Submitted:
27 November 2024
Posted:
28 November 2024
You are already at the latest version
Abstract
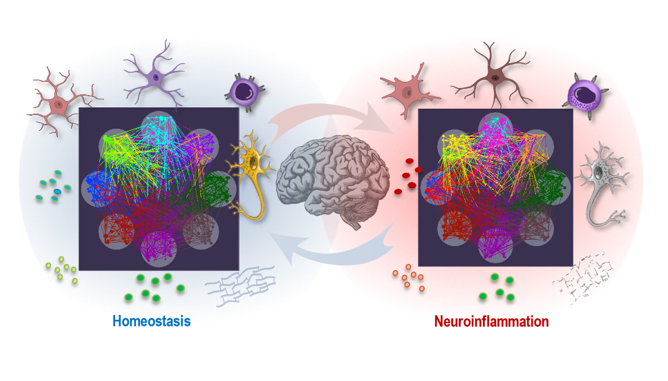
Neuroinflammation is a complex and multifaceted process that involves dynamic interactions among various cellular and molecular components. This sophisticated interplay supports both environmental adaptability and system resilience in the central nervous system (CNS) but may be disrupted during neuroinflammation. In this article, we first characterize the key players in the neuroimmune interactions, including microglia, astrocytes, neurons, immune cells, and essential signaling molecules such as cytokines, neurotransmitters, extracellular matrix (ECM) components, and neurotrophic factors. Under homeostatic conditions, these elements promote cellular cooperation and stability, whereas in neuroinflammatory states, they drive adaptive responses that may become pathological if dysregulated. We examine how neuroimmune interactions, mediated through these cellular actors and signaling pathways, create complex networks that regulate CNS functionality and responses to injury or inflammation. To further elucidate these dynamics, we provide insights into a multilayer network (MLN) approach, highlighting the interconnected nature of neuroimmune interactions under both inflammatory and homeostatic conditions. This perspective aims to enhance understanding of neuroimmune communication and the mechanisms underlying shifts from homeostasis to neuroinflammation. Applying MLN approach offers a more integrative view of CNS resilience and adaptability, helping to clarify inflammatory processes and identify novel intervention points within the layered landscape of neuroinflammatory responses.
Keywords:
1. Introduction
2. The Nervous System as a Dynamic Network: Cellular Interactions and Lifelong Adaptation
3. The Key Cellular and Molecular Players in Homeostatic and Pathological States
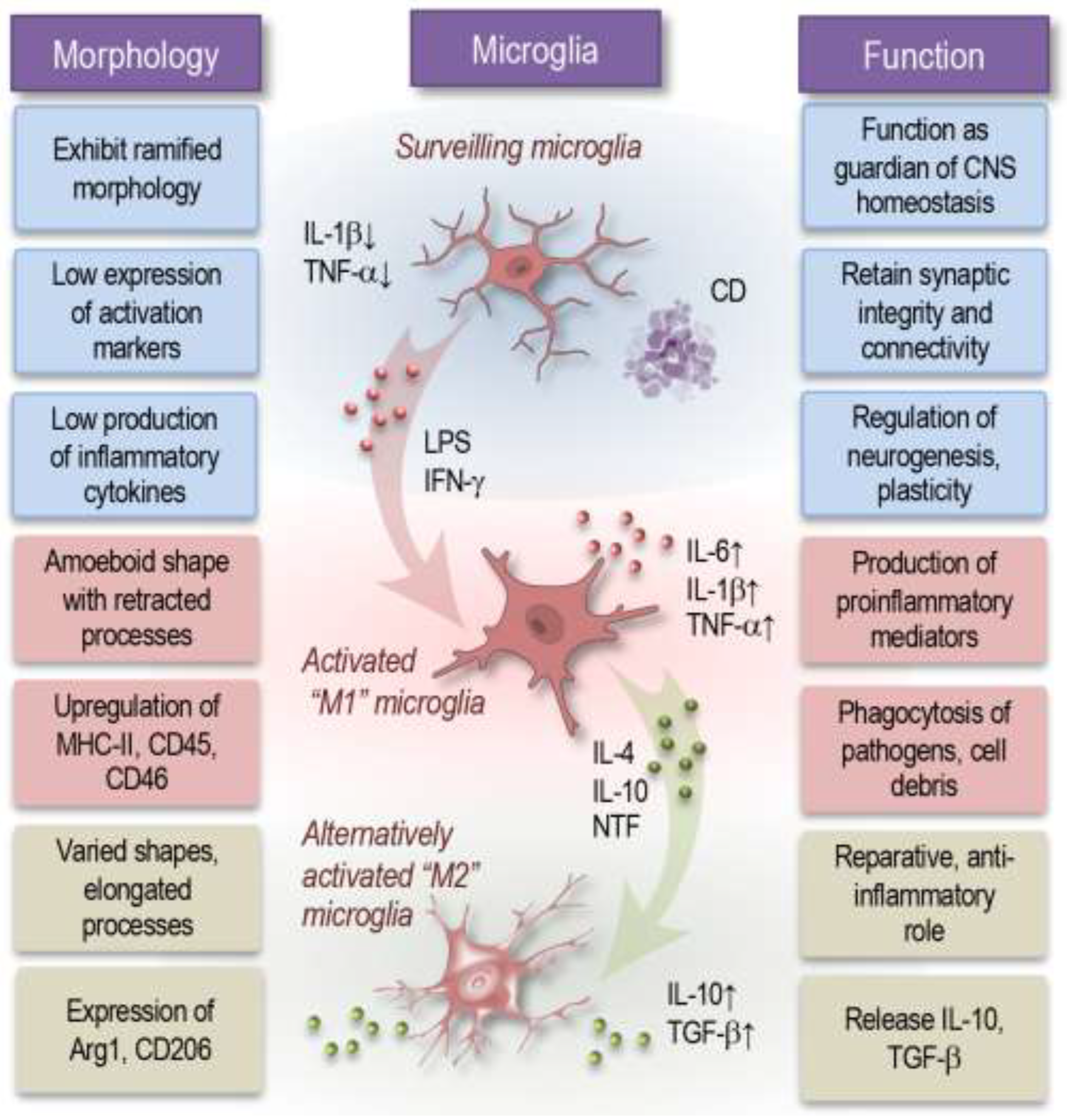
3.1. Microglia
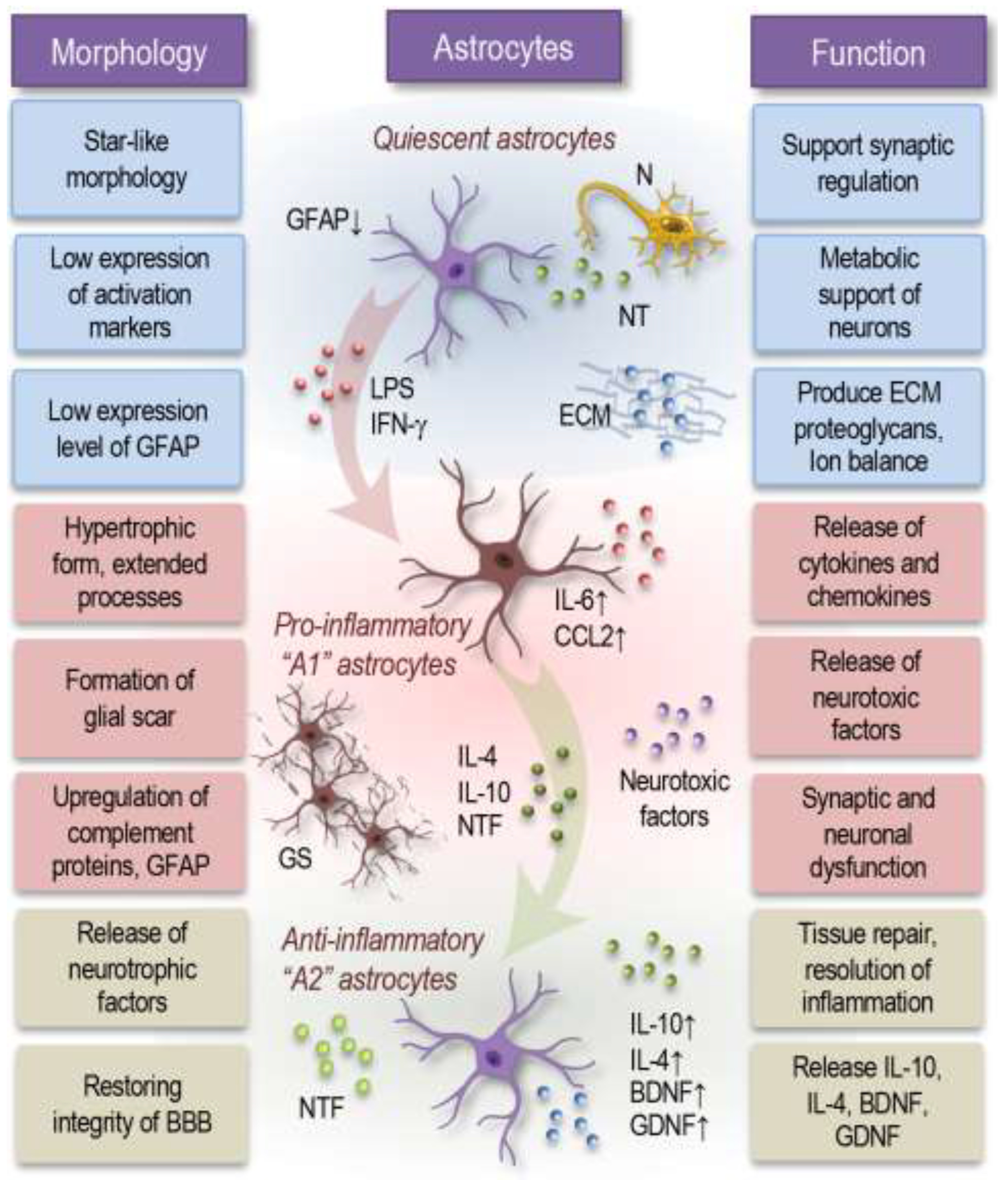
3.2. Astrocytes
3.3. Neurons
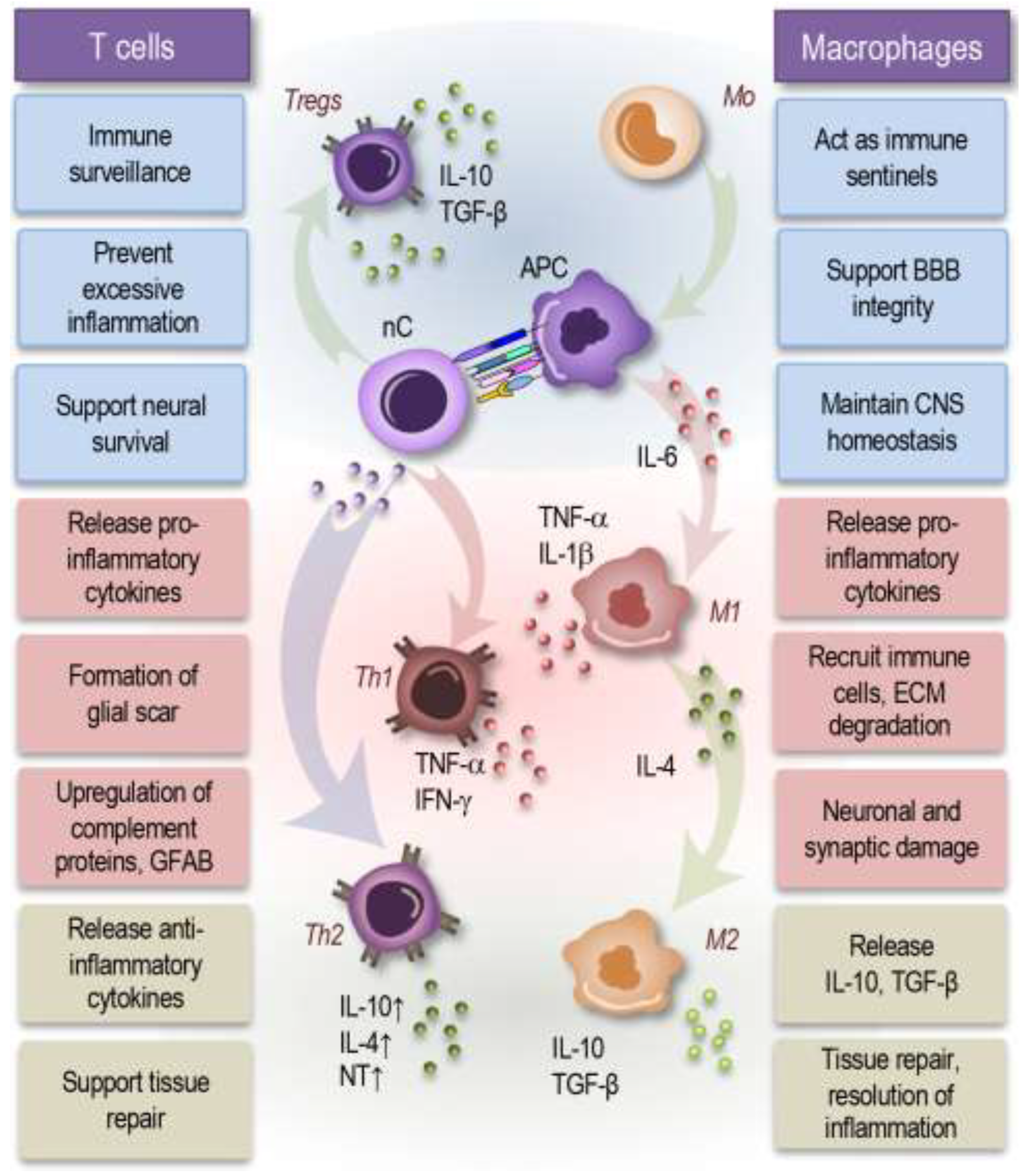
3.4. Immune cells
3.5. ECM
3.6. Cytokines
3.7. Neurotrophic Factors
3.8. Neurotransmitters
4. The Dynamic Interactions Between Neuroimmune Cells and Their Mediators
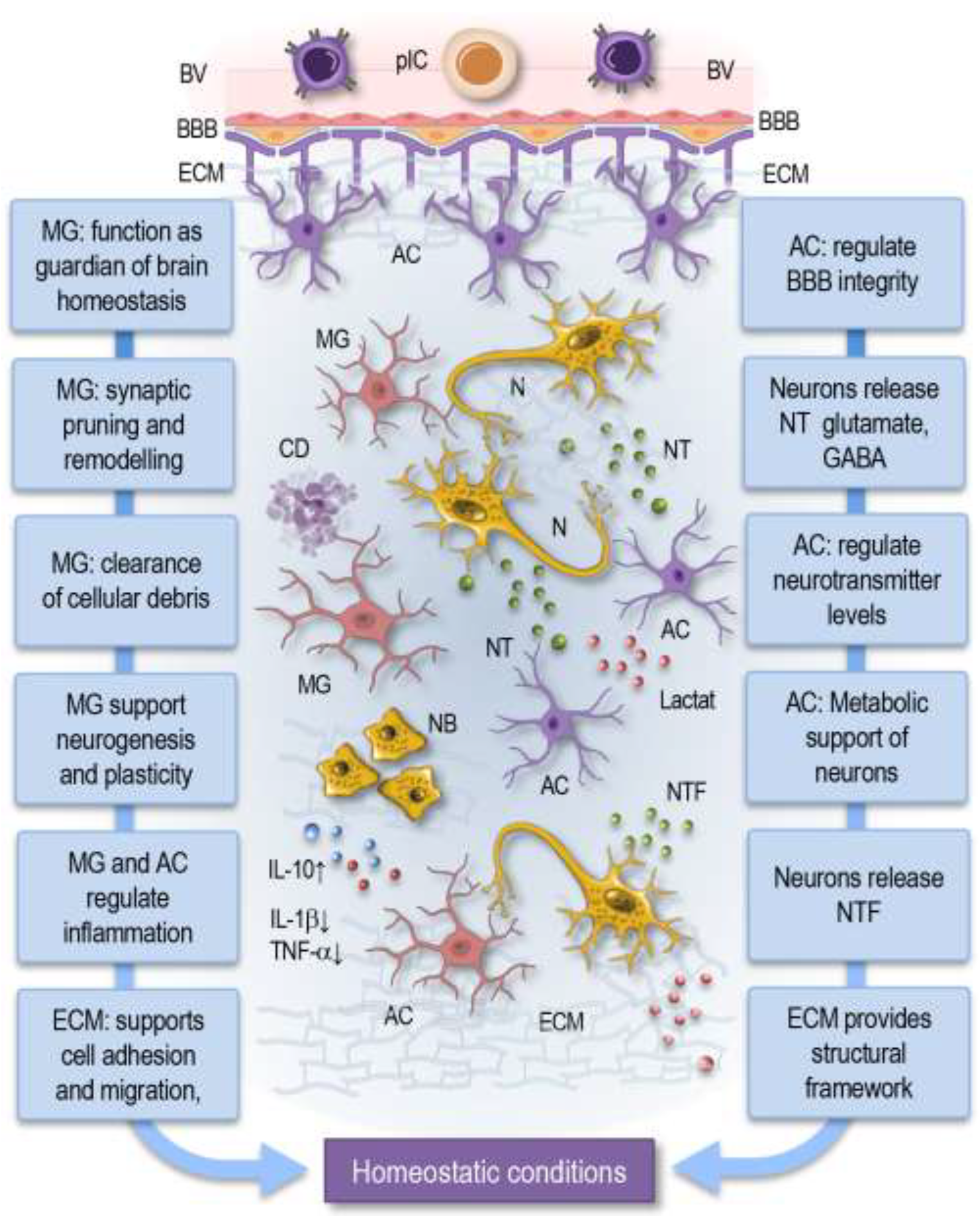
4.1. Homeostatic Conditions
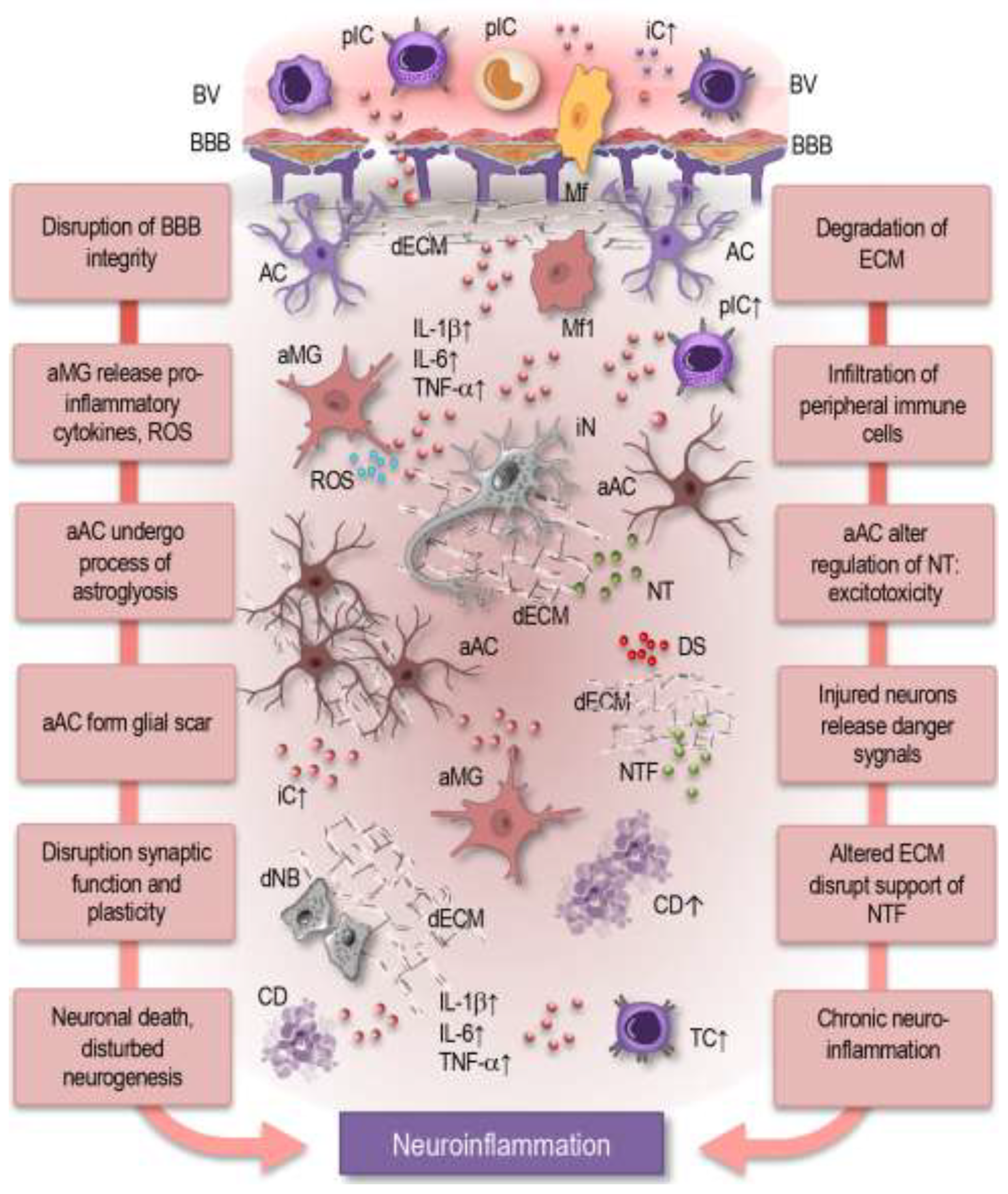
4.2. Neuroinflammatory Conditions
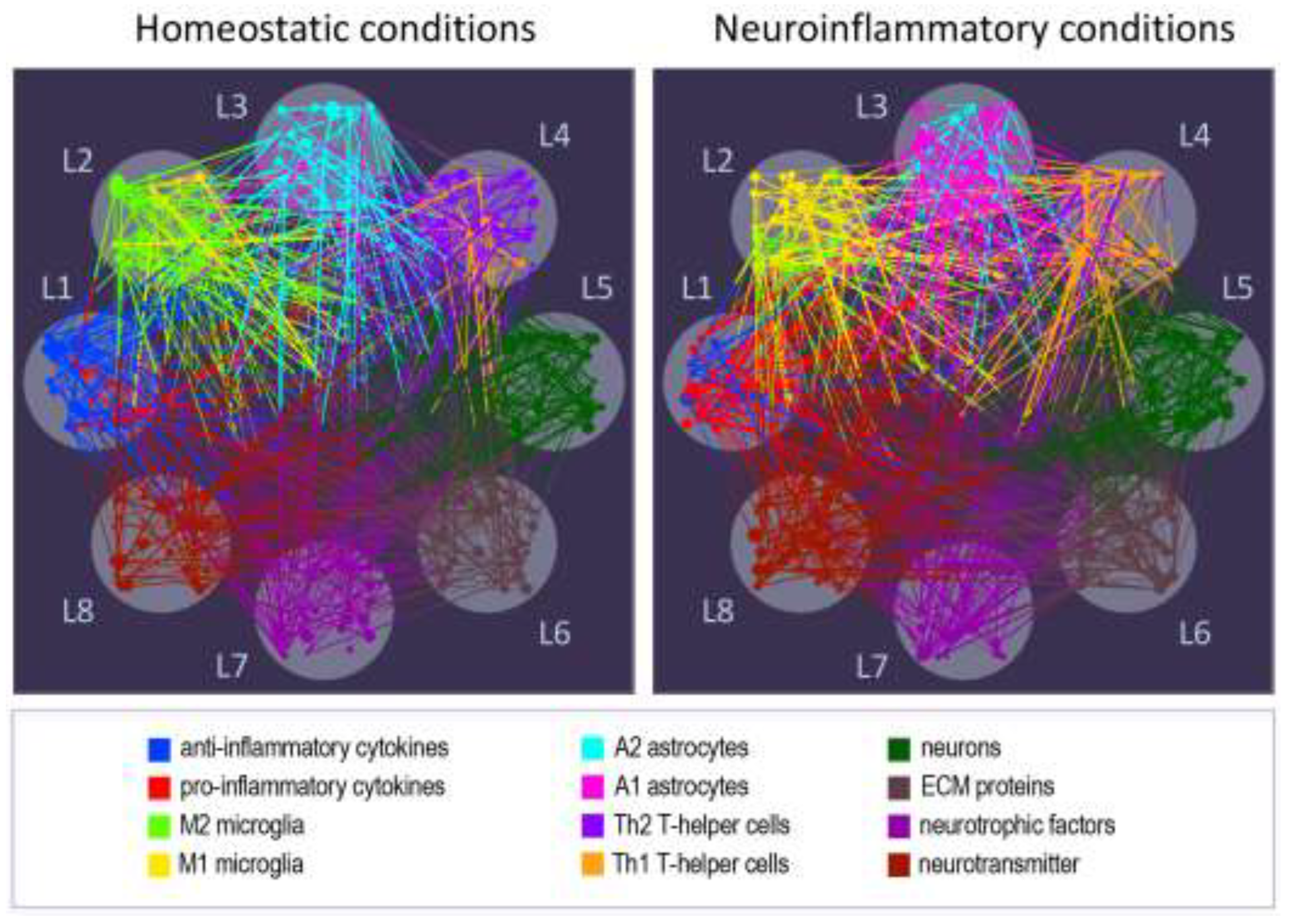
5. Understanding Complex Networks in Neuroimmune Interactions Through Multilayer Network Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nosi, D.; Lana, D.; Giovannini, M.G.; Delfino, G.; Zecchi-Orlandini, S. Neuroinflammation: Integrated Nervous Tissue Response through Intercellular Interactions at the "Whole System" Scale. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, S.; Müller, L.; Wenger, E.; Duzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci Biobehav Rev 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J Clin Invest 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: the devil is in the details. J Neurochem 2016, 139 Suppl 2, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 2013, 7, 6. [Google Scholar] [CrossRef]
- Li, H.; Ghorbani, S.; Ling, C.C.; Yong, V.W.; Xue, M. The extracellular matrix as modifier of neuroinflammation and recovery in ischemic stroke and intracerebral hemorrhage. Neurobiol Dis 2023, 186, 106282. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Lee, H.G.; Lee, J.H.; Flausino, L.E.; Quintana, F.J. Neuroinflammation: An astrocyte perspective. Sci Transl Med 2023, 15, eadi7828. [Google Scholar] [CrossRef]
