
Submitted:
08 November 2024
Posted:
12 November 2024
You are already at the latest version
Abstract
This study reports a 14-year-old male patient with multiple congenital anomalies, including hypospadias, complete cleft palate, and recurrent pneumonia. His clinical presentation includes significant physical and intellectual developmental delays, autism-like symptoms, and spastic diplegia. Whole-exome sequencing (WES) was performed due to these complex symptoms, revealing a novel heterozygous deletion on chromosome 10q24.31-q24.33. Laboratory findings indicated agammaglobulinemia, leading to prophylactic antibiotic therapy and immunoglobulin replacement. Additional imaging studies showed cystic malformation of the middle lobe of the right lung, sliding hiatal hernia with prolapse of the gastric mucosa, and brain anomalies consistent with Joubert syndrome. This case underscores the importance of genetic analysis in understanding the etiology of congenital anomalies and neurodevelopmental disorders, demonstrating that WES can provide critical insights into the genetic basis of complex clinical presentations. The identified chromosomal deletion contributes to the existing literature on genomic imbalances associated with similar phenotypes.
Keywords:
deletion
; copy number variation
; whole-exome sequencing
1. Introduction
The diagnostic potential for genetic disorders has been greatly increased by next-generation sequencing (NGS). A meta-analysis of 34,081 studies reveals a 36% diagnostic effectiveness for NGS in patients with neurodevelopmental disorders [1]. Chromosomal abnormalities are more prevalent than monogenic disorders in individuals with birth defects [2]. Whole-exome sequencing (WES) can discover 88% of pathogenic structural genomic variants (copy number variations, or CNVs) greater than three exons in length [1]. CNVs account for up to 35% of pathogenic variants in neurological patients, driven in part by genes such as PMP22, SMN1 and DMD [3]. While conventional cytogenetic techniques typically fail to identify CNVs, these variations are more prevalent than single nucleotide variants (SNVs) [4], and WES can detect SNVs, even at low-level mosaicism [5]. Consequently, WES offers a deeper genetic analysis for patients with unclassified congenital anomalies, and combining CNV and SNV searches enhances its diagnostic power [6,7]. Most CNV loci linked to neurodevelopmental disorders contain multiple genes highly expressed in the brain across developmental stages; the proteins they encode are involved in common pathogenic pathways [8]. Approximately 14.2% of multiple congenital anomalies and intellectual disabilities in patients are associated with CNVs over 400 kb [9]. Three patients had CNVs found in a recent WES investigation, which determined the etiology in 48.9% of instances [10].
This paper describes a 14-year-old male patient with congenital anomalies including hypospadias, complete cleft palate, and upper lip. The patient exhibited physical and intellectual developmental delays, autism-like symptoms, and cerebral palsy with spastic diplegia. The patient also had an immunodeficiency (agammaglobulinemia) and a history of recurrent pneumonias. This symptom complex led to WES analysis, revealing a previously unreported heterozygous deletion on the long arm of chromosome 10.
2. Case Presentation
A 14-year-old male (Figure 1) was admitted to the pediatric diagnostic department of the Russian Children’s Clinical Hospital, a branch of the Pirogov Russian National Research Medical University, for evaluation due to frequent pneumonia episodes. His mother has autoimmune thyroiditis. The child was born after the first pregnancy, complicated by two episodes of acute respiratory infection and hypertension in the mother. He was delivered at 40 weeks by vacuum extraction, with a birth weight of 3300 g and length of 52 cm. Apgar scores were 8/8.
From birth, the patient presented with multiple congenital anomalies, including hypospadias and complete clefts of the hard palate and upper lip. Psychomotor delay was noted from early infancy. Surgical correction of the cleft lip was performed at 1 year 4 months, and cleft palate repair at 5 years. Currently, he can walk with support, sits unaided, but does not speak or respond to simple commands. Diagnosed with cerebral palsy with spastic diplegia, his motor function level is classified as GMFCS III.
Over the past three years, he experienced approximately six pneumonia episodes.
In September 2023, at the age of 14, his parents brought him to the Russian Children’s Clinical Hospital for examination (hospitalized from 18.09.2023 to 08.10.2023). Upon admission, physical developmental delay was noted (height: 132 cm, <3rd percentile; weight: 24.5 kg, <3rd percentile, proportionate). Body temperature was normal. The patient did not speak, and did not follow simple commands, exhibiting profound intellectual disability. He could sit independently but required support to walk. Skin was pale without rashes; post-surgical scar on the lip was present. No edema; peripheral lymph nodes were unremarkable. Distal phalangeal deformities (clubbing) were noted. Respiration: 20/min, SpO₂: 98%. Nasal breathing was obstructed, with thick nasal discharge. Lung examination revealed vesicular breathing, diminished in the lower zones; no wheezing. Pulse: 90 bpm, regular rhythm, standing pulse rate: 90 bpm. Blood pressure (right arm): 100/70 mmHg. Heart sounds were rhythmic, clear, without murmurs. No appetite issues, occasional dysphagia with regurgitation. Abdomen: non-distended, soft, non-tender; liver and spleen not enlarged. Bladder and bowel function: incontinent. Stool: prone to constipation. Urination: free, painless, unchanged urine color, in diapers. The patient responded to auditory and visual stimuli.
Laboratory results showed agammaglobulinemia (IgG 0.31 g/L [normal 6.2–14.2], IgM 0.05 g/L [normal 0.5–1.7], IgA 0.01 g/L [normal 0.5–3]). Prophylactic therapy with trimethoprim-sulfamethoxazole and intravenous immunoglobulin replacement therapy were initiated.
