Submitted:
16 December 2024
Posted:
17 December 2024
You are already at the latest version
Abstract
Selective serotonin reuptake inhibitors (SSRIs) are a first line treatment for depressive and anxiety disorders. They function by selectively binding and blocking the serotonin transporter (SERT) and preventing reuptake of serotonin into the presynaptic terminal; however, the downstream changes and that determine the therapeutic effects remain largely unknown. There is growing evidence showing that serotonergic signalling and SERT blockade influences mitochondrial biogenesis and activity, suggesting that this may be an important downstream effect to consider in understanding the mechanism of action of these drugs. The aim of this study was to review the literature investigating mitochondrial biogenesis and activity as a result of SSRI treatment. Publications investigating SSRI treatment and mitochondrial function between 2007 and 2021 were identified by literature search using the PubMed database. Publications were categorised by whether mitochondrial biogenesis and activity was increased, decreased, restored following a disruption, or whether effects were mixed. Effects of SSRI treatment on mitochondrial biogenesis and function were dependent on the chosen dose and model. While cell culture based-studies showed mixed outcomes, the majority of animal studies showed that mitochondrial biogenesis and oxidative phosphorylation was increased, while oxidative stress was decreased. Collectively, the studies in this review suggest that SSRI treatment within the therapeutic dose range has the capacity to enhance mitochondrial biogenesis and function. Given that reduced oxidative capacity has been implicated in the pathophysiology of depressive and anxiety disorders, this may be important for understanding how SSRIs function to alleviate symptoms of these disorders.
Keywords:
Serotonin
; Mitochondria
; SSRI
; Neuropsychiatric Disorders
Introduction
Serotonergic signalling has a long-standing association with depressive and anxiety disorders, largely owing to its role in the regulation of mood. The serotonin transporter (SERT) functions to control synaptic concentrations of serotonin (5-hydroxytryptamine; 5-HT), and therefore has an essential role in the regulation of 5-HT signalling. SERT is also the target of selective serotonin reuptake inhibitors (SSRIs), a class of drugs included on the World Health Organisation List of Essential Medicines as a first line treatment for depressive and anxiety disorders (World Health Organisation 2021). They selectively bind and block SERT, preventing reuptake of 5-HT into the presynaptic terminal. In the short term, this reduces clearance of 5-HT from the synapse, promoting increased serotonergic signalling. However, most patients tend to experience a therapeutic delay, where the improvement of symptoms often occurs weeks to months after starting the medication (Trivedi et al. 2006). The cause of this delay remains largely unknown but may be attributed to downstream changes in 5-HT receptor expression and sensitivity (Commons and Linnros 2019). Additionally, a study by Erb et al. (2016) suggested that SSRI treatment promotes the translocation of G-proteins away from lipid rafts, allowing the activation of the cAMP signalling cascade and that this may account for the therapeutic delay.
Mitochondrial dysfunction is also implicated in the pathophysiology of numerous neuropsychiatric and neurodevelopmental disorders, and there is increasing evidence to suggest that signalling through multiple 5-HT receptors drives mitochondrial biogenesis (Valenti et al. 2017; Scholpa et al. 2018; Fanibunda et al. 2019). Interestingly, there is also some evidence to suggest that SERT may be found on the outer mitochondrial membrane; however, its function there remains unclear (Mukherjee et al. 1998; Haase et al. 2017). Given the gap in our understanding of the long-term changes that drive remission with antidepressant treatments, this begs the question of whether modulation of mitochondrial function by SSRIs treatment may influence therapeutic outcomes.
This review provides a systematic review of the literature investigating the effect of SSRI treatments on mitochondrial abundance and function, with a view to determining whether the literature is in consensus over whether these drugs are beneficial or detrimental to mitochondrial function. Understanding the relationship between serotonergic signalling and mitochondrial function may be important for understanding the pathophysiology of neuropsychiatric and neurodevelopmental disorders, and consequently this may highlight a pathway important for future dug development.
Methods
Publications studying SSRI treatment and mitochondrial abundance, biogenesis, or function between 1 January 2007 and 31 December 2021 were identified by literature search using the PubMed database; search terms were (“SSRI” OR “SELECTIVE SEROTONIN REUPTAKE INHIBITOR”) AND “MITOCHONDRIA”. Review articles, conference abstracts, and papers not relevant to this review were excluded. Studies of animals, cultured cells (human-and animal-derived), and isolated mitochondria were included, as well as studies of both chronic and one-off treatments.
There are many drugs that are classes as SSRIs, and all were eligible for inclusion in this review. Studies were read and the main findings were summarised. Mitochondria have a diverse range of cellular functions and as a result of this, the literature in this field employs a diverse range of experimental outputs to quantify mitochondrial biogenesis and function. To simplify this, studies were classified such that an “increase” in function refers to increased mitochondrial biogenesis, tricarboxylic acid (TCA) cycle activity, electron transport chain (ETC) activity, ATP production, oxygen consumption, or decreased reactive oxygen species (ROS) production. Studies were categorised by whether mitochondrial function or abundance was increased (↑) decreased (↓), mixed (↑↓), or neutral (↔). If mitochondrial function or abundance was returned to baseline with SSRI treatment following a disruption, studies were classified by whether the return to baseline was an increase (*↑) or a decrease (*↓). The search ended on 1 July 2022; all articles were screened for eligibility, briefly summarised and classified based on their findings twice by BT.
As mitochondrial function, serotonergic signalling, and SSRI efficacy also show sexual dimorphisms, publications utilising animal tissue or primary cultured cells were also analysed for sex bias. For this, studies were classified by the following categories: sex not stated, male, female, sexes combined (without sex comparisons), sex comparison (comparative data shown), and sex comparison (comparative data not shown). Papers that included both sexes but used them for separate experiments meaning that they could not be compared were included in the sexes combined category.
Risk of Bias Assessment
Table 1 shows a risk of bias assessment for this review as directed by the Risk of Bias in Systematic Reviews Tool (ROBIS).
Results
Study Selection
Literature search using the PubMed database identified 90 articles published between 1 January 2007 and 31 December 2021 which were then assessed for eligibility. Of these publications, 47 were excluded as they were either review articles (n = 7) or not relevant to this review (n = 40) (Figure 1). Of the 43 relevant publications identified, three studied isolated mitochondria (two of these also included cell culture experiments), nine studied cultured cells (primary or immortalised cell lines), and the remaining 31 were rodent studies. These studies included the following drugs: fluoxetine (30), norfluoxetine (1), fluvoxamine (2), paroxetine (6), sertraline (3), citalopram (4), dapoxetine (1), and vortioxetine (1) (Figure 2).
Study Outcomes Varied by Model
The findings of the reviewed studies pertaining to mitochondrial abundance or function were classified as increased, decreased, restored following a disruption, or neutral as indicated in Table 2 and Table 3. Studies showing that mitochondrial abundance or function was restored following a disruption were then further categorised by whether the restoration to baseline involved an increase or decrease in the measured parameters. In studies of isolated mitochondria, all three publications demonstrated that SSRI treatment was detrimental to mitochondrial function. For the nine studies using cultured cells, the outcomes were more varied – five studies demonstrated improved mitochondrial function, three showed decreased function, and one demonstrated mixed effects. Animal-based studies again showed a different pattern, with 25 out of 31 publications showing that SSRI treatment improved mitochondrial function, only three papers demonstrating decreased function, and three papers showing mixed effects (Figure 3).
Publications Were Biased Towards Studying Males
Sex bias in preclinical research is a well-documented issue; however, despite efforts by funding bodies, male bias in neuroscience research remains a significant issue (Beery and Zucker 2011; Coiro and Pollak 2019; Woitowich et al. 2020; Thorne et al. 2022a), male bias in biological research is a significant issue. Given that mitochondrial function and SSRI efficacy both show important sex differences (Ventura-Clapier et al. 2017; LeGates et al. 2019), the publications in this review that studied animals or cultured primary cells were also analysed for potential sex biases. Of the identified articles, only 8.1% addressed sex as a biological variable by comparing males and females. Seventy-eight percent studied exclusively males, 2.7% studied females, 11% did not state the sex of the animals and another 2.7% combined sexes without comparative analysis (Figure 4).
SSRI Treatments in Isolated Mitochondria
Three studies investigated the effects of SSRI treatments in isolated mitochondria. All three of these studies demonstrated some level of mitochondrial dysfunction with SSRI treatment; however, all of these studies also utilised high drug concentrations that are well above therapeutic ranges (Magalhães et al. 2017). Hroudova and Fisar (2010) investigated the effect of citalopram (among additional antidepressants and mood stabilisers) on the activity of citrate synthase and ETC complexes in mitochondria isolated from the pig brain cortex. The concentration of citalopram used in this study was very high (500 μM). While this concentration of citalopram did not influence complex IV activity, citrate synthase activity was increased and the activity of complexes I and II were decreased with treatment. Abdel-Razaq et al. (2011) showed that treatment with norfluoxetine (the active metabolite of fluoxetine) impaired mitochondrial function in isolated mitochondria and cultured cells. In isolated rat heart mitochondria, mitochondrial membrane potential (MMP) and oxygen consumption in the presence of ADP (state-3 respiration) were reduced with norfluoxetine treatment in a dose-dependent manner. Similarly, norfluoxetine reduced activity of ETC complexes I, II/III and IV in CHO cells also in a dose-dependent manner. Interestingly, complex IV activity was inhibited to the greatest degree, with activity being undetectable at a 50 μM dose. Li et al. (2012) investigated the effect of sertraline on isolated rat liver mitochondria and cultured primary rat hepatocytes. In isolated mitochondria, the activity of ETC complex I and ATP synthase were reduced with sertraline concentrations above 25 μM, while complex II, III, and IV activity was not influenced by sertraline treatment at any dose. The authors also demonstrated that cellular ATP was depleted when primary rat hepatocytes were treated with sertraline in doses at 37.5 μM and above. Similarly, uncoupling of oxidative phosphorylation (OXPHOS) was demonstrated with high doses.
Collectively these studies suggest that high dose SSRI exposure to isolated mitochondria may be toxic; however, the use of isolated mitochondria in these studies bypasses the mechanism by which these drugs function in vivo. As such, the toxicity and impairment to mitochondrial function shown in these studies are difficult to interpret without further in vivo studies.
Effects of SSRI Treatments on Cultured Cells
Studies that investigated the effects of SSRI treatments on cultured cells showed varied outcomes. Some suggested that treatment was detrimental to mitochondrial function, while others showed that activity was enhanced. Jeong et al. (2017) investigated the effect of dapoxetine, fluoxetine, and citalopram on glutamate-induced excitotoxicity in cultured rat primary hippocampal neurons. The authors showed that dapoxetine treatment (5 μM) prevented glutamate-induced Ca2+ increases, loss of mitochondrial membrane potential, and cell death. However, similar effects were not seen with fluoxetine and citalopram treatments.
