Submitted:
05 November 2024
Posted:
06 November 2024
You are already at the latest version
Abstract
Many children suffer from neurodevelopmental aberrations that have long-term effects. To un-derstand the consequences of pathological processes during particular periods in neurodevelop-ment, one has to understand the differences in the developmental timelines of brain regions. The cerebellum is one of the first brain structures to differentiate during development, but one of the last to achieve maturity. This relatively long period of development underscores its vulnerability to detrimental environmental exposures throughout gestation. Moreover, as postnatal functional-ity of the cerebellum is multifaceted, enveloping sensorimotor, cognitive, and emotional domains, prenatal disruptions in cerebellar development can result in a large variety of neurological and mental health disorders. Here, we review major intrauterine insults that affect cerebellar devel-opment in both humans and rodents, ranging from abuse of toxic chemical agents such as alcohol, nicotine, cannabis and opioids, to stress, malnutrition as well as infections. Understanding these pathological mechanisms in the context of the different stages of cerebellar development in hu-mans and rodents can help us to identify critical and vulnerable periods and thereby to prevent the risk of associated prenatal and early postnatal damage that can lead to lifelong neurological and cognitive disabilities. The scope of the review is to raise awareness and to provide information for expecting parents, obstetricians, and other healthcare professionals to eventually de-sign strategies for preventing or rescuing related neurodevelopmental disorders.
Keywords:
Cerebellum
; development
; alcohol
; intrauterine insults
; sleep
; stress
; malnutrition
; infection
; Nicotine
; motor memory
; smoking
; drug
; abuse
1. Introduction
Neurodevelopmental disorders, characterized by cognitive, neurological, and psychiatric complications, affect one in six children in industrialized nations, causing enduring consequences with extensive societal and economic ramifications [1]. Detrimental prenatal environments can modify brain development, potentially leading to neurodevelopmental disorders [2,3]. The intricate influence of environmental variables on human fetal neurodevelopmental conditions is yet to be precisely determined. The developmental timelines of brain regions vary, indicating that the type, severity, and timing of harmful exposures are crucial in determining resulting aberrations [4]. The cerebellar anlage appears in humans from embryonic day (E) 29 and in rodents from E8.5 onwards [5,6,7]. It grows rapidly during gestation and continues to mature until late postnatal life [8] with long-range connections from the cerebellum to the thalamus and cerebrum starting to develop prenatally and continuing to develop postnatally. Within the cerebellum, there are regional differences in the development of the anatomical local and long-range connections [9,10]. The differential spatiotemporal cerebellar development is causally related to a distinctive functional development. Shifts in neuroscience perspectives have reframed the cerebellum, traditionally overshadowed by the corticocentric viewpoint, acknowledging its role in both motor and non-motor functions with distinct cerebellar areas processing the diverse functions [11,12,13,14]. Current understandings affirm the cerebellum’s pivotal role in modulating motor control, precision, and learning, correlating cerebellar damages with substantial motor disorders [15] but Schmahmann’s elucidation of the “Cerebellar Cognitive Affective Syndrome” marked a paradigmatic shift, extending cerebellar functionality to encompass cognitive and affective domains [16]. As did the work of Andreasen and colleagues, who developed a model that postulates the emergence of ‘cognitive dysmetria’ with ‘poor mental coordination’ in case of disruption of the functional connectivity between prefrontal regions, the thalamic nuclei and the cerebellum [17,18]. Contemporary research corroborates the cerebellum’s integration in attention, behavior, cognition, and language, but also associates its aberrations with developmental anomalies like dyslexia, autism, and attention deficit hyperactive disorder [12,13,19].
All this underlines the cerebellum’s vulnerability to detrimental environmental exposures happening during different time points of gestation [20], highlighting the significance of studying the associated biomedical mechanisms and consequences underlying the diverse set of cerebellar neurodevelopmental problems [14].
2. Scope
The development of the cerebellum has been a focus of many outstanding lines of research and over the years multiple reviews have been published describing the development of the cerebellum and its cyto-architecture (for example [8,13,21,22,23]). Throughout prenatal development, the cerebellum can be affected by both genetic and intrauterine insults [20], often leading to poor neurodevelopmental outcomes. We are focusing our review on maternal insults during pregnancy that are known to affect a myriad of human mothers; these include maternal substance abuse, intrauterine stress-related events, undernutrition and infection such as chorioamnionitis (CA). The scope of the review is to summarize, compare and align human and rodent research such that we get a better understanding of the impacts of intrauterine insults on fetal and postnatal cerebellar development to provide information for expecting parents, obstetricians, and other healthcare professionals before, during and after pregnancy.
3. Cerebellar Anatomy
Regional differences in cerebellar vulnerability are underlined by anatomical differences. The cerebellar hemispheres, united by the vermis, can be divided into individual lobules I-X (reviewed in [24]), which are split into the anterior (lobule I-V), posterior (VI-X), and flocculonodular lobe by fissures (Figure 1a).
The anterior lobe together with lobules VIIIA and B have been suggested to be the sensorimotor cerebellum, and the posterior lobe together with the lateral lobules Crus 1 and 2 of lobule VII the cognitive cerebellum [27,28,29,30]. The cerebellum receives information via the three-layered and folded cerebellar cortex that surrounds the cerebellar nuclei (CN) (Figure 1). The sole output neurons of the cerebellar cortex are inhibitory Purkinje cells (PCs). They are in the middle PC layer (PCL). The expression patterns of different genes, proteins and enzymes, such as aldolase-C (Zebrin II/ZII) [31], the small heat shock protein (HSP)25 [32], GRID2 [33],phospholipase C, and beta4 [34], appear to reveal a reproducible pattern of parasagittal stripes of PCs, around which the rest of the cerebellar cortex is organized [35]. This anatomical and molecular differentiation is preserved across species and interestingly, the foundation for such a subdivision into individual cerebellar stripes and zones lies already in the early cerebellar embryology, including that of humans [36]. Mutations in those genes can lead to severe neurodevelopmental issues [37,38,39]. However, mapping the cellular, molecular and spatial composition of the human, macaque, marmoset and mouse cerebellum revealed differences between species [40,41,42]. In humans, cytoarchitecturally distinct regions and developmentally transient cell types have been discovered that are distinct from the mouse cerebellum [40]. Furthermore, primate-specific cell subtypes of PCs and molecular layer interneurons (MLIs) as well as region-specific gene expression profiles were found in primates compared to the mouse cerebellum [42]. In humans and primates, region-selective gene expression is predominantly observed in granule cells (GCs) and in PCs in rodents [41,42]. In rodents, it has been shown that ZII+ PC learning is expressed in upbound changes in so-called simple spike activity whereas ZII- zone learning as downbound [26]. The foundation for such differences in learning profiles relies on the expression profile of PC genes and not on synaptic inputs [26,43,44,45,46,47,48]. The modulation of simple spike firing of PCs is tuned by various inputs. Most of these inputs reside in the outermost layer of the cerebellar cortex, the so-called molecular layer (ML). The interneurons in this layer, the MLI's, have historically been divided into stellate and basket neurons, which inhibit PCs predominantly at their dendrites and soma, respectively. However, there are also MLIs that mainly inhibit other MLIs and disinhibit PCs [49]. PCs receive their excitatory inputs from climbing- (CFs), and parallel fibers (PFs). CFs arise in the inferior olive and PFs are the axons of GCs, located in the GC layer (GL), and which in turn receive input from mossy fibers (MFs). The GL also comprises inhibitory Golgi cells, and in the zebrin positive microzones of the nodule, uvular, and flocculus, excitatory unipolar brush cells (UBCs). All neurons in the GL increase the diversity of the MF information, which then ultimately is relayed via the PFs to the PCs. The PC axons project onto glutamatergic CN neurons that control the premotor and non-motor nuclei in the brainstem and diencephalon, as well as the gamma-aminobutyric acid (GABA)ergic local interneurons and projection neurons. The latter provide feedback to the inferior olive [50,51]. Even though the lamination and neuronal subtypes show similarities between humans and rodents, the ratio between the neuronal subtypes differs [52]. Furthermore, despite this uniform appearance with its distinct microzones, microcomplexes and micromodules (Figure 1b), one must remember that the cerebellum is very heterogenous in terms of the genetic and physiological composition within the microzones (Figure 1) [44].
4. Embryology of the Cerebellum
Each species exhibits particularities regarding gestation. Term parturition in women occurs around 280 days after the onset of their last menstrual period (Figure 2) [53].
In mouse and rat, gestation is shorter, lasting between 20 and 22 days [54]. Regarding the comparison between cerebellar brain development of rodents and humans, one must consider different aspects, since they do not run proportionally in parallel [22,55,56]. The developmental journey of the human cerebellum commences 29/30 days after conception [5,6,7] and ends around 2-3 year after birth (Figure 2). The rodent cerebellum starts to develop around E8.5 and ends around postnatal week 3 (Figure 3) [5,6,7]. There are two progenitor zones, the ventricular zone (VZ) and the rhombic lips (RLs), at the start of cerebellar development. Forty to forty-five days after conception, the human cerebellar VZ splits into a VZ and a subventricular zone [57]. The RLs expand spatiotemporal and promote growth and maintenance of the posterior lobe. In mouse the VZ does not split and RLs presence is transient [22]. The external granule layer (eGL) is a third progenitor zone later during development in both humans and rodents. Cerebellar maturation can be distinguished by a multifaceted, symbiotic cascade, encompassing gene expressions, electrical network interactions, and environmental factors, which are inherently interlinked with the evolution of the hindbrain (hb) and midbrain (mb). The transcription factors Otx2 and Gbx2, which are expressed anterior and posterior from the so-called isthmic organizer region between the mb and hb, are important for the general development of most of the rostral and caudal regions of the central nervous system [58]. Originating from the caudal-most primary neural tube vesicles, the rhombencephalon (hb) bifurcates into the metencephalon and myelencephalon. Segmented along the rostral-caudal axis into seven rhombomeres, the cerebellum’s emergence is facilitated by transcription factors and signalling molecules, leading to the formation of specialized epithelium, RL1, from the dorsal portions of the metencephalon [59] through bilateral expansion of the Alar plate [60]. The latter occurs in the presence of Gbx2 and absence of Otx2 and Hoxa2 [58,61,62]. Wnt family members, fibroblast growth factors (especially Fgf8 and Fgf17), En1-2, Lmx-1, and sonic-hedgehog (shh) play a major role in defining the isthmic organizer region and the anterior-posterior as well as the dorso-ventral patterning of cerebellar development [63,64,65]. Mutations in any of these genes lead to severe implications for cerebellar development or even death in rodents [66,67,68,69]. In rodents, around E9, cerebellar histogenesis starts. Just above the fourth ventricle the two germinative compartments of the RLs are formed, adjacent to the roof plate and the VZ placed in the inner side. The bulges grow and give rise to the unitary cerebellar plate comprising the vermis and hemispheres. The cerebellar medial regions expand, an orthogonal rotation happens, and the cerebellar wing-like anlagen transform into a homogeneous cylindric vermis at E15.5. Cerebellar size expansion and increased lobular complexity occurs from gestational week (gw) 20 in humans and from birth (postnatal day 0 (P0)) in mice [50].
4.1. Neurogenesis
In both humans and rodents, the RLs form the origin for all glutamatergic cerebellar neurons, whereas all GABAergic neurons as well as glia cells, oligodendrocytes and astrocytes originate from the VZ (Figure 2 and Figure 3) [22,23,57]. Birthdating studies showed that the projection neurons are produced first, at the onset of cerebellar neurogenesis.
4.1.1. Glutamatergic Neuron Development
In rodents, the RLs are defined by the expression of the mouse homolog of Drosophila atonal (ATOH1) transcription factor [72] forming the origin of all glutamatergic cerebellar neurons [73]. ATOH1 expression begins at E9.5 in mice and from E10.5 to E12.5 progenitors leaving the rostral RLs give rise to the large CN neurons, migrating to the surface of the cerebellar anlage, where they aggregate in the nuclear transitory zone. From there they move inward from the PC plate to form the four CN on both sides [74]. Progenitors migrating between E14 and E21 give rise to UBCs in distinct cerebellar areas [75]. Then GCs start proliferating in response to shh signalling from PCs [76]. The GC lineage arises already around E8.75. GC progenitors exit the upper RLs moving tangentially along the cerebellar surface, by E16 eventually covering the entire cerebellar anlage [77]. At least three transverse GC progenitor zones are identified by gene expression and birth-dating. Postmitotic GCs migrate from eGL to the inner GL guided by the Bergmann glia fibers, which are oriented in the same plane (Figure 3 top). Thereby the eGL topography is projected into the nascent inner GL [77]. After birth the GL is an 8 layered structure with another layer of proliferating granule precursor cells [77,78]. The proliferation window of murine GCs progenitors closes only at the end of the second postnatal week in rodents, when the last postmitotic GCs, from the deepest portion of the eGL, migrate inwardly to the nascent GL, marking the end of the eGL and ceasing ATOH1 expression [72,79]. Interestingly, recent studies showed that the cell-fate specification among the cerebellar VZ and RL is not absolute. A so-called posterior transitory zone expresses genes to develop bipotent progenitors for cerebellar glutamatergic neurons [80].
4.1.2. GABAergic Neuron Development
The VZ is defined by the pancreas transcription factor 1-a (Ptf1a), giving rise to GABAergic neurons. A Ptf1a-neurogenin 1/2 (Neurog1/2)- early B-cell factor 2 (EBF2) regulatory network is implicated in PC subtype specification [81]. In rodents, PCs are born between E10 and E13 and undergo terminal mitosis. Dividing VZ precursors emigrate into the cerebellar prospective white matter, via the cerebellar plate and form an array of clusters (E14-E18), which are suggested to aggregate into microzones or also called topographical organization centres (TOCs) (Figure 1). These TOCs are not only specific for ingrowing afferent precerebellar MF and CF inputs as well as interneurons, but also for subsets of glia cells and migrating GCs [82]. As the PC clusters disperse into parasagittal stripes (Figure 1), all components disperse with them, forming the adult cerebellar parasagittal architecture. MFs disconnect and form local connections with GCs within the zone. At least five molecularly distinct PC subgroups have been identified throughout development with distinctive levels of Foxp1 and Foxp2, respectively. Foxp1+/Foxp2+ PCs strongly express reelin receptors and lack Ebf2. Reelin controls PC migration [83,84,85,86]. Early born PCs are likely to become ZII+ during adulthood, while late-born PCs adopt the ZII− phenotype, which is in line with the high aldolase-C expression in the phylogenetically older vestibulocerebellum (Figure 1a) [87]. The postnatal development of PCs can be divided into different stages. The first distinction can be made between intrinsic maturation by PCs themselves, and guided maturation by stimulation of other cell types, such as GCs [88]. In rodents, intrinsic growing starts with a fast somatic growth from P0-P9 followed by a rapid dendritic growth from P9-P18 [89,90] with more processes growing outwards from the soma. During the second postnatal week, the processes become more complex by growing rapidly and increasing the number of branches. This is the start of the dendritic tree, which will be completed around postnatal week 4 in rodents [91]. CFs are also involved in the dendritic arborisation of PCs during these stages, presumably by stimulating PCs. This may explain why higher-order mammals, including humans, show both a higher level of persistent multiple CF innervation and a higher complexity of the dendritic trees of their PCs [92]. GABAergic interneurons in the CN are born between E10.5–E11.5, and Golgi cells at approximately E13.5–postnatally (peak around E14–E16) [23,93,94]. Late-born GABAergic interneurons, including stellate and basket cells, derive from secondary precursors in the prospective white matter at later stages (from E13 to P5 with a peak around birth) [95]. Thus, cerebellar neuronal subtypes depend on when and where they are generated from neural progenitors. Additionally, the cerebellum accommodates astrocytes, glia, and oligodendrocytes, the origins of which are not fully discovered [96]. The start – and end time as well as the total duration of the developmental timeline of cerebellar cell types display distinct susceptibilities to environmental insults and genetic mutations [97].
4.2. Embryology of the Precerebellar System
Interior olive neurons are derived from the dorsal neuroepithelium, the caudal hb (RL 6-8) at E9.5 to E11.5 in rodents. Olivocerebellar projections, the CF, are being formed at E17.5 [98] and already at the late embryonic stage, the olivocerebellar bundle shows an organized topographic projection pattern. Neurons in a particular subnucleus of the inferior olive project via their CFs to a specific part of the cerebellum. The bundles with CFs run contralaterally through the inferior cerebellar peduncle. Axons that leave the peduncle rostrally project to the vermis, whereas CFs innervating the other cerebellar areas pass through the more caudal parts of the inferior cerebellar peduncle [99]. During early development CF axons form a plexus with abundant branching, while in the second postnatal week axonal branches disappear and those so-called nest terminals grow into an entire CF terminal in the following weeks. The latter form the distinct one-to-one synaptic connection in rodents (Figure 3, for comparison in human, see recent [92]). MFs are derived from the dorsal neuroepithelium 1 domain of the caudal hb (RL 6-8) and are generated at slightly later stages (E10.5-E16.5) compared to CFs [100]. Initially axonal fibers of GCs that receive MF input form contacts with the soma of PCs, and only around P5-P15 they start to turn into the typical parallel fibers that establish synaptic contacts with the dendrites of PCs [91].
5. Extrinsic Deterrents Influencing Cerebellar Development
As mentioned under scope, multiple genetic and external conditions can significantly hinder both pre- and postnatal development of the cerebellum, resulting in impaired maturation and functionality. In this review we focus on main common maternal disruptors during prenatal stages including 1. teratogenic exposures during pregnancy to substances like alcohol, nicotine, cannabis and opioids; 2. increases in cortisol level (stress); 3. intrauterine growth restriction (IUGR) resulting from e.g., malnutrition; and 4. chorioamnionitis (CA) [2].
5.1. Maternal Substance (ab)use and Cerebellar Maturation
The cerebellum has been suggested to be sensitive to drugs with abuse liability, including alcohol, nicotine, cannabis and opioids , leading to decreased cerebellar volume, increased apoptosis, and behavioural differences [101,102,103,104,105,106,107]. The criticial periods of sensitivity to drugs of abuse coincide with the long cerebellar neuron proliferation and migration phase, which coincides with the embrionic and postnatal first three weeks of a rodents life and the third trimester of gestation until 1.5 years in humans [107,108]. It has been suggested that drugs act by mimcking or interfering with normal endogenous neurotransmitter-receptor interactions and thereby misleading the endogenous timing and sequence of endogenous neurotransmitter-receptor interactions.
5.1.1. Maternal Alcohol Consumption – Impact on Cerebellar Maturation
One of the primary challenges in studying the effects of maternal alcohol consumption on fetal cerebellar maturation in humans is the inherent limitations of fetal neuroimaging. These include a low signal-to-noise ratio in ultrasound imaging, motion artifacts in MRI scans, limited availability of postmortem data, and the absence of experimental human studies. Despite these obstacles, research in this area is important to understand the impact of alcohol exposure on cerebellar development. Excessive alcohol exposure during pregnancy results in children with fetal alcohol spectrum disorder (FASD), which usually comes with symptoms such as balance disturbances and impairment of motor skills [109,110]. Cerebellar degeneration might contribute to some of the cognitive disabilities that can be observed in children with FASD [110]. As the cerebellum undergoes rapid growth during the second and third trimesters of pregnancy [111], it is particularly vulnerable to alcohol exposure during this critical period [112]. Studies reveal that early cessation of alcohol use upon pregnancy recognition can mitigate some adverse outcomes, as no significant differences in fetal growth measures are observed between those who abstained and the non-exposed groups [113]. However, heavy alcohol consumption that continues after conception is associated with altered fetal cerebellar development, showing a reduced transcerebellar diameter [113]. Fetal neuroimaging findings on the cerebellar vermis measurements are mixed, with some studies showing decreased vermis lobules I-V sizes in children exposed to alcohol in utero, while others found no significant differences [114,115,116,117]. Inconsistent results are also noted in the literature regarding the impact of prenatal alcohol exposure on overall cerebellar development [118]. While some studies highlight smaller cerebellar volumes in alcohol-exposed groups [113], others report no significant reductions in cerebellar growth [2]. Furthermore, prenatal alcohol exposure has been linked to changes in cerebellar white matter integrity, potentially contributing to impaired motor and cognitive functions [119,120]. These neurodevelopmental disruptions are thought to arise from altered myelination [119] and impaired neural transmission in cerebellar tracts [121]. fMRI analysis also suggests distinct patterns of network activation regarding working memory in two differently alcohol-exposed groups, revealing dose and temporal sensitivity for alcohol [122]. Despite these findings, variability in outcomes across studies underscores the complex interplay of alcohol exposure patterns, individual differences, and methodological challenges in assessing the effects of alcohol on fetal brain development.
