Submitted:
02 December 2024
Posted:
03 December 2024
You are already at the latest version
Abstract
A large number of novel viruses potentially pathogenic for humans are discovered currently. Studying many of them by classical methods of virology is difficult due to the absence of a live viral particles or a sufficient amount of its genetic material. In this case, the use of modern methods of bioinformatics, synthetic and structural biology can help. Haseki tick virus (HSTV) is a recently discovered tick-borne unclassified ssRNA(+) virus. HSTV positive patients experienced fever and elevated temperature. However, at the moment, there is no information on the tertiary structure and functions of its proteins. In this work, we used AlphaFold 3 for annotation of HSTV protein functions, based on the principle that the tertiary structure of a protein is inextricably linked with its molecular function. We were the first to obtain models of tertiary structures and describe the putative functions of HSTV nonstructural proteins (NS3 helicase, NS3 protease, NS5 RNA-dependent RNA-polymerase, and NS5 methyltransferase). These proteins play a key role in the replication of the viral genome and can be targets for the development of direct-acting antiviral drugs.
Keywords:
Ixodid ticks
; tick-borne infection
; RNA viruses
; Flaviviridae
; orthoflavi-like viruses
; viral proteins
; protein structure
; AlphaFold 3
1. Introduction
Preventive measures against viral threats are ones of the most important issues of biological security. The high virulence and variability of many dangerous human viruses as well as their ubiquitous distribution make investigation of novel viruses an important stage in protection from infectious threats.
According to the International Committee on Taxonomy of Viruses (ICTV) report, 11 273 viral species, of which more than 3 000 are pathogenic to humans, have been identified by 2022 [1]. However, according to virologists’ suggestions, the number of virus species not studied by humans may exceed several millions, many of which may be pathogenic to humans. There are entire eras in human history when viral pandemics and epidemics led to mass infections and many deaths, as was the case with the COVID-19 pandemic [2]. According to annual WHO reports, viral infections account currently for 60 to 75% of infectious diseases. Furthermore, viruses exhibit sufficiently high genetic variability to overcome interspecies barriers or accumulate mutations that significantly increase their virulence. Therefore, investigation of novel, previously unidentified viruses is a key task of preventive measures against viral threats.
Many newly discovered viruses cannot be studied using classical virology techniques due to the laborious culture of novel viruses or the lack of a sufficient amount of their genetic material. In this case, an understanding of the action mechanisms of novel, potentially pathogenic human viruses and investigation of their biochemical composition, viral protein structures, and pathways of genetic information implementation in the cell may help in the development of effective therapeutic and prophylactic biologics. In this case, modern synthetic and structural biology techniques enable artificial production of recombinant proteins identical to natural viral ones and investigation of their structure and properties. However, X-ray structural analysis cannot be often used for proteins of novel viruses due to difficulties in producing protein crystals of sufficient size and requires additional approaches to phase restoration. A promising technique to study the structure and functions of novel unique viral proteins is a recently announced AlphaFold 3 neural network [3].
Haseki tick virus (HSTV) was first discovered in a retrospective study of blood sera from tick-bitten patients and Ixodes persulcatus ticks from various regions of the Russian Federation in 2019. There are reports of identification of viruses closely related to HSTV in Georgia and Poland [4]. The HSTV genome is represented by a single-stranded positive-sense RNA of approximately 16 000 nucleotides in size that encodes an approximately 5 100 amino acid polyprotein flanked by 5’- and 3’-untranslated regions. On the basis of the genome structure, this virus was initially classified as an unclassified orthoflavi-like virus of the Flaviviridae family. Many members of this family cause serious diseases in humans. HSTV-infected patients experienced fever and elevated temperature [5]. However, the ability of HSTV to cause symptomatic infection in humans requires further investigation. HSTV is also closely related to other recently identified unclassified pestiviruses: Bole tick virus 4 (China, Thailand, Slovakia, Romania, and Kenya), Trinbago virus (China, Trinidad and Tobago), and Dermacentor reticulatus pestivirus-like virus 1 (Croatia, Georgia, and Poland), which have been detected in Dermacentor reticulatus, Rhipicephalus sanguineus, Rhipicephalus turanicus, Hyalomma punctata, Hyalomma truncatum, Hyalomma rufipes, and Hyalomma dromedarii ticks [4,6,7,8,9,10]. In addition, Bole tick virus 4 was detected in skin and serum samples from tick-bitten patients presenting with both mild and severe symptoms [11]. Despite the ubiquitous distribution of HSTV-related viruses, information on the structure and function of their viral proteins is currently lacking. This poses a serious challenge to understanding the mechanisms and evolution of HSTV viral infection and precludes taxonomic identification of HSTV and establishment of continuous monitoring.
In this study, we used the AlphaFold 3 neural network and other bioinformatics methods to identify key HSTV nonstructural proteins in the polyprotein, which lacked annotated sequence homologs.
2. Results
2.1. Determination of Localization of Haseki Tick Virus Nonstructural Proteins
Nonstructural (NS) proteins of Flaviviridae family members interact with several host factors to form a membrane-bound replication complex where viral RNA is synthesized. Some NS proteins are transmembrane and anchored to the membrane of the endoplasmic reticulum. Other NS proteins are localized to the membrane via membrane-associated regions or viral protein cofactors [12]. We used a structural approach to search for NS proteins in the C-terminal region of the HSTV polyprotein (1 272–5 104 a.a.), comparing NS protein structures of Orthoflavivirus genus (NS2A, NS2B, NS3, NS4A, NS4B, NS5), Hepacivirus genus (NS2, NS3, NS4A, NS4B, NS5A, NS5B), and Pestivirus genus (NS2, NS3, NS4A, NS4B, NS5A, NS5B) viruses available in PDB.
2.1.1. Search for Putative HSTV Nonstructural Transmembrane Proteins
In the nonstructural region of the HSTV polyprotein, two transmembrane proteins (NSTR1, NSTR2) were found at positions 1 272–1 601 and 2 833–2 855, which may be associated with NS2A/NS2B and NS4A/NS4B proteins of Flaviviridae family members (Figure 1, Table S1). HSTV NSTR1 consists of four transmembrane domains of 21 a.a. (NSTR1-D1), 17 a.a. (NSTR1-D2), 24 a.a. (NSTR1-D3), and 24 a.a. (NSTR1-D4) in length and one 127 a.a. cytoplasmic domain (~14 kDa). Between NSTR1-D2 and NSTR1-D3, there is a 24 a.a. membrane-localized domain. In Flaviviridae family members, the NS2A protein is reported to consist of five transmembrane domains and be about 220 a.a. in length, in contrast to the four transmembrane domains in the HSTV polyprotein. The putative HSTV NSTR1 protein is preceded by a 17 a.a. membrane-bound region, the role of which we did not elucidate. A complex between NSTR1-D4 and the NSTR1 cytoplasmic domain may be a prototype of the NS2B protein that is a cofactor of the NS3 serine protease of Orthoflavivirus genus members. HSTV NSTR2 consists of four transmembrane domains of 23 a.a. (NSTR2-D1), 26 a.a. (NSTR2-D2), 18 a.a. (NSTR2-D3), and 20 a.a. (NSTR2-D4) in length, which are located one after another. In Flaviviridae family members, three transmembrane domains in the NS4A protein and four transmembrane domains in the NS4B protein have been reported, which is inconsistent with the data on four transmembrane domains in the HSTV sequence. We were unable to predict the tertiary structures of HSTV NSTR1 and HSTV NSTR2 due to the lack of structural data on NS2A and NS4A/NS4B proteins of Flaviviridae family members in PDB. In addition, at the HSTV polyprotein C-terminus (4 910–5 104 a.a.), there are four 20 a.a., 23 a.a., 34 a.a., and 20 a.a. transmembrane domains whose function is unknown (Figure 1, Table S1).
2.1.2. Search for putative HSTV nonstructural cytoplasmic proteins
- NS3 protein
All four Flaviviridae genera (Orthoflavivirus, Hepacivirus, Pegivirus, and Pestivirus) have the ~70 kDa NS3 protein in their genome structure. NS3 consists of two domains: the N-terminal domain of serine protease, which is a key protease in post-translational cleavage of the viral polyprotein, and the C-terminal domain of helicase, which plays an important role in unwinding of viral RNA duplexes during replication [13].
Putative NS3 was found in the HSTV polyprotein at positions 1 598–2 291 (Figure 1). The size of HSTV NS3 was 694 a.a. (~77 kDa). Putative HSTV NS3 consists of HSTV serine NS3 protease (NS3pro) of 204 a.a. in length (~22 kDa), located in the polyprotein at positions 1 598–1 801, and HSTV NS3 helicase (NS3hel) of 490 a.a. in length (~55 kDa), located in the polyprotein at positions 1 802–2 291. The sizes of HSTV NS3pro and HSTV NS3hel are consistent with those of similar viral proteins of Flaviviridae family members. The HSTV NS3pro amino acid sequence is most similar to that of Dengue virus NS3pro (UniProtKB accession number: Q91H74) and Zika virus NS3pro (UniProtKB accession number: H8XX12), but their amino acid sequence identity is 23% and 21%, respectively. Identity of the HSTV NS3pro amino acid sequence with that of other Flaviviridae family members is not more than 13% for Hepacivirus and 8% for Pestivirus. The HSTV NS3hel amino acid sequence is most similar to that of classical swine fever virus NS3hel (UniProtKB accession number: Q5U8X5), but their amino acid sequence identity is 28%. Homology between HSTV NS3hel and those from Ortoflavivirus (UniProtKB accession number: P14336) and Hepacivirus (UniProtKB accession number: Q9WMX2) is not more than 21%.
- Methyltransferase and RNA-dependent RNA-polymerase
Ortoflavivirus genus members possess a ~103 kDa NS5 protein consisting of two functional domains with methyltransferase (NS5MTase) and RNA-dependent RNA polymerase (NS5RdRp) activities, whereas Pegivirus, Pestivirus, and Hepacivirus lack NS5MTase, but have the phosphoprotein NS5A and NS5B that functions as RdRp [14]. NS5MTase is responsible for the formation of the RNA cap attached to the 5’-end of the viral RNA. NS5 MTase transfers a methyl group from an S-adenosyl-L-methionine donor to N-7-guanine and ribose 2’-OH of the first RNA nucleotide to form an S-adenosyl-L-homocysteine by-product. NS5A is a multifunctional phosphoprotein consisting of three domains (DI, DII, and DIII). The DI and DII domains are involved in genome replication, whereas DIII plays a role in virus assembly [15]. NS5RdRp synthesizes viral RNA [12].
In the HSTV polyprotein, putative HSTV NS5MTase was found at positions 3 445–3 676 a.a., and putative HSTV NS5RdRp was found at positions 3 999–4 709 a.a. (Figure 1). The NS5A phosphoprotein was not detected in HSTV. The size of HSTV NS5MTase and HSTV NS5RdRp was 231 a.a. (~26 kDa) and 711 a.a. (~82 kDa), respectively. Between HSTV NS5MTase and HSTV NS5RdRp, there is a 323 a.a. (~36 kDa) protein domain with unknown properties (NS5-X). This protein domain remains folded only when complexed with HSTV NS5MTase and HSTV NS5RdRp (Figure 1). The detected putative HSTV NS5MTase has no amino acid sequence homologs. The level of identity of HSTV NS5MTase with NS5MTase of Ortoflavivirus members is ~5%. The HSTV NS5RdRp amino acid sequence is most similar to that of bovine viral diarrhea virus (UniProtKB accession number: Q96662) and classical swine fever virus (UniProtKB accession number: Q5U8X5), but their amino acid sequence identity is only 23% and 22%, respectively. Homology between HSTV NS5RdRp and that of Ortoflavivirus (UniProtKB accession number: P14336) and Hepacivirus (UniProtKB accession number: Q9WMX2) members is not more than 17%.
