
Submitted:
17 December 2024
Posted:
18 December 2024
You are already at the latest version
Abstract
Polyhydroxybutyrate (PHB) is a biodegradable natural polymer produced by different prokaryotes as a valuable carbon and energy storage compound. Its biosynthesis pathway requires the sole expression of the phaCAB operon, although auxiliary genes play a role in controlling polymer accumulation, degradation, granules formation and stabilisation. Due to its biodegradability, PHB is currently regarded as a promising alternative to synthetic plastics for industrial/biotechnological applications. Azohydromonas lata strain H1 has been reported to accumulate PHB by using simple, inexpensive carbon sources. Here we present the first de novo genome assembly of the A. lata strain H1. The genome assembly is over 7.7Mb in size including a circular megaplasmid of approximately 456Kbp. In addition to the phaCAB operon, single genes ascribable to PhaC and PhaA functions and auxiliary genes were also detected. A comparative genomic analysis of the available genomes of the genus Azohydromonas revealed the presence of phaCAB and auxiliary genes in all Azohydromonas species investigated, suggesting the PHB production is a common feature of the genus. Based on sequence identity, we also suggest A. australica as the closest species to which the phaCAB operon of the strain H1 reported in 1998 is similar.
Keywords:
Bioplastic
; bacteria biopolymers
; bioeconomy
; polyhydroxyalkanoates
; PHA
; biodegradable
1. Introduction
The exponential increase in fossil-derived plastic waste and the growing demand for plastics [1,2] make urgency to replace petrochemical-based plastics with biodegradable polymers [3,4,5]. Polyhydroxyalkanoates (PHAs) are a group of that are biodegradable biopolymers, resembling synthetic plastics, produced by a range of diverse prokaryotes and accumulated in cytoplasm as granules reserve of carbon and energy [6,7,8]. The homopolymer polyhydroxybutyrate (PHB) is currently regarded as one of the most promising PHA for biotechnological applications [9]. Since the growing economic interest, PHBs producing bacteria are being increasingly studied. The Gram-negatives Cupriavidus necator (formerly Ralstonia eutropha) strain H16 (DSM 428; ATCC17699), Azohydromonas lata (formerly Alcaligenes latus) strain H1 (DSM1123; ATCC 29714) and Escherichia coli expressing the PHB biosynthetic genes from these strains [10] (Pena_2014) can accumulate large amounts of PHB and are considered the most promising systems for large scale PHB production? C. necator H16, in particular, represents the model organism for the study of PHB production [11,12]. Three main distinct pathways for PHB synthesis are described [7]; in C. necator three genes, usually organized in an operon (phaCAB), are deemed sufficient for PHB biosynthesis: PHA synthase (phaC), 3-chetothiolase (phaA) and acetoacetyl-CoA reductase (phaB). PhaCs (the crucial function common to all the known pathways) are generally categorized into 4 classes (I to IV) based on amino acid sequence, in vivo substrate specificities of the enzymes and the number/composition subunits forming the catalytic complex [13]. An additional class (class V) was recently proposed by Tan and colleagues, based on the PhaC of Janthinobacterium sp. [14]. Beyond the three main genes (phaA, phaB and phaC), many auxiliary genes have been identified and characterized to encode important functions in controlling polymer accumulation and degradation. These include: the regulatory gene phaR, the depolymerase phaZ, the extracellular oligomer hydrolase phaY, and the phasin phaP involved in granules formation/stabilisation [15,16,17]. With a view on circular economy, many studies target efficient and low-cost PHB production by using, in addition to native producers, bacteria (e.g., Escherichia coli) engineered with heterologous PHB genes/operons [11], with the objective of producing PHB efficiently from low-cost biomass (e.g., whey waste, starch, wastewater) substrates. Since the availability of the reference C. necator H16 genome [18,19], the phaCAB operon, auxiliary genes associated with PHB production and isologs of PhaA (beta-ketothiolases) and PhaB (reductases) with different substrate specificity have been characterized and studied in this organism [19,20]. However, equivalent considerations do not apply to the A. lata strain H1. Expanding the repertoire of known/characterized PHB genes could provide crucial insights for the optimization of biotechnological applications based on the genetic engineering of the pathway. Here, we present the first de novo genome assembly of A. lata strain H1, whose information might be exploitable for biotechnological applications [10,12]. Although the sequence of the phaCAB operon of A.lata H1 has been previously determined [21], we reasoned that the assembly of genome sequence could offer significant advances in the identification of genes associated with PHB production and offer a more complete representation of the organization of the PHB operon together with the number and configuration of auxiliary genes and potential isologs in A. lata. Moreover, our comparative genomic analyses of phaCAB and related genes within the genus Azohydromonas, could advance the general knowledge of genomic organization and conservation of these genes and inform the design of prospective biotechnological application.
2. Materials and Methods
2.1. Genome Sequencing, Assembly and Annotation
A.lata strain H1 was obtained from the Leibniz institute DSMZ-German Collection of Microorganisms and cell culture GmbH (Germany) (strain code DSM1123). Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen Inc., Hilden, Germany) Kit (QIAGEN. Italy) and purified using Amicon Ultra-0.5. MWCO 30 kDa (Millipore, Burlington, MS, USA) according to the manufacturer’s protocols. Purity and concentration were assessed by NanoDrop spectrophotometer and Qubit 3.0 (Thermo Scientific, Italy), respectively. Library was prepared from the purified bacteria genomic DNA using Illumina DNA Prep (Illumina, San Diego, CA, USA). The sequencing was carried out using Miseq Reagent Kit v2 (500 cycles) and the Illumina MiSeq platform (Illumina, San Diego, CA, USA). Adaptor sequences and low-quality regions were removed by using Trimmomatic with default parameters [22]. Overlapping paired-end reads were merged by Pear [23]. Genome assembly was performed by the Unicycler workflow, using the SPAdes assembler [24; GATK team]. The following kmers sizes were used: 27,53,71,87,99,111,119,127. Contigs shorter than 200 bp in size were discarded. The genome was deposited at NCBI under the GenBank accession number GCA_034427735.1. Gene annotation was performed by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [25]. Annotation of rRNA were manually refined by performing blastn (v2.9.0-2) [26] sequence similarity searches and manual alignment with the 5S, 16S and 23S rRNA genes from the draft genome assembly of the A. lata type strain NBRC 102462 (GenBank accession number GCA_001571085.1). CRISPR-Cas systems and CRISPR array(s) were annotated by CRISPRCasFinder with default parameters [27]. Potential self-targeting of spacers was assessed by blastn sequence similarity searches of the spacers’ sequences in the genome. Default parameters were used and only matches showing an e-value ≤ e-50 and identity ≥ 90% were considered. Spacers sequences were also searched in the NCBI nr/nt (https://ftp.ncbi.nlm.nih.gov/blast/db/) database to assess the presence of sequences of exogenous origin. Default parameters were used. CRISPR array subtype was inferred based on sequence similarity with the most similar sequence in the NCBI nr/nt database (https://ftp.ncbi.nlm.nih.gov/blast/db/). Candidate transposases, resistance genes and Toxin-Antitoxin (TA) systems were inferred according to the PGAP annotation. General features and genome statistics were computed using custom scripts written in Python (v 3.8.10) and R (v 4.2.0) and standard bash shell utilities. The circular map of the candidate plasmid sequence was generated with the web-based implementation of Proksee [28].
2.2. ANI and dDDH
Average Nucleotide Identity (ANI) and Digital DNA-DNA hybridization (dDDH) were computed by considering the complete collection of publicly available Azohydromonas genome assemblies accessible through NCBI Refseq (https://www.ncbi.nlm.nih.gov/assembly/?term=Azohydromonas) in December 2023 (Table S1). ANI values were computed by OrthoAniU (Lee et. al., 2016). dDDH values were determined by Genome-to-Genome-Distance-Calculator (GGDC-3.0) with recommended settings [29].
2.3. PHB Genes Detection and Class Designation of PhaCs
PHB genes were annotated by blastp sequence similarity searchers with PHB genes from C. necator H16 (GeneBank AM260479.1) [19]. Only blastp matches showing an e-value ≤ e-50 and identity ≥ 40% were considered. Candidate PhaC protein sequences from A. lata H1 were assigned to one of the five classes of PhaC enzymes based on similarity, as determined by blastp searches (with default parameters), with respect to an arbitrarily selected collection of representative sequence for every class (Table S2). Only matches with an e-value ≤ e-50 were considered, and the class was assigned based on the best blastp match. Candidate PhaC protein sequences were aligned by using the online version of Clustal Omega [30], the alignment was visualized with JalViewer [31].
3. Results
3.1. Genome Assembly of A. lata H1
The genome assembly of A. lata strain H1 is accessible at GenBank under the accession number GCA_034427735.1. The draft genome is over 7.7Mb in size and includes a circular megaplasmid of approximately 456Kbp (Figure 1 and GenBank: JAXOJX010000001.1).
The genome assembly consists of 302 scaffolds and the N50 value is 89,247 bp. Descriptive statistics are reported in Table 1.
