
Submitted:
30 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
The incidence of melanoma, the most lethal form of skin cancer, has increased due to ultraviolet exposure. The molecular characterization of melanomas has shown a high mutational burden led to the identification of some recurrent genetic alterations. BRAF gene is mutated in 40-50% of melanomas and its role in melanoma development is paramount. BRAF mutations confer constitutive activation of MAPK signalling. The large majority (abou 90%) of BRAF mutations occur at amino acid 600; the majority are BRAFV600E mutations and less frequently BRAFv600K, V600D and V600M. The introduction of drugs that directly target BRAF-mutant protein (BRAF inhibitors) and of agents that stimulate immune response through targeting of immune check inhibitor consistently improved the survival of melanoma BRAFV600-mutant patients with unresectable/metastatic disease. In parallel, studies in melanoma stage II-III patients with resectable disease have shown that adjuvant therapy with ICIs and/or targeted therapy improves PFS and RFS, but not OS compared to placebo; however, neoadjuvant therapy plus adjuvant therapy improved therapeutic response compared to adjuvant therapy alone.
Keywords:
skin tumors
; melanoma
; BRAF mutations
; MAPK
; immunotherapy
; targeted therapy
; genomic profiling
; tumor evolution
1. Introduction
Melanoma is an aggressive neoplasia originated from the malignant transformation of melanocytes. It is responsible for most of deaths related to skin tumors. Melanoma is a heterogeneous disease at both phenotypical and molecular levels. The 2018 World Health Organization (WHO) classification of skin tumors identified 9 different types of melanomas, differentiated for their epidemiology, clinical features and genomic alterations [1]. At etiological level, melanomas can be distinguished into two large groups, one related to sun exposure and another not related to sun exposure. Sun-exposed melanomas are characterized by their mutational signatures (related to ultraviolet damage) anatomic location and epidemiology and are subdivided by histopathologic degree of cumulative solar damage (CSD) of the surrounding skin into high-CSD melanomas (lentigo maligna and desmoplastic melanomas) and low-CSD melanomas (superficial spreading melanomas); the melanomas not related to sun exposure include acral melanomas, Spitz melanomas, mucosal melanomas, and uveal melanomas [1].
Numerous studies have characterized the molecular abnormalities observed in cutaneous melanomas. These studies have shown that cutaneous melanomas are characterized by recurrent genetic alterations occurring at the level of genes involved in RAS/MAPK/ pathways (BRAF, RAS, NF1), Telomerase (Telomerase Promoter), Cell cycle (RB1, CDKN2A), Apoptosis (TP53, MDM2), PTEN/PI3K/AKT pathway (PTEN, PI3K) and MITF (MITF) [2,3].
In 2015 a multiplatform analysis of a large cohort of cutaneous melanomas (mostly metastatic tumors) allowed the definition of four molecular subtypes, characterized by a distinct profile of genetic alterations: (a) BRAF-mutated subtype (52% of total) with frequent hot-spot mutations mostly occurring at the level of V600 and more rarely of V601 and rare non-spot mutations at the level of exon 11; hot-spot BRAF mutations are usually mutually exclusive with RAS mutations, while exon 11 BRAF mutations co-occurred with RAS hot-spot and NF1 mutations; (b) RAS-mutated subtype (about 30% of total) characterized by mutations at the level of RAS genes, much more frequently than HRAS or KRAS; (c) an NF1-mutated subtype (14% of total) characterized by loss-of-function NF1 mutations leading to MAPK activation; (d) a triple wild-type subtype (15% of total) characterized by absence of hot-spot BRAF, RAS and NF1 mutations and by the presence of KIT, GNAQ and TYRP1 mutations in 10-20% of cases [4].
Non-cutaneous melanomas display some remarkable differences in their genetic abnormalities compared to cutaneous melanomas. Acral melanomas display a profile of driver mutations similar to that observed in cutaneous melanomas, with BRAF being the most frequently mutated, followed by NRAS and NF1 mutations; TERT promoter mutations are less frequent than in cutaneous melanomas; acral melanomas have a higher frequency of triple-negative melanomas (45-58%) and, consequently exhibit a higher frequency of KIT, GNAQ and TYRP1 mutations compared with cutaneous melanomas [2-3].
Mucosal melanomas have a mutational profile characterized by KIT as the most frequent mutations (20-25% of cases), followed by BRAF and NRAS mutations (whose frequency is lower than that observed in cutaneous melanomas) and NF1 mutations; TERT promoter mutations are observed at lower frequency than in cutaneous melanomas; the mutational burden is lower in mucosal melanomas than in cutaneous melanomas [2-3].
Uveal melanomas are the most frequent tumors of the eye and display a genomic mutational profile different from cutaneous melanomas, with GNAQ/GNA11, BAP1 and SF3B1 being the genes most frequently mutated [2-3].
2. BRAF-Mutated Melanomas
The mitogen-activated protein (MAP) signaling pathway, also known as RAS/RAF/MEK/ERK regulates cell proliferation through a cascade of kinase phosphorylations. The process is initiated by binding of an extracellular ligand to a mitogenic transmembrane receptor, with consequent receptor dimerization and autophosphorylation and GDP conversion to GTP; then, the GTP-RAS complex activates BRAF which forms dimers and activates the cascade involving MEK 1-2 and ERK 1-2, leading finally to the activation of cell proliferation. BRAF is therefore a key serine/threonine protein kinase of the MAPK pathway. BRAF protein is encoded by the BRAF gene, also known as proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homology B.
BRAF protein belongs to the Raf kinase family of growth signal transduction kinases and is composed by 766-amino acids. This protein is composed of three conserved domains (CR) typical of the Raf kinase family members: conserved region 1 (CR1) contains a GTP-Ras, autoregulatory binding domain, conserved region 2 (CR2) is a serine-rich hinge region; conserved region 3 (CR3) contains a catalytic protein kinase domain involved in the phosphorylation of substrates at the level of specific consensus sequences. Particularly, the CR1 region exerts an auto-inhibitory activity on BRAF activation through an inhibition of the CR3 kinase domain; this region contains two subdomains, a Ras binding domain (RBD) located at 155-227 and involved in GTP-Ras binding and a Cystein-rich domain (CRD), a phorbol ester/DAG-binding zinc finger motif, located at 234-280, involved in B-Raf membrane docking after Ras binding. An initial N-terminal domain, called BRAF-specific region (BSR) and located at 10-145, together with CRD exerts an inhibitory regulation of BRAF activation [5]. The CR2 is a flexible linker region that contains a Raf phosphorylation site and a binding site for 14-3-3 protein which contributes to maintain Raf in its autoinhibited state [6]. The CR3 contains the BRAF enzymatic domain located at 457-717 and subdivided into two lobes connected by a short hinge region: the N-lobe located at 457-.530 is responsible for ATP binding; the C-lobe located at the active site is represented by a cleft located between the two lobes and the catalytic site involves and Asp residues at 576 in the C-lobe, located facing the cleft [7]. The CR3 contains some subregions playing an important role in enzyme physiology; furthermore, some of these subregions are altered in their function in consequence of cancer-related genetic alterations. The P-loop is located at residues 467-471 and is involved in stabilization of the non-transferable phosphate groups of ATP during enzymatic ATP binding. The catalytic loop at residues 574-581 and contributes to the enzymatic activity of the kinase domain consisting in the transfer of γ-phosphate of ATP to BRAF protein substrate. The activation loop is located at the level of residues 596-600 and is strongly bound to P-loop through hydrophobic interactions, thus triggering the shift of the enzyme to its active state.
Figure 1.
Schematic representation of the structure of BRAF protein. BRAF protein is composed by 766 amino acids. Several structural regions, functionally relevant were identified: BSR (BRAF-specific region); CR1 (Constant Region 1) containing two domains, RBD (Ras-binding domain) and CRD (Cystein-Rich Domain); CR2 (Constant Region 2); CR3 containing the kinase domain. Within the kinase domain is outlined the activation loop with its most frequent missense mutaions observed in melanomas.
Figure 1.
Schematic representation of the structure of BRAF protein. BRAF protein is composed by 766 amino acids. Several structural regions, functionally relevant were identified: BSR (BRAF-specific region); CR1 (Constant Region 1) containing two domains, RBD (Ras-binding domain) and CRD (Cystein-Rich Domain); CR2 (Constant Region 2); CR3 containing the kinase domain. Within the kinase domain is outlined the activation loop with its most frequent missense mutaions observed in melanomas.
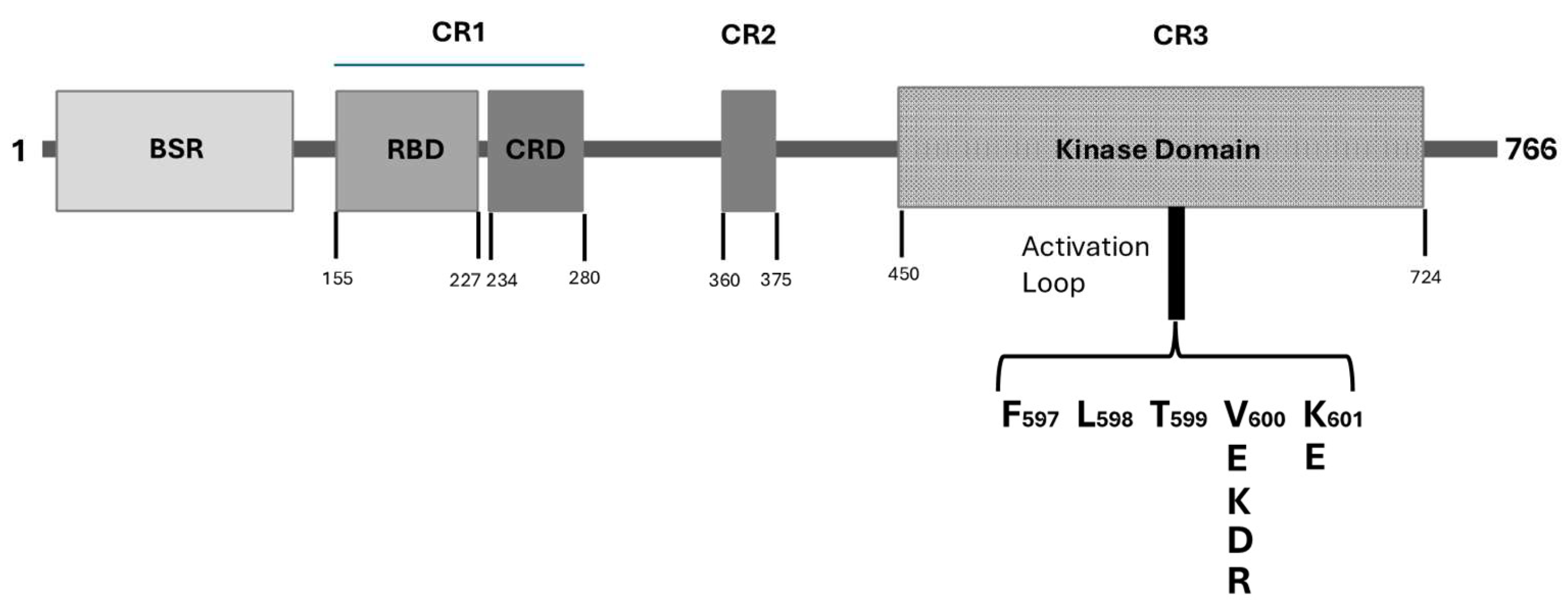
2.1. BRAF Mutations
BRAF is frequently mutated in several human cancers: particularly frequent are mutations in melanoma and thyroid cancer; less frequent in colorectal cancer, non-small cell lung cancer, glioma and bladder cancer [8]. BRAF mutations occurring in cancer are classified into four distinct molecular groups: class I mutations determine a high activation of Ras-independent monomeric BRAF; all these variants are missense mutations of Val600, an amino acid residue located in the activation loop; class II BRAF mutants signal constitutively active RAF dimers and determine a moderate or high Ras-independent activation of BRAF; class III BRAF mutations determine RAS-dependent activation of home and heterodimers and generate a loss-of-function BRAF variant; class IV mutations are related to fusion events involving the BRAF gene [8].
In melanomas the most frequent mutations are represented by class I mutations (about 75%), followed by class I and class III mutations (about 12% and 11%, respectively). Among class I mutations, the most frequent mutations are represented by V600E mutations, caused by a missense mutation determining the substitution of a Valine residue with a Glutamine residue. V600E mutants exhibit a strong BRAF activity estimated about 500 times higher than WT-BRAF; this mutant disrupts the interaction between the activation loop and the P-loop and thus determines the constitutive BRAF activation; the high BRAF activation directly signals to ERK through phosphorylation of MEK [9]. The second most common V600 mutant is V600K generated by a missense mutation leading to the substitution of Valine 600 with Arginine; V600K melanomas are usually observed in patients with a history of chronic sun damage and exposure and are associated with a poor prognosis; V600K melanomas are less dependent on the MAPK/ERK pathway, with a higher expression of PI3KB [10,11]. The variant V600R involving the substitution of Valine with Arginine and V600D involving the substitution of Valine with Spartic acid are more rarely observed in melanoma patients and display biological characteristics like those reported for V600K variant.
The second generation of ATP-competitive RAF inhibitors, called class I inhibitors, including Vemurafenib, Dabrafenib and Encorafenib are potent and selective inhibitors of BRAFV600 mutants [12]. These inhibitors, in addition to exert a potent inhibition of BRAFV600 mutants, induce also paradoxical activation of RAF signaling in BRAF-WT cells and tumors, including those with RAS mutations; this phenomenon is related to the differential effects of class I inhibitors on BRAF monomers (inhibitory) versus BRAF dimers (stimulatory) [13]. It is important to underline that the RAF inhibitor paradox represents also the molecular basis for the clinical success of regimens based on the association of a BRAF inhibitor with a MEK inhibitor: in fact, the early ERK activation induced by class I BRAF inhibitors through the paradox RAF inhibitor is inhibited by MEK inhibitors.
As above discussed, class II mutations are less activating than class I mutations and can be subdivided into class IIa and IIb, according to ntheir mutational profile, with L597 and K601 mutations of the activation loop observed in class IIa and with G476 and G469 of the glycine-rich region of kinase domain observed in class IIb mutations [14]. Melanomas bearing class II mutations are less sensitive to BRAF inhibitors than those with class I mutations; however, class II-mutant melanomas are sensitive to double inhibition with BRAF and MEK inhibitors [15]. Analysis of literature data have further supported the conclusion that melanomas bearing class II mutations are more sensitive to the combination of BRAF plus MEK inhibitors than to BRAF or MEK inhibitors alone [16].
