
Submitted:
25 November 2024
Posted:
27 November 2024
You are already at the latest version
Abstract
Macular telangiectasia type 2 (MacTel) is a slowly progressive macular disorder often diagnosed late due to the gradual onset of vision loss. Recent advances in diagnostic techniques have facilitated earlier detection, revealing MacTel is more common than initially thought. The disease is genetically complex, with multiple variants contributing incrementally to the overall risk. Familial occurrence of the disease prompted investigations of the genetic background of MacTel. In aim to raise the disease molecular milieu, a literature review of the clinical reports and publications investigating the genetic factors of MacTel was performed. To date, disease-associated variants were found in genes involved in amino acid (glycine/serine) metabolism and transport, urea cycle, lipid metabolism, and retinal vasculature and thickness. Variants in genes implicated in sphingolipid metabolism and fatty acid/steroid/retinol metabolism have been found in patients with neurological disorders who also have MacTel. Retinal metabolism involves complex biochemical processes essential for maintaining the high energy requirements of the retina. Genetic alternations can disrupt key metabolic pathways, leading to retinal cell degradation and the subsequent vision loss that characterizes several retinal disorders, including MacTel. This review article summarises genetic findings that may allow MacTel to be investigated further as an inherited retinal disorder.
Keywords:
retina
; macular disorder
; macular telangiectasia type 2
; MacTel
; complex disease
; genetic predisposition
; genomic biomarkers
1. Introduction
Macular telangiectasia type 2 (MacTel) is a bilateral retinal disease with a late onset. Symptoms typically appear over the age of 40 years. The disease affects people all over the world with no racial and no clear gender predilection [1,2]. It was originally thought to be a disease of the retinal vessels. However, with the development of diagnostic imaging techniques, it has been shown that it is primarily the consequence of neurodegenerative changes in the retina, with vascular changes appearing later in the disease development. Due to the low level of awareness of the disease, many patients with earlier stages of the disease remain undiagnosed. For the same reason, some patients with later stages of the disease are misdiagnosed with other macular disorders.
The diagnosis of MacTel is based on clinical examination and multimodal imaging diagnostics, e.g. color fundus photography, fundus autofluorescence, fluorescein angiography, optical coherence tomography (OCT), and OCT-angiography. The heterogeneity of the clinical presentation often makes diagnosis difficult and challenging. Typical clinical findings include reduced retinal transparency, crystalline deposits, mildy ectatic capillaries, blunted venules, photoreceptor loss, retinal pigment plaques, intraretinal cavities, lamellar or full-thickness macular holes, and neovascular complexes within the macula [3]. Observational studies have increased our knowledge of the disease. Vision loss primarily affects the patients’ ability to read, however, when the disease progresses, more widespread loss of vision may occur. Structural changes have been studied in detail with functional correlations. Furthermore, it was established that certain systemic conditions, such as diabetes mellitus type 2 (T2DM), hypertension, heart disease, and thyroid disease, were more prevalent in patients with MacTel [1,4,5,6,7,8,9,10]. Neurodegenerative changes are thought to be the primary manifestation affecting vision, with the concomitant occurrence of vascular aberrations, that may lead to neovascularisation as the disease progresses. A secondary vascular involvement in some cases could align with the increased occurrence of T2DM, hypertension, and coronary artery disease in the patients [11]. MacTel also co-occurs with neurological disorders, indicating a complex interplay of genetic variants and the involvement of different genes, leading to different neurological manifestations and phenotypes. For example, comorbidity with hereditary sensory and autonomic neuropathy type 1 (HSAN1) [12,13,14,15], and autosomal recessive spastic paraplegia type 56 (SPG56) [16] have been reported. Disease classification has evolved with the development of diagnostic techniques (Table 1). Originally, idiopathic juxtafoveolar retinal telangiectasis were classified into three groups, type 1, type 2 (with sub-types A and B), and type 3. Sub-type 2A (MacTel) was further divided into five stages [2]. Yannuzzi and colleagues revised the classification into non-proliferative and proliferative disease [17]. The MacTel Research Group recently developed an up-to-date classification of MacTel disease with 7 grades, by incorporating image analysis results from novel imaging modalities. A revised classification using all imaging modalities likely facilitates a better description of the grades and progression of MacTel and communications between clinical and basic science researchers [18]. There is currently no proven treatment for MacTel available. Intravitreal treatment with vascular endothelial growth factor inhibitors is available for exudative neovascularization [19], however, no treatment exists for neurodegenerative changes.
The prevalence of MacTel is estimated to be 0.0045–0.1% [20,21]. Advances in diagnostic approaches have made it possible to recognize milder, earlier forms of the disease that were overlooked in the past, hence the wide reported range in the prevalence of this disease. It is now generally accepted that MacTel is more common than originally thought. The apparent genetic penetrance of MacTel is important for gene discovery studies and for clinical risk assessment of affected individuals’ family members. It was calculated to be 0.35 by sibling analysis and 0.55 by parent analysis. Combining the sibling and parent analyses, the apparent penetrance was 0.38 [22]. The occurrence of the disease in multiple family members prompted several investigations of the genetic background of MacTel. The initial studies, analysing candidate genes, and potential disease-associated loci did not identify disease-associated genetic factors, indicating a more complex genetic background is responsible for the disease mechanism [23,24,25,26]. Further studies, utilizing GWAS, uncovered several genetic variants in a large cohort of patients [27,28]. Approaches used for the discovery of MacTel risk variants/gene candidates are revised in Figure 1. Although not characterized by any systemic pathology, the disease-associated metabolic profile has been described, outlining reduced serum serine and glycine levels, and altered lipid metabolism in MacTel patients [12,29]. These findings were consistent with genetic studies that identified variants in genes implicated in the glycine/serine metabolic pathway and lipid metabolism.
This article summarizes genetic alterations that have been associated with MacTel and reported up to August 1, 2024. Online libraries PubMed and Google Scholar were searched for the relevant literature. Gene names are consistent with the HUGO Gene Nomenclature Committee (HGNC) [30] and the National Center for Biotechnology Information (NCBI) [31] databases. The variants are represented with rs numbers and include information about their location on a level of a coding sequence, genome, or protein (GRCh38.p14) (Table S1–S4). Allele frequencies were collected from gnomAD for the total and European (Non-Finnish) ancestry groups [32]. Variant type, consequence, and clinical significance were obtained from NCBI databases ClinVar and dbSNP, and Ensembl Release 112 (May, 2024) [33].
2. Key Genes, Genomic Regions, and Metabolic Pathways Associated with MacTel
2.1. Familial Occurrence of MacTel
The involvement of genetic factors in MacTel pathogenesis has been proposed due to the clinical reports of the disease in monozygotic twins and family members (Table 2).
In the late 1970s, the disease was characterized as a distinct clinical entity for the first time. Hutton et al. (1978) examined four patients, two of whom were sisters (aged 46 and 56 years), with an unusual form of retinal telangiectasia [34]. Putteman et al. (1984) reported the familial occurrence of idiopathic familial juxtafoveolar retinal telangiectasia in several family members [35]. Chew et al. (1986) described the condition in the non-diabetic brother of a diabetic patient while reporting on five patients with parafoveal telangiectasies and mild diabetic retinopathy [6]. In an update of classification and follow-up study on idiopathic juxtafoveolar retinal telangiectasis, three of the 92 patients were siblings, and the disease was present in two of 89 families [2]. Isaacs and McAllister (1996) studied two sisters over an 8-year period with bilateral idiopathic juxtafoveolar retinal telangiectasis [36]. Oh and Park (1999) described vertical transmission in the 29-year-old daughter of a 58-year-old affected father [37]. At that time, the case was unique in vertical transmission of the disease and the fibrovascular tissue developed in the young proband. The authors assumed that inherited factors and T2DM have predisposed the proband to develop early and severe fibrovascular tissues. Another report on the vertical transmission of MacTel and T2DM in three families was published in 2012 [38]. Vertical transmission in families suggests a dominant inheritance [3]. In 2000, bilateral juxtafoveolar telangiectasis was reported in monozygotic twins (64 years old sisters) and raised the question of genetic influences in the pathogenesis of the disease [39]. Later reports of affected monozygotic twins were published in 2005 (68-year old sisters) [40], 2007 (63-year old sisters) [41] and 2009 (65/56-year old sisters, 56-year old brothers) [11] strengthening the theory of the genetic component of the disease.
