Submitted:
16 December 2024
Posted:
17 December 2024
You are already at the latest version
Abstract
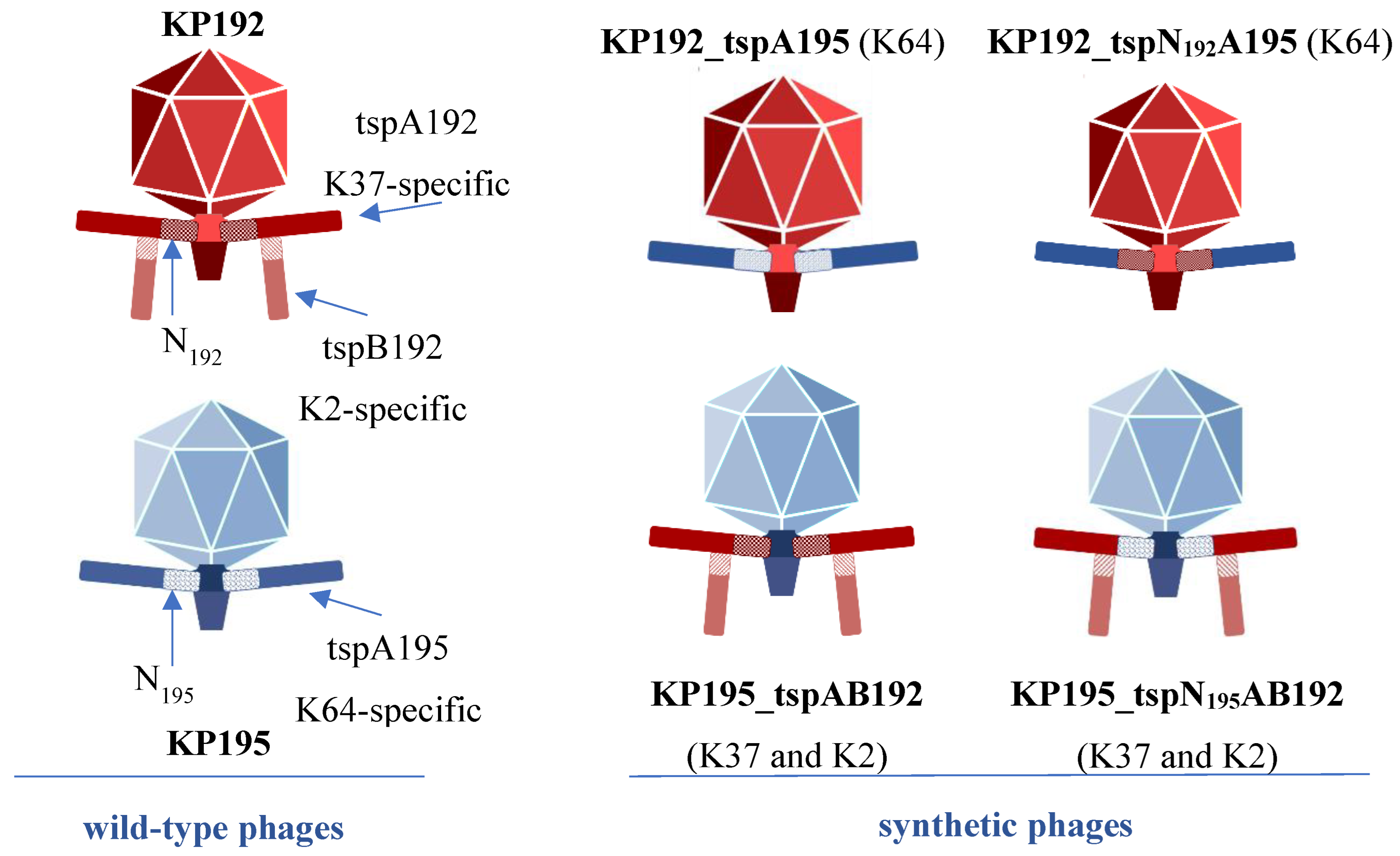
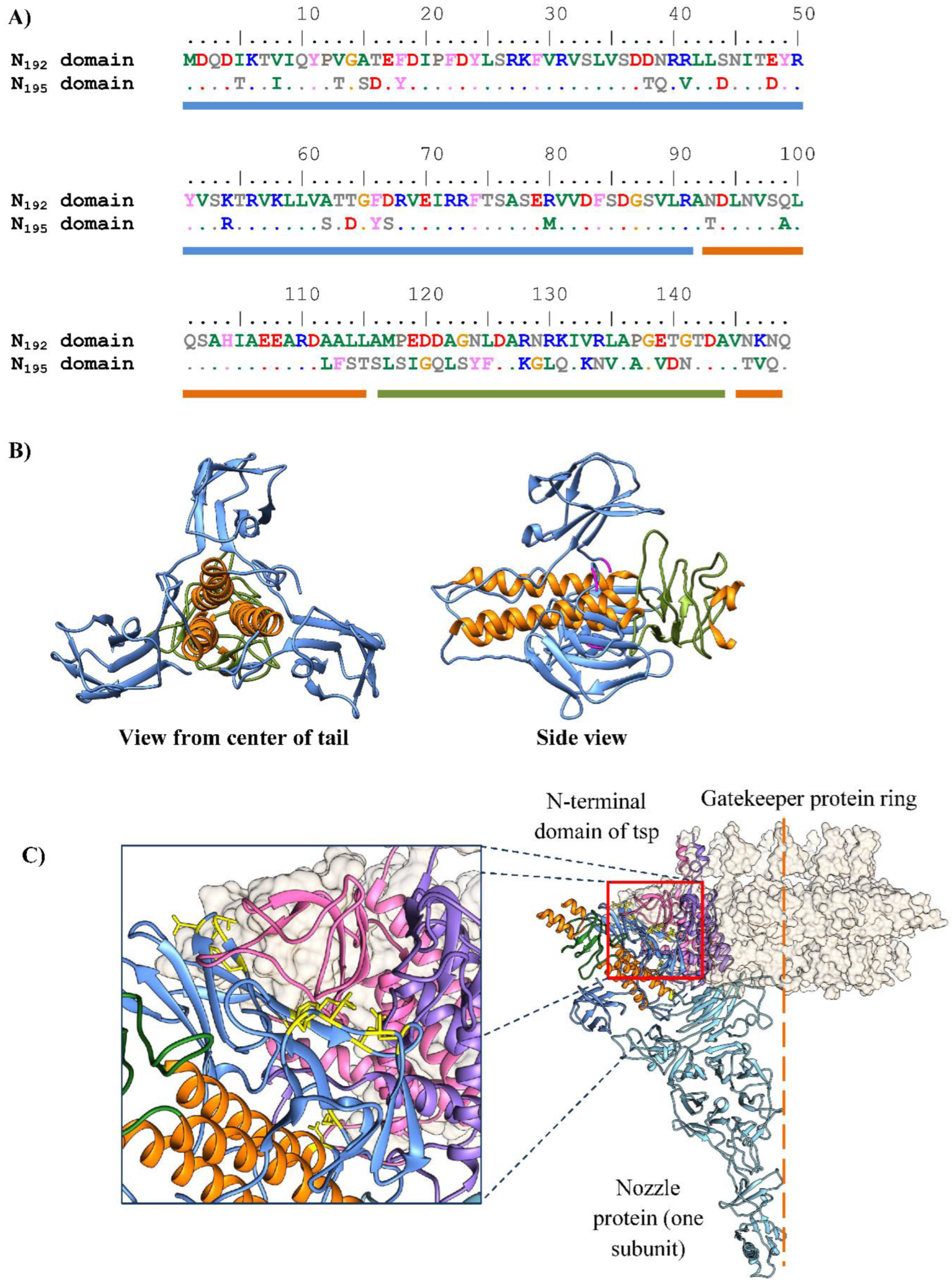
In this study, synthetic phages with swapped tailspike protein genes were constructed based on Przondovirus bacteriophages KP32_192 and KP32_195, which infect Klebsiella pneumoniae of different capsular serotypes. The specificity of these phages has changed in accordance with the genes of tailspike proteins. However, the reproduction efficiency of these phages strongly depended on the phage scaffold used, the sequence of the N-terminal domain of tail-anchored spikes, and the Klebsiella strain used. Moreover, phages with a non-native scaffold replicated on one of the strains more efficiently than wild-type phage with the same tailspikes. It has also been shown that the sequence of the N-terminal domain of tail-anchored spikes can influence the efficiency of phage adsorption. Therefore, it is necessary to consider not only the capsular specificity of tailspike proteins but also some other genes in the recipient phage genome when designing synthetic phages against the target bacterium.
