
Submitted:
07 November 2024
Posted:
08 November 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The origin of COVID-19 remains unclear despite extensive research. Theoretical models can simplify complex epigenetic landscapes by reducing vast methylation sites into manageable sets, revealing fundamental pathogen interactions that leap medical advances for the first time in tracing virus origin in the literature and practices. In our study, a max-logistic intelligence classifier analyzed 865,859 Infinium MethylationEPIC sites (CpGs), identifying eight CpGs that achieved 100% accuracy in distinguishing COVID-19 patients from other respiratory disease patients and healthy controls. One CpG, cg07126281, linked to the SAMM50 gene, shares genetic ties with rare infectious diseases like Sennetsu fever and glanders, suggesting a potential connection between COVID-19 and these diseases, possibly transmitted through contaminated seafood or glanders-infected individuals. Identifying such links among 865,859 CpG sites is challenging, with a random correlation probability of less than one in ten million. However, the likelihood of finding meaningful associations with rare diseases lowers this probability to one in one hundred million, reinforcing the credibility of our findings. These results highlight the importance of investigating seafood markets and global supply chains in tracing COVID-19's origins and emphasize the need for ongoing biosafety and biosecurity measures to prevent future outbreaks.
Keywords:
Biosafety and biosecurity
; DNA methylations
; site-site interaction effects
; rare diseases
; Sennetsu fever
; glanders
The resurgence of COVID-19 has become a growing concern, with the World Health Organization (WHO) warning of a summertime surge (UN News, 08/06/2024). While ongoing studies on the current state of COVID-19 are crucial, understanding the origins of the pandemic is equally important. Despite extensive research, the origin of COVID-19 remains elusive, with unknown factors behind its emergence contributing to trillions in economic losses and millions of deaths. This underscores the need for new scientific approaches to uncover the genomic and DNA methylation drivers of SARS-CoV-2 replication [1,2,3,4,5,6,7,8,9,10,11,12,13].
Our previous work achieved the highest accuracy in the literature by revealing significant deviations in Omicron’s gene-gene interactions compared to earlier variants. We hypothesized that Omicron might have been transmitted from COVID-19-infected animals back to humans, providing a new method for calculating the reproduction number (R0) and explaining Omicron’s high transmissibility [12]. Moreover, we discovered significant genomic differences between SARS-CoV-2 (NP/OP PCR swabs) and COVID-19 (blood samples) [10]. By using optimal interactive genomic biomarkers, we identified adverse effects on gene expression from the BNT162b2 vaccine in COVID-19-convalescent octogenarians [11] and from inactivated vaccines using data from GSE189263 [14].
Reliable biomarkers are essential for scientific progress, as they must exhibit consistent properties across various trials and cohorts, achieving an overall accuracy of 95% or higher. However, identifying such biomarkers is challenging. Many gene biomarkers identified in single trials fail to apply to other cohorts, resulting in low or zero commonality across different groups. This limitation has hindered medical advancements, wasted resources, and cost lives. For instance, well-known genes like BRCA1 and BRCA2 have shown low efficiency in breast cancer diagnosis at RNA level, as noted in published research [15]. These issues raise concerns about the validity of many published gene biomarkers, which may mislead research and obscure the truth. The limitations of current analysis methods and tools, particularly the reliance on fold-changes without considering gene interactions (synergy), further restrict their usefulness.
Methylation’s role in gene expression has become central in disease studies [13,16]. Errors in methylation could cause diseases, prompting research into COVID-19 at the DNA methylation level. The origin of SARS-CoV-2 (COVID-19) has puzzled the scientific community since its identification in December 2019. Initially considered an RNA virus [1,2,3,4,5,6,7,8,9,10,11,12], our research [13].suggested it may be better understood as a virus transcribing viral DNA into RNA, due to the long incubation period associated with MX1-related diseases. This discovery could significantly alter our understanding of viruses but does not resolve the fundamental question of SARS-CoV-2’s origin. Our research aims to identify optimal interactive DNA methylation markers for COVID-19 and investigate the origins of the virus, whether from humans, animals, or both. Our findings suggest that COVID-19 likely originated in humans rather than animals like bats or pangolins, which might have led previous research into a non-informative direction.
This research delves into the origins of COVID-19 by identifying a variant of Sennetsu fever and/or glanders, two rare diseases, as highly probable pathogens. This innovative approach opens new avenues for understanding how COVID-19 may have emerged and could guide future studies on both COVID-19 and similar diseases. It also highlights the broader implications for biosafety and biosecurity. The principle of Murphy’s law—suggesting that anything that can go wrong will—resonates with rare diseases like Sennetsu fever and glanders. If these diseases were to re-emerge as new viral variants, the consequences could be devastating. Our research leverages extreme value theory and max-logistic intelligence models to better understand these potential scenarios. For example, our findings on the CpG site cg07126281 (linked to the SAMM50 gene) suggest that SARS-CoV-2 may have been triggered by Sennetsu fever, possibly transmitted through consuming raw or undercooked gray mullet fish or other contaminated seafood. These findings suggest COVID-19 might have emerged through natural events, offering valuable insights into its origins and reinforcing the need for ongoing research into biosafety and biosecurity practices.
The complexity of uncovering such connections is immense. Each methylation site, or its corresponding gene, may be linked to one or multiple known or unknown pathogens. Theoretically, if we could account for all these associations, it would allow for a comprehensive understanding of potential pathogens. However, the probability of finding an exact correlation between a methylation site and a specific pathogen is incredibly low—less than one in ten million (a simple calculation using combinatorics with a total of 865,859). By refining our hypotheses and exploring further potential interactions, this probability decreases to one in a hundred million, underscoring the difficulty of such research. Despite these challenges, advanced mathematical models, while idealistic, could reduce the vast number of methylation sites into smaller, more focused sets. These models may reveal critical pathogen relationships and the biological mechanisms by which they cause disease. In doing so, they not only improve our understanding of pathogens but also provide essential insights into the processes that allow these pathogens to impact human health. Such methods boost confidence in the research, suggesting that the associations between methylation sites and pathogens identified in this study are highly credible.
Furthermore, using methylation for species identification or virus tracing is indeed an innovative concept. Although research in this area is still relatively limited, its theoretical basis and potential applications are quite promising. If our max-logistic intelligence method can be fully developed and validated, it could represent a significant breakthrough in biology.
The etiology insights from our findings include: 1) pursuing animal origins of COVID-19 may be a misguided direction; 2) SARS-CoV-2 might have been triggered by Sennetsu fever, transmitted through infected seafood, which calls for new measures of biosafety and biosecurity; 3) future research should focus on the most critical DNA methylation and RNA gene markers; 4) understanding these biomarkers could lead to effective antiviral drugs and therapies; and 5) new research directions are essential for studying future unknown X-viruses.
Method
In this study, we do not engage in clinical trials or laboratory experiments. Instead, our data is sourced from the Gene Expression Omnibus (GEO) database. Detailed information about the dataset is provided in the following section.
Subsequently, we offer a concise introduction to the mathematical model employed in our research. This model is closely related to the widely utilized logistic regression method frequently referenced in medical literature. Readers who find the mathematical details overwhelming may choose to skip Equation (4) without compromising their understanding of the overall model.
We employ the newly proven max-logistic intelligence regression classifier to differentiate between confirmed COVID-19 cases, healthy controls, and other COVID-19-free respiratory diseases. This novel method stands apart from traditional AI algorithms, classical statistics, and modern machine learning approaches like random forest, deep learning, and support vector machines [8,9,15,17]. Unlike other methods, max-logistic intelligence enhances interpretability, consistency, and robustness, essential for establishing causal relationships, as demonstrated in our prior research on COVID-19 and cancer biomarkers [8,9,10,11,12,13,15,17,18,19]. While it can be considered an AI or machine learning algorithm, this approach offers an explicit formula and clear interpretability. We illustrate this innovative procedure using DNA methylation beta values from COVID-19 positive and negative patients.
Suppose is the th individual patient’s COVID-19 status ( for COVID-19-free, for infected) and are the CpG beta values, with CpG sites in this study. Here, stands for the th type of beta values drawn based on different biological sampling methodologies. Note that most published works set , and hence the superscript can be dropped from the predictors. In this research paper, , as we have one methylation dataset analyzed in Section 3, and in the dataset, there are other ARIs (Acute Respiratory Infections) patients. Using a logit link (or any monotone link functions), we can model the risk probability of the th person’s infection status as:
or alternatively, we write
where is an intercept, is a observed vector, and is a coefficient vector which characterizes the contribution of each predictor (CpG site, in this study) to the risk.
Considering that there have been many variants of SARS-CoV-2 and multiple symptoms (subtypes) of COVID-19 diseases, it is natural to assume that the epigenetic structures of all subtypes can be different. Suppose that all subtypes of SARS-CoV-2 may be related to groups of CpG sites:
where is the th individual in the sample, and is the number of CpG sites in th group.
