Submitted:
27 October 2025
Posted:
28 October 2025
Read the latest preprint version here
Abstract
Epithelial–mesenchymal transition (EMT) and its reverse, mesenchymal–epithelial transition (MET), are core cellular processes that drive epithelial–mesenchymal plasticity (EMP) in cancer. EMP regulates how tumor cells switch between detached and proliferative states, affecting cancer progression, spread, and resistance to treatment. Cancer cells gain invasive traits during EMT, enabling their escape from the primary site. At the same time, MET allows seeded cancer cells at distant sites to reacquire epithelial characteristics necessary for growth at secondary locations. These transitions are regulated by interconnected signaling pathways such as TGF‑β, Wnt/β‑catenin, and Notch, coordinated by master transcription factors including Snail, ZEB, and Twist. The tumor microenvironment—particularly hypoxia, inflammatory cues, and microRNA regulators like the miR‑200 family—modulates these processes, fostering hybrid epithelial/mesenchymal phenotypes that underlie tumor heterogeneity and therapy resistance. Addressing EMP requires multi-level therapeutic strategies and smart targeting reprogramming of transitional states, especially by incorporating epigenetic modifiers into the treatment protocol. This review describes a Combination, Timing, and Sequencing (CTS) framework to leverage EMP dynamics to improve cancer treatment outcomes while minimizing toxicities.
Keywords:
epithelial-mesenchymal transition
; mesenchymal-epithelial transition
; cancer metastasis
; tumor invasion
; cellular plasticity
; transcription factors
; TGF-β signaling
; microRNA regulation
; therapeutic resistance
; cancer stem cells
; hybrid phenotypes
; personalized medicine
; targeted therapy
Introduction
Epithelial–mesenchymal transition (EMT) and its reverse process, mesenchymal–epithelial transition (MET), are key cellular plasticity regulators that underlie normal embryonic development and pathological conditions such as cancer. During EMT, polarized epithelial cells acquire a mesenchymal phenotype characterized by loss of cell–cell adhesion, reduced epithelial marker expression, and elevated production of mesenchymal proteins including N-cadherin and vimentin. These changes trigger several cascading effects. First, tumor cells with enhanced mesenchymal traits acquire a migratory phenotype, which drives tissue infiltration and metastasis, ultimately promoting disease progression [1,2]. EMT facilitates tumor invasion and metastasis, enabling cancer cells to disseminate from the primary tumor site to distant organs, a process critically linked with poor prognostic outcomes in various cancers [1,3]. Second, acquiring stem cell-like properties often accompanies this increased invasive capability. Third, EMT-associated mechanisms confer survival advantages to cancer cells under therapeutic pressure, resulting in drug resistance [4].
After establishing a niche at the distant site under a favorable microenvironment, cancer cells with mesenchymal characteristics need to undergo evolutionary reversion to epithelial cells to proliferate at the distant site [5,6]. This process occurs through mesenchymal-epithelial transition (MET), in which mesenchymal cells revert to a more epithelial-like state, gaining characteristics such as enhanced adhesion and reduced motility [6,7]. MET thus plays a critical role in establishing secondary tumors by restoring proliferative capabilities necessary for metastatic outgrowth at distant sites [3]. EMT and MET are not binary transitions but represent a spectrum of intermediate phenotypes that profoundly influence cancer cell behavior. This plasticity enables cancer cells to dynamically adapt to changing microenvironmental conditions and therapeutic pressures [8].
This review explores the dynamic interplay between EMT and MET in driving tumor invasion, stemness, and drug resistance. By examining the molecular triggers and functional consequences of these transitions, we explore the mechanisms underlying metastasis and therapeutic failure to identify potential strategies to target adaptive EMT/MET plasticity cancer cell populations and simultaneously prevent cancer dissemination [3,9,10].
1. Cancer Transition, Stemness, Progression, and Ecological Evolution
Epithelial-Mesenchymal Transition (EMT) is a fundamental cellular process in which epithelial cells lose their characteristic junctions and polarity, transitioning towards a more migratory and invasive mesenchymal phenotype. This transformation is crucial in various biological contexts, including embryogenesis, tissue repair, and notably, cancer metastasis [11]. During EMT, cells exhibit reduced epithelial markers such as E-cadherin while gaining mesenchymal markers, including N-cadherin and vimentin. Such changes facilitate enhanced migratory capabilities, enabling cells to detach from the primary tumor and invade surrounding tissues [12]. Evidence suggests that cells undergoing EMT may acquire stem-like properties, contributing to tumor heterogeneity and metastatic potential [13].
1.1. Primary Drivers of EMT
1.1.1. ROS - Hypoxia Stress
Reactive oxygen species (ROS) are a set of normal metabolic messenger substances that, depending on the metabolic dynamics, have a dual action of tumor promotion or apoptotic tumor suppression. Due to inefficient mitochondrial respiration consequent to oxidative stress, excessive ROS build-up sets in a cascade of events (Figure 1). Excessive ROS facilitates stabilization of hypoxia-inducible factor 1α (HIF-1α), which is otherwise expected to typically break after deactivating ROS. Accumulated HIF-1α promotes glycolysis, reducing oxidative phosphorylation and further increasing ROS production, setting up a vicious loop. With anaerobic glycolysis, the hydrogen ion pump is activated to reduce intracellular acidosis, and H+ ion diffuses extracellularly, producing extracellular acidosis. This increasing extracellular acidosis unwittingly promotes immunosuppression and inactivates several anticancer drugs. Intracellular HIF-1α thus accumulates in low oxygen conditions, impacting mitochondrial and glucose metabolism [14]. This stabilized HIF-1α re-enters the nucleus. Re-entered HIF-1α forms a transcription complex with the stable hypoxia-inducible factor 1 beta subunit (HIF-1β) to form a heterodimer, which binds to DNA motifs affecting the dozens of homeostatic genes, leading to cascading effects in cancer progression [15]. Hypoxia is a foundational stress that leads to core phenotypes developing multidirectional adaptive mechanisms, one of which is EMT. Downstream effects through HIF-1α are aberrant vasculature, metabolic reprogramming, the acquisition of cancer stem cell traits, and immune dysregulation, all of which play critical roles in cancer progression, EMT/metastasis, secondary mutations, heterogeneity, and resistance [16,17]. HIFs promote the expression of EMT-inducing transcription factors such as Snail and Twist. These transcription factors facilitate the downregulation of epithelial markers while enhancing the expression of mesenchymal proteins, thereby promoting cellular plasticity [18,19].
EMT-TME interaction further drives aggressive tumor phenotypes through upregulated angiogenic factors, HIF-1a, vessel over-pruning, myeloid cell recruitment, CAFs, increased co-option/mimicry, in turn promoting aberrant angiogenesis, tumor growth, EMT plasticity, and metastasis. Hypoxia, which stimulates β1 integrin, decreases adherens junction proteins and impacts pericyte coverage, promoting the invasiveness of the EMT cancer cells [20,21]. Tumor hypoxia also induces angiogenic dormancy, autophagy, lactate production, stem cell proliferation, and lymphangiogenesis, accompanying EMT, with increasing therapy resistance [22].
1.1.2. Metabolic and Mitochondrial Stress
The heterodimer of HIF-1α and HIF-1β (Figure 1) promotes the expression of a series of target genes, including glucose transporters (GLUTs) and the glycolysis enzyme lactate dehydrogenase A (LDHA), upon formation under hypoxia, initiating anaerobic glycolysis—the Warburg effect. LDHA accelerates the conversion of pyruvate to lactate, facilitating the proliferation of cancer cells [23]. Lactate, a byproduct of glycolysis, seeps into the tumor microenvironment (TME), resulting in an acidic pH. This process leads to a significant and diverse accumulation of immunosuppressive cells, which fosters genomic mutations and further fuels tumor growth, as well as the development of resistant phenotypic sub-clones (Figure 1) [24,25].
Accelerated mitochondrial biogenesis and metabolic pathways hasten cancer progression and EMT. Several studies have supported mitochondrial dysfunction, glycolysis, and TGF-β-induced EMT. Despite reprogramming all three glucose, amino acid, and lipid metabolic pathways, forming a network with intracellular signaling, proliferating cancer cells run out of nutrition, compelling them to undergo mesenchymal transition [26].
1.1.3. Inflammatory Stress and Role of TGFβ
An inflammatory microenvironment (iTME) stress drives carcinogenesis, immune escape, and progression, exacerbated by oxidative stress (ROS). A variety of transcription factors, kinases, and chemokines are involved in this process. The first and primary factor identified is TGF-β, which initially takes the role of tumor suppression, later plays a fundamental role in tumor progression, EMT, and stemness. The so-called poorly understood “TGF-β switch” leads to the induction of Snail and Smad pathways, leading to EMT (Figure 2). Over time, TGF-β acquires multiple tumor-promoting effects, including the induction of EMT, increasing the fibrosis/ rigidity of the ECM, and preventing immunological cross-talk in the ECM, thus forming a critical therapy target. iTME associated with cells like CAFs, TAMs, neutrophils, eosinophils, mast cells, T helper 17 (Th17), and Treg cells induce and sustain EMT by producing proinflammatory cytokines [27]. Inflammatory signals present in tumors, particularly cytokines such as IL-6 and IL-8, contribute to the induction of EMT by activating pathways like JAK/STAT and NF-κB, which have been implicated in maintaining mesenchymal traits and supporting tumor progression [19]. The interconnected resistance factors are promoted by NF-κB-induced inflammation, resulting in the acquisition of cancer stem cell properties and EMT, with invasion, angiogenesis, and metastasis [28].
1.1.4. Spatial Stress
ECM and the cell stromal communication system undergo stiffness, altered shear force, and increased pressure from increasing cell proliferation within a given space. This spatial stress is an additional factor that induces EMT in cancer cells. This is an example of mechanotransduction, where sensing and transmitting mechanical cues are converted to biochemical impulses, encouraging tumor invasion and metastases through EMT. However, the mechanism involved is not fully understood [29]. The reduction of spatial stress significantly impacts ECM softness and disrupts mechanical barriers [30].
1.1.5. Hypoxia - Metabolic – Inflammatory Stress and Cancer Immune Editing Loop
Immunoediting theory describes a three-step immunosurveillance, equilibrium, and immune escape by avoiding T cell homing [31]. During the immune editing phase, the process gives decipherable cues about the dialectics in cross-talk between malignant cells, ECM, soluble cues (cytokines/growth factors), and neighboring cells=. This sets up a multiple-pathway converging loop where EMT is initiated and sustained. Dynamic phenotypic plasticity and heterogeneity exist between epithelial cells and immune cells. EMT in cancer is reprogramming borrowed from the normal physiological embryonic harmonized process. Cytotoxic CD8+ T cells eliminate the potential proliferating cancer cells during immunosurveillance and, in equilibrium. These cells are reprogrammed into the immunosuppressive ones that stimulate EMT, immune exclusion/deviation, and let cancer cells acquire augmented stemness and therapy resistance [32,33]=. In various studies, the interplay between hypoxia and inflammation has been illustrated to maintain a state of "partial EMT," whereby cancer cells retain some epithelial characteristics while acquiring increased migratory capabilities, enhancing their metastatic potential [18,19].
Figure 1 depicts the various stress mechanisms that lead to the initiation and multipathway progression of cancer with the EMT.
1.1.6. Therapeutic Interventions – Evolutionary Mutational Predator - Prey Game
The remission outcome looks promising during and after anticancer therapy. However, resistance might have developed much earlier, and the predator-prey dynamic could already favor the “prey” (cancer). Within the tumor ecosystem, cancer cells exhibit ecological spatial and temporal Darwinian dynamics of natural selection, leading to diverse cell subpopulations. The evolutionary changes involve heritable genomic mutations such as chromosomal rearrangements, gene duplication, and aneuploidy. Unlike infectious organisms, cancer cells—accessing the vast information of the human genome—show remarkable adaptability, evolving alternative resistant pathways. Through combined ecological and evolutionary interactions, resistance to therapy develops. This “evolutionary rescue” can occur even in a small, rapidly declining cancer cell population, after eliminating the bulk of cells. This rescue results firstly from the preexistence of a small fraction of resistant cell population, secondly due to respite given for these cells to recover (for example, due to inappropriate timing and drug holidays), or thirdly due to exposure to sublethal drug concentrations that facilitate acclimatization (such as inadequate drug delivery to the tumor microenvironment). At the molecular level, membrane extrusion pumps may amplify suboptimal drug concentrations, leading to multidrug resistance (MDR). A classical example of success in addressing these issues is the consistent sequencing of multiple drugs in pediatric lymphoma to overcome MDR. Thus, incorporating eco-evolutionary principles in cancer therapy focuses on limiting proliferation, both with low-persisting cells or in advanced cases considered incurable. However, these eco-evolutionary aspects are less thoroughly explored in the literature [4].
1.2. Molecular Drivers of EMT
- 1.2.1 Key signaling pathways play critical roles in driving EMT processes. Among the most significant is the Transforming Growth Factor-beta (TGF-β) signaling pathway, which is a well-established inducer of EMT in various cancer types. TGF-β activates Smad proteins that enter the nucleus and regulate the transcription of EMT-related genes, subsequently leading to cytoskeletal reorganization and increased cellular motility [34,35].
Other influential pathways include Integrin, Hepatocyte Growth Factor (HGF), Epidermal Growth Factor (EGF), Tyrosine Kinase Receptors (TKR), Sonic Hedgehog - PTCH1(SHH), NOTCH, and the Wingless/Integrated (Wnt/β)-catenin pathway, which can all promote EMT by activating downstream transcription factors that compel the cells to acquire mesenchymal traits (Figure 2) [36,37].
- 1.2.2 Transcription factors, particularly those categorized as EMT-TFs (transcription factors), are instrumental in orchestrating the EMT process. The Snail family (SNAI1 and SNAI2), ZEB family (ZEB1 and ZEB2), and TWIST1 are prominent transcriptional regulators that inhibit the expression of E-cadherin and promote mesenchymal gene expression [38,39]. These EMT-TFs initiate the transcriptional changes required for EMT and are subject to extensive post-translational modifications, which can alter their activity and stability, thereby influencing the efficiency and timing of the transition [40]. For example, phosphorylation and ubiquitination can modulate the activity of these factors, enhancing or repressing their role in EMT [41]. Created with BioRender.com.
- 1.2.3 Role of MicroRNA: MicroRNA-200c (miR-200c) is increasingly recognized as a crucial miRNA molecule that plays a spectrum of roles in all aspects of EMT and cancer cell evolution. It has context-dependent actions where, in certain situations, it enhances apoptosis, tumor inhibition, reduces cellular inflammation, suppresses pyroptosis, etc, and in other situations, it has a pivotal complex role in promoting EMT. It can also play a role in modulating TME by M2 phenotypic macrophage polarization, density of TILs, expression of PDL1, CTLs exhaustion, and cancer cell exosomal load. MiR-200c can thus act as both a biomarker and a therapeutic target [10,42,43].
2. Mechanisms of MET
Mesenchymal-Epithelial Transition (MET) is the reverse process whereby mesenchymal cells regain epithelial characteristics. MET is vital for metastatic colonization, allowing cancer cells to proliferate at secondary sites [44]. Metastatic tumors maintain the primary tumor's histologic features by the MET process by regaining E-cadherin, β-catenin, and connexin, sometimes much more than the primary tumor. In this transition, cells re-establish cell-cell junctions and regain epithelial markers, providing an environment conducive to growth within new tissues [45]. The mesenchymal-to-epithelial transition (MET) represents the reversal of Epithelial–Mesenchymal Transition (EMT) and is facilitated by specific miRNAs. Agents facilitating MET can restore epithelial characteristics in cancer cells, potentially reducing their metastatic capabilities and enhancing treatment sensitivity. For example, compounds such as KLF4 have been shown to promote MET by downregulating EMT-inducing transcription factors, effectively reversing the mesenchymal phenotype of cancer cells [46]. Repressed E-cadherin expression, a hallmark of EMT, on restoration re-establishes cell-cell junctions and chromatin accessibility, impairing the migratory capabilities of cancer cells. Reversible surface E-cadherin and cellular gene expression dynamics are closely related to TGFβ treatment and its levels during the in vitro study [47].
