
Submitted:
28 November 2024
Posted:
29 November 2024
You are already at the latest version
Abstract
Age-related macular degeneration (AMD) is an eye disease that can lead to legal blindness and sight loss. The advanced form of AMD is categorized into dry and neovascular AMD. In neovascular AMD, the formation of new blood vessels disturbs the structure of the retina and induces an inflammatory response. Neovascular AMD is treated with antibodies and fusion proteins against vascular endothelial growth factor A (VEGFA) to inhibit neovascularization and delay sight loss. Nevertheless, a quarter of neovascular AMD patients do not respond to therapy. Many of these patients presented with subretinal fibrotic scars. Retinal pigment epithelium (RPE) cells, choroidal fibroblasts, and retinal glial cells play crucial roles in the formation of the fibrotic scar, as they can undergo a mesenchymal transition mediated by transforming growth factor beta and other molecules, leading to their transdifferentiation into myofibroblasts, which are critical players in subretinal fibrosis. The mesenchymal transition of retinal cells and dysfunction of the extracellular matrix, the two main aspects of fibrotic scar formation, are associated with impaired autophagy, an important player in AMD pathogenesis, but the causal relationship between autophagy and subretinal fibrosis is not known. This narrative/perspective review presents information on neovascular AMD, subretinal fibrosis, and autophagy and argues that impaired autophagy may be important for fibrosis-related resistance to anti-VEGFA therapy in neovascular AMD.
Keywords:
neovascular age-related macular degeneration
; subretinal fibrosis
; autophagy
; epithelial‒mesenchymal transition
; endothelial‒mesenchymal transition
; transforming growth factor beta 2
; extracellular matrix deposits
1. Introduction
Age-related macular degeneration is an eye disease whose incidence increases strongly with age and is an emerging problem in the healthcare system due to the aging of societies. It affects the macula, a small posterior object in the retina’s center containing the fovea, which is responsible for central, color, and high-resolution vision. This may lead to serious vision impairment and sight loss. The highest reported AMD incidence is in high-income countries, but it may result from a more accurate screening of the disease and a longer life expectancy in these countries. A recent meta-analysis revealed that, in 2020, 1.85 million individuals were projected to be blind due to AMD, and another 6.23 million presented moderate and severe vision impairment with visual acuity (VA) less than 6/18 to 3/60 and blindness (VA less than 3/60) caused by AMD [1]. These numbers imply the urgent need for novel treatment modalities, with dry AMD, the most common form of the disease, being untreatable at present. Usually, AMD is initiated as dry AMD but may progress to neovascular AMD, whose progression is faster, and vision loss is more likely in dry AMD than in dry AMD.
The early phase of AMD is characterized by pigmentation abnormalities in the retinal pigment epithelium (RPE) and the occurrence of drusen, which are extracellular objects composed of lipids and proteins [2]. In its advanced stage, AMD occurs in two clinically distinguishable forms: dry (nonexudative, atrophic) and neovascular (exudative, neovascular) AMD (Figure 1). Dry AMD is the initial form of the disease that may transform into neovascular AMD, accounting for 15–20% of all cases of advanced AMD [3]. The late form of dry AMD, geographic atrophy (GA), is associated with the death of photoreceptors and atrophy of supporting retinal pigment epithelium (RPE) cells, along with the choriocapillaris, and may lead to vision loss [4]. Despite many preclinical studies and several clinical trials, advanced dry AMD is currently not treatable. Although the results of the recent FILLY, OAKS and DERBY clinical trials with pegcetacoplan produced promising results and this drug was approved by the FDA, it should be stressed that it only delays GA development compared with untreated controls [5].
AMD is a complex disease with many potential risk factors, and the interplay between them contributes to AMD pathogenesis. These factors are genetic/epigenetic, environmental, and lifestyle factors, and many are related to oxidative stress. Moreover, the retina is characterized by high oxygen metabolism, which leads to the overproduction of reactive oxygen and nitrogen species (RONS) that may damage cellular components. Furthermore, aging, by definition the main risk factor for AMD, is also associated with oxidative stress. Although several pathways link AMD pathogenesis with oxidative stress, it is still not known whether stress is the cause or a consequence of AMD or both [6].
Although the diagnostic criteria and procedures are well established and the clinical picture of AMD is clear, it is still an undertreated disease. The main reason for this is the incomplete recognition of the mechanisms of AMD pathogenesis. This, in turn, is underlined by insufficient progress in experimental studies on the molecular basis of the disease, impeded by restricted access to human target material and the limited value of animal models of human AMD [7]. Consequently, there are neither AMD-specific preventive recommendations nor therapies for GA in Europe, and there are limited methods for preventing the formation of new vessels in the choroid in neovascular AMD [8]. However, the introduction of medicines against vascular endothelial growth factor A (VEGFA) and its receptor represents a breakthrough in AMD treatment, resulting in a significant delay in vision loss in neovascular AMD patients.
In some neovascular AMD cases, anti-VEGFA treatment is ineffective, but the reasons for this are not completely known [9]. In patients who experience recurrence of neovascularization after initial inhibition, alternative signaling pathways to VEGFA can be activated [10]. Moreover, neovascular AMD patients constitute a heterogeneous group of anatomical, morphological, and genetic cases, and such heterogeneity may influence the results of anti-VEGFA treatment [11]. Furthermore, the formation of new blood vessels may be associated with leakage and hemorrhage, resulting in an inflammatory response and the involvement of the outer retina in fibrosis [12]. On the other hand, subretinal fibrosis was observed after anti-VEGFA therapy in patients with myopathic choroidal neovascularization, with age, baseline best-corrected visual acuity (BCVA) and the macular integrity index (MI) as predictive risk factors [13,14]. Therefore, fibrosis can contribute to the pitfall of anti-VEGFA therapy. However, the MARINA study revealed that ranibizumab, an anti-VEGFA agent, reduced or delayed the progression of subretinal fibrosis in neovascular AMD patients [15]. The progression of neovascular AMD has several common features, such as aberrant wound healing, often resulting in fibrosis, and fibrotic scars are the endpoint of untreated exudative AMD [16].
Fibrosis can be defined as the progressive buildup of connective tissue induced by chronic inflammation or/and tissue injury, which may lead to irreversible damage to the organ where it occurs and possibly failure of this organ [17]. It may be a major cause of morbidity and mortality in a plethora of disorders [18]. Fibrosis may be responsible for a significant disease burden, as the prevalence of fibrosis-related disorders is estimated to be 5000 per 100,000 person-years [19]. Fibrosis can affect many organs, including the lungs, liver, intestine, kidney, heart, skin, and oral mucosa [20,21,22,23,24,25,26]. Independent of the cause of organ damage, a common feature of fibrosis is the activation of fibroblasts and the excessive deposition of extracellular matrix (ECM), including collagen and fibronectin [27].
Autophagy, a process of removing and recycling damaged or unneeded cellular components, plays a role in AMD pathogenesis [28,29]. It may be involved in drusen biogenesis, the senescence-associated secretory phenotype, inflammation, epithelial‒mesenchymal transition and other effects important in AMD pathogenesis. Autophagy is also reported to be impaired in fibrosis in several tissues and disorders, including pulmonary, neural, cardiac, hepatic, and renal diseases, but no experimental data show the involvement of autophagy in macular fibrosis in neovascular AMD [30]. In addition, autophagy can have both beneficial and detrimental effects on many processes; therefore, often, it cannot be classified into “friend/foe” categories and requires the determination of specific cellular conditions.
In general, fibrosis may occur at any age, but it is definitely prevalent in elderly individuals. Aging is the most serious risk factor for idiopathic pulmonary fibrosis (IPF) [31]. In several other interstitial diseases, the fibrotic response increases and worsens with age [32]. Age-related increases in fibrosis also occur in diseases other than lung disease [33]. Therefore, fibrosis may not be considered an aspect of pathology but also an element of biological aging, reflected in the concept of “fibroageing” [34].
In this narrative/perspective review, we present basic information on neovascular AMD and its treatment, discuss the clinical features and risk factors for macular fibrosis secondary to neovascular AMD, and consider the potential of autophagy in AMD-related fibrosis.
2. Neovascular AMD and Fibrosis
Although a fraction of neovascular AMD cases are usually initially diagnosed as dry AMD, the exact relationship between these two forms of AMD is not known, and in their advanced forms, they might be considered different diseases [35]. We and others have shown that neovascular AMD might be associated with increased mortality [36]. Recently, it was suggested that persistent central vitreomacular adhesion might be common for both AMD forms [37].
AMD is a complex disease characterized by the interplay of genetic/epigenetic, environmental, and lifestyle factors. Similar to general AMD, neovascular AMD risk factors can be divided into documented and putative factors. Advanced age is by definition the most serious risk factor for AMD, but there is no agreement in the determination of the age threshold for AMD, as 50--65 years is often mentioned, and the consensus on the Neovascular Age-Related Macular Degeneration Nomenclature Study Group mentions “beyond 50 years” [38]. Therefore, chronological aging may be an initial indicator of the real risk factor associated with aging, and consequently, biological aging better reflects this risk factor than its chronological counterpart. However, even biological aging cannot be considered an independent risk factor, as it depends on an individual’s genetic constitution, environmental conditions, and lifestyle. However, chronological aging cannot be ignored in AMD pathogenesis, but it must be combined with other factors to assess its potential as a risk factor. In addition to aging, smoking is the most consistently reported risk factor for AMD [39]. Some variants of the complement factor H (CFH), apolipoprotein (APOE), age-related maculopathy susceptibility 2 (ARMS2), and HtrA serine peptidase 1 (HTRA1) genes are the strongest genetic risk factors for AMD [40,41,42].
Neovascular AMD is characterized by disruption of Bruch’s membrane and the formation of macular neovascularization (MNV; also known as choroidal neovascularization (CNV)) membranes that are materials for the formation of new blood vessels growing beneath the macula. These vessels protrude from the choriocapillaris (CC) through Bruch’s membrane into the subretinal area. A decrease in blood supply caused by stenosis of CC large vessels may result in the loss of choroidal vasculature, which, along with defects in Bruch’s membrane, may initiate neovascular AMD [43]. The RPE remains integral in neovascular AMD, in contrast to the GA, and its cells produce angiogenic factors, including the VEGFA necessary for synthesizing new blood vessels from the CC [44].
Three subtypes of MNV and consequently neovascular AMD, I-III, can be distinguished depending on the localization of the origin of neovascularization [38]. They are type 1: sub-RPE occult MNV membrane (MNV1); type 2: classic subretinal membrane above the RPE (MNV2); and type 3: retinal angiomatous proliferation (MNV3) [38]. The HARBOR study revealed that MNV2 lesions were more often associated with fibrosis than were MNV1 lesions resulting from the more extensive neovascularization observed with MNV2 [45]. However, in each MNV subtype, newly formed blood vessels are fragile, not hermetic, and predisposed to cause leakage and hemorrhage beneath the RPE or photoreceptors, resulting in severe vision impairment or loss [46]. These devastating effects induce an inflammatory response with the release of stromal and immune cells, which induces the transition of the neovascular endothelial cluster to a fibrovascular membrane [46]. Therefore, fibrosis may be beneficial for the inhibition of leakage and exudation. However, excessive fibrosis may lead to a fibrotic scar, the main cause of sight loss in neovascular AMD. The development of submacular fibrosis is associated with lesions in the RPE and photoreceptors, causing irreversible vision impairments despite anti-VEGFA treatment [47,48].
The clinical definition of retinal fibrosis provided by the Neovascular Age-Related Macular Degeneration Nomenclature Study Group describes it as the build-up of tissue in any layer of the retina, including the subretinal space, RPE monolayer, or sub-RPE space [38] (Figure 2). However, this is rather a general description of fibrosis, and a more specific and more practical depiction describes macular fibrosis as a clearly marked, elevated structure of white–yellowish material within or under the retina that does not exhibit features of dehaemoglobinized blood or hard exudation in fundus examination [49,50]. The time course of macular fibrosis can be classified into three stages: minimal, prominent, and hyperreflective subretinal fibrosis [51].
No specific, i.e., independent of AMD, macular fibrotic risk factors have been identified thus far. However, some studies have reported that an increased incidence of macular fibrosis is correlated with some parameters of neovascular AMD and general ophthalmologic characteristics. For example, a large-cohort, multicenter study with neovascular AMD patients at 10 years reported that the independent factors associated with fibrosis were larger central subfield thickness variation, submacular hemorrhages, and worse baseline visual acuity [52]. Furthermore, type 2 MNV was significantly associated with mixed and subretinal fibrosis. Blocked fluorescence on fluorescein angiography suggests considerable basal damage, increased retinal thickness, foveal subretinal fluid, and subretinal hyperreflective material (SHRM) under the center of the fovea at baseline [53,54]. Although the precise specification of SHRM is unknown, it is considered a diagnostic marker for macular fibrosis [55].
3. Molecular Mechanisms of Submacular Fibrosis
The exact mechanism of the formation of the submacular fibrosis scar is unknown. It is accepted that it is the consequence of prolonged tissue damage and contains several sequential stages, including cellular death and inflammation, cell proliferation and tissue replacement, degradation of the extracellular matrix (ECM), and tissue remodeling [46]. The blood‒retina barrier (BRB) regulates the passage of molecules and immune cells from the systemic circulation to retinal compartments. The outer BRB consists of RPE cells, Bruch’s membrane and fenestrated endothelium [56]. However, the connections between choroidal endothelial cells can loosen during aging, leading to increased permeability and extravasation of macromolecules from choroidal capillaries [57]. The increased permeability may further lead to the accumulation of fluid and induce low-level inflammation in the surrounding tissue microenvironment due to infiltrating immune cells [58]. Fibrosis in neovascular AMD can result from hypoxia-driven angiogenesis resulting in fibrovascular scarring [58].
Disruption of Bruch’s membrane is a critical event in the formation of MNVs, as it allows choroidal endothelial cells to proliferate and penetrate to form neovasculature. The newly formed vessels are fragile and not hermetic, supporting further tissue damage and the release of inflammatory molecules. The subsequent interplay between various cell types, including myofibroblasts and immune cells, leads to excess ECM deposition and tissue remodeling, resulting in fibrotic healing [59]. Macular fibrosis may be formed in neovascular AMD from vascular damage during the process of the angiofibrotic switch [60].
The accumulation of differentiated myofibroblasts is a critical element in the development of subretinal fibrosis, as it leads to increased ECM deposition, tissue contraction, and impaired functions [61]. However, myofibroblasts are absent in normal macula, and their precursors in neovascular AMD have not been unequivocally identified, as original markers are lost in transdifferentiation [12]. Retinal pigment epithelial cells are the main candidates involved in subretinal fibrosis in neovascular AMD [62]. Under normal conditions, RPE cells do not proliferate due to spatial restrictions mediated by cadherins [63]. However, under certain circumstances, RPE cells may lose their epithelial phenotype and undergo mesenchymal transition, contributing to ECM deposition and MNV progression [64]. Therefore, RPE cells and their epithelial‒mesenchymal transition (EMT) can be critical for subretinal fibrosis [46].
EMT can be considered a continuum, whereby cells exhibit epithelial (E), intermediate (EM), and mesenchymal (M) phenotypes. As cells transition, they sequentially lose apicobasal polarity and cell‒cell adhesions, and they gain front‒back polarity and enhanced cell‒matrix interactions. The EMT regulators include the following transcription factors: SNAI1/2 (zinc finger protein SNAI1/2), ZEB1/2 (zinc finger E-box-binding homeobox 1/2), TWIST1 (twist-related protein 1), GRHL2 (grainyhead-like protein 2 homolog), OVOL1/2 (putative transcription factor Ovo-like 1), and PRRX1 (paired mesoderm homeobox protein 1), as well as miRNAs and other epigenetic control factors at the promoters of epithelial and mesenchymal genes [65].
EMT in RPE cells is associated with the loss of apicobasal polarity, translation to a fibroblastic morphology, and acquisition of migratory mesenchymal characteristics (Figure 3) [66].
Another source of myofibroblasts can be endothelial cells that undergo endothelial‒mesenchymal transition (EndT) [67]. Several other cell types, such as fibrocytes, pericytes, and myeloid cells, including macrophages, can be involved in fibrosis in neovascular AMD [68].
Inflammation is critical for neovascular AMD development [43]. Myeloid cells, including blood-derived macrophages and resident retinal microglia, are the main elements linking inflammation with neovascular AMD, as they stimulate the neuroinflammatory cycle, resulting in tissue damage and inducing the proliferation and differentiation of stromal cells via the production of proinflammatory and profibrotic mediators [69]. Microglia may express proinflammatory molecules, including interleukin-1 beta (IL1B), interleukin-6 (IL6), and tumor necrosis factor (TNF), as observed in Alzheimer’s disease [70]. If such release occurs at the site of retinal damage, it can exacerbate fibrosis.