Keywords:
1. Introduction
2. Methods
2.1. Phages, Bacterial and Yeast Strains
2.2. Culturing Conditions
2.3. Preparation of PCR Products for Assembly of Phage Genomes
2.4. Phage Genome Assembly in Yeast
2.5. Yeast Colony Screening
2.6. Isolation of a Yeast Centromeric Plasmid Containing the Bacteriophage Genome
2.7. Phage Genome “Rebooting”
2.8. Verification of Genome Assembly Accuracy and Genome Sequencing
2.9. Phage Propagation and Purification
2.10. Determination of Infectious Titer of Phage Samples
2.11. Determination of Pseudo-Physical Titer (TiterPP) of Phage Samples
2.12. Efficiency of Plating Determination
2.13. Bacterial Killing Assay
2.14. Determination of Phage Adsorption Efficiency
2.15. One-Step Growth Curves Determination
2.16. Protein Structure Modelling and Visualization
2.17. Bioinformatic Analysis of Differences Between KP192 and KP195 Genomes
3. Results
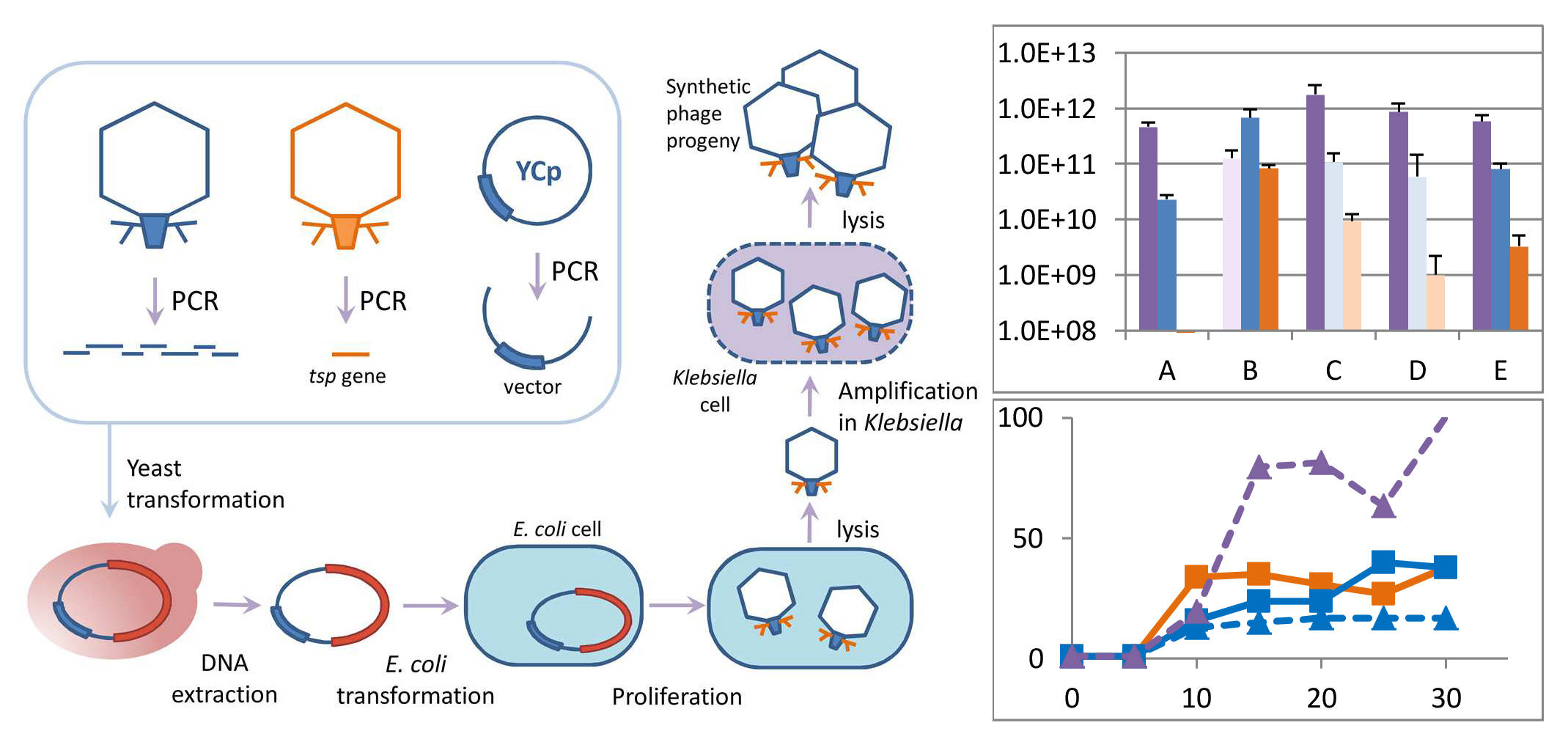
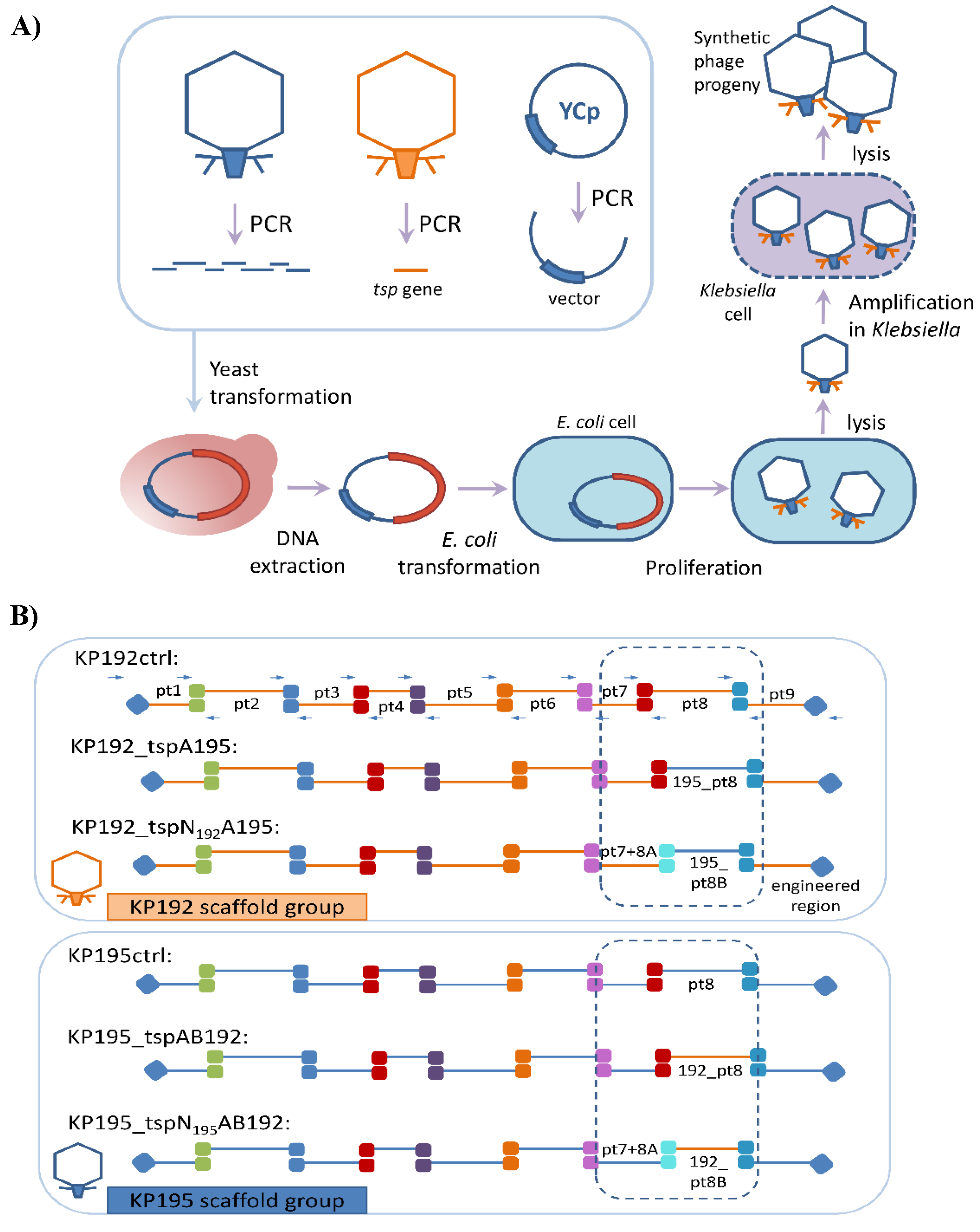
3.1. Study Design
3.2. Assembly of Synthetic Phage Genomes
3.3. “Rebooting” of Klebsiella Phage Genomes
3.4. Infectious Properties of Phages KP192, KP195_tspAB192 and KP195_tspN195AB192
3.5. Analysis of Differences Between the Genomes of Phages KP192 and KP195 That Potentially Affect the Efficiency of Reproduction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abedon ST. Bacteriophage Adsorption: Likelihood of Virion Encounter with Bacteria and Other Factors Affecting Rates. Antibiotics (Basel). 2023 Apr 7;12(4):723. [CrossRef]
- Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, Bodenstein SW, Evans DA, Hung CC, O’Neill M, Reiman D, Tunyasuvunakool K, Wu Z, Žemgulytė A, Arvaniti E, Beattie C, Bertolli O, Bridgland A, Cherepanov A, Congreve M, Cowen-Rivers AI, Cowie A, Figurnov M, Fuchs FB, Gladman H, Jain R, Khan YA, Low CMR, Perlin K, Potapenko A, Savy P, Singh S, Stecula A, Thillaisundaram A, Tong C, Yakneen S, Zhong ED, Zielinski M, Žídek A, Bapst V, Kohli P, Jaderberg M, Hassabis D, Jumper JM. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024 Jun;630(8016):493-500. [CrossRef]