- Matejuk, A.; Vandenbark, A.A.; Offner, H. Cross-Talk of the CNS With Immune Cells and Functions in Health and Disease. Front Neurol 2021, 12, 672455. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed]
- Tooley, U.A.; Bassett, D.S.; Mackey, A.P. Environmental influences on the pace of brain development. Nature Reviews Neuroscience 2021, 22, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Amanollahi, M.; Jameie, M.; Heidari, A.; Rezaei, N. The Dialogue Between Neuroinflammation and Adult Neurogenesis: Mechanisms Involved and Alterations in Neurological Diseases. Mol Neurobiol 2023, 60, 923–959. [Google Scholar] [CrossRef] [PubMed]
- Werker, J.F.; Hensch, T.K. Critical periods in speech perception: new directions. Annu Rev Psychol 2015, 66, 173–196. [Google Scholar] [CrossRef]
- Zocher, S.; Schilling, S.; Grzyb, A.N.; Adusumilli, V.S.; Bogado Lopes, J.; Gunther, S.; Overall, R.W.; Winter, Y.; Kempermann, G. Early-life environmental enrichment generates persistent individualized behavior in mice. Sci Adv 2020, 6, eabb1478. [Google Scholar] [CrossRef]
- Gervain, J. Plasticity in early language acquisition: the effects of prenatal and early childhood experience. Curr Opin Neurobiol 2015, 35, 13–20. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Rhea, E.M.; Reed, M.; Banks, W.A.; Erickson, M.A. The neurovascular extracellular matrix in health and disease. Exp Biol Med (Maywood) 2021, 246, 835–844. [Google Scholar] [CrossRef]
- Nithianantharajah, J.; Hannan, A.J. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci 2006, 7, 697–709. [Google Scholar] [CrossRef]
- van Praag, H.; Kempermann, G.; Gage, F.H. Neural consequences of environmental enrichment. Nat Rev Neurosci 2000, 1, 191–198. [Google Scholar] [CrossRef]
- Maharjan, R.; Diaz Bustamante, L.; Ghattas, K.N.; Ilyas, S.; Al-Refai, R.; Khan, S. Role of Lifestyle in Neuroplasticity and Neurogenesis in an Aging Brain. Cureus 2020, 12, e10639. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, C.; Parolisi, R.; Bonfanti, L. Brain Structural Plasticity: From Adult Neurogenesis to Immature Neurons. Front Neurosci 2020, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Cassilhas, R.C.; Tufik, S.; de Mello, M.T. Physical exercise, neuroplasticity, spatial learning and memory. Cell Mol Life Sci 2016, 73, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Goh, J.O.; Park, D.C. Neuroplasticity and cognitive aging: the scaffolding theory of aging and cognition. Restor Neurol Neurosci 2009, 27, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.M.M.; Meyer, K.A.; Santpere, G.; Gulden, F.O.; Sestan, N. Evolution of the Human Nervous System Function, Structure, and Development. Cell 2017, 170, 226–247. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M.E. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol 2017, 595, 1929–1945. [Google Scholar] [CrossRef]
- Arnò, B.; Grassivaro, F.; Rossi, C.; Bergamaschi, A.; Castiglioni, V.; Furlan, R.; Greter, M.; Favaro, R.; Comi, G.; Becher, B.; et al. Neural progenitor cells orchestrate microglia migration and positioning into the developing cortex. Nature Communications 2014, 5, 5611. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. Journal of Neuroinflammation 2021, 18, 258. [Google Scholar] [CrossRef]