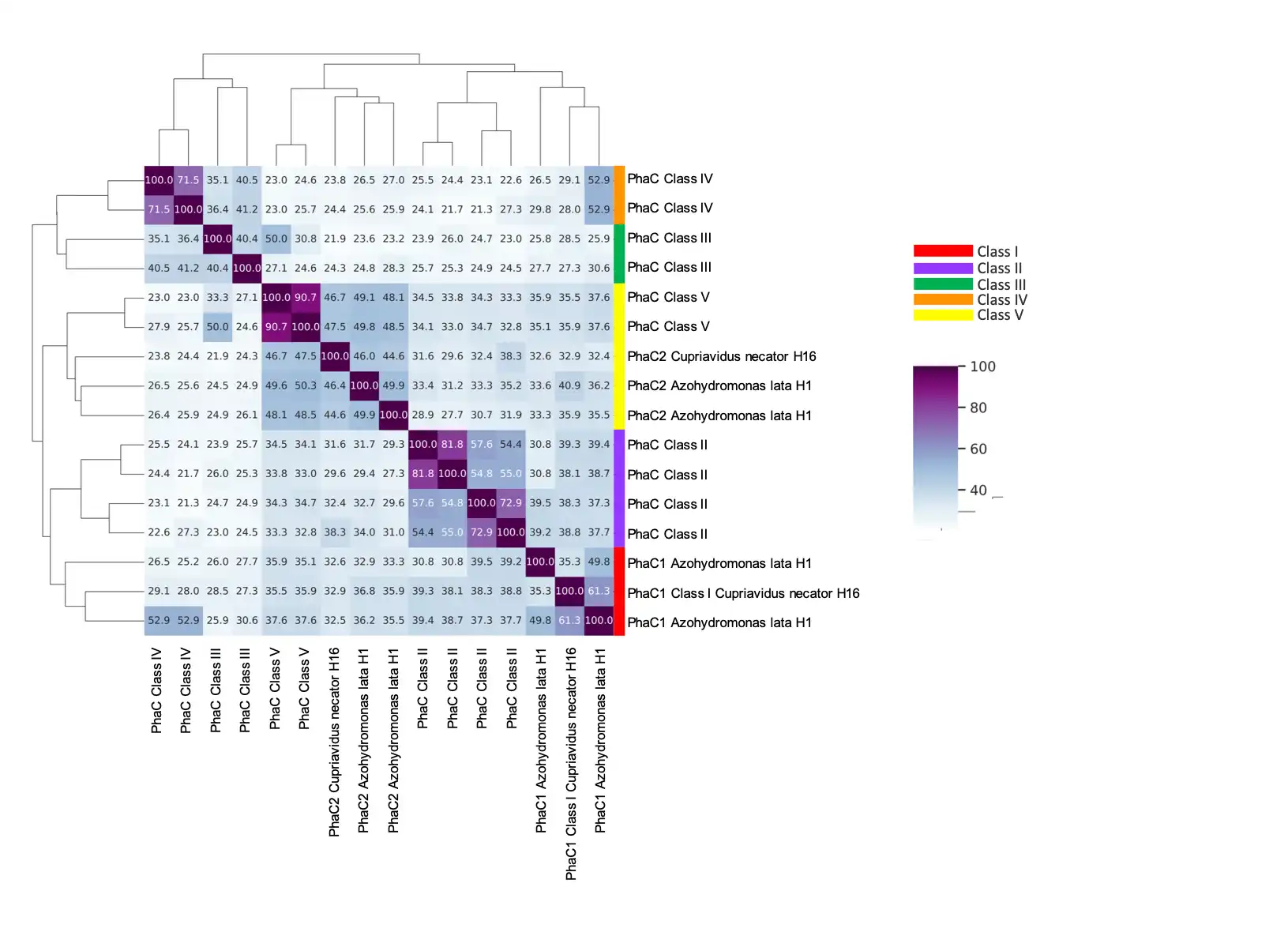
The G+C content is 65.6% for the megaplasmid and 68.7% for the remaining scaffolds. A total of 1 rRNA 5S, 3 rRNAs 16S, 2 rRNAs 23S, and 56 tRNAs genes were identified; none of these are localized in the megaplasmid. The PGAP annotation predicted 6945 protein coding genes, of which 408 on the megaplasmid; a putative function was assigned to 5933 proteins (megaplasmid 300), while 14,5% of the proteins (1012) were annotated as “hypothetical proteins”. A total of 49 genes encoding putative transposases were annotated by PGAP; 16 transposases were localized on the megaplasmid and 10 formed a potential cluster spanning approximately 100 Kb in size (Figure 1 and Figure 2). Based on the PGAP annotation, 35 transposases were assigned to 8 different families while 14 were not assigned to a family (Table 2, Table S3).
TA systems play a role in maintaining plasmids in bacteria cells [32]. They usually consist of two proteins (antitoxin and toxin) encoded by two juxtaposed genes, organized in operon with antitoxin gene more frequently upstream the toxin gene [33]. TA operons characterized by the toxin gene upstream the antitoxin have been recently reported [34]. Based on the nature (protein or RNA) and mode of action of antitoxins, TA systems have been classified into different classes [33] with type II systems being the most abundant and extensively studied. Candidate TA systems were manually searched in the DSM1123 megaplasmid inspecting candidate toxin and antitoxin genes, according to the PGAP annotation. Two distinct putative type II TAs were identified in the A. lata megaplasmid (Table 3).
The first system (system II-A hereafter) belongs to the RelE/ParE family and is composed of two protein coding genes: the toxin (locus tag SM757_01580) and the HigA antitoxin (locus tag SM757_01585). The second system (system II-B hereafter) has a similar configuration being formed by the RelE/ParE toxin (locus tag SM757_01815) and HigA antitoxin (locus tag SM757_01820). The system II-B includes an additional gene (locus tag SM757_01810) encoding for VapB antitoxin. Whereas three components type II TA systems have been previously described [33; Toxin-Antitoxin Database http://bioinfo-mml.sjtu.edu.cn/TADB2/index.php], to the best of our knowledge, this is the first reported instance of a potential VapB/RelE/HigA TA system. Interestingly, 10 Kbp upstream of system II-B the presence of a predicted antitoxin AbiEi domain protein (locus tag SM757_01845), which could represent a partial abortive type IV system (AbiEi/AbiEii) [35], was also noticed. All the candidate TAs co-localise in the same plasmid region adjacent to the cluster of the 10 predicted transposase genes described above (Figure 1).
3.2. Nucleotide Identity Levels in the Genus Azohydromonas
The current taxonomic classification of A. lata collocates the species in the family Alcaligenaceae, order Burkholderiales. Recently, Mogro and colleagues have published a phylogeny of the genus supporting the classification of Azohydromonas in the family Comamonadaceae [36]. Phenetic clustering of nucleotide identity levels among Azohydromonas genome assemblies as available in December 2023 (Table S1), indicates the reference strain A. lata NBRC102462 as the most similar sequence to A. lata H1 (Figure 3), with an ANI of 98.7%, which is well within the range observed between isolates of the same species [37]. At the species level A. aeria (84.5), A. australica (85.7), A. caseinilytica (84.6) and an uncultured Azohydromonas specimen (84.8) had the highest level of sequence identity with A. lata. Nearly identical patterns of genome identity levels were recovered also when similarity metrics based on dDDH [38] were considered (Figure 4).
Further, our ANI and dDDH analyses suggest high levels of sequence identity between A. sediminis (strains YIM 73032 and SYSU G00088) and Calidifontimicrobium sp strain SYSU G02091, but a relatively low (ANI~76%) level of sequence identity between these species and the other Azohydromonas species. In consideration of the fact that genome sequence identity levels ranging in between 72-75% are normally used to delineate a bacterial genus [37], these patterns of genome identity levels - along with the observation that A. sediminis strains present considerably smaller genomes compared other Azohydromonas (average size 3.6 Mb w.r.t 7.2Mb, Table S4)- might advocate reclassification of Calidifontimicrobium sp. strain SYSU G02091 under the genus Azohydromonas, or eventually suggest the classification of the three strains (YIM 73032, SYSU G00088 and SYSU G02091) under a genus different from Azohydromonas.
3.3. CRISPR-Cas Systems
Soil is the environment from which A. lata H1 has been isolated. In this complex environment exogenous genetic elements such as plasmids and phages, may represent a life threat (e.g., phages) or a metabolic burden (e.g., plasmids). Invasion of such elements is counteracted by the adaptive immunity defence mechanism CRISPR–Cas system. Studying the presence/absence of CRISPR–Cas systems in the genome allows to better delineate the strain adaptation in its natural habitat based on the balance between protection provided by CRISPR systems and their possible deleterious effects (e.g., self--targeting spacers), the role played by exogenous genetic elements (e.g., plasmids, phages, etc.) in strain evolution and the horizontal transfer of CRISPR system. A CRISPR array was identified in the draft assembly of the A. lata H1 genome (contig JAXOJX000000044 25075-26262) by CRISPRCasFinder [27]. However, no cas genes were annotated by PGAP. The identified CRISPR array contained 20 direct repeats (DRs) with the consensus DR sequence GTATTTCCCGCGCGAGCGGGGATAAACCG showing highest level of similarity with putative subtype I-E DRs in the genome assembly of Cronobacter sakazakii (GenBank accession number GCA_009648895.1). Sequence similarity searches throughout the genome of A. lata H1 did not indicate any potential self-targeting spacer. The candidate spacers sequences did not show any detectable protospacer by similarity with publicly available plasmids and/or phage sequences, suggesting that the spacers’ protospacers might not have been sequenced. Although we cannot exclude that the lack of a set of cas genes in the genome might result from incomplete assembly or inaccurate annotation, the observations reported above might be compatible with an exogenous origin of the identified CRISPR array. Alternatively, a CRISPR array not associated with known cas genes might indicate a possible role of the array beyond the adaptive immunity (e.g., regulatory function) [39] or association with Cas proteins yet to be identified.
