
Submitted:
13 December 2024
Posted:
16 December 2024
You are already at the latest version
Abstract
BC (BC) stemness contributes to aggressive primary tumor progression and increased propensity for metastasis. Additionally, BC stem-like cells (BCSCs) immensely contribute to drug resistance and relapse. Knowing the biological attributes of BCSCs is vital to employ efficacious therapies against them. In this review, we aim to cover the intrinsic characteristics of BCSCs, the autocrine signaling and paracrine activation by the available cytokines in the tumor microenvironment (TME) and opportunities for targeted therapies both intrinsic and extrinsic to BCSCs. Salient actionable molecular targets and vulnerabilities within the BCSCs and their surrounding bulk tumor and stromal cell compartments in triple-negative breast (TNBC) will be mostly discussed. Enhanced knowledge regarding the BCSCs and their microenvironment will enable us to effectively treat late stage and refractory BC.
Keywords:
BC stemness
; chemoresistance
; therapy failure
; CSC-directed therapy
Introduction
BC (BC) is a complex and heterogeneous disease characterized by the incessant proliferation of cancer cells in the breast parenchyma [1]. It is one of the most common cancers affecting women worldwide and has a significant morbidity and mortality [2]. However, it can also affect men, albeit less frequently [3]. BC can manifest as different subtypes having distinct molecular profile that dictate the treatment approaches accordingly [4].
Amongst the BC subtypes, triple-negative BC (TNBC) constitutes 10%–15% of BC cases, more frequently affecting younger African American (AA) and older European White (EW) or Caucasian women [5,6]. It is characterized by aggressive growth, propensity for early metastasis, and limited treatment options [7]. Despite advancements in cancer research and therapy, TNBC continues to present significant challenges, contributing to its higher rates of recurrence and mortality [8].
Heterogeneity in TNBC
TNBC is clinically classified by its lack of or low expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor2 (HER2) receptors. However, it is characterized by its high metastatic propensity and lethal prognosis. Although patients may initially respond to treatments and enter remission, the rate of recurrence is higher in TNBC than in other BC types (TNBC paradox). Both the metastatic and recurrent nature of TNBC can be attributed at least in part to BC stem-like cells (BCSCs). The observed heterogeneity led to the classification of TNBC further through gene expression profiling [9]. These original classifications have been newly refined and termed the TNBCtype-4 [10]. This is to reflect the biologically diverse cancers in TNBC that have specific transcriptional and immune cell infiltration profiles. The groupings include two basal-like (BL1, BL2), a mesenchymal (M), and a luminal androgen receptor (LAR) subtype [10]. Interestingly, recent studies have found expression profiles from different Lehmann classifications within the same tumor [11] indicating the heterogenous nature of TNBC. These might be useful in predicting a patient’s response to chemotherapeutic agents [10].
Patients often receive similar initial treatments regardless of TNBCtype-4 subtype status in the cancer clinic. These options consist of surgical resection of the tumor followed by an adjuvant cocktail of chemotherapeutic agents or neoadjuvant chemotherapy (NACT) such as taxanes and anthracyclines. These are excellent at controlling the levels of bulk tumor cells but with attendant severe toxicity [12]. However, such treatments are ineffective against BCSC subpopulation. BCSCs are inherently resistant but targeting the transcription factor (TF) sex determining region Y-box 2 (SOX2) and the efflux drug transporter ATP binding cassette type G2 (ABCG2) sensitized BCSCs to therapeutic agents [13,14]. Patients may achieve pathological complete response (pCR) with these regimens. When pCR is not accomplished using NACT, it results in minimal residual disease (MRD). In MRD, the BCSCs remain viable and can undergo multilineage differentiation and repopulate the tumor back with an aggressive, chemoresistant and highly metastatic phenotype [15]. Furthermore, in MRD state, some tumors can switch between the subtypes in TNBC-4type after NACT [16]. This plasticity could potentially be attributed to BCSCs, governing dedifferentiation and metastasis through a variety of mechanisms [17].
Although most often the subclassifications in TNBC serve as a norm to calculate the risk factors and response to chemotherapy for a given patient, some new metrics are formulated to use as inclusion criteria for adopting novel treatments. Novel approaches in subtyping can differentiate the extent of tumor infiltrating lymphocytes (TILs) [18]. Future directions aim to predict responses to immune checkpoint inhibitors (ICIs) and accentuate existing immune responses through them [19]. The KEYNOTE-355 trial showed that TNBC patients late in the disease course that exceeded a certain threshold of TILs and programmed death ligand1 (PD-L1) expression were likely to benefit from pembrolizumab, an ICI [20]. Regardless of subtype, the KEYNOTE-522 trial demonstrated efficacy of pembrolizumab in early TNBC patients to progress to pCR [21]. The results from these trials were encouraging and push forth a new wave of ICI therapy clinical trials in TNBC [22]. These trials may illustrate the need to further distinguish reliable TNBC subtypes to identify the tumors that are likely to respond to specific types of treatments. This is the aim of precision medicine for cancer therapy. Not every tumor cell expresses programmed death ligand1 (PD-L1), and these successful clinical trials are the only ones targeting subpopulations of tumor cells. Recent evidence suggests that immune checkpoints are under tight transcriptional control [23]. Currently, there is no evidence pointing to a direct link between immune checkpoints and BCSCs. However, due to the extent of plasticity in these subpopulations, it would not be surprising to discover one in the future.
Metastatic TNBC is characterized by drug resistance, recurrence, vasculogenic mimicry, neoangiogenesis and high mortality.
Through a variety of molecular mechanisms in metastatic TNBC (mTNBC), chemoresistance and relapses are observed. Drug resistance poses a significant problem when evaluating treatment options for mTNBC due to the ability of the cancer to evade NACT or targeted chemotherapy. One of the major mechanisms contributing to chemotherapeutic multidrug resistance (MDR) involves amplified expression of a specific protein superfamily, ATP binding cassette (ABC) transporters. These are a large superfamily of integral membrane proteins [24] that can transport various molecules and xenobiotics across biological membranes. Though such transport is energetically unfavorable, these proteins can mediate this by coupling the transport with adenosine triphosphate (ATP) hydrolysis [24,25,26]. When overexpressed, the ABC transporters can efflux out the chemotherapeutic drugs leading to a decrease in the intracellular concentration [27,28,29]. This negatively affects drug efficacy, and thus can lead to MDR. However, if a stable and cytotoxic intracellular level of a chemotherapeutic drug can be maintained, the resistance could be overcome [30]. Molecularly, the ABC superfamily is comprised of seven subfamily proteins [24] and some specific members within the ABC superfamily are found to contribute to this MDR [30]. In the ABC-B subfamily, ATP binding cassette group B1 (ABCB1) or P-glycoprotein (P-gp) has been demonstrated to contribute to MDR when overexpressed [28,30]. P-gp inhibitors are a potentially effective drug option for TNBC when combined with other targeted therapies. However, the recently developed P-gp inhibitors have failed due to adverse drug reactions and high toxicity [31]. A recent study showed that biocompatible nanocarriers could downregulate the expression of P-gp which could potentiate the chemotherapeutic response [32]. Therefore, utilizing this treatment modality appears to be promising.
Although drug efflux pumps are a major component of drug resistance, other mechanisms can promote drug resistance in mTNBC. Metabolic adaptations, signaling pathways, and presence of BCSCs have profound impact on drug resistance [33,34]. BCSCs are naturally endowed with ABCG2 drug efflux transporter or BC resistance protein (BCRP) and is a marker for BCSCs. This along with ABCB1 makes BCSCs resistant to most NACT. Additionally, physiological changes that occur within the tumor microenvironment (TME) affect drug resistance. TME hypoxia is one component that is beneficial to the growth of mTNBC because it allows cancer cells to thrive in a low pH or acidic microenvironment. This phenomenon not only promotes immune evasion by cancer cells but also increases the primary tumor progression and metastatic capability. Additionally, there is an upregulation of drug efflux pumps that compromises the uptake of chemotherapeutic drugs [35,36,37].
The drug efflux transporters ABCB1 and ABCG2 specifically have been targeted by tyrosine kinase inhibitors (TKIs) in several studies [38,39,40] and using small molecule inhibitors (SMI) such as Elacridar [40] to study MDR in tumors. Using small interfering RNA (siRNA) knockdown approach to downregulate ABC transporter expression has shown to induce chemosensitivity in MDA-MB-231 cells [41].
Although the TME hypoxia provides TNBC with drug resistant mechanisms and proliferation capabilities, malignant cells ultimately require oxygen and nutrients to survive and continue growth [42]. Therefore, the molecular mechanism of neoangiogenesis is essential for the survival of mTNBC. Neoangiogenesis is the generation of new blood vessels from the existing vasculature that involves the migration, replication and growth of endothelial cells [43]. Vascular endothelial growth factor (VEGF) is a potent angiogenic factor. There is an increased expression of VEGF and there are four isoforms of VEGF that regulate neoangiogenesis and lymphangiogenesis [43,44]. Specifically, in TNBC, the levels of VEGF are significantly higher compared to other types of BC [45,46]. Additionally, mTNBC can also assemble tumor vasculature through a process called vasculogenic mimicry (VM). In this process the TNBC develops new blood vessels as in neoangiogenesis, however the vasculature is lined by tumor cells rather than endothelial cells or a mixture of tumor and endothelial cells [47]. In VM, TNBC cells obtain the ability to anastomose with normal systemic circulation without the need for endothelial cells [47]. This provides nutrients for continued development and contributes to TNBC aggressiveness.
Given the listed molecular mechanisms and the various properties that metastatic TNBC can acquire, there is a high risk of recurrence as well as a high rate of mortality especially among young AA and EW patients [48,49]. Specific genetic markers and extrinsic factors potentially contribute to worst clinical outcomes [48,50]. In patients diagnosed with early-stage TNBC, up to 50% of them experience relapse [51,52,53]. In comparison, within the United States and Western countries, mTNBC has a 5-year survival rate between 4-20% [49,54,55]. Despite the high recurrence and mortality rate of mTNBC, currently innovative therapeutic strategies need to be developed to improve prognosis and outcome.
BCSCs and the BC Stemness Markers
BC stem-like cells (BCSCs) are a subpopulation of tumor cells that express stemness markers and possess self-renewal capacity. BCSCs are known to have a high level of cluster of differentiation (CD) CD44 antigen (CD44+) and a low level of CD24 (CD24-) [56]. BCSCs can initiate a tumor and enable primary tumor progression and metastasis. CD44 is the plasma membrane-resident adhesive receptor for hyaluronic acid and is involved in cell migration and extravasation [57]. CD24 is a small glycosylphosphatidylinositol-linked cell surface protein that functions as a ligand of P-selectin and regulates cell migration, invasion, and proliferation [58]. In the initial study involving BCSCs, Al-Hajj et al. demonstrated that a small subset of tumor cells with CD44+/CD24-/low lineage could generate tumors in immunodeficient mice [4]. In contrast, other tumor cells without CD44+/CD24-/low lineage failed to initiate tumor formation in similar cells [4]. Furthermore, CD44+/CD24-/low cells from human BC exhibited self-renewal, extensive proliferation, formation of clonal mammospheres, a hallmark of cancer stemness, and resistance to chemotherapy in vitro [4]. Another key hallmark study indicated the role of aldehyde dehydrogenases (ALDHs) in BCSCs in addition to being a marker for BCSCs [59]. ALDHs are enzymes responsible for detoxification via oxidation of intracellular aldehydes [60]. It is believed that ALDHs play a crucial role in maintenance but also in the differentiation of stem-like cells by metabolizing retinal into retinoic acid [60]. In the study by Charafe-Jauffret et. al., the stemness of BC cells was demonstrated by isolating ALDH marker-containing cells via a fluorescent ALDEFLUOR assay [59].
While CD44+/CD24-, and ALDHs are the salient biomarkers for BCSCs, other biomarkers such as C-X-C chemokine receptor4 (CXCR4), CD49f, CD133, and junctional adhesion molecule A (JAM-A) have also been identified [61,62]. The presence of CD49f was linked to tumor-initiating properties of BCSCs in mice along with enhanced drug resistance to paclitaxel and doxorubicin [63]. CD133, also referred to as prominin-1, is a cell surface glycoprotein detected in TNBC and BC gene1 (BRCA1)-deficient mouse tumors. Increased CD133 levels are associated with an adverse prognosis in patients diagnosed with invasive BC [63,64].
Characteristics of BCSCs in TNBC
BCSCs within the TNBC subtype drive high rates of tumorigenicity through the processes of self-renewal and differentiation into bulk tumor and stromal cells [65]. Many factors contribute to BC stemness. Firstly, TNBCs more commonly lose the negative regulation of the Wnt (wingless-related integration site) /β-catenin pathway, adenomatous polyposis coli (APC), ultimately dysregulating the normal cell division process. The BCSCs from TNBC appear to rely on ribosomal S6 kinases (RSKs) for proliferation and survival that is upregulated by the Y-box binding protein-1 (YB-1) oncogene [67], which has an elevated level of expression in aggressive cell lines [68].
The dysregulation of self-renewal in BCSCs, as well as the ability to asymmetrically divide, leads to tumor heterogeneity that gives rise to cells of highly diverse properties [69]. Importantly, the plasticity of BCSCs between epithelial and mesenchymal phenotypes allows cells to evade traditional chemo- and radiotherapies resulting in MRD that may be responsible for tumor relapse [70]. Many factors expressed at a notably higher level in TNBC have been suggested to be essential for the BC stemness, some of which include the c-KIT receptor tyrosine kinase, high mobility group A1 (HMGA1), delta N Isoform of Tumor Protein 63 (ΔNp63), and Kruppel-like factor5 (KLF5) [66,71]. OCT4, SOX2, KLF4 and c-MYC (OSKM) TFs are expressed in many high-grade TNBCs and help to maintain pluripotency and cancer stem cell (CSC) activity and poor differentiation [66,72]. Octamer binding transcription factor 4 (OCT4) specifically has been linked to both dysplasia and an increase in ALDH1 positivity, often marking a poor prognosis [66,70,72]. Meanwhile, SOX2 TF plays a role in the promotion of BCSC proliferation and metastasis [72].