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 2018, 98, 1170–1183. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Bispo, A.; Liu, J.; Sabu, S.; Wu, R.; DiBona, V.L.; Zheng, J.; Murugan, M.; Zhang, H.; Tang, Y.; et al. The GluN2A Subunit Regulates Neuronal NMDA receptor-Induced Microglia-Neuron Physical Interactions. Sci Rep 2018, 8, 828. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Pitossi, F.; Balschun, D.; Wagner, A.; del Rey, A.; Besedovsky, H.O. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci U S A 1998, 95, 7778–7783. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.A.; Cotman, C.W. Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev 2017, 34, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translational Neurodegeneration 2020, 9, 42. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, Y.S.; Kim, I.B. A Distinct Microglial Cell Population Expressing Both CD86 and CD206 Constitutes a Dominant Type and Executes Phagocytosis in Two Mouse Models of Retinal Degeneration. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Stratoulias, V.; Ruiz, R.; Kanatani, S.; Osman, A.M.; Keane, L.; Armengol, J.A.; Rodriguez-Moreno, A.; Murgoci, A.N.; Garcia-Dominguez, I.; Alonso-Bellido, I.; et al. ARG1-expressing microglia show a distinct molecular signature and modulate postnatal development and function of the mouse brain. Nat Neurosci 2023, 26, 1008–1020. [Google Scholar] [CrossRef]
- Atta, A.A.; Ibrahim, W.W.; Mohamed, A.F.; Abdelkader, N.F. Microglia polarization in nociplastic pain: mechanisms and perspectives. Inflammopharmacology 2023, 31, 1053–1067. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat Rev Neurol 2023, 19, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Kunchok, A.; Zekeridou, A.; McKeon, A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol 2019, 32, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol 2015, 32, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Brayman, V.L.; Taetzsch, T.; Miko, M.; Dahal, S.; Risher, W.C.; Valdez, G. Roles of the synaptic molecules Hevin and SPARC in mouse neuromuscular junction development and repair. Neurosci Lett 2021, 746, 135663. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.J.; Bindu, D.S.; Eroglu, C. Building and destroying synaptic bridges: How do Hevin/Sparcl1, SPARC, and MDGAs modify trans-synaptic neurexin-neuroligin interactions? Structure 2021, 29, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Tsirka, S.E. Inflammation modulates expression of laminin in the central nervous system following ischemic injury. J Neuroinflammation 2012, 9, 159. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 2006, 9, 260–267. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.V. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 2013, 33, 12870–12886. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer's Disease. J Neurosci 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Oliveira, C.R.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008, 156, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Yin, S.; Sun, Y.; Kou, L.; Zou, W.; Wang, Y.; Jin, Z.; Wang, T.; Xia, Y. Astrocyte-neuron communication through the complement C3-C3aR pathway in Parkinson’s disease. Brain, Behavior, and Immunity 2025, 123, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Fenn, A.M.; Godbout, J.P. 132. The role of astrocytes in IL-10 mediated regulation of inflammation within the CNS. Brain, Behavior, and Immunity 2013, 32, e38. [Google Scholar] [CrossRef]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef]