3.4. Resistance Proteins
As stated in the previous section, A.lata H1 was purified from soil samples collected in Berkeley University (California) before September 1977. Soil bacteria might be exposed to a variety of environmental stimuli, including competition with other microbial populations and/or exposure to chemical pollutants. Hence, the identification of candidate resistance genes might be an indication of the adaptation of the A. lata H1 to competitive habitats. A total of 11 ORFs possibly associated with resistance proteins were annotated by PGAP (Table 4).
None of these are localized on the megaplasmid. These candidate resistance genes can be broadly categorized in 6 different classes: heavy metals resistance (arsenical-cadmium (SM757_03940), cobalt-zinc-cadmium (SM757_03785), toxic metal(loid) resistances (4 genes tellurite TerB SM757_05525, SM757_19280, SM757_24300, SM757_24860), toxic compounds resistance (chromate resistance 3 genes, SM757_03450, SM757_03465, SM757_05070), glyoxalase/bleomycin/dioxygenase resistance (SM757_12920), and organic hydroperoxide resistance (SM757_22515). Although in the absence of an experimental validation predicted patterns of resistance cannot be confirmed, we speculate that the presence of genomic regions encoding for 6 different families/superfamilies of resistance genes suggests a possible wide resilience of A. lata H1 to different environments. This is consistent with the environmental origin of the strain.
3.5. Annotation of PHB Related Genes
The ability of the A. lata strain H1 to produce PHB is an attractive feature for biotechnological applications. Cloning and molecular analysis of the strain’s operon phaCAB has been previously reported by Choi et al. [21]. Here we report a detailed description of the phaCAB operon and auxiliary genes related to polyhydroxybutyrate production in the genome of A. lata H1. The model organism C. necator H16 was used as a query to identify candidate orthologous genes in the proteome of A.lata H1. A total of 27 PHB related genes were identified (Table 5 and Table S5). Of these, 4 were annotated as phaC, 1 as phaB and 6 as phaA. An arrangement of genes compatible with a phaCAB operon was identified at genomic coordinates JAXOJX010000042 5833-9548 as follows SM757_22330, poly(R)-hydroxyalkanoic acid synthase (phaC) (1611pb); SM757_22335, acetyl-CoA acetyltransferase (phaA) (1179bp); and SM757_22340 acetoacetyl-CoA reductase (phaB) (738bp); 3 extra phaCs (named phaC1 and phaC2 in coherence with C. necator H16 annotation) and 5 extra phaA (SM757_00745 on the megaplasmid) were predicted. These extra pha genes did not show an operon architecture (e.g phaCA) as previously described in other species [16,40]. The PHA synthase encoded by phaC1 gene in the phaCAB operon reported for C. necator H16 and A. lata H1, was already classified as class I [13] (Rhem, 2003). To establish the class of the predicted extra PhaCs, protein sequence similarity with respect to a selection of representative protein from class I to V PhaCs [14,41] was used (Table S2). Based on this analysis, the A. lata PhaC1s (SM757_22330 and SM757_05625) are putatively assigned to the class I while the PhaC2s (SM757_29010 and SM757_26710) were assigned to class V (Figure 5, Table S6).
Rhem and colleagues [13] identified 8 key amino acid residues (S260, C319, G322, D351, W425, D480, G507 and H508) by referring to C. necator H16 PhaC1 that are universally conserved in all PHA synthases suggesting an important role of those residues in protein function. The conservation of these aminoacids was more recently confirmed by Tan and colleagues also in Class V PhaCs [14]. We performed an equivalent analysis for both the C. necator H16 PhaC2 and the candidate extra PhaCs annotated in A. lata H1. All the 8 key residues, including the putative catalytic triad (C319, D480 and H508 based on C. necator H16 PhaC1), were conserved (Figure 6; Figure S1). Isologs of PhaA (beta-ketothiolases) and PhaB (reductases) with different substrate specificity have been previously reported in C. necator H16 [19,20]. Based on the PGAP annotation 7 candidate PhaA isologs (thiolases) were identified in A. lata H1, among these one (SM757_01140) is encoded on the megaplasmid (Table 5 and Table S5). Based on the same criteria, 3 candidates PhaB isologs (reductase) have been predicted (ORFs SM757_08215, SM757_31855 and SM757_23050). Interestingly, our sequence similarity analyses identified in the genome of A. lata H1 at least one candidate homolog for all of the auxiliary PHA genes described in the model C. necator H16, including: two phaY hydrolases (locus tags SM757_26695 and SM757_23895); two polyhydroxyalkanoate depolymerase phaZ (locus tags SM757_08600 and SM757_15980); one phaP phasin family protein (locus tag SM757_26850) and a phaR transcriptional regulator (locus tag SM757_25825).
3.6. PhaCAB and Auxiliary Genes in the Azohydromonas Genus
The same procedure used for the annotation of candidate pha genes in Azohydromas lata H1, was applied to the complete collection of publicly available Azohydromonas spp. genome assemblies (Table S1). Candidate phaA, phaB and phaC genes as well as auxiliary PHA genes and putative isologs were recovered in all the genomes considered. Potentially intact phaCAB operons were observed in: A. lata NBRC 102462, Calidifontimicrobium sp. SYSU G02091, A. sediminis (SYSU G00088 and YIM 73032), A. caseinilytica G-1-1-1-4, whereas in Azohydromonas aeria, Azohydromonas australica and the uncultured Azohydromoas sp. a partial phaCA operon was identified. In these last three cases however both genes were consistently localized at one of the extremities of a contig in all the genome assemblies, potentially suggesting possible issues in the assembly due to multiple copies of the operon. In line with this hypothesis, a complete candidate phaB gene was annotated in A. aeria and A. australica in a non-adjacent region with respect to the phaCA incomplete operon. Further, we observed that A. aeria and A. australica were also associated with an increased number of candidate PHB genes, auxiliary genes and isologs (Table S4). Conversely, and probably due to their reduced genome size, Calidifontimicrobium sp. SYSU G02091, A. sediminis strains SYSU G00088 and YIM 73032, displayed a more limited repertoire of candidate PHB related genes and isologs. The sequence of the phaC, phaA and phaB from the phaCAB operon, both annotated by our analyses in the genome of A.lata H1 and reported by Choi et. al. [21], were independently compared with candidate phaCAB genes from other Azohydromonas spp. As expected, high levels of similarity were observed between the A.lata H1 and the A. lata type strain NBRC102462 genes (range 99.1%-99.8%) while, the sequences reconstructed by Choi had the highest levels of similarity (range 94.3%-100%) with Azohydromonas australica DSM 1124 phaCAB genes (Table 6, Table 7 and Table 8). These results indicate that the phaCAB operon described by Choi et. al. was probably isolated from A. australica. It might be explained in the light of the recent reclassification/split of the presumed species of isolation, Alcaligenes latus strains IAM 12599T and IAM 12664 into 2 distinct species: A. lata and A. australica by Xie and colleagues [42].