In various cancers, silencing the Translationally Controlled Tumor Protein (TCTP) is critical for MET reversion. Normalizing the tumor microenvironment (TME) is a key strategy for cancer reversion, as it restores the functional relationships with normal cells. As recently reported, although poorly understood, senescent cells, considered as irreversibly phenotypic, can be reactivated to secrete promigratory cytokines or re-enter the cell cycle [48].
However, the exact molecular mechanism by which this happens is still elusive and less characterized compared to EMT. The re-repression by transcription factor GRHL2, activating E-cadherin and Claudin-4, and OVOL1/2 possibly drives towards MET. This drive towards MET may be complete or incomplete, giving rise to hybrid plasticity [49].
The dynamic interplay between EMT and MET illustrates the plasticity of cancer cells, enabling them to adapt to various microenvironments throughout cancer progression. Circulating tumor cells have epithelial and mesenchymal markers, indicating a hybrid population [50]. Thus, EMT, when the milieu is stressful (infiltration and metastasis), and MET, when the milieu is fertile (proliferation), are the foundational mechanisms of cancer cells for adaptation, progression, evolution, and survival. In vitro studies with the addition of bone morphogenetic protein 7 (BMP7), reduction of TGF-β, and hypoxia in MET are observed [51]. EMT-MET connections are separated in time and space, undergoing multiple pathophysiological and epigenetic changes, indicating whether MET is truly a reversal status of EMT or a transitional process [52]. Therefore, “Reversion” may be a more appropriate term (compared to “reversal”) since the cancer cells may not reach the truly original state by MET after EMT [53]. During these adaptations in the interim period, like EMT, MET re-differentiation can also increase the stemness, indicating that the MET status at the metastatic site may be more resistant than the primary, although of similar characteristics [50]. These lacunae in understanding EMT-MET plasticity need to be considered in the treatment strategy of facilitating MET.
3. EMT-MET Spectrum: Epithelial-Mesenchymal Plasticity (EMP) Characterization
The dynamic, pleiotropic spectrum of to-and-fro EMT-MET adaptations by the cancer cells is considered epithelial-mesenchymal plasticity (Figure 3). EMP bridges several milestones of cancer development, such as proliferation, basement degradation, EMT, stemness, invasion of surrounding tissue, extravasation into the vasculature, and dissemination through the circulatory system. This, in turn, leads to metastatic seeding, dormancy, MET, and proliferation at the favorable metastatic site, all leading to therapy resistance. The complexity of EMP, with only a minor number of cancer cells of the total population participating, is replete with several controversies due to a lack of irrefutable evidence. Additionally, several terms dot the literature analysis, like partial EMP (not committed to, thus exploiting both polar states), incomplete EMP (just short of ), both referring to not fully transitioning to either way, resulting in a hybrid state (coexpression of both) or metastable (relatively fixed somewhere in the EMP axis spectrum). A similar situation occurs in non-epithelial cancers, such as dedifferentiation and redifferentiation. The term transdifferentiation deals with the process of MET, with cells getting features of different lineages, e.g., upon specific targeting, breast cancer cells transformed into adipocytes [54].
These hybrid EMT states exhibit increasing stemness, plasticity, adaptability to varying microenvironmental conditions and therapeutic pressures, and enhanced metastatic potential. The interconnected roles of EMT-related transcription factors and cancer stem cell (CSC) markers (e.g., CD44, ALDH) hint at a complex regulatory network that underscores the plasticity and adaptability of carcinoma cells during tumor progression [44].
Identifying specific epithelial and mesenchymal markers is crucial in characterizing EMT and MET phenotypes. Epithelial markers such as E-cadherin, cytokeratins, and occludins are often diminished in advanced cancer stages, whereas mesenchymal markers, including vimentin, N-cadherin, and fibronectin, gain prominence [43,55]. Immunohistochemical profiling of these markers can provide insights into the EMT status of tumors, assisting in prognostic evaluations and therapeutic decisions [12,56]. Additionally, hybrid epithelial/mesenchymal (E/M) states, characterized by the co-expression of both epithelial and mesenchymal markers, have been linked to increased tumor aggressiveness and therapeutic resistance [57,58]. In summary, characterization of EMT and MET provides invaluable insights into cancer biology, particularly in the context of tumor invasion and metastasis (Figure 4).
Implications of EMP: 1. EMP helps the cancer cells to preserve original epithelial features, preserving both higher metastatic potential and stemness as demonstrated with single-cell sequencing, especially spatially localized to the leading edge of cancer [59]. 2. Therapy resistance can arise from reduced proapoptotic signals, immune checkpoint upregulation, improved DNA repair, enhanced suppression of immune cells, increased drug efflux, and cells switching to dormancy. The presence of EMP post-therapy compared to pre-therapy is a poor prognostic factor [54]. 3. The complex EMP regulatory network emphasizes the challenges of targeting EMT in therapeutic settings, as inhibiting one process may inadvertently affect the survival and proliferation of cancer stem cells [60,61]. To overcome such a possibility, epigenic regulators that target key steps, preventing the interconversion of cancer cells within EMP, would sensitize them to subsequent standard therapies or cause extreme EMT reversion to terminal differentiation/apoptosis [5]. 4. The greatest advantage of the MET reversion strategy is that it can improve the sensitivity before timed standard therapy CT/RT/Stereotactic Body Radiotherapy (SBRT), simultaneously targeting the progression in the proliferation stage [62]. 5. The milieu is as critical as cancer cell features in EMP and needs to be normalized for better therapy response. The stromal component plays a significant role in MET in addition to intrinsic factors [63]. 6. Therefore, the strategic therapy approach requires handling both the features in appropriate combination, timing, and sequence. Fundamentally, the drug that can sensitize should precede the next one that gets complemented in the scheduled protocol. Also, an exploratory analysis is required to identify the regimen with the least precipitation of partial EMP or DTPs.
Finally, the cancer is not just about EMT-MET, EMP plasticity, and resistance mechanisms. EMP is embedded in a complex network of cancer cell protective dynamic evolutions, each factor feeding on the others, forming a web of therapy escape resistant mechanisms (Figure 5).
4. Targeting EMT-MET Double-Bind – Combinations, Timing and Sequencing (CTS) Strategy
The dynamic interplay between EMT, which encourages metastases, and MET, leading to proliferation, represents a double-bind situation. Additionally, the spectrum of cells with a mix of epithelial and mesenchymal dichotomies, with their plasticity to adapt to the changing milieu and survive, represents a therapeutic opportunity, precision, and conundrum [64]. The deepest evasive action and most difficult to eliminate comes from DTPs waiting for the appropriate microenvironment condition to start reproliferating, much after the standard therapy is over and presumed cure is achieved [4]. Recent advances in incorporating epigenetic modifiers in the treatment protocol could disrupt this impasse. Therefore, the present authors emphasize that changing the paradigm of monitored therapy approach from MTD to Evolutionary therapy intervention during the course of therapy, as appropriate, becomes today's most advanced and fundamental reason for the cure of cancer. Appropriate non-cross-resistant combination therapies, in complementary timing and sequencing (CTS) strategy through personalized monitoring approaches, [65] exploiting tumor-specific transitional states (EMP) using epigenetic modifiers, state-of-the-art technologies, and tumor micro- and macro-environmental (systemic immunity) factors, is the foundation of this approach. Kwon et al (2021) also detail the right timing, combination, sequencing, and delivery centered around optimizing immunotherapy response. The authors highlight prior administration of IMMUNOTHERAPY when used with other conventional definitive combination therapies [66].
4.1. Inducing Mesenchymal-Epithelial Transition (MET) – Clinical Imperativeness and an Opportunity?
A central challenge in oncology is the remarkable plasticity of cancer cells, particularly their ability to switch between epithelial and mesenchymal states. The Epithelial-Mesenchymal Transition (EMT) is a cellular reprogramming that endows cancer cells with the aggressive traits responsible for the deadliest aspects of the disease. This transition facilitates invasion, dissemination, and acquiring cancer stem cell (CSC) properties. Critically, EMT is a primary driver of acquired resistance to a broad spectrum of therapies, including chemotherapy (CT), radiotherapy (RT), and targeted agents. Thus, EMT-MET plasticity accommodates the changing milieu, forming an excellent tool for cancer cell survival [67,68,69].
However, this plasticity could also be exploited as a key vulnerability. While EMT is essential for cancer cells to escape the primary tumor, the reverse process, the indispensable step of Mesenchymal-Epithelial Transition (MET), is required for these disseminated cells to colonize a distant organ and form a macroscopic metastasis [5]. This biological necessity provides a compelling therapeutic rationale: pharmacologically inducing MET can reprogram aggressive, mesenchymal-like cancer cells to roll back into a more differentiated, less motile, and therapeutically susceptible epithelial state. This strategy aims not to kill cancer cells directly during the EMT phase, but to strip them of their greater malignant capabilities by inducing MET, thereby inhibiting metastasis and inducing chemosensitization [70]. The pleotropic transitioning and interconversion unstable states open the door for sensitizing them to stress during progression or pushing them to the extreme EMP state (lethal EMT). The EMP cells can also be lead to terminal (trans) differentiation to adipocytes or apoptosis, as is demonstrated in breast and pancreatic cancers, respectively [5,71,72]. The possibility of enhancing metastatic growth with EMT reversing agents should be kept in mind, necessitating its usage within a clear therapeutic window [73], highlighting the importance of timingin the therapeutic protocol.
A growing body of preclinical and clinical evidence has identified several pharmacological agents capable of inducing MET, offering a new dimension to cancer therapy.
Literature analysis shows that there are broadly five therapeutic categories of epigenetic modifiers: 1. One that acts by preventing EMT and invasive ability [64]. 2. Those that sensitize the cancer cells to subsequent CT/RT/targeted therapy/immunotherapy, requiring proper timing and sequencing in any combination [74,75]. 3. Push the cancer cells to a lethal EMT/EMP state when the cancer cells enter the DTP state, senescence, or dormancy-reversion. 4. Keep the cancer cells frozen “locked up”, in “plastostatic state”, preventing them from progression or evolution to a more resistant phenotype [64,76]. 5. The fifth type is the least explored, reverting the cells to a normalized, relatively stable, or benign transdifferentiation state with the least potential to revert to a malignant state [4,76]. This emphasizes the mechanism-based classification in the present article, strategizing where EMs should be integrated in the CTS Strategy of MTD-EBT spectrum. This may be at some variance with the target strategy based on the EMT process [64].
4.2. Epigenetic Modifiers
a) Eribulin - A Clinical Proof-of-Concept: The most definitive clinical validation of therapeutic MET induction comes from studies of eribulin mesylate, an FDA-approved microtubule dynamics inhibitor used in metastatic breast cancer. Beyond its primary antimitotic function, eribulin possesses a unique, non-mitotic mechanism of action that reverses the EMT process.
Preclinical work in triple-negative breast cancer (TNBC) models demonstrated that eribulin treatment causes a profound phenotypic shift from a mesenchymal to an epithelial state. This is marked by the upregulation of epithelial markers like E-cadherin and the downregulation of mesenchymal markers such as vimentin and the key EMT-transcription factor ZEB1 [74]. This mechanism has been attributed to eribulin's ability to induce ZEB1-SWI/SNF-directed chromatin remodeling, effectively reprogramming the cell's epigenetic landscape [77].
Crucially, these preclinical findings have been strongly supported by studies in patient-derived orthotopic xenograft (PDOX) models, where eribulin treatment induced MET in tumors derived from TNBC patients [75]. By inducing MET, eribulin reduces the metastatic potential of cancer cells and resensitizes them to other therapeutic agents. This provides a strong rationale for using eribulin as prepriming therapy before the evolution of chemoresistance and metastatic progression [74,75].
b). Histone deacetylase inhibitors (HDACis) can reprogram gene expression to favor an epithelial state. The class I HDACi mocetinostat reverses ZEB1-associated drug resistance in pancreatic cancer models by restoring the expression of miR-203, a microRNA that targets and represses ZEB1 [69].
c). Direct EMT-TF Inhibitors: Preclinical small molecules have been designed to block the function of Snail transcription factors. GN25 prevents the interaction between Snail1 and p53, restoring p53's function and reducing tumor progression in K-Ras-mutated cancer models [78]. Other compounds, such as Co(III)-Ebox, prevent Snail1 from binding to the E-cadherin promoter, thereby blocking its primary repressive function [79]. In pancreatic adenocarcinoma, suppression of EMT with the nontoxic low-dose ML210 chemical compound in combination with gemcitabine prevented cell migration [80]. In an animal study, systemically tagged nanoparticle therapy after endosomal escape at low pH releases siDANCR, which is degraded by the RNA-Induced Silencing Complex (RISC) of cellular DANCR, preventing EMT and phosphorylation [81].
d). Repurposed Agents:
- Metformin improves insulin sensitivity, modulates metabolism, and inhibits EMT and reduces the metastatic potential of cervical cancer cells via inhibition of mTOR and TGF-β signaling [82].
-
Salinomycin reverses doxorubicin-induced EMT by downregulating mesenchymal markers and upregulating E-cadherin, and restores chemosensitivity [83].Salinomycin, an AMPK inhibitor, can cause mitochondrial dysfunction and induce autophagy to cause metabolic reprogramming and overcome CSCs' RT/CT resistance [84].
e). Targeted Therapies: Inhibitors of oncogenic pathways that drive EMT can also induce MET. For example, c-MET inhibitors such as crizotinib resensitize small-cell lung cancer cells to CT by blocking Met-dependent EMT [85].
f). miRNA-Based Therapeutics: The miR-200 family is a key regulator of the epithelial phenotype, functioning in a double-negative feedback loop with ZEB1/ZEB2. Restoring miR-200 function using therapeutic mimics can induce MET, upregulate E-cadherin, and reduce motility [86].
g). Multitargeted-Epigenetic-Therapy (MTET): This approach did not use EMT-inducing cytotoxic and cytostatic drugs. The strategy was to reverse the partial EMT to full MET status by targeting the SNAIL, SLUG, and Wnt pathways epigenetic modifiers. The mainstay was using a combination of polyphenols to modify epigenomic signatures. The other drugs were GnRH agonists to target HIF-1α and VEGF by reducing hypoxia, inhibit the Ras-mediated MAPK; growth hormone blocker somatostatin analogs to inhibit IGF-1 and TGFβ; quinolone-based drugs to inhibit Heat Shock Protein-90 (HSP-90) chaperones and PI3k/MAPK pathways; off-label drug Riluzole inhibiting NMDA coupling with PI3k; several small molecule TKI inhibitors; reversing EGFR resistance by C-Met inhibitors; MEK inhibitor, Selumetenib to activate Wnt pathways. The authors note that metformin should be used cautiously for fear of triggering PI3k, and celecoxib may induce EMT. Results as documented with tumor response markers, plasma VEGF, IGF-1, and TGF-beta 1, imaging, & CTC were promising without a decrease in quality of life [65].