- Garg, S.K.; Kipnis, J.; Banerjee, R. IFN-gamma and IL-4 differentially shape metabolic responses and neuroprotective phenotype of astrocytes. J Neurochem 2009, 108, 1155–1166. [Google Scholar] [CrossRef]
- Albini, M.; Krawczun-Rygmaczewska, A.; Cesca, F. Astrocytes and brain-derived neurotrophic factor (BDNF). Neurosci Res 2023, 197, 42–51. [Google Scholar] [CrossRef]
- Kuno, R.; Yoshida, Y.; Nitta, A.; Nabeshima, T.; Wang, J.; Sonobe, Y.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. The role of TNF-alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res 2006, 1116, 12–18. [Google Scholar] [CrossRef]
- Trinchero, M.F.; Giacomini, D.; Schinder, A.F. Dynamic interplay between GABAergic networks and developing neurons in the adult hippocampus. Curr Opin Neurobiol 2021, 69, 124–130. [Google Scholar] [CrossRef]
- Lovinger, D.M. Communication networks in the brain: neurons, receptors, neurotransmitters, and alcohol. Alcohol Res Health 2008, 31, 196–214. [Google Scholar]
- Accogli, A.; Addour-Boudrahem, N.; Srour, M. Neurogenesis, neuronal migration, and axon guidance. Handb Clin Neurol 2020, 173, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bury, L.A.; Chen, F.; Aldinger, K.A.; Miranda, H.C.; Wynshaw-Boris, A. Generation of advanced cerebellar organoids for neurogenesis and neuronal network development. Hum Mol Genet 2023, 32, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Peng, J.; Murugan, M.; Mo, M.; Lalani, A.; Xie, P.; Xu, P.; Margolis, D.J.; Wu, L.J. Regulation of Physical Microglia-Neuron Interactions by Fractalkine Signaling after Status Epilepticus. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Iovino, L.; Nosi, D.; Wenk, G.L.; Giovannini, M.G. The neuron-astrocyte-microglia triad involvement in neuroinflammaging mechanisms in the CA3 hippocampus of memory-impaired aged rats. Exp Gerontol 2016, 83, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Papadimitriou, C.H.; Friederici, A.D. Bridging the Gap Between Neurons and Cognition Through Assemblies of Neurons. Neural Comput 2022, 34, 291–306. [Google Scholar] [CrossRef]
- Opitz, B. Memory function and the hippocampus. Front Neurol Neurosci 2014, 34, 51–59. [Google Scholar] [CrossRef]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte-neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Song, I.; Dityatev, A. Crosstalk between glia, extracellular matrix and neurons. Brain Research Bulletin 2018, 136, 101–108. [Google Scholar] [CrossRef]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol 2015, 45, 180–191. [Google Scholar] [CrossRef]
- Gemechu, J.M.; Bentivoglio, M. T Cell Recruitment in the Brain during Normal Aging. Front Cell Neurosci 2012, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Ownby, R.L. Neuroinflammation and cognitive aging. Curr Psychiatry Rep 2010, 12, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Levite, M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Current Opinion in Pharmacology 2008, 8, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Ria, F.; Adorini, L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunology today 2000, 21, 141–147. [Google Scholar] [CrossRef]
- Mayne, K.; White, J.A.; McMurran, C.E.; Rivera, F.J.; de la Fuente, A.G. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front Aging Neurosci 2020, 12, 572090. [Google Scholar] [CrossRef]