4. Discussion
The increasing usage of plastic in many anthropic activities has determined a global environmental crisis of plastic pollution with serious risks for the animal and human health. PHA is a natural-based polymer suitable to produce bioplastic and represents an eco-friendly alternative to synthetic plastic. Here we report the first draft genome assembly of Azohydromonas lata H1, a PHB producing strain with potential biotechnological applications. Interestingly the genome assembly revealed the presence of a megaplasmid of 456 Kbp, not reported in the genome assembly of the reference strain A. lata NBRC 102462. The megaplasmid harbours two distinct type II TA systems (here named II-A and II-B). The II-B system is, to the best of our knowledge, the first report of a VapB/RelE/HigA three component system. Sequence similarity matrix based on ANI and dDDH confirmed the taxonomic assignment of A. lata within the genus Azohydromonas. However, patterns of genome sequence identity, coupled with the observation of substantial differences in the size of the genome, might advocate for a partial revision of the genus Azohydromonas itself. More specifically, A. sediminis (YIM 73032, SYSU G00088) and Calidifontimicrobium sp (SYSU G02091) presented levels of ANI around 76% with all the other Azohydromonas species, a value that is considered borderline for the delineation of bacterial genus; moreover, the size of the genome was significantly reduced in these species (~ 3.8Mb compared to ~7.4Mb of other Azohydromonas species, supplementary Table 4). Further, since the high level of sequence identity A. sediminis (YIM 73032, SYSU G00088) and Calidifontimicrobium sp. (SYSU G02091), they should be reclassified under the same species. By focusing on genes implicated in PHB production and granules formation, beyond the phaCAB operon, several auxiliary genes associated with PHB utilization and granules formation were identified in A.lata H1. These include phaR, phaP, phaY, as well as extra copies of phaA and phaC and a number of potential phaA and phaB isologs. In total, 4 distinct phaC (2 PhaC1 and 2 PhaC2 based on C. necator H16 annotation) genes were identified by our analyses. Based on sequence similarity, the 2 PhaC1 were putatively assigned to the class I and the 2 PhaC2 to class V potentially suggesting a broad substrate specificity for PHB production by A. lata H1, even though experimental validation is required to corroborate the preferable substrate of the isolate. Our analyses indicate the presence of PHB and related genes in all the Azohydromonas genome assemblies considered, suggesting that all the species of the genus Azohydromonas are endowed with the molecular machinery for PHB production. Interestingly, according to the observed patterns of PHB gene distribution, A. australica is the species with the largest repertoire of PHB genes within the genus Azohydromonas, and speculations purely based on gene dosage/gene number would suggest high levels of PHB production in this species. Consistent with this hypothesis and in consideration of our sequence similarity analyses, we suggest that the phaCAB described by Choi et.al. in 1998 [21] - which was isolated from a specimen with “high concentration with high productivity” of PHB - is assigned to A. australica and not A. lata H1. Results of this study in addition to the first draft genome sequence of A. lata strain H1 supplies a comprehensive delineation of the genetic repertoire of PHB genes in the genus Azohydromonas and underlines the importance of comparative genomics for informing the design of biotechnological application based on microbial species. By using modern approaches based on genome sequence and identity/similarity metrics we derive a more precise and unequivocal identification of the species of origin of the phaCAB operon originally described by Choi et. al. in 1998 [21], and we speculate that - at least in A. australica - increased PHB production might depend on PHB genes number/gene dosage, rather than on the optimization of the catalytic activity of a specific enzyme. This observation coupled with the widespread distribution of PHB related genes across the genus Azohydromonas, as evidenced by our analyses, prompts for further functional studies for a more accurate characterization of levels of PHB production, the underlying molecular pathways and potential biotechnological applications of Azohydromonas.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: MSA of candidate PhaC proteins aligned by using the online version of Clustal Omega and visualized with JalViewer; Table S1: Azohydromonas assemblies used for nucleotide identity level analysis (ANI and dDDH); Table S2: PhaCs protein sequences considered to assign class to the PhaCs of Azohydromonas lata H1; Table S3: Predicted transposases; Table S4: Genomic features among available assemblies of Azohydromonas genus; Table S5: PHB genes in A. lata H1; Table S6: Results from the blastp similarity search for the assignment of PhaC class based on identity values.
Author Contributions
Conceptualization, M.S.; Investigation, D.T., C.P. and P.D.; Methodology, D.T., L.T., G.C. and C.M.; Writing – original draft, D.T., P. D. and M.S.; Writing – review and editing, C.P., M.C., G.P. and M.S.; Supervision, C.P., M.C. and M.S.; Software, D.T. and P.D.; Formal analysis, L.T., M.O., C.C., G.C. and C.M; Data Curation, A.M., C.M., C.C. and M.O.; Visualization, A.M., C.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by ELIXIR-IT through the empowering project ELIXIRNextGenIT (Grant Code IR0000010 to G.P.).
Data Availability Statement
All sequence data are publicly available at NCBI under the GenBank accession number GCA_034427735.1. All data generated or analyzed during this study are included in the results All sequence data are publicly available at NCBI under the GenBank accession number GCA_034427735.1. All data generated or analyzed during this study are included in the results section or in the supplementary material of this paper. Scripts and auxiliary files generated for the current study are available at Zenodo in the repository at link.
Acknowledgments
We thank Vito Emanuele Carofiglio (EggPlant s.r.l.), Domenico Centrone (EggPlant s.r.l.) and Luca Sconosciuto (EggPlant s.r.l.) for providing A. lata strain H1 (DSM1123).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Priyadarshini, R.; Palanisami, T.; Pugazhendi, A.; Gnanamani, A.; Parthiba Karthikeyan, O. Editorial: Plastic to Bioplastic (P2BP): A Green Technology for Circular Bioeconomy. Front Microbiol 2022, 13, 851045, . [CrossRef]
- Zaman, A.; Newman, P. Plastics: are they part of the zero-waste agenda or the toxic-waste agenda? Sustainable Earth 2021, 4, . [CrossRef]
- Bhavsar, P.; Bhave, M.; Webb, H.K. Solving the plastic dilemma: the fungal and bacterial biodegradability of polyurethanes. World J Microbiol Biotechnol 2023, 39, 122, . [CrossRef]
- Pandey, P.; Dhiman, M.; Kansal, A.; Subudhi, S.P. Plastic waste management for sustainable environment: techniques and approaches. Waste Dispos Sustain Energy 2023, 1-18, . [CrossRef]
- Rosenboom, J.G.; Langer, R.; Traverso, G. Bioplastics for a circular economy. Nat Rev Mater 2022, 7, 117-137, . [CrossRef]
- Madison, L.L.; Huisman, G.W. Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol Mol Biol Rev 1999, 63, 21-53, . [CrossRef]
- Obruca, S.; Dvorak, P.; Sedlacek, P.; Koller, M.; Sedlar, K.; Pernicova, I.; Safranek, D. Polyhydroxyalkanoates synthesis by halophiles and thermophiles: towards sustainable production of microbial bioplastics. Biotechnol Adv 2022, 58, 107906, . [CrossRef]
- Obruca, S.; Sedlacek, P.; Slaninova, E.; Fritz, I.; Daffert, C.; Meixner, K.; Sedrlova, Z.; Koller, M. Novel unexpected functions of PHA granules. Appl Microbiol Biotechnol 2020, 104, 4795-4810, . [CrossRef]