The Wnt pathway is also heavily involved in stemness through its involvement in the promotion of epithelial-mesenchymal transition (EMT) when in an unfavorable environment, which promotes mesenchymal characteristics in BCSCs. Alongside many key EMT-promoting gene expression, an elevated expression of the frizzled (Fz) Wnt receptor is observed in TNBC. In collaboration with co-receptor low-density lipoprotein receptor-related protein6 (LRP6), Wnt induces T-cell factor (TCF)-dependent gene transcription and, therefore, downstream activation of receptor tyrosine kinase-like orphan receptor (ROR1). LRP6 is regulated by TF SOX9, which is also elevated in TNBC and in normal mammary stem cells. In studies targeting the LRP6 co-receptor, SUM-149 cells displayed lower CD44, cellular myelocytomatosis (c-MYC), and ALDHs, as well as increased CD24 expression, lowered EMT, and decreased self-renewal. ROR1 activation has also been linked to EMT-promotion associated with the upregulation of phosphatidyl-inositol (3) kinase/protein kinase B (PI3K/Akt) and cyclic AMP response element binding protein (CREB) pathways in studies with the MDA-MB-231 TNBC cell line. These studies also demonstrated that yes-associated protein1 (YAP1) also induces EMT [66]. Besides Wnt pathway, the Notch pathway may play a role in this phenotypic pattern. TNBC often displays increased neurogenic locus notch homolog protein4 (Notch4) expression, which regulates EMT with signal transducers and activators of transcription3 (STAT3) as well [66].
Resistance
Although radio- and chemotherapies are effective in reducing bulk tumor cells, BCSCs are more resistant to such treatments and are therefore left behind to proliferate, directly resulting in the recurrence of the tumor in a more drug-resistant form than the initial tumor. TNBC cell lines have displayed many distinctive traits to which this resistance may be attributed [73]. One of these common traits is the upregulation of the Hedgehog signaling pathway by an increased expression of Smoothened receptor (SMO) and induction of glioma-associated oncogene homolog1 (GLI1) [66], ultimately increasing expression of ABC transporters [74]. Eukaryotic translation initiation factor 4A1 (eIF4A1) is also suggested to regulate ABC transporter expression, as well as many other vital oncogenic proteins involved in both drug resistance and cancer stemness such as the pluripotency TFs (PTFs) SOX2, OCT4, NANOG and survivin [72,75]. Overexpression of WASP-family verprolin-homologous protein3 (WAVE3), a member of the (Wiskott–Aldrich syndrome protein) WASP/WAVE family of actin-cytoskeleton remodeling proteins, has been notably overexpressed in several TNBC lines as well as BCSCs. WAVE3 promotes cell migration, direction, and invasion and therefore is a strong indicator of TNBC as well as the tumor size, stage, and lymph node metastasis [68]. WAVE3 is responsible for both anti-apoptotic and chemoresistant properties in cancer cells, as well as translocation of the YB-1 TF to the nucleus [68]. Drug-resistant TNBCs have also possessed other common traits such as increased expression of Musashi RNA binding protein 1 (MSI1), OCT4, and CD44s, as well as elevated rates of fatty acid oxidation regulated by (Janus kinase) JAK/STAT3 pathway that may be critical in self-renewal and chemoresistance [67,76]. These anti-apoptotic properties and increased rates of DNA repair aid in radio-resistance are commonly shared among CSC subpopulations. In TNBC, irradiation has been shown to increase ALDEFLOUR+ BCSC populations in radio-resistant SUM 149 and SUM 159 cell lines. These SUM 159 cells have displayed increased expression of PTFs such as SOX2, OCT4, NANOG, KLF4, and c-MYC that may contribute to the ensuing reduction of reactive oxygen species (ROS) levels, the latter being highly indicative of a poor prognosis and high CSC activity [66]. Under hypoxic conditions, hypoxia inducible factor 1α (HIF-1α) is induced in TNBC and other cancers that indirectly reduces ROS by promoting the transcription of ALDH enzymes [77]. The elevated level of OCT4 promoted ALDH levels and decreased the pCR to radiotherapy [66]. This results in over 50% of TNBC cases displaying enhanced ALDH1 activity [77]. This leads to an increased drug detoxification via metabolic conversion, as well as overall decreased oxidative stress within the tumor cells [75], further contributing to a characteristically poor prognosis.
Metastasis
TNBC is highly metastatic and more often disseminates to the lungs, bones, and brain [70,78]. Much of this may be attributed to the previously mentioned process of EMT due to the invasive nature of mesenchymal-like BCSCs [66]. Under unfavorable conditions, such as exposure to neoadjuvant radio- and chemotherapies, or even under hypoxic conditions, EMT is induced in BCSCs, and metastatic attributes are increasingly expressed. Under hypoxic conditions, the TFs HIF-1α and hypoxia inducible factor 2α (HIF-2α) are upregulated and induce the Wnt signaling pathway and SOX2 and c-MYC-dependent gene transcription, respectively [66,77]. This supports EMT, pluripotency and metastasis. Certain transfer RNA-derived small non-coding RNAs (tDRs) are also involved in the regulation of hypoxia-induced chemoresistance in TNBC, including tDR-0009 that regulates EMT-involved STAT3 activation [70].
The C-X-C chemokine receptor 4 (CXCR4) is a marker for BCSCs and is involved in metastases of many solid tumor types. CXCR4 mediates directed cell migration in a C-X-C chemokine ligand 12 (CXCL12)-dependent manner [79,80]. CXCL12, the chemokine ligand for CXCR4, is highly enriched in predilection sites such as the lungs and bones. CXCR4 critically regulates the LIM and SH3 protein1 (LASP1) and argonaute2 (Ago2) interaction that upregulates the expression of C-C chemokine receptor 7 (CCR7) in the tumor cells [81]. Tumor cells endowed with an enhanced cell surface expression of CCR7 can metastasize to the sentinel or draining lymph nodes as they are highly enriched in C-C chemokine ligand 21 (CCL21), the ligand for CCR7. Both CXCR4 and LASP1 are upregulated in invasive BC, and LASP1 directly interacts with eIF4A1 and eukaryotic translation initiation factor 4B (eIF4B) to increase the translation of oncogenic mRNAs. The resulting oncoproteins such as Rho-associated coiled-coil containing protein kinase 1 (ROCK1), survivin, cyclin D1, and Mouse double minute 2 homolog (MDM2) are directly associated with cell proliferation, migration and invasion [82]. CXCR4-associated LASP1 also notably aids in the stabilization of the Snail1 TF responsible for reduced intercellular adhesion and increased migration through the induction of EMT [83].
Role of ABCG2 and CD133 in Regulating BC Stemness
BCSCs play a vital role in tumor initiation, aggressiveness, as well as therapeutic resistance. Of particular importance to BC stemness is the ABC transporter ABCG2 and the transmembrane glycoprotein CD133, also known as Prominin-1 [84]. Given the aggressive nature of TNBC, comprehending the roles that ABCG2 and CD133 play in the stemness of BCSCs is vital to improving patient outcomes and identifying new therapeutic strategies.
ABCG2 functions as an efflux pump that utilizes ATP hydrolysis to expel various substances such as xenobiotics from the cell. These transporters play a homeostatic and self-defensive role in regulating processes such as detoxification and drug resistance [84,85]. ABCG2 transcription is increased in hypoxic conditions via the binding of HIF-1α to a hypoxia response element (HRE) in the ABCG2 promoter [86], and has also been shown to be synergistically elevated by interleukin 6 (IL-6) and endoplasmic reticulum (ER) stress in plasma cells [87]. ABCG2 specifically is directly responsible for causing multidrug resistance (MDR) to a wide structural variety of anticancer drugs and is present in remarkably elevated levels in patients with TNBC who respond poorly to chemotherapy [84,85,88,89]. ABC transporters, and especially ABCG2, thus pose a significant obstacle to the ability of chemotherapeutic drugs to induce cytotoxicity in BCSCs, thus causing chemoresistance [84].
The primary role that ABCG2 plays in BC stemness is promoting drug resistance. ABCG2 expression increases dramatically both in response to the use of chemotherapeutic treatments as well as even prior to their use, indicating that it promotes drug resistance, aggressive tumor growth metastasis and relapses. One study found that inhibiting ABCG2 using the bromodomain extra-terminal domain (BET) inhibitor JQ1 led to the loss of cancer stemness in a TNBC model, and therefore resulted in better prognosis through restoration of sensitivity to chemotherapy [88]. The reported evidence further supports the use of ABCG2 as a biomarker for chemotherapy responses in BC patients [90,91,92].
CD133 is a well-known BCSC marker in TNBC and is often employed in isolating BCSCs. It is one of the major contributors to the regulation of BC stemness by promoting self-renewal, drug resistance, and metastasis. CD133 has an elevated expression in TNBC with a poorer prognosis [93]. It promotes the ability of TNBC cells to metastasize to distant organs [84,94]. BCSCs endowed with a high expression of CD133 have been shown to proliferate after neoadjuvant hormonal therapy in luminal BC [95]. Like ABCG2, CD133 overexpression has been associated with drug resistance, as well as repopulation of tumor cells, enhanced repair mechanisms that prevent cell death, and epigenetic changes [84,94].
Studies show that vasculogenic mimicry (VM) and CD133 expression are highly related and support primary tumor progression and relapse in TNBC [96]. Zhang et al. (2014) demonstrated that VM and CD133 are related through hypoxia that is often found in a rapidly growing tumor. Antiangiogenic agents can induce hypoxia, which increases the expression of EMT-TF twist family BHLH transcription factor 1 (Twist1). Twist1 further increases the population of CD133+ cells, which promotes the increase in VM channels, allowing the tumor cells to expand and regrow. CD133 was found to be strongly and significantly associated with N-cadherin, an important EMT marker [97]. This finding underscores the importance of CD133 in promoting the self-renewal as well as invasiveness of tumor cells [93,98]. Through its impact on stemness, CD133 has thus become a highly effective and specific target for therapeutic strategies in TNBC. Anti-CD133 antibodies have been used extensively in research to enhance chemotherapeutic drug delivery and even lead to the elimination of BCSCs [84,94,99].
ABCG2 and CD133 both play pivotal roles in regulating BC stemness in TNBC and may serve as effective targets for treatment. Their roles in drug resistance, self-renewal, and metastasis emphasize their importance and inclusion in therapeutic strategies. However, there is a need for further research into the predictive and prognostic value of both biomarkers.
The Role of the Hippo Pathway Downstream Effectors YAP and TAZ in BC Stem-Like Cells
Overview of the Hippo Pathway
The Hippo (or Salvador-Warts-Hippo) pathway is an evolutionarily conserved signaling pathway that regulates several processes related to cellular growth, including proliferation, survival, differentiation, regeneration, repair, and organ size [100,101,102,103,104]. It was named after the gene hpo, which codes a Drosophila melanogaster (fruit fly) kinase called “Hippo” that limits tissue growth [105,106,107]; mutations in this gene lead to uncontrolled proliferation and compromised apoptosis. The first components of this pathway were discovered in 1995 in D. melanogaster [108,109,110]. Since then, more than 30 other proteins have been identified as part of this pathway in both mammals and D. melanogaster [103]. The core of the mammalian Hippo pathway comprises a cascade of kinases with their associated scaffold proteins and culminates with the modulation of the cellular location of two transcriptional coactivators: Yes-associated protein (YAP), and transcriptional coactivator with PDZ-binding motif (TAZ), also known as WW-domain-containing transcription regulator 1 (WWTR1) [102,103,111,112,113]. One of the ways that the pathway can be mechanistically initiated is by thousand and one kinases 1-3 (TAOK 1-3), which phosphorylate and activate sterile 20-like kinases 1 and 2 (MST1/2) – mammalian homologs of Hippo [114,115]. Active MST1/2 phosphorylates the scaffold proteins Salvador homolog 1 (SAV1) and monopolar spindle 1 (mps1) binder 1 A and B (MOB1 A/B), which assist MST1/2 in the recruitment, phosphorylation, and activation of large tumor suppressor 1 and 2 (LATS1/2) – mammalian homologs of Warts [116,117,118]. Interestingly, TAOKs and MAP kinase kinase kinase kinases (MAP4Ks) can directly phosphorylate LATS1/2 [119], and MST1/2 [120] and LATS1/2 [121] can be activated by autophosphorylation as well. In turn, LATS1/2 phosphorylates cytoplasmic YAP/TAZ, causing its sequestration in the cytosolic compartment by binding to 14-3-3 proteins and posterior ubiquitination and degradation [122]. Thus, the central axis of the pathway is the kinases MST1/2 and LATS1/2, which directly regulate the physiological output of the pathway by controlling the nuclear translocation of YAP/TAZ, the “acting arms” or effectors of the cascade. The cytoplasmic retention and subsequent degradation of YAP and TAZ is regulated by the Hippo pathway in response to intrinsic and extrinsic cues and peripheral components to the pathway that relay signals to the core kinases [111]. These cues can range from physical (cell-cell contact, cell polarity, mechanical signals) to biochemical (soluble factors, G-protein-coupled receptors, stress signals, nutrient availability) and have been extensively reviewed elsewhere [110,111,119,123,124,125,126,127,128]. Upon translocation to the nucleus, YAP/TAZ activate transcriptional programs involved in cell proliferation and survival [113,128,129,130]. Therefore, the Hippo pathway acts as a tumor suppression network, and its dysfunction has been linked to the development of several types of cancer [102,131,132,133,134,135,136,137]. In recent years, further understanding of the Hippo pathway has identified its role in other cellular processes, including microRNA biogenesis [138,139], lymphatic vessel stability, angiogenesis, and hemodynamics [140,141,142,143], immunity [144,145,146,147], autophagy [148], and cell ploidy [149,150].
YAP and TAZ – The Acting Arms of the Hippo
YAP and TAZ are two related transcriptional coactivators encoded by paralogous genes with nearly 50% amino acid sequence similarity [151]. YAP was discovered by Sudol as a protein with the ability to interact with the Src homology 3 (SH3) domain of tyrosine kinases YES, SRC, and Abelson leukemia (ABL) [152], and TAZ was discovered by Yaffe as a novel 14-3-3 binding molecule [153]; however, they rose to prominence when their functions started to become clear after being identified as the mammal orthologs of Yorkie, the executor of the Hippo pathway in Drosophila [100]. Due to the lack of DNA-binding domains, they rely on other factors to exert their transcriptional regulation. Notably, they interact with members of the transcriptional enhancer factor (TEA)-domain (TEAD) family of DNA-binding factors (TEAD1-4) [154]. YAP/TAZ-TEAD complexes bind mostly to distant enhancer elements, and only a minute fraction binds to promoters [155]. YAP/TAZ plays an essential role in cell and tissue proliferation, growth, and apoptosis, and their paramount function is to regulate the growth of organs until they reach their intended size [129,156,157]. These transcription regulators are part of the complex machinery the cell employs to sense, communicate, and interact with its surroundings. Its activity on cells is influenced by mechanical factors such as cell shape and polarity, which is governed by cytoskeletal architecture [128,158]. These factors, alongside cell adhesions and matrix complexity, reflect the local stromal composition and the situation of the cells within the tissue microenvironment, and allow YAP/TAZ to act accordingly [159,160]. Similar to how YAP/TAZ reacts to extrinsic mechanical and physical cues, it responds to intrinsic metabolic and biochemical inputs such as glucose homeostasis [161,162,163] and lipid metabolism [164]. YAP/TAZ converts such mechanical and biochemical inputs into gene expression and biological responses.