- Ando H, Lemire S, Pires DP, Lu TK. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015 Sep 23;1(3):187-196. [CrossRef]
- Brzozowska E, Pyra A, Pawlik K, Janik M, Górska S, Urbańska N, Drulis-Kawa Z, Gamian A. Hydrolytic activity determination of Tail Tubular Protein A of Klebsiella pneumoniae bacteriophages towards saccharide substrates. Sci Rep. 2017 Dec 22;7(1):18048. [CrossRef]
- Carroll-Portillo A, Coffman CN, Varga MG, Alcock J, Singh SB, Lin HC. Standard Bacteriophage Purification Procedures Cause Loss in Numbers and Activity. Viruses. 2021 Feb 20;13(2):328. [CrossRef]
- Chen W, Xiao H, Wang L, Wang X, Tan Z, Han Z, Li X, Yang F, Liu Z, Song J, Liu H, Cheng L. Structural changes in bacteriophage T7 upon receptor-induced genome ejection. Proc Natl Acad Sci U S A. 2021 Sep 14;118(37):e2102003118. [CrossRef]
- Cuervo A, Fàbrega-Ferrer M, Machón C, Conesa JJ, Fernández FJ, Pérez-Luque R, Pérez-Ruiz M, Pous J, Vega MC, Carrascosa JL, Coll M. Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat Commun. 2019 Aug 20;10(1):3746. [CrossRef]
- Cui L, Watanabe S, Miyanaga K, Kiga K, Sasahara T, Aiba Y, Tan XE, Veeranarayanan S, Thitiananpakorn K, Nguyen HM, Wannigama DL. A Comprehensive Review on Phage Therapy and Phage-Based Drug Development. Antibiotics (Basel). 2024 Sep 11;13(9):870. [CrossRef]
- Dunne M, Prokhorov NS, Loessner MJ, Leiman PG. Reprogramming bacteriophage host range: design principles and strategies for engineering receptor binding proteins. Curr Opin Biotechnol. 2021 Apr;68:272-281. [CrossRef]
- Dunne M, Prokhorov NS, Loessner MJ, Leiman PG. Reprogramming bacteriophage host range: design principles and strategies for engineering receptor binding proteins. Curr Opin Biotechnol. 2021 Apr;68:272-281. [CrossRef]
