Submitted:
26 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
We have previously shown that 2-thiouridine (S2U), either as a single nucleoside or as an element of RNA chain, is effectively desulfurized under applied in vitro oxidative conditions. Chemically induced desulfuration of S2U resulted in two products: 4-pyrimidinone nucleoside (H2U) and uridine (U). Recently, we investigated whether desulfuration of S2U is a natural process that also occurs in cells exposed to oxidative stress or whether it only occurs in the test tube during chemical reaction with oxidants at high concentrations. Using different types of eukaryotic cells, such as baker's yeast, human cancer cells or modified HEK293 cells with impaired antioxidant system, we confirmed that 5-substituted 2-thiouridines are oxidatively desulfurized in the wobble position of the anticodon of some tRNAs. Quantitative LC-MS/MS-MRMhr analysis of nucleoside mixtures obtained from hydrolyzed tRNA revealed the presence of desulfuration products of mcm5S2U: mcm5H2U and mcm5U modifications. We also observed some amounts of immature cm5S2U, cm5H2U and cm5U products, which may indicate a disruption of an enzymatic modification pathway at the C5 position of 2-thiouridine. The observed process, which is triggered by oxidative stress in living cells, could impair the function of 2-thiouridine-containing tRNAs and alter the translation of genetic information.
Keywords:
tRNA damage
; desulfuration
; oxidative stress
; LC-MS/MS analysis
; 5-methylcarboxymethyl-2-thiouridine
; mcm5S2U
; 5-methylcarboxymethyl-4-pyrimidinone riboside
; mcmH2U
; 5-methylcarboxymethyl-uridine
; mcm5U
1. Introduction
Transfer RNAs (tRNAs) are important molecules in all living organisms that translate messenger RNA (mRNA) and deliver amino acids to the ribosome in the order specified by the genetic information. The biology of tRNA maturation and the principles of primary function in translation are well understood and belong to the so-called rudimentary knowledge. However, tRNA has once again become the important research target due to advances in new technologies, particularly new methods based on mass spectrometry, RNA sequencing and NMR spectroscopy. These new technologies offer the opportunity to expand the knowledge of new, unknown modifications in tRNA or in other RNA types, to follow the cellular dynamics of RNA modification profiles triggered in particular by stress, to discover cellular pathways and enzymes responsible for the introduction or removal of nucleoside modifications in RNA [1,2,3,4,5,6]. Our current knowledge of the enzymes that modify nucleosides in RNA is still incomplete, e.g. about 23% of human enzymes have not yet been identified and another 20% require further experimental validation [7]. This is why it is so important to advance research in this area.
TRNAs are the most abundant category of small non-coding RNA molecules in the cell and have the highest modification rates, which distinguishes them from other types of cellular RNAs. To date, about 170 different nucleoside modifications have been identified in RNA, and at least 120 modified nucleosides are found in tRNAs [8,9,10,11,12]. Modifications in tRNA can be classified according to their function, type and position within the tRNA structure. Modifications in the core region of tRNA, located within arms D and TΨC, improve and stabilize the tertiary structure of tRNA and maintain the balance between flexibility and stability of the L-form of the tRNA unit. Nucleoside modifications located in the anticodon-stem-loop domain modulate codon-anticodon interaction and play a regulatory function during decoding [13,14]. Modified nucleosides in tRNA can also be determinants for other components of the translational apparatus such as aminoacyl-tRNA synthetases (aaRSs) [15,16]. In addition, tRNA modifications can be recognition elements of ribonucleases, leading to the generation of tRNA fragments that influence multiple cellular processes [17].
Sulfur is an important component of various classes of biomolecules such as proteins, nucleic acids, sugars, sulfur-containing vitamins, e.g. thiamine, iron–sulfur cofactors for enzymes, a variety of sulfur-containing metabolites, etc. [18]. In nucleic acids, the sulfur substitution of an oxygen atom on the nucleobase or in a phosphate backbone leads to a variety of sulfur-modified nucleosides or nucleotides [19]. TRNA also contains sulfur-modified nucleosides, which are incorporated post-transcriptionally into the maturing tRNA chain by specific modifying enzymes. Depending on the position at which the modifications occur in the tRNA chain, they have different functions, such as stabilizing tRNA structures, enabling the identification of tRNAs by aaRSs, improving ribosomal binding to aminoacylated tRNAs, maintaining the reading frame, and ensuring correct codon-anticodon base pairing [20].
In general, sulfur-containing modifications of nucleosides are found at seven different positions in the tRNA chain. These are 8, 9, 32, 33, 34, 37 and 54 tRNA chain positions. However, the presence of modifications depends on the organism from which the tRNA originates. The modification at position 9 (S4U9) is only characteristic for the archaeon Thermoplasma acidophilum [21], the modification at position 33 (S2U33) is characteristic for Trypanosomatis. Bacterial and archaeal tRNAs contain five thio-nucleosides: 4-thiouridine at position 8 (S4U8), 2-thiocytidine at position 32 (S2C32), 5-methylaminomethyl-2-thiouridine, 5-carboxymethylaminomethyl-2-thiouridine or their 5-(c)mnm-S-geranyl-2-thiouridine derivatives at position 34 (mnm5S2U34, cmnm5S2U34, (c)mnm5geS2U34) and 2-methylthio-N6-isopentenyladenosine, 2-methylthio-N6-hydroxyisopentenyladenosine or 2-methylthio-N6-threonyl-carbamoyl adenosine at position 37 (mS2i6A37, m5S2io6A37, mS2t6A37) [22]. Some of the thermophilic bacteria have additional 5-methyl-2-thiouridine (m5S2U54) at position 54. Eukaryotic organisms contain sulfur-modified nucleosides at two positions 34 and 37 of the anticodon loop. There are 5-methylcarboxymethyl-2-thiouridine (mcm5S2U34) and derivatives that differ by the substituent at C5 and 2-methylthio-N6-hydroxyisopentenyladenosine (m5S2io6A37) or 2-methylthio-N6-threonyl-carbamoyladenosine at position 37 (mS2t6A37) [23,24].