The discovery of affected asymptomatic family members suggested the condition is more severe than previously thought. The identification of asymptomatic family members or members with early-stage disease is currently limited by the lack of non-invasive diagnostic techniques that can detect subtle changes, characteristic of early disease stages, and that might have been missed by biomicroscopic examination. In addition, studies support the hypothesis that patients with MacTel have a genetic predisposition for the disease. However, it has been suggested that a secondary factor contributes to the development of clinically evident features in monozygotic twins. Influence of environmental factors in combination with genetic factors could potentially explain the phenotypic discordance between monozygotic twins with differences in disease stage. The local, intraocular environment might also be important. A particular emphasis should be given to epigenetic variations, which could establish a pathogenic link between external and internal factors.
2.2. Candidate Gene-Screening Analysis
Barbazetto et al. (2008) investigated variants in the ATM gene, age-related macular degeneration (AMD)-associated variants in CFH and CFB genes, and 10q26 loci in a cohort of 30 MacTel patients [23]. Variations in the ATM were screened by a combination of denaturing high-performance liquid chromatography and direct sequencing. Major AMD-associated alleles in CFH, CFB, and 10q26 were screened by polymerase chain reaction-restriction fragment-length polymorphism. Screening of the ATM identified amino acid changes in 23 patients of European-American descent. The screening revealed a null allele rs587780612 (ATM) in 2 of 23 (8.7%) patients of European descent, previously disease-associated missense alleles rs4986761, rs1800057 (both in ATM) in 4 of 23 (17.4%) patients, as well as common missense alleles rs4986761, rs1800057, rs1801516, rs148993589, rs1800058, rs2234997, and rs3218695 (all in ATM) in 7 of 23 (30.4%) patients (Figure 2, Table S1). Interestingly, no variants were identified in the ATM gene in patients of Asian or Hispanic descent. The frequencies of the major AMD-associated alleles in CFH, CFB, and 10q26 loci in the MacTel cohort were similar to those in the ethnically matched general population. Between 26 and 57% MacTel patients of European-American ancestry included in the study carried ATM alleles that could be disease-associated. Therefore, certain variations in ATM may predispose to the development of MacTel [23].
Parmalee et al. (2010) searched for the gene(s) responsible for MacTel by a candidate-gene screening approach [25]. Candidate genes were selected based on the following criteria: 1) known to cause or be associated with diseases with phenotypes similar to MacTel, 2) genes with known function in the retinal vasculature or macular pigment transport, 3) genes that emerged from expression microarray data from mouse models designed to mimic MacTel phenotype characteristics and 4) genes expressed in the retina that are also related to T2DM or hypertension, which have increased prevalence in MacTel patients. Probands (8) from eight families with at least two affected individuals were screened by direct sequencing of 27 candidate genes, including AGGF1, ANG1, DKK1, FZD4, HIF1A, LRP5, NDP, PEDF, THBS1, TIE2, VHL, AGTRL1, APLN, CFB, LRG1, PLVAP, GTSP1, SCARB1, CCM2, IGFBP3, SSPN, TGFB2, AGTR1, ALDH3A2, OXGR1, SUCNR1, and VLDLR. Altogether, 23 nonsynonymous missense variants, 22 synonymous, and 61 intronic variants were identified. Of nonsynonymous variants, 12 missense variants were further screened by TaqMan assays in 400 MacTel cases and 368 controls for co-segregation with the disease and/or allele frequency analysis. None of these variants segregated with the disease. Only one variant, rs1695 (GTSP1), showed a trend toward significant frequency difference between MacTel cases and controls suggesting being a possible modifier, but not a causal gene for MacTel (Figure 2, Table S1). Furthermore, a variant rs61735304 (FZD4), previously suggested as a disease-causing variant in familial exudative vitreoretinopathy, was determined to be a rare benign polymorphism [25].
Szental et al. (2010) conducted a case-control study on 39 MacTel cases and 21 controls to determine whether two variants of the GSTP1 gene were associated with MacTel [26]. The Pi isoform of glutathione S-transferase (GSTP1) is a xanthophyll-binding protein (XBP). The uptake of xanthophyll pigment into the macula is thought to be facilitated by XBP and patients with MacTel have decreased macular pigment centrally. Two typical functional polymorphic sites, rs1695 and rs1138272, in GSTP1 were analysed. The study found no statistically significant associations between the variants and MacTel. However, a trend towards a greater frequency of the GG genotype of the rs1695 (GSTP1) in cases (3/39; 8%) could be significant (Figure 2, Table S1). The biological plausibility of impaired macular pigment uptake in MacTel makes GSTP1 an excellent candidate gene for further investigation in larger cohorts of patients and unaffected controls [26].
GSTP1 plays a crucial role in the detoxification of electrophilic compounds, including carcinogens, therapeutic drugs, environmental toxins, and products of oxidative stress, by conjugation with glutathione [42]. The rs1695 (c.313A>G) results in an amino acid substitution at position 105 of the GTSP1 protein (p.Ile105Val). This single nucleotide polymorphism (SNP) affects enzyme activity and specificity which may impact the detoxification efficiency [43,44]. Due to high metabolic demands and oxidative stress in the retina [45], the GSTP1 variants could be further investigated for their influence on retinal health.
2.3. Genome-Wide Linkage Analysis
As part of the MacTel project, 17 pedigrees with multiple affected family members, consistent with autosomal dominant segregation with reduced penetrance, were identified out of a large cohort of MacTel patients [24]. Genome-wide linkage analysis of seventeen families with a total of 71 individuals (including 45 affected or possibly affected) identified a single peak with a multi-point LOD score of 3.45 on chromosome 1 at 1q41-42 under a dominant model. Recombination mapping defined a minimal candidate region of 15.6 Mb (214.32 –229.92 Mb), encompassing the 1q41-42 linkage peak. Interestingly, Sanger sequencing of the top 14 positional candidate genes, including DISP1, TLR5, SUSD4, CAPN8, CAPN2, TP53BP2, FBXO28, DEGS1, NVL, CNIH4, WDR26, and CNIH3 and the micro RNA MIR320B2 under the linkage peak revealed no causal variants in these pedigrees (Figure 3) [24].
However, according to UCSC Genome Browser, this region contains more than 1700 transcripts (including gene isoforms and noncoding RNA transcripts) [46,47]. Therefore, the disease could be driven by variants in and/or aberrant expression of either of these coding and noncoding genes. Furthermore, it would be interesting to identify the minimal driver or initiation aberrations that together with passenger alterations contribute to different clinical courses of the disease.
2.4. Genome-Wide Association Studies
The first GWAS identified three genome-wide (GW) significant (P < 5 × 10-8) associations, i.e. rs477992 (PHGDH), rs715 (CPS1), and rs73171800 (ENSG00000271904; TMEM161B–MEF2C) [28]. The discovery stage included genotype data for 6,312,048 SNPs in 476 MacTel cases and 1,733 controls of European descent. Variants at six loci surpassed the GW significance threshold. Three were discounted (1p36.22, 3p24.1, and 7p21.3) after failing the technical validation step. The remaining three loci (1p12, 2q34, and 5q14.3) included 149 SNPs that reached GW significance. Seven of 149 SNPs, including rs539708, rs666930, rs483180 (all in PHGDH); rs715, rs4673553 (both in CPS1); rs17478824 (ENSG00000271904; TMEM161B–MEF2C), and rs73173548 (MIR9-2HG) were genotyped with TaqMan assays in the replication stage in an independent cohort of 172 MacTel cases and 1,134 controls (European descent) [28].
At locus 5q14.3 the strongest associated variant in the discovery stage was rs73171800 (ENSG00000271904; TMEM161B–MEF2C) among 116 SNPs in this region. The rs73171800 is in strong linkage disequilibrium (LD) with rs17478824 (ENSG00000271904; TMEM161B–MEF2C), rs73173548 (MIR9-2HG), rs2194025 (TMEM161B–MEF2C), and rs17421627 (MIR9-2HG) (all among GW significant SNPs at this locus). For further analysis in the replication stage in an independent cohort of MacTel patients, the rs17478824 (ENSG00000271904; TMEM161B–MEF2C) and rs73173548 (MIR9-2HG) variants were selected. The analyses in the independent cohort strongly confirmed that minor alleles of these two variants increased the risk of developing MacTel [28].
