
Submitted:
15 November 2024
Posted:
18 November 2024
You are already at the latest version
Abstract
Hutchinson-Gilford Progeria Syndrome (HGPS) is a pediatric condition characterized by clinical features that resemble accelerated aging. The abnormal accumulation of a toxic form of the lamin A protein known as progerin disrupts cellular functions, leading to various complications, including growth retardation, loss of subcutaneous fat, abnormal skin, alopecia, osteoporosis, and progressive joint contractures. Death primarily occurs as the result of complications from progressive atherosclerosis, especially from cardiac disease such as myocardial infarction or heart failure, or cerebrovascular disease like stroke. Despite the availability of lonafarnib, the only US Food and Drug Administration-approved treatment for HGPS, cardiovascular complications remain the leading cause of morbidity and mortality in affected patients. Defective angiogenesis- the process of forming new blood vessels from existing ones- plays a crucial role in the development of cardiovascular disease. A recent study suggests that Angiopoietin-2 (Ang2), a pro-angiogenic growth factor that regulates angiogenesis and vascular stability, may offer therapeutic potential for the treatment of HGPS. In this review, we describe the clinical features and key cellular processes impacted by progerin and discuss the therapeutic potential of Ang2 in addressing these challenges.
Keywords:
1. Introduction
2. Potential Therapeutic Roles of Angiopoietin-2 in HGPS
2.1. Atheroprotective role of Ang2
2.2. Alleviating Endothelial Dysfunction
2.3. The Possible Role in Valvogenesis and Cardiac Development
2.4. The Possible Role in Maintaining Lymphatic Vascular Integrity