Class III BRAF mutations have a different mutational profile involving N581 or D594 residues and are RAS-dependent, have low or absent BRAF kinase activity and cooperate with concurrent RAS or NF1 mutations; melanomas bearing these mutations are less sensitive to BRAF+MEK inhibitors than melanomas with class II mutations [14].
An additional event that can induce BRAF activation is represented by BRAF fusion events observed in a minority of melanoma patients; these fusion events usually are not associated with BRAF mutations and are highly heterogeneous at molecular level for the variability of BRAF fusion partners [17,18]. This molecular heterogeneity is reflected also by a concomitant phenotypic and clinical heterogeneity, with variable responses to BRAF/MEK inhibitors [19].
The analysis of the co-mutational profile showed that six different genes had mutual exclusivity with BRAF, including the driver genes NRAS and NF1, a pattern reflective of their complementary roles in the activation of the MAPK pathway; similarly, TLR4 and EGFR are both able to regulate the MAPK pathway and are also mutually exclusive with the BRAF mutation; in addition, also ARH, GAP21 and GABRA 6 genes are mutually exclusive with BRAF mutations [20].
The co-mutation profile of class III BRAF mutant melanomas differs substantially from that observed in class I BRAF mutant melanomas. In fact, NF1 loss-of-function mutations frequently co-occur with BRAF non-V600 class II mutations: in fact, melanomas harboring BRAF III non-V600 mutations have frequent co-occurring, NF1 loss-of-function mutations (67%) and RAS mutations (22%); furthermore, 15% of NF1 loss-of-function mutant melanomas harbor concomitant class III C600 BRAF mutations [21].
In some melanoma patients, BRAF copy number gains may be observed in addition to BRAF mutations. BRAF gene is located on chromosome 7q and gains of chromosome 7 are observed in about 50% of patients with primary cutaneous melanomas [22]. Maldonado and coworkers reported that 9 patients out of 19 with BRAF mutations had an increased 7q number; on the other hand, 15 patients out of 49 with WT-BRAF melanomas exhibited an increased 7q number [23]. In 7 of the 9 BRAF-mutant patients with 7q gain, the BRAF allelic ratio evaluation suggested a gain of the mutant BRAF allele [23]. Helias-Rodzewicz et al. have evaluated variations of BRAF mutant allele percentage and 7q copy number in 368 melanoma patients [24]. 38% of these patients displayed BRAF mutations, 66% with heterozygous allele frequency and 19% with BRAF-mutant allele frequency >60%; chromosome 7 polysomy was observed in 16% of BRAF-WT, 35% of BRAF-heterozygous and 54% of BRAF-mutant with allelic frequency >60% [24]. In parallel, 33 melanocytic nevi were explored, with 27 displaying V600E BRAF mutations, all without chromosome 7 gain [24]. Comparable observations were made by Stagni et al. who investigated 46 metastatic, BRAF-mutant melanoma patients before treatment with BRAF inhibitors and 50% of the displayed BRAF gains, mostly related to chromosome 7 gain; the analysis of BRAF-mutant allele frequency showed that 64% were heterozygous, while 12.5% and 23% of them showed a low and a high BRAF-mutant allele frequency; patients with high BRAF-mutant allele frequency have concomitant BRAF copy number gain [25]. Importantly, patients with heterozygous or high-BRAF-mutant allele frequency responded to treatment with BRAF inhibitors better than those with low BRAF-mutant allele frequency [25].
Birkeland and coworkers have explored the patterns of genomic evolution occurring in advanced melanoma, at the level of metastatic progression. An event observed in many melanoma patients at advanced stage consisted in a low-level of copy number gains of at least one BRAF-containing allele occurring in 78% of patients with BRAF-mutant melanomas, and in 15% of BRAF-WT melanomas; in all patients with BRAF-mutant melanomas, with only one exception, the gained BRAF allele was the mutated allele [25]. Whole genome duplication was an event observed in about 40% of patients and occurred later compared to BRAF copy number gain [26].
2.2. Role of BRAF Mutations in Melanomagenesis
Many studies have supported a key role of BRAF mutations in early stages of melanomagenesis. First, BRAF mutations, such as BRAFV600E were initially reported in 82% of melanocytic nevi, with N-RAS mutations present in a minority of cases [27]. This finding was confirmed in subsequent studies [28]. The BRAF mutation involves one of the two alleles in every melanocytic nevus, thus suggesting that a nevus derives from the outgrowth of a single melanocyte that acquired BRAF mutations [29]. This interpretation is directly supported by the immunostaining of melanocytic nevi with an antibody (VE1) specific for BRAFV600E mutation detecting this genetic abnormality in all the melanocytes composing a BRAF-mutated nevus [29]. A key role of BRAF mutations in nevi formation is directly supported by studies in animal models engineered to express BRAFV600E in the melanocyte lineage (transgenic mice expressing BRAFV600E under the control of the melanocyte-specific mtfa promoter): these animals developed patches of ectopic melanocytes (“fish nevi”) [30]. In TP53-deficient fish, BRAF-mutant-generated nevi progressed to melanomas [30].
The sequencing of melanoma-relevant genes in primary melanomas and their adjacent precursor lesions, including benign nevi areas and intermediate lesions to melanoma allowed to define the genetic evolution from nevi to melanomas [31]. Benign nevi lesions harbored BRAFV600E mutations, while intermediate lesions were enriched for NRAS and BRAFV600E mutations and additional driver mutations; intermediate lesions and melanomas in situ harbored TERT promoter mutations; biallelic CDKN2A inactivation, as well as PTEN and TP53 mutations emerged only in advanced melanomas [31]. A sequential genomic and transcriptomic analysis extending from nevi up to regional metastases allowed the definition of pathways activated ur disrupted during melanoma evolution; somatic alterations sequentially induced NAPK pathway activation, upregulation of telomerase, modulation of chromatin landscape, G1/S checkpoint override, ramp-up of MAPK signaling, disruption of the p53 pathway and activation of the PI3K pathway [32].
The study of Shain and coworkers provided evidence that copy number alterations represent a genetic abnormality mainly observed at late stages of melanoma evolution. Matsuta and coworkers observed chromosome 7 gains in 41% of a melanoma population mainly composed by primary melanomas [33]. Casorzo et al. observed the absence of chromosome 7 polysomy in melanocytic nevi; in nevus-associated melanomas, chromosome 7 gains were observed only in melanoma sectors of these lesions [34]. Udart et al. observed higher frequency of chromosome 7 polysomy in metastatic melanomas compared to primary melanomas (41% vs 15%, respectively) [35].
3. Therapy of BRAF-Mutant Melanomas
The progresses made in the understanding of the genetic alterations of melanomas have led to the development of numerous new drugs that have been introduced and evaluated in clinical trials.
Concerning BRAF-mutant melanomas, two types of new drugs were introduced in therapy in the last years, one represented by drugs directly and specifically targeting either BRAF mutant molecules or MEK and the other one represented by immune check inhibitors, drugs targeting molecules that negatively regulate the immune response. The introduction of these drugs has completely revolutionized the medical therapy of BRAF-mutant melanomas.
3.1. Adjuvant Therapy in Stage II Melanoma
It was estimated that about 15% of newly diagnosed melanomas are stage II tumors; about 7% of all melanomas are stage IIB and IIC melanomas, with an infiltration in dermis of >2mm or >4mm, respectively and with a consistent risk of recurrence. Outcomes for patients with stage II melanomas are highly heterogeneous, with a very low risk of death for patients at stage IIA (94% of survival at 10 years) to a moderate risk of death for patients at stage IIB (85% of survival at 10 years)> and at stage IIC (75% of survival at 10 years). Thus, there is a clear rationale for an adjuvant therapy in resected stage IIB/C melanoma patients. Obviously, this therapeutic choice has to take into account a risk/benefit analysis.
Two pivotal clinical trials have evaluated the safety and the effectiveness of adjuvant Pembrolizumab and adjuvant Nivolumab in resected stage IIB/C melanoma patients. (Table 1) The KEYNOTE-716 phase III randomized, double-blind trial explored the safety and the effectiveness of pembrolizumab in 976 melanoma patients, resected, with stage IIB/C cutaneous melanoma without lymph node regional involvement, randomly assigned to treatment with Pembrolizumab (487 patients) or to placebo (489 patients) [36]. The main endpoint of the study consisted in evaluation of the effect of treatment on RFS and DMFS. The median RFS was not reached in both groups; the estimated 36-month RFS rate was 76.2% for Pembrolizumab and 63.4% for placebo. The improvement of RFS in the Pembrolizumab group compared to the Placebo group was observed in both stage IIB (79.7% vs 66.5%, respectively) and IIC (71.4% vs 58%, respectively) [36]. The median DMFS was not reached in both groups; the estimated 36-month DMFS was 84.4% for Pembrolizumab and 74.7% for Placebo [36]. The improvement of DMFS was observed in both stage IIB (86.7% vs 78.9%) and stage IIC patients (80.9% vs 68.1%, respectively) [36]. The CHECK Mate 76K phase III, double-blind trial involved the enrollment of 790 melanoma patients with stage IIB/C disease, randomized 2:1 to treatment with Nivolumab or with Placebo. At 7.8 months of follow-up, Nivolumab improved both RFS and DMFS over Placebo [37]. At 12 months, RFS was 89% among patients treated with Nivolumab compared to 79% in the Placebo group; this improvement was related to a reduction of distant (4.9% vs 11.7%) and locoregional (2.1% vs 7.6%) recurrences compared to placebo. The RFS benefit due to Nivolumab was observed in both stage IIA and IIB patients [37]. Importantly, Nivolumab improved RFS both in BRAF-WT (91.2% vs 77.1%) and BRAFV600-mutant melanomas (87.3% vs 81.7%) [37]. Furthermore, Nivolumab improved DMFS rate compared to placebo: at 12 months, DMFS 92.3% in the Nivolumab group compared to 86.7% in the Placebo group [37]. This improvement in DMFS was particularly evident among stage IIC patients (87.9% vs 78.7%) [37]. Treatment-related grade 3-4 adverse events were observed in 10.3% (Nivolumab) and 2.3% (Placebo) of patients [37]. One treatment-related death (0.2%) occurred in patients treated with Nivolumab.
Recently, it was proposed a phase III clinical trial aiming to evaluate the safety and the efficacy of combined BRAF+MEK inhibitor (Encorafenib+Binimetinib) in resected stage II BRAFV600-mutant melanoma patients [38].
Interestingly, the phase II/III randomized trial DETECTION will explore circulating tumor DNA-guided therapy for stage IIB/IIC melanoma patients after surgical resection [39]. This clinical trial is based on several recent studies showing that circulating tumor DNA (ctDNA, the tumor-derived fraction of circulating free DNA in the blood) represents a suitable biomarker of active tumor disease in stage II/III melanoma patients [40,41]. A recent study based on the analysis of 90 stage IIA-IIID cutaneous melanoma patients showed that personalized, tumor-informed ctDNA analysis offers an additional tool to conventional monitoring for melanoma patients to detect molecular residual disease and was found to be prognostic [42]. Furthermore, longitudinal monitoring of ctDNA using a personalized assay is highly prognostic in stage II/III melanoma patients undergoing curative resection [43]. Thus, the DETECTION trial will involve melanoma patients with stage IIB/IIC disease with BRAF/NRAS/TERT promoter mutations, surgically resected; the patients are then analyzed for the presence of ctDNA and those positive will be randomized 1:1 to continue routing follow-up and therapy at choice of clinical investigatore or to undergo treatment with Nivolumab [39].
3.2. Aduvant Therapy in stage III Melanoma
The immunotherapy randomized phase III trial EORTC 18071 showed a better RFS and OS of stage III melanoma patients treated with Ipilimumab compared with placebo [44]. In the evaluation with 7 years of follow-up, the RFS (HR 0.75, p<0.001), DMFS (HR 0.76, p0.002) and oS (HR 0.73, p<0.002) showed a significant benefit in the Ipelimumab group compared with the placebo group [45].
These results have supported the approval of Ipilimumab as adjuvant therapy for stage III melanoma by US FDA. However, the high toxicity of Ipilimumab monotherapy greatly limited its current adoption in adjuvant setting. In the EORTC 1325/KN-054 trial, phase III, double blind, high-risk stage IIIA/B melanoma patients surgically resected were randomly assigned to receive 200 mg of Pembrolizumab or placebo intravenously every 3 weeks for a total of 18 doses (about 1 year). A five-year analysis of this study was reported in 2022 [46]. In the overall treated population, Pembrolizumab administration was associated with longer recurrence-free survival (RFS) than placebo (55% vs 39%, respectively) and longer metastasis-free survival (MFS) than placebo (60% vs 44%, respectively) [46]. In patients with BRAF-V600 mutations, the RFS at 5 years was 54% in the Pembrolizumab group and 35% in the placebo group [46]. Adverse events were rarely observed and limited to 9 patients among those treated with Pembrolizumab and 1 patient treated with placebo [46]. An analysis extended to seven-year follow-up confirmed the results observed in the previous analysis. Particularly, at 7 years, DMFS was 54% in the Pembrolizumab and 42% in the Placebo group; progression/recurrence-free survival 2 (PRSF2) was 61% in the Pembrolizumab group and 53% in the Placebo group [47].
The randomized phase III Check Mate 238 study compared the safety and the efficacy of Nivolumab and Ipilimumab in 906 melanoma patients with stage IIIB, IIIC or IV tumors surgically resected [48]. The initial results observed in this study showed a 12-month RFS of 70.5% in the Nivolumab group and 60.8% in the Ipilimumab group [48]. Furthermore, a lower rate of grade 3-4 adverse events was observed in the Nivolumab group compared to the Ipilimumab group [48]. Both patients with BRAF-WT and BRAF-mutant melanomas displayed benefit from Nivolumab therapy [48]. According to these results Nivolumab was approved by the US FDA in 2017. The analysis of recurrences of patients involved in this trial showed a rate of recurrence of 44% in Nivolumab group and 51% in the Ipilimumab group [49]. Nivolumab-treated patients with early or late recurrence benefitted from an Ipilimumab-based therapy or targeted therapy (BRAF and MEK inhibitors) in BRAF-mutant tumors [49]. In both groups of patients, biomarkers associated with improved RFS and OS were represented by higher levels of tumor mutational burden, tumor PD-L1, intratumoral CD8+ T cells and IFN-γ-associated gene signature, and lower levels of serum C-reactive protein levels [50].
