derived Neurotrophic Factor (BDNF) and phosphorylated Cyclic AMP Response Element-Binding Protein (pCREB) in animal models indicates potential cognitive benefits. This highlights the need for further research into the long-term effects and safety of Dioscorea nipponica and its primary compound, dioscin, particularly regarding their impact on human mood disorders.
Research involving Centipeda minima has highlighted its complex phytochemical composition, demonstrating significant anti-inflammatory effects through inhibiting various pathways, including NF-κB and iNOS expression. Li et al. observed a reduction in inflammatory mediators in both in vitro and in vivo settings, reinforcing its potential to develop comprehensive anti-inflammatory therapies against neuroinflammation [44]. However, the variability in phytochemical compositions underscores the need for future studies that systematically evaluate the contributions of individual components, which would enhance the reproducibility and standardization of therapeutic applications.
Atractylodis Rhizoma Alba has effectively suppressed inflammatory markers in LPS-stimulated BV-2 cells. The mechanisms involved include significant inhibition of NF-κB and Mitogen-Activated Protein Kinase (MAPK) signaling pathways, which are critical in the inflammatory response [45]. The promising results call for integrative approaches in clinical applications. Still, the lack of long-term studies means that further investigations are necessary to determine potential side effects and interactions with other therapeutic agents.
The potential of Vaccinium bracteatum in modulating neuroinflammation is also noteworthy, with studies showing significant reductions in pro-inflammatory cytokines and oxidative stress markers. The effective doses ranged from 1 to 20 μg/mL, illustrating its promise as an anti-inflammatory and antioxidant agent [46]. However, variability across studies necessitates additional research to clarify the underlying mechanisms and to confirm its efficacy across different biological models and clinical scenarios. Lonicera japonica also exhibits multifaceted anti-inflammatory properties, significantly reducing markers of inflammation in vitro [47]. However, the complex composition of this medicinal plant necessitates further exploration into which specific compounds contribute most significantly to its therapeutic effects. Future studies should aim to isolate these components, evaluate their respective contributions to enhancing anti-inflammatory responses, and determine optimal dosages for effective therapeutic outcomes.
In Eom et al.'s study, Bambusae caulis demonstrated promising reductions in key inflammatory markers and oxidative stress levels. The observed alterations in Heme Oxygenase-1 (HO-1) and Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) signaling pathways suggest that this plant may hold potential for developing new treatments for neuroinflammation [48]. However, the need for additional studies to identify specific active components and optimize therapeutic protocols remains a priority for future research.
Lastly, Zingiberis Rhizoma demonstrates substantial anti-inflammatory effects in vitro on LPS-stimulated BV-2 cells. The administration of 1, 5, or 10 μg/ml of ginger extract for 24 hours resulted in a marked reduction of Nitric Oxide (NO) and Prostaglandin E2 (PGE2) production, alongside decreased expression of COX-2 messengers Ribonucleic Acid (mRNA). Additionally, the treatment led to lower levels of pro-inflammatory cytokines and downregulated the phosphorylation of MAPK signaling molecules (Extracellular Signal-Regulated Kinase 1 and 2 [ERK1/2], p38 MAPK, and c-Jun N-terminal kinase [JNK]). Notably, there was also a reduction in NF-κB nuclear translocation, highlighting its potential in modulating neuroinflammatory pathways [49]. Given these promising findings, Zingiberis Rhizoma may be a valuable candidate for developing neuroinflammation interventions. However, further research is critical to optimize dosing strategies and clarify the underlying mechanisms of action to realize its therapeutic potential.
Overall, while these studies highlight the potential of medicinal plants in regulating neuroinflammation, further research efforts are essential to validate findings across different biological systems, establish optimal dosages, and explore long-term effects in clinical populations. Future research directions could significantly benefit from integrating various advanced methodologies. For instance, genetic studies can be employed to investigate the molecular pathways affected by phytochemicals found in these plants. By utilizing gene expression profiling and Ribonucleic Acid (RNA) sequencing techniques, researchers could elucidate how specific phytochemicals modulate inflammatory pathways at the genomic level, providing insights into their efficacy and mechanism of action.
Immunological assays are also crucial for comprehensively understanding the immune responses elicited by these medicinal plants. Techniques such as flow cytometry and Enzyme-Linked Immunosorbent Assay (ELISA) could quantify cytokine levels and other immune markers in response to treatment with various plant extracts. These essays could help identify the most effective extracts and dosages that reduce neuroinflammation and enhance neuroprotection. Additionally, investigating the immunomodulatory effects of these plants on various immune cell types, including microglia and astrocytes, could reveal how they influence the neuroinflammatory milieu in the CNS.
Molecular docking studies based on plants' phytochemical profiles could also provide valuable insights into their potential interactions with key molecular targets involved in neuroinflammation. Researchers can predict how phytochemicals bind to receptors or enzymes involved in inflammatory pathways by employing computational modeling. Such studies could facilitate the identification of promising compounds for further development and help optimize their structures to enhance efficacy and reduce potential side effects.
Moreover, exploring different extracts, such as aqueous, alcoholic, or supercritical fluid extracts, may reveal varying levels of bioactive compounds and their corresponding effects on neuroinflammation. Comparative studies on the efficacy of these extraction methods could uncover the most effective approaches to maximize the therapeutic potential of medicinal plants. Investigating the influence of specific extraction techniques on the stability and bioavailability of phytochemicals will also be necessary, as this could directly impact their clinical applicability.
Finally, integrating nanomedicine and nanocarriers into the research framework presents an exciting avenue for enhancing the delivery of medicinal plant extracts to treat neuroinflammation. Researchers could improve phytochemicals' solubility, stability, and bioavailability by employing nanoparticles as carriers. Formulating nanocarriers targeting neuroinflammatory sites in the CNS could significantly enhance therapeutic outcomes while minimizing systemic side effects. Furthermore, studies exploring the synergistic effects of combining multiple phytochemicals within nanocarriers could lead to more effective multi-targeted therapies for neuroinflammation.
A multifaceted research approach combining genetic studies, immunological assays, molecular docking, extraction method evaluations, and nanomedicine strategies will advance our understanding of medicinal plants for neuroinflammation. Such efforts can lead to the development of optimized therapeutic protocols that not only leverage the unique properties of each plant but also ensure their safe and practical application in clinical settings. By addressing these areas, future research can provide a more comprehensive understanding of the role of medicinal plants in mitigating neuroinflammation, ultimately contributing to innovative treatment strategies for related neurological disorders.
9. Conclusions
Exploring medicinal plants in neuroinflammation has yielded promising insights, revealing their potential as valuable therapeutic agents. Studies on Cleistocalyx nervosum var. paniala, Curcuma longa, and Cannabis sativa demonstrate their ability to modulate inflammatory pathways and provide symptomatic relief for neurodegenerative conditions. Each plant exhibits unique mechanisms of action, from suppressing pro-inflammatory markers to enhancing neuroprotective factors, underscoring their multifaceted therapeutic potential. Figure 3 illustrates the primary mechanisms by which medicinal plants affect neuroinflammation and associated neuroinflammatory disorders.
Despite these encouraging findings, challenges remain. Variability in effective doses, the need for standardized protocols, and the complexity of phytochemical compositions necessitate further research. Rigorous in vivo studies, comprehensive safety assessments, and standardized clinical trials are essential to validate these findings and establish these plants' clinical efficacy and safety profiles.
Future research should integrate personalized medicine approaches, tailoring treatments based on individual genetic, environmental, and lifestyle factors to enhance therapeutic efficacy and minimize adverse effects. Additionally, the principles of green health care—emphasizing sustainability, ecological balance, and the use of natural therapies—should guide the development and application of medicinal plant-based treatments. Employing advanced methodologies, including genetic studies to elucidate molecular pathways, immunological assays to evaluate immune responses, and molecular docking studies to predict interactions with key inflammatory targets, will further advance this field. Optimizing extraction methods and exploring nanomedicine for improved delivery could also enhance the therapeutic potential of these plants. In conclusion, a holistic and multifaceted research approach incorporating personalized medicine and green healthcare principles is crucial for advancing our understanding of medicinal plants in neuroinflammation.