The competing (risk) factor classifier is defined as:
where s are intercepts, is a observed vector, and is a coefficient vector which characterizes the contribution of each predictor in the group to the risk.
Remark 1.
In (3), is mainly related to the largest component CFj = , i.e., all components compete to take the most significant effect.
Remark 2.
Taking , (3) is reduced to the classical logistic regression, i.e., the classical logistic regression is a special case of the new classifier. Compared to black-box machine learning methods like random forests and deep learning, the model in (3) offers clear, interpretable signatures with selected CpG sites, bridging linear models and advanced machine learning. It retains key properties like interpretability, computability, predictability, and stability, similar to Zhang’s (2021) observation [17].
We have to choose a threshold probability value to decide a patient’s class label in practice. Following the general trend in the literature, we set the threshold to be 0.5. As such, if , the th individual is classified as being disease-free; otherwise, the individual is classified as having the disease.
With the above-established notations and the idea of a quotient correlation coefficient [20], Zhang (2021) [17] introduced a new machine learning classifier (in this paper, we term it as max-logistic intelligence classifier), the smallest subset and smallest number of signatures (S4), for . We extended the S4 classifier from to any as follows:
where is an indicative function, is defined in Equation (3), is the index set of all CpG sites, , are index sets corresponding to (2), is the union of , is the number of elements in , and are penalty parameters, and and are the final CpG set selected in the final classifiers and the number of final signatures.
Remark 3.
When the S4 classifier achieves 100% accuracy, it establishes bioequivalence and a unique DNA methylation geometry space, a feature not found in other classifiers [9]. This equivalence didn’t appear in any other approaches in the literature, which motivate us to term it as max-logistic intelligence.
Remark 4.
Remark 5.
Adjusting the threshold in (4) between 0 and 1 affects the intercepts in (3) but not the coefficients, leaving clustering and classification results unchanged [17], which makes calculating AUC unnecessary.
The Start of SARS-CoV-2: Data Descriptions, Results, and Interpretations
The Data
The COVID-19 dataset analyzed in this section is publicly accessible under the identifier GSE174818 [22]. In our previous study [13], we utilized this dataset and employed two well-established formulas for calculating methylation beta values: (1) M/(M + U) and (2) (M + 1)/(M + U + 2). These formulas are standard in DNA methylation analysis in addition to M/(M+U+a) [23], with a being a positive value (100 is often used in the literature) to avoid the denominator being zero, where beta values are derived from the intensity counts of methylated (M) and unmethylated (U) signals. While both formulas yielded a 100% accuracy rate, the resulting sets of CpG sites and the formulas themselves should be considered necessary conditions. However, they fall short of being sufficient or causal conditions due to the lack of evidence supporting their comprehensive applicability.
To tackle a complex problem like this, it is imperative to have two key elements: i) all necessary supporting data and information, and ii) an appropriate analytical approach. Unfortunately, the scarcity of comprehensive resources in the existing literature renders the first element incomplete, underscoring the critical importance of the second. This paper introduces a new formula for calculating beta values: (M + 100)/(M + U + 200), i.e., adding 100 to both the numerator and the denominator of M/(M+U+100) [23]. This novel transformation not only achieves compelling and interpretable results but also highlights a crucial point: even when all necessary resources and information are available, solving the problem remains unattainable without the correct analytical approach. Many biologists and statisticians believe that adding numbers like 1, 100, 200 to the numerator and denominator has no significant impact, which can be tested in routine statistical inferences. However, the effect is nonignorable regarding species identification or virus tracing that needs 100% accuracy, as demonstrated in our earlier work [13]. We further note in GSE174818 whose Excel table contains exactly 865,859 rows (CpG sites), there are some M and U values being either single digits or double digits, i.e., less than 100. Readers may ask why it is 100, how about 500, etc. We tried several other numbers, e.g., 50. The results didn’t lead to 100% accuracy with a single digit of CpG sites. Of course, more computer experiments can be explored with our model framework. However, we found adding 100 leads to meaningful information.
The Analysis
Solving S4 classifiers (4), we get Table 1 as follows.
In the table, the classifiers CF1, CF2 and CF3 in Equation (3) are defined as:
| CF1: | -27.7252 -92.8578*MFSD11 +20.3366*CAB39 +35.1956*SERPINB8 |
| CF2: | -78.6488 +55.57*SDK1 +42.3906*CAB39 -60.8459*SAMM50 |
| CF3: | -20.8835 -101.445* KCNAB1 -24.2856* ZNF280D +116.8975* RANP1 |
Then, 0.5 is the threshold for computing risk probability in the logistic regression function. CFmax is defined as the max(CF1, CF2, CF3). We note that the threshold 0.5 can be changed to any other value between 0 and 1, and the conclusions won’t be changed. Such a unique property can hardly be found in any other approaches, making this new approach the most robust one.
In the table, SDK1 (Sidekick Cell Adhesion Molecule 1) is a Protein Coding gene. Diseases associated with SDK1 include Immunodeficiency 11B With Atopic Dermatitis and Brugada Syndrome 9. Among its related pathways is Cell junction organization. MFSD11 (Major Facilitator Superfamily Domain Containing 11) is a Protein Coding gene. Diseases associated with MFSD11 include Atypical Chronic Myeloid Leukemia, Bcr-Abl1 Negative, and Leukemia Acute Myeloid. CAB39 (Calcium Binding Protein 39) is a Protein Coding gene. Among its related pathways are Innate Immune System and Endochondral ossification. Gene Ontology (GO) annotations related to this gene include binding and kinase binding. SERPINB8 (Serpin Family B Member 8) is a Protein Coding gene. Diseases associated with SERPINB8 include Peeling Skin Syndrome 5 and Peeling Skin Syndrome 4. Among its related pathways are Response to elevated platelet cytosolic Ca2+ and Dissolution of Fibrin Clot. Gene Ontology (GO) annotations related to this gene include serine-type endopeptidase inhibitor activity. SAMM50 (SAMM50 Sorting And Assembly Machinery Component) is a Protein Coding gene. Diseases associated with SAMM50 include glanders. Among its related pathways are Signaling by Rho GTPases and Transcriptional activation of mitochondrial biogenesis. KCNAB1 (Potassium Voltage-Gated Channel Subfamily A Regulatory Beta Subunit 1) is a Protein Coding gene. Diseases associated with KCNAB1 include Episodic Ataxia, Type 1 and Developmental And Epileptic Encephalopathy 32. Among its related pathways are Potassium Channels and Transmission across Chemical Synapses. Gene Ontology (GO) annotations related to this gene include voltage-gated potassium channel activity and NADPH binding. ZNF280D (Zinc Finger Protein 280D) is a Protein Coding gene. Diseases associated with ZNF280D include Borderline Glaucoma and Intellectual Developmental Disorder, X-Linked 109. RANP1 (RAN Pseudogene 1) is a Pseudogene. The above information is from genecards.org (Accessed on 08/02/2024).
Figure 1 presents critical site methylation levels and risk probabilities corresponding to different combinations in the dataset and Table 1. It can be seen that each plot shows a methylation signature pattern and functional effects of the sites/genes involved.
S4 classifiers can simultaneously perform clustering and classifications. In this analysis, there are seven clusters (classes). Figure 2 presents a Venn diagram showing all COVID-19-positive patients classifications.
From the figure, we can see that there are 22, 21, and 2 patients who can only be detected by CF1, CF2, and CF3, individually and respectively. 41, 8, and 1 patient can be simultaneously detected by CF1 and CF2, CF1 and CF3, and CF2 and CF3, respectively. Seven patients can be simultaneously detected by all three classifiers CF1, CF2, and CF3. These seven patients are all ICU patients, which strongly indicates that the eight identified CpG sites (genes) and their derived classifiers are informative.
Recall that diseases associated with SAMM50 include glanders. Among its related pathways are signaling by rho GTPases and transcriptional activation of mitochondrial biogenesis. SAMM50 is also associated with sennetsu fever https://diseases.jensenlab.org/Entity?documents=10&type1=9606&id1=ENSP00000345445&type2=-26&id2=DOID:0050485 (Accessed on 08/02/2024). From https://www.malacards.org/card/sennetsu_fever (Accessed on 08/02/2024), we can see among eight genes associated with sennetsu fever, four genes PDF, MTCH1, TIMM10, TIMM9 are all mitochondrial. These phenomena lead to the fact SAMM50 might have caused diseases of Sennetsu fever and/or glanders.
We next review two rare diseases: Sennetsu fever and glanders.