5-Substituted 2-thiouridines (R5S2U) are universal thio-modifications found in three domains of life. The modified R5S2U contain a sulfur atom instead of the oxygen atom at position 2 of the uracil ring and the additional substituent at position 5, the nature of which depends on the origin of the tRNA. Depending on the organism and subcellular localization of the tRNA, the substituent R5 can be hypermodified to methylaminomethyl- (mnm-), carboxymethylaminomethyl- (cmnm-) in bacteria and archaea, to methylcarboxymethyl- (mcm-) in the eukaryotic cytosol or to taurinomethyl- (τm-) in the mitochondria of mammals [19,24,25]. Several functions have been proposed for the R5S2U34 modification. The first important function, based on localization at the wobble position of the tRNA, is the recognition of the third nucleoside of the codon triplet in codon-anticodon interaction. The R5S2U units primarily facilitate the Watson-Crick base pairing with adenosine at the 3’-end of the 5’-NNA-3’ mRNA codons and restrict wobble base pairing with G at the third position of the 5'-NNG-3' codons. The other function, the sulfur atom at position C2 of the uracil ring, is the identity-promoting element in aminoacylation reactions [26] and increases translation efficiency at the ribosome by increasing the binding affinity of aminoacylated tRNAs to the A site of the ribosome as well as the GTP hydrolysis rate [27]. In addition, the R5S2U34 modification preserves translation fidelity by preventing +1 and +2 ribosome frame shifting [28,29]. In yeast Saccharomyces cerevisiae, the mcm5S2U occurs mainly at the wobble position of the tRNA specific for Lys, Glu or Gln (tRNAGlu, tRNALys and tRNAGln). As mentioned above, 5-substituted S2U is also found in human mitochondrial lysine specific tRNA (hmt-tRNALys ).
The 5-substituted 2-thiouridines have been the subject of interest of our group (researchers from CMMS PAS and TUL) for several years [31–35]. Earlier research by Corsaro and Pistara, carried out in the late 1990s, has shown that nucleosides containing the thiocarbonyl group are sensitive to oxidizing conditions despite their stabilizing properties [30]. Studying this modification, we discovered that the 2-thiouridine (S2U), alone or incorporated into an RNA oligonucleotide chain, was desulfured under oxidative conditions, i.e. the sulfur atom was removed from the S2U molecule, and the two reaction products deprived of the sulfur atom were: 4-pyrimidinone riboside (H2U) and uridine (U), Scheme 1 [31].
The ratio of S2U desulfuration products depended on the reaction conditions: pH, type of R5 substituent, and type and concentration of oxidizing reagent. The reaction carried out at the lower pH (6.6) produced mainly H2U (80% H2U and 20% U), while in the reaction carried out at the higher pH (7.6), the main product was uridine (80% U and 20% H2U) [31,32]. In addition, we have shown that the substituent R5, although not directly involved in the reaction with the oxidizing agent, strongly affects the pathway of desulfuration and the product ratio. When the substituent R5 was changed, the amount of R5H2U decreased in the following order: H>CH3O->CH3OC(O)CH2-> HOC(O)CH2NHCH2- ~ CH3NHCH2. This effect was observed within the physiological pH range (pH between 6.6 to 7.6), but was more pronounced at lower pH values (pH 6.6) [34]. Conformational analysis of the R5H2U desulfuration product showed that R5H2U predominantly adopts the C2'-endo conformation regardless of the nature of the R5 substituent, in contrast to the C3'-endo conformation of R5S2U [31,35]. Due to the altered pattern of hydrogen bonding, the nucleoside H2U forms a different base pairing with the nucleoside in the complementary RNA chain than S2U. 2-Thiouridines hybridize preferentially with adenosines (S2U-A), whereas wobble pairing with guanosine residues (S2U-G) is restricted due to the less effective hydrogen bonding between the N1H donor of guanine and the sulfur acceptor in 2-thiouracil. After the conversion of S2U to H2U, the situation is different. The H2U nucleoside hybridizes preferentially with guanosine because it has a different pattern of hydrogen bonds than the acceptors and donors of uridine and 2-thiouridine [35]. These results suggest that the conversion of R5S2U within the tRNA wobble position to its R5H2U product may be of great biological significance because the structural and functional features of the R5H2U unit are highly altered compared to the original R5S2U.
In the present study, we addressed the question of whether desulfuration of RS2U is possible in the natural tRNAs in living cells exposed to oxidative stress. To solve this issue, we used eukaryotic model cells with impaired antioxidant systems: the yeast Saccharomyces cerevisiae, human cancer cells and modified HEK293 cells with mutations in the SOD2 and Cat genes that resulted in suppression of the expression of catalase (Cat) and mitochondrial superoxide dismutase (MnSOD2), enzymes important for the cellular antioxidant system. To induce oxidative stress in the cells, we used stressors known from the literature: H2O2, NaAsO2 and NaClO [36]. First, we determined the maximum concentrations of oxidative reagents that can be applied in cell culture and do not lead to cell death, and we created cell culture conditions under oxidative stress. Subsequently, the tRNAs extracted from the cells cultured under oxidative stress were hydrolyzed to nucleosides and subjected to LC-MS/MS analysis to search for mcm5S2U-tRNA desulfuration products.