Supplementary Materials
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Author Contributions
Conceptualization, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; methodology, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; software, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; validation, S.M.B., M.T., and L.F.L.; formal analysis, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; investigation, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; resources, S.M.B., M.T., and L.F.L.; data curation, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; writing—original draft preparation, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; writing—review and editing, S.M.B., B.L.B., J.d.S.C.O., J.P., C.B.L., M.T., and L.F.L.; visualization, S.M.B., M.T., and L.F.L.; supervision, S.M.B., M.T., and L.F.L.; project administration, S.M.B., M.T., and L.F.L.; funding acquisition, S.M.B., M.T., and L.F.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We would like to express our sincere gratitude to Servier Medical Art for providing the medical figures used in the production of the images for this article. Their high-quality illustrations significantly enhanced the clarity and visual appeal of our work. Servier Medical Art is licensed under CC BY 4.0, allowing for their use with proper attribution. We appreciate their support and contribution to the advancement of scientific communication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Davinelli, S.; Maes, M.; Corbi, G.; Zarrelli, A.; Willcox, D.C.; Scapagnini, G. Dietary phytochemicals and neuro-inflammaging: from mechanistic insights to translational challenges. Immun Ageing 2016, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Xiao, Q.; Fan, H.J.; Xu, L.; Qin, S.C.; Yang, L.X.; Jin, X.M.; Xiao, B.G.; Zhang, B.; Ma, C.G.; et al. Wuzi Yanzong Pill relieves MPTP-induced motor dysfunction and neuron loss by inhibiting NLRP3 inflammasome-mediated neuroinflammation. Metab Brain Dis 2023. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. A Decade of Dedication: Pioneering Perspectives on Neurological Diseases and Mental Illnesses. Biomedicines 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Revolutionizing our understanding of Parkinson's disease: Dr. Heinz Reichmann's pioneering research and future research direction. Journal of neural transmission (Vienna, Austria : 1996) 2024. [CrossRef]
- Rink, C.; Khanna, S. Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid Redox Signal 2011, 14, 1889–1903. [Google Scholar] [CrossRef]
- Morozumi, T.; Preziosa, P.; Meani, A.; Albergoni, M.; Margoni, M.; Pagani, E.; Filippi, M.; Rocca, M.A. Influence of cardiorespiratory fitness and MRI measures of neuroinflammation on hippocampal volume in multiple sclerosis. J Neurol Neurosurg Psychiatry 2023. [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front Cell Neurosci 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Direito, R.; Barbalho, S.M.; Figueira, M.E.; Minniti, G.; de Carvalho, G.M.; de Oliveira Zanuso, B.; de Oliveira Dos Santos, A.R.; de Góes Corrêa, N.; Rodrigues, V.D.; de Alvares Goulart, R.; et al. Medicinal Plants, Phytochemicals and Regulation of the NLRP3 Inflammasome in Inflammatory Bowel Diseases: A Comprehensive Review. Metabolites 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, II; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int J Mol Sci 2022, 23. [CrossRef]
- de Lima, E.P.; Moretti, R.C., Jr.; Torres Pomini, K.; Laurindo, L.F.; Sloan, K.P.; Sloan, L.A.; Castro, M.V.M.; Baldi, E., Jr.; Ferraz, B.F.R.; de Souza Bastos Mazuqueli Pereira, E.; et al. Glycolipid Metabolic Disorders, Metainflammation, Oxidative Stress, and Cardiovascular Diseases: Unraveling Pathways. Biology 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Girotto, O.S.; Furlan, O.O.; Moretti Junior, R.C.; Goulart, R.A.; Baldi Junior, E.; Barbalho-Lamas, C.; Fornari Laurindo, L.; Barbalho, S.M. Effects of apples (Malus domestica) and their derivatives on metabolic conditions related to inflammation and oxidative stress and an overview of by-products use in food processing. Critical reviews in food science and nutrition 2024, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Valotto Neto, L.J.; Reverete de Araujo, M.; Moretti Junior, R.C.; Mendes Machado, N.; Joshi, R.K.; Dos Santos Buglio, D.; Barbalho Lamas, C.; Direito, R.; Fornari Laurindo, L.; Tanaka, M.; et al. Investigating the Neuroprotective and Cognitive-Enhancing Effects of Bacopa monnieri: A Systematic Review Focused on Inflammation, Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis. Antioxidants (Basel, Switzerland) 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Silveira Rossi, J.L.; Barbalho, S.M.; Reverete de Araujo, R.; Bechara, M.D.; Sloan, K.P.; Sloan, L.A.J.D.m.r.; reviews. Metabolic syndrome and cardiovascular diseases: Going beyond traditional risk factors. 2022, 38, e3502.
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Fabisiak, T.; Patel, M. Crosstalk between neuroinflammation and oxidative stress in epilepsy. Front Cell Dev Biol 2022, 10, 976953. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, A.; Torres-Paniagua, A.M.; Acero, G.; Díaz, G.; Gevorkian, G. Increased TSPO expression, pyroglutamate-modified amyloid beta (AβN3(pE)) accumulation and transient clustering of microglia in the thalamus of Tg-SwDI mice. J Neuroimmunol 2023, 382, 578150. [Google Scholar] [CrossRef]
- Laurindo, L.F.; de Carvalho, G.M.; de Oliveira Zanuso, B.; Figueira, M.E.; Direito, R.; de Alvares Goulart, R.; Buglio, D.S.; Barbalho, S.M. Curcumin-Based Nanomedicines in the Treatment of Inflammatory and Immunomodulated Diseases: An Evidence-Based Comprehensive Review. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- Tanaka, M.; Battaglia, S.; Giménez-Llort, L.; Chen, C.; Hepsomali, P.; Avenanti, A.; Vécsei, L. Innovation at the Intersection: Emerging Translational Research in Neurology and Psychiatry. Cells 2024, 13. [Google Scholar] [CrossRef]
- Tanaka, M.; Chen, C. Editorial: Towards a mechanistic understanding of depression, anxiety, and their comorbidity: perspectives from cognitive neuroscience. Frontiers in behavioral neuroscience 2023, 17, 1268156. [Google Scholar] [CrossRef] [PubMed]
- Hopper, A.T.; Campbell, B.M.; Kao, H.; Pintchovski, S.A.; Staal, R.G.W. Chapter Four - Recent Developments in Targeting Neuroinflammation in Disease. In Annual Reports in Medicinal Chemistry, Desai, M.C., Ed.; Academic Press: 2012; Volume 47, pp. 37-53.
- Liu, Y.; Yang, H.; Luo, N.; Fu, Y.; Qiu, F.; Pan, Z.; Li, X.; Jian, W.; Yang, X.; Xue, Q.; et al. An Fgr kinase inhibitor attenuates sepsis-associated encephalopathy by ameliorating mitochondrial dysfunction, oxidative stress, and neuroinflammation via the SIRT1/PGC-1α signaling pathway. J Transl Med 2023, 21, 486. [Google Scholar] [CrossRef] [PubMed]
- Bássoli, R.; Audi, D.; Ramalho, B.; Audi, M.; Quesada, K.; Barbalho, S.J.J.o.H.M. The Effects of Curcumin on Neurodegenerative Diseases: A Systematic Review. 2023, 42, 100771.
- Barbalho, S.M.; Direito, R.; Laurindo, L.F.; Marton, L.T.; Guiguer, E.L.; Goulart, R.d.A.; Tofano, R.J.; Carvalho, A.C.; Flato, U.A.P.; Capelluppi Tofano, V.A.J.A. Ginkgo biloba in the aging process: A narrative review. 2022, 11, 525.
- de Oliveira Zanuso, B.; Dos Santos, A.R.d.O.; Miola, V.F.B.; Campos, L.M.G.; Spilla, C.S.G.; Barbalho, S.M.J.E.g. Panax ginseng and aging related disorders: A systematic review. 2022, 161, 111731.
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer's disease. Mol Neurodegener 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Blank-Stein, N.; Mass, E. Macrophage and monocyte subsets in response to ischemic stroke. Eur J Immunol 2023, e2250233. [Google Scholar] [CrossRef]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: an overview. Journal of Nutritional Science 2016, 5, e47. [Google Scholar] [CrossRef]
- Park, K. The Role of Dietary Phytochemicals: Evidence from Epidemiological Studies. Nutrients 2023, 15, 1371. [Google Scholar] [CrossRef]
- Buglio, D.S.; Marton, L.T.; Laurindo, L.F.; Guiguer, E.L.; Araújo, A.C.; Buchaim, R.L.; Goulart, R.A.; Rubira, C.J.; Barbalho, S.M. The Role of Resveratrol in Mild Cognitive Impairment and Alzheimer's Disease: A Systematic Review. J Med Food 2022, 25, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Barbalho, S.M.; Bueno Ottoboni, A.M.M.; Fiorini, A.M.R.; Guiguer, E.L.; Nicolau, C.C.T.; Goulart, R.d.A.; Flato, U.A.P.J.C.r.i.f.s.; nutrition. Grape juice or wine: which is the best option? 2020, 60, 3876-3889.
- Barbalho, S.M.; Bueno Ottoboni, A.M.M.; Fiorini, A.M.R.; Guiguer, E.L.; Nicolau, C.C.T.; Goulart, R.d.A.; Flato, U.A.P.J.C.r.i.f.s.