Growth factors mediate the communication between molecules that interact in the process of fibrosis [71]. The main mediator is transforming growth factor beta-2 proprotein (TGFB2), which is involved in the mesenchymal transformation of macrophages and epithelial, endothelial and other types of cells [72]. TGFB2 was found in MNVs and retinal fibrosis scars, and its expression was positively correlated with scar severity (reviewed in [46]). In RPE cells, TGFB2 induces EMT, with the main involvement of mothers in the decapentaplegic homolog 3 (SMAD3) signaling pathway [73]. Another growth factor important for fibrosis is connective tissue growth factor (CTGF), which regulates mechanisms leading from wound healing to fibrosis [74]. The inhibition of CTGF caused reduced fibrosis in a mouse model of laser-induced MNV [75]. The same model was used to show that fibroblast growth factor 2 (FGF2), a member of the TFGF family, was reported to decrease subretinal fibrosis [76]. Platelet-derived growth factor (PDGF) is the key player in idiopathic pulmonary fibrosis but may play an important role in fibrosis in other parts of the human body [77]. Inhibition of PDGF through blockade of its receptor beta (PDGFRB) in a laser-induced MNV mouse model caused a decrease in MNV formation and reduced subretinal fibrosis [78]. However, the PDGFB inhibitor pegpleranib was discontinued in phase III due to a lack of efficacy [79].
Leaky vessels are associated with extravasation of fibrinogen from the vasculature [80]. Thus, fibrosis can be a consequence of aberrant wound healing, the early phase of which includes the formation of a provisional extracellular matrix containing fibrin, fibrinogen, laminin and fibronectin [81]. Interestingly, thrombin, an enzyme that converts fibrinogen to fibrin, was shown to reduce the transepithelial resistance of RPE cells, generate complement C3/C5 cleavage products, and increase the expression of connective tissue growth factor and VEGF [82]. Interference with thrombin action by anticoagulants may also delay the onset of neovascular AMD and help patients remain on anti-VEGF treatment for a longer time [82,83]. Moreover, the direct thrombin inhibitor dabigatran can reduce pulmonary fibrosis [84]. Excessive accumulation of collagen and other ECM components occurs in further stages of fibrosis. Fibroblasts occupy this provisional ECM and proliferate in response to activators produced by leukocytes that migrate into the wound and are retained by the ECM structure [81]. Extracellular matrix components, including fibronectin, collagen, and laminin, are important elements of MNV membranes, and their accumulation leads to retinal fibrosis [85]. Several other ECM components, including periostin, a secreted cell-adhesion protein that functions as a ligand for alpha-V/beta-3 and alpha-V/beta-5 integrins and therefore supports epithelial cell adhesion and migration, have been shown to play a role in MNVs and are considered antifibrotic targets in neovascular AMD therapy [86,87].
Studies on fibrotic scar regeneration in zebrafish have identified the WNT/catenin pathway as a regulator of collagen within the scar matrix [88]. Therefore, the WNT/catenin pathway may be important in fibrosis and may be a potential therapeutic target in neovascular AMD [89].
The destructive role of fibrosis in neovascular AMD implies its ability to target this disease, but currently, there is no approved antifibrotic treatment for neovascular AMD patients [46]. Clinicaltrials.gov lists only one clinical trial of a drug directed to reduce fibrosis in neovascular AMD (https://clinicaltrials.gov/search?cond=AMD%20-%20Age-related%20Macular%20Degeneration&term=Fibrosis, accessed on September 09, 2024). This trial aimed to evaluate the safety, tolerability, and clinical activity of RXI-109 administered by intravitreal injection to reduce the progression of subretinal fibrosis in subjects with advanced neovascular AMD, but no results have been published thus far (https://clinicaltrials.gov/study/NCT02599064?cond=AMD%20-%20Age-Related%20Macular%20Degeneration&term=Fibrosis&page=3&rank=26, accessed on September 09, 2024). RXI-109 targets and reduces the expression of connective tissue growth factor (CTGF), a key regulator of the scarring pathway [90]. Some ongoing trials support anti-VEGF treatment with other drugs, with fibrosis as one of the outcomes, as anti-VEGF monotherapy does not prevent the development of macular fibrosis, although the moment of initiation of treatment may matter in its prevention [60,91]. As mentioned above, many proteins and signaling pathways can regulate EMT and EndMT as well as the motility of myofibroblasts, contributing to retinal fibrosis. Therefore, these proteins/signaling pathways may be potential targets for antifibrotic therapy.
In summary, although the exact mechanism of submacular fibrosis is not known, the mesenchymal transformation of RPE cells, choroidal fibroblasts, and retinal glial cells to myofibroblasts through EMT or EndMT may be a crucial event in this process (Figure 4). Mechanistically, retinal EMT and EndMT in AMD may be driven by many signaling pathways, some of which are presented in Figure 4, and more details can be found in other reviews, e.g., [67,92,93]. These signaling pathways are mediated by a plethora of factors, including cytokines, growth factors, and EMC components, and TGFB2 is the master regulatory protein. Retinal fibrosis may be induced by low-level chronic inflammation or/and retinal injury, which is typical for advanced neovascular AMD, and autophagy impairment may exacerbate the formation of fibrotic lesions.
4. Autophagy in AMD
In autophagy, cells clear out damaged/dysfunctional or unneeded cellular material as well as invaders and their remnants, which is a core molecular pathway for the preservation of cellular and organismal homeostasis [94]. Autophagy may be realized in two ways: degradative autophagy or secretory autophagy. In the former, the material to clear out is degraded within the cell and possibly recycled; in the latter, it is removed from the cell. Degradative autophagy is classified into three categories: macroautophagy (hereafter referred to as autophagy if not specified otherwise), microautophagy, and chaperone-mediated autophagy (CMA). Both autophagy and secretory autophagy are initiated by the formation of an isolation membrane (phagophore), progressively encircling the material to be degraded or removed until its encapsulation and forming an autophagosome, which then fuses with either the lysosome (degradative autophagy) or the plasma membrane (secretory autophagy). The fusion of the autophagosome with the lysosome results in the autolysosome (autophagolysosome), in which cargo degradation occurs with the involvement of lysosomal enzymes. The products of degradation may be recycled and used in cell metabolism.
Microautophagy was the first discovered autophagy pathway and was initially thought to be the only constitutive autophagic pathway in all cells [95]. In microautophagy, the cargo is nonselectively sequestered by direct engulfment in deformations of the lysosomal membrane and is released into the lysosome for degradation (Figure 5). Unlike those in yeast, the mammalian mechanisms of microautophagy are poorly understood.
Macroautophagy and microautophagy nonspecifically degrade cellular waste, but CMA selectively targets soluble cytosolic proteins for degradation. The selective recognition of cargo is performed by cytosolic chaperones. The other essential difference between CMA and macroautophagy and microautophagy is that substrates are not engulfed but instead are transported through the lysosomal membrane in a receptor-mediated fashion [96] (Figure 5). Thus far, heat shock cognate 71 kDa protein (HSC70) is the only chaperone that has been shown to be involved in CMA, but it acts with its cochaperones, including HSC90 and HSC40 [96,97]. CMA may be induced by many factors, the first of which may induce conformational changes in the structure of cytosolic proteins [98].
Secretory autophagy may export proteins that lack the leader sequence, 16 to 20 aa at the N-terminus of some eukaryotic proteins, which determines their ultimate destination, but it may also be involved in extruding faulty cytoplasmic compounds, including mitochondria [99]. It may also impair macroautophagy [100].
Autophagosome synthesis involves a set of four functional groups of proteins, including the Unc-51-like autophagy-activating kinase 1 (ULK1) complex, class III phosphatidylinositol 3-kinase (PI3KC3), two ubiquitin-like proteins, microtubule-associated protein 1 light chain 3 (LC3), and autophagy-related protein 12 (ATG12), and the membrane cycling protein ATG9 [101]. This process is initiated by the nucleation of an isolation membrane (phagophore) and the activation of the ULK1 complex, which recruits ATG proteins. ULK1 stimulates PI3KC3 through the phosphorylation of ATG14 and Beclin-1 (BCN1), which supplement the membrane with phosphatidylinositol 3-phosphate (PI3P). ATG9 is translocated to the phagophore to provide lipids for its growth. ATG8 binds the phagophore through an E3 ubiquitin ligase-like reaction in which the ATG12/ATG5/ATG16 complex conjugates ATG8 to phosphatidylethanolamine (PE) [102].
The basic machinery of degradative autophagy consists of sequestosome 1 (SQSTM1/p62), optineurin (OPTN), ubiquilin 2 (UBQLN2), nibrin 1 (NBR1), WD repeat and FYVE domain containing 3 (ALFY), calcium binding and coiled-coil domain 2 (CALCOCO2), mechanistic target of rapamycin kinase (MTOR), and huntingtin (HTT) [103]. The formation of autolysosomes by the fusion of the autophagosome with the lysosome is a hallmark of degradative autophagy and is mediated by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins—a set of syntaxin 17 (STX17), synaptosome associated protein 29 (SNAP29), vesicle-associated membrane protein 7 (VAMP7) and VAMP8 [104]. This machinery targets its cargo via receptors/adaptors, which sense degradation signals on cargo proteins and bind LC3 and γ-aminobutyric acid receptor-associated protein (GABARAP) on autophagosomes [105].
Interleukins IL1B and IL18 were the first identified substrates for unconventional autophagy-based secretory pathways [106]. The secretory autophagy of IL1B starts with the formation of a complex with the tripartite motif containing 16 (TRIM16) protein and trafficking to an autophagy sequestration membrane [107], which is required for the lipidation of LC3-I to LC3-II. The IL1B-TRIM16 complex is subsequently bound by the SEC22 homolog B vesicle trafficking protein (SEC22B), which has a domain responsible for protein transport to the plasma membrane and a SNARE motif essential for the fusion of the secretory autophagosome with the membrane [17]. The R-SNARE SEC22B binds to the Qbc-SNAREs SNAP23 and SNAP29 on the plasma membrane. STX3 and STX4 mediate the formation of a SNARE complex on the plasma membrane that facilitates the fusion of the secretory autophagosome with the plasma membrane and IL-1B secretion.
The relationship between degradative and secretory autophagy is not completely clear. Functionally, secretory autophagy may take the role of its degradative counterpart when the fusion of the autophagosome with the lysosome is blocked. Both types of autophagy may depart from common precursors. Nevertheless, the secretory autophagosome may substantially differ from the degradative autophagosome, as its main role is trafficking and extracellular cargo export instead of degradation.
Another type of specialized autophagy in RPE cells is LC3-associated phagocytosis (LAP), which is considered the main mechanism responsible for the removal of shed photoreceptor outer segments (POSs) and the regeneration of part of the retinoid, effects crucial for the visual cycle [108]. LAP can be initiated by a morning burst of RPE phagocytosis, which coincides with the lipidation of LC3, which then associates with a single-membrane phagosome containing POSs (Figure 6). This association depends on ATG5 and BCN1 but is independent of the autophagy preinitiation complex.
Impaired autophagy is an important element of AMD pathogenesis, and its critical role in eye diseases is emerging [109]. The importance of both degradative and secretory autophagy in AMD pathogenesis was presented in three recent reviews [28,29,110].
Several studies on the relationship between autophagy and AMD have focused on oxidative stress as a factor in AMD pathogenesis. The importance of oxidative stress in AMD was highlighted in the previous sections, but many diseases are claimed to involve oxidative stress in their pathogenesis, but there are many pathways by which stress may be involved in AMD. The mutual relationship between autophagy and oxidative stress may help to identify a specific mechanism underlying the involvement of oxidative stress in AMD pathogenesis.
An increase in autophagosomes was observed in cultured human RPE from AMD donors compared with normal donors [111]. Moreover, the AMD RPE displayed an accumulation of lipid droplets and glycogen granules, disintegration of mitochondria, increased susceptibility to oxidative stress, increased levels of reactive oxygen species (ROS) under stress conditions, and reduced mitochondrial activity. Autophagy was impaired, and autophagic flux was reduced, as indicated by the reduced ratio of LC3-II/LC3-I and the inability to decrease SQSTM1 levels during starvation. Further studies on impaired autophagic pathways revealed expanded and ring-shaped LAMP1 (lysosomal associated membrane protein 1)-positive organelles in AMD RPE cells compared with the smaller discrete puncta observed in normal RPE cells. This work provides molecular details of the mechanism by which impaired autophagy may contribute to AMD pathogenesis.
Dysfunctional mitochondria may produce increased levels of ROS, which are a byproduct of the normal functioning of these organelles, as the complexes of the oxidative phosphorylation system (OXPHOS) produce ROS during their activity [112]. These ROS may damage biomolecules, including lipids, proteins, and nucleic acids, and autophagy may, at least in part, clear the consequences of the detrimental action of these ROS. Therefore, autophagy may decrease the consequences of physical and chemical factors on the retina, acting to protect against AMD induction and progression. Autophagy activation induced by pulses of light and phytochemicals has been shown to counteract oxidative stress in AMD [113]. These and other results support the conclusion that autophagy, heterophagy and mitophagy may exert protective effects against retinal damage and therefore prevent AMD [114]. Conversely, impaired autophagy may contribute to AMD pathogenesis.
The promotion of autophagy and phagocytosis through the activation of the peroxisome proliferator activated receptor alpha-transcription factor B/CD36 molecule (PPARα-TFEB/CD36) signaling pathway was reported to improve RPE cell survival under oxidative stress in cell culture and mouse AMD models [115]. We recently showed that the AKT serine/threonine kinase 2/sirtuin 5/TFEB 9AKT2/SIRT5/TFEB) pathway maintained autophagy, improved energy metabolism, and decreased the development of drusen, a key biomarker for the risk of developing neovascular AMD [116,117].
Owing to the potential importance of autophagy in AMD pathogenesis, it is considered a therapeutic target in AMD (reviewed in [118]). Moreover, in neovascular AMD, we can consider the dual effects of faricimab on VEGF and angiopoietin [119]. To support current intravitreal injections, other targets that suppress active neovascular membranes are needed. Therefore, the regulation of autophagy signaling has the potential to prevent fibrosis via improved energy metabolism. Despite active anti-VEGF treatment, some neovascular AMD patients develop fibrotic scars and are resistant to treatment, as shown in Figure 2. Anti-VEGF treatment responses might be improved once active autophagy is maintained as a supportive activity [115,116]. To prevent dry AMD progression, one can assume that it also prevents neovascular AMD development. However, recently reported actions to prevent geographic atrophy (GA) progression targeted to the complement system seem to be ineffective [4]. Autophagy and improved energy metabolism are thought to be important additive therapy targets.
Selective autophagy is the lysosomal degradation of selected intracellular components sequestered into autophagosomes, late endosomes, or lysosomes through the involvement of selective autophagy receptors, which interact with autophagy proteins via LC3-interacting region (LIR) motifs [120]. Deletion of the ATG5 and ATG7 genes was associated with an AMD-like phenotype in mice manifested by RPE thickening, hyper or hypotrophy, pigmentary abnormalities, and the accumulation of oxidized proteins [121]. Oxidative stress is a consistently reported secondary AMD risk factor and induces autophagy in RPE cells [122,123]. AMD progression is negatively correlated with autophagic flux [123,124].
Phagocytosis and recycling of the distal portions of the photoreceptor outer segments is a critical function of RPE cells, as a single RPE cell is in contact with ~30 rods and cones [125]. These processes, classified as heterophagy, require an efficient lysosomal degradation pathway, which decreases with age, contributing to AMD pathogenesis [126]. The autophagy receptor protein MTOR is a component of two complexes: mTORC1, which controls protein synthesis, cell growth, and proliferation, and mTORC2, which is a regulator of the actin cytoskeleton and promotes cell survival and cell cycle progression. We showed that defects in the MTORC1 signaling pathway gene are linked to neovascular AMD [127].