2.2. Tertiary Structure Models of Haseki Tick Virus Nonstructural Proteins
2.2.1. HSTV NS3 Protein
We generated a spatial structure model of putative HSTV NS3 with pLDDT = 73.59 (Figure S1). Pairwise alignment of the full-length HSTV NS3 with the NS3 crystal structures of Flaviviridae family members revealed a low level of structural similarity (Figure 2). The highest TM-score = 0.62 was found for Dengue virus NS3 (PDB ID: 2VBC) (Table S3). HSTV NS3pro and HSTV NS3hel are interconnected by a 13 a.a. flexible linker (Figure S2). This leads to instability of the spatial arrangement of HSTV NS3pro relative to HSTV NS3hel and, thus, to a low TM-score. Therefore, we further analyzed the spatial structures of HSTV NS3pro and HSTV NS3hel separately.
- HSTV NS3 protease
We could generate an HSTV NS3pro model with a high model confidence level (pLDDT = 70.40) (Figure S3). These data indicate that the HSTV NS3pro model may be used for functional analysis. The structure of NS3 protease was found to display the highest level of topological similarity to that of analogous proteins of Orthoflavivirus members, despite the fact that their amino acid sequence identity was less than 30% (Table 1, Table S4). The TM-score of HSTV NS3pro with DENV NS3pro and ZIKV NS3pro ranged from 0.70 to 0.79 and from 0.69 to 0.75, respectively, whereas the TM-score of HSTV NS3pro with HCV NS3pro varied from 0.60 to 0.69 (Figure S4).
The tertiary structure of HSTV NS3pro consists of two spatial domains (D1 and D2) composed of β-barrels. HSTV NS3pro D1 consists of seven antiparallel β-sheets (β1, β2, β3, β4, β5, β5, β7), and HSTV NS3pro D2 consists of eight antiparallel β-sheets (β1’, β2’, β3’, β4’, β5’, β6’, β7’, β8’). A small 5 a.a. α-helix is present between β3 and β4 in HSTV NS3pro D1, which is completely consistent with the tertiary structures of NS3pro of Flaviviridae family members from PDB. HSTV NS3pro D1 and D2 are connected by a 12 a.a flexible linker (Figures S5–S7). There are additional 9 and 13 a.a. insertions in HSTV NS3pro at amino acid positions 37–46 and 110–123, respectively. These insertions increase sizes of β4, β5, β10, and β11 sheets in HSTV NS3pro and partially form unstructured free loops.
We identified the active site of HSTV NS3pro (Figure 3a) similar to that of NS3pro in Orthoflavivirus genus members. The NS3pro active site in Orthoflavivirus genus members is formed by a catalytic His51–Asp75–Ser135(Gly133) triad, where these amino acids act as a nucleophile, base, and acid, respectively, upon peptide bond cleavage. Gly133 is not directly involved in the catalytic site, but, together with Ser135, forms a structural fold that acts as an anion pocket during nucleophilic attack. Topologically, HSTV NS3pro active site completely coincides with that of NS3pro in Orthoflavivirus genus members and is formed by a catalytic His55–Asp88–Ser163(Gly161) triad. Differences in the coordinates are caused by additional amino acid insertions in the HSTV NS3pro structure. The HSTV NS3pro active site is negatively charged and forms a charged pocket surrounded by hydrophobic regions (Figure 3b,c). In Orthoflaviviruses, NS3pro functions in a complex with NS2B that acts as a protease cofactor. In the complex, NS2B usually stabilizes the NS3pro active site and acts as an electron donor in peptide bond cleavage [16]. Simulation of the tertiary structure of a complex between HSTV NS3pro and the HSTV NSTR1 cytoplasmic domain revealed similarity to the topology of a NS2B–NS3pro complex in Orthoflavivirus genus members (Figure 3d). Upon simulation of the complex, the HSTV NS3pro cytoplasmic domain folds to a structure composed of three β-sheets interacting with the HSTV NS3pro surface (Figure S8).
- HSTV NS3 helicase
A tertiary structure model of HSTV NS3hel with pLDDT = 75.80 was generated using AlphaFold 3 (Figure S9). These data indicate that the HSTV NS3hel model may be used for functional analysis. HSTV NS3hel was found to have the highest topological similarity to Hepacivirus hominis NS3hel (TM-score: 0.66), despite low amino acid sequence identity (less than 30%). The TM-score of HSTV NS3hel with Dengue virus NS3hel and Zika virus NS3hel ranged from 0.60 to 0.63 and from 0.61 to 0.63, respectively, whereas the TM-score of HSTV NS3hel with classical swine fever virus NS3hel was 0.63 (Table 1, Table S5) (Figure S10).
The HSTV NS3hel structure has the shape of a flattened pyramid that can be divided into three domains: N-terminal domains 1 and 2 (D1, residues 1 802–1 962; D2, residues 1 963–2 131) and C-terminal domain 3 (D3, residues 2 132–2 291) (Figure S11). HSTV NS3hel D1 and D2 are tandem, conserved RecA-like domains with an α/β fold. HSTV NS3hel D1 consists of six parallel β-sheets (β1, β2, β3, β5, β6, β7) and one antiparallel β-sheet (β4), which are sandwiched by seven α-helices (α1–α7). Whereas HSTV NS3hel D2 contains six parallel β-sheets (β1’, β4’, β5’, β6’, β7’, β9’) and two antiparallel β-sheets (β2’ and β3’) sandwiched by four α-helices (α1’–α4’) (Figures S12–S14). In addition, a β-hairpin consisting of a pair of antiparallel β-sheets (β8A’–β8B’) protrudes from the HSTV NS3hel D2 domain and interacts with the HSTV NS3hel D3 domain. In particular, the β-hairpin is packed opposite a hydrophobic region formed by α1’’, α2’’, and the N-terminus of α5’’ of the HSTV NS3hel D3 domain (Figure 5a). The structure of the HSTV NS3hel D3 domain is very limitedly similar to that of NS3hel D3 in Flaviviridae family members, which is consistent with high variability of D3 in viral helicases (Table 1, Table S5). At polyprotein positions 1 869–1 887, HSTV NS3hel has an 18 a.a. insertion that forms a β3-sheet.
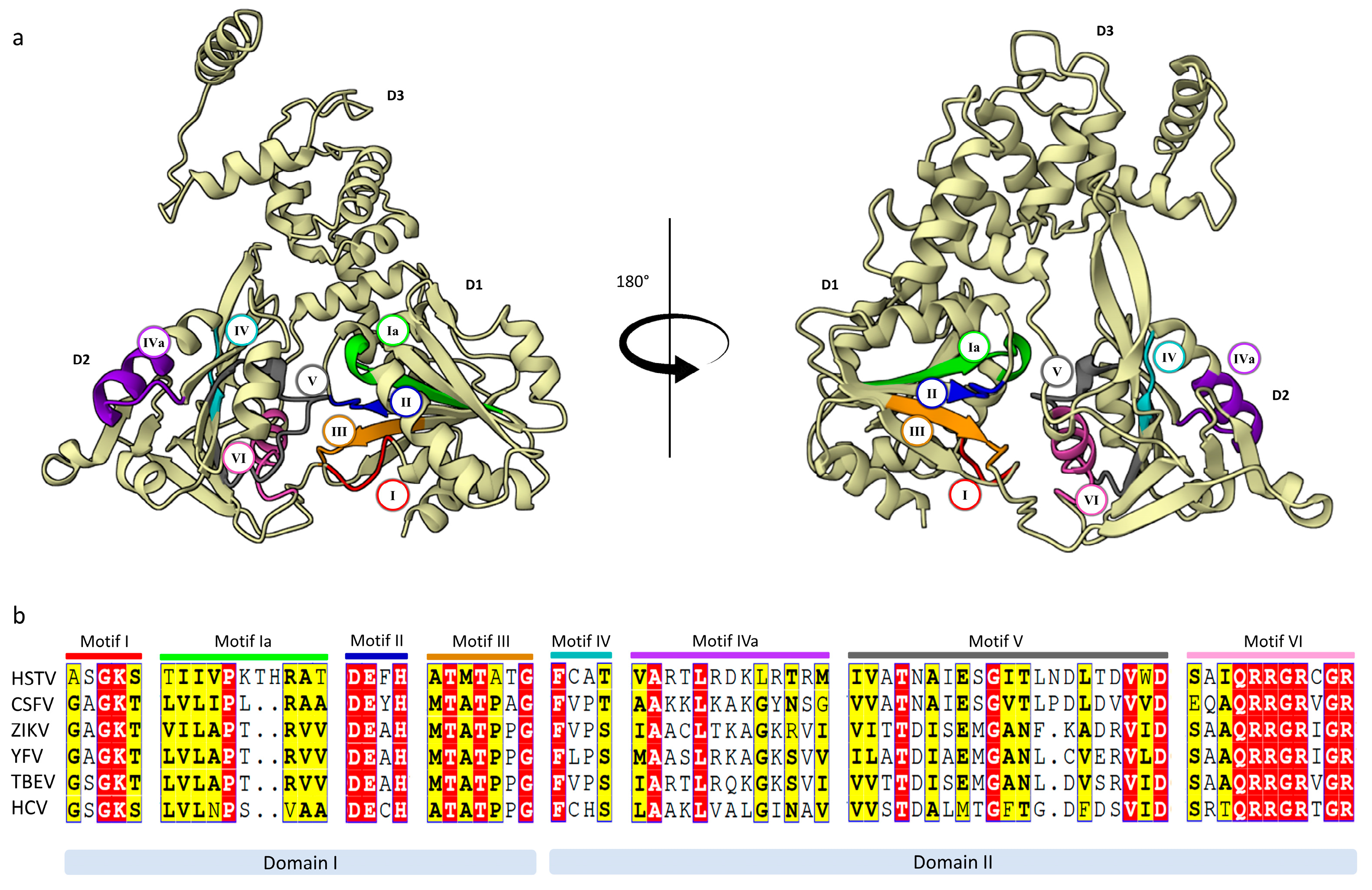
All motifs (I or Walker A, Ia, II or Walker B, III, IV, IVa, V, VI) characteristic of superfamily 2 helicases were found in HSTV NS3hel (Figure 4, S15) [13]. These motifs are located in a cleft between HSTV NS3hel D1 and D2 domains. The Walker A motif of HSTV NS3hel consists of a residue triad Gly1827, Lys1828, and Ser2079 and, together with the Walker B motif of HSTV NS3hel, consisting of residues Asp1930, Glu1931, and His1933, is responsible for binding to nucleotide triphosphatase (NTPase) and coordination of Mg2+ [17]. Motif III of HSTV NS3hel is composed of residues Thr1958 and Thr1960 and, together with Gln2108 of HSTV NS3hel motif VI, forms an exit channel for inorganic phosphate produced in hydrolysis [18]. Of critical importance for the NTPase activity is motif VI that is formed in HSTV NS3hel by residues Arg2109, Arg2110, Arg2112, and Arg2115 [13].
Figure 4.
Functional regions of HSTV NS3hel. (a) The overall tertiary structure of HSTV NS3hel with conservative motifs: motif I (red), motif Ia (green), motif II (blue), motif III (orange), motif IV (cyan), motif IVa (purple), motif V (grey), motif VI (pink); (b) Sequence alignment of the conservative motifs: red boxes - 100% aligned a.a. residues; yellow boxes - 80% aligned a.a. residues. Abbreviations: CSFV – classical swine fever virus; ZIKV – Zika virus; YFV – yellow fever virus; TBEV – tick-borne encephalitis virus; HCV – hepatitis C virus.
Figure 4.
Functional regions of HSTV NS3hel. (a) The overall tertiary structure of HSTV NS3hel with conservative motifs: motif I (red), motif Ia (green), motif II (blue), motif III (orange), motif IV (cyan), motif IVa (purple), motif V (grey), motif VI (pink); (b) Sequence alignment of the conservative motifs: red boxes - 100% aligned a.a. residues; yellow boxes - 80% aligned a.a. residues. Abbreviations: CSFV – classical swine fever virus; ZIKV – Zika virus; YFV – yellow fever virus; TBEV – tick-borne encephalitis virus; HCV – hepatitis C virus.