4.3. Two Concepts and One Strategy
The first conventional concept and foundation of modern anticancer therapy is to eliminate the maximum number of cancer cells in the shortest time, which is considered to have the maximum chance for a potential cure. The cancer therapies are administered at the maximum tolerated dose (MTD), foundational in phase I clinical trials, and translated into other phases. The two premises here are, one to eliminate the cancer cells before the resistance develops, and second, that resistant mutations are likely from a small rather than a large population of cancer cells. This time-tested strategy has been proven to cure cancer in specific populations of all cancers [4].
According to the second concept, the conventional MTD approach is the antithesis of Darwinian evolutionary dynamics. According to this proposal, the resistance trait is already encoded as partial resistance at low frequency in a subpopulation, albeit small. When it repopulates after MTD, it progresses into a whole resistance population. In colorectal cancer, a preexisting inherent resistant phenotype has been identified for checkpoint inhibitors, osimertinib, and BET inhibitors. In the second mechanism, cancer cells with features of phenotypic plasticity, having a spectrum of stem cells, dormant cells, or poly-aneuploid features, survive the MTD and transition into a more resistant state. Here, the evolutionary pressure ensuing MTD leads to acquired resistance where natural directional selection exerts development of increasingly resistant phenotypes that were not preexisting within the tumor cell population. In the evolutionary cascade, gene duplication, loss, and gain function are used by accessing the entire human genome epigenetically, even alien to the tissue of origin. This results in a small cancer cell population, scaling ecological recovery, and developing environmentally mediated drug resistance. The release from competition by the sensitive cancer cells, even normal cell pockets, and the cancer cell-modulated extracellular matrix facilitates the resistant phenotypes of cancer cell proliferation. Treatment based on evolutionary principles – evolutionary-based therapy (EBT) - is in the exploratory phase, the strategy of which is to remain ahead of the evolution. This approach involves using metronomic treatment to keep the proliferation in check and making dose adjustments to standard therapies while monitoring epigenetic markers as a dynamic treatment process. In advanced cancers, this will keep the patient with a good quality of life without toxicities, and no attempt is made to eradicate cancer cells, which the multidisciplinary group is evaluating [4]. The following perspectives deal with unifying these two concepts, based on the understanding that they are not mutually exclusive.
4.4. A Unified Perspective: Combinations, Timing, and Sequencing (CTS) Strategy
A cure is feasible with the MTD approach in the patient group where resistant traits are not already established. In other cases, combining both principles is necessary when resistant phenotypic evolution is possible. The current article proposes a CTS strategy to achieve that goal, supported by an understanding of cancer cell EMT-MET dynamics. MTD attempts to eliminate resistance before it develops, versus CTS, where sequencing takes out the resistant cell population one after another, with epigenetic reversion forming the backbone during treatment. (Figure 6) Memory CTLs and the immunity cycle. Immune editing theory. Three Foundations: Using maximum cell kill with minimal or acceptable toxicity, epigenetic reversions, and restoring immune editing. HIF-1α and TGFβ are the two primary theoretical targets, yet the limitations are that they also play a key role in normal homeostasis. Surgical excision of residual lesions or SBRT may help eliminate the core DTPs and stemness where necessary.
The three-pronged strategy of handling EMP, preventing EMT, focusing on EMP alterations, and encouraging MET reversion warrants caution since the entire gamut of the process can interfere with normal physiological homeostasis. One strategy would be to forestall the pivotal downstream oncogenic drivers by integrating targeted therapy with trimodal CT, RT, or immunotherapy [87].
4.5. Targeting Primary Mechanisms of EMP Resistance Network
a). Optimum Delivery of the Drug to the Intended Target: Due to deficient and aberrant vasculature, increased interstitial pressure, and the mechanotransduction barriers of the TME, effective drug delivery into the TME and the intracellular space is hindered. This leads to suboptimal response and subsequently encourages the evolution of resistant pathways. E.g., Taxanes, which bind to β-tubulin, and anthracyclines, which interact with topo-II, require an effective concentration at target sites (without ABC proteins reducing intracellular drug levels by efflux) [88].
b). Heterogeneity: The essence of EMP is its heterogeneity, within several other heterogeneous survival features of cancer (Figure 5) that need to be addressed with a tailored comprehensive approach, along with disruption-normalization of tumor-supporting TME. This heterogeneity of cancer requires using a non-cross-resistant combination in safe and optimal doses [88].
c). Plasticity: Three critical developments with EMP that need to be focused on in combination therapy are the inherent stemness, presence of reversible slow proliferating therapy evading drug-tolerant persisters (DTPs) progressing on Darwinian evolutionary mechanisms, and the transdifferentiation of malignant phenotypes. In prostate cancer, hormone-independent neuroendocrine phenotype development can be prevented by anti-IL-6 monoclonal antibody siltuximab and JAK kinase inhibitor ruxolitinib, operating upstream of STAT, laying the strategic foundation for intercepting EMP at junctional points. Transformed SCLCs in NSCLC respond better to taxanes than de novo ones and are resistant to immunotherapy [4,87]. DTPs are vulnerable to bromodomain and extraterminal domain (BET) protein inhibitors, especially on re-entering the cell cycle, inducing lethal apoptotic levels of ROS [89].
d). DTPs require special mention since they are the last barriers to therapy's success, being the most intransigent for elimination. To eliminate or subdue DTPs, the CTS strategy requires a different approach during the consolidation of the therapy phase, with a switch from the MTD schedule to specific epigenetic targeting.
e). Drug Efflux: Loss of stemness decreases the expression of ATP-binding cassette (ABC) proteins, reducing drug efflux and improving CT's efficacy. Therefore, the appropriate timing of anti-EMP combination therapy is critical to avoid the contrary result or inhibition of the normal healing process by EMT [90].
f). Stemness: EMP is one of the several reasons for stemness and requires calibrated sensitization and sequencing of the drug combination therapy schedule. In the planned sequencing, the targeted drug and epigenetic modifiers that tend to sensitize, irrespective of their independent action on cancer cells, deserve to precede the primary therapy or vice versa. Mesenchymal cells are sensitive to the AXL inhibitor SGI-7079. TGF-β inhibitor SB-431542 or receptor antibodies increase sensitivity to carboplatin. EMT signature showed better control with erlotinib in NSLC but not in other therapies. EMP showed resistance to EGFR and PI3K/AKT inhibitors irrespective of mutational status, but higher sensitivity was observed for CT [91].
4.6. Combinations, Timing, and Sequencing Strategy
In brief, selecting non-cross-resistant combination therapies, timing them to prevent resistance mechanisms from developing, and using innovative sequencing to structure previous treatments to sensitize cancer cells for the subsequent treatment—minimizing DTPs to manifest —becomes the key approach in CTS cancer management strategy.
Based on the above literature review, the proposed strategy has V phases that must be implemented systematically. The first is normalization of the vasculature, which will improve the delivery and reduce hypoxia to sensitize the cancer cells to the subsequent administration of MTD drugs in phase II priming therapy. (Table 1)
CTS Phase Ia – Primarily Vascular Normalization: Hypoxia is one of the foundational stresses for EMT initiation. Consequently, improving oxygen diffusion to sensitize the cells of EMT-MET plasticity would be the first step in initiating anticancer therapy (Figure 6) [92,93]. Tumor vascularization and oxygenation typically improve from day 1 of AAG administration. However, by Day 28, continued AAG administration leads to excessive pruning/regression of blood vessels due to anti-VEGF overaction, and the features of the aberrant vasculature, anoxia, and reproliferation of cancer cells return. This interval between the onset of normalization (around Day 2) and the excessive pruning of vessels with continued AAGs (around Day 28) is defined as a normalization time window. Firstly, this window opens up the vasculature for enhanced delivery of cytotoxic drugs & CTLs into the TME. Additionally, during this normalization window, the effectiveness of combinatorial radiation therapy and CT/immunotherapy is enhanced due to improved cancer cell kill effect, fixing the cytotoxic DNA damage. On stopping AAGs on Day 28, normalization of vascularization resumes because of the withdrawal effects as observed on imaging during drug holidays [35,43]. This second normalization window lasts about 3 to 4 weeks, and restarting AAGs at that time, the 3rd normalization window opens up. Further work is required to exploit this cyclical process indefinitely. Therefore, this approach becomes the first step of the prepriming phase for maximizing MTD response in Phase II [92]. Vascular normalization has multiple effects of reducing tumor invasion and metastasis, sensitizing cancer cells for subsequent CT, radiation therapy, and immunotherapy [94]; it acts as a stepping stone for tumor inhibitory TME transformation; immunotherapy further improves normalization, just like AAGs – vascular enhancement [95,96]. Certain local therapies and RT/CT accentuate VN, enhancing antigenicity and adjuvanticity [97,98].
CTS Phase 1b – Primarily to Cancer Cell Sensitization for Phase II: Epigenetic Modulation, reducing glycolysis in tumor cells, simultaneously making TILs nutrition-rich to improve the sensitivity of cancer cells, priming them for CT/RT that follow it. In EMP, more than 30 spectra of targetable genes involved with constant upregulation or downregulation may be a disadvantage in identifying clinically useful targets. Despite the significant possibilities, therapy should balance the context-dependent tumor suppressor and oncogene actions of miR-200c. In such situations, a comprehensive stepwise approach may be required. a) Prepriming combination therapy of antiangiogenics and miR-200c gene targeting, with other b) priming therapies, like reducing glycolysis of tumor cells, enhancing sensitivity to cisplatin-based CT. Microtubule-targeting CT drugs through class III β-tubulin (TUBB3) prevent resistance to 5-fluorouracil in ovarian cancer, improve the effects of PDL1 inhibitors immunotherapy, and targeted therapy like trastuzumab. Nanoparticles enhance the delivery and the concentration of miR-200c targeting drugs [42].
The most important study to handle the EMT-MET Double-Bind may be the pharmacological chromatin remodeling epigenetic mechanism of action of eribulin. Eribulin induces ZEB1-SWI/SNF-directed reversion of EMT to MET in triple-negative breast cancer ( especially with epithelial-mesenchymal heterogeneity), thus reducing the metastatic potential. However, this raises the theoretical possibility of a spurt in the proliferation of already seeded cancer cells in distant organs, which have now acquired and are armed with epithelial characteristics [60,61]. This possibility can be prevented by sequencing erubilin, which sensitizes MET-transformed cancer cells to the subsequent CT, preempting metastatic progression and chemoresistance [70].
CTS Phase 1c: The objective is to induce MET before the MTD schedule to bring the cells into an epithelial characteristics sensitive phase and have maximum cell kill without increased toxicity [87].
CTS Phase II – Primarily CT/TT/RT: This would be the standard CT/targeted therapy and RT/SBRT as priming therapies, preparing for the next phase, i.e., immunological targeting. The approach maximizes the cancer cell kill in MTD dose schedules, generates neoantigens to accelerate the immunity cycle, and long-term memory cells. Depending on the indication, SBRT alone or in a boost schedule improves the oxygenation by the 8th fraction in a schedule [99], in addition to significant cancer cell kill, mitigation of immune suppressive TME, and neoantigen generation. Cancer cells are made ready for immunotherapy by purging the immune suppressive elements by “unmasking the TME” and sensitizing the cancer cells further for maximum impact in this step (Figure 6). Other innovative local and intralesional therapies can be tested during this phase to improve antigenicity and adjuvanticity of the trimodal combination therapy of RT-CT-IMMUNOTHERAPY [98]. Radiation and CT over the decades have evolved into precise, personalized, having immunogenic targets for effective cancer cell kill with minimal toxicity [100]. The drugs facilitating macrophage-mediated (professional) phagocytosis, decreasing interstitial pressure by cytokine antagonists. The phagocytosis checkpoints immunotherapy, overcoming “don’t eat me” signals from cancer cells, complements T-cell responses, and is the new era in immunotherapy [101,102,104]. Even with decreased ISP and improved lymphatic channel patency, function may not recover. This recovery is essential for DCs to reach lymph nodes, and TGFβ blockers are known to restore lymphatic function, thus reestablishing the immunity cycle [92]. Additionally, CT/RT/SBRT during Phase II prepares the ground for subsequent immunotherapy/ex vivo vaccine/cell therapies/nanoparticle therapies in Phase III, acting as an in situ neoantigen generator [104,105,106].
Epigenetic Therapy (ET) strategic EMT inhibition also plays a role in this phase. Standard anticancer treatments, such as CT, targeted therapy, and radiation, can encourage type III EMT, resulting in immune evasion and anoikis resistance by Snail and Slug mediated inhibition of p53 induced apoptosis [107,108,109]. =After CT/RT, specifically DTPs are believed to constitute a reservoir, eventually leading to irreversible genetic mutations [110]=. RT is generally indicated in 50 to 70% of cancer patients and is known to induce EMT through TGF-β, Notch, and ERK pathways [111]. RT induced EMT can be overcome with timed epigenetic modifiers. Combining Icotinib intensity-modulated RT (Trial NCT01534585) in Nasopharyngeal carcinomas inhibits the EMT [112]. Vactosertib, a TGF-β/ALK5 inhibitor, reduces ROS oxidative stress, fibrosis, and stemness, consequently EMT, in addition to an independent antitumor effect [113]. DNA methyltransferase (DNMT) inhibitor, decitabine, sensitized cancer cells for RT, and resensitized ovarian cancer cells for cisplatin/carboplatin. Bromodomain protein 4 (BRD4) inhibitors improve sensitivity to chemo-RT and prevent the upregulation of PD-L1 by RT in NSLC [114]. The synergistic effect of the multimodal approach with advances in nanomaterial in situ vaccination technology awaits proper integration [105].
CTS Phase III - Primarily Immunotherapy: Strategically, at the beginning of this stage, cancer TME should be unmasked or stripped of its major protective shields and made ready for immune therapy manipulation. CT and RT, including SBRT, help improve immunotherapies' effect, although optimal dose, timing, and sequencing require further investigation [115,116]. Lesions no longer responding to immunotherapy combinations may have enhanced antitumor immunity, in situ vaccine effect, with re-oxygenation [9]. In the KEYNOTE-01 trial in non-small-cell lung cancer, even at a median of 9.5 months after RT, the IMMUNOTHERAPY group with a previous history of RT had longer progression-free and overall survival [117]. Incorporation of the Cyclical SBRT boost(s) 6 to 8 Gy/fraction in pulsed or PULSAR schedule will restore immunity cycle, memory cell bank creation/expansion as a deterrent for long-term recurrences, and enhance the immunotherapy effects, provided they precede (and not follow) each cycle of immunotherapy [117,118,119]. Professional Phagocytic agents will continue to improve TME [120]. ET approaches at his phase further accentuate immunotherapy effects [121].
CTS Phase IV: This is the consolidation therapy phase. Maintaining the soft ECM, encouraging immunological cross-talk, and unfavorable TME for recurrence is part of mopping up surviving cancer cells [30,122]. In addition to present-day standard chemo-immunotherapies, epigenetic modifiers, discussed above, during the maintenance therapy can play a significant role in mopping up the remaining cells/DCPs [89], or epigenetically inducing reversion/dedifferentiation or transdifferentiation. The essence of Phase IV is the elimination of DTPs, which are extreme EMTs persisting after TKIs or T-cell-centered immunotherapy within the population of MRD and drug withdrawal-associated dormancy. DTPs are expertly adaptive and intensely heterogeneous, with an intricate feedback ECM loop requiring an equally adaptive dosing schedule, epigenetic lethal targeting approaches, and immune-mediated eradication, thus exploiting their vulnerabilities [5,53,54]. Optimization and secondary targeting of TGF-β and ferroptosis is essential to reduce the long-term neurological, fibrosis in liver/ lung fibrosis, and activation of autoimmune disorders [64], which becomes critical since the EPs operate on the borderline of cancer-normal cell proliferation.
