Submitted:
02 November 2024
Posted:
05 November 2024
You are already at the latest version
Abstract
Estrogen receptor α (ERα) drives 2 out of 3 breast cancers and therefore ERα is a major therapeutic target for ER-positive breast cancer patients. Drugs that inhibit ERα activity or block estrogen synthesis in the body are currently being used in the clinic to treat ER-positive breast cancer and have been quite successful in controlling breast cancer progression for the majority of patients. However, ER-positive breast cancer often becomes resistant to these endocrine therapies, leading to endocrine-resistant metastatic breast cancer, a very aggressive cancer that leads to death. Recent large-scale genomic studies have revealed a series of activating somatic mutations in the ERα gene (ESR1) in endocrine-resistant metastatic breast cancer patients. Of these, Y537S and D538G mutations are found at a much higher rate in patients with metastatic breast cancer. Remarkably, these mutations produce an ERα with much higher transcriptional activity than wild type in the absence of estradiol, and traditional endocrine therapy has poor efficacy against ER mutants. Therefore, the development of new drugs that target ER mutants is an unmet clinical need for endocrine-resistant metastatic breast cancer. This review summarizes the recent preclinical and clinical trials targeting estrogen receptor mutant breast cancer.
Keywords:
Breast cancer
; Estrogen receptor mutants
; endocrine-resistant
; Y537S
; D538G
1. Introduction
Breast cancer is the most frequently diagnosed cancer in women and second leading cause of cancer death in women. There are more than 2.2 million new cases of breast cancer each year worldwide. It is predicted that the future burden of breast cancer is expected to increase to over 3 million new cases and 1 million deaths in 2040 [1]. Breast cancer is highly heterogenous with distinct molecular features and can be categorized as either positive or negative for the immunohistochemical expression of three hormone receptors: estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2); a cancer may be positive or negative for any of these, and those that are negative for all three are termed triple negative breast cancer (TNBC). Four main molecular subtypes are also used in the research settings of breast cancer: Luminal A, Luminal B, HER2 enriched and Triple negative/Basal like. Luminal A breast cancer is the most frequently diagnosed molecular subtype and makes up about 50 to 60% of all breast cancer cases [2]. Luminal A breast cancers express estrogen receptor and progesterone receptor and are therefore commonly called hormone receptor-positive (HR+) breast cancer. Luminal B breast cancers account for about 15 to 20% of all breast cancer cases. Luminal B breast cancer is also HR+ and can be either HER2 positive (HER2+) or HER2 negative (HER2-). Luminal B breast cancer expresses a high level of Ki-67. HER2-enriched breast cancers make up about 10 to 15% of all breast cancer cases and may be HR+ or HR-. TNBC represents 10% of all breast cancer cases.
In 1962, Jenson and Jacobsen were the first to describe that the biological effects of estrogen are arbitrated through a receptor that was subsequently identified by Toft and Gorski [3]. ERα was cloned in 1986 [4,5]. ERα is a member of the nuclear hormone receptor superfamily and acts as a transcription factor that regulates gene networks in various biological processes including cell growth and proliferation [6]. ERα activation is primarily regulated by estradiol, an 18-carbon steroid with an aromatic A-ring that is predominantly synthesized in the ovary. Estradiol exerts its actions by binding to ERα in the cytoplasm of the breast epithelial cell. Upon binding, ERα dimerizes and translocates from the cytosol to the nucleus, where ERα dimers can bind to specific estrogen response element (ERE) sequences in the genome and recruit coactivators (CoA) to form transcriptionally active complexes that directly drive the expression of genes that promote cell growth, proliferation and survival (Figure 1). Estrogen receptor complexes can also regulate gene transcription via tethering to other transcription factors (such as AP-1) without binding to ERE sequences (non-genomic effects). Tertiary ER-regulated genes lack ERE sequences, and the ER-mediated non-genomic signaling that occurs through protein-protein interactions with other transcription factors at their respective response elements affects a broad range of estrogen-regulated genes in breast epithelial cells [7].
2. ERα Domain Structure
ERα is encoded by the gene ESR1 on chromosome 6 (6q25.1) [8]. ERα comprises 595 amino acids and six structural domains, namely A to F (Figure 2) [9]. The amino-terminal A/B domain contains the transcriptional activation function (AF)-1 which interacts with coregulators and enhances gene transcription in the cell. The A/B domain contains serine phosphorylation sites at positions 104,106, 118 and 167 that are mainly modified in response to insulin-like growth factor 1, epidermal growth factor, and tumor growth factor α. Phosphorylation of Ser 118 can lead to ligand-independent activation through conformational changes in the A/B domain [10,11]. The C domain, also called the DNA binding domain (DBD), contains two zinc finger structures that interact with DNA and influence ER dimer formation. The C domain is highly conserved and responsible for binding to specific estrogen response element (ERE) regions of estrogen-regulated genes [12]. The D domain, also known as the hinge region, contains phosphorylation, acetylation, and sumoylation sites that regulate transcriptional activity [13,14]. Lysines 302 and 303 in the hinge region protect ERα from basal degradation [15]. The E domain, also called the ligand binding domain (LBD), contains 11 alpha-helices (H1, H3-12) in a globular assembly with a deep pocket as reported in 1997 with the first crystal structure of the ERα LBD with the natural ligand 17β estradiol [16]. In response to estradiol, helices 3, 4, 5 and 12 of the LBD form a cleft where coactivators containing the leucine-X-X-leucine-leucine (LxxLL) motif can bind to trigger gene transcription [17]. When partial agonists and antagonists bind, the LBD helix 12 shifts, altering the cleft and blocking interaction with the coactivator LxxLL motif [18]. Helix 12 repositioning in response to binding of fulvestrant leads to the degradation of ERα [19,20,21]. The 45 residue F domain, also known as the carboxyl-terminal domain, is unique to ERs among nuclear receptors. The ERα F domain has 45 amino acids. The F-domain is required for the partial agonist activity of tamoxifen and the effectiveness of E2-induced transcriptional activity function [22,23,24,25].
3. ERα in Human Breast Cancer
In normal breast tissue, ERα is expressed only around 10% of the epithelial cells however in the case of breast tumors, ERα is highly expressed (50% to 80%) [26]. ERα is a major oncogenic driver of breast cancer initiation and progression. Approximately 70% of breast cancers express ERα; since estrogen-mediated ERα signaling plays a central role in progression of these cancers, ERα is a prime therapeutic target for ER-positive breast cancer patients. In response to estradiol, ERα regulates several thousand genes in breast cancer cells that influence cancer cell growth and proliferation [27,28]. Genome-wide ERα chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) studies showed only a small fraction of ERα binding sites are distributed in the proximal promotor region: most ERα binding sites are located in the distal areas of ER target genes [28,29,30,31]. Brown and colleagues showed that ERα binding regions are highly enriched for the FOXA1 binding motif, and subsequently FOXA1 was identified as an important pioneer factor for ER-chromatin interactions in ER-positive breast cancer cells [32] and as necessary for E2-mediated gene expression [31]. Carroll and colleagues mapped the ERα binding sites in primary breast samples and showed that different ERα binding profiles are linked with different clinal outcomes of the breast cancer [33].
3.1. Endocrine Therapy in ER+ Breast Cancer
Endocrine therapy has been one of the most effective treatments for both early and advanced-stage ER+ breast cancer. Endocrine drugs inhibit ERα activity or block estrogen synthesis in the body. Currently, three types of drugs used in the clinic to treat ER+ breast cancer: selective estrogen receptor modulators, aromatase inhibitors, and selective estrogen receptor degraders.
Selective estrogen receptor modulators (SERMs) can act as estrogen agonists or antagonists depending on the target tissues. For example, tamoxifen is a classical SERM used widely to treat pre and postmenopausal women with ER+ breast cancer; it acts as an antagonist in breast tissue whereas it acts as an agonist in endometrium and bone [34,35]. Tamoxifen was developed in the late 1960s, its first clinical use was reported in 1971 [36], and it was approved by the Food and Drug Administration (FDA) in 1977. Tamoxifen is a non-steroidal drug and has been extensively used to treat ER+ breast cancer. The tamoxifen metabolite 4-hydroxytamoxifen (4-OHT) competes with estradiol (E2) at the ligand-binding site of ER, and 4-OHT-ER complexes that enter the nucleus bind to ERE regions and recruits corepressors, which lead to blocking a subset of E2-inducible genes and inhibits the growth of ER-containing breast cancer cells (Figure 3A). 4-OHT binding to the ERα LBD induces a different conformation than E2 binding, preventing recruitment of coactivators [37].