Ophthalmologic examination revealed background retinopathy and concomitant divergent strabismus. Hypomagnesemia was also noted (serum magnesium level: 0.61 mmol/L, normal range: 0.74–1.2 mmol/L).
WES results were received on 19.10.2023. The patient was also evaluated by a geneticist at the Research Centre for Medical Genetics, where a blood sample was taken for karyotyping and chromosomal microarray analysis (CMA).
Results of the chest computed tomography in the child revealed changes characteristic of cystic malformation of the middle lobe of the right lung, a hiatal hernia with prolapse of a part of the stomach, multiple subpleural cysts, and bronchiectasis (Figure 2A).
Results of the fibroesophagogastroduodenoscopy showed gastroesophageal reflux in the patient. High gastroesophageal reflux (up to the upper third of the esophagus), as well as a sliding hiatal hernia with prolapse of the gastric mucosa into the esophagus, were confirmed by radiography of the esophagus and stomach with barium contrast (Figure 2B). Based on the examination, a gastrostomy and Nissen fundoplication have been planned.
The small intestine biopsy characterizes signs of mildly active duodenitis, including deformed villi. The gastric biopsy reveals reactive gastropathy with foveolar hyperplasia of the glands. The lamina propria contains moderate lymphoplasmacytic infiltration with a mixture of neutrophils and eosinophils in both the stomach and small intestine. H. Pylori is not detected in Giemsa staining. The esophagus exhibits changes characteristic of esophagitis, with lymphocytic infiltration, mild basal layer hyperplasia, spongiosis, and increased height of connective tissue papillae.
Magnetic resonance imaging (MRI) of the brain showed brain anomalies consistent with Joubert syndrome. A cleft palate, an open nasopharyngeal fistula, and edema with polyposis of the nasal sinuses were identified (Figure 2C). The child has been consulted by a maxillofacial surgeon, and surgical correction of the cleft palate has been planned.
3. Genetic Testing
Genomic DNA was extracted from venous blood using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). DNA libraries were prepared from 500 ng of genomic DNA using the MGI Easy Universal DNA Library Prep Set (MGI Tech, Shenzhen, China) per manufacturer’s protocol. DNA fragmentation was performed using ultrasonication on a Covaris S-220 (Covaris, Inc., Woburn, MA, USA), resulting in an average fragment length of 250 bp. DNA library enrichment was done by pre-pooling using SureSelect Human All Exon v8 probes (Agilent Technologies, Santa Clara, CA, USA), covering the full human exome. DNA and library concentrations were measured with a Qubit Flex (Life Technologies, Carlsbad, CA, USA) using the dsDNA HS Assay Kit. Library quality was assessed using the Bioanalyzer 2100 with High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA). The libraries were circularized and sequenced in paired-end mode on a DNBSEQ G-400 platform using the DNBSEQ-G400RS High-throughput Sequencing Set PE100 (MGI Tech, Shenzhen, China), with an average coverage of 100×. FastQ files were generated using MGI Tech’s basecallLite software (ver. 1.0.7.84) from the manufacturer (MGI Tech, Shenzhen, China).
Bioinformatics quality control and sequencing data correction were performed using FastQC v0.11.9 [12] and bbduk v38.96 [13]. Alignment to the human reference genome GRCh37 was carried out with bwa-mem2 v2.2.1 [14]. Aligned files were converted to BAM format, sorted, and indexed with SAMtools v1.9 [15]. Duplicates were marked and quality metrics were collected with Picard v2.22.4 [16]. Variant calling was performed with bcftools v1.9 [17] and deepvariant v1.5.0 [18]. CNV analysis was conducted with CNVkit [19], ClinViewer [20], and annotated with AnnotSV [21]. Clinical significance assessment followed ACMG guidelines [22,23]. The identified variant was validated by CMA using Affimetrix CytoScan HD arrays on a venous blood sample.
A heterozygous deletion spanning 2.1 Mb (10q24.31-q24.33) on chromosome 10 was detected, involving 38 protein-coding genes, 12 of which (ARL3, CNNM2, CYP17A1, FGF8, GBF1, HPS6, LBX1, NFKB2, NT5C2, PITX3, SUFU, TRIM8) are disease-associated. Germline mutations in ARL3 are associated with Retinitis pigmentosa 83 (OMIM: 618173); CNNM2 is linked to Hypomagnesemia 6, renal (OMIM: 613882) and Hypomagnesemia, seizures, and impaired intellectual development 1 (OMIM: 616418). FGF8 mutations are linked to Hypogonadotropic hypogonadism 6 with or without anosmia (OMIM: 612702); GBF1 mutations associate with Charcot-Marie-Tooth disease, axonal, type 2GG (OMIM: 606483); NFKB2 mutations with Immunodeficiency, common variable, 10 (OMIM: 615577); PITX3 mutations with Anterior segment dysgenesis 1, multiple subtypes (OMIM: 107250) and Cataract 11, multiple types (OMIM: 610623). SUFU mutations are linked to Medulloblastoma (OMIM: 155255), familial meningiomas (OMIM: 607174), and Basal cell nevus syndrome 2 (OMIM: 620343). TRIM8 mutations are associated with Focal segmental glomerulosclerosis and neurodevelopmental syndrome (OMIM: 619428). Autosomal recessive syndromes and genes associated solely with this inheritance pattern are excluded, as no second clinically significant allele was detected.