The symptoms of Sennetsu fever may include a sudden high fever, headache, and muscle aches (myalgia) within a few weeks after the initial infection. In some cases, affected individuals may also experience nausea, vomiting, and loss of appetite (anorexia). A primary bacterial infectious disease that results in infection has a material basis in Neorickettsia Sennetsu, transmitted by ingesting raw or undercooked gray mullet fish infected with the trematodes. Sennetsu fever is a rare infectious disease from a group of diseases known as the human ehrlichioses. Researchers have known about ehrlichioses in animals for quite some time. For example, canine ehrlichiosis, an ehrlichial infection affecting dogs, was identified in 1935. On the other hand, in Japan, Sennetsu fever was the first identified human ehrlichiosis, and it was only discovered in 1954. Many ehrlichioses are transmitted through ticks; However, researchers still do not know the vector organism for Sennetsu fever. Although there is no consensus, many believe that the disease spreads through the consumption of uncooked fish. Hepatosplenomegaly (liver and spleen enlargement) is a common clinical finding in Sennetsu fever. In many cases, Sennetsu fever is associated with a drop in white blood cell (WBC) count and an increase in the concentration of liver enzymes (transaminases). Sennetsu fever is extremely rare, and so far, no cases of the disease have been reported outside Japan and Malaysia. Neoehrlichia mikurensis is a tickborne pathogen that occurs in many parts of Europe and Asia. It generally infects older or immunocompromised people. The above information was taken from https://www.malacards.org/card/sennetsu_fever https://rarediseases.org/rare-diseases/sennetsu-fever/ https://www.onio.com/article/five-rare-fevers-you-have-never-heard-of.html https://wwwnc.cdc.gov/travel/yellowbook/2020/travel-related-infectious-diseases/rickettsial-including-spotted-fever-and-typhus-fever-rickettsioses-scrub-typhus-anaplasmosis-and-ehr. (Accessed on 08/02/2024).
The authors [24] stated glanders are a highly contagious and often fatal zoonotic disease primarily of solipeds such as horses, mules, and donkeys. It was first described by the Greeks in 450–425 BC and again by the Romans in 400–500 AD. Throughout history, glanders have been known by other names, including equinia, malleus, droes, and farcy. Glanders is primarily characterized by ulcerating lesions of the skin and mucous membranes. Solipeds are the natural reservoir of Burkholderia mallei. Donkeys are prone to develop acute forms of glanders, while horses are more likely to develop chronic and latent diseases. Mules are susceptible to acute and chronic forms of the disease as well as latent infections. B. mallei, the etiological agent of glanders, is a Gram-negative, non-motile, facultative intracellular pathogen. At one time, B. mallei infections occurred worldwide, but over the last 100 years, the occurrence of glanders has decreased with the reduced economic reliance on solipeds as the primary mode of transportation, the implementation of testing all solipeds for glanders, and euthanizing those that are confirmed positive. The last naturally occurring human case in the United States was reported in 1934. Glanders in solipeds and humans have also been eradicated from Canada and Western Europe. However, sporadic infections of animals are still reported in Far East Asia, South America, Eastern Europe, North Africa, and the Middle East. Although human epidemics have not been recorded, isolated outbreaks in human populations and the deliberate use of B. mallei as a biological weapon have been documented [24].
COVID-19 symptoms of early infected patients (and even some patients now) are incredibly close to Sennetsu fever and glanders. However, COVID-19 and Sennetsu fever are different types of infectious diseases. We now interpret Table 1. In the table, CF2 is a combination of (SDK1, CAB39, SAMM50) with a specificity of 100% and a sensitivity of 68.63%. The negative coefficient sign (-60.8459) of SAMM50 tells that the higher the beta values (the methylation intensities), the lower the risk of COVID-19 positive. The functions of other genes can be interpreted similarly. However, this is not the whole story. The significant difference between our max-logistic intelligence model and results (Table 1) and other models lies in the interpretation of selected genes (CpG sites).
Before interpreting the results in Table 1, we discuss how site-site (gene-gene) interactions (synergy) are defined in this paper. They are characterized by their functions in classifiers. Such interactions are new to biological/medical/physical studies. At first glance, one may consider our model a variable selection model, like many other existing models in the literature. We argue that with the superior interpretability of the resulting classifiers and the mathematical equivalence (100% accuracy) between the classifiers and the diseases, we can infer that this model is a new kind of site-site interaction among CpG sites, and they can be used in exploring the causal relationship. Until now, no mathematical model has been able to characterize causal relationships fully.
Our earlier paper [17] mathematically proved that the classifiers lead to the smallest subset and number of signatures of our models (classifiers). In Ref [9], we established the geometry of genome space for COVID-19 classifiers, a finding with significant implications for disease classification. Such properties were not shown in the literature by other methods. With the mathematical equivalence and the geometry of genome space, we can explore the causal relationship between predictors (genes) and diseases and the gene-gene interactions (synergy).
Our models stand out due to their unique features, particularly the gene-gene interactions that are implied by their associated different coefficient signs and strengths. These features, which were missed in other existing models, contribute significantly to the robustness and accuracy of our classifiers.
The relationship between gene combinations and model performance can be likened to a basketball team where critical genes are players, and their combinations determine the team’s ability to score. In this analogy, a positive coefficient for a gene indicates it increases the likelihood of classification as COVID-19+, while a negative coefficient suggests the opposite. Imagine a team with CAB39 as Point Guard, SAMM50 as Shooting Guard, SDK1 as Center, MFSD11 as Power Forward, and SERPINB8 as Small Forward. The main scoring combinations are: (CF1: MFSD11, CAB39, SERPINB8), (CF2: SDK1, CAB39, SAMM50), and (CF3: KCNAB1, ZNF280D, RANP1). A negative sign for SAMM50 implies that decreasing its ball handling time (lowering its methylation level) increases scoring probability, akin to increasing the risk of COVID-19+. Conversely, increasing CAB39’s ball handling time improves the team’s scoring, analogous to raising the likelihood of COVID-19+ classification.
Unlike simple basketball plays, gene-gene interactions are more complex and can be seen as interactions that include playing time, coordination, and the entire stadium environment. Our models select genes and describe these intricate gene-gene interactions (synergy). These interactions are not merely regulatory or physical, as commonly discussed in the literature, but can be likened to quantum interference. In this analogy, subatomic particles in a probabilistic superposition state influence each other, affecting outcome probabilities when measured, similar to how gene interactions impact classification outcomes [25].
With the evidence of the very close symptoms between COVID-19 and Sennetsu fever and their shared SAMM50 gene, it may be safe to infer that COVID-19 was triggered by eating undercooked contaminated seafood, e.g., grey mullets infected with the trematodes, with its interactions with CAB39 (Calcium Binding Protein 39) and SDK1 (Sidekick Cell Adhesion Molecule 1). Recall that diseases associated with SDK1 include Immunodeficiency 11B With Atopic Dermatitis and Brugada Syndrome 9; the innate immune system and endochondral ossification are among CAB39’s related pathways, which makes CF2 a classifier of immune-related. The work [26] found that SARS-CoV-2 can infect hepatocytes and stimulate these cells to produce glucose through gluconeogenesis. As a result, the site-site interactions caused COVID-19 infection. Note that not all infected individuals in Table 1 were classified by CF2 (68.63% sensitivity), which again shows that COVID-19 only shares parts of the pathological features of Sennetsu fever at DNA methylation levels. Recall that the data GSE174818 was first public on May 21, 2021, and as such, COVID-19 has evolved from the first infected individuals in 2019. In other words, a 68.63% sensitivity (together with a 100% specificity) is already extremely significant, given SARS-CoV-2 is a very contagious virus. Given a 100% specificity, we note that any patient detected by any CFs is definitely COVID-19 positive. This unique property guarantees our finding to be meaningful.
In our previous work [13], we explored the genetic connections between COVID-19 and other infectious diseases and rare diseases, specifically SARS-CoV-1, MERS-CoV, subacute sclerosing panencephalitis (SSPE), and influenza. By identifying common genes that play crucial roles in these diseases, we established potential links between them and COVID-19. However, despite these connections, we were unable to conclude that these diseases (excluding SSPE) were the origins of the COVID-19 pandemic.
This paper presents a novel hypothesis by being the first to suggest that Sennetsu fever could potentially be the causative factor behind the onset of COVID-19.
One may question why the original paper of GSE174818 didn’t find these CpG sites, given that they are significant and lead to 100% accuracy in our new study. It is because of the limitation of analytical methods used in the original paper, even though the data contains critical information. Indeed, many literature analytical methods seldom yield 100% accuracy in published work, which makes our model and method desirable.
Heatmap Illustration
Although Figure 1 shows clear signature patterns of how eight CpG sites interact and synergies, reporting the methylation states (Beta values) for the eight CpG sites is crucial. This section presents the raw counts of methylated and unmethylated intensities, which is available in an Excel file (a link is provided in the Data Availability section). Additionally, we present heatmaps illustrating the eight CpG sites. These heatmaps will display both the Beta values (Figure 3) and the total intensity counts (sum of methylated and unmethylated intensities, Figure 4), better depicting the differential methylation and its biological significance.