YAP/TAZ shuttle between the cytosol and the nucleus according to their phosphorylation state [165], post-translational modifications [165,166], and by binding to other proteins to facilitate the movement [167]. Phosphorylation by LATS1/2 determines the subcellular localization of these transcription factors by creating a binding site for 14-3-3 proteins, sequestering YAP/TAZ complexed to 14-3-3 proteins in the cytosolic compartment [168], thus preventing their nuclear translocation. YAP/TAZ is then ubiquitinated and posteriorly degraded [169,170]. Interestingly, TAZ contains a second phosphodegron located in the N-terminal region that is not present in YAP [169,171], explaining in part the short half-life of this protein (around 2 h) [169] in contrast with the more stable and longer-lived YAP [172,173,174]. Although phosphodegron is found in YAP, its concentrations appear to be mainly regulated by nucleocytoplasmic shuttling [129]. Although the “canonical view” postulates that phosphorylated YAP is sequestered in the cytoplasm, other studies have challenged this: it was observed that phosphorylated YAP can be located in the nucleus [175,176] and that phosphorylation of YAP is needed but not sufficient for nuclear exclusion [176]. It is worth noting that although YAP and TAZ have almost identical functions, they usually act as separate proteins in monomeric or homodimeric form [177]. However, some isoforms of YAP can form heterodimers with TAZ [178,179].
YAP/TAZ and Cancer
Given the YAP/TAZ transcriptional functions related to genes that promote proliferation, survival, and growth, it is easy to understand why these molecules are ubiquitous in cancer. YAP/TAZ activation has been observed in a plethora of cancer types across murine models, human in-vitro models, and patients [180]. Once YAP/TAZ nuclear accumulation reaches a certain threshold, the overexpression of their target genes drives fundamental cancer phenotypic changes such as plasticity, drug resistance, uncontrolled cell proliferation, and metastasis [180,181]. Other “cancer enabler” attributes include stromal cell recruitment, inflammation, angiogenesis, and immune modulation [180,181,182].
Due to their physiological role, upregulating cell proliferation is perhaps one of the most straightforward effects of YAP/TAZ activation in cancer [183,184]. YAP/TAZ sustain aberrant proliferation by promoting the cell cycle and sustaining the expression of oncogenic, pro-mitotic, and DNA-replicating factors such as activator protein 1 (AP-1) or c-MYC [128,185,186,187,188]. Interestingly, AP-1 amplifies YAP/TAZ transcription, driving a positive feedback loop [186,189,190].
YAP/TAZ can enable cancer cells to escape diverse treatments by promoting resistance to cytotoxic and targeted regimes, hormonal therapy, immunotherapy, and radiotherapy [181,191,192,193,194]. YAP/TAZ has been linked to resistance to agents in diverse cytotoxic classes commonly used in various malignancies, including taxanes [195,196], anthracyclines [197], platinum agents [198] and antimetabolites [196]. Response to hormonal therapies can be under the influence of YAP/TAZ, as demonstrated by a study where tamoxifen-resistant MCF7 cells were re-sensitized by targeting YAP/TAZ with a knock-down approach [199]. The aberrant expression of YAP/TAZ is an important form of resistance to targeted therapy, which inhibits components within specific signaling pathways driving cell growth and survival [199,200]. YAP/TAZ has been found to confer resistance to epidermal growth factor receptor (EGFR) [201,202,203], mitogen-activated protein kinase (MAPK) [204,205] and HER2 [206] inhibitors.
Metastasis is a stressful and challenging journey for malignant cells in which they must circumvent immune surveillance, recruit local and systemic factors, and survive without anchoring molecules [207]. YAP/TAZ’s role in metastasis is well established, as gain of YAP/TAZ bestows non-metastatic cells with metastatic abilities, whereas YAP/TAZ downregulation hinders the metastatic potential [208]. Given YAP/TAZ link with mechanical cues and function as a response element to them, it is predictable that YAP/TAZ has a prominent role in cell migration and dissemination since these processes involve a journey through diverse mechanical situations such as compression, stretching, motility through distinct portions of extracellular matrix, adhesion, intravasation, circulation, extravasation and establishment within a new tissue with different composition and architecture [180]. In line with this, it has been found that YAP/TAZ contributes to different aspects of the metastatic process: cell motility, migration, and invasion [209,210,211,212,213], survival in the circulation [214,215], and vascular translocation [216,217,218].
YAP/TAZ as Drivers and Enhancers of BCSCs
YAP/TAZ can induce stemness characteristics in healthy cells, converting them into cells resembling tissue stem cells [219], and both factors have been linked to stemness in malignancy as well [220]. Although YAP and TAZ have overlapping functions, a careful literature review suggests that in BCSCs, TAZ functions primarily to mediate an aggressive BCSC aggressive phenotype (metastasis, chemoresistance) and, to a lesser extent, stimulate differentiated cells to acquire stem-like properties, while the YAP role in BCSCs leans toward the development and maintenance of the stemness state. However, this is not absolute. For instance, in BC, TAZ amplification can transform bulk tumor cells into cancer stem cells [192]. In a study, TAZ-mediated reprogramming of human mammary epithelial cells transformed them into experimental BCSCs displaying traits such as self-renewal, chemoresistance, epithelial-to-mesenchymal transition, and tumor-seeding capabilities [221]. TAZ has been identified as a central mediator of metastatic ability and chemoresistance of BCSCs, and the ability to replenish tumor cells [193]. Interestingly, TAZ upregulation in differentiated, non-tumorigenic BC cells induced their transformation to a migratory, tumorigenic phenotype, contrasting with how the loss of TAZ impaired metastatic ability and chemoresistance of BCSCs [193].
Similarly, the role of YAP in inducing and maintaining pluripotency has also been described. In one study, YAP knockdown led to a loss of embryonic stem cell pluripotency, while ectopic expression of YAP prevented embryonic stem cell differentiation in vitro and maintained stem cell-like characteristics even under differentiation conditions [222]. Consequently, it has been hypothesized that as YAP/TAZ induce differentiated cells to acquire cancer stemness, loss of YAP/TAZ could cause differentiation of BCSCs into a more differentiated and aggressive phenotype [180]. Further highlighting the role of YAP in BC stemness, it was discovered how YAP partners with serum response factor (SRF) and TEADs to increase the transcription output of Interleukin 6 (IL6) - an essential factor for the maintenance of BCSCs, and cancer stemness in general [223,224,225], and how the SRF-YAP-IL6 axis was required to maintain BC stemness [226].
The Receptor tyrosine kinase-like orphan receptor 1 (ROR1) is expressed in embryogenesis and cancer [227], but its expression becomes almost null in post-partum tissues, except in early B lymphocyte precursors [228]. In a study, the expression of ROR1 increased in BC after treatment with chemotherapy alongside the display of stemness characteristics. Interestingly, treatment with anti-ROR1 antibodies reversed cancer stemness and increased taxol sensitivity [229]. The authors observed a ROR1-dependent increase in the activation of YAP/TAZ that correlated with chemoresistance, indicating YAP/TAZ directly contributed to the ROR1-dependent chemoresistance. Thus, the stemness and chemoresistance observed in this study appeared to be under the direct influence of YAP/TAZ, which in turn was upregulated in cells with high ROR1 expression. Consequently, after anti-ROR1 treatment, chemoresistance, stemness, and YAP/TAZ activity were decreased [229].
YAP/TAZ Inhibition - A Promising Strategy to Curtail Cancer Stemness
The idea of targeting transcription factor co-activators has gained significant traction in oncology in the last decade, challenging the classical view that they are “undruggable”. Deregulation of transcription factors co-activators such as YAP/TAZ, c-MYC and β-catenin is at the core of the tumor initiation and progression [230].
As previously described, metastasis and drug resistance are among the main causes of death in BC patients. These characteristics are hallmarks of BCSCs [193,231], alongside the ability to replenish bulk tumor cells lost due to therapy. A novel approach to curtail these complications in patients could involve co-targeting the differentiated and stem cell compartments through YAP/TAZ inhibition. Some drugs have shown promising results in cancer: kinase inhibitors pazopanib and dasatinib, alongside statins, inhibited the nuclear translocation of YAP/TAZ, and pazopanib increased their proteasomal degradation, which translated into increased chemosensitivity in TNBC [232]. A pan-TEAD inhibitor, GNE-7883, was found to suppress cell proliferation in several cancer cell lines and demonstrated robust anti-tumor efficacy in murine modes. GNE-7883 treatment reversed resistance to Sotorasib, a KRAS G12C inhibitor [233]. Other YAP/TAZ inhibitor strategies are highlighted in Table 1 adapted from and reviewed in [234].
Factors Governing the Maintenance and Clonogenicity of BCSCs
As BCSCs are crucial for tumor initiation, progression, metastasis, and therapy resistance, understanding the factors governing their maintenance, clonogenicity, and paracrine interaction within the TME is vital in developing efficacious therapeutic strategies. The maintenance and clonogenicity of BCSCs is governed by a complex interplay of intrinsic and extrinsic factors, including the TME and hypoxic conditions. Some of the factors governing BCSC maintenance and clonogenicity include:
- Plasticity of BCSCs - BCSCs that underwent EMT exhibit enhanced invasive potential, enabling them to disseminate from primary tumors and form distant metastases, contributing to disease progression and poor prognosis. Additionally, such BCSCs display resistance to NACT and targeted therapies, due to their enhanced survival mechanisms and altered gene expression profiles through epigenetic adaptations [239]. The plasticity conferred by EMT enables BCSCs to adapt to changing microenvironments within the tumor and metastatic sites, facilitating tumor relapses. Targeting EMT and its associated signaling pathways may represent a promising therapeutic approach to restrict BCSCs to one state, which prevents plastic conversion to a more resistant form and improves treatment outcomes for BC patients. [240].
- Signaling pathways: Tumor cell signaling pathways such as Wnt, Notch, Hedgehog, and PI3K/Akt/mTOR (mammalian target of rapamycin), intricately regulate the behavior of BCSCs, dictating their self-renewal and differentiation capabilities [241]. Through a network of molecular interactions, these pathways regulate self-renewal, sustenance of cancer stemness, and survival of BCSCs. These signaling pathways prime and activate BCSCs for aggressive behaviors, fueling invasion, migration, and metastasis. By influencing the gene expression involved in cell fate determination and interactions with TME in a paracrine manner, these signaling cascades modulate the phenotypic and functional heterogeneity within BCSC populations [242,243]. Understanding the crosstalk between these pathways provides insights into the mechanisms underlying BC progression and offers potential co-targets for therapeutic intervention aimed at disrupting BCSC-mediated tumorigenesis and metastasis.
- Transcription factors: PTFs such as SOX2, OCT4 and NANOG serve as master regulators of cancer stemness in BCSCs, activating gene expression that sustains their self-renewal capacity [4]. These TFs exert control over critical cellular processes, including proliferation, differentiation, and survival, thereby contributing significantly to the clonogenicity and maintenance of BCSC populations within the tumors [244,245]. Their dysregulation or aberrant activity can drive therapy resistance and induce MRD, subsequent expansion and recurrence. Insights into the regulatory networks governed by these PTFs may provide valuable avenues for the development of novel strategies aimed at disrupting BCSC-mediated tumorigenesis and improving patient outcomes.
- Cytokines in the TME: Within the TME, the cytokine storm can drive the behavior of BCSCs, and they may oscillate between cancer stemness and bulk tumor cell states. Interleukins (ILs), such as IL-6 and IL-8 (CXCL8) C-X-C chemokine ligand 8, along with tumor necrosis factor a (TNF-a) and transforming growth factor b (TGF-b), represent key players in this regulatory network [80,246]. These cytokines from the TME exert a paracrine effect on BCSCs, influencing their survival, clonogenic expansion, survival, and migration. By engaging with specific receptors and initiating downstream signaling pathways, cytokines and chemokines modulate the gene expression associated with cancer stemness, plasticity, and chemoresistance in BCSCs [240]. Thus, the niche for BCSCs in the TME can foster their survival, clonogenicity and maintenance [247]. Co-targeting the cytokine signaling network may augment targeted therapies.
- Stromal cells within the TME: A dynamic interplay exists between stromal cell compartments comprising of cancer-associated fibroblasts (CAFs), endothelial cells, immune cells, acellular extracellular matrices (ECM) and the BCSCs [248]. CAFs, through the secretion of growth factors and cytokines, create a supportive niche for BCSCs, enhancing their maintenance and self-renewal capabilities [240]. Endothelial cells contribute to BCSC survival and proliferation by facilitating neoangiogenesis and providing nourishment. The immune cells, such as tumor-associated macrophages (TAM) and regulatory T lymphocytes (Treg), secrete factors that promote BCSC stemness [249]. This bi-directional communication between stromal and cancer cells (bulk tumor cells and BCSCs) enables the sustenance of cancer stemness and clonogenicity. Uncovering the supportive roles of the TME for BCSCs holds promise for developing novel therapeutic interventions aimed at disrupting BCSC-mediated tumorigenesis, metastasis, and drug resistance.
- Hypoxic TME: The role of hypoxia in BC has been discussed in earlier sections. HIFs activate a cascade of events within BCSCs, promoting their maintenance, enhancing their plasticity and promoting resistance to therapy. Through transcriptional activation of target genes involved in angiogenesis, metabolism, and cell survival, HIFs create a microenvironment conducive to BCSC survival and clonal expansion under hypoxic stress [250,251]. This hypoxia-driven adaptation confers a selective advantage to BCSCs, facilitating their persistence. Understanding the interplay between hypoxia, HIFs, and BCSC holds promising therapeutic potential for targeting aggressive and refractory tumors.
Previous studies show that HIF-1a knockdown suppressed the cancer stemness parameters in vitro [252]. One of the studies showed that Ganetespib, an inhibitor of heat shock protein 90 that stabilizes HIF-1 a, significantly reduced the levels of BCSCs in a TNBC mouse model `. These studies lend support to the idea of using HIF-inhibiting compounds to treat BCs.