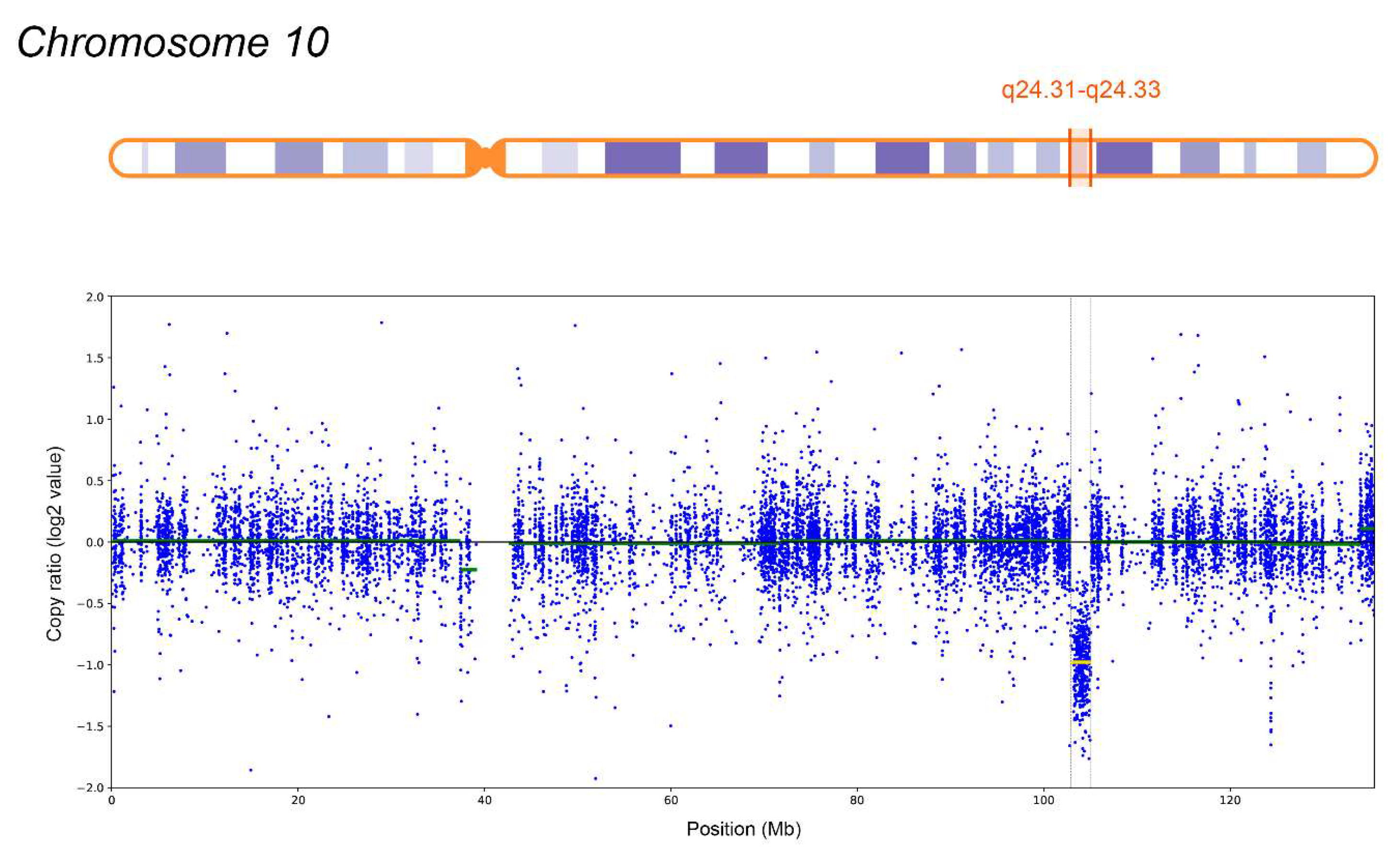
The identified CNV is not associated with known microdeletion syndromes and has not been reported in a healthy population. According to AnnotSV, GC content is 63.0% and 67.0% within ±100 bp of the CNV boundaries, suggesting potential non-allelic homologous recombination. The ExAC Z-score for deletions is lower than for duplications (1.093 and 1.384, respectively), indicating that the region is less tolerant to copy number increase. Based on ACMG criteria 2A (+1), 3C (+0.9), and 4L (+0.1), the deletion was classified as pathogenic (total score: 2). Validation by CMA (Figure 3): Molecular Karyotype (ISCN 2020): arr[GRCh37] 10q24.31-q24.33(102837530_105033440)x1 (~2.2 Mb), closely matching WES findings. The total length of regions of loss of heterozygosity, measuring 3 million bp or more, corresponds to the population rate (0.55%).