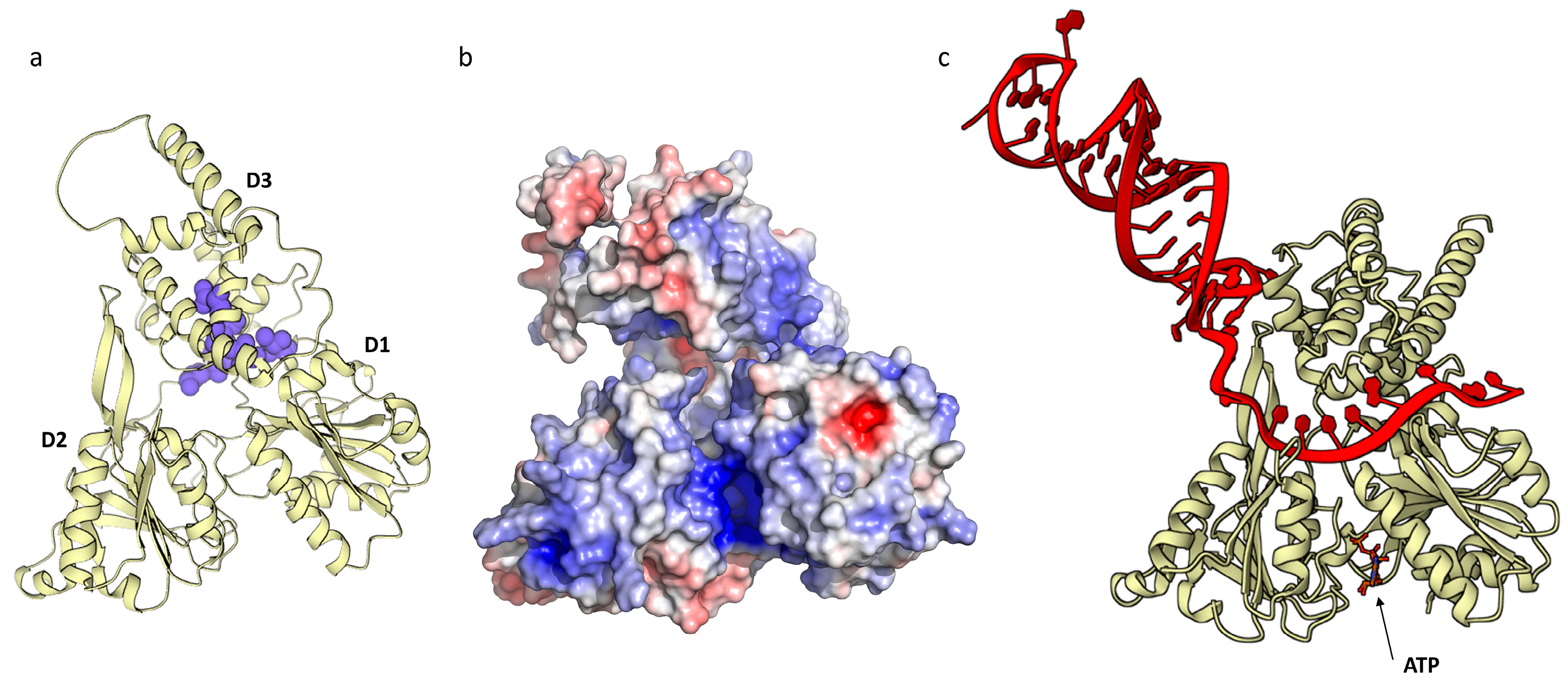
In the model of the HSTV NS3hel structure, a positively charged tunnel is clearly identified along the boundary of HSTV NS3hel D3, which directly interacts with HSTV NS3hel D1 and D2 (Figure 5b). The tunnel is lined with positively charged amino acids and is wide enough to accommodate a single-stranded nucleic acid passing through HSTV NS3hel D2 to D1. The positively-charged residues, most of which belong to HSTV NS3hel D1 and D2, presumably stabilize the nucleic acid sugar–phosphate backbone [13]. To assess the interaction of HSTV NS3hel with RNA and ATP, we built a model of a HSTV NS3hel–ATP–50-nucleotide random RNA complex in AlphaFold 3 (Figure 5c). The model shows that the RNA is indeed coordinated in the positively charged tunnel along the HSTV NS3hel D3 boundary.
Figure 5.
NS3 helicase HSTV tertiary structure: (a) hydrophobic clusters (blue) of HSTV NS3hel; (b) electrostatic surface potential of HSTV NS3hel; (c) tertiary structure of HSTV NS3hel (ivory) with RNA (red) and ATP. The positive surface potential is colored blue, the negative surface potential is colored red.
Figure 5.
NS3 helicase HSTV tertiary structure: (a) hydrophobic clusters (blue) of HSTV NS3hel; (b) electrostatic surface potential of HSTV NS3hel; (c) tertiary structure of HSTV NS3hel (ivory) with RNA (red) and ATP. The positive surface potential is colored blue, the negative surface potential is colored red.
2.2.2. HSTV NS5 RNA-Dependent RNA-Polymerase
A tertiary structure model of HSTV NS5RdRp had a high pLDDT = 87.41, despite the lack of amino acid sequence homologs (Figure S16). The highest TM-score = 0.72 was found for HSTV NS5RdRp with Dengue virus NS5RdRp (PDB ID: 7XD8) and Zika virus NS5RdRp (PDB ID: 5U0C). The structural similarity of HSTV NS5RdRp to NS5RdRp of Orthoflavivirus genus members and NS5RdRp of Pestivirus genus members was 0.66 to 0.72 and 0.66, respectively, whereas the TM-score for HSTV NS5RdRp with NS5RdRp of Hepacivirus hominis species was 0.63 (Table 2, Table S6) (Figure S17).
The spatial structure model of HSTV NS5RdRp adopts a right-hand shape with palm, finger, and thumb domains surrounding the active site, which is similar to that in all viral RdRps (Figure S18). The HSTV NS5RdRp finger domain consists of two regions: the first region is located in helices α1–8 and sheets β1–5 (residues 3 999–4 214), and the second region is located in helices α11–12 and sheets β7–8 (residues 4 273–4 316). The HSTV NS5RdRp palm domain is spread throughout HSTV NS5RdRp. The palm domain forms the RdRp core and is composed of helices α9–10 (residues 4 215–4 272), α15–16 (residues 4 317–4 453), and α13–14 as well as beta sheets β9–15 (Figures S19–S21). The HSTV NS5RdRp thumb domain is located at the C-terminal side of RdRp, consists of helices α15–27 (residues 4 454–4 709), and, together with the HSTV NS5RdRp palm domain, forms a dsRNA interaction channel (Figure S22).
In the HSTV NS5RdRp palm domain, we identified all the motifs characteristic of viral RdRps, which were located in positions of the spatial structure similar to those in other Flaviviridae family members (Figure S23). Given their spatial and amino acid similarity, we suggest that the identified HSTV NS5RdRp motifs perform functions similar to those in other Flaviviridae RdRps. HSTV NS5RdRp motifs D, E, and G are highly variable, but their spatial arrangement around the polymerase active site remains conserved. HSTV NS5RdRp motifs A–E are located within the most conserved palm domain, and HSTV NS5RdRp motifs F and G are located in the finger domain (Figure 6). HSTV NS5RdRp motifs A and C contain conserved aspartic acid residues (Asp4260, Asp4265, Asp4363 and Asp4364) that play a key role in the catalytic center activity, coordinating metal ions [19]. The HSTV NS5RdRp motif B includes conserved Ser4320 and Gly4321. Ser4320 is specific for RdRPs and forms hydrogen bonds with the 2’-hydroxyl group of ribose and Asp4265 of the HSTV NS5RdRp motif A. The HSTV NS5RdRp motif D starts after α14, mainly consists of unstructured loops, and forms an antiparallel β-structure with the HSTV NS5RdRp motif A. The HSTV NS5RdRp motif E interacts with the HSTV NS5RdRp motif C, and they both stabilize the de novo synthesized RNA product [20]. In the Flaviviridae family, the HSTV NS5RdRp motif E consists of a β-hairpin (β14–β15) located between the HSTV NS5RdRp palm and thumb domains [21,22]. Whereas, the HSTV NS5RdRp motif E contains a 20 a.a. insertion that forms a free loop. In addition, HSTV NS5RdRp exhibits two additional α-helices (α21 and α22) extending from a putative priming loop that is important for the enzymatic activity of RdRp because flavivirus HSTV NS5RdRps belong to primer-independent polymerases [23].
2.2.3. HSTV NS5 Methyltransferase
We modeled the structure of a HSTV polyprotein portion between putative HSTV NSTR2 and HSTV NS5RdRp (3 445–4 000 a.a.) to search for either the NS5A protein, which is present in Pestivirus, Hepacivirus, and Pegivirus members, or the NS5 methyltransferase domain, which is present in Ortoflavivirus. The generated model had a low pLDDT = 36.76 and a TM-score of < 0.30 (respective to known Flaviviridae NS5A structures) (). The protein model contained a highly structured and highly reliable N-terminal domain with a pLDDT = 71.10 and an extremely low pLDDT = 27.33 for an unstructured C-terminal domain. The HSTV N-terminal domain (Figure S25) had the highest structural similarity (TM-score = 0.77) to methyltransferase from Pyrococcus horikoshii (PDB ID: 1WY7). There was also similarity to methyltransferases of Ortoflavivirus members for which the TM-score ranged from 0.60 to 0.49. These data indicate that the modeled protein is HSTV methyltransferase (HSTV NS5MTase), despite the unusually distant location from NS5RdRp. Interestingly, the HSTV NS5MTase structure is highly identical to that in a recently discovered Alongshan virus (PDB ID: 8GY4) (Table 2, Table S7) (Figure 7a-b, Figure S26).
The tertiary structure of HSTV NS5MTase folds into a classical α/β/α structure where the central β-layer is surrounded by α-helices. The central β-layer consists of seven β-sheets (β1–β7) and contacts eight α-helices (α1–α8). The secondary structure of HSTV NS5MTase is as follows: α1–α2–α3–β1–α4–β2–α5–β3–α6–β4–α7–β5–α–β6–β7 (Figure 7c-d, Figure S27). Structural simulation of a complex of HSTV NS5MTase, HSTV NS5RdRp, and a protein between them (HSTV NS5-X) yields structured HSTV NS5-X (Figure 8a, Figures S28–S31). The tertiary structure of HSTV NS5-X exhibits eight α-helices and eleven β-sheets. Electrostatic potential analysis of the HSTV NS5MTase–NS5-X–NS5RdRp complex reveals that HSTV NS5-X interacts with HSTV NS5MTase to form a positively charged pocket (Figure 8b). Probably, HSTV NS5-X promotes stabilization of the de novo synthesized HSTV RNA and plays an auxiliary role in HSTV RNA capping.
3. Discussion
Hidden Markov model profiling is a common procedure for functional annotation of novel viral genes in metagenomes [24]. However, this procedure is not applicable to genomes at less than 30% amino acid identity compared with that of annotated genomes. Thus, numerous protein sequences remain functionally unannotated and unclassified. The introduction of AlphaFold 2 in 2020 and then AlphaFold 3 in May 2024 marked the birth of a new era of protein folds and virome functions due to application of deep learning and artificial intelligence algorithms in viral protein structure prediction [3]. Both multilayer neural networks use the primary and tertiary structures of proteins as input to model unknown structures. However, AlphaFold 3, unlike AlphaFold 2, is able to predict the 3D structure of biomolecular complexes of proteins, nucleic acids, and their ligands based solely on their linear sequences. This is a significant step toward understanding biomolecular interactions. In 2024, Hassabis and Jumper were awarded the Nobel Prize for developing artificial intelligence models to solve a problem in structural biology (https://www.nature.com/collections/edjcfdihdi) (accessed on October 9, 2024).