- Metabolic reprogramming: A subset of BCSCs (called energetic BCSCs) display an increase in glucose uptake, a high glycolytic rate through the Warburg effect that results in lactate accumulation, and a concurrent decrease in mitochondrial respiration [253]. Recent evidence suggests that BCSCs can alternate between glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) in the presence of oxygen, facilitating incessant tumor growth. This metabolic plasticity allows BCSCs to engage in OXPHOS generating ATP, thus promoting survival under conditions where glycolysis is impaired [254]. Interestingly, proliferative BCSCs prefer the OXPHOS metabolism, while quiescent BCSCs are dependent on glycolysis for their metabolism [255,256]. In addition, BCSCs have also been reported to rely on mitochondrial fatty acid oxi[28]dation as an alternative energy source to maintain their survival, self-renewal, and chemoresistance [257]. This metabolic adaptability makes them less vulnerable to many therapies targeting specific metabolic pathways. However, a combination therapy targeting more than one metabolic pathway may disrupt the availability of an array of metabolic mechanisms at the disposal of BCSCs.
To target BCSCs’ metabolic reprogramming abilities, a metabolic “two hit” strategy was proposed in which mitochondrial OXPHOS inhibitors first push CSCs toward glycolysis, which is when a glycolysis inhibitor would act as a second hit to firmly disrupt CSC metabolism [258]. De Francesco et al. (2018) proposed and demonstrated this strategy in BC cells with dodecyl(triphenyl)phosphonium (d-TPP), a mitochondrial inhibitor, along with glycolysis inhibitors vitamin C and OXPHOS inhibitor doxycycline [259]. The results showed effective BCSC suppression as well as seemingly selective toxicity only in cancer cells [259], suggesting promising potential for both the therapeutic strategy and TPP-based drugs.
Additional Strategies for Targeting BCSCs
Many strategies were previously outlined in the appropriate sections. Additional strategies are discussed here. In a clinical study, the ALDH1A1 gene expression data of 3455 patients was found to be negatively correlated with overall survival of BC patients [260,261]. Experimentally, it has been shown that SMI Nifuroxazide selectively inhibits ALDH1A1 in stem cells which contributes to the initiation and progression of melanoma [262]. Recently, it has also been identified that the disruptor of telomeric silencing 1 (DOT1L) protein is a key epigenetic regulator of ALDH1 in TNBC and its selective inhibition with SMI EPZ-5676 has shown reduced tumor growth in vivo [263]. Both the markers i.e., hyaluronan receptor (isoform CD44s) [264] and ALDHs are co-expressed over a small subset of stem-like cells which are highly disseminating in nature [4]. Another marker, the anti-apoptotic protein myeloid cell leukemia 1 (MCL1), is one of the key proteins involved in the survival of stem-like cells and in collaboration with c-MYC it promotes chemoresistance through oxidative phosphorylation [265]. A co-targeting approach involving MCL1 and c-MYC may be highly beneficial.
Genetic ablation and pharmacological targeting of the eukaryotic mRNA helicase, eIF4A1, is an effective strategy targeting TNBC cells and BCSCs. It has been shown that targeting eIF4A1 curtails metastasis by downregulation of the ABC drug transporter, along with other vital oncogenic proteins involved in survival and stemness such as SOX2, OCT4, baculoviral IAP repeat-containing 5 (BIRC5), and NANOG [72,266]. This brings a striking feature in explaining why targeting eIF4A1 controls stemness as well as drug resistance, though these are highly correlated [72,75].`
The C-X-C chemokine receptor 4 (CXCR4) is a G protein-coupled receptor and its perturbation leads to activation of the Gαi subunit and PI3K/mTORC1 axis promoting metastasis in TNBC [267] and hematological malignancies [268,269]. The PI3K/mTORC1 axis is upstream of eukaryotic initiation factor 4A1 (eIF4A1) and contributes to metastasis via activation of its downstream effectors [75,270]. Interestingly, it has been shown that enhanced expression of CXCR4 is correlated with poor outcomes in patients with TNBC [271]. CXCR4 gene silencing has been shown to increase the sensitivity of cisplatin in TNBC in vivo thereby lowering expression of p53 mutants and B-cell lymphoma-2 (Bcl-2) protein in mice [272]. CXCR4 signaling has been shown to maintain tamoxifen-resistant CSC population in tamoxifen-resistant MCF7 cells through aryl hydrocarbon receptor (AhR) signaling [273] and overexpression of CXCR4 was found to be significantly associated with distant metastasis indicating poor disease-free survival [274]. A CXCR4 antagonist in combination with an anti-microtubule showed promising results in initial trials [275], but further drug development is needed to optimize the use of CXCR4 as a BCSC target.
Targeting a variety of components in the TME that interact with BCSCs, as well as their receptors and downstream effectors, may further the effectiveness of existing therapies. For example, enhanced activation of C-X-C chemokine ligand 8 (CXCL8) occurs in response to the administration of PI3K inhibitors, leading to activation of the Janus kinase2 (JAK2) pathway and increased drug resistance [276]. Inhibition of CXCL8 or its chemokine receptors C-X-C chemokine receptor 1/2 (CXCR1) and CXCR2 can therefore enhance the efficacy of PI3K/mTORC1 inhibitors [276]. CXCR1 and CXCR2 further contribute to the directed migration of inflammatory immune cells that help BCSC survival; targeting them was shown to reduce the number of pro-tumor stromal cells in the TME [277]. Inhibition of both chemokine receptors via an SMI, Reparixin, led to a reduction of BCSC markers in HER2-negative patients and was well tolerated [278]. These promising results warrant further investigation with more aggressive BC phenotypes.
Paclitaxel was found to increase autocrine TGF-b signaling in BCSCs, enhancing their survival and chemoresistance [279]. It has been shown that inhibiting TGF-b itself as well as downstream factors such as cyclooxygenase 2 (COX-2) can prevent the induction and expansion of both CD44+/CD24-/low and ALDH+ BCSC populations [280]. It is important to anticipate specific drug escape routes in the TME in the context of the drug mechanism, as it may help boost treatment efficacy and mitigate the challenges of MDR.
BCSCs in Hormone Receptor-Positive BC
The presence of estrogen receptors (ER) and/or progesterone receptors (PR) along with the absence of the human epidermal growth factor receptor 2 (HER2) protein are categorized as Luminal A tumors in BC. Luminal B tumors also present ER and/or PR receptors but are HER2-positive[285]. BCSCs play a pivotal role in fueling the growth, survival, and metastatic spread of hormone-sensitive cancer cells[286]. The self-renewal of BCSCs is stimulated by the various cells and proteins of the tumor microenvironment (TME) and steroid hormone signaling[287]. Estrogen is a regulator of BCSCs through paracrine signaling involving fibroblast growth factor9 (FGF9). Estrogen binds to its receptor inducing secretion of FGF9 and T-box transcription factor3 (Tbx3) in surrounding non-BCSC cells. This expression of Tbx3 leads to further Wnt and FGF expression in a signaling cascade ultimately resulting in BCSC proliferation[288]. The Wnt/β-Catenin pathway is responsible for BC cell proliferation and stemness maintenance while the Wnt–PCP (planar cell polarity) and Wnt–Ca2+ noncanonical Wnt pathways are responsible for BC cell metastasis[289]. Estrogen can also increase the amount of BCSCs through contact-dependent signaling by upregulating Notch ligands in non-CSCs, which in turn stimulates the activity of Notch1 in BCSCs[290]. Progesterone also acts via paracrine signaling to upregulate the Notch pathway and NF-κB pathway through receptor activator of nuclear factor kappa-B ligand (RANK/RANKL) activation contributing to BCSC pool expansion[291]. HER2 overexpression comprises approximately 20% of BC cases and characterizes an aggressive subtype with a high rate of metastasis[292]. HER2 has been shown to regulate BCSCs through multiple pathways such as Notch allowing the cells to evade targeted therapies, undergo epithelial to mesenchymal transitioning, and invade surrounding cells[293].
Current understanding of the underlying mechanisms of hormone signaling in receptor-positive BC has led to the development of targeted therapies. Endocrine therapies typically target the estrogen pathways by depriving the tumor of estrogen or interfering with it signaling[294]. Tamoxifen and other selective estrogen receptor modulators (SERMs) competitively inhibit estrogen from binding to the estrogen receptor depriving the tumor of estrogen[295]. Selective estrogen receptor degraders (SERDs) such as Fulvestrant competitively inhibit estrogen to bind to ERs resulting in ER degradation[296]. Aromatase inhibitors deplete estrogen by inhibiting the enzyme aromatase necessary for estrogen biosynthesis[297]. The problem arises when BCSCs become resistant to estrogen therapies and play a primary role in decreased efficacy and metastatic relapse[298]. Hormone therapies have been seen to indirectly enrich BSCSs through a variety of mechanisms including upregulation of key elements involved in the transcriptional regulation of PTFs such as SOX2, OCT4, and NANOG[294]. Enhanced PI3K/AKT/mTOR signaling in BCSCs has also been implicated in their ability to survive, expand, metastasize, and resist treatments[299]. Mutations in the PI3K/AKT/mTOR pathway sustain the BCSC pool[300].
MicroRNAs (miRNAs) are small non-coding RNA segments that can silence target genes by initiating degradation of their messenger RNAs (mRNAs) or hindering translation[301]. Regulation of miRNAs through hormones contributes to the self-renewal of BCSCs and their resistance to targeted treatments[302]. The overexpression of miR-221 and miR-222 in hormone-positive BC downregulates ER which is linked to endocrine resistance[303]. HER2 can also be targeted in therapies. Trastuzumab, a monoclonal antibody that binds to an extracellular domain of the HER2 receptor inhibiting homodimerization and therefore signaling[304]. Additionally, lapatinib, a tyrosine kinase inhibitor, targets HER2 and EGFR through interactions at the ATP-binding site of the receptors[305]. However, mutations in the PI3K/AKT/mTOR pathway as well as loss of phosphatase and tensin homolog (PTEN) tumor suppressor involved with BCSC proliferation have also been implicated in Trastuzumab resistance[306]. Resistance to lapatinib stems from upregulation of miR-205-5p in BCSCs which increases EMT and metastatic potential[307]. Further understanding of the role of BCSCs in hormone-positive BC and factors that lead to therapy resistance is necessary to develop targeted therapies that can evade these mechanisms.
Conclusions
Though BCSCs are a small component of the tumors they significantly contribute to clonogenic replication through self-renewal. They contribute to MRD post-treatment and form a significant challenge in the clinic by displaying chemoresistance. A strategic approach is needed to synchronously target both the bulk tumor population and the BCSCs.
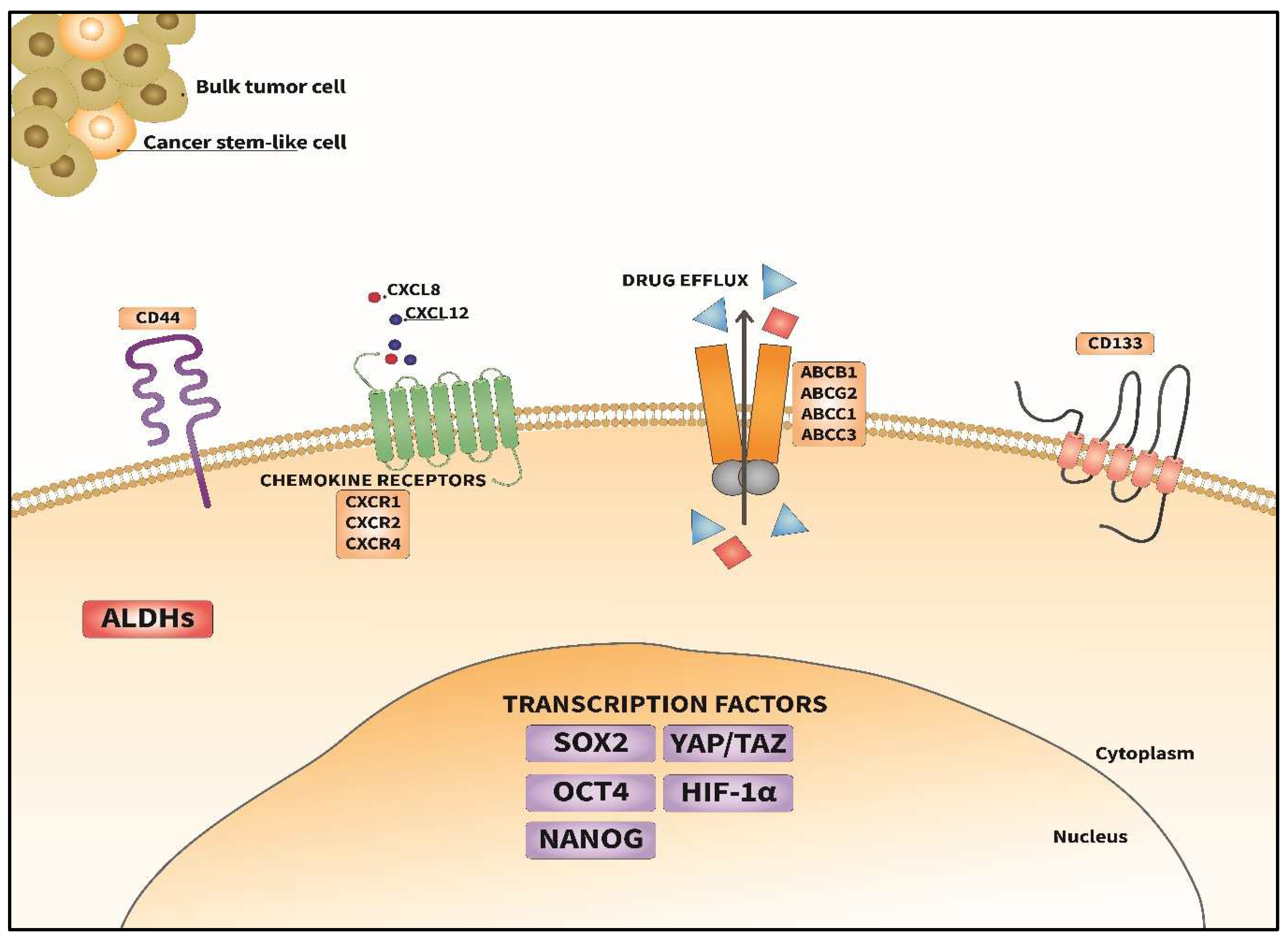
Figure 1.