In rodents, prenatal maternal ethanol administration limited to E8 or E9 leads to differences in cerebellar shape and decreased volume at E17 [123,124]. Although volumar changes are not visable anymore, shape differences together with bahavioural changes still exist in adult mice [125,126]. Ethanol administration at E7 does not alter cerebellar volumes at E17 [127]. The latter does not come as a major surprise, as the development of the cerebellum starts only at E8.5. Ethanol exposure for the first gw results in a decreased proportion of GABAa receptors (subunit ⍺1) in the adult cerebellum [128]. Alcohol administration during almost the whole pregnancy results in increased oxidative stress and apoptosis markers, as well as decreased GLUR1, PSD95 and ILK expression [125]. However, in another study, neither the amount of PCs decreases nor the cerebellar to body-weight ratio reduces at P10 when ethanol is administered during the third week of pregnancy [129], indicating that ethanol exposure has a higher impact on PCs neurogenesis than on PC differentiation. Yet another study revealed that alcohol exposure during both E12-19 and P2-9 resulted in decreased numbers of inhibitory interneurons in lobule II, lower numbers of PCs in lobules II, IV-V and IX, and decreased volumes of lobules II, IV-V, VI-VII, IX and X of the vermis at P16 [130]. Ethanol treatment before and during pregnancy as well as during lactation leads to morphological differences in the eGL, GCs and Bergmann glia in offspring [131]. Whether the morphological differences are caused by the maternal-, or prenatal ethanol exposure, or the combination of both, remains unclear. Postnatally, a ‘temporal window of vulnerability’ of the cerebellum for harmful effects of alcohol administration has been described. This window of vulnerability starts with birth of pups and ends with the start of the second postnatal week [132,133], a timeframe roughly corresponding to the beginning of the third trimester in humans. Alcohol administration for only 1 day during the first postnatal week results in reduced brain weight at P10, with the cerebellum being diminished the most [101]. The biggest loss of PCs after ethanol treatment occurs after ethanol consumption around P4-6, although lobule-specific sensitivity differences exist [134]. More specifically, alcohol administration between P2-P5 leads to reduced volumes of the GL and ML and a significant loss of PCs and GCs in all lobules of the vermis, except for the PCs of lobules VI and VII [101,135,136,137]. Ethanol-sensitivity of PCs in lobules I-V and IX decreases over time, whereas lobules VII vulnerability increases towards the end of the first postnatal week [133]. Also the numbers of MLIs decreases after ethanol exposure at P4-6. Nevertheless the amount of spontanous inhibitory synaptic currents as well as the hyperpolarization activated inward current (Ih) amplitudes in the remaining PCs increase [138]. Regarding external inputs, the CF distribution alters, as the CF terminals reduce in volume and immunostaining intensities relative to the PC volumes in P14 rats [139]. There is also a reduction in CF terminal volumes relative to PC volumes in P40 rats [140], suggesting decreased numbers of CF to PC connections. Ethanol exposure during both the first and second postnatal week, specifically P4-9, results in reduced overall cerebellar weight, reduced volumes of lobules I-IV, and IX-X, reduced numbers of GCs and PCs, increased microglia density and activated microglia [129,135,141,142]. Whether microglia are still affected by ethanol exposure later in life remains unclear. While Gursky et al. (2020) found increased microglia densities, Cealie and colleagues did not find significantly affected microglia morphology, density or microglia-PC interaction in lobule IV/V using in vivo two-photon imaging and fixed tissue [143,144]. Minor changes were found in the ML and PCL. The literature is inconsistent regarding the effects of alcohol exposure after the ‘window of vulnerability’, after the first postnatal week. No alterations in cerebellar weight, amount of PCs and GCs, or area loss in the GL or ML were found after alcohol exposure between P7 and P13 in most literature [132,133,134,139,140]. Hence three studies show effects of alcohol administration after P7. Two studies show CF distributional differences after ethanol exposure between P7-9 [139,140] and one study shows that ethanol administration during P10-12 can result in a thinner eGL and reduced numbers of GCs in the GL and a reduced size of the ML [145]. One of the open questions is what leads to the window of vulnerability to ethanol exposure and to what extent differential gene expression plays a role in that. The expression of some neurodevelopmental genes varies between the first and second postnatal weeks, and ethanol-induced alterations have been shown for those genes [133]. Genes involved in cerebellar vulnerability to ethanol are the nNOS gene and the CREB-gene. mRNA expression of the CREB-gene increases at P4 compared to P10 after ethanol exposure [133]. PCs are also much more sensitive to ethanol exposure at P4-9 when CREB is not expressed in PCs [141], suggesting that under control conditions the cAMP pathway plays a protective role in neonatal alcohol exposure during the first postnatal week. Mice with a mutation in the nNOS gene are more vulnerable to ethanol-induced behavioral changes and PC and GC losses [146]. Another pathway affected by ethanol exposure is the Wnt signaling pathway. As mentioned above, Wnt is important during cerebellar maturation and development. After maternal ethanol exposure from E6 until birth, 3 of the 84 genes related to the Wnt-pathway are down-regulated at P10 and 33 genes at P35 [147]. More specifically, Wnt5a, Fed 6, Didxc, Axis 2, Pzd4, Fzd6 and EP300 gene expression is decreased and Wnt5b gene expression is increased at P20 [147,148]. Moreover, other pathways affected by maternal ethanol exposure are insulin/IFG-1 and Notch signalling pathways at P30 [148]. Also postnatal ethanol exposure effects gene regulation. A recent study looking at alcohol induced transcriptomatic changes, found 2440 dysregulated genes one day after ethanol exposure at P4, and 1348 dysregulated genes one day after ethanol exposure at P4-5. These dysregulated genes included genes related to canonical pathways, diseased and biological functioning, microglial, astrocytes, oligodendrocyte lineage cells, and the cell cycle [149]. These findings may help to identify genes and pathways involved in fetal ethanol exposure-related diseases such as FASD. Ethanol exposure from P4-9 evokes gene expression changes in myelination-related genes which are reduced after ethanol administration [150] and neuro-inflammation related genes which are increased after ethanol administration [151,152]. In line with elevated neuroinflammation, activated microglia were found in lobules V and IX of the cerebellum, possibly through alterations in CX3CL1-CX3CR1 signalling [151,152]. The increase in neuroinflammation could also be due to the increased chemokine MCP-1 expression after ethanol exposure, as administration of an MPC-1 synthese inhibitor or MPC-1 receptor antagonist reduces the percentage activated microglia and decreases the expression of pro-inflammatory cytokines TNF-⍺ and IL-6 in the developing brain [153]. MCP-1 has been suggested to be crucial for ethanol-induced neurodegeneration. Caspase-3 levels, an apoptosis indicator, are increased after ethanol administration at P4 in the cerebellum [153,154] but are less elevated if ethanol is accompanied by an MPC-1 inhibitor and in MPC-1 deficient mice [153]. Similarilly to MPC-1 inhibitors, nicotinamide has also the ability to diminish neurodegeneration after ethanol exposure. Ieraci and Herrera et al. (2018) found increased levels of activated Caspase-3 and PARP-1 as well as increased neurodegeneration in lobules III, VI, IX and X after ethanol exposure at P4, with administration of nicotinamide reducing the ethanol induced neurodegeneration and apoptosis [154]. The specific PARP-1 inhibitor 3-ABA did not reduce Caspase-3 activation and neurodegeneration, suggesting that the working mechanisms of nicotinamide are mainly through reducing Caspase-3 activation. Other ethanol-induced effects on the cerebellum, such as neuroinflammation and degeneration, can be diminished by administration of a peroxisome prokiferation-activated receptor (PPAR)-γ agonists [136,151], or by low-intensity pulsed ultrasound exposure [145]. Whether these protective mechanisms also apply in humans, and if this might be interesting for treatment options in cases of alcohol-abuse during pregnancy, needs to be investigated.
Taken together, the literature is not consistent about the impact of maternal alcohol exposure on cerebellar maturation in human and rodents (for a recent systematic review combining multiple species, see [155]). Rodent studies revealed that there is a clear negative impact of alcohol usage on cerebellar development but that the effects depend on distinct periods of vulnerability. The developing cerebellum seems to be most vulnerable to alcohol administration during the first week of the rodents' postnatal life, which corresponds with the last trimester of pregnancy in humans. Within the cerebellum, and throughout the first postnatal week, alcohol-sensitivity between PCs located in the different lobules varies. Whether such periods of vulnerability with differential gene expression profiles and different treatment options also exist in humans needs to be determined. For an overview of the above mentioned studies, see Table S1.
5.1.2. Maternal Smoking – Impact on Cerebellar Maturation
Alterations in fetal brain maturation due to tobacco exposure have been observed in several human studies, including defective migration and maturation of PCs [105]. These developmental abnormalities suggest the association between maternal smoking and cerebellar malformations [156]. While some studies have not found significant differences in cerebellar volume [157], the broader body of research suggests that maternal smoking affects cerebellar function and structure in more subtle ways [158]. For instance, postmortem studies have revealed defective brain-derived neurotrophic factor (BDNF) expression in the cerebellar cortex [159], particularly in regions involved in respiratory control, further implicating tobacco exposure in impaired cerebellar development. Prenatally exposed children may also show altered brain activity, with greater recruitment of posterior brain regions, such as the cerebellum, possibly reflecting compensatory mechanisms for deficits in the prefrontal cortex [160,161]. Furthermore cerebellar nicotinic receptors have been implicated in the pathology of autism spectrum disorder [162,163,164,165]. These findings underscore the need for more research on the effects of nicotine, especially as newer nicotine products, such as e-cigarettes, gain popularity during pregnancy [166,167,168]. The long-term consequences of prenatal tobacco exposure on brain development remain a critical area of concern.
In rodents, a hetergenous population of nicotinic recetors (nAChRs) have been demonstred in GCs, the GL, the PCL, CN, and early during development (P0-P30) also on norepinephrin fibers [169,170,171,172], which suggests that nAChRs play a multitude of roles in cerebellar physiology. Activation of nAChRs elicit the release of GABA and norepinephrine in the developing and adult rat cerebellum [169,173,174]. Knocking out α7 nicotinic acetylcholine receptors in a mouse model has a significant effect on the proteome of the cerebellum, particularly with regards to myelin sheath formation, ion transport and glutamaterigc synapses [175]. α7 nicotinic acetylcholine receptors are mainly expresssed on PCs and already early during development. Between P3 and P5 there is moderate PC immunolabeling, which increases rapidly between P8 and P15 while at the same time it disappears from rostral lobules. The receptor localization seems to follow a columnar organization in areas where it is located. Finally, at P20, α7 subunit labeling is found again in all PCs, although with lower intensity. This distinct temporal and spatial distribution suggests that α7 receptor expression is developmentally regulated, with a time course that parallels the final differentiation of PCs [176]. α7 receptors are less sensitive to nicotine compared to other nicotine receptors [177]. The development of primary cerebellar neuroblasts in the eGL is mediated by α3-nAChR subunits [178]. Thereby nicotine can effect the general cerebellar develoment from early during development and indeed smoking is known to affect cerebellar development during different developmental stages. Many studies have investigated the impact of smoking or nicotine exposure on the maturing brain. Nicotine, which is the psychoactive drug in tobacco, is able to cross the placenta and for long periods of time it can even be found in higher concentrations in the fetus compared to the concentrations in the mother [179]. When the maturing embryo is exposed to nicotine it has an inhibitory effect on the development of stem cells [180]. Prenatally, nicotine exposure during almost the whole pregnancy results in a significant increase in the density of dying PCs and a reduced density of surviving PCs in adolescent and adult rats [181,182]. Moreover, glial fibrillary acidic protein (GFAP) immunoreactivity expression is significantly increased in the GL and white matter of the cerebellum in adolescent and adult rats [181,182]. The vulnerability to maternal nicotine exposure is exposure duration sensitive, with more severe histomorphological PC differences after 2 weeks of daily prenatal nicotine exposure compared to 1 week [183]. However, one study states that nicotine administration for the three prenatal weeks does not alter PC numbers in the cerebellar vermis at P10 [184]. Nicotine exposure in the first postnatal week increases apoptosis in the GL [183], and nicotine exposure in the first and second postnatal week reduces PCs in the cerebellar vermis in rats [135]. Although the evidence of nicotine-induced alterations in cerebellar development is not extensive, more research on other brain areas has pointed out the negative effects of pre- and postnatal nicotine exposure on cellular, molecular and behavioral levels [185,186,187,188]. Furthermore, nicotine exposure in adult rats increases apoptosis in the white matter of the cerebellum [189], highlighting the importance of future research to unravel the effects of maternal nicotine exposure on the development of the cerebellum. For an overview of the above-mentioned cerebellar studies, see Table S2.
Not only maternal nicotine exposure is harmful, also maternal smoking without nicotine can have negative effects on offspring. Studies show that maternal vaping without nicotine leads to neurological, behavioural, and epigenetic changes [190,191]. Regarding the cerebellum, maternal exposure to e-cigarettes, containing propylene glycol and vegetable glycerol, during the first three weeks of pregnancy in mice, results in increased pro-inflammatory cytokine IL-6 levels in the cerebellum of adolescent offspring [192]. Increased levels of cytokine IL-6 indicate an increase in neuroinflammation. With the high rise in e-cigarettes popularity, and the stigma of being ‘safer’ compared to tobacco smoking, more research is important to underline the consequences and risks of maternal e-cigarette usage for different brain areas.
5.1.3. Maternal Cannabinoid Usage – Impact on Cerebellar Maturation
Cannabis usage is the most commonly used illicit substance among pregnant women [193]. Δ9-tetrahydrocannabinol (THC) enters the fetus through the plasma with around 1/3 of the THC crossing the fetoplacental barrier [194], whereas postnatally it can enter the baby via breastmilk [195]. Prenatal cannabinoid exposure can cause preterm birth, leads to increase in admission in neonatal intensive care and results in behavioral and social deficits [196,197,198,199]. All of the previous are known to affect healthy cerebellar development. Regarding the direct effects of cannabinoids on the endogenous endocannabinoid system, the endogenous endocannabinoid system is comprised of endogenous endocannabinoids (eCB), the metabolic enzymes responsible for the formation and degradation of endocannabionids, and the cannabinoid receptors with interacting proteins. The endocannabinoid system exists from the earliest stage of pregnancy, in the perimplantated embryo and uterus, placenta and in the developing fetal brain [200,201,202]. There are two types of cannabinoid receptors, type 1 (CB1R) and type 2 (CB2R). At gw14 CB1R can be found in cerebellar cortex [200] and also later in life CB1R expression is widespread and high in the cerbellum of human and rodents [203]. The majority of receptors are located on the presynaptic side of terminals received by PCs and there is a moderate expression in the ML and low in the GL of the cerebellum [204]. CB1R are also located in mitochondria where they modulate energy homeostasis [205]. 2-Arachidonoylglycerol (2-AG) is the most abundant eCB in the cerebellum and diacylglycerol lipase α (DAGLα) is one of the major biosynthetic enzymes contributing to its production. Monoacylglycerol lipase (MAGL) is the key enzyme involved in 2-AG hydrolisis [206,207,208,209,210]. PCs play a key role in the production of 2-AG such that PCs are ready to activate CB1Rs in neurites approaching or traversing the PCL.
In humans, prenatal cannabis exposure has been associated with alterations in brain connectivity, particularly affecting regions such as the dorsolateral, medial, and superior frontal areas, insula, anterior temporal lobe, posterior cingulate cortex and cerebellum [211,212,213,214,215,216]. While some studies indicate that cannabis exposure during pregnancy may not lead to significant differences in cerebellar structure compared to controls, there is evidence that it can disrupt normal connectivity patterns [211,212,213,214,215,216,217]. For instance, increased connectivity between the hippocampus and cerebellum has been reported [218]. In addition,reduced connectivity between the caudate and cerebellar vermis, as well as hypo-connectivity between the anterior insula and cerebellum, has been observed in cannabis-exposed infants, suggesting that prenatal exposure to cannabis may impair the normal development of these networks. Functional MRI studies in young adults exposed to cannabis in utero show increased activity in the prefrontal cortex and decreased activity in the cerebellum, particularly during tasks involving working memory and response inhibition [160,219]. This altered activity may represent a compensatory mechanism, where increased prefrontal cortex recruitment compensates for cerebellar function deficits. These findings align with other studies that demonstrate increased activity in frontal regions and reduced cerebellar activity, highlighting how prenatal cannabis exposure can lead to long-term changes in brain function and connectivity [220]. Despite these findings, inconsistencies remain [221], with some studies reporting no significant differences in cerebellar volume or connectivity in cannabis-exposed individuals compared to controls. The variability in results may be due to differences in the timing and dosage of cannabis exposure, as well as the methods used to assess brain development [222]. These findings emphasize the need for further research, particularly in light of the increasing prevalence of cannabis use during pregnancy and its potential impact on fetal brain development [223].
In the mouse brain CB1 receptors are expressed from E11 (5-6 weeks old human embryo) with gradually increasing levels of both mRNA and receptor density throughout the prenatal period. At E17.5-P3 CB1R is prominently expressed in long-range axons in the brainstem and cerebellum and a role of eCB in the fasciculation of pontocerebellar, thalamocortical and subcortical axons has been suggested [224,225,226]. By P5 CB1R expression becomes low in the long-range axons and prominent in neurites of radially migrating GCs and in the parallel fibers in the anterior and central zones. During the first two weeks postnatally, CB1R expressing and differentiating GCs are located in the anterior and central vermis, and paravermis but not in other cerebellar zones [224]. MAGL expression is high in PCs lining the primary fissure, likely to cause higher rates of 2-AG hydrolisis. 2-AG hydrolisis dampens eCB signaling through CB1R in differenting GCs within lobes V-VI, likely revealing the specific eCB signaling within the anterior vermis [224]. CB knockouts show a reduction in the anterior cerebellar vermis size from the first postnatal week [224]. THC exposure from E5 to P20 leads to a reduction in glutamate transporter expressions of glial (GLAST) and neuronal (EAAC1) subtypes at P20, P30 and P70 [227]. One study shows that prenatal moderate levels of synthetic cannabinoid exposure from E3 until birth leads to a potential neuroprotective effect on the cerebellum in adolescent rats at P42 [228]. There is no change in the CB1R levels and AMPA-GluA1, whereas NMDA-GluN2A, which is associated with cytotoxic effects, is reduced. There is also a siginificant reduction in total MAO, which may link to the potential higher addiction chance in early adolescence [228]. Prenatal cannabinoid exposure can also reduce certain markers of oxidative stress in adolescent offsping [229,230]. Exposure to a low dosis of cannabis for 5 minutes a day from E5.5-17.5 resulted in an increased cerebellar volume in mice at P60 [231]. Maternal cannabis exposure from E5 until birth also changes intrinsic membrane properties by increasing for example the firing rate of PCs through altering the membrane excitability by modulation of ion channels in P50 old rats [232]. The eCB system plays an important role in cerebellar development as well its long-range connections with the MF and CF system and in PC cerebellar plasticity.
To summarize, cannabis abuse of high amounts during vulnerable periods during pregnancy might affect cerebellar development critically, leading to long-term effects. Moderate usage might play a neuroprotective role for certain cerebellar neurons.
5.1.4. Maternal Opioid Usage - Impact on Cerebellar Maturation
The current “opioid crisis” affects populations across continents and infants with prenatal opioid exposure are born almost every 15 minutes [233]. Incidences of opioid exposure are associated with an increase in the risk of perinatal problems such as neonatal abstinence syndrome (NAS), prematurity, and low birth weight. Opioids are known to cross the placenta and the blood-brain barrier as well as the mother's milk. Perinatal opioid exposure has a negative association with cognitive and motor outcomes persisting during school age [234]. High-affinity opioid binding sites are associated with the ML in humans and high levels of opioid receptors have been found on human GCs, suggesting opioid-dependent maturation [235,236,237,238,239]. Volumetric studies revealed that newborns with prenatal opioid exposure show in general smaller brain and basal ganglia volumes compared to population norms. Regarding cerebellar volume, multiple studies revealed a smaller cerebellar white matter and/or cerebellar volume [240] in the substance-exposed group compared to controls with the changes persisting until adolescence [241,242,243,244]. However, in most studies, the impacts of covariates such as smoking and or alcohol usage can hardly be measured because mothers in this group may often be polysubstance users.
In mice opioid receptors and peptides are widely expressed by developing cerebellar cells. Throughout development the opioid peptides and receptor expression changes considerably (for review read [107]), indicating endogenous opioid-dependent maturation. Opioid receptors appear in peripheral tissue around E9.5 and in neural tissues around E11.5 [245]. Heroine and morphine activate μ-opioid receptors, at high concentration also δ-, and κ-receptors. During development the eGL expresses μ-, and δ-, and a putative ξ-receptor but no κ-receptors, whereas the adult cerebellum does hardly express opioid receptors, only low levels of δ-receptor [246,247,248,249]. In line with the fact that early during development opioid peptides and receptors, such as μ-receptor, exist in the rodent cerebellum, different types of μ-drugs agonists lead to an inhibited growth of the cerebellum. The latter is most likely due to inhibition of GC neuroblast proliferation [246,250,251,252]. Methadone administration of mothers of dams during pregnancy and after birth leads to an attenuation of myelin development but an increased density and percentage of oligodendrocyte precursor cells and increased proliferative oligodendrocytes. Maternal methadone exposure furthermore leads to an increase in apoptosis of mature and myelinating oligodendrocytes at P7 in rat pups. Oligodendrocytes in the cerebellar white matter have been suggested to be more vulnerable to methadone than those in cerebral white matter, which is supported by the fact that the white matter of school aged children is only affected in the cerebellum but not cerebrum [253]. Opioid also affects the dendritic differentiation of GCs and PCs [254,255,256], furthermore supporting that throughout development endogenous opioids support GC and PC proliferation, differentiation, and maturation, hence after development, opioids fulfil a different or not such an important role anymore. Maternal opioid intake therefore may act through misleading the endogenous timing and sequence of endogenous neurotransmitter-receptor interactions, especially in eGL from E13 onwards in rodents.
5.2. Stress and Sleep and Cerebellar Development
5.2.1. Perinatal Stress - Impact on Cerebellar Maturation
Changes in the cortisol levels of mammals can affect the neuronal development of the unborn offspring significantly. Indeed, in humans maternal severe stress during pregnancy can influence fetal cerebellar development through a process known as 'fetal programming' [257,258]. This concept posits that variations in the intrauterine environment during critical fetal developmental phases can induce enduring changes in both the structure and function of the fetus, including the brain [259,260]. Such alterations arise when the fetus adapts or prepares for the expected postnatal environment based on these prenatal signals. Increased maternal anxiety can intensify the release of glucocorticoids and diminish the integrity of the placental barrier, facilitating greater glucocorticoid transmission to the fetus [261]. Additionally, heightened anxiety can reduce uterine blood flow, thereby negatively impacting fetal brain growth but not birth weight [260,261]. Such prenatal environmental changes may lead to cognitive, motor, and behavioral challenges in children [259]. Moreover, disruptions in neuroendocrine pathways like the hypothalamic-pituitary axis have been observed in offspring of mothers with heightened anxiety, suggesting potential long-term impacts on brain structure and function [258,262]. In a study by Buss et al. (2010), high pregnancy anxiety was associated with gray matter volume reductions in several brain regions, including the cerebellum [20], in offspring aged 6 to 9 years, as revealed by structural MRI scans [263]. Furthermore, prenatal exposure to maternal psychological stress is linked with increased sleep problems in toddlers and is associated with decreased fetal cerebellar-insular connectivity, although the specific mediating effects of fetal brain regions remain unidentified [263,264].
Similarly, in rats, stress can induce many cerebellar developmental abnormalities. For example, maternal stress at E7 and E14 results in decreased nuclear sizes of PCs and GCs, increased PC proliferation, increased density of PCs, reduced synapse-to-GC ratio, reduced GC-to-PC ratio, as well as decreased synaptophysin expression in the GCs during adolescence [265,266]. Likewise, maternal stress in mice during their third week of pregnancy results in long-lasting morphological differences in PCs located in the vermis of pups. The surface of the dendritic trees of the pups PCs increases during adolescence, but decreases during adulthood, accompanied by an increase in anxiety-related behaviour [267,268]. Prenatal stress induced by glucocorticoid administration at the end of the pregnancy results in reduced numbers of dendritic branches of the PCs and cerebellar weight in both adolescent and adult rats, increased levels of mGluR1 in adults [269], as well as irregularities in the eGL and PCL in juvenile rats [270]. Importantly, the negative impact of stress extends beyond pregnancy. For example, stress induced by daily corticosterone administration at P2-14 leads to a decrease in glucocorticoid receptor expression in the CN, while it impairs associative cerebellum-dependent learning later in life [271]. One hour of maternal separation between this same timeframe leads to decreased glucocorticoid receptors at P15, but increased glucocorticoid receptors later in life [272]. Along the same vein, stress induced by transportation of rodents during the 2nd postnatal week leads to changes in the excitability of CN neurons [273]. Overall, the evidence for the negative effects of stress during pregnancy and early postnatal life on development of the cerebellum is robust, but the specific impact depends on the precise period of stress induction and presumably also on the intensity thereof. For example, a relatively short period of 3 hours of maternal deprivation a day during the first two postnatal weeks increases neurogenesis, cell density in the GL of rats, it increases the mRNA and protein levels related to neuronal growth and survival, and it increases the myelinisation at short-term [274]. On the onder hand, 24 hours of maternal deprivation during the second postnatal week increases cell death in the GL and eGL [275], and reduces the numbers of astrocytes in the GL of male rats [276]. These findings may be explained by differences in neuronal vulnerability to stress during the first weeks postnatally [277,278]. Indeed, during the first 2 weeks after birth the stress-response may be relatively mild, which comes with decreased corticosterone levels and reduced levels of the adrenocorticotropic hormone (ACTH) and corticosterone release after mild stressful events [277,278]. This ‘stress-hyporesponsive period’ [219] is suggested to have a protective function for the maturing brain against elevated glucocorticoid levels [278]. However, during high stressful events, the sensitivity to stress is increased, indicating that this stress-hyporesponsive period is probably only a mechanism to protect the brain against mild stressors, i.e., not life-threatening situations [279]. In line, the administration of high doses of glucocorticoids for only one day during these two postnatal weeks is associated with increased degeneration in the eGL, whearas low doses is not [280]. Moreover, long-term effects in the GL are still visable in adult mice following a glucocorticiod injection at P7, however no differences in PC numbers in the PCL are identified. It will be interesting to gain a better understanding to what extent the beneficial impact of the stress-hyporesponsive period during early life exactly affects the cerebellum if there might be similar neonatal hyporesponsive periods regarding stress in humans as well (Table S3).