Figure 3 reveals no clear patterns, except for cg21872428 (RANP1) and cg25426982 (SERPINB8), where the mean beta values for non-COVID-19 patients are lower than those for COVID-19 patients. The mean beta values of cg07126281 (SAMM50) show that, except cells (3,1) and (5,1), the higher the methylation rates of cg07126281 (SAMM50), the lower the risk of COVID-19 positive, which is consistent with the prior interpretations regarding the negative coefficient sign of cg07126281 (SAMM50) in Table 1. This observation suggests that averaging may obscure the true effects and reinforces the importance of jointly studying interactions and synergies to uncover the underlying truths.
In Figure 4, the higher total intensity mean count (sum of methylated and unmethylated intensities) at cg07126281 (SAMM50) may be influenced by several factors. 1) CpG Site-Specific Regulation: Some CpG sites are located in biologically significant regions, such as promoters or enhancers of key immune-related genes. These regions may experience more dynamic methylation changes during immune activation in response to respiratory diseases, resulting in increased total intensity counts. 2) Inflammatory Response: Respiratory diseases, particularly viral infections like COVID-19, often trigger systemic inflammation that alters DNA methylation patterns in immune-related genes, leading to elevated methylation and unmethylation activities at CpG sites. 3) CpG Island Density: CpG sites within CpG islands, rich in CpG dinucleotides, are more likely to exhibit methylation changes, contributing to higher intensity counts. 4) Epigenetic Modulation by Disease: Severe respiratory diseases can modify gene expression by altering the epigenome, including DNA methylation patterns, reflecting disease-induced changes at key regulatory regions. 5) Cellular Heterogeneity in Whole Blood: The mixture of immune cell types in whole blood, each with distinct methylation patterns, can influence overall methylation intensity, especially with shifts in cell populations during immune responses. 6) CpG Site Polymorphism: Genetic variations near CpG sites, such as SNPs, can affect methylation status and intensity, resulting in higher counts.
Overall, the elevated total intensity at this CpG site suggests its critical role in immune or inflammatory pathways, making it a candidate for further functional studies.
Cohort-to-Cohort Cross-Validation
In this section, we use public data GSE193879 [3] (public on January 18, 2022) to cross-validate the findings presented in Table 1. GSE193879 used the same platform GPL21145 Infinium MethylationEPIC as GSE174818 used. In GSE193879 there were 43 MIS-C patients, 15 pediatric COVID-19 cases and 69 healthy controls. The significant difference is that the patients who participated in GSE174818 were adults in USA, while those who participated in GSE193879 were pediatric in Spain, which makes the comparisons indirect as pediatric patients’ immune systems can still be under development. Using the eight CpG sites in Table 1, we obtain the following Table 2 related to GSE193879.
Comparing Table 1 and Table 2, we can immediately see significant differences. The coefficient signs of SAMM50 in both tables are negative. The signs of other CpG sites are different from Table 1 to Table 2. This phenomenon reveals that SAMM50 played a fundamental biological role in early COVID-19 development regardless of whether the patients were adult or pediatric. Because the control groups in both tables (cohorts) are different, we cannot directly comment on other CpG sites’ accuracy and coefficient signs. A natural question arises: Are there some other genes beyond those in Table 1 that are also pivotal? Table 3 presents the performance of CpGs in Table 1.
In Table 3, ZNF71 (Zinc Finger Protein 71) is a Protein Coding gene. Diseases associated with ZNF71 include Myasthenic Syndrome, Congenital, 6, Presynaptic. Among its related pathways are Gene expression (Transcription). Gene Ontology (GO) annotations related to this gene include DNA-binding transcription factor activity. MMS19 (MMS19 Homolog, Cytosolic Iron-Sulfur Assembly Component) is a Protein Coding gene. Diseases associated with MMS19 include Progressive External Ophthalmoplegia With Mitochondrial DNA deletions, Autosomal Dominant 6 and Xeroderma Pigmentosum, Variant Type. Among its related pathways are Metabolism and Cytosolic iron-sulfur cluster assembly. Gene Ontology (GO) annotations related to this gene include binding and protein-macromolecule adaptor activity. MRPS35 (Mitochondrial Ribosomal Protein S35) is a Protein Coding gene. Among its related pathways are Mitochondrial translation and Metabolism of proteins. Gene Ontology (GO) annotations related to this gene include RNA binding and structural constituent of ribosome. SLC10A7 SLC10A7 (Solute Carrier Family 10 Member 7) is a Protein Coding gene. Diseases associated with SLC10A7 include Short Stature, Amelogenesis Imperfecta, And Skeletal Dysplasia With Scoliosis and Amelogenesis Imperfecta. Gene Ontology (GO) annotations related to this gene include symporter activity. The information above is from genecards.com (Accessed on 04/26/2024).
Again, SAMM50 in Table 3 shows the same sign as it is in Table 1 and Table 2. Recall that its related pathways are signaling by rho GTPases and transcriptional activation of mitochondrial biogenesis. SAMM50, MMS19, and MRPS35 are mitochondrial genes and core mitochondrial genes at the epigenetic level. In Table 3, MMS19 and MRPS35 were up-regulated, with SAMM50 being down-regulated with the infected. In the literature, core mitochondrial genes were found to be down-regulated during SARS-CoV-2 infection [27]. The authors derived the findings from mouse experiments, which can be different from humans, as shown in Table 3.
Discussions
Numerous genomic-level studies on COVID-19 have been published, each exploring the virus’s pathological causes from different perspectives. However, many of these studies face challenges in cross-validating results across different cohorts due to methodological limitations. An exception to this is our earlier work, which successfully cross-validated thirteen genes across fourteen cohort studies (whole blood samples and NP/OP PCR swabs samples) involving thousands of patients with diverse ethnicities, ages, and geographical regions [8,9,10]. This comprehensive study achieved nearly perfect performance and interpretability, a feat unmatched in the existing literature.
Most published studies focus on single-gene expression changes, neglecting the interaction effects between genes due to analytical limitations. This oversight often leads to inaccuracies and diminishes the practical utility of their findings. Our previous work discussed these limitations and proposed stringent criteria for defining critical differentially expressed genes (DEGs) [12,18,19]. These criteria are essential for improving the accuracy and relevance of genomic studies.
The Defense Advanced Research Projects Agency (DARPA) posed 23 biological-mathematical challenges, one of which, Challenge Fifteen, involves understanding the geometry of genome space. DARPA’s challenge seeks to revolutionize biological mathematics and strengthen U.S. defense capabilities. Our earlier research established the geometry of the COVID-19 genome space, possibly the first such work in the literature [8,9]. We further characterized the differences between the genome spaces of COVID-19 and SARS-CoV-2 [10], and established a link between Omicron infections in animals and humans [12]. Recently, we inferred that SARS-CoV-2 might be better understood as a DNA-transcribed RNA virus [13].
As our model and analysis method are significantly different from the biological, medical, and statistical literature, it is essential to compare with those mainstream. We discuss some technical constraints in the literature and their connections to our newly proposed max-logistic-intelligence classifiers.
Regarding microarray batch effects and data normalization in the literature, we recognize the critical importance of addressing batch effects in microarray data. In our discovery dataset (NCBI GEO Accession GSE174818), we calculated beta values using our innovative formula (M+100)/(M+U+200) from the intensity counts of methylated (M) and unmethylated (U) signals to ensure robust analysis. For cohort-to-cohort validation, we utilized the available beta values from the NCBI GEO dataset GSE193879, which includes pediatric COVID-19 patients, MIS-C patients, and healthy controls. While these datasets come from different sources and data types, traditional bioinformatics methods would typically require batch effect corrections.
However, our method (Equation (4)) is versatile enough to handle DNA methylation beta values generated by different platforms and data transformations and accommodate variations in viral strains over time without requiring batch correction. Many existing models struggle with heterogeneous populations and rely on batch effect adjustments, which can compromise inference accuracy. Our classifiers, on the other hand, achieved the highest accuracy for COVID-19 detection without adjusting for variables such as technical constraints, age, sex, or ethnicity.
We acknowledge that some public datasets lack comprehensive clinical and pathological information, limiting our ability to assess the prognostic value of the eight CpG sites. Nevertheless, our study uniquely applies a novel machine learning approach (max-logistic intelligence classifier), previously unused in infection studies. Furthermore, our findings were validated across diverse populations and ethnic groups, underscoring the potential of this approach for viral genetic tracing.
In earlier COVID-19 research, involving over thirteen cohorts, we consistently achieved high differential power without requiring batch effect corrections. This observation was also reflected in our published work on cancer [18].
We understand the importance of accounting for additional clinical parameters, such as comorbidities, sex, and age, which could potentially influence the methylation status of CpG sites. However, we emphasize that our method, specifically the approach detailed in Equation (4), achieves 100% accuracy without the inclusion of these clinical variables. This is because the clinical variables, while valuable for studying prognosis, are extrinsic factors. In contrast, the eight CpG sites we identified can be considered intrinsic variables with sufficient predictive power to diagnose the disease with complete accuracy and establish etiological links between COVID-19 infection and genetic variations.
