Submitted:
21 November 2024
Posted:
25 November 2024
You are already at the latest version
Abstract
The ATP-binding cassette (ABC) transporter superfamily, one of the largest membrane protein families, plays a crucial role in multidrug resistance (MDR) in cancer by mediating the efflux of various chemotherapeutic agents, thereby lowering their intracellular concentrations and diminishing therapeutic effectiveness. Beyond drug efflux, these transporters are also involved in vital biological processes, such as signal transduction in cancer. Over the past few decades, extensive structural and functional research has provided valuable insights into the broad substrate specificity and transport mechanisms of ABC transporters, leading to promising strategies for overcoming MDR. This review will provide a structural understanding of the interactions between ABC transporters and inhibitors to develop novel cancer therapeutics. Additionally, we focus on methods such as irradiation-based immune therapies, thermal therapies, nanomedicine, CRISPR-CAS, and natural therapies that can genetically modify ABC transporters to reduce their expression or reverse the drug efflux ability. Knowledge gained from these approaches can then be translated into the development of new cancer therapeutics that can combat chemotherapy resistance.
Keywords:
ABC transporters
; multidrug resistance (MDR)
; ABC transporter inhibitors
; cancer therapeutics
; drug efflux mechanism
Introduction
In the 21st century, cancer has become one of the leading causes of death globally, with the estimation that one in five men or women develop cancer in a lifetime [1,2]. According to the Global Cancer Observatory statistics, the total number of cancer cases (all types, in both sexes and ages) was recorded as 19, 976, 499, and deaths 9, 743,832 (48.7%) [3]. Over 90 % of these cancer-related deaths are attributed to metastatic cancer, which has been increasingly linked to multidrug resistance (MDR) [4,5,6,7,8]. Chemotherapy, often combined with surgery, immunotherapy, or radiotherapy, remains one of the most effective cancer treatments [9]. However, several studies have shown that ATP-binding cassette (ABC) transporters play a significant role in the active efflux of drugs from the cell and tend to be associated with conferring MDR in cancer cells, resulting in ineffective chemotherapy [10,11]. Therefore, in recent years, there has been a surge in the development of strategies to target ABC transporters in the treatment and prevention of cancers [12,13,14].
The general architecture of ABC transporters
A typical ABC transporter comprises a nucleotide-binding domain (NBD) that is responsible for ATP binding and hydrolysis and a transmembrane domain (TMD) that facilitates substrate export (away from NBDs) or substrate import (towards NBDs) (Figure 1a). Although ABC transporters share a fundamental architecture, they show considerable diversity in substrate specificity, transport mechanisms, and regulation. The NBDs are highly conserved hydrophilic and are located within the cytoplasm for ATP hydrolysis. NBDs comprise an alpha-helical domain, RecA-like domain, and beta domain, while the ATP binding occurs at the interface of two NBDs arranged in a head-to-tail orientation. These NBDs also include the Walker A motif (GXXGXGKS/T, X= any amino acid), which binds alpha and beta phosphates of ATP, while the glutamate and aspartate residues of Walker B (ΦΦΦΦD, Φ= hydrophobic) motifs coordinate with Mg2+ ion and water molecule to interact with the γ-phosphate group of ATP. The signature motif LSGGQ helps to orient the bound ATP molecule [15] (Figure 1b). In stark contrast to the NBDs, the TMDs are structurally diverse and recognize a wide range of substrates. They span the membrane and form channels to facilitate transport. Hence, the TMDs dictate the characteristics of the substrates that are to be transported [16,17] (Figure 1c).
Classification of ABC Transporters
The human ABC transporters are classified into seven subfamilies designated as ABCA to ABCG according to the similarity of their amino acid sequences [18]. Of these, five (subfamilies ABCA-ABCD and ABCG comprise transmembrane domains, while ABCE and ABCF lack transmembrane domains and are involved in cellular functions [19,20] (Figure 2). The human ABCA subfamily is the largest of all ABC transporters and encodes thirteen transporters. These transporters have remained comparatively understudied despite their role in lipid homeostasis and disease association [21,22]. The ABCB subfamily, arguably the most widely studied human ABC transporter, comprises ten transporters that can transport a wide range of substrates, including peptides, drugs, and ions. In this subfamily, ABCB1, also known as multidrug resistance (MDR) protein, is the first identified member and plays a crucial role in the cellular detoxification of cancer cells. To date, a wide range of anticancer drugs have been identified as substrates for ABCB1, and this continues to be a key focus for therapeutic/diagnostic development [23,24,25]. The ABCC subfamily contains twelve transporters that feature type I exporter fold collectively classified as MDR-associated proteins. These transporters have diverse functions, including ion transport, toxin efflux, drug metabolism as well as normal physiology [26,27,28]. Among these, ABCC1 is of considerable pharmacological and clinical importance as it is a major player in the MDR of cancer cells [29,30]. The ABCD subfamily contains four transporters, also known as adrenoleukodystrophy (ALD) transporters. These transporters are comparatively less studied and are mainly involved in fatty acid metabolism [31,32,33]. However, the role of ABCD transporters in cancer has remained inconclusive due to limited protein structural and topological information [34]. So far, five members have been found in the ABCG subfamily that assembles as homo/hetero dimers adopting a type II exporter fold [35,36]. These transporters are found to be involved in either sterol/lipid transport or the mediation of multidrug transport. ABCG2, also known as breast cancer resistance protein (BCRP), is of extreme physiological importance and transports a wide variety of drugs and affects their pharmacokinetics [37]. Due to its critical role in toxin efflux and drug resistance in cancer cells, it remains an attractive target for therapeutic development [38,39]. Among these forty-eight human ABC transporters with distinct functions, approximately thirteen are directly relevant to MDR (Table 1) [40]. MDR was first described over 50 years ago in a subline of HeLa cells resistant to actinomycin D, with subsequent studies showing cross-resistance across different cell lines [41]. The discovery of a transporter responsible for actively expelling drugs from resistant cells, called P-glycoproteins, altered drug permeability in Chinese hamster ovary and fibroblast cells [42]. In this review, we focus on structural details and inhibitor binding of the most clinically relevant ABC transporters, ABCB1, ABCB4, ABCB6, ABCC1, and ABCG2, that cause MDR during cancer therapy. We emphasize the latest novel inhibitors that are currently in preclinical and clinical studies, followed by proposing emerging strategies targeting ABC transporters to provide a broad perspective to understand the MDR in cancers.
ABCB1
ABCB1 is expressed at several blood-organ barriers and is pharmacologically important because its overexpression in certain tumors reduces the uptake of specific orally administered drugs [43,44,45,46]. ABCB1 is the first identified and most characterized MDR transporter and has been long recognized as a viable target to overcome cancer [47]. Due to the clinical significance of ABCB1, its structure and mechanism have been extensively investigated [48,49,50,51,52]. Although high-resolution x-ray crystallography structures of ABCB1 were available earlier, only using single particle cryoelectron microscopy (cryo-EM) it was possible to determine the structure of the human protein [51]. ABCB1 is a monomeric ABC transporter with pseudo-symmetric halves, each containing TMD & NBD (Figure 3a). The largely conserved NBDs dimerize to bind and hydrolyze ATP at their interface while the TMDs create a large cavity exceeding 6000 Å3 size through which substrates are transported [48]. ABCB1 has often been viewed as a “hydrophobic vacuum cleaner” as most of the substrates that pass through this large cavity are hydrophobic in nature [53,54]. Numerous structural and functional studies have revealed the molecular basis of ABCB1 poly specificity, allowing it to bind and efflux a diverse array of structurally dissimilar compounds [55,56]. Several studies have shown that hydrophobic residues that are located on the upper side of the pocket from TMDs (4&6, 10&12) play a critical role in substrate binding [48,54]. It is shown that different residues in the binding pocket present in TMDs may be involved in interaction with different substrates: For instance, the high-resolution structure of paclitaxel bound to ABCB1 depicts Q725, Q347, Q990 as interacting residues with the paclitaxel while the zosuquidar bound ABCB1 reveals M985, F982 as the interacting residues [51,57] (Figure 3a). Additionally, recent high-resolution cryo-em structures have shown that two inhibitor molecules can bind simultaneously within the cavity formed by the TMDs. One molecule occupies the central binding pocket, while the second binds to the access tunnel, which extends from the central binding pocket [54]. However, the exact purpose of ABCB1’s ability to bind two inhibitor molecules remains to be explored. Furthermore, the conserved glutamine Q475 in NBD1 coordinates with Mg2+ and gamma-phosphate of ATP thus, they play an important role in ATP hydrolysis and drug transport [52]. Among the therapeutic compounds that are transported by ABCB1, vinblastine and vincristine belonging to the vinca alkaloids family have been extensively studied and used in antitumor therapies [58,59]. In addition to these, a plethora of anticancer drugs (Table 2) have been tested to overcome the MDR in cancer. However, most of the inhibitory compounds that were developed showed promise in cellular assays but didn’t pass clinical trials due to selectivity, efficacy, and toxicity issues [60,61,62]. Nevertheless, these setbacks didn’t diminish interest in developing ABCB1 inhibitors, and research efforts are ongoing. This perseverance led to the discovery of the best-known small molecule inhibitor MRK16, which was developed against leukemia resistant to adriamycin and a monoclonal antibody UIC2 [54,57,63]. While the developed monoclonal antibody was not successful in clinical applications, the small molecule MRK16 was found to interfere only with the transport of bulky substrates like vincristine and not with flat molecules such as doxorubicin [51,54]. Consequently, many drug developers are now focusing on discovering new compounds that can bypass the activity of ABCB1. The details of this approach will be discussed under the emerging strategies section later in the review.
ABCB4
ABCB4 was originally identified as a sister protein to ABCB1, and these two proteins share 85% sequence similarity [64]. Various studies in the literature have shown that ABCB4 is capable of recognizing and transporting some ABCB1 substrates with low capacity [65,66,67] however, it is to be noted that this is likely not the physiologically relevant function of ABCB4. ABCB4 is expressed mainly in hepatocytes and is an active phospholipid translocator that is critical for bile generation [64,68,69,70]. The transport of phospholipids by ABCB4 is stimulated by bile salts exported by ABCB11, which in turn facilitates cholesterol export via ABCG5-ABCG8. Several mutations in the ABCB4 are associated with progressive familial intrahepatic cholestasis, a severe liver disease [71,72]. In addition to this, dysfunction and epigenetic silencing of ABCB4 is directly evident in hepatobiliary malignancies [73,74,75]. Despite its importance in health and diseases, so far only two high-resolution structures of ABCB4 are available [76,77], partly due to difficulties in obtaining suitable amounts of functional and pure ABCB4 [78]. ABCB4 consists of six intracellular domains and six extracellular loops separated by twelve TMDs and contains two NBDs. The TMD4 and TMD10 form a Y-shaped occluded cavity of ∼ 6500 Å3 at the center of the transporter, wherein the lipid phosphatidylcholine is bound [77] , resembling the structure of the ABCB1 transporter [51,54] (Figure 3b). The phosphatidylcholine binding pocket is above the residue W234 with its choline moiety stabilized by cation- π interactions with this residue. Azole antifungals, like posaconazole, inhibit ABCB4, reducing phosphatidylcholine secretion and increasing bile toxicity, which can lead to drug-induced liver injury (DILI) in susceptible patients [79,80].