In this study, we used AlphaFold 3, based on the principle that the tertiary structure of a protein is inextricably linked to its molecular function [25,26]. Using AlphaFold 3, we were able for the first time to annotate and model tertiary structures and characterize putative functions of NS proteins: NS3 helicase, NS3 protease, NS5 methyltransferase, and NS5 RNA-dependent RNA-polymerase of the recently discovered Haseki tick virus. In Flaviviridae members, the processes involved in virion morphogenesis are not fully understood, but interactions between structural and nonstructural proteins are known to be of critical importance [27]. NS3 and NS5 proteins play a key role in viral genome replication and may be targets for the development of direct-acting antiviral drugs [28,29]. In addition, we determined the localization of two HSTV membrane proteins (NSTR1 and NSTR2) that are putatively associated with NS2A/NS2B and NS4A/NS4B proteins that are viral cofactors and play an important role in viral RNA accumulation.
The primary structures of HSTV NS proteins lack homologs, so their identification in the HSTV polyprotein was difficult using homolog analysis methods. Despite a limited number of viral protein structures in the PDB and AlphaFold databases, the AlphaFold 3 neural network coped with the task of predicting HSTV NS proteins. The amino acid sequence sizes and molecular weights of the identified HSTV NS proteins are consistent with those of analogous proteins in Flaviviridae family viruses, whereas the HSTV genome size is approximately 1.5-fold larger. All the generated tertiary structures of HSTV proteins have a TM-score of > 0.5 compared with the tertiary structures of Flaviviridae viruses. This means that the protein structures have an approximately similar fold, and functional annotation of the proteins based on their structure is reasonable [30]. The tertiary structures of HSTV NS3hel, HSTV NS3pro, and HSTV NS5RdRp are most similar to those of Dengue virus proteins. Every year, millions of people are infected with Dengue virus through the bites of infected female Aedes mosquitoes. There is still no vaccine to protect against dengue fever and no specific antiviral drugs [31]. The structural similarity of HSTV NS and Dengue virus NS proteins may indicate the taxonomic unity of HSTV with Orthoflavivirus genus viruses. Whereas other unannotated viruses closely related to HSTV, such as Bole Tick Virus 4, Trinbago virus, and Dermacentor reticulatus pestivirus-like virus 1, were previously phylogenetically clustered as pesti-like viruses [4, 6-10].
NS3 and NS5 of Flaviviridae members form a multi-enzyme protein complex that is primarily involved in the synthesis of positive- and negative-sense viral RNA and its capping [32].
NS3 consists of NS3 helicase and NS3 protease [13]. In pestiviruses and hepaciviruses, the cleavage between NS2 and NS3 is catalyzed by NS2, and among NS3, NS4A, NS4B, NS5A, and NS5B is catalyzed by NS3 protease. Whereas in Orthoflaviviruses, NS3 protease acts together with NS2B. NS2B of Orthoflavivirus genus members is a small protein consisting of two domains. The N-terminal domain of NS2B is transmembrane and is involved in stabilization of the tertiary structure of NS3 protease. The soluble domain of NS2B is a cofactor of NS3 protease and forms the NS2B–NS3pro complex [33]. HSTV NS3 contains HSTV NS3pro and HSTV NS3hel domains, typical of Flaviviridae, connected by a flexible linker. HSTV NS3pro consists of two domains (D1 and D2) in the form of β-barrels connected by a flexible linker. The active site of NS3pro is formed by the triad His55–Asp88–Ser163(Gly161) located between the β-barrels. HSTV NS3pro complexed with the cytoplasmic domain of HSTV NSTR1 forms a closed conformation. This fact indicates that HSTV NSTR1 may be a cofactor of HSTV NS3pro, similar to Orthoflaviviruses. As previously shown using nuclear magnetic resonance, the complex of the NS2B cytoplasmic domain and NS3Pro of Orthoflaviviruses occurs primarily in a closed conformation, which provides valuable information on the conformational changes of proteases in the absence and presence of substrates and inhibitors that may be useful for the development of antiviral therapy [34]. The tertiary structure of HSTV NS3hel is typical of superfamily 2 helicases. HSTV NS3hel consists of three domains (D1–D3) and eight structural motifs (I, Ia, II, III, IV, IVa, V, and VI) situated in D1 and D2. For example, motifs I, or Walker A and II, or Walker B, III, and VI are responsible for ATP binding and hydrolysis. The secondary structure of motif I forms a phosphate loop allowing for the residues within the motif to bind the β-phosphate of bound nucleotide triphosphate [35]. Motif II is involved in magnesium ion coordination in the ATP-binding pocket. Motif III is located near the ATP hydrolysis active site. Motif III coordinates with motifs I, II, and VI and forms an RNA-binding cleft [36]. Motif VI is known as the arginine finger and stabilizes interactions between the residues within ATPase active site and the nucleic acid base of the bound nucleotide triphosphate molecule. Motifs Ia, IV, IVa, and V are responsible for interdomain interactions of NS3hel and binding of NS3hel to viral RNA [37]. The model of a produced biomolecular complex of HSTV NS3hel, ATP, and a HSTV RNA fragment shows that the viral RNA is indeed coordinated in a positively-charged tunnel along the boundary of HSTV NS3hel D3 that directly interacts with HSTV NS3hel D1 and D2. This fact confirms the correct functional annotation of NS3hel in the HSTV polyprotein.
RdRPs encoded by RNA viruses are a unique class of nucleic acid polymerases. RdRPs play a central role in viral genome replication and are therefore required for the viral life cycle in the host cell [38]. All RNA virus RdRPs adopt a right-hand shape with palm, finger, and thumb domains surrounding the active site [39]. The tertiary structure of HSTV NS5RdRp has an encircled human right-hand architecture, typical of RdRPs, and contains all structural motifs (A–F), despite an extremely low level of primary structure identity. Motifs A, B, C, and F directly interact with the NTP substrate and contain highly conserved residues. In contrast, motifs D, E, and G, located at the active site periphery, play primarily structural roles and are less conserved in their sequences [38]. Correct folding of motif E may affect the accuracy of RdRp function because it is located near the priming loop [40]. In HSTV NS5RdRp, we identified two additional α21 and α22 extending from the putative priming loop of HSTV NS5RdRp. We suggest that α21 and α22 may promote priming loop stabilization in the enzyme active site. In addition, the spatial model for this region has a low pLDTT confidence score, which may be due to the fact that the priming loop structure should be specific to the RNA of a particular virus.
Many viral genomes encode MTase domains whose primary role is to methylate the 5’-terminal cap structures of viral RNAs for RNA degradation protection and efficient genome translation. Some viral MTase domains are identical to cellular FtsJ/RrmJ-like MTases involved in cellular RNA modification, whereas other MTases found in (+)-RNA viruses belong to a separate Sindbis-like family [41]. NS5MTases from Orthoflavivirus genus members adopt an α/β/α topology, form, together with RdRp, the NS5 protein, and are unique in that they simultaneously possess guanylyltransferase (GTase), N7 MTase, and 2’-O-MTase activities [42]. We identified NS5MTase in the HSTV polyprotein, which has a characteristic α/β/α structure. However, NS5MTase and NS5RdRp in HSTV are separated by an unknown NS5-X domain that is folded only in a complex with NS5MTase and NS5RdRp. This is highly unusual for Orthoflavivirus genus members. To date, two modes of functional conformations of Orthoflavivirus NS5 have been identified by X-ray crystallography. The conformation similar to that of Japanese encephalitis virus NS5 typically has a fully folded NS5RdRP finger domain stabilized by intramolecular interactions of the MTase–RdRP complex, and the NS5RdRP ring finger tip is involved in interfacial interactions. In contrast, the conformation similar to that of Dengue virus NS5 has the NS5RdRP finger domain stabilized by NS5MTase, but without the NS5RdRP ring finger tip [38]. We suggest that the unusual arrangement of HSTV NS5MTase relative to HSTV NS5RdRp may be an example of a novel functional conformation of NS5 from Orthoflavivirus genus members. However, this requires additional structural evidence, such as X-ray crystallography of the HSTV NS5MTase/NS5-X/NS5RdRp complex.
Our results demonstrate the capabilities of the Alphafold 3 neural network for annotation of non-homologous viral genomes and prediction of novel viral protein folds, giving up the secrets of recently discovered unclassified viruses. Of course, computational structural methods still require protein structure confirmation by experimental methods, but they already enable rational design of proteins of novel viruses, increasing the likelihood of successful outcome of structural experiments.
4. Materials and Methods
4.1. HSTV Sequence
To annotate HSTV nonstructural proteins, we used the whole genome HSTV sequence under the accession number MW808978 from the NCBI GenBank (HSTV polyprotein sequence GenBank: UTQ11742).
4.2. Multiple Sequence Alignment (MSA) and Analysis
Closely related proteins with experimentally-solved spatial structures were searched with NCBI BLAST at the Protein Data Bank (PDB) using the blastp (protein–protein BLAST) algorithm (date of access: 01.09.2024). Multiple amino acid sequence alignments were performed using Many-against-Many-searching (MMSeqs2) [43]. Only amino acid sequences of proteins with tertiary structures annotated in PDB were used for MSA. We used HMMER v3.1b2 (date of access: 01.09.2024; https://www.ebi.ac.uk/Tools/hmmer/) [44] to search for protein domains in the Pfam database and NCBI Conserved Domains Database (CDD).
4.3. Search for Transmembrane Nonstructural Proteins
The search for transmembrane domains of nonstructural proteins in the polyprotein was performed using online services for membrane protein profile prediction: CCTOP (version v1.1.0; date of access: 01.09.2024; https://cctop.ttk.hu/job) [45] and TMHMM result (version 2.0, date of access: 01.09.2024; https://services.healthtech.dtu.dk/services/TMHMM-2.0/) [46].
4.4. Model Building Using AlphaFold 3 and Structural Alignments
Modeling of the spatial structures of putative HSTV proteins was performed using the AlphaFold 3 server (date of access: 01.09.2024; https://alphafoldserver.com/) [47]. The boundaries of putative HSTV nonstructural proteins in the polyprotein were determined by routine modeling of all possible protein structures in AlphaFold 3 and comparison of AlphaFold 3 structure models with all structures available in the PDB and AlphaFold databases using the FoldSeek server (access date: 01.09.2024; https://search.foldseek.com/) [48]. Spatial models for further analysis were selected based on the confidence coefficient for each amino acid with allowance for an AlphaFold 3 predicted local distance difference (pLDDT) scaled from 0 to 100, which estimates the difference in Cα interatomic distances between the reference and the predicted structures. Pairwise alignment of the spatial structures of viral proteins and generated spatial structure models of HSTV proteins was performed using a Pairwise Structure Alignment tool (date of access: 01.09.2024; https://www.rcsb.org/alignment;) with TM-align for pairwise structural alignment [49]. The level of topological similarity was evaluated based on the root mean square deviation (RMSD) coefficient and assessment of the number of superimposed atoms in structures (TM-score) on a scale from 0 to 1, where 1 indicates a perfect match between the predicted model and the reference structure. The alignment of secondary structures of HSTV viral proteins with those of Flaviviridae family members was visualized using the ESPript 3.0 software (date of access: 01.09.2024; https://espript.ibcp.fr/) [50]. Tertiary structure models of viral proteins were visualized using UCSF ChimeraX (Version 1.15rc) [51] and Mol* (date of access: 01.09.2024; https://molstar.org/) [52] software.
4.5. Protein Structure and Function Analysis
Hydrophobic regions of nonstructural protein models were generated using the ProteinTools server in the Hydrophobic clusters mode (date of access: 01.09.2024; https://proteintools.uni-bayreuth.de/clusters/) [53]. PyMol 3.0 with the Adaptive Poisson–Boltzmann Solver plug-in (https://apbs.readthedocs.io/en/latest/index.html) was used to calculate the electrostatic potential of generated nonstructural protein models.
