
Submitted:
10 December 2024
Posted:
11 December 2024
You are already at the latest version
Abstract
Mixed nBu4N+-substituted phosphomolybdovanadates of (nBu4N)2.5+xH0.5PMo12-xVxO40 (x = 1–4) gross composition have been prepared by a promising ecological method and investigated as catalysts for the transformation of 5-hydroxymethylfurfural. It has been shown that activation of vanadium(V) atoms in V2O5 using the H2O2-affected approach followed by their insertion into a Mo-containing framework provides a waste-free synthesis of mixed heteropoly anions. The resulting solids have been characterized by XRD, IR, ICP-AES, N2 adsorption-desorption, and TGA methods to estimate their structural and textural characteristics. The catalysts exhibit significant activity and good reusability (at least five times) in the oxidation of a biomass-derived precursor to produce 2,5-diformylfuran in yield above 90%.
Keywords:
2
; 5-diformylfuran
; 5-hydroxymethylfurfural
; heterogeneous catalysis
; heteropoly acid
; oxidation
; vanadium(V) activation
Introduction
The preparation of stable, environmentally friendly, and cost-effective heterogeneous catalysts for the production of important chemicals by transformation of biomass-derived compounds is an attractive area of research arising from modern green chemistry trends. Phosphomolybdates with intercalated vanadium(V) atoms of [PMo12–xVxO40](3+x)– composition, known as heteropoly acids (HPAs-x, x – vanadium(V) content), represent a promising type of polyoxometalates (POMs) with adjustable acid and oxidative properties [1,2]. Such Keggin-type POMs are currently being studied in various relevant areas of chemistry, such as fuel cells designing, preparing hybrid materials, desulfurization of petroleum products, synthesis of industrially valuable chemicals, and processing of plant raw materials [3,4,5,6,7,8].
The commonly used procedure for the synthesis of HPAs-x is an H+-triggered assembly of dissolved precursors according to the method of Tsigdinos and Hallada published in 1968 (Eq. (1)) [9]. Since this publication, the replacement of sodium molybdate and sodium orthophosphate with the corresponding acids has been the main change in subsequent (modern) syntheses. However, the application of NaVO3 as a vanadium(V) precursor is still widely used by researchers for the synthesis of mixed P-Mo-V POMs, while maintaining the need for the introduction of extraneous mineral acids and additional costs for subsequent purification.
Thus, the growing interest of researchers in the utilization of vanadium-containing POMs as chemical agents and catalysts in important industrial areas requires their preparation through green synthesis techniques. To achieve this goal, the application of the peroxide method for vanadium activation using V2O5 as an accessible precursor is a promising method, which excludes the insertion of any foreign components or strong aggressive acids [10].
5-Hydroxymethylfurfural (5-HMF) is a feedstock of natural origin that can be converted into a set of key intermediates for fine, pharmaceutical, and fragrance industries, among which 2,5-diformylfuran (DFF) deserves highlighting [11,12]. This compound is a versatile building block applied in the syntheses of non-toxic polymers with optical and electronic properties [13,14], heterocyclic ligands [15], fungicides [16], drugs [17], etc. As a result, the oxidation of 5-HMF is a synthetic route of interest, promising for testing the activity of compounds with oxidizing ability.
Besides the green synthesis of the catalyst, it is the activity and recoverability that are also important criteria for the successful realization of oxidative transformations. Among various means, an increase of vanadium(V) content, which is responsible for the redox ability, is the most promising way to enhance the catalytic activity and productivity of HPAs-x as environmentally friendly oxidation catalysts. The catalyst recoverability can be achieved by replacing H+ ions with large cations at high substitution degree [18,19]. At that, samples with substitution values close to, but not reaching complete substitution, are highly promising owing to possibility of their use not only in oxidative reactions, but also directly in one-pot processes as bifunctional catalysts [20]. For this reason, here we describe the preparation and characterization of forward-looking Keggin-type [PMo12-xVxO40](3+x)– heteropolyanions in the form of nBu4N+ acidic salts (TBA2.5+xHPA-x) with x in the range of 1–4 and their further investigation as heterogeneous catalysts for 5-HMF oxidation.
Experimental
Materials
85% H3PO4 (99.4%), V2O5 (99%), MoO3 (99%), and 30% H2O2 (high purity 8–4) were purchased from JSC Reakhim (Russia). Tetrabutylammonium bromide (nBu4NBr, 98%) was from JSC Vekton (Russia). Levulinic acid (LA, 98%), formic acid (FA, 98%), and 2,5-diformylfuran (DFF, 97%) were received from Sigma Aldrich. 5-Hydroxymethylfurfural (5-HMF, 97%), 5-formyl-2-furancarboxylic acid (FFCA, 98%), and 2,5-furandicarboxylic acid (FDCA, 99.5%) were purchased from Alfa Aesar.
Synthesis of H3+xPMo12-xVxO40 solutions
The synthesis of pure H3+xPMo12-xVxO40 solutions with x of 1–4 was carried out in three steps, including activation of V2O5 by an ecological acid-free peroxide method (Eq. (2)), preliminary formation of P-Mo Keggin framework and insertion of activated vanadium(V) atoms into the Keggin structure with obtaining a mixed-addenda anion.
The synthesis was carried out as follows: a sample of V2O5 was dissolved in distilled water (V1H2O) pre-cooled to 4–6°C. Solution of 30% H2O2 cooled to a similar temperature was gradually added to the resulting suspension upon continuous stirring, maintaining the temperature of the mixture below 15°C until complete V2O5 dissolving. The dark-orange solution of 0.0175 М H6V10O28 obtained was then stirred at room temperature until total O2 evolving and stabilized by Н3РО4 adding (V1H3PO4) according to Eq. (3). Note that this procedure, first applied in 1995 [21], increases the stability of the H6V10O28 solution up to several days, preventing the precipitation of a gel-like V2O5·nH2O and facilitating the large-scale synthesis of HPA-x solutions. The total volume of 85% H3PO4 was preliminary diluted to 2.5 M concentration and divided into 2 portions in accordance with the volume required to obtain a 0.0125 M solution of H9PV14O42.
At the next stage, a sample of MoO3 and the remaining amount of Н3РО4 (V2H3PO4) were introduced into a separate beaker with distilled water (V2H2O) and heated above 80°C. The solution of H9PV14O42 was added in portions to the suspension with stirring immediately after the appearance of a yellow colour. The resulting solution, after combining all components, was evaporated to the required volume and cooled to room temperature. The loadings of the required components (Table 1) were calculated using Eq. (4).

Table 1.
Components to synthesize 200 mL of 0.2 M HPA-x solution.
| Solution | Abbrev. | V2O5 | MoO3 | V1H3PO4 | V2H3PO4 | 30% H2O2 | V1H2O | V2H2O | ||
| [mol] | [g] | [mol] | [g] | [mL] | [mL] | [mol] | [L] | [L] | ||
| H4PMo11VO40 | HPA-1 | 0.02 | 0.44 | 63.3 | 1.16 | 14.84 | 0.3 | 0.23 | 0.79 | |
| H5PMo10V2O40 | HPA-2 | 0.04 | 0.40 | 57.6 | 2.28 | 13.72 | 0.6 | 0.46 | 0.72 | |
| H6PMo9V3O40 | HPA-3 | 0.06 | 0.36 | 51.8 | 3.44 | 12.56 | 0.9 | 0.69 | 0.65 | |
| H7PMo8V4O40 | HPA-4 | 0.08 | 0.32 | 46.1 | 4.57 | 11.43 | 1.2 | 0.91 | 0.58 | |
Synthesis of (nBu4N)2.5+xH0.5PMo12-xVxO40 solids
The desired (nBu4N)2.5+xH0.5PMo12-xVxO40 salts (TBA2.5+xHPA-x, x = 1–4) were obtained in the form of yellowish-orange powders by partial substitution of H+ ions in the prepared H3+xPMo12-xVxO40 with nBu4N+ cations. To do this, a sample of nBu4NBr was dissolved in distilled water with the formation of 0.25 M solution, which was then gradually introduced with continuous stirring into a 0.20 M HPA-x solution heated to 40°C. After the introduction had been completed, the heat was turned off and the slurry was stirred at room temperature overnight. At the final stage, the precipitate was filtered off, washed with deionized water, and dried in an oven at 80°С for 3 h.
Instrumental measurements
Powder X-ray diffraction (XRD) patterns were recorded at room temperature on a Stoe Stadi MP diffractometer using a Mo-Kα (λ = 0.70926 Å) radiation and re-counted on Cu-Kα (λ = 1.5418 Å) one. A bent Ge (111) Johann-type monochromator was used to form a primary beam. The recording was carried out in the transmission mode, for which a thin layer of the sample was clamped between two thin films. The signal was registered using a Dectris Mythen position-sensitive detector at the 2θ range of 2°–30°. The scanning step and accumulation time were 0.015° and 30 s, respectively.