Aromatase inhibitors target cytochrome p450 19A1 (aromatase), an enzyme involved in the bio-synthetic conversion of androgens into estrogens. Aromatase inhibitors block estrogen production in the body, which may stop the growth of ER-containing cancer cells that need estrogen to grow. In the presence of aromatase inhibitors, ERs are inactive in the cell (Figure 3B). Three aromatase inhibitors (letrozole, anastrozole, and exemestane) are currently approved by the FDA and used in the clinic to treat early locally advanced, and metastatic ER+ breast cancer [38] . They may also be used to help prevent breast cancer in patients who are at a high risk of developing it. Letrozole is a non-steroidal drug that targets the aromatase active site [39].
Selective estrogen receptor degraders (SERDs) fulvestrant and elascestrant are the only drugs approved by the FDA to treat advanced or metastatic ER+ breast cancer. SERDs act as ERα antagonists and also cause ERα degradation in cells. Fulvestrant is a steroid drug administered intramuscularly once monthly and was approved by the FDA in 2002. It is a 7α-alkylsulphinyl analogue of 17β-estradiol and acts as a pure antiestrogen that inhibits ERα dimerization and nuclear translocation and leads to accelerated ERα degradation through the proteasomal degradation pathway (Figure 3C) [40,41]. These effects completely block ERα induced transcriptional activity in breast cancer cells, which in turn inhibits tumor progression, invasion, angiogenesis, and metastasis [40,42,43,44]. Elacestrant, a non-steroidal small molecule drug, was approved by FDA in 2023 for the treatment of ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer. In an ER+ breast cancer cell line model, elacestrant induces ERα degradation through the proteasomal pathway, blocking the activity of E2-regulated genes associated with breast cancer cell growth and proliferation [45]. The antitumor effects of elacestrant have been shown in preclinical studies and clinical trials using several prognostic and predictive markers [45,46,47]. Elacestrant binds preferentially to ERα over ERβ with half-maximal inhibitory concentrations of 48 vs 870 nmol/L [48]. It has also demonstrated antitumor activity in ER-positive patient-derived xenograft (PDX) tumor models with ESR1 mutations and with models for cyclin-dependent kinase 4/6 (CDK4/6) inhibitor resistance [45,49].
3.2. Endocrine-Resistant Breast Cancer
Antiestrogen therapy has been one of the most successful therapeutic approaches in ER+ breast cancer. Most ER+ breast cancer patients initially responded well to endocrine therapy that attenuates ERα signaling, either by blocking the production of estrogens via aromatase inhibitors or antagonizing the activity of estrogens by competitive binding of ER antagonists such as tamoxifen and fulvestrant, a selective ERα degrader (SERD) [50,51,52,53,54]. Although estrogen-disrupting therapies are often effective both in the adjuvant and metastatic setting, patients relapse after prolonged endocrine therapy which remains a major clinical problem [55,56]. Several mechanisms of endocrine resistance have been identified, including loss of ERα expression, altered activity of ERα coactivators, and crosstalk with growth factor receptors such as HER2 and IGF1R [20,57,58,59,60,61,62,63,64]. Studies have shown that the growth factor-driven mitogenic pathway can drive ER-mediated gene transcription in the absence of estradiol. Furthermore, genomic alterations in the ESR1 gene itself represent a common mechanism of endocrine resistance. Indeed, recent large-scale genomic studies have revealed a series of activating somatic mutations in the ER gene (ESR1) in endocrine-resistant metastatic breast cancer (MBC) patients [65,66,67,68,69,70,71,72,73]. This review highlights the recent preclinical and clinical trials of drugs targeting estrogen receptor mutant breast cancer.
3.3. ESR1 Mutation
In 1997, Fuqua and colleagues were the first to identify an ESR1 mutation (Y537N) in the ligand binding domain of ERα in metastatic ER+ breast cancer patient samples. Nevertheless, the significant role of ESR1 mutations in endocrine resistance was not entirely recognized until 2013, when two independent studies confirmed that somatic ESR1 mutations are relatively common (10–50%) in endocrine therapy-resistant metastatic ER+ breast cancer patients [65,67]. These somatic mutations are found predominantly in the ERα ligand-binding domain (LBD), and the ERα mutations Y537S or D538G mutations that are present in up to 40% of endocrine-therapy-resistant metastatic breast cancer patients are a major mechanism of acquired resistance to hormonal therapies [69,74,75,76,77,78]. These mutations produce an ER with high transcriptional activity even in the absence of estradiol and with a weaker affinity for anti-estrogens such as tamoxifen and fulvestrant that allows cells to resist these drugs [69,74,75,76,79,80,81,82]. Through conformational biases, mutant ERα proteins induce transcriptional gene activities that are linked with aggressive disease [83,84]. For instance, structural studies indicate that the Y537S mutant stabilize the agonist conformation in the absence of ligand by forming a hydrogen bond between S537 and D351 that cannot form in WT ERα [85,86]. Y537S and D538G mutations promote constitutive binding of steroid receptor coactivator 3 (SRC3) to these ERα mutant receptors in the absence of ligand, with Y537S recruiting SRC3 coactivator with higher affinity than D538G. Ligand binding assays reveal that these mutants have weaker affinity than wild type for estradiol and for the antiestrogens tamoxifen and fulvestrant.
Studies in breast cancer cell lines demonstrated that Y537S and D538G mutations produce high levels of ERα transcription activities even in the absence of estradiol, leading to drug resistance.[65,86] Further, these mutant ERα produce neomorphic transcriptional activities that lead to the expression of genes associated with aggressive disease in genetically engineered breast cancer models [87]. Y537S and D538G mutant breast cancer cells show increased cell proliferation over wild type (WT) in response to insulin-like growth factor [88]. A genome-wide ERα binding study showed that cells expressing WT protein occupy a small number of DNA binding sites in the absence of estradiol compared to the ERα mutants, leading to distinct alterations in the ER binding pattern. Motif analysis revealed that the mutant-specific ER binding sites are highly enriched in ERE motifs whereas the WT sites are enriched in FOXA1 motifs, indicating that FOXA1 is essential for WT-specific ER DNA binding but not for mutant ER DNA binding. Transcriptional changes driven by mutant-specific ER DNA binding substantially promote a metastatic phenotype in mice [83], and these mutations are prognostic of poor outcomes in patients with metastatic disease [89].
3.4. Therapeutic Strategies for ESR1 Mutant Breast Cancer.
Endocrine therapy plays a central role in treating early and metastatic ER+ breast cancer. The management of ER+ metastatic breast cancer combines endocrine therapy with CDK4/6 inhibitors as the standard of care (SOC) treatment. Clinical studies showed that ESR1 mutations that promote ligand-independent activation (e.g. Y537S, D538G) occur predominantly in metastatic or advanced breast cancer patients who were previously treated with estrogen deprivation therapy, especially aromatase inhibitors. Therefore, estrogen deprivation therapy may not be effective in ESR1 mutant breast cancer. Indeed, clinical data suggest reduced efficacy of aromatase inhibitors compared with fulvestrant in patients who have ESR1 mutation in the tumor or circulating tumor DNA (ctDNA) [90]. As cancer cells die, ctDNA is released into plasma in small quantities and provides the opportunity to profile tumors for somatic mutations [91,92]. Blood-based ctDNA is preferred owing to greater sensitivity [92]. The advanced digital droplet PCR (ddPCR) method is a more sensitive method to identify ESR1 mutations in cell-free DNA separated from the plasma [93]. The PALOMA-3 multicenter double-blind, randomized phase 3 trial showed that fulvestrant in combination with palbociclib (CDK4/6 inhibitor) was linked with significant improvement in progression-free survival compared with fulvestrant plus placebo, regardless of the endocrine resistance, hormone-receptor expression level, and PIK3CA mutational status. This combination therapy is used as a therapeutic option for patients with recurrent hormone-receptor-positive, HER2-negative metastatic breast cancer that has progressed on previous endocrine therapy [94,95].
More recently, elacestrant has been approved by the FDA for the treatment of postmenopausal women with ER+, HER2- and ESR1 mutant metastatic or advanced breast cancer with disease progression following more than one line of endocrine therapy. Elacestrant is the first orally available small-molecule drug in the SERD class. A multinational phase 3 randomized study (ClinicalTrials.gov identifier: NCT03778931, EMERALD) showed that monotherapy with elacestrant dramatically decreases the risk of cancer progression compared with SOC endocrine therapy in patients with ER+ and HER2- metastatic breast cancer (with or without ESR1 mutations) who had progression after first-or second-line treatment with the combination of endocrine therapy and a CDK4/6 inhibitor [46]. The most common side effects to elacestrant were dyslipidemia, musculoskeletal pain, and nausea; elevated levels of triglycerides, AST, ALT, and creatinine; and reduced levels of haemoglobin and sodium. Other adverse effects such as decreased appetite, diarrhoea, headache, abdominal pain, constipation, hot flush, and dyspepsia were also observed in response to elacestrant treatment [96,97].
