Submitted:
26 November 2024
Posted:
28 November 2024
You are already at the latest version
Abstract
This article describes the basics of chemical thermodynamics and its application to the study of plant biomass and its main components, cellulose, hemicelluloses, lignin, etc. The energy potential of various biomass types, as well as biomass-based solid, liquid, and gaseous biofuels, is determined. A method of additive contributions of combustion enthalpies of main components is proposed to calculate the combustion enthalpy of biomass sample. It has been also established that the potential of thermal energy of the initial biomass is higher than the energy potential of secondary biofuels released from this biomass. The thermodynamic functions of plant biopolymers are calculated. Moreover, the thermodynamic stability of various crystalline allomorphs of cellulose and amorphous cellulose is studied. The melting enthalpies of crystallites with different types of crystalline structures are estimated. A thermochemical method for determining the degree of crystallinity of cellulose is proposed. The most important biomass components are cellulose and other polysaccharides. The thermodynamics of enzymatic hydrolysis of polysaccharides and their conversion into glucose is described. In addition, the thermodynamic analysis of the conversion process of glucose into bioethanol is performed. Considerable attention is also paid to the thermochemistry of cellulose alkalization, etherification, and esterification.
Keywords:
biomass
; cellulose
; hemicelluloses
; lignin
; biofuels
; chemistry
; energy potential
; thermochemistry
; chemical thermodynamics
1. Basics of Chemical Thermodynamics
Chemical thermodynamics is an interdisciplinary science that combines thermodynamics, physical chemistry, statistical and chemical physics, and some other scientific areas. The main tasks of chemical thermodynamics are to determine thermodynamic functions, study the transformations of various forms of energy during chemical reactions and phase transitions, predict the feasibility and direction of chemical processes, and study the ability of chemical systems to perform useful work [1,2]. A section of chemical thermodynamics is thermochemistry, which studies the thermal effects of chemical reactions and physicochemical processes.
Chemical thermodynamics considers isolated, closed, and open systems. In an isolated system, there is no exchange of energy and matter with the external environment. In a closed system, there is no exchange of matter, but energy exchange with the external environment is carried out. In an open system, the exchange of both matter and energy with the external environment is possible.
There are also laws of thermodynamics. The zeroth law of thermodynamics states that an isolated thermodynamic system spontaneously passes over time into a state of thermodynamic equilibrium.
The first law of thermodynamics states that the heat received by a system from outside is used to increase the internal energy of the system and to perform work by this system.
According to the second law of thermodynamics, heat or thermal energy cannot spontaneously pass from a cooler body to a hotter body. Another formulation of the second thermodynamic law is as follows: in a nonequilibrium isolated system, entropy increases and reaches a maximum when thermodynamic equilibrium is established. The consequence of the second law is that a perpetual motion machine of the second kind is impossible.
The third law of thermodynamics states that the entropy of any equilibrium system, as the temperature approaches absolute zero, ceases to depend on any state parameters and tends to a certain limit close to zero. Moreover, all processes near absolute zero that transfer the system from one equilibrium state to another occur without changing the entropy.
Chemical thermodynamics operates with several terms, parameters, and thermodynamic functions such as temperature (T), pressure (P), volume (V), work (A), heat (Q), kinetic energy (Ek), internal energy (U), enthalpy (H), entropy (S), free energy or potential of Helmholtz (F) and Gibbs (G), etc.
In the case of an isolated system, the change in internal energy is zero: ΔU=0.
For a reaction occurring in closed or open systems at constant temperature and volume, the thermal effect (∆Q) of this reaction is equal to the change in the internal energy (∆U) of the system, i.e.,
∆Q = ∆U,
If a reaction occurs in an open system at constant temperature and pressure, then from the first law of thermodynamics it follows:
∆Q = ∆U + Р∆V = (U2 + PV2) - (U1 + PV1),
Denoting U + PV = H, where H is called enthalpy, we obtain:
Thus, in an open system at P=const, the thermal effect of the reaction is equal to the enthalpy change.
∆Q = ∆Н,
As a result of a chemical reaction, enthalpy can decrease (∆rН < 0) when heat is released, or increase (∆rН > 0) when heat is absorbed from outside. According to the Lavoisier-Laplace law, the thermal effect of a forward reaction is always equal to the thermal effect of a reverse reaction with the opposite sign. The second law of thermochemistry, proposed by Hess, states that the thermal effect of a reaction depends only on the initial and final state and does not depend on the intermediate stages of the reaction. There is also another formulation of this law, namely: if a chemical process occurs in several stages, then the overall thermal effect of the process is equal to the algebraic sum of the thermal effects of the individual, intermediate stages.
Two important consequences follow from Hess's law. Firstly. The thermal effect of a chemical reaction (∆rН) is equal to the difference between the sums of the combustion enthalpies (∆cНr) of the starting reagents and the combustion enthalpies (∆cНp) of the reaction products, taking into account their stoichiometric coefficients:
Secondly. The thermal effect of a chemical reaction is equal to the difference between the sums of the formation enthalpies (∆fНp) of the reaction products and the formation enthalpies (∆fНr) of the starting reagents, taking into account their stoichiometric coefficients.
Numerical values of standard formation enthalpy for various substances under standard conditions (To=298.15 K, Po = 0.1 MPa) are given in reference books.
∆rН = ∑nr ∆cНr - ∑np ∆cНp,
∆rН = ∑np ∆fНp - ∑nr ∆fНr,
The concept of entropy was first introduced by Rudolf Clausius in the late 19th century. Entropy is considered a function of the state of a thermodynamic system, related to the reduced thermal energy:
From the second law of thermodynamics, it follows that the spontaneous transfer of heat from a hotter region to a colder one is determined by the increase in the entropy of the system.
S = Q/T,
According to the Ludwig Boltzmann concept, entropy is a function of system state, characterizing the degree of statistical disorder (W) in the arrangement of elements of a system, ions, atoms, molecules, etc.:
If at Р = constant the temperature rises, then the disorder in the arrangement of atoms or molecules increases, which leads to an increase in the entropy, namely:
where Cp is the specific heat capacity; and T2 >T1.
S = R lnW,
∆S = Cp ln(T2/T1),
If the open system at Р = const is in equilibrium state, then the following equality is true:
For example, at equilibrium between liquid and vapor at Tc, the change in entropy during vapor condensation (∆Sc) can be calculated if the condensation enthalpy (∆Нc) is known:
Similarly, one can find the change in entropy during melting at Tm of a crystalline substance:
∆Н = T∆S,
∆Sc = ∆Нc/Tc,
∆Sm = ∆Нm/Tm,
If a chemical reaction occurs, the change in entropy can be found using an equation similar to the Hess equation:
where Sp and Sr are entropy values for obtaining products and starting reagents, respectively.
∆rS = ∑np Sp - ∑nr Sr,
Numerical values of standard entropy for various substances under standard conditions (To=298.15 K, Po = 0.1 MPa) are given in reference books.
Chemical thermodynamics can be also used to predict the feasibility and direction of chemical processes. For this purpose, special thermodynamic functions of the system state are used, which reflect the influence of two factors on the process, energy and entropy. These special thermodynamic functions have the property that their sign is a criterion for the feasibility or non-feasibility of a spontaneous reaction.
For isothermal reactions occurring at constant volume, such a function is the Helmholtz free energy, which is calculated using the equation:
∆rF = ∆rU - T∆rS,
In the case of isothermal reactions occurring at constant pressure, a criterion for the feasibility of a spontaneous reaction is the Gibbs free energy or potential. The change in the Gibbs potential during the reaction is determined by the equation:
∆rG = ∆rH - T∆rS.
These special thermodynamic functions characterize the degree of non-equilibrium of the reaction system. The more negative their values are, the greater the deviation of the system from equilibrium and the greater the probability of the reaction being feasible. The feasibility of a reaction is facilitated by a decrease in the internal energy or enthalpy and an increase in the disorder of the system, characterized by the entropy factor.
Thus, the criterion for the feasibility of a reaction in systems at constant volume is ∆rF< 0. This can happen in the following cases:
∆rU< 0, while ∆rS > 0,
∆rU >0, and ∆rS >0, but T∆rS >∆rU,
∆rU< 0, while ∆rS< 0, but [∆rU] > [T∆rS]
For open systems proceeding at constant pressure, the criterion for reaction feasibility is ∆rG < 0. This can happen when:
∆rH< 0, while ∆rS >0
∆rH >0, and ∆rS >0, but T∆rS >∆rH
∆rH< 0, while ∆rS< 0, but [∆rH] > [T∆rS]
If the reaction is reversible and an equilibrium is established between the forward and reverse reactions, such an equilibrium is characterized by a constant Keq. According to Van't Hoff, the equilibrium constant of a chemical reaction is an exponential function of the thermodynamic potential:
Vice versa, if the value of the equilibrium constant is known, then the thermodynamic potential of the reversible reaction can be calculated, as follows:
at V = const, Keq = EXP (-∆rF/RT ),
at P= const, Keq = EXP (-∆rG/RT),
at V= const, ∆rF = -RT lnKeq,
at P= const, ∆rG = -RT lnKeq,
To describe the state of the multi-component system with a variable number of different components, a partial thermodynamic function of a component called chemical potential is used:
Such a function determines the change in thermodynamic potential when the number of components in the system changes. The importance of chemical potential is that one of the conditions of equilibrium in a system is the equality of the chemical potential of any component of the system in different phases and at different points of one phase. This function is widely used in physical chemistry to study the processes of dissolution, sorption, etc. For example, for a solution in equilibrium state at P and T = const, the following relationship exists:
where Ps is the vapor pressure of the solvent above the solution, and Po is the vapor pressure of the pure solvent.
μk = (δG/δNk)T, P, k ≠ i,
Δμs = RT ln(Ps/Po),
If Ps < Po the Δμs < 0, which is a criterion for good compatibility between the solvent and the polymer.
Thermodynamic analysis of nanoscale particles must take into account a great specific surface area (Ssp) of such particles and its change in various processes:
where σ is the specific surface energy of particles; Vm is the molar volume.
∆Gn = σVm ΔSsp,
Such nanoparticles tend to aggregate, which is thermodynamically favorable since this process leads to a decrease in the specific surface area (ΔSsp < 0) and provides the negative value of the thermodynamic potential (∆Gn < 0).
Another example is the Gibbs-Thomson equation, according to which the melting point decreases with decreasing size of nanocrystallites. It was found that ∆Gn depends on the temperature change, as follows: ∆Gn = ΔmH (ΔT/Tm). On the other hand, for nano- particles ΔSsp = 4/L. Substituting the right parts of ∆Gn and ΔSsp into eq. (21), the Gibbs-Thomson equation can be obtained:
where Tn and Tm are the melting points of nano-crystallites and macrocrystals, L is the size of nano-crystallite, and ΔmH is the melting enthalpy.
Tn = Tm (1 - 4 σVm/(L ΔmH)),
2. Methods for Determining the Thermodynamic Characteristics of Substances
Values of standard thermodynamic functions (TDFs), such as the standard formation enthalpy (ΔfHo) and entropy (So), for various substances, can be found in reference books. However, if reference data is not available, the values of these TDFs are determined experimentally. The standard formation enthalpy of an organic substance CxHyOz can be calculated from the experimentally determined combustion enthalpy (ΔcHo) of this substance at standard conditions (To=298.15 K, P=0.1 MPa). The combustion process of the substance is:
CxHyOz + (x + 0.25y - 0.5z) O2 → x CO2 + 0.5y H2O + ΔcHo(CxHyOz),
The standard formation enthalpy of this substance can be calculated, as follows:
where ΔfHo(CO2, g) = -393.51 kJ/mol and ΔfHo(H2O, l) = -285.83 kJ/mol are standard enthalpies of the formation of carbon dioxide and liquid water, respectively.
ΔfHo(CxHyOz) = x ΔfHo(CO2, g) + 0.5y ΔfHo(H2O, l) - ΔcHo(CxHyOz),
Precision oxygen bomb calorimeters of various designs can be used to burn samples [3]. Typically, it consists of a strong stainless-steel bomb, which is placed in a water calorimeter. A small sample (0.5-1 g) is put in a crucible inside the bomb and 1 ml of water is added. Oxygen is pumped into the bomb to provide a pressure of 2.5 to 3 MPa. After temperature equilibrium is established, the sample is ignited, the temperature rise is measured with an accuracy of ±0.001 K, and the combustion enthalpy is calculated. The true mass of the used sample is determined from the mass of the produced CO2. The corrections for ignition and forming of acid traces should be made. To adjust the enthalpy of combustion to standard conditions (To=298.15 K, P=0.1 MPa) the Washburn correction, as well as the correction for the change in the number of moles of gases before and after combustion are introduced. For each sample, several experiments are performed to obtain a reliable value of the combustion enthalpy and standard deviation.
To measure the specific heat capacity (Cp) of the dry sample, a scanning adiabatic vacuum calorimeter is used [3,4]. The experiments usually are carried out in the temperature range of 80-340 K. Below 80 K, Cp values can be estimated by the method of Kelly, et al. [5]. The accuracy of Cp determination is ±0.005 J/mol K. For each sample, several experiments should be performed to find the reliable Cp value and standard deviation.
The entropy value of the sample at temperature T is calculated, as follows:
If T is the standard temperature, To = 298.15 K, then the result of the calculation is the standard entropy of the sample, So.
Along with the bomb calorimeter and the scanning adiabatic calorimeter, other types of calorimeters are also used to study the thermochemical properties of samples. For example, to measure the enthalpy of sample wetting and dissolution, isothermal or adiabatic calorimeters, as well as microcalorimeters can be applied [6,7].
For example, to determine the wetting enthalpy of cellulose, a small sample (ca 1 g) is prepared. Before starting experiments, the air-dry sample is weighed into a special glass ampoule and dried in a vacuum at 378 K to a constant weight. The glass ampoule containing the dry sample is sealed and introduced into the calorimetric cell filled with distilled water. The calorimeter is thermostated at 298.15 K to achieve an equilibrium state. Thereafter, the sealed ampoule with the dry sample is broken to ensure that the sample is wetted with water. The released exothermic heat effect of wetting is measured with accuracy ±0.01 J. Several of the same samples usually are tested to calculate an average enthalpy value and standard deviation.
In addition to calorimetry, several other methods can be used to characterize samples. These methods may include X-ray scattering, NMR, FTIR-spectroscopy, DTA, electron microscopy, sorption, etc. [8,9,10,11].
This review article will show how thermochemical methods can be applied to the study of plant biomass and its main components, cellulose, hemicelluloses, lignin, etc. Considerable attention will be paid to the thermodynamics of physicochemical and chemical transformations of cellulose and nanocellulose, as well as other components of plant biomass. The thermodynamics of second solid, liquid, and gaseous biofuels released from the initial biomass was also considered.
3. Thermochemistry of Plant Biomass
There are many applications of chemical thermodynamics to various substances and diverse reaction classes (synthesis, decomposition, substitution, exchange, oxidation, reduction, etc.). Currently, due to the depletion of fossil fuel reserves and the harm that their use causes to the environment, much attention is paid to the use of renewable natural resources, including plant biomass.
The existence and further development of the present civilization require expanded consumption of energy, chemicals, and materials. Nowadays, the main energy sources are still fossil fuels, namely coal, petroleum, and natural gas [12]. However, the increased use of fossil fuels is causing acute environmental problems, since the combustion of such fuels is accompanied by the emission of carbon dioxide triggering the greenhouse effect and global warming.
An additional problem is that fossil resources are not reproduced in nature. Therefore, their reserves are exhausted and run down in an essentially permanent manner [13]. To eliminate the imbalance in the fossil sources, an increased utilization of alternative sources of energy and raw materials is required. Considerable attention in recent years has been given to plant biomass, which in contrast to fossil sources is continuously renewed in nature [14].
The total resources of plant biomass reach 1.5 trillion tons [15]. The potential resources of biomass among alternative and renewable energy sources is above 70% [16], while its share in the production of alternative energy exceeds 50% [17]. Despite advances in solar and wind energy, in many countries plant biomass still accounts for a significant share of energy production [17]. So in Africa, up to 60% of energy is generated from biomass. East Asia countries and China receive up to 25% of energy from biomass, while in Latin America, the share of biomass energy is 18%. Only in the USA, the European Union, and other developed regions and countries, the share of biomass in energy production is small and does not exceed 3%.
The plant biomass is formed in nature from carbon dioxide and water by photosynthesis absorbing solar energy. When the plant biomass is burned, it releases the accumulated solar energy in the form of heat, along with the release of used water and carbon dioxide stored in the biomass. Therefore, plant biomass is considered a CO2-neutral source of renewable energy [17,18].
Currently implemented technologies for the production of liquid biofuels are based on the transformation of carbohydrates into bioethanol and vegetable oils into biodiesel fuel. The main sources of these carbohydrates are juices of sugarcane, sugar beet, and sweet sorghum, as well as starches of corn, wheat, potatoes, and some other agricultural plants. Since these carbohydrates and vegetable oils are required in the food industry, their use for the production of biofuels is limited. Moreover, further expansion of the production to a higher volume of bioethanol and/or biodiesel will cause a shortage of land areas, exhaustion of the soil, excessive consumption of water and energy, deficits of food and feed products, and increase their prices [19].
An alternative way to obtain biofuels without competing with the food and feed industry is the use of inedible biomass, which is an abundant, renewable, and inexpensive plant material. This biomass type involves energy crops (e.g., miscanthus, switchgrass, Bermuda grass, etc.), forest residues (e.g., sawdust, twigs, shrubs, etc.), residues of agricultural plants (e.g. stalks, husks, cobs, etc.), residues of textile, pulp, and paper, municipal paper waste, etc. Moreover, huge amounts of algae can be used as appropriate feedstock for the production of bioenergy or chemicals. The total resources of inedible biomass accumulated annually are estimated at 10 billion tons, the combustion of which could potentially generate about 150 EJ of bio-energy [20].
The main components of plant biomass are cellulose, hemicelluloses, and lignin (Table 1).
Cellulose is a linear, stereoregular, semicrystalline polysaccharide consisting of anhydroglucose units (AGUs) having a “chair” conformation and linked to each other in long macromolecules by chemical β-1,4-glycosidic bonds in a head-to-tail manner [16,22,23,24,25]. Macromolecules of natural cellulose from various origins may include 2,000 to 30,000 AGUs. During the process of cellulose isolation from plant materials and cellulose modification, partial depolymerization of the macromolecules is observed. Each AGU of cellulose contains three hydroxyl functional groups: one primary and two secondary groups. Hydroxyl groups of adjacent chains are linked by strong hydrogen bonds, which leads to the formation of thread-like elementary nanofibrils and their bundles, called microfibrils, which contain nanocrystallites and non-crystalline domains. Besides, hydroxyl groups can be replaced by various substituents, resulting in the formation of cellulose derivatives, such as ethers, esters, etc. [26,27].