5. Conclusions
In this study, we, for the first time, structurally and functionally annotated NS3 helicase, NS3 protease, NS5 methyltransferase, and NS5 RNA-dependent RNA-polymerase of the novel Haseki tick virus, which have extremely low protein sequence identity coefficients compared with proteins from PDB. In addition, we proposed the arrangement of transmembrane proteins in the HSTV polyprotein and models of HSTV biomolecular complexes. The high structural alignment similarity of HSTV NS proteins to Orthoflavivirus NS proteins suggests a hypothesis of a possible common origin of these viruses and evolutionary or taxonomic unity. Future studies will be aimed at confirming the tertiary structures of HSTV NS proteins by X-ray crystallography to gain fundamental knowledge about the structure of HSTV and a targeted approach to the development of POC tests and biologics.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Putative HSTV nonstructural transmembrane proteins; Table S2: Putative cleavage sites of HSTV NS3pro; Figure S1: Tertiary structure model of HSTV NS3 in pLDDT color; Table S3: Comparison of amino acid sequences and tertiary structure of HSTV NS3 with NS3 of Flaviviridae family viruses; Figure S2: Tertiary structure model of HSTV NS3: NS3-helicase domain (green) and NS3-protease domain (red); Figure S3: Tertiary structure model of HSTV NS3 protease in pLDDT color; Table S4: Comparison of amino acid sequences and tertiary structure of HSTV NS3pro domain with NS3 protease domain of Flaviviridae family viruses; Figure S4: Imposition models of spatial structures HSTV NS3pro (ivory) with: (a) Dengue virus, PDB ID: 2FOM (blue); (b) Hepatitis C virus, PDB ID: 2F9U (green); (c) Classical swine fever virus, PDB ID: 5WX1 (red); Figure S5: The model of spatial structure HSTV NS3pro: α-helix (red), β-strand (green); Figure S6: Secondary structure of HSTV NS3pro.; Figure S7: Topology diagram of HSTV NS3pro: α-helix (red), β-strands (pink), N-amino-terminus, C-carboxyl-terminus; Figure S8: Model of spatial structure of HSTV NS3pro (ivory) in complex with NSTR1 extracellular domain (red); Figure S9: Tertiary structure model of HSTV NS3 helicase in pLDDT color; Table S5: Comparison of amino acid sequences and tertiary structure of HSTV NS3hel domains with NS3hel domains of Flaviviridae family viruses; Figure S1: Imposition models of HSTV NS3 helicase tertiary structures (ivory) with: (a) Dengue virus, PDB ID: 2JLS (blue); (b) Hepatitis C virus, PDB ID: 1A1V (green); (c) Classical swine fever virus, PDB ID: 4CBL (red); Figure S11: Domain organization of HSTV NS3 helicase spatial structure: domain 1 (green), domain 2 (blue), and domain 3 (red); Figure S12: The model of spatial structure of HSTV NS3hel: α-helix (red), β-strand (green); Figure S13: Secondary structure of HSTV NS3hel; Figure S15: Sequence alignment of HSTV NS3hel with NS3hel Dengue virus (2JLS), NS3hel Hepatitis C virus (1A1V), and NS3hel Classical swine fever virus (4CBL); Figure S16: Tertiary structure model of HSTV NS5RdRp in pLDDT color; Table S6: Comparison of amino acid sequences and tertiary structure of HSTV NS5 RdRp with NS5RdRp of Flaviviridae family viruses; Figure S17: Imposition models of HSTV NS5RdRp spatial structures (ivory) with: (a) Dengue virus, PDB ID: 7XD8 (blue); (b) Hepatitis C virus, PDB ID: 6GP9 (green); (c) Classical swine fever virus, PDB ID: 7EKJ (red); Figure S18: Domain organization of HSTV NS5RdRp spatial structure: thumb domain (red), palm domain (green), and fingers domain (blue); Figure S19: The model of spatial structure of HSTV NS5RdRp: α-helix (red), β-strand (green); Figure S20: Secondary structure of HSTV NS5RdRp; Figure S21: Topology diagram of HSTV NS5RdRp: α-helix (red), β-strands (pink), N-amino-terminus, C-carboxyl-terminus; Figure S22: Electrostatic surface potential of HSTV NS5RdRp; Figure S23: Sequence alignment of the HSTV NS5RdRp with NS5RdRp Dengue virus (7XD8), NS5RdRp Classical swine fever virus (7EKJ), and NS5RdRp Hepatitis C virus (6GP9); Figure S25: Tertiary structure model of HSTV NS5Mtase in pLDDT color; Table S7: Comparison of amino acid sequences and tertiary structure of HSTV NS5MTase with NS5MTase of Flaviviridae family viruses; Figure S26: Imposition models of HSTV NS5Mtase spatial structures (ivory) with: (a) Pyrococcus horikoshii, PDB ID: 1WY7 (magenta); (b) Dengue virus, PDB ID: 3P97 (blue); (c) Modoc Virus, PDB ID: 2WA1) (green); (d) Alongshan virus, PDB ID: 8GY4 (red); Figure S27: Secondary structure of HSTV NS5Mtase; Figure S28: Tertiary structure model of HSTV NS5-X in pLDDT color: (a) NS5-X monomer, (b) NS5-X dimer; Figure S29: Imposition models of HSTV NS5-X spatial structures (ivory) with NS5A zinc-binding domain of Hepatitis C virus, PDB ID: 1ZH1 (green): (a) NS5-X monomer, (b) NS5-X dimer; Figure S30: Tertiary structure model of HSTV NS5MTase-NS5-X-NS5RdRp in pLDDT color; Figure S31: Domain organization of HSTV NS5MTase-NS5-X-NS5RdRp spatial structure: NS5MTase (green), NS5-X (red), NS5RdRp (blue).
Author Contributions
Conceptualization, A.G.; methodology, A.G., I.O. and N.R.; software, I.O. and N.R.; validation, I.O., N.R. and D.A.; formal analysis, A.G.; investigation, A.G., I.O., N.R. and D.A.; resources, A.G.; data curation, A.G. and A.A.; writing—original draft preparation, A.G., I.O., N.R. and D.A.; writing—review and editing, A.G. and A.A.; visualization, I.O. and N.R; supervision, A.A.; project administration, A.A.; funding acquisition, A.G. and A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation (agreement No. 075-15-2021-1355 of October 12, 2021) as part of the implementation of certain activities of the Federal Scientific and Technical Program for the Development of Synchrotron and Neutron Research and Research Infrastructure.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zerbini, F.M.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Changes to Virus Taxonomy and the ICTV Statutes Ratified by the International Committee on Taxonomy of Viruses (2023). Arch Virol 2023, 168, 175. [CrossRef]
- Paul Chrystal The History of the World in 100 Pandemic; Pen and Sword History: Barnsley, United Kingdom, 2021; ISBN 9781399005432.
- Nomburg, J.; Doherty, E.E.; Price, N.; Bellieny-Rabelo, D.; Zhu, Y.K.; Doudna, J.A. Birth of Protein Folds and Functions in the Virome. Nature 2024, 633, 710–717. [CrossRef]
- Ergunay, K.; Bourke, B.P.; Reinbold-Wasson, D.D.; Nikolich, M.P.; Nelson, S.P.; Caicedo-Quiroga, L.; Vaydayko, N.; Kirkitadze, G.; Chunashvili, T.; Long, L.S.; et al. The Expanding Range of Emerging Tick-Borne Viruses in Eastern Europe and the Black Sea Region. Sci Rep 2023, 13, 19824. [CrossRef]
- Kartashov, M.Y.; Gladysheva, A. V.; Shvalov, A.N.; Tupota, N.L.; Chernikova, A.A.; Ternovoi, V.A.; Loktev, V.B. Novel Flavi-like Virus in Ixodid Ticks and Patients in Russia. Ticks Tick Borne Dis 2023, 14, 102101. [CrossRef]
- Shi, M.; Lin, X.-D.; Vasilakis, N.; Tian, J.-H.; Li, C.-X.; Chen, L.-J.; Eastwood, G.; Diao, X.-N.; Chen, M.-H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J Virol 2016, 90, 659–669. [CrossRef]
- Temmam, S.; Chrétien, D.; Bigot, T.; Dufour, E.; Petres, S.; Desquesnes, M.; Devillers, E.; Dumarest, M.; Yousfi, L.; Jittapalapong, S.; et al. Monitoring Silent Spillovers Before Emergence: A Pilot Study at the Tick/Human Interface in Thailand. Front Microbiol 2019, 10. [CrossRef]
- Zakham, F.; Albalawi, A.E.; Alanazi, A.D.; Truong Nguyen, P.; Alouffi, A.S.; Alaoui, A.; Sironen, T.; Smura, T.; Vapalahti, O. Viral RNA Metagenomics of Hyalomma Ticks Collected from Dromedary Camels in Makkah Province, Saudi Arabia. Viruses 2021, 13, 1396. [CrossRef]
- Bratuleanu, B.E.; Temmam, S.; Chrétien, D.; Regnault, B.; Pérot, P.; Bouchier, C.; Bigot, T.; Savuța, G.; Eloit, M. The Virome of Rhipicephalus , Dermacentor and Haemaphysalis Ticks from Eastern Romania Includes Novel Viruses with Potential Relevance for Public Health. Transbound Emerg Dis 2022, 69, 1387–1403. [CrossRef]
- Sameroff, S.; Tokarz, R.; Vucelja, M.; Jain, K.; Oleynik, A.; Boljfetić, M.; Bjedov, L.; Yates, R.A.; Margaletić, J.; Oura, C.A.L.; et al. Virome of Ixodes Ricinus, Dermacentor Reticulatus, and Haemaphysalis Concinna Ticks from Croatia. Viruses 2022, 14, 929. [CrossRef]
- Zhang, J.; Zheng, Y.-C.; Chu, Y.-L.; Cui, X.-M.; Wei, R.; Bian, C.; Liu, H.-B.; Yao, N.-N.; Jiang, R.-R.; Huo, Q.-B.; et al. Skin Infectome of Patients with a Tick Bite History. Front Cell Infect Microbiol 2023, 13. [CrossRef]