Key molecular factors that confer BCSCs their distinctive characteristics. This diagram illustrates the molecular components associated with cancer stemness in BC. Surface markers such as CD44s and CD133 are prominently expressed and are critical for cell identification and signaling. Chemokine receptors (CXCR1, CXCR2, CXCR4) interact with their cognate ligands, to regulate clonogenicity, self-renewal, migration and paracrine interactions in the TME. Drug efflux transporters (ABCB1, ABCG2, ABCC1, ABCC3) facilitate chemoresistance by actively pumping out therapeutic agents. Intracellularly, key transcription factors, including SOX2, OCT4, NANOG, YAP/TAZ, and HIF-1α, govern cancer stemness. Additionally, elevated ALDH activity marks BCSC expression and contributes to their maintenance and differentiation.
Figure 1.
Key molecular factors that confer BCSCs their distinctive characteristics. This diagram illustrates the molecular components associated with cancer stemness in BC. Surface markers such as CD44s and CD133 are prominently expressed and are critical for cell identification and signaling. Chemokine receptors (CXCR1, CXCR2, CXCR4) interact with their cognate ligands, to regulate clonogenicity, self-renewal, migration and paracrine interactions in the TME. Drug efflux transporters (ABCB1, ABCG2, ABCC1, ABCC3) facilitate chemoresistance by actively pumping out therapeutic agents. Intracellularly, key transcription factors, including SOX2, OCT4, NANOG, YAP/TAZ, and HIF-1α, govern cancer stemness. Additionally, elevated ALDH activity marks BCSC expression and contributes to their maintenance and differentiation.
Author Contributions
Conceptualization, D.R.; Writing—Original Draft Preparation, D.D., A.B., D.T., T.J., and D.R.; Writing—Review and Editing–J. F. and D.R.; Scientific input for figure–D.T., Funding Acquisition - D.R. and J. F.; Supervision and Co-ordination–D.R. We thank Sara Virginia Hernandez Mendoza for her skillful and dedicated artwork for all figures. All authors have read and agreed to the submitted version of the manuscript.
Funding
Funding for this body of work was provided by the National Cancer Institute/National Institute of Health (R211CA256462 and R01CA258682) and The University of Toledo startup funds (F110796) (to D.R.) and the Institutional Development Award (IDeA) from the NIGMS/NIH (1P20 GM135000-04) (to J. F.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Health.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AA | African American |
| ABCs | ATP binding cassettes |
| ABCB1 | ATP binding cassette B1 |
| ABCG2 | ATP binding cassette G2 |
| ABL | Abelson leukemia |
| Ago2 | Argonaute 2 |
| AhR | Aryl hydrocarbon receptor |
| ALDHs | Aldehyde dehydrogenases |
| ALDH1 | Aldehyde dehydrogenase 1 |
| ALDH1A1 | Aldehyde dehydrogenase 1A1 |
| APC | Adenomatous polyposis coli |
| AP-1 | Activator protein 1 |
| ATP | Adenosine triphosphate |
| BC | BC |
| BCRP | BC resistance protein |
| BCSCs | BC stem-like cells |
| BET | Bromodomain extra-terminal domain |
| BIRC5 | Baculoviral IAP repeat-containing 5 |
| BL1 | Basal-like 1 |
| BL2 | Basal-like 2 |
| BRCA1 | Breast cancer gene 1 |
| CAFs | Cancer-associated fibroblasts |
| CCL21 | C-C motif chemokine ligand 21 |
| CCR7 | C-C chemokine receptor type 7 |
| CD | Cluster of differentiation |
| CD24 | Cluster of differentiation 24 |
| CD44 | Cluster of differentiation 44 |
| CD49f | Cluster of differentiation 49f |
| CD133 | Cluster of differentiation 133 |
| CEACAM1 | carcinoembryonic antigen cell adhesion molecule 1 |
| COX-2 | Cyclooxygenase 2 |
| CREB | cyclic AMP response element-binding protein |
| CSC | Cancer stem cell |
| CXCR1 | C-X-C chemokine receptor 1 |
| CXCR2 | C-X-C chemokine receptor 2 |
| CXCR4 | C-X-C chemokine receptor 4 |
| CXCL8 | C-X-C motif chemokine ligand 8 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| c-MYC | cellular myelocytomatosis |
| DNA | Deoxyribonucleic acid |
| DOT1L | Disruptor of telomeric silencing 1 |
| d-TPP | Dodecyl(triphenyl)phosphonium |
| ECM | Extracellular matrix |
| eIF4A1 | Eukaryotic initiation factor 4A1 |
| eIF4B | Eukaryotic initiation factor 4B |
| EMT | Epithelial-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| ER | Estrogen receptor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal regulated kinase |
| EW | European White |
| FGF9 | Fibroblast growth factor9 |
| Fz | Frizzled receptor |
| GLI1 | Glioma-associated oncogene homolog 1 |
| HER2 | Human epidermal growth factor receptor2 |
| HIFs | Hypoxia Inducible factors |
| HIF-1a | Hypoxia inducible factor 1a |
| HIF-2a | Hypoxia inducible factor 2a |
| HMGA1 | High mobility group A1 |
| HRE | Hypoxia response element |
| ICIs | Immune checkpoint inhibitors |
| Ils | Interleukins |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IRF | Interferon regulatory factor |
| JAK | Janus kinase |
| JAK2 | Janus kinase2 |
| JAM-A | Junctional adhesion molecule A |
| KLF4 | Kruppel-like factor 4 |
| KLF5 | Kruppel-like factor 5 |
| LAR | Luminal androgen receptor |
| LASP1 | LIM and SH3 protein 1 |
| LATS1/2 | Large tumor suppressor 1 and 2 |
| LRP6 | Low-density lipoprotein receptor-related protein 6 |
| M | Mesenchymal |
| MAPK | Mitogen activated protein kinase |
| MAP4K | Mitogen activated protein kinase kinase kinase kinases |
| MCL1 | Myeloid cell leukemia 1 |
| MDM2 | Mouse double minute 2 homolog |
| MDR | Multidrug resistance |
| miRNAs | MicroRNAs |
| MOB1 A/B | Monopolar spindle (mps1) binder 1 A/B |
| MRD | Minimal residual disease |
| mRNAs | Messenger RNAs |
| MSI1 | Musashi RNA binding protein 1 |
| MST1/2 | Mammalian sterile 20-like kinase 1/2 |
| mTNBC | Metastatic triple-negative BC |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NACT | Neoadjuvant chemotherapy |
| NF-kB | Nuclear factor kappa of B lymphocytes |
| Notch 1-4 | Neurogenic locus notch homolog protein 1-4 |
| OCT4 | Octamer-binding transcription factor 4 |
| OSKM | OCT4, SOX2, KLF4, c-MYC |
| OXPHOS | Oxidative phosphorylation |
| PCP | planar cell polarity |
| pCR | Pathological complete response |
| PD-L1 | Programmed death ligand 1 |
| PI3K | Phosphatidylinositol-3-kinase |
| PR | Progesterone receptor |
| PTEN | Phosphate and tensin homolog |
| PTFs | Pluripotent transcription factors |
| P-gp | P-glycoprotein |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| RNA | Ribonucleic acid |
| ROCK1 | Rho-associated coiled-coil containing protein kinase 1 |
| ROR1 | Receptor tyrosine kinase-like orphan receptor 1 |
| ROS | Reactive oxygen species |
| RSKs | Ribosomal S6 kinases |
| SAV1 | Salvador homolog 1 |
| SERDs | Selective estrogen receptor degraders |
| SETRMs | Selective estrogen receptor modulators |
| SH3 | Src homology 3 |
| siRNA | Small interfering RNA |
| SMIs | Small molecule inhibitors |
| SMO | Smoothened receptor |
| SOX2 | SRY (sex determining region Y)-box2 |
| SOX9 | SRY (sex determining region Y)-box9 |
| SRF | Serum response factor |
| STAT3 | Signal transducers and activators of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TAOKs | Thousand and one kinases |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| Tbx3 | T-box transcription factor3 |
| TCF | T-cell factor |
| tDRs | Transfer RNA-derived small non-coding RNAs |
| TEAD 1-4 | Transcriptional enhancer associate domain 1-4 |
| TF | Transcription factors |
| TGF-b | Transforming growth factor b |
| TILs | Tumor infiltrating lymphocytes |
| TKIs | Tyrosine kinase inhibitors |
| TNBC | Triple-negative BC |
| TNF-a | Tumor necrosis factor a |
| TME | Tumor microenvironment |
| TPP | Target product profile |
| Tregs | Regulatory T cells |
| Twist1 | Twist family BHLH transcription factor 1 |
| VEGF | Vascular endothelial growth factor |
| VM | Vascular mimicry |
| WASP | Wiskott–Aldrich syndrome protein |
| WAVE3 | WASP-family verprolin-homologous protein |
| Wnt | Wingless-related integration site |
| WWTR1 | WW-domain-containing transcription regulator 1 |
| YAP | Yes-associated protein |
| YB-1 | Y-box binding protein 1 |
| (ΔNp63) | Delta N Isoform of Tumor Protein 63 |
References
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell stem cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. (Lond) 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A.; Eisner, J.R.; Parker, J.S.; Selitsky, S.R.; Park, B.H.; Elias, A.D.; Baskin-Bey, E.S.; Cardoso, F. Male breast cancer: a disease distinct from female breast cancer. Breast Cancer Res. Treat. 2019, 173, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.M.; Hoadley, K.A.; Troester, M.A. Race and Ancestry in Immune Response to Breast Cancer. Cancer Discov. 2022, 12, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PloS one 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef]
- Lee, J. Current Treatment Landscape for Early Triple-Negative Breast Cancer (TNBC). J. Clin. Med. 2023, 12. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Das, S.; Mukherjee, P.; Chatterjee, R.; Jamal, Z.; Chatterji, U. Enhancing Chemosensitivity of Breast Cancer Stem Cells by Downregulating SOX2 and ABCG2 Using Wedelolactone-encapsulated Nanoparticles. Mol. Cancer Ther. 2019, 18, 680–692. [Google Scholar] [CrossRef]
- Go, J.; Ahn, J.H.; Park, J.M.; Choi, S.B.; Kim, J.Y.; Park, H.S.; Kim, S.I.; Park, B.W.; Park, S. Distinct Prognosis of Minimal Residual Disease According to Breast Cancer Subtype in Patients with Breast or Nodal Pathologic Complete Response After Neoadjuvant Chemotherapy. Ann. Surg. Oncol. 2023, 30, 7060–7068. [Google Scholar] [CrossRef]
- Masuda, H.; Harano, K.; Miura, S.; Wang, Y.; Hirota, Y.; Harada, O.; Jolly, M.K.; Matsunaga, Y.; Lim, B.; Wood, A.L.; et al. Changes in Triple-Negative Breast Cancer Molecular Subtypes in Patients Without Pathologic Complete Response After Neoadjuvant Systemic Chemotherapy. JCO Precis. Oncol. 2022, 6, e2000368. [Google Scholar] [CrossRef]
- Davies, A.; Zoubeidi, A.; Beltran, H.; Selth, L.A. The Transcriptional and Epigenetic Landscape of Cancer Cell Lineage Plasticity. Cancer Discov. 2023, 13, 1771–1788. [Google Scholar] [CrossRef]
- Prado-Vázquez, G.; Gámez-Pozo, A.; Trilla-Fuertes, L.; Arevalillo, J.M.; Zapater-Moros, A.; Ferrer-Gómez, M.; Díaz-Almirón, M.; López-Vacas, R.; Navarro, H.; Maín, P.; et al. A novel approach to triple-negative breast cancer molecular classification reveals a luminal immune-positive subgroup with good prognoses. Sci. Rep. 2019, 9, 1538. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Savas, P.; Luen, S.J.; Virassamy, B.; Salgado, R.; Loi, S. Clinical Validity and Utility of Tumor-Infiltrating Lymphocytes in Routine Clinical Practice for Breast Cancer Patients: Current and Future Directions. Front. Oncol. 2017, 7, 156. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Dulal, D.; Boring, A.; Terrero, D.; Johnson, T.; Tiwari, A.K.; Raman, D. Tackling of Immunorefractory Tumors by Targeting Alternative Immune Checkpoints. Cancers 2023, 15, 2774. [Google Scholar] [CrossRef]
- Qu, C.; Cui, H.; Xiao, S.; Dong, L.; Lu, Q.; Zhang, L.; Wang, P.; Xin, M.; Zhi, H.; Liu, C.; et al. The landscape of immune checkpoint-related long non-coding RNAs core regulatory circuitry reveals implications for immunoregulation and immunotherapy responses. Commun. Biol. 2024, 7, 327. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Moitra, K.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Hum. Mutat. 2022, 43, 1162–1182. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Liang, X.J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.J.; Gong, L.H.; Zheng, F.Y.; Cheng, K.J.; Chen, Z.S.; Shi, Z. Triterpenoids as reversal agents for anticancer drug resistance treatment. Drug Discov. Today 2014, 19, 482–488. [Google Scholar] [CrossRef]
- Abd El-Aziz, Y.S.; Spillane, A.J.; Jansson, P.J.; Sahni, S. Role of ABCB1 in mediating chemoresistance of triple-negative breast cancers. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Sherlach, K.S.; Roepe, P.D. “Drug resistance associated membrane proteins”. Front. Physiol. 2014, 5, 108. [Google Scholar] [CrossRef]
- Dufour, R.; Daumar, P.; Mounetou, E.; Aubel, C.; Kwiatkowski, F.; Abrial, C.; Vatoux, C.; Penault-Llorca, F.; Bamdad, M. BCRP and P-gp relay overexpression in triple negative basal-like breast cancer cell line: a prospective role in resistance to Olaparib. Sci. Rep. 2015, 5, 12670. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Fathy Abd-Ellatef, G.E.; Gazzano, E.; Chirio, D.; Hamed, A.R.; Belisario, D.C.; Zuddas, C.; Peira, E.; Rolando, B.; Kopecka, J.; Assem Said Marie, M.; et al. Curcumin-Loaded Solid Lipid Nanoparticles Bypass P-Glycoprotein Mediated Doxorubicin Resistance in Triple Negative Breast Cancer Cells. Pharmaceutics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Bivona, T.G. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8. [Google Scholar] [CrossRef]