Infrared spectra of attenuated total reflection (IR-ATR) were obtained using a Cary 660 FTIR spectrometer (Agilent Technologies) equipped with a GladiATR ATR device (PIKE Technologies). The spectra were recorded in the range of 4000–350 cm–1 with a resolution of 4 cm–1 and an accumulation of 40 scans.
Elemental analysis of the samples and leaching of structural components (P, Mo, V) were investigated by inductively coupled plasma atomic emission spectrometry (ICP-AES) on a Perkin Elmer Optima 4300 DV optical emission spectrometer.
Surface area and porosity of solids were determined by the BET method from nitrogen physisorption using an ASAP-2400 instrument (Micromeritics, USA) at 77.4 K. Prior to the measurements, the samples were degassed at 80°C overnight.
Thermogravimetric analysis (TGA) was performed using a simultaneous thermal analyzer NETZSCH STA 449 C Jupiter in a temperature range of 30–600°C at a heating rate of 10 °C·min–1 under helium flow of 30 mL·min–1.
Hydrolytic stability tests
Hydrolytic stability tests were carried out in a 100 mL batch reactor equipped with a thermostat and reflux condenser. To perform the test, a sample of salt (400 mg) and distilled water (50 mL) were placed into the reactor and retained at boiling temperature and vigorous stirring (1000 rpm) for 3 h. After completion of treatment, the solid was separated by filtration and dried at 80°C for 3 h, whereas the mother liquor was analysed by ICP-AES for P, Mo, and V content. Each sample was subjected to sequential three-fold processing. To estimate their structural stability, after the 1st and 3rd runs, the precipitates were investigated by IR-ATR and XRD.
Catalytic experiments
Oxidation of 5-HMF was performed under atmospheric pressure of oxygen upon vigorous stirring (900 rpm) in a 50 mL jacketed glass reactor equipped with a thermostat and a substrate dosing system. A sample of 5-HMF (0.05 g, 0.39 mmol) dissolved in H2O (5 mL) was gradually introduced into a heated to 50–95°C reactor containing a slurry of catalyst in H2O (30 mL). The catalyst content was varied in the range of 0.0975–0.39 mmol. The reaction was carried out for 30–180 min followed by separation of the catalyst by centrifugation at 104 rpm for 20 min and analysis of the resulting products. Regeneration of catalysts was performed by heating the samples in oxygen atmosphere at 150°C for 3 h.
Analysis of products
Substrate conversion and distribution of the products were controlled by HPLC, GC, and GC-MS. For GC analysis, a Hromos GH-1000 chromatograph equipped with a flame ionization detector and a KB-5 capillary column (30 m × 0.32 μm × 0.25 μm) was applied. The analysis was carried out in the programmed mode (100°C – 1 min – 25 °C/min – 220°C – 10 min) with o-cresol as an internal standard. GC-MS analysis was performed using an Agilent Technologies 7000 GC/MS Triple Quad instrument fitted out with a GsBP1-MS capillary column. HPLC analysis was executed using a Shimadzu Prominence LC-20 instrument equipped with refractive index and diode array detectors. Measurements were implemented using a Rezex ROA-Organic Acid H+ column (Phenomenex, 300 × 5 mm). An aqueous solution of 0.005 M H2SO4 (HPLC grade) was used as a mobile phase at a flow rate of 0.5 mL·min–1 and a column temperature of 50°C.
Results and Discussion
Solutions of HPA-x with x of 1–4 were synthesized by careful reacting a phosphomolybdate solution, obtained by dissolving the structure-forming molybdenum(VI) oxide in H2O/H3PO4, with a solution of H9PV14O42, prepared separately by treatment of vanadium(V) oxide using H2O2/H3PO4 (Figure 1). Activation of vanadium by peroxide method according to Eqns. (2) and (3) with its subsequent insertion into Mo-containing framework allowed obtaining HPA-x solutions free of extraneous ions, part of which was subjected to slow rotary evaporation for preparing the model patterns. These model patterns, as with TBA2.5+xHPA-x (x = 1–4) salts obtained by precipitation with tetra-n-butylammonium cations, were then characterized by various techniques such as XRD, IR-ATR, TGA, N2 adsorption–desorption. The elemental composition of the obtained samples was confirmed by ICP-AES with a deviation between the obtained and calculated values of P, Mo, and V contents below 3 wt %.
IR-ATR investigations
In general, the formation of mixed [РМo12-хVхО40](3+x)– anions can be considered as the replacement of MoVI atoms with VV in the [РМo12О40]3– heteropolyanion, the structure of which can be represented as a central PO4 tetrahedron surrounded by twelve MoO6 octahedrons assembled into four Mo3O13 triads [22]. As a consequence, such anion contain twelve quasi-linear Mo-Ob-Mo bonds between octahedrons from different Mo3O13 triads, twelve Mo-Oc-Mo bonds between octahedrons within Mo3O13 sets, four P-Oa-{Mo3O13} bonds between central heteroatom and triads, and twelve terminal bonds Mo=Od. This leads to the formation of characteristic vibrations of the Keggin anions in the range of 700–1100 cm–1, which makes IR spectroscopy a convenient method for their characterization.
According to Figure 2, the spectra of HPA-x (x = 1–4) contain four recognized strong absorption peaks in the fingerprint region of the Keggin polyoxoanions, which could be attributed to the fundamental P-Oa, Mo-Od, Mo-Ob-Mo, and Mo-Oc-Mo asymmetric stretching vibrations. Compared with the IR spectrum of unsubstituted H3PMo12O40 acid [23], the obtained vibrations in mixed P-Mo-V patterns did not reveal a significant difference, suggesting the formation of the Keggin anions at preparing HPAs-x via “peroxide” technique. The shoulder observed for P-Oa-{Mo3O13} vibrations can be explained by the replacement of Mo atoms by V ones in the framework of HP-anions with formation of Mo-O-V linkages [24], its presence becoming more noticeable with raising substitution content. Vanadium insertion also affects on the charge and symmetry of anions, causing a slight shift of νas(Mo-Od) and νas(Mo-Oc-Mo) stretching vibrations and a band of νas(Mo-Ob-Mo) to lower and higher wavenumbers, correspondingly, compared to vanadium-free acid. The same effect was previously observed in the literature [8,25], confirming the retaining of the primary structure during the formation of mixed P-Mo-V anions. The similarity of (nBu4N)2.5+xH0.5PMo12-xVxO40 spectra at 1100–700 cm–1 region with those of corresponding model acids strongly confirms the retention of the primary structure during the precipitation.