Reddy et al. (2021) investigated the neuroprotective role of citalopram against the effect of mutant amyloid precursor protein (APP) in immortalised rat hippocampal cells (HT22 cells). This study showed that citalopram treatment enhanced mitochondrial biogenesis and mitophagy in control HT22 cells, and additionally rescued these functions as well as mitochondrial fragmentation caused impaired by mutant APP transfection.
Paroxetine Treatments
Three of the cell culture-based studies utilised paroxetine (Gerö et al. 2013; Steiner et al. 2015; Jeong et al. 2015), and each of these studies showed that paroxetine treatment had a beneficial effect on mitochondrial function. Gerö et al. (2013) showed that paroxetine reduced hyperglycaemia-induced mitochondrial ROS production in endothelial cells, having protective effects against oxidative damage to DNA and proteins. Interestingly, this effect was evident without any effect on electron transport or bioenergetics. Steiner et al. (2015) showed that paroxetine interacted with mitochondrial proteins in cultured rat primary cortical neurons where it had neuroprotective effects. Treatment was protective against nitrosative stress and mitochondrial swelling due to Ca2+ overload. Some of these effects were also seen with fluoxetine treatment, although to a lesser extent. Interestingly, these effects were also seen when there was a complete absence of SERT, suggesting that the protective effects were not all due to SERT binding. Finally, Jeong et al. (2015) showed that paroxetine treatment enhanced mitochondrial biogenesis in a human neuroblastoma cell line (SK-N-MC cells). The authors demonstrated a dose-dependent increase in mitochondrial mass, mtDNA copy number, glucose consumption, and cellular ATP levels (drug concentration between 2 and 6 μM); however, these effects were less evident with higher dose 10 μM paroxetine.
Fluoxetine Treatments
Four studies investigated how fluoxetine impacted mitochondrial dynamics in cultured cells, and three of these studies suggested that fluoxetine treatment impaired mitochondrial function. Han and Lee (2009) investigated the effect of fluoxetine (15 μM) treatment on MPP+-induced neurotoxicity in differentiated PC12 cells. While other classes of antidepressants had protective effects, the authors showed that fluoxetine (15 μM) enhanced the effects of MPP+, increasing DNA damage due to oxidative stress and exacerbating the loss of mitochondrial membrane potential. Similarly, Lee et al. (2010) found that fluoxetine treatment (15 μM) induced the loss of mitochondrial membrane potential, and increased ROS production in a dose-dependent manner in an ovarian cancer cell line (OVCAR-3 cells). Using Jurkat cells, HeLa cells, and human peripheral blood leukocytes, Charles et al. (2017) also showed that high dose fluoxetine treatment (40 μM) resulted in Ca2+ release from the endoplasmic reticulum, and consequent accumulation in the mitochondria, resulting in reduced O2 consumption and ATP production. In contrast to these studies, Chen et al. (2007) investigated the effect of fluoxetine treatment on mitochondrial trafficking in cultured primary rat hippocampal neurons. This study showed that 3 μM fluoxetine promoted trafficking of mitochondria towards the axon terminal. Although these findings do not specifically indicate enhanced or reduced mitochondrial function, this suggests that SSRI treatment may stimulate a redistribution of energy sources.
Although some of these studies explored mitochondrial function in non-neural cell lines and are therefore not relevant in the context of neuropsychiatric disorders and the mechanism of action of SSRIs, the systemic effects of drugs are important to consider for potential side effects. However, as there was substantial variety across the cell types, drugs, and doses, it is difficult to draw any meaningful conclusions from these studies as a whole.
Summarised Findings – Cells and Isolated Mitochondria
Publications in this review investigated the effect of SSRI treatment on mitochondrial abundance or activity in a range of tissues. Twenty three out of 31 animal studies investigated effects in the brain, and the most common brain region studied was the hippocampus (studied in 16 out of 23 publications that studied the brain). Given that SSRIs are prescribed for neuropsychiatric disorders, the effects on mitochondria in the brain are most relevant to consider. However, serotonin functions systemically and has a role in regulating a range of physiological functions including gastrointestinal motility, vasoconstriction, and platelet aggregation (Berger et al. 2009). As such, investigating the effect of SSRIs in a range of tissues is also relevant.
Animal studies were largely consistent in that the majority showed that mitochondrial function was either improved or normalised with SSRI treatment. There was only one study that suggested that SSRI treatment was detrimental to mitochondrial function, and in this study Ahmadian et al. (2017) showed that citalopram treatment (20 mg/kg) resulted in oxidative damage in the livers of rats. The authors corroborated these finding in vitro where they showed that 500 μM citalopram resulted in oxidative damage and collapse of the mitochondrial membrane potential in cultured rat hepatocytes. As this study focussed on the liver, it is less relevant in understanding a potential mechanism of actions for SSRIs as a treatment for neuropsychiatric disorders. Although hepatotoxicity is an important safety consideration for pharmaceuticals, the toxicity shown in this study can likely be attributed to the high doses used both in vivo and in vitro.
Two additional studies showed that fluoxetine treatment resulted in reduced mitochondrial activity in the brain; however, both of these studies showed that treatment normalised activity that was raised as a result of chronic unpredictable stress (CUS) paradigms (Wen et al. 2014; Emmerzaal et al. 2021). Wen et al. (2014) showed that 5 mg/kg fluoxetine restored the respiratory control ratio, ATP production, and antioxidant defence to baseline in the dorsal raphe of stressed animals. Emmerzaal et al. (2021) showed that complex III and IV activity were reduced with 15 mg/kg fluoxetine treatment in the frontal cortex of stressed animals; however, unstressed animals were not included in this study to determine the effect of the CUS paradigm alone on mitochondrial function. Although it was assumed that treatment restored ETC activity rather than exacerbating the effects of CUS, this is unclear.
There were three studies that suggested that SSRI treatment had mixed effects on mitochondrial function, and all three of these studies were fundamentally very different. In their proteomic study, Głombik et al. (2017) showed that fluoxetine treatment (10 mg/kg) had a varying effect on the expression of proteins associated with mitochondrial biogenesis and function in the hippocampus of prenatally stress rats. Of note, fluoxetine treatment resulted in the upregulation of DJ-1, a protein involved in sensing oxidative stress and stimulating mitochondrial biogenesis. Braz et al. (2020a) investigated the effect of fluoxetine treatment (10 mg/kg) in rats that were overfed in the first 21 days of life. The authors showed that overfeeding during this time resulted in decreased oxygen consumption and increased oxidative stress in brown adipose tissue, and these changes were ameliorated with fluoxetine treatment. Interestingly, fluoxetine treatment had a slightly different effect in normofed animals, resulting in increased oxygen consumption and increased oxidative stress in brown adipose tissue (BAT). In considering these studies incorporating BAT, it is also important to note that mitochondria serve a unique purpose in these cells relative to the rest of the body. BAT expresses the uncoupling protein-1 (UCP-1) which causes a proton leak across the inner mitochondrial membrane, facilitating the thermogenic properties of BAT. As such, measures of mitochondrial function that are employed throughout these papers have a unique meaning in this tissue type and should be interpreted as such (Lee et al. 2019).
The final study showing varied effects on mitochondrial function also demonstrated that these differences were sex dependent. As most of the studies in this review were conducted in male animals only, these findings were particularly interesting. Adzic et al. (2013) investigated the effect of fluoxetine treatment (5 mg/kg) alongside 21 days of isolation stress, and showed sexually dimorphic differences in mRNA expression and activity of ETC components in the brain as a result. Complex IV activity and mRNA expression of mitochondrially-encoded subunits for complex IV were increased in the prefrontal cortex (PFC) of fluoxetine treated control females, whereas no differences were seen in the PFC of male counterparts. They also showed that expression and activity were increased in the hippocampus of stressed fluoxetine treated females but decreased in the hippocampus of male counterparts. There were only two other studies in this review that included sex comparisons, and one of these was conducted by the same group as the study just described. This study built on their previous findings and showed that mitochondrial oestrogen receptor β has an important role in the sexually dimorphic nature of fluoxetine-induced changes in behaviour and complex IV activity. These findings are significant, as it has been demonstrated that sex differences in antidepressant effectiveness in humans can also be attributed to sex hormones (Adzic et al. 2017).
The third publication that incorporated sex as a biological variable also demonstrated interesting sex differences. In this study, Silva et al. (2018) showed that fluoxetine treatment (10 mg/kg) during the neonatal period improved mitochondrial function in the brainstem at adulthood in a sexually dimorphic manner. For males, fluoxetine treatment significantly increased citrate synthase activity, ADP-stimulated respiration, and the respiratory control ratio, while also reducing ROS production in the brainstem. Whereas for females, improvements in antioxidant capacity were seen without significant changes to respiratory capacity. There were six other publications by this group that were identified for this review, with these studies making up almost a quarter of animal studies. An additional four of these publications investigated the same paradigm described above and they all reached a similar consensus, showing that fluoxetine treatment during the first 21 days of life improved mitochondrial function at adulthood, with each of these studies exploring the effects in different tissues (da Silva et al. 2015b, a; Braz et al. 2016; Silva et al. 2018; Simões-Alves et al. 2018). As these effects were seen long after the treatment was stopped, this suggests that fluoxetine treatment during an important developmental stage may result in long term or permanent changes to mitochondrial bioenergetics. The other two studies by this group investigated how fluoxetine treatment from PND 39-59 influences mitochondrial function at adulthood following overfeeding during the neonatal period (Braz et al. 2020a, b). They showed that fluoxetine treatment during adolescence restores mitochondrial function and oxidative balance in the hypothalamus and brown adipose tissue following perturbations due to overfeeding during the neonatal period.
The most consistent findings across animal studies were that SSRI treatment enhanced ETC activity and OXPHOS and was also protective against oxidative stress. These findings were consistent in publications studying the effects of SSRIs alone as well as in chronic unpredictable stress and social isolation stress models. For example, Villa et al. (2016, 2017) showed that fluoxetine treatment (10 mg/kg) alone enhanced complex IV activity in mitochondria isolated from the frontal cortex (FC) and hippocampus of rats. Using stress-based models, Adzic et al. (2017) showed that reduced complex IV activity in the hippocampus of stressed females was restored with fluoxetine treatment, and Wen et al. (2014) showed that decreased ATP production in the DRN with chronic unpredictable stress was also restored with fluoxetine treatment. Considering oxidative stress, Tutakhail et al. (2019) showed that fluoxetine treatment (18 mg/kg) reduced ROS production in skeletal muscle, and Garabadu et al. (2015) showed that paroxetine (10 mg/kg) treatment rescued stress-induced oxidative damage in the brain. Interestingly, publications by Arafat and Shabaan (2020) and Shu et al. (2019) both showed that fluoxetine treatment rescued ultrastructural changes to mitochondria in the hippocampus resulting from stress-based paradigms, suggesting that the effect of SSRIs on mitochondrial health and function is broad.
Although the majority of animal studies showed that SSRI treatment increased mitochondrial activity or ameliorated mitochondrial function that was impaired by previous conditions, there were two publications that showed the opposite effect. Both of these studies showed that chronic unpredictable stress resulted in increased ETC activity or ATP production in the brain, and this condition was reversed with fluoxetine treatment (Wen et al. 2014; Emmerzaal et al. 2021). Taking these studies into account, it may be that SSRI treatment has the capacity to restore mitochondrial function whether this entails an increase or a decrease in activity. In saying this, the studies suggesting that treatment increases mitochondrial activity far outnumber those that suggest the opposite.