- Zheng, S.; Zou, Y.; Liang, J.Y.; Xiao, W.; Yang, A.; Meng, T.; Lu, S.; Luo, Z.; Xie, X. Identification and validation of a combined hypoxia and immune index for triple-negative breast cancer. Mol. Oncol. 2020, 14, 2814–2833. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.; Barsoum, I.; Kim, J.; Black, M.; Siemens, R.D. Mechanisms Of Hypoxia-Induced Immune Escape In Cancer And Their Regulation By Nitric Oxide. Redox Biol. 2015, 5, 417. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- D’Cunha, R.R.; Murry, D.J.; An, G. Nilotinib Alters the Efflux Transporter-Mediated Pharmacokinetics of Afatinib in Mice. J. Pharm. Sci. 2019, 108, 3434–3442. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.T.; Ganguly, S.S.; Bennett, H.; Friend, J.W.; Tepe, J.; Plattner, R. Imatinib reverses doxorubicin resistance by affecting activation of STAT3-dependent NF-κB and HSP27/p38/AKT pathways and by inhibiting ABCB1. PLoS One 2013, 8, e55509. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef]
- Sun, M.; Yang, C.; Zheng, J.; Wang, M.; Chen, M.; Le, D.Q.S.; Kjems, J.; Bunger, C.E. Enhanced efficacy of chemotherapy for breast cancer stem cells by simultaneous suppression of multidrug resistance and antiapoptotic cellular defense. Acta Biomater. 2015, 28, 171–182. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5. [Google Scholar] [CrossRef]
- Ray, A.; Dhar, S.; Ray, B.K. Control of VEGF expression in triple-negative breast carcinoma cells by suppression of SAF-1 transcription factor activity. Mol. Cancer Res. 2011, 9, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Dent, S.F. The role of VEGF in triple-negative breast cancer: where do we go from here? Ann. Oncol. 2009, 20, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.U.; Cho, H.M.; Das, R.; Gil-Henn, H.; Ramakrishnan, S.; Al Bayati, A.; Carroll, S.F.; Zhang, Y.; Sankar, A.P.; Elledge, C.; et al. Inhibition of Vasculogenic Mimicry and Angiogenesis by an Anti-EGFR IgG1-Human Endostatin-P125A Fusion Protein Reduces Triple Negative Breast Cancer Metastases. Cells 2021, 10. [Google Scholar] [CrossRef]
- Urru, S.A.M.; Gallus, S.; Bosetti, C.; Moi, T.; Medda, R.; Sollai, E.; Murgia, A.; Sanges, F.; Pira, G.; Manca, A.; et al. Clinical and pathological factors influencing survival in a large cohort of triple-negative breast cancer patients. BMC Cancer 2018, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.M.; Olopade, O.I. Epidemiology of Triple-Negative Breast Cancer: A Review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.L.; Updike, K.L.; Factor, R.E.; Henry, N.L.; Boucher, K.M.; Bernard, P.S.; Varley, K.E. A Multigene Assay Determines Risk of Recurrence in Patients with Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3466–3478. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Gradishar, W.J. Triple-Negative Breast Cancer: Current Practice and Future Directions. J. Oncol. Pract. 2017, 13, 301–303. [Google Scholar] [CrossRef] [PubMed]
- den Brok, W.D.; Speers, C.H.; Gondara, L.; Baxter, E.; Tyldesley, S.K.; Lohrisch, C.A. Survival with metastatic breast cancer based on initial presentation, de novo versus relapsed. Breast Cancer Res. Treat. 2017, 161, 549–556. [Google Scholar] [CrossRef]
- Almansour, N.M. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Hassn Mesrati, M.; Syafruddin, S.E.; Mohtar, M.A.; Syahir, A. CD44: A Multifunctional Mediator of Cancer Progression. Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Altevogt, P.; Sammar, M.; Hüser, L.; Kristiansen, G. Novel insights into the function of CD24: A driving force in cancer. Int. J. Cancer 2021, 148, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Chute, J.P.; Muramoto, G.G.; Whitesides, J.; Colvin, M.; Safi, R.; Chao, N.J.; McDonnell, D.P. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11707–11712. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Lim, C.L. Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investig. 2017, 4, 96. [Google Scholar] [CrossRef]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.K.; Kanojia, D.; Liu, X.; Singh, U.P.; Berger, F.G.; Wang, Q.; Chen, H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFβ signaling. Oncogene 2012, 31, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.M.; Jordan, C.T. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670–1673. [Google Scholar] [CrossRef]
- Dittmer, J. Breast cancer stem cells: Features, key drivers and treatment options. Semin. Cancer Biol. 2018, 53, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Reipas, K.; Hu, K.; Berns, R.; Firmino, N.; Stratford, A.L.; Dunn, S.E. Inhibition of RSK with the novel small-molecule inhibitor LJI308 overcomes chemoresistance by eliminating cancer stem cells. Oncotarget 2015, 6. [Google Scholar] [CrossRef]
- Bledzka, K.; Schiemann, B.; Schiemann, W.P.; Fox, P.; Plow, E.F.; Sossey-Alaoui, K. The WAVE3-YB1 interaction regulates cancer stem cells activity in breast cancer. Oncotarget 2017, 8, 104072–104089. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.Y.; Zhang, H.; Yang, J.; Chen, Y.; Lu, H. Asymmetric Cell Division and Tumor Heterogeneity. Front. Cell Dev. Biol. 2022, 10, 938685. [Google Scholar] [CrossRef]
- Sridharan, S.; Howard, C.M.; Tilley, A.M.C.; Subramaniyan, B.; Tiwari, A.K.; Ruch, R.J.; Raman, D. Novel and Alternative Targets Against Breast Cancer Stemness to Combat Chemoresistance. Front. Oncol. 2019, 9, 1003. [Google Scholar] [CrossRef]
- Banerjee, A.; Arvinrad, P.; Darley, M.; Laversin, S.A.; Parker, R.; Rose-Zerilli, M.J.J.; Townsend, P.A.; Cutress, R.I.; Beers, S.A.; Houghton, F.D.; et al. The effects of restricted glycolysis on stem-cell like characteristics of breast cancer cells. Oncotarget 2018, 9, 23274–23288. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Robeson, M.; Bastihalli-Tukaramrao, D.; Howard, C.M.; Subramaniyan, B.; Tilley, A.M.C.; Tiwari, A.K.; Raman, D. Targeting of the Eukaryotic Translation Initiation Factor 4A Against Breast Cancer Stemness. Front. Oncol. 2019, 9, 1311. [Google Scholar] [CrossRef]
- Raman, D.; Cimpean, A.M.; De Miglio, M.R. Editorial: Drug resistance in breast cancer - mechanisms and approaches to overcome chemoresistance. Front. Oncol. 2022, 12, 1080684. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Tiwari, A.K. Role of eIF4A1 in triple-negative breast cancer stem-like cell-mediated drug resistance. Cancer Rep. (Hoboken) 2022, 5, e1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.-J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e135. [Google Scholar] [CrossRef]
- Lee, K.-L.; Kuo, Y.-C.; Ho, Y.-S.; Huang, Y.-H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers 2019, 11, 1334. [Google Scholar] [CrossRef]
- Brandolini, L.; Cristiano, L.; Fidoamore, A.; De Pizzol, M.; Di Giacomo, E.; Florio, T.M.; Confalone, G.; Galante, A.; Cinque, B.; Benedetti, E.; et al. Targeting CXCR1 on breast cancer stem cells: signaling pathways and clinical application modelling. Oncotarget 2015, 6, 43375–43394. [Google Scholar] [CrossRef] [PubMed]
- Tilley, A.M.C.; Howard, C.M.; Sridharan, S.; Subramaniyan, B.; Bearss, N.R.; Alkhalili, S.; Raman, D. The CXCR4-Dependent LASP1-Ago2 Interaction in Triple-Negative Breast Cancer. Cancers 2020, 12, 2455. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Howard, C.M.; Tilley, A.M.C.; Sridharan, S. Cell-Cell Interaction | Chemokine Receptors. In Encyclopedia of Biological Chemistry III; 2021; pp. 699–710.
- Butt, E.; Howard, C.M.; Raman, D. LASP1 in Cellular Signaling and Gene Expression: More than Just a Cytoskeletal Regulator. Cells 2022, 11. [Google Scholar] [CrossRef]
- Howard, C.M.; Bearss, N.; Subramaniyan, B.; Tilley, A.; Sridharan, S.; Villa, N.; Fraser, C.S.; Raman, D. The CXCR4-LASP1-eIF4F Axis Promotes Translation of Oncogenic Proteins in Triple-Negative Breast Cancer Cells. Front. Oncol. 2019, 9, 284. [Google Scholar] [CrossRef]
- Subramaniyan, B.; Sridharan, S.; MHoward, C.; MC Tilley, A.; Basuroy, T.; de la Serna, I.; Butt, E.; Raman, D. Role of the CXCR4-LASP1 Axis in the Stabilization of Snail1 in Triple-Negative Breast Cancer. Cancers 2020, 12, 2372. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Krishnamurthy, T.P.; Sola, P. Targeted Drug Therapy to Overcome Chemoresistance in Triple-negative Breast Cancer. Curr. Cancer Drug Targets 2020, 20, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Ross, D.D. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin. J. Cancer 2012, 31, 73–99. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, N.; Morii, E.; Ikeda, J.-i.; Qiu, Y.; Mamato, S.; Tian, T.; Fukuhara, S.; Aozasa, K. Synergistic effect of interleukin-6 and endoplasmic reticulum stress inducers on the high level of ABCG2 expression in plasma cells. Lab. Investig. 2009, 89, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Oviedo, L.; Nuncia-Cantarero, M.; Morcillo-Garcia, S.; Nieto-Jimenez, C.; Burgos, M.; Corrales-Sanchez, V.; Perez-Peña, J.; Győrffy, B.; Ocaña, A.; Galán-Moya, E.M. Identification of a stemness-related gene panel associated with BET inhibition in triple negative breast cancer. Cell. Oncol. 2020, 43, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Tanić, N.; Prvanović, M.; Milovanović, Z.; Tanić, N. Friend or foe: ABCG2, ABCC1 and ABCB1 expression in triple-negative breast cancer. Breast Cancer 2021, 28, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Yang, C.; Han, L.; Liu, L.; Liu, Y.J. Expression of BCRP/ABCG2 Protein in Invasive Breast Cancer and Response to Neoadjuvant Chemotherapy. Oncol. Res. Treat. 2021, 45, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Palasuberniam, P.; Yang, X.; Kraus, D.; Jones, P.; Myers, K.A.; Chen, B. ABCG2 transporter inhibitor restores the sensitivity of triple negative breast cancer cells to aminolevulinic acid-mediated photodynamic therapy. Sci. Rep. 2015, 5, 13298. [Google Scholar] [CrossRef]
- Garrido-Cano, I.; Adam-Artigues, A.; Lameirinhas, A.; Blandez, J.F.; Candela-Noguera, V.; Rojo, F.; Zazo, S.; Madoz-Gúrpide, J.; Lluch, A.; Bermejo, B.; et al. miR-99a-5p modulates doxorubicin resistance via the COX-2/ABCG2 axis in triple-negative breast cancer: from the discovery to in vivo studies. Cancer Commun. 2022, 42, 1412–1416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, B.; Zhao, X.; Ma, Y.; Ji, R.; Gu, Q.; Dong, X.; Li, J.; Liu, F.; Jia, X.; et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol. Cancer 2014, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133hi cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef]
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef]
- Bock, C.; Kuhn, C.; Ditsch, N.; Krebold, R.; Heublein, S.; Mayr, D.; Doisneau-Sixou, S.; Jeschke, U. Strong correlation between N-cadherin and CD133 in breast cancer: role of both markers in metastatic events. J. Cancer Res. Clin. Oncol. 2014, 140, 1873–1881. [Google Scholar] [CrossRef]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef]
- Pantalacci, S.; Tapon, N.; Leopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003, 5, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes. Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Jho, E.H. The history and regulatory mechanism of the Hippo pathway. BMB Rep. 2018, 51, 106–118. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes. Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Boggiano, J.C.; Vanderzalm, P.J.; Fehon, R.G. Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Dev. Cell 2011, 21, 888–895. [Google Scholar] [CrossRef]
- Poon, C.L.; Lin, J.I.; Zhang, X.; Harvey, K.F. The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev. Cell 2011, 21, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.H.; Nousiainen, M.; Chalamalasetty, R.B.; Schafer, A.; Nigg, E.A.; Sillje, H.H. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 2005, 24, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Schmitz, D.; Hemmings, B.A. The human tumour suppressor LATS1 is activated by human MOB1 at the membrane. Biochem. Biophys. Res. Commun. 2006, 345, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Aylon, Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017, 24, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [PubMed]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004, 381, 453–462. [Google Scholar] [CrossRef]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Harvey, K.; Tapon, N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef]
- Li, F.L.; Fu, V.; Liu, G.; Tang, T.; Konradi, A.W.; Peng, X.; Kemper, E.; Cravatt, B.F.; Franklin, J.M.; Wu, Z.; et al. Hippo pathway regulation by phosphatidylinositol transfer protein and phosphoinositides. Nat. Chem. Biol. 2022, 18, 1076–1086. [Google Scholar] [CrossRef]
- Yu, F.X.; Guan, K.L. The Hippo pathway: regulators and regulations. Genes. Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Tan, Y.; Zhang, W.; Tang, X.; Nice, E.C.; Huang, C.; Aguilar, M. Hippo signaling in cancer: regulatory mechanisms and therapeutic strategies. Aust. J. Chem. 2023, 76, 399–412. [Google Scholar] [CrossRef]
- Mohajan, S.; Jaiswal, P.K.; Vatanmakarian, M.; Yousefi, H.; Sankaralingam, S.; Alahari, S.K.; Koul, S.; Koul, H.K. Hippo pathway: Regulation, deregulation and potential therapeutic targets in cancer. Cancer Lett. 2021, 507, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef]
- Lv, L.; Zhou, X. Targeting Hippo signaling in cancer: novel perspectives and therapeutic potential. MedComm (2020) 2023, 4, e375. [Google Scholar] [CrossRef] [PubMed]
- Han, Y. Analysis of the role of the Hippo pathway in cancer. J. Transl. Med. 2019, 17, 116. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef]