2.5. The Possible Role in Promoting White Adipose Tissue Hemostasis
2.6. The Possible Role in Facilitating Bone Wound Healing
2.7. The Possible Role in Enhancing Blood Flow Recovery in Ischemic Tissue
2.8. The Possible Neuroprotective Effects of Ang2

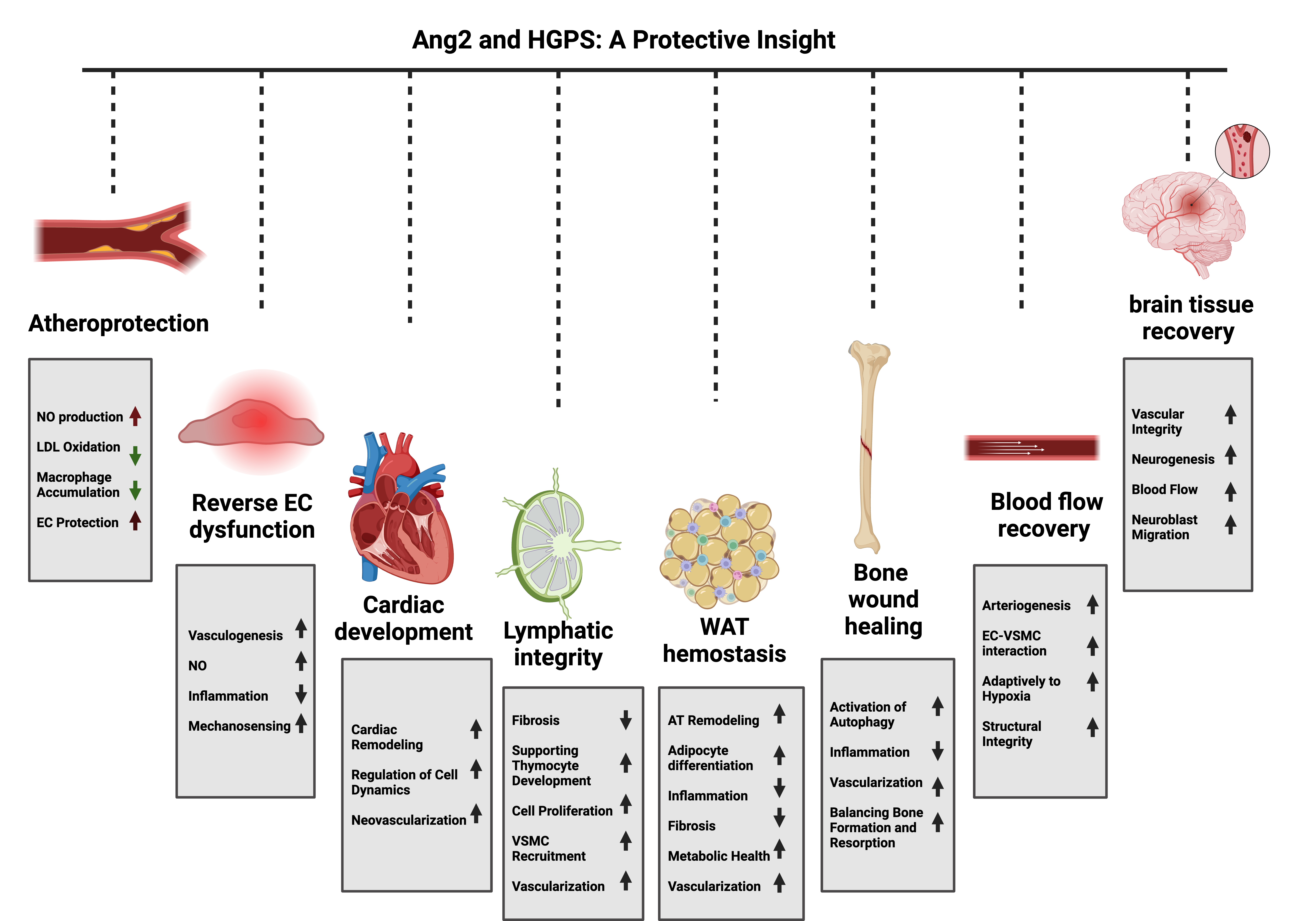
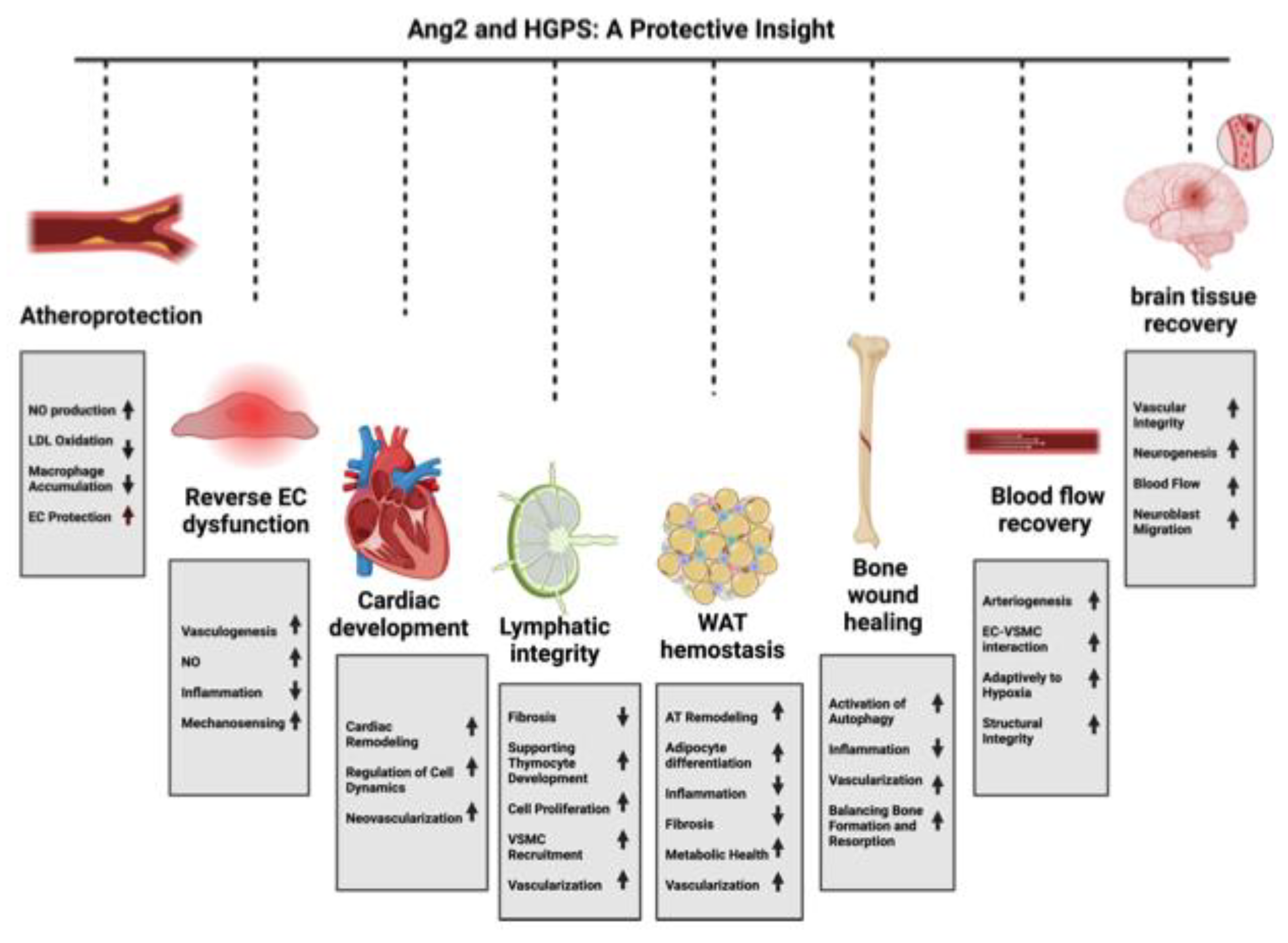
| Function/Role | Evidence | Mechanism | Outcome | References |
|---|---|---|---|---|
| Atheroprotection | Ang-2 reduces atherosclerotic lesion size, macrophage accumulation, and LDL oxidation; requires eNOS activity. | Reduces LDL oxidation and macrophage accumulation through eNOS activation | Ang2 protects against atherosclerosis by modulating LDL oxidation and inflammation | [30,33,39] |
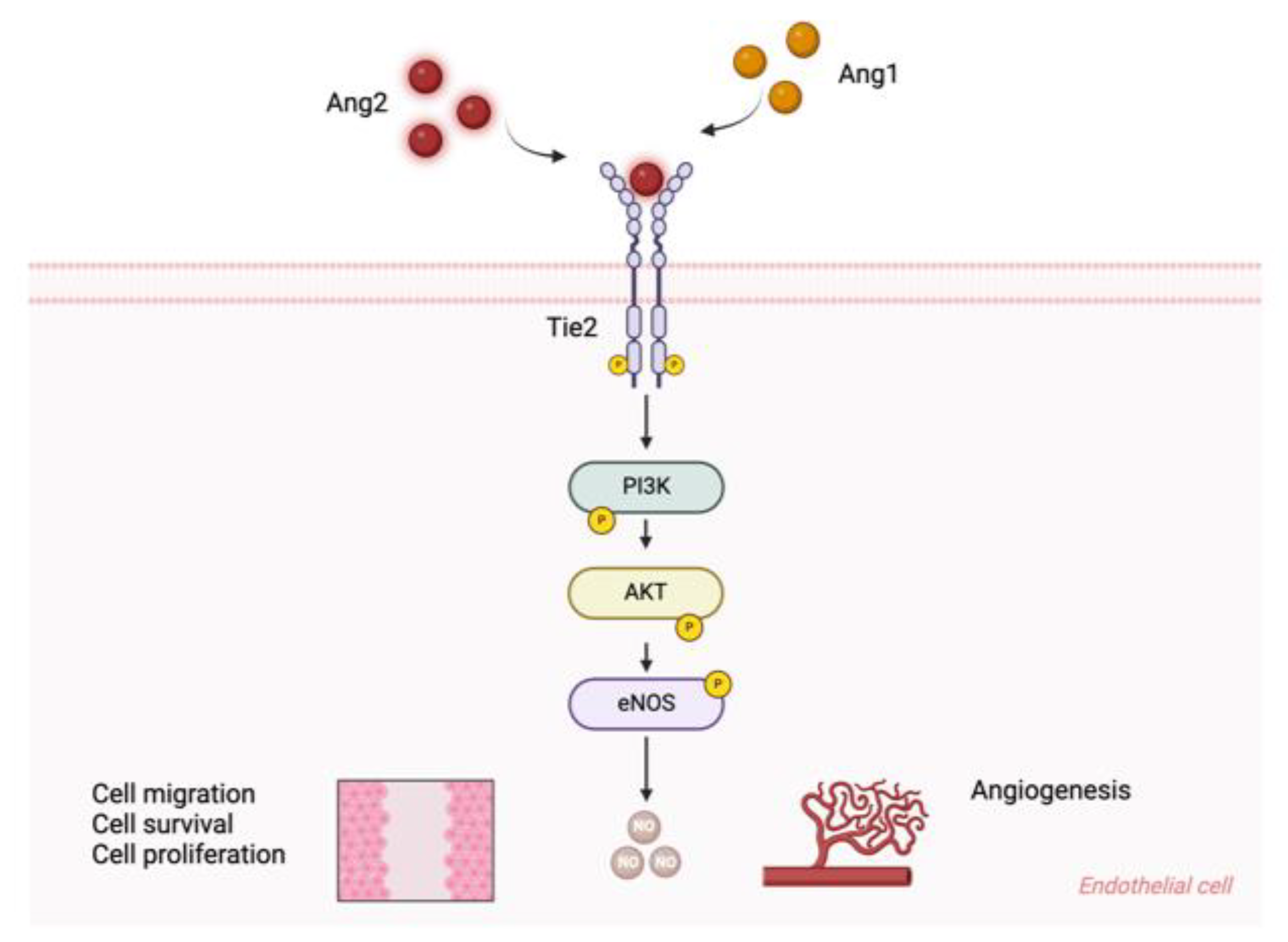
| Alleviating Endothelial Dysfunction | Ang2 induces Tie2 activation, enhances EC survival, migration, and tube formation | Activates Tie2-Akt signaling pathway | Promotes endothelial cell function and angiogenesis | [21,33,53,57] |
| Cardiac Development and Function | Ang2 knockdown mice show aortic valve stenosis and cardiac dysfunction; Ang2 administration improves heart function | Maintains cardiac redox balance and promotes neovascularization | Ang2 is crucial for cardiac development and function | [59,61,62,106] |
| Lymphatic Vascular Integrity | Ang2 deficiency in mice led to severe lymphatic dysfunction, abnormal vessel patterning, and chylous ascites | Maintains lymphatic integrity by activating the Tie2 receptor, promoting endothelial cell survival and proliferation, and regulating lymphatic vessel maturation and remodeling | Ang2 is essential for lymphatic vessel development and function | [64-66] |