- Alboni, S.; Maggi, L. Editorial: Cytokines as Players of Neuronal Plasticity and Sensitivity to Environment in Healthy and Pathological Brain. Front Cell Neurosci 2015, 9, 508. [Google Scholar] [CrossRef]
- Silvin, A.; Qian, J.; Ginhoux, F. Brain macrophage development, diversity and dysregulation in health and disease. Cellular & Molecular Immunology 2023, 20, 1277–1289. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 2013, 33, 13820–13833. [Google Scholar] [CrossRef]
- McKenna, F.; McLaughlin, P.J.; Lewis, B.J.; Sibbring, G.C.; Cummerson, J.A.; Bowen-Jones, D.; Moots, R.J. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol 2002, 132, 34–40. [Google Scholar] [CrossRef]
- Freire-Garabal, M.; Núñez, M.J.; Balboa, J.; López-Delgado, P.; Gallego, R.; García-Caballero, T.; Fernández-Roel, M.D.; Brenlla, J.; Rey-Méndez, M. Serotonin upregulates the activity of phagocytosis through 5-HT 1A receptors. British Journal of Pharmacology 2003, 139, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Suguro, T.; Watanabe, T.; Kanome, T.; Kodate, S.; Hirano, T.; Miyazaki, A.; Adachi, M. Serotonin acts as an up-regulator of acyl-coenzyme A:cholesterol acyltransferase-1 in human monocyte-macrophages. Atherosclerosis 2006, 186, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Reyes-García, M.G.; García-Tamayo, F. A neurotransmitter system that regulates macrophage pro-inflammatory functions. Journal of Neuroimmunology 2009, 216, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.S.; Heijnen, P.D.A.M.; Breur, M.; de Vries, H.E.; Tool, A.T.J.; Amor, S.; Dijkstra, C.D. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. Journal of Neuroinflammation 2014, 11, 23. [Google Scholar] [CrossRef]
- Chen, O.; Luo, X.; Ji, R.R. Macrophages and microglia in inflammation and neuroinflammation underlying different pain states. Med Rev (2021) 2023, 3, 381–407. [Google Scholar] [CrossRef]
- de Castro Brás, L.E.; Frangogiannis, N.G. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biology 2020, 91-92, 176–187. [Google Scholar] [CrossRef]
- Jang, D.G.; Sim, H.J.; Song, E.K.; Kwon, T.; Park, T.J. Extracellular matrixes and neuroinflammation. BMB Rep 2020, 53, 491–499. [Google Scholar] [CrossRef]
- Li, H.; Ghorbani, S.; Ling, C.-C.; Yong, V.W.; Xue, M. The extracellular matrix as modifier of neuroinflammation and recovery in ischemic stroke and intracerebral hemorrhage. Neurobiology of Disease 2023, 186, 106282. [Google Scholar] [CrossRef]
- Rauch, U. Brain matrix: Structure, turnover and necessity. In Proceedings of the Biochemical Society Transactions; 2007; pp. 656–660. [Google Scholar]
- Ghorbani, S.; Yong, V.W. The extracellular matrix as modifier of neuroinflammation and remyelination in multiple sclerosis. Brain 2021, 144, 1958–1973. [Google Scholar] [CrossRef]
- Iozzo, R.V. Basement membrane proteoglycans: from cellar to ceiling. Nature Reviews Molecular Cell Biology 2005, 6, 646–656. [Google Scholar] [CrossRef]