ABCB6
ABCB6 is a homodimeric transporter comprising a single TMD and NBD with an N-terminal extension (TMD0) with TMDs responsible for substrate translocation, while the NBDs are involved in ATP binding and hydrolysis [17]. The functional role of TMD0 in ABCB6 is not fully understood, but earlier studies indicate that TMD0 may perform the regulatory, trafficking, localization, and stability role of the transporter [81,82]. Since its first discovery in 1996 [83], ABCB6’s role has been shown in drug resistance [84,85] and toxic metal resistance [86] while primarily functioning as a porphyrin transporter [87,88,89]. Mutations in the ABCB6 gene are linked to tissue-specific disorders such as ocular coloboma [90], familial pseudohyperkalemia [91], porphyria, and dyschromatosis universalis hereditarian [92]. ABCB6 is demonstrated to transport a broad spectrum of substrates, including porphyrin metabolites [93], heavy metals [86], and organic compounds [94]. In addition to these, ABCB6 can interact with various anticancer drugs, including camptothecin, cisplatin, paclitaxel, doxorubicin, methotrexate, and vincristine [84,85,95,96,97]. The molecular mechanisms of transportation and substrate recognition for ABCB6 remained largely unclear until the first glimpse was provided by the cryo-EM structure very recently [98]. However, this study has a major limitation as it is mainly based on a 4.0-Å resolution model, which is borderline sufficient to identify side chains [98]. This is followed by the determination of a few more cryo-EM structures of ABCB6 in the apo-state, nucleotide, and cadmium ion bound (Cd(II)) state to shed light on the mechanism that ABCB6 mediates to transport a broad range of substrates [99,100,101,102]. The overall structure of ABCB6 shows an elongated shape with 130 Å in height, and the two NBDs, like two “legs,” are separated by 35 Å (Figure 3c). In the apo-state, ABCB6 adopts an inward-facing conformation, and the TMD region contains a loop acting like a gate named a “plug.” The substrate-binding cavity within the TMD is divided into two parts: 1) closed cavity and 2) open-form cavity, which is different when compared to most substrate-binding pockets found in other ABC transporters. The “plug” of ABCB6 comprises flexible residues that provide flexibility for multi-substrate binding, and in the inward position, the “plug” acts as an obstructing substrate and could be opened during ATP-driven conformational change in ABCB6. The “plug” together with residue W546, provides a hydrophobic environment for substrate translocation, and the “plug” divides TMDs into two pockets, with pocket one playing a role in substrate recruitment on the other and pocket 2 for the transport of substrate. Moreover, it was found that ABCB6 displayed resistance to anticancer drugs such as 5-fluorouracil (5-FU), SN-38, and vincristine [84]. Therefore, obtaining more structural information about the specific regions within the transmembrane domains (TMD) of ABCB6 is essential to gain mechanistic insights. This knowledge is crucial for designing targeted inhibitors or drugs that can effectively modulate ABCB6 activity.
ABCC1
ABCC1 is a single polypeptide chain consisting of TMDs and NBDs. It shares approximately 23% sequence identity with ABCB1 and exhibits a notable overlap in the range of substrates it transports [103]. Only one nucleotide-binding domain located on the NBD2 is responsible for hydrolyzing ATP and providing energy for translocation [104]. ABCC1 contains an N-terminal membrane-bound region (TMDo) domain that links to the transporter core through a Lo linker (Figure 3d). The loss or mutation of Lo linker results in protein misfolding and defective function [105]. So far, ABCC1 seems to have the broadest substrate specificity of any ABCC subfamily member, at least in vitro [106]. A key question in understanding the multidrug resistance of ABCC1 is how a single transporter can recognize a wide range of structurally diverse molecules. This has led to significant interest in identifying the atomic determinants responsible for its ability to recognize such a wide range of substrates. The first structural insights into ABCC1 were provided by the bovine ABCC1, which shares 91% amino acid identity with human ABCC1. Later, the cryo-EM structure of the human counterpart was determined [103,104]. Structural studies of this transporter reveal that it forms a single, bipartite substrate-binding site, which enables the transporter to accommodate and recognize a broad spectrum of substrates with varying chemical structures. The binding pocket of the ABCC1 transporter is formed by two distinct bundles: TM1 and TM2. The residues on the inner-facing side of these bundles differ significantly in their properties [104]. Positively charged residues are in TM1, while hydrophobic residues are found in TM2. The positively charged region typically interacts with the glutathione (GSH) moiety, while the hydrophobic region binds to the substrate [103]. The ABCB1 and ABCC1 are both ABC transporters that contribute to multidrug resistance but differ significantly in their functional properties [107]. ABCB1 primarily transports hydrophobic and weakly cationic compounds, while ABCC1 mainly transports organic acids. The translocation pathways of these transporters are chemically distinct, with ABCB1 having a hydrophobic pathway with acidic patches and ABCC1 having a basic pathway. ABCB1 uses a "hydrophobic vacuum" model, where substrates partition into the lipid bilayer before entering the transporter, while ABCC1 recruits substrates directly from the cytoplasm via its well-ordered transmembrane helices. ABCB1 substrate recognition is influenced by membrane partitioning, while ABCC1’s ability to recognize a wide range of substrates is due to the bipartite nature and flexibility of its substrate-binding site [51,54,103,104]. ABCB1 is shown to confer MDR to etoposide, daunorubicin, and vinblastine in ABCB1-transfected HEK293 cells [108]. Apart from a few reported inhibitors of ABCC1 (Table 2), there is a lack of potent inhibitors, highlighting the need for additional substrate-bound cryo-EM structures and biochemical validations. These are essential for achieving an atomic-level understanding of the poly-substrate specificity of ABCC1.
ABCG2
ABCG2 consists of six transmembrane helices and one ATP-binding site, with one nucleotide-binding domain (NBD) and one transmembrane domain (TMD) on each polypeptide chain. These chains dimerize to form a functional active transporter. [109] (Figure 3e). This transporter recognizes and transports a broad spectrum of substrates, primarily hydrophobic, polycyclic, and relatively flat molecules. The transporter also recognizes endogenous substrates as well as exogenous cytotoxic compounds [110,111] (Table 2). In colon cancer, ABCG2 was observed to confer resistance against various chemotherapy drugs, including topotecan, SN-38, mitoxantrone, doxorubicin, and several tyrosine kinases [111,112,113]. Owing to its clinical relevance, ABCG2 is listed on the US Food and Drug Administration and the European Medicines Agency lists of transporters to be checked for transporter-drug interactions [114]. Over the years, there have been many in vivo studies on the transporter that provided information on localization, physiological functions, trafficking, and substrate specificity [115,116,117]. However, the mechanistic understanding of ABCG2 was limited due to a lack of high-resolution structure, which is partly because of the challenges associated with its overexpression and functional purification. While the crystal structure [118] and low-resolution electron microscopy [119] imaging provided the first structural glimpse of the transporter, it was only after the determination of cryo-EM structure of human ABCC2 that the structural understanding became much clearer [109,120,121,122] (Figure 3e). These structures reveal an inwards-open confirmation of ABCG2 with the binding pocket formed between TM2 and TM5a domains. The transmembrane helices and intracellular loops of ABCG2 are shorter than B-subfamily ABC transporters, resulting in a smaller distance between the NBDs and the membrane. Thus, ABCG2 is more compact [109]. Two standout features of the ABCG2 structures are the presence of 1) a deep hydrophobic cavity within two TMDs and 2) an external cavity located below the external loop separated by a “leucine plug.”[109]. This structural arrangement suggests that ABCG2’s preference for flat substrates contrasts with ABCB1, which favors globular hydrophobic substrates [51]. As for its potential value in the treatment of cancer and pharmacotherapy, huge efforts have been invested over the past decades in the development of specific inhibitors against human ABCG2 [111,112,113,123]. The first and well-characterized ABCG2 inhibitor, fungal toxin fumitremorgin C (FTC), has been ruled out owing to its neurotoxicity [124] , while its derivative Ko143 was less toxic but lacked specificity [125]. Attempts have also been made to develop ABCG2 inhibitors derived from tariquidar, a third-generation ABCB1 inhibitor [126,127,128]. In this context, the structures of ABCG2 bound to the Ko143-derived inhibitors, and the tariquidar-derived inhibitor molecules have been determined [120]. It was shown that depending on their shape, the inhibitor molecules can symmetrically or asymmetrically occupy the binding pocket cavity within the TMDs. Additionally, febuxostat, an approved drug used in patients with hyperuricemia, was serendipitously identified as a strong ABCG2 inhibitor both in vitro and in vivo [129]. Furthermore, a recent study has demonstrated that two potential anti-cancer compounds are currently under clinical examination that could competitively inhibit both ABCG2 and ABCB1 [130]. Nonetheless, to the best of our knowledge, the clinical use of such drugs for ABCG2 inhibition has not yet been approved due to concerns about safety and specificity. This may have been a common reason responsible for the failure of the clinical development of ABCG2 inhibitors thus far. Therefore, much larger efforts are needed to understand the catalytic cycle of ABCG2 and in general other multidrug transporters in physiologically relevant conformational states, including their intermediates.
Mechanistic Overview of Drug Transport
The common structural features among the MDR-related ABC transporters feature a central substrate-binding pocket, which is remarkably flexible and can have the adaptability to ac- accommodate a wide range of substrates [131]. The universal mechanism of drug translocation by MDR-related ABC transporters is commonly explained by the "alternating access" model. According to this model, the transporter undergoes significant conformational changes between two states: 1) inward-facing and 2) outward-facing. In the inward-facing conformation, the binding sites are accessible to substrates and ATP. Upon ATP binding, the transporter undergoes a conformational shift to the outward-facing state, which allows the substrates to get expelled. Following substrate release, ATP hydrolysis triggers the return of the transporter to its inward-facing conformation. These conformational changes, driven by ATP binding and hydrolysis, enable the transporter to efficiently transport substrates in/out of the membrane, thereby contributing to multidrug resistance [132]. Although the “alternating access” model is universal among MDR-related ABC transporters, there are minor distinctions among them. In the case of ABCB1, the bound drug is sealed by TM4 & TM10, which prevents the drug from returning to the cytosol. The two halves of the ABCB1 transporter move closer together, collapsing the substrate pocket and transitioning to an outward-facing conformation, facilitating substrate release before ATP hydrolysis [51]. In the case of ABCB4, which employs a similar mechanism as ABCB1, the central binding pocket harbors a phospholipid instead of a drug. This makes the proposed mechanism for ABCB4 unique, as it involves relatively subtle adaptations that fine-tune a general multidrug export mechanism specifically for phospholipid transport [77]. Compared to ABCB1, ABCB6 exhibits adaptations in both substrate binding and the ATP-triggered transport pathway. The long hydrophobic "plug" helps to form a flexible cavity that accommodates substrate binding. Following substrate binding, the surrounding helices adjust, and the "plug" reduces interactions, allowing ABCB6 to shift to an ATP-sensitive conformation [99]. In contrast, ABCC1 undergoes a rearrangement of pocket residues upon substrate binding rather than NBD movement [103]. In the case of ABCG2, the substrate recruitment occurs via cytoplasm or directly through the membrane. The substrate first binds to cavity 1 of ABCG2, and upon ATP binding in NBDs, the substrate later moves to cavity two and then gets expelled out of the cells [109].