The focus of this review was to explore the effect of SSRI treatment on mitochondrial biogenesis and function in the brain with a view to understanding whether this may be important in the treatment of neuropsychiatric disorders. While the majority of reviewed studies investigated effects in the brain, there were ten publications that either focussed on effects in peripheral tissues or explored this in addition to brain tissue. In the brain, SSRIs prevent reuptake of 5-HT into the presynaptic terminal, resulting in increased synaptic 5-HT; however, the effect of SSRIs in peripheral tissues is less clear. It is also unclear whether the effects of SSRI treatment reflect increased extracellular 5-HT and resultant changes to 5-HT receptor activation, or changes to intracellular 5-HT. It is possible that SERT blockade results in decreased intracellular 5-HT, thus influencing processes such as protein serotonylation or intracellular 5-HT receptor activation (Muma and Mi 2015; Wang et al. 2016; Bader 2019; Tempio et al. 2020), both of which can have important effects on cell function.
Publications that looked at the effects of SSRIs in peripheral tissue studied the heart, liver, brown adipose tissue, and skeletal muscle (Figure 5). SSRI effects in these tissue types may be due to SERT binding as SERT expression has been demonstrated in the liver, skeletal muscle, and heart (Ni and Watts 2006; Chen et al. 2012; Al-Zoairy et al. 2017); however, whether SERT is expressed in BAT was not clear from a review of the literature. As such, there may be additional indirect mechanisms to consider. For example, serotonin influences energy expenditure in BAT by increasing sympathetic drive, so it is possible that the effects described in BAT may be attributed to this mechanism rather than SERT binding in BAT itself (Morrison et al. 2014). However, as previously discussed, mitochondrial function in BAT is complex to interpret, adding a further layer of complexity to understanding the relationship between 5-HT and mitochondrial function in this tissue.
Summarised Findings – Animal Studies
Discussion
This review suggests that SSRI treatment influences mitochondrial abundance and function; however, the nature of these effects differs substantially depending on the model chosen. Studies in isolated mitochondria suggested that SSRIs may be detrimental to mitochondrial function and cell culture-based studies showed mixed effects. Animal studies on the other hand showed that SSRI treatment mostly enhanced mitochondrial function, and this effect was seen across a variety of tissue types.
Animal Studies Were Most Relevant
Studies in this review include experiments using isolated mitochondria, cultured cells, and animals. None of these models are perfect for understanding how these drugs function in human physiology; however, all three provide a unique perspective for understanding the effect of SSRIs on mitochondrial function and abundance. Animal models are likely the most physiologically relevant as they allow the study of complex effects owing to interactions between different cell types and tissues. Most importantly, these studies allow molecular analyses to be conducted on brain tissue – something that cannot be easily done in humans. A drawback of animal studies is that rodent brains differ to human brains, meaning not all effects that are found in rats and mice will be relevant to humans. Cell culture models can overcome this issue as they allow the study of human cells; however, in vitro models are limited as they are reliant on immortalised cells or primary rodent cells and do not reflect the complex tissue environment and interactions between different cell types. Importantly, it is unclear how SERT blockade alters serotonergic signalling in vitro, as extracellular levels of 5-HT are defined by the cell culture conditions rather than being dependent on SERT activity.
Studies of isolated mitochondria are likely the least physiologically relevant, as these experiments bypass the mechanism by which drugs function in vivo - in the case of SSRIs, by binding and blocking synaptic SERT. However, SSRIs are taken up inside the cell and are found physically associated with mitochondria where they may be bound to SERT (Mukherjee et al. 1998; Haase et al. 2017). As such, studies of isolated mitochondria may be able to elucidate the subcellular effects of SSRIs and provide insight into the function of SERT on mitochondria as this is currently unclear.
Doses Were Variable
Studies in isolated mitochondria all employed high drug concentrations that were likely not physiologically relevant and mostly higher than those seen in cell culture-based studies. In this sense, it is unsurprising that all three studies suggested that SSRI treatment impaired ETC complex activity. Studies of isolated mitochondria have the potential to elucidate the connection between SERT, fluoxetine, and mitochondria; however, this would require the use of physiologically relevant doses. Therapeutic plasma concentrations for SSRIs range between 20-200 ng/mL for citalopram (Overø 1982), 30-120 ng/mL for paroxetine, and 50-500 ng/mL for fluoxetine (Magalhães et al. 2017). These drugs tend to accumulate at higher concentrations in the brain, for example, therapeutic fluoxetine concentrations sit around 20 times higher in the brain than in the plasma (Karson et al. 1993). Although it is difficult to directly translate in vitro concentrations to human doses, this suggests that many of the mitochondrial and cell culture-based studies use very high concentrations. In this sense, it is not surprising that many of these studies report toxic effects and suggest that SSRIs were detrimental to mitochondrial function, especially given that high drug doses increase the likelihood of off target effects.
In a 2016 review, it was proposed that fluoxetine treatment may have detrimental effects on mitochondrial function and that more research is needed to investigate the toxicological effects of fluoxetine (de Oliveira 2016). However, the studies presented here collectively suggest that SSRIs may have the opposite effect, enhancing mitochondrial function. Signalling through multiple 5-HT receptors has been shown to enhance mitochondrial biogenesis (Valenti et al. 2017; Simmons et al. 2019; Fanibunda et al. 2019), and this is likely an important consideration given that SERT blockade increases synaptic 5-HT thus potentially increasing post-synaptic 5-HT signalling.
In considering the effect of SSRIs on mitochondrial biogenesis and function, there are three main factors to consider – the association of SERT with mitochondria and its potential function there, the role of intracellular 5-HT, and serotonergic signalling as a promoter of mitochondrial biogenesis. A study investigating the localisation of radiolabelled fluoxetine in the rat brain demonstrated that 60-70% of fluoxetine was found in the mitochondrial/synaptosomal fraction (Mukherjee et al. 1998). Although it is unclear what the binding site for fluoxetine is on mitochondria, a more recent proteomic study demonstrated that SERT was associated with mitochondrial outer membrane proteins, suggesting that the mitochondrial binding site for fluoxetine may be SERT (Haase et al. 2017). The relationship between 5-HT and mitochondria extends beyond SERT, as monoamine oxidase-A, the enzyme responsible for breaking down 5-HT is also located on the outer mitochondrial membrane (Naoi et al. 2016). Also, 5-HT receptors 3 and 4 have been shown to be located intracellularly on the outer mitochondrial membrane of cardiomyocytes where they serve to regulate mitochondrial function and Ca2+ homeostasis (Wang et al. 2016). Similarly, 5-HT7 receptors have been localised to the mitochondrial membrane in SH-SY5Y cells where they may function to enhance ETC complex IV activity (Tempio et al. 2020). In this sense, it may also be important to consider the potential for SERT blockade to decrease intracellular concentrations of 5-HT, thus influencing these intracellular 5-HT receptors. This may also impact serotonylation of small G proteins, which results in constitutive activation and a myriad of downstream effects (Muma and Mi 2015).
In addition to this, there is convincing evidence that signalling through multiple 5-HT receptors stimulates mitochondrial biogenesis. Fanibunda et al. (2019) determined that signalling through 5-HT2A receptors in cultured cortical neurons results in increased oxygen consumption, ATP production, mtDNA mass, and antioxidant capacity. This effect was regulated by the transcriptional coactivator PGC-1α, which was upregulated by 5-HT signalling. 5-HT1F receptor agonists have also been shown to induce mitochondrial biogenesis in rodent models of kidney injury, spinal cord injury, and Parkinson’s disease (Scholpa et al. 2018; Gibbs et al. 2018; Simmons et al. 2019; Fanibunda et al. 2019). Additionally, stimulation of the 5-HT7 receptor has been shown to rescue impaired ATP production in a the brain of a rat model of Rett Syndrome, further supporting a potential role of serotonergic signalling in the regulation of mitochondrial bioenergetics (Valenti et al. 2017). However, it remains unclear whether these effects are mediated by 5-HT acting on cell surface receptors or via receptors on the outer mitochondrial membrane.
Non-SERT Related Effects of SSRIs
SSRIs are typically understood to exert their effects by binding and blocking SERT, preventing the reuptake of 5-HT into the presynaptic terminal and consequently altering serotonergic signalling. However, there is evidence from studies presented in this review that effects may be mediated by non-SERT related activity. Moriguchi et al. (2015) showed that treatment with fluvoxamine normalised ATP production in CaMKIV null mice and suggested that these effects were attributed to the sigma-1 receptor, rather than SERT binding. The authors showed that the same effect on ATP production was not seen when animals were treated with paroxetine, an SSRI without sigma-1 receptor binding capacity. Interestingly, Steiner et al. (2015) also showed that the neuroprotective effects of fluoxetine and paroxetine were maintained in SERT knockout mice, suggesting that these effects are likely driven by SERT-independent activity. Interestingly, these are not the only studies to suggest that SSRIs may elicit their effects by mechanisms unrelated to SERT binding. There is evidence to suggest that antidepressants function by binding TRKB receptors, facilitating their activation by brain-derived neurotrophic factor (Casarotto et al. 2021). Additionally, it has been shown that antidepressants accumulate in lipid rafts independent of SERT binding, and that the consequent translocation of G-proteins away from lipid rafts allows the activation of the cAMP signalling cascade (Erb et al. 2016). These studies collectively suggest that SSRIs may function by mechanisms additional to SERT binding, and this may be important for understanding the relationship between SSRIs and mitochondrial function, as well as for the development of new and effective antidepressants.
Sex Bias in the Literature
SSRIs are used to treat a range of psychological conditions, most notably depressive and anxiety disorders. Depressive and anxiety disorders are diagnosed in women at around twice the rate of men, and symptoms and comorbid conditions associated with these conditions also differ between sexes (Eid et al. 2019; Bangasser and Cuarenta 2021). Women diagnosed with MDD are more likely to experience comorbid anxiety-related disorders, gastrointestinal disturbances, and eating disorders, whereas men diagnosed with MDD are more likely to report substance abuse (Eid et al. 2019). Importantly, the efficacy of SSRIs also differs between men and women, and this should be an important factor that is considered in studies investigating the effects of these drugs. Fluoxetine treatment increased serum tryptophan (5-HT precursor) levels by 83% in women, compared with just 32% in men (Bano et al. 2004), and further to this, SSRIs are more effective in treating depression for women than men. The efficacy of SSRIs is reduced for women after menopause, but this is reversed with hormone replacement therapy, again suggesting a role for sex hormones (LeGates et al. 2019).
Considering that both the efficacy of SSRIs and the conditions that they treat differ between sexes, it is interesting that only three of the studies identified in this review addressed sex as a biological variable. This is especially important to consider as it is well recognised that many parameters of mitochondrial function differ between sexes (Thorne et al. 2022b), and all the studies in this review measure mitochondrial abundance or function as a treatment outcome. There is increasing recognition of the differences between male and female physiology and appreciation that findings in males cannot necessarily be extrapolated to females. This is supported by the finding that the three studies comparing males and females described in this review all identified interesting sex differences.