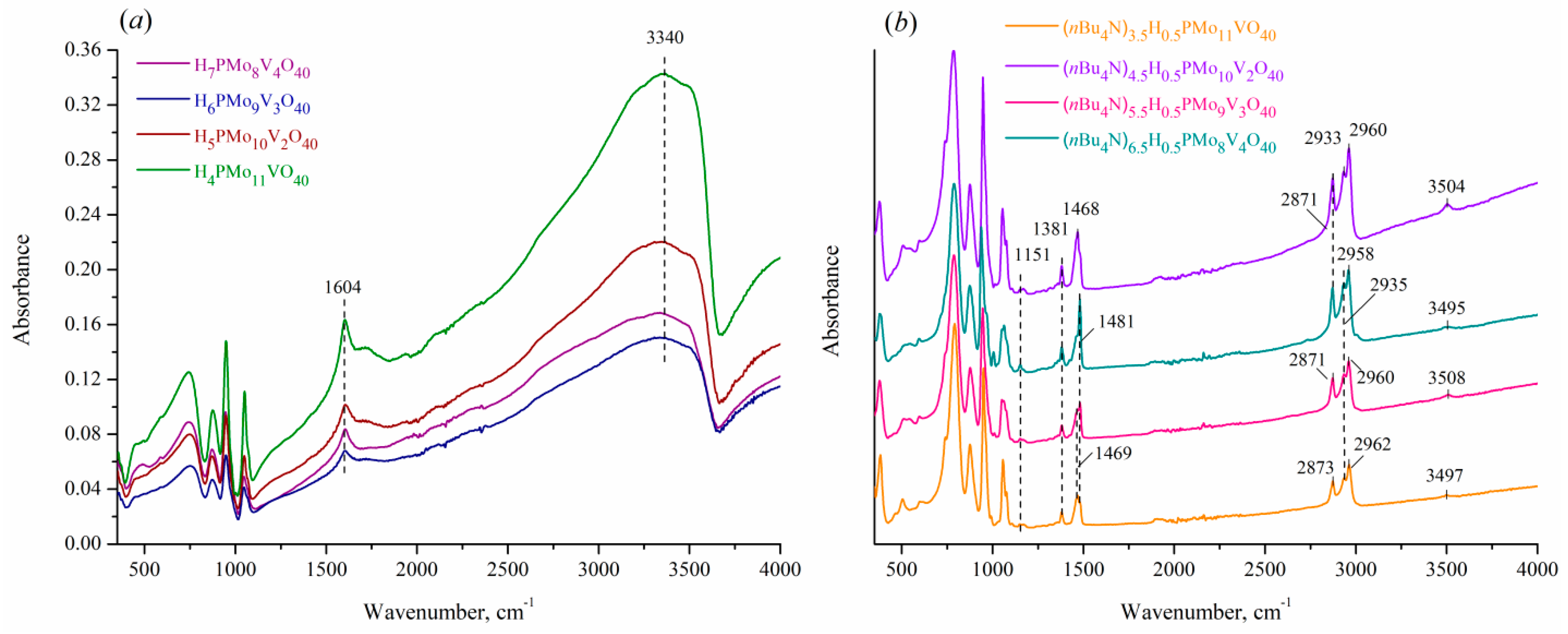
Expanded spectra of the prepared acids (Figure 3(a)) contain two additional bands at 3340 and 1604 cm–1 that can be attributed to hydrogen bonds in crystal water [26]. Insertion of nBu4N+ cations gives rise to new bands at 1481, 1468, and 1381 cm–1, as well as a set of peaks in the region 3000–2800 cm–1 (Figure 3(b)), which originate from asymmetric and/or symmetric bending and stretching vibrations of C–H, –CH2–, and –CH3 groups, respectively [27]. The peaks at 1153 and 1003 cm–1 can be ascribed to C–N stretching vibrations, whereas other characteristic peaks of nBu4N+ cation appear at 1000–600 cm–1 and overlap with polyanion fingerprint domain. Nevertheless, it allows supposing that nBu4N+ cation also maintains its structure in the obtained salts without degradation. The registered spectra are in good agreement with those described in the literature for sodium salts of HPAs-x with x of 1–3 [8,28].
Figure 2.
IR-ATR spectra of (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts and corresponding H3+xPMo12-xVxO40 acids with x of 1 (a), 2 (b), 3 (c), and 4 (d) in fingerprint region of the Keggin anion.
Figure 2.
IR-ATR spectra of (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts and corresponding H3+xPMo12-xVxO40 acids with x of 1 (a), 2 (b), 3 (c), and 4 (d) in fingerprint region of the Keggin anion.
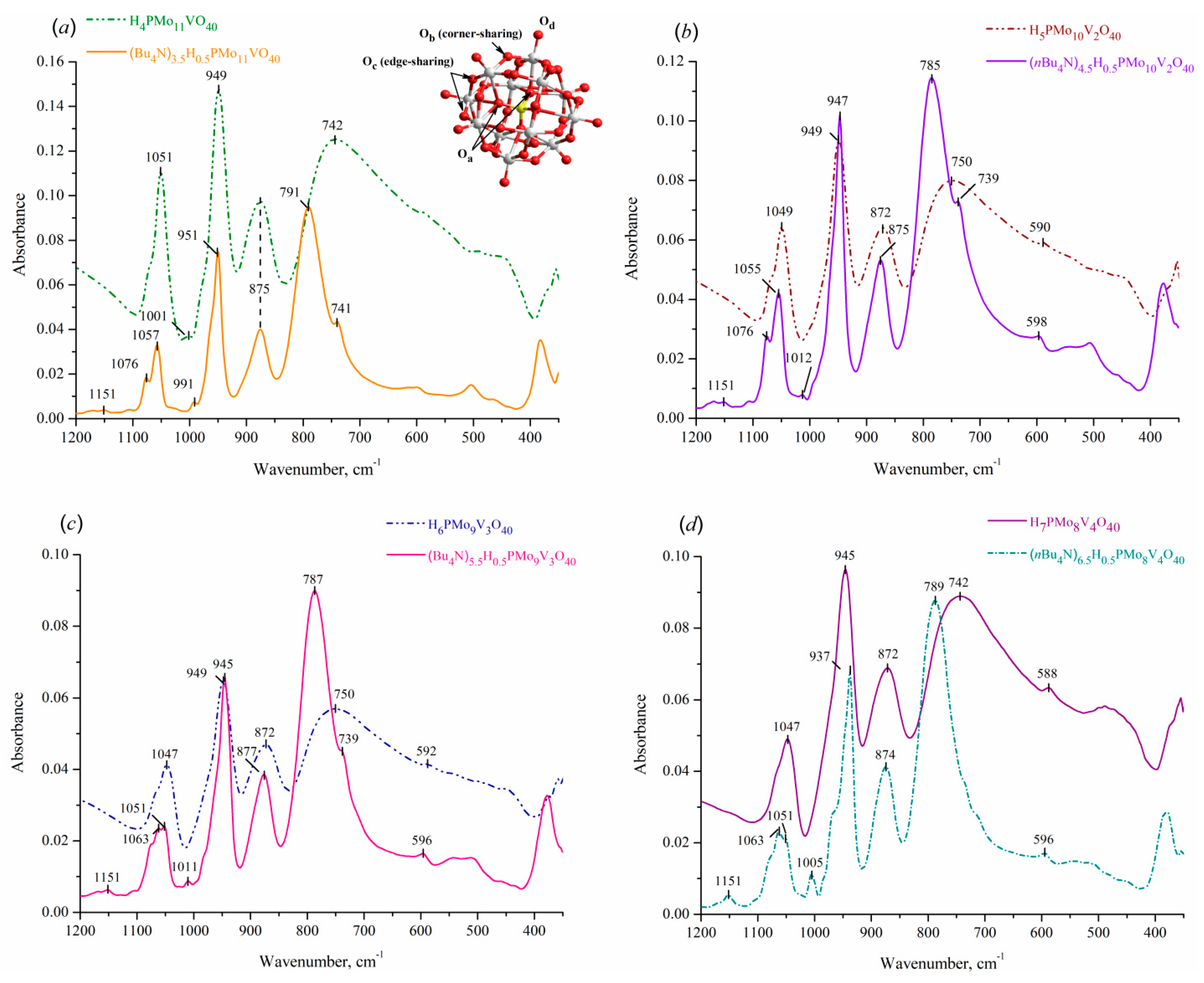
Figure 3.
Expanded IR-ATR spectra of (a) H3+xPMo12-xVxO40 acids and (b) (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts with x of 1–4.
Figure 3.
Expanded IR-ATR spectra of (a) H3+xPMo12-xVxO40 acids and (b) (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts with x of 1–4.
X-ray diffraction
To further investigate the obtained solids, the powder X-ray diffraction patterns were recorded in the 2θ range of 3°–40°. Figure 4 shows the diffraction patterns of HPA-x (x = 1–4) acids and the corresponding nBu4N+-substituted salts, each of which contains characteristic peaks in similar ranges of 2θ, with these ranges having a good convergence with those for unsubstituted H3PMo12O40 acid [8,29]. Some main peaks in the diffraction patterns keep safe after increasing the vanadium content and replacing protons with nBu4N+ ions, resulting in similarity of obtained profiles. The divergence between obtained patterns and literature data [30] can be attributed to the difference in content and type of cations and hydration level, which affect the symmetry and arrangement of elements in the unit cell. Redistribution of peak intensities watched with increasing x can be explained by the formation of several isomeric Keggin anions (anions with different values and positions of x). The presence of separate phases of addenda atoms (MoO3 and V2O5) is not observed.