5.2.2. Impact of Sleep Deprivation on Cerebellar Maturation
Both during late gestational periods and early postnatal periods, newborn humans and rodent pups sleep almost 80% of their time, and it has been suggested that active sleep periods play a major role in the functional development of the cerebellum. For example, given that the cerebellum receives a copy of motor commands as well as subsequent signals about sensory feedback during sleep periods with muscle twitches, the cerebellum may be entrained during sleep to develop predictive coding of movements [281,282]. During such active sleep synapses may be strengthened or weakened for the sensorimotor system to develop [283]. As a consequence, without sleep related twitching the cerebellum may not develop its distinct ability to process the motor commands and sensory feedback signals within the expected time period [284,285,286,287]. Therefore, it is not surprising that Tfap2b, which is a gene that acts during early embryonic stages, controls not only sleep in mice, but also affects functioning of GABAergic neurons in the cerebellum [288,289]. In this regard too, it will be interesting to find out which sleep-control genes and how sleep restriction affects development of the cerebellum and/or that of other brain regions [290]. Considering that sleep deprivation is a major stressor of pregnant mothers, a study testing the effects of sleep deprivation is overdue, both with regard to the development of the cerebellum and that of other brain regions [291,292].
5.3. Intrauterine Growth Restriction- Impacts on Cerebellar Maturation
IUGR affects around 10% of human pregnancies and is associated with long-term motor and cognitive problems [293,294]. The cause for IUGR can be maternal factors such as undernutrition and maternal smoking but also placental and cord abnormalities, as well as fetal factors such as congenital heart disease. Obviously from the description, since the causes for IUGR are heterogeneous, there is also overlap with the consequences described for the other insults in this review [295]. IUGR leads to a heterogeneous set of fetal clinical pathologies. Recent studies suggest that certain motor deficits in patients can result from abnormal cerebellar development due to IUGR [296,297].
In humans, instances of decreased cerebellar volume, or cerebellar hypoplasia, are frequently observed in fetuses experiencing preterm birth (i.e., babies born <37 weeks of gestation) and/or IUGR (also called fetal growth restriction: FGR) [298]. Alterations to typical cerebellar growth can occur because of influences that can be relatively direct or indirect [299]. The neuropathology underlying IUGR is intricate and unique compared to preterm infants without IUGR and term infants exposed to acute hypoxia. Research, spanning human imaging, post-mortem examinations, and animal models, often paints a picture of IUGR brains having diminished volume, compromised gray and white matter structures, and cellular anomalies. Specifically, gray matter regions exhibit fewer cells with a chaotic cortical configuration, whereas white matter appears immature with signs of inflammation and astrogliosis [298]. The structural connectivity, especially along motor and cortico-striatal-thalamic tracts, has been suggested to be altered in IUGR brains, correlating with adverse neurodevelopmental outcomes in affected children [300,301]. The risks of neurodevelopmental impairments in IUGR are modulated by the severity of growth restriction, its onset timing, the presence of relative “brain sparing” and gestational age at birth.
Rodent studies have shown that bilateral uterine vessel ligation to restrict blood flow to the fetus can be used as a model for IUGR. Artery ligation in mice from E12.5 days onwards leads to cerebellar changes in myelination especially when it is combined with hyperoxia [302]. Unfortunately, no specifics about the region of the cerebellum analysed or the neuron types affected by demyelination were given. Applying artery ligation at E18 in rats leads towards the end of the first postnatal week to a 30% increase in the width of the eGL, while there is no difference in the width of the proliferative zone or the proliferating marker Ki67 in GCs. The increase in eGL following artery ligation may be partly because Bergman glia cells and their fiber density become disorganized and decreased [303,304]. Indeed, since the Bergman glia fibers normally guide the migration of the GCs from the eGL to the GL during early development, any structural disturbance in the Bergman fibers may affect GC transfer [303,304]. Additionally, the expression of genes that are necessary for a healthy migration to the GL may be affected following uterine vessel ligation, which could further worsen the deficits in cerebellar development. GC defects in turn may affect normal PC development, as suggested by guinea pig studies [305]. Maternal and postnatal malnutrition from E0-P21 of the rat increases lipoperoxidation and decreases superoxide dismutase [306], which may lead to lipid oxidative damage. Malnutrition of fetus during the last 5 embryonic days leads to reduced levels of glutamic acid decarboxylase (GAD) only at P2, examined with high-performance liquid chromatography (HPLC) of the whole cerebellum [307]. Compared to controls, HPLC also revealed an increase in the amino acids, alanine and taurine. Thus, rodent data suggest that in babies with IUGR, the cerebellum is likely to be affected, since GCs cannot sufficiently migrate from eGL to GL, which in turn may impact development and myelination of PCs. However, one must consider the limitations and caveats of the uterine vessel ligation animal model, since human placental insufficiency usually develops more gradually across the different gestational periods with the consequence that cerebellar development can be affected differentially with potential for compensatory mechanisms to be engaged.
5.4. Intrauterine Infections and the Impact on Cerebellar Maturation
Chorioamnionitis (CA), is an infection of the chorion and amnion of the mother that can lead to a fetal inflammatory response, with adverse consequences for the developing fetal brain [308]. When brain inflammation is prolonged and/or becomes severe, it can exacerbate damage through further influx of cytokines, chemokines, and other inflammatory mediators released from glial cells. In humans, there is a strong causal link between CA, preterm brain injury, and the pathogenesis of severe postnatal neurological deficits, such as cerebral palsy [309]. When measured in premature infants, there is a significant association between exposure to CA and neurodevelopmental impairments from 18 to 30 months of corrected age [310,311,312], decreased cognitive performance at 5 years [309], and autism spectrum disorder [313]. The most frequent route that causes CA development in humans is the ascending microbial invasion from the lower genital tract. In a recent study conducted by Jain and colleagues [314], moderate to severe acute histological CA was found to elevate the risk of structural brain anomalies also in the cerebellum as seen on MRI, both directly and by prompting premature birth.
In mice, one model to induce CA is ureaplasma-induced perinatal inflammation at E13.5, which induces a significant decrease in calbindin-positive neurons as well as a moderate decrease in myelin basic protein (MBP) [315]. PCs are the major cell type being calbindin positive. Another mouse model for CA makes use of inflammation-induced encephalopathy of prematurity driven by systemic administration of pro-inflammatory IL-1β. This model has been shown to interfere with the physiological roles of microglia in the cerebellum; indeed, inducing inflammatory activation with this approach results in perturbed oligodendrocyte development and myelination in whole cerebellum lysates used for Western blotting [316]. IL-1β administration during the first postnatal week, a timing equivalent to the last trimester for brain development in humans, leads to specific reductions in gray and white matter volumes of the mouse cerebellar lobules, most specifically lobules I and II, and the nucleus interpositus from the second postnatal week onwards. These volume changes, which can be detected with MRI as of the second week, are preceded by reduced proliferation of OLIG2+ cells as well as reduced levels of MBP and myelin-associated glycoprotein (MAG). Moreover, the density of IBA1+ cerebellar microglia is increased both during the first postnatal week and P45, with evidence for increased microglial proliferation during the first two weeks postnatal. CA also induces a significant enrichment of pro-inflammatory markers in microglia from cerebellum and cerebrum, with the cerebellar microglia displaying a unique type I interferon signalling dysregulation. In summary, perinatal inflammation driven by systemic IL-1β leads to cerebellar volume deficits, especially in lobules I and II but also other lobules, which likely reflect oligodendrocyte pathology downstream of microglial activation [316].
6. Vulnerable and Critical Periods in Cerebellar Development Affected by Intrauterine Insults
Lifestyle choices and environmental exposures of mothers can impact the cerebellar development of fetuses, potentially causing lifelong consequences for the structure and function of the cerebellum leading to neurological motor and cognitive disabilities [2,20]. Abundant animal research has revealed the multifaceted negative effects of a hostile intrauterine environment on cerebellar development. Human studies in this area are limited, focusing often on postnatal cerebellar manifestations (e.g., neuroimaging measurements), while disregarding the impact of prenatal anomalies on individual cells, layers, different subareas of cerebellar development, and growth. Here we provide a perspective on the potential critical periods for a healthy cerebellar development during gestation, highlighting opportunities to prevent risks for mothers and health care specialists of perinatal cerebellar brain injury considering rodent and human research.
In human and rodent studies on developmental disorders caused by insults, the cerebellum is still a relatively neglected brain region and thus the effects of insults on cerebellar development are underestimated and sometimes even controversial. It has for example been shown that stress during the second and third gestational period leads to changes in maternal care later in life as well as epigenetic variations [317], suggesting an effect on cerebellar functionality. What this explicitly means for cerebellar development still needs to be determined.
When the impact of insults on cerebellar development is analysed, the focus lies often on PCs and GCs, since they form the sole output neurons of the cerebellar cortex and most of all neurons in the brain, respectively. We now know that depending on the time point, duration, and severity of the insult, different subareas, layers, and neuron types are affected. What makes the cerebellum different from other brain areas is that the critical periods are extended over the whole gestational period until 3-4 weeks postnatally in rodents and 3 years in humans. Considering cerebellar neurogenesis, proliferation and migration, most likely GABAergic interneuron proliferation is affected slightly later during gestation compared to excitatory CN neurons but there are hardly any studies looking at the effects of an insults on all individual neuron types. This is surprising since CN neurons will be important to study since excitatory CNs develop first within the cerebellum, they are the output neurons of the cerebellum and they are the relay structure of the whole cerebellar cortex, being of major importance for cerebellar-thalamo-cortical communication [318,319,320,321,322].
When interpreting results of our review, it is important to consider three key factors. First, it must be mentioned that all information provided in this review is dependent on the study design chosen by the investigators. Therefore, it is inevitable that there are differences in cerebellar modifications, such as cell type, and timeframes when comparing the studies within and between different intrauterine and postnatal insults. This means that information provided in this review must been seen as an overview; if different cell types are not mentioned to be affected by a specific intrauterine insult, this does not mean that they are unaffected. Additionally, in the cerebellum, a single neuron type could cause a chain reaction of malfunctioning cells due to the aforementioned developmental dependencies. Second, and as previously mentioned, there are differences in the development of the cerebellum as a whole, cerebellar subareas, and the molecular composition of cerebellar cell types comparing humans and rodents [40,41,42]. The folia complexity of human cerebellum is greater with enlarged hemispheres relative to the medial cerebellar vermis [22]. In humans, foliation and growth of the cerebellum takes place during gestation, whereas in rodents the largest growth is postnatal. Besides the surface area is greater and the neuronal subtype ratios differ significantly between individual cerebellar areas [52,323]. Next to that, the developmental timeline of individual cell types is different with slight regional differences between areas (Figure 1, Figure 2 and Figure 3 [9,23,57]). Third, the fact that there are regional differences in the vulnerability of the cerebellum to intrauterine and postnatal insults shows that all studies discussed in this review should be interpreted with care and that where necessary the lobules and ideally microzones should be reinvestigated and seen as individual functional regions. In most studies, it is not obvious, whether researchers selected cerebellar areas to be analysed a priori to the data gathering. One cannot see the cerebellum as one structure when it comes to vulnerability and functional development (see e.g., [26]) even though it has a uniform appearance during adulthood.
Unlike human research [20], research in rodents permits identification of specific time points in cerebellar development that appear most critical for insults following manipulation. Brain development in rodent pups generally resembles the development of human in that the orders of their different gestational periods correspond relatively well [324], but extrapolating rodent studies to the human situation remains difficult as the absolute and relative durations of the different stages vary substantially [22,57,325]. As a result, critical periods of cerebellar development that are most vulnerable to specific agents, vary among the different mammalian species. These differences must be considered when raising awareness and providing information for expecting parents, obstetricians, and other healthcare professionals to eventually design strategies for preventing or rescuing related neurodevelopmental disorders. Nevertheless, below we separately discuss the major mechanistic principles for the three prenatal trimesters in humans and discuss separately the first two postnatal weeks in rodents, where the development of the rodent cerebellum resembles the human cerebellum during the third gestational period. After we discuss how different subareas of the cerebellum might be affected by intrauterine insults.
6.1. First Trimester (Until gw13 Human, Until E12/13 Rodents)
Even though cerebellar development of mammals starts during the first trimester of pregnancy, hardly any study in humans or rodents focuses on the effects of intrauterine insults limited to this trimester. In humans, PCs are born within this period (beginning of 7th gw, rodents between E10-E13.5) until the PC plate is formed (13th gw). Also, the eGL starts to develop (10-11th gw humans, E12.5-16.5 rodents), and large as well as GABAergic CN neurons.
Vulnerability of the cerebellum to drugs with abuse liability has been studied in humans and rodents [101,102,103,104,105,106,107]. However human studies suffer from the fact that most studies rely on postmortem data or brain scans made after birth. Furthermore, most human mothers do not use excessive drugs only for a couple of days during pregnancy, but they continue throughout pregnancy, or they are polysubstance users. In rodents, maternal alcohol exposure for only one day at E8 or E9 leads to long-term effects on the cerebellar shape and short-term decreased volumes as well as a decreased proportion of GABA receptors, having a high impact on PC neurogenesis [123,125,126,127,128,129]. Maternal alcohol exposure from the first trimester onwards effects gene expression of genes related to Wnt signalling, with Wnt being important for cerebellar maturation and development [147]. eCB and opioid receptors are known to be expressed differently throughout development, and early heavy usage of cannabinoids and opioids will affect cerebellar development negatively. eCB and opioid receptors start being expressed around E11 (gw5-6) [224,225,226,245]. The eCB system plays an important role for cerebellar development and its long-range connections with the MF and CF [224,225,226]. Thereby manipulating the eCB system through excessive usage of drugs will likely affect cerebellar development and the synaptic inputs through the MF and CF system. Hence, prenatal moderate levels of synthetic cannabinoid exposure from E3-birth leads to a potential neuroprotective effect on the cerebellum [228,231]. Opioid receptors agonists during early pregnancy affect PC and GC proliferation negatively and can even lead to an attenuation of myelination. Maternal opioid intake can mislead the endogenous timing and sequence of neurotransmitter-receptor interactions, especially in the eGL from E13 rodents [246,247,248,249,250,251,252]. Maternal nicotine usage starting in the first week and ending in the last week of pregnancy has been shown to affect PC development [326,327] but a detailed analysis on the effects of nicotine usage during the first trimester has not been performed, yet. Stress has also been shown to affect the nuclear size of PCs and GCs, as well as the dendritic structure of CN neurons, when it occurs one day during the first and one day during the second gw in rodents [231,232]. Studies on the effects of sleep deprivation, IUGR or CA on cerebellar development solely during the first trimester in humans or until E13 in rodents has not been published, which is a gap in cerebellar research.
To conclude, for most studies the effects of insults on cerebellar development cannot be isolated and assigned to the first trimester. In rodents with genetic variations that affect the development of the cerebellar anlage, the mutation often leads to severe developmental problems, e.g. [74,79]. If insults are not deadly, developmental compensations, as is often seen in rodent research, might also occur, e.g. [328].
6.2. Second Trimester (gw13-26 Human, E13-Birth Rodents)
During the second trimester of pregnancy most cerebellar neuron types migrate. GCs differentiate from eGL and migrate to the GL along Bergman glia. UBCs and GABAergic interneurons develop and start migrating.
Excessive drug usage during the second trimester has also been shown to impact cerebellar development, especially if it occurs throughout pregnancy. In rodents, alcohol administration during a certain period of the second trimester is less harmful for PCs compared to other periods [129] although alcohol administration during the second trimester combined with the first and/or third trimester leads to changes in the GC, Bergmann glia and PC development [129,130,131]. Effects have been shown to be regional [130]. Nicotine, cannabis as well as opioid usage during the first and second trimester affects the cerebellum, specifically in terms of GC and PC survival and neuroinflammation [228,231,326,327,329]. Opioid for example is known to affect the dendritic differentiation of GCs and PCs [254,255,256], supporting that throughout development endogenous opioids maintain GC and PC proliferation, differentiation, and maturation. Maternal opioid intake therefore may act through misleading the endogenous timing and sequence of endogenous neurotransmitter-receptor interactions, especially in eGL from E13 in rodents. Maternal stress during only the third week of gestation affects the morphology of the PC dendrites in rodents [269]. IUGR and CA induced at E12.5 and E13.5 in rodents (end of first / beginning second trimester in humans), respectively, both lead to adverse effects on myelination and synaptogenesis [303,304,315,316,330]. CA furthermore has been shown to lead to a decrease in calbindin-positive neurons, which are PCs in the cerebellar cortex [315]. One must keep in mind that significant effects on the early development of one neuron type will most likely also affect the migration of the other cerebellar neurons [88,331]. Thereby insults lasting longer, such as often happens with CA, IUGR, as well as opioid, nicotine, or alcohol addicted mothers, lead to adverse effects that might be rather general with yet important implications.
To summarize, insults affecting rodents’ embryos throughout the last weeks before being born, they affect mainly Bergmann glia, PC, and GC development by defects seen in the migration, morphology, myelinization, and synaptogenesis. To our current knowledge, no clear regional differences in the vulnerability of the cerebellum have been described when harmful insults occur exclusively during the second trimester. Hence, we know that for example opioid receptors are not equally distributed across the rodent and human cerebellum and thereby will lead to differential effects when opioid receptor agonists or antagonists are used.
6.3. Third Trimester (gw27 - gw40 Human)
During the third trimester, the development of the cerebellum in humans is different from the development in rodents. The cerebellum grows and foliates, neurons proliferate and migrate. Substance (ab)use, stress, IUGR as well as CA in humans affect cerebellar development but insults are often not specific for the third trimester but happen throughout pregnancy.
Recently one study in non-human primates showed that CA during the third trimester leads to PCs loss and a disrupted maturation of GCs, with the PC loss being accompanied by decreased shh signalling from PCs to GC [332,333]. CA furthermore accelerated pre-oligodendrocyte maturation into myelinating oligodendrocytes, which is not in line with rodent research but in line with increased expression of MBP in the cerebellum of CA-exposed fetuses. These findings are also consistent with reported histopathological findings in individuals with autism and suggest a potential mechanism through which perinatal inflammation together with a change in the cerebellar excitation and inhibition balance through changes in myelination contributes to neurodevelopmental disorders in human.
6.4. Two Weeks Postnatal Rodents
The development of the rodent cerebellum during the first two weeks after birth (P0-P14) resembles the cerebellum of a last trimester embryo in humans. Hence, this period of cerebellar development in rodents happens postnatally and is not reliant on maternal blood supply and the placenta anymore. Alcohol administration for only one day during the first postnatal week significantly alters cerebellar weight at P10 [101]. Throughout the first week regional differences in the effects on volumes of the GL and ML as well as PC and GC loss have been shown. Ethanol exposure furthermore affects the CF to PC connections and microglia activation. Mainly lobules I-IV and IX-X are affected by ethanol [101,133,135,136,137,139,334], which corresponds with human studies showing a decrease in volumes of lobules I-V of the vermis [335]. Lobule VII is less vulnerable during the beginning of the first postnatal week compared to the end. After the first postnatal week the window of vulnerability for ethanol seems to close [132,134,139,140]. The CREB-gene is likely to play are role in protecting the cerebellum for ethanol-induced PC loss during this timeperiod [133,141]. Nicotine and opioid usage during these first two postnatal weeks affect both the GCs and PCs [135,183,253,254,255,256]. It has been shown for methadone that it negatively influences myelination and that it leads to an increase in apoptosis of mature and myelinating oligodendrocytes at P7 in rat pups [254,255,256]. The eCB and CB1R is prominently expressed in long-range axons of the pontocerebellum [224,225,226] during the first postnatal weeks and since eCB has been suggested to play a role in the fasciculation of pontocerebellar axons [224], usage of cannabis during that period or earlier can lead to detrimental effects on the timing and spatial distribution of postnatal MF spreading [336,337]. Since eCB also affect cerebellar PC plasticity significantly [207], a change in the timing and spatial distribution of eCB can thereby influence cerebellar development negatively. Also stress leads to time- and doses sensitive changes: mild stressors during the first and second postnatal week, also known as the ‘stress-hyporesponsive period’, induce GC proliferation, whereas more severe stress is related to degeneration in the GL [274,275,280,338]. In rodents, PCs seem less sensitive to stress during the first postnatal weeks. Regarding alcohol, the vulnerability seems to be more time-specific: where administration during the first week after birth leads to both PC and GC loss, administration during the second week rarely affects PCs and GCs.
To conclude, in rodents the first weeks postnatally are very vulnerable and lead to severe effects, however dependent on the insults there are certain critical periods where the cerebellum is more or less vulnerable. There are time periods in the rodents’ life where the cerebellum is less vulnerable to both stress and alcohol. Whether there is also such a period in human cerebellar development is unknown. What has been shown is that for example language and working memory impairments vary in the degree according to the duration and amount of alcohol exposure but the relation with cerebellar development is unstudied.
6.5. Regional Differences in How Intrauterine Insults Effect Cerebellar Development
A neglected area of research is to differentiate the effects of intrauterine insults on the highly heterogeneous individual subareas of the cerebellum (Figure 1). It has been shown that the anterior cerebellum, which connects most with the sensorimotor system matures before the posterior cerebellum and thus early onset insults may lead to more severe or more general motor deficits compared to late onset insults. Due to receptor and eCB location CB knock-out mice show a reduced size specifically in the anterior vermis of the cerebellum [224], also suggesting motor deficits. CB1R KO knock-out mice show impairments in fine motor forepaw coordination with no defects in gross motor coordination tasks, which is consistent with mild cerebellar ataxia [224]. Posterior cerebellar lesions may lead to more severe cognitive outcomes due to the connectivity of the posterior cerebellum with association cortices and because posterior cerebellar development follows that of the anterior cerebellum. Midline posterior vermal damage is more associated with behavioural dysregulations such as autism-like diseases. On another note, Wang and colleagues also mentioned that developmental diaschisis may evolve due to the deleterious effects of cerebellar lesions on the regions of the cerebral cortex to which the cerebellum projects [19,339,340,341,342,343,344]. In line with that cerebellar output controls the pattern of ongoing activity in subcortical projection neurons and aberrant development of those long-range connections, due to for example prenatal cannabis usage, can also lead to fine forepaw coordination impairments [214]. Whereas the gross anatomy of the cerebellum looks similar across species and within the cerebellum of one species, the microscopic anatomy does not. Within the cerebellum there is not only the classical functional subdivision into an anterior, posterior and flocculonodular lobe but also into individual lobules, the vermis and the hemisphere, microzones, microcomplexes and micromodules, all of which containing similar types of neurons that express distinct molecular markers with characteristic physiological properties [10,43,44,90]. Data suggest that distinct lobules and neuronal subtypes are affected differently depending on the insult happening, which may at least partially be explained by a difference in the developmental timeline between areas [9,22,320]. The development of these molecular TOC profiles is tightly regulated and the regional differences in genetic and thus molecular identity affect the intrinsic excitability as well the synaptic plasticity and thereby eventually also the functionality of lobes, lobules, areas, microzones, microcomplexes and micromodules as well as individual neuronal subtypes species specific. Ethanol leads to time-dependent and cerebellar lobule specific effects on PC development. As an example, whereas the literature is inconsistent about the vulnerability of lobule VI during the first postnatal week, PCs in lobule VII are only affected when exposed to ethanol at the end of this first week [134]. Moreover, lobules I-V and IX were more affected when exposed to ethanol at P4-5 compared to P6-7, suggesting the ethanol-vulnerability to decrease over time and being cerebellar region-specific. Together these data suggest regional differences and time-dependencies in the development of ethanol responsiveness in the cerebellum [10,24,259].