Author Contributions to the Field
Investigations using animal models are also strongly influenced by the small number of research groups working in this area. For example, Claudia Lagranha is listed as a corresponding author on seven of the animal studies identified in this review. Work by this author investigated fluoxetine treatment in the first 21 days of life and during adolescence, while the remaining animal studies in this review largely focussed on SSRI treatments at adulthood. The studies by this author suggest that 5-HT signalling may be important for regulating mitochondrial function in brain development, with these effects persisting to adulthood. This reflects that serotonin has a role in brain development, being important in the development of thalamocortical axons and for proper organisation of the somatosensory cortex in rodents (Gaspar et al. 2003). Interestingly, studies by this author showed that these effects on mitochondrial function are not limited to the brain, showing similar effects in the heart, skeletal muscle, and brown adipose tissue as well (da Silva et al. 2015b, a; Braz et al. 2016; Silva et al. 2018; Simões-Alves et al. 2018). This suggests that modulation of serotonergic signalling during early life stages may have important effects throughout many tissues.
Limitations
While comprehensive, this review has limitations which are attributed to the broadness of the study. A vast array of methods were employed by the studies in this review, and this variation was evident across chosen drugs, doses, treatment duration, method of administration, species, and cell type, as well as the range of assays investigating mitochondrial abundance and activity. While some focussed on mitochondrial abundance, others chose OXPHOS and ATP production or oxidative stress or calcium signalling. Categorising such a broad array of results as either an increase or decrease dismisses the number of pathways and functions that mitochondria serve to regulate, and thus minimises the true complexity of mitochondrial biology. However, the many functions of mitochondria are intrinsically linked and should also not be viewed in isolation; this review serves to give a more generalised overview of the effect of SSRIs on mitochondrial biology. Studies were also not weighted based on study design and methodology, this review was instead focussed on providing a broad perspective and determining whether the published literature reached consensus on the relationship between SSRI treatment and mitochondrial biogenesis and function. Lastly, it is a limitation that all analyses were conducted by the same author, and while articles were reviewed twice by this author to improve accuracy and reduce bias, this is still a limited approach.
Conclusions
This review incorporated a variety of studies to explore the effect of SSRI treatments on mitochondrial biogenesis and function. Of these, animal studies likely provide the best insight for understanding the effects of SSRIs, as studies in isolated mitochondria or cultured cells are unlikely to reflect the complexities of 5-HT physiology. These studies collectively suggest that SSRI treatment within the therapeutic dose range may enhance mitochondrial biogenesis, respiratory chain activity, and ATP production, while also being protective against oxidative damage. Whether this effect is consistent in humans is unclear, and this is largely due to difficulties with studying the human brain. However, imaging studies have identified reduced glucose metabolism in the brains of people with MDD (Videbech 2000), and shown that these changes can be reversed with paroxetine treatment (Kennedy et al. 2001). The mechanism by which SSRIs function remains to be fully elucidated; however, there is a growing body of evidence to suggest that their effect on mitochondrial function may be important. Within this, consideration should be given to both extracellular 5-HT signalling and the effect of 5-HT receptors and SERT on the mitochondrial membrane. While cell surface 5-HT receptors have been shown to promote mitochondrial biogenesis, the effect of intracellular 5-HT is less clear, and this should be explored.
Data Availability Statement
Further data is available from the corresponding author upon request.
Acknowledgments
This work was supported by Victoria University of Wellington and the Neurological Foundation of New Zealand.
Conflicts of Interest
The authors have no relevant financial or non-financial interests to disclose.
References
- Abdel-Razaq W, Kendall DA, Bates TE (2011) The effects of antidepressants on mitochondrial function in a model cell system and isolated mitochondria. Neurochem Res 36:327–338. [CrossRef]
- Adzic M, Lukic I, Mitic M, et al (2013) Brain region- and sex-specific modulation of mitochondrial glucocorticoid receptor phosphorylation in fluoxetine treated stressed rats: Effects on energy metabolism. Psychoneuroendocrinology 38:2914–2924. [CrossRef]
- Adzic M, Mitic M, Radojcic M (2017) Mitochondrial estrogen receptors as a vulnerability factor of chronic stress and mediator of fluoxetine treatment in female and male rat hippocampus. Brain Res 1671:77–84. [CrossRef]
- Ahmadian E, Eftekhari A, Fard JK, et al (2017) In vitro and in vivo evaluation of the mechanisms of citalopram-induced hepatotoxicity. Arch Pharm Res 40:1296–1313. [CrossRef]
- Al-Zoairy R, Pedrini MT, Khan MI, et al (2017) Serotonin improves glucose metabolism by Serotonylation of the small GTPase Rab4 in L6 skeletal muscle cells. Diabetol Metab Syndr 9:1. [CrossRef]
- Arafat EA, Shabaan DA (2020) Fluoxetine ameliorates adult hippocampal injury in rats after early maternal separation. A biochemical, histological and immunohistochemical study. Biotech Histochem 95:55–68. [CrossRef]
- Bader M (2019) Serotonylation: Serotonin Signaling and Epigenetics. Front Mol Neurosci 12:288. [CrossRef]
- Bangasser DA, Cuarenta A (2021) Sex differences in anxiety and depression: circuits and mechanisms. Nat Rev Neurosci 1–11. [CrossRef]
- Bano S, Akhter S, Afridi MI (2004) Gender based response to fluoxetine hydrochloride medication in endogenous depression. J Coll Physicians Surg Pak 14:161–165.
- Beery AK, Zucker I (2011) Sex Bias in Neuroscience and Biomedical Research. Neurosci Biobehav Rev 35:565–572. [CrossRef]
- Berger M, Gray JA, Roth BL (2009) The Expanded Biology of Serotonin. Annu Rev Med 60:355–366. [CrossRef]
- Braz GRF, da Silva AI, Silva SCA, et al (2020a) Chronic serotonin reuptake inhibition uncouples brown fat mitochondria and induces beiging/browning process of white fat in overfed rats. Life Sci 245:117307. [CrossRef]
- Braz GRF, Freitas CM, Nascimento L, et al (2016) Neonatal SSRI exposure improves mitochondrial function and antioxidant defense in rat heart. Appl Physiol Nutr Metab 41:362–369. [CrossRef]
- Braz GRF, Silva SC de A, Pedroza AA da S, et al (2020b) Fluoxetine administration in juvenile overfed rats improves hypothalamic mitochondrial respiration and REDOX status and induces mitochondrial biogenesis transcriptional expression. Eur J Pharmacol 881:173200. [CrossRef]
- Casarotto PC, Girych M, Fred SM, et al (2021) Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184:1299-1313.e19. [CrossRef]
- Charles E, Hammadi M, Kischel P, et al (2017) The antidepressant fluoxetine induces necrosis by energy depletion and mitochondrial calcium overload. Oncotarget 8:3181–3196. [CrossRef]
- Chen F, Danladi J, Ardalan M, et al (2018) A Critical Role of Mitochondria in BDNF-Associated Synaptic Plasticity After One-Week Vortioxetine Treatment. Int J Neuropsychopharmacol 21:603–615. [CrossRef]
- Chen S, Owens GC, Crossin KL, Edelman DB (2007) Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol Cell Neurosci 36:472–483. [CrossRef]
- Chen X, Margolis KJ, Gershon MD, et al (2012) Reduced serotonin reuptake transporter (SERT) function causes insulin resistance and hepatic steatosis independent of food intake. PloS One 7:e32511. [CrossRef]
- Coiro P, Pollak DD (2019) Sex and gender bias in the experimental neurosciences: the case of the maternal immune activation model. Transl Psychiatry 9:1–8. [CrossRef]
- Commons KG, Linnros SE (2019) Delayed Antidepressant Efficacy and the Desensitization Hypothesis. ACS Chem Neurosci 10:3048–3052. [CrossRef]
- da Silva AI, Braz GRF, Pedroza AA, et al (2015a) Fluoxetine induces lean phenotype in rat by increasing the brown/white adipose tissue ratio and UCP1 expression. J Bioenerg Biomembr 47:309–318. [CrossRef]
- da Silva AI, Braz GRF, Silva-Filho R, et al (2015b) Effect of fluoxetine treatment on mitochondrial bioenergetics in central and peripheral rat tissues. Appl Physiol Nutr Metab Physiol Appl Nutr Metab 40:565–574. [CrossRef]
- Daud FV, Murad N, Meneghini A, et al (2009) Fluoxetine effects on mitochondrial ultrastructure of right ventricle in rats exposed to cold stress. Rev Bras Cir Cardiovasc 24:173–179. [CrossRef]
- de Oliveira MR (2016) Fluoxetine and the mitochondria: A review of the toxicological aspects. Toxicol Lett 258:185–191. [CrossRef]
- Eid RS, Gobinath AR, Galea LAM (2019) Sex differences in depression: Insights from clinical and preclinical studies. Prog Neurobiol 176:86–102. [CrossRef]
- Emmerzaal TL, Jacobs L, Geenen B, et al (2021) Chronic fluoxetine or ketamine treatment differentially affects brain energy homeostasis which is not exacerbated in mice with trait suboptimal mitochondrial function. Eur J Neurosci 53:2986–3001. [CrossRef]
- Erb SJ, Schappi JM, Rasenick MM (2016) Antidepressants Accumulate in Lipid Rafts Independent of Monoamine Transporters to Modulate Redistribution of the G Protein, Gαs. J Biol Chem 291:19725–19733. [CrossRef]
- Fanibunda SE, Deb S, Maniyadath B, et al (2019) Serotonin regulates mitochondrial biogenesis and function in rodent cortical neurons via the 5-HT2A receptor and SIRT1-PGC-1 alpha axis. Proc Natl Acad Sci U S A 116:11028–11037. [CrossRef]
- Filipovic D, Costina V, Peric I, et al (2017) Chronic fluoxetine treatment directs energy metabolism towards the citric acid cycle and oxidative phosphorylation in rat hippocampal nonsynaptic mitochondria. Brain Res 1659:41–54. [CrossRef]
- Garabadu D, Ahmad A, Krishnamurthy S (2015) Risperidone Attenuates Modified Stress-Re-stress Paradigm-Induced Mitochondrial Dysfunction and Apoptosis in Rats Exhibiting Post-traumatic Stress Disorder-Like Symptoms. J Mol Neurosci MN 56:299–312. [CrossRef]
- Gaspar P, Cases O, Maroteaux L (2003) The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci 4:1002–1012. [CrossRef]
- Gerö D, Szoleczky P, Suzuki K, et al (2013) Cell-based screening identifies paroxetine as an inhibitor of diabetic endothelial dysfunction. Diabetes 62:953–964. [CrossRef]
- Gibbs WS, Collier JB, Morris M, et al (2018) 5-HT1F receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am J Physiol Renal Physiol 315:F1119–F1128. [CrossRef]
- Głombik K, Stachowicz A, Trojan E, et al (2017) Evaluation of the effectiveness of chronic antidepressant drug treatments in the hippocampal mitochondria - A proteomic study in an animal model of depression. Prog Neuropsychopharmacol Biol Psychiatry 78:51–60. [CrossRef]
- Haase J, Grudzinska-Goebel J, Muller HK, et al (2017) Serotonin Transporter Associated Protein Complexes Are Enriched in Synaptic Vesicle Proteins and Proteins Involved in Energy Metabolism and Ion Homeostasis. Acs Chem Neurosci 8:1101–1116. [CrossRef]
- Han YS, Lee CS (2009) Antidepressants reveal differential effect against 1-methyl-4-phenylpyridinium toxicity in differentiated PC12 cells. Eur J Pharmacol 604:36–44. [CrossRef]
- Hroudova J, Fisar Z (2010) Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuro Endocrinol Lett 31:336–342.