Preclinical mouse models of breast cancer studies (xenograft and patient-derived xenograft) showed that elacestrant prevents E2-induced tumor growth in ESR1 WT and mutants[45,49]. Furthermore, the anti-tumor activity of elacestrant translates into various PDX models representing intrinsic and acquired CDK4/6 inhibitor resistance [49]. Another study evaluated the efficacy of elacestrant alone or in combination with either palbociclib (CDK4/6 inhibitor) or everolimus (mTOR inhibitor) in a xenograft mouse model. Elacestrant alone attained a significant reduction in tumor growth, but combination treatment with palbociclib or everolimus produced even greater inhibition of tumor growth [43]. Furthermore, in two different PDX models harboring ESR1 mutations, combination treatment with elacestrant and palbociclib effectively stopped tumor growth [45,49].
4. SERDs in Clinical Trials
In addition to elacestrant, there are several promising oral SERDs in various stages of clinical development (Table 1). This review focuses on oral SERDs in Phase 3 trials.
4.1. Giredestrant (GDC-9545)
Giredestrant is an investigational nonsteroidal oral SERD that binds to the ligand-binding domain and triggers intranuclear ER immobilization before degradation [98]. Giredestrant retains efficacy with ER mutants Y537S and D538G. GDC-9545 induces rapid ERα degradation and anti-proliferation across an ER+ breast cancer cell line panel [99]. In a preclinical model, at low doses, GDC-9545 promotes tumor regressions either as a single agent or combined with a CDK4/6 inhibitor in an ESR1 Y537S mutant PDX and a wild-type ERα breast tumor model [99]. In the phase Ia/b GO39932 study (ClinicalTrials.gov identifier: NCT03332797), the efficacy and tolerability of giredestrant were studied in ER+ and HER2- locally advanced/ metastatic breast cancer patients who previously received endocrine therapy [98]. The primary analysis demonstrated that giredestrant is well tolerated and potentially clinically active as a single agent and in combination with palbociclib for the treatment of patients who have disease progression on prior endocrine therapy, including patients with ESR1 mutations. Hepatotoxicity was reported with giredestrant treatment. The phase II acelERA study (ClinicalTrials.gov identifier: NCT04576455) compares the efficacy and safety of giredestrant with physician's choice of endocrine monotherapy for ER+, HER2-negative (HER2–) breast cancer in the second or third line [100]. This study showed that giredestrant treatment improved progression-free survival (PFS) but was not statistically significant with ESR1 mutant tumors. Furthermore, giredestrant trended to a favorable benefit including patients with ESR1 mutations. Giredestrant is currently being investigated in phase III trials in ER+, HER2– breast cancer. This randomized, open-label multicenter study will evaluate the efficacy and safety of adjuvant giredestrant compared with endocrine therapy of physician's choice in participants with medium- and high-risk Stage I-III ER+ and HER2- breast cancer (ClinicalTrials.gov identifier: NCT04961996). Another study will evaluate the efficacy and safety of giredestrant compared with fulvestrant, both in combination with the physician's choice of a CDK4/6 inhibitor (ClinicalTrials.gov identifier: NCT06065748) in patients with ER+, HER2- advanced breast cancer who have developed resistance to adjuvant endocrine therapy. The results of these ongoing phase 3 trials are not yet available.
4.2. Imlunestrant (LY3484356)
Imlunestrant is an investigational orally administered SERD that was developed by Eli Lilly [101]. LY3484356 is a pure ERα antagonist with a highly potent and efficient degrader against wild-type and mutant ER. LY3484356 is also a potent inhibitor of ERα-mediated transcription in vitro and in vivo [102]. It inhibits cell proliferation in wild-type ERα and ESR1 mutant breast cancer cell lines. LY3484356 has maintained persistent target inhibition up to 96h after the last dose in ESR1 wild type and ESR1 Y537S mutant xenograft tumors [102]. LY3484356 exhibited substantial tumor growth inhibition and tumor regressions in wild-type ESR1 breast cancer xenograft as well as ESR1 mutant breast cancer PDX models [102]. Furthermore, LY3484356 has displayed additivity in combination with CDK4/6 inhibitors, mTOR inhibitors, and PIK3CA inhibitors in blocking cell proliferation as well as tumor growth inhibition in xenograft and PDX models of breast cancer . Phase 1a/1b EMBER (ClinicalTrials.gov identifier: NCT04188548) is a comprehensive, open-label, dose-escalation (phase 1a) trial of imlunestrant followed by several dose-expansion cohorts (phase 1b) examining imlunestrant as monotherapy and in combination with abemaciclib with or without aromatase inhibitors everolimus, or alpelisib in ER+ advanced breast cancer and endometrial endometrioid cancer. The overall results demonstrate that imlunestrant has an adaptable safety profile with antitumor activity in ER+/HER2- advanced breast cancer including in patients with baseline ESR1 mutations and fulvestrant- and/or CDK4/6 inhibitor- refractory disease [101]. Based on these positive results, imlunestran entered into phase 3 EMBER-3 trial (ClinicalTrials.gov identifier: NCT04975308); this study is currently evaluating imlunestrant compared to standard hormone therapy as well as imlunestrant plus abemaciclib in patients with ER+/HER2- locally advanced or metastatic breast cancer who had previously been treated with endocrine therapy.
4.3. Camizestrant (AZD-9833)
Camizestrant is an investigational oral SERD that was developed by AstraZeneca [103,104]. Camizestrant shows strong and selective ER degradation and significant antiproliferation activity in ESR1 wild-type and mutant breast cancer cell lines [105]. Camizestrant has demonstrated anti-cancer activity in fulvestrant-resistant ESR1 WT, Y537S, and D538G PDX models [105]. Camizestrant in combination with CDK4/6 inhibitors or PI3K/AKT/mTOR inhibitors showed enhanced efficacy in CDK4/6-sensitive and -resistant models [105,106]. Phase 1 trial SERENA-1 (ClinicalTrials.gov identifier: NCT03616587) has demonstrated that camizestrant is well tolerated and has improved clinical activity as monotherapy or in combination with CDK4/6 inhibitors in ER+/HER2- advanced breast cancer patients who had received one or more previous lines of endocrine therapy including CDK4/6 inhibitors [103]. In the randomized multicenter phase 2 trial SERENA-2 (ClinicalTrials.gov identifier: NCT04214288), the efficacy and safety profiles of oral camizestrant were compared with fulvestrant; camizestrant substantially improved progression-free survival (PFS) versus fulvestrant in post-menopausal patients with ER+ advanced breast cancer who had previously been treated with endocrine therapy. Notably, camizestrant remained more effective than fluvestrant in patients with ESR1 mutations [107]. SERENA-4 (ClinicalTrials.gov identifier: NCT04711252) is an ongoing phase 3 randomized, double-blind study that is assessing the efficacy and safety of camizestrant plus palbociclib versus anastrozole plus palbociclib as first-line therapy for patients with HR+/HER2- advanced breast cancer who have not established systemic treatment for advanced disease. SERENA-6 (ClinicalTrials.gov identifier: NCT04964934) is another ongoing phase 3 trial that evaluates the efficacy and safety of substituting from an aromatase inhibitor to camizestrant, but continuing the same CDK4/6 inhibitors, upon finding of ESR1 mutations in circulating tumor DNA before progression of the disease on first-line therapy for HR+/HER2- advanced breast cancer [104].