- McAdam, B.; Brennan Fournet, M.; McDonald, P.; Mojicevic, M. Production of Polyhydroxybutyrate (PHB) and Factors Impacting Its Chemical and Mechanical Characteristics. Polymers (Basel) 2020, 12, . [CrossRef]
- Pena, C.; Castillo, T.; Garcia, A.; Millan, M.; Segura, D. Biotechnological strategies to improve production of microbial poly-(3-hydroxybutyrate): a review of recent research work. Microb Biotechnol 2014, 7, 278-293, . [CrossRef]
- Reinecke, F.; Steinbuchel, A. Ralstonia eutropha strain H16 as model organism for PHA metabolism and for biotechnological production of technically interesting biopolymers. J Mol Microbiol Biotechnol 2009, 16, 91-108, . [CrossRef]
- Wang, B.; Sharma-Shivappa, R.R.; Olson, J.W.; Khan, S.A. Upstream process optimization of polyhydroxybutyrate (PHB) by Alcaligenes latus using two-stage batch and fed-batch fermentation strategies. Bioprocess Biosyst Eng 2012, 35, 1591-1602, . [CrossRef]
- Rehm, B.H. Polyester synthases: natural catalysts for plastics. Biochem J 2003, 376, 15-33, . [CrossRef]
- Tan, I.K.P.; Foong, C.P.; Tan, H.T.; Lim, H.; Zain, N.A.; Tan, Y.C.; Hoh, C.C.; Sudesh, K. Polyhydroxyalkanoate (PHA) synthase genes and PHA-associated gene clusters in Pseudomonas spp. and Janthinobacterium spp. isolated from Antarctica. J Biotechnol 2020, 313, 18-28, . [CrossRef]
- Brigham, C.J.; Reimer, E.N.; Rha, C.; Sinskey, A.J. Examination of PHB Depolymerases in Ralstonia eutropha: Further Elucidation of the Roles of Enzymes in PHB Homeostasis. AMB Express 2012, 2, 26, . [CrossRef]
- Kutralam-Muniasamy, G.; Marsch, R.; Perez-Guevara, F. Investigation on the Evolutionary Relation of Diverse Polyhydroxyalkanoate Gene Clusters in Betaproteobacteria. Journal of molecular evolution 2018, 86, 470-483, . [CrossRef]
- Zhao, H.; Wei, H.; Liu, X.; Yao, Z.; Xu, M.; Wei, D.; Wang, J.; Wang, X.; Chen, G.Q. Structural Insights on PHA Binding Protein PhaP from Aeromonas hydrophila. Scientific reports 2016, 6, 39424, . [CrossRef]
- Little, G.T.; Ehsaan, M.; Arenas-Lopez, C.; Jawed, K.; Winzer, K.; Kovacs, K.; Minton, N.P. Complete Genome Sequence of Cupriavidus necator H16 (DSM 428). Microbiol Resour Announc 2019, 8, . [CrossRef]
- Pohlmann, A.; Fricke, W.F.; Reinecke, F.; Kusian, B.; Liesegang, H.; Cramm, R.; Eitinger, T.; Ewering, C.; Potter, M.; Schwartz, E.; et al. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat Biotechnol 2006, 24, 1257-1262, . [CrossRef]
- Slater, S.; Houmiel, K.L.; Tran, M.; Mitsky, T.A.; Taylor, N.B.; Padgette, S.R.; Gruys, K.J. Multiple beta-ketothiolases mediate poly(beta-hydroxyalkanoate) copolymer synthesis in Ralstonia eutropha. J Bacteriol 1998, 180, 1979-1987, . [CrossRef]
- Choi, J.I.; Lee, S.Y.; Han, K. Cloning of the Alcaligenes latus polyhydroxyalkanoate biosynthesis genes and use of these genes for enhanced production of Poly(3-hydroxybutyrate) in Escherichia coli. Appl Environ Microbiol 1998, 64, 4897-4903, . [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114-2120, . [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614-620, . [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology: a journal of computational molecular cell biology 2012, 19, 455-477, . [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 2016, 44, 6614-6624, . [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 2004, 32, W20-25, . [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 2018, 46, W246-W251, . [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res 2023, 51, W484-W492, . [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Goker, M. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res 2022, 50, D801-D807, . [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology 2011, 7, 539, . [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189-1191, . [CrossRef]
- Qi, Q.; Kamruzzaman, M.; Iredell, J.R. The higBA-Type Toxin-Antitoxin System in IncC Plasmids Is a Mobilizable Ciprofloxacin-Inducible System. mSphere 2021, 6, e0042421, . [CrossRef]
- Qiu, J.; Zhai, Y.; Wei, M.; Zheng, C.; Jiao, X. Toxin-antitoxin systems: Classification, biological roles, and applications. Microbiological research 2022, 264, 127159, . [CrossRef]
- Boss, L.; Gorniak, M.; Lewanczyk, A.; Morcinek-Orlowska, J.; Baranska, S.; Szalewska-Palasz, A. Identification of Three Type II Toxin-Antitoxin Systems in Model Bacterial Plant Pathogen Dickeya dadantii 3937. International journal of molecular sciences 2021, 22, . [CrossRef]
- Hampton, H.G.; Jackson, S.A.; Fagerlund, R.D.; Vogel, A.I.M.; Dy, R.L.; Blower, T.R.; Fineran, P.C. AbiEi Binds Cooperatively to the Type IV abiE Toxin-Antitoxin Operator Via a Positively-Charged Surface and Causes DNA Bending and Negative Autoregulation. J Mol Biol 2018, 430, 1141-1156, . [CrossRef]
- Mogro, E.G.; Cafiero, J.H.; Lozano, M.J.; Draghi, W.O. The phylogeny of the genus Azohydromonas supports its transfer to the family Comamonadaceae. Int J Syst Evol Microbiol 2022, 72, . [CrossRef]
- Barco, R.A.; Garrity, G.M.; Scott, J.J.; Amend, J.P.; Nealson, K.H.; Emerson, D. A Genus Definition for Bacteria and Archaea Based on a Standard Genome Relatedness Index. mBio 2020, 11, . [CrossRef]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 2009, 106, 19126-19131, . [CrossRef]
- Devi, V.; Harjai, K.; Chhibber, S. CRISPR-Cas systems: role in cellular processes beyond adaptive immunity. Folia Microbiol (Praha) 2022, 67, 837-850, . [CrossRef]
- Wan, J.H.; Ng, L.M.; Neoh, S.Z.; Kajitani, R.; Itoh, T.; Kajiwara, S.; Sudesh, K. Complete genome sequence of Aquitalea pelogenes USM4 (JCM19919), a polyhydroxyalkanoate producer. Arch Microbiol 2023, 205, 66, . [CrossRef]
- Zain, N.A.; Ng, L.M.; Foong, C.P.; Tai, Y.T.; Nanthini, J.; Sudesh, K. Complete Genome Sequence of a Novel Polyhydroxyalkanoate (PHA) Producer, Jeongeupia sp. USM3 (JCM 19920) and Characterization of Its PHA Synthases. Curr Microbiol 2020, 77, 500-508, . [CrossRef]
- Xie, C.H.; Yokota, A. Reclassification of Alcaligenes latus strains IAM 12599T and IAM 12664 and Pseudomonas saccharophila as Azohydromonas lata gen. nov., comb. nov., Azohydromonas australica sp. nov. and Pelomonas saccharophila gen. nov., comb. nov., respectively. Int J Syst Evol Microbiol 2005, 55, 2419-2425, . [CrossRef]
Figure 1.
Circular map of the candidate plasmid generated with the web-based implementation of Proksee. Orange: CDS univocally annotated by PGAP; gray: CDS without a predicted function; blue: predicted transposases; green: TA system inferred proteins.
Figure 1.
Circular map of the candidate plasmid generated with the web-based implementation of Proksee. Orange: CDS univocally annotated by PGAP; gray: CDS without a predicted function; blue: predicted transposases; green: TA system inferred proteins.
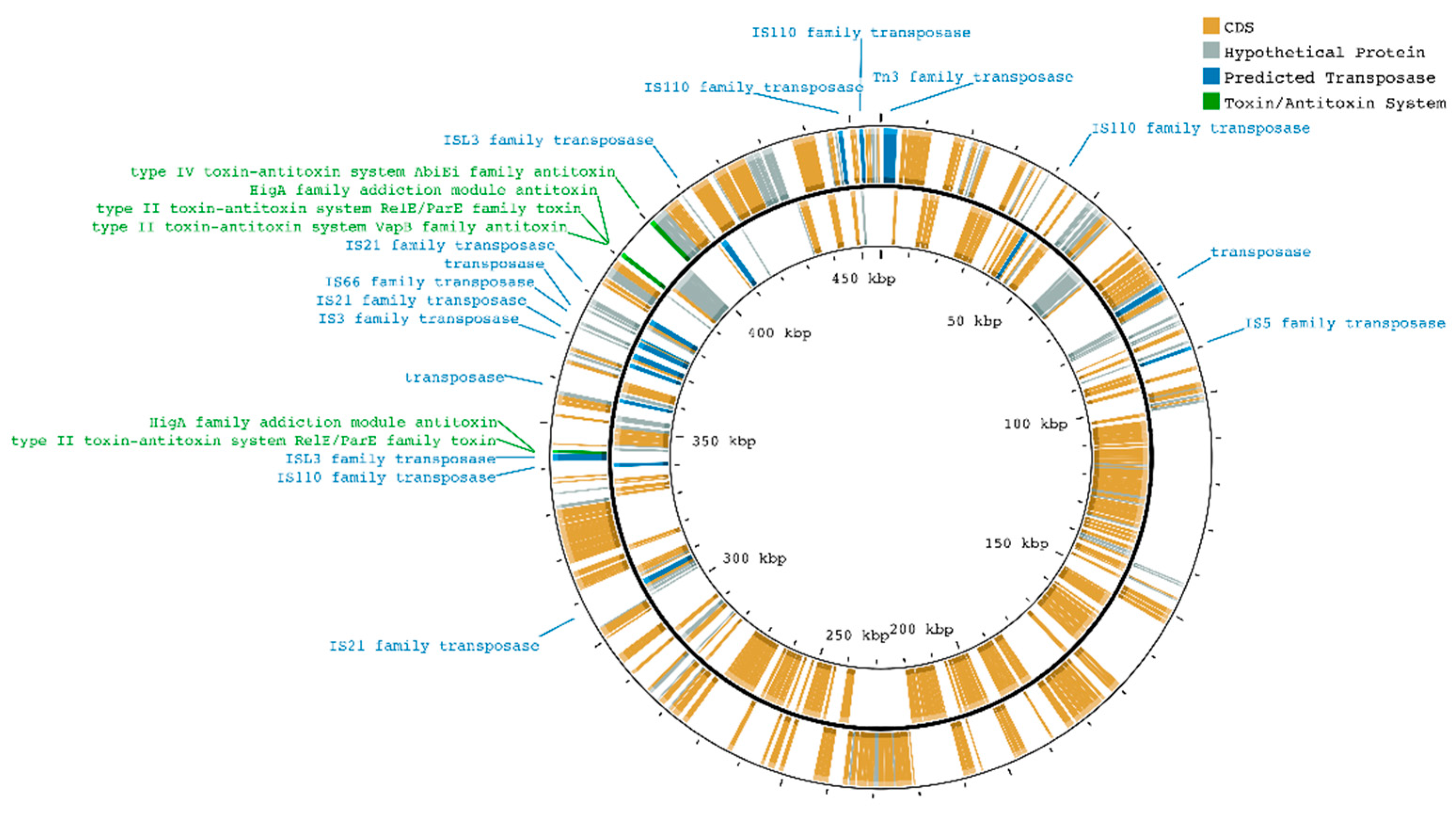
Figure 2.
Linear representation of the transposes localized on the megaplasmid. The red line corresponds to the plasmid reported in a linearized fashion while the blue bars spanning above are the projections of the predicted transposases on the plasmid. The potential cluster of transposases is visible on the last quarter (from 310 kbp to 410 kbp) of the plasmid from the IS21 family transposase (left side) to the ISL3 family transposase (right side).
Figure 2.
Linear representation of the transposes localized on the megaplasmid. The red line corresponds to the plasmid reported in a linearized fashion while the blue bars spanning above are the projections of the predicted transposases on the plasmid. The potential cluster of transposases is visible on the last quarter (from 310 kbp to 410 kbp) of the plasmid from the IS21 family transposase (left side) to the ISL3 family transposase (right side).