- Jeong I, Yang JS, Hong YJ, et al (2017) Dapoxetine induces neuroprotective effects against glutamate-induced neuronal cell death by inhibiting calcium signaling and mitochondrial depolarization in cultured rat hippocampal neurons. Eur J Pharmacol 805:36–45. [CrossRef]
- Jeong J, Park M, Yoon JS, et al (2015) Requirement of AMPK activation for neuronal metabolic-enhancing effects of antidepressant paroxetine. Neuroreport 26:424–428. [CrossRef]
- Karson CN, Newton JE, Livingston R, et al (1993) Human brain fluoxetine concentrations. J Neuropsychiatry Clin Neurosci 5:322–329. [CrossRef]
- Kennedy SH, Evans KR, Krüger S, et al (2001) Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry 158:899–905. [CrossRef]
- Khedr LH, Nassar NN, Rashed L, et al (2019) TLR4 signaling modulation of PGC1-α mediated mitochondrial biogenesis in the LPS-Chronic mild stress model: Effect of fluoxetine and pentoxiyfylline. Life Sci 239:116869. [CrossRef]
- Kumar P, Kumar A (2009) Possible role of sertraline against 3-nitropropionic acid induced behavioral, oxidative stress and mitochondrial dysfunctions in rat brain. Prog Neuropsychopharmacol Biol Psychiatry 33:100–108. [CrossRef]
- Kwatra M, Jangra A, Mishra M, et al (2016) Naringin and Sertraline Ameliorate Doxorubicin-Induced Behavioral Deficits Through Modulation of Serotonin Level and Mitochondrial Complexes Protection Pathway in Rat Hippocampus. Neurochem Res 41:2352–2366. [CrossRef]
- Lee CS, Kim YJ, Jang ER, et al (2010) Fluoxetine Induces Apoptosis in Ovarian Carcinoma Cell Line OVCAR-3 Through Reactive Oxygen Species-Dependent Activation of Nuclear Factor-κB. Basic Clin Pharmacol Toxicol 106:446–453. [CrossRef]
- Lee JH, Park A, Oh K-J, et al (2019) The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int J Mol Sci 20:4924. [CrossRef]
- LeGates TA, Kvarta MD, Thompson SM (2019) Sex differences in antidepressant efficacy. Neuropsychopharmacology 44:140–154. [CrossRef]
- Li Y, Couch L, Higuchi M, et al (2012) Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicol Sci 127:582–591. [CrossRef]
- Ludka FK, Dal-Cim T, Binder LB, et al (2017) Atorvastatin and Fluoxetine Prevent Oxidative Stress and Mitochondrial Dysfunction Evoked by Glutamate Toxicity in Hippocampal Slices. Mol Neurobiol 54:3149–3161. [CrossRef]
- Magalhães P, Alves G, Llerena A, Falcão A (2017) Therapeutic Drug Monitoring of Fluoxetine, Norfluoxetine and Paroxetine: A New Tool Based on Microextraction by Packed Sorbent Coupled to Liquid Chromatography. J Anal Toxicol 41:631–638. [CrossRef]
- Moriguchi S, Sakagami H, Yabuki Y, et al (2015) Stimulation of Sigma-1 Receptor Ameliorates Depressive-like Behaviors in CaMKIV Null Mice. Mol Neurobiol 52:1210–1222. [CrossRef]
- Morrison SF, Madden CJ, Tupone D (2014) Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab 19:741–756. [CrossRef]
- Mukherjee J, Das MK, Yang ZY, Lew R (1998) Evaluation of the binding of the radiolabeled antidepressant drug, F-18-fluoxetine in the rodent brain: An in vitro and in vivo study. Nucl Med Biol 25:605–610. [CrossRef]
- Muma NA, Mi Z (2015) Serotonylation and Transamidation of Other Monoamines. ACS Chem Neurosci 6:961–969. [CrossRef]
- Naoi M, Riederer P, Maruyama W (2016) Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: genetic and environmental factors involved in type A MAO expression. J Neural Transm 123:91–106. [CrossRef]
- Ni W, Watts SW (2006) 5-Hydroxytryptamine in the Cardiovascular System: Focus on the Serotonin Transporter (sert). Clin Exp Pharmacol Physiol 33:575–583. [CrossRef]
- Overø KF (1982) Kinetics of citalopram in man; plasma levels in patients. Prog Neuropsychopharmacol Biol Psychiatry 6:311–318. [CrossRef]
- Peric I, Costina V, Stanisavljevic A, et al (2018) Proteomic characterization of hippocampus of chronically socially isolated rats treated with fluoxetine: Depression-like behaviour and fluoxetine mechanism of action. Neuropharmacology 135:268–283. [CrossRef]
- Reddy AP, Sawant N, Morton H, et al (2021) Selective serotonin reuptake inhibitor citalopram ameliorates cognitive decline and protects against amyloid beta-induced mitochondrial dynamics, biogenesis, autophagy, mitophagy and synaptic toxicities in a mouse model of Alzheimer’s disease. Hum Mol Genet 30:789–810. [CrossRef]
- Scaini G, Santos PM, Benedet J, et al (2010) Evaluation of Krebs cycle enzymes in the brain of rats after chronic administration of antidepressants. Brain Res Bull 82:224–227. [CrossRef]
- Scholpa NE, Lynn MK, Corum D, et al (2018) 5-HT1F receptor-mediated mitochondrial biogenesis for the treatment of Parkinson’s disease. Br J Pharmacol 175:348–358. [CrossRef]
- Shu X, Sun Y, Sun X, et al (2019) The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis 10:1–16. [CrossRef]
- Silva TLA, Braz GRF, Silva SC de A, et al (2018) Serotonin transporter inhibition during neonatal period induces sex-dependent effects on mitochondrial bioenergetics in the rat brainstem. Eur J Neurosci 48:1620–1634. [CrossRef]
- Simmons EC, Scholpa NE, Cleveland KH, Schnellmann RG (2019) 5-HT1F Receptor Agonist Induces Mitochondrial Biogenesis and Promotes Recovery from Spinal Cord Injury. J Pharmacol Exp Ther. [CrossRef]
- Simões-Alves AC, Silva-Filho RC, Braz GRF, et al (2018) Neonatal treatment with fluoxetine improves mitochondrial respiration and reduces oxidative stress in liver of adult rats. J Cell Biochem 119:6555–6565. [CrossRef]
- Sonei N, Amiri S, Jafarian I, et al (2017) Mitochondrial dysfunction bridges negative affective disorders and cardiomyopathy in socially isolated rats: Pros and cons of fluoxetine. World J Biol Psychiatry 18:39–53. [CrossRef]
- Steiner JP, Bachani M, Wolfson-Stofko B, et al (2015) Interaction of Paroxetine with Mitochondrial Proteins Mediates Neuroprotection. Neurotherapeutics 12:200–216. [CrossRef]
- Tagashira H, Bhuiyan MS, Shioda N, Fukunaga K (2014) Fluvoxamine rescues mitochondrial Ca2+ transport and ATP production through σ(1)-receptor in hypertrophic cardiomyocytes. Life Sci 95:89–100. [CrossRef]
- Tempio A, Niso M, Laera L, et al (2020) Mitochondrial Membranes of Human SH-SY5Y Neuroblastoma Cells Express Serotonin 5-HT7 Receptor. Int J Mol Sci 21:. [CrossRef]
- Thorne BN, Ellenbroek BA, Day DJ (2022a) Sex bias in the serotonin transporter knockout model: Implications for neuropsychiatric disorder research. Neurosci Biobehav Rev 134:. [CrossRef]
- Thorne BN, Ellenbroek BA, Day DJ (2022b) The serotonin reuptake transporter modulates mitochondrial copy number and mitochondrial respiratory complex gene expression in the frontal cortex and cerebellum in a sexually dimorphic manner. J Neurosci Res 100:869–879. [CrossRef]
- Trivedi MH, Rush AJ, Wisniewski SR, et al (2006) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163:28–40. [CrossRef]
- Tutakhail A, Nazari QA, Khabil S, et al (2019) Muscular and mitochondrial effects of long-term fluoxetine treatment in mice, combined with physical endurance exercise on treadmill. Life Sci 232:116508. [CrossRef]
- Valenti D, de Bari L, Vigli D, et al (2017) Stimulation of the brain serotonin receptor 7 rescues mitochondrial dysfunction in female mice from two models of Rett syndrome. Neuropharmacology 121:79–88. [CrossRef]
- Ventura-Clapier R, Moulin M, Piquereau J, et al (2017) Mitochondria: a central target for sex differences in pathologies. Clin Sci Lond 131:803–822. [CrossRef]
- Videbech P (2000) PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta Psychiatr Scand 101:11–20. [CrossRef]
- Villa RF, Ferrari F, Bagini L, et al (2017) Mitochondrial energy metabolism of rat hippocampus after treatment with the antidepressants desipramine and fluoxetine. Neuropharmacology 121:30–38. [CrossRef]
- Villa RF, Ferrari F, Gorini A, et al (2016) Effect of desipramine and fluoxetine on energy metabolism of cerebral mitochondria. Neuroscience 330:326–334. [CrossRef]
- Wang Q, Zhang H, Xu H, et al (2016) 5-HTR3 and 5-HTR4 located on the mitochondrial membrane and functionally regulated mitochondrial functions. Sci Rep 6:1–10. [CrossRef]
- Wen L, Jin Y, Li L, et al (2014) Exercise prevents raphe nucleus mitochondrial overactivity in a rat depression model. Physiol Behav 132:57–65. [CrossRef]
- Woitowich NC, Beery A, Woodruff T (2020) A 10-year follow-up study of sex inclusion in the biological sciences. eLife 9:e56344. [CrossRef]
- World Health Organisation (2021) WHO model list of essential medicines: 22nd list (2021).
Figure 1.
PRISMA Flow Chart showing the number of records retrieved, screened and analysed for this review.
Figure 1.
PRISMA Flow Chart showing the number of records retrieved, screened and analysed for this review.