At locus 2q34 the strongest associated variant was rs715 (CPS1) of 21 SNPs discovered in this region. Together with another significant variant rs4673553 (CPS1), these two were selected for replication in the independent cohort. The major allele of rs715 (CPS1) confers risk for MacTel. Females were at nearly two times greater risk of MacTel for each additional copy of the rs715 (CPS1) risk allele [28].
At locus 1p12 the most significant variant was rs477992 (PHGDH), followed by rs478093 (PHGDH) of 12 significant SNPs at this locus. The signal was replicated with variant rs483180 (PHGDH) in the independent cohort. Minor alleles of rs477992 and rs478093 (both in PHGDH) confer risk for MacTel and were associated with decreased serine levels [28].
25 loci showed suggestive evidence for association with MacTel (1 × 10-5 > P ≥ 5 × 10-8). These loci were explored for overlap with genetic regions previously associated with serine and glycine levels, and the loci at 3q21.3 and 7p11.2 were identified. At 3q21.3 rs9820286 and rs9880406 (both between ALDH1L1–KLF15) are suggestively associated with MacTel, and association with MacTel for rs9880406 (ALDH1L1–KLF15) was replicated in the independent cohort. At 7p11.2 suggestive significance was found for variants rs4947534 and rs4948102 (both in PSPH) (the strongest signal at this locus). Variants selected for replication in the independent cohort were rs4535700 and rs11238389 (both in PSPH). The variants rs4947534, rs4948102, and rs11238389 (all in PSPH) are in perfect LD and are in high LD with rs4535700 (PSPH). The minor allele of these variants is associated with MacTel risk, and, similarly, the minor allele of rs4947534 (PSPH) is associated with reduced serine levels and increased homocysteine levels [28].
Identified genetic loci accounted for only 5% of the estimated heritability. By increasing the sample size, more disease-associated loci were discovered. The second GWAS included 1,067 MacTel cases and 3,799 controls (European descent, with a higher proportion of females) [27]. The extended study confirmed all three previously reported loci (1p12, 2q34, and 5q14.3) and identified seven novel GW significant loci (2p14, 3p24.1, 3p21.31, 7p11.2, 9p22.3, 10q21.3, and 19p13.2) and 7 novel suggestively significant loci (1q32.3, 2p23.2, 6q13, 8q24.21, 11p13, 17q25.1, and 19p13.2). After quality control and imputation, genotype data for 7,289,516 SNPs were included in GW association testing. MacTel consortium samples were genotyped on the Illumina Human Omni5-Exome-4 Array or the Illumina Global Screening Array; AREDs controls were genotyped on the Illumina Omni2.5 BeadChip Array, and the Australian Twinning controls on the Illumina Global Screening Array. Two GWAS were performed, one for the entire cohort (full-cohort GWAS) and one restricted to individuals of European descent (European-cohort GWAS). Eleven independent GW significant disease associations in ten regions were identified in the full-cohort analysis, of which seven results in six regions were preserved in the European cohort GWAS [27].
The study by Bonelli and colleagues further confirmed the involvement of specific metabolic pathways in MacTel pathogenesis (Figure 4). Variants were found within genes implicated in the glycine/serine metabolic pathway (PHGDH and PSPH), metabolite transport (SLC1A4 and SLC6A20), urea cycle (CPS1), retinal vasculature and thickness (MIR9-2HG) and other genes TTC39B, REEP3 or intergenic regions between SLC4A7–EOMES, and CERS4–FBN3 [27].
Variants at 2q34 and 5q14.3 are in LD with the previously identified tagging SNPs rs715 (CPS1) and rs73171800 (ENSG00000271904; TMEM161B–MEF2C), while the most significant SNP rs146953046 (PHGDH) in the 1p12 region is in low LD with, and independent from the previously identified SNP rs477992 (PHGDH). Conditional analysis at this locus revealed a second signal, where the most significant SNP, rs532303 (PHGDH), was in strong LD with the previously identified SNP rs477992 (PHGDH) [27].
Conditional regression analysis (GWAS conditioning on the metabolic polygenic risk scores) identified four GW significant peaks. Original signal rs73171800 (ENSG00000271904; TMEM161B–MEF2C) on locus 5q14.3 and three novel disease loci independent of endogenous serine biosynthetic capacity, including rs35356316 (EOMES–SLC4A4) at 3p24.1, rs36259 (CERS4), and rs4804075 (NFILZ) at 19p13.2. Genetically induced serine deficiency was suggested to be the primary causal metabolic driver of MacTel, with a contribution of T2DM genetic risk. Conversely, glycine, threonine, and retinal vascular traits were deemed unlikely to be causal for MacTel [48].
GWAS have significantly advanced our understanding of the genetic basis of many complex traits and diseases. However, they come with several limitations, including 1) the identification of common genetic variants usually having small effects on the disease, while they are not effective in identifying rare variants with larger impact on phenotype; 2) GWAS often identifies loci associated with traits, but not the specific causal variants; 3) complex traits and diseases are often influenced by multiple genetic and environmental factors, making it challenging for GWAS to capture the full genetic architecture. MacTel-associated genomic variants discovered through GWAS are shown in Figure 4, Table S.
2.5. Next-Generation Sequencing Studies
Genes and variants leading to reduced serine levels observed in patients with MacTel remain elusive. Whole exome sequencing data of 793 MacTel cases and 17,610 controls has been used to identify the disease-associated variants. The collapsing analysis identified 22 rare variants in PHGDH in 29 MacTel cases (Table S3). The resulting functional defects, induced by the most common missense variant rs139063843 (PHGDH) were strongly associated with the decrease in serine biosynthesis and accumulation of deoxysphingolipids (deoxySLs) in retinal pigmented epithelial (RPE) cells [49].
2.6. Implications of Risk-Associated Metabolic Pathways on MacTel Pathogenesis
The pathogenetic mechanism leading to the MacTel development is largely unknown. Most genetic studies clearly indicated that genetic differences in glycine/serine metabolism, urea cycle, and metabolite transport could constitute the possible background for MacTel development. In addition, differences in disease progression suggested that certain combinations of genetic aberrations in conjunction with lifestyle and other factors influence the trajectory of the disease severity and progression over time.
In addition to genetic studies, the causative disease mechanisms have also been investigated using other omics approaches (metabolomics, proteomics), and multiple models have been developed, including induced pluripotent stem cells, organoids, and animals (mouse and rat models) [50,51,52,53,54,55].