Mitochondrial dysfunctions are important in AMD pathogenesis, and dysfunctional mitochondria are removed from cells via mitophagy, a selective autophagy that prevents the vicious cycle of mitochondria, leading to the overproduction of RONS [128]. Mitophagy involves many proteins, including PINK1 (PTEN induced kinase 1), PRKN (parkin RBR E3 ubiquitin protein ligase), OPTN, AMBRA1 (autophagy and beclin 1 regulator 1), BNIP3 (BCL2 interacting protein 3), BNIP3L/NIX (BCL2 interacting protein 3 like) and FUNDC1 (FUN14 domain containing 1) [129,130]. We showed the upregulation of PINK1 and PRKN along with damaged mitochondria in mice bearing double knockout of the nuclear factor, erythroid 2 like 2 (NFE2L2) and peroxisome proliferator-activated receptor gamma coactivator 1-α (PPARGC1A, PGC-1α) genes, which caused an AMD-like pathological phenotype [131]. These results were confirmed in our subsequent work, which revealed the upregulation of LC3B [132]. Impaired mitochondria are cleared independently of the conjugation system needed to conjugate ATG8 to PE on autophagosome membranes, which is critical for macroautophagy [133]. Faulty mitochondria are extruded from cells via secretory autophagic pathways called Autophagic Secretion of Mitochondria (ASM). As impaired mitochondria may directly contribute to lipofuscin and drusen formation, ASM may improve the decrease in mitochondrial quality control in AMD [134].
Defects in autophagy may result in cellular senescence, another important element of AMD pathogenesis [135,136]. Lipofuscin granules, which contain mainly protein and lipids but also metals and sugars, accumulate in the RPE, but lipofuscin is a marker of cellular senescence [137]. The changes observed in RPE cells associated with AMD, such as DNA damage, telomere erosion, and metabolic alterations, may result in cellular senescence and the accumulation of senescent cells [138,139]. Impaired autophagy may contribute to such changes and potentiate senescence in AMD-affected eyes. On the other hand, senescent cells display impaired autophagy; therefore, autophagy and senescence form a feedback loop that is important in AMD pathogenesis. Therefore, senolytic agents, i.e., substances that eliminate senescent cells, are considered therapeutics for AMD [140]. Beta-crystallin A3 (CRYBA1/αB), a crystallin chaperone peptide, was shown to reduce senescence in experimental AMD [141]. Moreover, CRYBA1/βA3/A1-crystallin was shown to regulate autophagy in RPE cells via V-ATPase-MTORC1, and our study revealed that senescence might interact with EMT in the RPE cells of NFE2L2/PPARGC1A-double-knockout mice, which display an AMD-like phenotype [131,142].
Endoplasmic reticulum (ER) stress involves the accumulation of unfolded proteins in the ER following oxidative stress and metabolic and ischemic insult [143]. ER stress is reported to occur in AMD, as it is stimulated by many AMD pathogenesis factors [144,145]. Cells respond to ER stress via the unfolded protein response (UPR), and mitochondria are key players in the UPR [146]. The autophagy-ER-mitochondrion interplay in RPE cells has been reported in AMD [144,147].
Inflammation is consistently reported in AMD pathogenesis, and secretory autophagy is reported to export inflammatory mediators from RPE cells [43,148]. Moreover, secretory autophagy has been reported to be involved in the release of proinflammatory cytokines and components of the complement system, which are important players in AMD pathogenesis [149].
In summary, there are many pathways in which autophagy may be involved in AMD pathogenesis. These processes include senescence, mitochondrial quality control, inflammation, and interplay. Despite consistent reports indicating that autophagy is an important element of AMD pathogenesis, no ongoing clinical trials have targeted this process in AMD. Therefore, more preclinical studies on the role of autophagy in AMD are needed. However, autophagy is considered a “double-edged sword” in cellular homeostasis because of its ability to promote life and death [150]. Consequently, impaired autophagy may contribute to AMD pathogenesis, but the same may involve overactive autophagy, which may degrade/extrude the cellular components still needed for homeostasis.
5. Impaired Autophagy Related to Submacular Fibrosis
Few experimental studies on the role of autophagy in fibrosis are related to AMD. In fact, the entry “autophagy fibrosis AMD” or “autophagy fibrosis age-related macular degeneration” in PubMed returns only one experimental paper showing that the antifibrotic action of 3-methyladenine (3-MA) in a mouse model of subretinal fibrosis was attributed to the inhibition of the Pi3K (phosphatidylinositol-3-kinase)/AKT pathway rather than its PI3K-autophagy counterpart [151]. 3-MA is not only an autophagy inhibitor but also a selective inhibitor of PI3K. However, some features of autophagy and the fibrotic process can be common in different organs, tissues, and cells. The transition of epithelial cells to the mesenchymal state may be a prerequisite for fibrosis initiation in many organs. Moreover, EMT may occur in these organs through common factors, including growth factors.
As stated above, EMT is important in fibrosis, as it enables the mesenchymal transformation of RPE cells to myofibroblasts, which are crucial for fibrotic scar formation. Most studies on the role of autophagy in EMT have been performed in cancer, and this role is strongly context dependent, i.e., depends on the cancer type, stage of progression, type of cancer cells, and cellular environment. Although the formation of MNVs and cancer progression require angiogenesis, cancer cells have many specific characteristics that are not directly related to retinal cells. This specificity is underlined first by the genetic/epigenetic constitution of cancer cells and the resulting signaling pathways regulating their functions. Cancer is a “disease of genes” with a contribution of environmental/lifestyle factors to its pathogenesis, and AMD is a condition in which age is the dominant factor of its pathogenesis, with the considerable influence of environmental/lifestyle factors and rather minor, although not fully recognized, genetic/epigenetic factors. Therefore, the results from cancer research should not be directly translated into AMD. Moreover, although there are substantial differences between advanced dry AMD and neovascular AMD with systemic influence, they are eye diseases. Nevertheless, cancer cells represent a highly nonhomogeneous population, even if they are derived from the same tumor. Therefore, it is not surprising that autophagy is reported to stimulate and inhibit EMT in cancer, depending on various, not always identified, factors [152,153,154,155].
AMD is strongly genetically associated with the complement system. However, almost all treatments that attempt to prevent AMD progression fail once they are targeted to the complement system [4]. Large clinical trials may cover individual patient variations. We know that AMD is a very heterogeneous eye disease. Therefore, more individual patient-related imaging, blood biomarker and cadaver tissue sample data and analyses are needed to understand why some AMD patients develop fibrosis in the macula. In addition to cell culture models and animal models, large human material analyses with individual patients are needed to understand who ultimately develops fibrosis and blindness. Humans are the only individuals who develop real AMD, and all other models only mimic signs of AMD.
Many studies on the role of autophagy in fibrosis have also been performed in the pulmonary system [156]. Pulmonary fibrosis is induced by autophagy triggered by TGFB1 and is mediated by EMT [157]. However, TGFB1 also displays inflammatory properties, which are important in pulmonary fibrosis, but interleukin 37, which displays anti-inflammatory properties, decreases lung fibroblast proliferation and shows antifibrotic activity by enhancing autophagy and inhibiting TGFB1-induced inflammation [158]. However, autophagy may exert different, or even opposite, functions in pulmonary fibrosis in different cell types of the pulmonary system, including epithelial and endothelial cells, smooth muscle cell fibroblasts, and macrophages [159].
Mouse embryonic fibroblasts (MEFs) from wild-type and transgenic animals constitute an established model for investigating autophagy in cells with mesenchymal characteristics [160]. They may be combined with ARPE-19 cells to study EMT and its consequences in the retina. ATG7 is an autophagy protein essential for autophagosome formation, the function of the LC3 system, and ATG12 conjugation [161]. Feng et al. did not detect LC3-II in MEFs derived from ATG7 knockout mice, suggesting the inability of these cells to form autophagosomes and therefore perform autophagy [162]. The expression of SQSTM1 confirmed the inhibition of autophagic flux. Autophagy impairment was associated with increased mesenchymal markers, such as N-cadherin, vimentin, and alpha smooth muscle actin, suggesting that autophagy deficiency might lead to EMT in MEFs. Serum deprivation in WT MEFs, a condition that induces autophagy, resulted in SQSTM1 degradation, LC3-II accumulation, and decreased N-cadherin. Therefore, autophagy delays the mesenchymal process. TGFB2 induced the transdifferentiation of RPE cells into myofibroblasts, resulting in EMT. This process activates autophagy; therefore, the absence of autophagy might facilitate ETM. Twist-related protein 1 (TWIST1) is the main transcription factor involved in embryonic morphogenesis, and it activates mesenchymal markers and promotes EMT [163]. It was speculated that selective autophagy mediated by SQSTM1 might degrade TWIST1, opposing EMT. This speculation was confirmed by the observation that starvation induced TWIST1 binding to SQSTM1 and, eventually, the degradation of TWIST1. However, TGFB2 stimulation did not affect TWIST1-SQSTM1 binding but blocked this binding when autophagy occurred. Therefore, autophagy might prevent ETM induction by TGFB2. The production of stable ATG7 KD transfectants from the RPE resulted in impaired autophagy, resulting in the loss of the epithelial phenotype and acquisition of the mesenchymal phenotype by RPE cells. Therefore, functional autophagy is necessary for the maintenance of the epithelial phenotype by RPE cells. Treatment of ARPE-19 cells with TGFB2 and rapamycin, an autophagy activator, inhibited TGFB2-induced phosphorylation of MTOR and increased SQSTM1 degradation and LC3-II accumulation. Therefore, rapamycin induced autophagy by blocking the phosphorylation of MTOR by TGFB2. Elevated autophagic activity protected RPE cells from EMT stress and inhibited their migration and contraction. Although this study was performed in the context of proliferative vitreoretinopathy (PVR), a fibrous complication of intraocular surgery, the observed effects may lead to different outcomes, including retinal fibrosis in neovascular AMD [162].
Miao et al. performed another study aimed at PVR mechanisms and reported the colocalization of keratin 8 (KRT8) with LC3B, an autophagy marker, in subretinal and epiretinal membranes from PVR patients [164]. Additionally, TGFB2 induced KRT8 phosphorylation and autophagy in ARPE-19 cells. Moreover, these cells displayed an increase in ETM and autophagy markers, suggesting that TGFB2-induced ETM in RPE cells stimulated autophagy. Consequently, the suppression of autophagy inhibited ETM in RPE cells, as confirmed by both pharmacological and genetic studies. These results are not in complete agreement with those obtained by Feng et al. ¬, who drew two different conclusions on the role of autophagy in TGFB2-induced EMT and possibly fibrosis in retinal cells [162]. However, there are some significant differences between these two studies. Feng et al. used ATG7 KD transfectants, whereas Mioa et al. suppressed the expression of ATG5 and BCN1 with siRNAs. Moreover, Feng et al. induced autophagy through starvation, which is a natural way to switch on this process, whereas Miao et al. observed autophagy after induction of ETM by TGFB2. The different conditions of both studies and the complexity of the autophagic process make it very difficult to directly compare the outcomes of these two experiments. The problem of interpretation of the works of Feng et al. and Miao et al. will be addressed in the concluding section.
As mentioned in the previous section, beta-crystallin A3 (CRBA1, βA3/A1-crystallin) is important for lysosomal clearance in RPE cells. This protein is also important in the EMT of RPE cells [165]. CRBA1 was upregulated in polarized RPE cells but not in undifferentiated cells. Loss of CRBA1 in murine and human RPE cells was associated with upregulation of SNAIL and vimentin, downregulation of E-cadherin, and increased cell migration. Essentially, the same associations were observed in RPE cells isolated from samples from AMD patients compared with those from age-matched controls. The authors concluded that AMD might be initiated by defects in lysosomal clearance in the RPE and subsequent EMT of RPE cells to rescue the stress associated with clearance defects. Therefore, CRBA1 may be considered a target in AMD therapy to reverse EMT and prevent fibrosis.
Treatment of ARPE-19 cells with TGFB2 resulted in increased autophagic flux, as indicated by the expression of MLP3B (LC3-II) and SQSTM1 [166]. Furthermore, autophagy activation increased TGFB2-induced EMT, and autophagy inhibition attenuated EMT. Additionally, autophagy activation increased the migration and invasion of RPE cells, but the inhibition of autophagy reduced these phenomena. The authors concluded that autophagy might serve as a potential therapeutic target to attenuate EMT in intraocular fibrotic disorders.
The extracellular matrix may play multiple roles in AMD pathogenesis [167]. RPE cells are located between the neural retina and the choroid and are associated with Bruch’s membrane (BM)-ECM complex [168]. Damage to the RPE and choriocapillaris and inflammation in AMD may lead to the formation of an abnormal ECM [169]. Abnormal ECM results in altered RPE–choriocapillaris behavior, ultimately leading to atrophy of the retina, RPE, and choriocapillaris [170]. The loss of balance between the production and removal of ECM components may lead to the aggregation of ECM elements and their increased deposition, which can promote tissue fibrosis [171]. On the other hand, ECM components, including proteoglycans and active fragments, may stimulate or inhibit autophagy [172,173,174]. Therefore, the local environment of the retina, which is affected by the composition of the BM-ECM complex, may influence the formation of retinal fibrotic scars not only by providing collagen and other compounds that are essential for this process but also by modulating the EMT mediated by autophagy. Autophagy is not the only pathway in which the chemical and physical properties of the ECM regulate ETM [175].
The important role of autophagy in the regulation of fibrotic processes has been shown in other eye diseases. To identify the molecular mechanism underlying pterygium, an eye disease in which fibrosis is involved in its pathogenesis, a model of fibrosis was established by treating human primary conjunctival fibroblasts with TGFB1 [176]. The activation of autophagy slowed the progression of fibrosis in these fibroblasts, whereas the inhibition of autophagy increased fibrosis. Fibrosis is promoted by SQSTM1, which activates the protein kinase C-iota type (PRKCI)-nuclear factor kappa-light-chain-enhancer of activated B cells (NFKB) signaling pathway. This work provides a mechanism for fibrotic scar formation in the eye, which might not be limited to pterygium. The importance of autophagy in fibrosis has also been confirmed in another model of human pterygium in which limbal stem cell excision in combination with alkali burn was performed in rabbits to establish a model of limbal deficiency and conjunctival fibrovascular invasion [177]. Pirarubicin, an autophagy inducer, was used in that study. Graves’ orbitopathy (GO) is another eye disease in which fibrosis and orbital fibroblasts are involved [178]. Profibrotic stimulation with TGFB1 upregulated inositol-requiring enzyme 1 (IRE1), whereas silencing IRE1 suppressed fibrosis and autophagy responses in orbital fibroblasts obtained from GO patients [179]. Consequently, the inhibition of autophagy with bafilomycin A1 decreased the expression of profibrotic proteins. Therefore, IRE1 may be a mediator of the regulation of fibrosis by autophagy. The regulation of fibrosis by autophagy might also be important in corneal disorders [180]. 3-MA, an autophagy inhibitor, decreased the expression of the fibrosis-related proteins fibronectin and collagen alpha-1(I) chain (COL1A1) in the retina of a mouse diabetic model [181]. However, treatment with chloroquine, another autophagy inhibitor, did not affect fibrosis or the expression of apoptosis-related proteins. Therefore, autophagy may regulate fibrosis in diabetic retinopathy, but further research is needed to reveal the details of this regulation, especially in the context of the significance of autophagy phases (early vs. late).
In summary, despite the rich literature on the role of autophagy in fibrosis in cancer and pulmonary disorders, very few works are related to the importance of autophagy in fibrosis in AMD, and consequently, it is difficult to determine the mechanism underlying this phenomenon. However, when experimental data for AMD and certain common aspects of autophagy and fibrosis are combined, several mechanistic features related to the importance of autophagy in retinal fibrosis can be identified (Figure 7).
It may be assumed that the process occurring in the RPE and eventually resulting in subretinal fibrosis starts with EMT, which can be induced by various influences. Although the growth factor TGFB2 is most often used in experimental studies, many other factors may induce EMT in the RPE. Some of these factors may be linked to pathological processes in the eye, including AMD [182]. EMT may induce apoptosis if not already induced, but autophagy inhibits EMT. When autophagy is impaired, the EMT of RPE cells may lead to their transformation to myofibroblasts, which support fibrosis. As the ECM influences autophagy via many pathways, its abnormal function may result in autophagy impairment. Additionally, a dysfunctional ECM may deposit its components, including collagens I and IV, fibronectin, and laminins, at the site of fibrotic scar formation. In addition to RPE cells, choroidal fibroblasts and retinal glial cells may also transdifferentiate into myofibroblasts through EMT and EndMT, contributing to fibrosis. Independent of their origin, myofibroblasts may synthesize and release ECM components, modulating their contents and functions and consequently contributing to fibrotic scar formation [183]. This pathway involved in autophagy in fibrosis needs to be empirically verified, as no experiments on the role of autophagy in the formation of subretinal fibrotic scars in neovascular AMD have been performed thus far.