4.4. Vepdegestrant (ARV-471)
Vepdegestrant is an investigational, orally bioavailable proteolysis-targeting chimera (PROTAC) protein degrader that is designed to target and degrade ERα wild type and mutant proteins. In preclinical studies, vepdegestrant induced degradation of WT and mutant ERα (including Y537S and D538G) and antiproliferation in a panel of ER+ breast cancer cell lines [108]. Vepdegestrant exhibited dose-dependent tumor regressions in xenograft and PDX models of breast cancer including Y537S and palbociclib-resistant Y537S [108]. More importantly, a combination with small molecule inhibitors of CDK4/6 (palbociclib, abemaciclib, or ribociclib), PI3K (alpelisib or inavolisib) or mTOR (everolimus) produced robust tumor regressions in most cases [108]. Collectively, these preclinical studies suggest that dual pathway targeting by vepdegestrant and CDK4/6 or PI3K/mTOR signaling could result in better therapeutic outcomes for patients with advanced ER+/HER2− breast cancer [108]. Based on the encouraging preclinical data, a first-in-human Phase 1/2 study (ClinicalTrials.gov identifier: NCT04072952) of vepdegestrant monotherapy and in combination with palbociclib was conducted in ER+/HER2- breast cancer patients; vepdegestrant demonstrated antitumor activity and was tolerated in daily doses from 30 to 700 mg, with no dose-limiting toxicities [109]. Based on the results of the dose escalation study, vepdegestrant 200 and 500 mg were further assessed in VERITAC, the Phase 2 expansion cohort of the Phase 1/2 study. Preliminary results showed anti-tumor activity as well as a well-tolerated safety profile [110]. VERITAC-2 (ClinicalTrials.gov identifier: NCT05654623) is a phase 3 study that compares the efficacy and safety of vepdegestrant with fulvestrant in patients with ER+/HER2- advanced breast cancer after prior combination endocrine therapy and CDK4/6 inhibitor therapy [111].
4.5. Palazestrant (OP-1250)
Palazestrant is an investigational orally bioavailable SERD and ER antagonist that was developed by Olema Pharmaceuticals. In breast cancer cell lines model, palazestrant inhibits estrogen-induced transcriptional activity and blocks agonist activity on estrogen-induced genes as well as antiproliferative activity in WT and ESR1 mutant cells [112]. In preclinical studies, palazestrant demonstrated tumor regression in ER+ xenograft models, and CDK4/6 inhibitors enhance its efficacy [112]. In PDX models bearing the ESR1 Y537S mutant, palazestrant inhibits tumor growth at a 3 mg/kg dose and enhances tumor shrinkage at higher doses or combined with CDK4/6 inhibitors. Palazestrant outperformed elacestrant, a clinically FDA-approved drug for patients with ESR1 mutations [112]. In pharmacokinetic analyses of mouse xenograft studies, palazestrant displays excellent brain penetrance and an encouraging half-life [112]. In an intracranial xenograft study, treatment with 10 mg/kg palazestrant demonstrated tumor shrinkage and survival of all animals over a 100-day dosing interval, significantly surpassing the performance of fulvestrant and tamoxifen [112]. Based on the encouraging preclinical data, palazestrant entered into clinical trials. ClinicalTrials.gov identifier: NCT04505826 is a Phase 1 dose escalation and dose expansion and Phase 2 monotherapy to assess the dose-limiting toxicity (DLT), maximum tolerated dose (MTD) and/or recommended Phase 2 dose, to analyze the safety and pharmacokinetic profile, and to evaluation the preliminary anti-tumor activity of palazestrant as a single agent in adult with ER+/HER2- metastatic breast cancer or locally advanced breast cancer with and without ESR1 mutation. Palazestrant showed an acceptable safety profile, satisfactory pharmacokinetics and encouraging antitumor efficacy in patients with and without ESR1 mutation at the endorsed Phase 2 dose of 120 mg once a day. OPERA-01 (ClinicalTrials.gov identifier: NCT06016738) is a multicenter, randomized, phase 3 clinical trial comparing the efficacy and safety of palazestrant as a single agent to endocrine therapies (fulvestrant, anastrozole, letrozole, or exemestane) in patients with ER+, HER2– metastatic breast cancer that relapsed on 1-2 prior lines of endocrine therapies in combination with CDK4/6 inhibitor. This phase 3 trial was started last year and is still recruiting patients.
5. Conclusions and Future Directions
Endocrine therapies are effective both in the adjuvant and metastatic setting with ER+ breast cancer patients. However, a major clinical problem is that patients often relapse after prolonged endocrine therapy. One of the major mechanisms that drives endocrine-resistant metastatic breast cancers is constitutively active, somatic point mutations in the ligand binding domain of ERα. The most common ligand-binding domain point mutations are Y537S and D538G, which promote estrogen-independent ER transcriptional activity and decreased sensitivity to current endocrine drugs tamoxifen and fulvestrant. Recent drug discovery efforts for orally available SERDs have led to the identification of several novel investigational agents undergoing clinical evaluation. So far, oral SERD efforts for breast cancer have led to the authorization of elacestrant, the first oral SERD approved for treating metastatic, hormone-resistant breast cancer including ESR1 mutants. Tackling issues of endocrine resistance to various antiestrogens and anticancer agents will continue to be a critical challenge, and it seems likely that the development of effective combination therapy will be needed to completely combat resistance mechanisms that occur during treatment with new inhibitors. Indeed, next generation of oral SERDs are currently being evaluated with CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib). As an alternative, or a complement, inhibitors of PI3K, AKT and/or mTOR are being combined with SERDs, since abnormal activation of growth factor signaling cascades have been associated with endocrine therapy resistance.
It will also be important to identify and develop first-generation ESR1-mutant specific inhibitors because elacestrant and other clinical investigational SERDs exhibit lower activity against Y537S and D538G mutants than against wild type receptor. Drug discovery efforts that use novel and creative strategies to target these mutants will provide a promising new avenue for the direct pharmacological inhibition of ERα mutant proteins in endocrine therapy resistant metastatic breast cancer patients.
Acknowledgments
The author acknowledges funding support to M.P. from NIH grant R03 CA259664 and Cancer Prevention Research Institute of Texas grant RP220524. The author also thanks Dr. Kevin MacKenzie, Ph.D., for his critical reading of the manuscript.
Conflicts of Interest
The author declares no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish.
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Toft, D.; Gorski, J. A receptor molecule for estrogens: isolation from the rat uterus and preliminary characterization. Proceedings of the National Academy of Sciences of the United States of America 1966, 55, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor complementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef]
- Nilsson, S.; Makela, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol Rev 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Gosden, J.R.; Middleton, P.G.; Rout, D. Localization of the human oestrogen receptor gene to chromosome 6q24----q27 by in situ hybridization. Cytogenet Cell Genet 1986, 43, 218–220. [Google Scholar] [CrossRef]
- Green, S.; Kumar, V.; Krust, A.; Walter, P.; Chambon, P. Structural and functional domains of the estrogen receptor. Cold Spring Harb Symp Quant Biol 1986, 51 Pt 2, 751–758. [Google Scholar] [CrossRef]
- Sarwar, N.; Kim, J.S.; Jiang, J.; Peston, D.; Sinnett, H.D.; Madden, P.; Gee, J.M.; Nicholson, R.I.; Lykkesfeldt, A.E.; Shousha, S.; et al. Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr Relat Cancer 2006, 13, 851–861. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Finn, G.; Solodin, N.M.; Singarapu, K.K.; Sahu, S.C.; Markley, J.L.; Kadunc, K.J.; Ellison-Zelski, S.J.; Kariagina, A.; Haslam, S.Z.; et al. Regulation of estrogen receptor alpha N-terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol Cell Biol 2012, 32, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Klein-Hitpass, L.; Ryffel, G.U.; Heitlinger, E.; Cato, A.C. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res 1988, 16, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.C.; Corbo, L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol Endocrinol 2005, 19, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Pestell, R.; Curran, E.M.; Welshons, W.V.; Fuqua, S.A. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer research 2004, 64, 9199–9208. [Google Scholar] [CrossRef]