- Sen, T.; Samanta, S.K. Medicinal plants, human health and biodiversity: a broad review. Adv Biochem Eng Biotechnol 2015, 147, 59–110. [Google Scholar] [CrossRef] [PubMed]
- Chaachouay, N.; Zidane, L. Plant-Derived Natural Products: A Source for Drug Discovery and Development. Drugs and Drug Candidates 2024, 3, 184–207. [Google Scholar] [CrossRef]
- Pagotto, G.L.O.; Santos, L.; Osman, N.; Lamas, C.B.; Laurindo, L.F.; Pomini, K.T.; Guissoni, L.M.; Lima, E.P.; Goulart, R.A.; Catharin, V.; et al. Ginkgo biloba: A Leaf of Hope in the Fight against Alzheimer's Dementia: Clinical Trial Systematic Review. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Tyler, S.E.B.; Tyler, L.D.K. Pathways to healing: Plants with therapeutic potential for neurodegenerative diseases. IBRO Neurosci Rep 2023, 14, 210–234. [Google Scholar] [CrossRef]
- Suk, K. Regulation of neuroinflammation by herbal medicine and its implications for neurodegenerative diseases. A focus on traditional medicines and flavonoids. Neurosignals 2005, 14, 23–33. [Google Scholar] [CrossRef]
- Janpaijit, S.; Sillapachaiyaporn, C.; Theerasri, A.; Charoenkiatkul, S.; Sukprasansap, M.; Tencomnao, T. Cleistocalyx nervosum var. paniala Berry Seed Protects against TNF-α-Stimulated Neuroinflammation by Inducing HO-1 and Suppressing NF-κB Mechanism in BV-2 Microglial Cells. Molecules (Basel, Switzerland) 2023, 28. [Google Scholar] [CrossRef]
- Janpaijit, S.; Lertpatipanpong, P.; Sillapachaiyaporn, C.; Baek, S.J.; Charoenkiatkul, S.; Tencomnao, T.; Sukprasansap, M. Anti-neuroinflammatory effects of Cleistocalyx nervosum var. paniala berry-seed extract in BV-2 microglial cells via inhibition of MAPKs/NF-κB signaling pathway. Heliyon 2022, 8, e11869. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Lee, Y.S.; Yoon, D.; Kim, G.S.; Lee, D.Y. The ethanolic extract of Curcuma longa grown in Korea exhibits anti-neuroinflammatory effects by activating of nuclear transcription factor erythroid-2-related factor 2/heme oxygenase-1 signaling pathway. BMC complementary medicine and therapies 2022, 22, 343. [Google Scholar] [CrossRef]
- Eun, C.S.; Lim, J.S.; Lee, J.; Lee, S.P.; Yang, S.A. The protective effect of fermented Curcuma longa L. on memory dysfunction in oxidative stress-induced C6 gliomal cells, proinflammatory-activated BV2 microglial cells, and scopolamine-induced amnesia model in mice. BMC complementary and alternative medicine 2017, 17, 367. [Google Scholar] [CrossRef] [PubMed]
- Borgonetti, V.; Benatti, C.; Governa, P.; Isoldi, G.; Pellati, F.; Alboni, S.; Tascedda, F.; Montopoli, M.; Galeotti, N.; Manetti, F.; et al. Non-psychotropic Cannabis sativa L. phytocomplex modulates microglial inflammatory response through CB2 receptors-, endocannabinoids-, and NF-κB-mediated signaling. Phytotherapy research : PTR 2022, 36, 2246–2263. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Kim, Y.S.; Jakaria, M.; Yu, Y.J.; Ahn, J.Y.; Kim, I.S.; Choi, D.K. Dioscorea nipponica Makino Rhizome Extract and Its Active Compound Dioscin Protect against Neuroinflammation and Scopolamine-Induced Memory Deficits. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Zhou, Y.L.; He, D.H.; Liu, W.; Fan, X.Z.; Wang, Q.; Pan, H.F.; Cheng, Y.X.; Liu, Y.Q. Centipeda minima extract exerts antineuroinflammatory effects via the inhibition of NF-κB signaling pathway. Phytomedicine : international journal of phytotherapy and phytopharmacology 2020, 67, 153164. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.H.; Li, W.; Go, Y.; Oh, Y.C. Atractylodis Rhizoma Alba Attenuates Neuroinflammation in BV2 Microglia upon LPS Stimulation by Inducing HO-1 Activity and Inhibiting NF-κB and MAPK. International journal of molecular sciences 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Ma, S.X.; Ko, Y.H.; Seo, J.Y.; Lee, B.R.; Lee, T.H.; Kim, S.Y.; Lee, S.Y.; Jang, C.G. Vaccinium bracteatum Thunb. Exerts Anti-Inflammatory Activity by Inhibiting NF-κB Activation in BV-2 Microglial Cells. Biomolecules & therapeutics 2016, 24, 543–551. [Google Scholar] [CrossRef]
- Kwon, S.H.; Ma, S.X.; Hong, S.I.; Lee, S.Y.; Jang, C.G. Lonicera japonica THUNB. Extract Inhibits Lipopolysaccharide-Stimulated Inflammatory Responses by Suppressing NF-κB Signaling in BV-2 Microglial Cells. Journal of medicinal food 2015, 18, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.W.; Park, S.Y.; Kim, Y.H.; Seong, S.J.; Jin, M.L.; Ryu, E.Y.; Kim, M.J.; Lee, S.J. Bambusae Caulis in Taeniam modulates neuroprotective and anti-neuroinflammatory effects in hippocampal and microglial cells via HO-1- and Nrf-2-mediated pathways. International journal of molecular medicine 2012, 30, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.W.; Yoon, C.H.; Park, K.M.; Han, H.S.; Park, Y.K. Hexane fraction of Zingiberis Rhizoma Crudus extract inhibits the production of nitric oxide and proinflammatory cytokines in LPS-stimulated BV2 microglial cells via the NF-kappaB pathway. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association 2009, 47, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Kallstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-kappaB. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.E.; Walker, A.K.; Weickert, C.S. Neuroinflammation in schizophrenia: the role of nuclear factor kappa B. Transl Psychiatry 2021, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Motolani, A.; Campos, L.; Lu, T. The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer's Disease. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Barbalho, S.M.; Bueno Ottoboni, A.M.M.; Fiorini, A.M.R.; Guiguer, E.L.; Nicolau, C.C.T.; Goulart, R.d.A.; Flato, U.A.P.J.C.r.i.f.s.
- Sul, O.J.; Ra, S.W. Quercetin Prevents LPS-Induced Oxidative Stress and Inflammation by Modulating NOX2/ROS/NF-kB in Lung Epithelial Cells. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-kappaB-Mediated Neuroinflammation in Parkinson's Disease and Potential Therapeutic Effect of Polyphenols. Neurotox Res 2020, 37, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, X.; Zhu, G.; Liu, H.; Chen, J.; Wang, Y.; He, X. Quercetin inhibits TNF-alpha induced HUVECs apoptosis and inflammation via downregulating NF-kB and AP-1 signaling pathway in vitro. Medicine (Baltimore) 2020, 99, e22241. [Google Scholar] [CrossRef] [PubMed]
- Nennig, S.E.; Schank, J.R. The Role of NFkB in Drug Addiction: Beyond Inflammation. Alcohol Alcohol 2017, 52, 172–179. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther 2020, 5, 209. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front Mol Neurosci 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J Neuroinflammation 2019, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: a general review. Int J Neurosci 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Silva, M.E.; Rund, L.A.; Vailati-Riboni, M.; Pacheco, M.T.B.; Johnson, R.W. Copper-Binding Peptides Attenuate Microglia Inflammation through Suppression of NF-kB Pathway. Mol Nutr Food Res 2021, 65, e2100153. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: a concept in redox biology and medicine. Redox Biol 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Park, J.S.; Rustamov, N.; Roh, Y.S. The Roles of NFR2-Regulated Oxidative Stress and Mitochondrial Quality Control in Chronic Liver Diseases. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Seen, S. Chronic liver disease and oxidative stress - a narrative review. Expert Rev Gastroenterol Hepatol 2021, 15, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- A, N.K.; Sharma, R.P.; Colangelo, A.M.; Ignatenko, A.; Martorana, F.; Jennen, D.; Briede, J.J.; Brady, N.; Barberis, M.; Mondeel, T.; et al. ROS networks: designs, aging, Parkinson's disease and precision therapies. NPJ Syst Biol Appl 2020, 6, 34. [Google Scholar] [CrossRef]
- Badenetti, L.; Manzoli, R.; Rubin, M.; Cozza, G.; Moro, E. Monitoring Nrf2/ARE Pathway Activity with a New Zebrafish Reporter System. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D'Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef]
- Wei, Z.; Zhao, J.; Zhang, L.; Xia, M. Cell-Based Assays to Identify Modulators of Nrf2/ARE Pathway. Methods Mol Biol 2022, 2474, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Dordoe, C.; Wang, X.; Lin, P.; Wang, Z.; Hu, J.; Wang, D.; Fang, Y.; Liang, F.; Ye, S.; Chen, J.; et al. Non-mitogenic fibroblast growth factor 1 protects against ischemic stroke by regulating microglia/macrophage polarization through Nrf2 and NF-kappaB pathways. Neuropharmacology 2022, 212, 109064. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 2013, 100, 30–47. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J Adv Res 2021, 34, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front Cell Dev Biol 2022, 10, 826204. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Muramatsu, A.; Saito, R.; Asami, S.; Honda, T.; Hosoya, T.; Itoh, K.; Yamamoto, M.; Suzuki, T.; Dinkova-Kostova, A.T. C151 in KEAP1 is the main cysteine sensor for the cyanoenone class of NRF2 activators, irrespective of molecular size or shape. Sci Rep 2018, 8, 8037. [Google Scholar] [CrossRef] [PubMed]