The randomized phase III trial SWOG 1404 evaluated whether adjuvant Pembrolizumab (647 patients) improved RFS or OS in comparison with high-dose IFNα-2b for one year or Ipilimumab for up to three years (654 patients) in resected high-risk stage III melanoma patients [51]. At a median follow-up of 47.5 months, Pembrolizumab was associated with significantly longer RFS compared to the two other immunotherapies (HR 0.77, p<0.002); however, Pembrolizumab did not improve OS compared to the other two therapies [51]. Treatment-related adverse events (grade 3-5) were lower in patients treated with Pembrolizumab (19.5%) than in those treated with IFNα-2b (71.2%) or Ipilimumab (49.2%) [51]. A secondary analysis showed that the quality of life was significantly better among patients treated with Pembrolizumab compared to the two immunotherapy groups [52].
IMMUNED phase II randomized clinical trial compared adjuvant Nivolumab (1mg/kg) plus Ipilimumab (3mg(kg) versus Nivolumab versus Placebo in 167 patients with resected stage IV melanoma [53]. 4-year RFS was 64.2% in the Nivolumab plus Ipilimumab group, 31.4% in the Nivolumab group and 15% in the Placebo group; 4-year OS was 83.8% in the Nivolumab plus Ipilimumab group, 72.6% in the Nivolumab group and 63.1% in the placebo group [53]. Interestingly, patients with BRAFV600bnmutations benefitted from Nivolumab plus Ipilimumab more than BRAF-WT patients (HR 0.11 vs 0.44, p<0.019). Grade 3-4 adverse events were more frequent in the Nivolumab plus Ipilimumab group (71% vs 29%, respectively) [53].
However, the CheckMate 915 phase III double-blinded trial failed to show a benefit in RFS in patients receiving Ipilimumab (1mg/kg for 6 weeks) plus Nivolumab (240 ng every two weeks) or Nivolumab alone [53]. Probably, the lower dose of Ipilimumab adopted in this study may explain the different results observed in this study compared to the IMMUNED trial [54].
In BRAF-mutant melanoma stage III patients surgically resected was explored also the safety and the efficacy of BRAF inhibitors, associated with a MEK inhibitor. Thus, the COMBI-AD trial (NCT 01682083) evaluated 12 months of adjuvant therapy based on Dabrafenib plus Trametinib or on Placebo in patients with resected stage III melanoma with BRAF V600 mutations [55]. Particularly, in this double-blind, placebo-controlled, phase III trial, 870 patients with completely resected, stage III melanoma with BRAFV600E or BRAFV600K mutations received Dabrafenib at a dose of 150 mg twice daily plus Trametinib at a dose of 2mg once daily (combination therapy, 438 patients) or Placebo (432 patients) for 12 months [55]. Based on the results observed for RFS at the prespecified date, the use of Darafrenib plus Trametinib for stage III melanoma patients with BRAFV600 mutations was approved in many countries. Recently, the final results of this trial were reported with a follow-up of more than 8 years [56]. Relapse-free survival , as well as distant metastasis-free survival were significantly better for Dabrafenib plus Trametinib compared to Placebo; the analysis of overall survival showed that the risk of death was 20% lower in the Dabrafenib plus Trametinib group compared to Placebo, but this benefit was not significant; among patients with BRAFV600E mutations it was observed a 25% lower risk of death in the Dabrafenib plus trametinib group compared to Placebo, while in patients with BRAFV600K mutations the overall survival was slightly better in the Placebo than in the Placebo group than in the Dabrafenib plus Trametinib group [56]. Analysis of biomarkers correlating with response in these patients showed that: MAPK pathway genomic alterations at baseline did not affect treatment benefit or clinical outcome; an IFN-γ gene expression signature higher than the median was prognostic for prolonged RFS in both treatment groups; low tumor mutational burden was associated with longer RFS in the group of patients treated with Dabrafenib and Trametinib [57].
Adjuvant BRAF/MEK inhibitors and immunotherapy with anti-PD1 have become the standard of care for resected high-risk stage III melanoma patients. However, few studies have directly compared hand to hand BRAF/MEK inhibitors vs anti-PD1 agents in resected BRAF-mutant melanoma patients (Table 2). In this context, a recent study based on a nation-wide cohort in the Netherlands allowed the comparison for all resected high-risk stage III melanoma patients a comparison of outcomes between first-line BRAF/MEK inhibitors and anti-PD1, including 225 patients treated with BRAF/MEK inhibitors and 729 treated with anti-PD1 [58]. Through a propensity score matching, two similar groups of 213 patients were defined; after matching, the 1- and 2-year RFS, DMFS and OS rates were not significantly different between the two groups of patients [58]. These observations suggest similar outcomes between adjuvant BRAF/MEK inhibitors and anti-PD1 treatment in stage III melanoma [58].
A multicenter, retrospective cohort study, performed in 15 melanoma centers located in various countries, involved the evaluation of outcomes of 598 melanoma patients with resected stage III BRAFV600-mutant melanoma who received treatment based on either anti-PD1 agent (205 patients) or Dabrafenib plus Trametinib (393 patients). At a median follow-up of 33 months, the median RFS was 51 months in the Dabrafeib plus Trametinib group and 44.8 months with anti-PD1, with comparable OS and DMFS rates [59]. Among patients who experienced recurrence, the proportion of distant metastases was higher in the Dabrafenib plus Tramitinib group [59].
A multicenter real-world study of the German Dermatologic Cooperative Oncology Group explored RFS, overall and melanoma-specific survival (MSS) and the response to the subsequent treatment in 589 stage III melanoma patients undergoing adjuvant treatment with PD1 inhibitors or with BRAF+MEK inhibitors [60]. Among BRAF-mutant patients RFS at 24 months was 49% for PD1 and 67% for patients treated with targeted therapy; 24-month MSS was 87% for PD1 and 92% for targeted therapy group [60].
Melanoma recurrence occurs in a significant proportion of melanoma patients undergoing adjuvant treatment with anti-PD1 agents; some of these patients displayed an early recurrence during anti-PD1 treatment, while other patients displayed a late recurrence occurring after the end of the treatment with anti-PD1 [61]. In a group of BRAF-mutant patients with recurrence after anti-PD1 therapy, Owen and coworkers reported a response to BRAF/MEK inhibitors in 18/23 patients with early recurrence and in 9/10 with late recurrence [61]. Bhave et al. reported 85 BRAF-mutant melanoma patients who developed recurrent disease after adjuvant treatment with BRAF/MEK inhibitors [62]. Response to anti-PD1, combination Nivolumab/Ipilimumab, BRAF/MEK inhibitor rechallenge, and Ipilimumab monotherapy was 63%, 62%, 25% and 10%, respectively and 2-year OS was 84%, 92%, 49% and 45%, respectively [62].
Taylor and coworkers explored a group of 73 BRAF-mutant melanoma patients who received adjuvant therapy with anti-PD1 agent and who recurred: all these patients underwent local therapy, and a group of these patients (61 patients) received a “secong adjuvant” therapy with BRAF/MEK inhibitors and a second group (12 patients) no additional therapy [63]. RFS was significantly better among patients undergoing a second adjuvant treatment (30.8 vs 4 months), but overall survival was similar in the two groups of patients [63].
As above outlined, the randomized phase III clinical trials of adjuvant therapies have failed to show a significant benefit at the level of overall survival. In this context, a recent study presented at the ESMO Congress (Barcelona, Spain 13-17 September) reported the OS data in a large cohort of 1117 patients with stage III sentinel lymph node-positive cutaneous melanoma patients, subdivided into two cohorts one not receiving adjuvant treatments (506 patients) and the other one (611 patients) receiving adjuvant treatments with anti-PD1 or BRAF/MEK inhibitors; the 3-year OS rates were 80.9% and 80.1% in the cohorts of patients receiving or not adjuvant treatments, respectively [64].
Adjuvant immunotherapy or target therapy are costly and are both associated with potential for enduring life-long adverse events. Particularly, the studies with adjuvant anti-PD1 immunotherapy or targeted therapy with BRAF/MEK inhibitors have clearly shown a benefit at the level of RFS, but lack of OS benefit. This finding raises concerns about long-term efficacy of adjuvant therapy in stage II/III melanoma patients. These observations have also shown is not a strong surrogate for OS in the context of adjuvant therapy of melanoma patients [65]. Additional major concerns include the adverse events induced by adjuvant therapies and their high cost [65]. Therefore, individualized decision-making is of crucial importance before the definitive incorporation of adjuvant targeted therapy and immunotherapy as standard treatment for melanoma patients [65]. Therefore, an adequate risk-stratification of melanoma stage II/III patients is required for a guide treatment decision for these patients. In this context, a recent study developed and validated a novel model to predict RFS and MFS after sentinel lymph node biopsy in stage II/III melanoma patients; the development cohort consisted of 4071 patients and the validation cohort of 4822 patients. This model was based on six prognostic factors: sentinel node status, Breslow thickness, presence of ulceration, age at sentinel biopsy, primary tumor location, and maximum diameter of the largest sentinel node metastasis [66]. This model accurately predicted patient-specific risk probabilities for 5-year RFS and MFS, thus offering an important tool for clinical decision making when evaluating adjuvant treatments in patients with high-risk melanomas.
3.3. Neoadjuvant Therapy of Melanoma
In addition to adjuvant treatments, other studies have attempted a different approach consisting of treating melanoma patients with resectable disease with immunotherapy or targeted therapy before surgical resection and then to perform an adjuvant treatment.
A pivotal phase Ib trial (NCT 02434354) evaluated in 30 stage III/IV resectable melanoma patients the long-term outcomes following a treatment based on neoadjuvant (a single dose of 200 mg of Pembrolizumab) 3 weeks before surgical resection, followed by 1 year of adjuvant Pembrolizumab [67]. In a first report on this study, Huang et al showed that 8 of 27 treated patients exhibited a complete response or major pathological response after neoadjuvant therapy: thse rapid clinical responses were associated with accumulation of exhausted CD8+ cells in the tumor at 3 weeks; pretreatment immune signature was associated with clinical response [67]. In contrast, immune suppression and mutational escape correlated with resistance to the treatment [67]. The follow-up of 30 patients at 61.9 months, treated in the context of this trial, showed that: no deaths were observed among patients with complete or major pathological reponse, compared to a 5-year survival of 72.8% for the remainder of the cohort; 2 of 8 patients with major pathologica response relapsed; 8 of 22 patients with incomplete pathological response relapsed; the median ime to recurrence was 3.9 years for patients with ≤10% viable tumor and 0.6 years for patients with >10% viable tumor cells [68].
The NeoCombi phase II trial (NCT 01972347) evaluated the safety and the efficacy of neoadjuvant Dabrafenib plus Trametinib in 35 BRAF-mutant resectable stage IIIB-C melanoma patients; these patients received a treatment based on 12 weeks of neoadjuvant therapy, followed by 40 weeks of adjuvant therapy [69]. At resection, 86% of patients had a RECIST response, with 46% of complete pathological responses (pCR) [69]. Grade 3-4 adverse events were reported in 29% of patients [69]. In asubsequent study, a long-term evaluation of these patients at 5 years was made. Overall RFS was 40%: 53% in aptients with pCR and 28% in those with non-pCR; overall DMFS was 57%: 59% in patients with pCR and 55% in those with non-pCR; OS was 80%: 88% in patients achieving pCR and 71% in those non-achieving a pCR [69]. Overall recurrence was observed in 60% of patients: locoregional recurrence in 34% of patients and distant recurrence in 26% of patients [70].
The CombiNeo phase II trial evaluated Dabrafenib and Trametinib in 21 resectable IIIB-C or oligometastatic stage IV melanoma patients with BRAF mutations; patients were randomized to standard care (surgery and adjuvant therapy) or to neoadjuvant (4 weeks) and adjuvant therapy based on Dabrafenib plus Trametinib (44 weeks). The trial terminated early due to a markedly longer PFS and OS in the neoadjuvant group compared to the standard therapy group (for RFS, at 18.6 months 71% vs 0%, respectively; for OS, 19.7 months vs 2.9 months, respectively) [71].
The OPACIN and OPACIN-Neo trials evaluated the safety and the effectiveness of neoadjuvant therapy using Nivolumab and Ipilimumab in the treatment of high-risk stage III resectable melanoma patients [72]. In the OPACIN trial 20 patients were randomized to receive adjuvant-only treatment based on four cycles of Nivolumab plus Ipilimumab or neoadjuvant and adjuvant therapy, based on two cycles of Nivolumsb plus Ipilimumab [72]. The estimated 5-year RFS and OS rates for the neoadjuvant arm were 70% and 90% compared to 60% and 70% for the adjuvant arm [72]. The OPACIN-Neo trial evaluated different dosing schedules and identified that the most favorable was Ipilimumab (1mg/kg) combined with Novilumab (3mg/kg) every three weeks, resulting after a follow-up of 47 months in a 3-yr RFS and OS rates of 82% and 92%, respectively; for patients with a pathological response, the 3-yr RFS was 95% compared to 37% for patients not achieving a pathological response [72].
A personalized response-directed treatment after neoadjuvant treatment with Nivolumab and Ipilimumab was evaluated in the PRADO trial, an extension cohort of the OPACIN-Neo trial [73]. In this trial, patients. Achieving major pathological response after neoadjuvant therapy omitted surgical lymph node dissection and adjuvant therapy; patients with partial pathological response underwent surgical lymph node dissection only, whereas patients with pathological non-response underwent both surgical lymph node dissection and adjuvant therapy (according to their BRAF mutational status). The 24-month RFS and DMFS rates were 93% and 98% in patients with major pathological response, 64% and 64% in patients with partial pathological response and 71% and 76% in patients with pathological non-response [73].
In 2021, the International Neoadjuvant Melanoma Consortium reported a first pooled analysis from six clinical trials of anti-PD-1-based immunotherapy or BRAF/MEK-targetd therapy involving a total of 192 melanoma patients [74]. A complete pathological response (pCR) was observed in 40% of patients: 47% with targeted therapy and 33% with immunotherapy; pCR correlated with improved RFS and OS; In patients with pCR and treated with immunotherapy RFS at years was 96%, but lower among patients who achieved pCR with targeted therapy (79%) [74]. A more recent analysis reported the pooled analysis on 818 patients with stage ≥IIIB melanoma (77% of patients included in clinical trials and 23% real-world patients) [75]. Median follow-up was 3 years. Patients received neoadjuvant treatment with ICIs (610 patients: 169 PD1-alone, 351 PD1+Ipilimumab, 59 PD1+LAG-3, 27 PD1+other immunotherapy agents), or with targeted therapy (BRAF/MEK inhibitors, 88 patients) or with ICI plus targeted therapy (120 patients) [74]. The analysis of 3-year EFS was: 64% with PD1 alone, 76% with PD1+CTLA4, 82% with PD1+LAG-3, 37% with BRAF/MEK inhibitors and 72% with targeted therapy plus PD1 [75].