- Berry, N.B.; Fan, M.; Nephew, K.P. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol Endocrinol 2008, 22, 1535–1551. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Arao, Y.; Korach, K.S. The physiological role of estrogen receptor functional domains. Essays Biochem 2021, 65, 867–875. [Google Scholar] [CrossRef]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O'Malley, B.W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Molecular cell 2000, 5, 939–948. [Google Scholar] [CrossRef]
- Lonard, D.M.; O'Malley, B.W. Molecular Pathways: Targeting Steroid Receptor Coactivators in Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2016, 22, 5403–5407. [Google Scholar] [CrossRef]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proceedings of the National Academy of Sciences of the United States of America 1992, 89, 4037–4041. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.; Rientjes, J.M.; Stewart, A.F. Different positioning of the ligand-binding domain helix 12 and the F domain of the estrogen receptor accounts for functional differences between agonists and antagonists. EMBO J 1998, 17, 765–773. [Google Scholar] [CrossRef]
- Schwartz, J.A.; Zhong, L.; Deighton-Collins, S.; Zhao, C.; Skafar, D.F. Mutations targeted to a predicted helix in the extreme carboxyl-terminal region of the human estrogen receptor-alpha alter its response to estradiol and 4-hydroxytamoxifen. The Journal of biological chemistry 2002, 277, 13202–13209. [Google Scholar] [CrossRef]
- Koide, A.; Zhao, C.; Naganuma, M.; Abrams, J.; Deighton-Collins, S.; Skafar, D.F.; Koide, S. Identification of regions within the F domain of the human estrogen receptor alpha that are important for modulating transactivation and protein-protein interactions. Mol Endocrinol 2007, 21, 829–842. [Google Scholar] [CrossRef]
- Arao, Y.; Korach, K.S. The F domain of estrogen receptor alpha is involved in species-specific, tamoxifen-mediated transactivation. The Journal of biological chemistry 2018, 293, 8495–8507. [Google Scholar] [CrossRef]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proceedings of the National Academy of Sciences of the United States of America 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, V.; Robinson-Rechavi, M. Meta-analysis of estrogen response in MCF-7 distinguishes early target genes involved in signaling and cell proliferation from later target genes involved in cell cycle and DNA repair. BMC Syst Biol 2011, 5, 138. [Google Scholar] [CrossRef]
- Welboren, W.J.; Sweep, F.C.; Span, P.N.; Stunnenberg, H.G. Genomic actions of estrogen receptor alpha: what are the targets and how are they regulated? Endocr Relat Cancer 2009, 16, 1073–1089. [Google Scholar] [CrossRef]
- Palaniappan, M.; Edwards, D.; Creighton, C.J.; Medina, D.; Conneely, O.M. Reprogramming of the estrogen responsive transcriptome contributes to tamoxifen-dependent protection against tumorigenesis in the p53 null mammary epithelial cells. PloS one 2018, 13, e0194913. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nature genetics 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nature genetics 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Tamoxifen: a most unlikely pioneering medicine. Nature reviews. Drug discovery 2003, 2, 205–213. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer 2014, 21, R235–246. [Google Scholar] [CrossRef]
- Cole, M.P.; Jones, C.T.; Todd, I.D. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. British journal of cancer 1971, 25, 270–275. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Buschmann, M.; Wiegand, A.; Schnellbacher, K.; Bonn, R.; Rehe, A.; Trenk, D.; Jahnchen, E.; Roskamm, H. Comparison of the effects of two different galenical preparations of glyceryl trinitrate on pulmonary artery pressure and on the finger pulse curve. Eur J Clin Pharmacol 1993, 44, 451–456. [Google Scholar] [CrossRef]
- Nabholtz, J.M. Long-term safety of aromatase inhibitors in the treatment of breast cancer. Ther Clin Risk Manag 2008, 4, 189–204. [Google Scholar] [CrossRef]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. British journal of cancer 2004, 90 Suppl 1, S2–6. [Google Scholar] [CrossRef]
- Carlson, R.W. The history and mechanism of action of fulvestrant. Clin Breast Cancer 2005, 6 Suppl 1, S5–8. [Google Scholar] [CrossRef]
- Wardley, A.M. Fulvestrant: a review of its development, pre-clinical and clinical data. Int J Clin Pract 2002, 56, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, S.; Simigdala, N.; Ribas, R.; Schuster, E.; Leal, M.F.; Nikitorowicz-Buniak, J.; Rega, C.; Bihani, T.; Patel, H.; Johnston, S.R.; et al. Elacestrant demonstrates strong anti-estrogenic activity in PDX models of estrogen-receptor positive endocrine-resistant and fulvestrant-resistant breast cancer. NPJ Breast Cancer 2022, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.F.; Nicholson, R.I.; Bundred, N.J.; Anderson, E.; Rayter, Z.; Dowsett, M.; Fox, J.N.; Gee, J.M.; Webster, A.; Wakeling, A.E.; et al. Comparison of the short-term biological effects of 7alpha-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)-nonyl]estra-1,3,5, (10)-triene-3,17beta-diol (Faslodex) versus tamoxifen in postmenopausal women with primary breast cancer. Cancer research 2001, 61, 6739–6746. [Google Scholar]
- Bihani, T.; Patel, H.K.; Arlt, H.; Tao, N.; Jiang, H.; Brown, J.L.; Purandare, D.M.; Hattersley, G.; Garner, F. Elacestrant (RAD1901), a Selective Estrogen Receptor Degrader (SERD), Has Antitumor Activity in Multiple ER(+) Breast Cancer Patient-derived Xenograft Models. Clinical cancer research : an official journal of the American Association for Cancer Research 2017, 23, 4793–4804. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J Clin Oncol 2022, 40, 3246–3256. [Google Scholar] [CrossRef]
- Jager, A.; de Vries, E.G.E.; der Houven van Oordt, C.W.M.; Neven, P.; Venema, C.M.; Glaudemans, A.; Wang, Y.; Bagley, R.G.; Conlan, M.G.; Aftimos, P. A phase 1b study evaluating the effect of elacestrant treatment on estrogen receptor availability and estradiol binding to the estrogen receptor in metastatic breast cancer lesions using (18)F-FES PET/CT imaging. Breast cancer research : BCR 2020, 22, 97. [Google Scholar] [CrossRef]
- Garner, F.; Shomali, M.; Paquin, D.; Lyttle, C.R.; Hattersley, G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs 2015, 26, 948–956. [Google Scholar] [CrossRef]
- Patel, H.K.; Tao, N.; Lee, K.M.; Huerta, M.; Arlt, H.; Mullarkey, T.; Troy, S.; Arteaga, C.L.; Bihani, T. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast cancer research : BCR 2019, 21, 146. [Google Scholar] [CrossRef]
- Gombos, A. Selective oestrogen receptor degraders in breast cancer: a review and perspectives. Current opinion in oncology 2019, 31, 424–429. [Google Scholar] [CrossRef]
- Fanning, S.W.; Greene, G.L. Next-Generation ERalpha Inhibitors for Endocrine-Resistant ER+ Breast Cancer. Endocrinology 2019, 160, 759–769. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D.P.; Wardell, S.E. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Current opinion in pharmacology 2010, 10, 620–628. [Google Scholar] [CrossRef]
- McDonnell, D.P. The molecular pharmacology of estrogen receptor modulators: implications for the treatment of breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2005, 11, 871s–877s. [Google Scholar] [CrossRef]
- Puhalla, S.; Bhattacharya, S.; Davidson, N.E. Hormonal therapy in breast cancer: a model disease for the personalization of cancer care. Molecular oncology 2012, 6, 222–236. [Google Scholar] [CrossRef]
- Nardone, A.; De Angelis, C.; Trivedi, M.V.; Osborne, C.K.; Schiff, R. The changing role of ER in endocrine resistance. Breast 2015, 24 Suppl 2, S60–66. [Google Scholar] [CrossRef]
- Will, M.; Liang, J.; Metcalfe, C.; Chandarlapaty, S. Therapeutic resistance to anti-oestrogen therapy in breast cancer. Nature reviews. Cancer 2023, 23, 673–685. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annual review of medicine 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annual review of medicine 2015, 66, 111–128. [Google Scholar] [CrossRef]
- Morrison, G.; Fu, X.; Shea, M.; Nanda, S.; Giuliano, M.; Wang, T.; Klinowska, T.; Osborne, C.K.; Rimawi, M.F.; Schiff, R. Therapeutic potential of the dual EGFR/HER2 inhibitor AZD8931 in circumventing endocrine resistance. Breast cancer research and treatment 2014, 144, 263–272. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nature reviews. Clinical oncology 2011, 9, 16–32. [Google Scholar] [CrossRef]