| White Adipose Tissue Homeostasis | Ang2 overexpression improved WAT vascularization, glucose tolerance, and insulin sensitivity in mice | Enhances WAT vascularization and has anti-fibrotic effects | Ang2 protects against obesity-related inflammation and fibrosis | [77,78,80] |
| Bone Wound Healing | Ang2 treatment accelerated bone defect repair in rabbits and correlated with increased autophagy and vascularization | Activates autophagy and promotes bone repair | Enhanced bone healing and vascularization in bone defect | [87,90] |
| Blood Flow Recovery in Ischemia | Ang2 upregulation in hindlimb ischemia models aids in blood flow recovery and neovascularization | Promote arteriogenesis and capillary coverage | Improved blood flow recovery and neovascularization post-ischemia | [94] |
| Neuroprotection | High BBB leakage associated with low Ang2 expression; Ang2 improves BBB integrity and promotes neurogenesis post-stroke. | High BBB leakage associated with low Ang2 expression; Ang2 improves BBB integrity and promotes neurogenesis post-stroke. | Ang2 protects BBB integrity and promotes recovery after ischemic stroke. | [102-105] Mae et al., 2021; Liu et al., 2009; Marteau et al., 2013; Lv et al., 2022 |
3. Summary
Author Contributions
Data Availability Statement
Conflicts of Interest
References
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Barkan, R.; Gruenbaum, Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 2013, 12, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Cao, K.; Blair, C.D.; Faddah, D.A.; Kieckhaefer, J.E.; Olive, M.; Erdos, M.R.; Nabel, E.G.; Collins, F.S. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest 2011, 121, 2833–2844. [Google Scholar] [CrossRef]
- Gordon, L.B.; Brown, W.T.; Collins, F.S. Hutchinson-Gilford Progeria Syndrome. In GeneReviews((R)), Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Seattle (WA), 1993.