CTS Phase V - Presently Exploratory – Evolutionary EMP Intervention Strategy: Primarily, this is about abating inflammation and restoring immune-editing. This phase also focuses on DTPs, Dormancy, and Senescent cells that remain once the scheduled therapy course is over or stopped because of toxicity or progression. The objective is to keep them “locked up” in the nonproliferation phase/dormancy of EMP, targeting re-entering the cell cycle and encouraging reversion towards normalcy. Mechanistically, the strategy is to mitigate/prevent the ROS stress. Methodology involves specific epigenetic modifiers for lethal ROS-induced apoptosis [89], reducing inflammation, ECM suppleness, and encouraging immune cross-talk [30]. Epigenetic modifications in reducing inflammation and recurrence can be explored using senolytics/senostatics, although there could be doubt about their clinical effectiveness [12][124] and lifestyle approaches [125,126].
Other approaches are evolutionary-informed therapy (EIT) or the MTET strategy. The objective of adaptive therapy (dormancy therapy) is to prevent adaptive resistance, e.g., inhibiting DYRK1/2 kinases, forcing cancer cells into quiescence, and re-exposing them to CT on cell cycle re-entry, with the strategy of preventing the establishment of resistant clones These approaches are being explored in patients who have gone into remission or have exhausted the radical therapy options with appropriate immunological monitoring, with the primary objective of preventing the evolution and progression of resistance clones [4,48,65].
5. Limitations
These perspectives fall short of having a definitive outcome of a cure for cancer. Essentially, the reversion hypothesis rightly suggests that reversing certain epigenetic changes and pathways of transformed malignant cells may never restore them to their original state before carcinogenesis began. EMP finally leaves residual malignant features in one of the forms, such as DTPs cells, Dormancy, Senescent cells, or normalized (almost normal) cells, which can always have the potential to get reactivated to proliferate and induce recurrence with an evolutionarily more resistant population. In-depth understanding of EMP epigenetic interactions in the overall features of varied types of heterogeneity in cancer and targeting with epigenetic modulators would be the new frontier in oncology. These EMs can keep such cells locked up in a non-proliferation status, controlled under low ROS stress, or induce lethal intracellular ROS levels (e.g, BET inhibitors) - pushing the cells to apoptosis. Since these low-level proliferating or hibernating cells function at the margins of normal homeostasis, and if there is a need for long-term medications like any other chronic lifestyle disease, the drugs should have low toxicity levels. However, the CTS strategy can potentially take anticancer management, following the MTD schedule, to the deepest part, targeting DTPs, enforcing reversion/differentiation, and activating in situ vaccine effects. Also, one fundamental aspect of the CTS strategy in targeting EMP is effectively using the available therapeutic window [73]. Future developments can be incorporated, depending on the mechanism of action, within the structure of the CTS concept.
6. Summary
The interplay between epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET) is critical in cancer biology, influencing tumor progression, stemness, metastasis, and therapeutic responses. EMT equips cancer cells with increased invasion and motility, allowing them to detach from primary tumors and establish secondary growth sites. This transition contributes to the aggressive nature of tumors and is related to the acquisition of cancer stem cell-like properties, which often correlate with treatment resistance and poor clinical outcomes [127,128]. Conversely, MET is responsible for establishing the metastatic growth, suggesting that a nuanced understanding of these transitions is necessary to develop effective cancer therapies [129].
The dynamic nature of these processes implies that cancer cells can transition between epithelial and mesenchymal states in response to environmental stimuli, including cytokines and growth factors [128]. Such plasticity suggests that targeting pathways involved in these transitions could provide a dual approach to inhibit tumor invasion while promoting MET, potentially enhancing patient responses to existing therapies [130]. Changing contradictory dynamics of EMP, angiogenesis, and TME immune profile encapsulate a critical aspect of tumor biology. The timing of therapies within the available therapeutic window is vital. Vascular normalization, vascular promotion, EMP, and cancer cell sensitization just before the established accelerated repopulation is a challenge in a clinical setup, which mandates a careful strategy. One can extend the limited therapeutic window by properly selecting “drugs” and planned sequencing [73,116,131]. For example, cyclical AAGs can extend the normalization window [92]; Eribulin mesylate, which increases tumor vasculature (vascular promotion) and perfusion in animal models, also induces MET and sensitizes cancer cells if chemotherapy is administered subsequently [74,75,131].
7. Future Research Directions to Unravel Complexities and Improve Therapeutic Strategies
Future research must focus on elucidating the intricate regulatory networks that manage the dynamics of EMT and MET within the tumor microenvironment. Given the pivotal role of microRNAs, long non-coding RNAs, and various signaling pathways in these transitions [132], a comprehensive analysis of these components may reveal potential biomarkers for patient stratification and treatment monitoring. Enhanced understanding of the temporal changes in EMT and MET states can help identify new pharmacological targets, including agents that promote MET or inhibit EMT effectively [133].
Research should also leverage single-cell sequencing technologies and advanced imaging techniques to map the EMT-MET continuum across different cancer types. This will provide insights into the heterogeneity of tumor cell populations and the influence of varying microenvironments on these transitional states [134,135]. Furthermore, investigating the potential of combinatorial therapies that aim to inhibit EMT while facilitating MET could represent a transformative approach in managing aggressive cancers, ultimately enhancing the efficacy of existing treatments and improving patient prognoses [136,137]. An earlier version of this article is in preprint [138].
In conclusion, a deeper understanding of the interplay between EMT and MET, combined with advancements in research methodologies, holds the promise of unlocking new therapeutic opportunities that could significantly alter the current landscape of cancer treatment. Giordanengo, L et al (2025) highlight the detailed, intricate relationship between EMT and cancer progression spectrum, offering an exciting paradigm in therapeutic potential. Lack of routine adoption of EMT pathological characterization, integration into clinical practice, and absence of standardized protocols despite a vast volume of experimental research results and a promising list of drugs make its integration paramount [139]. A structured approach of a combination, timing, and sequencing (CTS) strategy template enunciated in the present article, incorporating the epigenetic modifiers (“currently the most clinically advanced strategy”) [64], is likely to be a major disruptor.
Not all the therapy approaches discussed in the present review are necessary for a given patient. However, the present article provides the perspectives of any selected treatment, as per the indications for a particular patient, vis-à-vis others in the combinations for maximum probability of control with the least toxicity. Therefore, the present article highlights how to improve the efficiency of combination therapies in relation to each other with appropriate timing and sequencing. The critical role of drugs in acting at multiple levels of the pathways, e.g., Eribulin mesylate, a MET inducer, chemosenitizer, and vascular promoter, or multispecific TGFβ blockers (eg, Vactosertib), has great potential [74,75,113,131]. Clinical trials are limited about EMT for its specific inhibition [139]. The proposed structured approach in the present article of individually effective treatments in combinations can be a guide for future systematic research.
References
- Kim-Fuchs C, Le C, Pimentel M, Shackleford D, Ferrari D, Angst E, et al. Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav Immun. 2014;40:40-47. [CrossRef]
- Fong M, Zhou W, Liu L, Alontaga A, Chandra M, Ashby J, et al. Breast-cancer-secreted mir-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17(2):183-194. [CrossRef]
- Al-bataineh M, Alzamora R, Ohmi K, Ho P, Marciszyn A, Gong F, et al. Aurora kinase a activates the vacuolar h+-atpase (v-atpase) in kidney carcinoma cells. Am J Physiol Renal Physiol. 2016;310(11):F1216-F1228. [CrossRef]
- Gatenby RA, Brown JS. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb Perspect Med. 2020 Nov 2;10(11):a040972. [CrossRef]
- Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019 May 6;49(3):361-374. [CrossRef]
- Liu X, Zhang Y, Wu X, Xu F, Ma H, Wu M, et al. Targeting ferroptosis pathway to combat therapy resistance and metastasis of cancer. Front Pharmacol. 2022;13:909821. [CrossRef]
- Ferguson R, Novosyadlyy R, Fierz Y, Alikhani N, Sun H, Yakar S, et al. Hyperinsulinemia enhances c-myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res. 2012;14(1):R14. [CrossRef]
- Eslami-S Z, Cortés-Hernández L, Glogovitis I, Antunes-Ferreira M, D'Ambrosi S, Kurma K, et al. In vitro cross-talk between metastasis-competent circulating tumor cells and platelets in colon cancer: a malicious association during the harsh journey in the blood. Front Cell Dev Biol. 2023;11:1209846. [CrossRef]
- Kogure A, Yoshioka Y, Ochiya T. Extracellular vesicles in cancer metastasis: potential as therapeutic targets and materials. Int J Mol Sci. 2020;21(12):4463. [CrossRef]
- Gregory P, Bracken C, Smith E, Bert A, Wright J, Roslan S, et al. An autocrine tgf-β/zeb/mir-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22(10):1686-1698. [CrossRef]
- Huang Y, Du J, Mi Y, Li T, Gong Y, Ouyang H, et al. Long non-coding rnas contribute to the inhibition of proliferation and emt by pterostilbene in human breast cancer. Front Oncol. 2018;8:629. [CrossRef]
- Wei L, Li K, Pang X, Guo B, Min S, Huang Y, et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J Exp Clin Cancer Res. 2016;35(1):148. [CrossRef]
- Feldker N, Ferrazzi F, Schuhwerk H, Widholz S, Guenther K, Frisch I, et al. Genome-wide cooperation of emt transcription factor zeb1 with yap and ap-1 in breast cancer. EMBO J. 2020;39(17):e103209. [CrossRef]
- G. Z. Qiu, M. Z. Jin, J. X. Dai, W. Sun, J. H. Feng, W. L. Jin, Tumorin the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies G. Z. Qiu, M. Z. Jin, J. X. Dai, W. Sun, J. H. Feng, W. L. Jin, Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies, Trends Pharmacol Sci 38(8)(2017) 669–686. [CrossRef]
- Montemagno C and Pagès G (2020) Resistance to Antiangiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 8:584. [CrossRef]
- Kathrine S. R, Thomas H L Y, Michail S. Chemoradiotherapy in Cancer Treatment: Rationale and Clinical Applications. Anticancer Research Jan 2021, 41 (1) 1-7. [CrossRef]
- Clarke J M, Hurwitz H I. Understanding and targeting resistance to antiangiogenic therapies. J Gastrointest Oncol 2013;4(3):253-263. [CrossRef]
- Ye X, Weinberg R. Epithelial--mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675-686. [CrossRef]
- Suarez-Carmona M, Bourcy M, Lesage J, Leroi N, Syne L, Blacher S, et al. Soluble factors regulated by epithelial--mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J Pathol. 2015;236(4):491-504. [CrossRef]
- Haibe Y, Kreidieh M, El Hajj H et al. Resistance Mechanisms to Antiangiogenic Therapies in Cancer. Front. Oncol. (2020) 10:221. [CrossRef]
- Ayoub NM, Jaradat SK, Al-Shami KM, Alkhalifa AE. Targeting Angiogenesis in Breast Cancer: Current Evidence and Future Perspectives of Novel Antiangiogenic Approaches. Front Pharmacol. 2022 Feb 25;13:838133. [CrossRef]
- Montemagno C and Pagès G (2020) Resistance to Antiangiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 8:584. 2020. [CrossRef]
- Wegiel B, Vuerich M, Daneshmandi Sand Seth P (2018) Metabolic Switch in the Tumor Microenvironment Determi[nes Immune Responses to Anticancer Therapy. Front. Oncol. (2018) 8:284. [CrossRef]
- Weihua Wu, Shimin Zhao, Metabolic changes in Cancer: beyond the Warburg effect, Acta Biochimica et Biophysica Sinica, Volume 45, Issue 1, January 2013, Pages 18–26. [CrossRef]
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016 Mar;41(3):211-218. Erratum in: Trends Biochem Sci. 2016 Mar;41(3):287. Erratum in: Trends Biochem Sci. 2016 Mar;41(3):287. https://doi.org/10.1016/j.tibs.2016.01.004. [CrossRef]
- Liaghat M, Ferdousmakan S, Mortazavi SH, Yahyazadeh S, Irani A, Banihashemi S, Seyedi Asl FS, Akbari A, Farzam F, Aziziyan F, Bakhtiyari M, Arghavani MJ, Zalpoor H, Nabi-Afjadi M. The impact of epithelial-mesenchymal transition (EMT) induced by metabolic processes and intracellular signaling pathways on chemo-resistance, metastasis, and recurrence in solid tumors. Cell Commun Signal. 2024 Dec 2;22(1):575. [CrossRef]
- Saitoh M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-β signaling during tumor progression. Cancer Sci. 2015 May;106(5):481-8. [CrossRef]
- Taniguchi, K., Karin, M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18, 309–324 (2018). [CrossRef]
- Horta CA, Doan K, Yang J. Mechanotransduction pathways in regulating epithelial-mesenchymal plasticity. Curr Opin Cell Biol. 2023 Dec;85:102245. [CrossRef]
- Zhao Y, Yu X, Li J. Manipulation of immune‒vascular cross-talk: new strategies towards cancer treatment. Acta Pharm Sin B. 2020 Nov;10(11):2018-2036. [CrossRef]
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004 Aug;21(2):137-48. PMID: 15308095. [CrossRef]
- Sistigu A, Di Modugno F, Manic G, Nisticò P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. 2017 Aug;36:67-77. [CrossRef]
- Romeo E, Caserta CA, Rumio C, Marcucci F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells. 2019 May 15;8(5):460. [CrossRef]
- Wang J, Zhu Y, Tan J, Meng X, Xie H, Wang R. Lysyl oxidase promotes epithelial-to-mesenchymal transition during paraquat-induced pulmonary fibrosis. Mol Biosyst. 2016;12(2):499-507. [CrossRef]
- Miyake Y, Nagaoka Y, Okamura K, Takeishi Y, Tamaoki S, Hatta M. SNAI2 is induced by transforming growth factor-β1, but is not essential for epithelial-mesenchymal transition in human keratinocyte HaCaT cells. Exp Ther Med. 2021 Oct;22(4):1124. [CrossRef]
- Deshmukh A, Vasaikar S, Tomczak K, Tripathi S, Hollander P, Arslan E, et al. Identification of emt signaling cross-talk and gene regulatory networks by single-cell rna sequencing. Proc Natl Acad Sci U S A. 2021;118(19):e2102050118. [CrossRef]
- Frey P, Devisme A, Schrempp M, Andrieux G, Boerries M, Hecht A. Canonical bmp signaling executes epithelial-mesenchymal transition downstream of snail1. Cancers (Basel). 2020;12(4):1019. [CrossRef]
- Yi JH, Zhang ZC, Zhang MB, He X, Lin HR, Huang HW, Dai HB, Huang YW. Role of epithelial-to-mesenchymal transition in the pulmonary fibrosis induced by paraquat in rats. World J Emerg Med. 2021;12(3):214-220. [CrossRef]
- Wang JQ, Yan FQ, Wang LH, Yin WJ, Chang TY, Liu JP, Wu KJ. Identification of new hypoxia-regulated epithelial-mesenchymal transition marker genes labeled by H3K4 acetylation. Genes Chromosomes Cancer. 2020 Feb;59(2):73-83. [CrossRef]
- Grasset EM, Dunworth M, Sharma G, Loth M, Tandurella J, Cimino-Mathews A, Gentz M, Bracht S, Haynes M, Fertig EJ, Ewald AJ. Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Sci Transl Med. 2022 Aug 3;14(656):eabn7571.