- Choi, H.J.; Joo, H.S.; Won, H.Y.; Min, K.W.; Kim, H.Y.; Son, T.; Oh, Y.H.; Lee, J.Y.; Kong, G. Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. Journal of the National Cancer Institute 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Agboke, F.A.; Cunliffe, H.E.; Ramos, P.; Jordan, V.C. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. European journal of cancer 2014, 50, 2866–2876. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, S.A.; Powter, B.; Balakrishnar, B.; Mok, K.; Soon, P.; Franken, A.; Neubauer, H.; de Souza, P.; Becker, T.M. Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability. Cells 2020, 9. [Google Scholar] [CrossRef]
- Gururaj, A.E.; Rayala, S.K.; Vadlamudi, R.K.; Kumar, R. Novel mechanisms of resistance to endocrine therapy: genomic and nongenomic considerations. Clinical cancer research : an official journal of the American Association for Cancer Research 2006, 12, 1001s–1007s. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nature genetics 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer research 2013, 73, 6856–6864. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature genetics 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer research 1997, 57, 1244–1249. [Google Scholar]
- Oesterreich, S.; Davidson, N.E. The search for ESR1 mutations in breast cancer. Nature genetics 2013, 45, 1415–1416. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.A. Estrogen receptor mutations and functional consequences for breast cancer. Trends in endocrinology and metabolism: TEM 2015, 26, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. Journal of cancer metastasis and treatment 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nature reviews. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- Carlson, K.E.; Choi, I.; Gee, A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry 1997, 36, 14897–14905. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, S.; Gustafsson, J.A. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocrine reviews 2019, 40, 1207–1249. [Google Scholar] [CrossRef] [PubMed]
- Zundelevich, A.; Dadiani, M.; Kahana-Edwin, S.; Itay, A.; Sella, T.; Gadot, M.; Cesarkas, K.; Farage-Barhom, S.; Saar, E.G.; Eyal, E.; et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast cancer research : BCR 2020, 22, 16. [Google Scholar] [CrossRef]
- Hancock, G.R.; Gertz, J.; Jeselsohn, R.; Fanning, S.W. Estrogen Receptor Alpha Mutations, Truncations, Heterodimers, and Therapies. Endocrinology 2024, 165. [Google Scholar] [CrossRef] [PubMed]
- Dustin, D.; Gu, G.; Beyer, A.R.; Herzog, S.K.; Edwards, D.G.; Lin, H.; Gonzalez, T.L.; Grimm, S.L.; Coarfa, C.; Chan, D.W.; et al. RON signalling promotes therapeutic resistance in ESR1 mutant breast cancer. British journal of cancer 2021, 124, 191–206. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Y.; Yates, M.E.; Tasdemir, N.; Bahreini, A.; Chen, J.; Levine, K.M.; Priedigkeit, N.M.; Nasrazadani, A.; Ali, S.; et al. Hotspot ESR1 Mutations Are Multimodal and Contextual Modulators of Breast Cancer Metastasis. Cancer research 2022, 82, 1321–1339. [Google Scholar] [CrossRef]
- Grinshpun, A.; Sandusky, Z.M.; Jeselsohn, R. The Clinical Utility of ESR1 Mutations in Hormone Receptor-Positive, HER2-Negative Advanced Breast Cancer. Hematology/oncology clinics of North America 2023, 37, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Grinshpun, A.; Chen, V.; Sandusky, Z.M.; Fanning, S.W.; Jeselsohn, R. ESR1 activating mutations: From structure to clinical application. Biochimica et biophysica acta. Reviews on cancer 2023, 1878, 188830. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer cell 2018, 33, 173–186. [Google Scholar] [CrossRef]
- Arnesen, S.; Blanchard, Z.; Williams, M.M.; Berrett, K.C.; Li, Z.; Oesterreich, S.; Richer, J.K.; Gertz, J. Estrogen Receptor Alpha Mutations in Breast Cancer Cells Cause Gene Expression Changes through Constant Activity and Secondary Effects. Cancer research 2021, 81, 539–551. [Google Scholar] [CrossRef]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; Nowak, J.; Sharma, S.K.; Hahm, J.B.; Kulp, K.; Hochberg, R.B.; Zhou, H.; Katzenellenbogen, J.A.; et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nature chemical biology 2008, 4, 241–247. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016, 5. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Norris, J.D.; Chang, C.Y. Neomorphic ERalpha Mutations Drive Progression in Breast Cancer and Present a Challenge for New Drug Discovery. Cancer cell 2018, 33, 153–155. [Google Scholar] [CrossRef]
- Li, Z.; Levine, K.M.; Bahreini, A.; Wang, P.; Chu, D.; Park, B.H.; Oesterreich, S.; Lee, A.V. Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells. Endocrinology 2018, 159, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol 2016, 2, 1310–1315. [Google Scholar] [CrossRef]
- Henry, N.L.; Somerfield, M.R.; Dayao, Z.; Elias, A.; Kalinsky, K.; McShane, L.M.; Moy, B.; Park, B.H.; Shanahan, K.M.; Sharma, P.; et al. Biomarkers for Systemic Therapy in Metastatic Breast Cancer: ASCO Guideline Update. J Clin Oncol 2022, 40, 3205–3221. [Google Scholar] [CrossRef]
- Bellward, G.D.; Norstrom, R.J.; Whitehead, P.E.; Elliott, J.E.; Bandiera, S.M.; Dworschak, C.; Chang, T.; Forbes, S.; Cadario, B.; Hart, L.E.; et al. Comparison of polychlorinated dibenzodioxin levels with hepatic mixed-function oxidase induction in great blue herons. J Toxicol Environ Health 1990, 30, 33–52. [Google Scholar] [CrossRef]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Wang, P.; Bahreini, A.; Gyanchandani, R.; Lucas, P.C.; Hartmaier, R.J.; Watters, R.J.; Jonnalagadda, A.R.; Trejo Bittar, H.E.; Berg, A.; Hamilton, R.L.; et al. Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clinical cancer research : an official journal of the American Association for Cancer Research 2016, 22, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidants and the central nervous system: some fundamental questions. Is oxidant damage relevant to Parkinson's disease, Alzheimer's disease, traumatic injury or stroke? Acta Neurol Scand Suppl 1989, 126, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.R.; Jhaveri, K.; Kalinsky, K.; Bardia, A.; Wander, S.A. Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer. Nature reviews. Clinical oncology 2024, 21, 743–761. [Google Scholar] [CrossRef]
- Bardia, A.; Cortes, J.; Bidard, F.C.; Neven, P.; Garcia-Saenz, J.; Aftimos, P.; O'Shaughnessy, J.; Lu, J.; Tonini, G.; Scartoni, S.; et al. Elacestrant in ER+, HER2- Metastatic Breast Cancer with ESR1-Mutated Tumors: Subgroup Analyses from the Phase III EMERALD Trial by Prior Duration of Endocrine Therapy plus CDK4/6 Inhibitor and in Clinical Subgroups. Clinical cancer research : an official journal of the American Association for Cancer Research 2024, 30, 4299–4309. [Google Scholar] [CrossRef]
- Bhatia, N.; Thareja, S. Elacestrant: a new FDA-approved SERD for the treatment of breast cancer. Med Oncol 2023, 40, 180. [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Bellet, M.; Turner, N.C.; Loi, S.; Bardia, A.; Boni, V.; Sohn, J.; Neilan, T.G.; Villanueva-Vazquez, R.; Kabos, P.; et al. Phase Ia/b Study of Giredestrant +/- Palbociclib and +/- Luteinizing Hormone-Releasing Hormone Agonists in Estrogen Receptor-Positive, HER2-Negative, Locally Advanced/Metastatic Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2024, 30, 754–766. [Google Scholar] [CrossRef]
- Liang, J.; Zbieg, J.R.; Blake, R.A.; Chang, J.H.; Daly, S.; DiPasquale, A.G.; Friedman, L.S.; Gelzleichter, T.; Gill, M.; Giltnane, J.M.; et al. GDC-9545 (Giredestrant): A Potent and Orally Bioavailable Selective Estrogen Receptor Antagonist and Degrader with an Exceptional Preclinical Profile for ER+ Breast Cancer. J Med Chem 2021, 64, 11841–11856. [Google Scholar] [CrossRef]
- Martin, M.; Lim, E.; Chavez-MacGregor, M.; Bardia, A.; Wu, J.; Zhang, Q.; Nowecki, Z.; Cruz, F.M.; Safin, R.; Kim, S.B.; et al. Giredestrant for Estrogen Receptor-Positive, HER2-Negative, Previously Treated Advanced Breast Cancer: Results From the Randomized, Phase II acelERA Breast Cancer Study. J Clin Oncol 2024, 42, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.L.; Lim, E.; Jeselsohn, R.; Ma, C.X.; Hamilton, E.P.; Osborne, C.; Bhave, M.; Kaufman, P.A.; Beck, J.T.; Manso Sanchez, L.; et al. Imlunestrant, an Oral Selective Estrogen Receptor Degrader, as Monotherapy and in Combination With Targeted Therapy in Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Phase Ia/Ib EMBER Study. J Clin Oncol 2024, JCO2302733. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.Z.B.; Shen, W; Mur, C; Barr, R; Kindler, L.J; Rubio, A; Bastian, J.A; Cohen, J.D; Mattioni, B.E; Yuen, E; Baker, T.K; Castanares, M.A; Fei, D; Manro, J.R; Lallena, M.J; Peng, S.B; de Dios, A. Preclinical characterization of LY3484356, a novel, potent and orally bioavailable selective estrogen receptor degrader (SERD) In Proceedings of the American Association for Cancer Research Annual Meeting, 2021; p. abstr. 1236.