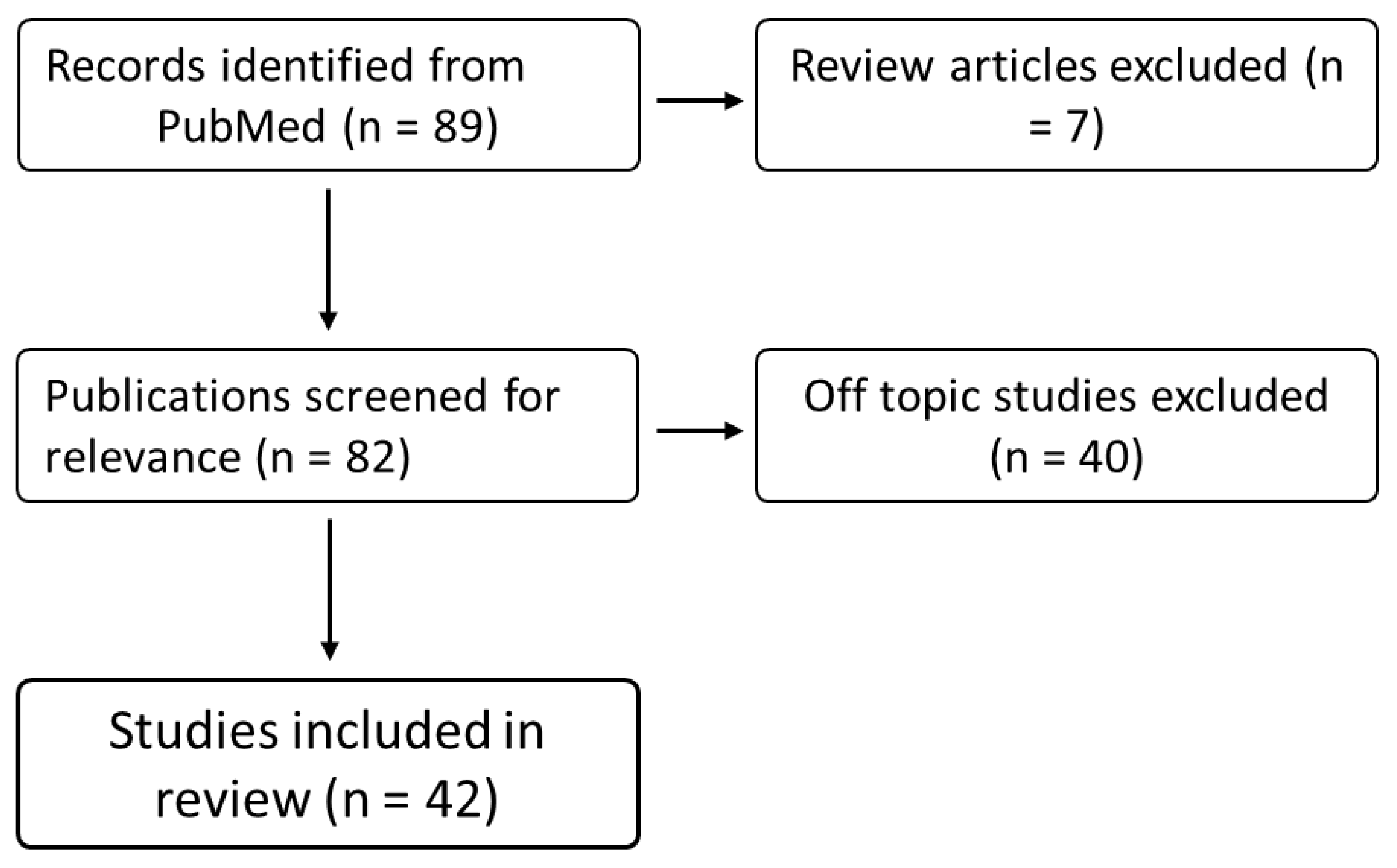
Figure 2.
Overview of drugs and doses in reviewed publications. (A) Publications investigating the effect of SSRI treatment on mitochondrial dynamics in isolated mitochondria, cultured cells, and animals between 2007 and 2021 (n = 40). (B) Publications investigating any drug classed as an SSRI were eligible for inclusion in this review. The most commonly used drug was fluoxetine (65% percent of publications), followed by paroxetine (13%), and citalopram (9%). (C, D) Doses and concentrations of SSRIs used in reviewed publications (all drugs combined).
Figure 2.
Overview of drugs and doses in reviewed publications. (A) Publications investigating the effect of SSRI treatment on mitochondrial dynamics in isolated mitochondria, cultured cells, and animals between 2007 and 2021 (n = 40). (B) Publications investigating any drug classed as an SSRI were eligible for inclusion in this review. The most commonly used drug was fluoxetine (65% percent of publications), followed by paroxetine (13%), and citalopram (9%). (C, D) Doses and concentrations of SSRIs used in reviewed publications (all drugs combined).

Figure 3.
Study outcomes by model used. Publications were classified based on whether SSRI treatment increased or decreased mitochondrial abundance or function (from baseline or a restoration), or whether the effects were mixed or neutral. Mitochondrial function was decreased in 100% of mitochondrial studies, 33% of cell culture studies, and 9.7% of animal studies. Function was increased in 56% of cell culture studies and 81% of animal studies. 11% of cell culture studies and 9.7% of animal studies showed a neutral or mixed effect.
Figure 3.
Study outcomes by model used. Publications were classified based on whether SSRI treatment increased or decreased mitochondrial abundance or function (from baseline or a restoration), or whether the effects were mixed or neutral. Mitochondrial function was decreased in 100% of mitochondrial studies, 33% of cell culture studies, and 9.7% of animal studies. Function was increased in 56% of cell culture studies and 81% of animal studies. 11% of cell culture studies and 9.7% of animal studies showed a neutral or mixed effect.
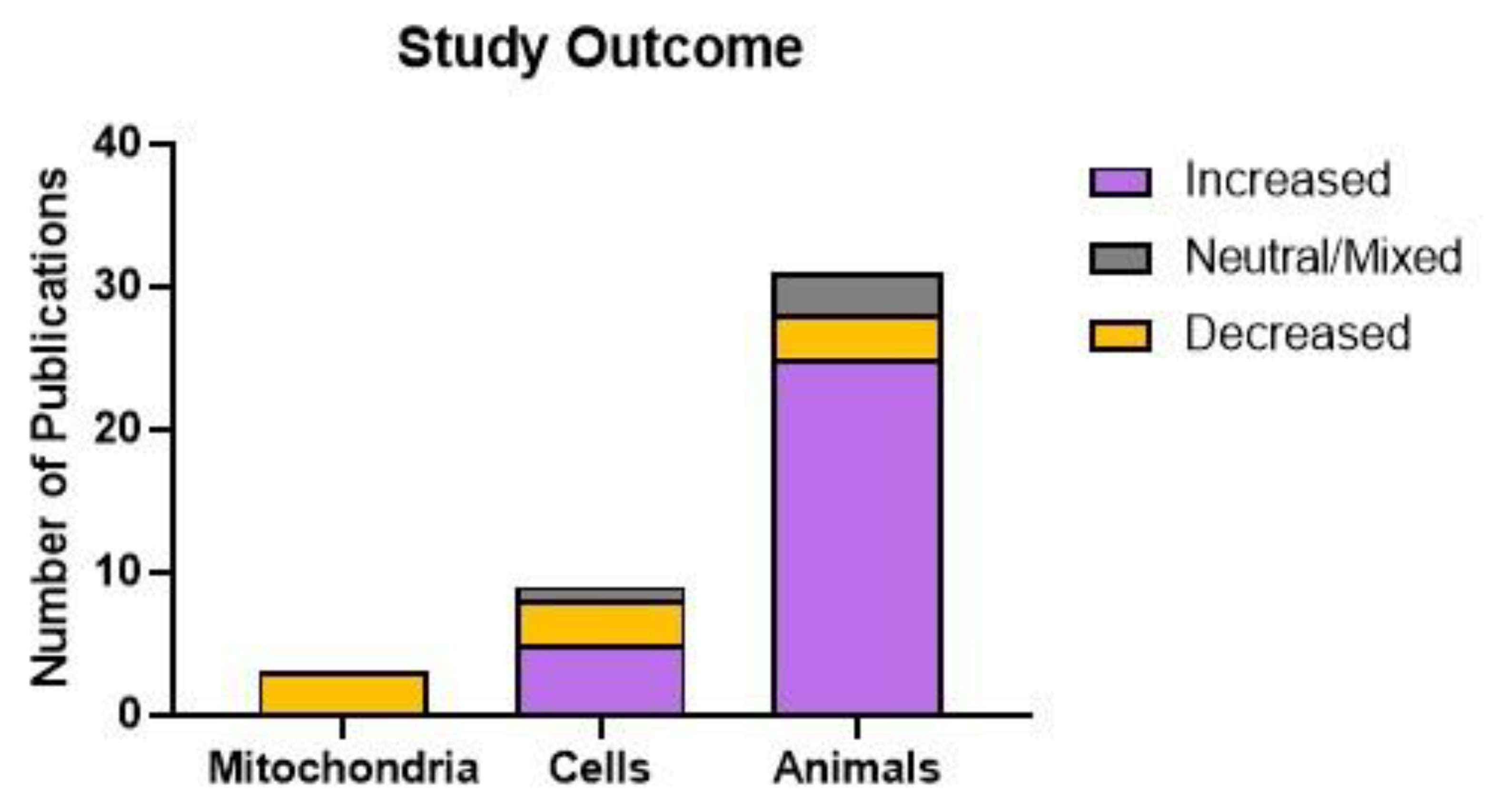
Figure 4.
Male bias in the reviewed publications. Publications included in this review were classified by whether they addressed sex as a biological variable. Studies of immortalised cell lines were excluded from this analysis.
Figure 4.
Male bias in the reviewed publications. Publications included in this review were classified by whether they addressed sex as a biological variable. Studies of immortalised cell lines were excluded from this analysis.