Investigation of textural characteristics
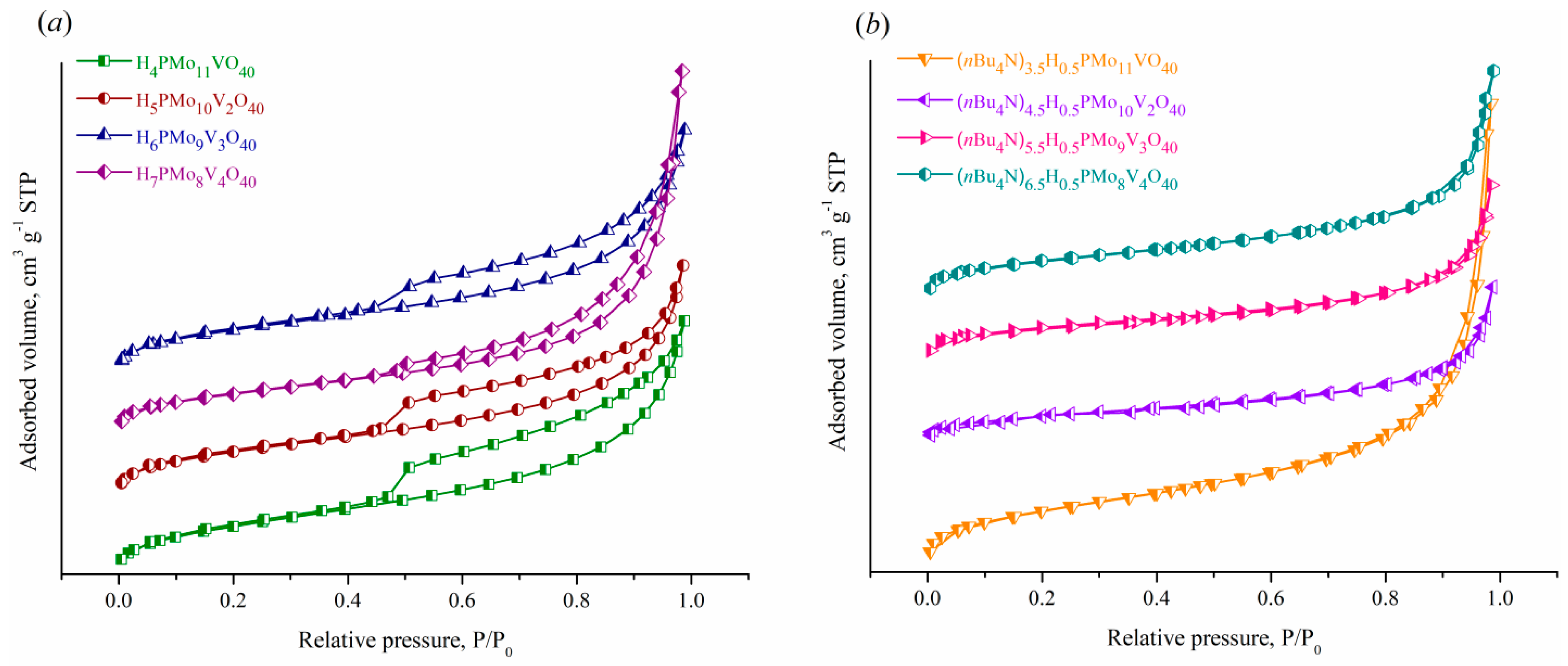
The textural properties of the samples obtained were characterized by low-temperature adsorption-desorption of nitrogen at 77.4 K (Figure 5, Table 2).
According to Figure 5(a), the HPAs-x have type II isotherms according to the IUPAC classification [31], characteristic of disperse macroporous and nonporous materials, with H3-shaped hysteresis loops usually attributed to slit-like materials. The adsorption isotherms of TBA2.5+xHPA-x acidic salts (Figure 5(b)) are S-shaped curves of the same type with a feebly marked capillary condensation hysteresis loops shifted to the region of Р/Р0 > 0.85, which suggests the presence of mesopores. All samples possess a low specific surface area in the range of 2–13 m2·g–1 with its values decreasing with passing from parental acids to the corresponding nBu4N+ salts as well as with increasing vanadium(V) content (Table 2).
Figure 5.
N2 adsorption-desorption isotherms of (a) H3+xPMo12-xVxO40 acids and (b) (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts with x of 1–4.
Figure 5.
N2 adsorption-desorption isotherms of (a) H3+xPMo12-xVxO40 acids and (b) (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts with x of 1–4.
Thermal analysis
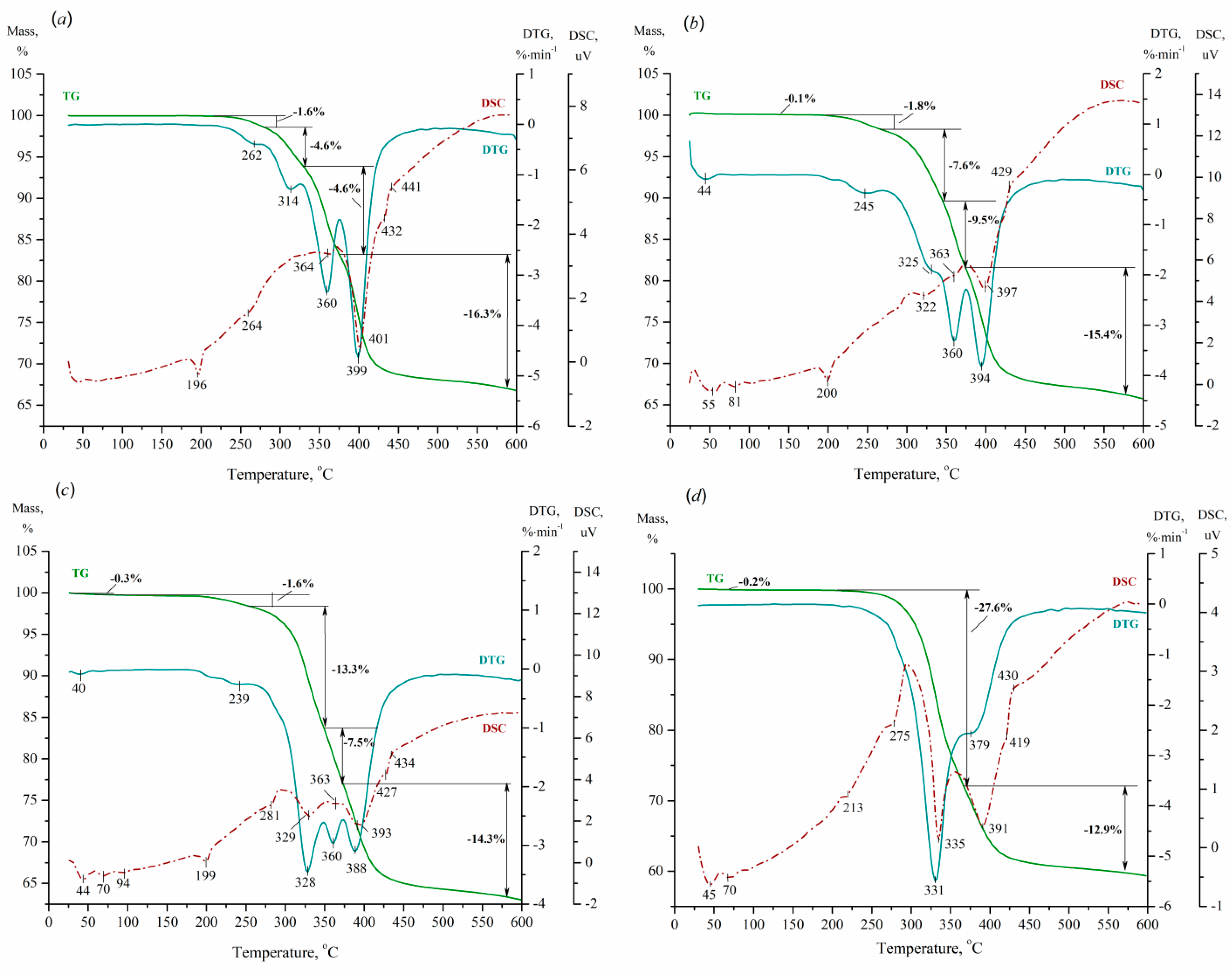
Figure 6 and Figure 7 depict the TG/DTG/DSC profiles of the parental acids and prepared nBu4N+ acidic salts, allowing one to estimate their thermal stability and degree of hydration.
According to Figure 6, the parental acids exhibit a continuous stepwise loss of weight in the range of room temperature (r.t.) to 419–458°C. The first weight loss occurs at heating the samples from r.t. up to 59–93°C, with a minimum on the DTG curves at 51–70°C. This stage can be assigned to the release of physisorbed water, with its duration and observed weight loss decreasing as x increases. The second stage proceeds to 89–144°C, with the detected loss of mass increasing in the range of 3.3–11.0% with rising charge of heteropoly anion. It can be attributed to the liberation of zeolitic water. The next thermal event is characterized by a weight damage of 2.0–5.1 wt % and occurs to 165–195°C. This can be ascribed to the elimination of crystallization (hydrogen-bonded to the acidic protons) water to form an anhydrous compound. The subsequent alteration can be ascribed to the loss of structural water (acidic protons) and the beginning of destruction of the Keggin framework with its total collapse to structure-forming oxides near 419–458°C (according to the exothermic peak on the DSC curves). The displacement of these temperatures to the low-temperature region compared to vanadium-free H3PMo12O40 acid (500°C) [8] seems to be due to reducing symmetry of the Keggin anions at the insertion of vanadium(V) atoms.