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int J Biol Sci 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Bidault, G.; Garcia, M.; Capeau, J.; Morichon, R.; Vigouroux, C.; Bereziat, V. Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells. Cells 2020, 9. [Google Scholar] [CrossRef]
- Herrera, M.D.; Mingorance, C.; Rodriguez-Rodriguez, R.; Alvarez de Sotomayor, M. Endothelial dysfunction and aging: an update. Ageing Res Rev 2010, 9, 142–152. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Orosz, Z.; Ungvari, Z. Altered mitochondrial energy metabolism may play a role in vascular aging. Med Hypotheses 2006, 67, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.M.; Yasmin; McEniery, C. M.; Maki-Petaja, K.M.; Booth, A.D.; Cockcroft, J.R.; Wilkinson, I.B. Isolated systolic hypertension is characterized by increased aortic stiffness and endothelial dysfunction. Hypertension 2007, 50, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Price, J.M.; Hellermann, A.; Hellermann, G.; Sutton, E.T. Aging enhances vascular dysfunction induced by the Alzheimer's peptide beta-amyloid. Neurol Res 2004, 26, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.; Dumitrica, D.M.; Tebeanu, E.; Sapundgieva, A.; Dragomir, L.; Cristea, I. [Age related macular degeneration and C-reactive protein]. Oftalmologia 2008, 52, 73–76. [Google Scholar]
- Sanada, M.; Taguchi, A.; Higashi, Y.; Tsuda, M.; Kodama, I.; Yoshizumi, M.; Ohama, K. Forearm endothelial function and bone mineral loss in postmenopausal women. Atherosclerosis 2004, 176, 387–392. [Google Scholar] [CrossRef]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Daly, C.; Pasnikowski, E.; Burova, E.; Wong, V.; Aldrich, T.H.; Griffiths, J.; Ioffe, E.; Daly, T.J.; Fandl, J.P.; Papadopoulos, N.; et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci U S A 2006, 103, 15491–15496. [Google Scholar] [CrossRef]
- Kim, I.; Kim, J.H.; Moon, S.O.; Kwak, H.J.; Kim, N.G.; Koh, G.Y. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3'-kinase/Akt signal transduction pathway. Oncogene 2000, 19, 4549–4552. [Google Scholar] [CrossRef]
- Teichert-Kuliszewska, K.; Maisonpierre, P.C.; Jones, N.; Campbell, A.I.; Master, Z.; Bendeck, M.P.; Alitalo, K.; Dumont, D.J.; Yancopoulos, G.D.; Stewart, D.J. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc Res 2001, 49, 659–670. [Google Scholar] [CrossRef]
- Siemerink, M.J.; Augustin, A.J.; Schlingemann, R.O. Mechanisms of ocular angiogenesis and its molecular mediators. Dev Ophthalmol 2010, 46, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Torres, J.; Calvo, C.J.; Llach, A.; Guzman-Martinez, G.; Caballero, R.; Gonzalez-Gomez, C.; Jimenez-Borreguero, L.J.; Guadix, J.A.; Osorio, F.G.; Lopez-Otin, C.; et al. Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations. Proc Natl Acad Sci U S A 2016, 113, E7250–E7259. [Google Scholar] [CrossRef] [PubMed]
- DeBusk, F.L. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr 1972, 80, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.B.; Baba, N.; Boesel, C.P. Cardiovascular abnormalities in progeria. Case report and review of the literature. Arch Pathol Lab Med 1981, 105, 384–386. [Google Scholar]
- Gerhard-Herman, M.; Smoot, L.B.; Wake, N.; Kieran, M.W.; Kleinman, M.E.; Miller, D.T.; Schwartzman, A.; Giobbie-Hurder, A.; Neuberg, D.; Gordon, L.B. Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 2012, 59, 92–97. [Google Scholar] [CrossRef]
- Stehbens, W.E.; Delahunt, B.; Shozawa, T.; Gilbert-Barness, E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc Pathol 2001, 10, 133–136. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-Garcia, O.; Dominguez-Perez, M.; Gonzalez-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front Pharmacol 2019, 10, 1568. [Google Scholar] [CrossRef]