- Fawcett, J.W.; Oohashi, T.; Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature Reviews Neuroscience 2019, 20, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and Function. Biomolecules 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J Cell Sci 2000, 113 Pt 2, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bai, Q.; Yong, V.W.; Xue, M. EMMPRIN Promotes the Expression of MMP-9 and Exacerbates Neurological Dysfunction in a Mouse Model of Intracerebral Hemorrhage. Neurochemical Research 2022, 47, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Doring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann Neurol 2012, 72, 419–432. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M. Extracellular matrix molecules and synaptic plasticity. Nature Reviews Neuroscience 2003, 4, 456–468. [Google Scholar] [CrossRef]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Aspberg, A.; Miura, R.; Bourdoulous, S.; Shimonaka, M.; Heinegård, D.; Schachner, M.; Ruoslahti, E.; Yamaguchi, Y.U. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proceedings of the National Academy of Sciences of the United States of America 1997, 94, 10116–10121. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Comeglio, P.; Herrera-Puerta, E.; Piaceri, I.; Nacmias, B.; Benelli, M.; Kelsey, G.; Maggi, M.; Gallina, P.; et al. Tumor Necrosis Factor alpha Influences Phenotypic Plasticity and Promotes Epigenetic Changes in Human Basal Forebrain Cholinergic Neuroblasts. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Kveler, K.; Starosvetsky, E.; Ziv-Kenet, A.; Kalugny, Y.; Gorelik, Y.; Shalev-Malul, G.; Aizenbud-Reshef, N.; Dubovik, T.; Briller, M.; Campbell, J.; et al. Immune-centric network of cytokines and cells in disease context identified by computational mining of PubMed. Nat Biotechnol 2018, 36, 651–659. [Google Scholar] [CrossRef]
- Goshen, I.; Kreisel, T.; Ben-Menachem-Zidon, O.; Licht, T.; Weidenfeld, J.; Ben-Hur, T.; Yirmiya, R. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 2008, 13, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Suyama, K.; Nishida, K.; Sei, Y.; Basile, A.S. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett 1996, 206, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Park, E.; You, B.; Jung, Y.; Park, A.R.; Park, S.G.; Lee, J.R. Neuronal synapse formation induced by microglia and interleukin 10. PLoS One 2013, 8, e81218. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef]
- Baune, B.T.; Ponath, G.; Rothermundt, M.; Riess, O.; Funke, H.; Berger, K. Association between genetic variants of IL-1beta, IL-6 and TNF-alpha cytokines and cognitive performance in the elderly general population of the MEMO-study. Psychoneuroendocrinology 2008, 33, 68–76. [Google Scholar] [CrossRef]
- Lewitus, G.M.; Pribiag, H.; Duseja, R.; St-Hilaire, M.; Stellwagen, D. An adaptive role of TNFalpha in the regulation of striatal synapses. J Neurosci 2014, 34, 6146–6155. [Google Scholar] [CrossRef]
- Bains, J.S.; Oliet, S.H. Glia: they make your memories stick! Trends Neurosci 2007, 30, 417–424. [Google Scholar] [CrossRef]
- Goshen, I.; Yirmiya, R. Interleukin-1 (IL-1): a central regulator of stress responses. Front Neuroendocrinol 2009, 30, 30–45. [Google Scholar] [CrossRef]
- Palasz, E.; Wilkaniec, A.; Stanaszek, L.; Andrzejewska, A.; Adamczyk, A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Fang, J. Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun 2006, 20, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Schnydrig, S.; Korner, L.; Landweer, S.; Ernst, B.; Walker, G.; Otten, U.; Kunz, D. Peripheral lipopolysaccharide administration transiently affects expression of brain-derived neurotrophic factor, corticotropin and proopiomelanocortin in mouse brain. Neurosci Lett 2007, 429, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.R.; Barrientos, R.M.; Ahrendsen, J.T.; Hoover, J.M.; Maier, S.F.; Patterson, S.L. Aging and infection reduce expression of specific brain-derived neurotrophic factor mRNAs in hippocampus. Neurobiol Aging 2012, 33, 832 e831–814. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Calabrese, F.; Rossetti, A.C.; Racagni, G.; Gass, P.; Riva, M.A.; Molteni, R. Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci 2014, 8, 430. [Google Scholar] [CrossRef]