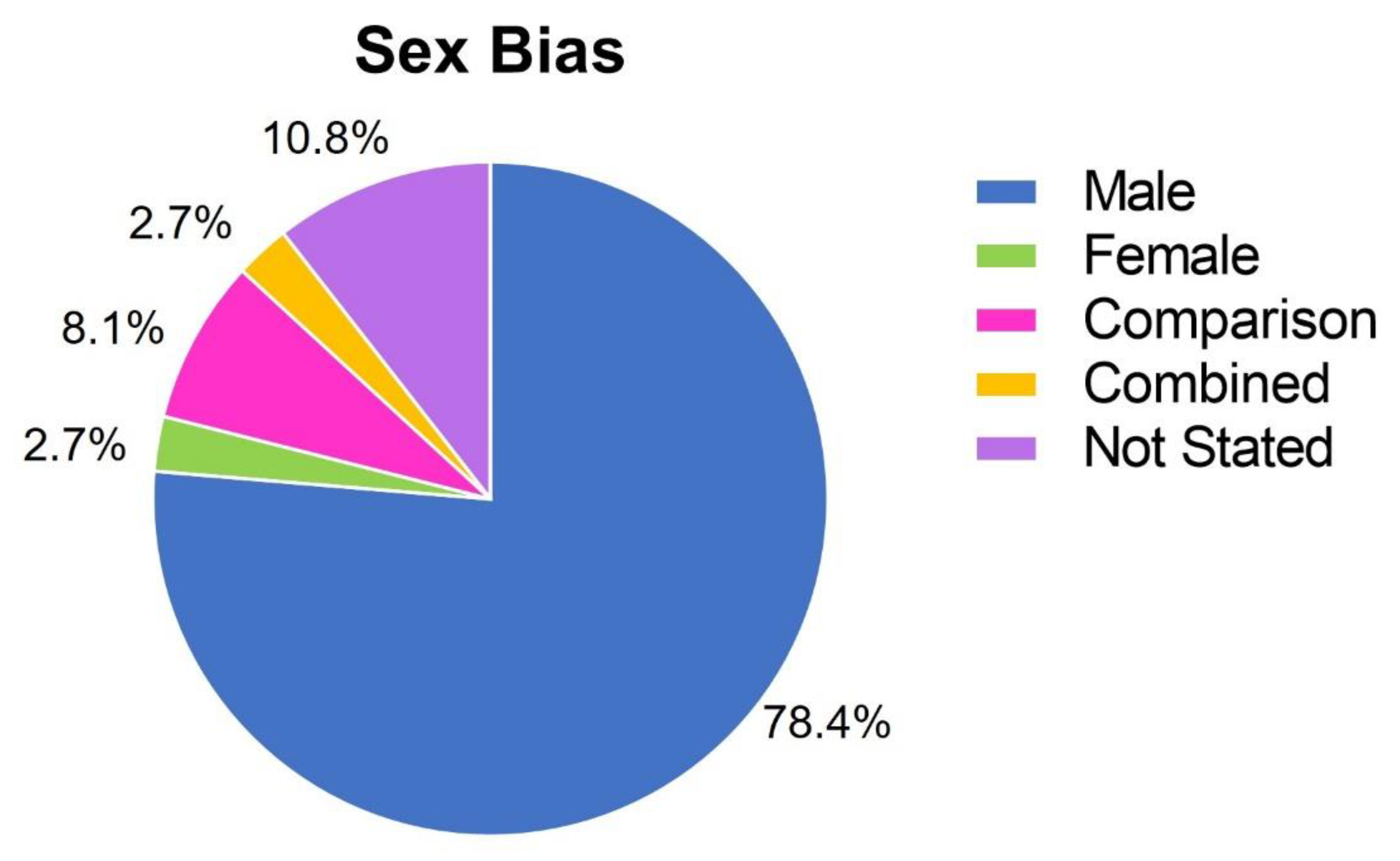
Figure 5.
Study outcomes by tissue type in animal studies. Publications of animal studies were classified by whether SSRI treatment increased or decreased mitochondrial function, or whether effects were mixed or neutral. Mitochondrial function was increased in 20 of 23 studies in the brain, all three studies of the heart, one of two studies in the liver and brown adipose tissue, and both studies of skeletal muscle. Function was decreased in one study of the brain and one of the liver, and neutral or mixed effects were found in two brain studies, and one study of brown adipose tissue.
Figure 5.
Study outcomes by tissue type in animal studies. Publications of animal studies were classified by whether SSRI treatment increased or decreased mitochondrial function, or whether effects were mixed or neutral. Mitochondrial function was increased in 20 of 23 studies in the brain, all three studies of the heart, one of two studies in the liver and brown adipose tissue, and both studies of skeletal muscle. Function was decreased in one study of the brain and one of the liver, and neutral or mixed effects were found in two brain studies, and one study of brown adipose tissue.
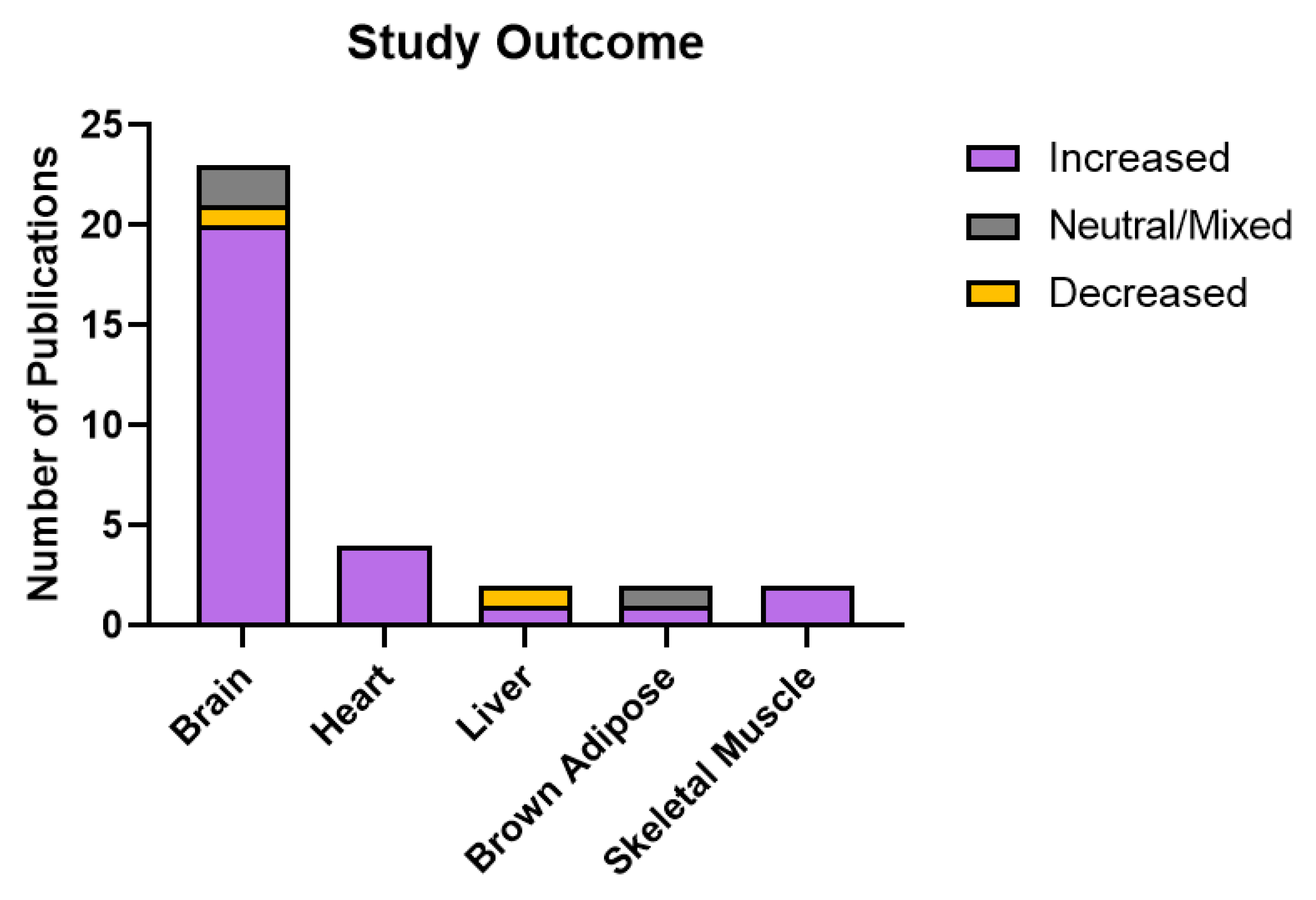

Table 1.
Risk of bias. Judgement of bias risk in this systematic review using the ROBIS tool.
| Domain | Concern | Rationale for Concern |
|---|---|---|
| Concerns regarding specification of study eligibility criteria |
Year of publication, language | Scientific studies published outside of the date range or in languages other than English may have been missed. |
| Concerns regarding methods used to identify and/or select studies |
Database Search | It is possible that the database searched did not contain an exhaustive list of the published literature in this field. |
| Concerns regarding methods used to collect data and appraise studies |
Single author review | Studies were identified and reviewed twice by one author, and this may have resulted in bias and/or error. |
| Concerns regarding the synthesis and findings |
Primary study quality and bias; diversity of methods used in primary studies. | Primary studies were not classified based on methodological robustness – each study was equally weighted with regard to findings. Primary studies used a wide variety of methods to determine mitochondrial function |
Table 2.
Publications studying the effect of SSRIs in isolated mitochondria and cultured cells. Summarised findings of publications investigating the effects of SSRI treatment on mitochondrial dynamics in studies of cultured cells and isolated mitochondria. Mitochondrial function or abundance was classified as increased (↑), decreased (↓), or neutral (↔).Animal studies.
Table 2.
Publications studying the effect of SSRIs in isolated mitochondria and cultured cells. Summarised findings of publications investigating the effects of SSRI treatment on mitochondrial dynamics in studies of cultured cells and isolated mitochondria. Mitochondrial function or abundance was classified as increased (↑), decreased (↓), or neutral (↔).Animal studies.
| Model/Sex | Treatment | Findings | Study |
|---|---|---|---|
| Pig brain mitochondria | 500 μM Citalopram | ↓ Citalopram inhibited complex I and II activity. | (Hroudova and Fisar 2010) |
| Rat heart mitochondria, CHOβ2SPAP cells | 10-50 μM Norfluoxetine | ↓ Norfluoxetine caused a decrease in MMP, complex I/II/III/IV activity and reduced O2 consumption. Effects were in cells and isolated mitochondria. | (Abdel-Razaq et al. 2011) |
| Primary rat hepatocytes; isolated liver mitochondria/Male | 12.5-100 μM Sertraline; 0.5 - 24 hours | ↓ Sertraline impaired complex I and ATP synthase but not other ETC complexes; uncoupled OXPHOS in mitochondria. Showed ATP depletion in cells. | (Li et al. 2012) |
| Rat primary hippocampal neuron cultures/ Male |
3 μM Fluoxetine | ↔ Treatment promoted anterograde axonal transport of mitochondria in hippocampal neurons. | (Chen et al. 2007) |
| PC12 Cells | 15 μM Fluoxetine, 24 hours | ↓ Fluoxetine had an additive effect with rotenone and MPP+ toxicity. Induced mitochondrial membrane permeability change and oxidative stress. Reduced cell viability. | (Han and Lee 2009) |
| OVCAR-3 and SK-OV-3 Cells | 15 μM Fluoxetine, 24 hours | ↓ Treatment induced activation of apoptotic proteins, cell death, ROS formation, loss of MMP, and cytochrome c release. | (Lee et al. 2010) |
| bEnd.3 and EA.hy926 Cells | 10 μM Paroxetine, 3 days | ↑ Paroxetine reduced hyperglycaemia-induced mitochondrial ROS formation, DNA damage, and protein oxidation without influencing electron transport or cellular bioenergetics. | (Gerö et al. 2013) |
| SK-N-MC Cells | 2-10 μM Paroxetine | ↑ Dose-dependent increase in mitochondrial biogenesis, mtDNA copy number, TFAM/PGC1a mRNA expression, ATP levels, and glucose uptake. | (Jeong et al. 2015) |
| Sprague-Dawley Rat, SERT Knockout mouse, human primary neuronal cultures; Sprague Dawley Rats | 0.01-10 μM Paroxetine/ Fluoxetine; 10 mg/kg, 10 or 28 days |
↑ SSRIs identified as protective against oxidative stress. Paroxetine and fluoxetine protected against Tat-induced neurotoxicity (paroxetine to a greater extent). Paroxetine stimulated proliferation of NPCs and generation of newborn neurons. Inhibited Ca2+-induced swelling in brain mitochondria. | (Steiner et al. 2015) |
| Jurkat and HeLa Cells; Patient PBMCs | 40 μM Fluoxetine | ↓ Decreased oxygen consumption, ATP content with fluoxetine treatment. | (Charles et al. 2017) |
| Rat primary hippocampal neuron cultures/Not Stated | 5 μM Dapoxetine, Fluoxetine, Citalopram | ↑ Dapoxetine treatment inhibited glutamate-induced Ca2+ increase, mitochondrial depolarisation, and cell death. Effects of citalopram and fluoxetine less pronounced. | (Jeong et al. 2017) |
| HT22 Cells | 20 μM Citalopram | ↑ Citalopram treatment enhanced mitochondrial biogenesis and mitophagy in HT22 cells. Treatment restored impaired mitochondrial dynamics in cells transfected with mutant APP. | (Reddy et al. 2021) |
Table 3.