- Hamilton, E.; Oliveira, M.; Turner, N.; Garcia-Corbacho, J.; Hernando, C.; Ciruelos, E.M.; Kabos, P.; Ruiz-Borrego, M.; Armstrong, A.; Patel, M.R.; et al. A phase I dose escalation and expansion trial of the next-generation oral SERD camizestrant in women with ER-positive, HER2-negative advanced breast cancer: SERENA-1 monotherapy results. Ann Oncol 2024, 35, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Huang-Bartlett, C.; Kalinsky, K.; Cristofanilli, M.; Bianchini, G.; Chia, S.; Iwata, H.; Janni, W.; Ma, C.X.; Mayer, E.L.; et al. Design of SERENA-6, a phase III switching trial of camizestrant in ESR1-mutant breast cancer during first-line treatment. Future Oncol 2023, 19, 559–573. [Google Scholar] [CrossRef]
- Lawson, M.; Cureton, N.; Ros, S.; Cheraghchi-Bashi, A.; Urosevic, J.; D'Arcy, S.; Delpuech, O.; DuPont, M.; Fisher, D.I.; Gangl, E.T.; et al. The Next-Generation Oral Selective Estrogen Receptor Degrader Camizestrant (AZD9833) Suppresses ER+ Breast Cancer Growth and Overcomes Endocrine and CDK4/6 Inhibitor Resistance. Cancer research 2023, 83, 3989–4004. [Google Scholar] [CrossRef]
- Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J Med Chem 2020, 63, 14530–14559. [Google Scholar] [CrossRef]
- Oliveira, M.e.a. Camizestrant, a next generation oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: Results of the randomized, multi-dose Phase 2 SERENA-2 trial. 2023; pp. abstr. GS3-02.
- Gough, S.M.; Flanagan, J.J.; Teh, J.; Andreoli, M.; Rousseau, E.; Pannone, M.; Bookbinder, M.; Willard, R.; Davenport, K.; Bortolon, E.; et al. Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models. Clinical cancer research : an official journal of the American Association for Cancer Research 2024, 30, 3549–3563. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.V. , V; Han,H.S; Ranciato,J. et al. First-in-human safety and activity of ARV-471, a novel PROTAC® estrogen receptor degrader, in ER+/HER2- locally advanced or metastatic breast cancer. 2022; pp. abstr. PD13-08.
- Schott, A.F.e.a. ARV-471, a PROTAC estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: phase 2 expansion (VERITAC) of a phase 1/2 study. 2023; pp. abstr. GS3-03.
- Hamilton, E.P.; Ma, C.; De Laurentiis, M.; Iwata, H.; Hurvitz, S.A.; Wander, S.A.; Danso, M.; Lu, D.R.; Perkins Smith, J.; Liu, Y.; et al. VERITAC-2: a Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER2- advanced breast cancer. Future Oncol 2024, 20, 2447–2455. [Google Scholar] [CrossRef]
- Parisian, A.D.; Barratt, S.A.; Hodges-Gallagher, L.; Ortega, F.E.; Pena, G.; Sapugay, J.; Robello, B.; Sun, R.; Kulp, D.; Palanisamy, G.S.; et al. Palazestrant (OP-1250), A Complete Estrogen Receptor Antagonist, Inhibits Wild-type and Mutant ER-positive Breast Cancer Models as Monotherapy and in Combination. Mol Cancer Ther 2024, 23, 285–300. [Google Scholar] [CrossRef]
Figure 1.
Estradiol -mediated activation of estrogen receptor signaling in mammary epithelial cells. Estradiol (E2) stimulates estrogen receptor α, leading to dimerization, translocation from cytosol to nucleus, and binding to estrogen response elements (ERE) in the genome. These estrogen receptor (ER) dimers recruit other transcription factors (TFs) and coactivators (CoA) to form transcriptionally active complexes at gene enhancers, leading to increased ER-dependent transcription which drive cell growth, proliferation and survival.
Figure 1.
Estradiol -mediated activation of estrogen receptor signaling in mammary epithelial cells. Estradiol (E2) stimulates estrogen receptor α, leading to dimerization, translocation from cytosol to nucleus, and binding to estrogen response elements (ERE) in the genome. These estrogen receptor (ER) dimers recruit other transcription factors (TFs) and coactivators (CoA) to form transcriptionally active complexes at gene enhancers, leading to increased ER-dependent transcription which drive cell growth, proliferation and survival.
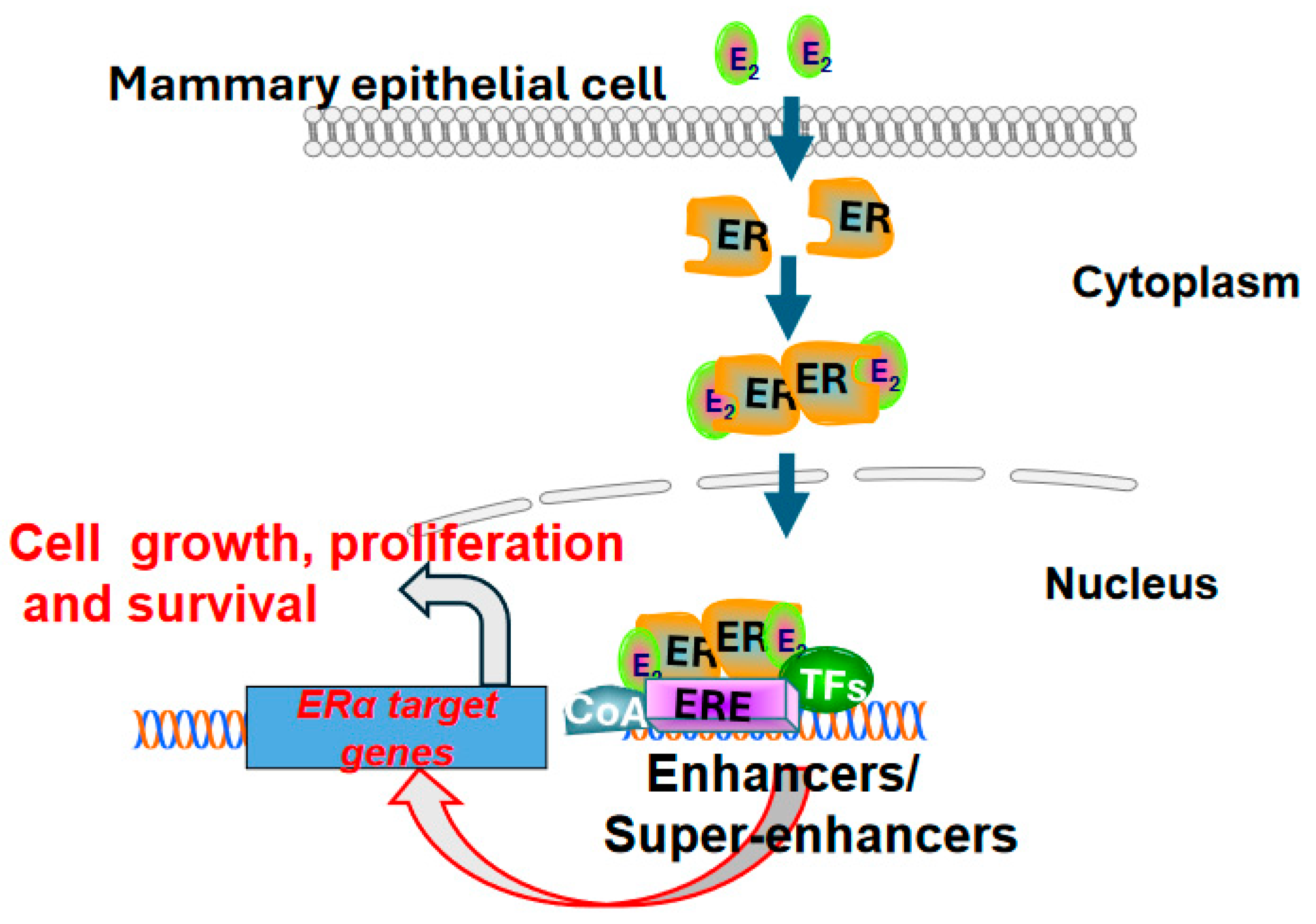
Figure 2.
Schematic representation of estrogen receptor α domain structure.