6. Conclusions, Outstanding Questions, and Perspectives
Retinal fibrosis may be the cause of resistance to anti-VEGFA therapy and the reason for irreversible sight loss in neovascular AMD patients. Therefore, identifying adjuvant therapies to support anti-VEGFA treatment is particularly important. A better understanding of the molecular mechanisms underlying fibrosis in neovascular AMD is needed. EMT in RPE cells and EndMT in choroidal fibroblasts and retinal glial cells are essential for the transdifferentiation of these cells to myofibroblasts, which are directly involved in the formation of fibrotic lesions. TGFB2 may be the master regulator of EMT/EndMT, but other proteins, including VEGFA, can also play a role in this process. Autophagy may be stimulated by EMT and then inhibit EMT, although not all results concerning the association of autophagy with EMT in the retina and other tissues are consistent. Therefore, the relationship between autophagy and EMT/EndMT should be addressed in future research.
In addition to EMT/EndMT, aberrant ECM function is essential for the formation of subretinal fibrotic scars, which contain several ECM components, including collagen and fibronectin. ECM components also play important roles in the regulation of autophagy, as several ECM-derived proteoglycans and proteins, including decorin, biglycan, endorepellin, endostatin, collagen VI, and plasminogen kringle 5, are inducers of autophagy [184]. However, some other ECM components, including laminin α2, perlecan, and lumican, suppress autophagy. The ECM can direct autophagy by interacting with numerous receptors, interplaying with their coreceptors and adhesion molecules. Therefore, impaired ECM functions may result in impaired autophagy. The building components of the fibrotic scar provided by the ECM may be degraded by RPM cells via the LAP mechanism, such as the LAP of POS.
Therefore, key events in the formation of subretinal scars are EMT/EndMT in RPE cells/choroid fibroblasts and retinal glial cells and aberrant functioning of the ECM, resulting in excessive deposits of its compounds in the outer retina. Autophagy in RPE cells may contribute to this process, as it may be stimulated by mesenchymal transitions and regulated by ECM components. However, the role of autophagy in EMT/EndMT in the retina is not completely clear, as it may be dependent on the cellular context. Two seminal works on the role of autophagy in EMT in RPE cells by Feng et al. and Miao et al. did not yield consistent results [162,164]. Nevertheless, they both underscore the importance of autophagy in EMT and point to the possibility of a protective role of autophagy against ETM. Therefore, impaired autophagy may be associated with mesenchymal transition, which may initiate and support the transdifferentiation of epithelial endothelial cells to myofibroblasts and the formation of subretinal fibrotic lesions.
Low-level chronic inflammation and tissue injury may be primary factors that initiate subretinal fibrosis. The former may be the source of growth factors and cytokines stimulating ETM/EndMT (Figure 4), and the latter may be a bud for fibrotic scar formation, as this process can be seen as aberrant wound healing. Both chronic inflammation and injury of the outer retina are typical conditions associated with advanced neovascular AMD. Moreover, this disease is strongly related to dysfunctional ECM [185]. Therefore, events related to neovascular AMD may initiate fibrotic scar formation. Two outstanding questions may be addressed on this point. First, why do only some but not all patients with neovascular AMD develop fibrosis, despite an advanced form of this disease being associated with extensive MNV? Second, both chronic inflammation and tissue injury occur in other retinal diseases and disorders of other eye structures, and the question is whether neovascular AMD has distinguishing features from other eye diseases to promote fibrosis [186,187].
AMD is strongly genetically associated with the complement system. However, almost all treatments attempting to prevent AMD progression fail once the complement system is targeted [4]. Large clinical trials may cover individual patient variations. AMD is a very heterogeneous eye disease. Therefore, more individual patient-related imaging, blood biomarker, and cadaver tissue sample data and analyses are needed to understand why some AMD patients develop fibrosis in the macula.
Autophagy plays an important role in AMD pathogenesis and is reported to be altered during fibrosis in many organs [29,188]. However, changes in autophagy cannot be unequivocally assessed as detrimental or beneficial. Impaired autophagy may contribute to neovascular AMD pathogenesis and reportedly has a protective effect against EMT in RPE cells. Consequently, neovascular AMD opens an avenue for EMT activation in the RPE and fibrosis. However, autophagy strongly depends on the cellular context, and its function may change even in the same tissue conditions, as it is engaged in almost all, if not all, cellular processes [189]. Moreover, many aspects of autophagy await a molecular explanation, e.g., the interplay, if any, between degradative and secretory autophagy, which may be crucial for determining the role of autophagy in AMD pathogenesis [28]. Additionally, the relationship between macroautophagy and LC3-associated phagocytosis may be important for the formation of fibrotic scars in neovascular AMD; thus, this relationship should be addressed in future research. In the same context of removing scar components, microautophagy warrants further study. In general, there are many outstanding questions concerning the role of autophagy and the mechanism of its action in RPE cells and AMD that should be answered before its potential in fibrotic scar formation in neovascular AMD can be precisely determined.
A combination of omics data is needed to clarify the role of autophagy in the prevention of fibrosis. It includes studies of autophagy–associated gene variants, RNA transcripts, proteins, metabolites, macular cell types, spatial cells, and cell‒cell interactions linked to AMD pathology. Answers are needed to determine why AMD patients have different phenotypes, progression rates and treatment responses. Autophagy-related multiomic biomarkers provide novel opportunities to create an AMD biomarker map and a macular cell atlas to further understand AMD pathogenesis and progression. Intravitreal injections heavily load patients and clinics. Thus far, novel drug formulations and long-lasting drug release systems might change current treatment protocols [190].
The key role of TGFB2 in fibrotic lesion formation highlights the importance of growth factors in this process (Figure 4) and suggests their therapeutic potential in neovascular AMD. RBM-007, an aptamer directed against FGF2, inhibited FGF2-induced angiogenesis, laser-induced MNVs, and MNVs with fibrosis in animal models of neovascular AMD [76]. Therefore, compounds displaying antiangiogenic and antifibrotic effects should be further investigated as therapeutics independent of or adjuvants to anti-VEGFA treatment.
The resistance of neovascular AMD with fibrotic scars to anti-VEGFA therapy presents yet another question: can the process of intraocular injection of VEGFA antibodies contribute to fibrotic scar formation or progression? Studies performed thus far do not unequivocally answer this question.
Given that fibrosis has been investigated as a potential therapeutic target in several diseases and is the cause of the most serious consequences of neovascular AMD, it might be associated with the consideration of autophagy in AMD therapy, but preclinical studies and clinical trials are needed to confirm this finding.
Author Contributions
Conceptualization, J.B. and K.K.; writing—original draft preparation, J.B., E.P., H.H., and K.K.; writing—review and editing, X.X.; visualization, X.X.; supervision, X.X.; project administration, X.X.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
None
Conflicts of Interest
Hanna Helotera is an employee of Roche. The remaining authors declare no conflicts of interest.
References
- Global estimates on the number of people blind or visually impaired by age-related macular degeneration: a meta-analysis from 2000 to 2020. Eye (Lond) 2024. [CrossRef]
- Fine, S.L.; Berger, J.W.; Maguire, M.G.; Ho, A.C. Age-related macular degeneration. The New England journal of medicine 2000, 342, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat Rev Dis Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.M.; Bhardwaj, S.; Barclay, C.; Gaspar, L.; Schwartz, J. Global Burden of Dry Age-Related Macular Degeneration: A Targeted Literature Review. Clinical therapeutics 2021, 43, 1792–1818. [Google Scholar] [CrossRef]
- Patel, S.B.; Heier, J.S.; Chaudhary, V.; Wykoff, C.C. Treatment of geographic atrophy: an update on data related to pegcetacoplan. Curr Opin Ophthalmol 2024, 35, 64–72. [Google Scholar] [CrossRef]
- Vatner, S.F.; Zhang, J.; Oydanich, M.; Berkman, T.; Naftalovich, R.; Vatner, D.E. Healthful aging mediated by inhibition of oxidative stress. Aging research reviews 2020, 64, 101194. [Google Scholar] [CrossRef]
- Jonathan, R.; Paul, P.; Sophie, P.; Hollingsworth, T.J.; Ilyse, K.; Monica, M.J. An Overview of Age-Related Macular Degeneration: Clinical, Pre-Clinical Animal Models and Bidirectional Translation. In Preclinical Animal Modeling in Medicine, Enkhsaikhan, P., Joseph, F.P., Lu, L., Eds.; IntechOpen: Rijeka, 2021. [Google Scholar]
- Roodhooft, J. No efficacious treatment for age-related macular degeneration. Bull Soc Belge Ophtalmol 2000, 276, 83–92. [Google Scholar] [PubMed]
- Sharma, D.; Zachary, I.; Jia, H. Mechanisms of Acquired Resistance to Anti-VEGF Therapy for Neovascular Eye Diseases. Investigative ophthalmology & visual science 2023, 64, 28. [Google Scholar] [CrossRef]
- Leitch, I.M.; Gerometta, M.; Eichenbaum, D.; Finger, R.P.; Steinle, N.C.; Baldwin, M.E. Vascular Endothelial Growth Factor C and D Signaling Pathways as Potential Targets for the Treatment of Neovascular Age-Related Macular Degeneration: A Narrative Review. Ophthalmol Ther 2024, 13, 1857–1875. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Z.; Webster, K.A.; Paulus, Y.M. Treatment Strategies for Anti-VEGF Resistance in Neovascular Age-Related Macular Degeneration by Targeting Arteriolar Choroidal Neovascularization. Biomolecules 2024, 14, 252. [Google Scholar] [CrossRef] [PubMed]
- Little, K.; Ma, J.H.; Yang, N.; Chen, M.; Xu, H. Myofibroblasts in macular fibrosis secondary to neovascular age-related macular degeneration - the potential sources and molecular cues for their recruitment and activation. EBioMedicine 2018, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Park, K.H.; Woo, S.J. SUBRETINAL FIBROSIS AFTER ANTIVASCULAR ENDOTHELIAL GROWTH FACTOR THERAPY IN EYES WITH MYOPIC CHOROIDAL NEOVASCULARIZATION. Retina (Philadelphia, Pa.) 2016, 36, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhao, X.; Li, S.; Sun, L.; Xin, W.; Wang, Z.; Zhang, A.; Zhang, J.; Ding, X. Risk factors for subretinal fibrosis after anti-VEGF treatment of myopic choroidal neovascularisation. British Journal of Ophthalmology 2021, 105, 103. [Google Scholar] [CrossRef]
- Kaiser, P.K.; Blodi, B.A.; Shapiro, H.; Acharya, N.R. Angiographic and optical coherence tomographic results of the MARINA study of ranibizumab in neovascular age-related macular degeneration. Ophthalmology 2007, 114, 1868–1875. [Google Scholar] [CrossRef]
- Lehmann, G.L.; Benedicto, I.; Philp, N.J.; Rodriguez-Boulan, E. Plasma membrane protein polarity and trafficking in RPE cells: past, present and future. Experimental eye research 2014, 126, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Montesi, S.B.; Désogère, P.; Fuchs, B.C.; Caravan, P. Molecular imaging of fibrosis: recent advances and future directions. The Journal of clinical investigation 2019, 129, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M. Mechanisms of fibrosis. Seminars in cell & developmental biology 2020, 101, 77. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.F. Targeting metabolic dysregulation for fibrosis therapy. Nature reviews. Drug discovery 2020, 19, 57–75. [Google Scholar] [CrossRef]
- D’Haens, G.; Rieder, F.; Feagan, B.G.; Higgins, P.D.R.; Panés, J.; Maaser, C.; Rogler, G.; Löwenberg, M.; van der Voort, R.; Pinzani, M.; et al. Challenges in the Pathophysiology, Diagnosis, and Management of Intestinal Fibrosis in Inflammatory Bowel Disease. Gastroenterology 2022, 162, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu Rev Physiol 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 2021, 18, 151–166. [Google Scholar] [CrossRef]
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: mechanisms, diagnosis and therapeutic approaches. Nature reviews. Cardiology 2021, 18, 479–498. [Google Scholar] [CrossRef]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu Rev Pathol 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.W.; Shih, Y.H.; Fuh, L.J.; Shieh, T.M. Oral Submucous Fibrosis: A Review on Biomarkers, Pathogenic Mechanisms, and Treatments. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Talbott, H.E.; Mascharak, S.; Griffin, M.; Wan, D.C.; Longaker, M.T. Wound healing, fibroblast heterogeneity, and fibrosis. Cell stem cell 2022, 29, 1161–1180. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: from mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Kaarniranta, K. Secretory autophagy: a turn key for understanding AMD pathology and developing new therapeutic targets? Expert opinion on therapeutic targets 2022, 26, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2022, 1–13. [Google Scholar] [CrossRef]
- Li, Y.; Liu, R.; Wu, J.; Li, X. Self-eating: friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics 2020, 10, 7993–8017. [Google Scholar] [CrossRef]
- Koudstaal, T.; Wijsenbeek, M.S. Idiopathic pulmonary fibrosis. La Presse Médicale 2023, 52, 104166. [Google Scholar] [CrossRef]
- Cottin, V.; Wollin, L.; Fischer, A.; Quaresma, M.; Stowasser, S.; Harari, S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev 2019, 28. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.L.; Miao, H.; Wang, Y.N.; Liu, F.; Li, P.; Zhao, Y.Y. TGF-β as A Master Regulator of Aging-Associated Tissue Fibrosis. Aging and disease 2023, 14, 1633–1650. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Fibroageing: An aging pathological feature driven by dysregulated extracellular matrix-cell mechanobiology. Aging research reviews 2021, 70, 101393. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. Advances in the management of macular degeneration. F1000Prime Rep 2014, 6, 29. [Google Scholar] [CrossRef]
- Blasiak, J.; Watala, C.; Tuuminen, R.; Kivinen, N.; Koskela, A.; Uusitalo-Järvinen, H.; Tuulonen, A.; Winiarczyk, M.; Mackiewicz, J.; Zmorzyński, S.; et al. Expression of VEGFA-regulating miRNAs and mortality in neovascular AMD. Journal of cellular and molecular medicine 2019, 23, 8464–8471. [Google Scholar] [CrossRef] [PubMed]
- Bakaliou, A.; Georgakopoulos, C.; Tsilimbaris, M.; Farmakakis, N. Posterior Vitreous Detachment and Its Role in the Evolution of Dry to Neovascular Age Related Macular Degeneration. Clin Ophthalmol 2023, 17, 879–885. [Google Scholar] [CrossRef]
- Spaide, R.F.; Jaffe, G.J.; Sarraf, D.; Freund, K.B.; Sadda, S.R.; Staurenghi, G.; Waheed, N.K.; Chakravarthy, U.; Rosenfeld, P.J.; Holz, F.G.; et al. Consensus Nomenclature for Reporting Neovascular Age-Related Macular Degeneration Data: Consensus on Neovascular Age-Related Macular Degeneration Nomenclature Study Group. Ophthalmology 2020, 127, 616–636. [Google Scholar] [CrossRef]
- Kulkarni, A.; Banait, S. Through the Smoke: An In-Depth Review on Cigarette Smoking and Its Impact on Ocular Health. Cureus 2023, 15, e47779. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.L.; Quinn, J.; Xue, K. Interactions between Apolipoprotein E Metabolism and Retinal Inflammation in Age-Related Macular Degeneration. Life (Basel) 2021, 11. [Google Scholar] [CrossRef]