- Ahmed, A.; Fujisawa, T.; Niu, X.L.; Ahmad, S.; Al-Ani, B.; Chudasama, K.; Abbas, A.; Potluri, R.; Bhandari, V.; Findley, C.M.; et al. Angiopoietin-2 confers Atheroprotection in apoE-/- mice by inhibiting LDL oxidation via nitric oxide. Circ Res 2009, 104, 1333–1336. [Google Scholar] [CrossRef]
- Gete, Y.G.; Koblan, L.W.; Mao, X.; Trappio, M.; Mahadik, B.; Fisher, J.P.; Liu, D.R.; Cao, K. Mechanisms of angiogenic incompetence in Hutchinson-Gilford progeria via downregulation of endothelial NOS. Aging Cell 2021, 20, e13388. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J Clin Invest 2019, 129, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Vakili, S.; Izydore, E.K.; Losert, L.; Cabral, W.A.; Tavarez, U.L.; Shores, K.; Xue, H.; Erdos, M.R.; Truskey, G.A.; Collins, F.S.; et al. Angiopoietin-2 reverses endothelial cell dysfunction in progeria vasculature. Aging Cell 2024, e14375. [Google Scholar] [CrossRef] [PubMed]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Babaei, S.; Teichert-Kuliszewska, K.; Zhang, Q.; Jones, N.; Dumont, D.J.; Stewart, D.J. Angiogenic actions of angiopoietin-1 require endothelium-derived nitric oxide. Am J Pathol 2003, 162, 1927–1936. [Google Scholar] [CrossRef]
- Lemieux, C.; Maliba, R.; Favier, J.; Theoret, J.F.; Merhi, Y.; Sirois, M.G. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood 2005, 105, 1523–1530. [Google Scholar] [CrossRef]
- Celletti, F.L.; Waugh, J.M.; Amabile, P.G.; Brendolan, A.; Hilfiker, P.R.; Dake, M.D. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med 2001, 7, 425–429. [Google Scholar] [CrossRef]
- Nykanen, A.I.; Pajusola, K.; Krebs, R.; Keranen, M.A.; Raisky, O.; Koskinen, P.K.; Alitalo, K.; Lemstrom, K.B. Common protective and diverse smooth muscle cell effects of AAV-mediated angiopoietin-1 and -2 expression in rat cardiac allograft vasculopathy. Circ Res 2006, 98, 1373–1380. [Google Scholar] [CrossRef]
- Yu, H.; Moran, C.S.; Trollope, A.F.; Woodward, L.; Kinobe, R.; Rush, C.M.; Golledge, J. Angiopoietin-2 attenuates angiotensin II-induced aortic aneurysm and atherosclerosis in apolipoprotein E-deficient mice. Sci Rep 2016, 6, 35190. [Google Scholar] [CrossRef]
- Kozakova, M.; Palombo, C. Vascular Ageing and Aerobic Exercise. Int J Environ Res Public Health 2021, 18. [Google Scholar] [CrossRef]
- Anderson, T.J.; Gerhard, M.D.; Meredith, I.T.; Charbonneau, F.; Delagrange, D.; Creager, M.A.; Selwyn, A.P.; Ganz, P. Systemic nature of endothelial dysfunction in atherosclerosis. Am J Cardiol 1995, 75, 71B–74B. [Google Scholar] [CrossRef]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial-Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front Cardiovasc Med 2018, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Haga, J.H.; Li, Y.S.; Chien, S. Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech 2007, 40, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol 2018, 15, 555–565. [Google Scholar] [CrossRef]
- Jiang, Y.; Ji, J.Y. Progerin-Induced Impairment in Wound Healing and Proliferation in Vascular Endothelial Cells. Front Aging 2022, 3, 844885. [Google Scholar] [CrossRef]
- Wang, C.; Baker, B.M.; Chen, C.S.; Schwartz, M.A. Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol 2013, 33, 2130–2136. [Google Scholar] [CrossRef]
- Decker, M.L.; Chavez, E.; Vulto, I.; Lansdorp, P.M. Telomere length in Hutchinson-Gilford progeria syndrome. Mech Ageing Dev 2009, 130, 377–383. [Google Scholar] [CrossRef]
- Lenain, C.; de Graaf, C.A.; Pagie, L.; Visser, N.L.; de Haas, M.; de Vries, S.S.; Peric-Hupkes, D.; van Steensel, B.; Peeper, D.S. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res 2017, 27, 1634–1644. [Google Scholar] [CrossRef]