- Fournet-Fayard S, Joly B, Forestier C. Transformation of wild type Klebsiella pneumoniae with plasmid DNA by electroporation.Journal of Microbiological Methods 1995 November; 24(1):49-54. [CrossRef]
- Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, Merryman C, Young L, Noskov VN, Glass JI, Venter JC, Hutchison CA 3rd, Smith HO. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008 Feb 29;319(5867):1215-20. [CrossRef]
- Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992 Mar 25;20(6):1425. [CrossRef]
- Gietz RD. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol Biol. 2014;1163:33-44. [CrossRef]
- Guo F, Liu Z, Fang PA, Zhang Q, Wright ET, Wu W, Zhang C, Vago F, Ren Y, Jakana J, Chiu W, Serwer P, Jiang W. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc Natl Acad Sci U S A. 2014 Oct 28;111(43):E4606-14. [CrossRef]
- Jaschke PR, Lieberman EK, Rodriguez J, Sierra A, Endy D. A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology. 2012 Dec 20;434(2):278-84. [CrossRef]
- Kemp P, Garcia LR, Molineux IJ. Changes in bacteriophage T7 virion structure at the initiation of infection. Virology. 2005 Sep 30;340(2):307-17. [CrossRef]
- Krishna R, Wang J, Ahern W, Sturmfels P, Venkatesh P, Kalvet I, Lee GR, Morey-Burrows FS, Anishchenko I, Humphreys IR, McHugh R, Vafeados D, Li X, Sutherland GA, Hitchcock A, Hunter CN, Kang A, Brackenbrough E, Bera AK, Baek M, DiMaio F, Baker D. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science. 2024 Apr 19;384(6693):eadl2528. [CrossRef]
- Larionov V, Kouprina N, Graves J, Resnick MA. Highly selective isolation of human DNAs from rodent-human hybrid cells as circular yeast artificial chromosomes by transformation-associated recombination cloning. Proc Natl Acad Sci U S A. 1996 Nov 26;93(24):13925-30. [CrossRef]
- Latka A, Leiman PG, Drulis-Kawa Z, Briers Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front Microbiol. 2019 Nov 15;10:2649. [CrossRef]