Figure 6.
TG/DTG/DSC profiles of H3+xPMo12-xVxO40 acids with x of 1 (a), 2 (b), 3 (c), and 4 (d).
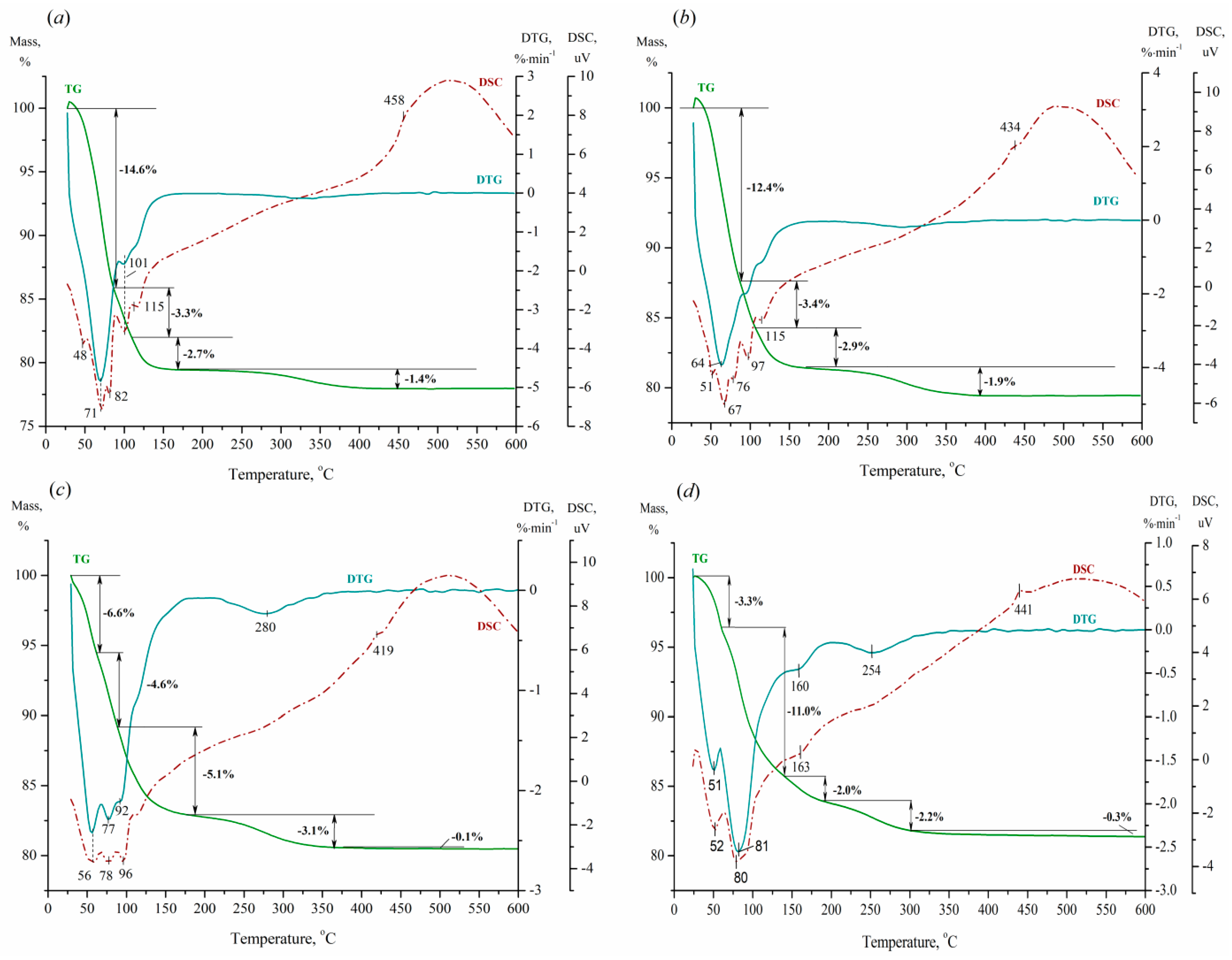
According to Figure 7, the TG curves of TBA2.5+xHPA-x acidic salts have a relatively constant value to 185–210°C, after which they continuously decrease up to 430–440°C. The observed decrease in the TG curves is reflected by a large and broad endothermic peak on the DTG curves, which contains from 2 to 4 marked minima depending on the salt composition. This stepwise loss of weight can be assigned to the decomposition and removal of nBu4N+ cations, as well as to the same processes discussed above for the parental acids. Additional peaks are present in the DSC profiles up to 215°C, indicating minor intermediate rearrangements, presumably due to three-dimensional nBu4N+ cations. It can be concluded that the prepared salts are stable up to at least 370°C without loss of nBu4N+ cations and degradation of the Keggin anions. It is obvious that the replacement of H+ ions with nBu4N+ cations leads to an increase in thermal stability.
Figure 7.
TG/DTG/DSC profiles of (nBu4N)2.5+xH0.5PMo12-xVxO40 salts with x of 1 (a), 2 (b), 3 (c), and 4 (d).
Figure 7.
TG/DTG/DSC profiles of (nBu4N)2.5+xH0.5PMo12-xVxO40 salts with x of 1 (a), 2 (b), 3 (c), and 4 (d).
Investigation of hydrolytic stability
The stability of the catalyst under reaction conditions is an important criterion for the successful implementation of the process, as well as proof of the heterogeneous nature of the reaction. The study of the synthesized TBA2.5+xHPA-x salts for stability and solubility was performed in 3 cycles by hydrothermal treatment at boiling temperature for 3 h. After completion of each treatment, the solid was separated and investigated by IR-ATR and XRD, whereas the mother liquor was analysed by ICP-AES for P, Mo and V content.
According to the data obtained, the solubility of salts after the first treatment cycle changed in the following series: TBA6.5HPA-4 (0.6%) < TBA5.5HPA-3 (0.8%) < TBA3.5HPA-1 (1.4%) < TBA4.5HPA-2 (2.2%). After the second cycle of treatment, the solubility of the samples was: TBA6.5HPA-4 (0.2%) < TBA5.5HPA-3 (0.3%) < TBA3.5HPA-1 (0.8%) < TBA4.5HPA-2 (1.2%). After the third treatment cycle, the solubility of TBA4.5HPA-2 salt has continued to decrease and amounted to 1.0%; for the others salts, the total leaching of the components was less than 0.1%, which confirms their heterogeneous nature.
Characterization of spent salts by XRD and IR-ATR (Fig. S1 and S2) showed significant structural stability of nBu4N+-containing acidic salts. The IR spectra and diffraction patterns of fresh salts and their spent samples were similar, with for TBA6.5HPA-4 and TBA5.5HPA-3 these being almost identical. For these samples, no perceivable differences in both peak intensities and their position, as well as no disappearance of peaks or the appearance of new bands, were observed in the obtained spectra. This confirms the preservation of the Keggin structure in spent solids and implies that (nBu4N)2.5+хH0.5PMo12-хVхO40 with x of 3 and 4 are stable and reusable water-insoluble compounds. As a consequence, owing to the presence of vanadium(V) atoms capable of oxidation, these salts can potentially be used as catalysts for the transformation of organic compounds.
Catalytic tests
The catalytic activity of the obtained salts was studied in the oxidation reaction of 5-HMF as a model substrate. This reaction was disclosed to result in a preferred production of DFF with some formation of 5-formyl-2-furancarboxylic (FFCA), levulinic (LA), and formic (FA) acids as by-products. The influence of several parameters on 5-HMF conversion and DFF selectivity, such as temperature, substrate-to-catalyst molar ratio, and reaction time, was investigated and presented in Figure 8.
The first series of experiments was aimed at assessing the effect of temperature (Figure 8(a)). It was found that increasing the reaction temperature in the range of 50–95°C had a positive action on substrate conversion, raising it from 35 to 70%. The use of TBA6.5HPA-4 salt leads to slightly superior values compared to TBA5.5HPA-3, evidencing that the content of vanadium has a beneficial effect on the transformation of 5-HMF. Besides higher conversion, TBA6.5HPA-4 catalyst also provides the greater yield toward the desired product (DFF), which some rises with increasing temperature. Selectivity of products changes differently that can be associated with varying in the contribution of processes of DFF overoxidation and 5-HMF hydration. The data obtained show that a temperature of 80°C provides the best relationship between the conversion of 5-HMF and the selectivity of DFF relative to other products.