Hemicelluloses are highly amorphous heteropolysaccharides [16,22,23,28,29]. In the cell walls of plant fibers, hemicelluloses fulfill the function of binder between cellulose fibrils and lignin. The chemical structure of hemicelluloses consists of chains of a variety of pentoses or hexoses units. Various plants and also its tissues contain different types of hemicelluloses. Agricultural and herbaceous plants, as well as hardwoods, contain mainly xylan, pentosan-type hemicellulose, while softwoods are enriched with mannan, which is hexosan-type hemicellulose. Hemicelluloses are heteropolymers. Xylan, for example, can be present in the form of glucuronoxylan (in hardwoods) or arabinoxylan (in grasses and grains), while mannan presents in the form of glucomannan (in softwoods) or galactomannan (in carob tree). The polysaccharide complex of biomass containing cellulose and hemicelluloses is called holocellulose.
Lignin is an aromatic, amorphous, and hydrophobic polymer [16,22,23,30,31]. It is a complex biopolymer of phenylpropane units, which are linked to each other with a variety of different chemical bonds with the formation three-dimensional network. Due to such structural organization, lignin is resistant to some chemical reagents and enzymes. This biopolymer consists of three main phenylpropane units such as guaiacyl (G), syringyl (S), and hydroxyphenyl (H). The relative ratios of these units can vary for different biomass sources. Lignin of softwoods is mainly of G-type, whereas lignin of hardwoods contains a mixture of G-and S-units. Lignin of herbaceous plants consists usually of all three types of phenylpropane units.
In addition to the main components, biomasses may contain inorganic substances (ash), organic extractives (lipids, waxes, and resins), starch, pectin, etc. If the initial biomass is subjected to chemical, physicochemical, or biological treatment, its chemical composition changes.
The chemical composition directly affects the calorific value of biomass. Increased content of lignin, lipids, resins, and waxes contributes to achieving a high calorific value of biomass [22,23,32]. This is due to the increased value of combustion enthalpy of lignin, ΔcH from -25 to -26 MJ/kg [33] compared with polysaccharides, ΔcH from -17.4 to -17.6 MJ/kg [3]. In addition, extractive substances of biomass, lipids, waxes, and resins have ΔcH from -36 to -38 MJ/kg [20,34,35,36], which is much higher than that combustion enthalpy of polymeric components of the biomass, lignin, and polysaccharides.
Numerous experiments on measuring the combustion enthalpy or gross heating value (GHV) have shown that for dry wood samples of different species, the difference in the calorific values is small [37,38]. Softwood samples have an average value of ΔcH = -20.5 MJ/kg, while hardwood samples have an average value of ΔcH = -19.7 MJ/kg [38] (Table 2). Forest residues have a lower GHV with ΔcH = -18.8 MJ/kg [39]. The calorific value of biomass of cereal crops and herbaceous plants turned out to be lower than that of woody biomass [20,40]. The lowest GHV was observed for paper waste containing a large amount of inorganic fillers [20,22,23], while olive samples having increased content of lignin and lipids are distinguished by quite high value of combustion enthalpy (Table 2).
There are several known methods for extracting energy from biomass. The most well-known method is the use of biomass as solid biofuel. In addition, biomass can be a feedstock for obtaining secondary biofuels from it, such as biochar, bio-oil, bioethanol, biodiesel, and biogas.
However, the direct use of biomass as a solid fuel raises several problems. The initial biomass is a non-dense and heterogeneous material consisting of pieces of various sizes [18]. Moreover, it can contain moisture and inorganic substances. These features of the initial biomass worsen its fuel properties, since moisture and inorganic admixtures decrease the calorific value, and the low bulk density of the initial biomass declines the density of thermal energy, which significantly reduces the productivity of furnaces.
To improve the fuel performance, the initial biomass should be demineralized, dried, and densified into briquets and pellets [41,42,43,44,45]. For example, it was shown that the initial unpressed Switchgrass biomass has unsatisfactory characteristics: bulk density, BD = 98 kg/m3, and thermal energy density, ED = 1.8 GJ/m3, while after pelletization these features increased 5 times [46]. When plastic waste is used as a binder, these features of the pelletized solid biofuel increase by 8 times, which is also accompanied by a rise in the GHV of the pellets.
Along with experimental methods, numerous attempts have been made to calculate the combustion heat of biomass. The most common approach is to use several correlation equations to calculate the calorific value of each type of biomass [32,33,47,48,49,50]. For the calculation, diverse variables were used such as ash content and moisture; individual biomass components, such as lignin; volatile matter and fixed carbon; various physical properties, etc. In addition, correlation equations based on the determination of the elemental composition of biomass were proposed [47,51,52,53]. However, since the amount of plant biomass species is unlimited, the number of correlation equations can also be unlimited. In addition, the articles [32,33,47] do not provide a critical analysis of the various mathematical models and correlation equations, so it is impossible to identify the equation that is best suited for specific biomass types. Analyzing calculation results of GHV for various biomasses [32,48], Park and co-authors [50] concluded that the discrepancies between calculations and experimental data are high, over 10%.
To improve the accuracy of calculations, it is necessary to use another method than finding correlations with some randomly selected biomass characteristics. In our studies, we used a method based on the rule of additive contributions [20]:
where Ci is the percentage content of components (see e.g., Table 3); ΔcH i is GHV of the component of the biomass (Table 4).
ΔcH = 0.01 ∑ Ci ΔcH i,
The results revealed that the calculation method based on the rule of additive contributions gives values of combustion enthalpy for biomass samples that are very close to the experimental data (Table 3). More detailed studies have shown that the average deviation between calculated and experimental results does not exceed 3% [54].
4. Thermal Energy Potential of Secondary Biofuels
Plant biomass is also a feedstock for producing secondary biofuels, such as biochar, bio-oil, and biogas. For this purpose, the initial biomass is pyrolyzed, i.e. subjected to heat treatment in the absence or lack of oxygen [16,22,55,56].
Slow pyrolysis of biomass is carried out with a heating rate below 60 degrees/min at temperatures of about 500oC and residence time of 10-20 min with the formation of biochar, bio-oil, and biogas in approximately the same amounts. If slow pyrolysis is processed at high temperatures, from 600 to 1000oC, then strong gasification of biomass with a predominant hydrogen fraction occurs [57,58]. Hydrothermal gasification of biomass, especially in the presence of a catalyst, significantly increases the hydrogen content in biogas.
Fast pyrolysis is performed at temperatures from 500 to 650oC for short contact time. This pyrolysis type produces usually 60% bio-oil, 20% biochar, and 20% biogas.
In addition, flash pyrolysis is known, proceeding at temperatures higher than 650oC with a contact time of less than 1 sec. The main product of flash pyrolysis is biogas.
It was found that biochar has GHV ca -27 MJ/kg [22]. The bio-oil is a dark and thick liquid containing a complex mixture of water and various organic substances (tar, hydrocarbons, furan derivatives, aldehydes, ketones, phenols, organic acids, methanol, etc.) with GHV ca -16 MJ/kg [22].
The biogas is a mixture, containing CO2, CO, H2, and CH4, with a GHV ca -10 MJ/m3 [16,22]. This biogas is formed as a result of the following processes [16]:
- 1).
- Oxidation of biochar at increased temperature
C + 0.5 O2 → CO with ΔrH (T) = -103.9 kJ/mol
C + O2 → CO2 with ΔrH (T) = -370.1 kJ/mol
- 2).
- Reduction reactions of gases at increased temperature
C + CO2 2CO with ΔrH (T) = 162.4 kJ/mol
C + H2O CO + H2 with ΔrH (T) = 131.9 kJ/mol
CO + H2O CO2 + H2 with ΔrH (T) = -30.5 kJ/mol
C + 2H2 CH4 with ΔrH (T) = -65 kJ/mol
The average gasification temperature is 800oC or 1073 K [57,58]. Therefore, instead of the standard enthalpies of reactions, ΔrHo, at 298.15 K, the enthalpies of these reactions, ΔrH (T), at a temperature of T = 1073 K should be used. The calculated values of ΔrH (T) are shown near the equation of the corresponding reaction.
To implement endothermic reactions that have positive values of reaction enthalpy, they must absorb thermal energy from the outside. Exothermic reactions can occur spontaneously without heat absorption from the outside, but often a temperature rise is necessary to increase the rate of such reactions.
Using slow pyrolysis of 100 kg woody biomass, the following yield of secondary biofuel was determined: 30 kg biochar, 18.1 kg bio-oil, and 14 m3 biogas [59]. It can be calculated that the total content of thermal energy (TEC) of such biofuel will be TEC = - 1.24 GJ, which is only 62% of the thermal energy content in 100 kg initial biomass equal to -2 GJ.
Another study [57] found that the yield of biogas after the gasification of pine wood was ca 37 m3 from 100 kg of wood. In this case, the TEC of such biogas volume does not exceed 20% of the TEC in 100 kg initial wood.
Biogas enriched with methane can be obtained by anaerobic digestion [60]. This process is performed at a pH 6-7 under the action of mesophilic bacteria at a temperature of 30-40 °C or thermophilic bacteria at a temperature of 50-60 °C. As a feedstock for biogas production, MSW, waste paper, residue of pulp and paper, agricultural waste, etc. can be used [16,60,61]. Lignocellulosic biomass is pretreated to loosen its physical structure and reduce the content of lignin hindering the bioconversion process. The pretreated biomass is diluted with water to prepare aqueous dispersion for anaerobic digestion.
The process of anaerobic biodegradation of organic substances usually takes 3 - 4 weeks and occurs in several stages. In the first stage, called hydrolysis, the complex organic molecules (СM) break down into simple molecules (SM). At the second stage, called acidogenesis, deeper biodegradation occurs with the release of volatile products and the formation of carboxylic acids (CA). In the third stage, called acetogenesis, simple molecules and carboxylic acids are converted to acetic acid (AA). In the last stage, called methanogenesis, acetic acid is converted to biogas (BG) containing mainly methane and carbon dioxide.
Thus, the anaerobic digestion process of biomass can be expressed, as follows:
CM → SM → CA → AA → BG
For example, in the first stage, cellulose and other C6-polysaccharides split into monomeric glucose (GL). Further, glucose forms carboxylic acids and acetic acid, which is transformed into biogas.
-(C6 H10O5-)n + n H2O → nС6H12O6
nС6H12O6 → 3n CH3COOH
3n CH3COOH → 3n CH4 + 3n CO2
Total process: -(C6H10O5-)n + n H2O → 3n CH4 + 3n CO2
The theoretical yield of this biogas from 1 t cellulose is 830 m3 and their thermal energy content TEC = -14.9 GJ, ca 86% from the TEC of initial cellulosic feedstock with TEC = - 17.4 GJ. However, due to increased cellulose crystallinity, the actual yield of biogas and its TEC from cellulose are 1.5 - 2 times lower than the theoretical values. An even lower biogas yield is observed during the anaerobic digestion of lignocellulosic raw materials.
To extract energy from plant biomass, it can be also hydrolyzed and transformed into monosaccharides, mainly glucose, followed by fermentation to produce bioethanol.
Features of the enzymatic hydrolysis of various plant materials have been discussed in many studies [62,63,64,65,66]. To implement the effective hydrolysis of biomass, enzyme preparations were used, which include at least three types of specific enzymes, such as endo-1,4-β-glucanases, exo-1,4-β-glucanases, and β-glucosidases. These enzymes act synergistically because endo-acting enzymes generate new chain ends for the exo-acting enzymes, which release the oligosaccharides that are converted into glucose by β-glucosidases [64].
The enzymatic hydrolysis of biomass is usually carried out at a temperature of 50 °C and pH 5 using a dose of enzyme preparation of 10 to 40 mg of protein per 1 g of substrate. It has been found that to achieve maximum glucose concentration during enzymatic hydrolysis, the optimal loading of the cellulose-containing substrate in the aqueous enzyme system should be at least 150 g/L [62,63,64]. At a higher substrate loading, enzymatic hydrolysis ceases due to a significant reduction in the mass transfer and inhibition of cellulolytic enzymes by a large amount of formed glucose [67].
In addition, it was established that the initial plant biomass exhibits high resistance to enzymatic cleavage due to the compact structure of the plant material and the presence of lignin and other non-cellulosic components hindering the hydrolysis process. Therefore, the plant biomass is pretreated to loosen its physical structure, eliminate non-cellulosic components, and increase cellulose accessibility to cellulolytic enzyme molecules. To reduce biomass recalcitrance, various pretreatment methods can be used such as steam explosion, acid hydrolysis, alkaline extraction, oxidation, and combined methods [68,69,70,71,72,73,74]. After the acidic pretreatment of the biomass, predominantly hemicelluloses are removed, which leads to an increase in the cello-lignin content. During alkaline and oxidative pretreatments, partial removal of hemicelluloses and lignin from the biomass occurs, which is accompanied by an increase in the cellulose content in the pretreated biomass and a reduction of biomass recalcitrance to enzymatic hydrolysis.
Combined pretreatment methods are considered the most effective for removing non-cellulosic components from biomass and improving enzymatic hydrolyzability. For example, 1 kg of switchgrass biomass was treated with dilute nitric acid and then with dilute alkali solution [46]. The result was 410 g of cellulose-enriched pre-treated biomass (PTB). After enzymatic hydrolysis of PTB, 320 g of glucose was obtained. In the fermentation stage, glucose produces 163 g bioethanol with a thermal energy content of TEC = -4.9 MJ, which however is only 27% of the TEC in 1 kg initial biomass equal to -18.3 MJ.
Special plant types containing vegetable oil are well known such as soybeans, rapeseed, olives, sunflower, palms, etc. However, these oil plants are in demand in the food industry, and their use for energy production is limited. For this purpose, it is desirable to use non-edible lipid sources such as camelina, jatropha, pongamia legume, crabapple, jojoba, castor oil plant, tung, algae, etc. Tall oil and waste cooking and frying oils are considered suitable feedstocks for biofuel production.
Though vegetable oils, technical oils, and other lipids have a high calorific value (-35 to -40 MJ/kg), they are less suitable as diesel fuel due to relatively low cetane number, low volatility, high flash point, and increased viscosity [22]. To improve fuel characteristics, lipids are subjected to transesterification. This process is carried out by reacting triglycerides (TGL) of lipids with alcohols, usually with methanol (MET), in the presence of catalysts. The final products are glycerol (GL) and ester of methanol and fatty acid (e.g. linoleic acid) used as a biodiesel fuel (BDF):
The glycerol fraction is separated from biodiesel fuel and the excess alcohol is removed by distillation or partial evaporation [16].
C3H5(O2CR)3 + 3CH3OH → 3RCO2CH3 + C3H5(OH)3
The following standard formation enthalpies were found or determined from combustion enthalpies of the starting reagents and products of this reaction (Table 5).
Using values of standard enthalpies, the enthalpy of the transesterification reaction was calculated as follows:
As a result, it was found that the enthalpy of transesterification reaction ΔrHo = -363 kJ/mol. Thus, this reaction is exothermic and can proceed spontaneously without a temperature increase. However, to speed up the process, it is heated to 50-70oC [16,75].
ΔrHo = ΔfHo(GL) + 3ΔfHo(BDF) - ΔfHo(TGL) - 3ΔfHo(MET),
As with other types of biofuels, the TEC of biodiesel fuel can be also compared to the TEC of initial feedstock. Let's say, for example, that the source of vegetable oil is fallen olive fruits. From one ton of these fruits, 230 kg of oil can be extracted and subjected to transesterification. The calculations showed that the resulting biodiesel fuel will have a thermal energy content TEC= -7.4 GJ, which is however significantly less than the amount of energy TEC= -26. 1 GJ that can be obtained by burning 1 ton of fallen olives.
Although biomass is considered CO2 neutral and the secondary biofuels extracted from it are CO2 tolerant, the increasing greenhouse effect requires a reduction in CO2 concentration in the atmosphere. Therefore, the process of converting carbon dioxide into biomethane, known as the Sabatier reaction, has attracted considerable attention [76,77].
The Sabatier reaction is expressed by the following equation:
CO2 + 4H2 ⇌ CH4 + 2H2O
Thus, it is the hydrogenation process of carbon dioxide. However, at normal or slightly increased temperatures and pressures, the rate of direct reaction probable is negligible, and the process is shifted to the left. To ensure the conversion of CO2 to CH4, the reaction should be carried out at elevated temperatures and pressures in the presence of a catalyst [78,79,80]. Studies have shown that a high yield of synthetic methane, over 90% of the theoretical yield. can be achieved if the reaction temperature is about 400oC (673.15 K) and the pressure is about 3 MPa [79]. As catalysts, Ni and Ru on alumina and some others are used [76,80].
The Sabatier reaction most likely occurs in two stages. In the first stage, carbon dioxide is reduced with hydrogen to carbon monoxide, and in the second stage, carbon monoxide is converted to methane.
CO2 + H2 ⇌ CO + H2O ΔrHo = 41.2 kJ/mol
CO + 3H2 ⇌ CH4 + H2O ΔrHo = -206.2 kJ/mol
Total: CO2 + 4H2 ⇌ CH4 + 2H2O ΔrHo = -165 kJ/mol
It was found that the Sabatier reaction is exothermic [76]. Several references also indicated that the standard enthalpy of the Sabatier reaction, ΔrHo = -165 kJ/mol CH4 when formed water is in the gaseous (vapor) state [76,81,82,83,84,85,86]. The standard Gibbs potential of this reaction was also estimated [85]. Unfortunately, in the case, when the formed water vapor condenses into liquid, the calculation of the standard enthalpy and Gibbs potential of the reaction was not performed. However, thermodynamic analysis requires calculating three thermodynamic functions, enthalpy, entropy, and Gibbs potential, not only for standard temperature and pressure but also for real conditions occurring at high temperature and pressure. For this purpose, a complete thermodynamic analysis of the Sabatier reaction was carried out under standard and real reaction conditions [87].
Using reference books, the standard enthalpies of formation (ΔfHo) and the standard enthalpies (So) for reagents and products of the Sabatier reaction were found (Table 6) and used for further calculations.
Thermodynamic analysis in the case of the gaseous (vapor) phase state of the formed water
The standard enthalpy of the Sabatier reaction was calculated using the equation:
Similarly, the standard entropy of this reaction was calculated, as follows:
ΔrHo(g) = ΔfHo(CH4, g) + 2ΔfHo(H2O, g) - ΔfHo(CO2, g) - 4ΔfHo(H2, g),
ΔrSo(g) = So(CH4, g) + 2So(H2O, g) - So(CO2, g) - 4So(H2, g),
In addition, the Gibbs potential of the reaction was found:
where To = 298.15 K is the standard temperature.
ΔrGo(g) = ΔrHo(g) -To ΔrSo(g),
Substituting the values of the required TDFs of reagents and products from Table 6 into these equations, the standard TDFs of the Sabatier reaction were obtained (Table 7).