2. Results and Discussion
2.1. Characteristics of Cells Used in Research
In the first part of the studies, we used three laboratory strains of yeast Saccharomyces cerevisiae 1) INVSc1, the commercial, fast-growing, diploid strain designed primarily for protein expression [MATa his3D1 leu2 trp1-289 ura3-52 MAT his3D1 leu2 trp1-289 ura3-52], 2) M3, the wild-type strain [MATa lys2 his4 trp1ade2 leu2 ura3], and 3) M3Δsod1, the isogenic to M3 parent strain [MATa lys2 his4 trp1ade2 leu2 ura3 Δsod1::kanMX4] with deletion of the copper-zinc superoxide dismutase gene [37,38]. Yeast is a very useful and practical model microorganism for tRNA research because its culture is simple, inexpensive and allows suitable amounts of biomass to be obtained in a short time. The yeast S. cerevisiae contains three types of tRNA with the modification mcm5S2U at the wobble position of the anticodon. These are tRNALys, tRNAGlu and tRNAGln, which represent a useful model for the study of intracellular R5S2U desulfuration. A description of yeast strains and culture conditions can be found in Supplementary Material, Table S1.
In the next experiments, we examined human cancer cells, both adherent and growing in suspension, derived from eight human cancer cell lines (HeLa, K562, MOLT-4, A431, A375, A549, U87 MG, MCF-7) and the human HEK293 cell line as a model of normal cells. A description of all cell lines and culture conditions can be found in Supplementary Material, Table S2. Cancer cells served as model cells with intracellular oxidative stress [39,40]. Cancer cells usually show reduced activity of antioxidant enzymes compared to normal cells. Reduced activity of superoxide dismutases (Cu-ZnSOD1, MnSOD2) and catalase (Cat), which is found in many types of cancer cells, leads to the accumulation of ROS (reactive oxygen species) in the cells, which in turn leads to intracellular oxidative stress and its consequences for biomolecules.
The third cell type used in the studies were HEK293 cells that were genetically modified using the CRISPR/Cas9 gene editing system to damage the human SOD1, SOD2 and Cat genes.
2.1.1. Application of the CRISPR/Cas9 Gene Editing System for Genome Modification of Human HEK293 Cells
The CRISPR (clustered regularly interspaced short palindromic repeats) is an effective and precise system for genome editing in living cells that is used in many scientific fields. The CRISPR/Cas9 system consists of two main components, the guide RNA (sgRNA), which recognizes the target DNA region of interest and directs there the Cas nuclease for gene editing, and the CRISPR-associated (Cas) nuclease, which creates double-stranded breaks at a site 3 base pairs upstream of the protospacer adjacent motif (PAM). Subsequently, the double-strand breaks are repaired by cellular DNA repair mechanisms, non-homologous end joining or homology-directed repair, leading to mutations/translocations/deletions of nucleotides in the target gen and finally to lack of expression. The sgRNA consists of two parts: the crispr RNA (crRNA), a 17-20 nucleotide sequence complementary to the target DNA, and a tracrRNA, which serves as a binding scaffold for the Cas9 nuclease.
We used the CRISPR/Cas9 system to introduce mutations in the human genes for catalase (Cat) and cytoplasmic (Cu-Zn-SOD1) or mitochondrial (Mn-SOD2) superoxide dismutases to inhibit their expression in the studied cells. These three enzymes are an important part of the human antioxidant system, which is responsible for neutralizing reactive oxygen species in the cells and protecting the cells from oxidative stress and free radicals. Superoxide dismutase catalyzes the conversion of the toxic superoxide anion O2•‒ into oxygen and hydrogen peroxide. Catalase is responsible for the decomposition of hydrogen peroxide into water and oxygen.
The Edit-R, a commercial predesigned, synthetic, sgRNAs were used, three variants per one gene, targeting three human genes: Cat, catalase gene [GenBank: AY545477,target sequence: sg1: 16228-16247, sg2: 15200-15219, sg3: 24427-24446]; SOD1, superoxide dismutase 1 gene [GenBank: AY835629, target sequence: sg1: 7730-7749, sg2: 139-158, sg3: 4248-4267] and SOD2, superoxide dismutase 2 gene [GenBank: AY26790, target sequence sg1: 528-547, sg2: 476-495, sg3: 452-471] and the Edit-R sgRNA targeting the human PPIB, Peptidylprolyl isomerase B gene [GenBank: AY962310,], as a positive control for gene editing, TrueCut Cas9 v2 nuclease (5 µg/µL), and Lipofectamine CRISPRMAX transfection reagent. The sequences of Edit-R sgRNAs and PCR primers are listed in Table S3. After transfection, culture of HEK293 cells was continued under optimal conditions according to the protocol described in the Materials and Methods. After the next 3 days, cells were harvested, counted and diluted in fresh medium to the density 1 cell per 100 µL and transferred into 96-well plates to generate single-cell clones. After the next 3 weeks of growth, the first selection of cells was performed and those cells in the wells of the 96-well plate were selected that showed characteristics of a single cell origin, i.e. they grew in a single location, not scattered over the entire surface of the well. These cells were selected and transferred to 48-well plates, then to 24-well plates, then to 6-well plates and finally to culture flasks. Supplementary Table S4 provides a simple calculation of the efficiency of the CRISPR/Cas9 procedure and clone selection.