- Duan, Y.; Zeng, M.; Jiang, B.; Zhang, W.; Wang, M.; Jia, R.; Zhu, D.; Liu, M.; Zhao, X.; Yang, Q.; et al. Flavivirus RNA-Dependent RNA Polymerase Interacts with Genome UTRs and Viral Proteins to Facilitate Flavivirus RNA Replication. Viruses 2019, 11, 929. [CrossRef]
- Du Pont, K.E.; McCullagh, M.; Geiss, B.J. Conserved Motifs in the Flavivirus <scp>NS3 RNA</Scp> Helicase Enzyme. WIREs RNA 2022, 13. [CrossRef]
- Li, R.; Niu, Z.; Liu, Y.; Bai, X.; Wang, D.; Chen, C. Crystal Structure and Cap Binding Analysis of the Methyltransferase of Langat Virus. Antiviral Res 2022, 208, 105459. [CrossRef]
- Chen, S.; Harris, M. NS5A Domain I Antagonises PKR to Facilitate the Assembly of Infectious Hepatitis C Virus Particles. PLoS Pathog 2023, 19, e1010812. [CrossRef]
- Shiryaev, S.A.; Cieplak, P.; Cheltsov, A.; Liddington, R.C.; Terskikh, A. V. Dual Function of Zika Virus NS2B-NS3 Protease. PLoS Pathog 2023, 19, e1011795. [CrossRef]
- Benarroch, D.; Selisko, B.; Locatelli, G.A.; Maga, G.; Romette, J.-L.; Canard, B. The RNA Helicase, Nucleotide 5′-Triphosphatase, and RNA 5′-Triphosphatase Activities of Dengue Virus Protein NS3 Are Mg2+-Dependent and Require a Functional Walker B Motif in the Helicase Catalytic Core. Virology 2004, 328, 208–218. [CrossRef]
- Luo, D.; Xu, T.; Watson, R.P.; Scherer-Becker, D.; Sampath, A.; Jahnke, W.; Yeong, S.S.; Wang, C.H.; Lim, S.P.; Strongin, A.; et al. Insights into RNA Unwinding and ATP Hydrolysis by the Flavivirus NS3 Protein. EMBO J 2008, 27, 3209–3219. [CrossRef]
- Lu, G.; Gong, P. A Structural View of the RNA-Dependent RNA Polymerases from the Flavivirus Genus. Virus Res 2017, 234, 34–43. [CrossRef]
- Wu, J.; Liu, W.; Gong, P. A Structural Overview of RNA-Dependent RNA Polymerases from the Flaviviridae Family. Int J Mol Sci 2015, 16, 12943–12957. [CrossRef]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D.; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Structural Basis for RNA Replication by the Hepatitis C Virus Polymerase. Science (1979) 2015, 347, 771–775. [CrossRef]
- Shu, B.; Gong, P. Structural Basis of Viral RNA-Dependent RNA Polymerase Catalysis and Translocation. Proceedings of the National Academy of Sciences 2016, 113. [CrossRef]
- Krejčová, K.; Krafcikova, P.; Klima, M.; Chalupska, D.; Chalupsky, K.; Zilecka, E.; Boura, E. Structural and Functional Insights in Flavivirus NS5 Proteins Gained by the Structure of Ntaya Virus Polymerase and Methyltransferase. Structure 2024, 32, 1099-1109.e3. [CrossRef]
- Zayed, A.A.; Lücking, D.; Mohssen, M.; Cronin, D.; Bolduc, B.; Gregory, A.C.; Hargreaves, K.R.; Piehowski, P.D.; White III, R.A.; Huang, E.L.; et al. Efam: An e Xpanded, Metaproteome-Supported HMM Profile Database of Viral Protein Fam Ilies. Bioinformatics 2021, 37, 4202–4208. [CrossRef]
- Durairaj, J.; Waterhouse, A.M.; Mets, T.; Brodiazhenko, T.; Abdullah, M.; Studer, G.; Tauriello, G.; Akdel, M.; Andreeva, A.; Bateman, A.; et al. Uncovering New Families and Folds in the Natural Protein Universe. Nature 2023, 622, 646–653. [CrossRef]
- Barrio-Hernandez, I.; Yeo, J.; Jänes, J.; Mirdita, M.; Gilchrist, C.L.M.; Wein, T.; Varadi, M.; Velankar, S.; Beltrao, P.; Steinegger, M. Clustering Predicted Structures at the Scale of the Known Protein Universe. Nature 2023, 622, 637–645. [CrossRef]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of Assembly: Roles of Flaviviridae Non-Structural Proteins in Virion Morphogenesis. Nat Rev Microbiol 2008, 6, 699–708. [CrossRef]
- Dietz, C.; Maasoumy, B. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection—From Drug Discovery to Successful Implementation in Clinical Practice. Viruses 2022, 14, 1325. [CrossRef]
- Goh, J.Z.H.; De Hayr, L.; Khromykh, A.A.; Slonchak, A. The Flavivirus Non-Structural Protein 5 (NS5): Structure, Functions, and Targeting for Development of Vaccines and Therapeutics. Vaccines (Basel) 2024, 12, 865. [CrossRef]
- Xu, J.; Zhang, Y. How Significant Is a Protein Structure Similarity with TM-Score = 0.5? Bioinformatics 2010, 26, 889–895. [CrossRef]
- Sinha, S.; Singh, K.; Ravi Kumar, Y.S.; Roy, R.; Phadnis, S.; Meena, V.; Bhattacharyya, S.; Verma, B. Dengue Virus Pathogenesis and Host Molecular Machineries. J Biomed Sci 2024, 31, 43. [CrossRef]
- Osawa, T.; Aoki, M.; Ehara, H.; Sekine, S. Structures of Dengue Virus RNA Replicase Complexes. Mol Cell 2023, 83, 2781-2791.e4. [CrossRef]
- Dubrau, D.; Tortorici, M.A.; Rey, F.A.; Tautz, N. A Positive-Strand RNA Virus Uses Alternative Protein-Protein Interactions within a Viral Protease/Cofactor Complex to Switch between RNA Replication and Virion Morphogenesis. PLoS Pathog 2017, 13, e1006134. [CrossRef]
- Teramoto, T.; Choi, K.H.; Padmanabhan, R. Flavivirus Proteases: The Viral Achilles Heel to Prevent Future Pandemics. Antiviral Res 2023, 210, 105516. [CrossRef]
- Saraste, M.; Sibbald, P.R.; Wittinghofer, A. The P-Loop — a Common Motif in ATP- and GTP-Binding Proteins. Trends Biochem Sci 1990, 15, 430–434. [CrossRef]
- Banroques, J.; Doère, M.; Dreyfus, M.; Linder, P.; Tanner, N.K. Motif III in Superfamily 2 “Helicases” Helps Convert the Binding Energy of ATP into a High-Affinity RNA Binding Site in the Yeast DEAD-Box Protein Ded1. J Mol Biol 2010, 396, 949–966. [CrossRef]
- Sampath, A.; Xu, T.; Chao, A.; Luo, D.; Lescar, J.; Vasudevan, S.G. Structure-Based Mutational Analysis of the NS3 Helicase from Dengue Virus. J Virol 2006, 80, 6686–6690. [CrossRef]
- Yang, J.; Jing, X.; Yi, W.; Li, X.-D.; Yao, C.; Zhang, B.; Zheng, Z.; Wang, H.; Gong, P. Crystal Structure of a Tick-Borne Flavivirus RNA-Dependent RNA Polymerase Suggests a Host Adaptation Hotspot in RNA Viruses. Nucleic Acids Res 2021, 49, 1567–1580. [CrossRef]
- Bruenn, J.A. A Structural and Primary Sequence Comparison of the Viral RNA-Dependent RNA Polymerases. Nucleic Acids Res 2003, 31, 1821–1829. [CrossRef]
- Selisko, B.; Papageorgiou, N.; Ferron, F.; Canard, B. Structural and Functional Basis of the Fidelity of Nucleotide Selection by Flavivirus RNA-Dependent RNA Polymerases. Viruses 2018, 10, 59. [CrossRef]
- Mushegian, A. Methyltransferases of Riboviria. Biomolecules 2022, 12, 1247. [CrossRef]
- Jia, H.; Zhong, Y.; Peng, C.; Gong, P. Crystal Structures of Flavivirus NS5 Guanylyltransferase Reveal a GMP-Arginine Adduct. J Virol 2022, 96. [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat Biotechnol 2017, 35, 1026–1028. [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER Web Server: 2018 Update. Nucleic Acids Res 2018, 46, W200–W204. [CrossRef]
- Dobson, L.; Reményi, I.; Tusnády, G.E. CCTOP: A Consensus Constrained TOPology Prediction Web Server. Nucleic Acids Res 2015, 43, W408–W412. [CrossRef]
- Möller, S.; Croning, M.D.R.; Apweiler, R. Evaluation of Methods for the Prediction of Membrane Spanning Regions. Bioinformatics 2001, 17, 646–653. [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [CrossRef]
- van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and Accurate Protein Structure Search with Foldseek. Nat Biotechnol 2024, 42, 243–246. [CrossRef]
- Bittrich, S.; Segura, J.; Duarte, J.M.; Burley, S.K.; Rose, Y. RCSB Protein Data Bank: Exploring Protein 3D Similarities via Comprehensive Structural Alignments. Bioinformatics 2024, 40. [CrossRef]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res 2014, 42, W320–W324. [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Science 2018, 27, 14–25. [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res 2021, 49, W431–W437. [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProteinTools: A Toolkit to Analyze Protein Structures. Nucleic Acids Res 2021, 49, W559–W566. [CrossRef]
Figure 1.
Haseki tick virus genome structure and chain topology of the translated single polyprotein (nonstructural part).
Figure 1.
Haseki tick virus genome structure and chain topology of the translated single polyprotein (nonstructural part).
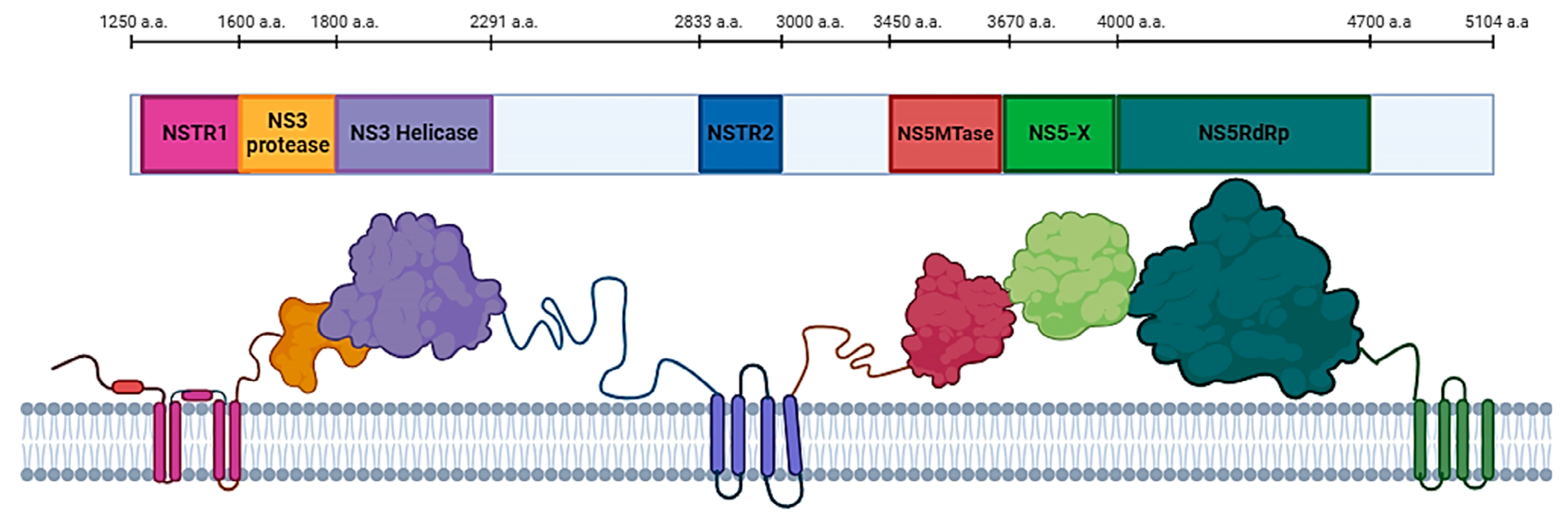
Figure 2.
Imposition models of NS3 HSTV tertiary structures (ivory) with: (a) Dengue virus, PDB ID: 2VBC (blue); (b) Hepatitis C virus, PDB ID: 2F9U (green); (c) Classical swine fever virus, PDB ID: 5WX1 (red).
Figure 2.
Imposition models of NS3 HSTV tertiary structures (ivory) with: (a) Dengue virus, PDB ID: 2VBC (blue); (b) Hepatitis C virus, PDB ID: 2F9U (green); (c) Classical swine fever virus, PDB ID: 5WX1 (red).