- Mojiri, A.; Walther, B.K.; Jiang, C.; Matrone, G.; Holgate, R.; Xu, Q.; Morales, E.; Wang, G.; Gu, J.; Wang, R.; et al. Telomerase therapy reverses vascular senescence and extends lifespan in progeria mice. Eur Heart J 2021, 42, 4352–4369. [Google Scholar] [CrossRef]
- Sato, T.N.; Tozawa, Y.; Deutsch, U.; Wolburg-Buchholz, K.; Fujiwara, Y.; Gendron-Maguire, M.; Gridley, T.; Wolburg, H.; Risau, W.; Qin, Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Puri, M.C.; Rossant, J.; Alitalo, K.; Bernstein, A.; Partanen, J. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. EMBO J 1995, 14, 5884–5891. [Google Scholar] [CrossRef] [PubMed]
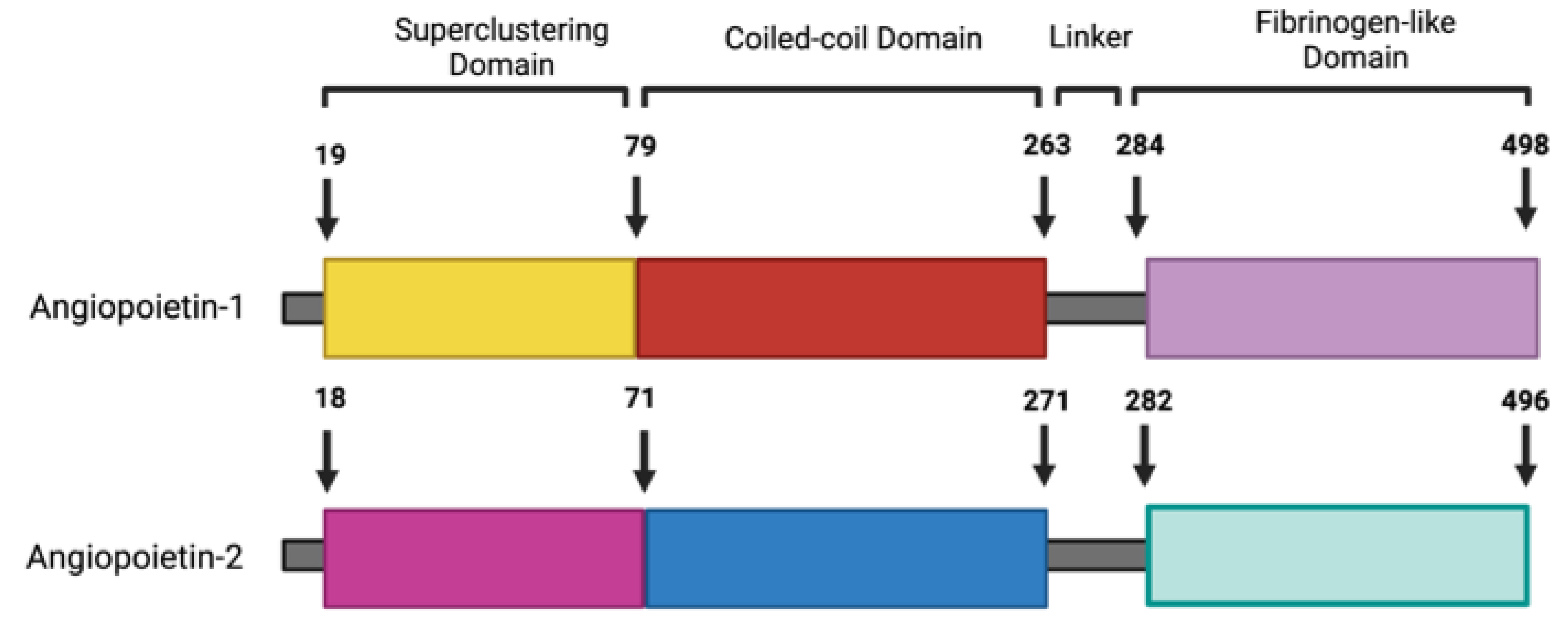
- Fiedler, U.; Krissl, T.; Koidl, S.; Weiss, C.; Koblizek, T.; Deutsch, U.; Martiny-Baron, G.; Marme, D.; Augustin, H.G. Angiopoietin-1 and angiopoietin-2 share the same binding domains in the Tie-2 receptor involving the first Ig-like loop and the epidermal growth factor-like repeats. J Biol Chem 2003, 278, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest 2012, 122, 1991–2005. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.T.; Khankin, E.V.; Karumanchi, S.A.; Parikh, S.M. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol 2009, 29, 2011–2022. [Google Scholar] [CrossRef]
- Asahara, T.; Chen, D.; Takahashi, T.; Fujikawa, K.; Kearney, M.; Magner, M.; Yancopoulos, G.D.; Isner, J.M. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res 1998, 83, 233–240. [Google Scholar] [CrossRef]
- Prakash, A.; Gordon, L.B.; Kleinman, M.E.; Gurary, E.B.; Massaro, J.; D'Agostino, R., Sr.; Kieran, M.W.; Gerhard-Herman, M.; Smoot, L. Cardiac Abnormalities in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA Cardiol 2018, 3, 326–334. [Google Scholar] [CrossRef]
- Labbe, P.; Munoz Goyette, V.; Thorin-Trescases, N.; Villeneuve, L.; Desanlis, I.; Delwarde, C.; Shi, Y.F.; Martel, C.; Yu, C.; Alikashani, A.; et al. Angiopoietin-like 2 is essential to aortic valve development in mice. Commun Biol 2022, 5, 1277. [Google Scholar] [CrossRef]
- Labbe, P.; Martel, C.; Shi, Y.F.; Montezano, A.; He, Y.; Gillis, M.A.; Higgins, M.E.; Villeneuve, L.; Touyz, R.; Tardif, J.C.; et al. Knockdown of ANGPTL2 promotes left ventricular systolic dysfunction by upregulation of NOX4 in mice. Front Physiol 2024, 15, 1320065. [Google Scholar] [CrossRef]
- Li, C.; Matsushita, S.; Li, Z.; Guan, J.; Amano, A. c-kit Positive Cardiac Outgrowth Cells Demonstrate Better Ability for Cardiac Recovery Against Ischemic Myopathy. J Stem Cell Res Ther 2017, 7. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Schroll, M.; Fenzl, F.; Lederer, E.M.; Hartinger, R.; Arnold, R.; Cagla Togan, D.; Guo, R.; Liu, S.; Petry, A.; et al. Inflammation and Fibrosis in Progeria: Organ-Specific Responses in an HGPS Mouse Model. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Thurston, G.; Hackett, S.F.; Renard, R.; Wang, Q.; McClain, J.; Martin, C.; Witte, C.; Witte, M.H.; Jackson, D.; et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell 2002, 3, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, M.; Hunter, R.; Bernas, M.; Gale, N.; Yancopoulos, G.; Erickson, R.; Witte, M. Defective remodeling and maturation of the lymphatic vasculature in Angiopoietin-2 deficient mice. Dev Biol 2008, 319, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.P.; Chen, S.H.; Trinh, J.; Kim, H.; Coomber, B.L.; Dumont, D.J. Differential response of lymphatic, venous and arterial endothelial cells to angiopoietin-1 and angiopoietin-2. BMC Cell Biol 2007, 8, 10. [Google Scholar] [CrossRef]