Incorporating extrinsic variables, such as age or comorbidities, would not enhance the model’s predictive capacity. In fact, it could obscure the underlying biological truths by introducing factors unrelated to the core genetic mechanisms at play. From a mathematical standpoint, when a model achieves 100% accuracy, adding additional variables does not improve its predictive ability but could instead dilute its clarity. In such cases, the existing model can still be applied to explore subtypes associated with specific clinical variables if needed without compromising its foundational predictive strength.
Correction for multiple testing and false positives is often a significant challenge for model builders. However, these concerns are not relevant to our novel model (Equation (4)). As demonstrated in the discovery dataset (GSE174818) and Table 1, each of the individual classifiers achieved 100% specificity, eliminating the possibility of false positives. In the Venn diagram, the intersection of all three individual classifiers identifies the same seven ICU patients, making our model uniquely reliable and irreplaceable.
Regarding multiple testing, it’s important to highlight that our model represents a new AI-based approach that differs fundamentally from traditional statistical methods. Unlike models that rely on individual CpG site fold changes and the need for multiple hypothesis testing to determine significance, our model focuses on the interactions and synergies among a minimal set of CpG sites. By studying these interactions, we avoid the pitfalls of traditional statistical approaches, including the correction for multiple comparisons, making our model both innovative and robust.
This AI-driven model offers a fresh perspective, bypassing the ‘nightmare’ problems typically encountered in traditional approaches, such as false positives and the complexities of multiple testing corrections. Its strength lies in identifying synergistic patterns rather than isolated changes, resulting in a more accurate and biologically meaningful prediction system.
In this paper, we investigate what may have triggered the onset of COVID-19, using the same dataset (GSE174818) as in our previous study, but with a different focus [13]. Our earlier work identified specific CpG sites and genes, such as MX1, which are associated with diseases like influenza and subacute sclerosing panencephalitis (SSPE). SSPE, a severe neurological disorder with a long incubation period, could be a significant concern for COVID-19 [13]. This highlights the urgent need to explore the potential of MX1 in COVID-19 research.
We also identified 31 CpG sites that regulate the SAMM50 gene, but cg07126281 plays a crucial role in COVID-19 infections. Additionally, literature shows that tumors can arise from epigenetic dysregulation, leading to inherited altered cell fates [28]. While CF3 has a specificity of 100%, its sensitivity is only 17.65%, detecting just two patients. The RANP1 gene, a non-protein-coding pseudogene, also requires special attention. Its positive coefficient suggests that an increase in its CpG site methylation could trigger COVID-19.
The precise origin of COVID-19 remains elusive despite extensive scientific research. Though inherently idealized, theoretical models can reduce the complexity of the epigenetic landscape by condensing a vast number of methylation sites into smaller, more interpretable subsets, thereby highlighting key pathogen interactions and underlying biological processes. Traditional species identification relies on nucleic acid sequences or protein sequences, but methylation offers another layer of genetic information. Methylation profiles could potentially serve as a “fingerprint” for specific species. In this study, we utilized a max-logistic intelligence classifier to analyze 865,859 critical Infinium MethylationEPIC sites (CpGs), ultimately identifying eight CpGs that achieved 100% accuracy in distinguishing COVID-19 patients from other respiratory diseases and healthy controls. Notably, one of these sites, cg07126281, is associated with the SAMM50 gene, which shares genetic links with rare infectious diseases such as Sennetsu fever and glanders. This raises the intriguing possibility of a connection between COVID-19 and variants of these rare diseases, potentially transmitted through contaminated seafood or contact with individuals infected by glanders.
The task of identifying such associations among methylation sites is inherently challenging, given the sheer volume of over 865,859 CpG sites and the extensive list of known and unknown pathogens. The probability of randomly identifying a meaningful correlation between a pathogen and a CpG site is less than one in ten million (calculated using combinatorics). However, hypotheses connecting COVID-19 with rare diseases significantly lower this probability to one in one hundred million, thereby strengthening the plausibility of these associations. Our findings emphasize the critical importance of investigating seafood markets and global supply chains in tracing the origins of COVID-19, while underscoring the need for continuous biosafety and biosecurity measures to prevent future pandemics.
In their review paper, Sepsis Therapies: Learning from 30 Years of Failure in Translational Research to Propose New Leads [29], the authors highlighted that the use of inappropriate animal models and patient selection criteria contributed to the failure of many sepsis therapies. These same issues may have impacted research on other infectious diseases, including COVID-19, which is the focus of this paper.
However, we propose that an additional, potentially decisive factor has been overlooked in many studies and clinical trials: the efficiency of the analytical methods employed. In particular, the lack of attention to appropriate data transformation and the use of robust mathematical models has often hindered the ability to draw meaningful conclusions. This paper demonstrates that more accurate and valuable insights can be obtained by applying rigorous analytical techniques, ultimately leading to the discovery of the cause of COVID-19.
Again, our mathematical model and analysis method are significantly different from the literature approaches. As a result, it is legitimate that our findings are significantly different from the published work but yet verified [30,31], among many others, which makes our results a significant factor in unlocking how COVID-19 started. One may further question that our data was from 2021, and then the conclusion may not be directly linked to the original virus. We note that the COVID-19-infected individuals in GSE174818 still had the earliest SARS-CoV-2 variant, i.e., the CpGs carried the original information.
Finally, we reflect on Murphy’s law: “Anything that can go wrong will go wrong at the worst possible time.” Rare diseases like Sennetsu fever and glanders can cause enormous losses if they reemerge as new virus variants. The COVID-19 pandemic exemplifies this principle, as described by extreme value theory and our max-logistic intelligence model. These observations confirm Murphy’s law and emphasize the importance of preparedness and innovative research approaches.
Conclusions
The cause of COVID-19 and the drivers of SARS-CoV-2 replication remain unclear. Our research offers an extremely promising clue, suggesting that the virus may have been triggered by the rare Sennetsu fever, possibly transmitted through consuming raw or undercooked gray mullet fish infected with trematodes or by individuals infected with glanders. This finding naturally connects to the seafood markets and supply chains, offering new etiology insights into the origins of SARS-CoV-2. This breakthrough provides a critical clue in the quest to understand the origins of COVID-19 and underscores the importance of continued investigation. Our findings and any new findings will set new study protocols for studying future rare infectious diseases and biosafety and biosecurity measures for managing the diseases.
Data Availability
The datasets are publicly available. The data links are stated in Section Data Description. Computing outputs are in a supplementary file available online https://pages.stat.wisc.edu/~zjz/SARS-CoV-2origins.zip. Readers can also make an email request to the author in case the link is not working. The results presented in this paper are verifiable by simply checking the Excel sheets and formulas in the file.
Competing Interests
The Authors declare no Competing Financial or Non-Financial Interests.
Statement of ethics
The authors conducted research based on published work. The new research does not need IRB approval and a statement of ethics.
Limitation statements
Our results, though computational, are based on rigorous mathematical proofs of biological equivalence. The concepts of site-site, gene-gene, and site-site interactions represent a revolutionary leap in medical research. Given the perfect performance, our findings warrant deeper microbiological and laboratory investigation. In the literature, we didn’t find any other rare infectious diseases that had genetic variation links to the CpGs identified in this paper, which may need further investigation. The dataset collected in the USA is unique; other regions lacked key CpGs like cg07126281 due to different platforms, hindering discovery. However, we achieved consistent results in our other work with 14 global datasets.
Author Contributions
ZZ conceived and wrote this article. ZZ did all the data analysis.
References
- Callaway, E. The quest to find genes that drive severe covid. Nature 2021, 595, 346–348. [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature, pages 474-477, 2021. [CrossRef]
- Davalos V, García-Prieto CA, Ferrer G, Aguilera-Albesa S et al. Epigenetic profiling linked to multisystem inflammatory syndrome in children (MIS-C): A multicenter, retrospective study. EClinicalMedicine 2022 Aug;50:101515. PMID: 35770252.