Changing the reaction time in the range of 0.5–3 h (Figure 8(b)) showed that although the substrate conversion increased over time for both catalysts, the selectivity of the products progressed various. The best yield of the target product for TBA6.5HPA-4 is achieved after 1.5 hours and then practically does not change, while for TBA5.5HPA-3 a similar yield value is observed only after 2 hours.
Estimating the influence of catalyst loading showed the predominant formation of DFF in the entire studied range, with an increase in catalyst content leading to a considerable raise of 5-HMF conversion (Figure 8(c)). It was found that the yields of FA and LA reduced monotonically with increasing catalyst amount, which indicates rising the rate of the target reaction and predominating oxidation process over the hydration of 5-HMF. On the other hand, the application of high catalyst loading resulted in increasing DFF overoxidation with the formation of 2,5-furandicarboxylic acid (FDCA) as another by-product.
Figure 8.
Effect of reaction temperature (a), process time (b), and catalyst loading (c) on 5-HMF conversion and product selectivity (a) or DFF yield (b, c). Reaction conditions: (a) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (10 mol%) in H2O (15 mL), 3 h, 0.1 MPa O2; (b) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (10 mol%) in H2O (15 mL), 80°C, 0.1 MPa O2; (c) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (10–50 mol%) in H2O (15 mL), 80°C, 0.1 MPa O2, 1.5 h (for TBA6.5HPA-4) or 2 h (for TBA5.5HPA-3).
Figure 8.
Effect of reaction temperature (a), process time (b), and catalyst loading (c) on 5-HMF conversion and product selectivity (a) or DFF yield (b, c). Reaction conditions: (a) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (10 mol%) in H2O (15 mL), 3 h, 0.1 MPa O2; (b) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (10 mol%) in H2O (15 mL), 80°C, 0.1 MPa O2; (c) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (10–50 mol%) in H2O (15 mL), 80°C, 0.1 MPa O2, 1.5 h (for TBA6.5HPA-4) or 2 h (for TBA5.5HPA-3).
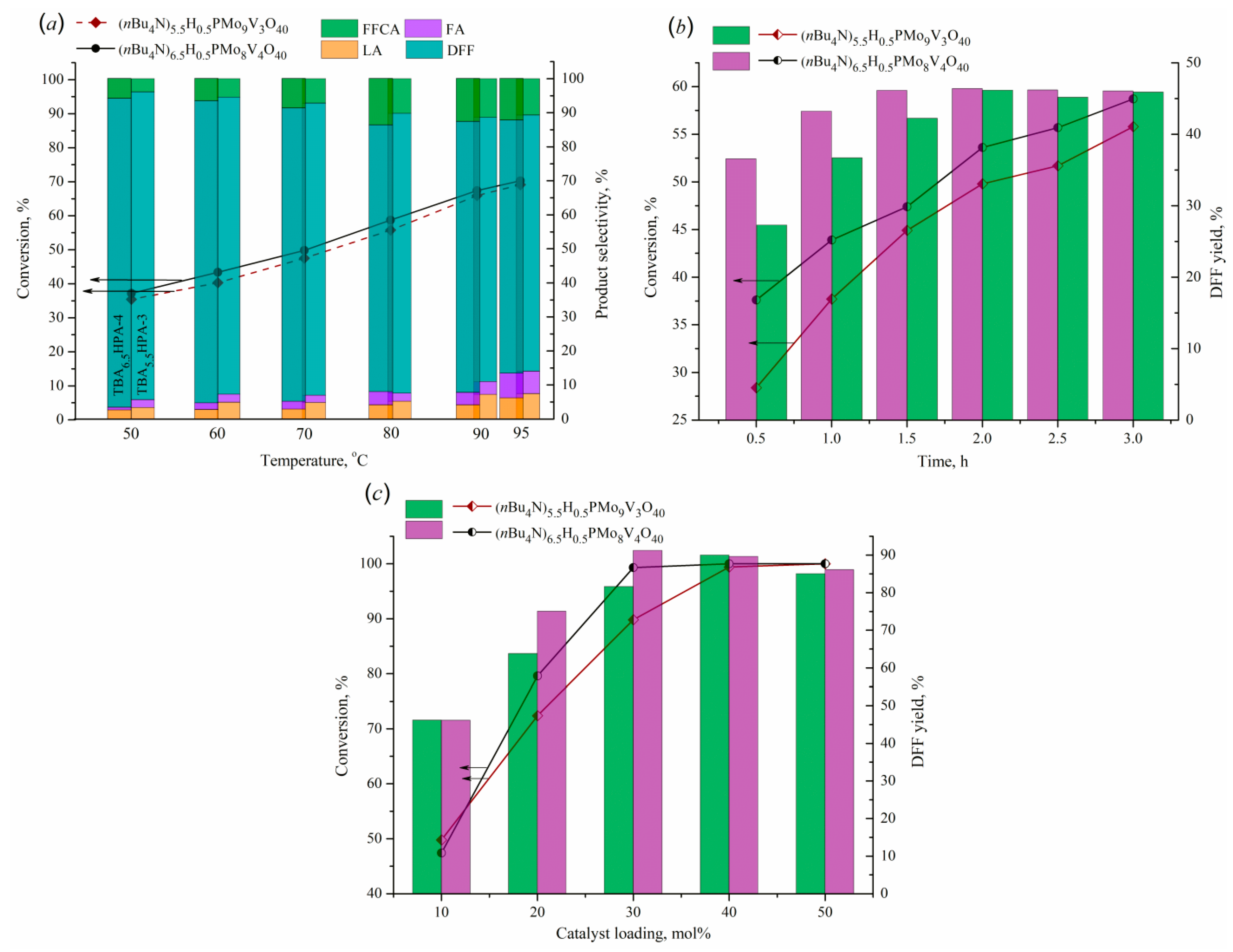
According to the data obtained, total substrate conversion and DFF selectivity of 91.2% are achieved in oxygen atmosphere (0.1 MPa) for 1.5 h at 80°C in the presence of TBA6.5HPA-4 catalyst at its loading of 30 mol %. The content of by-products is 2.6, 2.3 and 3.8% for LA, FA, and FFCA, respectively. Similar result in DFF yield of 90.9% can be obtained in the presence of TBA5.5HPA-3 catalyst, but this requires the application of a larger catalyst load (40 mol %) and an increase in reaction time to 2 h. The reached conversion and selectivity values are close to those for state-of-the-art solid catalysts studied in the oxidation of 5-HMF under similar conditions with water as a solvent [32].
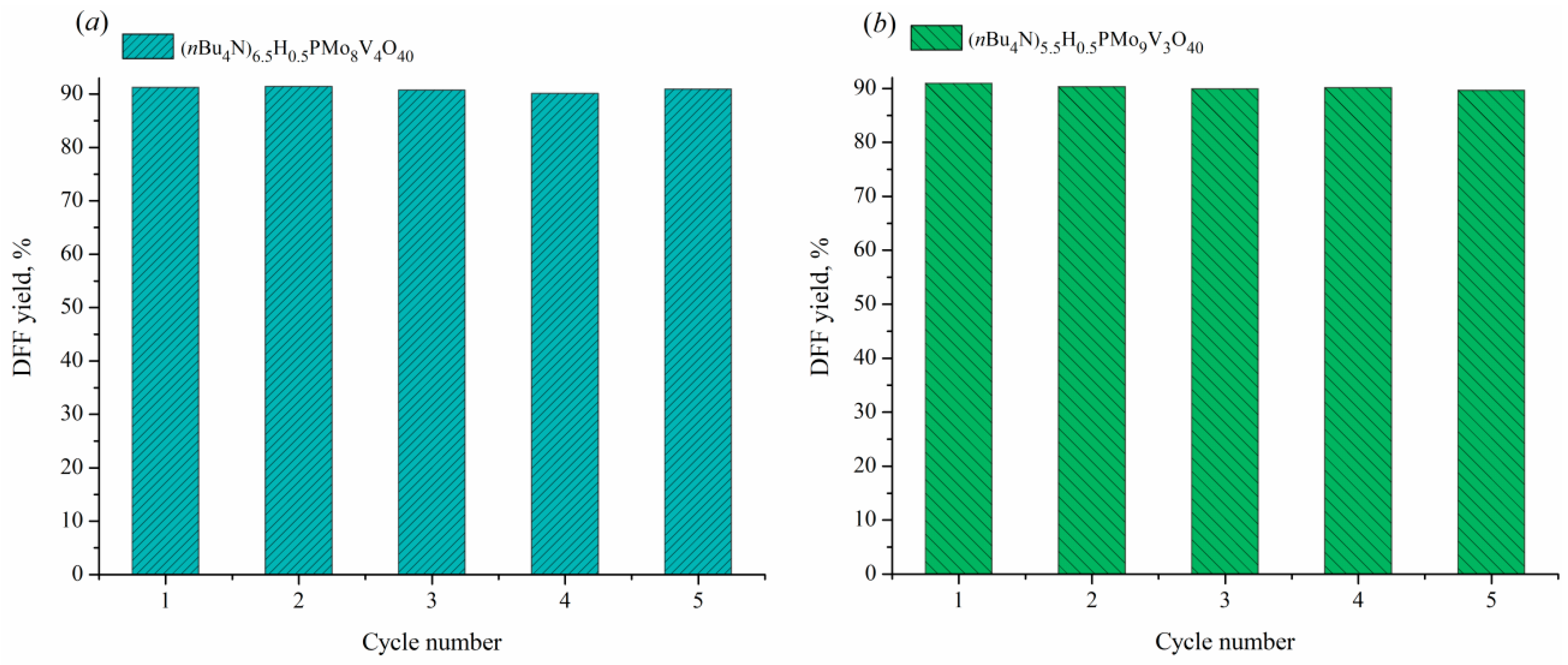
Investigation of catalyst reusability
The reusability of TBA2.5+xHPA-x (x = 3, 4) was investigated in 5 cycles of 5-HMF oxidation (Fig. (9)). After each reaction, the spent catalyst was separated by centrifugation and calcined at 150°C for 3 h in oxygen atmosphere. Both catalysts were found to demonstrate high stability and retention of catalytic activity during repeated application with the yield spread below 1.3%. The XRD patterns and IR spectra of the spent catalysts after 5 runs were identical to those of the samples after threefold hydrothermal treatment.