Initial selection of the clones obtained was performed by Western blot method using primary antibodies specific for SOD1, SOD2 or Cat antigens, to detect cells not expressing the target genes. For HEK293ΔSOD1 cells, 96 clones were tested, all of which expressed the SOD1 gene (0 positive clones, SOD1-sgRNA/Cas9 efficiency 0%). For HEK293ΔSOD2 cells, 109 clones were tested, 5 clones did not express the SOD2 gene (SOD2-sgRNA/Cas9 efficiency 4.6%). For HEK293ΔCat cells, 20 clones were tested, 19 clones did not express the Cat gene (Cat-sgRNA/Cas9 efficiency 95%), data summarized in Table S4. Figure 1 shows the Western blotting analysis of HEK293ΔSOD1, HEK293ΔSOD2 and HEK293ΔCat positive clones. The positive clones selected in this way were then subjected to a T7 endonuclease assay and genomic DNA sequencing, which confirmed the presence of mutations in the target genes resulting from the CRISPR/Cas, Figures S1 and S2. As the result of these experiments, we selected 5 clones of HEK293 cells with deletion of the SOD2 gene (HEK293ΔSOD2) and 19 clones with deletion of the Cat gene (HEK293ΔCat). Unfortunately, despite our efforts, all clones with suspected damage to the SOD1 gene continued to express SOD1 protein, which was confirmed by the immunoblotting method with antibodies specific for SOD1 antigen. Therefore, the subsequent R5S2U desulfuration studies were performed without HEK293ΔSOD1 cells, which were not available at that time.
2.2. Optimization of Yeast Cell Culture Conditions under Oxidative Stress
2.2.1. Evaluation of Yeast Viability in the Presence of Oxidative Stress-Inducing Reagents
The yeast cells (InvSc1, M3 and M3Δsod1 strains) were cultured under optimal growth conditions until they reached an exponential growth phase (OD600 =~0.6-0.8). In this growth phase, the cells are healthiest, divide intensively and the number of cells increases rapidly. The yeast cells were exposed to oxidizing agents such as H2O2 (at concentration 0, 5, 10, 25, 50, 75, 100 mM), NaAsO2 (at concentration 0, 10, 20, 40, 60, 80, 100 mM) or NaClO (at concentration 0, 3, 4, 5, 7, 10 mM) for the 1 or 2 hours, to see how they grow under stress conditions and to determine the range of oxidant concentrations in which complete cell damage and cell death do not occur. The following tests were performed to evaluate the viability of yeast cells in the presence of oxidizing agents: the spotting test and the colony forming unit (CFU) test. In addition, live and dead cell staining tests were initially performed (methylene blue staining or propidium iodide with fluorescein diacetate staining), but these tests were abandoned because strains M3 and M3Δsod1 were only slightly stained. Figure 2 shows the example of the spotting test used to evaluate the sensitivity of the tested yeast strains to hydrogen peroxide. The remaining results can be found in supplementary Figure S3. The detailed protocol for the yeast viability tests can be found in the Materials and Methods. The so-called “safe concentrations” of oxidative stress reagents at which yeast viability was at least 50% are listed in Table 1. The determined conditions were used in further studies.
2.2.2. Determination of the ROS Level in Yeast Cells after Incubation with H2O2, NaAsO2 or NaClO
The yeast cells were grown under optimal conditions until the exponential growth phase was reached (OD600=~0.6-0.8). For the measurement of intracellular ROS, the 2,7-dichloro-dihydrofluorescein diacetate (DCFDA) assay was performed with a non-fluorescent precursor of 2,7-dichloro-dihydrofluorescein. Detailed instructions can be found in the Materials and Methods. After the cells reached the appropriate cell density, they were incubated with the oxidizing compounds for 1 or 2 hours, washed with PBS buffer, and the fluorescence of the samples was measured using a plate reader. PBS buffer and untreated yeast cells were used as a negative control (no ROS). The level of ROS was measured in whole, intact cells (which were not lysed) suspended in PBS buffer. The results of the ROS analysis are shown in Figure 3 and Figure S4. The results show that reactive oxygen species (ROS) are formed in yeast cells under the influence of the oxidizing agents used. The highest concentration of ROS was observed in yeast cells exposed to H2O2 or NaAsO2.
The ROS level induced by NaClO was lower (1-2%) in both yeast strains (M3 and M3Δsod1), Figure S4. Care should be taken to read the counted ROS within the so-called “safe concentrations” of oxidizing agents, Table 1. The significant increase in ROS concentration visible in both graphs in Figure 3 means that the yeast cells were damaged by the oxidizing agents used at higher concentrations (H2O2 > 25 mM, NaAsO2 > 40 mM).