- Sebio, A.; Lenz, H.J. Molecular Pathways: Hippo Signaling, a Critical Tumor Suppressor. Clin. Cancer Res. 2015, 21, 5002–5007. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: a signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Chaulk, S.G.; Lattanzi, V.J.; Hiemer, S.E.; Fahlman, R.P.; Varelas, X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 2014, 289, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Sabine, A.; Bovay, E.; Demir, C.S.; Kimura, W.; Jaquet, M.; Agalarov, Y.; Zangger, N.; Scallan, J.P.; Graber, W.; Gulpinar, E.; et al. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Investig. 2015, 125, 3861–3877. [Google Scholar] [CrossRef]
- Wang, L.; Luo, J.Y.; Li, B.; Tian, X.Y.; Chen, L.J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, Y.; Ai, D.; Yao, L.; Jiang, H. The role of the Hippo pathway in heart disease. FEBS J. 2022, 289, 5819–5833. [Google Scholar] [CrossRef]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, Y.; Yin, F.; Yu, J.; Silverman, N.; Pan, D. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 2016, 164, 406–419. [Google Scholar] [CrossRef]
- Wang, S.; Xie, F.; Chu, F.; Zhang, Z.; Yang, B.; Dai, T.; Gao, L.; Wang, L.; Ling, L.; Jia, J.; et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKvarepsilon-mediated phosphorylation. Nat. Immunol. 2017, 18, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Li, X.; Zhou, D.; Geng, J.; Chen, L. Role of Hippo signaling in regulating immunity. Cell Mol. Immunol. 2018, 15, 1003–1009. [Google Scholar] [CrossRef]
- Mia, M.M.; Singh, M.K. Emerging roles of the Hippo signaling pathway in modulating immune response and inflammation-driven tissue repair and remodeling. FEBS J. 2022, 289, 4061–4081. [Google Scholar] [CrossRef]
- Wang, D.; He, J.; Huang, B.; Liu, S.; Zhu, H.; Xu, T. Emerging role of the Hippo pathway in autophagy. Cell Death Dis. 2020, 11, 880. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Q.; Liu, Q.; Li, Y.; Sun, X.; Hong, L.; Ji, S.; Liu, C.; Geng, J.; Zhang, W.; et al. Hippo Signaling Suppresses Cell Ploidy and Tumorigenesis through Skp2. Cancer Cell 2017, 31, 669–684 e667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Guan, K.L. Hippo pathway key to ploidy checkpoint. Cell 2014, 158, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Sancisi, V. YAP and TAZ Are Not Identical Twins. Trends Biochem. Sci. 2021, 46, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 1994, 9, 2145–2152. [Google Scholar] [PubMed]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef] [PubMed]
- Battilana, G.; Zanconato, F.; Piccolo, S. Mechanisms of YAP/TAZ transcriptional control. Cell Stress. 2021, 5, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes. Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, F.; Zhang, Q.; Chen, Y.; Wang, B.; Jiang, J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev. Cell 2008, 14, 377–387. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.; Sheetz, M. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 265–275. [Google Scholar] [CrossRef]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604 e595. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. Elife 2016, 5. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, J.; Jho, E.H. Role of the Hippo pathway and mechanisms for controlling cellular localization of YAP/TAZ. FEBS J. 2022, 289, 5798–5818. [Google Scholar] [CrossRef]
- Yan, F.; Qian, M.; He, Q.; Zhu, H.; Yang, B. The posttranslational modifications of Hippo-YAP pathway in cancer. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129397. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.T.; Yamashita, K.; Sakurai, N.; Ohno, S. The Epithelial Circumferential Actin Belt Regulates YAP/TAZ through Nucleocytoplasmic Shuttling of Merlin. Cell Rep. 2017, 20, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFbeta-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes. Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef]
- Huang, W.; Lv, X.; Liu, C.; Zha, Z.; Zhang, H.; Jiang, Y.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J. Biol. Chem. 2012, 287, 26245–26253. [Google Scholar] [CrossRef]
- Miranda, M.Z.; Bialik, J.F.; Speight, P.; Dan, Q.; Yeung, T.; Szaszi, K.; Pedersen, S.F.; Kapus, A. TGF-beta1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017, 292, 14902–14920. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Tian, X.; Zhang, C.; Miao, Y.; Wang, Y.; Peng, Y.; Qiu, S.; Wang, H.; Cui, J.; Cao, L.; et al. YAP ISGylation increases its stability and promotes its positive regulation on PPP by stimulating 6PGL transcription. Cell Death Discov. 2022, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, F.; Qiao, Y.; Zheng, W.; Liu, Y.; Chen, Y.; Wu, Q.; Liu, X.; Zhu, G.; Chen, Y.; et al. TFCP2 Is Required for YAP-Dependent Transcription to Stimulate Liver Malignancy. Cell Rep. 2017, 21, 1227–1239. [Google Scholar] [CrossRef]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Jia, Z.; Yang, X. Cysteine residues are essential for dimerization of Hippo pathway components YAP2L and TAZ. Sci. Rep. 2018, 8, 3485. [Google Scholar] [CrossRef] [PubMed]
- Ben, C.; Wu, X.; Takahashi-Kanemitsu, A.; Knight, C.T.; Hayashi, T.; Hatakeyama, M. Alternative splicing reverses the cell-intrinsic and cell-extrinsic pro-oncogenic potentials of YAP1. J. Biol. Chem. 2020, 295, 13965–13980. [Google Scholar] [CrossRef]
- Vrbsky, J.; Vinarsky, V.; Perestrelo, A.R.; De La Cruz, J.O.; Martino, F.; Pompeiano, A.; Izzi, V.; Hlinomaz, O.; Rotrekl, V.; Sudol, M.; et al. Evidence for discrete modes of YAP1 signaling via mRNA splice isoforms in development and diseases. Genomics 2021, 113, 1349–1365. [Google Scholar] [CrossRef]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: modulation, function and therapeutic approaches. Nat. Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef]
- Ortega, A.; Vera, I.; Diaz, M.P.; Navarro, C.; Rojas, M.; Torres, W.; Parra, H.; Salazar, J.; De Sanctis, J.B.; Bermudez, V. The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises. Int. J. Mol. Sci. 2021, 23. [Google Scholar] [CrossRef]
- Luo, J.; Zou, H.; Guo, Y.; Tong, T.; Chen, Y.; Xiao, Y.; Pan, Y.; Li, P. The oncogenic roles and clinical implications of YAP/TAZ in breast cancer. Br. J. Cancer 2023, 128, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, B.K.; Kanchwala, M.; Cai, J.; Wang, L.; Xing, C.; Zheng, Y.; Pan, D. YAP/TAZ drives cell proliferation and tumour growth via a polyamine-eIF5A hypusination-LSD1 axis. Nat. Cell Biol. 2022, 24, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.H.; Yang, D.; Lim, D.S.; Wang, C.Y.; Guan, K.L. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes. Dev. 2020, 34, 72–86. [Google Scholar] [CrossRef]
- Luo, J.; Deng, L.; Zou, H.; Guo, Y.; Tong, T.; Huang, M.; Ling, G.; Li, P. New insights into the ambivalent role of YAP/TAZ in human cancers. J. Exp. Clin. Cancer Res. 2023, 42, 130. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, X.; Wang, X.; Liu, H.; Geck, R.C.; Tewari, A.K.; Xiao, T.; Font-Tello, A.; Lim, K.; Jones, K.L.; et al. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nat. Cell Biol. 2021, 23, 1187–1198. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.A.; Squatrito, M.; Northcott, P.; Awan, A.; Holland, E.C.; Taylor, M.D.; Nahle, Z.; Kenney, A.M. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene 2012, 31, 1923–1937. [Google Scholar] [CrossRef]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef]
- Song, S.; Honjo, S.; Jin, J.; Chang, S.S.; Scott, A.W.; Chen, Q.; Kalhor, N.; Correa, A.M.; Hofstetter, W.L.; Albarracin, C.T.; et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin. Cancer Res. 2015, 21, 2580–2590. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, Q.; Liu, A.M.; Tang, C.; Gong, Y.; Bian, J.; Luk, J.M.; Xu, Z.; Chen, J. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol. Rep. 2013, 29, 840–846. [Google Scholar] [CrossRef]
- Song, J.; Xie, L.X.; Zhang, X.Y.; Hu, P.; Long, M.F.; Xiong, F.; Huang, J.; Ye, X.Q. Role of YAP in lung cancer resistance to cisplatin. Oncol. Lett. 2018, 16, 3949–3954. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jang, S.K.; Hong, S.E.; Park, C.S.; Seong, M.K.; Kim, H.A.; Park, K.S.; Kim, C.H.; Park, I.C.; Jin, H.O. Knockdown of YAP/TAZ sensitizes tamoxifen-resistant MCF7 breast cancer cells. Biochem. Biophys. Res. Commun. 2022, 601, 73–78. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Arang, N.; Wang, Z.; Costea, D.E.; Feng, X.; Goto, Y.; Izumi, H.; Gilardi, M.; Ando, K.; Gutkind, J.S. EGFR Regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun. Biol. 2021, 4, 1237. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.F.; Tseng, Y.C.; Nguyen, P.A.; Li, Y.C.; Ho, C.C.; Wu, C.W. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef]
- Lin, C.H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 2015, 26, 3946–3953. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–2450. [Google Scholar] [CrossRef]
- Nallet-Staub, F.; Marsaud, V.; Li, L.; Gilbert, C.; Dodier, S.; Bataille, V.; Sudol, M.; Herlyn, M.; Mauviel, A. Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J. Invest. Dermatol. 2014, 134, 123–132. [Google Scholar] [CrossRef]
- Liu, J.; Ye, L.; Li, Q.; Wu, X.; Wang, B.; Ouyang, Y.; Yuan, Z.; Li, J.; Lin, C. Synaptopodin-2 suppresses metastasis of triple-negative breast cancer via inhibition of YAP/TAZ activity. J. Pathol. 2018, 244, 71–83. [Google Scholar] [CrossRef]
- Mason, D.E.; Collins, J.M.; Dawahare, J.H.; Nguyen, T.D.; Lin, Y.; Voytik-Harbin, S.L.; Zorlutuna, P.; Yoder, M.C.; Boerckel, J.D. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J. Cell Biol. 2019, 218, 1369–1389. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.C.; Kang, J.H.; Hamza, B.; King, E.M.; Lamar, J.M.; Manalis, S.R.; Hynes, R.O. YAP Enhances Tumor Cell Dissemination by Promoting Intravascular Motility and Reentry into Systemic Circulation. Cancer Res. 2020, 80, 3867–3879. [Google Scholar] [CrossRef] [PubMed]
- Sharif, G.M.; Schmidt, M.O.; Yi, C.; Hu, Z.; Haddad, B.R.; Glasgow, E.; Riegel, A.T.; Wellstein, A. Cell growth density modulates cancer cell vascular invasion via Hippo pathway activity and CXCR2 signaling. Oncogene 2015, 34, 5879–5889. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Urtatiz, O.; Van Raamsdonk, C.D. Oncogenic G Protein GNAQ Induces Uveal Melanoma and Intravasation in Mice. Cancer Res. 2015, 75, 3384–3397. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Rouse, C.; Xu, X.; Wang, J.; Onaitis, M.W.; Pendergast, A.M. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight 2016, 1, e89647. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Fujimura, A.; Di Biagio, D.; Frasson, C.; Bresolin, S.; Soligo, S.; Basso, G.; Bicciato, S.; Rosato, A.; et al. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell stem cell 2016, 19, 725–737. [Google Scholar] [CrossRef]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef]
- Shen, H.; Chen, Y.; Wan, Y.; Liu, T.; Wang, J.; Zhang, Y.; Wei, L.; Hu, Q.; Xu, B.; Chernov, M.; et al. Identification of TAZ-Dependent Breast Cancer Vulnerabilities Using a Chemical Genomics Screening Approach. Front. Cell Dev. Biol. 2021, 9, 673374. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes. Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef]
- Balamurugan, K.; Mendoza-Villanueva, D.; Sharan, S.; Summers, G.H.; Dobrolecki, L.E.; Lewis, M.T.; Sterneck, E. C/EBPdelta links IL-6 and HIF-1 signaling to promote breast cancer stem cell-associated phenotypes. Oncogene 2019, 38, 3765–3780. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Wei, H. Interleukin 6 signaling maintains the stem-like properties of bladder cancer stem cells. Transl. Cancer Res. 2019, 8, 557–566. [Google Scholar] [CrossRef]
- Kim, T.; Yang, S.J.; Hwang, D.; Song, J.; Kim, M.; Kyum Kim, S.; Kang, K.; Ahn, J.; Lee, D.; Kim, M.Y.; et al. A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat. Commun. 2015, 6, 10186. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Goodpaster, T.; Randolph-Habecker, J.; Hoffstrom, B.G.; Jalikis, F.G.; Koch, L.K.; Berger, C.; Kosasih, P.L.; Rajan, A.; Sommermeyer, D.; et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin. Cancer Res. 2017, 23, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J. ROR1: an orphan becomes apparent. Blood 2022, 140, 1583–1591. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Ghia, E.M.; Huang, J.; Wu, L.; Zhang, J.; Lam, S.; Lei, Y.; He, J.; Cui, B.; et al. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc. Natl. Acad. Sci. USA 2019, 116, 1370–1377. [Google Scholar] [CrossRef]
- Tao, Z.; Wu, X. Targeting Transcription Factors in Cancer: From “Undruggable” to “Druggable”. Methods Mol. Biol. 2023, 2594, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Hagenbeek, T.J.; Zbieg, J.R.; Hafner, M.; Mroue, R.; Lacap, J.A.; Sodir, N.M.; Noland, C.L.; Afghani, S.; Kishore, A.; Bhat, K.P.; et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat. Cancer 2023, 4, 812–828. [Google Scholar] [CrossRef]
- Thrash, H.L.; Pendergast, A.M. Multi-Functional Regulation by YAP/TAZ Signaling Networks in Tumor Progression and Metastasis. Cancers 2023, 15, 4701. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes. Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Lakhani, N.J.; McKean, M.; Lingaraj, T.; Victor, L.; Sanchez-Martin, M.; Kacena, K.; Malek, K.S.; Santillana, S. A phase 1, first-in-human study of IK-930, an oral TEAD inhibitor targeting the Hippo pathway in subjects with advanced solid tumors. J. Clin. Oncol. 2022, 40, TPS3168–TPS3168. [Google Scholar] [CrossRef]