The SWOG S1801 study was a phase II, randomized Clinical trial enrolling patients with surgically resectable, Stage IIIB to IVC melanoma patients, in two groups: one receiving a regimen of three doses of preoperative Pembrolizumab, followed by surgical resection and subsequent 15 post-operative Pembrolizumab doses and the other one receiving adjuvant-only treatment with Pembrolizumab [76]. After a median follow-up of 14.7 months, the neoadjuvant group comprising a total of 154 patients exhibited a significantly longer event-free survival (EFS) in comparison with the adjuvant-only group involving a total of 159 patients (EFS at 2 years 72% in the neoadjuvant group and 49% in the adjuvant-only group) [76]. The subanalysis of patients according to BRAF mutational status showed: in the BRAF-mutant, EFS at 2 years, 74% in the neoadjuvant group and 55% in the adjuvant-only group [76]. This observation suggests a potentially better benefit in the BRAF-mutant patients compared to those BRAF-WT [76]. It is important to note that 16 patients randomized to the neoadjuvant treatment did not undergo surgery [75]. Pathology reports on surgical specimens showed that 21% of patients in the neoadjuvant arm displayed a complete pathological response [76].
Another recent study compared neoadjuvant and adjuvant therapy in melanoma patients with resectable disease. Thus. Blank et al. in the NCT 04949113 randomized phase III trial randomly assigned patients with resectable stage III melanoma to two cycles of neoadjuvant Ipilimumab plus Nivolumab followed by surgery followed by 12 cycles of adjuvant Nivolumab [77]. Only patients in the neoadjuvant group with a partial response or nonresponse received adjuvant treatment which consisted of 11 cycles of Nivolumab for patients BRAF-WT and Dabrafenib plus Trametinib for patients BRAF-mutated [77]. In the neoadjuvant group, 59% of patients had a major pathological response and 8% had a partial respoisne [77]. The estimated EFS at 12 months was 83.7% for the neoadjuvant group and 57.2% for the adjuvant-only group; in the BRAFV600E or BRAFV600K mutant patients, the estimated EFS at 12 months was 83.5% in the neoadjuvant group and 52.2% in the adjuvant-only group; in the BRAF-WT patients, EFS was 83.9% among the neoadjuvant group and 62.4% in the adjuvant-only group [77]. Adverse events of grade 3-4 related to systemic treatment were 29% in the neoadjuvant group and 15% in the adjuvant-only group [77]. At 18 months of follow-up, EFS was 80.8% and DMFS was 85.7% in the neoadjuvant group, compared to an EFS of 53.9% and DMFS of 62.4% in the adjuvant-only group [78].
Sub study 02C of the phase 1-2 KEYMAKER-U02 trial (NCT 04303169) is evaluating neoadjuvant Pembrolizumab with or without investigational agents followed by adjuvant Pembrolizumab for stage IIIB-C melanoma patients. This study involved 5 arms: arm1 Pembrolizumab plus Vibostilimab (anti-TIGIT) (at 18-months, RFS 90% and EFS 81%); arm2 Pembrolizumab plus Gebasaxturev (Cosaxievirus A21) (at 18-months, RFS 90% and EFS 72%); arm3 Pembrolizumab alone (at 18-months, RFS 82% and EFS 80%); arm4 Pembrolizumab plus MK-4830 (anti-ILT4) (at 18-months, EFS 78%); arm5 Pembrolizumab plus Favezilimab (anti-LAG-3) (at 6-months, RFS 93% and EFS 92%) [79].
3.4. Adjuvant Vaccination Studies
Several recent studies have explored different melanoma vaccination approaches in the adjuvant setting.
In this context, a recent study reported the clinical evaluation of multipeptide vaccines in melanoma patients in adjuvant setting [80]. In afirst study, vaccination with a cocktail of 12 melanoma peptides restricted by class I HLA molecules (12MP) plus a tetanus toxoid helper peptide induced CD8+ cell response to these 12MP in 100% of treated patients [80]. In an initial study, vaccination with a cocktail of 12 melanoma peptides restricted by class I HLA molecules (12MP) plus a tetanus toxoid helper peptide induced CD8+ cel, response to these 12MP in 100% of treated patients [81]. Subsequently, it was developed a vaccine comprising 6 melanoma peptides presented by class II HLA-DR molecules, whose injection induced CD4+ T-cell responses in most of melanoma patients and induced a clinical response in some patients [82].
Starting from the two multipeptide vaccinations, it was developed a multicenter, randomized, phase II trial to evaluate whether the combined vaccination with 12MP and 6MP would enhance CD8+ T-cell response to 12Mp and would improve clinical outcomes [80]. In this trial, it was adopted also a low-dose of cyclophosphamide treatment to reduce regulatory T cells and to improve T cell response to vaccination [80]. The study involved the enrollment of four arms of patients: A+B treated with 12MP+Tetanoid Toxin Peptide (TTP) and in arm B also pre-treated with cyclophosphamide; arm C+D 12MP+6MP, in arm D also pre-treated with cyclophosphamide [80]. The analysis of long-ter OS showed: median OS for 12MP+TTP 12.9 years and for MP12+MP6 not reached; OS rate estimates at 5, 10 and 15 years for MP12+MP6 were 74%, 68% and 61%, respectively and for MP12+TTP 68%, 56% and 45%, respectively [80]. For indidual study arms, the best RFS and OS data were observed for arm D and the less favorable for arm A [80]. The most significant and durable benefit deriving from the 12MP-6MP vaccination was observed in male patients [80].
Recent studies based on the use of adjuvant dendritic cell therapy in stage IIIB/C melanoma patients failed to show a significant improvement over placebo. Thus, in the MIND-DC randomized phase III trial, 148 patients with resected stage IIIB/C melanoma were randomized to adjuvant treatment with nDCs (autologous CD1c+ conventional and plasmocytoid dendritic cells loaded with tumor antigens) or placebo [83]. The 2-year RFS rate was 36.8% in the nDC treatment group and 46.9% in the control group; median RFS was 12.7 months in the nDC group and 19.9 months in the placebo group [83]. In conclusion, this study provided evidence that, while adjuvant nDC treatment in stage IIIB/C melanoma patients induced specific immune responses and was well tolerated, no benefit in RFS was observed [83].
mRNA-4157 is an mRNA-based cancer vaccine. When administered it will produce one of several dozen possible proteins commonly found in cancer patients. Particularly, mRNA-4157 targets up to 34 patient-specific tumor neoantigens to induce T-cell responses and potentiate antitumor activity. A recent randomized phase IIb study enrolled 157 completely resected stage IIIB-IV melanoma patients for adjuvant treatment with mRNA-4157 plus Pembrolizumab (107 patients) or Pembrolizumab monotherapy (50 patients). With a median follow-up of 23-24 months, RFS was longer with combination vs monotherapy (HR 0.51). At 18 months, RFS and death event rates were 89% and 22% for combination therapy compared to 62% and 40% for monotherapy [84]. An updated analysis of the clinical results observed in the rial KEYNOTE-942 showed that at a follow-up of 2.5 years: the RFS rate was 74.8% in the combo arm and 55.&% in the Pembrolizumab alone; in the combo arm there was a 49% risk reduction in recurrence and/or death compared to Pembrolizumab alone; there was a sustained improvement in DMFS in combo arm versus Pembrolizumab (HR 0.384); OS rate was 96% in combo arm and 90.2% in the Pembrolizumab arm; RFS benefit was observed in tumor burden high and TMB non-high melanomas [85]. Given the results obtained in the KEYNOTE-942 study, it was proposed the INTerpath-011 randomized controlled trail designed to evaluate the efficacy and safety of Pembrolizumab plus mRNA-4157 versus Pembrolizumab plus placebo in patients with high-risk stage II-IV melanoma [86].
The exploration of 4 resected non small cell lung cancer patients and 12 melanoma patients undergoing treatment with mRNA-4157 alone (NSCLC) or in combination with Pembrolizumab (melanoma9 allowed the study of the mechanisms underlying the immunogenicity of mRNA-4157 [87]. mRNA-4157 induced neoantigen T-cell responses and expression of cytotoxic CD8 and CD4 T cells. Particularly, mRNA-4157 induced consistent de novo and potentiated pre-exixsting T-cell responses to targeted neoantigens [87]. It is important to note that while mRNA-4157 is able to expand de novo T-cell clones, check point inhbitors act only on pre-existing T cells that may suboptimally primed. The response of individual patients to mRNA-4157 immunotherapy was variable and is associated to their pretreatment immunological status, with the identification of low immune responders (characterized by an increased frequency of naïve memory cells, but a lower frequency of effector memory and terminally differentiated effector cells) and high immune responders (higher proportion of effector memory CD8 T, T helper 1 and regulatornT-cell ratio and CD4 with cytotoxic potential) [87].
3.5. Immunotherapy and targeted therapy of metastatic melanoma
Many studies have explored the safety and the efficacy of various immunotherapeutic treatments in melanoma patients with stage III/ unresectable or metastatic disease (Table 3).
In this context, an international phase III, multicenter, randomized trial comparatively assessed in unresectable/metastatic melanoma patients, Nivolumab plus Ipilimumab versus Nivolumab alone or Ipilimumab alone [88]. 315 unresectable/metastatic III/IV melanoma patients were randomly assigned to one of these three treatments: Nivolumab plus Ipilimumab (Nivolumab 1mg/kg plus Ipilimumab 3mg/kg every 3 weeks, followed by Nivolumab 3mg/kg every 2 weeks); Nivolumab 3mg/kg every two weeks plus placebo; Ipilimumab 3mg/kg for four doses [88]. Treatment was continued until disease progression or unacceptable toxic effects. Randomization was stratified according to BRAF mutational status, metastasis stage and PD-L1 expression in the tumor. A final analysis of this trial was carried out with a minimum follow-up of 10 years. The median OS was 71.9 months with Nivolumab plus Ipilimumab, 36.9 months with Nivolumab and 19.9 months with Ipilimumab [88]. The hazard ratio for death was 0.53 for Nivolumab plus Ipilimumab compared to Ipilimumab alone and 0.63 for Nivolumab compared to Ipilimumab alone [88]. Median melanoma-specific survival was 120 months with Nivolumab plus Ipilimumab, 49.4 months with Nivolumab alone and 21.9 months with Ipilimumab alone [88]. Importantly, in patients with or without BRAF mutations Nivolumab plus Ipilimumab significantly improved OS and MSS compared to Ipilimumab alone [88]. It is important to note that in patients with BRAFV600 mutations the difference in OS between Nivolumab plus Ipilimumab and Nivolumab alone was more pronounced then in BRAF-WT patients (56% vs 42% compared to 50% and 45%, respectively) [88]. The peculiar sensitivity of BRAFV600 mutant melanoma to Nivolumab plus Ipilimumab seems to be related to over-representation in these tumors of interleukin-17 type helper T (TH17) gene expression signatures; high TH 17 signatures and neutrophils and predicted clinical responses to Nivolumab plus Ipilimumab but not to Nivolumab alone or Ipilimumab alone [89]. The presence of a progression-free survival at 3 years predicted long-term survival [88]. Grade 3 or 4 treatment-related adverse events occurred in 59%, 23% and 28% of the patients in The Nivolumab plus Ipilimumab, Nivolumab alone and Ipilimumab alone groups, respectively [88].
Several studies have supported the efficacy of Nivolumab plus Ipilimumab in the treatment of melanoma patients with brain metastases. The phase II clinical study Check Mate 204 evaluated the safety and the efficacy of Nivolumab plus Ipilimumab in 94 melanoma patients with brain metastases [90]. The patients were treated with a protocol similar to that adopted by Wolchol et al. [88]. 26% of patients achieved a CR, 30% a PR [90]. A long-term evaluation of this study involved a total of 119 melanoma patients with brain metastases, subdivided into a group of asymptomatic (101 patients) and a group of symptomatic (18 patients) and showed in asymptomatic patients an ORR of 53.5%, with a 36-month intracranial PFS of 54% and OS of 71.9%; in symptomatic patients, PFS was 18.9% and OS 36.6% [91].
The efficacy of Nivolumab plus Ipilimumab in the treatment of melanoma patients with brain metastases was confirmed in a real-world cohort of 79 patients, with ORR of 46.9% and a CRR of 16.5%; during a 5-year follow-up mOS was not reaches [92]. The NIBIT-M2 phase III study confirmed these results reporting a 4-year OS of 42.8% in melanoma patients with brain metastases [93].
Other studies have specifically addressed the treatment of melanoma BRAFV600 mutant patients with brain metastases. The SECOMBIT phase II trial explored the optimal sequential treatment for metastatic melanoma patients: BRAF/MEK inhibitors in first line, followed by immunotherapy or immunotherapy in first line, followed by BRAF/MEK inhibitors [94]. Thus, the Secombit trial involved the enrollment of 206 patients who were randomized across the three treatment arms: arm A (Encorafenib plus Binimetinib until progressive disease followed by Nivolumab plus Ipilimumab); arm B (Nivolumab plus Ipilimumab until progressive disease, followed by Encorafenib plus Binimetinib); arm C “sandwich” (Encorafenib plus Binimetinib for 8 weeks, followed by Nivolumab plus Ipilimumab until progressive disease, followed by Encorafenib plus Binemitinib) [94]. AT 4-year of follow-up, PFS rates for arm A, B and C were 34%, 55% and 54%, respectively; the OS rates at 3 and 4 years were 53% and 46% for arm A, 64% and 64% for arm B and 61% and 59% for arm C [94]. The results of this study clearly supported the sequence immunotherapy first, followed by targets therapy as the best therapeutic approach for metastatic melanoma patients [94]. In the SECOMBIT trial, the large majority of melanoma patients do not have brain metastases. Thus, the occurrence of brain metastases was explored during the follow-up of patients enrolled in the SECOMBIT trial: 23/69 in arm A, 11/69 in arm B and 9/68 in arm C [95]. At a median follow-up of 56 months, the 60-month brain metastasis free survival rates were 56% for arm A, 80% for arm B and 85% for arm C [95].
The DREAMSeq trial ECOG-ACRIN EA 6134 involved the treatment of metastatic BRAFV600-mutant melanoma patients with two different sequences of BRAF/MEK inhibitors and Nivolumab/Ipilimumab; patients treated first with Nivolumab/Ipilimumab and then Dabrafenib/Trametinib had a better PFS and OS compared to those treated first with Dabrafenib and Trametinib and then with Nivolumab/Ipilimumab [96].