- Latka A, Lemire S, Grimon D, Dams D, Maciejewska B, Lu T, Drulis-Kawa Z, Briers Y. Engineering the Modular Receptor-Binding Proteins of Klebsiella Phages Switches Their Capsule Serotype Specificity. mBio. 2021 May 4;12(3):e00455-21. [CrossRef]
- Leiman PG, Chipman PR, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell. 2004 Aug 20;118(4):419-29. [CrossRef]
- Lenneman BR, Fernbach J, Loessner MJ, Lu TK, Kilcher S. Enhancing phage therapy through synthetic biology and genome engineering. Curr Opin Biotechnol. 2021 Apr;68:151-159. [CrossRef]
- Marinelli LJ, Piuri M, Swigonová Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One. 2008;3(12):e3957. [CrossRef]
- Meile S, Du J, Staubli S, Grossmann S, Koliwer-Brandl H, Piffaretti P, Leitner L, Matter CI, Baggenstos J, Hunold L, Milek S, Guebeli C, Kozomara-Hocke M, Neumeier V, Botteon A, Klumpp J, Marschall J, McCallin S, Zbinden R, Kessler TM, Loessner MJ, Dunne M, Kilcher S. Engineered reporter phages for detection of Escherichia coli, Enterococcus, and Klebsiella in urine. Nat Commun. 2023 Jul 20;14(1):4336. [CrossRef]
- Moraru C, Varsani A, Kropinski AM. VIRIDIC-A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses. 2020 Nov 6;12(11):1268. [CrossRef]
- Morozova V, Babkin I, Kozlova Y, Baykov I, Bokovaya O, Tikunov A, Ushakova T, Bardasheva A, Ryabchikova E, Zelentsova E, Tikunova N. Isolation and Characterization of a Novel Klebsiella pneumoniae N4-like Bacteriophage KP8. Viruses. 2019 Dec 2;11(12):1115. [CrossRef]
- Patro LPP, Sudhakar KU, Rathinavelan T. K-PAM: a unified platform to distinguish Klebsiella species K- and O-antigen types, model antigen structures and identify hypervirulent strains. Sci Rep. 2020 Oct 7;10(1):16732. [CrossRef]
- Pyra A, Brzozowska E, Pawlik K, Gamian A, Dauter M, Dauter Z. Tail tubular protein A: a dual-function tail protein of Klebsiella pneumoniae bacteriophage KP32. Sci Rep. 2017 May 22;7(1):2223. [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; pp. 187–303.