- Boareto M, Jolly M, Goldman A, Pietilä M, Mani S, Sengupta S, et al. Notch-jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J R Soc Interface. 2016;13(118):20151106. [CrossRef]
- Guo, H.; Zhang, N.; Huang, T.; Shen, N. MicroRNA-200c in Cancer Generation, Invasion, and Metastasis. Int. J. Mol. Sci. 2025, 26, 710. [CrossRef]
- Zhang L, Hung GC, Meng S, Evans R, Xu J. LncRNA MALAT1 Regulates Hyperglycemia Induced EMT in Keratinocyte via miR-205. Noncoding RNA. 2023 Feb 11;9(1):14. [CrossRef]
- Chang H, Liu Y, Xue M, Liu H, Du S, Zhang L, et al. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016;44(6):2514-2527. [CrossRef]
- Tong J, Shen Y, Zhang Z, Hu Y, Zhang X, Han L. Apigenin inhibits epithelial-mesenchymal transition of human colon cancer cells through nf-κb/snail signaling pathway. Biosci Rep. 2019;39(5):BSR20190452. [CrossRef]
- Subbalakshmi AR, Sahoo S, McMullen I, Saxena AN, Venugopal SK, Somarelli JA, Jolly MK. KLF4 Induces Mesenchymal-Epithelial Transition (MET) by Suppressing Multiple EMT-Inducing Transcription Factors. Cancers (Basel). 2021 Oct 13;13(20):5135. [CrossRef]
- Johnson KS, Hussein S, Chakraborty P, Muruganantham A, Mikhail S, Gonzalez G, Song S, Jolly MK, Toneff MJ, Benton ML, Lin YC, Taube JH. CTCF Expression and Dynamic Motif Accessibility Modulates Epithelial-Mesenchymal Gene Expression. Cancers (Basel). 2022 Jan 1;14(1):209. [CrossRef]
- Škarková A, Bizzarri M, Janoštiak R, Mašek J, Rosel D, Brábek J. Educate, not kill: treating cancer without triggering its defenses. Trends Mol Med. 2024 Jul;30(7):673-685. [CrossRef]
- Ribatti D, Tamma R, Annese T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl Oncol. 2020 Jun;13(6):100773. [CrossRef]
- Liao TT, Yang MH. Revisiting epithelial-mesenchymal transition https://doi.org/10.1002/1878-0261.12096in cancer metastasis: the connection between epithelial plasticity and stemness. Mol Oncol. 2017 Jul;11(7):792-804. [CrossRef]
- Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Baba H, Mori M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010 Feb;101(2):293-9. [CrossRef]
- Moustakas A, de Herreros AG. Epithelial-mesenchymal transition in cancer. Mol Oncol. 2017 Jul;11(7):715-717. [CrossRef]
- Pensotti A, Bizzarri M, Bertolaso M. The phenotypic reversion of cancer: Experimental evidences on cancer reversibility through epigenetic mechanisms (Review). Oncol Rep. 2024 Mar;51(3):48. [CrossRef]
- Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019 Dec;19(12):716-732. [CrossRef]
- Addison JB, Voronkova MA, Fugett JH, Lin CC, Linville NC, Trinh B, Livengood RH, Smolkin MB, Schaller MD, Ruppert JM, Pugacheva EN, Creighton CJ, Ivanov AV. Functional Hierarchy and Cooperation of EMT Master Transcription Factors in Breast Cancer Metastasis. Mol Cancer Res. 2021 May;19(5):784-798. [CrossRef]
- Yu L, Kuang LY, He F, Du LL, Li QL, Sun W, Zhou YM, Li XM, Li XY, Chen DJ. The Role and Molecular Mechanism of Long Nocoding RNA-MEG3 in the Pathogenesis of Preeclampsia. Reprod Sci. 2018 Dec;25(12):1619-1628. [CrossRef]
- Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, Bar-Sagi D, Stanger BZ. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell. 2018 Jun 18;45(6):681-695.e4. [CrossRef]
- Knutsen E, Das Sajib S, Fiskaa T, Lorens J, Gudjonsson T, Mælandsmo GM, Johansen SD, Seternes OM, Perander M. Identification of a core EMT signature that separates basal-like breast cancers into partial- and post-EMT subtypes. Front Oncol. 2023 Dec 4;13:1249895. [CrossRef]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, Deschler DG, Varvares MA, Mylvaganam R, Rozenblatt-Rosen O, Rocco JW, Faquin WC, Lin DT, Regev A, Bernstein BE. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017 Dec 14;171(7):1611-1624.e24. [CrossRef]
- Cardoso IIV, Rosa MN, Moreno DA, Tufi LMB, Ramos LP, Pereira LAB, Silva L, Galvão JMS, Tosi IC, Lengert AVH, Da Cruz MC, Teixeira SA, Reis RM, Lopes LF, Pinto MT. Cisplatin-resistant germ cell tumor models: An exploration of the epithelial-mesenchymal transition regulator SLUG. Mol Med Rep. 2024 Dec;30(6):228. [CrossRef]
- Liu D, Skomorovska Y, Song J, Bowler E, Harris R, Ravasz M, Bai S, Ayati M, Tamai K, Koyuturk M, Yuan X, Wang Z, Wang Y, Ewing RM. ELF3 is an antagonist of oncogenic-signalling-induced expression of EMT-TF ZEB1. Cancer Biol Ther. 2019;20(1):90-100. [CrossRef]
- Sugandha Bhatia, James Monkman, Alan Kie Leong Toh, Shivashankar H. Nagaraj, Erik W. Thompson; Targeting epithelial–mesenchymal plasticity in cancer: clinical and preclinical advances in therapy and monitoring. Biochem J 1 October 2017; 474 (19): 3269–3306. [CrossRef]
- Del Pozo Martin Y, Park D, Ramachandran A, Ombrato L, Calvo F, Chakravarty P, Spencer-Dene B, Derzsi S, Hill CS, Sahai E, Malanchi I. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015 Dec 22;13(11):2456-2469. [CrossRef]
- Thompson, E.W., Redfern, A.D., Brabletz, S. et al. EMT and cancer: what clinicians should know. Nat Rev Clin Oncol 22, 711–733 (2025). [CrossRef]
- Nezami, M. , Hager, S. and Garner, J. (2015) EMT and Anti-EMT Strategies in Cancer. Journal of Cancer Therapy, 6, 1013-1019. [CrossRef]
- Kwon M, Jung H, Nam GH, Kim IS. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J Control Release. 2021 Mar 10;331:321-334. [CrossRef]
- Sánchez-Tilló E, Lázaro A, Torrent R, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29(24):3490–3500. [CrossRef]
- Connolly EP, Sun Y, Hei TK. Eribulin mesylate promotes a mesenchymal-epithelial transition (MET) and effectively radio-sensitizes triple-negative breast cancer cells. Cancer Res. 2015;75(9 Suppl):P6-02-04. [CrossRef]
- Meidhof S, Brabletz S, Stemmler MP, et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. 2015;7(6):832–847. [CrossRef]
- Bagheri M, Mohamed GA, Mohamed Saleem MA, Ognjenovic NB, Lu H, Kolling FW, Wilkins OM, Das S, LaCroix IS, Nagaraj SH, Muller KE, Gerber SA, Miller TW, Pattabiraman DR. Pharmacological induction of chromatin remodeling drives chemosensitization in triple-negative breast cancer. Cell Rep Med. 2024 Apr 16;5(4):101504. [CrossRef]
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massagué J. TGF-β Tumor Suppression through a Lethal EMT. Cell. 2016 Feb 25;164(5):1015-30. [CrossRef]
- Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, Bantug G, Morini MF, Wang J, Hess C, Christofori G. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell. 2019 Jan 14;35(1):17-32.e6. [CrossRef]
- Voon DC, Huang RY, Jackson RA, Thiery JP. The EMT spectrum and therapeutic opportunities. Mol Oncol. 2017 Jul;11(7):878-891. [CrossRef]
- Yoshida T, Ozawa Y, Kimura T, et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from EMT to MET. Br J Cancer. 2014;110(6):1497–1505.
- Lim HI, Yamamoto J, Inubushi S, Nishino H, Tashiro Y, Sugisawa N, Han Q, Sun YU, Choi HJ, Nam SJ, Kim MB, Lee JS, Hozumi C, Bouvet M, Singh SR, Hoffman RM. A Single Low Dose of Eribulin Regressed a Highly Aggressive Triple-negative Breast Cancer in a Patient-derived Orthotopic Xenograft Model. Anticancer Res. 2020 May;40(5):2481-2485. [CrossRef]
- Pensotti A, Bizzarri M, Bertolaso M. The phenotypic reversion of cancer: Experimental evidences on cancer reversibility through epigenetic mechanisms (Review). Oncol Rep. 2024 Mar;51(3):48. [CrossRef]
- Sánchez-Tilló E, Lázaro A, Torrent R, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29(24):3490–3500. [CrossRef]
- Lee SH, Shen GN, Jung YS, et al. Antitumor effect of novel small chemical inhibitors of Snail-p53 binding in K-Ras-mutated cancer cells. Oncogene. 2010;29(32):4576–4587. [CrossRef]
- Vistain LF, Yamamoto N, Rathore R, Cha P, Meade TJ. Targeted Inhibition of Snail Activity in Breast Cancer Cells by Using a Co(III) -Ebox Conjugate. Chembiochem. 2015 Sep 21;16(14):2065-72. [CrossRef]
- Takemura, K.; Ikeda, K.; Miyake, H.; Sogame, Y.; Yasuda, H.; Okada, N.; Iwata, K.; Sakagami, J.; Yamaguchi, K.; Itoh, Y.; et al. Epithelial–Mesenchymal Transition Suppression by ML210 Enhances Gemcitabine Anti-Tumor Effects on PDAC Cells. Biomolecules 2025, 15, 70. [CrossRef]
- Vaidya AM, Sun Z, Ayat N, Schilb A, Liu X, Jiang H, Sun D, Scheidt J, Qian V, He S, Gilmore H, Schiemann WP, Lu ZR. Systemic Delivery of Tumor-Targeting siRNA Nanoparticles against an Oncogenic LncRNA Facilitates Effective Triple-Negative Breast Cancer Therapy. Bioconjug Chem. 2019 Mar 20;30(3):907-919. [CrossRef]
- Cheng, K.; Hao, M. Metformin Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells. Int. J. Mol. Sci. 2016, 17, 2000. [CrossRef]
- Zhou Y, Liang C, Xue F, Chen W, Zhi X, Feng X, Bai X, Liang T. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the β-catenin/TCF complex association via FOXO3a activation. Oncotarget. 2015 Apr 30;6(12):10350-65. [CrossRef]
- Lyakhovich A, Lleonart ME. Bypassing Mechanisms of Mitochondria-Mediated Cancer Stem Cells Resistance to Chemo- and Radiotherapy. Oxid Med Cell Longev. 2016;2016:171634. [CrossRef]
- Cañadas I, Rojo F, Taus Á, Arpí O, Arumí-Uría M, Pijuan L, Menéndez S, Zazo S, Dómine M, Salido M, Mojal S, García de Herreros A, Rovira A, Albanell J, Arriola E. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res. 2014 Feb 15;20(4):938-50. [CrossRef]
- Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. 2013 Feb 15;132(4):745-54. [CrossRef]
- Bhat GR, Sethi I, Sadida HQ, Rah B, Mir R, Algehainy N, Albalawi IA, Masoodi T, Subbaraj GK, Jamal F, Singh M, Kumar R, Macha MA, Uddin S, Akil ASA, Haris M, Bhat AA. Cancer cell plasticity: from cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance. Cancer Metastasis Rev. 2024 Mar;43(1):197-228. [CrossRef]
- Jiang M, Wang J, Li Y, Zhang K, Wang T, Bo Z, Lu S, Rodríguez RA, Wei R, Zhu M, Nicot C, Sethi G. EMT and cancer stem cells: Drivers of therapy resistance and promising therapeutic targets. Drug Resist Updat. 2025 Nov;83:101276. [CrossRef]
- Chen M, Mainardi S, Lieftink C, Velds A, de Rink I, Yang C, Kuiken HJ, Morris B, Edwards F, Jochems F, van Tellingen O, Boeije M, Proost N, Jansen RA, Qin S, Jin H, Koen van der Mijn JC, Schepers A, Venkatesan S, Qin W, Beijersbergen RL, Wang L, Bernards R. Targeting of vulnerabilities of drug-tolerant persisters identified through functional genetics delays tumor relapse. Cell Rep Med. 2024 Mar 19;5(3):101471. [CrossRef]
- Huang Y, Hong W, Wei X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J Hematol Oncol. 2022 Sep 8;15(1):129. [CrossRef]
- Celià-Terrassa T, Kang Y. How important is EMT for cancer metastasis? PLoS Biol. 2024 Feb 7;22(2):e3002487. [CrossRef]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalisation of the vasculature is needed to treat cancer and other diseases. Physiol Rev. 2011 Jul;91(3):1071-121. [CrossRef]
- Jain RK. Normalisation of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005 Jan 7;307(5706):58-62. [CrossRef]
- Yang T, Xiao H, Liu X, Wang Z, Zhang Q, Wei N, Guo X. Vascular Normalization: A New Window Opened for Cancer Therapies. Front Oncol. 2021 Aug 12;11:719836. [CrossRef]
- Liu Z, Zhao Q, Zheng Z, Liu S, Meng L, Dong L, Jiang X. Vascular normalization in immunotherapy: A promising mechanisms combined with radiotherapy. Biomed Pharmacother. 2021 Jul;139:111607. [CrossRef]
- Magnussen AL, Mills IG. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br J Cancer. 2021 Aug;125(3):324-336. [CrossRef]
- Guo Z, Lei L, Zhang Z, Du M, Chen Z. The potential of vascular normalization for sensitization to radiotherapy. Heliyon. 2024 Jun 8;10(12):e32598. [CrossRef]