- Fao, L.; Mota, S.I.; Rego, A.C. Shaping the Nrf2-ARE-related pathways in Alzheimer's and Parkinson's diseases. Ageing Res Rev 2019, 54, 100942. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol 2015, 225, R83–99. [Google Scholar] [CrossRef]
- Laurindo, L.F.; de Maio, M.C.; Minniti, G.; de Goes Correa, N.; Barbalho, S.M.; Quesada, K.; Guiguer, E.L.; Sloan, K.P.; Detregiachi, C.R.P.; Araujo, A.C.; et al. Effects of Medicinal Plants and Phytochemicals in Nrf2 Pathways during Inflammatory Bowel Diseases and Related Colorectal Cancer: A Comprehensive Review. Metabolites 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L., K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, JD.; Wang, XJ.; Tang, X. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Research 2013, 73. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Liu, J. Relationship between oxidative stress and nuclear factor-erythroid-2-related factor 2 signaling in diabetic cardiomyopathy (Review). Exp Ther Med 2021. [Google Scholar] [CrossRef]
- Mohan, S.; Gupta, D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed Pharmacother 2018, 108, 1866–1878. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. Pathobiology of tobacco smoking and neurovascular disorders: untied strings and alternative products. Fluids Barriers CNS 2015, 12, 25. [Google Scholar] [CrossRef]
- Cumaoglu, A.; Agkaya, A.O.; Ozkul, Z. Effect of the Lipid Peroxidation Product 4-Hydroxynonenal on Neuroinflammation in Microglial Cells: Protective Role of Quercetin and Monochloropivaloylquercetin. Turk J Pharm Sci 2019, 16, 54–61. [Google Scholar] [CrossRef]
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Chu, S.F.; Zhang, Z.; Zhou, X.; He, W.B.; Chen, C.; Luo, P.; Liu, D.D.; Ai, Q.D.; Gong, H.F.; Wang, Z.Z.; et al. Ginsenoside Rg1 protects against ischemic/reperfusion-induced neuronal injury through miR-144/Nrf2/ARE pathway. Acta Pharmacol Sin 2019, 40, 13–25. [Google Scholar] [CrossRef]
- Ambrozova, N.; Ulrichova, J.; Galandakova, A. Models for the study of skin wound healing. The role of Nrf2 and NF-kappaB. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2017, 161, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev 2012, 64, 1055–1081. [Google Scholar] [CrossRef]
- Tao, S.; Liu, P.; Luo, G.; Rojo de la Vega, M.; Chen, H.; Wu, T.; Tillotson, J.; Chapman, E.; Zhang, D.D. p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol Cell Biol 2017, 37. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal 2010, 13, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Y.; Jiang, X.; Du, H.; Shi, Y.; Xiu, M.; Liu, Y.; He, J. Natural products targeting Nrf2/ARE signaling pathway in the treatment of inflammatory bowel disease. Biomed Pharmacother 2023, 164, 114950. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.Y.; Fu, M.D.; Liu, K.; Duan, X.C. Therapeutic effect of Keap1-Nrf2-ARE pathway-related drugs on age-related eye diseases through anti-oxidative stress. Int J Ophthalmol 2021, 14, 1260–1273. [Google Scholar] [CrossRef]
- Wang, Q.; Botchway, B.O.A.; Zhang, Y.; Liu, X. Ellagic acid activates the Keap1-Nrf2-ARE signaling pathway in improving Parkinson's disease: A review. Biomed Pharmacother 2022, 156, 113848. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun Biol 2021, 4, 576. [Google Scholar] [CrossRef]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci 2022, 291, 120111. [Google Scholar] [CrossRef]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Pi, J.; Zhang, Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol 2022, 54, 102389. [Google Scholar] [CrossRef]
- Liu, S.; Pi, J.; Zhang, Q. Mathematical modeling reveals quantitative properties of KEAP1-NRF2 signaling. Redox Biol 2021, 47, 102139. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front Neurosci 2020, 14, 267. [Google Scholar] [CrossRef]
- Cores, Á.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim Biophys Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef]
- He, W.J.; Lv, C.H.; Chen, Z.; Shi, M.; Zeng, C.X.; Hou, D.X.; Qin, S. The Regulatory Effect of Phytochemicals on Chronic Diseases by Targeting Nrf2-ARE Signaling Pathway. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Amoroso, R.; Maccallini, C.; Bellezza, I. Activators of Nrf2 to Counteract Neurodegenerative Diseases. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Jentho, E.; Weis, S. DAMPs and Innate Immune Training. Front Immunol 2021, 12, 699563. [Google Scholar] [CrossRef]
- Jiang, L.; Shao, Y.; Tian, Y.; Ouyang, C.; Wang, X. Nuclear Alarmin Cytokines in Inflammation. J Immunol Res 2020, 2020, 7206451. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Caroen, S.; Reid, T. What Exactly Is Inflammation (and What Is It Not?). Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V.; Lyamzaev, K.G. Innate Immunity and Phenoptosis. Biochemistry (Mosc) 2022, 87, 1634–1639. [Google Scholar] [CrossRef]
- Gu, Q.; Zou, J.; Zhou, Y.; Deng, Q. Mechanism of inflammasomes in cancer and targeted therapies. Front Oncol 2023, 13, 1133013. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Vande Walle, L.; Lamkanfi, M. Therapeutic modulation of inflammasome pathways. Immunol Rev 2020, 297, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hauenstein, A.V. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol Aspects Med 2020, 76, 100889. [Google Scholar] [CrossRef]
- Chen, C.; Xu, P. Activation and Pharmacological Regulation of Inflammasomes. Biomolecules 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Mathur, A.; Ngo, C.; Man, S.M. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol Rev 2018, 82. [Google Scholar] [CrossRef]
- Deng, Z.; Lu, L.; Li, B.; Shi, X.; Jin, H.; Hu, W. The roles of inflammasomes in cancer. Front Immunol 2023, 14, 1195572. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Fu, Y.; Tian, D.; Yan, W. The contrasting roles of inflammasomes in cancer. Am J Cancer Res 2018, 8, 566–583. [Google Scholar] [PubMed]
- Christgen, S.; Kanneganti, T.D. Inflammasomes and the fine line between defense and disease. Curr Opin Immunol 2020, 62, 39–44. [Google Scholar] [CrossRef]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Inflammasomes. Curr Biol 2020, 30, R689–r694. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chai, J. Assembly and Architecture of NLR Resistosomes and Inflammasomes. Annu Rev Biophys 2023, 52, 207–228. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation - a two-way street. Mol Med 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid Med Cell Longev 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Sun, D.; Xu, J.; Zhang, W.; Song, C.; Gao, C.; He, Y.; Shang, Y. Negative regulator NLRC3: Its potential role and regulatory mechanism in immune response and immune-related diseases. Front Immunol 2022, 13, 1012459. [Google Scholar] [CrossRef]
- Olona, A.; Leishman, S.; Anand, P.K. The NLRP3 inflammasome: regulation by metabolic signals. Trends Immunol 2022, 43, 978–989. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, C.; Vong, C.T.; Tao, H.; Li, H.; Wang, S.; Wang, Y. Rhein regulates redox-mediated activation of NLRP3 inflammasomes in intestinal inflammation through macrophage-activated crosstalk. Br J Pharmacol 2022, 179, 1978–1997. [Google Scholar] [CrossRef] [PubMed]
- Perri, A. The NLRP3-Inflammasome in Health and Disease. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Duez, H.; Pourcet, B. Nuclear Receptors in the Control of the NLRP3 Inflammasome Pathway. Front Endocrinol (Lausanne) 2021, 12, 630536. [Google Scholar] [CrossRef]
- Seoane, P.I.; Lee, B.; Hoyle, C.; Yu, S.; Lopez-Castejon, G.; Lowe, M.; Brough, D. The NLRP3-inflammasome as a sensor of organelle dysfunction. J Cell Biol 2020, 219. [Google Scholar] [CrossRef]
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch Pharm Res 2021, 44, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J Cell Sci 2017, 130, 3955–3963. [Google Scholar] [CrossRef]
- Bulte, D.; Rigamonti, C.; Romano, A.; Mortellaro, A. Inflammasomes: Mechanisms of Action and Involvement in Human Diseases. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu Rev Immunol 2023, 41, 301–316. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front Immunol 2018, 9, 2305. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, C.; Xu, J.; Ma, S.; Jia, H.; Yan, M.; An, F.; Zhou, Y.; Qi, J.; Bian, H. Race between virus and inflammasomes: inhibition or escape, intervention and therapy. Front Cell Infect Microbiol 2023, 13, 1173505. [Google Scholar] [CrossRef]
- Anderson, F.L.; Biggs, K.E.; Rankin, B.E.; Havrda, M.C. NLRP3 inflammasome in neurodegenerative disease. Transl Res 2023, 252, 21–33. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol 2021, 18, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, Q.; Xu, G.; Xiao, X.; Bai, Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front Immunol 2022, 13, 1109938. [Google Scholar] [CrossRef] [PubMed]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front Immunol 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and Non-Canonical Activation of NLRP3 Inflammasome at the Crossroad between Immune Tolerance and Intestinal Inflammation. Front Immunol 2017, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front Immunol 2020, 11, 211. [Google Scholar] [CrossRef]