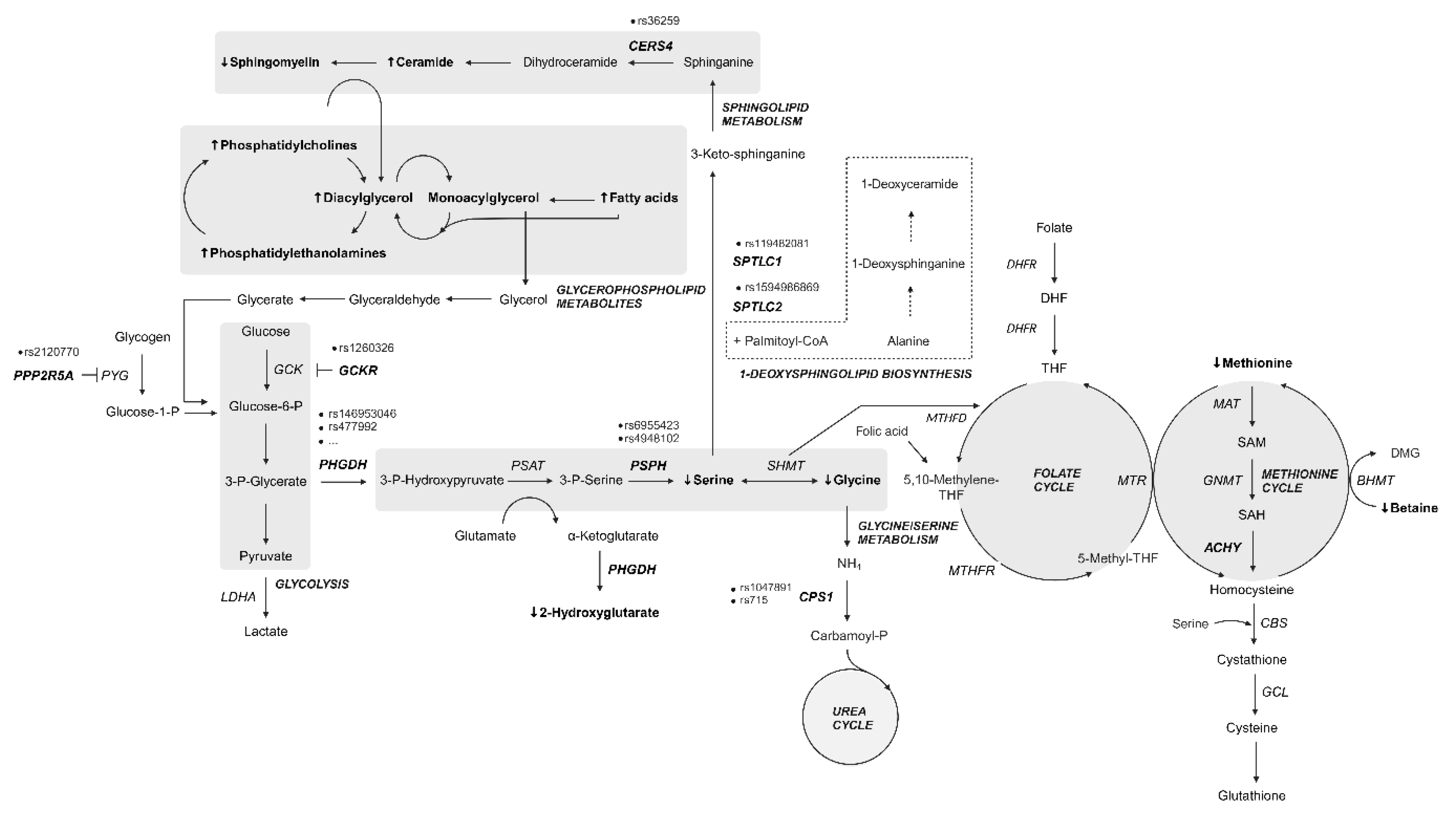
For example, a metabolomics study of 60 MacTel cases and 58 control subjects confirmed reduced serum serine and glycine levels in patients, and altered lipid metabolism with elevated phosphatidylethanolamines, long-chain fatty acids, diacylglycerols, and monoacylglycerols [29]. Furthermore, they also found out that methionine, betaine, and urea cycle intermediates were decreased. This finding prompted further investigation, focusing on key metabolites of methionine and one-carbon metabolism, such as homocysteine, S-adenosylmethionine, S-adenosylhomocysteine as well as total glutathione levels. Methylation-associated metabolite (MAM) levels were investigated in 29 MacTel patients, revealing that homocysteine could be an important metabolite discerning MacTel patients from other diseases with specific metabolite-methylation patterns [56]. In another study, utilizing cross-platform analyses of associations between genetic effects and blood levels of 174 metabolites, the researchers found that an increase of 1 standard deviation in serine levels via a genetic score reduced the likelihood of MacTel development by 95%, indicating that even slight changes in the key metabolite concentrations could have profound effects on particular cell types [57].
Research at the proteomic level further unveiled the aberrations in the protein expression levels directly in the vitreous, confirming the involvement of several metabolic pathways directly or indirectly involved in the amino acid and lipid metabolism, including the glycolytic anaplerotic reactions [58].
Overall, these and other studies indicated the strong link between MacTel, altered serine metabolism, and elevated levels of atypical deoxySLs. Some studies have already established genetic predisposition to altered amino acid homeostasis and identified variants that could be responsible for reduced serine levels in patients [12,27,28,49]. The functional variants in PHGDH identified in MacTel patients may result in decreased serine biosynthesis and accumulation of deoxySLs [49].
Serine is considered a conditionally essential amino acid for some tissues that have a high demand for protein synthesis, organelle turnover, and lipid homeostasis. Phosphoglycerate dehydrogenase (PHGDH) catalyses the first committed step of the de novo serine biosynthesis from 3-phosphoglycerate [59]. Interestingly, the ability of cells to synthesize serine from glycolytic intermediates, renders serine a non-essential amino acid, however, in recent years, its importance has been recognized by studies of malignant diseases (for review see [60,61]). It has been firmly established that serine is an important metabolite intertwining several essential metabolic pathways, including glycolysis, folate cycle, and one-carbon metabolism as well as the metabolism of sphingolipids, phospholipids, and sulphur-containing amino acids [59,62]. Serine is involved in cysteine biosynthesis via the transsulfurylation pathway; cysteine in turn is essential for the production of glutathione. Glutathione and glutathione-associated enzyme systems in the retina are one of the most important antioxidants that minimize the damage caused by photooxidation products [63].
Prolonged serine deficiency leads to the deregulation of amino acid homeostasis, which can result in the generation of non-canonical sphingolipids [64]. Glycine, alanine, cysteine, and threonine share structural similarities with alanine and when serine is deficient, serine palmitoyltransferase (SPT) can produce cytotoxic deoxySLs using alanine or glycine as a substrate for condensation with palmitoyl-CoA [15,64,65,66]. DeoxySLs were shown to be responsible for photoreceptor cell death in retinal organoids [12].
Studies also indicated that variants in metabolite transporters may be implicated in the pathological complexity of MacTel. Indeed, the SLC1A4 gene (protein ASCT1) encodes sodium-dependent neutral amino acids transporter, which transports serine, alanine, cysteine, proline, hydroxyproline, and threonine, whereas XTRP3 (gene SLC6A20) mediates the sodium- and chloride-dependent uptake of imino acids, such as proline, as well as glycine and N-methylated amino acids. Variations that impair the function of such transporters could further deregulate amino acid homeostasis in susceptible cells with specific requirements for certain amino acids, such as Müller glia and RPE [12,49,67].
2.7. Association of MacTel with Comorbidities
MacTel co-occurs with some neurological and metabolic disorders. Therefore, genetic variants in one gene or interactions between variants in several genes together with environmental factors could contribute to the development of primary and associated diseases. Such variants were found in MacTel patients in sub-units (SPTLC1 and SPTLC2) of the first enzyme in the ceramide/sphingolipid pathway serine palmitoyltransferase (SPT), and cytochrome P450 CYP2U1, implicated in oxidation and fatty acid metabolism (Table S4). SPT mutations are known to cause HSAN1, a rare condition that causes nerve damage in the extremities. The hallmark of the disease is loss of pain and temperature sensation in the distal parts of the lower limbs. The autonomic disturbances, if present, manifest as sweating abnormalities. Comparison of enzymatic properties in eleven SPTLC1 and six SPTLC2 isoforms revealed distinct associations between the mutant proteins and clinical phenotypes as well as the severity of the symptoms. Both isoforms are ubiquitously expressed and are involved in the condensation of palmitoyl-CoA and L-serine in the de novo synthesis of sphingolipids. Interestingly, most mutants showed increased activity and affinity for alanine and diminished specificity for serine, resulting in the overproduction of deoxysphingolipids, detectable in the plasma of patients. Three mutant isoforms, p.Ser331Phe, p.Ser331Tyr (SPTLC1), and p.Ile504Phe (SPTLC2), produced canonical C18-sphingolipids, as well as anomalous C20-sphingolipids [68].
SPT variants, p.Cys133Tyr (SPTLC1) and p.Ser384Phe (SPTLC2), which cause HSAN1, were identified in MacTel patients (9/11 patients with HSAN1) [12] and have been reported in two generations of a single-family [14]. An association of the variant in SPTLC2 with MacTel has also been reported in a family member, whose family has multiple members afflicted with HSAN1 [15]. Nevertheless, the link between HSAN1 and MacTel appears to be more complex and cannot be explained by the genetic variants alone, as patients with HASN1 (age 35–71 years) with variants in SPT showed no clinical signs of MacTel [13]. A particular phenotype with MacTel was observed in the case of a neurologically asymptomatic young boy presenting with an unusual phenotype of CYP2U1-related macular dystrophy. Whole-exome sequencing identified a disease-causing pathogenic variant in CYP2U1, causing SPG56 [16].