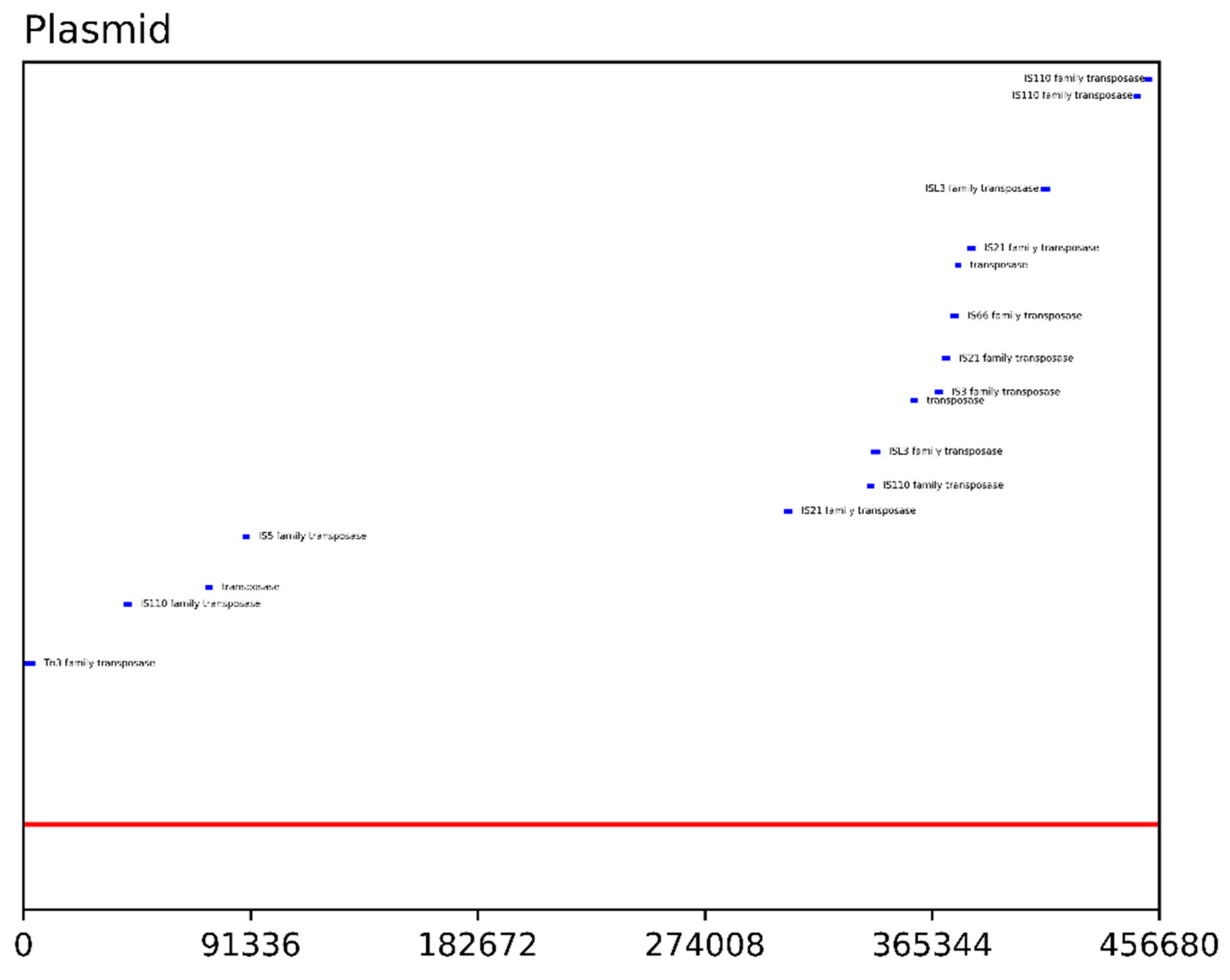
Figure 3.
Heatmap of the ANI values. The heatmap is calculated by OrthoAniU between each pair of Azohydromonas isolates assemblies including self-comparison (main diagonal). Darker shades of the color indicate higher ANI values.
Figure 3.
Heatmap of the ANI values. The heatmap is calculated by OrthoAniU between each pair of Azohydromonas isolates assemblies including self-comparison (main diagonal). Darker shades of the color indicate higher ANI values.
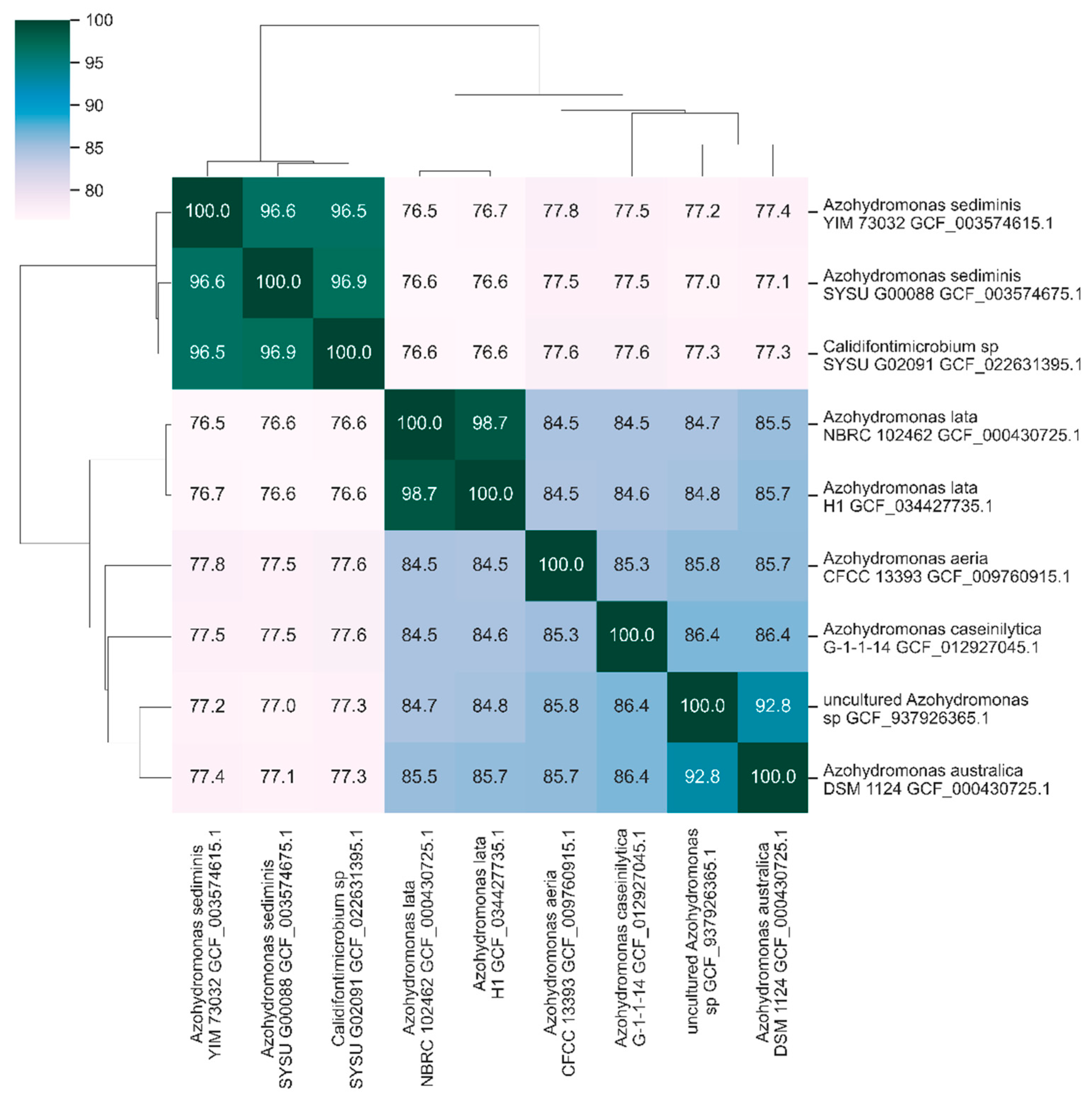
Figure 4.
Heatmap of the dDDH values. The heatmap has been computed by Genome-to-Genome-Distance-Calculator (GGDC-3.0) with recommended settings between each pair of Azohydromonas isolates assemblies. Self-comparison is on the main diagonal and darker color suggests higher dDDH values.
Figure 4.
Heatmap of the dDDH values. The heatmap has been computed by Genome-to-Genome-Distance-Calculator (GGDC-3.0) with recommended settings between each pair of Azohydromonas isolates assemblies. Self-comparison is on the main diagonal and darker color suggests higher dDDH values.