Thermodynamic analysis in the case of the liquid phase state of the formed water
The calculations were performed using similar equations with the difference that standard TDF of liquid (l) water was used instead of gaseous (g) water.
The calculation results are presented in Table 7. When the resulting water is liquid, the exothermic thermal effect of the reaction is higher than in the case of gaseous water. The same relationship is observed for the Gibbs potential of the Sabatier reaction. A more negative value of the Gibbs potential means that the process that forms liquid water is more favorable than the process that produces gaseous water (vapor).
Despite the negative value of the Gibbs potential, the process of carbon dioxide hydrogenation under standard conditions, To= 298.15 K and Po = 0.1 MPa, cannot be implemented, due to kinetic limitations [80]. As the temperature rises, the reaction rate increases, especially in the presence of a catalyst [76,78,79,80]. In addition, the elevation of pressure shifts the equilibrium of the reaction to the right and increases the methane yield [85,86].
Thermodynamic analysis of catalyzed Sabatier reaction at high temperatures and pressures
These calculations were carried out for real conditions of the Sabatier reaction, Tr = 673.15 K and Pr = 3 MPa in the catalyst presence, when the reaction rate is high enough to ensure a high yield of the formed methane. Under these conditions, the water is in a gaseous phase state. As is known, enthalpy is almost independent of pressure and the main influence on the enthalpy value is exerted by temperature. The enthalpy value of a gaseous substance at pressure Pr and temperature Tr can be found using the following equation:
Unlike enthalpy, the entropy depends on both pressure and temperature. The value of enthalpy at pressure Pr = 3 MPa and standard temperature To = 298.15 K was calculated using the equation:
where standard pressure Po=0.1 MPa, and R=8.3145 (J/mol K) is the gas constant.
S(Pr,To) = So(g) - R ln(Pr/Po),
The calculated values of S(Pr,To) at Pr = 3 MPa for the studied substances are shown in Table 8.
Further, the entropy value of the gaseous substance at pressure Pr and temperature Tr was calculated, as follows:
where Cp is the specific heat capacity of gaseous reagents and products.
The calculation results of TDFs are shown in Table 9.
Using these TDFs of gaseous substances, the enthalpy, entropy, and Gibbs potential of the Sabatier reaction at high temperature and pressure were obtained [87] (Table 10).
These results showed that the Sabatier reaction is highly exothermic also at increased temperature and pressure, while reaction entropy is negative. In addition, the Gibbs potential under these conditions is negative. Thus, the process of methane synthesis from carbon dioxide and hydrogen at elevated temperature Tr = 673.15 K and pressure Pr =3 MPa is energetically and thermodynamically favorable. Using the Van 't Hoff equation, it was found that the equilibrium constant of the reaction Keq is 2.04 x 103 [87]. Such a value of the equilibrium constant indicates that under real conditions the equilibrium of the Sabatier reaction is shifted to the right with the formation of methane and water vapor.
Calculations also showed that heating a compressed initial gas mixture from To to Tr requires the expenditure of 61.1 kJ/mol of thermal energy, which is 3 times less than the thermal energy released by the exothermic Sabatier reaction.
The Sabatier process continues to be improved. New designs of CO2 hydrogenation reactors with an improved heat exchange have been proposed [76,83,88]. In addition, more efficient catalytic systems and the removal of resulting water vapor allow for optimization of reaction conditions and enhance methane yield [76,80,88,89]. The thermal energy of the Sabatier process can be utilized to heat the initial gas mixture and electrolyze water [90]. Moreover, constructions are being developed that combine a CO2 hydrogenation reactor with a fuel combustion furnace [76], an electrolyzer [76,90], or an electrochemical cell [81,82].
5. Thermodynamic Characteristics for Main Components of Biomass
5.1. Thermodynamic Characteristics of Cellulose Samples
Standard combustion enthalpies (ΔcHo) of various semicrystalline cellulose Iβ samples, including cellulose isolated from wood, cotton, bast cellulose fibers, and some other biomass types, were determined in [3,7,20,49,54,91,92,93,94]. It was established that the ΔcHo value of these samples varies within small limits of ± 0.08 MJ/kg and its average value is ΔcHoav -17.41 MJ/kg or -2820.42 kJ/mol. Then, according to eq. (23), we can calculate that the standard enthalpy of cellulose formation is, on average ΔfHoav = -969.79 kJ/mol. However, detailed studies have shown, that there is still a small but significant difference in the thermodynamic characteristics between cellulose Iβ samples with different crystallinity [20].
Crystallinity is an important structural characteristic of cellulose, determining a complex of its physical, physiochemical, and chemical properties such as specific gravity, heat capacity, thermal expansion coefficient, sorption properties, hydrolyzability, etc. [24]. However, there is a problem associated with the lack of an accurate method for determining the degree of crystallinity of cellulose. There are only approximate methods for estimating the so-called crystallinity index (CrI) [11,95,96,97,98]. Moreover, different methods such as wide-angle X-ray scattering (WAXS), solid-state 13C-NMR, FTIR, Raman spectroscopy, etc. give different CrI values for the same cellulose sample; therefore, it is not clear which value of CrI should be preferred [99,100,101]. Even the same method, e.g., WAXS, also gives different CrI values for the same sample if different measurement techniques were used to separate X-ray scattering from the crystalline and amorphous domains [95,96,101]. For example, using different WAXS techniques, it was found that the CrI of the MCC Avicel sample varies from 49 to 82%, while the CrI of the cotton linter ranges from 55 to 87% [101].
Large discrepancies in the values of CrI when they are estimated by different methods or different measurement techniques of the same method are due to the use of different calculation equations, mathematical and structural models, and software. Secondly, this is due to the different experimental conditions of different methods. In addition, there are no standard protocols for preparing cellulose samples to determine their crystallinity. Thus, it is impossible to conclude which of the currently used methods and/or techniques for measuring cellulose crystallinity is the most suitable [101].
Therefore, a new method that works on other principles is advisable to develop. In our studies, a thermochemical method was proposed based on measuring the enthalpy of wetting (ΔwHo) of various cellulose samples [8,102]. It has been established that the interaction of cellulose with liquid water is accompanied by an exothermic thermal effect, i.e., the enthalpy of wetting. The higher the degree of amorphousness of the sample, the greater the exothermic value of the enthalpy of wetting [102]. As a result, the degrees of amorphicity (Y) and crystallinity (X) of cellulose samples were determined by a direct thermochemical method, as follows:
where ΔwHo is the enthalpy of wetting for the dry sample; and ΔwHoam = -168 kJ/kg DM or -27.2 kJ/mol AGUs is the wetting enthalpy for completely amorphous cellulose in the dry state [102].
Y = ΔwHo/ΔwHoam,
X = 1 - (ΔwHo/ΔwHoam),
The results have shown that the thermochemical method provides the determination degrees of amorphicity and crystallinity of cellulose samples with a standard deviation of no more than ±0.01 (Table 11).
It can be noted that the thermochemical method is direct, simple, fast, accurate, reliable, and reproducible. To measure the enthalpy of wetting any precise calorimeter can be used. It does not require special models, complex software, and calculations. This method does not have special requirements for the shape and size of the samples. They do not need to be milled or pressed. It is possible to use cellulose samples with different morphology and types of crystal structure (I, II, III, or IV) in the form of pieces, fibers, or powders. There are only two main conditions for preparing cellulose samples for testing: they must be chemically pure and completely dry.
After measuring the degree of crystallinity by a thermochemical method, it is possible to study how this structural characteristic affects the thermodynamic functions of cellulose samples.
Studies have shown that with a change in the degree of crystallinity (X), there is a tendency for a noticeable change in the TDFs of cellulose Iβ samples, which can be expressed with the following linear equations:
where ΔcHoam, ΔfHoam, and Soam are standard enthalpies of combustion and formation of AMC, and Soam is the standard entropy of AMC; coefficient K = 37. 2 kJ/mol, while coefficient k = 10.0 J/mol K.
ΔcHo = ΔcHoam + K X,
ΔfHo = ΔfHoam - K X,
So = Soam - k X,
Structural studies have shown that crystallites of celluloses of plants and tunicate have predominantly the allomorphic type Iβ with a monoclinic crystalline cell, while crystallites of algal and bacterial cellulose are characterized predominantly by the unstable allomorphic type Iα with a monoclinic crystalline cell [103,104].
After the physicochemical modification of cellulose, three additional crystalline allomorphs, II, III, and IV, were obtained [103,104,105,106,107]. Samples containing cellulose II crystallites were prepared by alkaline treatment of natural cellulose or by regeneration from cellulose solutions. Cellulose samples with crystalline allomorph III are obtained from I or II samples by treatment with ethylenediamine or liquid ammonia. Samples with crystalline form IV are usually prepared by heating cellulose III samples in hot glycerol. Different crystalline allomorphs of cellulose have different parameters of crystalline unit cells [103,104,105]. Along with various crystalline allomorphs, amorphous cellulose is known, which is produced by dry grinding of semicrystalline cellulose samples, by saponification of amorphous cellulose acetate in a non-aqueous medium, and some other methods [108].
Due to the existence of various crystalline allomorphs (CAs) and amorphous cellulose, various studies have been conducted to evaluate their phase stability. The amorphous phase state is considered to be labile since amorphous cellulose can easily crystallize into any crystalline allomorph under certain conditions. This conclusion is also supported by the results of thermodynamic studies [3]. In the case of crystalline allomorphs of cellulose, the problem of the relative stability of the phase state has not been completely solved and remains open. The study of thermodynamic characteristics gave grounds to believe that the phase state of CA II is more stable than CA I [8]. In addition, it was found that CA III is more reactive than CA II [106]. However, other studies concluded that the phase state stability of different crystalline allomorphs cannot be estimated with a reasonable degree of accuracy [3,107].
Therefore, additional studies were conducted for detailed investigations of the thermodynamic characteristics of various crystalline allomorphs of cellulose and amorphous cellulose [20,104]. For this purpose, cellulose samples having different crystalline allomorphs and different degrees of crystallinity (X) were prepared, and their standard TDFs were determined [20,104] (Table 13).
The results showed that an increase in the degree of crystallinity of the samples with the same type of crystalline allomorph leads to a linear change in the TDFs similar to that observed for samples of cellulose Iβ according to eq. (35)-(37). With a decreasing degree of crystallinity (X), the linear dependences of TDF converge to the same values corresponding to X = 0. This indicates that the amorphous phase in cellulose samples with different crystalline allomorphs has similar thermodynamic characteristics. On the other hand, linear extrapolation of these dependencies to values corresponding to X = 1 yields TDFs for different crystalline allomorphs (Table 14). In addition, the values of melting enthalpy (ΔmHo) of cellulose crystallites with different crystalline allomorphs were calculated.
The standard Gibbs potential of formation (ΔfGo) of crystalline allomorphs was also calculated using the equation:
where So is standard entropy, while ∑ Si is the sum of standard entropies of carbon atoms (graphite), molecules of H2 and O2 needed for forming one mole of AGUs.
ΔfGo = ΔfHo - To (So - ∑ 𝑆𝑖)
Based on the obtained results, the thermodynamic stability of crystalline allomorphs Iβ, II, III, IV and AMC was estimated. It can be assumed that the relative thermodynamic stability of cellulose crystalline allomorphs and AMC decreases in the following order:
On the other hand, the relative reactivity of the different crystalline allomorphs and AMC will decrease in the reverse order:
CII > CIV ≥ CI > CIII > AmC
AMC > CIII > CI ≥ CIV > CII
The obtained thermodynamic characteristics allow to explain the phase transitions and reactivity of cellulose samples with different crystalline allomorphs and AMC. Amorphous cellulose, AMC, is the most unstable and the most reactive, so it can crystallize into any crystalline allomorph under certain conditions [3,108]. For example, under the influence of moisture, amorphized cellulose recrystallizes into the most stable CA II. It is also known that after mercerization and regeneration from solutions, the structure of cellulose Iβ always transforms into CA II. Thus, among the various crystalline allomorphs, CA II is probably the most stable, while CA III is the most labile. It was found that high-temperature treatment of the sample with CA III at 480–490 K triggers the reverse transformation of CA III into CA Iβ [109], whereas, after treatment in glycerol above 533 K, CA III is transformed into CA IV [110].
As is known, the supramolecular architecture of cellulose consists of elementary nanofibrils and their bundles called microfibrils [24,111]. Moreover, each nanofibril is built of rod-like nanocrystallites with lateral size of 3-20 nm and length of 50-500 nm, as well as noncrystalline (amorphous) nanodomains. Due to this nanostructure, cellulose exhibits several specific properties.
The following example can be used to illustrate these specific properties. In plant biomass (e.g., softwood, SW) nano-fibrils are surrounded by an amorphous ligno-hemicellulose matrix and are separated from each other. During delignification at elevated temperatures in the cooking solution, the matrix is removed. This ensures direct contact of nano-fibrils and leads to aggregation by co-crystallization of adjacent nano-crystallites, which leads to an increase in the lateral size (L) of crystallites in isolated cellulose. The process of spontaneous aggregation and co-crystallization of nano-crystallites is thermodynamically favorable since it leads to a decrease in the specific surface area (Ssp) and a negative value of the thermodynamic Gibbs potential according to eq. (21) (Table 15).
Another example is when nano-crystallites III are transformed to crystallites IV after treatment in glycerol at temperature above 533 K. As is known, the theoretical equilibrium melting point of cellulose macrocrystals is estimated at 750 K [112]. However, nano-crystallites can melt at much lower temperatures due to the dependence of melting point on the lateral size of nano-crystallites by the Gibbs-Thomson equation (22) [113].
Calculations showed that the equilibrium melting point of cellulose crystallites reduces and reaches 533 K for nanocrystallites with a lateral size of less than 4 nm [112]. Thus, the mechanism of the transformation of nano-crystallites III into crystallites IV consists of reducing the melting point of small nano-crystallites III (L = 3.3 nm) to processing temperature (533 K) in plasticizing glycerol medium, amorphization of these crystallites, the transition of plasticized amorphized cellulose in the viscoelastic state, and finally the crystallization of viscoelastic amorphized cellulose into relatively large crystallites IV (L = 6.0 nm) that are also more thermodynamic stable than crystallites of CIII.
A characteristic feature of cellulose nanocrystals is also an increase in solubility (SB) and a decrease in alkali concentration (C) for mercerization with a decrease in their lateral size, L [112], which is consistent with Gibbs-Thomson theory [114].
where index n is related to nano-crystallites, while index m to macrocrystals.
ln SBn = ln SBm + (4σVm/L RT),
ln Cn = ln Cm - (4σVm/L RT),
5.2. Thermodynamic Characteristics of Non-Cellulosic Components of Biomass
Non-cellulosic components of biomass include mainly hemicelluloses, starch, pectin, lignin, and extractives. Hemicelluloses can consist of C-6 polysaccharides called hexosans (mannans, glucans, galactans, etc.) and C-5 polysaccharides called pentosans (xylans, arabans, etc.). The article [115] notes that the gross heating value (GHV) of amorphized hexoses and amorphized cellulose should be close, while the GHV of pentoses is higher than hexoses. Other studies [54] showed that the standard enthalpy of combustion of samples of hexosans is on average ΔcHo = -2833.6 kJ/mol (GHV = -17.49 MJ/kg), and samples of pentosans ΔcHo = -2348.0 kJ/mol (GHV = -17.79 MJ/kg) (Table 16).
Based on the chemical structure, it can be concluded that the GHV of starch and hexoses must be identical [115]. But, in reality, this is not the case, since different starch types differ from each other in amylose content and degree of crystallinity [116,117]. Therefore, thermodynamic characteristics should be determined for specific types of starch. For example, it was found that for corn starch, the enthalpy of combustion ΔcHo = -2836.6 kJ/mol, and the enthalpy of formation ΔfHo = -953.6 kJ/mol.
The pectin sample showed the value of combustion enthalpy lower than hexosans but higher than pentosans [118].
After studying various lignin samples, it was found that the average GHV of lignin from coniferous trees is -26.8 MJ/kg, and for lignin from deciduous trees and grass plants -25.3 MJ/kg [115]. The values of combustion enthalpy ΔcHo = -5172.4 kJ/mol of lignin samples from softwood and ΔcHo = -4984.1 kJ/mol of lignin samples from hardwood obtained in [54,118] correspond to the results of [115] if calculate the combustion enthalpy in MJ/kg.
The article [115] reports that the GHV of extractive substances is not constant and can vary widely depending on the plant species and the extraction method. For example, the GHV of extractives from coniferous wood species was in the range from -30 to -39 MJ/kg. To improve the reliability of the results, the calorific values of resin acids (e.g., abietic acid, etc.), which constitute the main part of the extractives in coniferous wood, were studied [54,118]. The results showed that the value of combustion enthalpy of these components is higher than -11500 kJ/mol (-38 MJ/kg), while their formation enthalpy is above -570 kJ/mol (Table 16).
6. Thermodynamic of Chemical Reactions of Cellulose and other Polysaccharides
6.1. Thermodynamics of Bioethanol Production
Bioethanol is produced by fermentation of glucose solutions, usually using the yeast Saccharomyces cerevisiae [119,120]. The glucose substrate required for fermentation is obtained by enzymatic hydrolysis of carbohydrates, starch, and cellulose. In the USA, bioethanol is produced primarily from corn starch [22], with an annual production volume of over 15 giga gallons [121].
The production process involves the extraction and purification of starch from corn kernels, dissolution of the starch at elevated temperatures to gelatinization, hydrolysis of the starch gel with α-amylase and then glucoamylase, yeast fermentation of the formed glucose to produce a dilute ethanol solution, followed by distillation and dehydration of the ethanol.
To hydrolyze starch, gel-like starch solutions with a concentration of 150 to 200 g/L usually are used [122]. The starch gel is hydrolyzed by α-amylase to liquefy the gel and facilitate the subsequent transformation of starch to glucose [123]. Since α-amylase is an endo-acting enzyme, it catalyzes the hydrolysis of inner α-1, 4-glycosidic and branched α-1,6-glycosidic bonds with the formation of oligomeric products, mainly, dextrin and maltose [124,125]. Further, these oligomers are hydrolyzed by the exo-enzyme, glucoamylase, resulting in their conversion into glucose [126].
Next, the resulting glucose solution is fermented, usually with S. Cerevisiae yeast [119,120], to produce a diluted alcohol solution with a concentration of no more than 100 g/L to prevent yeast inhibition [127,128]. To obtain absolute alcohol, the diluted alcohol solution was distilled and dehydrated.
Despite the progress in hydrolysis and bioethanol production, thermodynamical aspects of various steps of these processes have not been studied sufficiently. It is not clear, which step is exothermic and which is endothermic. What determines the feasibility of these processes, enthalpy or entropy? To solve these problems a thermodynamic analysis should be performed.
Enzymatic hydrolysis of a starch gel with the formation of glucose solution occurs, as follows:
The standard TDFs of the hydrolysis reaction were calculated using the following equations:
where H, GS, and SG denote hydrolysis, glucose solution, and starch gel, respectively.