The final list of genes included in the deletion region: ACTR1A, ARL3, ARMH3, AS3MT, BORCS7, BORCS7-ASMT, BTRC, C10orf95, CNNM2, CUEDC2, CYP17A1, DPCD, ELOVL3, FBXL15, FBXW4, FGF8, GBF1, HPS6, KCNIP2, KCNIP2-AS1, LBX1, LBX1-AS1, LDB1, LINC01514, LINC02681, LOC107984265, MFSD13A, MIR146B, MIR3158-1, MIR3158-2, NFKB2, NOLC1, NPM3, NT5C2, OGA, PITX3, POLL, PPRC1, PSD, RPARP-AS1, RPEL1, SFXN2, SUFU, TLX1, TLX1NB, TRIM8, WBP1L (total of 47); of which 37 encode proteins. Genes associated with diseases were identical to those found on WES. The haploinsufficiency genes are ACTR1A, CNNM2, LDB1, NFKB2, PPRC1, PSD, SUFU, TRIM8, FGF8, WBP1L, BTRC, LBX1, NT5C2, PITX3 (according to LOEUF, pLI, pHI, %HI), and the triplosensitive genes are LDB1 and TRIM8 (according to pTS). In the UK Biobank v2, 6 deletions and 36 duplications were detected in this region. In gnomAD SVs v4.1.0, the 5 largest deletions with a frequency of 0.000008 (per allele) ranged in size from 340 to 2041.53 kb, 4 of which had a LOF effect, and 1 was located in the intron of CNNM2. Despite the detection of 569 deletions and only 266 duplications, the largest 10 deletions were in the range of 0.158-0.745 Mb. This region also contained 239 insertions (ranging from 50 bp to 6017 bp) and 5 inversions.
The identified CNV has been registered in ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/variation/3342255/ (available online)
4. Discussion
Despite the uneven coverage of target regions, unlike the uniform coverage achieved with whole genome sequencing (WGS), it is recommended to perform CNV analysis on exome data before resorting to WGS, which is approximately four times more expensive [24,25]. For example, among 3040 patients who underwent WES, CNVs were found in 2.4% of cases, varying from a few exons of a single gene (1.4 Mb) to several genes (21.6 Mb) [26]. It is easier to identify large deletions on WES: heterozygous states exhibit runs of homozygosity, while homozygous states show no reads in the area. Duplications, especially when the syndrome has not been previously described and is not obvious from the clinical picture, are more challenging, as are instances of random read removal preceding the creation of the .bam file, when the variant allele frequency may change and abnormal genotypes may not be suspected. This limitation still exists; in one of the latest studies using WES data, 171 CNVs were identified, of which 140 were deletions and 15 duplications, while others represented more complex structural variants [27]. Analysis of WES data using ExomeDepth established that the sensitivity of the method is 89% for detecting large deletions and 65% for duplications, with a diagnostic value of 1.6%, which was comparable to CMA [28]. The CNVkit used in our practice differs from ExomeDepth and its counterparts by integrating sequence data from both target and non-target regions, thus providing a more comprehensive and accurate identification of CNVs.
The malformation "split hand/foot malformation" (SHFM) is defined by median clefts of the hands and feet, aplasia and/or hypoplasia of the phalanges, metacarpal and metatarsal bones, as well as syndactyly. This rare condition occurs in 1 in 8500–25000 newborns, yet accounts for 15% of all limb developmental disorders [29]. The most well-known condition associated with the locus 10q24 is SHFM type 3 (SHFM3) [30,31], which is characterized by an increase in tandem genomic repeats of size 325–570 kb involving at least the gene DACTYLIN (FBXW4) [30]. The etiology of SHFM3 is associated with disruptions in the apical ectodermal ridge of the limbs. The SHFM3 locus on 10q24 is conserved among vertebrates from zebrafish to humans, especially the region from TLX1 to FGF8, and contains numerous regulatory elements active in mouse embryos. In the human genome, the highest density of conserved non-coding elements is found in BTRC. It is claimed that duplication of the first exon of the BTRC gene is responsible for the formation of the SHFM3 phenotype, possibly through cis- or trans-acting effects on genes or regulatory sequences involved in limb development [32]. There are described cases of SHFM3 that are not caused by duplications; for example, an 80.2 kb microdeletion in the 10q24.32 region was found in a girl with absence of the right radius and thumb, shortening of the right ulna, hypoplasia of the left thenar, and a small apical defect in the interventricular septum. This deletion included part of the DPCD gene (exons 2–6), all of MIR3158-1, MIR3158-2, and part of the FBXW4 gene (exons 4–9). The CNV was inherited from an asymptomatic mother [33]. An analysis of the DECIPHER database conducted by Li CF et al. identified two male patients with deletions in the 10q24.31–q24.32 region. The CNV of the first patient measured 546.42 kb and included 13 genes from LINC01514 to part of FBXW4, with the patient having a cleft palate and lip, intellectual disability, microphthalmia, kidney pathology, vesicoureteral reflux, pulmonary artery stenosis, sensorineural hearing loss, short stature, and abdominal obesity. The deletion size of the second patient was 389.02 kb, including genes from BTRC to part of FBXW4, and the patient had aortic dilation, Pierre Robin sequence, median cleft palate, kidney hypoplasia, and a secondary atrial septal defect [32]. One patient with bilateral radial dysplasia had a substitution g.103380009A>G [hg19], or FBXW4(NM_022039.4):c.1301+4493T>C (6 intron), which had unclear clinical significance [33].