Translational role and Therapeutic Targeting Strategies for ABC Transporters
The highly conserved ABC transporters express in a tissue-specific manner, and their role in tumors is well documented [133,134] . According to the Gene Expression Atlas and the Human Protein Atlas, it is well-known that ABC transporters overexpressed in a variety of cancers, for example, ABCB1, ABCC1, and ABCG2 overexpression has been shown in acute myelocytic leukemia (AML), breast cancer, thyroid cancer, ovarian cancer, and multiple myeloma [135]. Overexpression of ABCB1 was also observed in melanoma and other multiple cancer cells. Research studies also confirmed that ABC transporters are one of the potential reasons for the development of MDR [136,137]. Despite their role in drug resistance, several ABC transporter inhibitors have been developed, particularly against ABCB1, ABCG2, and ABCC1. However, clinical studies failed with first- and second-generation ABC transporter inhibitors [138,139]. Recently Friedenberg et al., showed the beneficial effects of combination therapy using ABC transporter inhibitors. Studies revealed the use of valspodar (PSC-833) along with doxorubicin, vincristine, and dexamethasone versus valspodar alone in Phase III studies [140]. Development of third-generation inhibitors such as elacridar (GF120918), tariquidar (XR9576), zosuquidar (LY335979), and dofequidar evaluated as improved selective inhibitors against ABC transporters [55]. Reports suggested that Phase III clinical studies were carried out using tariquidar in combination with paclitaxel along with either carboplatin or vincristine in patients with non-small-cell lung cancer [141,142]. However, this, along with other clinical studies performed at the National Cancer Institute (NIH, Bethesda, USA) in Tariquidar, suggest that there is still optimism that therapeutic modulation of MDR mediated by ABC transporters may be beneficial to patients. Overall, these inhibitors showed limited clinical success; this topic has been extensively reviewed elsewhere [143,144,145]. Multiple reasons were recognized for the failure of these ABC inhibitors in clinical trials, such as side effects due to high drug toxicity, development of MDR due to multifactorial mechanisms, substrate overlap, and impaired drug clearance. For example, the inhibitor toxicity developed on ABC transporters could also cause the inhibition of metabolic cytochrome p450 enzyme function. Cytochrome P450 enzymes are becoming targets of ABC transporter inhibitors, particularly, P-gp inhibitors tested in combination with anticancer drugs were shown toxicity at tolerable doses. The latest P-gp inhibitors have also been reported to exhibit toxicity by inhibiting metabolic cytochrome p450 enzymes [139,146] [147,148,149,150]. Clinical trial investigations were terminated when using tariquidar, P-gp inhibitor (Third generation) combined with first-line chemotherapy in non-small cell lung cancer patients [143,144,145]. To overcome these limitations, there is a need to develop more effective combinational therapies to overcome MDR resistance and chemotherapeutic failures for effective cancer treatments [55,145,151].
Novel Therapeutic Strategies Targeting ABC Transporters
Novel therapeutic strategies are developed to treat multidrug resistance (MDR) in diseases, especially cancer and autoimmune diseases. Widely used therapeutic approaches are Light-based therapies coupled with inhibitors, nanomedicine-based therapies, RNA interference, and immunotherapy (Figure 4). Despite having advantages and disadvantages over different therapeutic approaches, we are highlighting some of them to discuss here. The current review addresses the advantages of applying novel therapeutic strategies coupled with ABC transporter inhibitors to overcome MDR.
Light-Based Therapies Coupled with ABC Transporter Inhibitors
Light-based therapies have been widely used as approved therapy against cancers across the globe [152]. The synergistic role of light irradiation and drug release or activation in killing cancer cells has shown promising outcomes in preclinical studies. This approach is extensively used in treating cancers, such as bladder, head & neck, and skin cancers [152]. The light-based therapies include photothermal therapy, photoimmunotherapy, and Photodynamic therapy (PDT). However, each method has its advantages and disadvantages [152,153,154,155,156]. In photothermal therapy, infrared radiation is used to generate heat on the targeted tissues to kill cancer cells. The limitation of this therapy is that the radiation can damage the targeted tissue and trigger further inflammation. With photoimmunotherapy, the radiation is combined with heat sensor-conjugated antibody specific to target protein expressed in cancer cells [153,154,155]. PDT utilizes light irradiation with a non-toxic photosensitizer agent that targets tumor cells and kills them by generating ROS [156]. These strategies are also being used for diagnostic and treatment purposes in the clinic. For example, benzoporphyrin derivative is an FDA-approved photosensitizer for age-related molecular degeneration and cancer, and it is also used as a conjugate with the ABCB1 monoclonal antibody UIC2 to target resistant cells [157]. PDT is the most advanced light-based treatment against cancers, and they are also very effective for all types of tumors associated with MDR. Coupling ABC transporter inhibitors with PDT may be a more effective approach to treating tumor-associated MDR. Recent studies identified new photosensitizer substrates redaporfin for P-gp, BPD for ABCG2 and P-gp, and rose Bengal for P-gp, ABCG2, and MRP1 [148,150,152]. It is shown that fumitremorgin [158], a P-gp inhibitor, significantly blocked the transport facilitated by ABCG2 and P-gp of rose Bengal and BPD. MCF-7/VP cells were found to have reduced intracellular accumulation of rose bengal, which was restored with the MRP1 inhibitor (MK571) [159]. This study provides novel insights that the known temoporfin, talaporfin sodium, methylene blue, and indocyanine green are not the substrates of ABCG2, P-gp, or MRP1, indicating that identifying the specific ABC transporter substrate is important for the success of photodynamic therapy. Together, coupling ABC transporter inhibitors and light energy with specific photosensitizers may provide more effective treatment by limiting the exportation of drugs from the target cells.
DNA-Based Therapeutics
Single-stranded DNA aptamers are highly selective towards specific proteins and are used to conjugate drugs [160]. A DNA aptamer conjugated with doxorubicin was developed against ABCG2 protein and assessed for its role in resistant breast cancer cells and found to potentiate the uptake and cytotoxicity of doxorubicin [161]. Further, DNA aptamers were designed against ABCG2-glycosylated extracellular regions using molecular engineering methods. The combination of glycosylated DNA aptamer with a monoclonal anti-ABCG2 complex was shown to block ABCG2-mediated doxorubicin transport and, thus, reverse MDR in resistant HepG2 liver cancer cells [162]. Besides single-stranded DNA aptamers. DNA-capped quantum dots encapsulated in gold nanoparticles carrying doxorubicin were used to target ABCC1 mRNA expression, which remarkably increased the efficacy of the drug in resistant cancer cells [163]. Another study designed a catalytic DNA or DNAzyme to target ABCB1 mRNA, which further significantly decreased drug resistance in MCF breast cancer cells [164]. However, for all these studies further clinical trials are needed to prove their in vivo efficacy.
Nanomedicine Approaches
The nanoparticle (NP) based medicinal approach is becoming popular in target cell drug delivery. Due to their unique characteristics, such as increased drug stability, sustained drug release, and extended life toward target drug delivery. Nanomedicine is a great choice for combating MDR in chemotherapy. NPs ranging as low as 10 nm to 200 nm size particles, coupled with chemotherapeutic agents, were delivered selectively to target cancer cells. NPs coupled with ABC transporter inhibitors have shown progressive results toward drug stability and delivery approaches. Several drugs already in the clinic with nano-formulations (such as Lipusu, Doxil, Abraxane, and Genoxil-PM) are encouraging to fight against MDR in cancers [156,165,166]. Recent studies evaluated testing nanomedicine platforms like mesoporous silica (MSN), Poly-lactic nanoparticles (PLA), and Graphene Quantum Dots (GQDs) along with selected drugs to inhibit MDR1(P-glycoprotein) and MRP1 [165,167,168]. PLA, MSN, and GQDs were reported to be nontoxic in MDA-MB-231 cancer cells. It’s been reported that MRP1 overexpression in MDR and targeting MRP1 via nanomedicine might be a useful way to overcome the MDR [136]. These studies revealed that PLA exhibits moderate inhibition of doxorubicin efflux by MRP1, and MSN is reported to be a strong inhibitor for doxorubicin efflux by MRP1. Doxorubicin is a chemotherapeutic drug used in multiple cancer treatments. NPs coated with chemotherapeutic agents alone couldn’t overcome MDR in the tumor environment, whereas combinational therapy includes, ABC transporters and chemotherapeutic agents might be effective in preventing long-term MDR in chemoresistance diseases.
RNA Interference and Gene Editing
RNA interference (RNAi) is a biological process that can be used to regulate target gene expression. RNAi regulate gene expression by interfering with messenger RNA (mRNA). Research studies suggested that RNAi has become a promising tool for gene therapy to reverse MDR mediated by ABC transporters. Currently, RNA interference to ABC transporters will be carried out in multiple ways, such as small interfering RNA (siRNA), short hairpin RNA (shRNA), and microRNA (miRNA) [169,170,171]. However, each of them has advantages and disadvantages in transiently regulating gene expression. Importantly, clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas9) show permanent regulation of gene expression in a specific target-oriented way. Here, we discuss the application of miRNAs and CRISPR/Cas9 regulation of ABC transporter gene expression
miRNA Targeting ABC Transporters
Small noncoding RNA molecules of 20-30 nucleotides, referred to as microRNAs, exhibit hairpin structures. miRNAs turned into important modulators contributing to the adaptive regulation of ABC transporters. ABC transporter family member P-gp, encoded by the ABCB1 gene, became the most targeted gene using miRNA [171,172]. Since ABCB1 is also known as the MDR1 gene, most of the researchers focused on regulating ABCB1 gene expression, and here, we are emphasizing research work carried out targeting the ABCB1 gene using miRNA [170,171] (Table 3).
CRISPR/Cas9 Targeting ABC Transporter
Researchers identified clustered regulatory interspaced short palindromic repeats/(CRISPR)-associated protein 9 (CRISPR/Cas9) system provides more precise gene editing technology at affordable cost. The mechanism behind this gene editing can be simply explained as the CRISPR/Cas9 system specifically targets and cleaves a specific DNA sequence, which is performed by Cas9 nuclease and interlinked with single guide RNA (sgRNA) [173,174,175]. By using CRISPR/Cas9 technology, the ABC transporter gene family is targeted for gene regulation. The principal ideology of using CRISPR/Cas9 mediated target of the ABC transporter family was to reduce drug efflux as well as to sensitize cancer cells to chemotherapeutic drugs. Here, providing several research studies showed effective gene regulation of ABC transporters using CRISPR/Cas9 technology. Research studies showed that CRISPR/cas9 was used to reverse the resistance of ovarian cancer cells to doxorubicin. Research studies also reported that CRISPR/Cas9-derived knockout of ABCB1 can improve the response to temozolomide, carmustine, and lomustine in gliobastoma multiforme cell model [176]. It is reported that targeting the knockdown of the ABCB1 gene using CRISPR/Cas9 technology significantly increased the accumulation of doxorubicin inside cells and enhanced chemosensitivity in HCT-8/v cancer cells [177,178], showing doxorubicin cell death increased upon CRISPR/Cas9 knockout of MDR1 in doxorubicin-resistant breast cancer cells [179,180]. Targeting the ABCB1 gene using CRISPR/Cas9 technology in ovarian cancer cells leads to increased sensitivity for chemotherapeutic drugs [176,181]. Given the many studies that are currently progressing, it is hoped that CRISPR/Cas9 technology will find solutions to overcome ABC transporter-mediated MDR and chemotherapeutic resistance. Also, the way will be opened for the clinical use of this technology to overcome drug resistance in cancers.