3. Conclusions
MacTel is a clinically heterogeneous and genetically complex macular disorder. The path to understanding the genetic complexity of the disease has not yet been paved. Translating this knowledge into clinically relevant information is a daunting task due to the MacTel phenotype complexity and the myriads of alterations that can affect the delicate expression and function of genes. Based on the current state of research (summarized in Figure 5), it appears that the combined aberrations in serine metabolic pathways, as well as in sphingolipid, and possibly also urea and cholesterol metabolism may contribute to the initiation and development of the disease. Studies have shown that the most important genetic changes, driving the course of the disease are most likely different variants in genes. It could also be possible that the disease is not caused by rare genetic variants, but by the particular combination of common genetic variants in certain genes of key metabolic pathways associated with MacTel pathogenesis. In addition to the variants in protein-coding genes, the variants in non-coding RNA molecules, that are involved in fine-tuning of the gene expression, could also influence the delicate metabolite balance in the cells. Epigenetic changes, such as DNA methylation and histone modifications, which can also aberrantly accumulate in response to oxidative stress, environmental, and other factors, might further contribute to neurodegeneration and abnormalities of MacTel. Taken together, these genetic and epigenetic factors, individual lifestyle factors, including quality of nutrition could account for differences in symptoms, time of onset, and disease severity.
It should be noted that systemic serine and glycine deficiency has already been strongly associated with metabolic syndrome and various macular as well as peripheral nerve disorders [12,69]. Emerging evidence clearly indicated the link between diabetic polyneuropathy and serine deficiency in diabetic mouse models, possibly due to altered glucose metabolism, leading to altered levels of 3-phosphoglycerate, the main serine precursor [69,70]. Interestingly, low serine levels and higher toxic deoxySLs have been associated with HSAN1 and patients with HSAN1 often present with MacTel, however, patients with a primary diagnosis of MacTel rarely have HSAN1. It is not yet clear, whether peripheral neuropathy may also be present in the primary MacTel background. Therefore, it appears that an imbalance of metabolites in specific cell types could underlie the disease and cause different symptoms in lieu of individual genetic and epigenetic frameworks. In the future, it would also be interesting to explore the specific metabolic requirements of particular cell types, as research indicated that some cells are particularly sensitive to metabolite fluctuations. The apparent functional dependence of supportive cells to retinal neurons on serine homeostasis may suggest that L-serine supplementation could be used to slow and possibly partially ameliorate vision loss in MacTel patients, as well as patients with other complex diseases, who are at risk for serine deficiency. In addition, serine supplementation could reduce oxidative stress and neurotoxicity of aberrant metabolic by-products and improve the antioxidant capacity of the retina.
In summary, understanding the underlying mechanisms and genetic background of MacTel will enable better management of the patients, early diagnosis, and the development of potential personalized treatments in the future.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1. MacTel-associated genetic alternations identified by a candidate-gene screening approach [23,25,26].; Table S2. MacTel-associated genomic variants discovered through genome-wide association studies [27,28,44].; Table S3. The 22 unique rare variants in PHDGH detected in 29 MacTel cases [45].; Table S4. Genetic alternations in patients with MacTel co-occurring with other neurological disorders [12,13,14,15,16].
Author Contributions
Literature review, writing of the original manuscript, figures and tables preparation, editing (A.K.), review and critical assessment of clinical part and terminology of the article (M.U., D.D.D.), review and critical assessment of biochemical and genetic part and terminology of the article (N.D., P.H.), conceptualization (A.K., N.D.), funding acquisition (N.D., M.U.). All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was supported by the Slovenian Research and Innovation Agency (ARIS) grant P1-0390, Junior Researcher funding to A.K., and the University Medical Centre Ljubljana Tertiary Research and Development Project 20230137.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Clemons TE, Gillies MC, Chew EY, Bird AC, Peto T, Figueroa MJ; et al. Baseline characteristics of participants in the natural history study of macular telangiectasia (MacTel) MacTel Project Report No. 2. Ophthalmic epidemiology. 2010;17(1):66-73. [CrossRef]
- Gass JD, Blodi BA. Idiopathic juxtafoveolar retinal telangiectasis. Update of classification and follow-up study. Ophthalmology. 1993;100(10):1536-46. [CrossRef]
- Charbel Issa P, Gillies MC, Chew EY, Bird AC, Heeren TF, Peto T; et al. Macular telangiectasia type 2. Progress in retinal and eye research. 2013;34:49-77. [CrossRef]
- Balaskas K, Leung I, Sallo FB, Clemons TE, Bird AC, Peto T. Associations between autofluorescence abnormalities and visual acuity in idiopathic macular telangiectasia type 2: MacTel project report number 5. Retina (Philadelphia, Pa). 2014;34(8):1630-6. [CrossRef]
- Clemons TE, Gillies MC, Chew EY, Bird AC, Peto T, Figueroa M; et al. The National Eye Institute Visual Function Questionnaire in the Macular Telangiectasia (MacTel) Project. Investigative ophthalmology & visual science. 2008;49(10):4340-6.
- Clemons TE, Gillies MC, Chew EY, Bird AC, Peto T, Wang JJ; et al. Medical characteristics of patients with macular telangiectasia type 2 (MacTel Type 2) MacTel project report no. 3. Ophthalmic epidemiology. 2013;20(2):109-13.
- Heeren TFC, Chew EY, Clemons T, Fruttiger M, Balaskas K, Schwartz R; et al. Macular Telangiectasia Type 2: Visual Acuity, Disease End Stage, and the MacTel Area: MacTel Project Report Number 8. Ophthalmology. 2020;127(11):1539-48.
- Peto T, Heeren TFC, Clemons TE, Sallo FB, Leung I, Chew EY; et al. CORRELATION OF CLINICAL AND STRUCTURAL PROGRESSION WITH VISUAL ACUITY LOSS IN MACULAR TELANGIECTASIA TYPE 2: MacTel Project Report No. 6-The MacTel Research Group. Retina (Philadelphia, Pa). 2018;38 Suppl 1(Suppl 1):S8-s13.
- Vujosevic S, Heeren TFC, Florea D, Leung I, Pauleikhoff D, Sallo F; et al. SCOTOMA CHARACTERISTICS IN MACULAR TELANGIECTASIA TYPE 2: MacTel Project Report No. 7-The MacTel Research Group. Retina (Philadelphia, Pa). 2018;38 Suppl 1:S14-s9.
- Zhang Q, Wang RK, Chen CL, Legarreta AD, Durbin MK, An L; et al. Swept Source Optical Coherence Tomography Angiography of Neovascular Macular Telangiectasia Type 2. Retina (Philadelphia, Pa). 2015;35(11):2285-99.
- Gillies MC, Zhu M, Chew E, Barthelmes D, Hughes E, Ali H; et al. Familial asymptomatic macular telangiectasia type 2. Ophthalmology. 2009;116(12):2422-9.
- Gantner ML, Eade K, Wallace M, Handzlik MK, Fallon R, Trombley J; et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. The New England journal of medicine. 2019;381(15):1422-33.
- Rodrigues FG, Pipis M, Heeren TFC, Fruttiger M, Gantner M, Vermeirsch S; et al. Description of a patient cohort with Hereditary Sensory Neuropathy type 1 without retinal disease Macular Telangiectasia type 2 - implications for retinal screening in HSN1. Journal of the peripheral nervous system : JPNS. 2022;27(3):215-24.
- Triplett J, Nicholson G, Sue C, Hornemann T, Yiannikas C. Hereditary sensory and autonomic neuropathy type IC accompanied by upper motor neuron abnormalities and type II juxtafoveal retinal telangiectasias. Journal of the peripheral nervous system : JPNS. 2019;24(2):224-9.
- Wilson LMQ, Saba S, Li J, Prasov L, Miller JML. Specific Deoxyceramide Species Correlate with Expression of Macular Telangiectasia Type 2 (MacTel2) in a SPTLC2 Carrier HSAN1 Family. Genes. 2023;14(4).
- El Matri K, Falfoul Y, Habibi I, Chebil A, Schorderet D, El Matri L. Macular Dystrophy with Bilateral Macular Telangiectasia Related to the CYP2U1 Pathogenic Variant Assessed with Multimodal Imaging Including OCT-Angiography. Genes. 2021;12(11).
- Yannuzzi LA, Bardal AM, Freund KB, Chen KJ, Eandi CM, Blodi B. Idiopathic macular telangiectasia. Archives of ophthalmology (Chicago, Ill : 1960). 2006;124(4):450-60.