The literature presents cases of deletions of the long arm of chromosome 10 with close coordinates. A patient with a deletion of 3.79 Mb in the 10q24.2-q24.32 region was born with a left-sided cleft lip, nystagmus, and clubfoot. Subsequently, breathing problems, a congenital type I posterior urethral valve, vesicoureteral reflux of grades II-III, as well as polycystic and hypoplastic kidneys were diagnosed, eventually requiring peritoneal dialysis at the age of 4. The patient also experienced delayed descent of the testicles, significant vision loss, and hypoplasia of the corpus callosum [34]. Another patient with a 5.54 Mb deletion in 10q24.31–q25.1 exhibited bilateral cleft lip and palate, as well as Dandy-Walker malformation with bilateral ventriculomegaly. Postnatal MRI of the brain showed absence of the lower part of the cerebellar vermis, enlarged fourth ventricle opening into the midline retrocerebellar cystic space, increased posterior cranial fossa, and anomalies corresponding to lobar holoprosencephaly. The patient experienced developmental delays, spastic quadriplegia, and malnutrition, requiring the placement of a gastrojejunostomy tube and ventriculoperitoneal shunting. Despite rehabilitative therapy and surgical interventions, by 22 months, the patient could not sit up or turn over independently and exhibited delays in speech and motor skills [35].
In the DECIPHER clinical data database, smaller deletions fully encompassed within the deletion region are described. Among them, excluding other genetic variants, there were 3 girls (the age of only one is known: 6 years) and 6 boys (from infancy to 19 years), while the phenotype was unspecified in 1 girl and 1 boy. Nervous system disorders were found in five patients and manifested as global developmental delays (2/7), intellectual disability (2/7), absence/delay in speech (2/7), ataxia together with generalized hypotonia (2/7), and seizures with frontoparietal polymicrogyria (1/7). Two patients exhibited short stature, and one of them also had abdominal obesity with microphthalmia, cleft lip and palate, short phalanges, pulmonary artery stenosis, kidney anomalies, vesicoureteral reflux, and sensorineural hearing loss. Strabismus along with a high-arched palate was found in one patient, and MRI of the brain showed anomalies in cortical gyration, pyramidal signs, hypoplasia of the cerebellar vermis, brain atrophy, lobar holoprosencephaly, and partial agenesis of the corpus callosum. One patient had only joint contracture in the upper limbs.
FGF8 (fibroblast growth factor 8) regulates processes of cell proliferation, differentiation, and migration. It is necessary for the normal formation of the brain, eyes, ears, and limbs, as well as for the development of the neural system associated with the release of gonadotropin-releasing hormone [36]. According to epidemiological data from 23 regions of the Russian Federation from 2011 to 2018, cleft lip/palate occurs in 8.58 per 10,000 newborns (compared to 8.42 according to EUROCAT) [37], making it the most common craniofacial defect overall [38], and the risk of stillbirth for patients with this defect is relatively low: 10 in 1000 pregnancies [39]. However, establishing the causes of non-syndromic orofacial clefts is more challenging; WES managed this in 10% of a cohort of 84 patients [40]. We believe that the cleft lip and palate in our patient, along with the disorder of sexual development, are caused by the haploinsufficiency of FGF8 [41]. It is hypothesized that the ACTR1A gene may be associated with intellectual disability since the dynactin it encodes functions within the presynaptic membrane to promote synaptic stability [42].
Loss-of-function mutations in the NFKB2 gene, especially in its C-terminal domain, play a key role in the development of primary immunodeficiencies. The most frequently described amino acid residue variations occur at Arg853, leading to the formation of premature termination codons, while the transcripts are predicted to escape nonsense-mediated decay mechanisms. The truncated proteins lack phosphorylation sites necessary for interaction with NIK, resulting in the protein p100 not being converted to p52. Consequently, this disrupts the non-canonical NF-κB pathway required for normal B-cell function and antibody production, leading to a clinical picture of common variable immunodeficiency in early childhood [43]. One of the main complaints in the patient was recurrent pneumonias, likely of aspiration nature, although immunoglobulin levels A, M, and G were significantly reduced. We suspect that the deletion of the NFKB2 gene is the cause of the patient’s immunodeficiency.
5. Conclusions
Establishing an accurate genetic diagnosis in patients with multiple congenital anomalies and intellectual disabilities often poses a complex clinical challenge. Thus, our case demonstrates the importance of employing a comprehensive genetic approach (WES and CMA). The identified heterozygous deletion on chromosome 10q24.31-q24.33 is newly described and clinically significant, explaining the phenotype of this patient.