Figure 9.
The change of DFF yield during 5 runs in the presence of TBA6.5HPA-4 (a) and TBA5.5HPA-3 (b). Conditions: (a) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (30 mol%) in H2O (15 mL), 80°C, 1.5 h, 0.1 MPa O2; (b) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (40 mol%) in H2O (15 mL), 80°C, 2 h, 0.1 MPa O2.
Figure 9.
The change of DFF yield during 5 runs in the presence of TBA6.5HPA-4 (a) and TBA5.5HPA-3 (b). Conditions: (a) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (30 mol%) in H2O (15 mL), 80°C, 1.5 h, 0.1 MPa O2; (b) 5-HMF (0.39 mmol) in H2O (5 mL), catalyst (40 mol%) in H2O (15 mL), 80°C, 2 h, 0.1 MPa O2.
Conclusions
Obtaining solid HPAs-x with a high content of vanadium, stable under reaction conditions, is of significant practical interest for catalysis and organic synthesis. This work shows that the application of an environmental strategy based on H2O2-affected activation of vanadium(V) atoms in the form of V2O5 with their subsequent insertion into Mo-containing Keggin frameworks allows obtaining foreign impurity-free H3+xPMo12-xVxO40 solutions with x from 1 to 4. The following partial substitution of protons in these solutions with tetra-n-butylammonium cations of large ionic radius leads to heterogenization of HPAs-x with forming mixed-addenda water-insoluble (nBu4N)2.5+xH0.5PMo12-xVxO40 salts. According to the physicochemical characterization, this modification keeps untouched the Keggin anion and results in obtaining materials stable up to at least 370°C and possessing significant hydrolytic stability and low solubility in water, especially for salts with x of 3 and 4. The catalytic potential of the obtained salts with higher stability was demonstrated in the oxidation of biomass-based 5-HMF into DFF. The presence of vanadium(V) atoms in the Keggin framework has been shown to play a crucial role to proceed the oxidation process. The obtained (nBu4N)2.5+xH0.5PMo12-xVxO40 salts with x of 3 and 4 were found to exhibit high activity in the formation of DFF with selectivity of higher 90% at total substrate conversion. These heterogenized salts can be recycled and reused at preservation of catalytic activity for at least five cycles. This work demonstrates the potential prospects of solid nBu4N+ salts of HPAs-x as green, water-insoluble oxidative catalysts to prepare valuable chemicals from biomass-based resources.
Supplementary Materials
The following addition information are available, Figure S1: Comparison of powder XRD patterns (Cu-Kα) of initial (nBu4N)2.5+xH0.5PMo12-xVxO40 salts and corresponding samples after threefold hydrothermal treatment for x of 1 (a), 2 (b), 3 (c), and 4 (d); Figure S2: Comparison of IR-ATR spectra of initial (nBu4N)2.5+xH0.5PMo12-xVxO40 salts and corresponding samples after threefold hydrothermal treatment for x of 1 (a), 2 (b), 3 (c), and 4 (d) in fingerprint region of the Keggin anion.
Funding
This work was supported by the Russian Science Foundation, project 24-13-00406.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhizhina, E.G. and Odyakov, V.F., Appl. Catal. A: Gen., 2009, vol. 358, pp. 254–258. [CrossRef]
- Heravi, M.M. and Bamoharram, F.F., Heteropolyacids as Highly Efficient and Green Catalysts Applied in Organic Transformations; Elsevier, 2022, p. 61. [CrossRef]
- Singh, R., Shah, A.A., Potter, A., Clarkson, B., Creeth, A., Downs, C., and Walsh, F.C., J. Power Sources, 2012, vol. 201, p. 159–163. [CrossRef]
- Clauβnitzer, J., Bertleff, B., Korth, W., Albert, J., Wasserscheid, P., and Jess, A., Chem. Eng. Technol., 2020, vol. 43, no. 3, pp. 465–475. [CrossRef]
- Ma, S., Bao, W., Liu, B., Zhang, C., Wang, C., Liu, Y., Guo, H., Pan, Y., Sun, D., and Lu, Y., Appl. Surf. Sci., 2022, vol. 606, art. 154781. [CrossRef]
- Woźniak Budych, M.J., Staszak, K., Bajek, A., Pniewski, F., Jastrząb, R., Staszak, M., Tylkowski, B., and Wieszczycka, K., Coord. Chem. Rev., 2023, vol. 493, art. 215306. [CrossRef]
- Li, J., Xu, J., Li, X., Gao, W., Wang, L., Wu, L., Lee, M., and Li, W., Soft Matter., 2016, vol. 12, pp. 5572–5580. [CrossRef]
- Vilanculo, C.B., da Silva, M.J., Rodrigues, A.A., Ferreira, S.O., and da Silva, R.C., RSC Adv., 2021, vol. 11, pp. 24072–24085. [CrossRef]
- Tsigdinos, G.A. and Hallada, C.J., Inorg. Chem., 1968, vol. 7, no. 3, pp. 437–441. [CrossRef]
- Odyakov, V.F. and Matveev, K.I., SU Patent 1782934 A1, 1992.
- van Putten, R.-J., van der Waal, J.C., de Jong, E., Rasrendra, C.B., Heeres, H.J., and de Vries, J.G., Chem. Rev., 2013, vol. 113, no. 3, pp. 1499–1597. [CrossRef]
- Delidovich, I., Hausoul, P.J.C., Deng, L., Pfutzenreuter, R., Rose, M., and Palkovits, R., Chem. Rev., 2016, vol. 116, no. 3, pp. 1540–1599. [CrossRef]
- Amarasekara, A.S., Green, D., and Williams, L.D., Eur. Polym. J., 2009, vol. 45, no. 2, pp. 595–598. [CrossRef]
- Ma, J., Wang, M., Du, Z., Chen, C., Gao, J., and Xu, J., Polym. Chem., 2012, vol. 3, pp. 2346–2349. [CrossRef]
- Richter, D.T. and Lash, T.D., Tetrahedron Lett., 1999, vol. 40, no. 37, pp. 6735–6738. [CrossRef]
- del Poeta, M., Schell, W.A., Dykstra, C.C., Jones, S., Tidwell, R.R., Czarny, A., Bajic, M., Kumar, A., Boykin, D., and Perfect, J.R., Antimicrob. Agents Chemother., 1998, vol. 42, no. 10, pp. 2495–2502. [CrossRef]
- Hopkins, K.T., Wilson, W.D., Bender, B.C., McCurdy, D.R., Hall, J.E., Tidwell, R.R., Kumar, A., Bajic, M., and Boykin, D.W., J Med. Chem., 1998, vol. 41, no. 20, pp. 3872–3878. [CrossRef]
- Kozhevnikov I.V. Catalysts for fine chemical synthesis. Vol. 2. Catalysis by polyoxometalates, Chichester: John Wiley & Sons Ltd., 2002.