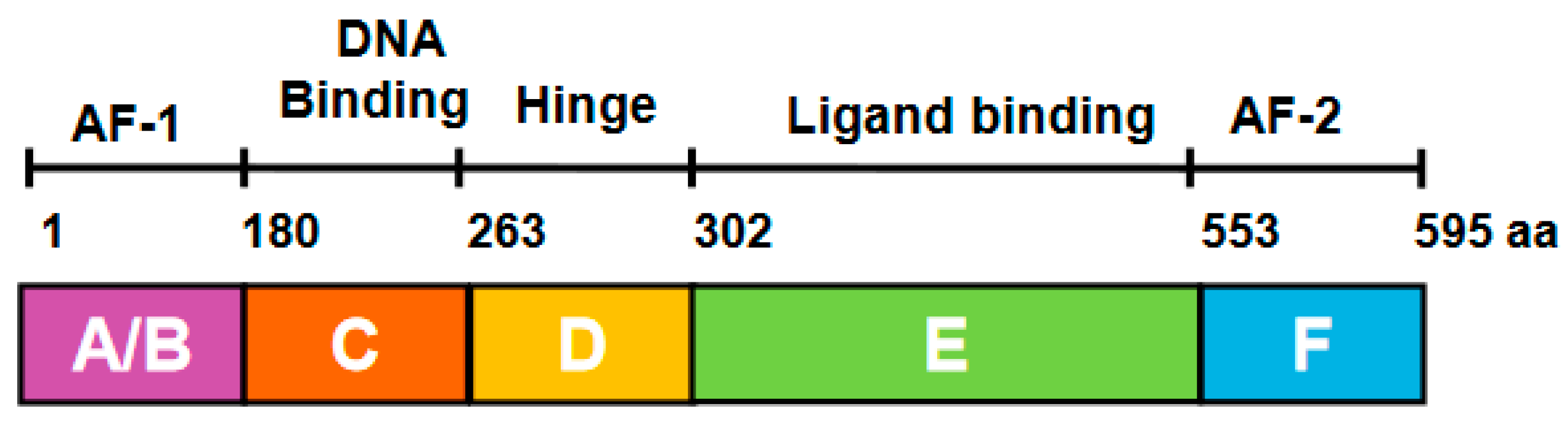
Figure 3.
Strategies to therapeutically inhibit estrogen receptor α signaling in ER+ breast cancer cell. (A) 4OH tamoxifen competitively blocks the binding of estradiol to estrogen receptor α. The 4OH tamoxifen and estrogen receptor α complex translocates from cytosol to nucleus, binds to estrogen response elements (ERE) in the genome, and recruits corepressors (CoR) to inhibit E2 induced transcription in ER+ breast cancer cells. (B) Aromatase inhibitors inhibit estradiol production by impeding the aromatization of testosterone to estradiol. The loss of estradiol renders estrogen receptor α inactive during treatment with aromatase inhibitors. (C) Selective estrogen receptor degraders (SERDs) such as fulvestrant block estrogen signaling by inducing conformations that result in the selective degradation of estrogen receptor α via proteasomal pathways.
Figure 3.
Strategies to therapeutically inhibit estrogen receptor α signaling in ER+ breast cancer cell. (A) 4OH tamoxifen competitively blocks the binding of estradiol to estrogen receptor α. The 4OH tamoxifen and estrogen receptor α complex translocates from cytosol to nucleus, binds to estrogen response elements (ERE) in the genome, and recruits corepressors (CoR) to inhibit E2 induced transcription in ER+ breast cancer cells. (B) Aromatase inhibitors inhibit estradiol production by impeding the aromatization of testosterone to estradiol. The loss of estradiol renders estrogen receptor α inactive during treatment with aromatase inhibitors. (C) Selective estrogen receptor degraders (SERDs) such as fulvestrant block estrogen signaling by inducing conformations that result in the selective degradation of estrogen receptor α via proteasomal pathways.
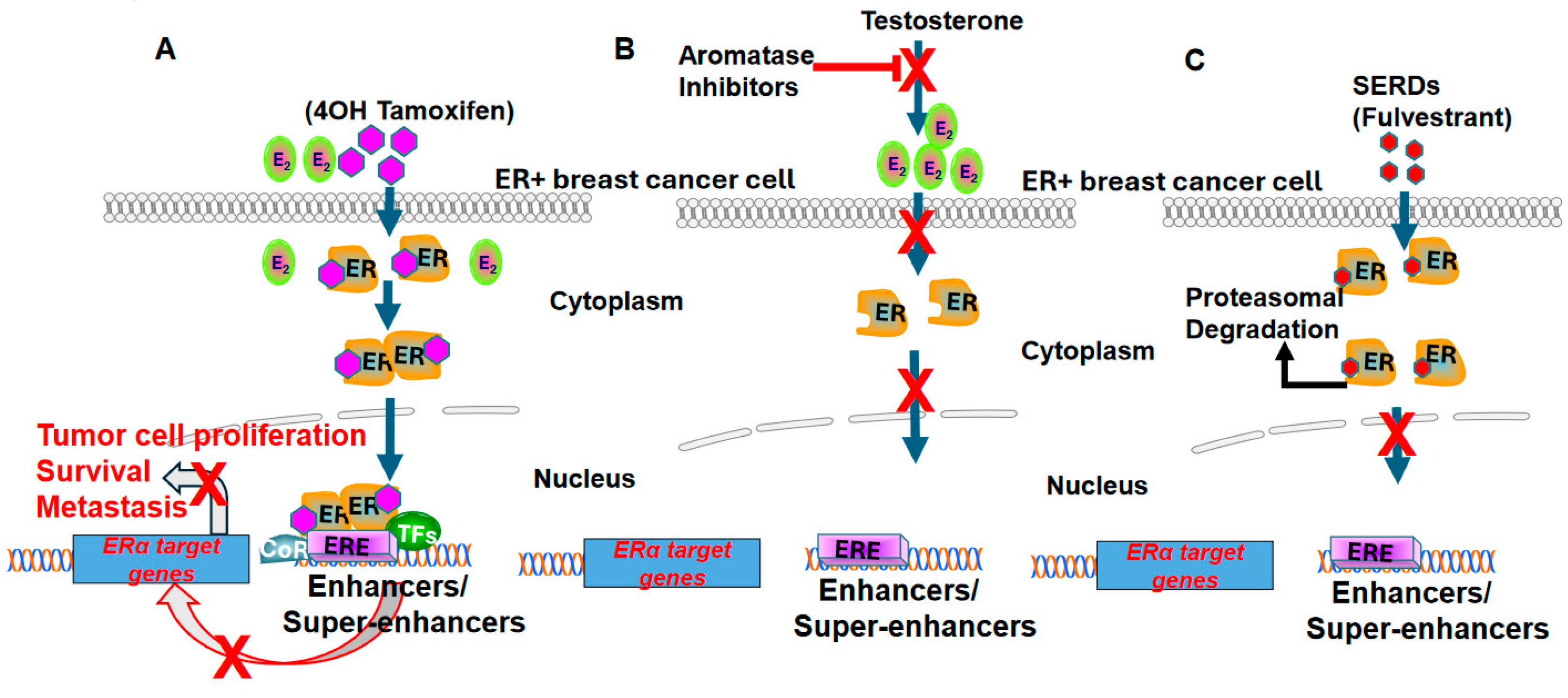

Table 1.
Next-Generation ER-Targeting Drugs in Clinical Trials.
| Study Drug | Mode of Action | Clinical Stage of Development | Status | Clinical Trial Identifier | Sponsor/Source |
|---|---|---|---|---|---|
| G1T48 | SERD | Phase 1 | Completed | NCT03455270 | G1 Therapeutics, Inc. |
| D-0502 | SERD | Phase 1 | Completed | NCT03471663 | InventisBio Co., Ltd |
| SIM0270 | SERD | Phase 1 | Recruiting | NCT05293964 | Jiangsu Simcere Pharmaceutical Co., Ltd. |
| H3B-654 | Covalent ERα antagonist |
Phase 1 | Completed | NCT03250676 | Eisai Inc. |
| ZN-c5 | SERD | Phase1/2 | Completed | NCT03560531 | Zeno Alpha Inc |
| GDC-9545 | SERD | Phase 3 | Recruiting | NCT06065748 | Hoffmann-La Roche |
| LY3484356 | SERD | Phase 3 | Recruiting | NCT05514054 | Eli Lilly and Company |
| AZD9833 | SERD | Phase 3 | Active, not recruiting | NCT04964934 | AstraZeneca |
| OP-1250 | SERD | Phase 3 | Recruiting | NCT06016738 | Olema Pharmaceuticals, Inc. |
| ARV-471 (PF- 07850327) |
ER PROTAC degrader |
Phase 3 | Recruiting | NCT05514054 | Pfizer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