- Duarte Azevedo, M.; Sander, S.; Tenenbaum, L. GDNF, A Neuron-Derived Factor Upregulated in Glial Cells during Disease. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Singh, G.; Sikder, A.; Phatale, V.; Srivastava, S.; Singh, S.B.; Khatri, D.K. Therapeutic potential of GDNF in neuroinflammation: Targeted delivery approaches for precision treatment in neurological diseases. Journal of Drug Delivery Science and Technology 2023, 87, 104876. [Google Scholar] [CrossRef]
- Deak, F.; Sonntag, W.E. Aging, synaptic dysfunction, and insulin-like growth factor (IGF)-1. J Gerontol A Biol Sci Med Sci 2012, 67, 611–625. [Google Scholar] [CrossRef]
- Tumati, S.; Burger, H.; Martens, S.; van der Schouw, Y.T.; Aleman, A. Association between Cognition and Serum Insulin-Like Growth Factor-1 in Middle-Aged & Older Men: An 8 Year Follow-Up Study. PLoS One 2016, 11, e0154450. [Google Scholar] [CrossRef]
- Wen, Y.; Dong, Z.; Liu, J.; Axerio-Cilies, P.; Du, Y.; Li, J.; Chen, L.; Zhang, L.; Liu, L.; Lu, J.; et al. Glutamate and GABAA receptor crosstalk mediates homeostatic regulation of neuronal excitation in the mammalian brain. Signal Transduction and Targeted Therapy 2022, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Platel, J.C.; Lacar, B.; Bordey, A. GABA and glutamate signaling: homeostatic control of adult forebrain neurogenesis. J Mol Histol 2007, 38, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Llansola, M.; Arenas, Y.M.; Sancho-Alonso, M.; Mincheva, G.; Palomares-Rodriguez, A.; Doverskog, M.; Izquierdo-Altarejos, P.; Felipo, V. Neuroinflammation alters GABAergic neurotransmission in hyperammonemia and hepatic encephalopathy, leading to motor incoordination. Mechanisms and therapeutic implications. Front Pharmacol 2024, 15, 1358323. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Balzano, T.; Castro, M.C.; Llansola, M.; Felipo, V. The Dual Role of the GABA(A) Receptor in Peripheral Inflammation and Neuroinflammation: A Study in Hyperammonemic Rats. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.; Al-Ayadhi, L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. Journal of Neuroinflammation 2014, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L. The network takeover. Nature Physics 2012, 8, 14–16. [Google Scholar] [CrossRef]
- Mheich, A.; Wendling, F.; Hassan, M. Brain network similarity: methods and applications. Netw Neurosci 2020, 4, 507–527. [Google Scholar] [CrossRef]
- Seguin, C.; Sporns, O.; Zalesky, A. Brain network communication: concepts, models and applications. Nature Reviews Neuroscience 2023, 24, 557–574. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Müller, L.; Rauskolb, S.; Sendtner, M.; Deutschbein, T.; Pawelec, G.; Muller, V. Network topology dynamics of circulating biomarkers and cognitive performance in older Cytomegalovirus-seropositive or -seronegative men and women. Immun Ageing 2019, 16, 31. [Google Scholar] [CrossRef]
- Boccaletti, S.; Bianconi, G.; Criado, R.; Del Genio, C.I.; Gomez-Gardenes, J.; Romance, M.; Sendina-Nadal, I.; Wang, Z.; Zanin, M. The structure and dynamics of multilayer networks. Phys Rep 2014, 544, 1–122. [Google Scholar] [CrossRef]