- Shen J, Zhou J, Chen GQ, Xiu ZL. Efficient Genome Engineering of a Virulent Klebsiella Bacteriophage Using CRISPR-Cas9. J Virol. 2018 Aug 16;92(17):e00534-18. [CrossRef]
- Strathdee SA, Hatfull GF, Mutalik VK, Schooley RT. Phage therapy: From biological mechanisms to future directions. Cell. 2023 Jan 5;186(1):17-31. [CrossRef]
- Wang C, Wang S, Jing S, Zeng Y, Yang L, Mu Y, Ding Z, Song Y, Sun Y, Zhang G, Wei D, Li M, Ma Y, Zhou H, Wu L, Feng J. Data-Driven Engineering of Phages with Tunable Capsule Tropism for Klebsiella pneumoniae. Adv Sci (Weinh). 2024 Sep;11(33):e2309972. [CrossRef]

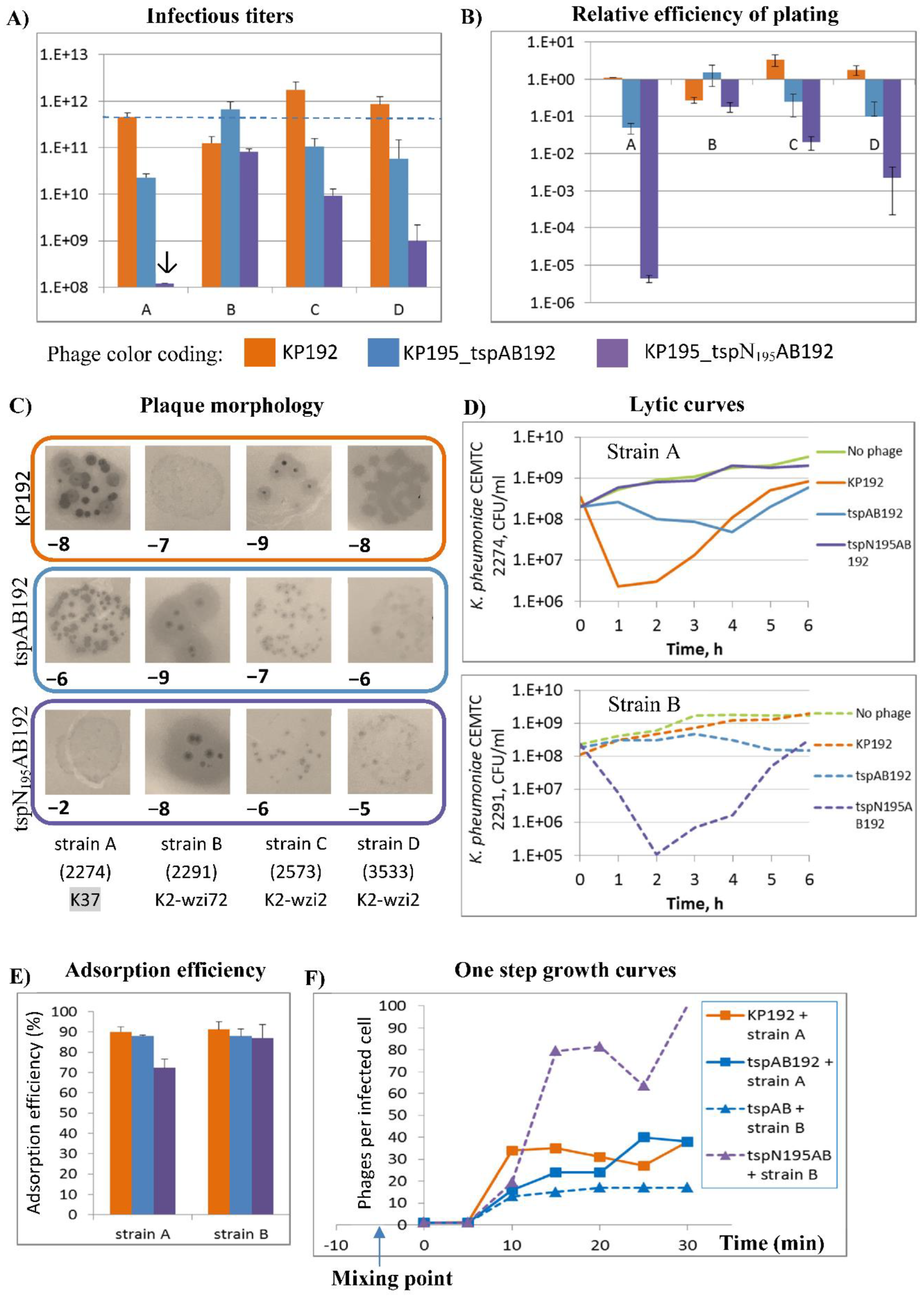
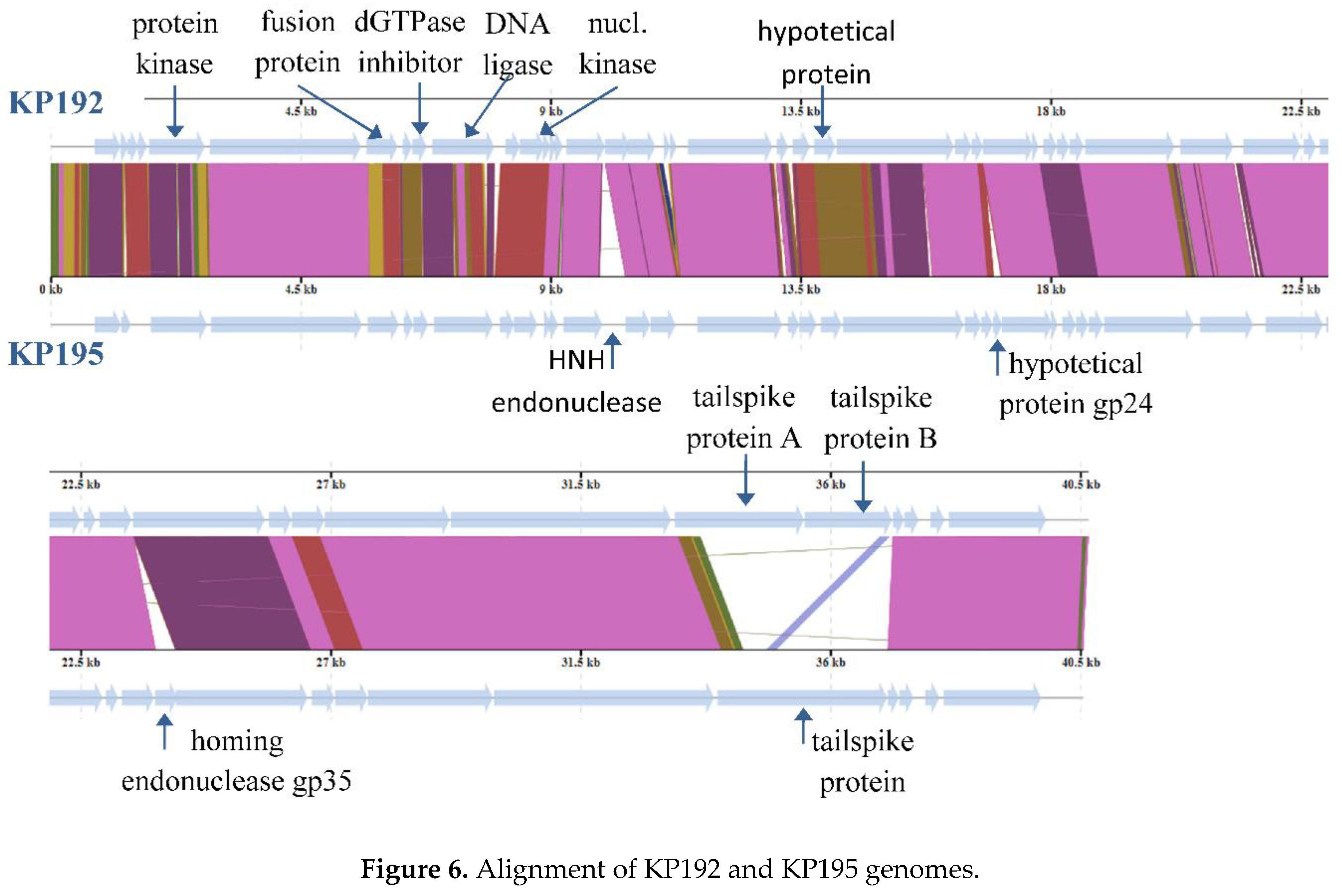

| Product name | Aminoacid identity | Locus tag1 | Note |
|---|---|---|---|
| Protein kinase | 81% | HOT22_gp03, HOT24_gp04 | The differences are located in two regions |
| Fusion protein | 77% | HOT22_gp05, HOT24_gp06 | The differences are located in N-terminal region |
| dGTPase inhibitor | 66% | HOT22_gp07, HOT24_gp08 | |
| DNA ligase | 79% | HOT22_gp08, HOT24_gp09 | The differences are located in two regions |
| Nucleotide kinase | 79% | HOT22_gp10, HOT24_gp11 | |
| HNH endonuclease | n/a | HOT24_gp14 | The gene is absent in KP192 phage |
| Hypothetical protein | 80% | HOT22_gp18, HOT24_gp20 | |
| DNA polymerase | 93% | HOT22_gp19, HOT24_gp21 | The enzyme of KP195 phage contains an insert near the 520 aa residue. |
| Hypotetical protein | n/a | HOT24_gp24 | The gene is absent in KP192 phage |
| Homing endonuclease | n/a | HOT24_gp35 | The gene is absent in KP192 phage |
| Tailspike protein A | 19% | HOT22_gp35, HOT24_gp41 | |
| Tailspike protein B | n/a | HOT22_gp36 | The gene is absent in KP195 phage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