-C6H10O5- x m H2O → C6H12O6 x (m-1) H2O
ΔrHo(H) = ΔfHo(GS) - ΔfHo(SG),
ΔrSo(H) = So(GS) - So(SG),
ΔrGo(H) = ΔrHo(H) -To ΔrSo(H),
The values of standard thermodynamic functions of GS and SG can be experimentally determined or found in reference books.
The results presented in Table 17 show that the standard thermodynamic functions for the hydrolysis reaction of starch gel have the same values regardless of the gel concentration. Since the Gibbs potential (ΔrGo) of this reaction has a negative value, the hydrolysis process is thermodynamically favorable. The entropy of this reaction increases due to the cleavage of long chains and the conversion of a high-molecular polysaccharide into a low-molecular product such as glucose. The enthalpy of hydrolysis reaction (ΔrHo) is exothermic, but its value is too small. Moreover, the absolute value of enthalpy is significantly less than the value of temperature-entropy factor (ToΔrSo). Thus, the hydrolysis reaction of starch gel with the formation of the glucose solution is determined mainly by the contribution of the temperature-entropy factor to the Gibbs potential. Therefore the equilibrium constant of the hydrolysis process has a moderate value: Keq = 2.65 x 103.
Glucose solution required to bioethanol production can be obtained also by enzymatic hydrolysis of cellulose [62,64,129]. Production of glucose from non-edible cellulose substrates has been regarded as a promising way to obtain this valuable bioproduct without competing with the food industry.
For the hydrolysis of cellulose, enzyme preparations are used, which include at least three types of specific enzymes such as endo-1,4-β-glucanases, exo-1,4-β-glucanases, and β-glucosidases [64,128]. Numerous studies have shown that when using cellulose substrates, a problem arises due to their low enzymatic digestibility. This is explained by the crystallinity of cellulose, which is considered the most important structural factor hindering enzymatic hydrolysis [64,128,130]. Therefore, a thermodynamic analysis was carried out to study the thermodynamic mechanism of enzymatic hydrolysis of cellulose substrates with different crystallinity [131].
An attempt to perform a thermodynamic study of the enzymatic hydrolysis of completely crystalline and completely amorphous cellulose at temperatures from 273 to 373 K using a small mass ratio of an aqueous catalyst system to cellulose (SCR) to obtain saturated glucose solutions followed by their dilution was made in [132]. However, this theoretical study did not take into consideration such specific features of enzymatic hydrolysis as the negative impact of cellulose crystallinity on the hydrolysis process, limited cellulose accessibility, and cessation of enzymatic hydrolysis at small SCR, low hydrolysis rate below room temperature and the inactivation of cellulolytic enzymes at temperatures above 328 K. These factors should be taken into consideration when studying the thermodynamics of the real process of enzymatic hydrolysis of cellulose.
When a wet semicrystalline cellulose substrate is treated with an aqueous enzyme system, the accessible amorphous domains of cellulose and the outer paracrystalline surface layers of the crystallites are hydrolyzed and form a final glucose solution containing n moles of H2O per mole of glucose (GL), while the internal highly ordered crystalline domains (CrD) remain unhydrolyzed. This process for one mole of AGUs of cellulose can be expressed, as follows:
where A is the accessibility degree of cellulose, which is a function of cellulose amorphicity (Y), crystallinity (X), and paracrystallinity (p) degrees: A = Y + pX.
-C6H10O5- x m H2O + (An + A - m) H2O → A (GL x n H2O) + (1-A) CrD
After the determination of standard TDFs of the wet cellulose substrates (CW), glucose solutions (GL x n H2O), and highly ordered crystalline domains (CrD), the TDFs of the hydrolysis reaction were calculated [131]:
ΔrHo(H) = (1-A) ΔfHo(CrD) + A ΔfHo(GL x n H2O) - ΔfHo(CW) - (An + A - m) ΔfHo(H2O, l),
ΔrSo(H) = (1-A) So(CrD) + A So(GL x n H2O) – So(CW) - (An + A - m) So(H2O,
ΔrGo(H) = ΔrHo(H) – To ΔrSo(H),
The obtained results showed [131] (Table 18) that the enthalpy of cellulose hydrolysis is small exothermic, like the hydrolysis enthalpy of starch. The value of the temperature-entropy factor (ToΔrSo) is significantly higher than the absolute value of the hydrolysis enthalpy. Therefore, the contribution of this factor to the negative Gibbs potential is predominant. In addition, with an increase in cellulose accessibility degree, the negative value of the Gibbs potential increases. Thus, the enzymatic hydrolysis of amorphous cellulose (AMC) is the most favorable. This is confirmed by the fact that the equilibrium constant of the hydrolysis reaction of AMC, Keq = 5.82 x 104, is more than a thousand times greater than that of semicrystalline cellulose substrates. In addition, the enzymatic hydrolysis of high-ordered crystalline domains of cellulose cannot be performed since the Gibbs potential of this process is close to zero.
Glucose solutions obtained after enzymatic hydrolysis of starch or cellulose can be used to produce bioethanol by yeast fermentation. The fermentation process can be described by the following equation:
To avoid producing an alcohol solution with a concentration higher than 100 g/L, which inhibits yeast, the concentration of the glucose solution should not exceed 196 g/L. In this case, the number of water molecules n per one mole of glucose is ca 51.
0.5 C6H12O6 x n H2O → CO2 + C2H6O x 0.5 n H2O
The standard TDFs of the fermentation process (F) of glucose solution (GS) conversion into ethanol solution (ES) were calculated using the following equations:
where ΔfHo(CO2) and So(CO2) are the standard formation enthalpy and entropy of carbon dioxide; ΔfHo(ES) and So(ES) are the standard formation enthalpy and entropy of ethanol solution; while ΔfHo(GS) and So(GS) are the standard formation enthalpy and entropy of glucose solution.
ΔrHo(F) = ΔfHo(CO2) + ΔfHo(ES) - 0.5 ΔfHo(GS),
ΔrSo(F) = So(CO2) + So(ES) - 0.5 So(GS),
ΔrGo(F) = ΔrHo(F) -To ΔrSo(F),
From the obtained results (Table 19) it follows, that the process of bioethanol production by fermentation of a glucose solution has a negative value of the Gibbs potential. In addition, the enthalpy of this process is quite exothermic and, together with the temperature-entropy factor, contributes to the negative Gibbs potential. Thus, the process of glucose conversion into bioethanol is both energetically and entropically favorable. In addition, it was found that the equilibrium constant of this process is very large: Keq =1.35 x 1020. Thus, the reaction equilibrium is strongly shifted towards the formation of bioethanol.
6.2. Thermochemistry of Cellulose Alkalization and Etherification
Treatment of cellulose with solutions of hydroxides of alkali metals is one of the most common methods of modification of this biopolymer. Currently, treatment with aqueous solutions of sodium hydroxide is used for the improvement of luster, hygroscopic properties, and dyeing of cellulose fibers and fabrics, for cellulose activation and refining, as well as in the production of cellulose ethers [133,134,135,136].
It has been established that after cellulose treatment with solutions of sodium hydroxide having a concentration of less than 10-11%, the crystalline structure of cellulose I remains unchanged [137]. However, when cellulose is alkalized with 16-20% alkali solutions the hydrated hydroxide ions penetrate between [10] planes of the crystalline lattice of CA I and transform it into the swollen crystalline lattice of alkali cellulose (AlC) containing one molecule of NaOH and three molecules of H2O per one anhydroglucose unit (AGU) in crystalline domains. Cellulose treatment with more concentrated sodium hydroxide solutions transforms the crystalline lattice of CA I into lattices of alkali celluloses containing one NaOH molecule and different numbers of water molecules n per one AGU [137]. For example, cellulose alkalization with 35-40% NaOH causes the formation of a crystalline lattice of AlC with the composition AGUNaOHH2O, i.e., containing no more than one molecule of H2O per AGU. During alkalization, the partial decrystallization of cellulose is observed.
After washing various alkali celluloses with water, hydroxide ions are replaced by water molecules resulting in the formation of the crystalline lattice of hydrate-cellulose, which after drying turns completely into the crystalline lattice of CA II [138]. It was also established that cellulose alkalization is accompanied by an exothermic heat effect, enthalpy of alkalization, ΔalHo [137] (Table 20).
The results show that the production of alkaline celluloses AlC 4, 5, and 6 is accompanied by a greater exothermic thermal effect than the production of alkaline celluloses AlC 1, 2, and 3.
Another significant thermodynamic characteristic is the standard enthalpy of the formation of alkali cellulose from one AGU and alkali solution containing m moles H2O per one mole of NaOH. Following Hess’s law, the standard enthalpy of the formation of alkali cellulose after the alkalization of cotton cellulose (CC) can be calculated, as follows:
where ΔfH (CC) = -969 kJ/mol is the standard enthalpy of formation of one mole of AGUs of original cotton cellulose [20], ΔfH (H2O) = -285.83 kJ/mol is the standard enthalpy of formation of liquid water, ΔfHo(Alkali) is the standard enthalpy of formation of alkali solutions [137].
ΔfHo(AlC) = ΔfHo (CC) + ΔfHo(Alkali) – (m-n) ΔfHo(H2O) + ΔalHo,
From the presented results (Table 20) it follows that the enthalpy of the formation of various alkaline celluloses is exothermic. Thus, the process of AlCs formation is energetically advantageous. In addition, since the exothermic enthalpy of alkalization to obtain AlC 4-6 has the greatest value, these alkaline celluloses must be more reactive than AlC 1-3. For this reason, the stage of cellulose alkalization before etherification is carried out using 30-40% alkali solutions, which provide alkaline celluloses AlC 4-6 with increased reactivity.
Consider as an example the process of production of methylcellulose (MC) [137]. The first stage of this process is cellulose alkalization, e.g., with a 40% NaOH solution at room temperature, after which the excess alkali is removed to obtain a mass ratio of alkali to cellulose of 3:1. The resulting alkali cellulose such as AlC 6 is kept for 1-2 h, and then methylated with methyl chloride:
C6H7O2 (OH)3·NaOH · H2O + (x-1) NaOH + x CH3Cl → C6H7O2 (OH)3-x (OCH3)x + x NaCl + (1+x) H2O
where x is the degree of substitution (DS) of MC. The DS of the samples, MC-1 and MC-2 was set to 1.3 and 1.8, respectively.
Taking into consideration the etherification process, the enthalpy of the methylation reaction of AlC 6 can be calculated using the equation:
where ΔfHo(AlC 6) is the standard enthalpy of formation of AlC 6), while ΔfHo(NaCl), ΔfHo(NaOH, ΔfHo(CH3Cl), and ΔfHo(H2O) are the standard enthalpies of formation of the corresponding substances.
ΔrHo = ΔfHo(MC) + xΔfHo(NaCl) + (1+x) ΔfHo(H2O) - ΔfHo(AlC 6) - (x-1) ΔfHo(NaOH) – x ΔfHo(CH3Cl),
As a result, the following values of enthalpy of the methylation reaction of AlC 6 to obtain methylcellulose samples were found [137].
Since the enthalpy values of the methylation reaction of AlC 6 were quite exothermic, this means that the methylation reaction is energetically favorable. Moreover, obtaining methylcellulose with a higher degree of substitution is preferable.
For MC 1 (DS = 1.3): ΔrHo (MC 1) = -184 kJ/mol.
For MC 2 (DS = 1.8): ΔrHo (MC 2) = -287 kJ/mol.
6.3. Thermochemistry of Cellulose Esterification
Among diverse cellulose derivatives, two derivatives, namely the acetates, and nitrates, are of particular importance. To obtain cellulose acetates, the starting cellulose material is placed in a suitable organic liquid and treated with an anhydride of acetic acid in the presence of a small amount of a catalyst [139,140]. The esterification process is heterogeneous if cellulose and its ester do not dissolve in the organic liquid. When the organic liquid is a solvent for cellulose and its ester, the reaction is carried out under homogeneous conditions. The heterogeneous acetylation of cellulose proceeds in the solid phase according to the laws of topochemical reactions: rapidly in amorphous domains and more slowly inside the cellulose crystallites. On the contrary, the homogeneous acetylation proceeds fairly uniformly in liquid media that cause swelling or/and dissolution of cellulose.
Studies have shown that replacing cellulose hydroxyls with acetate groups leads to a significant increase in the hydrophobicity of the resulting derivative [141]. In addition, when moving from a monoester to a triester, a noticeable increase in hydrophobicity is observed, since in this case all three hydroxyls in the repeating cellulose unit are replaced by non-polar groups. Due to their performance properties, cellulose acetates can be used to produce cigarette filters, separating membranes, optical films, self-cleaning materials, protective coatings, anti-fog goggles, and other products [141,142,143,144].
Another significant cellulose esters are nitrates. To produce nitrocelluloses the starting cellulose material can be treated with mixtures of nitric with sulfuric, or phosphoric acids [145,146]. In addition, mixtures of nitric acid with anhydrides of phosphoric or acetic acids also can be used for nitration. Nevertheless, in industry, only mixtures of nitric with sulfuric acid are applied to synthesize nitrocelluloses. Cellulose nitrates are used for the production of plastics, membranes, films, protective coatings, adhesives, varnishes and enamels, smokeless powder, rocket fuels, explosives, etc. [147,148,149].
Although the chemistry of esterification reactions of cellulose has been studied quite well, their thermochemical aspects are largely unexplored. Since only a small research number has been devoted to this topic [150,151], the specificity of the thermochemistry of cellulose esterification remained unclear. Therefore, further research was undertaken with samples of cellulose acetates (AC) and nitrates (NC) with different degrees of substitution (DS) [20,152]. The thermochemical characteristics of these samples are presented in Table 21.
For comparison with the obtained TD characteristics, some literature data are shown in Figure 1 and Figure 2. As can be seen, the experimental results [20,152] are confirmed by the literature data [150,151].
From the linear dependences of TDFs on DS, the TD characteristics of cellulose esters can be calculated using the following correlation equations.
For cellulose nitrates
where Kc(NC) = 48 and Kf(NC) = 96
ΔcHo(NC) = ΔcHo(AMC) + DS Kc(NC),
ΔfHo(NC) = ΔfHo(AMC) + DS Kf(NC),
For cellulose acetates
where Kc(AC) = 889 and Kf(AC)= 186
ΔcHo(AC) = ΔcHo(AMC) - DS Kc(AC),
ΔfHo(AC) = ΔfHo(AMC) - DS Kf(AC),
Structural studies indicate that as a result of esterification, cellulose esters with an amorphized structure are formed [141,153]. Thus, the process of cellulose esterification is accompanied by decrystallization (melting) of cellulose crystallites and subsequent esterification of amorphized cellulose (AMC) with the formation of amorphous esters. This process can be expressed by the following TD equation:
where ΔrHo(СС) and ΔrHo(AMC) are enthalpies of esterification of cotton cellulose (CC) and amorphized cellulose (AMC), respectively; ΔmHo =37 (kJ/mol) is the melting enthalpy of cellulose I crystallites, while X=0.7 is the average crystallinity degree of CC sample.
ΔrHo(СС) = ΔrHo(AMC) + X ΔmHo,
The enthalpy of nitration of AMC can be expressed, as follows:
On the other hand, the enthalpy of acetylation of AMC was calculated using the following equation:
The formation enthalpies of AMC, NC, and AC samples with various DS, reagents (HNO3 and acetic anhydride, AcAn), as well as low-molecular-weight products of esterification (H2O and acetic acid, AcAc), were determined experimentally or found in reference books.
ΔrHo(AMC)NC = ΔfHo(NC) + DS [ΔfHo(H2O) - ΔfHo(HNO3)] - ΔfH(AMC),
ΔrHo(AMC)AC = ΔfHo(AC) + DS [ΔfHo(AcAc) - ΔfHo(AcAn)] - ΔfHo(AMC),
As can be seen from the results (Table 22), the nitration reaction of cellulose to DS of 1.5 is endothermic. In this case, the only possibility for the feasibility of nitration reaction is that the absolute value of the temperature-entropy factor must be greater than the absolute value of reaction enthalpy, so that the Gibbs potential of this reaction becomes negative, namely:
where ΔrG is the Gibbs potential of cellulose nitration, and T is the reaction temperature.
[TΔrS] > [ΔrH] and ΔrG = (ΔrH -TΔS) < 0
Thus, the nitration process of cellulose with a weak nitrating system to DS up to 1.5 can only be implemented if the temperature-entropy factor is positive and makes a predominant contribution to the Gibbs potential.
When cellulose is nitrated with concentrated nitrating systems to DS above 1.5, the reaction enthalpy becomes exothermic. In this case, the feasibility of the nitration process may depend on both the contribution of exothermic enthalpy and temperature-entropy factor to the Gibbs potential.
Unlike nitration, the acetylation of cellulose is always an exothermic process, regardless of the achieved DS value (Table 23). The more DS, the higher the exothermic heat effect of this reaction. Besides, an increase in exothermic reaction entropy and positive temperature-entropy factor will facilitate the implementation of the acetylation process.
The final conclusion is that the esterification of cellulose to high degrees of substitution is advantageous because it is an energetically favorable process. The results also showed that cellulose acetylation is accompanied by the release of significantly more thermal energy than the cellulose nitration process (Figure 3).
7. Discussion
As is known, renewable plant biomass is considered CO2-neutral. Therefore, its combustion does not increase the concentration of this greenhouse gas in the atmosphere. It should also be noted that the volume of carbon dioxide formed during the combustion of 1 t biomass (ca 900 m3 CO2) is two times less than when burning 1 t such solid fossil fuel as coal (ca 1870 m3 CO2) and even 1.7 times less when burning 1 t of liquid or gaseous fossil fuels (ca 1600 m3 CO2). In addition, CO2 released during the combustion of plant biomass can be converted into gaseous biofuel, for example, bio-methane, using the Sabatier reaction.
Nevertheless, increased amounts of moisture and mineral impurities (ash) reduce the energy potential of biomass. Examples of such types of biomasses are fresh wood with high moisture content and office paper or rice husks with high ash content. On the other hand, it has been established that lipids, organic extractive substances, and lignin have increased calorific value, so their high content in biomass contributes to the extraction of elevated amounts of thermal energy during biomass combustion.
An additional problem is that the initial plant biomass is a loose and non-uniform material consisting of pieces of different sizes with low bulk density. These features of the biomass decline the density of thermal energy (ED), significantly reducing the productivity of the furnaces. To improve the fuel characteristics, the initial plant biomass should be demineralized, dried, and compacted. For example, if pelletize dried Switchgrass (SG) biomass with the addition of various binders such as starch and waste plastics, polystyrene (PS), and polyethylene (PE), then the density of thermal energy can be increased by 5-8 times (Figure 4). In terms of ED and GHV, biomass pelletized together with plastic binders is not inferior to coal, a traditional solid fossil fuel.