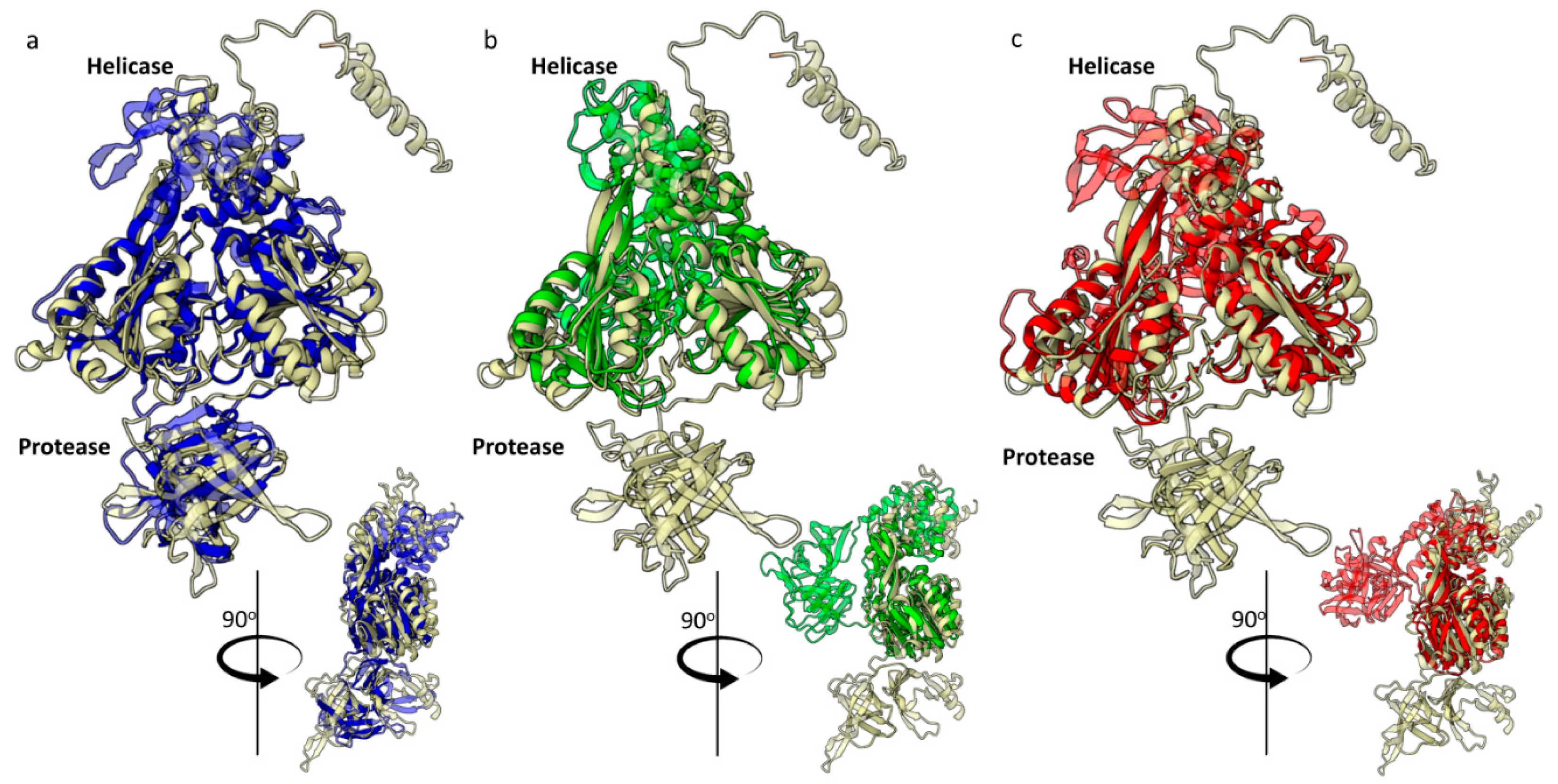
Figure 3.
HSTV NS3 protease tertiary structure. (a) Catalytic site (red) and key amino acids of HSTV NS3pro; (b) Hydrophobic clusters (blue) of HSTV NS3pro; (c) Electrostatic surface potential of HSTV NS3pro; (d) Imposition models of spatial structure of HSTV NS3pro (ivory) in complex with NSTR1 extracellular domain (red) and Zika virus (PDB ID: 5H6V) NS3pro (grey) in complex with NS2B cofactor (blue). The positive surface potential is colored blue, the negative surface potential is colored red.
Figure 3.
HSTV NS3 protease tertiary structure. (a) Catalytic site (red) and key amino acids of HSTV NS3pro; (b) Hydrophobic clusters (blue) of HSTV NS3pro; (c) Electrostatic surface potential of HSTV NS3pro; (d) Imposition models of spatial structure of HSTV NS3pro (ivory) in complex with NSTR1 extracellular domain (red) and Zika virus (PDB ID: 5H6V) NS3pro (grey) in complex with NS2B cofactor (blue). The positive surface potential is colored blue, the negative surface potential is colored red.
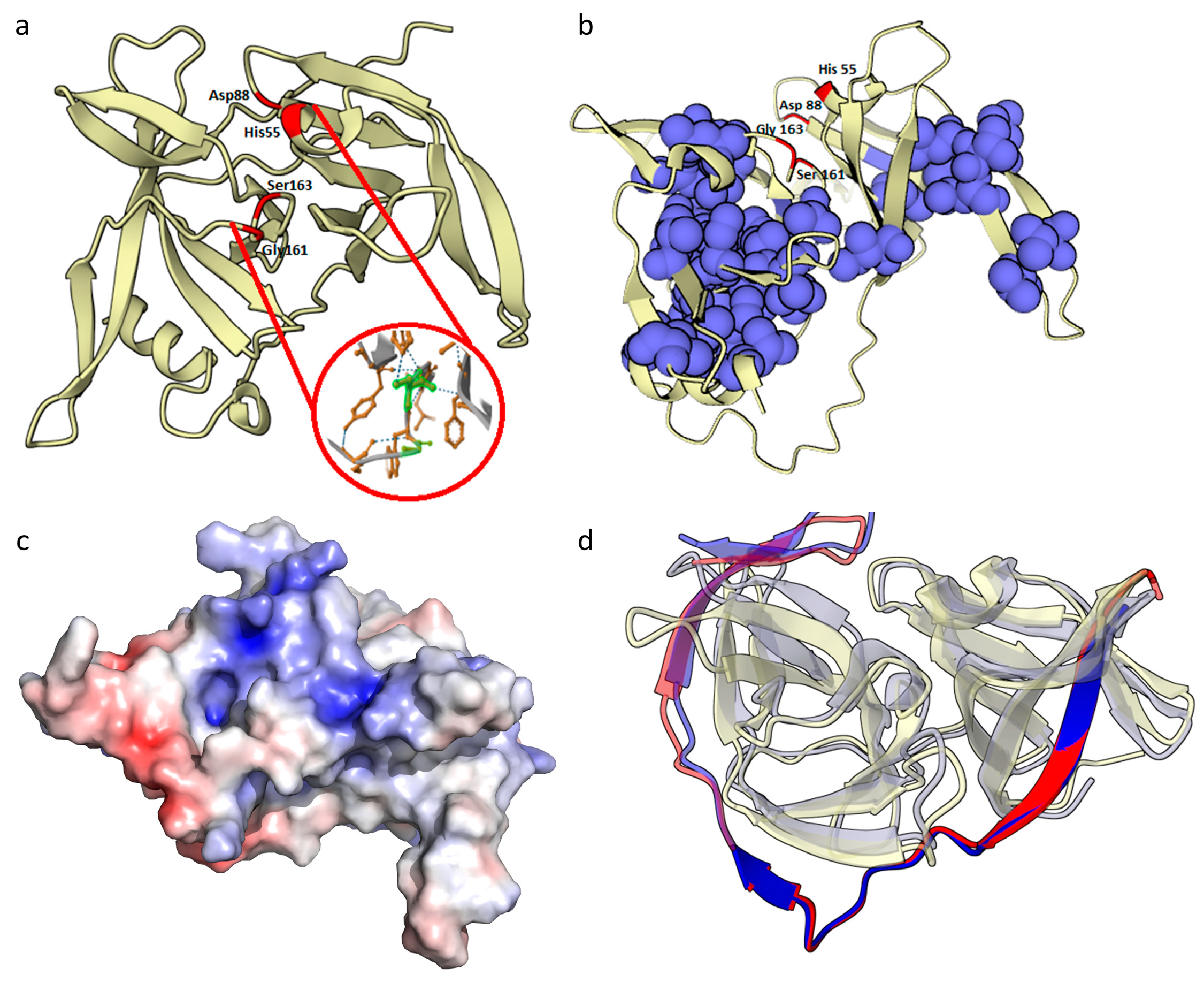
Figure 6.
Functional regions of HSTV NS5RdRp. (a) The overall tertiary structure of HSTV NS5RdRp with catalytic motifs: motif A (blue), motif B (orange), motif C (magenta), motif D (black), motif E (yellow), motif F (green), motif G (red), priming loop (PL) (cyan). (b) Sequence alignment of the HSTV NS5RdRp motifs: red boxes - 100% aligned a.a. residues, yellow boxes - 80% aligned a.a. residues. Abbreviations: CSFV – classical swine fever virus; ZIKV – Zika virus; YFV – yellow fever virus; TBEV – tick-borne encephalitis virus; HCV – hepatitis C virus.
Figure 6.
Functional regions of HSTV NS5RdRp. (a) The overall tertiary structure of HSTV NS5RdRp with catalytic motifs: motif A (blue), motif B (orange), motif C (magenta), motif D (black), motif E (yellow), motif F (green), motif G (red), priming loop (PL) (cyan). (b) Sequence alignment of the HSTV NS5RdRp motifs: red boxes - 100% aligned a.a. residues, yellow boxes - 80% aligned a.a. residues. Abbreviations: CSFV – classical swine fever virus; ZIKV – Zika virus; YFV – yellow fever virus; TBEV – tick-borne encephalitis virus; HCV – hepatitis C virus.
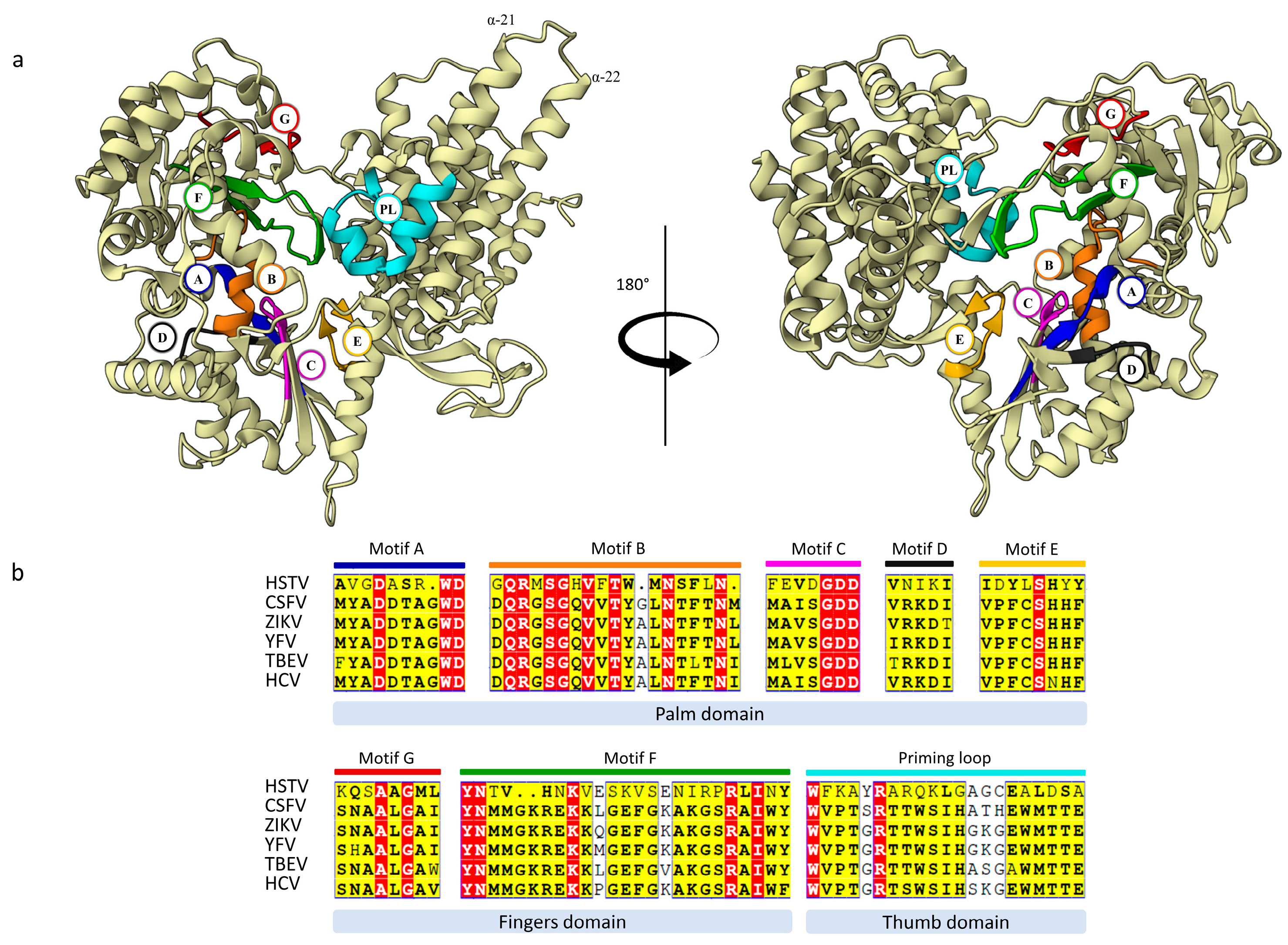
Figure 7.