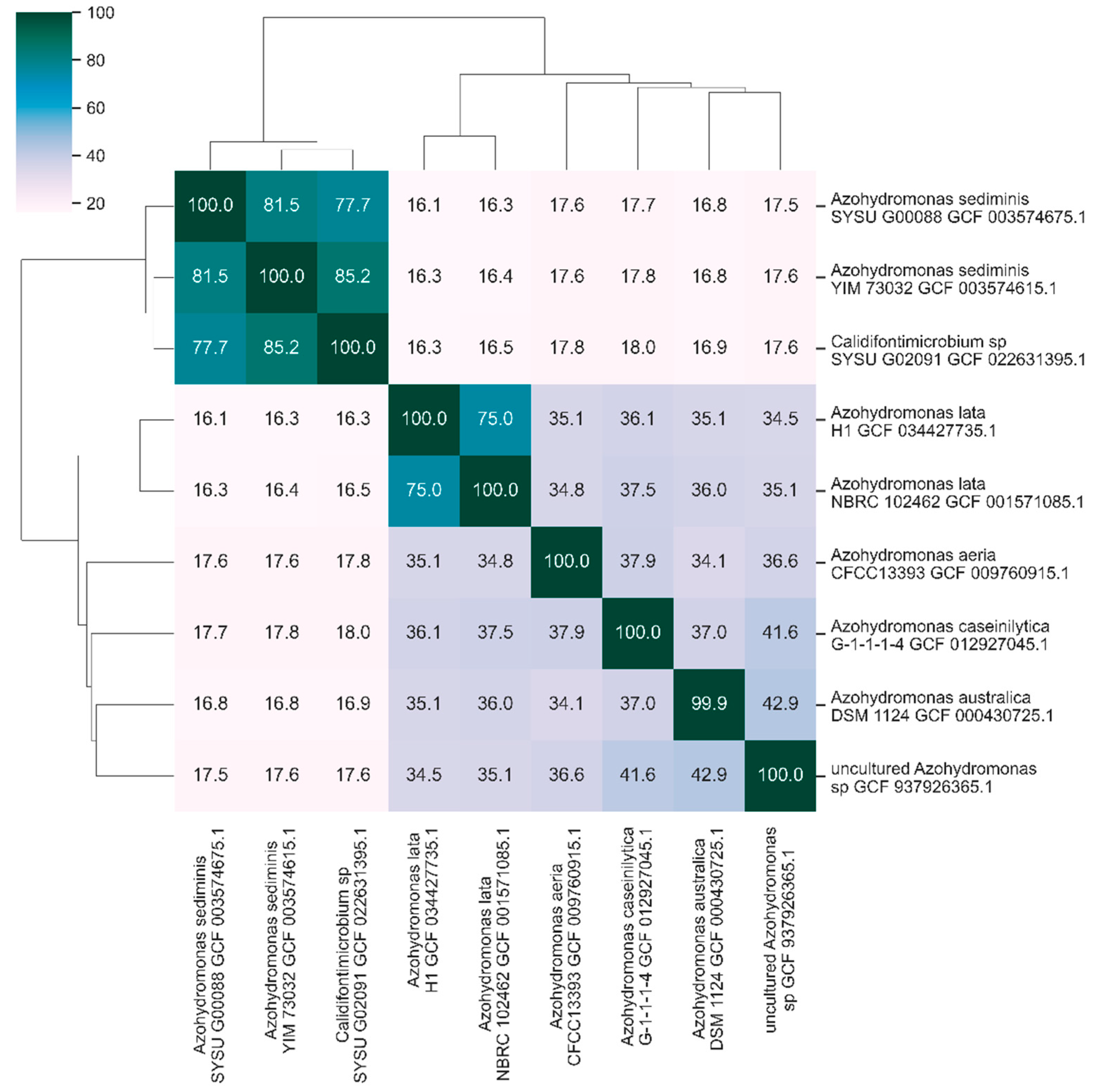
Figure 5.
Heatmap of blastp sequence similarity analysis among collection of representative PhaCs classes (I-V). Darker shades of the color mark higher identity values. Self-comparison occupies the main diagonal.
Figure 5.
Heatmap of blastp sequence similarity analysis among collection of representative PhaCs classes (I-V). Darker shades of the color mark higher identity values. Self-comparison occupies the main diagonal.
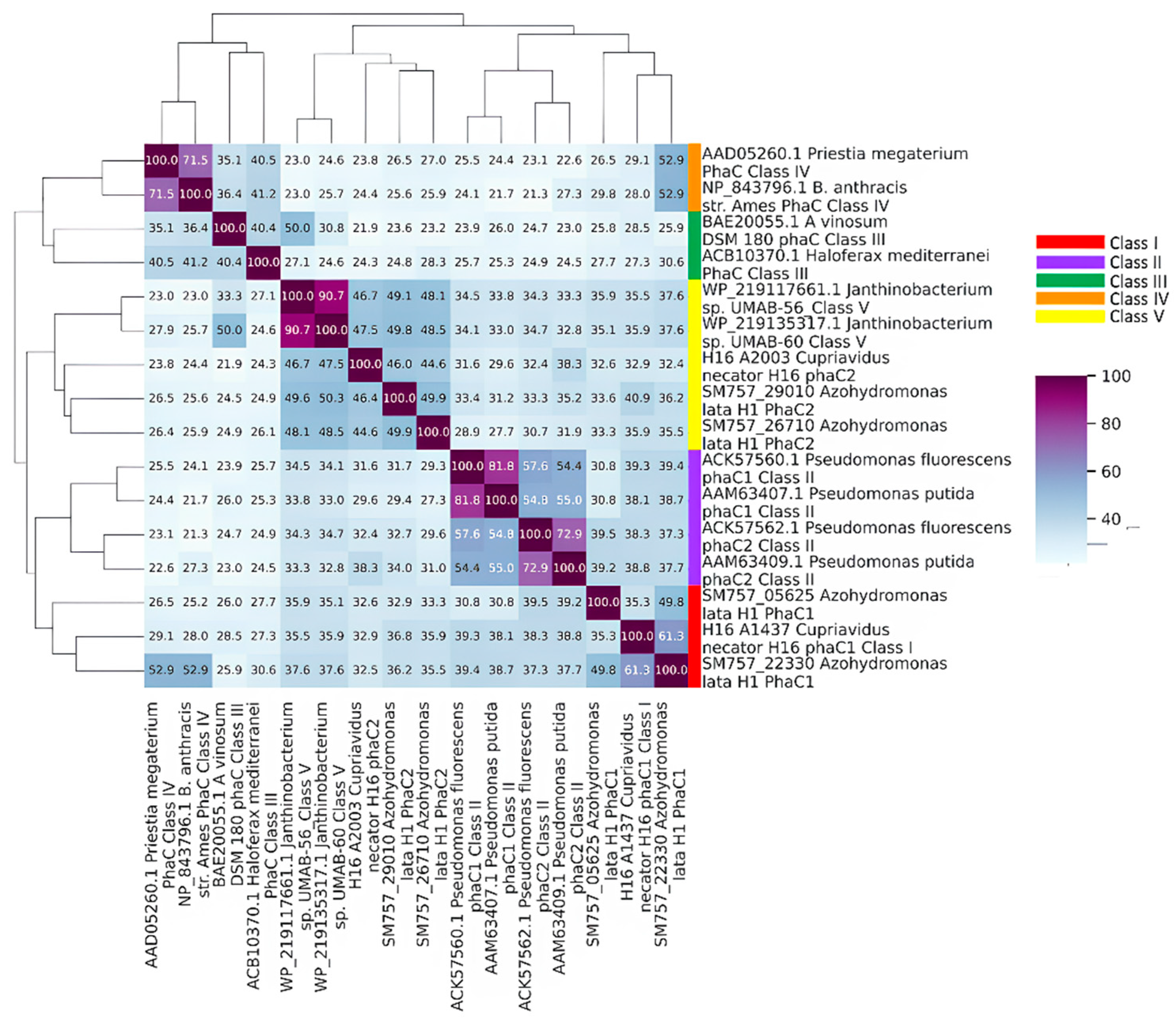
Figure 6.
Portion of the MSA of candidate PhaC proteins computed by the online version of Clustal Omega and visualized with JalViewer. Colored columns are the 8 conserved residues. Residues of the catalytic triad are highlighted with red stars on the top of the corresponding column.
Figure 6.
Portion of the MSA of candidate PhaC proteins computed by the online version of Clustal Omega and visualized with JalViewer. Colored columns are the 8 conserved residues. Residues of the catalytic triad are highlighted with red stars on the top of the corresponding column.


Table 1.
General features of the genome.
| Feature | Chromosome(s) | Megaplasmid |
|---|---|---|
| Size (bp) | 7328099 | 456680 |
| G+C ratio (%) | 68.7 | 65.6 |
| Percentage coding | 88.24 | 81.41 |
| tRNA | 56 | 0 |
| rRNA 5S, 16S, 23S | 1, 3, 2 | 0 |
| Transposases | 33 | 16 |
| Total number of CDSs | 6537 | 408 |
| No. of CDSs with assigned function | 5633 | 300 |
| CDSs with unknown function | 904 | 108 |
Table 2.
Predicted transposases.
| Transposase Family | Chromosome(s) | Plasmid |
|---|---|---|
| Tn3 | 2 | 1 |
| IS110 | 1 | 4 |
| transposase | 11 | 3 |
| IS5 | 1 | 1 |
| IS21 | 1 | 3 |
| ISL3 | 3 | 2 |
| IS3 | 1 | 1 |
| IS66 | 6 | 1 |
| IS630 | 7 | 0 |
Table 3.