- Furet, P.; Bordas, V.; Le Douget, M.; Salem, B.; Mesrouze, Y.; Imbach-Weese, P.; Sellner, H.; Voegtle, M.; Soldermann, N.; Chapeau, E.; et al. The First Class of Small Molecules Potently Disrupting the YAP-TEAD Interaction by Direct Competition. ChemMedChem 2022, 17, e202200303. [Google Scholar] [CrossRef] [PubMed]
- Macleod, A.R. Abstract ND11: The discovery and characterization of ION-537: A next generation antisense oligonucleotide inhibitor of YAP1 in preclinical cancer models. Cancer Res. 2021, 81, ND11–ND11. [Google Scholar] [CrossRef]
- Kotiyal, S.; Bhattacharya, S. Breast cancer stem cells, EMT and therapeutic targets. Biochem. Biophys. Res. Commun. 2014, 453, 112–116. [Google Scholar] [CrossRef]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef]
- Zeng, Z.; Fu, M.; Hu, Y.; Wei, Y.; Wei, X.; Luo, M. Regulation and signaling pathways in cancer stem cells: implications for targeted therapy for cancer. Mol. Cancer 2023, 22, 172. [Google Scholar] [CrossRef] [PubMed]
- Manni, W.; Min, W. Signaling pathways in the regulation of cancer stem cells and associated targeted therapy. MedComm (2020) 2022, 3, e176. [Google Scholar] [CrossRef]
- Mirzaei, S.; Paskeh, M.D.A.; Entezari, M.; Mirmazloomi, S.r.; Hassanpoor, A.; Aboutalebi, M.; Rezaei, S.; Hejazi, E.S.; Kakavand, A.; Heidari, H.; et al. SOX2 function in cancers: Association with growth, invasion, stemness and therapy response. Biomed. Pharmacother. 2022, 156, 113860. [Google Scholar] [CrossRef] [PubMed]
- Rasti, A.; Mehrazma, M.; Madjd, Z.; Abolhasani, M.; Saeednejad Zanjani, L.; Asgari, M. Co-expression of Cancer Stem Cell Markers OCT4 and NANOG Predicts Poor Prognosis in Renal Cell Carcinomas. Sci. Rep. 2018, 8, 11739. [Google Scholar] [CrossRef]
- Chen, W.; Qin, Y.; Liu, S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin. Transl. Med. 2018, 7, 27. [Google Scholar] [CrossRef]
- Fico, F.; Santamaria-Martínez, A. The Tumor Microenvironment as a Driving Force of Breast Cancer Stem Cell Plasticity. Cancers 2020, 12, 3863. [Google Scholar] [CrossRef]
- Huang, M.; Li, Y.; Zhang, H.; Nan, F. Breast cancer stromal fibroblasts promote the generation of CD44+CD24- cells through SDF-1/CXCR4 interaction. J. Exp. Clin. Cancer Res. 2010, 29, 80. [Google Scholar] [CrossRef]
- Li, F.; Xu, J.; Liu, S. Cancer Stem Cells and Neovascularization. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhi, W.I.; Lu, H.; Samanta, D.; Chen, I.; Gabrielson, E.; Semenza, G.L. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 2016, 7, 64527–64542. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Lu, H.; Samanta, D.; Salman, S.; Lu, Y.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc. Natl. Acad. Sci. USA 2018, 115, E9640–E9648. [Google Scholar] [CrossRef]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. U S A 2016, 113, E2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Ordaz-Ramos, A.; Tellez-Jimenez, O.; Vazquez-Santillan, K. Signaling pathways governing the maintenance of breast cancer stem cells and their therapeutic implications. Front. Cell Dev. Biol. 2023, 11, 1221175. [Google Scholar] [CrossRef]
- Wei, Q.; Qian, Y.; Yu, J.; Wong, C.C. Metabolic rewiring in the promotion of cancer metastasis: mechanisms and therapeutic implications. Oncogene 2020, 39, 6139–6156. [Google Scholar] [CrossRef]
- Gao, X.; Dong, Q.Z. Advance in metabolism and target therapy in breast cancer stem cells. World J. Stem Cells 2020, 12, 1295–1306. [Google Scholar] [CrossRef]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Ózsvári, B.; Sotgia, F.; Lisanti, M.P. Dodecyl-TPP Targets Mitochondria and Potently Eradicates Cancer Stem Cells (CSCs): Synergy With FDA-Approved Drugs and Natural Compounds (Vitamin C and Berberine). Front. Oncol. 2019, 9, 615. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Wu, S.; Xue, W.; Huang, X.; Yu, X.; Luo, M.; Huang, Y.; Liu, Y.; Bi, Z.; Qiu, X.; Bai, S. Distinct prognostic values of ALDH1 isoenzymes in breast cancer. Tumor Biol. 2015, 36, 2421–2426. [Google Scholar] [CrossRef] [PubMed]
- Sarvi, S.; Crispin, R.; Lu, Y.; Zeng, L.; Hurley, T.D.; Houston, D.R.; von Kriegsheim, A.; Chen, C.H.; Mochly-Rosen, D.; Ranzani, M.; et al. ALDH1 Bio-activates Nifuroxazide to Eradicate ALDH(High) Melanoma-Initiating Cells. Cell Chem. Biol. 2018, 25, 1456–1469.e1456. [Google Scholar] [CrossRef]
- Kurani, H.; Razavipour, S.F.; Harikumar, K.B.; Dunworth, M.; Ewald, A.J.; Nasir, A.; Pearson, G.; Van Booven, D.; Zhou, Z.; Azzam, D.; et al. DOT1L Is a Novel Cancer Stem Cell Target for Triple-Negative Breast Cancer. Clin. Cancer Res. 2022, 28, 1948–1965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Brown, R.L.; Wei, Y.; Zhao, P.; Liu, S.; Liu, X.; Deng, Y.; Hu, X.; Zhang, J.; Gao, X.D.; et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes. Dev. 2019, 33, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e637. [Google Scholar] [CrossRef]
- Sridharan, S.; Srivastava, S.; Terrero, D.; Malla, S.; Tiwari, A.K.; Raman, D. Abstract 4479: Targeting of eIF4A1 curtails lung metastases in triple-negative breast cancer. Cancer Res. 2023, 83, 4479–4479. [Google Scholar] [CrossRef]
- Yi, T.; Zhai, B.; Yu, Y.; Kiyotsugu, Y.; Raschle, T.; Etzkorn, M.; Seo, H.C.; Nagiec, M.; Luna, R.E.; Reinherz, E.L.; et al. Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Dulal, D.; Johnson, T.; Raman, D. Role of CXCL12/CXCR4 Axis in the Pathogenesis of Hematological Malignancies. 2024.
- Mehrpouri, M. The contributory roles of the CXCL12/CXCR4/CXCR7 axis in normal and malignant hematopoiesis: A possible therapeutic target in hematologic malignancies. Eur. J. Pharmacol. 2022, 920, 174831. [Google Scholar] [CrossRef] [PubMed]
- Dillenburg-Pilla, P.; Patel, V.; Mikelis, C.M.; Zárate-Bladés, C.R.; Doçi, C.L.; Amornphimoltham, P.; Wang, Z.; Martin, D.; Leelahavanichkul, K.; Dorsam, R.T.; et al. SDF-1/CXCL12 induces directional cell migration and spontaneous metastasis via a CXCR4/Gαi/mTORC1 axis. Faseb j 2015, 29, 1056–1068. [Google Scholar] [CrossRef]
- Chu, Q.D.; Panu, L.; Holm, N.T.; Li, B.D.; Johnson, L.W.; Zhang, S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J. Surg. Res. 2010, 159, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Peng, X.; Li, X.; Yang, P.; Xie, L.; Li, Y.; Du, C.; Zhang, G. Correction: Silencing of CXCR4 sensitizes triple-negative breast cancer cells to cisplatin. Oncotarget 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Hartung, A.; Bouchez, L.C.; Walker, J.R.; Reddy, V.A.; Cho, C.Y.; Schultz, P.G. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. Br. J. Cancer 2012, 107, 43–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: a systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, P.A.; Simon, S.P.; Martin, M.; Gil-Martin, M.; Pardo, P.G.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide (a CXCR4 antagonist) plus eribulin in HER2 negative metastatic breast cancer: Dose-response analysis of efficacy from phase I single-arm trial. J. Clin. Oncol. 2020, 38, e15209–e15209. [Google Scholar] [CrossRef]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Müller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/mTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Perez, R.P.; Yardley, D.; Han, L.K.; Reuben, J.M.; Gao, H.; McCanna, S.; Butler, B.; Ruffini, P.A.; Liu, Y.; et al. A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res. 2020, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Tian, J.; Hachim, M.Y.; Hachim, I.Y.; Dai, M.; Lo, C.; Raffa, F.A.; Ali, S.; Lebrun, J.J. Cyclooxygenase-2 regulates TGFβ-induced cancer stemness in triple-negative breast cancer. Sci. Rep. 2017, 7, 40258. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, W.; Guo, H.; Zhang, Y.; He, Y.; Lee, S.H.; Song, X.; Li, X.; Guo, Y.; Zhao, Y.; et al. NOTCH1 Signaling Regulates Self-Renewal and Platinum Chemoresistance of Cancer Stem-like Cells in Human Non-Small Cell Lung Cancer. Cancer Res. 2017, 77, 3082–3091. [Google Scholar] [CrossRef]
- Vorobyeva, A.; Bezverkhniaia, E.; Konovalova, E.; Schulga, A.; Garousi, J.; Vorontsova, O.; Abouzayed, A.; Orlova, A.; Deyev, S.; Tolmachev, V. Radionuclide Molecular Imaging of EpCAM Expression in Triple-Negative Breast Cancer Using the Scaffold Protein DARPin Ec1. Molecules 2020, 25, 4719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiong, X.; Sun, Y. Functional characterization of SOX2 as an anticancer target. Signal Transduct. Target. Ther. 2020, 5, 135. [Google Scholar] [CrossRef]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef]
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumor Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Gupta, P.B.; Rudnick, J.A.; Caballero, S.; Keller, P.J.; Lander, E.S.; Kuperwasser, C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21737–21742. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Harrison, H.; Simões, B.M.; Rogerson, L.; Howell, S.J.; Landberg, G.; Clarke, R.B. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. 2013, 15, R21. [Google Scholar] [CrossRef]
- Chen, B.; Ye, P.; Chen, Y.; Liu, T.; Cha, J.H.; Yan, X.; Yang, W.H. Involvement of the Estrogen and Progesterone Axis in Cancer Stemness: Elucidating Molecular Mechanisms and Clinical Significance. Front. Oncol. 2020, 10, 1657. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Zlobin, A.; Osipo, C. Notch-EGFR/HER2 Bidirectional Crosstalk in Breast Cancer. Front. Oncol. 2014, 4, 360. [Google Scholar] [CrossRef]
- Leung, E.Y.; Askarian-Amiri, M.E.; Sarkar, D.; Ferraro-Peyret, C.; Joseph, W.R.; Finlay, G.J.; Baguley, B.C. Endocrine Therapy of Estrogen Receptor-Positive Breast Cancer Cells: Early Differential Effects on Stem Cell Markers. Front. Oncol. 2017, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Ojo, D.; Wei, F.; Liu, Y.; Wang, E.; Zhang, H.; Lin, X.; Wong, N.; Bane, A.; Tang, D. Factors Promoting Tamoxifen Resistance in Breast Cancer via Stimulating Breast Cancer Stem Cell Expansion. Curr. Med. Chem. 2015, 22, 2360–2374. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-Q.; Sun, H.; Xie, Y.; Patel, H.; Bo, L.; Lin, H.; Chen, Z.-S. Therapeutic evolution in HR+/HER2- breast cancer: from targeted therapy to endocrine therapy. Front. Pharmacol. 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.; Braybrooke, J.; Gray, R.; Hills, R.K.; Liu, Z.; Pan, H.; Peto, R.; Dodwell, D.; McGale, P.; Taylor, C.; et al. Aromatase inhibitors versus tamoxifen in premenopausal women with oestrogen receptor-positive early-stage breast cancer treated with ovarian suppression: a patient-level meta-analysis of 7030 women from four randomised trials. Lancet Oncol. 2022, 23, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Ramkairsingh, M.; Lin, X.; Kapoor, A.; Major, P.; Tang, D. The Central Contributions of Breast Cancer Stem Cells in Developing Resistance to Endocrine Therapy in Estrogen Receptor (ER)-Positive Breast Cancer. Cancers 2019, 11, 1028. [Google Scholar] [CrossRef]
- Kolev, V.N.; Wright, Q.G.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR Dual Inhibitor VS-5584 Preferentially Targets Cancer Stem Cells. Cancer Research 2015, 75, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Lukong, K.E. Breast Cancer Stem-Like Cells in Drug Resistance: A Review of Mechanisms and Novel Therapeutic Strategies to Overcome Drug Resistance. Front. Oncol. 2022, 12, 856974. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Quintero, B. Cell-free microRNAs in blood and other body fluids, as cancer biomarkers. Cell Prolif. 2016, 49, 281–303. [Google Scholar] [CrossRef]
- Finlay-Schultz, J.; Sartorius, C.A. Steroid hormones, steroid receptors, and breast cancer stem cells. J. Mammary Gland. Biol. Neoplasia 2015, 20, 39–50. [Google Scholar] [CrossRef]
- Petri, B.J.; Klinge, C.M. Regulation of breast cancer metastasis signaling by miRNAs. Cancer Metastasis Rev. 2020, 39, 837–886. [Google Scholar] [CrossRef]
- Maadi, H.; Soheilifar, M.H.; Choi, W.S.; Moshtaghian, A.; Wang, Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers 2021, 13, 3540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhang, Y.-K.; Kathawala, R.J.; Chen, Z.-S. Repositioning of Tyrosine Kinase Inhibitors as Antagonists of ATP-Binding Cassette Transporters in Anticancer Drug Resistance. Cancers 2014, 6, 1925–1952. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef] [PubMed]
- De Cola, A.; Lamolinara, A.; Lanuti, P.; Rossi, C.; Iezzi, M.; Marchisio, M.; Todaro, M.; De Laurenzi, V. MiR-205-5p inhibition by locked nucleic acids impairs metastatic potential of breast cancer cells. Cell Death Dis. 2018, 9, 821. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Selected YAP/TAZ inhibition strategies.
| Drug | Target | Preclinical References | Clinical Trials |
|---|---|---|---|
| Verteporfin (Visudyne) | YAP/TAZ interaction with TEAD | [235] | Phase 1/2 EGFR-mutated glioblastoma NCT04590664 |
| IK-930 | TEAD palmitoylation inhibitor YAP/TAZ interaction with TEAD |
[236] First-in-human trial |
Phase 1 Epithelioid hemangioendothelioma and mesothelioma NCT05228015 |
| 0GNE-7883 | Pan-TEAD inhibitor | [233] | - |
| IAG933 | YAP/TAZ interaction with TEAD | [237] | Phase 1 Mesothelioma NCT04857372 |
| ION537 | Anti-YAP Antisense Oligonucleotide | [238] | Completed phase 1 NCT04659096 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