- Dite GS, Murphy NM, and Allman R. Development and validation of a clinical and genetic model for predicting risk of severe COVID-19. Epidemiology and Infection, 149:e162, 2021. [CrossRef]
- Konigsberg IR, Barnes B, Campbell M, Davidson E, Zhen Y, Pallisard O, Boorgula MP, Cox C, Nandy D, Barnes KC, et al. Host methylation predicts SARS-CoV-2 infection and clinical outcome. Commun Med (Lond). 2021;1(1):42. [CrossRef]
- Melms, J.C., Biermann, J., Huang, H. et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021). [CrossRef]
- Pairo-Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in COVID-19. Nature, 591:92-98, 2021. [CrossRef]
- Zhang Z. Five critical genes related to seven COVID-19 subtypes: A data science discovery. Journal of Data Science, 19(1):142-150, 2021. [CrossRef]
- Zhang Z. The existence of at least three genomic signature patterns and at least seven subtypes of COVID-19 and the end of the disease. Vaccines, 10, 761, 2022. [CrossRef]
- Zhang Z. Genomic Biomarker Heterogeneities Between SARS-CoV-2 and COVID-19. Vaccines 2022, 10(10), 1657; [CrossRef]
- Zhang Z, Genomic Transcriptome Benefits and Potential Harms of COVID-19 Vaccines Indicated from Optimized Genomic Biomarkers. Vaccines 2022, 10(11), 1774. [CrossRef]
- Zhang, Z. Omicron’s Intrinsic Gene-Gene Interactions Jumped Away from Earlier SARS-CoV-2 Variants and Gene Homologs between Humans and Animals. Advances in Biomarker Sciences and Technology 2023. [CrossRef]
- Zhang, Z. The Initial COVID-19 Reliable Interactive DNA Methylation Markers and Biological Implications. Biology. 2024; 13(4):245. [CrossRef]
- Zhang Y, Guo X, Li C, Kou Z et al. Transcriptome Analysis of Peripheral Blood Mononuclear Cells in SARS-CoV-2 Naïve and Recovered Individuals Vaccinated with Inactivated Vaccine. Front Cell Infect Microbiol 2021;11:821828.
- Zhang Z. Lift the veil of breast cancers using 4 or fewer critical genes. Cancer Informatics, 21:1-11, 2022. [CrossRef]
- Phillips, T. (2008) The role of methylation in gene expression. Nature Education 1(1):116.
- Zhang Z. Functional effects of four or fewer critical genes linked to lung cancers and new sub-types detected by a new machine learning classifier. Journal of Clinical Trials, 11:S14:100001, 2021. https://www.longdom.org/open-access/functional-effects-of-four-or-fewer-critical-genes-linked-to-lung-cancers-and-new-subtypes-detected-by-a-new-machine-learning-clas-88321.html.
- Liu, Y., Xu, Y., Li, X., Chen, M., Wang, X., Zhang, N., Zhang, H., Zhang, Z. (2023), Towards Precision Oncology Discovery: Four Less Known Genes and Their Unknown Interactions as Highest-Performed Biomarkers for Colorectal Cancer. npj Precision Oncology. https://www.nature.com/articles/s41698-024-00512-1.
- Liu Y, Zhang H, Xu Y, Liu, Y-Z, Al-Adra DP, Yeh MM, et al. (2023), Five Critical Gene-based Biomarkers with Optimal Performance for Hepatocellular Carcinoma, Cancer Informatics, 22. [CrossRef]
- Zhang, Z. Quotient correlation: A sample based alternative to Pearson’s correlation. Ann. Stat. 2008, 36, 1007–1030 . [CrossRef]
- Carapito R, Li R, Helms J, Carapito C et al. Identification of driver genes for critical forms of COVID-19 in a deeply phenotyped young patient cohort. Sci Transl Med 2022 Jan 19;14(628):eabj7521.
- Balnis J, Madrid A, Hogan KJ, Drake LA et al. Blood DNA methylation and COVID-19 outcomes. Clin Epigenetics 2021 May 25;13(1):118. PMID: 34034806.
- Du, P., Zhang, X., Huang, CC. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 587 (2010). [CrossRef]
- Van Zandt KE, Greer MT, Gelhaus HC. Glanders: an overview of infection in humans. Orphanet J Rare Dis. 2013 Sep 3;8:131. [CrossRef]
- Wright G. What is quantum interference? https://www.techtarget.com/whatis/definition/quantum-interference.
- Barreto EA, Cruz AS, Veras FP, Martins R, Bernardelli RS, Paiva IM, et al. COVID-19-related hyperglycemia is associated with infection of hepatocytes and stimulation of gluconeogenesis. Proc Natl Acad Sci U S A. 2023 May 23;120(21):e2217119120 . [CrossRef]
- Guarnieri JW, Dybas JM, Fazelinia H, Kim MS, Frere J, Zhang Y, Soto Albrecht Y, Murdock DG, Angelin A, Singh LN, Weiss SL, Best SM, Lott MT, Zhang S, Cope H, Zaksas V, Saravia-Butler A, Meydan C, Foox J, Mozsary C, Bram Y, Kidane Y, Priebe W, Emmett MR, Meller R, Demharter S, Stentoft-Hansen V, Salvatore M, Galeano D, Enguita FJ, Grabham P, Trovao NS, Singh U, Haltom J, Heise MT, Moorman NJ, Baxter VK, Madden EA, Taft-Benz SA, Anderson EJ, Sanders WA, Dickmander RJ, Baylin SB, Wurtele ES, Moraes-Vieira PM, Taylor D, Mason CE, Schisler JC, Schwartz RE, Beheshti A, Wallace DC. Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts. Sci Transl Med. 2023 Aug 9;15(708):eabq1533. [CrossRef]
- Parreno V, Loubiere V, Schuettengruber B, Fritsch L, Rawal CC, Erokhin M, Győrffy B, Normanno D, Di Stefano M, Moreaux J, Butova NL, Chiolo I, Chetverina D, Martinez AM, Cavalli G. Transient loss of Polycomb components induces an epigenetic cancer fate. Nature. 2024 Apr 24. [CrossRef]
- Cavaillon JM, Singer M, Skirecki T. Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol Med. 2020 Apr 7;12(4):e10128. [CrossRef]
- Liu, W.J., Liu, P., Lei, W. et al. Surveillance of SARS-CoV-2 at the Huanan Seafood Market. Nature 631, 402–408 (2024). [CrossRef]
- Crits-Christoph A, Levy JI, Pekar JE, Goldstein SA, Singh R, Hensel Z, Gangavarapu K, Rogers MB, Moshiri N, Garry RF, Holmes EC, Koopmans MPG, Lemey P, Popescu S, Rambaut A, Robertson DL, Suchard MA, Wertheim JO, Rasmussen AL, Andersen KG, Worobey M, Débarre F. Genetic tracing of market wildlife and viruses at the epicenter of the COVID-19 pandemic. Cell. 2024 Sep 19;187(19):5468-5482.e11. [CrossRef]
Figure 1.
COVID-19 classifiers in Table 1: Visualization of site-site relationship and site-risk probabilities. Note that 0.5 is the probability threshold.
Figure 1.
COVID-19 classifiers in Table 1: Visualization of site-site relationship and site-risk probabilities. Note that 0.5 is the probability threshold.
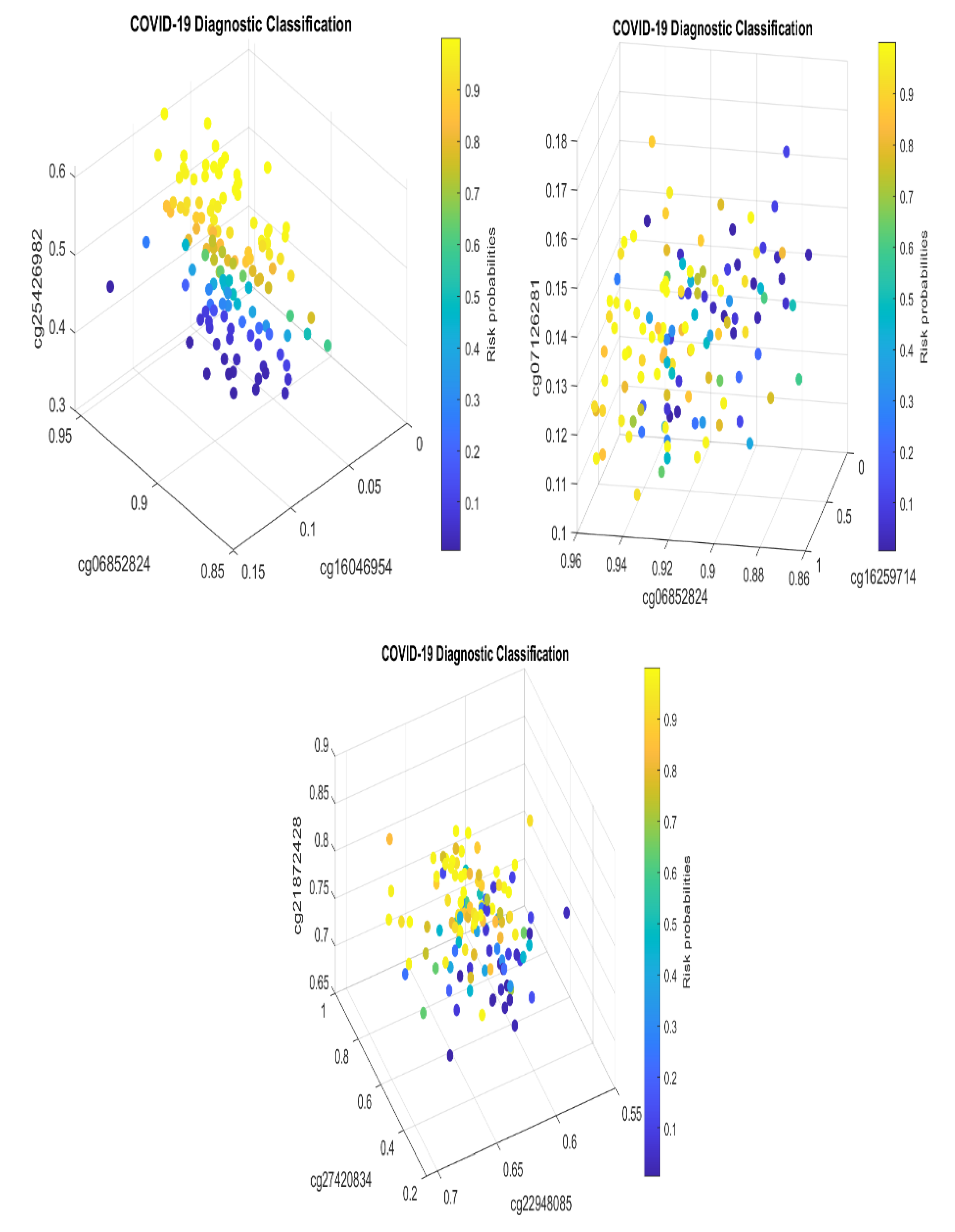
Figure 2.