- Zito, G.; Buscetta, M.; Cimino, M.; Dino, P.; Bucchieri, F.; Cipollina, C. Cellular Models and Assays to Study NLRP3 Inflammasome Biology. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Mei, X. Role of NLRP3 Inflammasomes in Neuroinflammation Diseases. Eur Neurol 2020, 83, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.Q.; Le, W. NLRP3 Inflammasome-Mediated Neuroinflammation and Related Mitochondrial Impairment in Parkinson's Disease. Neurosci Bull 2023, 39, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhang, L.; Yang, X. Interaction between autophagy and the NLRP3 inflammasome in Alzheimer's and Parkinson's disease. Front Aging Neurosci 2022, 14, 1018848. [Google Scholar] [CrossRef] [PubMed]
- Soraci, L.; Gambuzza, M.E.; Biscetti, L.; Laganà, P.; Lo Russo, C.; Buda, A.; Barresi, G.; Corsonello, A.; Lattanzio, F.; Lorello, G.; et al. Toll-like receptors and NLRP3 inflammasome-dependent pathways in Parkinson's disease: mechanisms and therapeutic implications. J Neurol 2023, 270, 1346–1360. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Ng, W.L.; Goh, S.Y.; Gulam, M.Y.; Wang, L.F.; Tan, E.K.; Ahn, M.; Chao, Y.X. Targeting the inflammasome in Parkinson's disease. Front Aging Neurosci 2022, 14, 957705. [Google Scholar] [CrossRef] [PubMed]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpedek-Kaminska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer's Disease Treatment. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Severini, C.; Barbato, C.; Di Certo, M.G.; Gabanella, F.; Petrella, C.; Di Stadio, A.; de Vincentiis, M.; Polimeni, A.; Ralli, M.; Greco, A. Alzheimer's Disease: New Concepts on the Role of Autoimmunity and NLRP3 Inflammasome in the Pathogenesis of the Disease. Curr Neuropharmacol 2021, 19, 498–512. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The Role of NLRP3 Inflammasome in Alzheimer's Disease and Potential Therapeutic Targets. Front Pharmacol 2022, 13, 845185. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, J.; Wei, S.; Deng, J.; Feng, X.; Liu, S.; Liu, M. A novel strategy for bioactive natural products targeting NLRP3 inflammasome in Alzheimer's disease. Front Pharmacol 2022, 13, 1077222. [Google Scholar] [CrossRef]
- Deng, C.; Cai, X.; Jin, K.; Wang, Q. Editorial: The NLRP3 inflammasome-mediated neuroinflammation and its related mitochondrial impairment in neurodegeneration. Front Aging Neurosci 2022, 14, 1118281. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, H.; Wang, D.; Guan, R.; Zou, Y.; Li, M.; Zhang, J.; Chen, J. NLRP3 deficiency protects against hypobaric hypoxia induced neuroinflammation and cognitive dysfunction. Ecotoxicol Environ Saf 2023, 255, 114828. [Google Scholar] [CrossRef]
- Yu, Q.; Zhao, T.; Liu, M.; Cao, D.; Li, J.; Li, Y.; Xia, M.; Wang, X.; Zheng, T.; Liu, C.; et al. Targeting NLRP3 Inflammasome in Translational Treatment of Nervous System Diseases: An Update. Front Pharmacol 2021, 12, 707696. [Google Scholar] [CrossRef]
- Herrera-deGuise, C.; Serra-Ruiz, X.; Lastiri, E.; Borruel, N. JAK inhibitors: A new dawn for oral therapies in inflammatory bowel diseases. Front Med (Lausanne) 2023, 10, 1089099. [Google Scholar] [CrossRef]
- Luo, Y.; Alexander, M.; Gadina, M.; O'Shea, J.J.; Meylan, F.; Schwartz, D.M. JAK-STAT signaling in human disease: From genetic syndromes to clinical inhibition. J Allergy Clin Immunol 2021, 148, 911–925. [Google Scholar] [CrossRef]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front Pharmacol 2022, 13, 821344. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, C.; Chen, Z.; Wang, X.; Hong, F.; Hao, D. Regulation of the JAK/STAT signaling pathway in spinal cord injury: an updated review. Front Immunol 2023, 14, 1276445. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Low, J.T.; Silke, J.; O'Reilly, L.A. Digesting the Role of JAK-STAT and Cytokine Signaling in Oral and Gastric Cancers. Front Immunol 2022, 13, 835997. [Google Scholar] [CrossRef] [PubMed]
- Durham, G.A.; Williams, J.J.L.; Nasim, M.T.; Palmer, T.M. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol Sci 2019, 40, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Bakay, M.; Hakonarson, H. SOCS-JAK-STAT inhibitors and SOCS mimetics as treatment options for autoimmune uveitis, psoriasis, lupus, and autoimmune encephalitis. Front Immunol 2023, 14, 1271102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gibson, S.A.; Benveniste, E.N.; Qin, H. Opportunities for Translation from the Bench: Therapeutic Intervention of the JAK/STAT Pathway in Neuroinflammatory Diseases. Crit Rev Immunol 2015, 35, 505–527. [Google Scholar] [CrossRef]
- Shih, P.Y.; Li, C.J.; Yong, S.B. Emerging trends in clinical research on Janus kinase inhibitors for atopic dermatitis treatment. Int Immunopharmacol 2023, 124, 111029. [Google Scholar] [CrossRef]
- Agashe, R.P.; Lippman, S.M.; Kurzrock, R. JAK: Not Just Another Kinase. Mol Cancer Ther 2022, 21, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Philips, R.L.; Wang, Y.; Cheon, H.; Kanno, Y.; Gadina, M.; Sartorelli, V.; Horvath, C.M.; Darnell, J.E., Jr.; Stark, G.R.; O'Shea, J.J. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 2022, 185, 3857–3876. [Google Scholar] [CrossRef]
- Somade, O.T.; Oyinloye, B.E.; Ajiboye, B.O.; Osukoya, O.A. Syringic acid demonstrates an anti-inflammatory effect via modulation of the NF-κB-iNOS-COX-2 and JAK-STAT signaling pathways in methyl cellosolve-induced hepato-testicular inflammation in rats. Biochem Biophys Rep 2023, 34, 101484. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zhang, Y.; Zhu, L.; Zhang, Y.; Song, J. The role of JAK/STAT signaling pathway in cerebral ischemia-reperfusion injury and the therapeutic effect of traditional Chinese medicine: A narrative review. Medicine (Baltimore) 2023, 102, e35890. [Google Scholar] [CrossRef] [PubMed]
- Puigdevall, L.; Michiels, C.; Stewardson, C.; Dumoutier, L. JAK/STAT: Why choose a classical or an alternative pathway when you can have both? J Cell Mol Med 2022, 26, 1865–1875. [Google Scholar] [CrossRef]
- Sabaawy, H.E.; Ryan, B.M.; Khiabanian, H.; Pine, S.R. JAK/STAT of all trades: linking inflammation with cancer development, tumor progression and therapy resistance. Carcinogenesis 2021, 42, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Sarapultsev, A.; Gusev, E.; Komelkova, M.; Utepova, I.; Luo, S.; Hu, D. JAK-STAT signaling in inflammation and stress-related diseases: implications for therapeutic interventions. Mol Biomed 2023, 4, 40. [Google Scholar] [CrossRef]
- Cardona, K.; Medina, J.; Orrego-Cardozo, M.; Restrepo de Mejía, F.; Elcoroaristizabal, X.; Naranjo Galvis, C.A. Inflammatory gene expression profiling in peripheral blood from patients with Alzheimer's disease reveals key pathways and hub genes with potential diagnostic utility: a preliminary study. PeerJ 2021, 9, e12016. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Singh, M.K.; Shyam, H.; Mishra, A.; Kumar, S.; Kumar, A.; Kushwaha, J. Role of JAK/STAT in the Neuroinflammation and its Association with Neurological Disorders. Ann Neurosci 2021, 28, 191–200. [Google Scholar] [CrossRef]
- Oh, S.L.; Zhou, M.; Chin, E.W.M.; Amarnath, G.; Cheah, C.H.; Ng, K.P.; Kandiah, N.; Goh, E.L.K.; Chiam, K.H. Alzheimer's Disease Blood Biomarkers Associated With Neuroinflammation as Therapeutic Targets for Early Personalized Intervention. Front Digit Health 2022, 4, 875895. [Google Scholar] [CrossRef]
- Varma, V.R.; Desai, R.J.; Navakkode, S.; Wong, L.W.; Anerillas, C.; Loeffler, T.; Schilcher, I.; Mahesri, M.; Chin, K.; Horton, D.B.; et al. Hydroxychloroquine lowers Alzheimer's disease and related dementias risk and rescues molecular phenotypes related to Alzheimer's disease. Mol Psychiatry 2023, 28, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Rusek, M.; Smith, J.; El-Khatib, K.; Aikins, K.; Czuczwar, S.J.; Pluta, R. The Role of the JAK/STAT Signaling Pathway in the Pathogenesis of Alzheimer's Disease: New Potential Treatment Target. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Nevado-Holgado, A.J.; Ribe, E.; Thei, L.; Furlong, L.; Mayer, M.A.; Quan, J.; Richardson, J.C.; Cavanagh, J.; Consortium, N.; Lovestone, S. Genetic and Real-World Clinical Data, Combined with Empirical Validation, Nominate Jak-Stat Signaling as a Target for Alzheimer's Disease Therapeutic Development. Cells 2019, 8. [Google Scholar] [CrossRef]