Publications studying SSRIs in animals. Summarised findings of publications investigating the effects of SSRI treatment on mitochondrial dynamics in animal studies. Mitochondrial function or abundance was classified as increased (↑), increased/restored following a disruption (*↑), decreased (↓), or decreased/restored following a disruption (*↓).
Table 3.
Publications studying SSRIs in animals. Summarised findings of publications investigating the effects of SSRI treatment on mitochondrial dynamics in animal studies. Mitochondrial function or abundance was classified as increased (↑), increased/restored following a disruption (*↑), decreased (↓), or decreased/restored following a disruption (*↓).
| Model/Sex | Treatment | Findings | Study |
| Wistar rats/ Male |
0.75 mg/kg Fluoxetine, 40 days | *↑ Fluoxetine prevented mitochondrial cristolysis in the heart under cold stress. | (Daud et al. 2009) |
| Wistar rats/ Male |
5, 10 mg/kg Sertraline, 14 days | *↑ Sertraline normalised electron transport complex activity and oxidative stress in the brains of a rat model of Huntington’s Disease. | (Kumar and Kumar 2009) |
| Wistar rats/Male | 10 mg/kg Paroxetine, 15 days | ↑ Paroxetine treatment increased citrate synthase and succinate dehydrogenase activity in the prefrontal cortex and hippocampus, but not the cerebellum. | (Scaini et al. 2010) |
| Wistar rats/Male and Female | 5 mg/kg Fluoxetine, 21 days | ↓↑ Males and females respond differently to fluoxetine treatment following chronic stress. ETC complex IV mRNA expression and activity were altered depending on sex, treatment, and brain region. | (Adzic et al. 2013) |
| Sprague-Dawley Rats/Male | 5 mg/kg Fluoxetine, 18 days | *↓ Chronic unpredictable stress resulted in increased ATP production and antioxidant defence in the DRN. Changes were normalised by exercise and fluoxetine treatment. | (Wen et al. 2014) |
| ICR Mice/Wistar Rat primary cardiomyocyte cultures | 1 mg/kg Fluvoxamine, 4 weeks; 5 μM in culture | *↑ Fluvoxamine treatment rescued ATP production in the hearts of mice that had undergone transverse aortic constriction. A similar effect was seen in cultured rat primary cardiomyocytes, whereby fluvoxamine treatment rescued Ca2+ mobilisation and ATP production in cardiomyocytes treated with angiotensin II to promote hypertrophy. | (Tagashira et al. 2014) |
| CaMKIV Null Mice/Male | 2.5 mg/kg Fluvoxamine, 1 mg/kg Paroxetine, 14 days | *↑ Fluvoxamine normalised ATP production in the hippocampus of CaMKIV null mice. Suggested that these changes were attributed to the sigma-1 receptor rather than altered serotonergic signalling, as treatment with paroxetine, an SSRI lacking sigma-1 receptor affinity, did not show the same effects. | (Moriguchi et al. 2015) |
| Wistar Rats/Male | 10 mg/kg Fluoxetine, first 21 days of life | ↑ Increased O2 consumption and citrate synthase activity, reduced ROS production in skeletal muscle and hypothalamus with fluoxetine treatment at adulthood. | (da Silva et al. 2015b) |
| Charles Foster Rats/Male | 10 mg/kg Paroxetine, 24 days | *↑ Paroxetine ameliorated stress-induced oxidative damage in the brain; no effect on OXPHOS. | (Garabadu et al. 2015) |
| Wistar Rats/Male | 10 mg/kg Fluoxetine, first 21 days of life | ↑ Fluoxetine increased mitochondrial respiration and proton leak increased expression of UCP1, decreased ROS production in brown adipose tissue at adulthood. | (da Silva et al. 2015a) |
| Wistar Rats/Male | 10 mg/kg fluoxetine, first 21 days of life | ↑ Neonatal fluoxetine treatment increased mitochondrial respiratory capacity and membrane potential and decreased ROS production in the heart at adulthood. | (Braz et al. 2016) |
| Sprague-Dawley Rats/Male | 10 mg/kg Fluoxetine, 21 days | ↑ Enhanced complex IV activity in non-synaptic mitochondria and synaptic “heavy” mitochondria isolated from the FC of rats. | (Villa et al. 2016) |
| Wistar Rats/Male | 5 mg/kg Sertraline | *↑ Combined sertraline and narinign treatment restored mitochondrial dysfunction and oxidative stress in the hippocampus following doxorubicin exposure. | (Kwatra et al. 2016) |
| Sprague-Dawley Rat Hepatocytes/ Not Stated |
20 mg/kg in rats; 500 μM Citalopram in isolated hepatocytes | ↓ In vivo experiments showed that treatment caused oxidative damage in the liver, and in vitro experiments showed that this dose caused oxidative damage and collapse of the mitochondrial membrane potential. | (Ahmadian et al. 2017) |
| Sprague-Dawley Rats/Male | 10 mg/kg Fluoxetine, 21 days | ↑ Enhanced complex IV, succinate dehydrogenase, and glutamate dehydrogenase activity non-synaptic mitochondria isolated from the hippocampus of rats. | (Villa et al. 2017) |
| Wistar Rats/Male | 7.5 mg/kg Fluoxetine | *↑ Treatment rescued decreased complex II activity in the brains and hearts of rats affected by social isolation stress. Oxidative damage, collapse of the mitochondrial membrane potential, and reduced ATP production were rescued in the brains only. | (Sonei et al. 2017) |
| Wistar Rats/Male | 15 mg/kg Fluoxetine, 21 days | ↑ Fluoxetine upregulated mitochondrial proteins related to OXPHOS and TCA cycle in the hippocampus. Upregulation of subunits for complexes I, II and III and ATP synthase. | (Filipovic et al. 2017) |
| Swiss Mice/Male | 10 mg/kg Fluoxetine (1 dose for acute, 7 days for chronic) | *↑ Chronic but not acute treatment was protective against oxidative stress and collapse of the mitochondrial membrane potential due to glutamate excitotoxicity in the hippocampus. | (Ludka et al. 2017) |
| Sprague-Dawley Rats/Male | 10 mg/kg Fluoxetine, 21 days | ↑↓ Proteomic study – upregulation and downregulation of a variety of proteins involved with mitochondrial dynamics, function, and maturation in the hippocampus with fluoxetine treatment. | (Głombik et al. 2017) |
| Wistar Rats/Male and Female | 5 mg/kg Fluoxetine, 21 days | ↑ Fluoxetine treatment increased complex IV activity in the hippocampus of control males and stressed females. | (Adzic et al. 2017) |
| Sprague-Dawley Rats/Male | Vortioxetine 1.6 g/kg in food, Fluoxetine 160 mg/L in drinking water, 7 days | ↑ Vortioxetine treatment increased number of mitochondria in total neuropil and axon terminals in the hippocampus. No change with fluoxetine treatment. | (Chen et al. 2018) |
| Wistar Rats/Male | 15 mg/kg Fluoxetine, 21 days | *↑ Fluoxetine treatment restored decreased expression of proteins involved with mitochondrial transport, Krebs cycle, and OXPHOS in the hippocampus following chronic stress. | (Peric et al. 2018) |
| Wistar Rats/Male | 10 mg/kg Fluoxetine, first 21 days of life | ↑ Increased oxygen consumption in the livers of fluoxetine treated animals at adulthood, reduced oxidative stress. Increased resistance to mPTP opening. | (Simões-Alves et al. 2018) |
| Wistar Rats/Male and Female | 10 mg/kg Fluoxetine, 21 days | ↑ Improved mitochondrial bioenergetics in the brainstem of fluoxetine treated males, improved antioxidant defence in the brainstem of treated females. | (Silva et al. 2018) |
| BALB/cJ Mice/Male | 18 mg/kg Fluoxetine, 6 weeks | ↑ Increased citrate synthase and complex IV activity and decreased ROS production in skeletal muscle in fluoxetine treated, exercising mice. | (Tutakhail et al. 2019) |
| C57/BL6J Mice/Male; primary cultured astrocytes | 10 mg/kg Fluoxetine, 28 days; 10 μM in culture | *↑ Mitochondrial structure in the hippocampus disrupted following stress, restored with fluoxetine. Treatment also promoted mitophagy in primary astrocytes. | (Shu et al. 2019) |
| Wistar Rats/Male | 10 mg/kg Fluoxetine, 30 days | *↑ Fluoxetine ameliorated reduced mtDNA copy number and mRNA expression of Ppargc1a, Tfam, Nrf1 in the hippocampus of stressed animals. | (Khedr et al. 2019) |
| Wistar Rats/Male | 10 mg/kg Fluoxetine, 21 days | ↑↓ Fluoxetine treatment (PND 39-59) resulted in increased oxygen consumption and decreased oxidative damage in brown adipose tissue of rats overfed as neonates. In normofed rats, oxygen consumption was also increased with fluoxetine treatment, but there was increased oxidative damage. | (Braz et al. 2020a) |
| Albino Rats/Male | 5 mg/kg Fluoxetine, 7 days | *↑ Fluoxetine ameliorated ultrastructural changes to mitochondria in the hippocampus of pups exposed to maternal separation stress. | (Arafat and Shabaan 2020) |
| Wistar Rats/Male | 10 mg/kg Fluoxetine, 21 days | ↑ Fluoxetine administration (PND 39-59) in rats overfed as neonates restored mitochondrial function, oxidative balance, and mitochondrial biogenesis in the hypothalamus. | (Braz et al. 2020b) |
| Ndufs4GT/GT Mice/Male | 15 mg/kg Fluoxetine, 21 days | *↓ Fluoxetine treatment reduced complex III and IV activity in the FC following chronic unpredictable stress. | (Emmerzaal et al. 2021) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