2.2.3. Monitoring the Reduction in the Amount of mcm5S2U-tRNA in Yeast Treated with H2O2, the γ-Toxin Assay
We confirmed the reduction of mcm5S2U-tRNA with the mcm5S2U modification in the wobble position using a γ toxin assay [41]. Gamma toxin from the yeast Kluyveromyces lactis is an enzyme that specifically recognizes the mcm5S2U in the wobble position of tRNA and cleaves the tRNA at this site (between nucleosidr at 34 and 35 position in the anticodon loop). If mcm5S2U is desulfured, it is not recognized by γ-toxin and not hydrolyzed. The recombinant γ-toxin was overexpressed in bacterial strain BL21(DE3) and purified by affinity chromatography on Ni-NTA agarose according to the protocol described in the Materials and Methods, yielding several micrograms of active enzyme. For the experiments we used the yeast Saccharomyces cerevisiae, the commercial strain INVSc1, which was the most resistant to H2O2 compared to the other two strains (IC50=50 mM, Table 1). Total cellular RNA isolated from yeast exposed to oxidative stress (H2O2) during culture was reacted with γ-toxin. Hydrolysis of mcm5S2U-tRNA was monitored by Northern blot hybridization of the resulting full-length tRNA and tRNA cleavage products with complementary DNA probe labeled with the 32P isotope at the 5’ end. As the result of the assay, two bands were observed on the membrane: (i) the band corresponding to the full-length tRNA (tRNA that does not contain mcm5S2U modification, not recognized and not hydrolyzed by γ-toxin) and the second (ii) band corresponding to the half fragment of the mcm5S2U-tRNA (after hydrolysis by γ-toxin), Figure 4B. 25SrRNA was used as a reference RNA.
The second assay used to quantitatively assess the reduction of mcm5S2U-tRNA was qRT-PCR with specific primers (shown schematically in Figure 4A). 25S rRNA, which did not contain mcm5S2U modification, was used as a negative control for the tRNA hydrolysis reaction catalyzed by γ-toxin Figure 4C.
Based on the results of qRT-PCR, we estimated that the use of H2O2 (up to 50 mM) during culture of yeast cells resulted in a loss of approximately 80% of the input mcm5S2U-tRNAGlu, which could indicate oxidative desulfuration of mcm5S2U-tRNA or disruption of cellular pathways (tRNA-modifying enzymes) specific for the introduction of mcm5S2U modification into the wobble position of the anticodon of tRNAGlu by oxidative stress. After the application of 100 mM H2O2 during yeast culture, we observe almost 100% loss/damage of mcm5S2U-tRNA, but this result is most likely due to yeast cell death and the direct effect of hydrogen peroxide on tRNA. This result was confirmed by the Northern blot method, which allowed the identification of uncleaved tRNA and the mcm5S2U-tRNAGlu cleavage product by γ toxin. Another interesting result was found in cells that were not treated with H2O2 during culture. About 40% of the analyzed tRNAGlu from not treated yeast was not hydrolyzed by γ toxin, which may indicate that this tRNA also does not contain the mcm5S2U modification at the wobble position of the anticodon, Figure 4C.
2.3. Optimization of Culture Conditions for Human Cancer Cells under Oxidative Stress
2.3.1. Evaluation of Human Cells Viability and Intracellular ROS Level
Human cancer cells were subjected to exogenous oxidative stress by exposing to H2O2, NaAsO2 or NaClO stressors during culture. The MTT assay was performed to evaluate cell viability under oxidative stress conditions and IC50 values were determined for all cells tested, Table 2, Figure 5 and Figure S6.
HEK293 cells (IC50=14.5 µM) and their mutant variants HEK293ΔSOD2 and HEK293ΔCat (IC50~10 µM) were the most sensitive to oxidizing reagents, especially H2O2, while MOLT-4 and K562 suspension cells (IC50 > 200 µM) were the least sensitive. The other compounds NaClO and NaAsO2, used at the indicated concentrations were not as lethal to most cells as H2O2. The IC50 determined for NaAsO2 was ~200 µM and for NaClO was ~100 µM for almost all cells tested. These conditions were used for further research.
In addition, we have confirmed for all oxidizing compounds used: H2O2, NaAsO2 or NaClO, that they cause the formation of ROS in cells (2,7 dichloro-dihydrofluorescein diacetate assay), Figure S7. When the concentration of the oxidizing reagent increases during incubation with the cells, the level of intracellular ROS also increases. Most ROS are formed in the cells treated with H2O2, the other compounds cause the approximately 20-80 times lower amount of ROS (corresponding values for NaAsO2 and NaClO). The ROS concentration in the cells changes over time, initially increasing and peaking after 1-2 hours, and then decreasing again. This phenomenon can be explained by the action of the cellular antioxidant system.
Due to the natural intracellular oxidative stress that exists in cancer cells, we evaluated and compared ROS levels in cancer cells that were not treated with oxidizing compounds using the fluorescein 2,7-dichloro-dihydro-diacetate assay. For this experiment, we selected HEK293, HeLa, A375, A549, A431 and MCF-7 cells and performed the assay with the same number of cells (25 x 103 cells/well). The cells were incubated with 2,7-dichloro-dihydrofluorescein diacetate for 30 minutes, washed, spread on the wells of a 96-well plate and the fluorescence values were measured after 1, 2, 4 and 6 hours of incubation at 37 °C, 5% CO2. The fluorescence values obtained for the different cell lines were compared with the fluorescence determined for the HEK293 cells as a baseline value. We found that all cancer cells tested had some level of intercellular ROS above the level found in HEK293 cells, Figure S8.