- Porro, C.; Cianciulli, A.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Curcumin Regulates Anti-Inflammatory Responses by JAK/STAT/SOCS Signaling Pathway in BV-2 Microglial Cells. Biology (Basel) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Gibson, S.A.; Buckley, J.A.; Qin, H.; Benveniste, E.N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol 2018, 189, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against α-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J Neurosci 2016, 36, 5144–5159. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther 2021, 6, 402. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Y.; Zhang, X.; Gu, X.; Mao, Y.; Peng, B. The origin and repopulation of microglia. Developmental neurobiology 2022, 82, 112–124. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. Journal of neuroinflammation 2021, 18, 1–16. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia biology: one century of evolving concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Sánchez-Castillo, A.I.; Sepúlveda, M.R.; Marín-Teva, J.L.; Cuadros, M.A.; Martín-Oliva, D.; González-Rey, E.; Delgado, M.; Neubrand, V.E. Switching roles: beneficial effects of adipose tissue-derived mesenchymal stem cells on microglia and their implication in neurodegenerative diseases. Biomolecules 2022, 12, 219. [Google Scholar] [CrossRef]
- Vidal-Itriago, A.; Radford, R.A.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Frontiers in immunology 2022, 13, 997786. [Google Scholar] [CrossRef] [PubMed]
- Gullotta, G.S.; Costantino, G.; Sortino, M.A.; Spampinato, S.F. Microglia and the blood–brain barrier: An external player in acute and chronic neuroinflammatory conditions. International Journal of Molecular Sciences 2023, 24, 9144. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Yeo, I.-J.; Hong, J.-T.; Eo, S.-K.; Lee, D.; Kim, K. Side-Chain Immune Oxysterols Induce Neuroinflammation by Activating Microglia. International Journal of Molecular Sciences 2023, 24, 15288. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Che, J.; Zhang, J. Emerging non-proinflammatory roles of microglia in healthy and diseased brains. Brain Research Bulletin 2023, 199, 110664. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, J.; Xu, Z.; Yan, H.; Tang, B.; Liu, C.; Chen, C.; Meng, Q. Microglia-containing human brain organoids for the study of brain development and pathology. Molecular Psychiatry 2023, 28, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annual review of immunology 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Matsudaira, T.; Prinz, M. Life and death of microglia: Mechanisms governing microglial states and fates. Immunology Letters 2022, 245, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wright-Jin, E.C.; Gutmann, D.H. Microglia as dynamic cellular mediators of brain function. Trends in Molecular Medicine 2019, 25, 967–979. [Google Scholar] [CrossRef]
- Costa, J.; Martins, S.; Ferreira, P.A.; Cardoso, A.M.; Guedes, J.R.; Peça, J.; Cardoso, A.L. The old guard: Age-related changes in microglia and their consequences. Mechanisms of ageing and development 2021, 197, 111512. [Google Scholar] [CrossRef] [PubMed]
- Al-Onaizi, M.; Al-Khalifah, A.; Qasem, D.; ElAli, A. Role of microglia in modulating adult neurogenesis in health and neurodegeneration. International journal of molecular sciences 2020, 21, 6875. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Marrocco, F.; Limatola, C. Microglial cells: sensors for neuronal activity and microbiota-derived molecules. Frontiers in immunology 2022, 13, 1011129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Liu, T.; Mao, Y.; Peng, B. Novel microglia-based therapeutic approaches to neurodegenerative disorders. Neuroscience bulletin 2023, 39, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Taso, O.; Wang, R.; Bayram, S.; Graham, A.C.; Garcia-Reitboeck, P.; Mallach, A.; Andrews, W.D.; Piers, T.M.; Botia, J.A. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Human molecular genetics 2020, 29, 3224–3248. [Google Scholar] [CrossRef]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araújo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What do microglia really do in healthy adult brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef]
- Var, S.R.; Strell, P.; Johnson, S.T.; Roman, A.; Vasilakos, Z.; Low, W.C. Transplanting microglia for treating CNS injuries and neurological diseases and disorders, and prospects for generating exogenic microglia. Cell transplantation 2023, 32, 09636897231171001. [Google Scholar] [CrossRef]
- Guzmán-Ruiz, M.A.; Guerrero-Vargas, N.N.; Lagunes-Cruz, A.; González-González, S.; García-Aviles, J.E.; Hurtado-Alvarado, G.; Mendez-Hernández, R.; Chavarría-Krauser, A.; Morin, J.P.; Arriaga-Avila, V. Circadian modulation of microglial physiological processes and immune responses. Glia 2023, 71, 155–167. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.L.; Nissen, J.C. The Pathological Activation of Microglia Is Modulated by Sexually Dimorphic Pathways. International Journal of Molecular Sciences 2023, 24, 4739. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translational neurodegeneration 2020, 9, 42. [Google Scholar] [CrossRef]
- Cianciulli, A.; Calvello, R.; Ruggiero, M.; Panaro, M.A. Inflammaging and brain: Curcumin and its beneficial potential as regulator of microglia activation. Molecules 2022, 27, 341. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.F.; Elbadry, A.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The dual face of microglia (M1/M2) as a potential target in the protective effect of nutraceuticals against neurodegenerative diseases. Frontiers in Aging 2023, 4, 1231706. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Valle, M.S.; Russo, A.; Malaguarnera, L. The interplay between ghrelin and microglia in neuroinflammation: implications for obesity and neurodegenerative diseases. International Journal of Molecular Sciences 2022, 23, 13432. [Google Scholar] [CrossRef]
- Liu, L.; Tong, F.; Li, H.; Bin, Y.; Ding, P.; Peng, L.; Liu, Z.; Dong, X. Maturation, morphology, and function: the decisive role of intestinal flora on microglia: a review. Journal of Integrative Neuroscience 2023, 22, 70. [Google Scholar] [CrossRef]
- Ng, P.Y.; McNeely, T.L.; Baker, D.J. Untangling senescent and damage-associated microglia in the aging and diseased brain. The FEBS journal 2023, 290, 1326–1339. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Wu, L.J. How microglia sense and regulate neuronal activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Hristovska, I.; Robert, M.; Combet, K.; Honnorat, J.; Comte, J.; Pascual, O. Sleep decreases neuronal activity control of microglial dynamics in mice. Nature communications 2022, 13, 6273. [Google Scholar] [CrossRef]
- Ikegami, A.; Kato, D.; Wake, H. Microglial process dynamics depend on astrocyte and synaptic activity. Nagoya Journal of Medical Science 2023, 85, 772. [Google Scholar]
- Ahn, K.; Lee, S.J.; Mook-Jung, I. White matter-associated microglia: New players in brain aging and neurodegenerative diseases. Ageing Res Rev 2022, 75, 101574. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Bougea, A.; Hatzimanolis, A.; Scarmeas, N.; Papageorgiou, S.G. Unraveling the Potential Underlying Mechanisms of Mild Behavioral Impairment: Focusing on Amyloid and Tau Pathology. Cells 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Qiao, O.; Ji, H.; Zhang, Y.; Zhang, X.; Zhang, X.; Liu, N.; Huang, L.; Liu, C.; Gao, W. New insights in drug development for Alzheimer's disease based on microglia function. Biomed Pharmacother 2021, 140, 111703. [Google Scholar] [CrossRef]
- Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nuñez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I.; et al. Transgenic Mouse Models of Alzheimer's Disease: An Integrative Analysis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Fujikawa, R.; Tsuda, M. The Functions and Phenotypes of Microglia in Alzheimer’s Disease. Cells 2023, 12, 1207. [Google Scholar] [CrossRef]
- Guan, Y.-H.; Zhang, L.-J.; Wang, S.-Y.; Deng, Y.-D.; Zhou, H.-S.; Chen, D.-Q.; Zhang, L.-C. The role of microglia in Alzheimer's disease and progress of treatment. Ibrain 2022, 8, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Dong, Y.; Ma, J.; Pan, R.; Liao, Y.; Kong, X.; Li, X.; Li, S.; Chen, P.; Wang, L.; et al. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer's disease pathology. Sci Adv 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer's Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Malvaso, A.; Gatti, A.; Negro, G.; Calatozzolo, C.; Medici, V.; Poloni, T.E. Microglial Senescence and Activation in Healthy Aging and Alzheimer's Disease: Systematic Review and Neuropathological Scoring. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, X. Different phenotypes of microglia in animal models of Alzheimer disease. Immun Ageing 2022, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Xu, Y.; Sun, L.; Hashimoto, M.; Wei, J. Microglial response to aging and neuroinflammation in the development of neurodegenerative diseases. Neural Regen Res 2024, 19, 1241–1248. [Google Scholar] [CrossRef]