Author Contributions
Conceptualization, A.A.B.; software, O.N.S.; data curation, O.P.P.; formal analysis, A.A.B.; resources, Yu.S.L., T.V.K., I.S.K., A.A.I. and S.S.V.; writing—original draft preparation, A.A.B. and Yu.S.L.; writing—review and editing, D.O.K.; supervision. D.O.K.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. Ethical review and approval by the Ethics Committee were not required for the clinical case presentation, in accordance with institutional requirements.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The sequence data are generated from patient sample and therefore are available under restricted access.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wortmann, S.B.; Oud, M.M.; Alders, M.; Coene, K.L.M.; Van Der Crabben, S.N.; Feichtinger, R.G.; Garanto, A.; Hoischen, A.; Langeveld, M.; Lefeber, D.; et al. How to Proceed after “Negative” Exome: A Review on Genetic Diagnostics, Limitations, Challenges, and Emerging New Multiomics Techniques. J of Inher Metab Disea 2022, 45, 663–681. [Google Scholar] [CrossRef]
- Feldkamp, M.L.; Carey, J.C.; Byrne, J.L.B.; Krikov, S.; Botto, L.D. Etiology and Clinical Presentation of Birth Defects: Population Based Study. BMJ 2017, j2249. [Google Scholar] [CrossRef]
- Truty, R.; Paul, J.; Kennemer, M.; Lincoln, S. E.; Olivares, E.; Nussbaum, R.L.; Aradhya, S. Prevalence and Properties of Intragenic Copy-Number Variation in Mendelian Disease Genes. Genetics in Medicine 2019, 21, 114–123. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Structural Variation in the Human Genome and Its Role in Disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef]
- Wright, C.F.; Prigmore, E.; Rajan, D.; Handsaker, J.; McRae, J.; Kaplanis, J.; Fitzgerald, T.W.; FitzPatrick, D.R.; Firth, H.V.; Hurles, M.E. Clinically-Relevant Postzygotic Mosaicism in Parents and Children with Developmental Disorders in Trio Exome Sequencing Data. Nat Commun 2019, 10, 2985. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Cisarova, K.; Niel-Butschi, F.; Mittaz-Crettol, L.; Fodstad, H.; Superti-Furga, A. CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations. Genes 2021, 12, 1427. [Google Scholar] [CrossRef]
- Tilemis, F.-N.; Marinakis, N.M.; Veltra, D.; Svingou, M.; Kekou, K.; Mitrakos, A.; Tzetis, M.; Kosma, K.; Makrythanasis, P.; Traeger-Synodinos, J.; et al. Germline CNV Detection through Whole-Exome Sequencing (WES) Data Analysis Enhances Resolution of Rare Genetic Diseases. Genes 2023, 14, 1490. [Google Scholar] [CrossRef]
- Azidane, S.; Gallego, X.; Durham, L.; Cáceres, M.; Guney, E.; Pérez-Cano, L. Identification of Novel Driver Risk Genes in CNV Loci Associated with Neurodevelopmental Disorders. Human Genetics and Genomics Advances 2024, 5, 100316. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A Copy Number Variation Morbidity Map of Developmental Delay. Nat Genet 2011, 43, 838–846. [Google Scholar] [CrossRef]
- Wayhelova, M.; Vallova, V.; Broz, P.; Mikulasova, A.; Smetana, J.; Dynkova Filkova, H.; Machackova, D.; Handzusova, K.; Gaillyova, R.; Kuglik, P. Exome Sequencing Improves the Molecular Diagnostics of Paediatric Unexplained Neurodevelopmental Disorders. Orphanet J Rare Dis 2024, 19, 41. [Google Scholar] [CrossRef]
- Belova, V.; Pavlova, A.; Afasizhev, R.; Moskalenko, V.; Korzhanova, M.; Krivoy, A.; Cheranev, V.; Nikashin, B.; Bulusheva, I.; Rebrikov, D.; et al. System Analysis of the Sequencing Quality of Human Whole Exome Samples on BGI NGS Platform. Sci Rep 2022, 12, 609. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2017; Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 14 October 2023).
- Bushnell, B. BBMap: a fast, accurate, splice-aware aligner. 2014. Available online: https://github.com/BioInfoTools/BBMap (accessed on 14 October 2023).
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Broad Institute. Picard Toolkit. 2014. Available online: https://broadinstitute.github.io/picard/ (accessed on 14 October 2023).
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P. T.; et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat Biotechnol 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Macnee, M.; Pérez-Palma, E.; Brünger, T.; Klöckner, C.; Platzer, K.; Stefanski, A.; Montanucci, L.; Bayat, A.; Radtke, M.; Collins, R.L.; et al. CNV-ClinViewer: Enhancing the Clinical Interpretation of Large Copy-Number Variants Online. Bioinformatics 2023, 39, btad290. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An Integrated Tool for Structural Variations Annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genetics in Medicine 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Testard, Q.; Vanhoye, X.; Yauy, K.; Naud, M.-E.; Vieville, G.; Rousseau, F.; Dauriat, B.; Marquet, V.; Bourthoumieu, S.; Geneviève, D.; et al. Exome Sequencing as a First-Tier Test for Copy Number Variant Detection: Retrospective Evaluation and Prospective Screening in 2418 Cases. J Med Genet 2022, 59, 1234–1240. [Google Scholar] [CrossRef]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are Whole-Exome and Whole-Genome Sequencing Approaches Cost-Effective? A Systematic Review of the Literature. Genetics in Medicine 2018, 20, 1122–1130. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical Application of Whole-Exome Sequencing across Clinical Indications. Genetics in Medicine 2016, 18, 696–704. [Google Scholar] [CrossRef]