- De Domenico, M. Multilayer modeling and analysis of human brain networks. Gigascience 2017, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, M. More is different in real-world multilayer networks. Nature Physics 2023, 19, 1247–1262. [Google Scholar] [CrossRef]
- Kivelä, M.; Arenas, A.; Barthelemy, M.; Gleeson, J.P.; Moreno, Y.; Porter, M.A. Multilayer networks. Journal of Complex Networks 2014, 2, 203–271. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: understanding the cell's functional organization. Nat Rev Genet 2004, 5, 101–113. [Google Scholar] [CrossRef]
- de Arruda, G.F.; Rodrigues, F.A.; Moreno, Y. Fundamentals of spreading processes in single and multilayer complex networks. Physics Reports 2018, 756, 1–59. [Google Scholar] [CrossRef]
- Zitnik, M.; Li, M.M.; Wells, A.; Glass, K.; Morselli Gysi, D.; Krishnan, A.; Murali, T.M.; Radivojac, P.; Roy, S.; Baudot, A.; et al. Current and future directions in network biology. Bioinform Adv 2024, 4, vbae099. [Google Scholar] [CrossRef]
- Buphamalai, P.; Kokotovic, T.; Nagy, V.; Menche, J. Network analysis reveals rare disease signatures across multiple levels of biological organization. Nat Commun 2021, 12, 6306. [Google Scholar] [CrossRef]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabasi, A.L. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef]
- Sharma, A.; Menche, J.; Huang, C.C.; Ort, T.; Zhou, X.; Kitsak, M.; Sahni, N.; Thibault, D.; Voung, L.; Guo, F.; et al. A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma. Hum Mol Genet 2015, 24, 3005–3020. [Google Scholar] [CrossRef]
- Bergthaler, A.; Menche, J. The immune system as a social network. Nat Immunol 2017, 18, 481–482. [Google Scholar] [CrossRef]
- Rieckmann, J.C.; Geiger, R.; Hornburg, D.; Wolf, T.; Kveler, K.; Jarrossay, D.; Sallusto, F.; Shen-Orr, S.S.; Lanzavecchia, A.; Mann, M.; et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol 2017, 18, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.; Jeong, H.; Barabasi, A.L. Error and attack tolerance of complex networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Artime, O.; Grassia, M.; De Domenico, M.; Gleeson, J.P.; Makse, H.A.; Mangioni, G.; Perc, M.; Radicchi, F. Robustness and resilience of complex networks. Nature Reviews Physics 2024, 6, 114–131. [Google Scholar] [CrossRef]
- Liu, X.; Maiorino, E.; Halu, A.; Glass, K.; Prasad, R.B.; Loscalzo, J.; Gao, J.; Sharma, A. Robustness and lethality in multilayer biological molecular networks. Nat Commun 2020, 11, 6043. [Google Scholar] [CrossRef] [PubMed]
- Bellingeri, M.; Bevacqua, D.; Scotognella, F.; Alfieri, R.; Cassi, D. A comparative analysis of link removal strategies in real complex weighted networks. Sci Rep 2020, 10, 3911. [Google Scholar] [CrossRef] [PubMed]
- Bellingeri, M.; Bevacqua, D.; Scotognella, F.; Cassi, D. The heterogeneity in link weights may decrease the robustness of real-world complex weighted networks. Sci Rep 2019, 9, 10692. [Google Scholar] [CrossRef]
- Newman, M.E.J. The structure and function of complex networks. SIAM Review 2003, 45, 167–256. [Google Scholar] [CrossRef]
- Buldyrev, S.V.; Parshani, R.; Paul, G.; Stanley, H.E.; Havlin, S. Catastrophic cascade of failures in interdependent networks. Nature 2010, 464, 1025–1028. [Google Scholar] [CrossRef]
- Vespignani, A. Complex networks: The fragility of interdependency. Nature 2010, 464, 984–985. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