- Sun, N.; Victor, M.B.; Park, Y.P.; Xiong, X.; Scannail, A.N.; Leary, N.; Prosper, S.; Viswanathan, S.; Luna, X.; Boix, C.A.; et al. Human microglial state dynamics in Alzheimer's disease progression. Cell 2023, 186, 4386–4403.e4329. [Google Scholar] [CrossRef]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer's disease brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Chernyaeva, L.; Ratti, G.; Teirilä, L.; Fudo, S.; Rankka, U.; Pelkonen, A.; Korhonen, P.; Leskinen, K.; Keskitalo, S.; Salokas, K.; et al. Reduced binding of apoE4 to complement factor H promotes amyloid-β oligomerization and neuroinflammation. EMBO Rep 2023, 24, e56467. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42:Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer's Mouse Model. J Neurosci 2020, 40, 1956–1974. [Google Scholar] [CrossRef]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M.; et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 2022, 185, 4153–4169.e4119. [Google Scholar] [CrossRef]
- Qiao, W.; Chen, Y.; Zhong, J.; Madden, B.J.; Charlesworth, C.M.; Martens, Y.A.; Liu, C.C.; Knight, J.; Ikezu, T.C.; Kurti, A.; et al. Trem2 H157Y increases soluble TREM2 production and reduces amyloid pathology. Mol Neurodegener 2023, 18, 8. [Google Scholar] [CrossRef]
- Prakash, P.; Manchanda, P.; Paouri, E.; Bisht, K.; Sharma, K.; Wijewardhane, P.R.; Randolph, C.E.; Clark, M.G.; Fine, J.; Thayer, E.A.; et al. Amyloid β Induces Lipid Droplet-Mediated Microglial Dysfunction in Alzheimer's Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Kellogg, C.M.; Pham, K.; Machalinski, A.H.; Porter, H.L.; Blankenship, H.E.; Tooley, K.; Stout, M.B.; Rice, H.C.; Sharpe, A.L.; Beckstead, M.J.; et al. Microglial MHC-I induction with aging and Alzheimer's is conserved in mouse models and humans. bioRxiv 2023. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong choline supplementation ameliorates Alzheimer's disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, E.K.J.; Boer, G.J. Parkinson's Disease: A Tale of Many Players. Med Princ Pract 2023, 32, 155–165. [Google Scholar] [CrossRef]
- Giannakopoulou, K.-M.; Roussaki, I.; Demestichas, K. Internet of Things Technologies and Machine Learning Methods for Parkinson’s Disease Diagnosis, Monitoring and Management: A Systematic Review. Sensors 2022, 22, 1799. [Google Scholar] [CrossRef] [PubMed]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson's Disease: A Multisystem Disorder. Neurosci Bull 2023, 39, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; MacAskill, M.; Pascoe, M.; Anderson, T.; Heron, C.L. Dimensions of apathy in Parkinson's disease. Brain Behav 2023, 13, e2862. [Google Scholar] [CrossRef]
- Cattaneo, C.; Jost, W.H. Pain in Parkinson's Disease: Pathophysiology, Classification and Treatment. J Integr Neurosci 2023, 22, 132. [Google Scholar] [CrossRef]
- Herz, D.M.; Brown, P. Moving, fast and slow: behavioural insights into bradykinesia in Parkinson's disease. Brain 2023, 146, 3576–3586. [Google Scholar] [CrossRef]
- Arioli, M.; Cattaneo, Z.; Rusconi, M.L.; Blandini, F.; Tettamanti, M. Action and emotion perception in Parkinson's disease: A neuroimaging meta-analysis. Neuroimage Clin 2022, 35, 103031. [Google Scholar] [CrossRef]
- Dirkx, M.F.; Bologna, M. The pathophysiology of Parkinson's disease tremor. J Neurol Sci 2022, 435, 120196. [Google Scholar] [CrossRef] [PubMed]
- Dietrichs, E.; Alves, G.; Benjaminsen, E.; Johansen, K.K.; Tysnes, O.B. Treatment of motor symptoms in Parkinson's disease. Tidsskr Nor Laegeforen 2023, 143. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin Geriatr Med 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Hicks, A.R.; Reynolds, R.H.; O'Callaghan, B.; García-Ruiz, S.; Gil-Martínez, A.L.; Botía, J.; Plun-Favreau, H.; Ryten, M. The non-specific lethal complex regulates genes and pathways genetically linked to Parkinson's disease. Brain 2023, 146, 4974–4987. [Google Scholar] [CrossRef]
- Sabogal-Guáqueta, A.M.; Marmolejo-Garza, A.; de Pádua, V.P.; Eggen, B.; Boddeke, E.; Dolga, A.M. Microglia alterations in neurodegenerative diseases and their modeling with human induced pluripotent stem cell and other platforms. Prog Neurobiol 2020, 190, 101805. [Google Scholar] [CrossRef]
- Kelly, R.; Joers, V.; Tansey, M.G.; McKernan, D.P.; Dowd, E. Microglial Phenotypes and Their Relationship to the Cannabinoid System: Therapeutic Implications for Parkinson's Disease. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia in Neurodegenerative Events-An Initiator or a Significant Other? Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Tofaris, G.K. Initiation and progression of α-synuclein pathology in Parkinson's disease. Cell Mol Life Sci 2022, 79, 210. [Google Scholar] [CrossRef] [PubMed]
- Toni, M. Special Issue "Neurobiology of Protein Synuclein". Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson's Disease. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Basellini, M.J.; Kothuis, J.M.; Comincini, A.; Pezzoli, G.; Cappelletti, G.; Mazzetti, S. Pathological Pathways and Alpha-Synuclein in Parkinson's Disease: A View from the Periphery. Front Biosci (Landmark Ed) 2023, 28, 33. [Google Scholar] [CrossRef]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson's disease. Mol Neurodegener 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Y.; Wang, C.; Han, T.; Liu, H.; Sun, L.; Hong, J.; Hashimoto, M.; Wei, J. The reciprocal interactions between microglia and T cells in Parkinson’s disease: a double-edged sword. Journal of Neuroinflammation 2023, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.Q.; Zheng, R.; Liu, Y.; Ruan, Y.; Lin, Z.H.; Xue, N.J.; Chen, Y.; Zhang, B.R.; Pu, J.L. Parkin regulates microglial NLRP3 and represses neurodegeneration in Parkinson's disease. Aging Cell 2023, 22, e13834. [Google Scholar] [CrossRef] [PubMed]
- Bo, X.; Xie, F.; Zhang, J.; Gu, R.; Li, X.; Li, S.; Yuan, Z.; Cheng, J. Deletion of Calhm2 alleviates MPTP-induced Parkinson's disease pathology by inhibiting EFHD2-STAT3 signaling in microglia. Theranostics 2023, 13, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Basurco, L.; Abellanas, M.A.; Ayerra, L.; Conde, E.; Vinueza-Gavilanes, R.; Luquin, E.; Vales, A.; Vilas, A.; Martin-Uriz, P.S.; Tamayo, I.; et al. Microglia and astrocyte activation is region-dependent in the α-synuclein mouse model of Parkinson's disease. Glia 2023, 71, 571–587. [Google Scholar] [CrossRef]
- Yang, J.; Hamade, M.; Wu, Q.; Wang, Q.; Axtell, R.; Giri, S.; Mao-Draayer, Y. Current and Future Biomarkers in Multiple Sclerosis. International Journal of Molecular Sciences 2022, 23, 5877. [Google Scholar] [CrossRef]
- Charabati, M.; Wheeler, M.A.; Weiner, H.L.; Quintana, F.J. Multiple sclerosis: Neuroimmune crosstalk and therapeutic targeting. Cell 2023, 186, 1309–1327. [Google Scholar] [CrossRef] [PubMed]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in multiple sclerosis. WIREs Mech Dis 2023, 15, e1583. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Ysrraelit, M.C. Multiple Sclerosis and Aging: The Dynamics of Demyelination and Remyelination. ASN Neuro 2022, 14, 17590914221118502. [Google Scholar] [CrossRef] [PubMed]
- Ellen, O.; Ye, S.; Nheu, D.; Dass, M.; Pagnin, M.; Ozturk, E.; Theotokis, P.; Grigoriadis, N.; Petratos, S. The Heterogeneous Multiple Sclerosis Lesion: How Can We Assess and Modify a Degenerating Lesion? Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Dai, C.; Zhou, X.; Barnes, J.A.; Chen, X.; Wang, Y.; Yuan, L.; Shingu, T.; Heimberger, A.B.; Chen, Y.; et al. Qki is an essential regulator of microglial phagocytosis in demyelination. J Exp Med 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, F.; Sun, M.; Wu, N.; Liu, B.; Yi, X.; Ge, R.; Fan, X. Microglia in the context of multiple sclerosis. Front Neurol 2023, 14, 1157287. [Google Scholar] [CrossRef]
- Yu, Z.; Fang, X.; Liu, W.; Sun, R.; Zhou, J.; Pu, Y.; Zhao, M.; Sun, D.; Xiang, Z.; Liu, P.; et al. Microglia Regulate Blood-Brain Barrier Integrity via MiR-126a-5p/MMP9 Axis during Inflammatory Demyelination. Adv Sci (Weinh) 2022, 9, e2105442. [Google Scholar] [CrossRef]
Figure 1.
Microglia have M1 and M2 phenotypes depending on conditions. In a homeostatic state, they support neuronal health through synaptic pruning and clearing misfolded proteins. Under stress, they shift to the pro-inflammatory M1 phenotype, releasing reactive oxygen species (ROS) and cytokines that can damage neurons. When exposed to Transforming Growth Factor Beta (TGF-β), Interleukin (IL)-4, IL-10, or IL-13, they switch to the anti-inflammatory M2 phenotype, aiding in phagocytosis, Extracellular Matrix (ECM) rebuilding, and neuronal survival.
Figure 1.
Microglia have M1 and M2 phenotypes depending on conditions. In a homeostatic state, they support neuronal health through synaptic pruning and clearing misfolded proteins. Under stress, they shift to the pro-inflammatory M1 phenotype, releasing reactive oxygen species (ROS) and cytokines that can damage neurons. When exposed to Transforming Growth Factor Beta (TGF-β), Interleukin (IL)-4, IL-10, or IL-13, they switch to the anti-inflammatory M2 phenotype, aiding in phagocytosis, Extracellular Matrix (ECM) rebuilding, and neuronal survival.
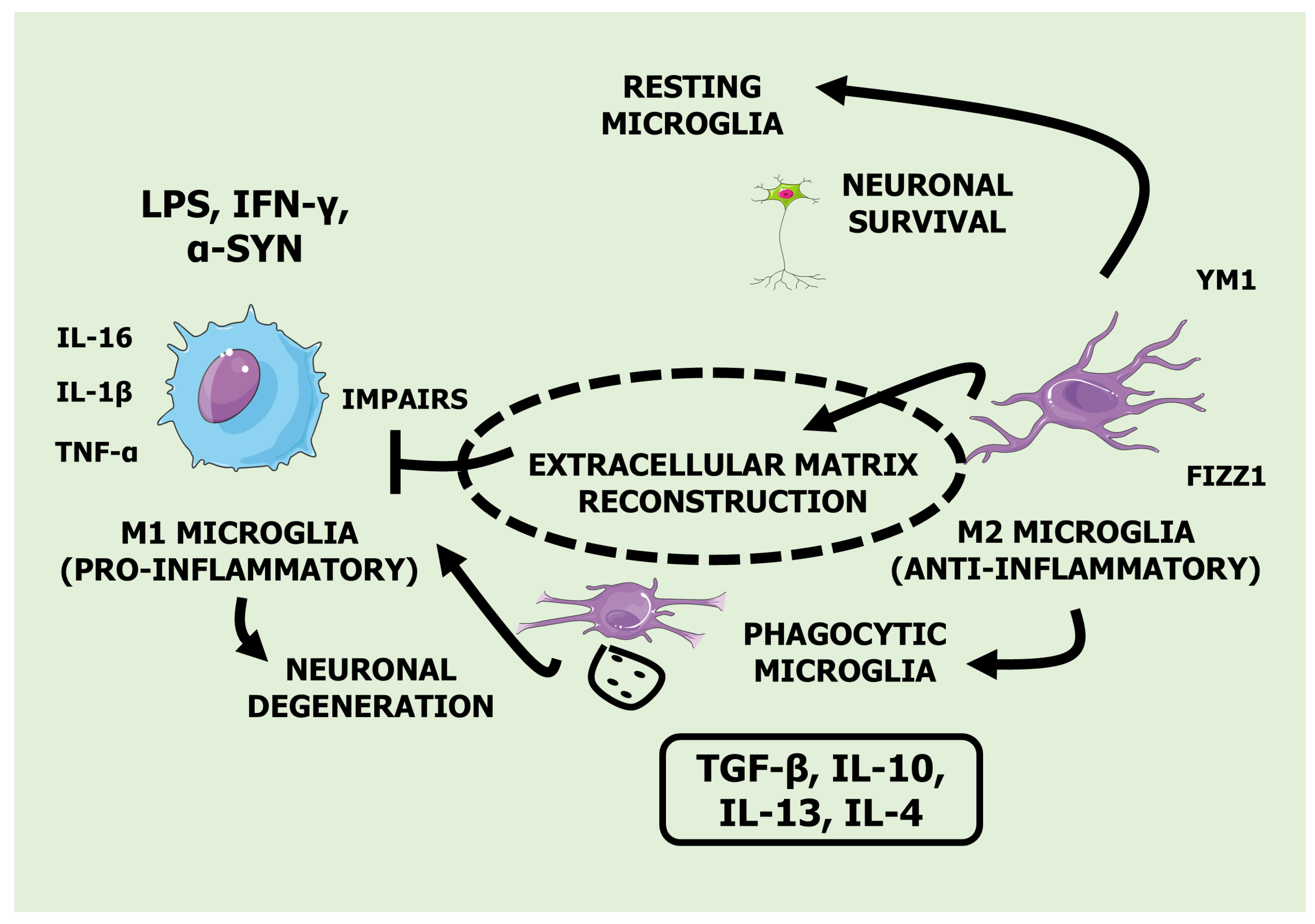
Figure 3.
Key Actions of Medicinal Plants in Countering Neuroinflammation and Related Disorders.


Table 1.
Medicinal Plants Effects on Neuroinflammation: In Vitro and In Vivo Models, Effective Dosages, Molecular Mechanisms, Clinical Implications, and Study Limitations.
Table 1.