Venn diagram of COVID-19 subtypes classified by AI-type classifiers.
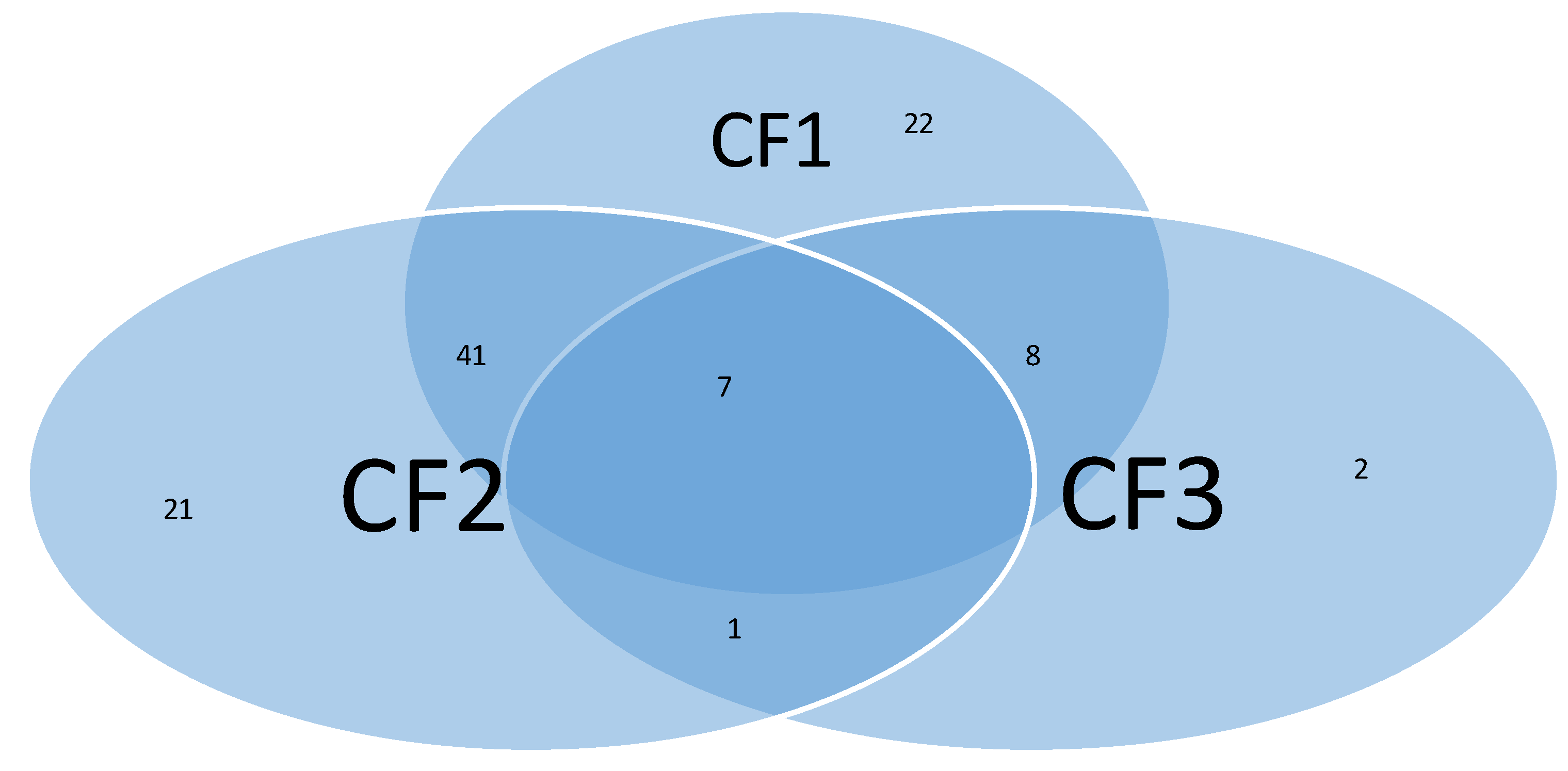
Figure 3.
Mean beta values of all patients. The cell (0,0) corresponds to patients with other diseases. The cells (1,1) to (7,1) correspond to Venn Diagram in Figure 2 and eight CpGs in Table 1.
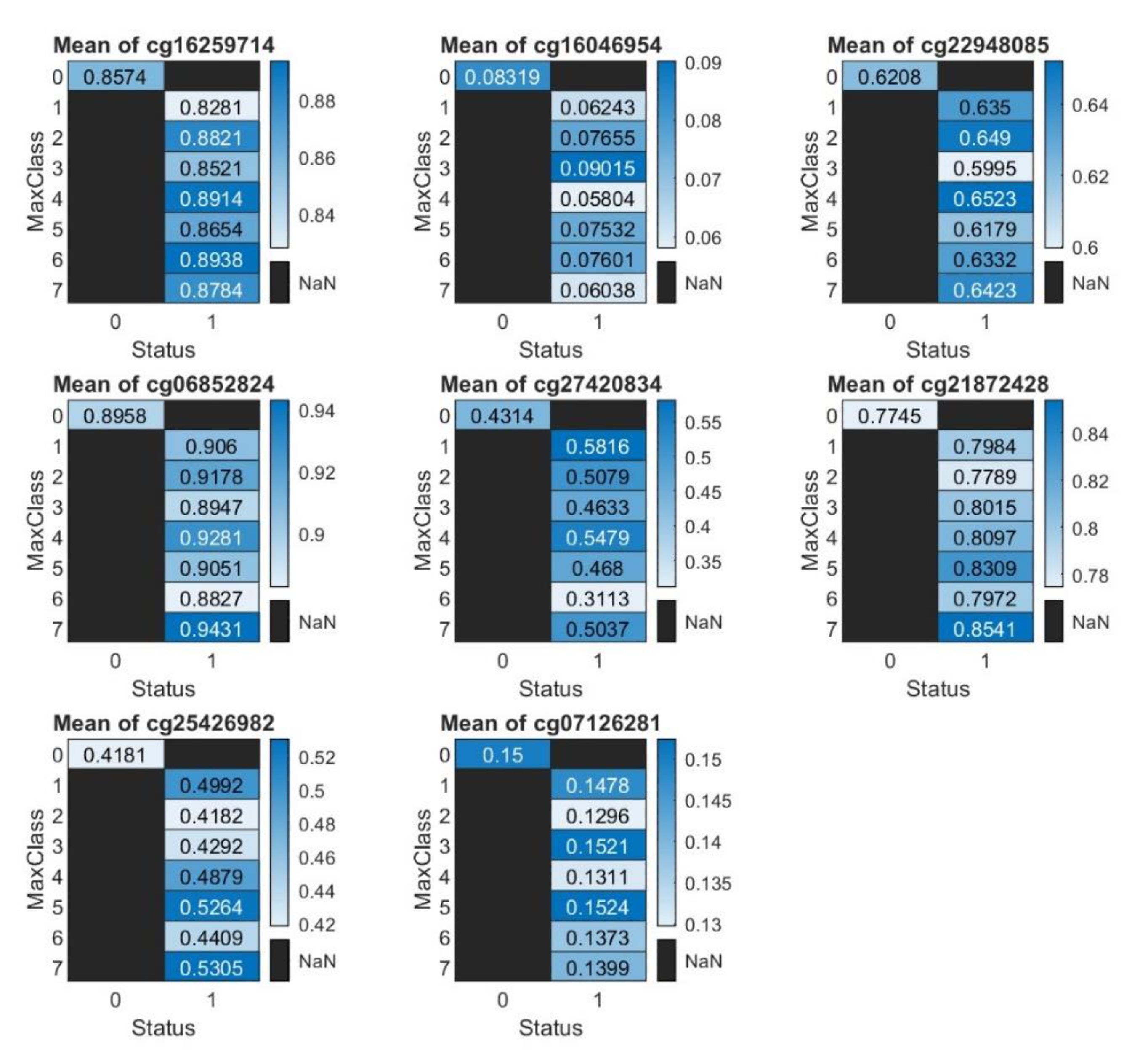
Figure 4.