What also matters when it comes to regional differences is whether the insult enters the cerebellum via e.g. the blood vessels (Figure 1), the blood-brain barrier, or via breastmilk. Dependent on whether the insult effects the general cerebellum or only locally some areas close to e.g. a blood vessel, this will entail regional differences in vulnerability (Figure 1). As an example, IUGR is not only linked to oxidative stress and neuroinflammation in the fetus’ later in life, furthermore, it is linked to placental insufficiency which in turn can induce chronic fetal hypoxia [297]. Due to the distinct vesselation, hypoxia may affect cerebellar regions differently and lead to region-specific cerebellar oxidative stress, cytokine imbalance, redox dysregulation [345], region-specific deficits in neuronal organization and/or axonal injury [297]. Oxidative stress and inflammation in the fetus are associated with impaired neurodevelopment and neurodevelopmental diseases such as autism spectrum [346]. Thus, next to the timing, and duration of the specific intrauterine insult to enter the cerebellum, also the way of administration is of importance when providing case-specific information for expecting parents, obstetricians, and other healthcare professionals to eventually design strategies for preventing or rescuing related neurodevelopmental disorders.
7. Conclusion
In summary, the cerebellum is an early-developing brain region that undergoes rapid growth during gestation and continues to mature into late postnatal life in both humans and rodents. Despite its important role, it is often neglected in developmental research on intrauterine insults. When studied, cerebellar development is frequently treated as homogeneous, overlooking its complex and region-specific maturation. There is no single critical period applicable to all intrauterine insults; instead, each insult must be evaluated individually. For example, the development of opioid receptors creates an early gestational vulnerability in cerebellar development, whereas ethanol exposure has a more severe impact during the third trimester or postnatally. Therefore, the critical periods of vulnerability in both humans and rodents depend on the timing, duration, location, and severity of each specific insult. Recognizing these nuances is essential for understanding and mitigating the effects of intrauterine challenges on cerebellar development.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Overview table summarizing literature on timepoints, exposure methods and effects of prenatal and early postnatal alcohol exposure on cerebellar outcome measures in rodents; Table S2: Overview table summarizing literature on timepoints, exposure methods and effects of prenatal and early postnatal nicotine exposure on cerebellar outcome measures in rodents.; Table S3: Overview table summarizing literature on timepoints, exposure methods and effects of prenatal and early postnatal stress exposure on cerebellar outcome measures in rodents.
Author Contributions
Conceptualization, J.A.W.W., J.D., C.B.C.; Writing – Original Draft Preparation, J.A.W.W., J.D., B.E.C.A.W., C.B.C.; Writing – Review & Editing, J.A.W.W., J.D., B.E.C.A.W, C.I.D.Z., C.B.C.; Supervision, J.D., C.B.C.; Funding acquisition, J.D., C.I.D.Z., C.B.C.
Funding
This study was enabled by funding from the Netherlands Organization for Scientific Research (NWO STEM - VBT 2021 19224 and NWO 863.14.005, C.B.C; NWO-ALW 824.02.001, C.I.D.Z.), the Dutch Organization for Medical Sciences (ZonMW 91120067, C.I.D.Z.), Medical Neuro-Delta (MD 01092019-31082023, C.I.D.Z.), INTENSE LSH-NWO (TTW/00798883, C.I.D.Z.), ERC-adv (GA-294775, C.I.D.Z.) and ERC-POC (nrs. 737619 and 768914, C.I.D.Z.), the NIN Vriendenfonds for Albinism (C.I.D.Z.), the Dutch NWO Gravitation Program, and the Dutch Brain Interface Initiative (DBI2, C.I.D.Z.).
Institutional Review Board Statement
Not applicable.
Acknowledgments
We would like to thank F.A.H. Ooms, the Cerebellar Coordination and Cognition group, the UMC Utrecht Brain Center, Division Woman and Baby, Department of Neonatology for their contribution to useful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dietrich, K.N.; Eskenazi, B.; Schantz, S.; Yolton, K.; Rauh, V.A.; Johnson, C.B.; Alkon, A.; Canfield, R.L.; Pessah, I.N.; Berman, R.F. Principles and Practices of Neurodevelopmental Assessment in Children: Lessons Learned from the Centers for Children’s Environmental Health and Disease Prevention Research. Environ Health Perspect 2005, 113, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Koning, I. V; Dudink, J.; Groenenberg, I.A.L.; Willemsen, S.P.; Reiss, I.K.M.; Steegers-Theunissen, R.P.M. Prenatal Cerebellar Growth Trajectories and the Impact of Periconceptional Maternal and Fetal Factors. Hum Reprod 2017, 32, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Mwaniki, M.K.; Atieno, M.; Lawn, J.E.; Newton, C.R. Long-Term Neurodevelopmental Outcomes after Intrauterine and Neonatal Insults: A Systematic Review. The Lancet 2012, 379, 445–452. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.J.; Meaney, M.J. Fetal Origins of Mental Health: The Developmental Origins of Health and Disease Hypothesis. American Journal of Psychiatry 2017, 174, 319–328. [Google Scholar] [CrossRef]
- Carletti, B.; Rossi, F. Neurogenesis in the Cerebellum. Neuroscientist 2008, 14, 91–100. [Google Scholar] [CrossRef]
- Hausmann, B.; Mangold, U.; Sievers, J.; Berry, M. Derivation of Cerebellar Golgi Neurons from the External Granular Layer: Evidence from Explantation of External Granule Cells in Vivo. J Comp Neurol 1985, 232, 511–522. [Google Scholar] [CrossRef]
- Haldipur, P.; Bharti, U.; Alberti, C.; Sarkar, C.; Gulati, G.; Iyengar, S.; Gressens, P.; Mani, S. Preterm Delivery Disrupts the Developmental Program of the Cerebellum. PLoS One 2011, 6, e23449. [Google Scholar] [CrossRef]
- van Essen, M.J.; Nayler, S.; Becker, E.B.E.; Jacob, J. Deconstructing Cerebellar Development Cell by Cell. PLoS Genet 2020, 16, e1008630. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Osório, C.; White, J.J.; van Zoomeren, S.; van der Stok, H.; Xiong, B.; Nettersheim, I.H.; Mak, W.A.; Runge, M.; Fiocchi, F.R.; et al. Differential Spatiotemporal Development of Purkinje Cell Populations and Cerebellum-Dependent Sensorimotor Behaviors. Elife 2021, 10. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Gornati, S. V.; Canto, C.B.; Libster, A.M.; Schonewille, M.; De Zeeuw, C.I.; Hoebeek, F.E. Activity of Cerebellar Nuclei Neurons Correlates with ZebrinII Identity of Their Purkinje Cell Afferents. Cells 2021, 10, 2686. [Google Scholar] [CrossRef]
- Badura, A.; Verpeut, J.L.; Metzger, J.W.; Pereira, T.D.; Pisano, T.J.; Deverett, B.; Bakshinskaya, D.E.; Wang, S.S.-H. Normal Cognitive and Social Development Require Posterior Cerebellar Activity. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Bruchhage, M.M.K.; Bucci, M.-P.; Becker, E.B.E. Cerebellar Involvement in Autism and ADHD. Handb Clin Neurol 2018, 155, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Sathyanesan, A.; Zhou, J.; Scafidi, J.; Heck, D.H.; Sillitoe, R. V; Gallo, V. Emerging Connections between Cerebellar Development, Behaviour and Complex Brain Disorders. Nat Rev Neurosci 2019, 20, 298–313. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.E.; Rey Hipolito, A.G.; Kim, L.H.; Kizek, D.J.; Perez, R.M.; Lin, T.; Sillitoe, R. V. Glutamatergic Cerebellar Neurons Differentially Contribute to the Acquisition of Motor and Social Behaviors. Nat Commun 2023, 14, 2771. [Google Scholar] [CrossRef]
- Manto, M.; Bower, J.M.; Conforto, A.B.; Delgado-García, J.M.; da Guarda, S.N.F.; Gerwig, M.; Habas, C.; Hagura, N.; Ivry, R.B.; Mariën, P.; et al. Consensus Paper: Roles of the Cerebellum in Motor Control—The Diversity of Ideas on Cerebellar Involvement in Movement. The Cerebellum 2012, 11, 457–487. [Google Scholar] [CrossRef]
- Schmahmann, J.D. Dysmetria of Thought: Clinical Consequences of Cerebellar Dysfunction on Cognition and Affect. Trends Cogn Sci 1998, 2, 362–371. [Google Scholar] [CrossRef]
- Andreasen, N.C.; Paradiso, S.; O’Leary, D.S. “Cognitive Dysmetria” as an Integrative Theory of Schizophrenia: A Dysfunction in Cortical-Subcortical-Cerebellar Circuitry? Schizophr Bull 1998, 24, 203–218. [Google Scholar] [CrossRef]
- Andreasen, N.C.; Pierson, R. The Role of the Cerebellum in Schizophrenia. Biol Psychiatry 2008, 64, 81–88. [Google Scholar] [CrossRef]
- Wang, S.S.-H.; Kloth, A.D.; Badura, A. The Cerebellum, Sensitive Periods, and Autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef]
- Koning, I. V; Tielemans, M.J.; Hoebeek, F.E.; Ecury-Goossen, G.M.; Reiss, I.K.M.; Steegers-Theunissen, R.P.M.; Dudink, J. Impacts on Prenatal Development of the Human Cerebellum: A Systematic Review. J Matern Fetal Neonatal Med 2017, 30, 2461–2468. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y. Signaling Pathways in Cerebellar Granule Cells Development. Am J Stem Cells 2019, 8, 1–6. [Google Scholar] [PubMed]
- Haldipur, P.; Millen, K.J.; Aldinger, K.A. Human Cerebellar Development and Transcriptomics: Implications for Neurodevelopmental Disorders. Annu Rev Neurosci 2022, 45, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Leto, K.; Arancillo, M.; Becker, E.B.E.; Buffo, A.; Chiang, C.; Ding, B.; Dobyns, W.B.; Dusart, I.; Haldipur, P.; Hatten, M.E.; et al. Consensus Paper: Cerebellar Development. Cerebellum 2015, 1–40. [Google Scholar] [CrossRef]
- Apps, R.; Hawkes, R. Cerebellar Cortical Organization: A One-Map Hypothesis. Nat Rev Neurosci 2009, 10, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Dudink, J.; Faneyte, S.J.; Hoebeek, F.E. Causes and Consequences of Structural Aberrations in Cerebellar Development. In Factors Affecting Neurodevelopment; Elsevier, 2021; pp. 371–382.
- De Zeeuw, C.I. Bidirectional Learning in Upbound and Downbound Microzones of the Cerebellum. Nat Rev Neurosci 2021, 22, 92–110. [Google Scholar] [CrossRef]
- Grodd, W.; Hülsmann, E.; Lotze, M.; Wildgruber, D.; Erb, M. Sensorimotor Mapping of the Human Cerebellum: FMRI Evidence of Somatotopic Organization. Hum Brain Mapp 2001, 13, 55–73. [Google Scholar] [CrossRef]
- Kelly, R.M.; Strick, P.L. Cerebellar Loops with Motor Cortex and Prefrontal Cortex of a Nonhuman Primate. J Neurosci 2003, 23, 8432–8444. [Google Scholar] [CrossRef]
- Schmahmann, J.D. The Cerebellum and Cognition. Neurosci Lett 2019, 688, 62–75. [Google Scholar] [CrossRef]
- Kansal, K.; Yang, Z.; Fishman, A.M.; Sair, H.I.; Ying, S.H.; Jedynak, B.M.; Prince, J.L.; Onyike, C.U. Structural Cerebellar Correlates of Cognitive and Motor Dysfunctions in Cerebellar Degeneration. Brain 2017, 140, 707–720. [Google Scholar] [CrossRef]
- Brochu, G.; Maler, L.; Hawkes, R. Zebrin II: A Polypeptide Antigen Expressed Selectively by Purkinje Cells Reveals Compartments in Rat and Fish Cerebellum. J Comp Neurol 1990, 291, 538–552. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Krueger-Naug, A.M.; Currie, R.W.; Hawkes, R. Constitutive Expression of the 25-KDa Heat Shock Protein Hsp25 Reveals Novel Parasagittal Bands of Purkinje Cells in the Adult Mouse Cerebellar Cortex. J Comp Neurol 2000, 416, 383–397. [Google Scholar] [CrossRef]
- Hashizume, M.; Miyazaki, T.; Sakimura, K.; Watanabe, M.; Kitamura, K.; Kano, M. Disruption of Cerebellar Microzonal Organization in GluD2 (GluRδ2) Knockout Mouse. Front Neural Circuits 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Marzban, H.; Watanabe, M.; Hawkes, R. Complementary Stripes of Phospholipase Cbeta3 and Cbeta4 Expression by Purkinje Cell Subsets in the Mouse Cerebellum. J Comp Neurol 2006, 496, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.; Hawkes, R. Zones and Stripes. In Essentials of Cerebellum and Cerebellar Disorders A Primer For Graduate Students Second Edition; 2023; pp. 99–106.
- Ament, S.A.; Cortes-Gutierrez, M.; Herb, B.R.; Mocci, E.; Colantuoni, C.; McCarthy, M.M. A Single-Cell Genomic Atlas for Maturation of the Human Cerebellum during Early Childhood. Sci Transl Med 2023. [Google Scholar] [CrossRef]
- Coarelli, G.; Wirth, T.; Tranchant, C.; Koenig, M.; Durr, A.; Anheim, M. The Inherited Cerebellar Ataxias: An Update. J Neurol 2023, 270, 208–222. [Google Scholar] [CrossRef]
- Selimi, F.; Lohof, A.M.; Heitz, S.; Lalouette, A.; Jarvis, C.I.; Bailly, Y.; Mariani, J. Lurcher GRID2-Induced Death and Depolarization Can Be Dissociated in Cerebellar Purkinje Cells. Neuron 2003, 37, 813–819. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Yoshioka, Y.; Suzuki, K.; Miyazaki, T.; Koura, M.; Saigoh, K.; Kajimura, N.; Monobe, Y.; Kusunoki, S.; Matsuda, J.; et al. A New Mouse Allele of Glutamate Receptor Delta 2 with Cerebellar Atrophy and Progressive Ataxia. PLoS One 2014, 9, e107867. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Thomson, Z.; Phelps, I.G.; Haldipur, P.; Deng, M.; Timms, A.E.; Hirano, M.; Santpere, G.; Roco, C.; Rosenberg, A.B.; et al. Spatial and Cell Type Transcriptional Landscape of Human Cerebellar Development. Nat Neurosci 2021, 24, 1163–1175. [Google Scholar] [CrossRef]
- Zhong, S.; Wang, M.; Huang, L.; Chen, Y.; Ge, Y.; Zhang, J.; Shi, Y.; Dong, H.; Zhou, X.; Wang, B.; et al. Single-Cell Epigenomics and Spatiotemporal Transcriptomics Reveal Human Cerebellar Development. Nat Commun 2023, 14, 7613. [Google Scholar] [CrossRef]
- Hao, S.; Zhu, X.; Huang, Z.; Yang, Q.; Liu, H.; Wu, Y.; Zhan, Y.; Dong, Y.; Li, C.; Wang, H.; et al. Cross-Species Single-Cell Spatial Transcriptomic Atlases of the Cerebellar Cortex. Science (1979) 2024, 385. [Google Scholar] [CrossRef]
- Voerman, S.; Urbanus, B.H.A.; Schonewille, M.; White, J.J.; De Zeeuw, C.I. Postsynaptic Plasticity of Purkinje Cells in Mice Is Determined by Molecular Identity. Commun Biol 2022, 5. [Google Scholar] [CrossRef] [PubMed]
- Voerman, S.; Broersen, R.; Swagemakers, S.M.A.; De Zeeuw, C.I.; van der Spek, P.J. Plasticity Mechanisms of Genetically Distinct Purkinje Cells. BioEssays 2024, 46. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, Y.; Broussard, G.J.; Kislin, M.; Yuede, C.M.; Zhang, S.; Dietmann, S.; Gabel, H.; Zhao, G.; Wang, S.S.-H.; et al. Transcriptomic Mapping Uncovers Purkinje Neuron Plasticity Driving Learning. Nature 2022, 605, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science (1979) 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Kozareva, V.; Martin, C.; Osorno, T.; Rudolph, S.; Guo, C.; Vanderburg, C.; Nadaf, N.; Regev, A.; Regehr, W.G.; Macosko, E. A Transcriptomic Atlas of Mouse Cerebellar Cortex Comprehensively Defines Cell Types. Nature 2021, 598, 214–219. [Google Scholar] [CrossRef]
- Zhou, H.; Lin, Z.; Voges, K.; Ju, C.; Gao, Z.; Bosman, L.W.; Ruigrok, T.J.; Hoebeek, F.E.; De Zeeuw, C.I.; Schonewille, M. Cerebellar Modules Operate at Different Frequencies. Elife 2014, 3, e02536. [Google Scholar] [CrossRef]
- Lackey, E.P.; Moreira, L.; Norton, A.; Hemelt, M.E.; Osorno, T.; Nguyen, T.M.; Macosko, E.Z.; Lee, W.-C.A.; Hull, C.A.; Regehr, W.G. Specialized Connectivity of Molecular Layer Interneuron Subtypes Leads to Disinhibition and Synchronous Inhibition of Cerebellar Purkinje Cells. Neuron 2024, 112, 2333–2348.e6. [Google Scholar] [CrossRef]
- Wang, X.; Novello, M.; Gao, Z.; Ruigrok, T.J.H.; De Zeeuw, C.I. Input and Output Organization of the Mesodiencephalic Junction for Cerebro-cerebellar Communication. J Neurosci Res 2022, 100, 620–637. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Hoebeek, F.E.; Bosman, L.W.J.; Schonewille, M.; Witter, L.; Koekkoek, S.K. Spatiotemporal Firing Patterns in the Cerebellum. Nat Rev Neurosci 2011, 12, 327–344. [Google Scholar] [CrossRef]
- Lange, W. Cell Number and Cell Density in the Cerebellar Cortex of Man and Some Other Mammals. Cell Tissue Res 1975, 157. [Google Scholar] [CrossRef]
- Jukic, A.M.; Baird, D.D.; Weinberg, C.R.; McConnaughey, D.R.; Wilcox, A.J. Length of Human Pregnancy and Contributors to Its Natural Variation. Hum Reprod 2013, 28, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Mukherjee, S.; Wilburn, A.N.; Cates, C.; Lewkowich, I.P.; Deshmukh, H.; Zacharias, W.J.; Chougnet, C.A. Pulmonary Consequences of Prenatal Inflammatory Exposures: Clinical Perspective and Review of Basic Immunological Mechanisms. Front Immunol 2020, 11, 1285. [Google Scholar] [CrossRef]
- Cappelletti, M.; Presicce, P.; Kallapur, S.G. Immunobiology of Acute Chorioamnionitis. Front Immunol 2020, 11, 649. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain Development in Rodents and Humans: Identifying Benchmarks of Maturation and Vulnerability to Injury across Species. Prog Neurobiol 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Haldipur, P.; Aldinger, K.A.; Bernardo, S.; Deng, M.; Timms, A.E.; Overman, L.M.; Winter, C.; Lisgo, S.N.; Razavi, F.; Silvestri, E.; et al. Spatiotemporal Expansion of Primary Progenitor Zones in the Developing Human Cerebellum. Science 2019, 366, 454–460. [Google Scholar] [CrossRef]
- Li, J.Y.; Joyner, A.L. Otx2 and Gbx2 Are Required for Refinement and Not Induction of Mid-Hindbrain Gene Expression. Development 2001, 128, 4979–4991. [Google Scholar] [CrossRef]
- Beckinghausen, J.; Sillitoe, R. V. Insights into Cerebellar Development and Connectivity. Neurosci Lett 2019, 688, 2–13. [Google Scholar] [CrossRef]
- Wingate, R.J.T. The Rhombic Lip and Early Cerebellar Development. Curr Opin Neurobiol 2001, 11, 82–88. [Google Scholar] [CrossRef]
- Millet, S.; Campbell, K.; Epstein, D.J.; Losos, K.; Harris, E.; Joyner, A.L. A Role for Gbx2 in Repression of Otx2 and Positioning the Mid/Hindbrain Organizer. Nature 1999, 401, 161–164. [Google Scholar] [CrossRef]
- Broccoli, V.; Boncinelli, E.; Wurst, W. The Caudal Limit of Otx2 Expression Positions the Isthmic Organizer. Nature 1999, 401, 164–168. [Google Scholar] [CrossRef]
- Chizhikov, V. V.; Lindgren, A.G.; Mishima, Y.; Roberts, R.W.; Aldinger, K.A.; Miesegaes, G.R.; Currle, D.S.; Monuki, E.S.; Millen, K.J. Lmx1a Regulates Fates and Location of Cells Originating from the Cerebellar Rhombic Lip and Telencephalic Cortical Hem. Proceedings of the National Academy of Sciences 2010, 107, 10725–10730. [Google Scholar] [CrossRef] [PubMed]
- Corrales, J.D.; Rocco, G.L.; Blaess, S.; Guo, Q.; Joyner, A.L. Spatial Pattern of Sonic Hedgehog Signaling through Gli Genes during Cerebellum Development. Development 2004, 131, 5581–5590. [Google Scholar] [CrossRef] [PubMed]
- Belzunce, I.; Belmonte-Mateos, C.; Pujades, C. The Interplay of Atoh1 Genes in the Lower Rhombic Lip during Hindbrain Morphogenesis. PLoS One 2020, 15, e0228225. [Google Scholar] [CrossRef] [PubMed]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 Induces Cerebral Small-Vessel Disease. J Clin Invest 2014, 124, 4877–4881. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Lehmann, O.J.; Hudgins, L.; Chizhikov, V. V; Bassuk, A.G.; Ades, L.C.; Krantz, I.D.; Dobyns, W.B.; Millen, K.J. FOXC1 Is Required for Normal Cerebellar Development and Is a Major Contributor to Chromosome 6p25.3 Dandy-Walker Malformation. Nat Genet 2009, 41, 1037–1042. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Timms, A.E.; Thomson, Z.; Mirzaa, G.M.; Bennett, J.T.; Rosenberg, A.B.; Roco, C.M.; Hirano, M.; Abidi, F.; Haldipur, P.; et al. Redefining the Etiologic Landscape of Cerebellar Malformations. Am J Hum Genet 2019, 105, 606–615. [Google Scholar] [CrossRef]
- Thomas, K.R.; Musci, T.S.; Neumann, P.E.; Capecchi, M.R. Swaying Is a Mutant Allele of the Proto-Oncogene Wnt-1. Cell 1991, 67, 969–976. [Google Scholar] [CrossRef]
- Lowenstein, E.D.; Cui, K.; Hernandez-Miranda, L.R. Regulation of Early Cerebellar Development. FEBS J 2023, 290, 2786–2804. [Google Scholar] [CrossRef]
- Martinez, S.; Andreu, A.; Mecklenburg, N.; Echevarria, D. Cellular and Molecular Basis of Cerebellar Development. Front Neuroanat 2013, 7. [Google Scholar] [CrossRef]
- Akazawa, C.; Ishibashi, M.; Shimizu, C.; Nakanishi, S.; Kageyama, R. A Mammalian Helix-Loop-Helix Factor Structurally Related to the Product of Drosophila Proneural Gene Atonal Is a Positive Transcriptional Regulator Expressed in the Developing Nervous System. Journal of Biological Chemistry 1995, 270, 8730–8738. [Google Scholar] [CrossRef]
- Hoshino, M.; Nakamura, S.; Mori, K.; Kawauchi, T.; Terao, M.; Nishimura, Y. V.; Fukuda, A.; Fuse, T.; Matsuo, N.; Sone, M.; et al. Ptf1a, a BHLH Transcriptional Gene, Defines GABAergic Neuronal Fates in Cerebellum. Neuron 2005, 47, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Machold, R.; Fishell, G. Math1 Is Expressed in Temporally Discrete Pools of Cerebellar Rhombic-Lip Neural Progenitors. Neuron 2005, 48, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sekerková, G.; Ilijic, E.; Mugnaini, E. Time of Origin of Unipolar Brush Cells in the Rat Cerebellum as Observed by Prenatal Bromodeoxyuridine Labeling. Neuroscience 2004, 127, 845–858. [Google Scholar] [CrossRef]
- Chizhikov, V.; Millen, K.J. Development and Malformations of the Cerebellum in Mice. Mol Genet Metab 2003, 80, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Consalez, G.G.; Goldowitz, D.; Casoni, F.; Hawkes, R. Origins, Development, and Compartmentation of the Granule Cells of the Cerebellum. Front Neural Circuits 2021, 14. [Google Scholar] [CrossRef]
- Komuro, H.; Yacubova, E.; Yacubova, E.; Rakic, P. Mode and Tempo of Tangential Cell Migration in the Cerebellar External Granular Layer. The Journal of Neuroscience 2001, 21, 527–540. [Google Scholar] [CrossRef]
- Helms, A.W.; Gowan, K.; Abney, A.; Savage, T.; Johnson, J.E. Overexpression of MATH1 Disrupts the Coordination of Neural Differentiation in Cerebellum Development. Molecular and Cellular Neuroscience 2001, 17, 671–682. [Google Scholar] [CrossRef]
- Wizeman, J.W.; Guo, Q.; Wilion, E.M.; Li, J.Y. Specification of Diverse Cell Types during Early Neurogenesis of the Mouse Cerebellum. Elife 2019, 8. [Google Scholar] [CrossRef]
- Zordan, P.; Croci, L.; Hawkes, R.; Consalez, G.G. Comparative Analysis of Proneural Gene Expression in the Embryonic Cerebellum. Developmental Dynamics 2008, 237, 1726–1735. [Google Scholar] [CrossRef]
- Miterko, L.N.; Sillitoe, R. V.; Hawkes, R. Zones and Stripes: Development of Cerebellar Topography. In Handbook of the Cerebellum and Cerebellar Disorders: Second Edition: Volume 3; 2021; Vol. 3.