Overall, these multifaceted approaches targeting ABC transporters provide significant potential to overcome MDR in cancer, making treatment more effective and adaptable (Figure 4).
Conclusions, Open Questions, and Future Perspectives
With significant advancements in structural biology techniques, our understanding of ABC transporter topology, ligand binding, and the substrate transport cycle has greatly improved. This review thoroughly discusses these topics and highlights their role in cancer-MDR. However, several mechanistic insights into these transporters remain elusive. For example, the role of the flexible linker region (50-100 amino acids), which typically connects the first NBD to the second TMD, remains enigmatic for most ABC transporters [182,183]. In the case of ABCB1, it is shown that shortening the linker region reduces the confirmational flexibility, affecting the substrate transport [184]. Nevertheless, the precise role of the central linker region warrants further investigation in future research. Similarly, the role of TMD0 exclusively found in the ABCB family, which typically links TMD1 by a flexible linker, remains largely unknown [103,185,186,187,188]. Although it appears that TMD0 doesn’t influence the substrate binding and transport, it may have a potential contribution to the occurrence of MDR. Hence, dedicated research is needed to understand the comprehensive role of TMD0 in ABC transporters. Another important area for investigation is specific lipid interactions of ABC transporters, particularly regarding both lipid transport and structural insights, as these remain challenging. Although the advent of AlphaFold [189] has made valuable availability of the structures of human ABC transporters for three-dimensional analysis and structure-based in silico drug design. These predicted models, which currently represent single states, offer limited insight into small-molecule, lipid, and protein interactions. However, the latest version of AlphaFold3, which has the capability to predict bimolecular interactions, needs to be thoroughly evaluated as part of future projects focused on ABC transporters [190]. Taken together, all these highlight the need for more exploratory studies that elucidate the mechanisms of ABC transporters, and particularly the MDR-related ones.
References
- Jokhadze, N., A. Das, and D.S. Dizon, Global cancer statistics: A healthy population relies on population health. CA Cancer J Clin, 2024. 74(3): p. 224-226. [CrossRef]
- Siegel, R.L., et al., Cancer statistics, 2023. CA Cancer J Clin, 2023. 73(1): p. 17-48.
- Ferlay J, E.M., Lam F, Laversanne M, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. . 2024.
- Ahmed, S., et al., Anticancer Potential of Furanocoumarins: Mechanistic and Therapeutic Aspects. Int J Mol Sci, 2020. 21(16). [CrossRef]
- Housman, G., et al., Drug resistance in cancer: an overview. Cancers (Basel), 2014. 6(3): p. 1769-92. [CrossRef]
- Mansoori, B., et al., The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull, 2017. 7(3): p. 339-348. [CrossRef]
- Cancer multidrug resistance. Nature Biotechnology, 2000. 18(10): p. IT18-IT20.
- Emran, T.B., et al., Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front Oncol, 2022. 12: p. 891652nce in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front Oncol, 2022. 12: p. 891652.
- Debela, D.T., et al., New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Medicine, 2021. 9: p. 20503121211034366. [CrossRef]
- Fletcher, J.I., et al., ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer, 2010. 10(2): p. 147-56. [CrossRef]
- Cui, Q., et al., Gaseous signaling molecules and their application in resistant cancer treatment: from invisible to visible. Future Med Chem, 2019. 11(4): p. 323-336. [CrossRef]
- Doyle, L.A., et al., A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A, 1998. 95(26): p. 15665-70. [CrossRef]
- Allikmets, R., et al., A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res, 1998. 58(23): p. 5337-9.
- Giacomini, K.M., et al., New and Emerging Research on Solute Carrier and ATP Binding Cassette Transporters in Drug Discovery and Development: Outlook From the International Transporter Consortium. Clin Pharmacol Ther, 2022. 112(3): p. 540-561. [CrossRef]
- Stockner, T., R. Gradisch, and L. Schmitt, The role of the degenerate nucleotide binding site in type I ABC exporters. FEBS Lett, 2020. 594(23): p. 3815-3838. [CrossRef]
- Rosenberg, M.F., et al., Three-dimensional Structures of the Mammalian Multidrug Resistance P-glycoprotein Demonstrate Major Conformational Changes in the Transmembrane Domains upon Nucleotide Binding*. Journal of Biological Chemistry, 2003. 278(10): p. 8294-8299. [CrossRef]
- Thomas, C., et al., Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett, 2020. 594(23): p. 3767-3775. [CrossRef]
- Borst, P. and R.O. Elferink, Mammalian ABC transporters in health and disease. Annu Rev Biochem, 2002. 71: p. 537-92. [CrossRef]
- Murina, V., et al., ABCF ATPases Involved in Protein Synthesis, Ribosome Assembly and Antibiotic Resistance: Structural and Functional Diversification across the Tree of Life. J Mol Biol, 2019. 431(18): p. 3568-3590. [CrossRef]
- Navarro-Quiles, C., E. Mateo-Bonmatí, and J.L. Micol, ABCE Proteins: From Molecules to Development. Front Plant Sci, 2018. 9: p. 1125. [CrossRef]
- Fitzgerald, M.L., Z. Mujawar, and N. Tamehiro, ABC transporters, atherosclerosis and inflammation. Atherosclerosis, 2010. 211(2): p. 361-70.
- Xie, T., et al., Cryo-EM structures of the human surfactant lipid transporter ABCA3. Sci Adv, 2022. 8(14): p. eabn3727. [CrossRef]
- Dietrich, C.G., A. Geier, and R.P. Oude Elferink, ABC of oral bioavailability: transporters as gatekeepers in the gut. Gut, 2003. 52(12): p. 1788-95. [CrossRef]
- Durmus, S., J.J. Hendrikx, and A.H. Schinkel, Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv Cancer Res, 2015. 125: p. 1-41.
- (USFDA), U.F.A.C.D.E.R., Invitro drug interaction studies—cytochrome P450 enzyme- and transporter-mediated drug interactions: guidance for industry. Guidance Doc., US FDA., 2020.
- Wang, J.Q., et al., Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist Updat, 2021. 54: p. 100743. [CrossRef]
- Chen, Z.S., et al., Characterization of the transport properties of human multidrug resistance protein 7 (MRP7, ABCC10). Mol Pharmacol, 2003. 63(2): p. 351-8. [CrossRef]
- Hopper-Borge, E., et al., Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res, 2009. 69(1): p. 178-84.
- Leslie, E.M., R.G. Deeley, and S.P. Cole, Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol, 2005. 204(3): p. 216-37. [CrossRef]
- Wang, L., et al., Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. eLife, 2020. 9: p. e56451. [CrossRef]
- Quazi, F., S. Lenevich, and R.S. Molday, ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun, 2012. 3: p. 925. [CrossRef]
- van Roermund, C.W., et al., The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. Faseb j, 2008. 22(12): p. 4201-8.
- van Roermund, C.W., et al., Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid β-oxidation. Biochim Biophys Acta, 2011. 1811(3): p. 148-52.
- Xu, D., et al., Cryo-EM structure of human lysosomal cobalamin exporter ABCD4. Cell Research, 2019. 29(12): p. 1039-1041.
- Liang, X., et al., p62/mTOR/LXRα pathway inhibits cholesterol efflux mediated by ABCA1 and ABCG1 during autophagy blockage. Biochem Biophys Res Commun, 2019. 514(4): p. 1093-1100. [CrossRef]
- Hegyi, Z. and L. Homolya, Functional Cooperativity between ABCG4 and ABCG1 Isoforms. PLoS One, 2016. 11(5): p. e0156516.
- Leslie, E.M., R.G. Deeley, and S.P.C. Cole, Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicology and Applied Pharmacology, 2005. 204(3): p. 216-237.
- Mo, W. and J.T. Zhang, Human ABCG2: structure, function, and its role in multidrug resistance. Int J Biochem Mol Biol, 2012. 3(1): p. 1-27.
- Szakács, G., et al., The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME–Tox). Drug Discovery Today, 2008. 13(9): p. 379-393. [CrossRef]
- Kadioglu, O., et al., Effect of ABC transporter expression and mutational status on survival rates of cancer patients. Biomed Pharmacother, 2020. 131: p. 110718. [CrossRef]
- Goldstein, M.N., I.J. Slotnick, and L.J. Journey, In vitro studies with HeLa cell line sensitive and resistant to actinomycin D. Ann N Y Acad Sci, 1960. 89: p. 474-83. [CrossRef]
- Juliano, R.L. and V. Ling, A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta, 1976. 455(1): p. 152-62. [CrossRef]
- Fromm, M.F., Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci, 2004. 25(8): p. 423-9. [CrossRef]
- Wishart, D.S., et al., DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res, 2006. 34(Database issue): p. D668-72. [CrossRef]
- Giacomini, K.M., et al., Membrane transporters in drug development. Nature Reviews Drug Discovery, 2010. 9(3): p. 215-236. [CrossRef]
- Robey, R.W., et al., Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer, 2018. 18(7): p. 452-464. [CrossRef]
- Gottesman, M.M. and V. Ling, The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett, 2006. 580(4): p. 998-1009. [CrossRef]
- Aller, S.G., et al., Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science, 2009. 323(5922): p. 1718-22.
- Jin, M.S., et al., Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature, 2012. 490(7421): p. 566-569. [CrossRef]
- Kodan, A., et al., Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc Natl Acad Sci U S A, 2014. 111(11): p. 4049-54. [CrossRef]
- Alam, A., et al., Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science, 2019. 363(6428): p. 753-756. [CrossRef]
- Kim, Y. and J. Chen, Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science, 2018. 359(6378): p. 915-919. [CrossRef]
- Waghray, D. and Q. Zhang, Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. Journal of Medicinal Chemistry, 2018. 61(12): p. 5108-5121. [CrossRef]
- Nosol, K., et al., Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc Natl Acad Sci U S A, 2020. 117(42): p. 26245-26253. [CrossRef]
- Dash, R.P., R. Jayachandra Babu, and N.R. Srinivas, Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur J Drug Metab Pharmacokinet, 2017. 42(6): p. 915-933. [CrossRef]
- Srinivas, N.R., Understanding the role of tariquidar, a potent Pgp inhibitor, in combination trials with cytotoxic drugs: What is missing? Cancer Chemother Pharmacol, 2016. 78(5): p. 1097-1098.
- Alam, A., et al., Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc Natl Acad Sci U S A, 2018. 115(9): p. E1973-e1982. [CrossRef]
- Martino, E., et al., Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg Med Chem Lett, 2018. 28(17): p. 2816-2826. [CrossRef]
- Zhou, X., et al., Double-sides sticking mechanism of vinblastine interacting with α,β-tubulin to get activity against cancer cells. J Biomol Struct Dyn, 2019. 37(15): p. 4080-4091.
- Crowley, E., C.A. McDevitt, and R. Callaghan, Generating inhibitors of P-glycoprotein: where to, now? Methods Mol Biol, 2010. 596: p. 405-32.
- Hyafil, F., et al., In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res, 1993. 53(19): p. 4595-602.