- Tian, J., Fang, C., Cheng, M., Wang, X., Chemical Engineering and Technology, 2011, vol. 34, no. 3, pp. 482–486. [CrossRef]
- Gromov, N.V., Medvedeva, T.B., Taran, O.P., Timofeeva, M.N., Parmon, V.N., Catalysis in Industry, 2021. vol. 13, no. 1, pp. 73–80. [CrossRef]
- Matveev, K.I., Odjakov, V.F., Zhizhina, E.G., Motornyj, Ju.A., and Lange, S.A., RU Patent 1669109 C, 1995.
- Pope, M.T., Heteropoly and Isopoly Oxometalates, Berlin: Springer-Verlag, 1983.
- Javidi, J., Esmaeilpour, M., Rahiminezhad, Z., and Dodeji, F.M., J. Clust. Sci., 2014, vol. 25, no. 6, p. 1511. [CrossRef]
- Rocchiccioli-Deltcheff, C. and Fournier, M., J. Chem. Soc. Faraday Trans., 1991, vol. 87, pp. 3913–3920. [CrossRef]
- Huang, T., Tian, N., Wu, Q., Yan, Y., and Yan, W., Mater. Chem. Phys., 2015, vol. 165, pp. 34–38. [CrossRef]
- Vakulenko, A., Dobrovolsky, Y., Leonova, L., Karelin, A., Kolesnikova, A., and Bukun, N., Solid State Ion., 2000, vol. 136, pp. 285–290. [CrossRef]
- Sutjahja, I.M., Wonorahardjo, S., and Wonorahardjo, S., Inorganics, 2020, vol. 8, no. 9, art. 51. [CrossRef]
- Boudjema, S., Rabah, H., and Choukchou-Braham, A., Acta. Phys. Pol. A, 2017, vol. 132, pp. 469–472. [CrossRef]
- Liu, Y., Zhu, L., Tang, J., Liu, M., Cheng, R., and Hu, C., ChemSusChem, 2014, vol. 7, no. 12, pp. 3541–3547. [CrossRef]
- Zhang, J., Tang, Y., Li, G., and Hu, C., Appl. Catal. A: Gen., 2005, vol. 278, no. 2, pp. 251–261. [CrossRef]
- Thommes, M., Kaneko, K., Neimark, A.V., Olivier, J.P., Rodriguez-Reinoso, F., Rouquerol, J., and Sing, K.S.W., Pure Appl. Chem., 2015, vol. 87, no. 9–10, pp. 1051–1069. [CrossRef]
- Ventura, M., Dibenedetto, A., and Aresta, M., Inorganica Chim. Acta, 2018, vol. 470, pp. 11–21. [CrossRef]
Figure 1.
Route of (nBu4N)2.5+xH0.5PMo12-xVxO40 synthesis.
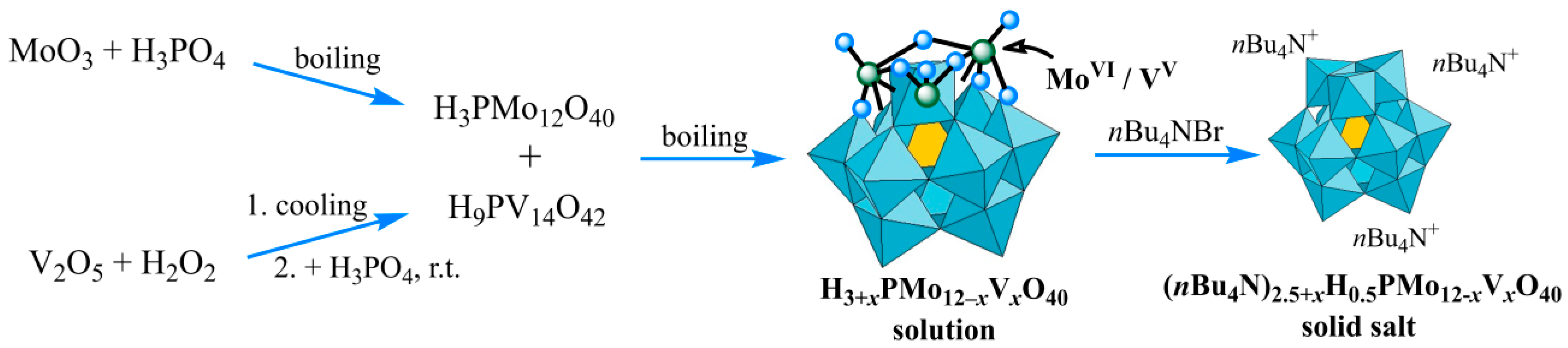
Figure 4.
Powder XRD patterns (Cu-Kα) of (a) H3+xPMo12-xVxO40 acids and (b) (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts with x of 1–4.
Figure 4.
Powder XRD patterns (Cu-Kα) of (a) H3+xPMo12-xVxO40 acids and (b) (nBu4N)2.5+xH0.5PMo12-xVxO40 acidic salts with x of 1–4.
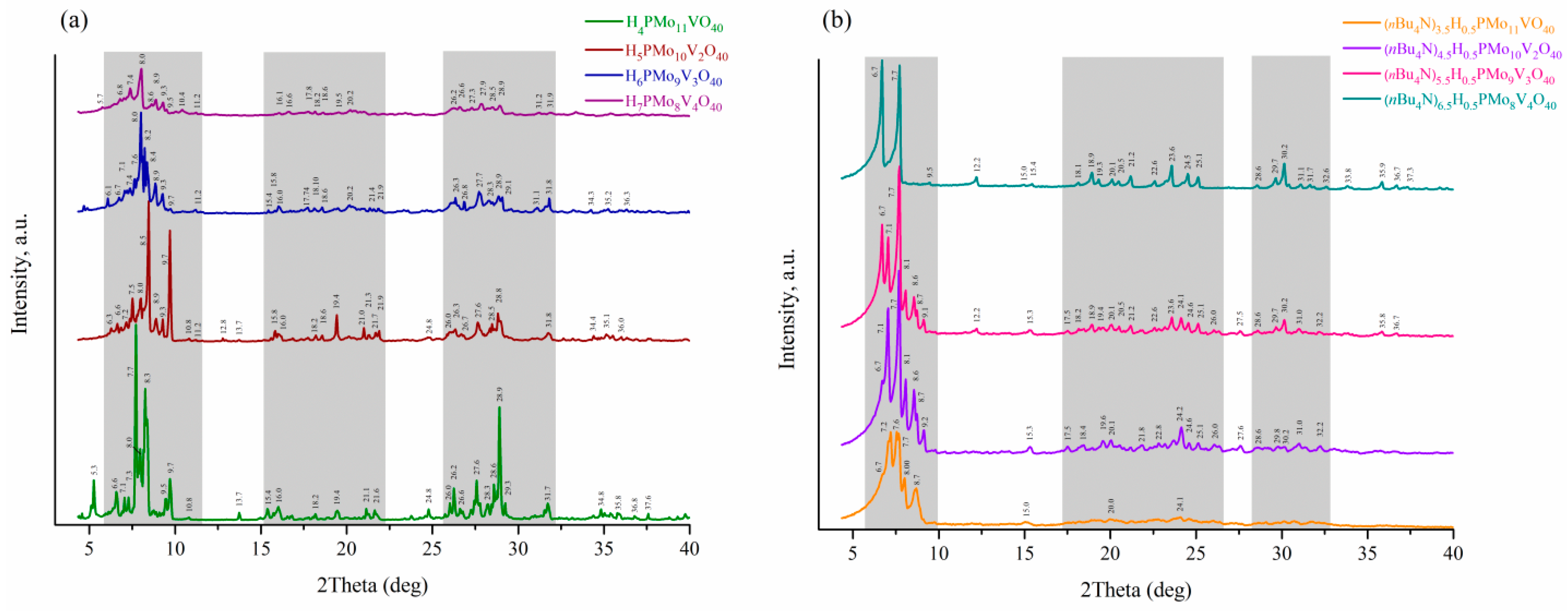
Table 2.
Textural characteristics of prepared samples.
| Sample | SBETa | Pore volumeb | Pore diameterc |
| [m2 g–1] | [cm3 g–1] | [Å] | |
| HPA-1 | 12.45 | 0.018 | 57.5 |
| HPA-2 | 11.63 | 0.016 | 56.3 |
| HPA-3 | 10.92 | 0.017 | 61.8 |
| HPA-4 | 8.40 | 0.023 | 110.5 |
| TBA3.5HPA-1 | 7.13 | 0.024 | 134.1 |
| TBA4.5HPA-2 | 2.37 | 0.007 | 125.8 |
| TBA5.5HPA-3 | 3.74 | 0.009 | 94.8 |
| TBA6.5HPA-4 | 4.60 | 0.012 | 101.4 |
aBET surface area. bSingle point total pore volume. cAverage pore diameter by the BET method.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