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and Yotari Disrupt the Disabled Gene and Produce a Reeler -like Phenotype in Mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef]
- Howell, B.W.; Hawkes, R.; Soriano, P.; Cooper, J.A. Neuronal Position in the Developing Brain Is Regulated by Mouse Disabled-1. Nature 1997, 389, 733–737. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; G. Miao, G.; Chen, S.-C.; Scares, H.D.; Morgan, J.I.; Curran, T. A Protein Related to Extracellular Matrix Proteins Deleted in the Mouse Mutant Reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-H.; Marzban, H.; Croci, L.; Consalez, G.G.; Hawkes, R. Purkinje Cell Subtype Specification in the Cerebellar Cortex: Early B-Cell Factor 2 Acts to Repress the Zebrin II-Positive Purkinje Cell Phenotype. Neuroscience 2008, 153, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tran-Anh, K.; Hirata, T.; Sugihara, I. Striped Distribution Pattern of Purkinje Cells of Different Birthdates in the Mouse Cerebellar Cortex Studied with the Neurog2-CreER Transgenic Line. Neuroscience 2021, 462, 122–140. [Google Scholar] [CrossRef]
- van der Heijden, M.E.; Lackey, E.P.; Perez, R.; Ișleyen, F.S.; Brown, A.M.; Donofrio, S.G.; Lin, T.; Zoghbi, H.Y.; Sillitoe, R. V Maturation of Purkinje Cell Firing Properties Relies on Neurogenesis of Excitatory Neurons. Elife 2021, 10. [Google Scholar] [CrossRef]
- McKay, B.E.; Turner, R.W. Physiological and Morphological Development of the Rat Cerebellar Purkinje Cell. J Physiol 2005, 567, 829–850. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Schonewille, M. Lobule-Related Action Potential Shape- and History-Dependent Current Integration in Purkinje Cells of Adult and Developing Mice. Cells 2023, 12. [Google Scholar] [CrossRef]
- Kapfhammer, J.P. Cellular and Molecular Control of Dendritic Growth and Development of Cerebellar Purkinje Cells. Prog Histochem Cytochem 2004, 39, 131–182. [Google Scholar] [CrossRef]
- Busch, S.E.; Hansel, C. Climbing Fiber Multi-Innervation of Mouse Purkinje Dendrites with Arborization Common to Human. Science (1979) 2023, 381, 420–427. [Google Scholar] [CrossRef]
- Sultan, F.; Czubayko, U.; Thier, P. Morphological Classification of the Rat Lateral Cerebellar Nuclear Neurons by Principal Component Analysis. Journal of Comparative Neurology 2003, 455, 139–155. [Google Scholar] [CrossRef]
- Leto, K.; Carletti, B.; Williams, I.M.; Magrassi, L.; Rossi, F. Different Types of Cerebellar GABAergic Interneurons Originate from a Common Pool of Multipotent Progenitor Cells. The Journal of Neuroscience 2006, 26, 11682–11694. [Google Scholar] [CrossRef] [PubMed]
- Leto, K.; Bartolini, A.; Yanagawa, Y.; Obata, K.; Magrassi, L.; Schilling, K.; Rossi, F. Laminar Fate and Phenotype Specification of Cerebellar GABAergic Interneurons. The Journal of Neuroscience 2009, 29, 7079–7091. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, P.; Parras, C.; Guillemot, F.; Rossi, F.; Wassef, M. Origins and Control of the Differentiation of Inhibitory Interneurons and Glia in the Cerebellum. Dev Biol 2009, 328, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje Cell Death in the Cerebellum. Prog Neurobiol 2003, 70, 473–507. [Google Scholar] [CrossRef]
- Chedotal, A.; Sotelo, C. Early Development of Olivocerebellar Projections in the Fetal Rat Using CGRP Immunocytochemistry. European Journal of Neuroscience 1992, 4, 1159–1179. [Google Scholar] [CrossRef]
- Sugihara, I. Microzonal Projection and Climbing Fiber Remodeling in Single Olivocerebellar Axons of Newborn Rats at Postnatal Days 4–7. Journal of Comparative Neurology 2005, 487, 93–106. [Google Scholar] [CrossRef]
- Rodriguez, C.I.; Dymecki, S.M. Origin of the Precerebellar System. Neuron 2000, 27, 475–486. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Marcussen, L.; West, J.R. A Single Day of Alcohol Exposure During the Brain Growth Spurt Induces Brain Weight Restriction and Cerebellar Purkinje Cell Loss. Alcohol 1989, 7, 107–114. [Google Scholar] [CrossRef]
- Sim, M.E.; Lyoo, I.K.; Streeter, C.C.; Covell, J.; Sarid-Segal, O.; Ciraulo, D.A.; Kim, M.J.; Kaufman, M.J.; Yurgelun-Todd, D.A.; Renshaw, P.F. Cerebellar Gray Matter Volume Correlates with Duration of Cocaine Use in Cocaine-Dependent Subjects. Neuropsychopharmacology 2007, 32, 2229–2237. [Google Scholar] [CrossRef]
- Manto, M.; Perrotta, G. Toxic-Induced Cerebellar Syndrome: From the Fetal Period to the Elderly. In; 2018; pp. 333–352.
- Dow-Edwards, D.L.; Benveniste, H.; Behnke, M.; Bandstra, E.S.; Singer, L.T.; Hurd, Y.L.; Stanford, L.R. Neuroimaging of Prenatal Drug Exposure. Neurotoxicol Teratol 2006, 28, 386–402. [Google Scholar] [CrossRef]
- Falk, L.; Nordberg, A.; Seiger, Å.; Kjældgaard, A.; Hellström-Lindahl, E. Smoking during Early Pregnancy Affects the Expression Pattern of Both Nicotinic and Muscarinic Acetylcholine Receptors in Human First Trimester Brainstem and Cerebellum. Neuroscience 2005, 132, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Lavezzi, A.M.; Corna, M.F.; Repetti, M.L.; Matturri, L. Cerebellar Purkinje Cell Vulnerability to Prenatal Nicotine Exposure in Sudden Unexplained Perinatal Death. Folia Neuropathol 2013, 4, 290–301. [Google Scholar] [CrossRef]
- Hauser, K.F.; Khurdayan, V.K.; Goody, R.J.; Nath, A.; Saria, A.; Pauly, J.R. Selective Vulnerability of Cerebellar Granule Neuroblasts and Their Progeny to Drugs with Abuse Liability. The Cerebellum 2003, 2, 184–195. [Google Scholar] [CrossRef]
- Bayer, S.A.; Altman, J.; Russo, R.J.; Zhang, X. Timetables of Neurogenesis in the Human Brain Based on Experimentally Determined Patterns in the Rat. Neurotoxicology 1993, 14. [Google Scholar]
- Riley, E.P.; McGee, C.L. Fetal Alcohol Spectrum Disorders: An Overview with Emphasis on Changes in Brain and Behavior. Exp Biol Med 2005, 230, 357–365. [Google Scholar] [CrossRef]
- Jaatinen, P.; Rintala, J. Mechanisms of Ethanol-Induced Degeneration in the Developing, Mature, and Aging Cerebellum. Cerebellum 2008, 7, 332–347. [Google Scholar] [CrossRef]
- Coviello, C.; Keunen, K.; Kersbergen, K.J.; Groenendaal, F.; Leemans, A.; Peels, B.; Isgum, I.; Viergever, M.A.; de Vries, L.S.; Buonocore, G.; et al. Effects of Early Nutrition and Growth on Brain Volumes, White Matter Microstructure, and Neurodevelopmental Outcome in Preterm Newborns. Pediatr Res 2018, 83, 102–110. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Shaikh, A.G. Alcohol Toxicity in the Developing Cerebellum. Diagnostics 2024, 14, 1415. [Google Scholar] [CrossRef]
- Handmaker, N.S.; Rayburn, W.F.; Meng, C.; Bell, J.B.; Rayburn, B.B.; Rappaport, V.J. Impact of Alcohol Exposure After Pregnancy Recognition on Ultrasonographic Fetal Growth Measures. Alcohol Clin Exp Res 2006, 30, 892–898. [Google Scholar] [CrossRef]
- Astley, S.J.; Aylward, E.H.; Olson, H.C.; Kerns, K.; Brooks, A.; Coggins, T.E.; Davies, J.; Dorn, S.; Gendler, B.; Jirikowic, T.; et al. Magnetic Resonance Imaging Outcomes From a Comprehensive Magnetic Resonance Study of Children With Fetal Alcohol Spectrum Disorders. Alcohol Clin Exp Res 2009, 33, 1671–1689. [Google Scholar] [CrossRef]
- Autti-Rämö, I.; Autti, T.; Korkman, M.; Kettunen, S.; Salonen, O.; Valanne, L. MRI Findings in Children with School Problems Who Had Been Exposed Prenatally to Alcohol. Dev Med Child Neurol 2002, 44, 98. [Google Scholar] [CrossRef] [PubMed]
- Sowell, E.R.; Jernigan, T.L.; Mattson, S.N.; Riley, E.P.; Sobel, D.F.; Jones, K.L. Abnormal Development of the Cerebellar Vermis in Children Prenatally Exposed to Alcohol: Size Reduction in Lobules I-V. Alcohol Clin Exp Res 1996, 20, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Archibald, S.L.; Fennema-Notestine, C.; Gamst, A.; Riley, E.P.; Mattson, S.N.; Jernigan, T.L. Brain Dysmorphology in Individuals with Severe Prenatal Alcohol Exposure. Dev Med Child Neurol 2001, 43, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E. V; Moore, E.M.; Lane, B.; Pohl, K.M.; Riley, E.P.; Pfefferbaum, A. Graded Cerebellar Lobular Volume Deficits in Adolescents and Young Adults with Fetal Alcohol Spectrum Disorders (FASD). Cerebral Cortex 2020, 30, 4729–4746. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Meintjes, E.M.; Molteno, C.D.; Spottiswoode, B.S.; Dodge, N.C.; Alhamud, A.A.; Stanton, M.E.; Peterson, B.S.; Jacobson, J.L.; Jacobson, S.W. White Matter Integrity of the Cerebellar Peduncles as a Mediator of Effects of Prenatal Alcohol Exposure on Eyeblink Conditioning. Hum Brain Mapp 2015, 36, 2470–2482. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Chong, S.; Tieng, Q.M.; Mardon, K.; Galloway, G.J.; Kurniawan, N.D. Radiological Studies of Fetal Alcohol Spectrum Disorders in Humans and Animal Models: An Updated Comprehensive Review. Magn Reson Imaging 2017, 43, 10–26. [Google Scholar] [CrossRef]
- du Plessis, L.; Jacobson, J.L.; Jacobson, S.W.; Hess, A.T.; van der Kouwe, A.; Avison, M.J.; Molteno, C.D.; Stanton, M.E.; Stanley, J.A.; Peterson, B.S.; et al. An In Vivo 1 H Magnetic Resonance Spectroscopy Study of the Deep Cerebellar Nuclei in Children with Fetal Alcohol Spectrum Disorders. Alcohol Clin Exp Res 2014, 38, 1330–1338. [Google Scholar] [CrossRef]
- Diwadkar, V.A.; Meintjes, E.M.; Goradia, D.; Dodge, N.C.; Warton, C.; Molteno, C.D.; Jacobson, S.W.; Jacobson, J.L. Differences in Cortico-striatal-cerebellar Activation during Working Memory in Syndromal and Nonsyndromal Children with Prenatal Alcohol Exposure. Hum Brain Mapp 2013, 34, 1931–1945. [Google Scholar] [CrossRef]
- Parnell, S.E.; O’Leary-Moore, S.K.; Godin, E.A.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic Resonance Microscopy Defines Ethanol-Induced Brain Abnormalities in Prenatal Mice: Effects of Acute Insult on Gestational Day 8. Alcohol Clin Exp Res 2009, 33, 1001–1011. [Google Scholar] [CrossRef]
- Parnell, S.E.; Holloway, H.T.; O’Leary-Moore, S.K.; Dehart, D.B.; Paniaqua, B.; Oguz, I.; Budin, F.; Styner, M.A.; Johnson, G.A.; Sulik, K.K. Magnetic Resonance Microscopy-Based Analyses of the Neuroanatomical Effects of Gestational Day 9 Ethanol Exposure in Mice. Neurotoxicol Teratol 2013, 39, 77–83. [Google Scholar] [CrossRef]
- Fish, E.W.; Holloway, H.T.; Rumple, A.; Baker, L.K.; Wieczorek, L.A.; Moy, S.S.; Paniagua, B.; Parnell, S.E. Acute Alcohol Exposure during Neurulation: Behavioral and Brain Structural Consequences in Adolescent C57BL/6J Mice. Behavioural Brain Research 2016, 311, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.W.; Wieczorek, L.A.; Rumple, A.; Suttie, M.; Moy, S.S.; Hammond, P.; Parnell, S.E. The Enduring Impact of Neurulation Stage Alcohol Exposure: A Combined Behavioral and Structural Neuroimaging Study in Adult Male and Female C57BL/6J Mice. Behavioural Brain Research 2018, 338, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Godin, E.A.; O’Leary-Moore, S.K.; Khan, A.A.; Parnell, S.E.; Ament, J.J.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic Resonance Microscopy Defines Ethanol-Induced Brain Abnormalities in Prenatal Mice: Effects of Acute Insult on Gestational Day 7. Alcohol Clin Exp Res 2010, 34, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Bhalla, R.; Cowin, G.; Stimson, D.H.R.; Song, X.; Chong, S.; Jackson, A.; Trigg, W.J.; Tieng, Q.M.; Mardon, K.; et al. GABAa Receptor Density Alterations Revealed in a Mouse Model of Early Moderate Prenatal Ethanol Exposure Using [18F]AH114726. Nucl Med Biol 2020, 88–89, 44–51. [Google Scholar] [CrossRef]
- Marcussen, B.L.; Goodlett, C.R.; Mahoney, J.C.; West, J.R. Developing Rat Purkinje Cells Are More Vulnerable to Alcohol-Induced Depletion During Differentiation Than During Neurogenesis. Alcohol 1994, 11, 147–156. [Google Scholar] [CrossRef]
- Nirgudkar, P.; Taylor, D.H.; Yanagawa, Y.; Valenzuela, C.F. Ethanol Exposure during Development Reduces GABAergic/Glycinergic Neuron Numbers and Lobule Volumes in the Mouse Cerebellar Vermis. Neurosci Lett 2016, 632, 86–91. [Google Scholar] [CrossRef]
- González-Burgos, I.; Alejandre-Gómez, M. Cerebellar Granule Cell and Bergmann Glial Cell Maturation in the Rat Is Disrupted by Pre- and Post-natal Exposure to Moderate Levels of Ethanol. International Journal of Developmental Neuroscience 2005, 23, 383–388. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Eilers, A.T. Alcohol-Induced Purkinje Cell Loss with a Single Binge Exposure in Neonatal Rats: A Stereological Study of Temporal Windows of Vulnerability. Alcohol Clin Exp Res 1997, 21, 738–744. [Google Scholar] [CrossRef]
- Karaçay, B.; Li, S.; Bonthius, D.J. Maturation-Dependent Alcohol Resistance in the Developing Mouse: Cerebellar Neuronal Loss and Gene Expression during Alcohol-Vulnerable and -Resistant Periods. Alcohol Clin Exp Res 2008, 32, 1439–1450. [Google Scholar] [CrossRef]
- Hamre, K.M.; West, J.R. The Effects of the Timing of Ethanol Exposure during the Brain Growth Spurt on the Number of Cerebellar Purkinje and Granule Cell Nuclear Profiles; 1993; Vol. 17;
- Chen, W.-J.A.; Parnell, S.E.; West, J.R.; Chen, W.-J.A.; Parnell, S.E.; Neonatal, J.R.W. Neonatal Alcohol and Nicotine Exposure Limits Brain Growth and Depletes Cerebellar Purkinje Cells; 1998; Vol. 15;
- Kane, C.J.M.; Phelan, K.D.; Han, L.; Smith, R.R.; Xie, J.; Douglas, J.C.; Drew, P.D. Protection of Neurons and Microglia against Ethanol in a Mouse Model of Fetal Alcohol Spectrum Disorders by Peroxisome Proliferator-Activated Receptor-γ Agonists. Brain Behav Immun 2011, 25. [Google Scholar] [CrossRef]
- Lee, Y.; Rowe, J.; Eskue, K.; West, J.R.; Maier, S.E. Alcohol Exposure on Postnatal Day 5 Induces Purkinje Cell Loss and Evidence of Purkinje Cell Degradation in Lobule I of Rat Cerebellum. Alcohol 2008, 42, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Light, K.E.; Hayar, A.M.; Pierce, D.R. Electrophysiological and Immunohistochemical Evidence for an Increase in GABAergic Inputs and HCN Channels in Purkinje Cells That Survive Developmental Ethanol Exposure. Cerebellum 2015, 14, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Pierce, D.R.; Hayar, A.; Williams, D.K.; Light, K.E. Developmental Alterations in Olivary Climbing Fiber Distribution Following Postnatal Ethanol Exposure in the Rat. Neuroscience 2010, 169, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Pierce, D.R.; Hayar, A.; Williams, D.K.; Light, K.E. Olivary Climbing Fiber Alterations in PN40 Rat Cerebellum Following Postnatal Ethanol Exposure. Brain Res 2011, 1378, 54–65. [Google Scholar] [CrossRef]
- Todd, D.; Clapp, M.; Dains, P.; Karacay, B.; Bonthius, D.J. Purkinje Cell-Specific Deletion of CREB Worsens Alcohol-Induced Cerebellar Neuronal Losses and Motor Deficits. Alcohol 2022, 101, 27–35. [Google Scholar] [CrossRef]
- Gursky, Z.H.; Johansson, J.R.; Klintsova, A.Y. Postnatal Alcohol Exposure and Adolescent Exercise Have Opposite Effects on Cerebellar Microglia in Rat. International Journal of Developmental Neuroscience 2020, 80, 558–571. [Google Scholar] [CrossRef]
- Cealie, M.K.Y.; Douglas, J.C.; Swan, H.K.; Vonkaenel, E.D.; McCall, M.N.; Drew, P.D.; Majewska, A.K. Developmental Ethanol Exposure Impacts Purkinje Cells but Not Microglia in the Young Adult Cerebellum. Cells 2024, 13. [Google Scholar] [CrossRef]
- Cealie, M.K.Y.; Douglas, J.C.; Le, L.H.D.; Vonkaenel, E.D.; McCall, M.N.; Drew, P.D.; Majewska, A.K. Developmental Ethanol Exposure Has Minimal Impact on Cerebellar Microglial Dynamics, Morphology, and Interactions with Purkinje Cells during Adolescence. Front Neurosci 2023, 17. [Google Scholar] [CrossRef]
- Bolbanabad, H.M.; Anvari, E.; Rezai, M.J.; Moayeri, A.; Kaffashian, M.R. Amelioration of Cerebellar Dysfunction in Rats Following Postnatal Ethanol Exposure Using Low-Intensity Pulsed Ultrasound. J Chem Neuroanat 2017, 81, 71–75. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Winters, Z.; Karacay, B.; Bousquet, S.L.; Bonthius, D.J. Importance of Genetics in Fetal Alcohol Effects: Null Mutation of the NNOS Gene Worsens Alcohol-Induced Cerebellar Neuronal Losses and Behavioral Deficits. Neurotoxicology 2015, 46, 60–72. [Google Scholar] [CrossRef]
- Gundogan, F.; Tong, M.; Chen, W.C.; Kim, C.; Nguyen, Q.-G.; Yu, R.; de la Monte, S.M. Chronic Prenatal Ethanol Exposure Disrupts WNT Signaling In Adolescent Cerebella. J Clin Exp Pathol 2013, 03. [Google Scholar] [CrossRef]
- Tong, M.; Ziplow, J.; Chen, W.C.; Nguyen, Q.-G.; Kim, C.; De La Monte, S.M. 2013.