Initial plant biomass can be also a feedstock for producing secondary biofuels, such as biochar, bio-oil, bioethanol, biodiesel fuel, and biogases. From the chart (Figure 5), one can understand how profitable it is to produce various types of secondary biofuels. The following secondary biofuels were analyzed: biochar (PBC), bio-oil (PBO), and biogas (PBG) of slow pyrolysis of biomass; biogas produced by biomass gasification (GBG); biogas of anaerobic digestion of biomass (ABG); bioethanol (BET) and biodiesel fuel (BDF).
The analysis of thermal energy yield from initial biomass (IBM) and secondary biofuels shows that it is most advantageous to produce biochar of slow pyrolysis and, maybe, biogas by anaerobic digestion of cellulose or other polysaccharides. However, in general, one can conclude that if the ultimate goal is to obtain the maximum amount of thermal energy, then it is more profitable to directly burn the initial biomass (preferably in the form of pellets) than to burn such an amount of secondary solid, liquid or gaseous biofuel that can be extracted from this biomass.
As known, the most important component of biomass is cellulose. It is a semicrystalline biopolymer, many properties of which depend on crystallinity. Unfortunately, currently, existing research methods allow to determine only an estimated characteristic of cellulose crystallinity, the so-called crystallinity index. Therefore, to determine the crystallinity degree, a new thermochemical method was developed based on the measurement of the enthalpy of wetting of cellulose samples. This new method provides the determination degree of cellulose crystallinity with a standard deviation of no more than ±0.01. The thermochemical method is direct, simple, fast, accurate, reliable, and reproducible. It does not require special models, complex software, and calculations. In addition, this method does not have special requirements for the shape, size, morphology, and type of crystalline allomorph of cellulose samples.
In addition to determining the degree of crystallinity, a thermochemical method was used to characterize the thermodynamic stability of cellulose crystallites with different types of crystalline allomorphs (CAs), Iβ, II, III, IV, and amorphous cellulose (AMC). The results showed that the relative thermodynamic stability of cellulose CAs and AMC decreases in the following order:
On the other hand, the relative reactivity decreases in the reverse order:
The obtained thermodynamic characteristics allow to explain the phase transitions and reactivity of AMC and cellulose samples with different crystalline allomorphs.
CII > CIV ≥ CI > CIII > AmC
AMC > CIII > CI ≥ CIV > CII
When discussing the thermodynamic properties of cellulose nanocrystallites, the Gibbs-Thomson theory should be used. This theory allows one to explain various specific features of the nanocrystallites such as their spontaneous aggregation, a decrease in the melting temperature and an increase in solubility with a decrease in the crystallites size, the phase transition of small nanocrystallites of CA III into larger crystallites of CA IV during high-temperature treatment, and other properties.
To study the thermodynamics of chemical reactions involving biomass and its components, it is necessary to know the standard thermodynamic functions (TDFs) such as the enthalpy of formation (ΔfHo) and the entropy (So) for the starting materials, reagents, and reaction products. The enthalpy of the formation of the main components of plant biomass, cellulose, lignin, hemicelluloses, and other organic substances, can be determined experimentally by studying the enthalpy of combustion (ΔcHo):
where ΔfHo(CO2,g) = -393.51 kJ/mol and ΔfHo(H2O,l) = -285.83 kJ/mol are standard enthalpies of the formation of carbon dioxide and liquid water, respectively.
ΔfHo(CxHyOz) = x ΔfHo(CO2, g) + 0.5y ΔfHo(H2O, l) - ΔcHo(CxHyOz),
To determine the standard entropy of a substance, measurements of its specific heat capacity are usually carried out using a scanning vacuum adiabatic calorimeter in the temperature range from 0 to standard temperature, 298.15 K.
Values of standard TDFs for cellulose and other components of plant biomass can be also found in scientific literature. In addition, standard TDFs for most reagents and reaction products are given in handbooks on chemical thermodynamics.
These TDFs are used to calculate the enthalpy (ΔrHo) and entropy (ΔrSo) values of diverse chemical reactions. For example, let a chemical reaction occur between a feedstock (F) and a reagent (R) to form a reaction product (P):
F + n R → m P
Then, the change in reaction enthalpy will be:
while the change in reaction entropy:
ΔrHo = m ΔfHo(P) - n ΔfHo (R) - ΔfHo(F),
ΔrSo = m So(P) - n So (R) - So(F),
The Gibbs potential of the reaction is calculated as follows
ΔrGo = ΔrHo - T ΔrSo,
Such a potential is used to predict the feasibility of a chemical reaction, namely, if the Gibbs potential has a negative value, then the reaction can occur, and in the case of a positive value of this potential, the reaction cannot proceed. Almost all reactions considered in this review article are feasible and therefore have a negative Gibbs potential.
In addition, the Gibbs potential determines the value of the equilibrium constant (Keq) of reversible reactions:
where EXP is the Euler's number “e”.
Keq = EXP (-ΔrGo/RT),
The more negative the value of ΔrGo, the greater the value of the equilibrium constant, and when the value of ΔrGo reaches -100, the reaction becomes practically irreversible. A typical example of such an irreversible reaction is the conversion of glucose solution into bioethanol with ΔrGo = -114.9 kJ/mol, the equilibrium constant of which has a very large value, Keq = 1.35 x 1020 (see section 6.1).
On the other hand, the value of Gibbs potential for the reaction of enzymatic hydrolysis of starch is not so large, ΔrGo = -19.54 kJ/mol; and therefore, the equilibrium constant of this process has a moderate value of Keq = 2.64 x 103, which indicates the possibility of reversibility of this reaction and a decrease in the yield of produced glucose compared to the theoretical value. The reaction of enzymatic hydrolysis of cellulose is also partially reversible.
Another important characteristic is the temperature coefficient of the reaction, which depends on the reaction enthalpy:
This coefficient allows to recommend whether to increase the reaction temperature or not. If the reaction is endothermic, then with increasing temperature the coefficient KТ will increase, which improves the implementation of such a reaction. However, for exothermic reactions, an increase in temperature will cause a decrease in the coefficient KT, which means a deterioration in the implementation of such a reaction.
KT = EXP (-ΔrHo/RT),
An example of an endothermic reaction is the process of hydrothermal gasification of biochar obtained from plant biomass by the slow pyrolysis method:
It can be calculated that with an increase in the temperature of the gasification process from 973 to 1273 K, the value of the temperature coefficient will increase by 47 times, which should improve the yield of formed syngas. Further, this syngas can be used, e.g., for the synthesis of artificial hydrocarbon fuel by the Fischer-Tropsch method.
Almost all reactions involving cellulose considered in this review article are exothermic (Table 24). Therefore, an increase in the temperature of the cellulose reactions reduces the thermal coefficient, which indicates a deterioration in the implementation of these reactions. For this reason, cellulose reactions occur at normal or moderate temperatures, usually in the range of 293 to 333 K.
8. Conclusions
This review article shows the possibility of applying chemical thermodynamics to the study of plant biomass and its main components: cellulose, hemicelluloses, lignin, etc. The energy potential of various biomass types is determined. It is established that dry and demineralized biomass has a gross heating value (GHV) in the range from -17 to -21 MJ/kg. However, the GHV of biomass enriched with lignin, lipids, and other organic extractives can be significantly higher. To improve fuel properties and especially the density of thermal energy, the initial plant biomass should be densified and converted into pellets with the addition of various binders. It is shown that pelletized biomass is not inferior to fossil coal in its fuel properties, and when burned, it emits almost two times less carbon dioxide. In addition, it should be taken into account that biomass is considered CO2-neutral and renewable, unlike fossil fuels. To calculate the combustion enthalpy of biomass, a method of additive contributions of combustion enthalpies of main components is proposed.
In addition to solid fuel, biomass can also be used as a raw material for the production of secondary biofuels such as biochar, bio-oil, biodiesel fuel, bioethanol, and biogases. However, it was shown that if the ultimate goal is to obtain the maximum amount of thermal energy, then it is more profitable to directly burn the biomass, preferably in pelletized shape, than to burn such an amount of secondary solid, liquid, or gaseous biofuel that can be extracted from starting amount of this biomass.
Using methods of chemical thermodynamics, the thermodynamic functions of cellulose and its derivatives were calculated. A new thermochemical method for the determination crystallinity degree of cellulose samples was also developed. In addition, the relative thermodynamic stability of different crystalline allomorphs of cellulose was estimated. It was found that the most stable allomorph is cellulose II, while the most labile is cellulose III, as well as amorphous cellulose. When studying the specific thermodynamic properties of cellulose nanocrystallites, the Gibbs-Thomson theory should be used.
Thermodynamic features of cellulose chemical reactions such as enzymatic hydrolysis, bioethanol production, alkalization, etherification, and esterification were analyzed. The thermodynamic functions of these reactions, as well as the equilibrium constants and temperature coefficients, were determined. Since most cellulose reactions are exothermic, increasing temperature is unfavorable for them. For this reason, these reactions should be carried out at normal and moderate temperatures.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are included in this article, see also: https://weizmann.academia.edu/MichaelIoelovich/Papers. Additional inquiries should be addressed to the corresponding author.
Conflicts of Interest
Author Michael Ioelovich is employed by the company Designer Energy Ltd. The author declares no conflicts of interest, namely, no personal or collaborative conflicts, no professional or commercial conflicts, or any other conflicts of interest.
References
- Koper, G.J.M. An Introduction to Chemical Thermodynamics, 2nd ed.; VSSD: Leiden, The Netherlands, 2008; 212 p.
- Klotz, I.M., Rosenberg, R.M. Chemical Thermodynamics, 7th ed.; Basic Concepts and Methods. Whiley & Sons Inc: Hoboken, USA, 2008; 588 p.
- Goldberg, R.N.; Schliesser, J.; Mittal, A.; et al. A thermodynamic investigation of the cellulose allomorphs: cellulose(am), cellulose I‚(cr), cellulose II(cr), and cellulose III(cr). J. Chem. Thermodyn. 2015, 81, 184-226. [CrossRef]
- Rabinovich, I.B.; Nistratov, V.P.; Fedoseev, V.B; et al. Low-temperature specific heat and thermodynamic functions of diethylzinc. J. Phys. Chem. 1988, 62, 1349-1352.
- Kelley, K.K.; Parks, G.S.; Huffman, H.M. A new method for extrapolating specific heat curves of organic compounds below the temperature of liquid air. J. Phys. Chem. 1929, 33, 1802–1805. [CrossRef]
- Harjunen, P.; Lehto, V.P.; Koivisto, M.; Levonen, E.; Paronen, P.; Järvinen, K. Determination of amorphous content of lactose samples by solution calorimetry. Drug Dev. Ind. Pharm. 2004, 30, 809–815. [CrossRef]
- Uryash, V.F.; Larina, V.N.; Kokurina, N.Yu.; Novoselova, N.V. Thermochemical characteristics of cellulose and its mixtures with water. J. Phys. Chem. 2010, 84, 1023-1029. [CrossRef]
- Ioelovich, M. Progress in Characterization of Cellulose and Cellulose Esters. Eliva Press: Chisinau, Moldova, 2023, 70 p.
- Ioelovich, M. Features of water vapor sorption by cellulose materials. SITA, 2022, 24, 21-31. [CrossRef]
- Manikanika, L.Ch. Extraction of cellulosic fibers from the natural resources: A short review. Materials Today: Proceedings 2022, 48, 1265-1270. [CrossRef]
- Rongpipi, S.; Ye, D.; Gomez, E.D.; Gomez, E.W. Progress and opportunities in the characterization of cellulose - An important regulator of cell wall growth and mechanics. Front. Plant Sci. 2019, 9, 1–28.
- Ang, T-Z.; Salem, M.; Kamarol, M.; et al., A comprehensive study of renewable energy sources: Classifications, challenges and suggestions. Energy Strategy Reviews 2022, 43, 100939, 1-27. [CrossRef]
- Lee, S.; Speight, J.G.; Loyalka, S.K. Handbook of Alternative Fuel Technologies, 1-st ed.; CRC Press: Boca Raton, USA, 2007; 568 p.
- Rashmi, R.; Tripti Tripathi, T.; Pandey, S.; Kumar, S. Transforming lignocellulosic biomass into biofuels: recent innovations in pretreatment and bioconversion techniques. IJRASET 2024, 12, 1079-1089. [CrossRef]
- Klemm, D.; Heublein, B.; Fink H.-P.; Bohn, A. Cellulose: fascinating biopolymer and sustainable raw material. Angew. Chem. 2005, 44, 2-37.
- Tursi, A. A review on biomass: importance, chemistry, classification, and conversion. Biofuel Research Journal, 2019, 22, 962-979. [CrossRef]
- Saidur, R.; Abdelaziz, E.A.; Demirbas, A.; et al. A review on biomass as a fuel for boilers. Renewable Sustainable Energy Reviews, 2011, 15, 2262-2289. [CrossRef]
- Ioelovich, M. High-energy fuel pellets. Sci. Environ. 2020, 3, 147–152.
- Čuček, L.; Martin, M.; Grossmann, I.E.; Kravanja, Z. Energy, water and process technologies integration for the simultaneous production of ethanol and food from the entire corn plant. Comp. Chem. Eng. 2011, 35, 1547-1557. [CrossRef]
- Ioelovich, M. Chemical thermodynamics of biomass, cellulose, and cellulose derivatives. World J. Adv. Res. Reviews 2024, 24, 1295–1338. [CrossRef]
- Gautam, S.P.; Bundela, P.S.; Pandey, A.K. A review on systematic study of cellulose. J. Appl. Natural Sci. 2010, 2, 330-343. [CrossRef]
- Ioelovich, M. Recent findings and the energetic potential of plant biomass as a renewable source of biofuels – a review. Bioresources 2015, 10, 1879-1914. [CrossRef]
- Ioelovich, M. Plant Biomass as a Renewable Source of Biofuels and Biochemicals, LAP: Saarbrücken, Germany, 2013; 52 p.
- Ioelovich, M. Cellulose: Nanostructured Natural Polymer. LAP: Saarbrücken, Germany, 2014; 88 p.
- Ioelovich, M. Models of supramolecular structure and properties of cellulose. J. Polym. Sci. 2016, 58, 925-943. [CrossRef]
- Gomri, C.; Cretin, M.; Semsarilar, M. Recent progress on chemical modification of cellulose nanocrystal (CNC) and its application in nanocomposite films and membranes-A comprehensive review. Carbohydrate Polymers 2022, 294, 119790, 1-31. [CrossRef]
- Aziz, T.; Farid, A.; Haq, F.; et al. A review on the modification of cellulose and its applications. Polymers 2022, 14, 3206, 1-34. [CrossRef]
- Rao, J.; Lv, Z.; Chen, G.; Peng, F. Hemicellulose: structure, chemical modification, and application. Prog. Polym. Sci., 2023, 140, 101675, 1-15. [CrossRef]
- He, Y.; Liu, Y.; Zhang, M. Hemicellulose and unlocking potential for sustainable applications in biomedical, packaging, and material sciences: a narrative review. Int. J. Biolog. Macromol. 2024, 280, 135657, 1-23. [CrossRef]
- Zhang, Y.; Naebe, M. Lignin: a review on structure, properties, and applications as a light-colored UV absorber. ACS Sustainable Chem. Eng. 2021, 9, 1427–1442. [CrossRef]
- Shorey, R.; Salaghi, A.; Fatehi, P. Valorization of lignin for advanced material applications: a review. RSC Sustain. 2024, 2, 804–831. [CrossRef]
- Demibras, A. Relationships between lignin contents and heating values of biomass. Energy Conversion and Management 2001, 42, 183-188. [CrossRef]
- Demibras, A. Higher heating values of lignin types from wood and non-wood lignocellulosic biomasses. Energy Sources, Part A, 2017, 39, 592–598.
- Abed, S.M.; Ali, A.H.; Noman, A.; et al. Structured lipids: enzymatic synthesis, health benefits and nutraceutical characteristics - a review. Int. J. Res. in Agricultural Sci. 2016, 3, 2348 – 3997.
- Gravalos, I.; Gialamas, T.; Koutsofitis, Z.; et al. Energetic study of animal fats and vegetable oils using combustion bomb calorimeter. J. Agricult. Machin. Sci. 2008, 4, 69-74.
- Suharto, S.; Ahyati, A.E. The properties of vegetable cooking oil as a fuel and its utilization in a modified pressurized cooking stove. IOP Conference Series: Earth Env. Sci. 2018, 105, 012047, 1-10. [CrossRef]
- Cichy,W; Witchak, M.; Walkowiak, M. Fuel properties of woody biomass from pruning operations in fruit orchards. Bioresources, 2017, 12, 6458-6470. [CrossRef]
- Makimuk, Y.V.; Ponomarev, D.A.; Kursevich, V.N.; Fesko, V.V. Combustion heat of wood fuel. Forestry J. 2017, 4, 116-129.
- Nurek, T.; Gendek A.; Roman, K. Forest residues as a renewable source of energy: elmental composition and physical properties. Boresources 2019, 14, 6-20.
- Shojaeiarani, J.; Baiwa, D.S.; Baiwa, S.G. Properties of densified solid fuels in relation to chemical composition, moisture content and bulk density. Boresources 2019, 14, 4996-5015.
- Tumuluru, J.S. Pelleting of pine and switchgrass blends: Effect of process variables and blend ratio on the pellet quality and energy consumption. Energies 2019, 12, 1198, 1-26. [CrossRef]
- Ioelovich, M. High-energy fuel pellets. SITA 2021, 23, 136-142.
- Jagtap, A.; Kalbande, S. Pelletization process for the production of fuel pellets from various surplus biomass: a review. Int. J. Env. & Climate Change 2023, 13, 200-207. [CrossRef]
- Elniski, A.; Dongre, P.; Bujanovic, B.M. Lignin use in enhancing the properties of willow pellets. Forests 2023, 14, 2041, 1-19. [CrossRef]
- Wu, J. How to make wood pellets—Small pellet mill and pellet plant. G. Energy 2024, 2, 1–5.
- Ioelovich, M. Analysis of energy potential of switchgrass biomass. Biomass 2024, 4, 1-11. [CrossRef]
- Vargas-Moreno, J.M.; Callejón-Ferre, A.J.; Pérez-Alonso, J.; Velázquez-Martí, B. A review of the mathematical models for predicting the heating value of biomass materials. Renew. Sustain. Energy Reviews 2012, 16, 3065-3083. [CrossRef]
- Domingos, I.; Ayata, U.; Ferreira, J.; et al. Calorific power improvement of wood by heat treatment and its relation to chemical composition. Energies 2020, 13, 5322. [CrossRef]
- Maksimuk, Y.; Antonova, Z.; Krouk, V.; et al. Prediction of higher heating value (HHV) based on the structural composition for biomass. Fuel 2021, 299, 120860, 1-7. [CrossRef]
- Park, S.; Kim, S.Y.; Kim, H.E.; et al. Calorific value prediction model using structure composition of heat-treated lignocellulosic biomass. Energies 2023, 16, 7896, 1-15. [CrossRef]
- Parikh, J.; Channiwala, S.A.; Ghosal, G.K. A correlation for calculating HHV from proximate analysis of solid fuels. Fuel 2005, 84, 487-494. [CrossRef]
- Suris, A.L. Heat of combustion of liquid halogen-organic compounds. Chem. Petrol. Eng. 2007, 43, 20–22. [CrossRef]
- Maksimuk, Y.; Krouk, V.; Antonova, Z. Calculation of the heat of combustion of wood fuel by elemental composition. Forestry J. 2016, 6, 110–121.