- Appleton, E.; Hassan, J.; Hak, C.C.W.; Sivamanoharan, N.; Wilkins, A.; Samson, A.; Ono, M.; Harrington, K.J.; Melcher, A.; Wennerberg, E. Kickstarting Immunity in Cold Tumours: Localised Tumour Therapy Combinations With Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 754436. [CrossRef]
- Shibamoto Y, Miyakawa A, Otsuka S, Iwata H. Radiobiology of hypofractionated stereotactic radiotherapy: what are the optimal fractionation schedules? J Radiat Res. 2016 Aug;57 Suppl 1(Suppl 1):i76-i82. [CrossRef]
- Chen HHW, Kuo MT. Improving radiotherapy in cancer treatment: Promises and challenges. Oncotarget. 2017 Jun 8;8(37):62742-62758. [CrossRef]
- Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004 Oct;4(10):806-13. [CrossRef]
- Lecoultre M, Chliate S, Espinoza FI, Tankov S, Dutoit V, Walker PR. Radio-chemotherapy of glioblastoma cells promotes phagocytosis by macrophages in vitro. Radiother Oncol. 2024 Jan;190:110049. [CrossRef]
- Ura B, Di Lorenzo G, Romano F, Monasta L, Mirenda G, Scrimin F, Ricci G. Interstitial Fluid in Gynecologic Tumors and Its Possible Application in the Clinical Practice. Int J Mol Sci. 2018 Dec 12;19(12):4018. [CrossRef]
- Feng K, Zhang X, Li J, Han M, Wang J, Chen F, Yi Z, Di L, Wang R. Neoantigens combined with in situ cancer vaccination induce personalized immunity and reshape the tumor microenvironment. Nat Commun. 2025 May 31;16(1):5074. [CrossRef]
- Liu, N., Wang, X., Wang, Z. et al. Nanomaterials-driven in situ vaccination: a novel frontier in tumor immunotherapy. J Hematol Oncol 18, 45 (2025). [CrossRef]
- Kerr MD, McBride DA, Chumber AK, Shah NJ. Combining therapeutic vaccines with chemo- and immunotherapies in the treatment of cancer. Expert Opin Drug Discov. 2021 Jan;16(1):89-99. [CrossRef]
- Bangarh R, Saini RV, Saini AK, Singh T, Joshi H, Ramniwas S, Shahwan M, Tuli HS. Dynamics of epithelial-mesenchymal plasticity driving cancer drug resistance. Cancer Pathog Ther. 2024 Jul 6;3(2):120-128. [CrossRef]
- Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009 Sep;27(9):2059-68. [CrossRef]
- Fontana R, Mestre-Farrera A, Yang J. Update on Epithelial-Mesenchymal Plasticity in Cancer Progression. Annu Rev Pathol. 2024 Jan 24;19:133-156. [CrossRef]
- Maria L. Lotsberg. Decoding cancer’s camouflage: epithelial-mesenchymal plasticity in resistance to immune checkpoint blockade. [CrossRef]
- Srinivasan D, Balakrishnan R, Chauhan A, Kumar J, Girija DM, Shrestha R, Shrestha R, Subbarayan R. Epithelial-Mesenchymal Transition in Cancer: Insights Into Therapeutic Targets and Clinical Implications. MedComm (2020). 2025 Aug 29;6(9):e70333. [CrossRef]
- Wang X, Xue X, Pang M, Yu L, Qian J, Li X, Tian M, Lyu A, Lu C, Liu Y. Epithelial-mesenchymal plasticity in cancer: signaling pathways and therapeutic targets. MedComm (2020). 2024 Aug 1;5(8):e659. [CrossRef]
- Park J, Choi J, Cho I, Sheen YY. Radiotherapy-induced oxidative stress and fibrosis in breast cancer are suppressed by vactosertib, a novel, orally bioavailable TGF-β/ALK5 inhibitor. Sci Rep. 2022 Sep 27;12(1):16104. [CrossRef]
- Wang Y, Han Y, Jin Y, He Q, Wang Z. The Advances in Epigenetics for Cancer Radiotherapy. Int J Mol Sci. 2022 May 18;23(10):5654. [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Martínez-Pérez, A.; Rodrigo, J.P.; García-Pedrero, J.M.; Gonzalez, S. Chemo-Immunotherapy: A New Trend in Cancer Treatment. Cancers 2023, 15, 2912. [CrossRef]
- Lussier, D.M.; Alspach, E.; Ward, J.P.; Miceli, A.P.; Runci, D.; White, J.M.; Mpoy, C.; Arthur, C.D.; Kohlmiller, H.N.; Jacks, T.; et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proc. Natl. Acad. Sci. USA 2022, 119, e2102611118. [CrossRef]
- Breen WG, Leventakos K, Dong H, Merrell KW. Radiation and immunotherapy: emerging mechanisms of synergy. J Thorac Dis. 2020 Nov;12(11):7011-7023. [CrossRef]
- He K, Barsoumian HB, Sezen D, Puebla-Osorio N, Hsu EY, Verma V, Abana CO, Chen D, Patel RR, Gu M, Cortez MA, Welsh JW. Pulsed Radiation Therapy to Improve Systemic Control of Metastatic Cancer. Front Oncol. 2021 Aug 23;11:737425. [CrossRef]
- Spaas M, Lievens Y. Is the Combination of Immunotherapy and Radiotherapy in Non-small Cell Lung Cancer a Feasible and Effective Approach? Front Med (Lausanne). 2019 Nov 7;6:244. [CrossRef]
- Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J, Zhao P, Fang L, Shi Y, Wang P. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct Target Ther. 2023 Mar 7;8(1):104. [CrossRef]
- Joshi S, Durden DL. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J Oncol. 2019 Feb 3;2019:5245034. [CrossRef]
- Zhang M, Zhang B. Extracellular matrix stiffness: mechanisms in tumor progression and therapeutic potential in cancer. Exp Hematol Oncol. 2025 Apr 10;14(1):54. [CrossRef]
- Short S, Fielder E, Miwa S, von Zglinicki T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine. 2019 Mar;41:683-692. [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All that Glitters Really Gold? Cancers 2021, 13, 723. [CrossRef]
- Marino P, Mininni M, Deiana G, Marino G, Divella R, Bochicchio I, Giuliano A, Lapadula S, Lettini AR, Sanseverino F. Healthy Lifestyle and Cancer Risk: Modifiable Risk Factors to Prevent Cancer. Nutrients. 2024 Mar 11;16(6):800. [CrossRef]
- Liu S, Jiang A, Tang F, Duan M, Li B. Drug-induced tolerant persisters in tumor: mechanism, vulnerability and perspective implication for clinical treatment. Mol Cancer. 2025 May 24;24(1):150. [CrossRef]
- Guo F, Kerrigan B, Yang D, Hu L, Shmulevich I, Sood A, et al. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J Hematol Oncol. 2014;7:19. [CrossRef]
- Yamamoto M, Sakane K, Tominaga K, Gotoh N, Niwa T, Kikuchi Y, et al. Intratumoral bidirectional transitions between epithelial and mesenchymal cells in triple-negative breast cancer. Cancer Sci. 2017;108(6):1210-1222. [CrossRef]
- Chen T, You Y, Jiang H, Wang ZZ. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol. 2017 Dec;232(12):3261-3272. [CrossRef]
- Yeh YH, Wang SW, Yeh YC, Hsiao HF, Li TK. Rhapontigenin inhibits TGF-β-mediated epithelial-mesenchymal transition via the PI3K/AKT/mTOR pathway and is not associated with HIF-1α degradation. Oncol Rep. 2016 May;35(5):2887-95. [CrossRef]
- Gómez-Escudero, J.; Berlana-Galán, P.; Guerra-Paes, E.; Torre-Cea, I.; Marcos-Zazo, L.; Carrera-Aguado, I.; Cáceres-Calle, D.; Sánchez-Juanes, F.; Muñoz-Félix, J.M. Vascular Disruption Therapy as a New Strategy for Cancer Treatment. Int. J. Mol. Sci. 2025, 26, 10085. [CrossRef]
- Sim HJ, Kim MR, Song MS, Lee SY. Kv3.4 regulates cell migration and invasion through TGF-β-induced epithelial-mesenchymal transition in A549 cells. Sci Rep. 2024 Jan 28;14(1):2309. [CrossRef]
- Jia D, Jolly MK, Boareto M, Parsana P, Mooney SM, Pienta KJ, Levine H, Ben-Jacob E. OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget. 2015 Jun 20;6(17):15436-48. [CrossRef]
- Mishra VK, Johnsen SA. Targeted therapy of epigenomic regulatory mechanisms controlling the epithelial to mesenchymal transition during tumor progression. Cell Tissue Res. 2014 Jun;356(3):617-30. [CrossRef]
- Zheng X, Dai F, Feng L, Zou H, Feng L, Xu M. Communication Between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights Into Cancer Progression. Front Oncol. 2021 Apr 21;11:617597. [CrossRef]
- Franssen L, Chaplain M. A mathematical multi-organ model for bidirectional epithelial-mesenchymal transitions in the metastatic spread of cancer. bioRxiv. 2019. [CrossRef]
- Lin WH, Chang YW, Hong MX, Hsu TC, Lee KC, Lin C, Lee JL. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT-MET switch and cancer metastasis. Oncogene. 2021 Jan;40(4):791-805. [CrossRef]
- Swamy, K.; Arakeri, G.; Basavalingaiah S, A. Combination, Timing, and Sequencing (CTS) Therapy Strategy for Unmasking Cancer for Immunotherapy. Preprints 2024, 2024111830. [CrossRef]
- Giordanengo, L.; Proment, A.; Botta, V.; Picca, F.; Munir, H.M.W.; Tao, J.; Olivero, M.; Taulli, R.; Bersani, F.; Sangiolo, D.; et al. Shifting Shapes: The Endothelial-to-Mesenchymal Transition as a Driver for Cancer Progression. Int. J. Mol. Sci. 2025, 26, 6353. [CrossRef]
Figure 1.
In a normoxic situation, Enzyme prolyl hydroxylases (PHDs) hydroxylate HIF-1α to relieve the free radicals superoxide anion (O2·−), among others, generated by ROS stress. Von Hippel-Lindau (VHL) protein marks for degradation. In a hypoxic environment, PHDs are inhibited, HIF-1 alpha stabilizes without degradation, and accumulates in the cytosol. Stabilized HIF-1α enters the nucleus, forming a dimer with HIF-1β, and that binds to DNA motifs, influencing numerous homeostatic genes and leading to cascading effects in cancer progression. (1) Shift to anaerobic metabolism—Warburg effect: lactate production alters pH levels, resulting in acidity. (2) Vascular Endothelial Growth Factor (VEGF) transcription stimulates angiogenesis, resulting in abnormal neoangiogenesis, exacerbating hypoxia, and establishing a vicious HIF-1α and proangiogenic factors cycle. (3) Activation of genomic mutations that drive cancer progression and the formation of diverse clones and subclones with phenotypically resistant populations of cancer cells. (4) Accumulation of various types of immunosuppressive cells, cancer-associated fibroblasts (CAFs), and cytokines in the tumor microenvironment (TME). (5) Epithelial-mesenchymal transition leading to the other pathway of stemness, metastases, and resistance. Created with BioRender.com.
Figure 1.
In a normoxic situation, Enzyme prolyl hydroxylases (PHDs) hydroxylate HIF-1α to relieve the free radicals superoxide anion (O2·−), among others, generated by ROS stress. Von Hippel-Lindau (VHL) protein marks for degradation. In a hypoxic environment, PHDs are inhibited, HIF-1 alpha stabilizes without degradation, and accumulates in the cytosol. Stabilized HIF-1α enters the nucleus, forming a dimer with HIF-1β, and that binds to DNA motifs, influencing numerous homeostatic genes and leading to cascading effects in cancer progression. (1) Shift to anaerobic metabolism—Warburg effect: lactate production alters pH levels, resulting in acidity. (2) Vascular Endothelial Growth Factor (VEGF) transcription stimulates angiogenesis, resulting in abnormal neoangiogenesis, exacerbating hypoxia, and establishing a vicious HIF-1α and proangiogenic factors cycle. (3) Activation of genomic mutations that drive cancer progression and the formation of diverse clones and subclones with phenotypically resistant populations of cancer cells. (4) Accumulation of various types of immunosuppressive cells, cancer-associated fibroblasts (CAFs), and cytokines in the tumor microenvironment (TME). (5) Epithelial-mesenchymal transition leading to the other pathway of stemness, metastases, and resistance. Created with BioRender.com.
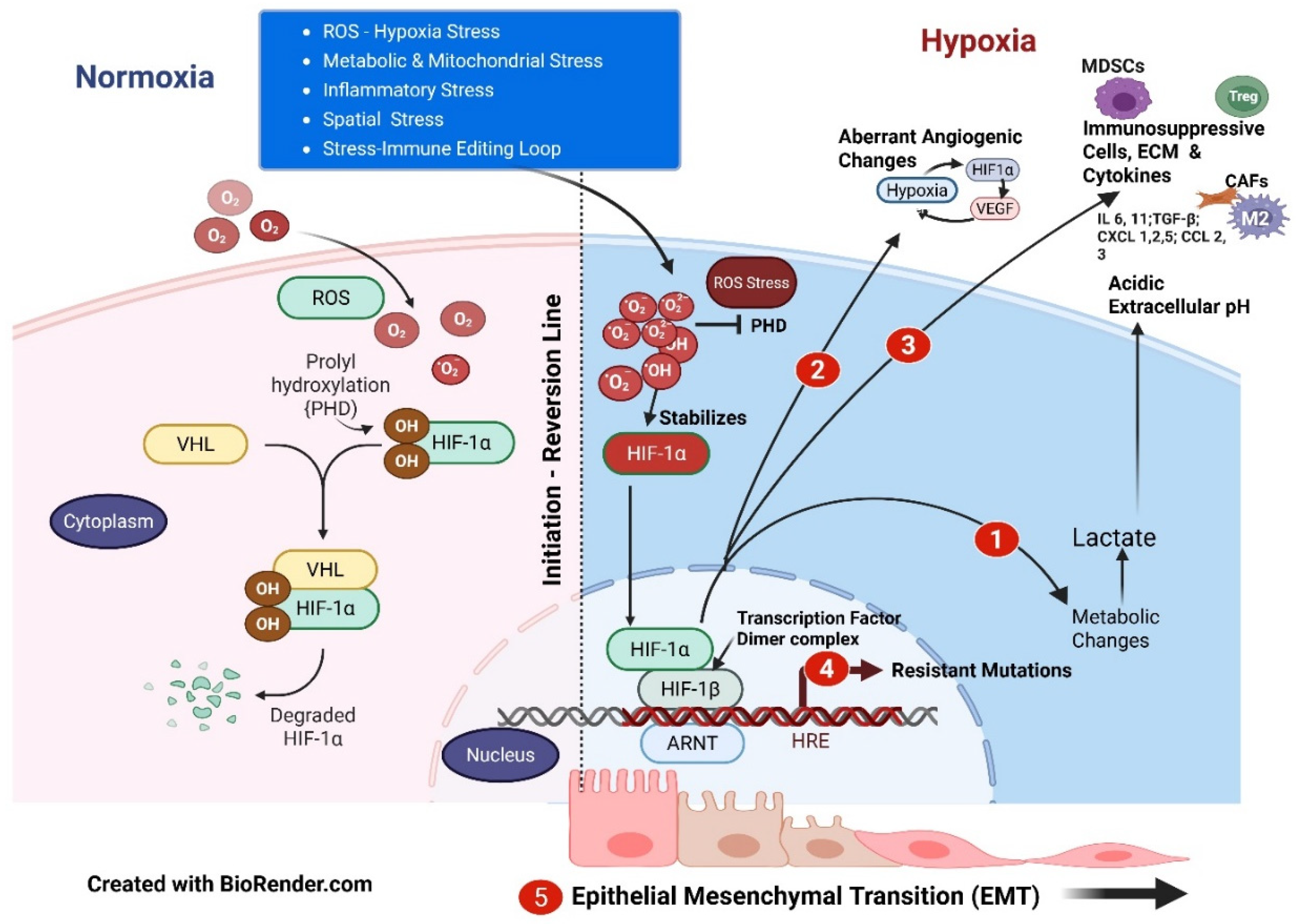
Figure 2.
Signaling pathways of TGFβ activation, epithelial-mesenchymal transition (EMT), and Progression. ECM = Extracellular Matrix; TGFβ = Transforming Growth Factor-Beta; TKR = Tyrosine Kinase Receptors; WNT = "Wingless/Integrated" family of genes; SHH = Sonic Hedgehog - PTCH1 = Patched-1 Protein - SMO = Smoothened, frizzled family receptor; IL = Interleukin; GP = Glycoprotein; Downstream = Transcription Factors (see Text). Created with BioRender.com.
Figure 2.
Signaling pathways of TGFβ activation, epithelial-mesenchymal transition (EMT), and Progression. ECM = Extracellular Matrix; TGFβ = Transforming Growth Factor-Beta; TKR = Tyrosine Kinase Receptors; WNT = "Wingless/Integrated" family of genes; SHH = Sonic Hedgehog - PTCH1 = Patched-1 Protein - SMO = Smoothened, frizzled family receptor; IL = Interleukin; GP = Glycoprotein; Downstream = Transcription Factors (see Text). Created with BioRender.com.