The TROCOTEL, a multicentre, open-label, single-arm, phase II study, involved a cohort of BRAFV600 mutant melanomas and a BRAF-WT cohort; both groups of patients had melanoma with CNS metastases [97]. Patients with BRAF-mutant melanomas received Atezolizumab, Vemurafenib and Cobimetinib, while BRAF-WT received Atezolizumab and Cobimetinib [97]. The cohort of BRAF-WT patients was stopped after first 15 patients [97]. Intracranial ORR was 42% in BRAF-mutant melanomas and and 27% in BRAF-WT melanomas [97].
The treatment based on Nivolumab and Ipilimumab is associated with higher incidence of treatment-related adverse events mainly induced by Ipilimumab. Therefore, some recent studies have explored to replace anti-CTLA-4 agents with anti-lymphocyte-activation gene 3 (LAG-3). In the phase II-III, double-blind, randomized RELATIVITY-047 trial Relatlimab (anti-LAG-3) and Nivolumab as a fixed dose as compared with Nivolumab alone was evaluated in melanoma patients with unresectable or metastatic disease [98]. The mPFS was 10.1 month with Relatlimab plus Nivolumab and 4.6 months with Nivolumab alone; at 12 months, PFS was 47.7% with Relatlimab plus Nivolumab and 36% with Nivolumab alone [98]. Grade 3-4 adveres events were observed in 18.9% of patients in the Relatlimab plus Nivolumab group and in 9.7% of patients treated with Nivolumab alone [98]. At 19.8 months of follow-up, the estimated OS was not reached among patients treated with Relatlimab plus Nivolumab and 34.1 months among patients treated with Nivolumab alone [99]. Importantly, both BRAFV600 -mutant anf BRAF-WT melanoma patients benefited from Relatlimab plus Nivolumab treatment compared to Nivolumab alone (HR 0.76 and 0.83, respectively) [99]. An update of this study at 3 years of follow-up continued to show a benefit of Relatlimab plus Nivolumab for that concerns PFS, ORR, OS and MSS (melanoma-specific survival) [100].
The exploration of the immunological response to Nivolumab plus Relatlimib showed: higher IFN-γ level increases over baseline in Nivolumab plus Relatlimab compared to Nivolumab alone [101]; decreased sLAG-3 levels in patients treated with Nivolumab plus Relatlimab [101]; higher baseline PD1+CD8+ and ICOS1+CD8+ T cells in responders to Nivolumab plus Relatlimab [102]; better response to Nivolumab plus Relatlimab and Nivolumab alone in patients exhibiting higher LAG-3 tumor expression [101].
Long et al. have performed an indirect treatment comparison between RELATIVITY-047 and CheckMate 067 trial data using patient level data from each trail and reache the conclusion that the two different treatments used in these two different studies induced similar PFS, ORR, OS and MSS; subgroup comparison showed larger numerical differences favoring Nivolumab plus Ipilimumab with acral melanoma and with BRAF-mutant melanomas; Nivolumab plut Ralatlimab was associated with lower grade 3-4 adverse events compared to Nivolumab plus Ipilimumab [103].
The phase I/IIa, open-label RELATIVITY-020 trial part D assessed safety and efficacy of Nivolumab and Relatlimab in 518 melanoma patients who have progressed during or within 3 months of 1 (D1) or >1 anti-PD1-containing regimes [104].The ORR was 12% in D1 and 9.2% in D2; the median duration of response was not reached in D1 and 12.8 months in D2; mPFS was 2.1 months in D1 and 3.2 months in D2; the 6-months PFS rate was 29.5 % in D1 and 27.7% in D2; grade 3-4 adverse events were 15% in D1 and 12.8% in D2 [104].
Relativity 048 is a phase I/II nonrandomized trial evaluating immune-oncology triplets, including Nivolumab+Relatlimab+Ipilimumab in various solid tumors. A recent study reported the preliminary results observed in a cohort of 46 melanoma patients with advanced disease. Median follow-up was 49.4 months, 8.7% had acral cutaneous melanoma, 50% were BRAF-positive, 79.9% were LAG-3 positive, 26.1% were tumor PD-L1-positive and 6.5% received prior adjuvant therapy. An ORR of 58.7% and an OS rate of 71.7% were observed [105]. Grade 3-4 adverse events occurred in 95% of patients [105]. Given the typology of treated patients, this drug triplet seemed to display an encouraging efficacy.
Other studies have explored new drug combinations for the treatment of patients with BRAF-mutant melanomas unresectable or metastatic.
Thus, Dummer evaluated Spartalizumab (an anti-PD1 antibody) in combination with Dabrafenib/trametinib in the treatment of BRAFV600-mutant unresectable melanoma patients. However, the results of this study failed to show any significant improvement of ORR and PFS rate in patients treated with Spartalizumab plus Dabrafenib/Trametinib, compared to Dabrafenib/Trametinib alone [106].
The Columbus trial involved the enrollment of 577 BRAFV600-mutant melanoma patients with unresectable metastatic disease randomily assigned to Encorafenib plus Binimetinib, Vemurafenib or Encorafenib [107]. Compared with Vemurafenib, Encorafenib plus Binimatinib extended PFS (14.9 vs 7.3 months) and mOS (33.6 vs 16.9 months); the drug combination was well tolerated and the rate of druig discontinuation was relatively low (10% vs 14%, respectively) [107]. The data observed in this first report were confirmed in two other reports performed with a follow-up of 5 [108] or 7 years [109]. Particularly, at 7 years of follow-up, PFS and OS rates weer 21.2% and 27.4% in the Encorafenib plus Binimetinib group and 6.4% and 18.2% in the Vemurafenib group, respectively; median melanoma-specific survival was 36.8 months in the Encorafenib plus Trametinib arm and 19.3 months in the Vemurafenib arm [109].
In the part 2 of the phase III COLUMBUS trial, Encorafenib (at 300 mg) plus Binimetinib was compared to Encorafenib alone (at 300 mg); the mPFS was 12.9 months for Encorafenib plus Binimetinib compared to 9.2 months for Encorafenib; the ORR was 68% for Encorafenib plus Binimitinib and 51% for Encorafenib alone [110].
The possible benefit deriving from an induction treatment with targeted therapy with BRAF + MEK inhibitors (Encorafenib and Binimetinib) prior to a combined immunotherapy with Nivolumab plus Ipilimumab in patients with advanced BRAFV600 mutant melanoma was explored in the EORTC phase II randomized EBIN study [111]. However, the results of this study failed to show any significant improvement of PFS in the group of patients pretreated with Encorafenib plus Benitinib and the with Nivolumab plus Ipilimumab, compared to those treated with Nivolumab plus Ipilimumab alone [111].
3.6. Adoptive Therapy with Tumor Infiltrating T Lymphocytes (TIL) in Melanoma Patients who Have Failed Immunotherapy and/or Targeted Therapy Treatments
Effective treatments are very limited for patients with advanced (unresectable or metastatic) melanoma who have progressed after immune checkpoint inhibitors and targeted therapies.
Recent studies have shown that adoptive cell therapy with TIL has consistently shown efficacy in these refractory/relapsing melanoma patients. Adoptive cell therapy with TIL offers a potential therapeutic option for metastatic melanoma patients: this immunotherapy is based on the extraction of a fragment of tumor followed by expansion under culture conditions that are permissive for the expansion of a polyclonal population of T lymphocytes, allowing the generation of a large number of T cells to be infused back into the patients. The advantage of this therapy consists in the generation of T lymphocytes whose cytotoxic potential is potentiated during ex vivo expansion, that can address the large repertoire of individual neoantigens expressed on melanoma cells.

Table 4.
Adoptive therapy with TIL in melanoma patients with ICI refractory disease.
| Clinical trial | Patients | Treatment | PFS | OS | Safety |
Parameters correlating with response |
|---|---|---|---|---|---|---|
| C-144-01 NCT 02360579 Nonrandomized phase II |
153 advanced melanoma, ICI refractory | Lifileucel (autologous TIL) >1x109 cells |
ORR 31.4% |
mOS 13.9 months 1yr 54% 2yr 33.9% 3yr 28.3% 4yr 22.2% |
Grade 3-4 100% |
Few responses in patients with high TMB and brain and liver metastases |
| M14 TIL NCT 092278887 |
168 advanced melanoma (86% ICI refractory) 84 TILs 84 Ipilimumab (Ipi) |
Autologous TILs At least 5x109 cells |
TIL 7.2 months Ipi 3.1 months |
mOS TIL 25.8 months Ipi 18.9 months |
Grade 3-4 TIL 100% Ipi 57% |
Not Reported |
Two clinical studies have contributed to the approval of Lifileucel (LN-144), an autologous TIL therapy that uses tumor-tissue T-cells capable of recognizing tissue antigens and being expanded ex vivo maintaining the heterogeneity repertoire of T cells using a centralized manufacturing process, for the treatment of metastatic melanoma patients. A phase II C-144-01 trial evaluated the safety and the efficacy of Lifileucel in patients with advanced melanoma who have progressed on ICI and BRAF inhibitors. A first report of this study involved 66 patients infused with >1x109 TIL cells; Lifileucel induced a significant anitutmor response with an ORR of 36%, with 2 complete responses and 22 partial responses disease control rate of 80% and with a median duration of response not reached after 18.7 months of follow-up [112]. A later report on this trial involved 153 melanoma patients with ICI or BRAF/MEK resistant disease; treatment with Lifileucel was associated 1,2,3 and 4-year os rates of 53%, 33.9%, 28.4% and 21.9% and with an ORR of 31.4% and with the median duration of response not reached [113]. The highest $-year survival rates were observed in patients with the more pronounced tumor responses (68.2%) [113]. Analysis of individual patients showed that few patients with brain or liver metastases or with high tumor burdens respond to treatment with TILs [113].
A second study (NCT 02278887) involved the evaluation of 168 melanoma patients with advanced disease (86% refractory to ICI treatment) to randomized treatment with Ipilimumab or with autologous TIL (at least 5x109 TILS); infusion of TILs was preceded by lymphodepleting chemotherapy and followed by high-dose interleukin-2 infusions [114]. Median PFS was 7.2 months in the TIL group and 3.1 moths in the Ipilimumab group; ORR was 49% in the TIL group and 21% in the Ipilimumab group; median-OS was 25.8 months in the TIL group and 18.9 months in the Ipilimumab group [114]. Grade 3-4 treatment-related adverse events were observed in 100% of patients undergoing treatment with TILs and 57% of those treated with Ipilimumab [114].
In February 2024 Lifileucel received accelerated approval by FDA based on objective response rates and duration of responses conferring substantial evidence of effectiveness in a population of melanoma patients with a high unmet medical need.
Given the results observed in the C-14401 trial, Lifileucel was evaluated in melanoma patients with unresectable/metastatic disease, untreated with ICIs and treated with BRAF/MEK inhibitors if BRAF-mutated, in the context of the IOV-COM-202 clinical study [115]. In the cohort 2A, 22 patients were enrolled and treated with a therapeutic regimen, consisting of Pembrolizumab, nonmyeloablative lymphodepletion, a single infusion of Lifileucel (1x109 – 150x109 cells) and Pembrolizumab until disease progression [115]. 36% of patients had BRAF mutations. ORR was 63.6% (22.7% CR and 40.9% PR). At a median follow-up of 17.2 months, duration of response was not reached [115]. In a significant proportion of responding patients, responses are maintained for ≥12 months. Most common grade 3-4 adverse events were thrombocytopenia, neutropenia and anemia [115].
A recent study performed a systematic review and meta-analysis of the most relevant studies involving TIL therapy in advanced melanoma and reached several important conclusions: no difference was found in median OS between studies with prior anti-PD1 oranti-PDL1 treatment and without; ORR was 34% and 44% for the studies with or without prior anti-PD1 or anti-PDL1 treatment, respectively [116]. The pooled CRR was 10% [116]. These observations reinforce the evidence that TIL should be considered as a treatment of choice in second line for metastatic melanoma patients failing after anti-PD1/PDL1 therapy [116].
4. Conclusions
In the last two decades there was a tremendous progress in the definition of the molecular alterations undelying the development of melanoma, one of the most malignant skin tumors. One of the most recurrent gentic alterations observed in melanoma are mutations of the BRAF protein kinase, whose constitutive activation plays a key role in the early stages of development of melanomas. The definition of these genetic alterations have fostered the development of new therapeutic approaches and were based either in the direct targeting of the mutated BRAF protein and of other constituents of the MAPK pathway or the targeting of the immune response, particularly at the level of immune check inhibitors. These two categories of drugs have led to a consistent improvement of the therapy of melanoma patients, BRAF-WT ot BRAF-mutant, with an advanced stage of development. Particularly, these progresses have led to a significant improvcement of OS of melanoma patients with unresectable or metastatic disease, with the definition of two immunotherapy treatments, based on the double targeting of PD1 and CTLA-4 or PD1 and LAG-3, that now represent the standard of care for these patients. The survival curves of these patients displayed stable tails that have plateaued over time, supporting the presumption that patients on the tail are in fact cured of their disease.
Treatment protocols based first on the use of immunotherapeutic agents and then of BRAF/MEK inhibitors are under evaluation and future studies will determine whether these switching therapy approaches may further improve the outcomes of these patients.
Patients progressing after anti-PD1-based therapy and targeted agents have limited therapeutic options. Therefore, there are no treatment options with approval based on data from patients with advanced melanoma who have progressed after one line of ICI therapy in BRAF-WT patients or two lines of therapy for BRAFV600 mutation-positive tumors. However, recent studies have shown that a part of these patients may significantly benefit from adoptive immunotherapy with TIL. Thus, Lifileucel, a TIL-based therapy, was recently approved for these patients.
The risk of relapse and the prognosis of stage II and III melanomas with resectable disease is highly heterogenous, with a part of patients having a consistent risk of relapse. Thus, numerous studies have explored the effect of adjuvant therapies based on ICIs or targeted therapy (in BRAF-mutant melanomas) and have shown a consistent improvement of PFS and RFS but not of OS compared to placebo. However, recent studies have shown that neoadjuvant therapy plus adjuvant therapy based on ICIs or targeted therapy, may induce a significant therapeutic improvement compared to adjuvant therapy in terms of PFS and RFS, suggesting a possible improvement also at the level of OS. Furthermore, additional studies will be required to determine the optimal protocols of neoadjuvant/adjuvant therapy for BRAF-WT and BRAF-mutant stage II-III melanomas. In parallel, additional criteria for a better risk stratification of stage II-III melanoma patients have been defined and offer the tool for a selection of patients idoneal for neoadjuvant/adjuvant treatments. In this context, it is important to note that neoadjuvant treatments offer an additional important parameter for the prognostic evaluation of stage II/III melanoma patients based on the pathological evaluation at the moment of tumor surgical resection. In fact, patients who achieve major pathological responses display unprecented and lasting survival benefit, while those with partial or no responses show a lower survival benefit and will need alternative therapeutic approaches.