- Pan, Y.; Fu, Y.; Baird, P.N.; Guymer, R.H.; Das, T.; Iwata, T. Exploring the contribution of ARMS2 and HTRA1 genetic risk factors in age-related macular degeneration. Progress in retinal and eye research 2023, 97, 101159. [Google Scholar] [CrossRef] [PubMed]
- Roshanshad, A.; Moosavi, S.A.; Arevalo, J.F. Association of the Complement Factor H Y402H Polymorphism and Response to Anti-Vascular Endothelial Growth Factor Treatment in Age-Related Macular Degeneration: An Updated Meta-Analysis. Ophthalmic Res 2024, 67, 358–386. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cellular and molecular life sciences: CMLS 2016, 73, 1765–1786. [Google Scholar] [CrossRef]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Molecular aspects of medicine 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Adrean, S.D.; Morgenthien, E.; Ghanekar, A.; Ali, F.S. Subretinal Fibrosis in HARBOR Varies by Choroidal Neovascularization Subtype. Ophthalmol Retina 2020, 4, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Tenbrock, L.; Wolf, J.; Boneva, S.; Schlecht, A.; Agostini, H.; Wieghofer, P.; Schlunck, G.; Lange, C. Subretinal fibrosis in neovascular age-related macular degeneration: current concepts, therapeutic avenues, and future perspectives. Cell and tissue research 2022, 387, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Shapiro, H.; Tuomi, L.; Webster, M.; Elledge, J.; Blodi, B. Characteristics of patients losing vision after 2 years of monthly dosing in the phase III ranibizumab clinical trials. Ophthalmology 2011, 118, 523–530. [Google Scholar] [CrossRef]
- Bachmeier, I.; Armendariz, B.G.; Yu, S.; Jäger, R.J.; Ebneter, A.; Glittenberg, C.; Pauleikhoff, D.; Sadda, S.R.; Chakravarthy, U.; Fauser, S. Fibrosis in neovascular age-related macular degeneration: A review of definitions based on clinical imaging. Survey of ophthalmology 2023, 68, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, I.; Armendariz, B.G.; Yu, S.; Jäger, R.J.; Ebneter, A.; Glittenberg, C.; Pauleikhoff, D.; Sadda, S.R.; Chakravarthy, U.; Fauser, S. Corrigendum to “Fibrosis in neovascular age-related macular degeneration: a review of definitions based on clinical imaging” [Surv Ophthalmol 68 (2023) 835-848/5]. Survey of ophthalmology 2024. [CrossRef] [PubMed]
- Rogers, A.H.; Martidis, A.; Greenberg, P.B.; Puliafito, C.A. Optical coherence tomography findings following photodynamic therapy of choroidal neovascularization. American journal of ophthalmology 2002, 134, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.; Cozzi, E.; Airaldi, M.; Nassisi, M.; Viola, F.; Aretti, A.; Milella, P.; Giuffrida, F.P.; Teo, K.C.Y.; Cheung, C.M.G.; et al. Ten-Year Incidence of Fibrosis and Risk Factors for Its Development in Neovascular Age-Related Macular Degeneration. American journal of ophthalmology 2023, 252, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Daniel, E.; Pan, W.; Ying, G.S.; Kim, B.J.; Grunwald, J.E.; Ferris, F.L., 3rd; Jaffe, G.J.; Toth, C.A.; Martin, D.F.; Fine, S.L.; et al. Development and Course of Scars in the Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2018, 125, 1037–1046. [Google Scholar] [CrossRef]
- Teo, K.Y.C.; Joe, A.W.; Nguyen, V.; Invernizzi, A.; Arnold, J.J.; Barthelmes, D.; Gillies, M. PREVALENCE AND RISK FACTORS FOR THE DEVELOPMENT OF PHYSICIAN-GRADED SUBRETINAL FIBROSIS IN EYES TREATED FOR NEOVASCULAR AGE-RELATED MACULAR DEGENERATION. Retina (Philadelphia, Pa.) 2020, 40, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, S.; Nittala, M.G.; Verma, A.; Fitzgerald, M.E.C.; Velaga, S.B.; Bhisitkul, R.B.; Sadda, S.R. Subretinal hyperreflective material in regions of atrophy and fibrosis in eyes with neovascular age-related macular degeneration. Can J Ophthalmol 2024. [CrossRef] [PubMed]
- O’Leary, F.; Campbell, M. The blood‒retina barrier in health and disease. Febs j 2023, 290, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Grebe, R.; Bhutto, I.A.; Edwards, M.; McLeod, D.S.; Lutty, G.A. Albumen Transport to Bruch’s Membrane and RPE by Choriocapillaris Caveolae. Investigative ophthalmology & visual science 2016, 57, 2213–2224. [Google Scholar] [CrossRef]
- Heloterä, H.; Kaarniranta, K. A Linkage between Angiogenesis and Inflammation in Neovascular Age-Related Macular Degeneration. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. The Journal of biological chemistry 2022, 298, 101530. [Google Scholar] [CrossRef]
- Roberts, P.K.; Zotter, S.; Montuoro, A.; Pircher, M.; Baumann, B.; Ritter, M.; Hitzenberger, C.K.; Schmidt-Erfurth, U. Identification and Quantification of the Angiofibrotic Switch in Neovascular AMD. Investigative ophthalmology & visual science 2019, 60, 304–311. [Google Scholar] [CrossRef]
- Saika, S.; Yamanaka, O.; Sumioka, T.; Miyamoto, T.; Miyazaki, K.; Okada, Y.; Kitano, A.; Shirai, K.; Tanaka, S.; Ikeda, K. Fibrotic disorders in the eye: targets of gene therapy. Progress in retinal and eye research 2008, 27, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sheng, X.; Ding, Q.; Wang, Y.; Zhao, J.; Zhang, J. Subretinal fibrosis secondary to neovascular age-related macular degeneration: mechanisms and potential therapeutic targets. Neural Regeneration Research 2025, 20, 378–393. [Google Scholar] [CrossRef]
- Yang, X.; Chung, J.Y.; Rai, U.; Esumi, N. Cadherins in the retinal pigment epithelium (RPE) revisited: P-cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PloS one 2018, 13, e0191279. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial–Mesenchymal Transition and Proliferation of Retinal Pigment Epithelial Cells Initiated upon Loss of Cell‒Cell Contact. Investigative ophthalmology & visual science 2010, 51, 2755–2763. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial–mesenchymal plasticity in cancer. Nat Med 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial–mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell‒cell contact. Investigative ophthalmology & visual science 2010, 51, 2755–2763. [Google Scholar] [CrossRef]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Gill, K.; Yoo, H.-S.; Chakravarthy, H.; Granville, D.J.; Matsubara, J.A. Exploring the role of granzyme B in subretinal fibrosis of age-related macular degeneration. Front Immunol 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Wieghofer, P.; Hagemeyer, N.; Sankowski, R.; Schlecht, A.; Staszewski, O.; Amann, L.; Gruber, M.; Koch, J.; Hausmann, A.; Zhang, P.; et al. Mapping the origin and fate of myeloid cells in distinct compartments of the eye by single-cell profiling. The EMBO journal 2021, 40, e105123. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 2015, 3, 136. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: novel roles and mediators. Frontiers in pharmacology 2014, 5, 123. [Google Scholar] [CrossRef]
- Grafe, I.; Alexander, S.; Peterson, J.R.; Snider, T.N.; Levi, B.; Lee, B.; Mishina, Y. TGF-β Family Signaling in Mesenchymal Differentiation. Cold Spring Harbor perspectives in biology 2018, 10. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Tanaka, T.; Yamanaka, O.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Yoo, J.; Flanders, K.C.; et al. Smad3 is required for dedifferentiation of retinal pigment epithelium following retinal detachment in mice. Lab Invest 2004, 84, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, N.; Chu, H.Y.; Yu, Y.; Zhang, Z.-K.; Zhang, G.; Zhang, B.-T. Connective Tissue Growth Factor: From Molecular Understandings to Drug Discovery. Frontiers in cell and developmental biology 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Daftarian, N.; Rohani, S.; Kanavi, M.R.; Suri, F.; Mirrahimi, M.; Hafezi-Moghadam, A.; Soheili, Z.S.; Ahmadieh, H. Effects of intravitreal connective tissue growth factor neutralizing antibody on choroidal neovascular membrane-associated subretinal fibrosis. Experimental eye research 2019, 184, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Nonaka, Y.; Futakawa, S.; Imai, H.; Akita, K.; Nishihata, T.; Fujiwara, M.; Ali, Y.; Bhisitkul, R.B.; Nakamura, Y. Anti-Angiogenic and Anti-Scarring Dual Action of an Anti-Fibroblast Growth Factor 2 Aptamer in Animal Models of Retinal Disease. Molecular therapy. Nucleic acids 2019, 17, 819–828. [Google Scholar] [CrossRef]
- Arai, T.; Hirose, M.; Kagawa, T.; Hatsuda, K.; Inoue, Y. Platelet-derived growth factor can predict survival and acute exacerbation in patients with idiopathic pulmonary fibrosis. Journal of Thoracic Disease 2022, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Noda, K.; Murata, M.; Wu, D.; Kanda, A.; Ishida, S. Blockade of Platelet-Derived Growth Factor Signaling Inhibits Choroidal Neovascularization and Subretinal Fibrosis in Mice. Journal of clinical medicine 2020, 9. [Google Scholar] [CrossRef]
- Armendariz, B.G.; Chakravarthy, U. Fibrosis in age-related neovascular macular degeneration in the anti-VEGF era. Eye (Lond) 2024. [CrossRef] [PubMed]
- Brown, L.F.; Dvorak, A.M.; Dvorak, H.F. Leaky vessels, fibrin deposition, and fibrosis: a sequence of events common to solid tumors and to many other types of disease. Am Rev Respir Dis 1989, 140, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Potter-Perigo, S. The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol 2011, 301, G950–955. [Google Scholar] [CrossRef] [PubMed]
- Akter, T.; Annamalai, B.; Obert, E.; Simpson, K.N.; Rohrer, B. Dabigatran and Neovascular AMD, Results From Retinal Pigment Epithelial Cell Monolayers, the Mouse Model of Choroidal Neovascularization, and Patients From the Medicare Data Base. Front Immunol 2022, 13, 896274. [Google Scholar] [CrossRef] [PubMed]
- Heloterä, H.; Siintamo, L.; Kivinen, N.; Abrahamsson, N.; Aaltonen, V.; Kaarniranta, K. Analysis of prognostic and predictive factors in neovascular age-related macular degeneration Kuopio cohort. Acta Ophthalmologica 2024, 102, 703–713. [Google Scholar] [CrossRef]
- Shea, B.S.; Probst, C.K.; Brazee, P.L.; Rotile, N.J.; Blasi, F.; Weinreb, P.H.; Black, K.E.; Sosnovik, D.E.; Van Cott, E.M.; Violette, S.M.; et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Pouw, A.E.; Greiner, M.A.; Coussa, R.G.; Jiao, C.; Han, I.C.; Skeie, J.M.; Fingert, J.H.; Mullins, R.F.; Sohn, E.H. Cell-Matrix Interactions in the Eye: From Cornea to Choroid. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Nakama, T.; Yoshida, S.; Ishikawa, K.; Kobayashi, Y.; Zhou, Y.; Nakao, S.; Sassa, Y.; Oshima, Y.; Takao, K.; Shimahara, A.; et al. Inhibition of choroidal fibrovascular membrane formation by new class of RNA interference therapeutic agent targeting periostin. Gene Ther 2015, 22, 127–137. [Google Scholar] [CrossRef]
- Yoshida, S.; Umeno, Y.; Haruta, M. Periostin in Eye Diseases. Advances in experimental medicine and biology 2019, 1132, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Wehner, D.; Tsarouchas, T.M.; Michael, A.; Haase, C.; Weidinger, G.; Reimer, M.M.; Becker, T.; Becker, C.G. Wnt signaling controls pro-regenerative Collagen XII in functional spinal cord regeneration in zebrafish. Nat Commun 2017, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jiang, N.; Zhang, Y.; Ye, S.; Liang, X.; Wang, X.; Lin, X.; Zong, R.; Chen, H.; Liu, Z. Fenofibrate Inhibits Subretinal Fibrosis Through Suppressing TGF-β—Smad2/3 signaling and Wnt signaling in Neovascular Age-Related Macular Degeneration. Frontiers in pharmacology 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.; Gagne, N.; Hafiz, G.; Mir, T.; Survi, M.; Barefoot, L.; Cardia, J.; Bulock, K.; Pavco, P.A.; Campochiaro, P.A. A Phase 1/2 multidose, dose escalation study to evaluate RXI-109 administered by intravitreal injection to reduce the progression of subretinal fibrosis in subjects with advanced neovascular AMD (NVAMD). Investigative ophthalmology & visual science 2017, 58, 3210–3210. [Google Scholar]
- Bloch, S.B.; Lund-Andersen, H.; Sander, B.; Larsen, M. Subfoveal fibrosis in eyes with neovascular age-related macular degeneration treated with intravitreal ranibizumab. American journal of ophthalmology 2013, 156, 116–124e111. [Google Scholar] [CrossRef]
- Higashijima, F.; Hasegawa, M.; Yoshimoto, T.; Kobayashi, Y.; Wakuta, M.; Kimura, K. Molecular mechanisms of TGFβ-mediated EMT of retinal pigment epithelium in subretinal fibrosis of age-related macular degeneration. Frontiers in Ophthalmology 2023, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, C.; Zhang, J.; Xu, G.-T.; Zhang, J. Molecular pathogenesis of subretinal fibrosis in neovascular AMD focusing on epithelial–mesenchymal transformation of retinal pigment epithelium. Neurobiology of Disease 2023, 185, 106250. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. The EMBO journal 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Klionsky, D.J.; Shen, H.-M. The emerging mechanisms and functions of microautophagy. Nature Reviews Molecular Cell Biology 2023, 24, 186–203. [Google Scholar] [CrossRef]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science (New York, N.Y.) 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science (New York, N.Y.) 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc Am Thorac Soc 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Liang, W.; Sagar, S.; Ravindran, R.; Najor, R.H.; Quiles, J.M.; Chi, L.; Diao, R.Y.; Woodall, B.P.; Leon, L.J.; Zumaya, E.; et al. Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat Commun 2023, 14, 5031. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell research 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends in cell biology 2017, 27, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Cafiso, D.C.; Tamm, L.K. Mechanisms of SNARE proteins in membrane fusion. Nature Reviews Molecular Cell Biology 2024, 25, 101–118. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nature cell biology 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- DuPont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. The EMBO journal 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Claude-Taupin, A.; Bissa, B.; Jia, J.; Gu, Y.; Deretic, V. Role of autophagy in IL-1β export and release from cells. Seminars in cell & developmental biology 2018, 83, 36–41. [Google Scholar] [CrossRef]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Petrovski, G.; Kauppinen, A. The Nobel Prized cellular target autophagy in eye diseases. Acta Ophthalmol 2017, 95, 335–336. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Koskela, A.; Blasiak, J.; Kaarniranta, K. Autophagy in drusen biogenesis secondary to age-related macular degeneration. Acta Ophthalmol 2024. [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell death & disease 2018, 8, e2537–e2537. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Frontiers in physiology 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Ferrucci, M.; Biagioni, F.; Berti, C.; Bumah, V.V.; Busceti, C.L.; Puglisi-Allegra, S.; Lazzeri, G.; Frati, A.; Fornai, F. Autophagy Activation Promoted by Pulses of Light and Phytochemicals Counteracting Oxidative Stress during Age-Related Macular Degeneration. Antioxidants 2023, 12, 1183. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. Antioxidative Role of Heterophagy, Autophagy, and Mitophagy in the Retina and Their Association with the Age-Related Macular Degeneration (AMD) Etiopathogenesis. Antioxidants 2023, 12, 1368. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Yu, X.; Zhu, J.; Zeng, J.; Kuang, X.; Zhang, Y.; Tang, S.; Zhang, Q.; Yan, J.; Shen, H. Gastrodin ameliorates oxidative stress-induced RPE damage by facilitating autophagy and phagocytosis through PPARα-TFEB/CD36 signaling pathway. Free Radic Biol Med 2024, 224, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sharma, R.; Bammidi, S.; Koontz, V.; Nemani, M.; Yazdankhah, M.; Kedziora, K.M.; Stolz, D.B.; Wallace, C.T.; Yu-Wei, C.; et al. The AKT2/SIRT5/TFEB pathway as a potential therapeutic target in nonneovascular AMD. Nat Commun 2024, 15, 6150. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Koskela, A.; Blasiak, J.; Kaarniranta, K. Autophagy in drusen biogenesis secondary to age-related macular degeneration. Acta Ophthalmol 2024, 102, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Vessey, K.A.; Jobling, A.I.; Greferath, U.; Fletcher, E.L. Pharmaceutical therapies targeting autophagy for the treatment of age-related macular degeneration. Current Opinion in Pharmacology 2024, 76, 102463. [Google Scholar] [CrossRef]