- Lemire, G.; Sanchis-Juan, A.; Russell, K.; Baxter, S.; Chao, K.R.; Singer-Berk, M.; Groopman, E.; Wong, I.; England, E.; Goodrich, J.; et al. Exome Copy Number Variant Detection, Analysis, and Classification in a Large Cohort of Families with Undiagnosed Rare Genetic Disease. The American Journal of Human Genetics 2024, 111, 863–876. [Google Scholar] [CrossRef]
- Marchuk, D.S.; Crooks, K.; Strande, N.; Kaiser-Rogers, K.; Milko, L.V.; Brandt, A.; Arreola, A.; Tilley, C.R.; Bizon, C.; Vora, N.L.; et al. Increasing the Diagnostic Yield of Exome Sequencing by Copy Number Variant Analysis. PLoS ONE 2018, 13, e0209185. [Google Scholar] [CrossRef]
- Holder-Espinasse, M.; Jamsheer, A.; Escande, F.; Andrieux, J.; Petit, F.; Sowinska-Seidler, A.; Socha, M.; Jakubiuk-Tomaszuk, A.; Gerard, M.; Mathieu-Dramard, M.; et al. Duplication of 10q24 Locus: Broadening the Clinical and Radiological Spectrum. Eur J Hum Genet 2019, 27, 525–534. [Google Scholar] [CrossRef]
- de Mollerat, X.J.; Gurrieri, F.; Morgan, C.T.; Sangiorgi, E.; Everman, D.B.; Gaspari, P.; Amiel, J.; Bamshad, M.J.; Lyle, R.; Blouin, J.L.; et al. A Genomic Rearrangement Resulting in a Tandem Duplication Is Associated with Split Hand-Split Foot Malformation 3 (SHFM3) at 10q24. Human Molecular Genetics 2003, 12, 1959–1971. [Google Scholar] [CrossRef]
- Dimitrov, B.I.; De Ravel, T.; Van Driessche, J.; De Die-Smulders, C.; Toutain, A.; Vermeesch, J.R.; Fryns, J.P.; Devriendt, K.; Debeer, P. Distal Limb Deficiencies, Micrognathia Syndrome, and Syndromic Forms of Split Hand Foot Malformation (SHFM) Are Caused by Chromosome 10q Genomic Rearrangements. Journal of Medical Genetics 2010, 47, 103–111. [Google Scholar] [CrossRef]
- Li, C.; Angione, K.; Milunsky, J. Identification of Critical Region Responsible for Split Hand/Foot Malformation Type 3 (SHFM3) Phenotype through Systematic Review of Literature and Mapping of Breakpoints Using Microarray Data. Microarrays 2015, 5, 2. [Google Scholar] [CrossRef]
- Vergult, S.; Hoogeboom, A.J.M.; Bijlsma, E.K.; Sante, T.; Klopocki, E.; De Wilde, B.; Jongmans, M.; Thiel, C.; Verheij, J.B.G.M.; Perez-Aytes, A.; et al. Complex Genetics of Radial Ray Deficiencies: Screening of a Cohort of 54 Patients. Genetics in Medicine 2013, 15, 195–202. [Google Scholar] [CrossRef]
- Hoefele, J.; Gabert, M.; Heinrich, U.; Benz, K.; Rompel, O.; Rost, I.; Klein, H.-G.; Kunstmann, E. A Novel Interstitial Deletion of 10q24.2q24.32 in a Patient with Renal Coloboma Syndrome. European Journal of Medical Genetics 2012, 55, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Peltekova, I.T.; Hurteau-Millar, J.; Armour, C.M. Novel Interstitial Deletion of 10q24.3–25.1 Associated with Multiple Congenital Anomalies Including Lobar Holoprosencephaly, Cleft Lip and Palate, and Hypoplastic Kidneys. American J of Med Genetics Pt A 2014, 164, 3132–3136. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. New Developments in the Biology of Fibroblast Growth Factors. WIREs Mechanisms of Disease 2022, 14, e1549. [Google Scholar] [CrossRef] [PubMed]
- Demikova, N.S.; Podolnaya, M.A.; Lapina, A.S. Analysis of the congenital malformations epidemiology in the regions of the Russian Federation. Medical Genetics 2020, 19(7), 31–32. (In Russ.).
- Mossey, P.A.; Modell, B. Epidemiology of Oral Clefts 2012: An International Perspective. In Frontiers of Oral Biology; Cobourne, M. T., Ed.; S. Karger AG, 2012; Vol. 16, pp 1–18.
- Heinke, D.; Nestoridi, E.; Hernandez-Diaz, S.; Williams, P.L.; Rich-Edwards, J.W.; Lin, A.E.; Van Bennekom, C.M.; Mitchell, A.A.; Nembhard, W.N.; Fretts, R.C.; et al. Risk of Stillbirth for Fetuses With Specific Birth Defects. Obstetrics & Gynecology 2020, 135, 133–140. [Google Scholar]
- Basha, M.; Demeer, B.; Revencu, N.; Helaers, R.; Theys, S.; Bou Saba, S.; Boute, O.; Devauchelle, B.; Francois, G.; Bayet, B.; et al. Whole Exome Sequencing Identifies Mutations in 10% of Patients with Familial Non-Syndromic Cleft Lip and/or Palate in Genes Mutated in Well-Known Syndromes. J Med Genet 2018, 55, 449–458. [Google Scholar] [CrossRef]
- Trarbach, E.B.; Abreu, A.P.; Silveira, L.F.G.; Garmes, H.M.; Baptista, M.T.M.; Teles, M.G.; Costa, E.M.F.; Mohammadi, M.; Pitteloud, N.; Mendonca, B.B.; et al. Nonsense Mutations in FGF8 Gene Causing Different Degrees of Human Gonadotropin-Releasing Deficiency. The Journal of Clinical Endocrinology & Metabolism 2010, 95, 3491–3496. [Google Scholar]
- Eaton, B.A.; Fetter, R.D.; Davis, G.W. Dynactin Is Necessary for Synapse Stabilization. Neuron 2002, 34, 729–741. [Google Scholar] [CrossRef]
- Aird, A.; Lagos, M.; Vargas-Hernández, A.; Posey, J.E.; Coban-Akdemir, Z.; Jhangiani, S.; Mace, E.M.; Reyes, A.; King, A.; Cavagnaro, F.; et al. Novel Heterozygous Mutation in NFKB2 Is Associated With Early Onset CVID and a Functional Defect in NK Cells Complicated by Disseminated CMV Infection and Severe Nephrotic Syndrome. Front. Pediatr. 2019, 7, 303. [Google Scholar] [CrossRef]
Figure 1.