Author Contributions
GC and EP were involved in researching, writing and editing the manuscript. UT was involved in conceptualization, organization, researching and editing the manuscript. All authors have read and agree to the published version of the manuscript.
Funding
“This research received no external funding”.
Institutional Review Board Statement
“Not applicable”.
Informed Consent Statement
“Not applicable.”.
Acknowledgments
None.
Conflicts of Interest
“The authors declare no conflicts of interest.”.
References
- Elder, D.E.; Bastian, B.C.; Cree, J.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization classification of cutaneous, mucosal and uveal melanoma. Arch Pathol Lab Med 2020, 144, 500–522. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Melanoma genetic abnormalities, tumor progression, clonal evolution and tumor initiating cells. Med Sci 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.T.; Yu, S.; Khale Ke, C.L.; Cheng, S.T. The genomic landscape of melanoma and its therapeutic implications. Genes 2023, 14, 1021. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [CrossRef] [PubMed]
- Trebino, T.E.; Markusic, B.; Nan, H.; Banerjee, S.; Wang, Z. Unveiling the domain-specific and Ras isoform-specifric details of BRAF kinase regulation. eLife 2023, 12, RP88836. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Gonzalez-Del Pino, G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF-MEK-14-3-3 complex. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Martinez-Fiesco, J.A.; Durrant, D.E.; Morrison, D.K.; Zhang, P. Structural insights into the BRAF monomer to dimer transition mediated by RAS binding. Nat Commun 2022, 13, 486. [Google Scholar] [CrossRef]
- Kazandjian, S.; Rousselle, E.; Danker, M.; Cescon, D.W.; Spreafico, A.; Ma, K.; Kavan, P.; Batist, G.; Rosae, A. The clinical, genomic, and transcriptomic landscape of BRAF mutant cancers. Cancers 2024, 16, 445. [Google Scholar] [CrossRef]
- Wan, P.; Garnett, M.; Roe, S.M.; Lee, S.; Malescu-Wan, P.; Good, V.M.; Cancer Genome Project, Jones, C. M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Nepote, A.; Avallone, G.; Ribero, S.; Cavallo, F.; Roccuzzo, G.; Mastorino, L.; Conforti, C.; Paruzzo, L.; Poletto, S.; Carnevale Schienca, F. ; Current controversies and challenges on BRAF V660K-mutant cutaneous melanoma. J Clin Med 2022, 11, 828. [Google Scholar] [CrossRef]
- Zengarini, C.; Mussi, M.; Veronesi, G.; Alessandrini, A.; Lambertini, M.; Dika, E. BRAF V600K vs BRAF V600E: a comparison of clinical and dermoscopic characteristics and response to immunotherapies and targeted therapies. Clin Exp Dermatol 2022, 47, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, A.J.; Chen, Z.; Rosen, N.; Solit, D.B. BRAF – a tumor-agnostic drug target with lineage-specific dependencies. Nat Rev Clin Oncol 2024, 21, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M. , Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Dahlman, K.B. Class matters: sensitivity of BRAF-mutant melanoma to MAPK inhibition. Clin Cancer Res 2018, 24, 6107–6109. [Google Scholar] [CrossRef]
- Dankner, M.; Lajoie, M.; Moldoveanu, D.; Nguyen, T.T.; Savage, P.; Rajkumar, S.; Huang, X.; Lvova, M.; Protopopov, A.; Vuzmann, D.; et al. Dual MAPK inhibition is an effective therapeutic strategy for a subset of class II BRAF mutant melanomas. Clin Cancer Res 2018, 24, 6483–6494. [Google Scholar] [CrossRef]
- Dankner, M.; Wang, Y.; Fazeldad, R.; Johnson, B.; Nebhan, C.A.; Dagogo-Jack, I.; Myall, M.J.; Richtig, G.; Bracht, J.; Gerlinger, M.; et al. Clinical activity of mitogen-activated protein kinase-targeted therapies in patients with non-V600 BRAF-mutant tumors. JCO Precis Oncol 2022, 6, e2200107. [Google Scholar] [CrossRef]
- Turner, J.A.; Bemis, J.; Bagby, S.; Capasso, A.; Yacob, B.; Chimed, T.S.; Van Gulick, R.; Lee, H.; Tobin, R.; Tentler, J.J.; et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene 2019, 38, 1296–1308. [Google Scholar] [CrossRef]
- Botton, T.; Televich, E.; Mishra, V.; Zhang, T.; Shain, A.H.; Berquel, C.; Gagnon, A.; Judson, R.L.; Ballotti, R.; Ribas, A. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep 2019, 29, 573–588. [Google Scholar] [CrossRef]
- Moran, J.; Le, L.; Nardi, V.; Golas, J.; Farahani, A.; Signorelli, S.; Onozato, M.L.; Foreman, R.K.; Duncan, L.; Lawrence, D.P.; et al. Identification of fusions with potential clinical significance in melanoma. Modern Pathol 2022, 35, 1837–1847. [Google Scholar] [CrossRef]
- Birkealv, S.; Harland, M.; Matuyama, L.; Rashid, M.; Mehta, I.; Laye, J.P.; Haase, K.; Mell, T.; Iyer, V.; Robles-Espinoza, C.D.; et al. Mutually exclusive genetic interactions and gene essentiality shape the genomic landscape of primary melanoma. J Pathol 2023, 259, 56–68. [Google Scholar] [CrossRef]
- Rajkumar, S.; Berry, D.; Heney, K.A.; Strong, C.; Ramsay, L.A.; Lajoie, M.; Alkallas, R.; Nguyen, T.T.; Thomson, C.; Adams, M.A.; et al. Melanomas with concurrent BRAF non-p.V600 and NF1 loss-of-function mutations are targeatable by BRAF/MEK inhibitor combination therapy. Cell Rep 2022, 39, 110634. [Google Scholar] [CrossRef] [PubMed]
- Bastian, B.C.; Le Boit, P.E.; Hamm, H.; Brocker, E.B.; Pinkel, D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res 1998, 58, 2170–2175. [Google Scholar] [PubMed]
- Maldonado, J.L.; Fridlyand, J.; Patel, H.; Jain, A.N.; Busam, K.; Kageshita, T.; Ono, T.; Albertson, D.G.; Pinkel, D.; Bastian, B.C. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst 2003, 95, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Hélias-Rodzewicz, Z.; Frunck-Brentano, E.; Baudoux, L.; Jung, C.K.; Zimmermann, U.; Marin, C.; Clerici, T.; Le Gall, C.; Peschaud, F.; Taly, V.; Saiag, P.; Emile, J.F. Variations in BRAF mutant allele percentage in melanomas. BMC Cancer 2015, 15, 497. [Google Scholar] [CrossRef]
- Stagni, C.; Zamuner, C.; Elefanti, L. , Zanin, T.; Del Bianco, P.; Sommariva, A.; Fabozzi, A.; Pigozzo, J.; Mocellin, S.; Montesco, M.C.; et al. BRAF gene copy number and mutant allele frequency correlate with time to progression in metastatic melanoma patients treated with MAPK inhibitors. Mol Cancer Ther 2018, 17, 1332–1340. [Google Scholar] [CrossRef]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; Lonning, P.E.; et al. Patterns of genomic evolution in advanced melanoma. Nat Commun 2018, 9, 2665. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat Genet 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Colebatch, A.J.; Ferguson, P.; Newell, F.; Kazakoff, S.H.; Witkowski, T.; Dobrovic, A.; Johansson, P.A.; Saw, R.; Strecth, J.R.; et al. Molecular genomic profiling of melanocytic nevi. J Invest Dermatol 2019, 139, 1762–1768. [Google Scholar] [CrossRef]
- Yeh, I.; von Deimling, A.; Bastian, B.C. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J Natl Cancer Inst 2013, 105, 917–919. [Google Scholar] [CrossRef]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopanl, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.; et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol 2005, 15, 249–254. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Tavelich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Joseph, N.M.; Benhamida, J.; Liu, S.; Prow, T.; Ruben, B.; North, J.; Pincus, L.; Yeh, I.; et al. Genomic and transcriptomic analysis reveals incrementsl disruption of key signaling pathways during melanoma evolution. Cancer Cell 2018, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Matsuta, M.; Imamura, Y.; Matsuta, M.; Sasaki, K.; Kon, S. Detection of numerical chromosomal aberrations in malignant melanomas using fluorescence in situ hybridization. J Cutan Pathol 1997, 24, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Casorzo, L.; Luzzi, C.; Nardacchione, A.; Picciotto, F.; Pisacana, A.; Risio, M. Fluorescence in situ hybridization (FISH) evaluation of chromosome 6, 7, 9 and 10 throughout human melanocytic tumorigenesis. Melanoma Res 2005, 15, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Udart, M.; Utikal, J.; Krahn, G.M.; Peter, R.U. Chromosome 7 aneusomy. A marker for metastatic melanomas? Expression of the epidermal growth factor receptor gene and chromosome 7 aneusomy in nevi, primary malignant melanomas and metastases. Neoplasia 2001, 3, 245–254. [Google Scholar] [CrossRef]
- Luke, J.J.; Ascierto, P.A.; Khattak, M.A.; de la Cruz Merino, L.; Del Vecchio, M.; Rutkowski, P.; Spagnolo, F.; Machiewicz, J.; Chiarion-Sileni, V.; Kirkwood, J.M.; et al. Pembrolizumab versus placebo as adjuvant therapy in resected stage IIB or IIC melanoma: final analysis of distant metastasis-free survival in the phase III KEYNOTE-716 study. J Clin Oncol 2024, 42, 1619–1624. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Del Vecchio, M.; Weber, J.; Hoeller, C.; Grob, J.J.; Mohr, P.; Loquai, C.; Dutriaux, C.; Chiarion-Sileni, V.; Machiewicz, J.; et al. Adjuvant nivolumab in resected stage IIB/C melanoma: primary results from the randomized, phase 3 CHECK Mate 76K trial. Nat Med 2023, 20, 2835–2843. [Google Scholar] [CrossRef]
- Akkoi, A.; Hauschild, A.; Long, G.; Mandala, M.; Kicinski, M.; Govaerts, A.S.; Klauck, I.; Ouali, M.; Longan, P.C.; Eggermont, A. COLUMBUS-AD: a phase III study of adjuvant encorafenib+binimetinib in resected stage IIB/IIC BRAF V600-mutated melanoma. Future Oncol 2023, 19, 2017–2027. [Google Scholar] [CrossRef]
- Lee, R.; Rothwell, D.G.; Jackson, R.; Smith, N.; Wong, S.Q.; Kelso, N.; Burghel, G.; Hewitt, C.; Clarke, H.; Michell, J.; et al. DETECTION phase II/III trial: circulating tumor DNA-guided therapy for stage IIB/C melanoma after surgical resection. J Clin Oncol 2022, 40 (suppl.16), TPS9603. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; Hatzimihalis, A.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann Oncol 2019, 30, m–804. [Google Scholar] [CrossRef]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fishjer, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Loriogan, P.; et al. Circulating tumor DNA predicts xurvival in patients with resected high-risk stage II/III melanoma. Ann Oncol 2018, 29, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Brunsgaard, E.K.; Kuzel, T.; Tan, A.; Rush Medical College; Rush Unive Med Center. Detection of molecular residual disease in stage II and III melanoma utilizing circulating tumor DNA. J Clin Oncol 2024, 42 (suppl.16). [Google Scholar] [CrossRef]
- Kolliapra, R.; Budde, G.; Aushev, V.N.; Brunsgaard, E.K.; Kuzel, T.; O’Donoughe, C.; Riddell, T.; Palsuledesai, C.C.; Krainock, M.; Liu, M.C.; et al. Logitudinal circulating tumor DNA monitoring for detection of molecular residual disease in patients with surgically resected stage II/III melanoma. J Clin Oncol 2023, 40 (suppl.16), 9582. [Google Scholar] [CrossRef]
- Eggermont, A.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomized, double-blind, phase I trial. Lancet Oncol 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Eggermont, A.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of stage III melanoma: long-term follow-up results of the European Organization for Research and Treatment of Cancer 18071 double-blind phase 3 randomised trial. Eur J Cancer 2019, 119, 1–10. [Google Scholar] [CrossRef]
- Eggermont, A.; Kicinski, M.; Blank, C.; Mandala, M.; Long, G.; Atkinson, V.; Dalle, S.; Haydon, A.; Mechcheryakov, A.; Khattak, A.; et al. Five-year analysis of adjuvant pembrolizumab or placebo in stage III melanoma. N Engl J Med Evid 2022, 1. [Google Scholar] [CrossRef]