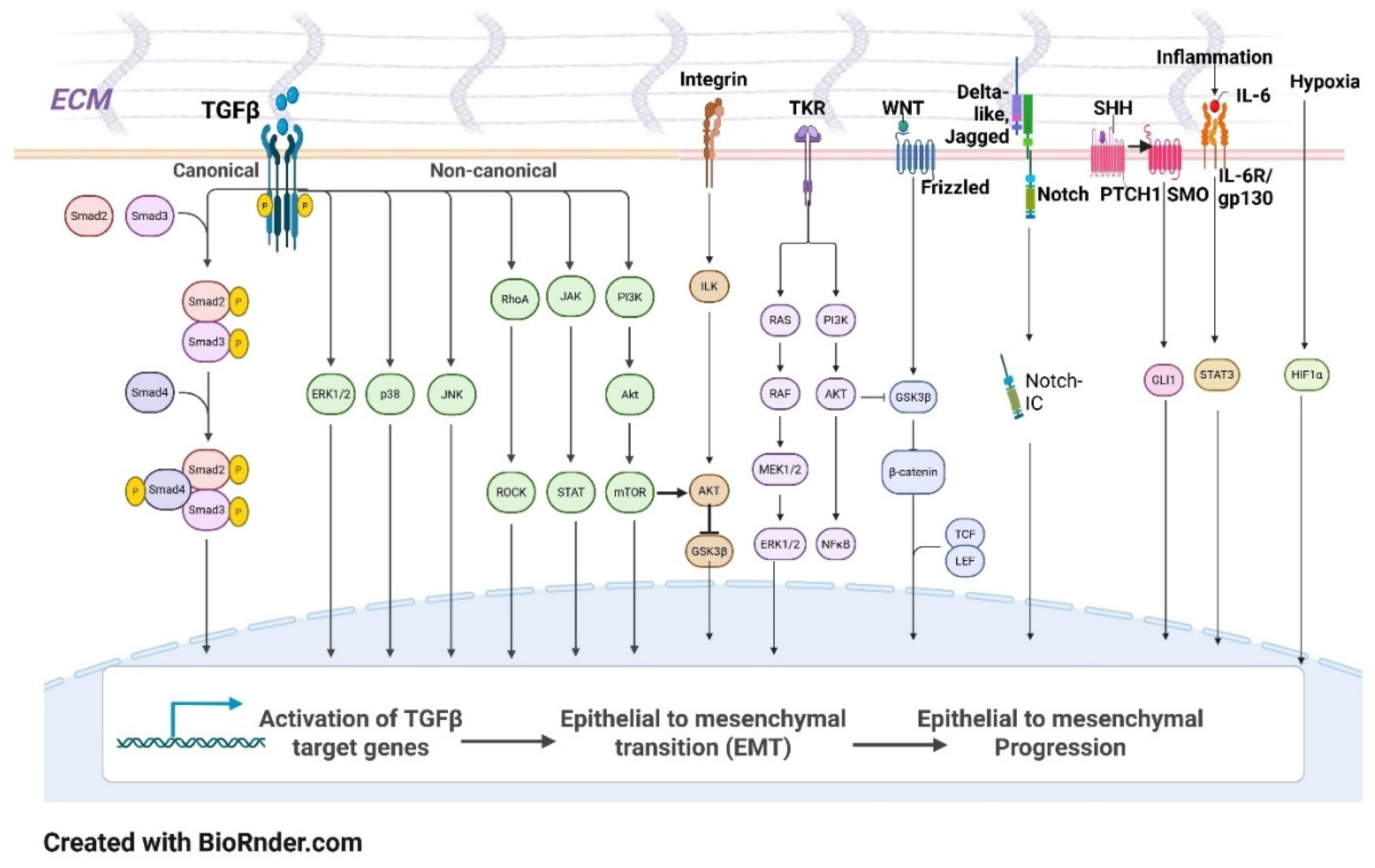
Figure 3.
Epithelial-Mesenchymal Transition (EMT) – Mesenchymal Epithelial Transition (MET) cancer cell pathways of initiation, invasion, dissemination, and colonization, and development of epithelial-mesenchymal plasticity (EMP). DMC = DNA methylation changes; CSC = Cancer Stem Cell; CC = Cell competition – Spatial stress; MCC = Mutated Colorectal Cancer Oncogene; TGFβ = Transforming Growth Factor; DTPs = Drug-Tolerant Persister cells. Created with BioRender.com.
Figure 3.
Epithelial-Mesenchymal Transition (EMT) – Mesenchymal Epithelial Transition (MET) cancer cell pathways of initiation, invasion, dissemination, and colonization, and development of epithelial-mesenchymal plasticity (EMP). DMC = DNA methylation changes; CSC = Cancer Stem Cell; CC = Cell competition – Spatial stress; MCC = Mutated Colorectal Cancer Oncogene; TGFβ = Transforming Growth Factor; DTPs = Drug-Tolerant Persister cells. Created with BioRender.com.
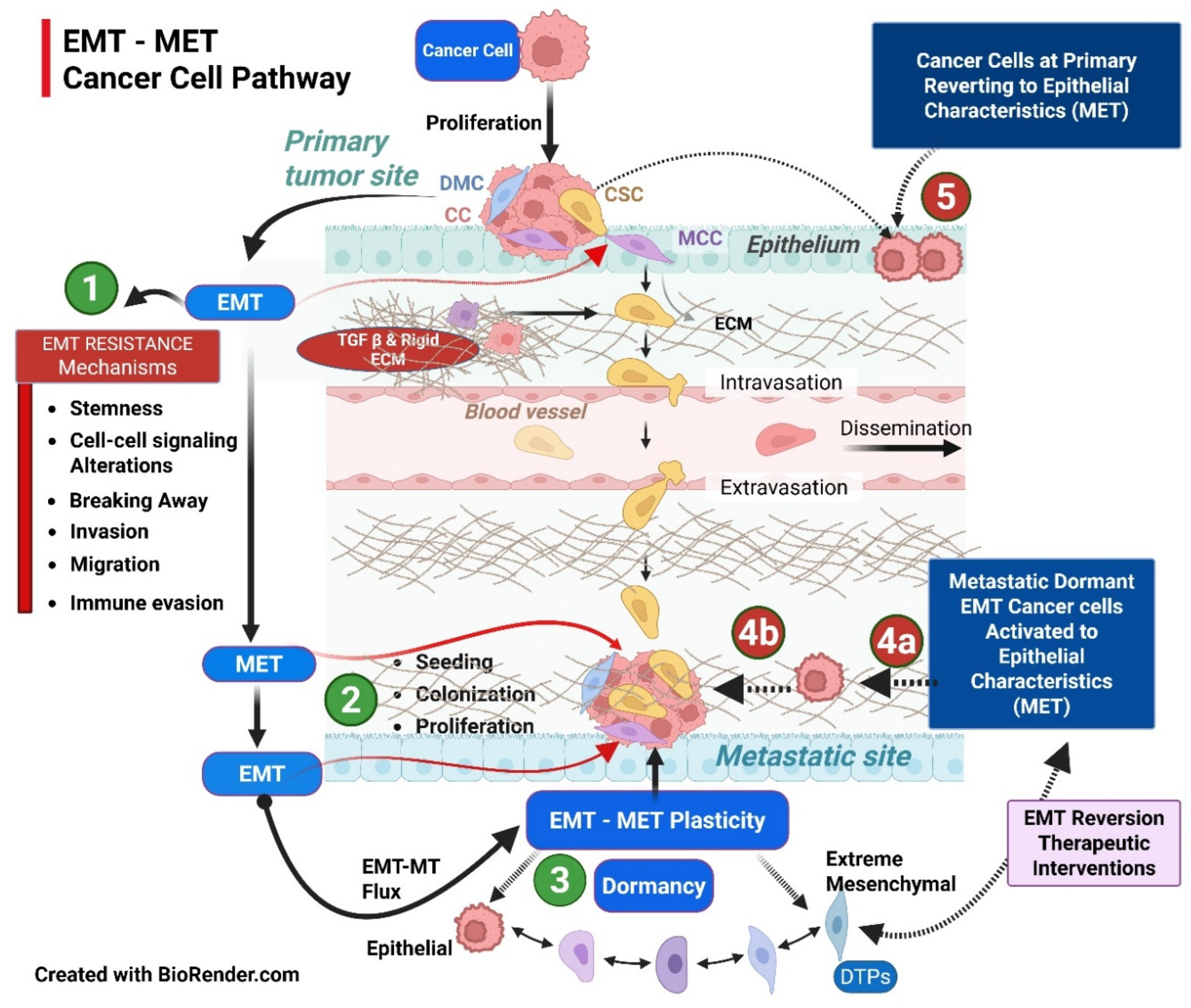
Figure 4.
Morphology of Epithelial-Mesenchymal Plasticity (EMP) and opportunity for restoration of mesenchymal cells with epithelial characteristics. Identifiable targets for Epigenetic modifiers in restoring cell junctions and signaling pathways. TGFβ = Transforming Growth Factor-Beta; TKR = Tyrosine Kinase Receptors; WNT "Wingless/Integrated" family of genes; SHH = Sonic Hedgehog - PTCH1 = Patched-1 Protein - SMO = Smoothened, frizzled family receptor; IL = Interleukin – GP = Glycoprotein. Created with BioRender.com.
Figure 4.
Morphology of Epithelial-Mesenchymal Plasticity (EMP) and opportunity for restoration of mesenchymal cells with epithelial characteristics. Identifiable targets for Epigenetic modifiers in restoring cell junctions and signaling pathways. TGFβ = Transforming Growth Factor-Beta; TKR = Tyrosine Kinase Receptors; WNT "Wingless/Integrated" family of genes; SHH = Sonic Hedgehog - PTCH1 = Patched-1 Protein - SMO = Smoothened, frizzled family receptor; IL = Interleukin – GP = Glycoprotein. Created with BioRender.com.
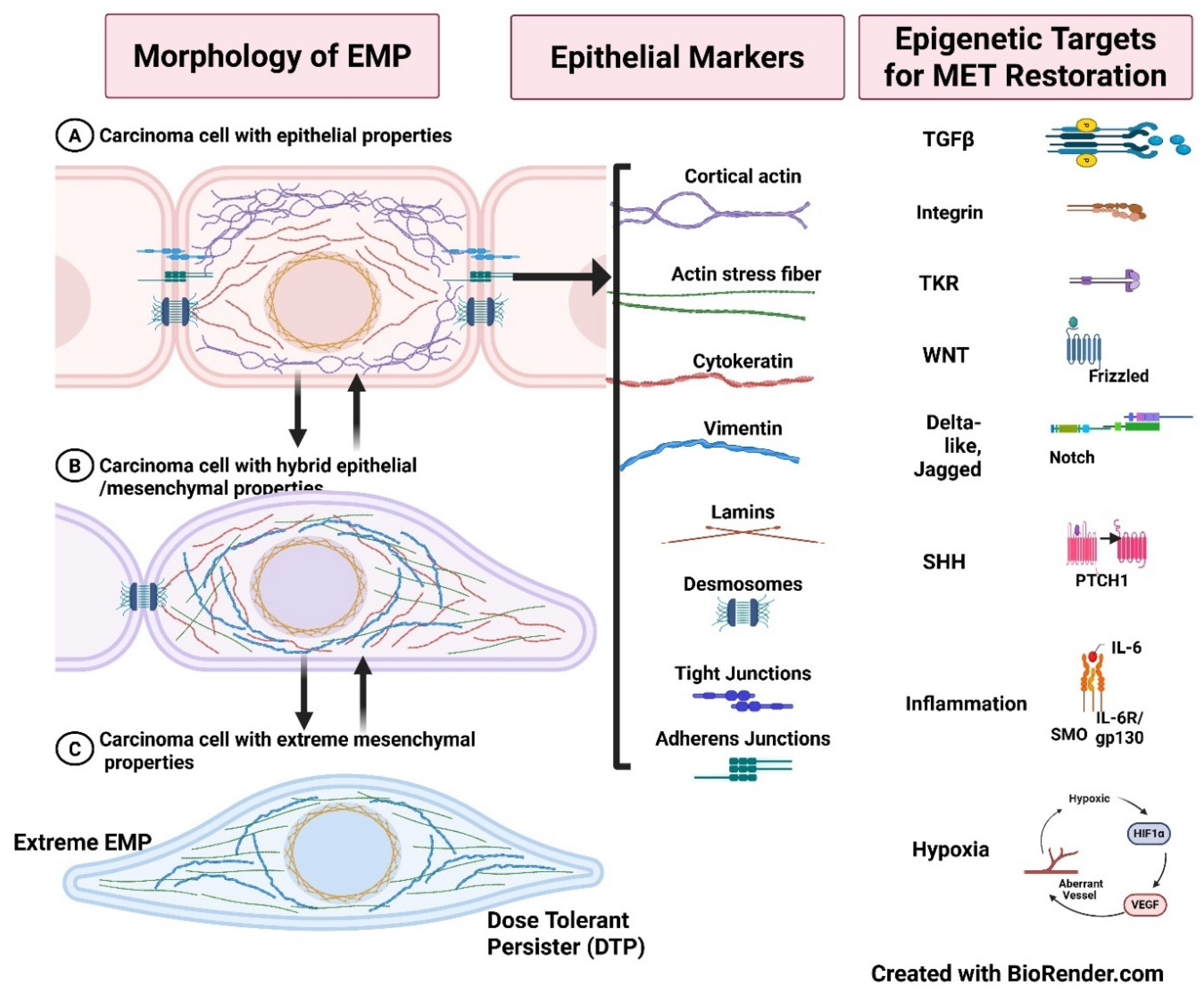
Figure 5.
Cancer Heterogeneity Matrix and web of pathways (one pathway nurturing the other in propagating the tumor) is responsible for resistance. Epithelial-mesenchymal transition (EMT) and plasticity (EMP) remain at the core of the primary and secondary resistance. The treatment strategies should encompass EMP and the entire gamut of cancer cell protective shields in a structured feedback-analytical approach. Limited pathway targeted approaches will herald the increasing evolutionary resistance [4]. Created with BioRender.com.
Figure 5.
Cancer Heterogeneity Matrix and web of pathways (one pathway nurturing the other in propagating the tumor) is responsible for resistance. Epithelial-mesenchymal transition (EMT) and plasticity (EMP) remain at the core of the primary and secondary resistance. The treatment strategies should encompass EMP and the entire gamut of cancer cell protective shields in a structured feedback-analytical approach. Limited pathway targeted approaches will herald the increasing evolutionary resistance [4]. Created with BioRender.com.