- Chew EY, Peto T, Clemons TE, Sallo FB, Pauleikhoff D, Leung I; et al. Macular Telangiectasia Type 2: A Classification System Using MultiModal Imaging MacTel Project Report Number 10. Ophthalmology science. 2023;3(2):100261.
- Toygar O, Guess MG, Youssef DS, Miller DM. Long-term Ootcomes of Intravitreal Bevacizumab Therapy for Subretinal Neovascularization Secondary to Idiopathic Macular Telangiectasia Type 2. Retina (Philadelphia, Pa). 2016;36(11):2150-7.
- Aung KZ, Wickremasinghe SS, Makeyeva G, Robman L, Guymer RH. The prevalence estimates of macular telangiectasia type 2: The Melbourne Collaborative Cohort Study. Retina (Philadelphia, Pa). 2010;30(3):473-8.
- Klein R, Blodi BA, Meuer SM, Myers CE, Chew EY, Klein BE. The prevalence of macular telangiectasia type 2 in the Beaver Dam eye study. American journal of ophthalmology. 2010;150(1):55-62.e2.
- Ronquillo CC, Wegner K, Calvo CM, Bernstein PS. Genetic Penetrance of Macular Telangiectasia Type 2. JAMA ophthalmology. 2018;136(10):1158-63.
- Barbazetto IA, Room M, Yannuzzi NA, Barile GR, Merriam JE, Bardal AM; et al. ATM gene variants in patients with idiopathic perifoveal telangiectasia. Investigative ophthalmology & visual science. 2008;49(9):3806-11.
- Parmalee NL, Schubert C, Figueroa M, Bird AC, Peto T, Gillies MC; et al. Identification of a potential susceptibility locus for macular telangiectasia type 2. PLoS ONE. 2012;7(8):e24268.
- Parmalee NL, Schubert C, Merriam JE, Allikmets K, Bird AC, Gillies MC; et al. Analysis of candidate genes for macular telangiectasia type 2. Molecular vision. 2010;16:2718-26.
- Szental JA, Baird PN, Richardson AJ, Islam FM, Scholl HP, Charbel Issa P; et al. Analysis of glutathione S-transferase Pi isoform (GSTP1) single-nucleotide polymorphisms and macular telangiectasia type 2. International ophthalmology. 2010;30(6):645-50.
- Bonelli R, Jackson VE, Prasad A, Munro JE, Farashi S, Heeren TFC; et al. Identification of genetic factors influencing metabolic dysregulation and retinal support for MacTel, a retinal disorder. Communications biology. 2021;4(1):274.
- Scerri TS, Quaglieri A, Cai C, Zernant J, Matsunami N, Baird L; et al. Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nature genetics. 2017;49(4):559-67.
- Bonelli R, Woods SM, Ansell BRE, Heeren TFC, Egan CA, Khan KN; et al. Systemic lipid dysregulation is a risk factor for macular neurodegenerative disease. Scientific reports. 2020;10(1):12165.
- Seal RL, Braschi B, Gray K, Jones TEM, Tweedie S, Haim-Vilmovsky L; et al. Genenames.org: The HGNC resources in 2023. Nucleic acids research. 2023;51(D1):D1003-d9.
- Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC; et al. Database resources of the national center for biotechnology information. Nucleic acids research. 2022;50(D1):D20-d6.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-43.
- Harrison PW, Amode MR, Austine-Orimoloye O, Azov Andrey G, Barba M, Barnes I; et al. Ensembl 2024. Nucleic acids research. 2023;52(D1):D891-D9.
- Hutton WL, Snyder WB, Fuller D, Vaiser A. Focal parafoveal retinal telangiectasis. Archives of ophthalmology (Chicago, Ill : 1960). 1978;96(8):1362-7.
- Putteman A, Toussaint D, Graff E, Verougstraete C. [Idiopathic familial juxtafoveolar retinal telangiectasias]. Bulletin de la Societe belge d’ophtalmologie. 1984;209:81-90.
- Isaacs TW, McAllister IL. Familial idiopathic juxtafoveolar retinal telangiectasis. Eye (London, England). 1996;10 ( Pt 5):639-42.
- Oh KT, Park DW. Bilateral juxtafoveal telangiectasis in a family. Retina (Philadelphia, Pa). 1999;19(3):246-7.
- Delaere L, Spielberg L, Leys AM. Vertical transmission of macular telangiectasia type 2. Retinal cases & brief reports. 2012;6(3):253-7.
- Menchini U, Virgili G, Bandello F, Malara C, Rapizzi E, Lanzetta P. Bilateral juxtafoveolar telangiectasis in monozygotic twins. American journal of ophthalmology. 2000;129(3):401-3.
- Siddiqui N, Fekrat S. Group 2A idiopathic juxtafoveolar retinal telangiectasia in monozygotic twins. American journal of ophthalmology. 2005;139(3):568-70.
- Hannan SR, Madhusudhana KC, Rennie C, Lotery AJ. Idiopathic juxtafoveolar retinal telangiectasis in monozygotic twins. The British journal of ophthalmology. 2007;91(12):1729-30.
- Lin H, Wu H, Li H, Song A, Yin W. The essential role of GSTP1 I105V polymorphism in the prediction of CDNB metabolism and toxicity: In silico and in vitro insights. Toxicology in vitro : An international journal published in association with BIBRA. 2023;90:105601.
- Chen YC, Tzeng CH, Chen PM, Lin JK, Lin TC, Chen WS; et al. Influence of GSTP1 I105V polymorphism on cumulative neuropathy and outcome of FOLFOX-4 treatment in Asian patients with colorectal carcinoma. Cancer science. 2010;101(2):530-5.
- Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: Relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19(2):275-80.
- Pan WW, Wubben TJ, Besirli CG. Photoreceptor metabolic reprogramming: Current understanding and therapeutic implications. Communications biology. 2021;4(1):245.
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D; et al. The UCSC Table Browser data retrieval tool. Nucleic acids research. 2004;32(Database issue):D493-6.
- Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M; et al. The UCSC Genome Browser database: 2023 update. Nucleic acids research. 2023;51(D1):D1188-d95.
- Bonelli R, Ansell BRE, Lotta L, Scerri T, Clemons TE, Leung I; et al. Genetic disruption of serine biosynthesis is a key driver of macular telangiectasia type 2 aetiology and progression. Genome medicine. 2021;13(1):39.
- Eade K, Gantner ML, Hostyk JA, Nagasaki T, Giles S, Fallon R; et al. Serine biosynthesis defect due to haploinsufficiency of PHGDH causes retinal disease. Nature metabolism. 2021;3(3):366-77.
- Baumann B, Sterling J, Song Y, Song D, Fruttiger M, Gillies M; et al. Conditional Müller Cell Ablation Leads to Retinal Iron Accumulation. Investigative ophthalmology & visual science. 2017;58(10):4223-34.
- Byrne LC, Khalid F, Lee T, Zin EA, Greenberg KP, Visel M; et al. AAV-mediated, optogenetic ablation of Müller Glia leads to structural and functional changes in the mouse retina. PLoS ONE. 2013;8(9):e76075.
- Eade KT, Ansell BRE, Giles S, Fallon R, Harkins-Perry S, Nagasaki T; et al. iPSC-derived retinal pigmented epithelial cells from patients with macular telangiectasia show decreased mitochondrial function. The Journal of clinical investigation. 2023;133(9).
- Hua J, Guerin KI, Chen J, Michán S, Stahl A, Krah NM; et al. Resveratrol inhibits pathologic retinal neovascularization in Vldlr(-/-) mice. Investigative ophthalmology & visual science. 2011;52(5):2809-16.
- Thomas ED, Timms AE, Giles S, Harkins-Perry S, Lyu P, Hoang T; et al. Cell-specific cis-regulatory elements and mechanisms of non-coding genetic disease in human retina and retinal organoids. Developmental cell. 2022;57(6):820-36.e6.
- Zhao M, Andrieu-Soler C, Kowalczuk L, Paz Cortés M, Berdugo M, Dernigoghossian M; et al. A new CRB1 rat mutation links Müller glial cells to retinal telangiectasia. The Journal of neuroscience : The official journal of the Society for Neuroscience. 2015;35(15):6093-106.