- Eggermont, A.; Kicinski, M.; Blank, C.; Mandala, M.; Long, G.; Atkinson, V.; Dalle, S.; Haydon, A.; Mechcheryakov, A.; Khattak, A.; et al. Seven-year analysis of adjuvant pembrolizumab versus placebo in stage III melanoma in the EORTC1325/KEYNOTE-054 trial. Eur J Cancer 2024, 211, 114327. [Google Scholar] [CrossRef]
- Weber, J.; Del Vecchio, H.J.; Mandala, M.; Gogas, A.M.; Arance, C.L.; Cowey, C.L.; Dalle, S.; Schenkler, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; Grob, J.J. Adjuvant Nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, H.J.; Gogas, A.M.; Arance, C.L.; Cowey, C.L.; Dalle, S.; Schenkler, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Outcomes with postrecurrence systemic therapy following adjuvant checkpoint inhibitor treatment for resected melanoma in Check Mate 238. J Clin Oncol 2024, in press. [Google Scholar] [CrossRef]
- Larkin, J.; Del Vecchio, M.; Mandal, M.; Gogas, H.; Arance Fernandez, A.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage III/IV melanoma: 5-year efficacy and biomarker results from CheckMate 238. Clin Cancer Res 2023, 29, 3352–3361. [Google Scholar] [CrossRef]
- Grossmann, K.F.; Othus, M.; Patel, S.P.; Trhini, A.A.; Sondak, V.K.; Knopp, M.V.; Petrella, T.M.; Truong, T.G.; Khushalani, N.; Cohen, J.V.; et al. Adjuvant pembrolizumab versus IFNα2b or ipilimumab in resected high-risk melanoma. Cancer Discov 2022, 12, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Linger, J.M.; Darke, A.; Othus, M.; Truong, T.G.; Kushalani, N.; Kendra, K.; Lewis, K.D.; Faller, B.; Funchain, P.; Buchbinder, E.I.; et al. Effectiveness of adjuvant pembrolizumab vs high-dose interferon or ipilimumab for quality-of-life outcomes in patients with resected melanoma: secondary analysis of the SWOG S1404 randomized clinical trial. JAMA Oncol 2023, 9, 251–260. [Google Scholar]
- Livingstone, E.; Zimmer, L.; Hassel, J.C.; Fluck, M.; Eigentler, T.K.; Loquai, C.; Haferkamp, S. .; Haferkamp, S.; Gutzmer, R.; Meier, F.; Mohr, P.; et al. Adjuvant nivolumab plus ipilimumab or nivolumab alone versus placebo in patients with resected stage IV melanoma with no evidence of disease: final results of a randomized, double-blind, phase 2 trial. Lancet 2022, 400, 1117–1129. [Google Scholar] [PubMed]
- Weber, J.S.; Schadedndorf, D.; Del Vecchio, M.; Larkin, J.; Atkinson, V.; Schenker, M.; Pigozzo, J.; Gogas, H.; Dalle, S.; Meyer, N.; et al. Adjuvant therapy of nivolumab combined with ipilimumab versus nivolumab alone in patients with resected stage IIIB-D or stage IV melanoma (CheckMate 915). J Clin Oncol 2023, 41, 517–527. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinomi, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med 2017, 377, 3813–3823. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Kirkwood, J.M.; Atkinson, V.; Mandalà, M.; Merelli, B.; Chiarion-Sileni, V.; Nyakas, M.; Haydon, A.; et al. Final results for adjuvant dabrafenib plus trametinib in stage III melanoma. N Engl J Med 2024. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitener, D.; Santinami, M.; Atkinson, V.; Mandalà, M.; et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAFV600-mutant, stage III melanoma (COMBI-AD): exploratory biomarker analyses from a randomized, phase 3 trial. Lancet Oncol 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Bloem, M.; de Meza, M.M.; Aarts, M.; Van Den Berkmortel, F.; Blank, C.; Blokx, W.; Boers-Sondersen, M.; Bonenkamp, H.J.; de Groot, J.W.; Haanen, J.; et al. Ajuvant BRAF/MEK versus anti-PD-1 in BRAF-mutant melanoma: propensity score-matched recurrence-free, distant metastasis-free, an overall survival. J Clin Oncol 2023, 42 (suppl. 16), 9573. [Google Scholar] [CrossRef]
- Bai, X.; Shaheen, A.; Grieco, C.; D’Arienzo, P.; Mina, F.; Czapla, J.; Lawless, A.; Bongiovanni, E.; Santaniello, U.; Zappi, H.; et al. Dabrafenib plus trametinib versus anti-PD-1 monotherapy ad adjuvant therapy in BRAF V600-mutant stage III melanoma after definitive surgery: a multicenter, retrospective cohort study. Lancet 2023, 65, 102290. [Google Scholar]
- Lodde, G.C.; Hassel, J.; Wulfken, L.M.; Meier, F.; Mohr, P.; Kahler, K.; Hauschild, A.; Schilling, B.; Loquai, C.; Berking, C.; et al. Adjuvant treatment and outcome of stage III melanoma patients: results of a multicenter real-world German Dermatologic Cooperative Oncology Group (DeCOG) study. Eur J Cancer 2023, 191, 112957. [Google Scholar] [CrossRef]
- Owen, C.N.; Shoushtari, A.N.; Chauhan, D.; Palmieri, D.J.; Lee, B.; Rohaan, M.W.; Mangana, J.; Atkinson, V.; Zaman, F.; Young, A.; et al. Management of early melanoma recurrence despite adjuvant anti-PD1 antibody therapy. Ann Oncol 2020, 31, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Bhave, P.; Pallan, L.; Long, G.V.; Menzies, A.M.; Atkinson, V.; Cohen, J.V.; Sullivan, R.J.; Chiarion-Sileni, V.; Nyakas, M.; Khler, K.; et al. Melanoma recurrence patterns and management after adjuvant targeted therapy: a multicentre analysis. Br J Cancer 2021, 124, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M. McKeown, J.; Dimitriou, F.; Jacques, S.K.; Zimmer, L.; Allayous, C.; Yeoh, H.L.; Haydon, A.; Ressler, J.M.; Galea, C.; et al. Efficacy and safety of “second adjuvant” therapy with BRAF/MEK inhibitors after local therapy for recurrent melanoma following adjuvant PD-1 based immunotherapy. Eur J Cancer 2024, 199, 113561. [Google Scholar] [PubMed]
- Helgadottir, H.; Ni, L.; Ullenhag, G.J.; Felkenius, J.; Mikiver, R.; Bagge, R.O.; Isaksson, K. Survival before and after the introduction of adjuvant treatment in stage III melanoma: a nationwide registry-based study. ESMO 2024, abst. 1079MO.
- Ochenduszko, S.; Puskulluoglu, M.; Pacholczak-Madej, R.; Ruiz-Millo, O. Adjuvant anti-PD1 immunotherapy of resected skin melanoma: an example of non-personalized medicine with no overall survival benefit. Crit Rev Oncol/Hematol 2024, 202, 104443. [Google Scholar] [CrossRef]
- Stassen, R.C.; Maas, C.; van der Veldt, A.; Lo, S.N.; Saw, R.; Varey, A.; Scolyer, R.; Long, G.V.; Thompson, J.F.; Rutkowski, P.; et al. Development and validation of novel model predict recurrence-free survival and melanoma-specific survival after sentinel lymph node biopsy in retrospective, multicentre analysis. Lancet Oncol 2024, 25, 509–517. [Google Scholar] [CrossRef]
- Huang, A.; Orlowski, R.; Xu, X.; Mick, R.; George, S.; Yan, P.; Manne, S.; Kraye, A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 2019, 25, 454–461. [Google Scholar] [CrossRef]
- Sharon, C.E.; Tortorello, C.N.; Ma, K.L.; Huang, A.S.; Xu, X.; Giles, L.R.; McGettigan, S.; Kreider, K.; Schuchter, L.M.; Mathew, A.J.; et al. Long-term outcomes to neoadjuvant pembrolizumab based on pathological response for patients with resectable stage III/IV cutaneous melanoma. Ann Oncol 2023, 34, 806–812. [Google Scholar] [CrossRef]
- Long, G.V.; Saw, R.P.; Lo, S.; Nieweg, O.E.; Shannon, K.F.; Gonzalez, M.; Gumisnki, A.; Lee, J.; Lee, H.; Ferguson, P.M.; et al. Neodajuvant dabrafenib combined with trametinib for resectable, stage IIIB-C, BRAFV600 mutation-positive melanoma (NeoCombi): a single-arm, open-label, single-centre, phase 2 trial. Lancet Oncol 2019, 20, 961–971. [Google Scholar] [CrossRef]
- Menzies, A.M.; Lo, S.; Saw, R.P.; Gonzalez, M.; Ching, S.; Nieweg, O.E.; Shannon, K.F.; Ferguson, P.M.; Lee, J.; Emmett, L.; et al. Five-year analysis of neoadjuvant dabrafenib and trametinib for stage III melanoma. Ann Oncol 2024, 35, 739–746. [Google Scholar] [CrossRef]
- Amaria, R.N.; Prieto, P.A.; Tetzlaff, M.; Reuben, A.; Andrews, M.C.; Ross, M.J.; Gliza, I.C.; Cormier, J.; HJwu, W.J.; Tawbi, H.; et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: a single-centre, open-label, randomized, phase 2 trial. Lancet Oncol 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Versluis, J.M.; Menzies, A.M.; Sikorska, K.; Rozeman, E.A.; Saw, R.; van Houdt, W.J.; Eriksson, H.; Klop, W.; ching, S.; van Thienen, J.V.; et al. Survival update of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanomas in the OpACIN and OpACIN-neo trials. Ann Oncol 2023, 34, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Reijers, I.I.; Menzies, A.; van Akkoi, A.; Versluis, J.; van den Heuvel, N.; Saw, R.; Pennington, T.; Kapiteijen, E.; van der Veidt, A.; Suijkerbuijk, K.; et al. Personalized response-directed surgery and adjuvant therapy after neoadjuvant ipilimumab and nivolumab in high-risk stage III melanoma: the PRADO trial. Nat Med 2022 28, 1178–1188. [CrossRef]
- Menzies, A.M.; Amaria, R.; Rozeman, E.; Huang, A.C.; Telzlaff, M.T.; van de Wiel, B.; Lo, S.; Tarhini, A.A.; Burton, E.; Pennington, T.E.; et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the international Neoadjuvant Melanoma Consortium (INMC). Nat Med 2021, 27, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Blank, C.U.; Amaria, R.N.; Hieken, T.J.; Sandhu, S.K.; Barros, M.J.; Mitchell, T.C.C.; Eroglu, Z.; Samoylenko, I.V.; Rutkowski, P.; et al. Long-term survival with neoadjuvant therapy in melanoma: updated pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Ann Oncol 2024, 35(S2), LBA41. [Google Scholar] [CrossRef]
- Patel, S.; Olhus, M.; Chen, Y.; Wright, G.P.; Yosl, K.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.; Fecher, L.A.; Truong, T.G.; et al. Neoadjuvant-adjuvant or adjuvant-only pembrolizumab in advanced melanoma. N Engl J Med 2023, 388, 813–823. [Google Scholar] [CrossRef]
- Bank, C.U.; Lucas, M.W.; Scolyer, R.A.; van de Wiel, B.A.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.; Lijnsvelt, J.; Maher, N.G.; et al. Neoadjuvant nivolumab and ipilimumab in resectable stage III melanoma. N Engl J Med 2024, in press. [Google Scholar]
- Lucas, M.W.; Menzies, A.M.; Lopez-Yurda, M.; Scolyer, R.A.; van de Wiel, B.; Saw, R.; van Houdt, W.; Maher, N.; Torres Acosta, A.; Boers-Sonderen, M.; et al. Distantmetastasis-free survival of neoadjuvant nivolumab plus ipilimumab versus adjuvant nivolumab in resectable, macroscopic stage III melanoma. Ann Oncol 2024, 35(S2), LBA42. [Google Scholar]
- Long, G.V.; Robert, C.; Hill, A.G.; Marqueste, C.G.; Portnoy, D.; Shapira, R.; Cohen, J.E.; Khattak, M.E.; Lebbe, C.; Menzies, A.M.; et al. KEYMAKER-U02 substudy 02C: neoadjuvant pembrolizumab (pembro) and investigational agents followed by adjuvant pembro for stage IIIB-D melanoma. Ann Oncol 2024, 35. [Google Scholar] [CrossRef]
- Slinguff, C.L.; Petroni, G.; Chianese-Bullock, K.; Smolkin, M.E.; Hibbitts, S.; Murphy, C.; Joahnsen, N.; Grosh, W.W.; Yamschichov, G.V.; Neese, P.Y.; et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin Cancer Res 2007, 13, 6386–6395. [Google Scholar] [CrossRef]
- Hu, Y.; Kim, H.; Blackwell, C.; Slingluff, C.L. Long-term outcomes of helper peptide vaccination for metastatic melanoma. Ann Surg 2015, 262, 456–464. [Google Scholar] [CrossRef]
- Ninmer, E.K.; Zhu, H.; Chianese, Bullock, K. ; van Mehren, M.; Haas, N.B.; Ross, M.I.; Dengel, L.T.; Sling luff, C.L. Multipeptide vaccines for melanoma in the adjuvant setting: long-term survival outcomes and post-hoc analysis of a randomized phase II trial. Nat Commun 2024, 15, 2570. [Google Scholar] [CrossRef] [PubMed]
- Bal, K.F.; Schreibelt, G.; Bloemendal, M.; van Willigen, W.; Hans-de Bree, S.; de Goede, A.; de Boer, A.; Bos, K.; Dulveman-de Boer, J.; Olde Nordekamp, M.; et al. Adjuvant dendritic cell therapy in stage IIIB/C melanoma: the MIND-DC randomized phase III trial. 2024, 15, 1632.