- Khachigian, L.M.; Liew, G.; Teo, K.Y.C.; Wong, T.Y.; Mitchell, P. Emerging therapeutic strategies for unmet need in neovascular age-related macular degeneration. Journal of translational medicine 2023, 21, 133. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Mechanisms of Selective Autophagy. Annu Rev Cell Dev Biol 2021, 37, 143–169. [Google Scholar] [CrossRef]
- Zhang, Y.; Cross, S.D.; Stanton, J.B.; Marmorstein, A.D.; Le, Y.Z.; Marmorstein, L.Y. Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Molecular vision 2017, 23, 228–241. [Google Scholar] [PubMed]
- Abokyi, S.; To, C.-H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxidative medicine and cellular longevity 2020, 2020, 7901270–7901270. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Piippo, N.; Korhonen, E.; Hytti, M.; Kinnunen, K.; Kaarniranta, K.; Kauppinen, A. Oxidative Stress is the Principal Contributor to Inflammasome Activation in Retinal Pigment Epithelium Cells with Defunct Proteasomes and Autophagy. Cell Physiol Biochem 2018, 49, 359–367. [Google Scholar] [CrossRef]
- LaVail, M.M. Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science (New York, N.Y.) 1976, 194, 1071–1074. [Google Scholar] [CrossRef]
- Blasiak, J.; Sobczuk, P.; Pawlowska, E.; Kaarniranta, K. Interplay between aging and other factors of the pathogenesis of age-related macular degeneration. Aging research reviews 2022, 81, 101735. [Google Scholar] [CrossRef]
- Paterno, J.J.; Koskela, A.; Hyttinen, J.M.T.; Vattulainen, E.; Synowiec, E.; Tuuminen, R.; Watala, C.; Blasiak, J.; Kaarniranta, K. Autophagy Genes for Neovascular Age-Related Macular Degeneration in a Finnish Case‒Control Study. Genes 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xiao, L.; Zhang, Z.; Wang, Y.; Kouis, P.; Rasmussen, L.J.; Dai, F. Effects of reactive oxygen species and mitochondrial dysfunction on reproductive aging. Frontiers in cell and developmental biology 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America 2014, 111, E4439–4448. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox biology 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sridevi Gurubaran, I.; Viiri, J.; Koskela, A.; Hyttinen, J.M.T.; Paterno, J.J.; Kis, G.; Antal, M.; Urtti, A.; Kauppinen, A.; Felszeghy, S.; et al. Mitophagy in the Retinal Pigment Epithelium of Dry Age-Related Macular Degeneration Investigated in the NFE2L2/PGC-1α(-/-) Mouse Model. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Tan, H.W.S.; Lu, G.; Dong, H.; Cho, Y.L.; Natalia, A.; Wang, L.; Chan, C.; Kappei, D.; Taneja, R.; Ling, S.C.; et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat Commun 2022, 13, 3720. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.L.; Grune, T.; Hohn, A. Mitochondrial contribution to lipofuscin formation. Redox biology 2017, 11, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cellular and molecular life sciences: CMLS 2020, 77, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.D.; Narita, M. Autophagy at the intersection of aging, senescence, and cancer. Molecular Oncology 2022, 16, 3259–3275. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Hyttinen, J.; Ryhanen, T.; Viiri, J.; Paimela, T.; Toropainen, E.; Sorri, I.; Salminen, A. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front Biosci (Elite Ed) 2010, 2, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Cenci, S. Autophagy and Protein Secretion. J Mol Biol 2020, 432, 2525–2545. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J Neuroinflammation 2021, 18, 32. [Google Scholar] [CrossRef]
- Chung, H.; Kim, C. Nutlin-3a for age-related macular degeneration. Aging 2022, 14, 5614–5616. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Reddy, S.T.; Hinton, D.R.; Kannan, R. Mechanisms of RPE senescence and potential role of αB crystallin peptide as a senolytic agent in experimental AMD. Experimental eye research 2022, 215, 108918. [Google Scholar] [CrossRef]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M.; et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduction and Targeted Therapy 2023, 8, 352. [Google Scholar] [CrossRef]
- Bilbao-Malavé, V.; González-Zamora, J.; de la Puente, M.; Recalde, S.; Fernandez-Robredo, P.; Hernandez, M.; Layana, A.G.; Saenz de Viteri, M. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Age Related Macular Degeneration, Role in Pathophysiology, and Possible New Therapeutic Strategies. Antioxidants (Basel, Switzerland) 2021, 10. [CrossRef]
- Kheitan, S.; Minuchehr, Z.; Soheili, Z.S. Exploring the cross talk between ER stress and inflammation in age-related macular degeneration. PloS one 2017, 12, e0181667. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Experimental & molecular medicine 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; DuPont, N.; Castillo, E.F.; Deretic, V. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun 2013, 5, 471–479. [Google Scholar] [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cellular and Molecular Life Sciences 2021, 78, 4487–4505. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: a double-edged sword. Science (New York, N.Y.) 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Bo, Q.; Shen, M.; Xiao, M.; Liang, J.; Zhai, Y.; Zhu, H.; Jiang, M.; Wang, F.; Luo, X.; Sun, X. 3-Methyladenine Alleviates Experimental Subretinal Fibrosis by Inhibiting Macrophages and M2 Polarization Through the PI3K/Akt Pathway. Journal of ocular pharmacology and therapeutics: the official journal of the Association for Ocular Pharmacology and Therapeutics 2020, 36, 618–628. [Google Scholar] [CrossRef]
- Bao, Y.; Ding, Z.; Zhao, P.; Li, J.; Chen, P.; Zheng, J.; Qian, Z. Autophagy inhibition potentiates the anti-EMT effects of alteronol through TGF-β/Smad3 signaling in melanoma cells. Cell death & disease 2020, 11, 223. [Google Scholar] [CrossRef]
- Chen, H.-T.; Liu, H.; Mao, M.-J.; Tan, Y.; Mo, X.-Q.; Meng, X.-J.; Cao, M.-T.; Zhong, C.-Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial–mesenchymal transition and its application in cancer therapy. Molecular cancer 2019, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Gundamaraju, R.; Lu, W.; Paul, M.K.; Jha, N.K.; Gupta, P.K.; Ojha, S.; Chattopadhyay, I.; Rao, P.V.; Ghavami, S. Autophagy and EMT in cancer and metastasis: Who controls whom? Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2022, 1868, 166431. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Liu, S. The role of epithelial–mesenchymal transition and autophagy in pancreatic ductal adenocarcinoma invasion. Cell death & disease 2023, 14, 506. [Google Scholar] [CrossRef]
- Hill, C.; Wang, Y. Autophagy in pulmonary fibrosis: friend or foe? Genes & diseases 2022, 9, 1594–1607. [Google Scholar] [CrossRef]
- Yue, Y.L.; Zhang, M.Y.; Liu, J.Y.; Fang, L.J.; Qu, Y.Q. The role of autophagy in idiopathic pulmonary fibrosis: from mechanisms to therapies. Ther Adv Respir Dis 2022, 16, 17534666221140972. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Baek, A.R.; Lee, J.H.; Jang, A.S.; Kim, D.J.; Chin, S.S.; Park, S.W. IL-37 Attenuates Lung Fibrosis by Inducing Autophagy and Regulating TGF-β1 Production in Mice. Journal of immunology (Baltimore, Md.: 1950) 2019, 203, 2265–2275. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Qiu, T.; Liu, W.; Yao, P. Autophagy, an important therapeutic target for pulmonary fibrosis diseases. Clinica Chimica Acta 2020, 502, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol 2005, Chapter 28, Unit 28.21. [CrossRef]
- Nitta, A.; Hori, K.; Tanida, I.; Igarashi, A.; Deyama, Y.; Ueno, T.; Kominami, E.; Sugai, M.; Aoki, K. Blocking LC3 lipidation and ATG12 conjugation reactions by ATG7 mutant protein containing C572S. Biochemical and biophysical research communications 2019, 508, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhao, X.; Guo, Q.; Feng, Y.; Ma, M.; Guo, W.; Dong, X.; Deng, C.; Li, C.; Song, X.; et al. Autophagy resists EMT process to maintain retinal pigment epithelium homeostasis. International Journal of Biological Sciences 2019, 15, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Giardina, B. Circulating tumor cells and cancer stem cells: A role for proteomics in defining the interrelationships between function, phenotype and differentiation with potential clinical applications. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2013, 1835, 129–143. [Google Scholar] [CrossRef]
- Miao, Q.; Xu, Y.; Yin, H.; Zhang, H.; Ye, J. KRT8 phosphorylation regulates the epithelial–mesenchymal transition in retinal pigment epithelial cells through autophagy modulation. Journal of cellular and molecular medicine 2020, 24, 3217–3228. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Shang, P.; Terasaki, H.; Stepicheva, N.; Hose, S.; Yazdankhah, M.; Weiss, J.; Sakamoto, T.; Bhutto, I.A.; Xia, S.; et al. A Role for βA3/A1-Crystallin in Type 2 EMT of RPE Cells Occurring in Dry Age-Related Macular Degeneration. Investigative ophthalmology & visual science 2018, 59, Amd104–amd113. [Google Scholar] [CrossRef]
- Wu, J.; Chen, X.; Liu, X.; Huang, S.; He, C.; Chen, B.; Liu, Y. Autophagy regulates TGF-β2-induced epithelial–mesenchymal transition in human retinal pigment epithelium cells. Molecular medicine reports 2018, 17, 3607–3614. [Google Scholar] [CrossRef]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Archives of ophthalmology (Chicago, Ill.: 1960) 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed]
- Booij, J.C.; Baas, D.C.; Beisekeeva, J.; Gorgels, T.G.; Bergen, A.A. The dynamic nature of Bruch’s membrane. Progress in retinal and eye research 2010, 29, 1–18. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.S.; Grebe, R.; Bhutto, I.; Merges, C.; Baba, T.; Lutty, G.A. Relationship between RPE and Choriocapillaris in Age-Related Macular Degeneration. Investigative ophthalmology & visual science 2009, 50, 4982–4991. [Google Scholar] [CrossRef]
- George, S.M.; Lu, F.; Rao, M.; Leach, L.L.; Gross, J.M. The retinal pigment epithelium: Development, injury responses, and regenerative potential in mammalian and nonmammalian systems. Progress in retinal and eye research 2021, 85, 100969. [Google Scholar] [CrossRef]
- Migneault, F.; Hébert, M.J. Autophagy, tissue repair, and fibrosis: a delicate balance. Matrix Biol 2021, 100-101, 182-196. [CrossRef]
- Neill, T.; Kapoor, A.; Xie, C.; Buraschi, S.; Iozzo, R.V. A functional outside-in signaling network of proteoglycans and matrix molecules regulating autophagy. Matrix Biol 2021, 100-101, 118-149. [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Decoding the Matrix: Instructive Roles of Proteoglycan Receptors. Biochemistry 2015, 54, 4583–4598. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Tredup, C.; Gubbiotti, M.A.; Iozzo, R.V. Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology. Febs j 2017, 284, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial–Mesenchymal Transition. Frontiers in cell and developmental biology 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Cai, Y.; Huang, J.; He, X.; Han, W.; Chen, W. Impairment of autophagy promotes human conjunctival fibrosis and pterygium occurrence by enhancing the SQSTM1-NF-κB signaling pathway. Journal of molecular cell biology 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.J.; Rong, S.S.; Xu, Y.S.; Zheng, L.B.; Qiu, W.Y.; Zhang, X.; Jiang, L.J.; Duan, R.P.; Tian, T.; Yao, Y.F. Anti-fibrosis potential of pirarubicin by inducing apoptotic and autophagic cell death in rabbit conjunctiva. Experimental eye research 2020, 200, 108215. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, S.; Fan, W.; Rokohl, A.C.; Ju, S.; Ju, X.; Guo, Y.; Heindl, L.M. Recent advances in graves ophthalmopathy medical therapy: a comprehensive literature review. International Ophthalmology 2023, 43, 1437–1449. [Google Scholar] [CrossRef]
- Lee, C.E.; Kim, J.Y.; Yoon, J.S.; Ko, J. Role of Inositol-Requiring Enzyme 1 and Autophagy in the Pro-Fibrotic Mechanism Underlying Graves’ Orbitopathy. Yonsei Med J 2024, 65, 397–405. [Google Scholar] [CrossRef]
- Martin, L.M.; Jeyabalan, N.; Tripathi, R.; Panigrahi, T.; Johnson, P.J.; Ghosh, A.; Mohan, R.R. Autophagy in corneal health and disease: A concise review. Ocul Surf 2019, 17, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.W.; Yu, W.; Wang, Y.N.; Zhang, X.Y.; Song, S.Q.; Gong, S.Y.; Meng, L.Y.; Gan, C.; Liu, B.J.; Gong, Q. Effects of autophagy inhibitor 3-methyladenine on a diabetic mice model. International journal of ophthalmology 2023, 16, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Koskela, A.; Pawlowska, E.; Liukkonen, M.; Ruuth, J.; Toropainen, E.; Hyttinen, J.M.T.; Viiri, J.; Eriksson, J.E.; Xu, H.; et al. Epithelial–Mesenchymal Transition and Senescence in the Retinal Pigment Epithelium of NFE2L2/PGC-1α Double Knock-Out Mice. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- Acosta, A.C.; Joud, H.; Sun, M.; Avila, M.Y.; Margo, C.E.; Espana, E.M. Keratocyte-Derived Myofibroblasts: Functional Differences With Their Fibroblast Precursors. Investigative ophthalmology & visual science 2023, 64, 9–9. [Google Scholar] [CrossRef]
- Schaefer, L.; Dikic, I. Autophagy: Instructions from the extracellular matrix. Matrix Biology 2021, 100-101, 1-8. [CrossRef]
- García-Onrubia, L.; Valentín-Bravo, F.J.; Coco-Martin, R.M.; González-Sarmiento, R.; Pastor, J.C.; Usategui-Martín, R.; Pastor-Idoate, S. Matrix Metalloproteinases in Age-Related Macular Degeneration (AMD). International journal of molecular sciences 2020, 21, 5934. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M. Fibrosis and diseases of the eye. The Journal of clinical investigation 2007, 117, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Hanumunthadu, D. Inflammatory eye disease: An overview of clinical presentation and management. Clin Med (Lond) 2022, 22, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Zehender, A.; Li, Y.-N.; Lin, N.-Y.; Stefanica, A.; Nüchel, J.; Chen, C.-W.; Hsu, H.-H.; Zhu, H.; Ding, X.; Huang, J.; et al. TGFβ promotes fibrosis by MYST1-dependent epigenetic regulation of autophagy. Nat Commun 2021, 12, 4404. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy participates in, well, just about everything. Cell Death & Differentiation 2020, 27, 831–832. [Google Scholar] [CrossRef]
- Pollari, V.; Reay, M.; Leino, L.; Assmuth, T.; Wirman, L.; Toropainen, E.; Kaarniranta, K. Preclinical in vivo tolerability assessment of silica as an intravitreal drug delivery vehicle. Investigative ophthalmology & visual science 2024, 65, 6118–6118. [Google Scholar]
Figure 1.
Age-related macular degeneration (AMD). Fundus images of the healthy macula (A), the macula of an intermediate dry AMD (B), late dry AMD (geographic atrophy, C), and neovascular AMD. The yellow arrow indicates drusen, the black arrow indicates retinal atrophy, and the white arrow indicates hemorrhages. Subretinal hemorrhage in neovascular AMD results in disturbances or loss of central vision symbolically presented in E.
Figure 1.
Age-related macular degeneration (AMD). Fundus images of the healthy macula (A), the macula of an intermediate dry AMD (B), late dry AMD (geographic atrophy, C), and neovascular AMD. The yellow arrow indicates drusen, the black arrow indicates retinal atrophy, and the white arrow indicates hemorrhages. Subretinal hemorrhage in neovascular AMD results in disturbances or loss of central vision symbolically presented in E.
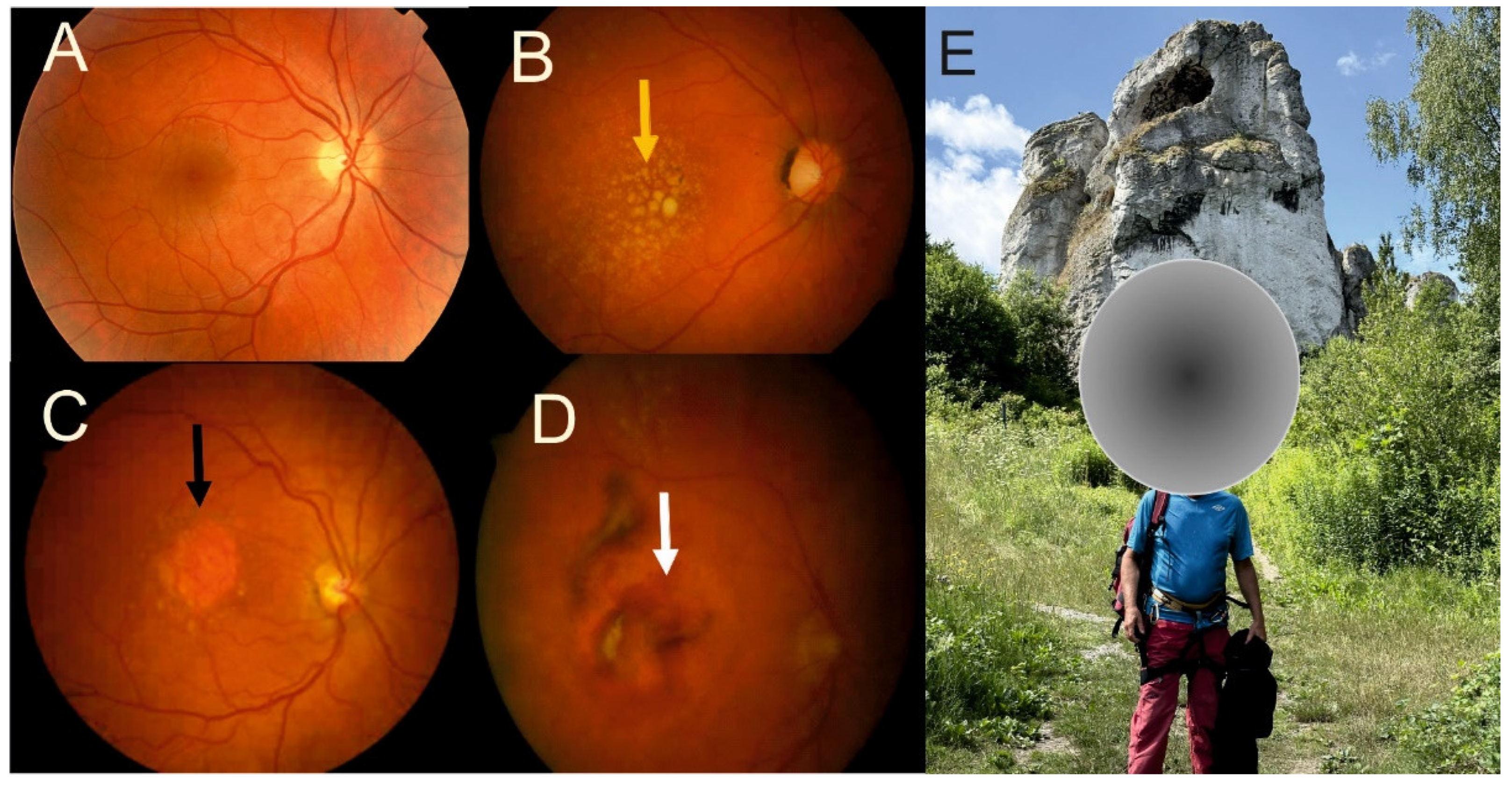
Figure 2.