2.4. Identification of R5S2U Desulfuration Products by LC-MS/MS (MRMhr) Analysis
2.4.1. R5S2U Desulfuration in Yeast tRNAGlu
The yeast tRNA specific for glutamic acid, tRNAGlu, contains the modified nucleoside mcm5S2U at the wobble position of the anticodon. Based on our previous observations of the desulfuration of 2-thiouridine under oxidative conditions, we decided to verify whether such desulfuration process occurs in the natural tRNA in yeast cells exposed to oxidative stress during their growth. For this purpose, we used the highly sensitive and highly selective LC-MS/MS (MRMhr) technique (QTRAP 6500+ mass spectrometer coupled to Exion LC System, Sciex, USA) and synthetic mcm5S2U, mcm5H2U, mcm5U, cm5S2U, cm5H2U and cm5U as nucleoside standards for LC-MS/MS analysis. After culture the yeast (S. cerevisiae, M3 and M3Δsod1 strains) in the presence of hydrogen peroxide (0-100 mM H2O2) the specific tRNAGlu was isolated from total cellular RNA by binding to the complementary biotinylated DNA probe, attached to the streptavidin-agarose resin. The resulting pure tRNAGlu was subjected to simultaneous hydrolysis by two nucleases: Benzonase (endonuclease from Serratia marcescens) and Phosphodiesterase I from Crotalus adamanteus venom (5'-exonuclease), and subsequently dephosphorylated by alkaline phosphatase CIAP. The resulting nucleoside mixture was analyzed by LC-MS/MS as described in detail in the Materials and Methods and in Table S7. Almost all nucleosides of interest (mcm5S2U, mcm5H2U, mcm5U, cm5S2U, cm5H2U and cm5U) were detected in samples analyzed, but the amount of nucleosides identified was dependent on the yeast strain from which the tRNAGlu was derived (M3 or its sod1 deletion variant, M3Δsod1) and on the oxidative conditions to which the yeast was exposed. The most abundant in the nucleosides mixture were the 5-substituted 2-thiouridines: mcm5S2U and cm5S2U, but mcm5H2U and mcm5U were also detected, which are probably products of oxidative desulfuration of mcm5S2U. The cm5H2U nucleoside was only detected in the samples from yeast after application of 100 mM H2O2 during growth. However, this result does not indicate desulfuration in living cells, as yeast belonging to M3 and M3Δsod1 strains do not survive under 100 mM H2O2 conditions. It can be assumed that the low percentage of identified mcm5H2U and cm5H2U in other samples (0-25 µM H2O2 M3; 0-5% M3Δsod1) could be due to the sensitivity of the analyzed H2U derivatives and the possible decay in yeast cells and/or during mass analysis. The percentages shown in the graphs indicate the percentage of the individual modified nucleosides in the nucleoside mixture. 100% is the sum of all 2-thiouridine derivatives tested.
First, we found certain amounts of mcm5H2U (4% M3; 2% M3Δsod1) and mcm5U (11% M3; 30% M3Δsod1) nucleosides in the so-called non-oxidatively treated cells, which could be due to the action of intracellular ROS in the yeast cells.
The nucleoside mixture derived from tRNAGlu isolated from M3 strain, contained the mcm5S2U, the content of which decreased from 40% to 31% with increasing H2O2 concentration (0-25 mM) used during yeast culture. At the same time, small amounts of mcm5H2U (4-5%) and mcm5U (11-21%) were detected. The levels of cm5S2U (41-43%) and cm5U (2-3%) remained fairly constant regardless of the H2O2 concentration used during culture.
The nucleoside mixture from tRNAGlu isolated from M3Δsod1 deletion variant at safe-concentration of 0-5 mM H2O2 contained constant level of mcm5S2U (26%), small amounts of mcm5H2U (2%) and the relatively high constant proportion of mcm5U (30%). After exceeding the safe H2O2 concentration during yeast culture (10-100 mM H2O2), the mcm5S2U content decreases rapidly by 26-12% with a simultaneous increase in mcm5U (31-46%) and especially mcm5H2U (3-30%). This effect is due to the direct action of hydrogen peroxide on tRNA and means that the tRNAGlu was probably directly damaged by the oxidizing agent because it damaged the yeast cells (the H2O2 concentration used was too high, above the "safe concentration"). The obtained results are shown in Figure 6 and Figure S12.
From the results obtained, we can conclude that R5S2U desulfuration occurs in yeast and the main reaction product is R5U, while the second desulfuration product, R5H2U, is present but maintained at the similarly low level in all samples analyzed.
2.4.2. Search for mcm5S2U Desulfuration Products in the tRNA Fraction of Cancer Cells
The fraction of small cellular RNAs (< 200 nt) was isolated from total cellular RNA of the investigated cancer cells cultured without or in the presence of oxidizing reagents (H2O2, NaAsO2 or NaClO at IC50 concentrations, Table 2). This approach is described in the literature as one of the methods to obtain tRNA for research, as tRNAs represent the vast majority of low molecular weight RNAs [41]. Our tRNA isolation protocol has been described in detail in the Materials and Methods. In brief, the cellular RNA fraction was fractionated on an Agilent SEC-3, 300Å HPLC column (150 mm × 7.8 mm) coupled with an FPLC AKTA purifier: Box-900, pH/C-900, UV-900, P-900, Frac 920 system, and the low molecular weight RNA fraction (< 200 nt) was separated from the large cellular RNAs, such us rRNAs and mRNAs. We chose this method to isolate the tRNA fraction containing not only the one specific tRNA, as was in yeast case, but the mixture of all cellular tRNAs, because the amount of specific tRNA isolated from mammalian cells is much smaller compared to the tRNA from yeast cells and may not be sufficient for further applications. The isolated tRNAs were subjected to nucleolytic hydrolysis (Benzonase, Phosphodiesterase 1) and dephosphorylation by alkaline phosphatase (CIAP) as described above. The resulting nucleoside mixture was analyzed by LC-MS/MS (ZenoTOF 7600 mass spectrometer coupled with ExionAC LC system, Sciex, USA) for the presence of mcm5S2U-tRNA desulfuration products. The LC-MS/MS experiments were repeated independly at least three times The results of the analysis can be found in Figure 7 and Figure 8. Similar to the tRNA analysis of yeast, the percentages shown in the graphs indicate the proportion of the individual modified nucleosides in the nucleoside mixture. 100% is the sum of all 2-thiouridine derivatives tested.