- Ioelovich, M. Thermodynamics of biomass-based solid fuels. Acad. J. Polym. Sci. 2018, 2, 1-9. [CrossRef]
- Lu, X.; Gu, X.; A review on lignin pyrolysis: pyrolytic behavior, mechanism, and relevant upgrading for improving process efficiency. Biotechnol. Biofuels and Bioprod. 2022, 15, 106, 1-43. [CrossRef]
- Villora-Picó, J.J.; González-Arias, J.; Baena-Moreno, F.M.; Reina, T.R. Renewable carbonaceous materials from biomass in catalytic processes: a review. Materials 2024, 17, 565, 1-32.
- Alvarado-Flores, J.J.; Alcaraz-Vera, J.V.; Ávalos-Rodríguez, M.L.; et al. Thermochemical production of hydrogen from biomass: pyrolysis and gasification. Energies 2024, 17, 537, 1-21. [CrossRef]
- Song, H.; Yang, G.; Xue, P.; et al. Recent development of biomass gasification for H2-rich gas production. Appl. Energy Combust. Sci. 2022, 10, 100059, 1-16. [CrossRef]
- Damartzis, T; Zabaniotou, A. Thermochemical conversion of biomass to second generation biofuels through integrated process design—a review. Renew. Sustain. Energy Reviews 2011, 15, 366-378. [CrossRef]
- Ioelovich, M. Energy potential of natural, synthetic polymers and waste materials - a review. Acad. J. Polym. Sci. 2018, 1, 555553, 1-15. [CrossRef]
- Hasan, S.; Alam, M.; Akter, A.; et al. Biogas production from cafeteria waste by anaerobic digestion. BIO Web Conf. 2023, 62, 03001, 1-9. [CrossRef]
- Amândio, M.S.T.; Rocha, J.M.S.; Xavier, A.M.R.B. Enzymatic hydrolysis strategies for cellulosic sugars production to obtain bioethanol from eucalyptus globulus bark. Fermentation 2023, 9, 241. [CrossRef]
- Reis, C.E.R.; Junior, N.L.; Bento, H.B.; et al. Process strategies to reduce cellulase enzyme loading for renewable sugar production in biorefineries. Chem. Eng. J. 2023, 451,138690. [CrossRef]
- Ioelovich, M. Thermodynamics of enzymatic hydrolysis of cellulose. World J. Adv. Res. Rev. 2024, 21, 577–586. [CrossRef]
- Da Silva, A.S.; Espinheira, R.P.; Teixeira, R.S.S.; de Souza, M.F.; Ferreira-Leitão, V.; Bon, E.P.S. Constraints and advances in high-solids enzymatic hydrolysis of lignocellulosic biomass: A critical review. Biotechnol. Biofuels 2020, 13, 58. [CrossRef]
- Ioelovich, M. Preparation, characterization and application of amorphized cellulose—A review. Polymers 2021, 13, 4313.
- Xiao, Z.; Zhang, X.; Gregg, D.J.; Saddler, J.N. Effects of sugar inhibition on cellulases and beta-glucosidase during enzymatic hydrolysis of softwood substrates. Appl. Biochem. Biotechnol. 2004, 113–116, 1115–1126.
- Ansanay, Y.; Kolar, P.; Sharma-Shivappa, R.; Cheng, J.; Arellano, C. Pretreatment of switchgrass for production of glucose via sulfonic acid-impregnated activated carbon. Processes 2021, 9, 504. [CrossRef]
- Mosier, N.; Wyman, C.; Dale, B.; Elander, R.; Lee, Y.Y.; Holtzapple, M.; Ladisch, M. Features of promising technologies for pretreatment of lignocellulosic biomass. Bioresour. Technol. 2005, 96, 673–686. [CrossRef]
- Kumar, P.; Barrett, D.; Delwiche, M.; Stroeve, P. Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind. Eng. Chem. Res. 2009, 48, 3713–3729. [CrossRef]
- Wyman, C.E.; Balan, V.; Dale, B.E.; Elander, R.T.; Falls, M.; Hames, B.; Holtzapple, M.T.; Ladisch, M.R.; Lee, Y.Y.; Mosier, N.; et al. Comparative data on effects of leading pretreatments and enzyme loadings and formulations on sugar yields from different switchgrass sources. Bioresour. Technol. 2011, 102, 11052–11062. [CrossRef]
- Ioelovich, M. Effect of chemical pretreatments on composition and enzymatic digestibility of plant biomass. Res. Rev. J. Chem. 2014, 3, 23–31.
- Xu, H.; Che, X.; Ding, Y.; Kong, Y.; Li, B.; Tian, W. Effect of crystallinity on pretreatment and enzymatic hydrolysis of lignocellulosic biomass based on multivariate analysis. Bioresour. Technol. 2019, 279, 271–280. [CrossRef]
- Zhang, H.; Han, L.; Dong, H. An insight to pretreatment, enzyme adsorption and enzymatic hydrolysis of lignocellulosic biomass: Experimental and modeling studies. Renew. Sustain. Energy Rev. 2021, 140, 110758. [CrossRef]
- Karmakar, R.; Kundu, K.; Rajor, A. Fuel properties and emission characteristics of biodiesel produced from unused algae grown in India. Petroleum Sci. 2018, 15, 385–395. [CrossRef]
- Navarro, J.C.; Centeno, M.A.; Laguna, O.H.; Odriozola, J.A. Policies and motivations for the CO2 valorization through the Sabatier reaction using structured catalysts: a review of the most recent advances. Catalysts, 2018; 8, 578: 1-25. [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.J.; et al. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; et al. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Advances, 2012, 2, 2358–2368. [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; et al. Review on methanation – from fundamentals to current projects. Fuel, 2016, 166, 276–296. [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 methanation: the effect of catalysts and reaction conditions. Energy Procedia, 2017, 105, 2022 – 2027. [CrossRef]
- Kubota, J.; Okumura, T.; Hayashi. R. Methane synthesis from CO2 and H2O using a phosphate-based electrochemical cell at 210–270 °C with oxide-supported Ru catalysts. Sustain. Energy Fuels 2022, 6, 1362-1372.
- Sagara, R.; Hayashi, R.; Hirata, A.; et al. Methane synthesis from CO2 and H2O using electrochemical cells with polymer electrolyte membranes and Ru catalysts: a comparative study to a phosphate-based electrolyte cell. Sustain. Energy Fuels 2023, 7, 5336-5341.
- Held, M.; Schollenberger, D.; Sauerschell, S.; et al. Power-to-gas: CO2 methanation concepts for SNG production at the Engler-Bunte-Institute. Chem. Ing. Tech. 2020, 92, 595-602.
- Koschany, F.; Schlereth, D.; Hinrichsen, O. On the kinetics of the methanation of carbon dioxide on coprecipitated NiAl(O)x. Applied Catalysis B: Environment. 2016, 181, 504-516.
- Falbo, L.; Martinelli, M.; Viscontia, C.G.; et al. Kinetics of CO2 methanation on a Ru-based catalyst at process conditions relevant for Power-to-Gas applications. Applied Catalysis B: Environment. 2018, 225, 354-363. [CrossRef]
- Miguel, C.V.; Mendes, A.; Madeira, L.M. Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J. CO2 Utilization 2018, 25, 128-136. [CrossRef]
- Ioelovich, M. Thermodynamic analysis of methane synthesis by hydrogenation of carbon dioxide. World J. Adv. Res. Rev. 2024, 24, 927–931. [CrossRef]
- Tommasi, M.; Degerli, S.N.; Ramis, G.; Rossetti, I. Advancements in CO2 methanation: A comprehensive review of catalysis, reactor design and process optimization. Chem. Eng. Res. and Design 2024, 201, 457-482. [CrossRef]
- Faria, A.C.; Miguel, C.V.; Madeira, L.M. Thermodynamic analysis of the CO2 methanation reaction with in situ water removal for biogas upgrading. J. CO2 Utilization 2018, 26, 271-280. [CrossRef]
- Schmidt, J. Molecules, mass, and finding the right energy balance: A deeper look into the methanation process. TES 2024, 19, 1-3.
- Ioelovich, M. Energy potential and natural and synthetic polymers and waste materials – a review. Acad. J. Polym. Sci. 2018, 1, 1-15. [CrossRef]
- Shafizadeh, F.; DeGroot, W.F. Combustion Characteristics of Cellulosic Fuels, Acad. Press: New York, USA, 1976; 1-17.
- Colbert, J.C.; Xiheng, H.; Kirklin, D.R. Enthalpy of combustion of microcrystalline cellulose. J. Res. Nat. Bur. Stand. 1981, 86, 655–1650. [CrossRef]
- Kienzle, E.; Schrag, I.; Butterwick, R.; Opitz. B. Calculation of gross energy in pet foods: new data on heat combustion and fibre analysis in a selection of foods for dogs and cats. J. Anim. Physiol. Anim. Nutr. 2001, 85, 148–157. [CrossRef]
- Park, S.; Baker, J.O.; Himmel, M.E.; et al. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3, 1–10. [CrossRef]
- Terinte, N.; Ibbett R.; Schuster, K.C. Overview on native cellulose and microcrystalline cellulose I structure studied by X-ray diffraction (WAXD): comparison between measurement techniques. Lenzing. Ber. 2011, 89, 118–131.
- Madhushani, W.H.; Priyadarshana, R.W.I.B.; Ranawana, S.R.W.; et al. Determining the crystallinity index of cellulose in chemically and mechanically extracted banana fiber for the synthesis of nanocellulose. J. Nat. Fibers 2022, 19, 7973–7981. [CrossRef]
- Leong, S.L.; Tiong, S.I.X.; Siva, S.P.; et al. Morphological control of cellulose nanocrystals via sulfuric acid hydrolysis based on sustainability considerations: An overview of the governing factors and potential challenges. J. Environ. Chem. Eng. 2022, 10, 1–20. [CrossRef]
- French, A.D. Increment in evolution of cellulose crystallinity analysis. Cellulose 2020, 27, 5445–5448. [CrossRef]
- Jang, S.-K.; Jeong, H.; Choi, I.-G. The effect of cellulose crystalline structure modification on glucose production from chemical: composition-controlled biomass. Sustainability 2023, 15, 5869, 1-12. [CrossRef]
- Salem, K.S.; Kasera, N.K.; Rahman, M.A.; et al. Comparison and assessment of methods for cellulose crystallinity determination. Chem. Soc. Rev. 2023, 18, 1-30. [CrossRef]
- Ioelovich, M. Application of thermochemical method to determine the crystallinity degree of cellulose materials. Appl. Sci. 2023, 13, 2387, 1-11. [CrossRef]
- Zugenmaier. P. Crystalline cellulose and cellulose derivatives. Characterization and structures; Springer-Verlag: Berlin, Heidelberg, Germany, 2008; 101-174.
- Ioelovich, M. Study of thermodynamic properties of various allomorphs of cellulose. ChemXpess 2016, 9, 259-265.
- Zugenmaier, P. Conformation and packing of various crystalline cellulose fibers. Progress in Polym. Sci. 2001, 26, 341-1417. [CrossRef]
- Zhe, L.; Xun, Z.; Guihua, Y.; Keiji, T.; Feng, X. Nanocrystals of cellulose allomorphs have different adsorption of cellulase and subsequent degradation. Ind. Crops and Products 2018, 112, 541-549.
- Srivastava, D.; Ahopelto, J.; Karttunen, A. Thermodynamic properties of crystalline cellulose allomorphs studied with dispersion-corrected density functional methods. Molecules 2022, 27, 6240, 1-11. [CrossRef]
- Ioelovich, M. Preparation, characterization and application of amorphized cellulose - a review. Polymers 2021, 13, 1-21.
- Wada, M. In situ observation of the crystalline transformation from cellulose IIII to Iβ. Macromolecules 2001, 34, 3271-3275.
- Kulshreshth K. A review of the literature on the formation of cellulose IV, its structure, and its significance in the technology of rayon manufacture. J. Text. Inst. 1979, 70, 13-18.
- Ioelovich, M. Distinctive features of cellulose nanocrystallites. SITA 2023, 25, 132-143. [CrossRef]
- Ioelovich, M. Peculiarity of phase transitions of cellulose nanocrystallites. ChemExpress 2016, 9, 1-14.
- Kaptay, G. The Gibbs equation versus the Kelvin and the Gibbs-Thomson equations to describe nucleation and equilibrium of nano-materials. J. Nanosci. Nanotech. 2011, 12, 1-9. [CrossRef]
- Perez, M. Gibbs-Thomson effect in phase transformations. Scripta Materialia 2005, 52, 709-712. [CrossRef]
- Maksimuk, Y.; Antonava, Z.; Krouk, V.; Korsakova, A.; Kursevich, V. Prediction of higher heating value (HHV) based on the structural composition for biomass. Fuel 2021, 299, 120860, 1-7. [CrossRef]
- Matveev, Y.I.; Van Soest, J.J.C.; Neiman, C.; et al. The relationship between thermodynamic and structural properties of low and high amylose maize starches. Carbohyd. Polym. 2001, 44, 151-160. [CrossRef]
- Vilaplana, F.; Hasjim, J.; Gilbert, R.G. Amylose content in starches: toward optimal definition and validating experimental methods. Carbohyd. Polym. 2012, 88,103–111. [CrossRef]
- Ioelovich, M. Comparison of methods for calculation of combustion heat of biopolymers. American J. Appl. Sci. Eng. & Technol. 2016, 1, 63-67.
- Salam, M.A.; Mushtaque, A.; Khan, S.; et al. The effect of glucose, temperature and pH on bioethanol production by Saccharomyces Cerevisiae. J. Popul. Therap. & Clinic Pharmacol. 2024, 31, 656-667.
- Ma, H.; Wang, Y.; Lv, P.; Zhou, J.; Gao, M.; et al. Ethanol production from a mixture of waste tissue paper and food waste through saccharification and mixed-culture fermentation. Fermentation 2024, 10, 194, 1-16. [CrossRef]
- Shukla, A.; Kumar, D.; Girdhar, M.; et al. Strategies of pretreatment of feedstocks for optimized bioethanol production: distinct and integrated approaches. Biotech. Biofuels & Bioprod. 2023, 16, 44, 1-33. [CrossRef]
- Krajang, M.; Malairuang, K.; Sukna, J.; et al. Single-step ethanol production from raw cassava starch using a combination of raw starch hydrolysis and fermentation, scale-up from 5-L laboratory and 200-L pilot plant to 3000-L industrial fermenters. Biotechnol. Biofuels 2021. 14, 68, 1-15. [CrossRef]
- Acosta-Pavas, J.C.; Alzate-Blandon, L.; Ruiz-Colorado, A.A. Enzymatic hydrolysis of wheat starch for glucose syrup production. Revista DYNA 2020, 87,173-182. [CrossRef]
- Xu, Q.-S.; Yan, T.-S.; Feng, J.-X. Efficient hydrolysis of raw starch and ethanol fermentation: a novel raw starch-digesting glucoamylase from Penicillium oxalicum. Biotechnol Biofuels 2016, 9, 216, 1-18. [CrossRef]
- Sharma, A.; Satyanarayana, T. Microbial acid-stable α-amylases: characteristics, genetic engineering and applications. Process Biochem. 2013, 48, 201–211.
- Marín-Navarro, J.; Polaina, J. Glucoamylases: structural and biotechnological aspects. Appl Microbiol Biotechnol. 2011, 89, 1267–1273. [CrossRef]
- Casey, G.P.; Ingledew. W.M.M. Ethanol tolerance in yeasts. CRC Critical Reviews in Microbiology 1986, 13, 219–280.
- Yaverino-Gutiérrez, M.A.; Wong, A.Y.C.-H.; Ibarra-Muñoz, L.A.; Chávez, A.C.F.; Sosa-Martínez, J.D.; Tagle-Pedroza, A.S.; Hernández-Beltran, J.U.; Sánchez-Muñoz, S.; Santos, J.C.d.; da Silva, S.S.; et al. Perspectives and progress in bioethanol processing and social economic impacts. Sustainability 2024, 16, 608, 1-31. [CrossRef]
- Al-Mardeai, S.; Elnajjar, E.; Hashaikeh, R.; Kruczek, B.; Van der Bruggen, B.; Al-Zuhair, S. Membrane bioreactors: a promising approach to enhanced enzymatic hydrolysis of cellulose. Catalysts 2022, 12, 1121, 1-26. [CrossRef]
- Vasi´c, K.; Knez, Ž.K.; Leitgeb, M. Bioethanol production by enzymatic hydrolysis from different lignocellulosic sources. Molecules 2021, 26, 753, 1-23.
- Ioelovich, M. Specific features of enzymatic hydrolysis of cellulose. World J. Adv. Res. & Reviews 2024, 22, 381–392. [CrossRef]
- Popovic, M.; Woodfield, B.F.; Hansen, L.D. Thermodynamics of hydrolysis of cellulose to glucose from 0 to 100 °C: cellulosic biofuel applications and climate change implications. J. Chem. Thermodynamics 2019, 128, 244-250. [CrossRef]
- Valášek, P.; Müller, M.; Šleger, V.; et al. Influence of alkali treatment on the microstructure and mechanical properties of Coir and Abaca fibers. Materials 2021, 14, 2636, 1-20. [CrossRef]
- Shahril, S.M.; Ridzuan, M.J.M.; Abdul-Majid, M.S.; et al Alkali treatment influence on cellulosic fiber from Furcraea foetida as potential reinforcement of polymeric composites. J. Mater. Res. Technol. 2022, 19, 2567-2583.
- Tatsumi, D.; Kanda, A.; Kondo, T. Characterization of mercerized cellulose nanofibrils prepared by aqueous counter collision process. J. Wood Sci. 2022, 68, 13, 1-9. [CrossRef]
- Yokota, S.; Nishimoto, A.; Kondo, T. Alkali-activation of cellulose nanofibrils to facilitate surface chemical modification under aqueous conditions. J. Wood Sci. 2022, 68, 14, 1-7. [CrossRef]
- Ioelovich, M. Thermochemistry of alkalization and etherification of cellulose. World J. Adv. Res. Reviews 2023, 20, 1166–1174. [CrossRef]
- Ferro, M.; Mannu, A.; Panzeri, W.; et al. An integrated approach to optimizing cellulose mercerization. Polymers 2020, 12, 1559, 1-16. [CrossRef]
- Filho, G.R.; Monteiro D.S.; Da Silva Meireles, C.; et al. Synthesis and characterization of cellulose acetate produced from recycled newspaper. Carbohydr. Polym. 2008, 73, 74-82. [CrossRef]
- Wolfs, J.; Meier M.A.R. A more sustainable synthesis approach using the DBU/CO2 switchable solvent system. Green Chem. 2021, 23, 4410-4420.