TA systems predicted on the megaplasmid.
| Locus Tag | Product |
|---|---|
| SM757_01580* | type II TA system RelE/ParE toxin |
| SM757_01585* | HigA addiction module antitoxin |
| SM757_01810** | type II TA system VapB antitoxin |
| SM757_01815** | type II TA system RelE/ParE toxin |
| SM757_01820** | HigA addiction module antitoxin |
| SM757_01845 | type IV TA system AbiEi antitoxin |
| * System II-A | |
| **System II-B | |
Table 4.
Regions putatively encoding for resistance proteins.
| Locus Tag | Accession of contig | Product |
|---|---|---|
| SM757_03450 | JAXOJX010000003.1 | chromate resistance protein |
| SM757_03465 | JAXOJX010000003.1 | chromate resistance protein |
| SM757_03785 | JAXOJX010000003.1 | cobalt-zinc-cadmium resistance protein |
| SM757_03940 | JAXOJX010000003.1 | ArsI/CadI family heavy metal resistance metalloenzyme |
| SM757_05070 | JAXOJX010000004.1 | chromate resistance protein |
| SM757_05525 | JAXOJX010000005.1 | TerB tellurite resistance protein |
| SM757_12920 | JAXOJX010000019.1 | glyoxalase/bleomycin resistance/dioxygenase protein |
| SM757_19280 | JAXOJX010000033.1 | TerB tellurite resistance protein |
| SM757_22515 | JAXOJX010000042.1 | organic hydroperoxide resistance protein |
| SM757_24300 | JAXOJX010000049.1 | TerB tellurite resistance protein |
| SM757_24860 | JAXOJX010000051.1 | TerB tellurite resistance protein |
Table 5.
Pha related functions predicted in the genome.
|
ALH1 locus tag |
ORF length | CONTIG | ALH1 predicted function | ORTHOLOGOUS | ||
| Locus tag | Gene(s) | function | ||||
| SM757_22330 | 1611 | JAXOJX010000042* | class I poly(R)-hydroxyalkanoic acid synthase | H16_A1437 | phaC1 | Poly(3-hydroxybutyrate) polymerase |
| SM757_05625 | 1752 | JAXOJX010000005 | class I poly(R)-hydroxyalkanoic acid synthase | H16_A1437 | phaC1 | Poly(3-hydroxybutyrate) polymerase |
| SM757_29010 | 1884 | JAXOJX010000076 | poly-beta-hydroxybutyrate polymerase N-terminal domain-containing protein | H16_A2003 | phaC2 | Poly(3-hydroxybutyrate) polymerase |
| SM757_26710 | 1767 | JAXOJX010000060 | alpha/beta fold hydrolase | H16_A2003 | phaC2 | Poly(3-hydroxybutyrate) polymerase |
| SM757_22335 | 1179 | JAXOJX010000042* | acetyl-CoA C-acetyltransferase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_28485 | 1185 | JAXOJX010000072 | beta-ketothiolase BktB | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_00745 | 1185 | JAXOJX010000001 | acetyl-CoA C-acyltransferase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_27765 | 1209 | JAXOJX010000067 | acetyl-CoA C-acyltransferase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_19820 | 1179 | JAXOJX010000035 | acetyl-CoA C-acyltransferase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_22610 | 1185 | JAXOJX010000043 | acetyl-CoA C-acyltransferase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_24435 | 1215 | JAXOJX010000049 | 3-oxoadipyl-CoA thiolase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_07035 | 1206 | JAXOJX010000007 | 3-oxoadipyl-CoA thiolase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_09505 | 1206 | JAXOJX010000012 | 3-oxoadipyl-CoA thiolase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_09155 | 1206 | JAXOJX010000011 | 3-oxoadipyl-CoA thiolase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_01140 | 1176 | JAXOJX010000001 | thiolase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_31645 | 1206 | JAXOJX010000098 | 3-oxoadipyl-CoA thiolase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_19540 | 1203 | JAXOJX010000034 | acetyl-CoA C-acyltransferase | H16_A1438 | phaA | Acetyl-CoA acetyltransferase |
| SM757_22340 | 738 | JAXOJX010000042* | acetoacetyl-CoA reductase | H16_A1439; H16_A2002; H16_A2171 | phaB1; phaB2; phaB3 | Acetoacetyl-CoA reductase |
| SM757_08215 | 750 | JAXOJX010000009 | 3-oxoacyl-ACP reductase FabG | H16_A1439; H16_A2002; H16_A2171 | phaB1; phaB2; phaB3 | Acetoacetyl-CoA reductase |
| SM757_31855 | 741 | JAXOJX010000100 | 3-oxoacyl-ACP reductase FabG | H16_A1439; H16_A2002; H16_A2171 | phaB1; phaB2; phaB3 | Acetoacetyl-CoA reductase |
| SM757_23050 | 747 | JAXOJX010000044 | 3-oxoacyl-ACP reductase FabG | H16_A2002; H16_A2171 | phaB2; phaB3 | Acetoacetyl-CoA reductase |
| SM757_08600 | 1344 | JAXOJX010000010 | polyhydroxyalkanoate depolymerase | H16_A1150; H16_A2862; H16_B0339; H16_B1014 | phaZ1; phaZ2; phaZ3; phaZ5 | intracellular poly(3-hydroxybutyrate |
| SM757_15980 | 1002 | JAXOJX010000025 | PHB depolymerase family esterase | H16_B2401 | phaZ7 | Poly(3-hydroxybutyrate) depolymeras |
| SM757_26695 | 2253 | JAXOJX010000060 | 3-hydroxybutyrate oligomer hydrolase protein | H16_A2251 | phaY1 | D-(-)-3-hydroxybutyrate hydrolase |
| SM757_23895 | 870 | JAXOJX010000047 | alpha/beta hydrolase | H16_A1335 | phaY2 | D-(-)-3-hydroxybutyrate hydrolase |
| SM757_26850 | 558 | JAXOJX010000062 | phasin family protein | H16_A1381 | phaP1 | Phasin (PHA-granule associated protein) |
| SM757_25825 | 594 | JAXOJX010000056 | polyhydroxyalkanoate synthesis repressor PhaR | H16_A1440 | phaR | transcriptional regulator |
Table 6.
Nucleotide identity among phaC from Choi et. al. and Azohydromonas phaC genes.
| Genus or Species (strain) | Identity (%) | Alignment length | Mismatches | E-value |
|---|---|---|---|---|
| australica (DMS1124) | 100.000 | 1611 | 0 | 0.0 |
| Uncultured Azohydromonas sp. | 94.727 | 1612 | 83 | 0.0 |
| aeria (CFCC 13393) | 91.646 | 1616 | 128 | 0.0 |
| lata (NBRC102462) | 90.627 | 1611 | 151 | 0.0 |
| lata (H1) | 90.503 | 1611 | 153 | 0.0 |
| caseinilytica (G-1-1-1-4) | 90.526 | 1615 | 145 | 0.0 |
| sediminis (SYSU G00080) | 80.455 | 1627 | 280 | 0.0 |
| Calidifontimicrobium sp (SYSU G02091) | 80.086 | 1627 | 286 | 0.0 |
Table 7.
Nucleotide identity among phaA from Choi et. al. and Azohydromonas phaA genes.
| Genus or Species (strain) | Identity (%) | Alignment length | Mismatches | E-value |
|---|---|---|---|---|
| australica (DMS1124) | 100.00 | 337 | 0 | 0.0 |
| Uncultured Azohydromonas sp. | 96.777 | 1179 | 38 | 0.0 |
| caseinilytica (G-1-1-1-4) | 96.438 | 1179 | 42 | 0.0 |
| lata (NBRC 102462) | 92.881 | 1180 | 82 | 0.0 |
| lata (H1) | 92.373 | 1180 | 88 | 0.0 |
| sediminis (YIM 73032) | 84.338 | 1187 | 170 | 0.0 |
| sediminis (SYSU G00080) | 84.233 | 1186 | 173 | 0.0 |
| Calidifontimicrobium sp (SYSU G02091) | 83.825 | 1187 | 176 | 0.0 |
| aeria (CFCC 13393) | 80.086 | 128 | 16 | 0 |
Table 8.
Nucleotide identity among phaB from Choi et. al. and Azohydromonas phaB genes.
| Genus or Species (strain; accession) | Identity (%) | Alignment length | Mismatches | E-value | ||
|---|---|---|---|---|---|---|
| caseinilytica (G-1-1-1-4) | 94.851 | 738 | 38 | 0.0 | ||
| aeria (CFCC13393; WP_157271469.1_7046) | 94,580 | 738 | 40 | 0.0 | ||
| australica (DSM 1124) | 94,309 | 738 | 42 | 0.0 | ||
| aeria (CFCC13393; WP_157272394.1_2294) | 94,038 | 738 | 44 | 0.0 | ||
| lata (H1) | 93.496 | 738 | 48 | 0.0 | ||
| lata (NBRC 102462) | 93.360 | 738 | 49 | 0.0 | ||
| Calidifontimicrobium sp (SYSU G02091) | 84.409 | 744 | 104 | 0.0 | ||
| sediminis (SYSU G00080) | 84.430 | 745 | 102 | 0.0 | ||
| sediminis (YIM 73032) | 84.161 | 745 | 104 | 0.0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