- Mazereeuw-Hautier, J.; Wilson, L.C.; Mohammed, S.; Smallwood, D.; Shackleton, S.; Atherton, D.J.; Harper, J.I. Hutchinson-Gilford progeria syndrome: clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature. Br J Dermatol 2007, 156, 1308–1314. [Google Scholar] [CrossRef]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002, 109, 1345–1350. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Robbins, A.L.; Savage, D.B. The genetics of lipid storage and human lipodystrophies. Trends Mol Med 2015, 21, 433–438. [Google Scholar] [CrossRef]
- Unger, R.H. The physiology of cellular liporegulation. Annu Rev Physiol 2003, 65, 333–347. [Google Scholar] [CrossRef]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. Adv Exp Med Biol 2017, 960, 161–196. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.M.; LaDana, C.; Wu, D.; Cao, K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging (Albany NY) 2013, 5, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat Commun 2020, 11, 481. [Google Scholar] [CrossRef]
- Cao, Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest 2007, 117, 2362–2368. [Google Scholar] [CrossRef]
- An, Y.A.; Sun, K.; Joffin, N.; Zhang, F.; Deng, Y.; Donze, O.; Kusminski, C.M.; Scherer, P.E. Angiopoietin-2 in white adipose tissue improves metabolic homeostasis through enhanced angiogenesis. Elife 2017, 6. [Google Scholar] [CrossRef]
- Sun, K.; Wernstedt Asterholm, I.; Kusminski, C.M.; Bueno, A.C.; Wang, Z.V.; Pollard, J.W.; Brekken, R.A.; Scherer, P.E. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A 2012, 109, 5874–5879. [Google Scholar] [CrossRef]
- Fang, J.; Dagenais, S.L.; Erickson, R.P.; Arlt, M.F.; Glynn, M.W.; Gorski, J.L.; Seaver, L.H.; Glover, T.W. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet 2000, 67, 1382–1388. [Google Scholar] [CrossRef]
- Cederberg, A.; Gronning, L.M.; Ahren, B.; Tasken, K.; Carlsson, P.; Enerback, S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 2001, 106, 563–573. [Google Scholar] [CrossRef]
- Xue, Y.; Cao, R.; Nilsson, D.; Chen, S.; Westergren, R.; Hedlund, E.M.; Martijn, C.; Rondahl, L.; Krauli, P.; Walum, E.; et al. FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc Natl Acad Sci U S A 2008, 105, 10167–10172. [Google Scholar] [CrossRef]
- Fernandez-Palazzi, F.; McLaren, A.T.; Slowie, D.F. Report on a case of Hutchinson-Gilford progeria, with special reference to orthopedic problems. Eur J Pediatr Surg 1992, 2, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Ozonoff, M.B.; Clemett, A.R. Progressive osteolysis in progeria. Am J Roentgenol Radium Ther Nucl Med 1967, 100, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Pendas, A.M.; Zhou, Z.; Cadinanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodriguez, F.; Tryggvason, K.; et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002, 31, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999, 147, 913–920. [Google Scholar] [CrossRef]
- ElHawary, H.; Baradaran, A.; Abi-Rafeh, J.; Vorstenbosch, J.; Xu, L.; Efanov, J.I. Bone Healing and Inflammation: Principles of Fracture and Repair. Semin Plast Surg 2021, 35, 198–203. [Google Scholar] [CrossRef]
- Yin, J.; Gong, G.; Sun, C.; Yin, Z.; Zhu, C.; Wang, B.; Hu, Q.; Zhu, Y.; Liu, X. Angiopoietin 2 promotes angiogenesis in tissue-engineered bone and improves repair of bone defects by inducing autophagy. Biomed Pharmacother 2018, 105, 932–939. [Google Scholar] [CrossRef]
- Hausman, M.R.; Schaffler, M.B.; Majeska, R.J. Prevention of fracture healing in rats by an inhibitor of angiogenesis. Bone 2001, 29, 560–564. [Google Scholar] [CrossRef]