HSTV NS5Mtase structure. (a-b) Imposition models of HSTV NS5MTase spatial structure (ivory) with: (a) NS5Mtase Pyrococcus horikoshii, PDB ID: 1WY7 (magenta); (b) NS5Mtase Dengue virus, PDB ID: 3P97 (blue); (c) The model of HSTV NS5MTase spatial structure: α-helix (red), β-strand (green); (d) Topology diagram of HSTV NS5MTase: α-helix (red), β-strands (pink), N-amino-terminus, C-carboxyl-terminus.
Figure 7.
HSTV NS5Mtase structure. (a-b) Imposition models of HSTV NS5MTase spatial structure (ivory) with: (a) NS5Mtase Pyrococcus horikoshii, PDB ID: 1WY7 (magenta); (b) NS5Mtase Dengue virus, PDB ID: 3P97 (blue); (c) The model of HSTV NS5MTase spatial structure: α-helix (red), β-strand (green); (d) Topology diagram of HSTV NS5MTase: α-helix (red), β-strands (pink), N-amino-terminus, C-carboxyl-terminus.
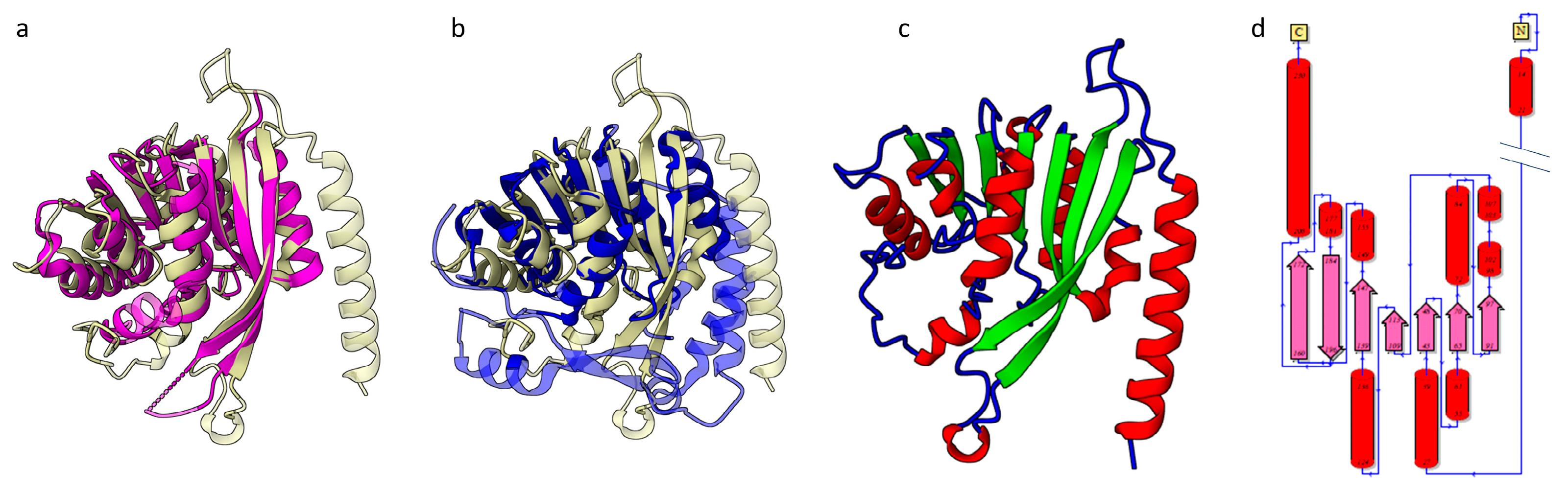
Figure 8.
Spatial structure of HSTV NS5Mtase in complex with HSTV NS5-X and HSTV NS5RdRp. (a) Imposition model of HSTV NS5MTase-NS5-X-NS5RdRp spatial structure with NS5A zinc-binding domain of Hepatitis C virus (green) (PDB ID: 1ZH1); (b) Electrostatic surface potential of HSTV NS5Mtase-NS5-X-NS5RdRp. The positive surface potential is colored blue, the negative surface potential is colored red.
Figure 8.
Spatial structure of HSTV NS5Mtase in complex with HSTV NS5-X and HSTV NS5RdRp. (a) Imposition model of HSTV NS5MTase-NS5-X-NS5RdRp spatial structure with NS5A zinc-binding domain of Hepatitis C virus (green) (PDB ID: 1ZH1); (b) Electrostatic surface potential of HSTV NS5Mtase-NS5-X-NS5RdRp. The positive surface potential is colored blue, the negative surface potential is colored red.
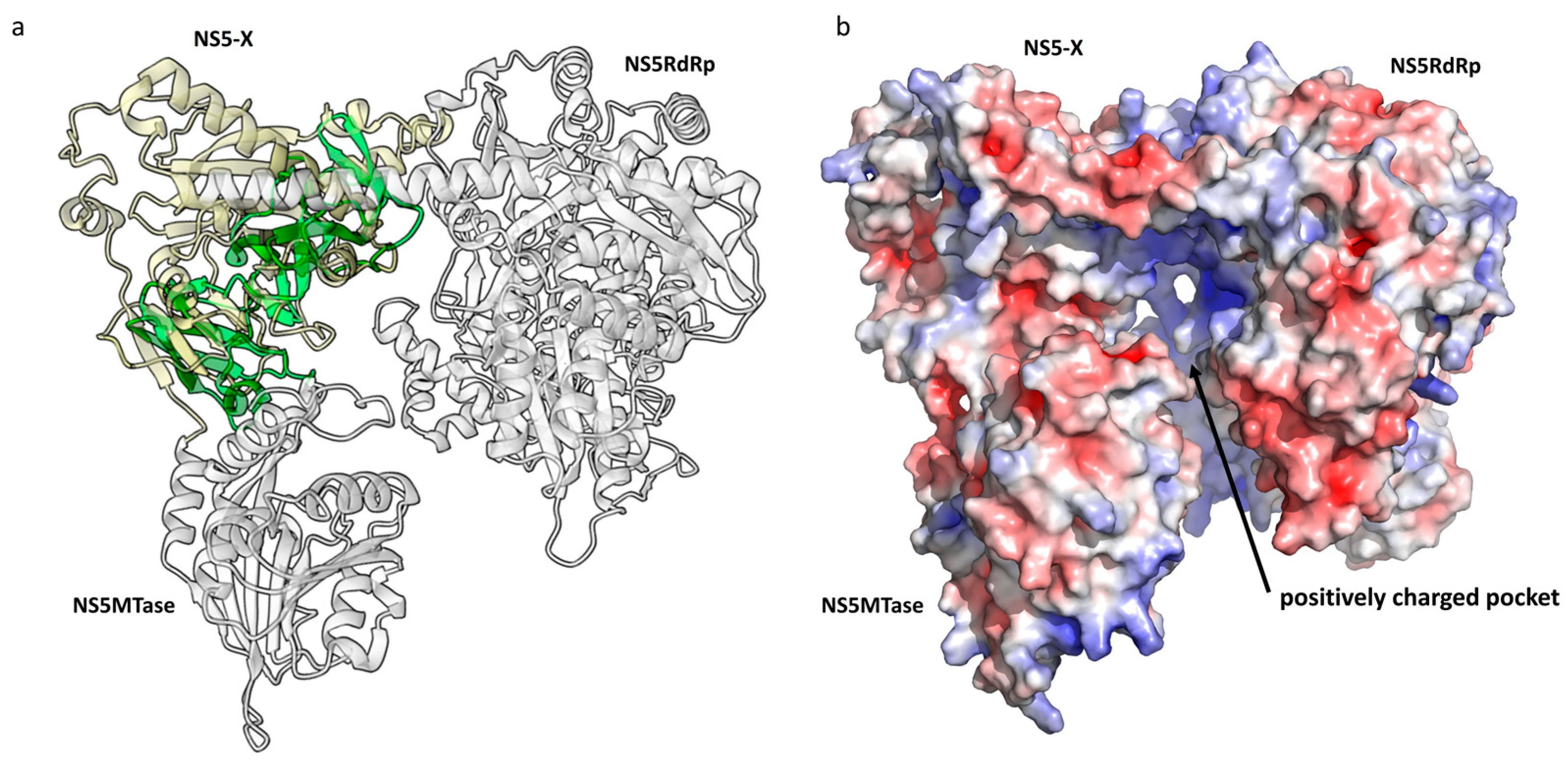

Table 1.
Comparison of amino acid sequences and tertiary structure of HSTV NS3 domains with NS3 domains of Flaviviridae family viruses.
Table 1.
Comparison of amino acid sequences and tertiary structure of HSTV NS3 domains with NS3 domains of Flaviviridae family viruses.
| PDB ID | Name of virus | TM-score | RMSD, Å | Aligned residues, a.a. |
1Amino acid sequence identity1, % |
|---|---|---|---|---|---|
| NS3pro HSTV | |||||
| 2FOM | Dengue virus | 0.79 | 2.33 | 137 | 23 |
| 2F9U | Hepatitis C virus | 0.69 | 4.64 | 118 | 17 |
| 5WX1 | Classical swine fever virus | 0.60 | 3.22 | 138 | 22 |
| NS3hel HSTV | |||||
| 1A1V | Hepatitis C virus | 0.66 | 3.46 | 328 | 20 |
| 2JLS | Dengue virus | 0.63 | 4.38 | 310 | 14 |
| 4CBL | Classical swine fever virus | 0.63 | 4.45 | 283 | 18 |
| D1-D2 NS3hel HSTV | |||||
| 1A1V | Hepatitis C virus | 0.76 | 2.45 | 268 | 21 |
| 2JLS | Dengue virus | 0.74 | 3.26 | 258 | 17 |
| 4CBL | Classical swine fever virus | 0.61 | 05.02 | 183 | 15 |
| D3 NS3hel HSTV | |||||
| 1A1V | Hepatitis C virus | 0.30 | 6.16 | 47 | 5 |
| 2JLS | Dengue virus | 0.36 | 5.7 | 57 | 5 |
| 4CBL | Classical swine fever virus | 0.39 | 5.79 | 59 | 8 |
1Amino acid sequence identity was calculated relative to the aligned residues in the tertiary structure.
Table 2.
Comparison of amino acid sequences and tertiary structure of NS5RdRp and NS5MTase of HSTV with NS5RdRp and NS5MTase of viruses of the Flaviviridae family.
Table 2.
Comparison of amino acid sequences and tertiary structure of NS5RdRp and NS5MTase of HSTV with NS5RdRp and NS5MTase of viruses of the Flaviviridae family.
| PDB ID | Name of virus | TM-score | RMSD, Å | Aligned residues, a.a. |
1Amino acid sequence identity, % |
|---|---|---|---|---|---|
| NS5RdRp | |||||
| 7XD8 | Dengue Virus | 0.72 | 3.26 | 498 | 13 |
| 7EKJ | Classical swine fever virus | 0.66 | 3.54 | 476 | 18 |
| 6GP9 | Hepatitis C virus | 0.63 | 3.85 | 431 | 14 |
| NS5MTase | |||||
| 1WY7 | Pyrococcus horikoshii | 0.77 | 3.02 | 164 | 22 |
| 3P97 | Dengue virus | 0.60 | 4.18 | 142 | 14 |
| 2WA1 | Modoc Virus | 0.59 | 3.69 | 142 | 12 |
| 8GY4 | Alongshan virus | 0.57 | 3.89 | 140 | 10 |
1Amino acid sequence identity was calculated relative to the aligned residues in the tertiary structure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