- Cripe, L.D., et al., Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood, 2010. 116(20): p. 4077-85. [CrossRef]
- Mechetner, E.B. and I.B. Roninson, Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc Natl Acad Sci U S A, 1992. 89(13): p. 5824-8. [CrossRef]
- Morita, S.Y. and T. Terada, Molecular mechanisms for biliary phospholipid and drug efflux mediated by ABCB4 and bile salts. Biomed Res Int, 2014. 2014: p. 954781. [CrossRef]
- Smith, A.J., et al., MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J Biol Chem, 2000. 275(31): p. 23530-9. [CrossRef]
- Sticova, E. and M. Jirsa, ABCB4 disease: Many faces of one gene deficiency. Annals of Hepatology, 2020. 19(2): p. 126-133. [CrossRef]
- Morita, S.Y., et al., Bile salt-stimulated phospholipid efflux mediated by ABCB4 localized in nonraft membranes. J Lipid Res, 2013. 54(5): p. 1221-30. [CrossRef]
- Smit, J.J., et al., Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell, 1993. 75(3): p. 451-62. [CrossRef]
- Boyer, J.L., Bile formation and secretion. Compr Physiol, 2013. 3(3): p. 1035-78. [CrossRef]
- Fagerberg, L., et al., Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics, 2014. 13(2): p. 397-406. [CrossRef]
- de Vree, J.M., et al., Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A, 1998. 95(1): p. 282-7. [CrossRef]
- Oude Elferink, R.P. and C.C. Paulusma, Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflugers Arch, 2007. 453(5): p. 601-10.
- Tougeron, D., et al., ABCB4/MDR3 gene mutations and cholangiocarcinomas. J Hepatol, 2012. 57(2): p. 467-8.
- Mhatre, S., et al., Common genetic variation and risk of gallbladder cancer in India: a case-control genome-wide association study. Lancet Oncol, 2017. 18(4): p. 535-544. [CrossRef]
- Kiehl, S., et al., ABCB4 is frequently epigenetically silenced in human cancers and inhibits tumor growth. Sci Rep, 2014. 4: p. 6899. [CrossRef]
- Olsen, J.A., et al., Structure of the human lipid exporter ABCB4 in a lipid environment. Nat Struct Mol Biol, 2020. 27(1): p. 62-707(1): p. 62-70. [CrossRef]
- Nosol, K., et al., Structures of ABCB4 provide insight into phosphatidylcholine translocation. Proc Natl Acad Sci U S A, 2021. 118(33). [CrossRef]
- Ishigami, M., et al., ATPase activity of nucleotide binding domains of human MDR3 in the context of MDR1. Biochim Biophys Acta, 2013. 1831(4): p. 683-90. [CrossRef]
- Mahdi, Z.M., et al., Role of Multidrug Resistance Protein 3 in Antifungal-Induced Cholestasis. Mol Pharmacol, 2016. 90(1): p. 23-34. [CrossRef]
- Frampton, J.E. and L.J. Scott, Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs, 2008. 68(7): p. 993-1016.
- Kiss, K., et al., Role of the N-terminal transmembrane domain in the endo-lysosomal targeting and function of the human ABCB6 protein. Biochem J, 2015. 467(1): p. 127-39. [CrossRef]
- Fukuda, Y., et al., Conserved intramolecular disulfide bond is critical to trafficking and fate of ATP-binding cassette (ABC) transporters ABCB6 and sulfonylurea receptor 1 (SUR1)/ABCC8. J Biol Chem, 2011. 286(10): p. 8481-8492.
- Boswell-Casteel, R.C., Y. Fukuda, and J.D. Schuetz, ABCB6, an ABC Transporter Impacting Drug Response and Disease. Aaps j, 2017. 20(1): p. 8. [CrossRef]
- Minami, K., et al., Expression of ABCB6 is related to resistance to 5-FU, SN-38 and vincristine. Anticancer Res, 2014. 34(9): p. 4767-73.
- Yasui, K., et al., Alteration in copy numbers of genes as a mechanism for acquired drug resistance. Cancer Res, 2004. 64(4): p. 1403-10. [CrossRef]
- Rakvács, Z., et al., The human ABCB6 protein is the functional homologue of HMT-1 proteins mediating cadmium detoxification. Cell Mol Life Sci, 2019. 76(20): p. 4131-4144. [CrossRef]
- Burke, M.A. and H. Ardehali, Mitochondrial ATP-binding cassette proteins. Transl Res, 2007. 150(2): p. 73-80. [CrossRef]
- Krishnamurthy, P. and J.D. Schuetz, The role of ABCG2 and ABCB6 in porphyrin metabolism and cell survival. Curr Pharm Biotechnol, 2011. 12(4): p. 647-55. [CrossRef]
- Krishnamurthy, P., T. Xie, and J.D. Schuetz, The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther, 2007. 114(3): p. 345-58. [CrossRef]
- Wang, L., et al., ABCB6 mutations cause ocular coloboma. Am J Hum Genet, 2012. 90(1): p. 40-8. [CrossRef]
- Andolfo, I., et al., Missense mutations in the ABCB6 transporter cause dominant familial pseudohyperkalemia. Am J Hematol, 2013. 88(1): p. 66-72. [CrossRef]
- Zhang, C., et al., Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J Invest Dermatol, 2013. 133(9): p. 2221-8. [CrossRef]
- Krishnamurthy, P.C., et al., Identification of a mammalian mitochondrial porphyrin transporter. Nature, 2006. 443(7111): p. 586-9. [CrossRef]
- Polireddy, K., et al., A novel flow cytometric HTS assay reveals functional modulators of ATP binding cassette transporter ABCB6. PLoS One, 2012. 7(7): p. e40005. [CrossRef]
- Park, S., et al., Gene expression profiling of ATP-binding cassette (ABC) transporters as a predictor of the pathologic response to neoadjuvant chemotherapy in breast cancer patients. Breast Cancer Res Treat, 2006. 99(1): p. 9-17. [CrossRef]
- Januchowski, R., et al., Microarray-based detection and expression analysis of ABC and SLC transporters in drug-resistant ovarian cancer cell lines. Biomed Pharmacother, 2013. 67(3): p. 240-5. [CrossRef]
- Varatharajan, S., et al., ATP-binding casette transporter expression in acute myeloid leukemia: association with in vitro cytotoxicity and prognostic markers. Pharmacogenomics, 2017. 18(3): p. 235-244. [CrossRef]
- Wang, C., et al., Cryo-electron microscopy structure of human ABCB6 transporter. Protein Sci, 2020. 29(12): p. 2363-2374. [CrossRef]
- Song, G., et al., Molecular insights into the human ABCB6 transporter. Cell Discovery, 2021. 7(1): p. 55. [CrossRef]
- Kim, S., et al., Structural Insights into Porphyrin Recognition by the Human ATP-Binding Cassette Transporter ABCB6. Mol Cells, 2022. 45(8): p. 575-587.
- Lee, S.S., et al., W546 stacking disruption traps the human porphyrin transporter ABCB6 in an outward-facing transient state. Commun Biol, 2023. 6(1): p. 960. [CrossRef]
- Choi, S.H., et al., Cryo-EM structure of cadmium-bound human ABCB6. Communications Biology, 2024. 7(1): p. 672. [CrossRef]
- Johnson, Z.L. and J. Chen, Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell, 2017. 168(6): p. 1075-1085.e9. [CrossRef]
- Conseil, G., et al., Structure-guided probing of the leukotriene C(4) binding site in human multidrug resistance protein 1 (MRP1; ABCC1). Faseb j, 2019. 33(10): p. 10692-10704. [CrossRef]
- Bakos, E., et al., Characterization of the amino-terminal regions in the human multidrug resistance protein (MRP1). J Cell Sci, 2000. 113 Pt 24: p. 4451-61. [CrossRef]
- Slot, A.J., S.V. Molinski, and S.P. Cole, Mammalian multidrug-resistance proteins (MRPs). Essays Biochem, 2011. 50(1): p. 179-207. [CrossRef]
- Seelig, A., A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem, 1998. 251(1-2): p. 252-61. [CrossRef]
- Yin, J.Y., et al., Characterization and analyses of multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphisms in Chinese population. Pharmacogenet Genomics, 2009. 19(3): p. 206-16.
- Taylor, N.M.I., et al., Structure of the human multidrug transporter ABCG2. Nature, 2017. 546(7659): p. 504-509. [CrossRef]
- Kowal, J., et al., Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J Mol Biol, 2021. 433(13): p. 166980.
- Toyoda, Y., T. Takada, and H. Suzuki, Inhibitors of Human ABCG2: From Technical Background to Recent Updates With Clinical Implications. Front Pharmacol, 2019. 10: p. 208. [CrossRef]
- Houghton, P.J., et al., Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res, 2004. 64(7): p. 2333-7. [CrossRef]
- Candeil, L., et al., ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int J Cancer, 2004. 109(6): p. 848-54. [CrossRef]
- Hillgren, K.M., et al., Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther, 2013. 94(1): p. 52-63. [CrossRef]
- Tamaki, A., et al., The controversial role of ABC transporters in clinical oncology. Essays Biochem, 2011. 50(1): p. 209-32. [CrossRef]
- Robey, R.W., et al., ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med Chem, 2010. 10(8): p. 625-33. [CrossRef]
- Zhou, S., et al., The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nature Medicine, 2001. 7(9): p. 1028-1034.
- Lee, J.Y., et al., Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature, 2016. 533(7604): p. 561-4.
- Rosenberg, M.F., et al., Three-dimensional structure of the human breast cancer resistance protein (BCRP/ABCG2) in an inward-facing conformation. Acta Crystallogr D Biol Crystallogr, 2015. 71(Pt 8): p. 1725-35. [CrossRef]
- Jackson, S.M., et al., Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nature Structural & Molecular Biology, 2018. 25(4): p. 333-340. [CrossRef]
- Manolaridis, I., et al., Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature, 2018. 563(7731): p. 426-430.
- Yu, Q., et al., Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. Nature Communications, 2021. 12(1): p. 4376.
- Puentes, C.O., et al., Solid phase synthesis of tariquidar-related modulators of ABC transporters preferring breast cancer resistance protein (ABCG2). Bioorg Med Chem Lett, 2011. 21(12): p. 3654-7. [CrossRef]
- Allen, J.D., et al., Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther, 2002. 1(6): p. 417-25.