Mean total intensity counts of all patients. The cell (0,0) corresponds to patients with other diseases. The cells (1,1) to (7,1) correspond to Venn Diagram in Figure 2 and eight CpGs in Table 1.
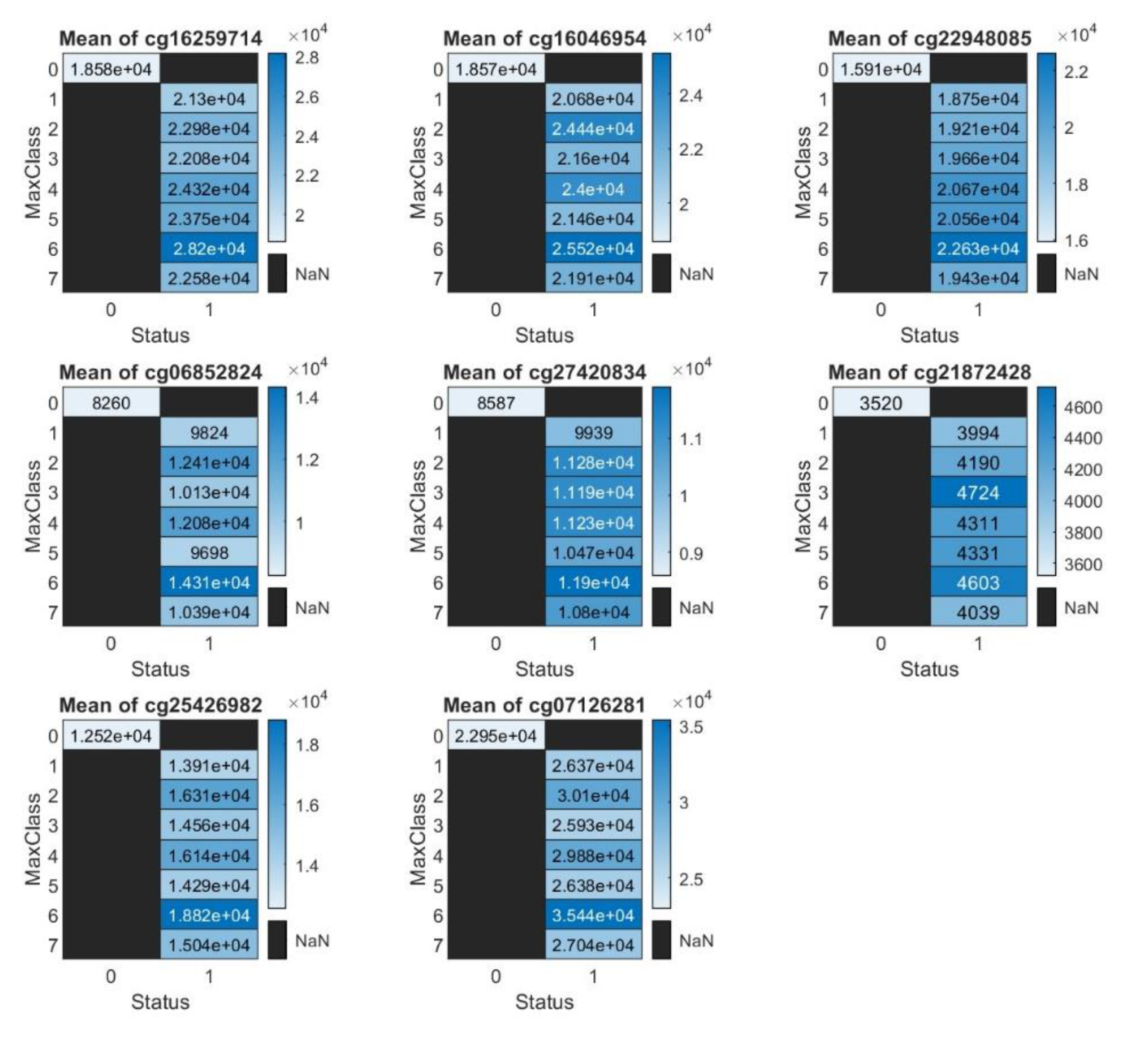

Table 1.
Performance of individual classifiers and max-logistic intelligence classifiers using blood sampled data GSE174818 to classify hospitalized COVID-19 patients and other types of patients (as control) into their respective groups. CF1, 2, 3 are three different classifiers. CFmax = max(CF1-3) is the max-logistic intelligence classifier. The numbers are fitted coefficient values.
Table 1.
Performance of individual classifiers and max-logistic intelligence classifiers using blood sampled data GSE174818 to classify hospitalized COVID-19 patients and other types of patients (as control) into their respective groups. CF1, 2, 3 are three different classifiers. CFmax = max(CF1-3) is the max-logistic intelligence classifier. The numbers are fitted coefficient values.
| sites | gene | CF1 | CF2 | CF3 | CFmax |
| Intercept | -27.7252 | -78.6488 | -20.8835 | ||
| cg16259714 | SDK1 | 55.57 | |||
| cg16046954 | MFSD11 | -92.8578 | |||
| cg06852824 | CAB39 | 20.3366 | 42.3906 | ||
| cg25426982 | SERPINB8 | 35.1956 | |||
| cg07126281 | SAMM50 | -60.8459 | |||
| cg22948085 | KCNAB1 | -101.445 | |||
| cg27420834 | ZNF280D | -24.2856 | |||
| cg21872428 | RANP1 | 116.8975 | |||
| Accuracy | % | 81.25 | 75 | 34.38 | 100 |
| Sensitivity | % | 76.47 | 68.63 | 17.65 | 100 |
| Specificity | % | 100 | 100 | 100 | 100 |
Table 2.
Performance of individual classifiers and max-logistic intelligence classifiers using blood sampled data GSE193879 to classify pediatric COVID-19 patients and other types of patients and healthy (as control) into their respective groups. CF1, 2 are two different classifiers. CFmax = max(CF1-2) is the max-logistic intelligence classifier. The numbers are fitted coefficient values.
Table 2.
Performance of individual classifiers and max-logistic intelligence classifiers using blood sampled data GSE193879 to classify pediatric COVID-19 patients and other types of patients and healthy (as control) into their respective groups. CF1, 2 are two different classifiers. CFmax = max(CF1-2) is the max-logistic intelligence classifier. The numbers are fitted coefficient values.
| sites | gene | CF1 | CF2 | CFmax |
| Intercept | 3.1477 | 8.8827 | ||
| cg16046954 | MFSD11 | 73.5632 | ||
| cg25426982 | SERPINB8 | -19.5833 | ||
| cg07126281 | SAMM50 | -115.236 | -110.706 | |
| cg27420834 | ZNF280D | 7.9757 | 18.9041 | |
| Accuracy | % | 88.19 | 85.83 | 87.40 |
| Sensitivity | % | 73.33 | 33.33 | 86.67 |
| Specificity | % | 90.18 | 92.86 | 87.50 |
Table 3.
Performance of individual classifiers and max-logistic intelligence classifiers using blood sampled data GSE193879 to classify hospitalized COVID-19 patients and other types of patients (as control) into their respective groups. CF1, 2 are two different classifiers. CFmax = max(CF1-2) is the max-logistic intelligence classifier. The numbers are fitted coefficient values.
Table 3.
Performance of individual classifiers and max-logistic intelligence classifiers using blood sampled data GSE193879 to classify hospitalized COVID-19 patients and other types of patients (as control) into their respective groups. CF1, 2 are two different classifiers. CFmax = max(CF1-2) is the max-logistic intelligence classifier. The numbers are fitted coefficient values.
| sites | gene | CF1 | CF2 | CFmax |
| Intercept | 35.2131 | 15.351 | ||
| cg12654612 | ZNF71 | 53.162 | ||
| cg19770550 | MMS19 | 218.9845 | ||
| cg03841686 | MRPS35 | 101.8423 | ||
| cg25042073 | SLC10A7 | -54.9763 | ||
| cg06852824 | CAB39 | -23.5245 | ||
| cg07126281 | SAMM50 | -73.7777 | ||
| Accuracy | % | 95.28 | 92.13 | 98.43 |
| Sensitivity | % | 73.33 | 33.33 | 100.00 |
| Specificity | % | 98.21 | 100.00 | 98.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