Medicinal Plants Effects on Neuroinflammation: In Vitro and In Vivo Models, Effective Dosages, Molecular Mechanisms, Clinical Implications, and Study Limitations.
| Plants | Models | Interventions | Mechanisms | Clinical Implications | Limitations | Ref |
|---|---|---|---|---|---|---|
| Cleistocalyx nervosum var. paniala (Ferulic acid, aurentiacin, brassitin, ellagic acid, alpinetin and resveratrol) |
TNF-α-stimulated BV-2 cells in vitro | 5, 10 or 25 μg/mL CNSE incubated for 24 h in vitro | ↓ COX-2 activation, ↓ iNOS function, ↓ TNF-α, IL-6 and IL-1β mRNA expression, ↓ p38MAPK and ERK 1/2 phosphorylation, ↓ NF-κB activation, ↓ p65 and IκB phosphorylation, ↑ HO-1 induction (in vitro) | Potential for new anti-inflammatory agents targeting neurodegenerative diseases. Could pave the way for natural, multi-targeted treatments. | Findings are promising but confined to cellular models; lacking animal studies and in-depth safety profiles. | [38] |
| LPS-stimulated BV-2 cells in vitro | 1, 5, 10, 25, 50 or 100 μg/mL CNSE incubated for 24 h in vitro | ↓ NO production, ↓ iNOS mRNA expression, ↓ TNF-α, IL-6 and IL-1β mRNA expression, ↓ MAPK phosphorylation, ↓ p-JNK, p-ERK 1/2 and p-p38 levels, ↓ NF-κB activation (in vitro) | Could contribute to the development of targeted anti-inflammatory therapies with fewer side effects. | Effects are robust in vitro but lack corroboration in animal models and human trials. | [39] | |
| Curcuma longa (Curcumin, demethoxycurcumin and bisdemethoxycurcumin) |
LPS-stimulated BV-2 cells in vitro | 12.5, 25, 50, 100, 150 or 200 μg/mL CLE incubated for 24 h in vitro | ↓ NO production, ↓ PGE2 production, ↓ iNOS and COX-2 expression, ↓ TNF-α, IL-6 and IL-1β mRNA expression, ↓ NF-κB activation, ↓ IκB-α phosphorylation and degradation, ↓ p65 nuclear translocation, ↓ MAPK (p38, ERK, and JNK) phosphorylation, ↑ HO-1 expression, ↑ Nrf2 nuclear translocation (in vitro) | Could enhance treatments for neuroinflammation and oxidative stress-related disorders, offering a natural alternative to synthetic drugs. | Effective dosing varies widely, and there is limited evidence of efficacy in diverse populations or clinical settings. | [40] |
| LPS-stimulated BV-2 cells in vitro and scopolamine-induced male ICR mice in vivo | 1, 10, 50 or 50 μg/mL FCL incubated for 24 h in vitro and 50, 100 and 200 mg/kg FCL in vivo | ↓ NO production, ↓ PGE2 production, ↓ iNOS and COX-2 expression, ↑ AP-1 inhibition, ↓ NF-κB activation, ↓ p-MAPKs, ↑ AChE inhibition (in vitro) and ↑ pCREB and BDNF expression (in vivo) | May offer new avenues for treating cognitive deficits and memory impairments associated with neurodegenerative conditions. | Discrepancies between in vitro and in vivo findings highlight a need for more consistent research methodologies. | [41] | |
|
Cannabis sativa (Cannabidiol, cannabigerol, cannabidiolic acid, tetrahydrocannabinol, β-caryophyllen, caryophyllene-oxide, α-Humulene and apigenin) |
LPS-stimulated BV-2 cells in vitro | 1 μg/mL CSE incubated for 24 h in vitro | ↓ TNF-α, IL-6 and IL-1β production, ↑ AEA and 2-AG expression, ↓ JNK and p38 activation, ↓ NF-κB nuclear translocation, ↓ ROS production (in vitro) | Could be a cornerstone for novel treatments targeting neuroinflammation and chronic pain, with potential applications in psychiatric and neurological disorders. | Clinical evidence is sparse, and variability in cannabis strains and compounds makes standardization difficult. | [42] |
| Dioscorea nipponica (Dioscin) |
LPS-stimulated BV-2 cells in vitro and scopolamine-induced male C57BL mice in vivo | 10, 20, 50 or 100 μg/mL dioscin incubated for 24 h in vitro and 60 mg/kg dioscin in vivo | ↓ iNOS and COX-2 expression, ↓ NO and PGE2 production, ↓ TNF-α, IL-6 and IL-1β mRNA expression, ↓ NF-κB nuclear translocation, ↓ IκB phosphorylation, ↓ p65 nuclear translocation (in vitro) and ↑ BDNF and pCREB expression (in vivo) | May support treatments aimed at improving cognitive functions and mood disorders by targeting neuroinflammatory pathways. | More extensive studies are required to confirm efficacy, dosage safety, and long-term impacts. | [43] |
|
Centipeda minima (Chlorogenic acid, caffeic acid, rutin, isochlorogenic acid A, isochlorogenic acid B, isochlorogenic acid C and 6-O-angeloylplenolin) |
LPS-stimulated BV-2 cells in vitro and LPS-stimulated male C57BL/6J mice in vivo | 2, 4 or 6 μg/mL ECM incubated for 24 h in vitro and 100, 200 mg/kg ECM in vivo | ↓ NF-κB nuclear translocation, ↓ IκB phosphorylation, ↓ COX-2 and iNOS expression, ↓ NO and PGE2 production, ↓ NOX proteins (in vitro) and ↓ NO, PGE2, TNF-α, IL-6 and IL-1β production, ↓ NF-κB nuclear translocation, ↓ iNOS, COX-2 and NOX2 and NOX4 expression (in vivo) | Potential to develop comprehensive anti-inflammatory therapies targeting multiple pathways involved in neuroinflammation. | Variability in phytochemical compositions can complicate standardization and reproducibility. | [44] |
|
Atractylodis Rhizoma Alba (Atractylenolide I, atractylenolide III, and atractylodin) |
LPS-stimulated BV-2 cells in vitro | 10, 50, or 100 μg/mL ARAE incubated for 24 h in vitro | ↓ NO production, ↓ TNF-α, IL-6 and IL-1β mRNA expression, ↓ iNOS and COX-2 expression, ↑ HO-1 mRNA expression, ↓ NF-κB activity, ↓ MAPK, p38, ERK and JNK activation (in vitro) | May contribute to integrative approaches for treating neuroinflammation and related conditions. | Lack of long-term studies and clinical trials limits understanding of potential side effects and interactions. | [45] |
|
Vaccinium bracteatum (Quercetin, chrysin, apigenin, kaempferol, and lutelin) |
LPS-stimulated BV-2 cells in vitro | 1, 2,5, 5, 10 or 20 µg/ml VBME incubated for 24 h in vitro | ↓ NO and PGE2 production, ↓ iNOS and COX-2 expression, ↓ NF-κB p65 nuclear translocation, ↓ TNF-α, IL-6 and IL-1β levels, ↓ ROS production (in vitro) | May inspire new anti-inflammatory and antioxidant treatments with fewer side effects. | Variability in results across different studies and cell models necessitates further investigation. | [46] |
|
Lonicera japonica (Chlorogenic acid,chlorogenic acid,caffeic acid,cryptochlorogenic acid,artichoke,isochlorogenic acid A,isochlorogenic acid B,isochlorogenic acid C,rutin,hibisin and loganin) |
LPS-stimulated BV-2 cells in vitro | 0.5, 5, 2.5, 5 or 10 µg/mL LJ incubated for 24 h in vitro | ↓ NO and PGE2 production, ↓ iNOS and COX-2 mRNA expression, ↓ TNF-α, IL-1β, MCP-1 and MMP-9 production, ↓ ROS levels, ↓ p38 MAPKs, ERK 1/2, JNK and PI3K/Akt phosphorylation, ↓ JAK1/STAT1/3 phosphorylation, ↓ NF-κB nuclear translocation (in vitro) | Could lead to new treatments targeting both neuroinflammation and related oxidative stress. | Complex composition requires more research to determine the most effective components and dosages. | [47] |
|
Bambusae caulis ( (-)-7'-epi-lyoniresinol 4,9'-di-O-β-D-glucopyranoside (7), (-)-lyoniresinol 4,9'-di-O-β-D-glucopyranoside (8) and bambulignan A) |
LPS-stimulatred BV-2 cells and glutamate-stimulated hippocampal HT22 cells in vitro | 10, 20, 40, 60 or 80 μg/ml BCE incubated for 24 h in vitro | ↓ NO, TNF-α, IL-1β and IL-6 levels, ↓ iNOS and COX-2 expression, ↓ ROS production, ↑ HO-1 mRNA expression, ↑ Nrf2 nuclear translocation (in vitro) | Potential for advancing treatments against neuroinflammation and oxidative damage through modulation of key inflammatory and oxidative pathways. | Additional studies required to delineate specific active components and refine therapeutic protocols. | [48] |
| Zingiberis Rhizoma (Gingerols and shogaol) |
LPS-stimulated BV-2 cells in vitro | 1, 5 or 10 μg/ml GHE incubated for 24 h in vitro | ↓ NO and PGE2 production, ↓ COX-2 mRNA expression, ↓ TNF-α and IL-1β production, ↓ MAPK molecules, ERK1/2, p38 MAPK, and JNK phosphorylation, ↓ NF-κB nuclear translocation (in vitro) | Promising candidate for developing interventions targeting neuroinflammatory processes and related molecular pathways. | Further research is essential to optimize dosing strategies and elucidate mechanistic details. | [49] |
Abbreviations: ↑, increase; ↓, Decrease; 2-AG, 2-Arachidonoylglycerol; AChE, Acetylcholinesterase; AEA, Anandamide; AP-1, Activator Protein 1; ARAE, Atractylodis Rhizoma Alba Ethanolic Extract; BCE, Bambusae Caulis In Taeniam Ethyl Acetate; BDNF, Brain-Derived Neurotrophic Factor; CSE, Cannabis Sativa Extract; CLE, Curcuma Longa Extract; CNSE, Cleistocalyx Nervosum Ethanolic Seed Extract; COX-2, Cyclooxigenase 2; pCREB, cAMP Response Element Binding protein; ECM, Ethanol Extract of C. Minima; ERK, Extracellular Signal–Regulated Kinase; FCL, Fermented Curcuma Longa; GHE, Ginger Hexan Extract; HO-1, Heme Oxygenase 1; IL-1β, Interleukin 1β; IL-6, Interleukin 6; iNOS, inducible Nitric Oxide Synthase; Iκbα, Nuclear Factor Of Kappa Light Polypeptide Gene Enhancer In B-Cells Inhibitor, Alpha; JAK/STAT, Janus Kinase/Signal Transducer And Activator of Transcription; JNK, C-Jun N-Terminal Kinase; LJ, Lonicera Japonica; LPS, Lipopolysaccharide; MAPK, Mitogen-Activated Protein Kinase; MCP-1, Monocyte Chemoattractant Protein-1; MMP-9, Matrix Metalloproteinase 9; mRNA, messenger Ribonucleic Acid; NO, Nitric Oxide; NOX, NADPH Oxidase; Nrf2, Erythroid-Derived Nuclear Factor; NF- Κb, Nuclear Factor Kappa B; PGE2, Prostaglandin E2; PI3K, Phosphatidylinositol 3-Kinase; ROS, Reactive Oxygen Species; TNF-α, Tumor Necrosis Factor-Alpha; VBME, Vaccinium Bracteatum Methanol Extract.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