- Pauleikhoff L, Wingert V, Grünert SC, Lange C, Hannibal L, Bucher F. Methylation-associated Pathways in Macular Telangiectasia Type 2 and Ophthalmologic Findings in Patients with Genetic Methylation Disorders. Retina (Philadelphia, Pa). 2024;44(6):1052-62.
- Lotta LA, Pietzner M, Stewart ID, Wittemans LBL, Li C, Bonelli R; et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nature genetics. 2021;53(1):54-64.
- Len AC, Powner MB, Zhu L, Hageman GS, Song X, Fruttiger M; et al. Pilot application of iTRAQ to the retinal disease Macular Telangiectasia. Journal of proteome research. 2012;11(2):537-53.
- Holeček, M. Serine Metabolism in Health and Disease and as a Conditionally Essential Amino Acid. Nutrients. 2022;14(9).
- Pan S, Fan M, Liu Z, Li X, Wang H. Serine, glycine and one-carbon metabolism in cancer (Review). International journal of oncology. 2021;58(2):158-70.
- Shunxi W, Xiaoxue Y, Guanbin S, Li Y, Junyu J, Wanqian L. Serine Metabolic Reprogramming in Tumorigenesis, Tumor Immunity, and Clinical Treatment. Advances in nutrition (Bethesda, Md). 2023;14(5):1050-66.
- Zhou X, Tian C, Cao Y, Zhao M, Wang K. The role of serine metabolism in lung cancer: From oncogenesis to tumor treatment. Frontiers in genetics. 2022;13:1084609.
- Brodzka S, Baszyński J, Rektor K, Hołderna-Bona K, Stanek E, Kurhaluk N; et al. The Role of Glutathione in Age-Related Macular Degeneration (AMD). International journal of molecular sciences. 2024;25(8).
- Cordes T, Kuna RS, McGregor GH, Khare SV, Gengatharan J, Muthusamy T; et al. 1-Deoxysphingolipid synthesis compromises anchorage-independent growth and plasma membrane endocytosis in cancer cells. Journal of lipid research. 2022;63(10):100281.
- Bejaoui K, Uchida Y, Yasuda S, Ho M, Nishijima M, Brown RH, Jr.; et al. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. The Journal of clinical investigation. 2002;110(9):1301-8.
- Rosarda JD, Giles S, Harkins-Perry S, Mills EA, Friedlander M, Wiseman RL; et al. Imbalanced unfolded protein response signaling contributes to 1-deoxysphingolipid retinal toxicity. Nature communications. 2023;14(1):4119.
- Handzlik MK, Metallo CM. Sources and Sinks of Serine in Nutrition, Health, and Disease. Annual review of nutrition. 2023;43:123-51.
- Bode H, Bourquin F, Suriyanarayanan S, Wei Y, Alecu I, Othman A; et al. HSAN1 mutations in serine palmitoyltransferase reveal a close structure-function-phenotype relationship. Human molecular genetics. 2016;25(5):853-65.
- Handzlik MK, Gengatharan JM, Frizzi KE, McGregor GH, Martino C, Rahman G; et al. Insulin-regulated serine and lipid metabolism drive peripheral neuropathy. Nature. 2023;614(7946):118-24.
- Holeček, M. Role of Impaired Glycolysis in Perturbations of Amino Acid Metabolism in Diabetes Mellitus. International journal of molecular sciences. 2023;24(2).
Figure 1.
Candidate MacTel risk variants and genes uncovered using listed approaches. Indicated are number of studies (N) and number of cases and/or controls (bold). *ATM sequence variants could be disease-associated [23]; *GSTP1 is a possible modifier, but not a causal gene for MacTel [25]; trend towards greater frequency of the GG genotype in cases [26].
Figure 1.
Candidate MacTel risk variants and genes uncovered using listed approaches. Indicated are number of studies (N) and number of cases and/or controls (bold). *ATM sequence variants could be disease-associated [23]; *GSTP1 is a possible modifier, but not a causal gene for MacTel [25]; trend towards greater frequency of the GG genotype in cases [26].
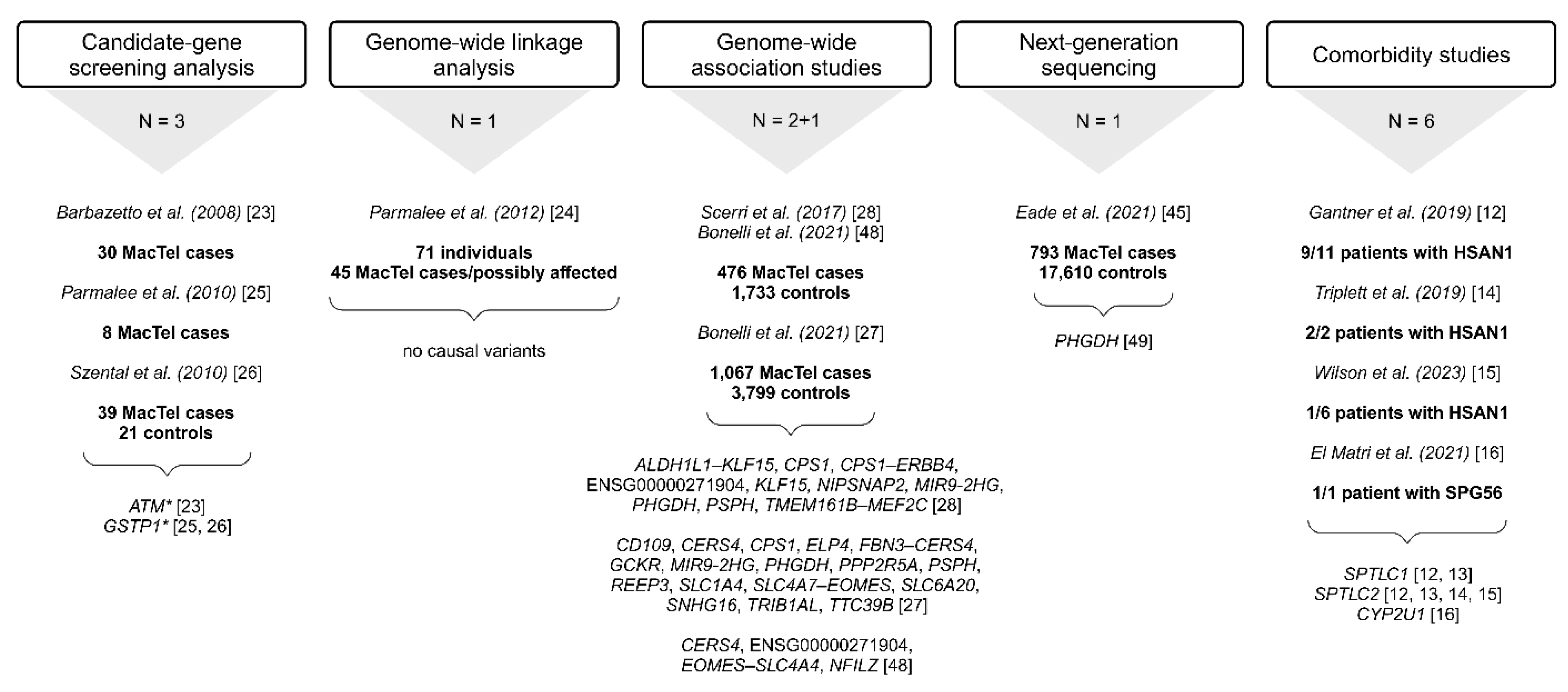
Figure 2.
AMD-associated variants in the ATM and variants in the GSTP1 found in MacTel patients through candidate-gene screening approaches [23,25,26]. *ATM sequence variants could be disease-associated [23]; *GSTP1 is a possible modifier, but not a causal gene for MacTel [25]; trend towards greater frequency of the GG genotype in cases [26]. .
Figure 2.
AMD-associated variants in the ATM and variants in the GSTP1 found in MacTel patients through candidate-gene screening approaches [23,25,26]. *ATM sequence variants could be disease-associated [23]; *GSTP1 is a possible modifier, but not a causal gene for MacTel [25]; trend towards greater frequency of the GG genotype in cases [26]. .