- Ioelovich, M. Adjustment of hydrophobic properties of cellulose materials. Polymers 2021, 13, 1241, 1-11. [CrossRef]
- Tan, J.; Liang, Y.; Sun, L.; Yang, Z.; Xu, J.; Dong, D.; Liu, H. Degradation characteristics of cellulose acetate in different aqueous conditions. Polymers 2023, 15, 4505, 1-13. [CrossRef]
- Bracciale, M.P.; De Caprariis, B.; Musivand, S.; Damizia, M.; De Filippis, P. Chemical recycling of cellulose acetate eyewear industry waste by hydrothermal treatment. Ind. Eng. Chem. Res. 2024, 63, 12, 5078–5088. [CrossRef]
- Fischer, S.; Thümmler, K.; Volkert, B.; et al. Properties and applications of cellulose acetate. Macromolec. Sympos. 2008, 262, 89-96.
- Panchenko, O.; Tutova, O. Problems and advances in production of nitrate cellulose. Chem. Plant Mater. 2005, 3, 85–88.
- Stovbun, S.V.; Nikolskiy, S.N.; Melnikov, V.P.; et al. Chemical physics of cellulose nitration. J. Physic. Chem. 2016, 10, 245–259. [CrossRef]
- Mattar, H.; Baz, Z.; Saleh, A.; et al. Nitrocellulose: structure, synthesis, characterization, and applications. Water, Energy, Food, and Env. J. 2020, 1, 1-15.
- Morris, E.; Pulham, C.R.; Morrison, C.A. Structure and properties of nitrocellulose: approaching 200 years of research. RSC Adv. 2023, 13, 32321-32333. [CrossRef]
- Rizkiansyah, R.R.; Mardiyati, Y.; Hariyantob, A.; Dirgantarac, T. Selecting appropriate cellulose morphology to enhance the nitrogen content of nitrocellulose. RSC Adv. 2024, 14, 28260–28271.
- Jessup, R.S.; Prosen, E. Heats of combustion and formation of cellulose and nitrocellulose (cellulose nitrate). Nat Bur. Stand. 1950, 44, 387-393. [CrossRef]
- Larina V.N.; Uryash, V.F.; Kushch, D.S. Thermochemical characteristics of cellulose acetates with different degrees of substitution. J. Phys. Chem. 2012; 86, 1776–1778.
- Ioelovich, M. Study of heat effects of topochemical esterification of cellulose. World J. Adv. Res. Reviews 2024, 23, 1232–1241 . [CrossRef]
- Trache, D.; Khimeche, K.; Mezroua, A.; Benziane, M. Physicochemical properties of microcrystalline nitrocellulose from alpha grass fibers and its thermal stability. J. Therm. Anal. Calorim. 2016, 124, 1485–1496.
Figure 1.
Dependence of standard formation enthalpy on the degree of substitution of NC samples.
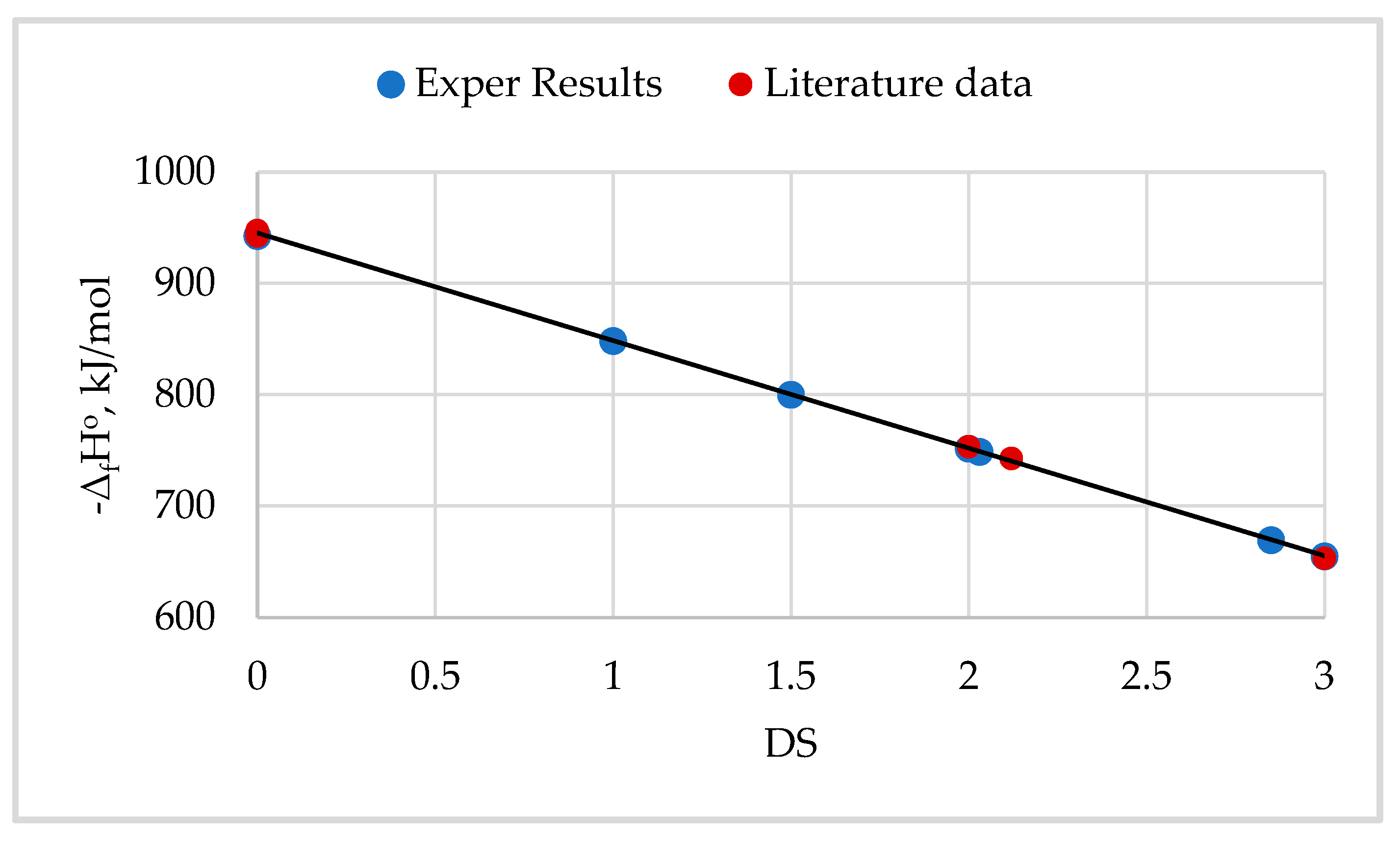
Figure 2.
Dependence of standard formation enthalpy on the degree of substitution of AC samples.
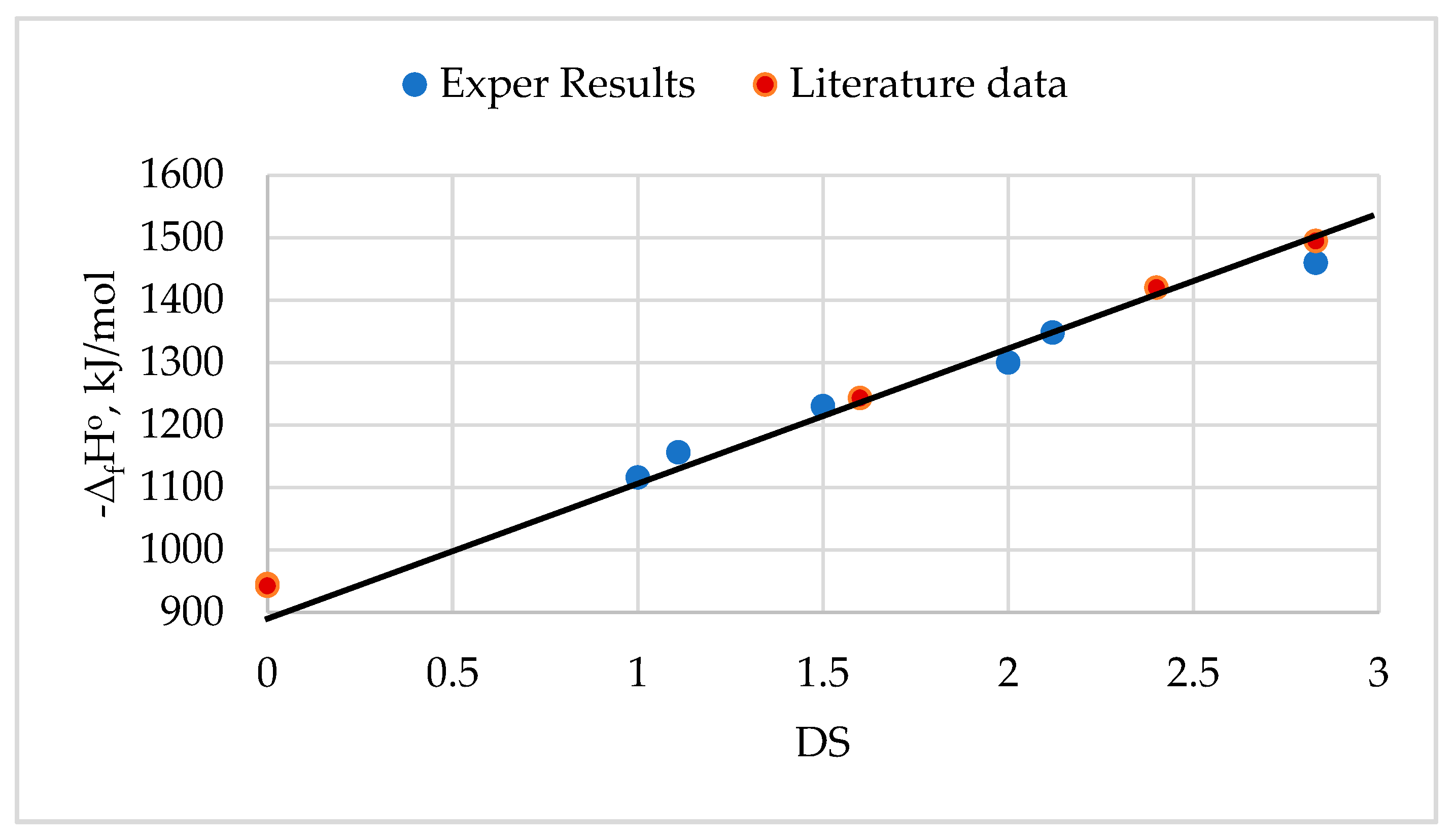
Figure 3.
Dependence of reaction enthalpy on the degree of substitution for nitro- (NC) and acetate (AC) celluloses.
Figure 3.
Dependence of reaction enthalpy on the degree of substitution for nitro- (NC) and acetate (AC) celluloses.
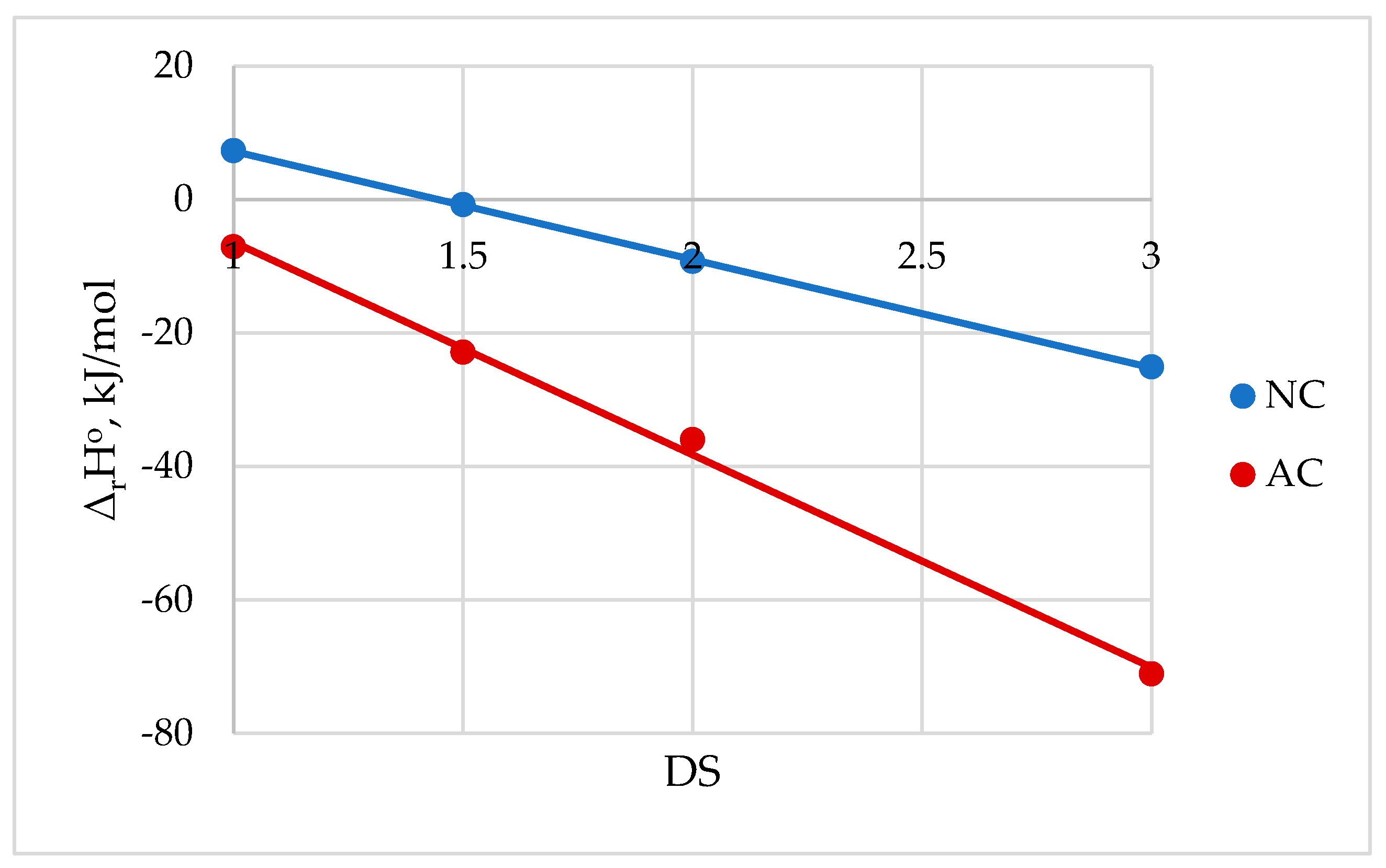
Figure 4.
The density of thermal energy of initial and pelletized SG biomass.
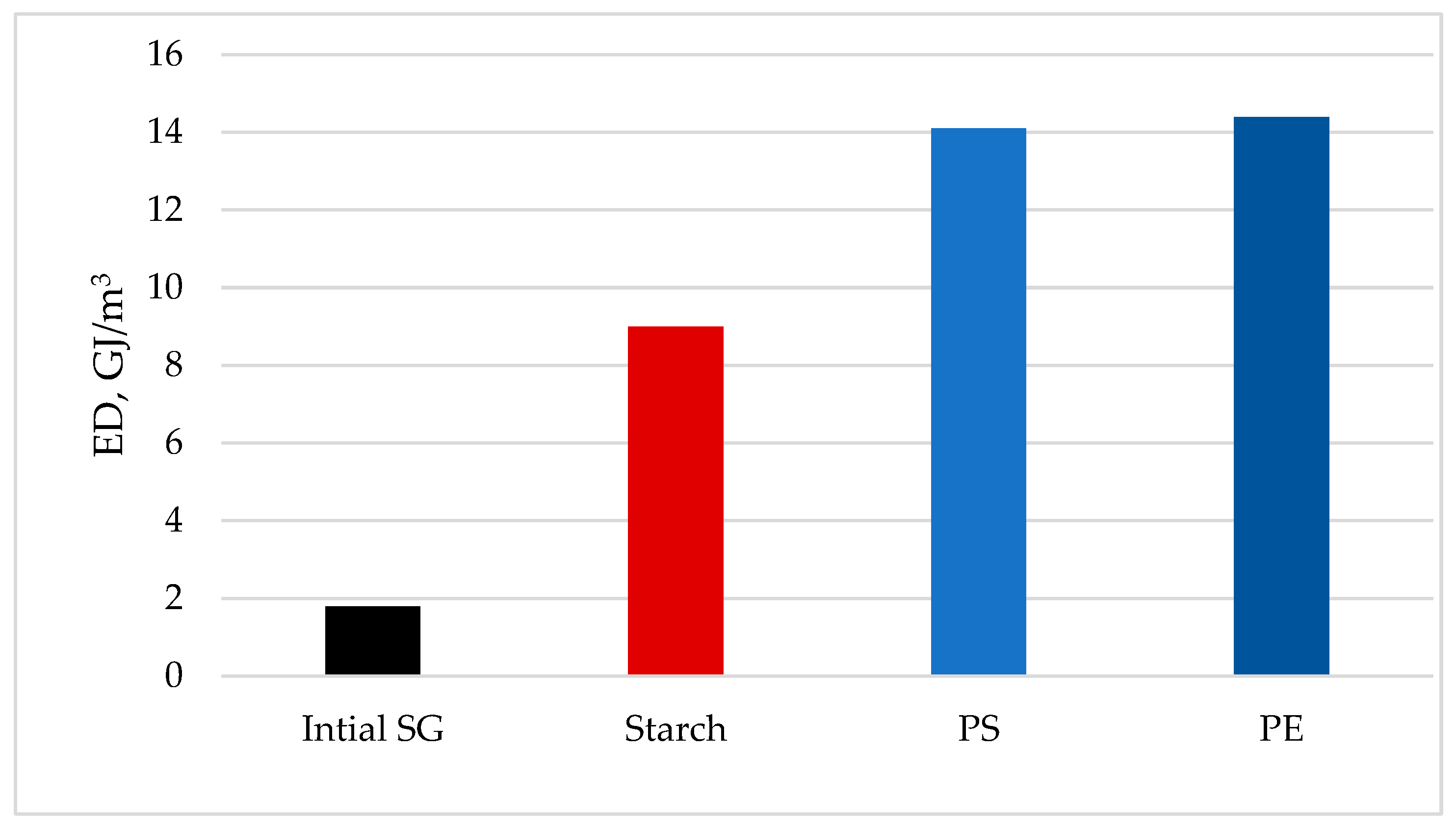
Figure 5.
Average thermal energy yield (EY) of various secondary biofuels from initial biomass.