- Weidner, L.D., et al., The Inhibitor Ko143 Is Not Specific for ABCG2. J Pharmacol Exp Ther, 2015. 354(3): p. 384-93. [CrossRef]
- Ochoa-Puentes, C., et al., Benzanilide-Biphenyl Replacement: A Bioisosteric Approach to Quinoline Carboxamide-Type ABCG2 Modulators. ACS Med Chem Lett, 2013. 4(4): p. 393-6. [CrossRef]
- Roe, M., et al., Reversal of P-glycoprotein mediated multidrug resistance by novel anthranilamide derivatives. Bioorg Med Chem Lett, 1999. 9(4): p. 595-600. [CrossRef]
- Köhler, S.C. and M. Wiese, HM30181 Derivatives as Novel Potent and Selective Inhibitors of the Breast Cancer Resistance Protein (BCRP/ABCG2). J Med Chem, 2015. 58(9): p. 3910-21. [CrossRef]
- Miyata, H., et al., Identification of Febuxostat as a New Strong ABCG2 Inhibitor: Potential Applications and Risks in Clinical Situations. Front Pharmacol, 2016. 7: p. 518. [CrossRef]
- Ji, N., et al., VS-4718 Antagonizes Multidrug Resistance in ABCB1- and ABCG2-Overexpressing Cancer Cells by Inhibiting the Efflux Function of ABC Transporters. Front Pharmacol, 2018. 9: p. 1236. [CrossRef]
- Alam, A. and K.P. Locher, Structure and Mechanism of Human ABC Transporters. Annu Rev Biophys, 2023. 52: p. 275-300. [CrossRef]
- Jones, P.M. and A.M. George, The Switch and Reciprocating Models for the Function of ABC Multidrug Exporters: Perspectives on Recent Research. International Journal of Molecular Sciences, 2023. 24(3): p. 2624. [CrossRef]
- Yuan, T., et al., Oxidative stress-mediated up-regulation of ABC transporters in lung cancer cells. J Biochem Mol Toxicol, 2022. 36(8): p. e23095. [CrossRef]
- Sajid, A., H. Rahman, and S.V. Ambudkar, Advances in the structure, mechanism and targeting of chemoresistance-linked ABC transporters. Nature Reviews Cancer, 2023. 23(11): p. 762-779. [CrossRef]
- Lainey, E., et al., Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle, 2012. 11(21): p. 4079-4092. [CrossRef]
- Mohammad, I.S., W. He, and L. Yin, Insight on Multidrug Resistance and Nanomedicine Approaches to Overcome MDR. Crit Rev Ther Drug Carrier Syst, 2020. 37(5): p. 473-509. [CrossRef]
- Pote, M.S. and R.N. Gacche, ATP-binding cassette efflux transporters and MDR in cancer. Drug Discovery Today, 2023. 28(5): p. 103537. [CrossRef]
- Cui, H., et al., ABC Transporter Inhibitors in Reversing Multidrug Resistance to Chemotherapy. Curr Drug Targets, 2015. 16(12): p. 1356-71. [CrossRef]
- Falasca, M. and K.J. Linton, Investigational ABC transporter inhibitors. Expert Opin Investig Drugs, 2012. 21(5): p. 657-6666. [CrossRef]
- Friedenberg, W.R., et al., Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95). Cancer, 2006. 106(4): p. 830-838. [CrossRef]
- Kelly, R.J., et al., A Pharmacodynamic Study of Docetaxel in Combination with the P-glycoprotein Antagonist Tariquidar (XR9576) in Patients with Lung, Ovarian, and Cervical Cancer. Clinical Cancer Research, 2011. 17(3): p. 569-580. [CrossRef]
- Sun, Y.-L., et al., Reversal of MRP7 (ABCC10)-Mediated Multidrug Resistance by Tariquidar. PLOS ONE, 2013. 8(2): p. e55576. [CrossRef]
- Tamaki, A., et al., The controversial role of ABC transporters in clinical oncology. Essays in Biochemistry, 2011. 50: p. 209-232. [CrossRef]
- Xiao, H., et al., Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front Pharmacol, 2021. 12: p. 648407. [CrossRef]
- Yu, M., A. Ocana, and I.F. Tannock, Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: why has it failed to provide clinical benefit? Cancer Metastasis Rev, 2013. 32(1-2): p. 211-27.
- Krapf, M.K., et al., Synthesis and biological evaluation of quinazoline derivatives - A SAR study of novel inhibitors of ABCG2. Eur J Med Chem, 2019. 161: p. 506-525. [CrossRef]
- Mohajeri, M. and A. Sahebkar, Protective effects of curcumin against doxorubicin-induced toxicity and resistance: A review. Crit Rev Oncol Hematol, 2018. 122: p. 30-51. [CrossRef]
- Omori, M., et al., Inhibitors of ABCB1 and ABCG2 overcame resistance to topoisomerase inhibitors in small cell lung cancer. Thorac Cancer, 2022. 13(15): p. 2142-2151. [CrossRef]
- Yarla, N.S., Bioactive Flavonoids as ABC Transporters Inhibitors for Reversion of Multidrug Resistance in Cancer. Journal of Marine Science: Research & Development, 2013. 4: p. 1-2.
- van der Noord, V.E., et al., Systematic screening identifies ABCG2 as critical factor underlying synergy of kinase inhibitors with transcriptional CDK inhibitors. Breast Cancer Res, 2023. 25(1): p. 51.
- Liu, L., et al., IGHMBP2-related clinical and genetic features in a cohort of Chinese Charcot-Marie-Tooth disease type 2 patients. Neuromuscul Disord, 2017. 27(2): p. 193-199. [CrossRef]
- Wang, C., et al., Near-infrared light induced in vivo photodynamic therapy of cancer based on upconversion nanoparticles. Biomaterials, 2011. 32(26): p. 6145-54. [CrossRef]
- Bui, K.C., et al., X-ray-irradiated K562 feeder cells for expansion of functional CAR-T cells. Biochem Biophys Rep, 2023. 33: p. 101399. [CrossRef]
- Du, J., et al., Comparative analysis of the immune responses in cancer cells irradiated with X-ray, proton and carbon-ion beams. Biochem Biophys Res Commun, 2021. 585: p. 55-60. [CrossRef]
- Kawanishi, M., M. Fujita, and K. Karasawa, Combining Carbon-Ion Irradiation and PARP Inhibitor, Olaparib Efficiently Kills BRCA1-Mutated Triple-Negative Breast Cancer Cells. Breast Cancer (Auckl), 2022. 16: p. 11782234221080553. [CrossRef]
- Zhao, D., et al., Application of MOF-based nanotherapeutics in light-mediated cancer diagnosis and therapy. J Nanobiotechnology, 2022. 20(1): p. 421. [CrossRef]
- De Vera, A.A., et al., Immuno-oncology agent IPI-549 is a modulator of P-glycoprotein (P-gp, MDR1, ABCB1)-mediated multidrug resistance (MDR) in cancer: In vitro and in vivo. Cancer Lett, 2019. 442: p. 91-103.
- Bihorel, S., et al., Modulation of the brain distribution of imatinib and its metabolites in mice by valspodar, zosuquidar and elacridar. Pharm Res, 2007. 24(9): p. 1720-8. [CrossRef]
- Mousavi, S.H., et al., Direct toxicity of Rose Bengal in MCF-7 cell line: role of apoptosis. Food Chem Toxicol, 2009. 47(4): p. 855-9. [CrossRef]
- He, J., et al., Recent progress of aptamer‒drug conjugates in cancer therapy. Acta Pharm Sin B, 2023. 13(4): p. 1358-1370. [CrossRef]
- Ma, W., et al., Self-Assembled Multivalent Aptamer Drug Conjugates: Enhanced Targeting and Cytotoxicity for HER2-Positive Gastric Cancer. ACS Appl Mater Interfaces, 2023. 15(37): p. 43359-43373. [CrossRef]
- Chang, C.H., et al., Combining ABCG2 Inhibitors with IMMU-132, an Anti-Trop-2 Antibody Conjugate of SN-38, Overcomes Resistance to SN-38 in Breast and Gastric Cancers. Mol Cancer Ther, 2016. 15(8): p. 1910-9.
- Pieper, S., et al., Incorporation of doxorubicin in different polymer nanoparticles and their anticancer activity. Beilstein J Nanotechnol, 2019. 10: p. 2062-2072. [CrossRef]
- Lage, H., Gene Therapeutic Approaches to Overcome ABCB1-Mediated Drug Resistance. Recent Results Cancer Res, 2016. 209: p. 87-94.
- Varlas, S., T.J. Neal, and S.P. Armes, Polymerization-induced self-assembly and disassembly during the synthesis of thermoresponsive ABC triblock copolymer nano-objects in aqueous solution. Chem Sci, 2022. 13(24): p. 7295-7303. [CrossRef]
- Yadav, P., S.V. Ambudkar, and N. Rajendra Prasad, Emerging nanotechnology-based therapeutics to combat multidrug-resistant cancer. J Nanobiotechnology, 2022. 20(1): p. 423. [CrossRef]
- Kunjachan, S., et al., Multidrug resistance: Physiological principles and nanomedical solutions. Adv Drug Deliv Rev, 2013. 65(13-14): p. 1852-1865. [CrossRef]
- de la Puente, P. and A.K. Azab, Nanoparticle delivery systems, general approaches, and their implementation in multiple myeloma. Eur J Haematol, 2017. 98(6): p. 529-541. [CrossRef]
- Guo, Z., et al., The novel ABC transporter ABCH1 is a potential target for RNAi-based insect pest control and resistance management. Scientific Reports, 2015. 5(1): p. 13728. [CrossRef]
- Hokaiwado, N., et al., RNAi-based drug discovery and its application to therapeutics. IDrugs, 2008. 11(4): p. 274-8.
- Uchino, K., T. Ochiya, and F. Takeshita, RNAi therapeutics and applications of microRNAs in cancer treatment. Jpn J Clin Oncol, 2013. 43(6): p. 596-607. [CrossRef]
- Tong, A.W., Small RNAs and non-small cell lung cancer. Curr Mol Med, 2006. 6(3): p. 339-49.
- Cho, S.W., et al., Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res, 2014. 24(1): p. 132-41. [CrossRef]
- Zhang, X.-H., et al., Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Molecular Therapy - Nucleic Acids, 2015. 4: p. e264. [CrossRef]
- Wegler, C., et al., Expanding the Efflux In Vitro Assay Toolbox: A CRISPR-Cas9 Edited MDCK Cell Line with Human BCRP and Completely Lacking Canine MDR1. Journal of Pharmaceutical Sciences, 2021. 110(1): p. 388-396. [CrossRef]
- Radtke, L., et al., CRISPR/Cas9-induced knockout reveals the role of ABCB1 in the response to temozolomide, carmustine and lomustine in glioblastoma multiforme. Pharmacol Res, 2022. 185: p. 106510. [CrossRef]
- Wang, H., et al., Infection risk in autoimmune hematological disorders with low-dose rituximab treatment. J Clin Lab Anal, 2020. 34(10): p. e23455.
- Wang, X. and M. Hong, Protein Kinases and Cross-talk between Post-translational Modifications in the Regulation of Drug Transporters. Mol Pharmacol, 2023. 103(1): p. 9-20. [CrossRef]
- Chen, M.L., et al., Physiological expression and function of the MDR1 transporter in cytotoxic T lymphocytes. J Exp Med, 2020. 217(5). [CrossRef]
- Liang, C., et al., Development and Characterization of MDR1 (Mdr1a/b) CRISPR/Cas9 Knockout Rat Model. Drug Metab Dispos, 2019. 47(2): p. 71-79. [CrossRef]
- Norouzi-Barough, L., et al., CRISPR/Cas9, a new approach to successful knockdown of ABCB1/P-glycoprotein and reversal of chemosensitivity in human epithelial ovarian cancer cell line. Iran J Basic Med Sci, 2018. 21(2): p. 181-187.
- Robert, X. and P. Gouet, Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Research, 2014. 42(W1): p. W320-W324. [CrossRef]
- Hrycyna, C.A., et al., Structural Flexibility of the Linker Region of Human P-Glycoprotein Permits ATP Hydrolysis and Drug Transport. Biochemistry, 1998. 37(39): p. 13660-13673. [CrossRef]
- Esser, L., et al., Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity*♦. Journal of Biological Chemistry, 2017. 292(2): p. 446-461. [CrossRef]
- Wang, J., et al., Placing steroid hormones within the human ABCC3 transporter reveals a compatible amphiphilic substrate-binding pocket. The EMBO Journal, 2023. 42(17): p. e113415. [CrossRef]
- Johnson, Z.L. and J. Chen, ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell, 2018. 172(1): p. 81-89.e10. [CrossRef]
- Kroll, T. , et al., Structure and Function of Hepatobiliary ATP Binding Cassette Transporters. Chemical Reviews, 2021. 121(9): p. 5240-5288. [CrossRef]
- Bakos, É., et al., Functional Multidrug Resistance Protein (MRP1) Lacking the N-terminal Transmembrane Domain*. Journal of Biological Chemistry, 1998. 273(48): p. 32167-32175. [CrossRef]
- Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589. [CrossRef]
- Abramson, J., et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024. 630(8016): p. 493-500. [CrossRef]
- Olsen, J.A., et al., Structure of the human lipid exporter ABCB4 in a lipid environment. Nature Structural & Molecular Biology, 2020. 27(1): p. 62-70. [CrossRef]
- Haenisch, S., A.N. Werk, and I. Cascorbi, MicroRNAs and their relevance to ABC transporters. Br J Clin Pharmacol, 2014. 77(4): p. 587-96. [CrossRef]
- Szczepanek, J., M. Skorupa, and A. Tretyn, MicroRNA as a Potential Therapeutic Molecule in Cancer. Cells, 2022. 11(6): p. 1008. [CrossRef]
Figure 1.