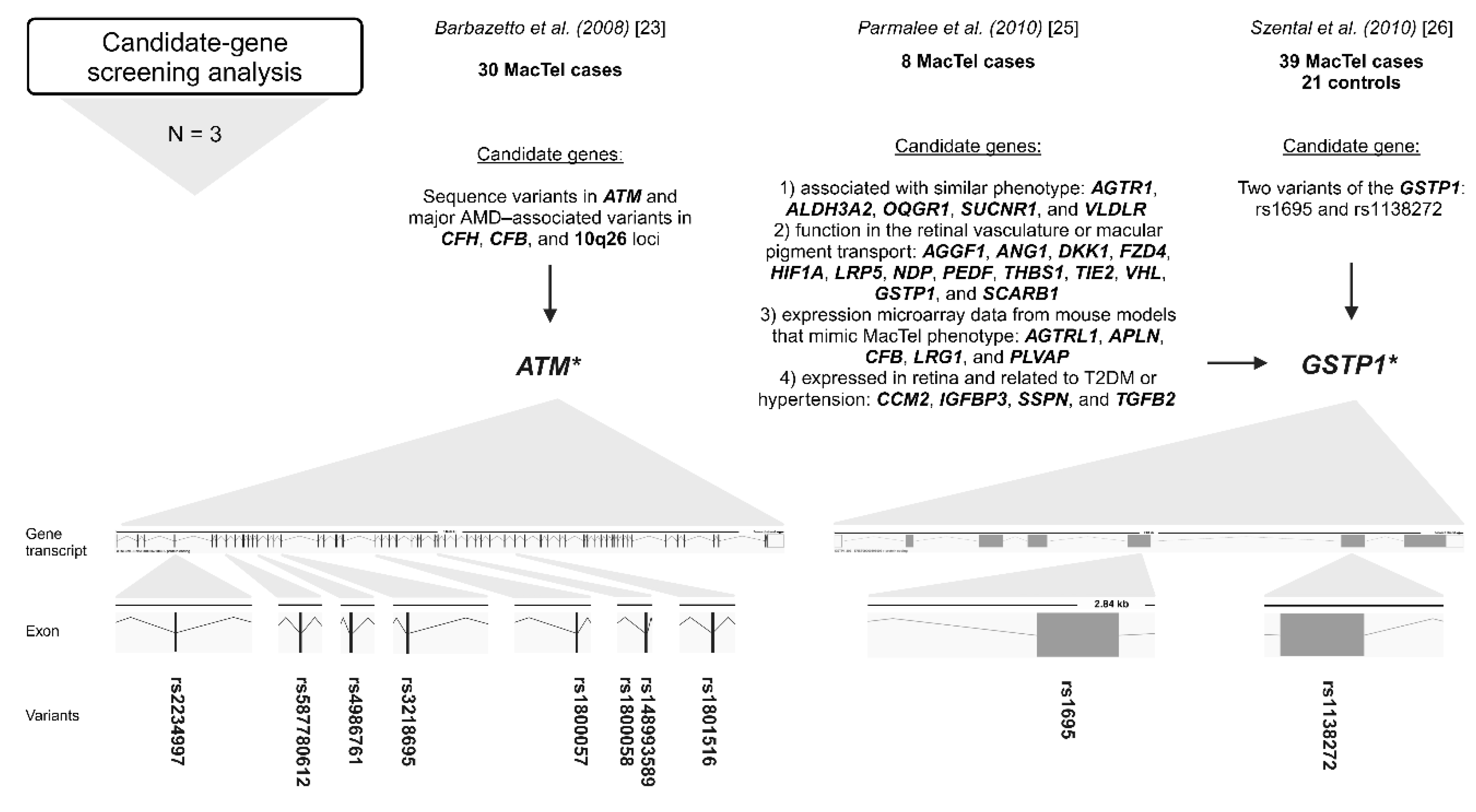
Figure 3.
Overview of the 16.5 Mb region at 1q41-42 investigated in genome-wide linkage analysis [24]. Marked genes: candidate genes screened for disease-associated variants [24]. .

Figure 4.
Identification of MacTel risk genes through genome-wide association studies [27,28]. Bold: the most significant variant at the locus; *Variants genotyped in the replication stage [28].

Figure 5.
Summary of metabolic pathway genes and metabolites altered in patients with MacTel. Bold, italic: genes with variants most commonly reported in MacTel patients; Bold, ↑: elevated serum metabolites [29]; Bold, ↓: reduced serum metabolites [29]; dashed border: alternative pathway.

| Gass and Blodi (1993) [2] | Yannuzzi et al. (2006) [17] | Chew et al. (2023) [18] |
|---|---|---|
| Group 1 1A: Visible and exudative IJRT 1B: Visible, exudative, and focal IJRT |
||
| Group 2 2A: Occult and nonexudative IJRT Stage 1: Diffuse hyperfluorescence Stage 2: Reduced parafoveolar retinal Stage 2: transparency Stage 3: Dilated right-angled venules Stage 4: Intraretinal pigment clumping Stage 5: Vascular membranes |
Non-proliferative perifoveal telangiectasia Proliferative perifoveal telangiectasia |
Grade 0: No EZ Break/No Pigmentation/No OCT HR Grade 1: Noncentral EZ Break/No pigment/No OCT HR Grade 2: Central EZ break/No pigment/No OCT HR Grade 3: Noncentral pigment/No, non-central, or central EZ/No Grade 3: OCT HR Grade 4: OCT HR/EZ break (either central or noncentral)/No Grade 4: pigment Grade 5: Central pigment/no exudative neovascularization/EZ Grade 5: present or not gradable Grade 6: Neovascularization (exudative) ± central pigment |
| 2B: Juvenile occult familial IJRT | ||
| Group 3 3A: Occlusive IJRT 3B: Occlusive IJRT with CNS vasculopathy |
Indicated in grey: classification of MacTel. Abbreviations: idiopathic juxtafoveolar retinal telangiectasis (IJRT); ellipsoid zone (EZ); optical coherence tomography (OCT); hyper-reflectivity (HR); central nervous system (CNS).
Table 2.
Clinical reports of MacTel in family studies and associated primary conditions [2,6,11,34,35,36,37,38,39,40,41].
| Affected individuals (age (years)) | Associated primary condition | Reference |
|---|---|---|
| Sisters (461 and 562) | 1Blurring of vision; 2Slightly distorted vision after a car accident | [34] |
| Several family members | / | [35] |
| Brothers (651 and /) | 1T2DM, mild nonproliferative DR, systemic hypertension, asteroid hyalosis, pigment epithelial hyperplasia, and mild macular edema | [6] |
| 3/92 patients were siblings the disease is present in 2/89 families |
14/92 patients had hypertension, other accompanying diseases were polycythemia vera, coronary artery disease, and borderline diabetes in one case each, two had coronary artery disease, and one had renal insufficiency associated with Alport’s disease | [2] |
| Sisters (491 and 482) | 1Reduced vision; 2Blurring of distance vision | [36] |
| Daughter (291) of the affected father (582) | 1T2DM, decreased vision; 2Macular edema | [37] |
| Monozygotic twins, sisters (641,2) | 1,2Vision loss | [39] |
| Monozygotic twins, sisters (681/2) | 1Bilateral metamorphopsia; 2T2DM, amblyopia | [40] |
| Monozygotic twins, sisters (631/2) | 1Metamorphopsia, subretinal NV; 2Metamorphopsia | [41] |
| Daughters (41 and 45) of affected mother (681) Brother (61) of the affected sister (742) Monozygotic twins, sisters (563) Monozygotic twins, brothers (564,5) |
1Blurring of vision; 2Blurring of vision, T2DM; 3Bilateral blurred vision, phototherapeutic keratectomy; 4Decrease of vision, developed T2DM; 5T2DM | [11] |
| Son (451) of the affected mother (792) Son (623) of the affected mother (764) Sister (655) and brother (786) |
1Reading difficulties. metamorphopsia, T2DM, mild DR, underwent coronary bypass at age of 40; 2Loss of vision, maculopathy of unknown origin, T2DM, DR; 3Visual loss, obesity, arterial hypertension, phlebothrombotic event at age of 57, hyperhomocysteinemia, antiphospholipid syndrome, T2DM; 4T2DM, reading difficulties, maculopathy in the absence of DR; 5Metamorphopsia, T2DM; 6T2DM | [38] |
Superscript numbers indicate the associated primary condition in the affected individual (family member). Abbreviations: diabetes mellitus type 2 (T2DM); diabetic retinopathy (DR); neovascularization (NV).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