- Holloway, K.N.; Douglas, J.C.; Rafferty, T.M.; Majewska, A.K.; Kane, C.J.M.; Drew, P.D. Ethanol-Induced Cerebellar Transcriptomic Changes in a Postnatal Model of Fetal Alcohol Spectrum Disorders: Focus on Disease Onset. Front Neurosci 2023, 17. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedz-Massey, V.M.; Douglas, J.C.; Rafferty, T.; Kane, C.J.M.; Drew, P.D. Ethanol Effects on Cerebellar Myelination in a Postnatal Mouse Model of Fetal Alcohol Spectrum Disorders. Alcohol 2021, 96, 43–53. [Google Scholar] [CrossRef]
- Drew, P.D.; Johnson, J.W.; Douglas, J.C.; Phelan, K.D.; Kane, C.J.M. Pioglitazone Blocks Ethanol Induction of Microglial Activation and Immune Responses in the Hippocampus, Cerebellum, and Cerebral Cortex in a Mouse Model of Fetal Alcohol Spectrum Disorders. Alcohol Clin Exp Res 2015, 39, 445–454. [Google Scholar] [CrossRef]
- Kane, C.J.M.; Douglas, J.C.; Rafferty, T.; Johnson, J.W.; Niedzwiedz-Massey, V.M.; Phelan, K.D.; Majewska, A.K.; Drew, P.D. Ethanol Modulation of Cerebellar Neuroinflammation in a Postnatal Mouse Model of Fetal Alcohol Spectrum Disorders. J Neurosci Res 2021, 99, 1986–2007. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, H.; Xu, M.; Frank, J.A.; Luo, J. Role of MCP-1 and CCR2 in Ethanol-Induced Neuroinflammation and Neurodegeneration in the Developing Brain. J Neuroinflammation 2018, 15. [Google Scholar] [CrossRef]
- Ieraci, A.; Herrera, D.G. Nicotinamide Inhibits Ethanol-Induced Caspase-3 and PARP-1 Over-Activation and Subsequent Neurodegeneration in the Developing Mouse Cerebellum. Cerebellum 2018, 17, 326–335. [Google Scholar] [CrossRef]
- Leung, E.C.H.; Jain, P.; Michealson, M.A.; Choi, H.; Ellsworth-Kopkowski, A.; Valenzuela, C.F. Recent Breakthroughs in Understanding the Cerebellum’s Role in Fetal Alcohol Spectrum Disorder: A Systematic Review. Alcohol 2024, 119, 37–71. [Google Scholar] [CrossRef]
- Ekblad, M.; Korkeila, J.; Parkkola, R.; Lapinleimu, H.; Haataja, L.; Lehtonen, L. Maternal Smoking during Pregnancy and Regional Brain Volumes in Preterm Infants. Journal of Pediatrics 2010, 156. [Google Scholar] [CrossRef]
- Zou, R.; Boer, O.D.; Felix, J.F.; Muetzel, R.L.; Franken, I.H.A.; Cecil, C.A.M.; El Marroun, H. Association of Maternal Tobacco Use During Pregnancy With Preadolescent Brain Morphology Among Offspring. JAMA Netw Open 2022, 5, e2224701. [Google Scholar] [CrossRef]
- Bublitz, M.H.; Stroud, L.R. Maternal Smoking During Pregnancy and Offspring Brain Structure and Function: Review and Agenda for Future Research. Nicotine & Tobacco Research 2012, 14, 388–397. [Google Scholar] [CrossRef]
- Roza, S.J.; Verburg, B.O.; Jaddoe, V.W. V.; Hofman, A.; Mackenbach, J.P.; Steegers, E.A.P.; Witteman, J.C.M.; Verhulst, F.C.; Tiemeier, H. Effects of Maternal Smoking in Pregnancy on Prenatal Brain Development. The Generation R Study. European Journal of Neuroscience 2007, 25, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.S.; Mohamed, F.B.; Carmody, D.P.; Bendersky, M.; Patel, S.; Khorrami, M.; Faro, S.H.; Lewis, M. Response Inhibition among Early Adolescents Prenatally Exposed to Tobacco: An FMRI Study. Neurotoxicol Teratol 2009, 31, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.A.; Fried, P.A.; Cameron, I.; Smith, A.M. The Long-Term Effects of Prenatal Nicotine Exposure on Response Inhibition: An FMRI Study of Young Adults. Neurotoxicol Teratol 2013, 39, 9–18. [Google Scholar] [CrossRef]
- Martin-Ruiz, C.M.; Lee, M.; Perry, R.H.; Baumann, M.; Court, J.A.; Perry, E.K. Molecular Analysis of Nicotinic Receptor Expression in Autism. Molecular Brain Research 2004, 123, 81–90. [Google Scholar] [CrossRef]
- Lee, M. Nicotinic Receptor Abnormalities in the Cerebellar Cortex in Autism. Brain 2002, 125, 1483–1495. [Google Scholar] [CrossRef]
- Turner, J.R.; Kellar, K.J. Nicotinic Cholinergic Receptors in the Rat Cerebellum: Multiple Heteromeric Subtypes. The Journal of Neuroscience 2005, 25, 9258–9265. [Google Scholar] [CrossRef]
- Graham, A.; Court, J.A.; Martin-Ruiz, C.M.; Jaros, E.; Perry, R.; Volsen, S.G.; Bose, S.; Evans, N.; Ince, P.; Kuryatov, A.; et al. Immunohistochemical Localisation of Nicotinic Acetylcholine Receptor Subunits in Human Cerebellum. Neuroscience 2002, 113, 493–507. [Google Scholar] [CrossRef]
- Ekblad, M.O. Association of Smoking During Pregnancy With Compromised Brain Development in Offspring. JAMA Netw Open 2022, 5, e2224714. [Google Scholar] [CrossRef]
- Bhandari, N.R.; Day, K.D.; Payakachat, N.; Franks, A.M.; McCain, K.R.; Ragland, D. Use and Risk Perception of Electronic Nicotine Delivery Systems and Tobacco in Pregnancy. Women’s Health Issues 2018, 28, 251–257. [Google Scholar] [CrossRef]
- Stroud, L.R.; Papandonatos, G.D.; Borba, K.; Kehoe, T.; Scott-Sheldon, L.A.J. Flavored Electronic Cigarette Use, Preferences, and Perceptions in Pregnant Mothers: A Correspondence Analysis Approach. Addictive Behaviors 2019, 91, 21–29. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K.T.; Leslie, F.M. Developmental Regulation of Nicotinic Acetylcholine Receptor-mediated [ 3 H]Norepinephrine Release from Rat Cerebellum. J Neurochem 2003, 84, 952–959. [Google Scholar] [CrossRef]
- Didier, M.; Berman, S.A.; Lindstrom, J.; Bursztajn, S. Characterization of Nicotinic Acetylcholine Receptors Expressed in Primary Cultures of Cerebellar Granule Cells. Molecular Brain Research 1995, 30, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R.; Kellar, K.J. Nicotinic Cholinergic Receptors in the Rat Cerebellum: Multiple Heteromeric Subtypes. The Journal of Neuroscience 2005, 25, 9258–9265. [Google Scholar] [CrossRef] [PubMed]
- Tribollet, E.; Bertrand, D.; Marguerat, A.; Raggenbass, M. Comparative Distribution of Nicotinic Receptor Subtypes during Development, Adulthood and Aging: An Autoradiographic Study in the Rat Brain. Neuroscience 2004, 124, 405–420. [Google Scholar] [CrossRef]
- Rossi, D.J.; Hamann, M.; Attwell, D. Multiple Modes of GABAergic Inhibition of Rat Cerebellar Granule Cells. J Physiol 2003, 548, 97–110. [Google Scholar] [CrossRef]
- De Filippi, G.; Baldwinson, T.; Sher, E. Evidence for Nicotinic Acetylcholine Receptor Activation in Rat Cerebellar Slices. Pharmacol Biochem Behav 2001, 70, 447–455. [Google Scholar] [CrossRef]
- Caban, K.M.; Seßenhausen, P.; Stöckl, J.B.; Popper, B.; Mayerhofer, A.; Fröhlich, T. Proteome Profile of the Cerebellum from A7 Nicotinic Acetylcholine Receptor Deficient Mice. Proteomics 2024, 24. [Google Scholar] [CrossRef]
- de Toro, E.D.; Juíz, J.M.; Smillie, F.I.; Lindstrom, J.; Criado, M. Expression of α 7 Neuronal Nicotinic Receptors during Postnatal Development of the Rat Cerebellum. Developmental Brain Research 1997, 98, 125–133. [Google Scholar] [CrossRef]
- Role, L.W.; Berg, D.K. Nicotinic Receptors in the Development and Modulation of CNS Synapses. Neuron 1996, 16, 1077–1085. [Google Scholar] [CrossRef]
- Opanashuk, L.A.; Pauly, J.R.; Hauser, K.F. Effect of Nicotine on Cerebellar Granule Neuron Development. European Journal of Neuroscience 2001, 13, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Deacon, A.; Suter, M.A.; Aagaard, K.M. Understanding the Placental Biology of Tobacco Smoke, Nicotine, and Marijuana (THC) Exposures During Pregnancy. Clin Obstet Gynecol 2022, 65, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Lee, S.M.; Ko, E.B.; Go, R.E.; Jeung, E.B.; Kim, M.S.; Choi, K.C. Inhibitory Effects of Cigarette Smoke Extracts on Neural Differentiation of Mouse Embryonic Stem Cells. Reproductive Toxicology 2020, 95, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Abou-Donia, M.B.; Khan, W.A.; Dechkovskaia, A.M.; Goldstein, L.B.; Bullman, S.L.; Abdel-Rahman, A. In Utero Exposure to Nicotine and Chlorpyrifos Alone, and in Combination Produces Persistent Sensorimotor Deficits and Purkinje Neuron Loss in the Cerebellum of Adult Offspring Rats. Arch Toxicol 2006, 80, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, A.; Dechkovskaia, A.M.; Sutton, J.M.; Chen, W.C.; Guan, X.; Khan, W.A.; Abou-Donia, M.B. Maternal Exposure of Rats to Nicotine via Infusion during Gestation Produces Neurobehavioral Deficits and Elevated Expression of Glial Fibrillary Acidic Protein in the Cerebellum and CA1 Subfield in the Offspring at Puberty. Toxicology 2005, 209, 245–261. [Google Scholar] [CrossRef]
- Huang, L.Z.; Abbott, L.C.; Winzer-Serhan, U.H. Effects of Chronic Neonatal Nicotine Exposure on Nicotinic Acetylcholine Receptor Binding, Cell Death and Morphology in Hippocampus and Cerebellum. Neuroscience 2007, 146, 1854–1868. [Google Scholar] [CrossRef]
- Chen, W.J.A.; Edwards, R.B. Prenatal Nicotine Exposure Does Not Cause Purkinje Cell Loss in the Developing Rat Cerebellar Vermis. Neurotoxicol Teratol 2003, 25, 633–637. [Google Scholar] [CrossRef]
- Polli, F.S.; Ipsen, T.H.; Caballero-Puntiverio, M.; Østerbøg, T.B.; Aznar, S.; Andreasen, J.T.; Kohlmeier, K.A. Cellular and Molecular Changes in Hippocampal Glutamate Signaling and Alterations in Learning, Attention, and Impulsivity Following Prenatal Nicotine Exposure. Mol Neurobiol 2020, 57, 2002–2020. [Google Scholar] [CrossRef]
- Nakauchi, S.; Su, H.; Trang, I.; Sumikawa, K. Long-Term Effects of Early Postnatal Nicotine Exposure on Cholinergic Function in the Mouse Hippocampal CA1 Region. Neurobiol Learn Mem 2021, 181. [Google Scholar] [CrossRef]
- Roy, T.S.; Sabherwal, U. Effects of Prenatal Nicotine Exposure on the Morphogenesis of Somatosensory Cortex. Neurotoxicol Teratol 1994, 16, 411–421. [Google Scholar] [CrossRef]
- Chan, Y.L.; Saad, S.; Pollock, C.; Oliver, B.; Al-Odat, I.; Zaky, A.A.; Jones, N.; Chen, H. Impact of Maternal Cigarette Smoke Exposure on Brain Inflammation and Oxidative Stress in Male Mice Offspring. Sci Rep 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wallauer, M.M.; Huf, F.; Tortorelli, L.S.; Rahmeier, F.L.; Carvalho, F.B.; Meurer, R.T.; da Cruz Fernandes, M. Morphological Changes in the Cerebellum as a Result of Ethanol Treatment and Cigarette Smoke Exposure: A Study on Astrogliosis, Apoptosis and Purkinje Cells. Neurosci Lett 2018, 672, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Maternal E-Cigarette Exposure Results in Cognitive and Epigenetic Alterations in Offspring in a Mouse Model. Chem Res Toxicol 2018, 31, 601–611. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Neurological Effects in the Offspring after Switching from Tobacco Cigarettes to E-Cigarettes during Pregnancy in a Mouse Model. Toxicological Sciences 2019, 172, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Church, J.S.; Chace-Donahue, F.; Blum, J.L.; Ratner, J.R.; Zelikoff, J.T.; Schwartzer, J.J. Neuroinflammatory and Behavioral Outcomes Measured in Adult Offspring of Mice Exposed Prenatally to E-Cigarette Aerosols. Environ Health Perspect 2020, 128. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Jew, C.P.; Lu, H.-C. Lasting Impacts of Prenatal Cannabis Exposure and the Role of Endogenous Cannabinoids In the Developing Brain. Future Neurol 2011, 6, 459–480. [Google Scholar] [CrossRef]
- Hutchings, D.E.; Martin, B.R.; Gamagaris, Z.; Miller, N.; Fico, T. Plasma Concentrations of Delta-9-Tetrahydrocannabinol in Dams and Fetuses Following Acute or Multiple Prenatal Dosing in Rats. Life Sci 1989, 44, 697–701. [Google Scholar] [CrossRef]
- Presence of Δ 9 -Tetrahydrocannabinol in Human Milk. New England Journal of Medicine 1982, 307, 819–820. [CrossRef]
- Hayer, S.; Mandelbaum, A.D.; Watch, L.; Ryan, K.S.; Hedges, M.A.; Manuzak, J.A.; Easley, C.A.; Schust, D.J.; Lo, J.O. Cannabis and Pregnancy: A Review. Obstet Gynecol Surv 2023, 78, 411–428. [Google Scholar] [CrossRef]
- Gunn, J.K.L.; Rosales, C.B.; Center, K.E.; Nuñez, A.; Gibson, S.J.; Christ, C.; Ehiri, J.E. Prenatal Exposure to Cannabis and Maternal and Child Health Outcomes: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e009986. [Google Scholar] [CrossRef]
- Metz, T.D.; Borgelt, L.M. Marijuana Use in Pregnancy and While Breastfeeding. Obstetrics & Gynecology 2018, 132, 1198–1210. [Google Scholar] [CrossRef]
- Klebanoff, M.A.; Wilkins, D.G.; Keim, S.A. Marijuana Use during Pregnancy and Preterm Birth: A Prospective Cohort Study. Am J Perinatol 2021, 38, e146–e154. [Google Scholar] [CrossRef] [PubMed]
- Habayeb, O.M.H.; Taylor, A.H.; Bell, S.C.; Taylor, D.J.; Konje, J.C. Expression of the Endocannabinoid System in Human First Trimester Placenta and Its Role in Trophoblast Proliferation. Endocrinology 2008, 149, 5052–5060. [Google Scholar] [CrossRef]
- Paria, B.C.; Dey, S.K. Ligand-Receptor Signaling with Endocannabinoids in Preimplantation Embryo Development and Implantation. Chem Phys Lipids 2000, 108, 211–220. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Berrendero, F.; Hernández, M.L.; Ramos, J.A. The Endogenous Cannabinoid System and Brain Development. Trends Neurosci 2000, 23, 14–20. [Google Scholar] [CrossRef]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the Pharmacology and Signal Transduction of the Human Cannabinoid CB1 and CB2 Receptors. Mol Pharmacol 1995, 48, 443–450. [Google Scholar]
- Takahashi, K.A.; Linden, D.J. Cannabinoid Receptor Modulation of Synapses Received by Cerebellar Purkinje Cells. J Neurophysiol 2000, 83, 1167–1180. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 Receptors Regulate Neuronal Energy Metabolism. Nat Neurosci 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes That Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem Biol 2007, 14, 1347–1356. [Google Scholar] [CrossRef]
- Carey, M.R.; Myoga, M.H.; McDaniels, K.R.; Marsicano, G.; Lutz, B.; Mackie, K.; Regehr, W.G. Presynaptic CB1 Receptors Regulate Synaptic Plasticity at Cerebellar Parallel Fiber Synapses. J Neurophysiol 2011, 105, 958–963. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.; Johnson, M.; Melvin, L.; de Costa, B.; Rice, K. Characterization and Localization of Cannabinoid Receptors in Rat Brain: A Quantitative in Vitro Autoradiographic Study. The Journal of Neuroscience 1991, 11, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Groen, B.G.S.; Lynn, A.B.; De Costa, B.R.; Richfield, E.K. Neuronal Localization of Cannabinoid Receptors and Second Messengers in Mutant Mouse Cerebellum. Brain Res 1991, 552, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid Receptor Localization in Brain. Proceedings of the National Academy of Sciences 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Grewen, K.; Salzwedel, A.P.; Gao, W. Functional Connectivity Disruption in Neonates with Prenatal Marijuana Exposure. Front Hum Neurosci 2015, 9. [Google Scholar] [CrossRef]
- Richardson, K.A.; Hester, A.K.; McLemore, G.L. Prenatal Cannabis Exposure - The “First Hit” to the Endocannabinoid System. Neurotoxicol Teratol 2016, 58, 5–14. [Google Scholar] [CrossRef]
- Calvigioni, D.; Hurd, Y.L.; Harkany, T.; Keimpema, E. Neuronal Substrates and Functional Consequences of Prenatal Cannabis Exposure. Eur Child Adolesc Psychiatry 2014, 23, 931–941. [Google Scholar] [CrossRef]
- Díaz-Alonso, J.; Aguado, T.; Wu, C.-S.; Palazuelos, J.; Hofmann, C.; Garcez, P.; Guillemot, F.; Lu, H.-C.; Lutz, B.; Guzmán, M.; et al. The CB 1 Cannabinoid Receptor Drives Corticospinal Motor Neuron Differentiation through the Ctip2/Satb2 Transcriptional Regulation Axis. The Journal of Neuroscience 2012, 32, 16651–16665. [Google Scholar] [CrossRef]
- Huizink, A.C. Prenatal Cannabis Exposure and Infant Outcomes: Overview of Studies. Prog Neuropsychopharmacol Biol Psychiatry 2014, 52, 45–52. [Google Scholar] [CrossRef]
- Fried, P.A.; Watkinson, B.; Gray, R. Differential Effects on Cognitive Functioning in 9- to 12-Year Olds Prenatally Exposed to Cigarettes and Marihuana. Neurotoxicol Teratol 1998, 20, 293–306. [Google Scholar] [CrossRef]
- Motamedi, S.; Amleshi, R.S.; Javar, B.A.; Shams, P.; Kohlmeier, K.A.; Shabani, M. Cannabis during Pregnancy: A Way to Transfer an Impairment to Later Life. Birth Defects Res 2023, 115, 1327–1344. [Google Scholar] [CrossRef]
- Thomason, M.E.; Palopoli, A.C.; Jariwala, N.N.; Werchan, D.M.; Chen, A.; Adhikari, S.; Espinoza-Heredia, C.; Brito, N.H.; Trentacosta, C.J. Miswiring the Brain: Human Prenatal Δ9-Tetrahydrocannabinol Use Associated with Altered Fetal Hippocampal Brain Network Connectivity. Dev Cogn Neurosci 2021, 51, 101000. [Google Scholar] [CrossRef]
- Smith, A.M.; Fried, P.A.; Hogan, M.J.; Cameron, I. Effects of Prenatal Marijuana on Response Inhibition: An FMRI Study of Young Adults. Neurotoxicol Teratol 2004, 26, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Bossong, M.G.; Jansma, J.M.; van Hell, H.H.; Jager, G.; Oudman, E.; Saliasi, E.; Kahn, R.S.; Ramsey, N.F. Effects of Δ9-Tetrahydrocannabinol on Human Working Memory Function. Biol Psychiatry 2012, 71, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Jew, C.P.; Lu, H.-C. Lasting Impacts of Prenatal Cannabis Exposure and the Role of Endogenous Cannabinoids In the Developing Brain. Future Neurol 2011, 6, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Jutras-Aswad, D.; DiNieri, J.A.; Harkany, T.; Hurd, Y.L. Neurobiological Consequences of Maternal Cannabis on Human Fetal Development and Its Neuropsychiatric Outcome. Eur Arch Psychiatry Clin Neurosci 2009, 259, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Calvigioni, D.; Hurd, Y.L.; Harkany, T.; Keimpema, E. Neuronal Substrates and Functional Consequences of Prenatal Cannabis Exposure. Eur Child Adolesc Psychiatry 2014, 23, 931–941. [Google Scholar] [CrossRef]