ABC transporter general architecture. (a) Schematic of ABC transporter domain architecture, with arrows indicating the direction of transport. (b) NBD of ABC transporters. Shown here is NBD of ABCB1 (PDB-7A69) with the conserved ATPase domains separately labeled and colored. The NBD is oriented to show the NBD surface that constitutes the NBD-NBD interface. (c) Top view (over the membrane panel) of arrangement of transmembrane helices in ABC transporters. Shown here are both drug free (PDB- 7A65) and drug bound states (PDB-7A69) of ABCB1. Abbreviations: NBD, nucleotide binding domain; TMD, transmembrane domain.
Figure 1.
ABC transporter general architecture. (a) Schematic of ABC transporter domain architecture, with arrows indicating the direction of transport. (b) NBD of ABC transporters. Shown here is NBD of ABCB1 (PDB-7A69) with the conserved ATPase domains separately labeled and colored. The NBD is oriented to show the NBD surface that constitutes the NBD-NBD interface. (c) Top view (over the membrane panel) of arrangement of transmembrane helices in ABC transporters. Shown here are both drug free (PDB- 7A65) and drug bound states (PDB-7A69) of ABCB1. Abbreviations: NBD, nucleotide binding domain; TMD, transmembrane domain.
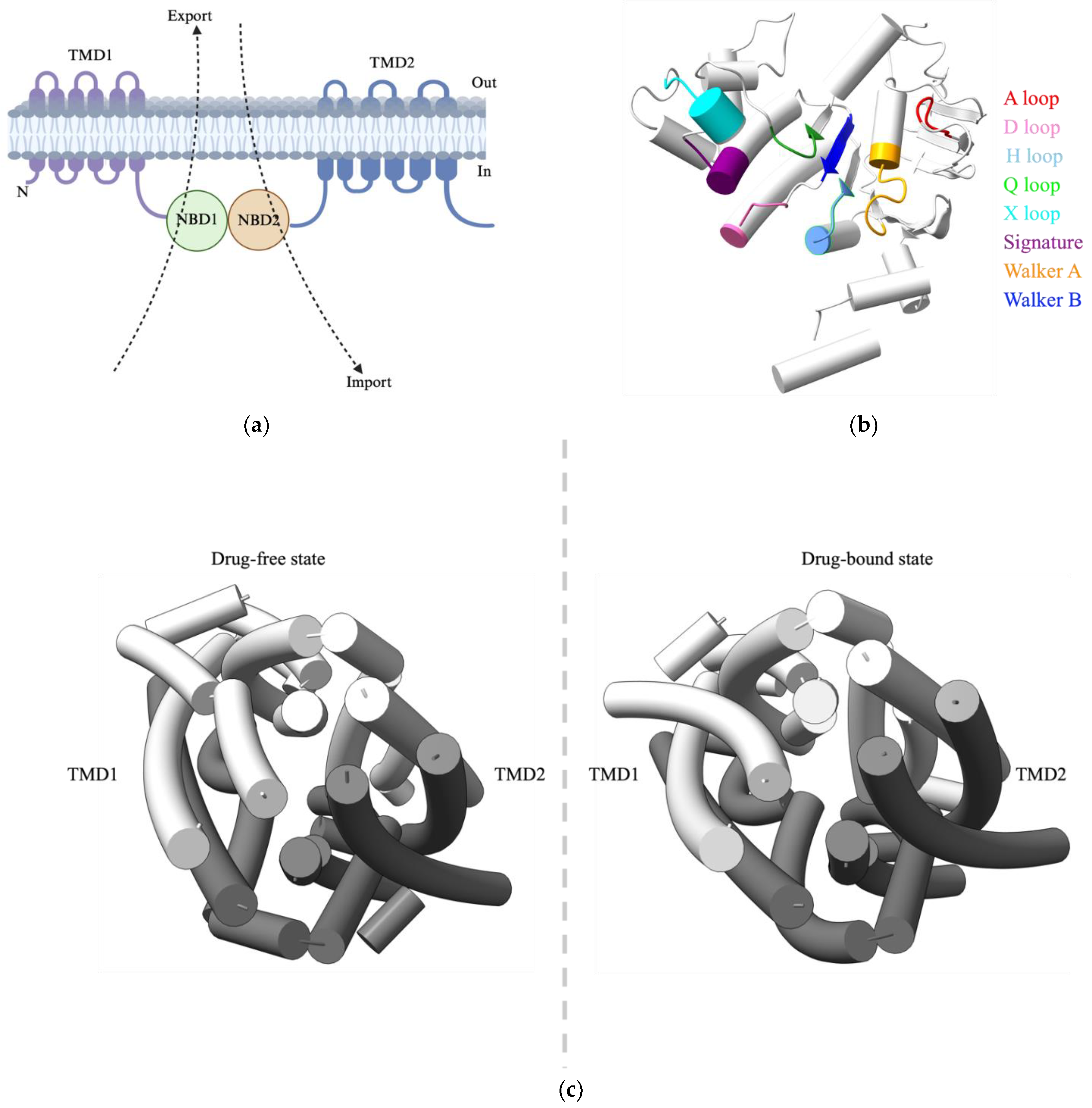
Figure 2.
Topological structures of ABC transporters. ABCA, ABCB, ABCC, ABCD and ABCG having both nuclear binding domain (NBDs) and transmembrane binding domains (TMDs). ABCE have only NMDs and don’t have TMDs, also their topological structures are unclear.
Figure 2.
Topological structures of ABC transporters. ABCA, ABCB, ABCC, ABCD and ABCG having both nuclear binding domain (NBDs) and transmembrane binding domains (TMDs). ABCE have only NMDs and don’t have TMDs, also their topological structures are unclear.
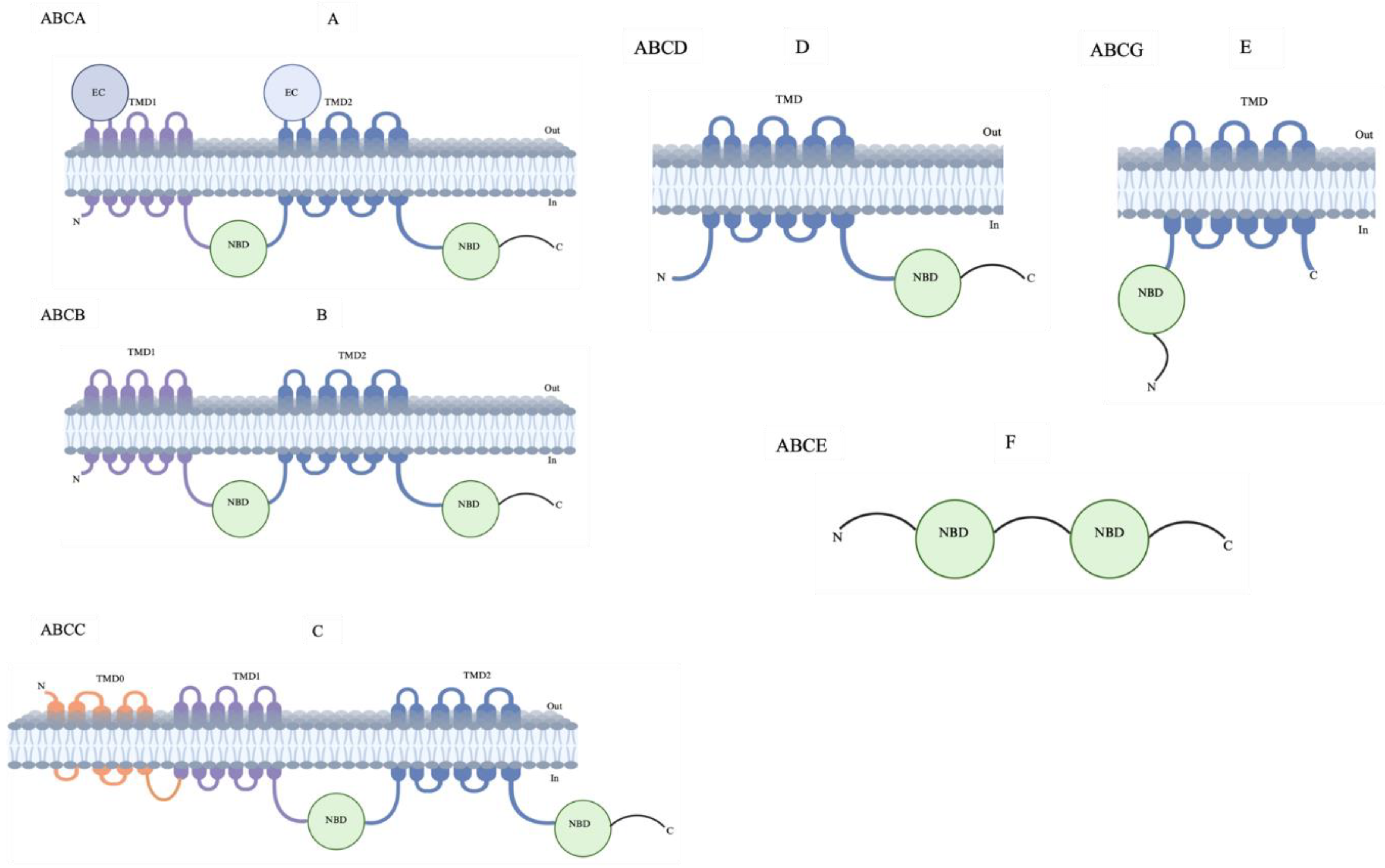
Figure 3.
Overview of major ABC transporters that mediate cancer MDR. Structures and topologies are derived from protein data bank (PDB) for the five ABC transporters bound to inhibitors/substrates. A; ABCB1: Zosuquidar- PDB ID- 7A6F. B; ABCB4:PC6 (phosphatidyl choline): - PDB ID- 7NIV. C; ABCB6: HEME- PDB ID- 7DNZ. D; ABCC1: LTX (leukotriene A4 conjugated with glutathione)- PDB ID- 5UJA. ABCG2: BWQ (derivative of fumitremorgin C-related inhibitor Ko143)- PDB ID- 6FFC. Area outlined red dashed lines in each transporter indicate appropriate location of drug-binding cavity. Figures are generated using UCSF Chimera X: Tools for structure building and analysis.
Figure 3.