First, we examined the content of modified nucleosides (mcm5S2U, mcm5H2U, mcm5U, cm5S2U, cm5H2U, cm5U) in the tRNA from cancer cells that were not exposed to exogenous oxidative stress, the so-called untreated cells. LC-MS/MS analysis revealed that in addition to the modified 2-thiouridines (mcm5S2U and cmS2U), which occur in the highest quantities (29-53% and 33-57%, respectively), their desulfuration products (mcm5H2U, mcm5U, cm5U) are also present at the level dependent on cell line examined, Figure 7AB. The highest content of mcm5H2U was identified in leukemic cells (MOLT-4 and K562, 11% and 6% respectively) growing in suspension and in adherent melanoma cells (A375, 5%). The highest level of mcm5U was found in HeLa cells (37%), which distinguishes the cells from other cell lines where the average content of mcm5U was 4-8%. The cm5H2U modification was not identified in tRNA from cancer cells that were not subjected to oxidative treatment, which could be due to the low levels of this nucleoside, which are below the detection limit.
Next, we compared the effect of three oxidizing agents (H2O2, NaAsO2 and NaClO, used individually in IC50 concentration, Table 2) on the formation of 2-thiouridine desulfuration products in the low-weight (< 200 nt) cellular RNA fraction. Figure 8 shows the presence of desulfuration products in the three selected cell lines (HEK293, HeLa and A375). As expected, different oxidizing agents had different effects on the cells and at the same time the effect depended on the individual sensitivity of the cells tested. In HEK293 unmodified cells, the significant decrease in mcm5S2U content was observed after the application of oxidizing agents (from 43%, in untreated cells to the level of 24-20% in cells treated with H2O2, or NaAsO2 or NaClO, respectively). At the same time, the increase in mcm5S2U desulfuration products, mcm5H2U (from 2% in untreated cells to 6-14%) and mcm5U (from 5% in untreated cell to 11-12%), was observed. A significant increase in mcm5H2U concentration (from 2% in untreated cells to 14-16%) was also observed in Hela cells, especially after the cells were treated with H2O2 or NaAsO2. In A375 cells, the significant decrease in the amount of mcm5S2U was observed (from 33% in untreated cells to 21%), leading to the increase in the amount of mcm5U (from 8% to 16%). However, the second desulfuration product remained at an approximately constant level (4-5%, similar as in the untreated cells). Interestingly, the cm5H2U modification, which was not present in untreated cells, was detected in all tested cells treated with H2O2, NaAsO2 and NaClO at the level of 3-5%.
We also performed a time-dependent experiment in which cells were exposed to H2O2-induced oxidative stress (in IC50 concentration, Table 2) for 2, 4 or 6 hours (data not shown). LC-MS/MS analysis showed that the highest decrease in mcm5S2U level and appearance of 2-thiouridine desulfuration products in the samples occurred after 2-4 hours of cell culture under oxidative stress conditions. Afterwards, the cells returned to the initial state, which can be explained by the activity of the cellular antioxidant system and the decomposition of the oxidizing agent. This effect was also dependent on the individual sensitivity of the cells of the respective cell line.
2.4.3. Analysis of HEK293ΔSOD2 and HEK293ΔCat tRNA Fraction Content
The experiment aimed to test the extent to which 2-thiouridine desulfuration occurs in cells with a partially damaged antioxidant system compared to unmodified HEK293 cells. For this purpose, cells were selected that showed no expression of the Cat or SOD2 gene, Figure 1. The detailed protocol is described in the Materials and Methods, in section 2.1.1 and suplementary Figure S2. Unmodified HEK293 and modified HEK293ΔSOD2 (clones 1-1C6; 2-1 H12; 3-1D8) and HEK293ΔCat (clones 3-1E1; 2-2F7) cells obtained by the CRISPR/Cas9 gene editing protocol were exposed to oxidative stres during their growth (0-50 µM H2O2, IC50=14,5 µM H2O2 for HEK293; IC50=10 µM H2O2 for HEK293 mutation variants, Table 2). The same procedure as described above was used to isolate the low-weight fraction of cellular RNAs. The isolated tRNAs were subjected to nucleolytic hydrolysis (Benzonase, Phosphodiesterase 1) and dephosphorylation by alkaline phosphatase (CIAP). The resulting nucleoside mixture was analyzed by LC-MS/MS (ZenoTOF 7600 mass spectrometer coupled with ExionAC LC system, Sciex, USA) for the presence of mcm5S2U-tRNA desulfuration products. The synthetic nucleosides: mcm5S2U, mcm5H2U, mcm5U, cm5S2U, cm5H2U and cm5U served as nucleoside standards for LC-MS/MS analysis. The LC-MS/MS experiment was repeated twice independently. The results are shown in Figure 9.
The tRNA fraction isolated from unmodified HEK293 cells was characterized by the high content of mcm5S2U (76-80%), accompanied by the low content of cm5S2U (4%). In the presence of H2O2, the level of mcm5S2U decreased slightly, while the gradual increase in mcm5H2U (0-9%), the putative desulfuration product, was observed. After the application of 50 µM H2O2, the content of mcm5H2U increased to 15%, which is probably due to the direct effect of peroxide on tRNA in damaged cells.