Fibrosis in neovascular age-related macular degeneration (AMD). Fluorescein angiography (FAG) revealed early macular neovascular membrane (white arrows) with early fluorescein filling (20 and 30 sec) and late (5 min) staining with leakage. The FAG image corresponds to an early optical coherent tomography (OCT) image with retinal pigment epithelial (RPE) detachment (green arrow). Four years after 20 intravitreal injections, the disease progressed to late fibrotic neovascular AMD (red arrow). The green lines indicate degenerated RPE layers. MNV – macular neovascularization.
Figure 2.
Fibrosis in neovascular age-related macular degeneration (AMD). Fluorescein angiography (FAG) revealed early macular neovascular membrane (white arrows) with early fluorescein filling (20 and 30 sec) and late (5 min) staining with leakage. The FAG image corresponds to an early optical coherent tomography (OCT) image with retinal pigment epithelial (RPE) detachment (green arrow). Four years after 20 intravitreal injections, the disease progressed to late fibrotic neovascular AMD (red arrow). The green lines indicate degenerated RPE layers. MNV – macular neovascularization.
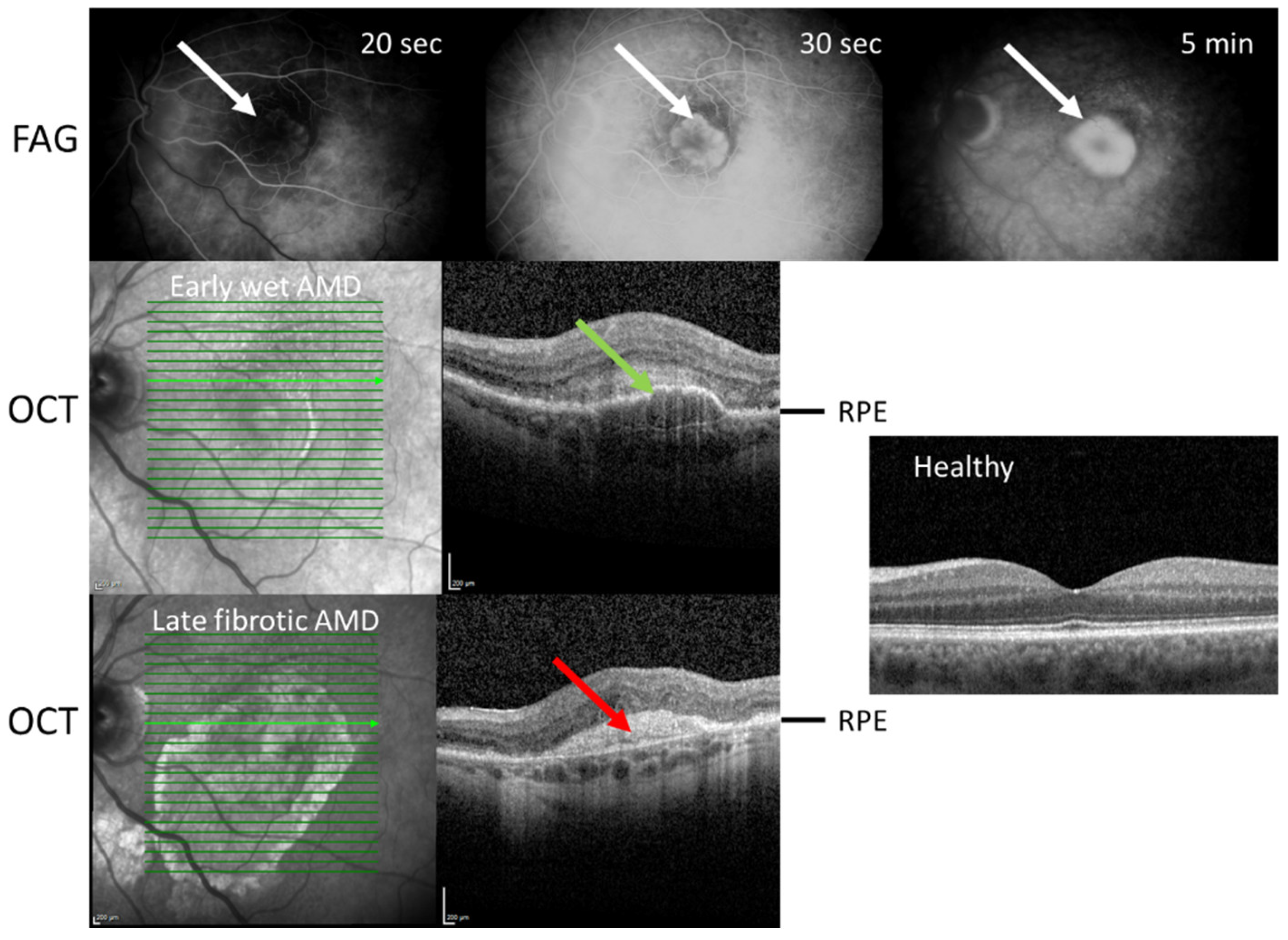
Figure 3.
Epithelial‒mesenchymal transition (EMT) in retinal pigment epithelial (RPE) cells. During EMT, cuboidal, polarized, tightly adjacent, and nonmotile RPE cells transdifferentiate into spindle-shaped mesenchymal cells that acquire migratory and contractile properties and lose mutual contact. Created with BioRender.com.
Figure 3.
Epithelial‒mesenchymal transition (EMT) in retinal pigment epithelial (RPE) cells. During EMT, cuboidal, polarized, tightly adjacent, and nonmotile RPE cells transdifferentiate into spindle-shaped mesenchymal cells that acquire migratory and contractile properties and lose mutual contact. Created with BioRender.com.
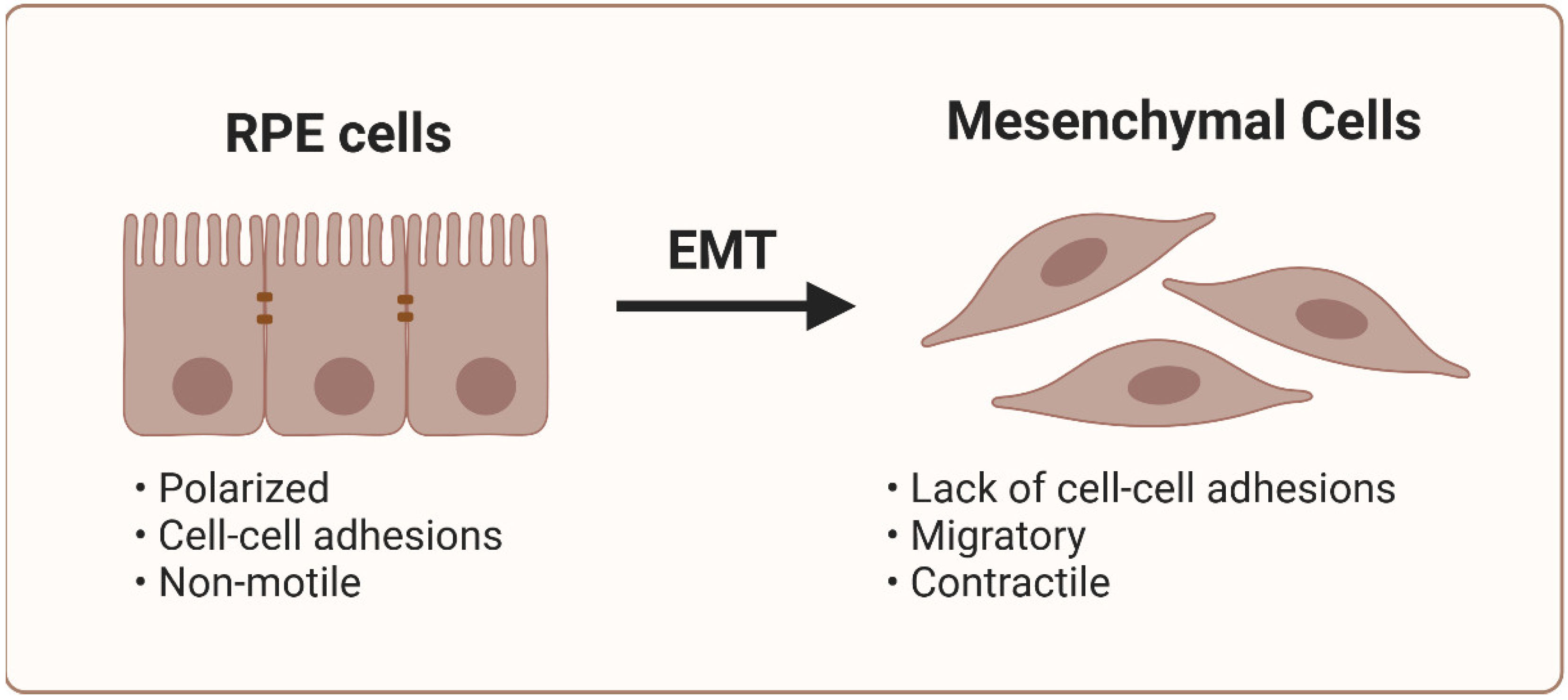
Figure 4.
Signaling pathways and mechanistic drivers involved in mesenchymal‒epithelial transition (EMT) and endothelial‒mesenchymal transition (EndMT) in neovascular age-related macular degeneration (AMD). The action of numerous extracellular cytokines and receptor tyrosine kinases (RTKs) activates the EMT of retinal pigment epithelial cells and the EndMT of choroidal endothelial cells. TGFB2 is the master regulator that activates the canonical SMAD and noncanonical signaling pathways. The WNT/CTNB1 (catenin beta-1) signaling pathway also plays a key role in both EMT and EndMT. The activation of EMT and EndMT results in the upregulation of several transcription factors (TFs) encoding mesenchymal genes and the downregulation of TFs encoding epithelial and endothelial genes. This ultimately leads to the transdifferentiation of epithelial and endothelial cells to myofibroblasts and excessive extracellular matrix deposition, which occurs in subretinal fibrotic lesions in AMD. JAGGED/NOTCH – protein Jagged1/2/neurogenic locus notch homolog protein 1, YAP – transcription activator yes 1; PI3K – phosphatidylinositol 3-kinase; AKT – AKT serine/threonine kinase 1; MAPK – mitogen-activated protein kinase; MTOR – mechanistic/mammalian target of rapamycin. Created with BioRender.com.
Figure 4.
Signaling pathways and mechanistic drivers involved in mesenchymal‒epithelial transition (EMT) and endothelial‒mesenchymal transition (EndMT) in neovascular age-related macular degeneration (AMD). The action of numerous extracellular cytokines and receptor tyrosine kinases (RTKs) activates the EMT of retinal pigment epithelial cells and the EndMT of choroidal endothelial cells. TGFB2 is the master regulator that activates the canonical SMAD and noncanonical signaling pathways. The WNT/CTNB1 (catenin beta-1) signaling pathway also plays a key role in both EMT and EndMT. The activation of EMT and EndMT results in the upregulation of several transcription factors (TFs) encoding mesenchymal genes and the downregulation of TFs encoding epithelial and endothelial genes. This ultimately leads to the transdifferentiation of epithelial and endothelial cells to myofibroblasts and excessive extracellular matrix deposition, which occurs in subretinal fibrotic lesions in AMD. JAGGED/NOTCH – protein Jagged1/2/neurogenic locus notch homolog protein 1, YAP – transcription activator yes 1; PI3K – phosphatidylinositol 3-kinase; AKT – AKT serine/threonine kinase 1; MAPK – mitogen-activated protein kinase; MTOR – mechanistic/mammalian target of rapamycin. Created with BioRender.com.
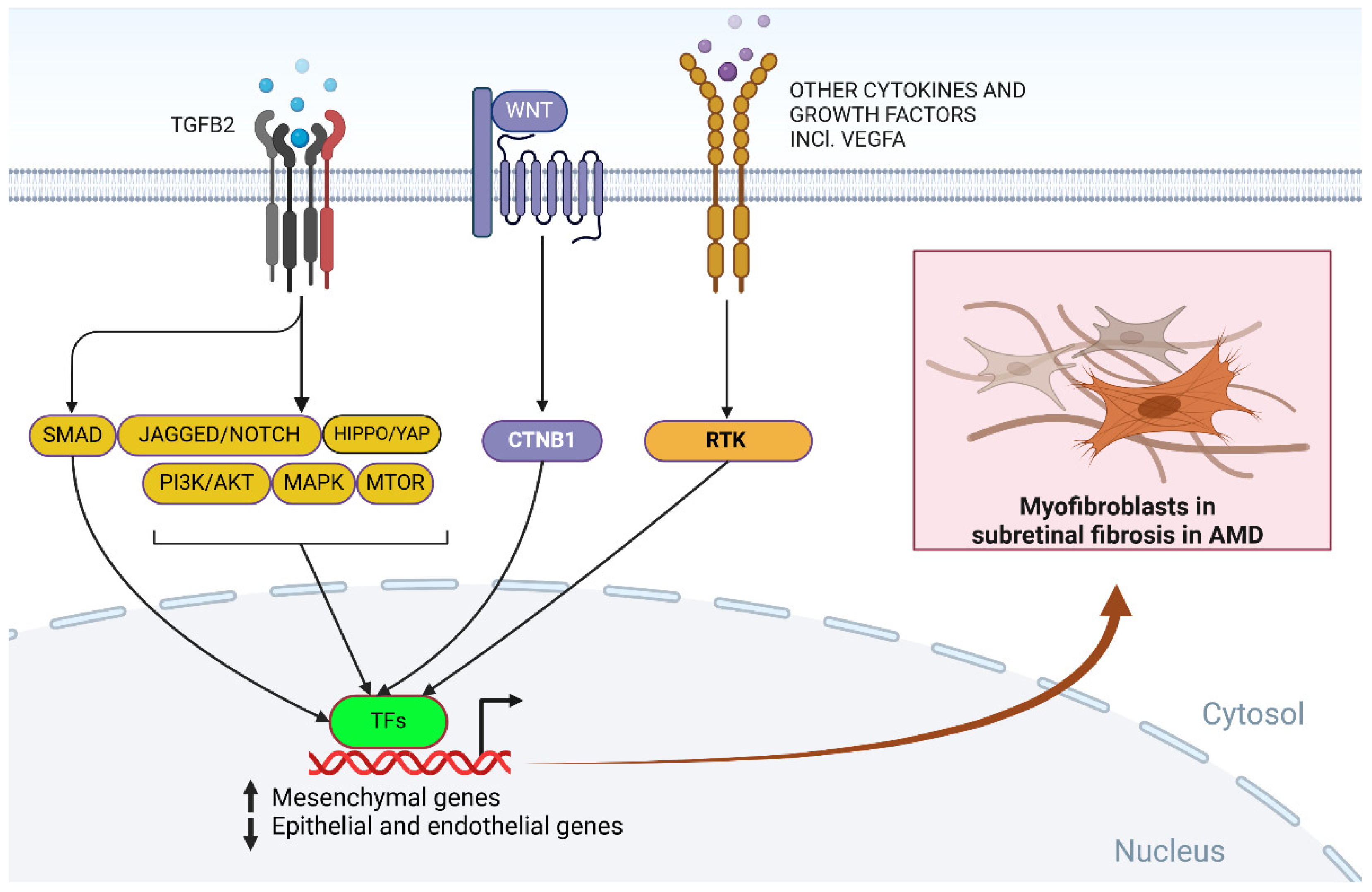
Figure 5.