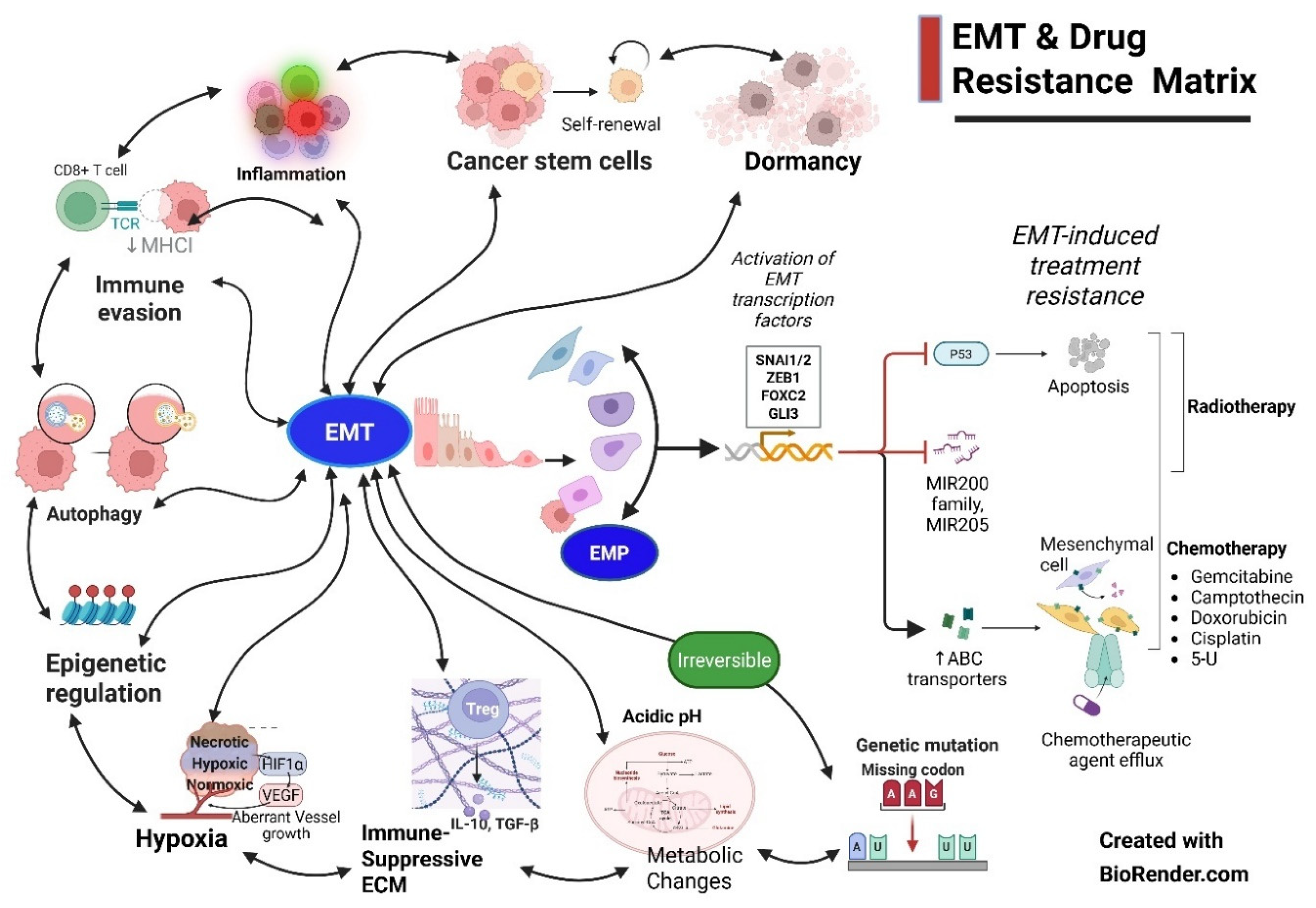
Figure 6.
Smart Targering with Combination, Timing, and Sequencing (CTS) strategy. Starting the treatment with antiangiogenics to normalize the vasculature (1a), along with epigenetic modifiers to sensitize the subsequent CT (1b), and inducing the more sensitive phase of MET reversion (1c). This is followed by cytotoxic cell kill strategies with the maximum tolerated dose (MTD) (2). With enhanced vasculature and depletion of immunosuppressive TME cells, the stage is set for optimized immunotherapy response (3). Inhibitory (a) and (b) represent preventing cancer progression to EMT by timely CTS intervention 1, 2, and 3. The most difficult to eliminate extreme mesenchymal state or drug-tolerant persister cells, i.e, DTPs, require surgery/SBRT (4) and epigenetic modifiers with specific strategies (5). Created with BioRender.com.
Figure 6.
Smart Targering with Combination, Timing, and Sequencing (CTS) strategy. Starting the treatment with antiangiogenics to normalize the vasculature (1a), along with epigenetic modifiers to sensitize the subsequent CT (1b), and inducing the more sensitive phase of MET reversion (1c). This is followed by cytotoxic cell kill strategies with the maximum tolerated dose (MTD) (2). With enhanced vasculature and depletion of immunosuppressive TME cells, the stage is set for optimized immunotherapy response (3). Inhibitory (a) and (b) represent preventing cancer progression to EMT by timely CTS intervention 1, 2, and 3. The most difficult to eliminate extreme mesenchymal state or drug-tolerant persister cells, i.e, DTPs, require surgery/SBRT (4) and epigenetic modifiers with specific strategies (5). Created with BioRender.com.
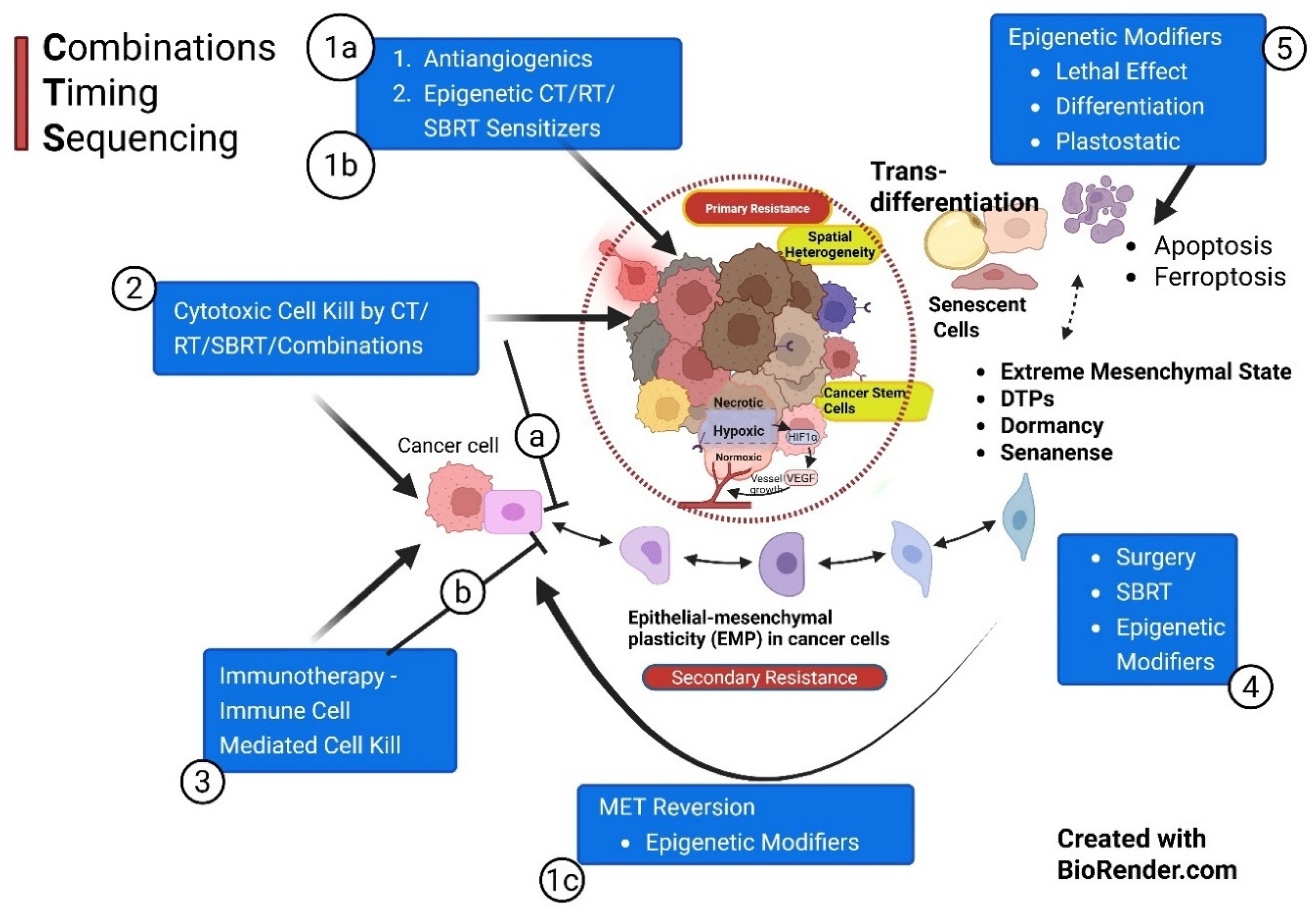

Table 1.
Summary of Combinations, Timing, and Sequencing Strategy (CTS) strategy.
| Phases in Sequence | Strategy & Mechanisms |
Timing | Objective | References |
|---|---|---|---|---|
| Phase I a |
Prepriming therapy Primarily Vascular Normalization a. With AAGs / Small molecules to initiate vascular normalization & hypoxia targeting (3 weeks on 3 weeks off) |
Start: Day 0 |
1. Normalization of vascular structure is Essential for effective subsequent chemo-immunotherapy delivery. It also improves sensitivity to RT/SBRT. 2. Improve by adding newer strategies/drugs for vascular normalization. |
Vascular Normalization: a. Goel S et al, 2011 [92] b. Jain RK et al 2005 [93] c. Ting Yang et al, 2021 [94] d. Zijing Liu, et al 2021 [95] e. Magnussen AL et al, 2021. [96] f. Zhili Guo, et al, 2024. [97] |
| Phase 1b |
Prepriming therapy: Primarily, Epigenetic Therapy as a CT/RT sensitizer a. With epigenetic modifiers for subsequent CT/ RT in Phase II. Eg. Eribulin b. Reducing Drug Efflux |
Starts in the 1st week of AAGs (> Day 2) |
1. Anticancer standard therapy has been RT and CT for decades, evolving with Maximum Tolerated Doses (MTD). EMs change the landscape by reversing epigenetic changes, sensitizing cancer cells for CT/RT. 2. EMs help achieve effective drug concentration in cancer cells 3. Prevent EMT transformation and ensuing stemness |
Epigenetic Modifiers: a. Guo H et al., 2025 [42]. b. Cardoso IIV et al. 2024 [60] c. Liu D et al, 2019 [61] d. Bagheri M et al., 2024 [70] b. Huang Y et al., 2022. [90] d. Connolly EP et al., 2015 [68] e. Meidhof S, et al., 2015 [69] f. Yoshida T et al., 2014, [74] g. HYE IN LIM et al., 2020, [75] |
| Phase 1c |
Prepriming therapy: Primarily, Epigenetic Therapy as MET Inducer – MET Reversion Push the cells to EMT cells to gain Epithelial features using the MET pathway |
Before the start of CT/RT – schedule to be optimized | This facilitates Mesenchymal cells to acquire more susceptible epithelial characteristics and reduce stemness.. |
MET Iducers .KLF4: Subbalakshmi A et al, 2021 [46] .E-cadherin & TGFβ: Johnson K, et al., 2022 [47] .Target EMT signaling: Ribatti et al, 2020 [49] |
| Phase II |
Priming therapy: Primarily, selective cancer cell kill and interstitial pressure reduction. a. MTD strategy: Cytotoxic therapy and activation of cytotoxic lymphocytes to “unmask” the cancer for concurrent or subsequent immunotherapy even in cold and PDL1-negative tumors. b. Professional Phagocytosis c. Decrease ISP d. Antigenicity & Adjuvanticity f. Lymphatic normalization g. In situ vaccination h. Nanoparticle therapy i. In vitro vaccine therapy j. Epigenetic therapy (ET) |
Start CT/RT (SBRT) in the 1st week of AAGs (> Day 2) | Cancer cell lysis (preferably by ICD); Create waves of neoantigen generation; Immune suppressive/exhausted cell depletion in TME; decrease ISP; enhanced vascularity (vascular promotion), improved lymphatic drainage for neoantigen presentation in the lymph nodes, and reinvigorating the Immunity cycle; enhanced fresh TME CTLs infiltration. | .EMP Prevention: Bhat R et al, 2024 [87] .RT/CT: Chen HW. 2017 [100] . Phagocytosis - Marc Lecoultre et al, 2024. [102] . ISP -Carl-Henrik Heldin, et al., 2004, [101] - Blendi Ura, et al., 2018 [103] . Lymphatic normalization Goel S et al, 2011 [92] . Antigenicity & Adjuvanticity -Appleton, E., et al 2021. [98] . In situ Vaccination: Feng K, et al, 2025, [104] . Nanoparticle therapy - Naimeng Liu, et al [105] .Vaccine therapy -Matthew D Kerr, et al., 2022, [106] . Epigenetic Therapy integration: -Bangarh R et al. 2024 [107] -Kurrey NK et al. 2009 [108] -Fotana R et al, 2024 [109] |
| Phase III |
Primary therapy Primarily Immune Modulation a. When Cancer Cells and TME are primed and Ready to initiate with immunotherapy. Additionally; b. Timed/Pulsed SBRT/SBRT Boost c. Immune adjuvants d. Phagocytosis checkpoints e. In situ vaccination f. Nanoparticle therapy g. Epigenetic therapy |
2 to 3 weeks after Neoadjuvant CT/RT (SBRT) Or after Surgery |
1. Optimize the immunotherapy schedule by starting immunotherapy when cancer cells are unmasked and CCME modulated for maximum response & least toxicity. 2. Integrated Boost/Pulsed SBRT for dynamic generation of contemporary neoantigen for in-situ vaccination effect and to improve memory cell pool. |
.Immunotherapy Optimization: -Sordo-Bahamonde et al., 2023, [115] -Lussier, DM et al., 2022 [116] . Timed SBRT: -Breen, WG et al., 2020 [117] -He K et al., 2021 [118]. - Mathieu Spaas, et al., 2019 [119] . Oxygenation: -Shibamoto, Y et al., 2016 [99] . In situ vaccination: -Feng K. et al, 2025; 104] -Kewen He. et al, 2021. [118] . Nanoparticle-Immunotherapy: - Naimeng Liu et al., 2025 [105] . Epigenetic therapy: Shweta Josh, et al., 2019. [121] |
| Phase IV |
Post-Primary therapy: Primarily about eliminating MRD/Dormancy/DTPs/ Senescence a. Consolidation Therapy of Immunotherapy Effects, Normalization of ECM and Immune Activated TILs. b. Anti-evolutionary resistance strategy & epigenetic modifiers, e.g., (BET) protein inhibitors c. Normalized soft ECM |
2 to 3 weeks After Primary Therapy. Maintenance Immunotherapy / Targeted therapy as per the guidelines |
1. Design the maintenance therapy with the least long-term side effects. 2. Develop anticancer/repurposed drugs suitable for long-term medications to prevent the recurrence/ eliminate dormant cells, like any other chronic disease. |
. Supple ECM: - Zhao Y, et al, 2020 [30] -Targeting TGFβ, Matrix metallo-proteinases, Integrins, etc.: -Meiling Zhang, et al, 2025 [122] .Targeting DTPs: -Wei Lu et al, 2019 [5] -Williams ED et al, 2019 [54] -BET inhibitors: - Chen M et al, 2024 [89] .Epigenetic Reversion -Pensotti A et al., 2024 [53] . Monitoring: -Nezami et al., 2015 [65] |
| Phase V |
Probative therapy: Primarily to abate inflammation and restore immune editing. Presently Exploratory – Evolutionary Therapy Strategy /EMP Intervention Strategy. Reprogramming of the ECM/Cancer reversion. Prevents late recurrences. Monitoring is done with epigenetic markers. |
Starts from the point when patients are apparently cured/ unacceptable toxicity/ Progression |
1. To keep the ECM supple. Secondly, to target HIF1-α and ROS to flip towards normalization of the ECM and eliminate dormant cells. 2. Reducing inflammation by maintenance therapy, senolytics, and lifestyle modifications or combinations thereof. 3. Evolutionary Infomed Therapy (EIT) or MTET strategy. |
.Reducing ROS/ HIF-1α: -Mengnuo Chen, et al 2024 [89] .Senescence: -Škarková A et al.,2024 [48] . Senolytics: -Susan Short, et al, 2019 [123] . Lifestyle: -Pasquale Marino. et al 2024 [125] -Shujie Liu, et al, 2025 [126] . Evolutionary Therapies: - Gatenby, et al, 2020 [4] - Nezami. et al., 2015. [65] - Škarková A, et al, 2024 [48] . Monitoring: -Nezami. Et al 2015 [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