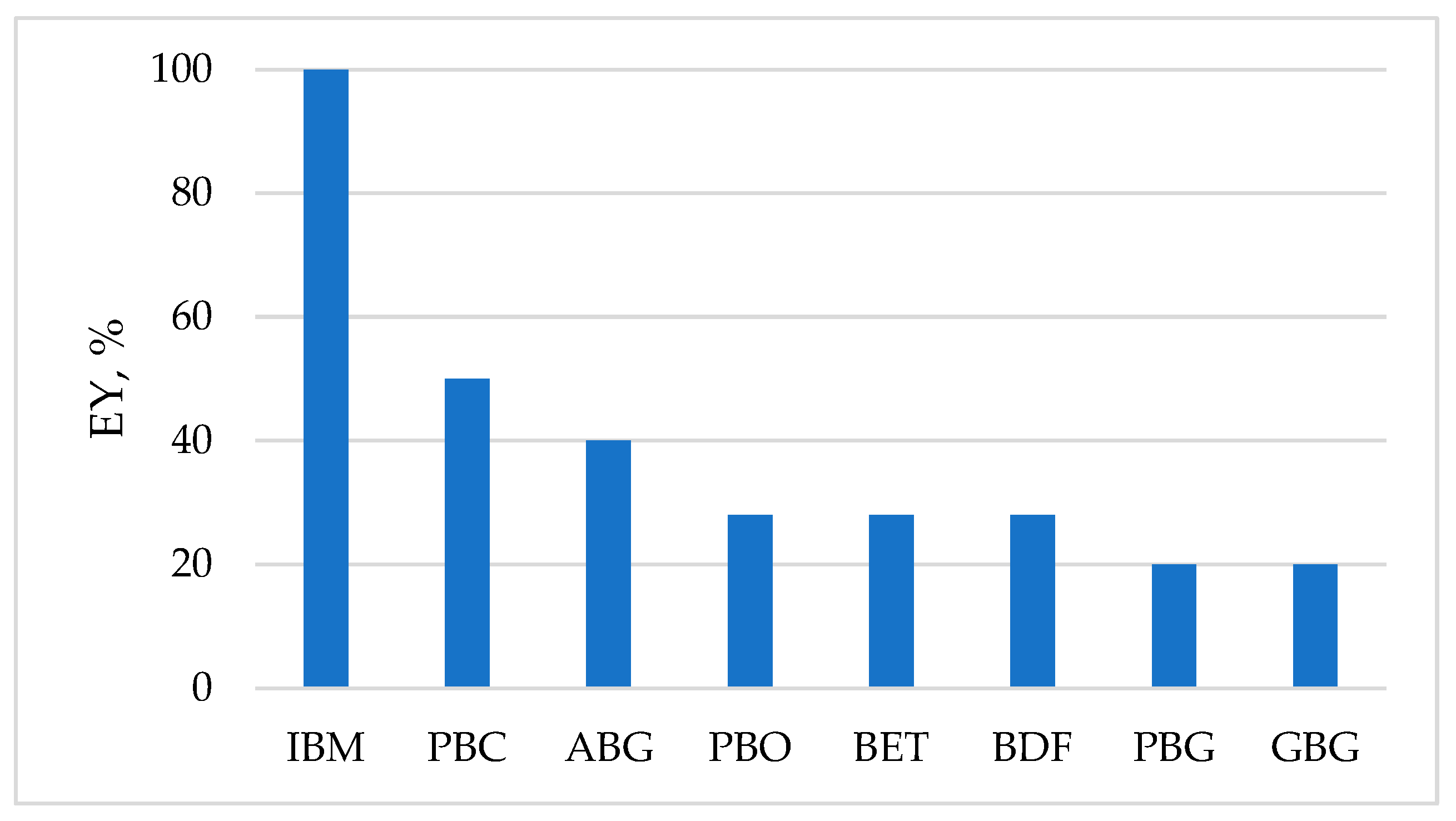

| Biomass | Cellulose,% | *Hemi,% | Lignin, % |
|---|---|---|---|
| Cotton linter | 95 | 2 | nd |
| Flax | 71 | 21 | 2 |
| Jute | 70 | 15 | 13 |
| Softwood | 47 | 22 | 27 |
| Hardwood | 46 | 25 | 24 |
| Bamboo | 40 | 26 | 21 |
| Bagasse | 38 | 25 | 20 |
| Miscanthus | 39 | 27 | 20 |
| Switchgrass | 38 | 30 | 20 |
| Corn stover | 36 | 30 | 20 |
| Wheat straw | 35 | 30 | 18 |
| Olive pomace | 23 | 24 | 34 |
| Olive husk | 24 | 24 | 48 |
| Nutshells | 27 | 27 | 35 |
| Waste office paper | 63 | 5 | 1 |
| Waste cardboard | 60 | 15 | 11 |
* Hemi denotes “Hemicelluloses”; nd denotes “not determined”.
| Biomass | -ΔcH, MJ/kg |
|---|---|
| Office paper waste | 12.5 |
| Rice straw | 15.7 |
| Rice husk | 16.5 |
| Wheat straw | 17.1 |
| Corn stover | 17.2 |
| Switchgrass | 18.3 |
| Miscanthus | 18.6 |
| Forest residues | 18.8 |
| Hardwood | 19.7 |
| Softwood | 20.5 |
| Olive pomace | 22.3 |
| Fallen olives | 26.1 |
Table 3.
Calculated (ΔcHc) and experimental (ΔcHe) values for some plant biomasses*.
| Biomass | H,% | L, % | E, % | A, % | -ΔcHc, MJ/kg | -ΔcHe, MJ/kg |
|---|---|---|---|---|---|---|
| Softwood | 69 | 27 | 3 | 1 | 20.4 | 20.5 |
| Hardwood | 71 | 24 | 3 | 1 | 19.5 | 19.7 |
| Switchgrass | 68 | 20 | 4 | 6 | 18.4 | 18.3 |
| Corn stover | 66 | 20 | 2 | 8 | 17.3 | 17.2 |
| Office paper waste | 68 | 1 | 1 | 30 | 12.5 | 12.5 |
* H, L, E, and A denote holocellulose (cellulose and hemi complex), lignin, extractives, and ash.
| Component | -ΔcH i, MJ/kg |
|---|---|
| Holocellulose | 17.5 |
| Lignin of SW | 26.8 |
| Lignin of HW, HP & AR | 25.0 |
| Extractives | 37.0 |
| Ash | 0 |
* SW, HW, HP, and AR are softwood, hardwood, herbaceous plants, and agricultural residues, respectively.
Table 5.
Standard enthalpies of reagents and products.
| Substance | -ΔcHo, kJ/mol | -ΔfHo, kJ/mol |
|---|---|---|
| Methanol | 726.5 | 238.7 |
| Glycerol | 1664.5 | 659.4 |
| Triglyceride | 33364 | 3071.7 |
| Biodiesel fuel | 11172 | 1163.8 |
Table 6.
Standard thermodynamic functions (TDFs).
| Substance | ΔfHo, kJ/mol | So, J/mol K |
|---|---|---|
| CO2 gas | -393.51 | 213.6 |
| H2 gas | 0 | 130.6 |
| CH4 gas | -74.85 | 186.19 |
| H2O gas (vapor) | -241.84 | 188.74 |
| H2O liquid | -285.83 | 69.96 |
Table 7.
TDFs of the Sabatier reaction at standard conditions: at To = 298.15 K and Po = 0.1 MPa.
| TDF | Values for the Case of Gaseous H2O Formation | Values for the Case of Liquid H2O Formation |
|---|---|---|
| ΔrHo (g), kJ/mol | -165.00 | -253.00 |
| ΔrSo(g), J/mol K | -172.33 | -409.89 |
| ΔrGo (g), kJ/mol | -113.62 | -130.79 |
Table 8.
Entropy values of substances at Pr=3 MPa and To = 298.15 K.
| Substance | S(Pr,To), J/mol K |
|---|---|
| CO2 gas | 185.32 |
| H2 gas | 102.32 |
| CH4 gas | 157.91 |
| H2O gas | 160.46 |
Table 9.
Values of TDFs at Tr = 673.15 K and Pr = 3 MPa.
| Substance | ΔfH(Pr,Tr), kJ/mol | S(Pr,Tr), J/mol K |
|---|---|---|
| CO2 gas | -378.21 | 218.49 |
| H2 gas | 10.56 | 125.50 |
| CH4 gas | -59.80 | 190.31 |
| H2O gas | -228.89 | 188.50 |
Table 10.
TDFs of the reaction at Tr=673.15 K and Pr=3 MPa.
| TDFs | Values |
|---|---|
| ΔrH(Pr,Tr), kJ/mol | -181.61 |
| ΔrS(Pr,Tr), J/mol K | -149.02 |
| Tr ΔrS(Pr,Tr), kJ/mol | -100.30 |
| ΔrG(Pr,Tr), kJ/mol | -81.31 |
Table 11.
Wetting enthalpy and structural characteristics of cellulose samples.
| Samples | CA* | -ΔwHo, kJ/mol | X | Y |
|---|---|---|---|---|
| MCC | Iβ | 6.8 ± 0.1 | 0.75 ± 0.01 | 0.25 ± 0.01 |
| Cotton cellulose (CC) | Iβ | 7.9 ± 0.1 | 0.71 ± 0.01 | 0.29 ± 0.01 |
| Kraft cellulose (KC) | Iβ | 9.6 ± 0.1 | 0.65 ± 0.01 | 0.35 ± 0.01 |
| Sulfite cellulose | Iβ | 10.1 ± 0.1 | 0.63 ± 0.01 | 0.37 ± 0.01 |
| Mercerized CC | II | 12.2 ± 0.1 | 0.55 ± 0.01 | 0.45 ± 0.01 |
| Mercerized KC | II | 12.7 ± 0.1 | 0.53 ± 0.01 | 0.47 ± 0.01 |
| Rayon cellulose fibers | II | 16.9 ± 0.1 | 0.38 ± 0.01 | 0.62 ± 0.01 |
* CA denotes the type of crystalline allomorph.
Table 12.
Standard TDFs of some cellulose Iβ samples [20].
Table 12.
Standard TDFs of some cellulose Iβ samples [20].
| Samples | X | -ΔcHo, kJ/mol | -ΔfHo, kJ/mol | So, J/mol K |
|---|---|---|---|---|
| *CR Iβ | 1 | 2810.6 | 979.6 | 180.0 |
| MCC | 0.75 ± 0.01 | 2819.8±1.8 | 970.4±1.8 | 182.5 ±1.5 |
| Cotton cellulose (CC) | 0.71 ± 0.01 | 2821.2±1.7 | 969.0±1.7 | 182.9 ±1.9 |
| Kraft cellulose (KC) | 0.65 ± 0.01 | 2823.6±2.2 | 966.6±2.2 | 183.5 ±2.0 |
| Sulfite cellulose (SC) | 0.63 ± 0.01 | 2823.8 ±2.0 | 966.4 ±2.0 | 183.7 ±2.2 |
| Milled CC | 0.28 ± 0.01 | 2837.4±2.1 | 952.8±2.1 | 187.2 ±1.8 |
| *AMC | 0 | 2847.8 | 942.4 | 190.0 |
* CR Iβ and AC denote Iβ crystallites and amorphous cellulose.
Table 13.
Standard TDFs cellulose samples with various CA.
| CA | X | -ΔcHo, kJ/mol | -ΔfHo, kJ/mol | So, J/mol K |
|---|---|---|---|---|
| CII | 0.55±0.01 | 2823.3±1.8 | 966.9±1.8 | 184.1 ±2.2 |
| CII | 0.53±0.01 | 2824.2±2.3 | 966.3±2.3 | 184.4±2.1 |
| CII | 0.38±0.01 | 2830.0±1.7 | 960.2±1.7 | 186.0±2.0 |
| CIII | 0.37±0.01 | 2836.4±2.3 | 953.8±2.3 | 186.5 ±2.2 |
| CIII | 0.35±0.01 | 2837.0±2.0 | 953.2±2.0 | 186.7±2.0 |
| CIV | 0.60±0.01 | 2825.0±2.1 | 965.2±2.1 | 184.1±2.3 |
| CIV | 0.57±0.01 | 2826.1±1.9 | 964.1±1.9 | 184.4 ±2.4 |
| AMC | 0 | 2847.8 | 942.4 | 190.0 |
| CA | -ΔcHo, kJ/mol | -ΔfHo, kJ/mol | ΔmHo, kJ/mol | ΔfGo, kJ/mol |
|---|---|---|---|---|
| Iβ | 2810.8 | 979.6 | 37.2 | 676.4 |
| II | 2803.3 | 986.9 | 44.5 | 683.7 |
| III | 2817.0 | 973.2 | 30.8 | 670.0 |
| IV | 2809.8 | 980.4 | 38.0 | 677.2 |
| AMC | 2847.8 | 942.4 | 0 | 639.2 |
| Samples | L, nm | Ssp (m2/m3) | ΔGn/σ (kJ m2/J mol) | AD, % |
|---|---|---|---|---|
| Natural cellulose of SW | 3.5 | 1.1 x 109 | 0 | 0 |
| Organosolv cellulose | 4.2 | 9.5 x 108 | -15 | 20 |
| Sulfite cellulose | 6.1 | 6.5 x 108 | -45 | 74 |
| Kraft cellulose | 7.0 | 5.7 x 108 | -53 | 100 |
| Component | -ΔcHo, kJ/mol | -ΔfHo, kJ/mol |
|---|---|---|
| Corn starch | 2836.6 ± 2.0 | 953.6 ± 2.0 |
| Hexosans of SW | 2833.6 ± 2.4 | 956.6 ± 2.4 |
| Pentosans of HW | 2348.0 ± 2.3 | 762.9 ± 2.3 |
| Citrus pectin | 2416.5 ± 1.8 | 1087.9 ± 1.8 |
| Lignin of SW | 5172.4 ± 2.1 | 585.4 ± 2.1 |
| Lignin of HW | 4984.1 ± 2.2 | 695.0 ± 2.2 |
| Abietic acid | 11580.1 ± 2.1 | 577.6 ± 2.1 |
| Isopimaric acid | 11568.1 ± 2.0 | 589.5 ± 2.0 |
| Levopimaric acid | 11571.0 ± 2.2 | 586.6 ± 2.2 |
Table 17.
TDFs of the hydrolysis reaction of starch gel.
| SG, g/L | GS, g/L | ΔrHo, kJ/mol | ToΔrSo, kJ/mol | ΔrGo, kJ/mol |
|---|---|---|---|---|
| 150 | 169.5 | -2.37 | 17.17 | -19.54 |
| 170 | 192.3 | -2.37 | 17.17 | -19.54 |
| 200 | 227.3 | -2.37 | 17.17 | -19.54 |
Table 18.
TDFs of the hydrolysis reaction of cellulose substrates.
| Cellulose | X | A | ΔrHo, kJ/mol | ToΔrSo, kJ/mol | ΔrGo, kJ/mol |
|---|---|---|---|---|---|
| MCC | 0.75 | 0.36 | -0.21 | 6.08 | -6.29 |
| Cotton cellulose | 0.71 | 0.43 | -0.22 | 7.01 | -7.23 |
| Kraft cellulose | 0.65 | 0.49 | -0.50 | 8.18 | -8.68 |
| Sulfite cellulose | 0.63 | 0.53 | -0.50 | 8.70 | -9.20 |
| AMC | 0 | 1.0 | -3.34 | 23.86 | -27.20 |
Table 19.
TDFs of the process of glucose conversion into ethanol (ET).
| TDFs | Value |
|---|---|
| ΔrHo(F), kJ/mol ET | -40.77 |
| ToΔrSo(F), kJ/mol ET | 74.13 |
| ΔrGo(F), kJ/mol ET | -114.90 |
Table 20.
Enthalpy of alkalization and formation of various alkalicelliloses.
| NaOH, % | m | n | AlC label | -ΔalHo, kJ/mol | -ΔfHo, kJ/mol |
|---|---|---|---|---|---|
| 16 | 11.7 | 4 | AlC 1 | 24.1 | 2605 |
| 18 | 10.1 | 4 | AlC 2 | 24.3 | 2605 |
| 20 | 8.9 | 4 | AlC 3 | 24.4 | 2605 |
| 30 | 5.2 | 2 | AlC 4 | 25.6 | 2030 |
| 35 | 4.1 | 1 | AlC 5 | 25.7 | 1742 |
| 40 | 3.3 | 1 | AlC 6 | 25.8 | 1738 |
Table 21.
Standard enthalpies of combustion and formation of AC and NC samples.
| Sample | DS | -ΔcHo, kJ/mol | -ΔfHo, kJ/mol |
|---|---|---|---|
| AMC | 0 | 2847 | 942 |
| NC-1 | 0.97 | 2800.8 | 851.2 |
| NC-2 | 2.03 | 2752.3 | 748.6 |
| NC-3 | 2.85 | 2714.8 | 669.2 |
| AC-1 | 1.11 | 3833.7 | 1148.3 |
| AC-2 | 2.12 | 4731.8 | 1326.2 |
| AC-3 | 2.80 | 5337.2 | 1463 |
Table 22.
Standard enthalpies of cellulose nitration [20].
Table 22.
Standard enthalpies of cellulose nitration [20].
| Sample | DS | ΔrHo, kJ/mol |
|---|---|---|
| Mono-nitrocellulose | 1 | 7.3 |
| 1.5-substituted NC | 1.5 | -0.8 |
| Dinitrocellulose | 2 | -9.3 |
| Trinitrocellulose | 3 | -25.1 |
Table 23.
Standard enthalpies of cellulose acetylation [20].
Table 23.
Standard enthalpies of cellulose acetylation [20].
| Sample | DS | ΔrHo, kJ/mol |
|---|---|---|
| Monoacetate cellulose | 1 | -7.1 |
| 1.5-substituted AC | 1.5 | -22.9 |
| Diacetate cellulose | 2 | -38.6 |
| Triacetate cellulose | 3 | -71.1 |
Table 24.
Enthalpy and temperature coefficient of some cellulose reactions.
| Reaction | -ΔrHo, kJ/mol | KT (298.15 K) | KT (343.15 K) |
|---|---|---|---|
| Enzymatic hydrolysis AMC | 3.34 | 3.85 | 3.22 |
| Bioethanol production | 40.77 | 1.39 x 107 | 1.61 x 106 |
| Alkalization with 20% NaOH | 24.4 | 1.88 x 104 | 5.18 x 103 |
| Alkalization with 40% NaOH | 25.8 | 3.31 x 104 | 8.46 x 103 |
| Methylation of CC to SD = 2 | 26.3 | 4.05 x 104 | 1.01 x 104 |
| Nitration of CC to DS = 2 | 9.2 | 40.90 | 25.14 |
| Acetylation of CC to DS = 2 | 38.6 | 5.79 x 106 | 7.51 x 105 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