- Lienau, J.; Schell, H.; Duda, G.N.; Seebeck, P.; Muchow, S.; Bail, H.J. Initial vascularization and tissue differentiation are influenced by fixation stability. J Orthop Res 2005, 23, 639–645. [Google Scholar] [CrossRef]
- Lienau, J.; Schmidt-Bleek, K.; Peters, A.; Haschke, F.; Duda, G.N.; Perka, C.; Bail, H.J.; Schutze, N.; Jakob, F.; Schell, H. Differential regulation of blood vessel formation between standard and delayed bone healing. J Orthop Res 2009, 27, 1133–1140. [Google Scholar] [CrossRef]
- Goto, T.; Fujioka, M.; Ishida, M.; Kuribayashi, M.; Ueshima, K.; Kubo, T. Noninvasive up-regulation of angiopoietin-2 and fibroblast growth factor-2 in bone marrow by pulsed electromagnetic field therapy. J Orthop Sci 2010, 15, 661–665. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef]
- Mandriota, S.J.; Pyke, C.; Di Sanza, C.; Quinodoz, P.; Pittet, B.; Pepper, M.S. Hypoxia-inducible angiopoietin-2 expression is mimicked by iodonium compounds and occurs in the rat brain and skin in response to systemic hypoxia and tissue ischemia. Am J Pathol 2000, 156, 2077–2089. [Google Scholar] [CrossRef]
- Tressel, S.L.; Kim, H.; Ni, C.W.; Chang, K.; Velasquez-Castano, J.C.; Taylor, W.R.; Yoon, Y.S.; Jo, H. Angiopoietin-2 stimulates blood flow recovery after femoral artery occlusion by inducing inflammation and arteriogenesis. Arterioscler Thromb Vasc Biol 2008, 28, 1989–1995. [Google Scholar] [CrossRef]
- Scholz, D.; Ziegelhoeffer, T.; Helisch, A.; Wagner, S.; Friedrich, C.; Podzuweit, T.; Schaper, W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol 2002, 34, 775–787. [Google Scholar] [CrossRef]
- Silvera, V.M.; Gordon, L.B.; Orbach, D.B.; Campbell, S.E.; Machan, J.T.; Ullrich, N.J. Imaging characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndrome. AJNR Am J Neuroradiol 2013, 34, 1091–1097. [Google Scholar] [CrossRef]
- Wang, J.; Yu, Q.; Ma, X.; Yuan, Z.; Mao, J. Hutchinson-Gilford progeria syndrome complicated with stroke: A report of 2 cases and literature review. Front Pediatr 2022, 10, 1056225. [Google Scholar] [CrossRef]
- Pantoni, L.; Garcia, J.H. Pathogenesis of leukoaraiosis: a review. Stroke 1997, 28, 652–659. [Google Scholar] [CrossRef]
- Procter, T.V.; Williams, A.; Montagne, A. Interplay between Brain Pericytes and Endothelial Cells in Dementia. Am J Pathol 2021, 191, 1917–1931. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab 2012, 32, 1139–1151. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Mae, M.A.; He, L.; Nordling, S.; Vazquez-Liebanas, E.; Nahar, K.; Jung, B.; Li, X.; Tan, B.C.; Chin Foo, J.; Cazenave-Gassiot, A.; et al. Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss. Circ Res 2021, 128, e46–e62. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Zhang, R.L.; Hozeska-Solgot, A.; Gregg, S.C.; Buller, B.; Lu, M.; Zhang, Z.G. Angiopoietin 2 mediates the differentiation and migration of neural progenitor cells in the subventricular zone after stroke. J Biol Chem 2009, 284, 22680–22689. [Google Scholar] [CrossRef] [PubMed]
- Marteau, L.; Valable, S.; Divoux, D.; Roussel, S.A.; Touzani, O.; MacKenzie, E.T.; Bernaudin, M.; Petit, E. Angiopoietin-2 is vasoprotective in the acute phase of cerebral ischemia. J Cereb Blood Flow Metab 2013, 33, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.L.; Du, Y.T.; Chen, X.; Lei, Y.; Sun, F.Y. Neuroprotective Effect of Angiopoietin2 Is Associated with Angiogenesis in Mouse Brain Following Ischemic Stroke. Brain Sci 2022, 12. [Google Scholar] [CrossRef]
- Nykanen, A.I.; Tikkanen, J.M.; Krebs, R.; Keranen, M.A.; Sihvola, R.K.; Sandelin, H.; Tuuminen, R.; Raisky, O.; Koskinen, P.K.; Lemstrom, K.B. Angiogenic growth factors in cardiac allograft rejection. Transplantation 2006, 82, S22–S24. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