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long. G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-5147 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomized, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Khattak, A.M.; Carlino, M.S.; Meniawy, T.; Taylor, M.H. ; Ansstas, G,M Kim, K.B.; McKean, M., Sullivan, R.J., Faries, M.B., Eds.; et al. Individualized neoantigen therapy mRNA-4157 (V940) plus pembrolizumab in resected melanoma: 3-year update from the mRNA-4157-P201 (KEYNOTE-942) trial. J Clin Oncol 2024, 16 (suppl.16), LBA9512. [Google Scholar]
- Weber, J.S.; Luke, J.J.; Khattak, A.M.; Carlino, M.S.; Meehan, R.S.; Brown, M.; Zhang, J.; Krepler, C.; Duic, J.P.; Long, G.V.; et al. INTerpath-001: pembrolizumab with V940 (mRAN-4157) versus pembrolizumab with placebo for adjuvant treatment of high-risk stage II-IV melanoma. J Clin Oncol 2024, 42 (suppl.16), TPS9616. [Google Scholar] [CrossRef]
- Gainor, J.F.; Patel, M.R.; Weber, J.S.; Gutierrez, M.; Bauman, J.E.; Clarke, J.M.; Julian, R.; Scott, A.J.; Geiger, J.L.; Kirtane, K.; et al. T-cell responses to individualized neoantigen therapy mRNA-4157 (V940) alone or in combination with pembrolizumab in the phase 1 KEYNOTE-603 study. Cancer Discov 2024, in press. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, PO.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. Final, 10-year outcomes with Nivolumab plus Ipilimumab in advanced melanoma. N Engl J Med 2024, in press. [Google Scholar] [CrossRef]
- Varaljai, R.; Zimmer, L.; Al-Matary, Y.; Kaptein, P.; Albrecht, L.J.; Shannan, B.; Brase, J.C.; Gusenleitner, D.; Amaral, T.; Wyss, N.; et al. Interleukin 17 signaling supports clinical benefit of dual CTLA-4 and PD-1 checkpoint inhibition in melanoma. Nat Cancer 2023, 4, 1292–1308. [Google Scholar] [CrossRef]
- Tawbi, H.; Forsyth, P.; Algazi, A.; Hamid, O.; Maschos, S.J.; Khoshalani, N.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in melanoma mestastatic to the brain. N Engl J Med 2018, 379, 722–730. [Google Scholar] [CrossRef]
- Tawbi, H.; Forsyth, P.; Algazi, A.; Hamid, O.; Lao, C.D.; Mochos, S.J.; Atkins, M.B.; Lewis, K.; Postow, M.A.; Thomas, R.P.; et al. Long-term outcomes of patients with active melanoma brain metastases treated with combination nivolumab plus ipilimumab (CheckMate 204): final results of an open-label, multicentre, phase 2 study. Lancet Oncol 2021, 22, 1692–1704. [Google Scholar] [CrossRef]
- Kattenoj, K.D.; Moberg, C.L.; Gulbrandt, L.M.; Friis, R.B.; Mapendano, C.K.; Petersen, S.K.; Ruhlmann, C.H.; Svane, I.M.; Donia, M.; et al. Efficacy of Ipilimumab and Nivolumab in patients with melanoma and brain metastases-a Danish real-world cohort. Cancers 2024, 16, 2559. [Google Scholar] [CrossRef]
- Di Giacomo, A.M.; Chiarion-Sileni, V.; Del Vecchio, M.; Ferrucci, P.F.; Guida, M.; Quaglino, P.; Guidoboni, M.; Marchetti, P.; Simonetti, E.; Santangelo, F.; et al. Nivolumab plus ipilimumab in melanoma patients with asymptomatic brain metastases: 7-year outcomes and quality of life from the multicenter phase III NIBIT-M2 trial. Eur J Cancer 2024, 199, 113531. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Casula, M.; Bulgarelli, J.; Pisano, M.; Piccinini, C.; Piccin, L.; Cossu, A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M. , et al. Sequential immunotherapy and targeted therapy for metastatic BRAF V600 mutated melanoma: 4-year survival and biomarkers evaluation from the phase III SECOMBIT trial. Nat Commun 2024, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M.; Rutkowski, P.; Ferraresi, V.; Arance, A.; Guida, M.; Maiello, E.; Gogas, H.; et al. Sequencing of checkpoint or BRAF/MEK inhibitors on brain metastases in melanoma. NEJM Evid 2014, 3, EVIUDoa2400087. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination dabrafenib and trametinib versus combination nivolumab and ipilimumab for patients with advanced BRAF-mutant melanoma: yhe DREAMseq trial-ECOG-ACRIN EA6134. J Clin Oncol 2023, 41, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Queirolo, P.; Duhard, P.G.; Hu, Y.; Wang, D.; de Azevedo, S.J.; Robert, C.; Ascierto, P.A.; Chiarion-Sileni, V.; Pronzato, P.; et al. Atezolizumab, vemurafenib, and cobimetinib in patients with melanoma with CNS metastases (TRICOTEL): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2023, 24, e461–e471. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo-Gutierrez, E.; Rutkopwski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med 2022, 386, e461–e471. [Google Scholar] [CrossRef]
- Long, G.V.; Hodi, S.; Lipson, E.J.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Salman, P.; Castillo-Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; et al. Overall survival and response with nivolumab and relatlimab in advanced melanoma. NEJM Evid 2023, 2. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Hodi, S.; Schadendorf, D.; Ascierto, P.A.; Mammala, L.; Castillo-Gutierrez, E.; Rutkowski, P.; Gogas, H.; Lao, C.D.; Menez, J.; et al. Nivolumab (NIVO) plus relatlimab (RELA) in previously untreated metastatic or unresectable melanoma (RELATIVITY-047): overall survival (OS) and melanoma specific survival (MSS) outcomes at 3 years. J Clin Oncol 2024, 42 (suppl. 16), 9524. [Google Scholar] [CrossRef]
- Dolfi, S.; Tang, T.; Long, G.; Ascierto, P.; Hodi, S.; Lipson, E.; Schadendorf, D.; Wojcik, J.; Postelnek, J.; Wang, Y.; et al. Biomarker analyses of baseline tumor specimens and on-treatment changes in sera samples of patients enrolled in the RELATIVITY-047 trial to characterize LAG-3 biology. J Immunother Cancer 2023, 10 (suppl.2), 606. [Google Scholar]
- Lipson, E.J.; Dolfi, S.; Tang, H.; Gogas, H.; Tawbi, H.A.; Hodi, F.S.; Ascierto, P.A.; Gutierrez, E.C.; Schadendorf, D.; Medina Soto, F.A.; et al. Unraveling relatlimab (RELA)-specific biology using biomarker analyses in patients with advanced melanoma treated with nivolumab (NIVO)+RELA or NIVO alone in RELATIVITY-047. Annals Oncol 2023, 34 (suppl. S2), LBAS1. [Google Scholar] [CrossRef]
- Long, G.V.; Lipson, E.J.; Hodi, S.; Ascierto, P.A.; Larkin, J.; Lao, C.; Grob, J.J.; Ejzykowicz, F.; Moshyk, A.; Garcia-Horton, V.; et al. First-line nivolumab plus relatlimab versus nivolumab plus ipilimumab in advanced melanoma: an indirect treatment comparison using RELATIVITY-047 and CheckMate 067 trial data. J Clin Oncol 2024, in press. [CrossRef]
- Ascierto, P.A.; Lipson, E.J. , Dummer, R.; Larkin, J.; Long, G.V.; Sanborn, R.E.; Chiarion-Sileni, V.; Dreno, B.; Dalle, S.; Schadendorf, D.; et al. Nivolumab and relatlimab in patients with advanced melanoma that had progressed on anti-programmed death-1/programmed death ligand 1 therapy: results from the phase I/IIa RELATIVITY-020 trial. J Clin Oncol 2023, 41, 2724–2735. [Google Scholar] [PubMed]
- Ascierto, P.A.; Dummer, R.; Gaudy-Marqueste, C.; Bowyer, S; Lipson, E. J.; Ghisoni, E.; Middleton, M.R.; Ratto, B.; Jackson, W.J.; Cheong, A.; et al. Efficacy and safety of triplet nivolumab, relatlimab and ipilimumab (NIVO+RELA+IPI) in advanced melanoma: results from RELATIVITY-048. J Clin Oncol 2024, 42 (suppl.16). [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Robert, C.; Tawby, H.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized phase III trial evaluating spartalizumab plus dabrafenib and trametinib for BRAFV600-mutant unersectable or metastatic melanoma. J Clin Oncol 2022, 40, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: landmark analysis of a randomized phase 3 trial of encorafenib plus binimetinib vs vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. Eur J Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsova, I.; Liszkay, G.; et al. COLUMBUS 5-year update: a randomized, open-label, phase III trial of encorafenib pluis binimetinib versus vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. J Clin Oncol 2022, 40, 4178–4188. [Google Scholar] [CrossRef]
- Schadendorf, D.; Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Grroot, J.W.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Karjsova, I.; et al. COLUMBUS 7-year update: a randomized, open-label, phase III trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600E/K-mutant melanoma. EUR J Cancer 2024, 204, 114073. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R. Contribution of MEK inhibition to BRAF/MEK inhibitor combination treatment of BRAF-mutant melanoma: part 2 of the randomized, open-label phase III COLUMBUS trial. J Clin Oncol 2023, 2023. 41, 4621–4631. [Google Scholar] [CrossRef]
- Robert, C.; Dutriaux, C.; Oppong, F.; Kicinski, M.; Routier, E.; Neidhardt, E.M.; Durand, X.; Barooudjian, B.; Saiag, P.; Gaudy-Marqueste, C.; et al. Combination of encorafenib and binemitinib followed by ipilimumab and nivolumab versus ipilimumab and nivolumab in patients with advanced BRAF-V600E/K-mutated melanoma: the primary analysis of an EORTC randomized phase II study (EBIN). J Clin Oncol 2024, 42 (suppl.16), LBA9503. [Google Scholar] [CrossRef]
- Samaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a tumorinfiltrating lymphocyte therapy, in metastatic melanoma. J Clin Oncol 2021, 39, 2656–2666. [Google Scholar]
- Medina, T.; Chesney, J.A.; Whitman, E.; Kluger, H.; Thomas, S.; Sarnaik, A.A.; Kirkwood, J.M. , Larkin, J.; Weber, J.; Hamid, O.; et al. Long-term efficacy and safety of lifileucel tumor-infiltrating lymphocyte (TIL) cell tehrapy in patients with advanced melanoma: a 4-year analysis of the C-144-01 study. J Immunother Cancer 2023, 11 (suppl. 1), A1–A1731. [Google Scholar]
- Rohaan, M.W.; Borch, T.H.; van den Bergh, G.H.; Met, O.; Kessels, R.; Geukes Foppen, M.H.; Granhoj, J.S.; Nuijen, B.; Mijenhuis, C.; Jedema, I.; et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced lymphoma. N Engl J Med 2022, 387, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Gogas, H.; Hong, Y.K.; In, G.K.; de Speville Uribe, B.D.; Furness, A.; Castano, A.G.; Haeflinger, S.; He, K.; Medina, T.; et al. Efficacy and safety of Lifileucel, an autologous tumor-infiltrating lymphocyte cell therapy, and pembrolizumab in patients with immune checkpoint inhibitor-naïve unresectable or metastatic melanoma: updated results from IOV-COM-202 cohort 1A. J Clin Oncol 2024, 42 (suppl. 16), 9505. [Google Scholar] [CrossRef]
- Martin-Lluesma, S.; Svane, I.M.; Dafni, U.; Vervita, K.; Karlis, D.; Dimopoulou, G.; Tsourti, Z.; Rohaan, M.W.; Haanen. J.B.; Coukos, G. Efficacy of TIL therapy in advanced cutaneous melanoma in the current immune-oncology era: updated systematic review and meta-analysis. Ann Oncol 2024, 35, 860–872. [Google Scholar] [CrossRef] [PubMed]
Table 1.
Adjuvant phase clinical studies involving stage II melanoma patients.
| Clinical trial | Patients | Treatment | RFS | DMFS | Safety | Parameters correlating with response |
|---|---|---|---|---|---|---|
| KEYNOTE-716 Phase III double-blind, randomized |
976 stage IIB/C 487 (Pembro) 489 (Placebo) |
Adjuvant Pembroluzumab (PE) vs Placebo (PL) | 36-months All 76.2%(PE) 63.4%(PL) IIB 79.7%(PE) 66.5% (PL) IIC 72.4%(PE) 58% PL) |
36-months All 84.4%(PE) 74.7%(PL) IIB 86.7%(PE) 78.9%(PL) IIC 80.9%(PE) 68.1%(PL) |
Grade 3-4 17.2%(PE) 5.1% (PL) |
Not Reported |
| CHECK MATE 76k Phase III double-blind, randomized |
790 stage IIB/C 526 (Nivo) 264 (Placebo) |
Adjuvant Nivolumab (NI) vs Placebo (PL) |
12 months All 89%(NI) 79% (PL) BRAF-WT 91.2% (NI) 77.1% (PL) BRAF-mut 87.3% (NI) 81.7% (PL) |
12 months All 92.3%(NI) 86.7%(PL) IIC 87.9%(NI) 78.7%(PL) |
Grade 3-4 10.3% (NI) 2.3% (PL) |
Higher IFN-γ signature and % CD8+ cells |
Table 2.
and targeted therapy (Dabrafenib plus Trametinib) as adjuvant therapy in resected stage III BRAFV600 – mutant melanoma patients.
Table 2.
and targeted therapy (Dabrafenib plus Trametinib) as adjuvant therapy in resected stage III BRAFV600 – mutant melanoma patients.
| Clinical study | Patients | Treatment | RFS | DMFS | OS | Rate of recurrence |
|---|---|---|---|---|---|---|
| Bai et al. Multicenter, retrospective cohort study |
598 stage III BRAF-mutant melanoma | 393 pts Dabrafenib plus Trametinib (DT) 205 pts anti-PD1 (PD1) |
At 33 months DT 51 months PD1 44.8 months |
Not reported | At 3 years DT 74.4% PD1 77.9% |
Progression DT 45% PD1 27.7% Distant Metastases DT 20% PD1 26% |
| Bloem et al. Dutch Melanoma Treatment Registry Nation-wide cohort |
416 Two groups of 213 propensity score-matched stage IIIB BRAF-mutant |
213 pts DT 213 pts anti-PD1 |
At 2 years DT 80.4% PD1 85.1% |
At 2 years DT 84.1% PD1 82.1% |
At 2 years DT80.4% PD1 85.1% |
DT 34% PD1 30% |
Table 3.
Clinical studies in metastatic melanoma patients involving a long-term evaluation.
| Clinical trial | Patients | Treatment | PFS | OS | Safety |
Parameters correlating with response |
|---|---|---|---|---|---|---|
| CHECK MATE 067 NCT 01844505 Phase III, randomized |
945 metastatic 314 (Nivo+Ipi) 316 (Nivo) 315 (ipi) |
Nivo+Ipi Nivo Ipi Follow-up 10 years |
Nivo+Ipi 11.5mo Nivo 6.9mo Ipi 2.9mo |
OS at 10-yr Nivo+Ipi 71.9mo Nivo 36.9mo Ipi 19.9mo MSS Nivo+Ipi >120mo Nivo 49.4mo Ipi 21.9mo |
Grade 3-4 Nivo+Ipi 59% Nivo 23% Ipi 29% |
BRAF-mut respond to Nivo+Ipi better than BRAF-WT Response to Nivo-Ipi is associated with TH17 signatures |
| CHECK MATE 204 NCT 02320058 Phase II open-label, multicentre |
119 with brain metastases 101 asymptomatic (cohort A) 18 symptomatic (cohort B) |
Nivo+Ipi 36 months follow-up |
36-mo intracranial Cohort A 57.4% Cohort B 18.9% |
Cohort A 71.9% Cohort B 36.6% |
Grade 3-4 5% |
Not Reported |
| SECOMBIT NCT 02631447 Phase II, randomized |
206 patients with BRAFV600 metastatic ArmA (69) ArmB (69) Arm C (68) |
Arm A Enco+Bini→Nivo+Ipi Arm B Nivo+Ipi→Enco+Bini Arm C Enco+Bini 8wk; Nivo+Ipi→Enco+Bini |
At 4 years Arm A 29% Arm B 55% Arm C 54% |
At 4 years Arm A 46% Arm B 64% Arm C 59% |
Not Reported | Improved OS in patients with JAK mutations and low IFN-γ serum levels |
| RELATIVITY-047 NCT 03470922 Phase II-III Double blind, randomized |
714 metastatic Nivo+Rela (355) Nivo (359) |
Nivo+Rela Nivo |
At 5 years Nivo+Rela 48./% Nivo 39.4% |
At 5 years Nivo+Rel 48.7% Nivo 39.4% |
Grade 3-4 Nivo+Ipi 22% Nivo 12% |
Improved response to Nivo+Rela in high baseline PD1+CD8+ and ICOS1+CD8+ T cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