Patient's appearance. Photo from the family archive.

Figure 2.
(A) Chest computed tomography scan showing cystic malformation of the middle lobe of the right lung, hiatal hernia with gastric prolapse, subpleural cysts, and bronchiectasis. (B) Esophageal and gastric fluoroscopy with barium contrast demonstrating a sliding hiatal hernia with prolapse of the gastric mucosa. (C) Brain magnetic resonance imaging indicating anomalies consistent with Joubert syndrome, cleft palate, and edema with polyposis of the paranasal sinuses.
Figure 2.
(A) Chest computed tomography scan showing cystic malformation of the middle lobe of the right lung, hiatal hernia with gastric prolapse, subpleural cysts, and bronchiectasis. (B) Esophageal and gastric fluoroscopy with barium contrast demonstrating a sliding hiatal hernia with prolapse of the gastric mucosa. (C) Brain magnetic resonance imaging indicating anomalies consistent with Joubert syndrome, cleft palate, and edema with polyposis of the paranasal sinuses.
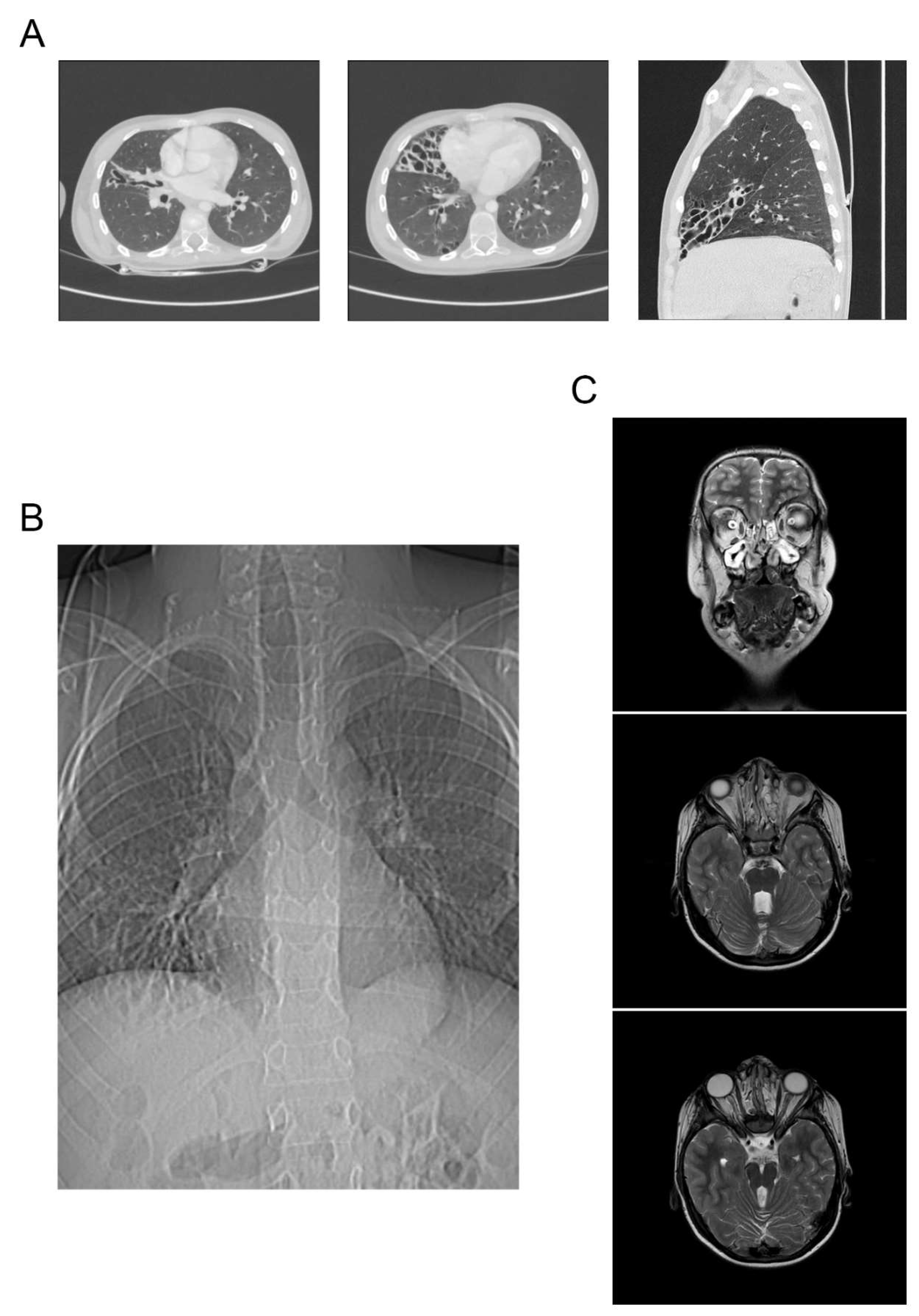
Figure 3.
Region of heterozygous copy loss arr[GRCh37] 10q24.31-q24.33(102837530_105033440)x1 (~2.2 Mb) of patient N., displayed in the IGV genome browser (data from whole exome sequencing).
Figure 3.
Region of heterozygous copy loss arr[GRCh37] 10q24.31-q24.33(102837530_105033440)x1 (~2.2 Mb) of patient N., displayed in the IGV genome browser (data from whole exome sequencing).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