- Martinez, L.R.; Black, K.C.; Webb, B.T.; Bell, A.; Baygani, S.K.; Mier, T.J.; Dominguez, L.; Mackie, K.; Kalinovsky, A. Components of Endocannabinoid Signaling System Are Expressed in the Perinatal Mouse Cerebellum and Required for Its Normal Development. eNeuro 2020, 7, ENEURO.0471–19.2020. [Google Scholar] [CrossRef]
- Wu, C.; Zhu, J.; Wager-Miller, J.; Wang, S.; O’Leary, D.; Monory, K.; Lutz, B.; Mackie, K.; Lu, H. Requirement of Cannabinoid CB 1 Receptors in Cortical Pyramidal Neurons for Appropriate Development of Corticothalamic and Thalamocortical Projections. European Journal of Neuroscience 2010, 32, 693–706. [Google Scholar] [CrossRef]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid Signaling Controls Pyramidal Cell Specification and Long-Range Axon Patterning. Proceedings of the National Academy of Sciences 2008, 105, 8760–8765. [Google Scholar] [CrossRef]
- Suárez, I.; Bodega, G.; Rubio, M.; Fernández-Ruiz, J.J.; Ramos, J.A.; Fernández, B. Prenatal Cannabinoid Exposure Down- Regulates Glutamate Transporter Expressions (GLAST and EAAC1) in the Rat Cerebellum. Dev Neurosci 2004, 26, 45–53. [Google Scholar] [CrossRef]
- Pinky, P.D.; Majrashi, M.; Fujihashi, A.; Bloemer, J.; Govindarajulu, M.; Ramesh, S.; Reed, M.N.; Moore, T.; Suppiramaniam, V.; Dhanasekaran, M. Effects of Prenatal Synthetic Cannabinoid Exposure on the Cerebellum of Adolescent Rat Offspring. Heliyon 2021, 7, e06730. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, J. van; Talhout, R.; Vleeming, W.; Opperhuizen, A. Contribution of Monoamine Oxidase (MAO) Inhibition to Tobacco and Alcohol Addiction. Life Sci 2006, 79, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Day, N.L.; Goldschmidt, L.; Thomas, C.A. Prenatal Marijuana Exposure Contributes to the Prediction of Marijuana Use at Age 14. Addiction 2006, 101, 1313–1322. [Google Scholar] [CrossRef]
- Benevenuto, S.G.M.; Domenico, M.D.; Yariwake, V.Y.; Dias, C.T.; Mendes-da-Silva, C.; Alves, N. de O.; Caumo, S.E. da S.; Vasconcellos, P.; Morais, D.R.; Cardoso, M.S.; et al. Prenatal Exposure to Cannabis Smoke Induces Early and Lasting Damage to the Brain. Neurochem Int 2022, 160, 105406. [Google Scholar] [CrossRef]
- Shabani, M.; Hosseinmardi, N.; Haghani, M.; Shaibani, V.; Janahmadi, M. Maternal Exposure to the CB1 Cannabinoid Agonist WIN 55212–2 Produces Robust Changes in Motor Function and Intrinsic Electrophysiological Properties of Cerebellar Purkinje Neurons in Rat Offspring. Neuroscience 2011, 172, 139–152. [Google Scholar] [CrossRef]
- Honein, M.A.; Boyle, C.; Redfield, R.R. Public Health Surveillance of Prenatal Opioid Exposure in Mothers and Infants. Pediatrics 2019, 143. [Google Scholar] [CrossRef]
- Yeoh, S.L.; Eastwood, J.; Wright, I.M.; Morton, R.; Melhuish, E.; Ward, M.; Oei, J.L. Cognitive and Motor Outcomes of Children With Prenatal Opioid Exposure A Systematic Review and Meta-Analysis. JAMA Netw Open 2019, 2. [Google Scholar] [CrossRef]
- Kim, E.; Clark, A.L.; Kiss, A.; Hahn, J.W.; Wesselschmidt, R.; Coscia, C.J.; Belcheva, M.M. μ- and κ-Opioids Induce the Differentiation of Embryonic Stem Cells to Neural Progenitors. Journal of Biological Chemistry 2006, 281, 33749–33760. [Google Scholar] [CrossRef]
- Schadrack, J.; Willoch, F.; Platzer, S.; Bartenstein, P.; Mahal, B.; Dworzak, D.; Wester, H.J.; Zieglgänsberger, W.; Tölle, T.R. Opioid Receptors in the Human Cerebellum: Evidence from [11C] Diprenorphine PET, MRNA Expression and Autoradiography. Neuroreport 1999, 10. [Google Scholar] [CrossRef]
- Platzer, S.; Winkler, A.; Schadrack, J.; Dworzak, D.; Tölle, T.R.; Zieglgänsberger, W.; Spanagel, R. Autoradiographic Distribution of μ-, δ- and Κ1-Opioid Stimulated [35S]Guanylyl-5’-O-(γ-Thio)-Triphosphate Binding in Human Frontal Cortex and Cerebellum. Neurosci Lett 2000, 283. [Google Scholar] [CrossRef]
- Hiller, J.M.; Fan, L.Q. Laminar Distribution of the Multiple Opioid Receptors in the Human Cerebral Cortex. Neurochem Res 1996, 21. [Google Scholar] [CrossRef] [PubMed]
- Kinney, H.C.; White, W.F. Opioid Receptors Localize to the External Granular Cell Layer of the Developing Human Cerebellum. Neuroscience 1991, 45. [Google Scholar] [CrossRef]
- McPherson, C.; Haslam, M.; Pineda, R.; Rogers, C.; Neil, J.J.; Inder, T.E. Brain Injury and Development in Preterm Infants Exposed to Fentanyl. Annals of Pharmacotherapy 2015, 49, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Rubic, M.; Seah, J.; Rae, C.; Wright, I.M.R.; Kaltenbach, K.; Feller, J.M.; Abdel-Latif, M.E.; Chu, C.; Oei, J.L.; et al. Do Maternal Opioids Reduce Neonatal Regional Brain Volumes? A Pilot Study. Journal of Perinatology 2014, 34, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Sirnes, E.; Oltedal, L.; Bartsch, H.; Eide, G.E.; Elgen, I.B.; Aukland, S.M. Brain Morphology in School-Aged Children with Prenatal Opioid Exposure: A Structural MRI Study. Early Hum Dev 2017, 106–107, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, E.; Slinning, K.; Moe, V.; Due-Tønnessen, P.; Fjell, A.; Walhovd, K.B. Neuroanatomical Characteristics of Youths with Prenatal Opioid and Poly-Drug Exposure. Neurotoxicol Teratol 2018, 68. [Google Scholar] [CrossRef]
- Walhovd, K.B.; Moe, V.; Slinning, K.; Due-Tønnessen, P.; Bjørnerud, A.; Dale, A.M.; van der Kouwe, A.; Quinn, B.T.; Kosofsky, B.; Greve, D.; et al. Volumetric Cerebral Characteristics of Children Exposed to Opiates and Other Substances in Utero. Neuroimage 2007, 36. [Google Scholar] [CrossRef]
- Zhu, Y.; Hsu, M.S.; Pintar, J.E. Developmental Expression of the μ, κ and δ Opioid Receptor MRNAs in Mouse. Journal of Neuroscience 1998, 18. [Google Scholar] [CrossRef]
- Hauser, K.F.; Houdi, A.A.; Turbek, C.S.; Elde, R.P.; Maxson, W. Opioids Intrinsically Inhibit the Genesis of Mouse Cerebellar Granule Neuron Precursors in Vitro: Differential Impact of μ and δ Receptor Activation on Proliferation and Neurite Elongation. European Journal of Neuroscience 2000, 12. [Google Scholar] [CrossRef]
- Zagon, I.S.; Gibo, D.M.; McLaughlin, P.J. Zeta (ξ), a Growth-Related Opioid Receptor in Developing Rat Cerebellum: Identification and Characterization. Brain Res 1991, 551. [Google Scholar] [CrossRef]
- Zagon, I.S.; Verderame, M.F.; McLaughlin, P.J. The Biology of the Opioid Growth Factor Receptor (OGFr). Brain Res Rev 2002, 38. [Google Scholar] [CrossRef] [PubMed]
- Abeyta, A.; Dettmer, T.S.; Barnes, A.; Vega, D.; Carta, M.; Gallegos, N.; Raymond-Stintz, M.; Savage, D.D.; Valenzuela, C.F.; Saland, L.C. Delta Opioid Receptor Localization in the Rat Cerebellum. Brain Res 2002, 931. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J. Methadone and Brain Development. Experientia 1977, 33. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J. Morphine and Brain Growth Retardation in the Rat. Pharmacology 1977, 15. [Google Scholar] [CrossRef]
- Lorber, B.A.; Freitag, S.K.; Bartolome, J. V. Effects of Beta-Endorphin on DNA Synthesis in Brain Regions of Preweanling Rats. Brain Res 1990, 531. [Google Scholar] [CrossRef]
- Gibson, J.M.; Chu, T.; Zeng, W.; Wethall, A.C.; Kong, M.; Mellen, N.; Devlin Phinney, L.A.; Cai, J. Perinatal Methadone Exposure Attenuates Myelination and Induces Oligodendrocyte Apoptosis in Neonatal Rat Brain. Exp Biol Med 2022, 247. [Google Scholar] [CrossRef]
- Hauser, K.F.; Gurwell, J.A.; Turbek, C.S. Morphine Inhibits Purkinje Cell Survival and Dendritic Differentiation in Organotypic Cultures of the Mouse Cerebellum. Exp Neurol 1994, 130. [Google Scholar] [CrossRef]
- Hauser, K.F.; McLaughlin, P.J.; Zagon, I.S. Endogenous Opioid Systems and the Regulation of Dendritic Growth and Spine Formation. Journal of Comparative Neurology 1989, 281. [Google Scholar] [CrossRef]
- Hauser, K.F.; McLaughlin, P.J.; Zagon, I.S. Endogenous Opioids Regulate Dendritic Growth and Spine Formation in Developing Rat Brain. Brain Res 1987, 416. [Google Scholar] [CrossRef]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of Stress throughout the Lifespan on the Brain, Behaviour and Cognition. Nat Rev Neurosci 2009, 10, 434–445. [Google Scholar] [CrossRef]
- Glover, V.; O’Donnell, K.J.; O’Connor, T.G.; Fisher, J. Prenatal Maternal Stress, Fetal Programming, and Mechanisms Underlying Later Psychopathology—A Global Perspective. Dev Psychopathol 2018, 30, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Letourneau, N.; Lebel, C. Prenatal Maternal Anxiety and Children’s Brain Structure and Function: A Systematic Review of Neuroimaging Studies. J Affect Disord 2018, 241, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.-N.; Chen, Y.-P.; Dong, Y.-P.; Shuai, H.-L.; Xiao, X.-M.; Reichetzeder, C.; Hocher, B. Late Gestational Maternal Serum Cortisol Is Inversely Associated with Fetal Brain Growth. Neurosci Biobehav Rev 2012, 36, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Eberle, C.; Fasig, T.; Brüseke, F.; Stichling, S. Impact of Maternal Prenatal Stress by Glucocorticoids on Metabolic and Cardiovascular Outcomes in Their Offspring: A Systematic Scoping Review. PLoS One 2021, 16, e0245386. [Google Scholar] [CrossRef]
- Dunkel Schetter, C.; Tanner, L. Anxiety, Depression and Stress in Pregnancy. Curr Opin Psychiatry 2012, 25, 141–148. [Google Scholar] [CrossRef]
- Buss, C.; Davis, E.P.; Muftuler, L.T.; Head, K.; Sandman, C.A. High Pregnancy Anxiety during Mid-Gestation Is Associated with Decreased Gray Matter Density in 6–9-Year-Old Children. Psychoneuroendocrinology 2010, 35, 141–153. [Google Scholar] [CrossRef]
- van den Heuvel, M.I.; Hect, J.L.; Smarr, B.L.; Qawasmeh, T.; Kriegsfeld, L.J.; Barcelona, J.; Hijazi, K.E.; Thomason, M.E. Maternal Stress during Pregnancy Alters Fetal Cortico-Cerebellar Connectivity in Utero and Increases Child Sleep Problems after Birth. Sci Rep 2021, 11, 2228. [Google Scholar] [CrossRef]
- Ulupinar, E.; Yucel, F. Prenatal Stress Reduces Interneuronal Connectivity in the Rat Cerebellar Granular Layer. In Proceedings of the Neurotoxicology and Teratology; May 2005; Vol. 27; pp. 475–484. [Google Scholar]
- Ulupinar, E.; Yucel, F.; Ortug, G. The Effects of Prenatal Stress on the Purkinje Cell Neurogenesis. Neurotoxicol Teratol 2006, 28, 86–94. [Google Scholar] [CrossRef]
- Pascual, R.; Ebner, D.; Araneda, R.; Urqueta, M.J.; Bustamante, C. Maternal Stress Induces Long-Lasting Purkinje Cell Developmental Impairments in Mouse Offspring. Eur J Pediatr 2010, 169, 1517–1522. [Google Scholar] [CrossRef]
- Pascual, R.; Valencia, M.; Bustamante, C. Purkinje Cell Dendritic Atrophy Induced by Prenatal Stress Is Mitigated by Early Environmental Enrichment. Neuropediatrics 2015, 46, 37–43. [Google Scholar] [CrossRef]
- Pascual, R.; Santander, O.; Cuevas, I.; Valencia, M. Prenatal Glucocorticoid Administration Persistently Increased the Immunohistochemical Expression of Type-1 Metabotropic Glutamate Receptor and Purkinje Cell Dendritic Growth in the Cerebellar Cortex of the Rat. Rom J Morphol Embryol 2017, 58, 67–72. [Google Scholar] [PubMed]
- Rivas-Manzano, P.; Ramírez-Escoto, M.M.; Rosa-Rugerio, C.D. la; Rugerio-Vargas, C.; Ortiz-Hernández, R.; Torres-Ramírez, N. Argentic Staining Reveals Changes in Cerebellar Tissue Organisation by Prenatal Glucocorticoid Administration in Rats. Histol Histopathol 2021, 36, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Wilber, A.A.; Lin, G.L.; Wellman, C.L. Neonatal Corticosterone Administration Impairs Adult Eyeblink Conditioning and Decreases Glucocorticoid Receptor Expression in the Cerebellar Interpositus Nucleus. Neuroscience 2011, 177, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Wilber, A.A.; Wellman, C.L. Neonatal Maternal Separation Alters the Development of Glucocorticoid Receptor Expression in the Interpositus Nucleus of the Cerebellum. International Journal of Developmental Neuroscience 2009, 27, 649–654. [Google Scholar] [CrossRef]
- Schneider, E.R.; Civillico, E.F.; Wang, S.S.-H. Calcium-Based Dendritic Excitability and Its Regulation in the Deep Cerebellar Nuclei. J Neurophysiol 2013, 109, 2282–2292. [Google Scholar] [CrossRef]
- Miki, T.; Yokoyama, T.; Kusaka, T.; Suzuki, S.; Ohta, K.; Warita, K.; Wang, Z.-Y.; Ueki, M.; Sumitani, K.; Bellinger, F.P.; et al. Early Postnatal Repeated Maternal Deprivation Causes a Transient Increase in OMpg and BDNF in Rat Cerebellum Suggesting Precocious Myelination. J Neurol Sci 2014, 336, 62–67. [Google Scholar] [CrossRef]
- Zhang, L.-X.; Levine, S.; Dent, G.; Zhan, Y.; Xing, G.; Okimoto, D.; Gordon, M.K.; Post, R.M.; Smith, M.A. Maternal Deprivation Increases Cell Death in the Infant Rat Brain; 2002; Vol. 133;
- Llorente-Berzal, A.; Manzanedo, C.; Daza-Losada, M.; Valero, M.; López-Gallardo, M.; Aguilar, M.A.; Rodríguez-Arias, M.; Miñarro, J.; Viveros, M.-P. Sex-Dependent Effects of Early Maternal Deprivation on MDMA-Induced Conditioned Place Preference in Adolescent Rats: Possible Neurochemical Correlates. Toxicology 2013, 311, 78–86. [Google Scholar] [CrossRef]
- Schmidt, M.; Enthoven, L.; Van Der Mark, M.; Levine, S.; De Kloet, E.R.; Oitzl, M.S. The Postnatal Development of the Hypothalamic-Pituitary-Adrenal Axis in the Mouse. International Journal of Developmental Neuroscience 2003, 21, 125–132. [Google Scholar] [CrossRef]
- Sapolsky’, R.M.; Meaney’, M.J. Maturation of the Adrenocortical Stress Response: Neuroendocrine Control Mechanisms and the Stress Hyporesponsive Period; 1986; Vol. 11;
- Schmidt, M. V. Stress-Hyporesponsive Period. In Stress: Physiology, Biochemistry, and Pathology Handbook of Stress Series, Volume 3; Elsevier, 2019; pp. 49–56 ISBN 9780128131466.
- Noguchi, K.K.; Walls, K.C.; Wozniak, D.F.; Olney, J.W.; Roth, K.A.; Farber, N.B. Acute Neonatal Glucocorticoid Exposure Produces Selective and Rapid Cerebellar Neural Progenitor Cell Apoptotic Death. Cell Death Differ 2008, 15, 1582–1592. [Google Scholar] [CrossRef]
- Blumberg, M.; Dooley, J.; Sokoloff, G. The Developing Brain Revealed during Sleep. Curr Opin Physiol 2020, 15. [Google Scholar] [CrossRef]
- Canto, C.B.; Bauer, S.; Hoogland, T.M.; Hoedemaker, H.H.; Geelen, C.; Loyola, S.; Miaja, P.; Zeeuw, C.I. De Sleep-State Dependent Cerebellar Processing in Adult Mice. bioRxiv, 5647. [Google Scholar] [CrossRef]
- Tagliazucchi, E.; Laufs, H. Decoding Wakefulness Levels from Typical FMRI Resting-State Data Reveals Reliable Drifts between Wakefulness and Sleep. Neuron 2014, 82, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, L.; Yang, G.; Gan, W.-B. REM Sleep Selectively Prunes and Maintains New Synapses in Development and Learning. Nat Neurosci 2017, 20, 427–437. [Google Scholar] [CrossRef]
- Dooley, J.C.; Sokoloff, G.; Blumberg, M.S. Movements during Sleep Reveal the Developmental Emergence of a Cerebellar-Dependent Internal Model in Motor Thalamus. Curr Biol 2021, 31, 5501–5511.e5. [Google Scholar] [CrossRef]
- Mukherjee, D.; Sokoloff, G.; Blumberg, M.S. Corollary Discharge in Precerebellar Nuclei of Sleeping Infant Rats. Elife 2018, 7. [Google Scholar] [CrossRef]
- Crapse, T.B.; Sommer, M.A. Corollary Discharge across the Animal Kingdom. Nat Rev Neurosci 2008, 9, 587–600. [Google Scholar] [CrossRef]
- Hu, Y.; Korovaichuk, A.; Astiz, M.; Schroeder, H.; Islam, R.; Barrenetxea, J.; Fischer, A.; Oster, H.; Bringmann, H. Functional Divergence of Mammalian TFAP2a and TFAP2b Transcription Factors for Bidirectional Sleep Control. Genetics 2020, 216, 735–752. [Google Scholar] [CrossRef]
- Hu, Y.; Bringmann, H. Tfap2b Acts in GABAergic Neurons to Control Sleep in Mice. Sci Rep 2023, 13, 8026. [Google Scholar] [CrossRef]
- Pardo, G.V.E.; Goularte, J.F.; Hoefel, A.L.; de Castro, A.L.; Kucharski, L.C.; da Rosa Araujo, A.S.; Lucion, A.B. Effects of Sleep Restriction during Pregnancy on the Mother and Fetuses in Rats. Physiol Behav 2016, 155, 66–76. [Google Scholar] [CrossRef]
- Canto, C.B.; Onuki, Y.; Bruinsma, B.; van der Werf, Y.D.; De Zeeuw, C.I. The Sleeping Cerebellum. Trends Neurosci 2017, 40, 309–323. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Canto, C.B. Sleep Deprivation Directly Following Eyeblink-Conditioning Impairs Memory Consolidation. Neurobiol Learn Mem 2020, 170, 107165. [Google Scholar] [CrossRef]
- Vollmer, B.; Edmonds, C.J. School Age Neurological and Cognitive Outcomes of Fetal Growth Retardation or Small for Gestational Age Birth Weight. Front Endocrinol (Lausanne) 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, J.; Schleger, F.; Keune, J.; Wiechers, C.; Pauluschke-Froehlich, J.; Weiss, M.; Conzelmann, A.; Brucker, S.; Preissl, H.; Kiefer-Schmidt, I. Impact of Intrauterine Growth Restriction on Cognitive and Motor Development at 2 Years of Age. Front Physiol 2018, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Chu, A. Intrauterine Growth Restriction: Hungry for an Answer. Physiology (Bethesda) 2016, 31, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fu, W.; Liu, J. Neurodevelopment in Children with Intrauterine Growth Restriction: Adverse Effects and Interventions. J Matern Fetal Neonatal Med 2016, 29, 660–668. [Google Scholar] [CrossRef]
- Miller, S.L.; Huppi, P.S.; Mallard, C. The Consequences of Fetal Growth Restriction on Brain Structure and Neurodevelopmental Outcome. J Physiol 2016, 594, 807–823. [Google Scholar] [CrossRef]