Outline of the degradative and secretory autophagy pathways. Both types of autophagy are initiated by the formation of the isolation membrane (phagophore), which grows to form an autophagosome to encapsulate the material to be degraded or secreted (cargo). The role of a phagophore in secretory autophagy may differ from that of its degradative counterpart. In macroautophagy, the autophagosome fuses with the lysosome to form an autolysosome in which the cargo is degraded by lysosomal enzymes. A variant of macroautophagy may occur with the involvement of a late endosome (dark blue circle) and the formation of an amphisome. In chaperone-mediated autophagy, the cargo carries a specific motif, exemplified here by a protein with the lysine–phenylalanine–glutamic acid–arginine–glutamine sequence recognized by a lysosomal membrane-bound protein with the involvement of a chaperone, exemplified here by heat shock cognate 71 kDa protein (HSC70). In microautophagy, the cargo is directly engulfed by a lysosome and degraded. In secretory autophagy, the autophagosome is different from its degradative counterpart and fuses with the plasma membrane to extrude the cargo. Created with BioRender.com.
Figure 5.
Outline of the degradative and secretory autophagy pathways. Both types of autophagy are initiated by the formation of the isolation membrane (phagophore), which grows to form an autophagosome to encapsulate the material to be degraded or secreted (cargo). The role of a phagophore in secretory autophagy may differ from that of its degradative counterpart. In macroautophagy, the autophagosome fuses with the lysosome to form an autolysosome in which the cargo is degraded by lysosomal enzymes. A variant of macroautophagy may occur with the involvement of a late endosome (dark blue circle) and the formation of an amphisome. In chaperone-mediated autophagy, the cargo carries a specific motif, exemplified here by a protein with the lysine–phenylalanine–glutamic acid–arginine–glutamine sequence recognized by a lysosomal membrane-bound protein with the involvement of a chaperone, exemplified here by heat shock cognate 71 kDa protein (HSC70). In microautophagy, the cargo is directly engulfed by a lysosome and degraded. In secretory autophagy, the autophagosome is different from its degradative counterpart and fuses with the plasma membrane to extrude the cargo. Created with BioRender.com.
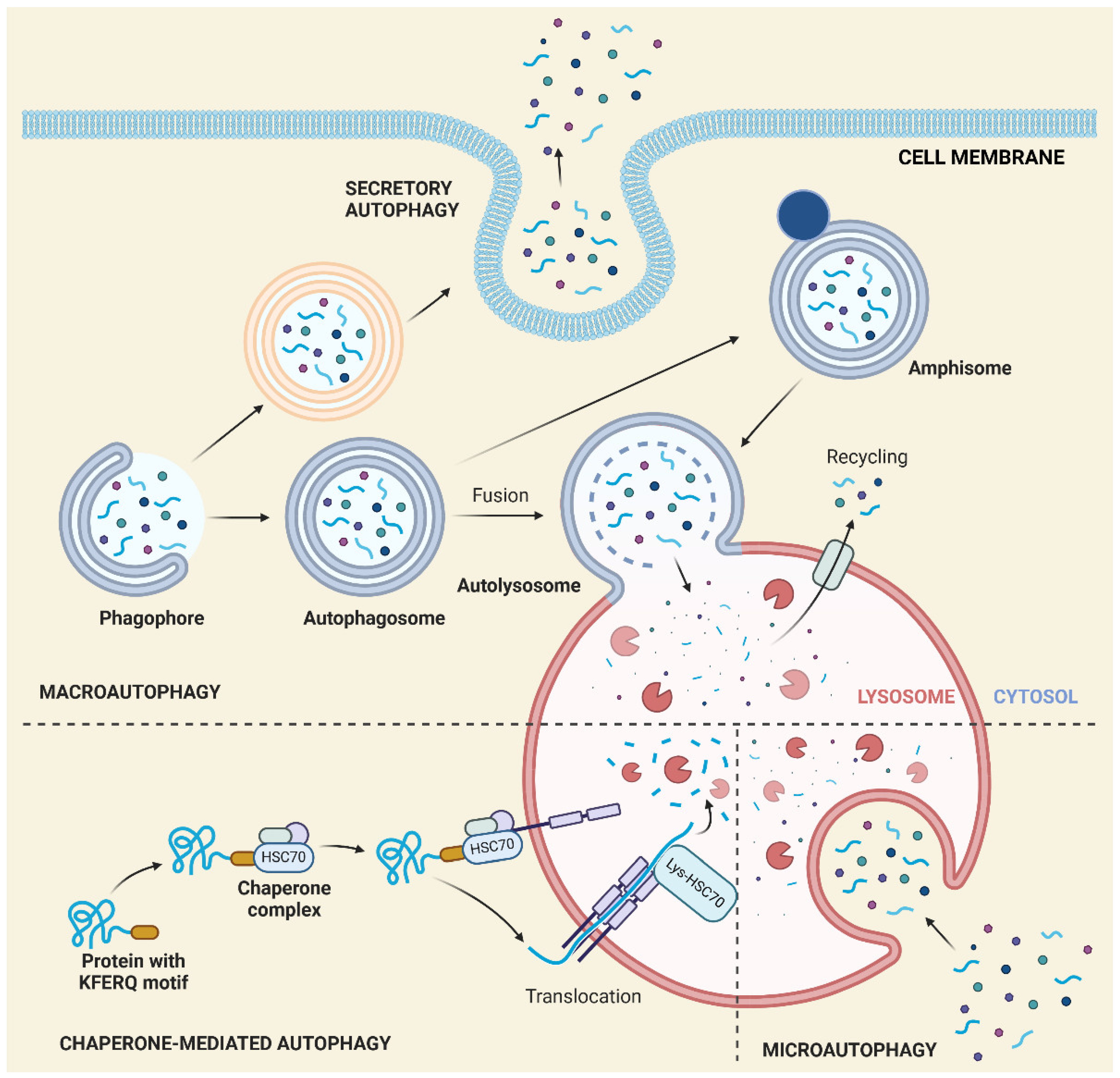
Figure 6.
LC3-associated phagocytosis (LAP) of shed photoreceptor (blue – rods, red – cons) segments (POS) in retinal pigment epithelium (RPE) cells. Phagosomes containing POSs are bound by the lipidated microtubule-associated protein 1A/1B light chain 3ALC3 (LC3) with the involvement of several autophagy proteins not presented in this figure. Created with BioRender.com.
Figure 6.
LC3-associated phagocytosis (LAP) of shed photoreceptor (blue – rods, red – cons) segments (POS) in retinal pigment epithelium (RPE) cells. Phagosomes containing POSs are bound by the lipidated microtubule-associated protein 1A/1B light chain 3ALC3 (LC3) with the involvement of several autophagy proteins not presented in this figure. Created with BioRender.com.
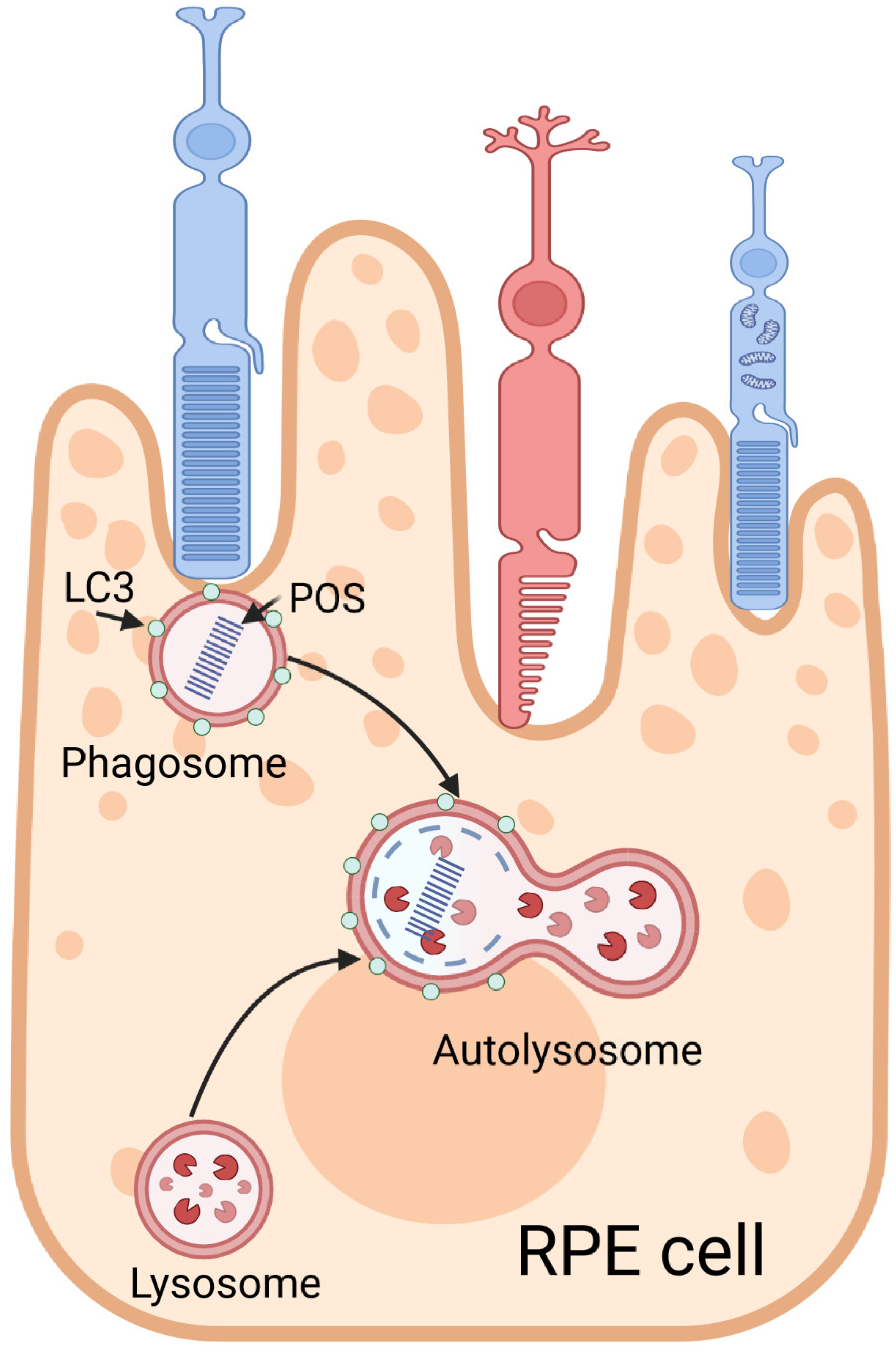
Figure 7.
Potential role of autophagy in subretinal fibrosis in neovascular age-related macular degeneration (AMD). Retinal pigment epithelium (RPE) cells may be stimulated to undergo epithelial‒mesenchymal transition (EMT) by low-level chronic inflammation or severe tissue injury, both of which are typical for neovascular AMD, resulting in transdifferentiation to myofibroblasts, which support fibrosis. The extracellular matrix (ECM) may regulate autophagy, and when dysfunctional, it may deposit ECM proteins to a retinal scar. Myofibroblasts may change the composition of the ECM and contribute to its dysfunction. Beta-crystallin A3 (CRBA1) may positively regulate autophagy and support the epithelial phenotype of RPE cells, preventing myofibroblast transformation and, consequently, fibrosis. The signaling pathways regulating EMT are presented in Figure 4. Subretinal fibrosis may also be promoted by the endothelial‒mesenchymal transition of choroidal fibroblasts and retinal glial cells. Created with BioRender.com.
Figure 7.
Potential role of autophagy in subretinal fibrosis in neovascular age-related macular degeneration (AMD). Retinal pigment epithelium (RPE) cells may be stimulated to undergo epithelial‒mesenchymal transition (EMT) by low-level chronic inflammation or severe tissue injury, both of which are typical for neovascular AMD, resulting in transdifferentiation to myofibroblasts, which support fibrosis. The extracellular matrix (ECM) may regulate autophagy, and when dysfunctional, it may deposit ECM proteins to a retinal scar. Myofibroblasts may change the composition of the ECM and contribute to its dysfunction. Beta-crystallin A3 (CRBA1) may positively regulate autophagy and support the epithelial phenotype of RPE cells, preventing myofibroblast transformation and, consequently, fibrosis. The signaling pathways regulating EMT are presented in Figure 4. Subretinal fibrosis may also be promoted by the endothelial‒mesenchymal transition of choroidal fibroblasts and retinal glial cells. Created with BioRender.com.
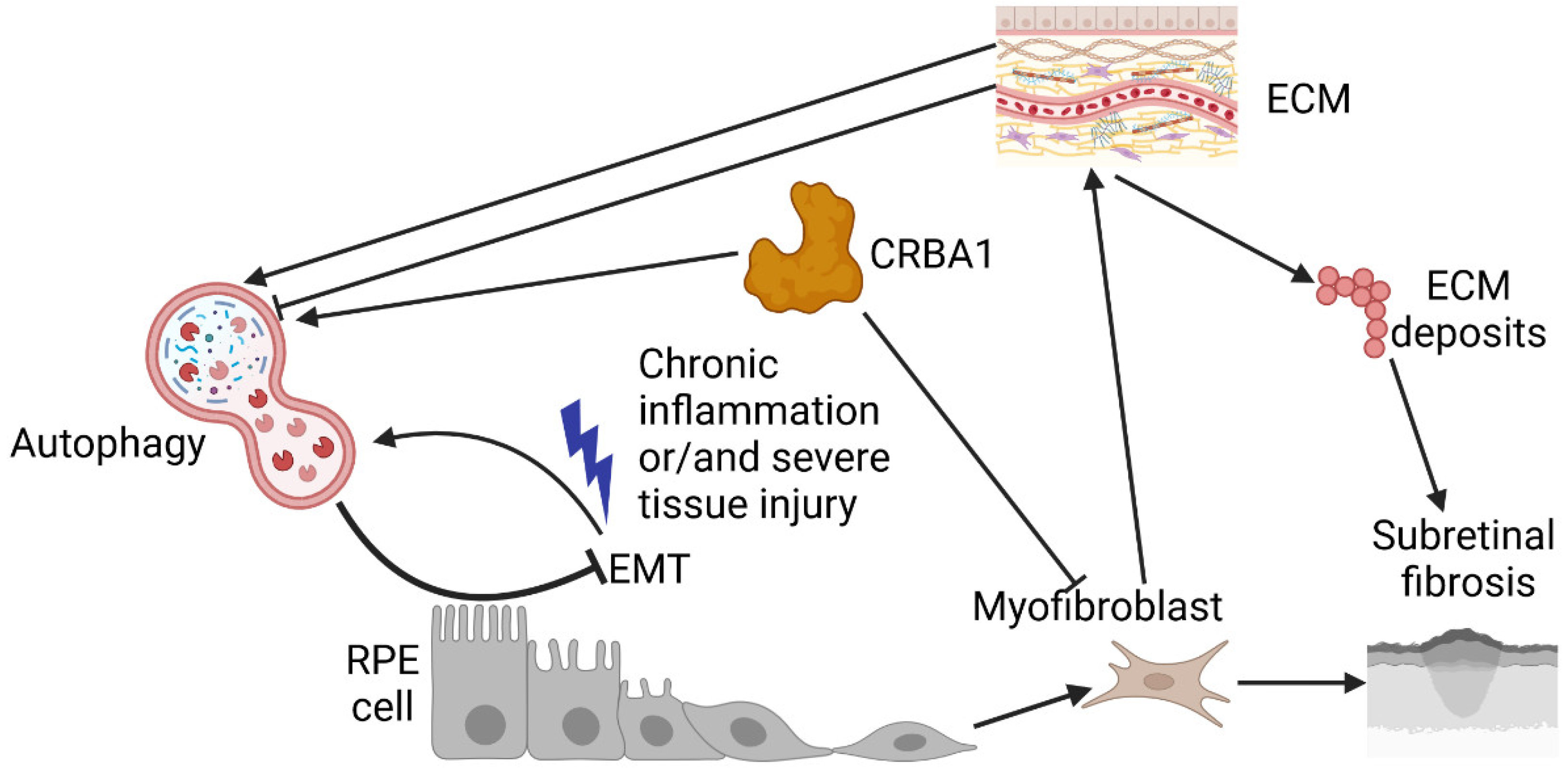
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