Overview of major ABC transporters that mediate cancer MDR. Structures and topologies are derived from protein data bank (PDB) for the five ABC transporters bound to inhibitors/substrates. A; ABCB1: Zosuquidar- PDB ID- 7A6F. B; ABCB4:PC6 (phosphatidyl choline): - PDB ID- 7NIV. C; ABCB6: HEME- PDB ID- 7DNZ. D; ABCC1: LTX (leukotriene A4 conjugated with glutathione)- PDB ID- 5UJA. ABCG2: BWQ (derivative of fumitremorgin C-related inhibitor Ko143)- PDB ID- 6FFC. Area outlined red dashed lines in each transporter indicate appropriate location of drug-binding cavity. Figures are generated using UCSF Chimera X: Tools for structure building and analysis.
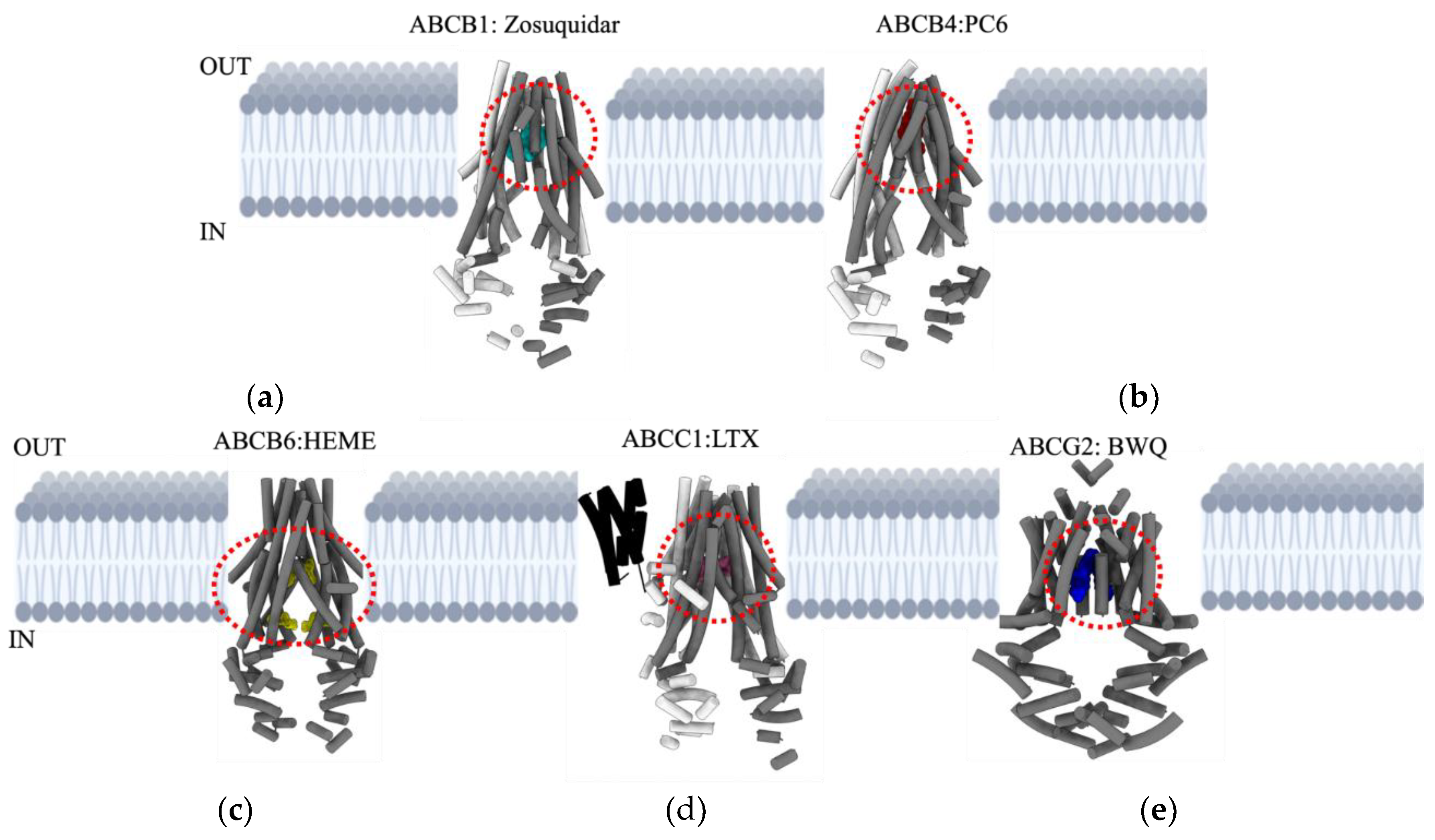
Figure 4.
Schematic illustrating the emerging therapies targeting major ABC transporters that mediate cancer MDR.
Figure 4.
Schematic illustrating the emerging therapies targeting major ABC transporters that mediate cancer MDR.

Table 1.
List of chemo-resistant human ABC transporters.
| Name | Function | Molecular Weight kDa | Disease Association |
|---|---|---|---|
| ABCA2 | Lipid transporter | 269.8 | IDPOGSA |
| ABCA3 | Phospholipid transporter | 191.4 | SMDP3 |
| ABCB1 | Transports drugs & phospholipids | 141.4 | MDR |
| ABCB2 | Peptide transporter | 80.9 | BLS1 |
| ABCB5 | Multidrug exporter | 138.6 | MDR |
| ABCC1 | Exporter of anions and xenobiotics | 171.5 | MDR |
| ABCC2 | Transporter of bile acid conjugates | 174.2 | MDR |
| ABCC3 | Transporter of bile acid conjugates | 169.3 | MDR |
| ABCC4 | Transporter of metabolites | 149.5 | MDR |
| ABCC5 | Transporter of amnio acid metabolites | 160.6 | MDR |
| ABCC6 | Exporter of xenobiotics | 164.9 | PXE |
| ABCC10 | Transporter of glutathione conjugate | 161.6 | MDR |
| ABCG2 | Drug efflux pump | 72.3 | MDR |
Abbreviations: IDPOGSA, intellectual developmental disorder with poor growth with or without seizures or ataxia, SMDP3, pulmonary surfactant metabolism dysfunctions, MDR, multidrug resistance, BLS1, bare lymphocyte syndrome 1, PXE, pseudoxanthoma elasticum.
Table 2.
List of key residues within the binding pocket, substrates, and inhibitors of major ABC transporters that mediate cancer MDR. The residues that are involved in both substrate & inhibitor binding are shown in red.
Table 2.
List of key residues within the binding pocket, substrates, and inhibitors of major ABC transporters that mediate cancer MDR. The residues that are involved in both substrate & inhibitor binding are shown in red.
| Transporters | Key residues within the binding pocket | Mediate through/ substrates | Current Drugs/Inhibitors under use or under clinical trials |
|---|---|---|---|
| ABCB1 | Y310, I340, F343, S344, Q347, Q725, Q946, Y953, F983, M9846, A987, Q990 [51] | A wide range of substrates. For example: Paclitaxel, Vincristine, Doxorubicin | Verapamil, Tariquidar, SelonsertibTepotinib, PND-1186, MK-8776, Rydapt, Erdafitinib, CGM097, Poziotinib, synthetic peptides, Nanobody nb-592, Biricodar, laniquidar, valspodar, querectin |
| ABCB4 | A737, G954, P726, T775, A286, R47, D243, S346, S 320, Q855, S978, A953, F357, A 346.[191] | Primarily involved in lipid transport. Depending on the need sometimes also mediates doxorubicin. | Curcumin |
| ABCB6 | T320, S322, T323, W546, A 492, G588, A681.[99] | Primarily involved in porphyrin transport. Depending on the need sometimes also mediates cisplatin, arsenic trioxide, vincristine, camptothecin, CPt-11, 5-Fu, SN-38, Doxorubicin, methotrexate, topotecan. | No known inhibitors so far. |
| ABCC1 | K332, H335, L381, F385, Y440, T550, W553, F594, M1092, R1196, Y1242, N1244, W1245, R1248. [103] | Transports leukotriene. Depending on the need sometimes also mediates vincristine, vinblastine, Doxorubicin, methotrexate, daunorubicin, etoposide, betulin | MK571, Flavopiridol, Apigenin, Chalcone, Kaempferol, Morin, Quercetin, GSK1940529A, Indomethacin, verapamil and its derivatives, macrocyclic peptides (CP11), sipholenol A, |
| ABCG2 | L405, F432, T435, N436, F439, S440, V442, T542, V546, M549 [109]. | Transports drugs. Such as topotecan, mitoxantrone, SN-38, OTS964, GSK1070916, Tivantinib, Pevonedistat, Imatinib. | KO143, FTC, Selonsertib, TAE684, M3814, CC-671, Antibody & Nanobody: 5D3, Nb8, Nb17, Nb96. Tyrosine kinase inhibitors (Dacomitinib, pozoiotinib, venetoclax, olmutinib, gefitinib, quizartinib, novobiocin. |
Table 3.
List of miRNAs targeting major ABC transporters that mediate cancer MDR. The miRNA targets referenced from Haenisch et al. and Szczepanek et al. [192,193].
| miRNA target | Targeting ABC transporter | Tested in |
|---|---|---|
| miR137 | ABCB1 | Human breast cancer cells MCF-7 |
| miR145 | ABCB1 | Human colon carcinoma cells, Caco-2 |
| miR200c | ABCB1 | Human breast cancer cells, MCF-7 |
| miR331-5p | ABCB1 | Chronic myelogenous leukaemia cells K562 |
| miR451 | ABCB1 | Human breast cancer cells, MCF-7 |
| miR1253 | ABCB1 | Human breast cancer cells, MDA-MB-231 |
| miR-271 | ABCB1 | Human oesophageal squamous cell lines ECA109 and TE-14 |
| ABCB1 | Human gastric cancer cell line MKN45 | |
| ABCB1 | Human Ovarian cancer cell line A2780 | |
| miR138 | ABCB1 | Human Leukaemia cell line HL-60 |
| miR296 | ABCB1 | Human oesophageal squamous cell lines ECA109 |
| miR451 | ABCB1 | Ovarian cancer cell line A2780 |
| miR223 | ABCB4 | Human liver cancer cell line |
| miR34a | ABCB4 | Human liver cancer cell line |
| miR1 | ABCB4 | Human liver cancer cell line |
| miR9 | ABCB4 | Human liver cancer cell line |
| miR449 | ABCB4 | Human liver cancer cell line |
| miR370 | ABCB6 | Human lung, liver cancer cell lines |
| miR27a | ABCB6 | Human lung, liver cancer cell lines |
| miR16 | ABCB6 | Human lung, liver cancer cell lines |
| miR181a | ABCB6 | Human lung, liver cancer cell lines |
| miR506 | ABCC1 | Human breast cancer cell line |
| miR27a | ABCC1 | Human breast, lung and cancer cell line |
| miR122 | ABCC1 | Human breast, lung and cancer cell line |
| miR370 | ABCC1 | Human breast, lung and cancer cell line |
| miR16 | ABCC1 | Human breast, lung and cancer cell line |
| miR328 | ABCG2 | Human breast, lung and cancer cell line |
| miR451 | ABCG2 | Human breast, lung and cancer cell line |
| miR506 | ABCG2 | Human breast, ovarian cancer cell line |
| miR34a | ABCG2 | Human breast, ovarian cancer cell line |
| miR30a | ABCG2 | Human breast, ovarian cancer cell line |
| miR9 | ABCG2 | Human breast, ovarian cancer cell line |
| miR138 | ABCG2 | Human breast, ovarian cancer cell line |
| miR204 | ABCG2 | Human breast, ovarian cancer cell line |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