- Olivier, P.; Baud, O.; Bouslama, M.; Evrard, P.; Gressens, P.; Verney, C. Moderate Growth Restriction: Deleterious and Protective Effects on White Matter Damage. Neurobiol Dis 2007, 26, 253–263. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain Injury in Premature Infants: A Complex Amalgam of Destructive and Developmental Disturbances. Lancet Neurol 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Eixarch, E.; Muñoz-Moreno, E.; Bargallo, N.; Batalle, D.; Gratacos, E. Motor and Cortico-Striatal-Thalamic Connectivity Alterations in Intrauterine Growth Restriction. Am J Obstet Gynecol 2016, 214, 725.e1–725.e9. [Google Scholar] [CrossRef]
- Fischi-Gomez, E.; Muñoz-Moreno, E.; Vasung, L.; Griffa, A.; Borradori-Tolsa, C.; Monnier, M.; Lazeyras, F.; Thiran, J.-P.; Hüppi, P.S. Brain Network Characterization of High-Risk Preterm-Born School-Age Children. Neuroimage Clin 2016, 11, 195–209. [Google Scholar] [CrossRef]
- Chang, J.L.; Bashir, M.; Santiago, C.; Farrow, K.; Fung, C.; Brown, A.S.; Dettman, R.W.; Dizon, M.L.V. Intrauterine Growth Restriction and Hyperoxia as a Cause of White Matter Injury. Dev Neurosci 2018, 40, 344–357. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Fattakhov, N.; Buddington, R.K.; Chizhikov, V. V Intrauterine Growth Restriction Compromises Cerebellar Development by Affecting Radial Migration of Granule Cells via the JamC/Pard3a Molecular Pathway. Exp Neurol 2021, 336, 113537. [Google Scholar] [CrossRef] [PubMed]
- McDougall, A.R.A.; Wiradjaja, V.; Azhan, A.; Li, A.; Hale, N.; Wlodek, M.E.; Hooper, S.B.; Wallace, M.J.; Tolcos, M. Intrauterine Growth Restriction Alters the Postnatal Development of the Rat Cerebellum. Dev Neurosci 2017, 39, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Tolcos, M.; McDougall, A.; Shields, A.; Chung, Y.; O’Dowd, R.; Turnley, A.; Wallace, M.; Rees, S. Intrauterine Growth Restriction Affects Cerebellar Granule Cells in the Developing Guinea Pig Brain. Dev Neurosci 2018, 40, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Bonatto, F.; Polydoro, M.; Andrades, M.É.; Júnior, M.L.C. da F.; Dal-Pizzol, F.; Rotta, L.N.; Souza, D.O.; Perry, M.L.; Moreira, J.C.F. Effects of Maternal Protein Malnutrition on Oxidative Markers in the Young Rat Cortex and Cerebellum. Neurosci Lett 2006, 406, 281–284. [Google Scholar] [CrossRef]
- Chanez, C.; Rabin, O.; Heroux, M.; Giguere, J.F. Cerebral Amino Acid Changes in an Animal Model of Intrauterine Growth Retardation. Metab Brain Dis 1993, 8, 61–72. [Google Scholar] [CrossRef]
- Tita, A.T.N.; Andrews, W.W. Diagnosis and Management of Clinical Chorioamnionitis. Clin Perinatol 2010, 37, 339–354. [Google Scholar] [CrossRef]
- Ylijoki, M.; Lehtonen, L.; Lind, A.; Ekholm, E.; Lapinleimu, H.; Kujari, H.; Haataja, L. Chorioamnionitis and Five-Year Neurodevelopmental Outcome in Preterm Infants. Neonatology 2016, 110, 286–295. [Google Scholar] [CrossRef]
- Hendson, L.; Russell, L.; Robertson, C.M.T.; Liang, Y.; Chen, Y.; Abdalla, A.; Lacaze-Masmonteil, T. Neonatal and Neurodevelopmental Outcomes of Very Low Birth Weight Infants with Histologic Chorioamnionitis. J Pediatr 2011, 158, 397–402. [Google Scholar] [CrossRef]
- Nasef, N.; Shabaan, A.E.; Schurr, P.; Iaboni, D.; Choudhury, J.; Church, P.; Dunn, M.S. Effect of Clinical and Histological Chorioamnionitis on the Outcome of Preterm Infants. Am J Perinatol 2013, 30, 59–68. [Google Scholar] [CrossRef]
- Pappas, A.; Kendrick, D.E.; Shankaran, S.; Stoll, B.J.; Bell, E.F.; Laptook, A.R.; Walsh, M.C.; Das, A.; Hale, E.C.; Newman, N.S.; et al. Chorioamnionitis and Early Childhood Outcomes among Extremely Low-Gestational-Age Neonates. JAMA Pediatr 2014, 168, 137–147. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Sullivan, N.R.; Soul, J.S.; Robertson, R.L.J.; Moore, M.; Ringer, S.A.; Volpe, J.J.; du Plessis, A.J. Positive Screening for Autism in Ex-Preterm Infants: Prevalence and Risk Factors. Pediatrics 2008, 121, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.G.; Willis, K.A.; Jobe, A.; Ambalavanan, N. Chorioamnionitis and Neonatal Outcomes. Pediatr Res 2022, 91, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Normann, E.; Lacaze-Masmonteil, T.; Eaton, F.; Schwendimann, L.; Gressens, P.; Thébaud, B. A Novel Mouse Model of Ureaplasma-Induced Perinatal Inflammation: Effects on Lung and Brain Injury. Pediatr Res 2009, 65, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Van Steenwinckel, J.; Fleiss, B.; Scheuer, T.; Bührer, C.; Faivre, V.; Lemoine, S.; Blugeon, C.; Schwendimann, L.; Csaba, Z.; et al. A Unique Cerebellar Pattern of Microglia Activation in a Mouse Model of Encephalopathy of Prematurity. Glia 2022, 70, 1699–1719. [Google Scholar] [CrossRef]
- Gudsnuk, K.M.A.; Champagne, F.A. Epigenetic Effects of Early Developmental Experiences. Clin Perinatol 2011, 38, 703–717. [Google Scholar] [CrossRef]
- Broersen, R.; Canto, C.B.; De Zeeuw, C.I. Cerebellar Nuclei: Associative Motor Learning in Zebrafish. Current Biology 2023, 33, R867–R870. [Google Scholar] [CrossRef]
- Canto, C.B.; Broersen, R.; De Zeeuw, C.I. Intrinsic Excitement in Cerebellar Nuclei Neurons during Learning. Proceedings of the National Academy of Sciences 2018, 115, 9824–9826. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Gornati, S. V; Canto, C.B.; Libster, A.M.; Schonewille, M.; De Zeeuw, C.I.; Hoebeek, F.E. Activity of Cerebellar Nuclei Neurons Correlates with ZebrinII Identity of Their Purkinje Cell Afferents. Cells 2021, 10. [Google Scholar] [CrossRef]
- Witter, L.; Canto, C.B.; Hoogland, T.M.; de Gruijl, J.R.; De Zeeuw, C.I. Strength and Timing of Motor Responses Mediated by Rebound Firing in the Cerebellar Nuclei after Purkinje Cell Activation. Front Neural Circuits 2013, 7, 133. [Google Scholar] [CrossRef]
- Hoogland, T.M.; De Gruijl, J.R.; Witter, L.; Canto, C.B.; De Zeeuw, C.I. Role of Synchronous Activation of Cerebellar Purkinje Cell Ensembles in Multi-Joint Movement Control. Curr Biol 2015, 25, 1157–1165. [Google Scholar] [CrossRef]
- Lange, W. Regional Differences in the Distribution of Golgi Cells in the Cerebellar Cortex of Man and Some Other Mammals. Cell Tissue Res 1974, 153, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling Transformations of Neurodevelopmental Sequences across Mammalian Species. The Journal of Neuroscience 2013, 33, 7368–7383. [Google Scholar] [CrossRef] [PubMed]
- Haldipur, P.; Dang, D.; Millen, K.J. Embryology. Handb Clin Neurol 2018, 154, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Abou-Donia, M.B.; Khan, W.A.; Dechkovskaia, A.M.; Goldstein, L.B.; Bullman, S.L.; Abdel-Rahman, A. In Utero Exposure to Nicotine and Chlorpyrifos Alone, and in Combination Produces Persistent Sensorimotor Deficits and Purkinje Neuron Loss in the Cerebellum of Adult Offspring Rats. Arch Toxicol 2006, 80, 620–631. [Google Scholar] [CrossRef]
- Abdel-Rahman, A.; Dechkovskaia, A.M.; Sutton, J.M.; Chen, W.C.; Guan, X.; Khan, W.A.; Abou-Donia, M.B. Maternal Exposure of Rats to Nicotine via Infusion during Gestation Produces Neurobehavioral Deficits and Elevated Expression of Glial Fibrillary Acidic Protein in the Cerebellum and CA1 Subfield in the Offspring at Puberty. Toxicology 2005, 209, 245–261. [Google Scholar] [CrossRef]
- Wojcinski, A.; Morabito, M.; Lawton, A.K.; Stephen, D.N.; Joyner, A.L. Genetic Deletion of Genes in the Cerebellar Rhombic Lip Lineage Can Stimulate Compensation through Adaptive Reprogramming of Ventricular Zone-Derived Progenitors. Neural Dev 2019, 14, 4. [Google Scholar] [CrossRef]
- Al-Amri, I.S.; Kadim, I.T.; Alkindi, A.; Khalaf, S.K.; Hamaed, A.; Al-Yaqoobi, S.S.; Al-Harrasi, A.S.; Al-Hashmi, S.A.; Al-Maasbi, A.A.; Al-Harthy, R.S.; et al. Effects of Prenatal Tobacco Smoke on Cerebellum Histomorphological Changes at Critical Developmental Stages in CD-1 Mice. International Journal of Morphology 2021, 39, 318–326. [Google Scholar] [CrossRef]
- Chang, J.L.; Bashir, M.; Santiago, C.; Farrow, K.; Fung, C.; Brown, A.S.; Dettman, R.W.; Dizon, M.L.V. Intrauterine Growth Restriction and Hyperoxia as a Cause of White Matter Injury. Dev Neurosci 2018, 40, 344–357. [Google Scholar] [CrossRef]
- van der Heijden, M.E.; Sillitoe, R. V. Interactions Between Purkinje Cells and Granule Cells Coordinate the Development of Functional Cerebellar Circuits. Neuroscience 2021, 462, 4–21. [Google Scholar] [CrossRef]
- Newman, J.; Tong, X.; Tan, A.; Paiva, V.N. De; Presicce, P.; Kannan, P.S.; Williams, K.; Damianos, A.; Newsam, M.T.; Benny, M.; et al. Single-Nucleus RNA-Seq Reveals Disrupted Cell Maturation by Chorioamnionitis in the Preterm Cerebellum of Nonhuman Primates. bioRxiv, 4997. [Google Scholar] [CrossRef]
- Newman, J.; Tong, X.; Tan, A.; Yeasky, T.; De Paiva, V.N.; Presicce, P.; Kannan, P.S.; Williams, K.; Damianos, A.; Tamase Newsam, M.; et al. Chorioamnionitis Accelerates Granule Cell and Oligodendrocyte Maturation in the Cerebellum of Preterm Nonhuman Primates. J Neuroinflammation 2024, 21, 16. [Google Scholar] [CrossRef]
- Topper, L.A.; Baculis, B.C.; Valenzuela, C.F. Exposure of Neonatal Rats to Alcohol Has Differential Effects on Neuroinflammation and Neuronal Survival in the Cerebellum and Hippocampus. J Neuroinflammation 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.L.; Crocker, N.; Mattson, S.N.; Riley, E.P. Neuroimaging and Fetal Alcohol Spectrum Disorders. Dev Disabil Res Rev 2009, 15, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kalinovsky, A.; Boukhtouche, F.; Blazeski, R.; Bornmann, C.; Suzuki, N.; Mason, C.A.; Scheiffele, P. Development of Axon-Target Specificity of Ponto-Cerebellar Afferents. PLoS Biol 2011, 9, e1001013. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, K.W.S.; Zhang, L.-L. Ontogeny of Afferents to the Fetal Rat Cerebellum. Cells Tissues Organs 1992, 145, 17–23. [Google Scholar] [CrossRef]
- Roque, A.; Lajud, N.; Valdez, J.J.; Torner, L. Early-Life Stress Increases Granule Cell Density in the Cerebellum of Male Rats. Brain Res 2019, 1723. [Google Scholar] [CrossRef]
- Stoodley, C.J. Distinct Regions of the Cerebellum Show Gray Matter Decreases in Autism, ADHD, and Developmental Dyslexia. Front Syst Neurosci 2014, 8, 92. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Schmahmann, J.D. Functional Topography of the Human Cerebellum. Handb Clin Neurol 2018, 154, 59–70. [Google Scholar] [CrossRef]
- Stoodley, C.J. The Cerebellum and Cognition: Evidence from Functional Imaging Studies. The Cerebellum 2012, 11, 352–365. [Google Scholar] [CrossRef]
- D’Mello, A.M.; Stoodley, C.J. Cerebro-Cerebellar Circuits in Autism Spectrum Disorder. Front Neurosci 2015, 9, 408. [Google Scholar] [CrossRef]
- Stoodley, C.J. The Cerebellum and Neurodevelopmental Disorders. Cerebellum 2016, 15. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Chilingaryan, G.; Guizard, N.; Robertson, R.L.; Du Plessis, A.J. Cerebellar Injury in the Premature Infant Is Associated with Impaired Growth of Specific Cerebral Regions. Pediatr Res 2010, 68. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Li, Z.Q.; Sun, Y.; Wang, T.; Wan, C.L.; Li, X.W.; Zhao, X.Z.; Feng, G.Y.; Li, Sh.; St Clair, D.; et al. The Role of Pro-Inflammatory Factors in Mediating the Effects on the Fetus of Prenatal Undernutrition: Implications for Schizophrenia. Schizophr Res 2008, 99, 48–55. [Google Scholar] [CrossRef]
- Walker, C.K.; Krakowiak, P.; Baker, A.; Hansen, R.L.; Ozonoff, S.; Hertz-Picciotto, I. Preeclampsia, Placental Insufficiency, and Autism Spectrum Disorder or Developmental Delay. JAMA Pediatr 2015, 169, 154. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Majrashi, M.; Ramesh, S.; Govindarajulu, M.; Bloemer, J.; Fujihashi, A.; Crump, B.R.; Hightower, H.; Bhattacharya, S.; Moore, T.; et al. Assessment of the Cerebellar Neurotoxic Effects of Nicotine in Prenatal Alcohol Exposure in Rats. Life Sci 2018, 194, 177–184. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Anatomy of the human and rodent cerebellum. (a) An unfolded view of the human (top) and rodent (bottom) cerebellum. (Top) The three cerebellar zones (right hemisphere: medial, intermediate and lateral) are interconnected with different parts of the cerebellar nuclei (left hemisphere), indicated with the same color-coding. The areas of the cortex served by the cerebellar arteries are indicated on the left (adapted from [25]). (Bottom) Schematic of the mouse cerebellum with sagittal microzones visualised based on the expression levels of aldolase-C (also known as zebrin II (ZII); right hemisphere). Different shades of grey indicate how strong (dark grey=strong ZII+, white=ZII-) ZII is expressed. (b) The cytoarchitectures of neighbouring microzones such as those indicated in (a, bottom) are almost identical (adapted with permission from [26]). The microzones, microcomplexes and micromodules are all build up in a comparable manner. Individual zones communicate with each other through e.g. parallel fibers of granule cells that cross the zones/complexes/modules.
Figure 1.
Anatomy of the human and rodent cerebellum. (a) An unfolded view of the human (top) and rodent (bottom) cerebellum. (Top) The three cerebellar zones (right hemisphere: medial, intermediate and lateral) are interconnected with different parts of the cerebellar nuclei (left hemisphere), indicated with the same color-coding. The areas of the cortex served by the cerebellar arteries are indicated on the left (adapted from [25]). (Bottom) Schematic of the mouse cerebellum with sagittal microzones visualised based on the expression levels of aldolase-C (also known as zebrin II (ZII); right hemisphere). Different shades of grey indicate how strong (dark grey=strong ZII+, white=ZII-) ZII is expressed. (b) The cytoarchitectures of neighbouring microzones such as those indicated in (a, bottom) are almost identical (adapted with permission from [26]). The microzones, microcomplexes and micromodules are all build up in a comparable manner. Individual zones communicate with each other through e.g. parallel fibers of granule cells that cross the zones/complexes/modules.
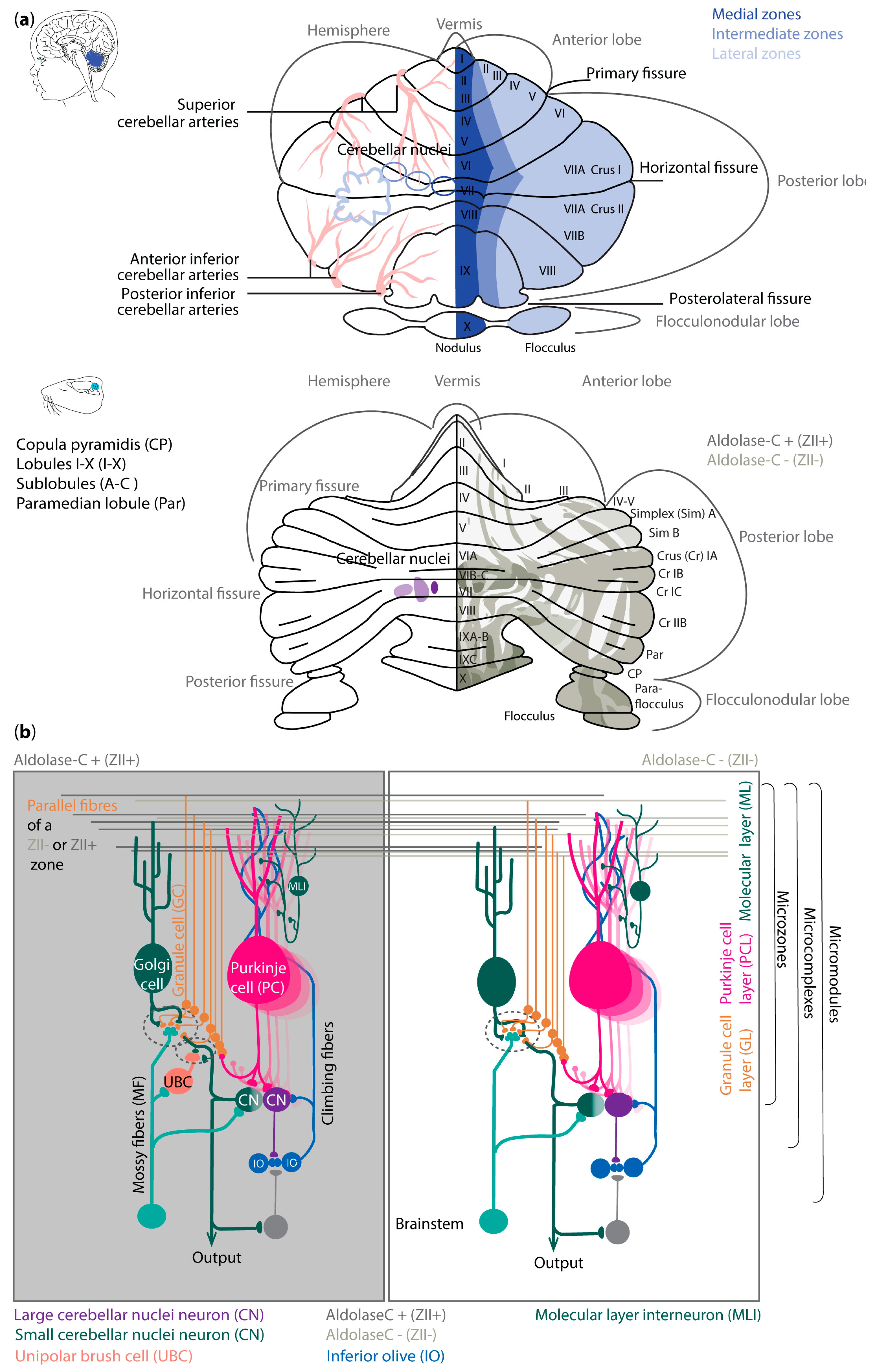
Figure 2.
Developmental timeline of the human cerebellum. Top row: Sagittal sections of the outline of the human cerebellum at 5 time points during development, starting from gestational week (gw) 11 (adapted from [20]) with below different aspects of the development being highlighted. Bottom row: The timeline of the development of individual cerebellar cell types. The duration and timing of development is indicated with a color-coded bar with a distinct bar length. Excitatory (+) granule cells (GC, orange) derive from the rhombic lips (RL, red). Inhibitory (-) GABAergic Purkinje cells (PC, pink) derive from the ventricular zone (VZ, green).
Figure 2.
Developmental timeline of the human cerebellum. Top row: Sagittal sections of the outline of the human cerebellum at 5 time points during development, starting from gestational week (gw) 11 (adapted from [20]) with below different aspects of the development being highlighted. Bottom row: The timeline of the development of individual cerebellar cell types. The duration and timing of development is indicated with a color-coded bar with a distinct bar length. Excitatory (+) granule cells (GC, orange) derive from the rhombic lips (RL, red). Inhibitory (-) GABAergic Purkinje cells (PC, pink) derive from the ventricular zone (VZ, green).
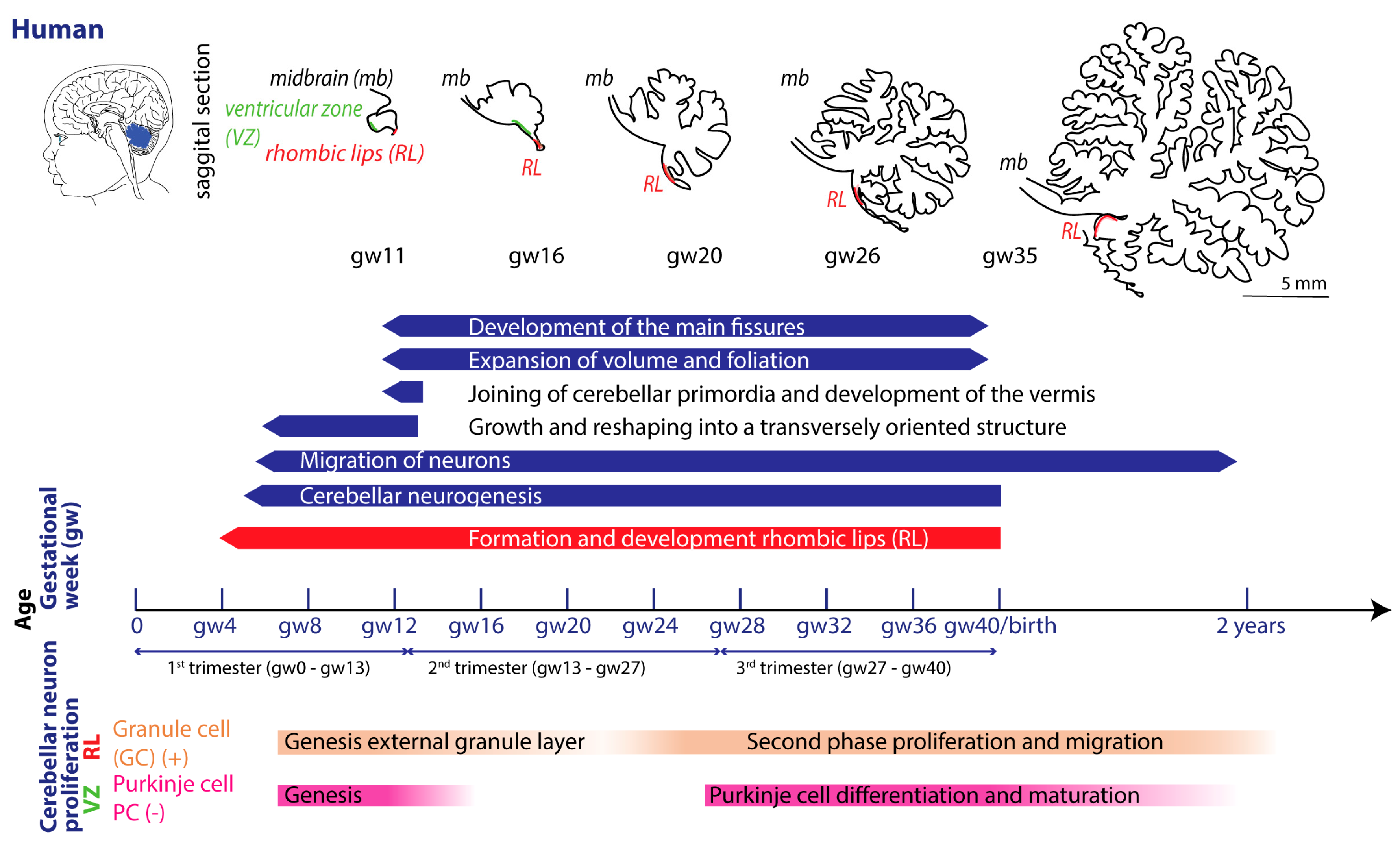
Figure 3.
Developmental timeline of the rodent cerebellum. Top row: The first two outlines show the differentiation of the hind- and midbrain (mb) and the relative location of the cerebellar anlage [70]. Embryonic day (E)18.5, postnatal ~(P)4 and P21 show the outlines of cerebellar midsagittal cuts (adapted from [71]). Arrowheads indicate the migration direction of individual neuron types. At ~P4 and P21 also a zoom-in of the cerebellar cortex is presented to show the location of individual cell types in the (external) granule layer (GL), Purkinje cell layer (PCL) and molecular layer (ML). The inputs of the two precerebellar systems, the climbing fibers (CF) and mossy fibers (MF), and the output to the cerebellar nuclei (CN) is also indicated. Below, the timeline of the development of the cerebellum and the timeline of the development of individual cerebellar cell types is drawn.
Figure 3.
Developmental timeline of the rodent cerebellum. Top row: The first two outlines show the differentiation of the hind- and midbrain (mb) and the relative location of the cerebellar anlage [70]. Embryonic day (E)18.5, postnatal ~(P)4 and P21 show the outlines of cerebellar midsagittal cuts (adapted from [71]). Arrowheads indicate the migration direction of individual neuron types. At ~P4 and P21 also a zoom-in of the cerebellar cortex is presented to show the location of individual cell types in the (external) granule layer (GL), Purkinje cell layer (PCL) and molecular layer (ML). The inputs of the two precerebellar systems, the climbing fibers (CF) and mossy fibers (MF), and the output to the cerebellar nuclei (CN) is also indicated. Below, the timeline of the development of the cerebellum and the timeline of the development of individual cerebellar cell types is drawn.
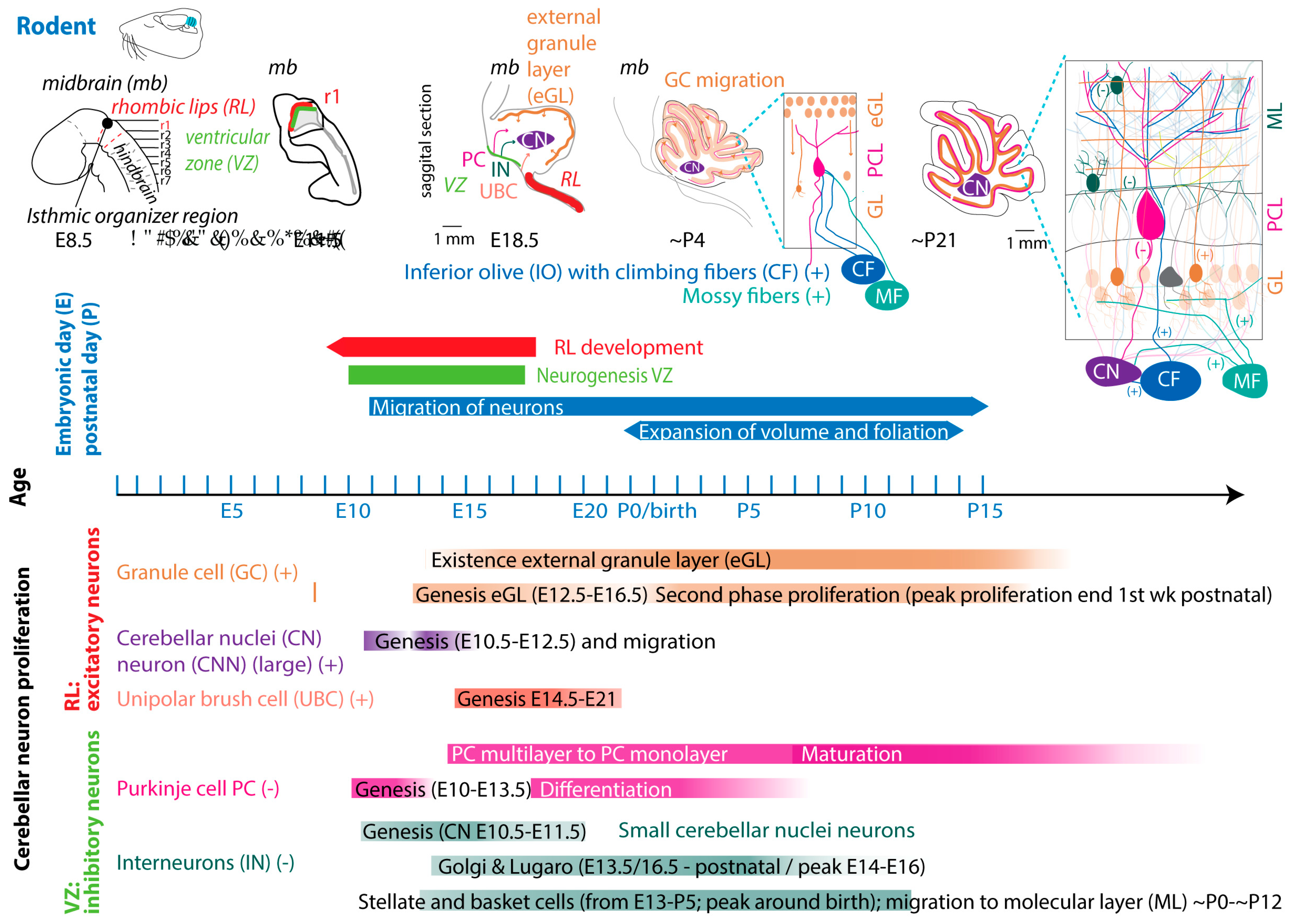
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