HEK293ΔCat delection variants: 3-1E1 clone, characterized by a deletion of 14 bp in CRISPR target sequence) and 2-2F7 clone, without sequence changes in the target sequence (changes probably occur outside the sequenced target sequence) Figure S2, do not express the Cat gene, Figure 1. The tRNA fraction isolated from these cells contains the high content of mcm5S2U (89% in untreated cells), the low level of cm5S2U (1-6%). After the application of 5 µM H2O2 to the culture, the level of mcm5S2U rapidly decreases to 60%, which is accompanied by an increase in the amount of mcm5H2U (from 7% to 24%) and mcm5U (from 2% to 12%). Further increasing the H2O2 concentration (10-50 µM) in the cell culture does not lead to the increase in the amount of 2-thouridine desulfuration products.
HEK293ΔSOD2 deletion variants: 3-1D8 clone, characterized by a deletion of 6 bp and numerous mutations outside the target sequence; 1-1C6 clone, characterized by at least twelve point mutations and an insertion outside the target sequence; 2-1H12 clone, characterized by numerous mutations inside and outside the target sequence, do not express the SOD2 gene, Figure 1. The tRNA fraction isolated from these cells contains the high level of mcm5S2U (57- 83% in untreated cells), the low level of cm5S2U (2-5%) depending on the clone. After application of H2O2 to the culture, we observed the decrease in the amount of mcm5S2U (from 83 to 70%) with a simultaneous increase in the amount of mcm5H2U (from 7% to 16%) and mcm5U (from 7% to 11%) only in the case of 1-1C6 clone. In the other two clones (3-1D8 and 2-1H12) the situation was reversed. We observe the increase in the amount of mcm5S2U and the decrease in mcm5H2U, which is difficult to explain at this point.
In summary, the expected major changes in the course of the 2-thiouridine desulfuration process did not occur in HEK293 mutant variants with inactive Cat or SOD2 genes. The slight increase in the amount of mcm5S2U desulfuration products was observed, but this was dependent on the clone studied. Our observation can be explained by the fact that only individual Cat or SOD2 genes were silenced in the cells. Knock out all three genes in the one type of cells would most likely be fatal for the cells. In our case the genes coding for the other enzymes of the antioxidant system remained active and the antioxidant enzymes reduced the effect of oxidative stress on the cells.
3. Materials and Methods
3.1. Chemical Synthesis of a R5-Substituted-2-Thiouridines and Derivatives
Nucleoside standards (mcm5S2U, mcm5H2U, mcm5U, cmS2U, cm5H2U, cm5U, S2U, H2U, U) for LC-MS/MS analysis were synthesized at Lodz University of Technology (TUL) according to previously established and published protocols. The structures of the products were confirmed by NMR spectroscopy. 5-Methylcarboxymethyl-2-thiouridine (mcm5S2U) was synthesized using the "silyl method" of N-glycosidic bond formation between 5-ethylcarboxymethyl-2-thiouracil and 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose. [41] followed by transesterification of the ethyl ester performed in a 0.1M solution of sodium methoxide in methanol. 5-Methylcarboxymethyluridine (mcm5U) was prepared by the malonylation of the protected 5-bromouridine, followed by decarboxylation of the 5-malonyl moiety of the nucleoside. [42]. Desulfuration of the protected mcm5S2U was carried out with 3-chloroperbenzoic acid (mCPBA) to give mcm5H2U. [34] The ester groups of the modified nucleosides were hydrolyzed under basic conditions (in 0.1M KOH aqueous solution) to appropriate acids to give cm5S2U, cm5U, cm5H2U [34].
3.2. Protocols for Yeast
3.2.1. Yeast Cell Culture
Yeast cells were cultured overnight in a flask containing 50 mL of liquid YPD medium (1% yeast extract; 2% peptone; 2% glucose) in an incubator (30 °C) with vigorous shaking (200-250 rpm). The next day, fresh YPD medium (200 mL) was inoculated with 1% inoculum from the overnight culture and the yeast culture was continued in an Erlenmeyer flask for the next 3-4 hours at 30 °C, until the optical density of the cells at λ=600 nm (OD600) reached the value of 0.8-1.0. Then the oxidizing reagent was added directly to the medium at different concentrations (H2O2: 0, 5, 10, 25, 50, 75, 100 mM; NaAsO2: 0, 10, 20, 40, 60, 80, 100 mM; NaClO: 0, 10, 20, 40, 60, 80, 100 mM), and the yeast grew was continued under the same temperature and agitation conditions for 1 or 2 hours. After completion of the culture, the yeast cells were harvested by centrifugation (6000 rpm, 10 min at 4 °C), washed, counted and subjected to further analysis or frozen at -80 °C until further use.
3.2.2. Yeast Viability Assay
To determine the viability of the yeast cells, the spotting assay and the colony-forming unit (CFU) assay were performed according to the protocol of Kwolek-Mirek et al. [44]. Yeast grew in the liquid YPD medium under the conditions described above. When the culture reached the exponential growth phase (OD600=0.8-1.0) the yeast cells were exposed to oxidative stress. After 1 or 2 hours of oxidative stress, the cells were centrifuged (6000 rpm, 10 min at 4 °C), washed in sterile PBS buffer, counted, and diluted to 107, 106, 105, 104, 103 cells/mL. For the spotting assay, 5 µl of each suspension was spotted onto solid YPD medium (containing 1.5% agar) and incubated at 30 °C for 48 hours, then the growth of yeast was observed and compared. For the colony-forming unit assay, 100 µl of the 103 dilution was applied to solid YPD medium and incubated at 30 °C for 48 hours. The colonies that grew on the plate were counted and compared with others plates.
3.2.3. Yeast Intracellular ROS Level
To determine the intracellular ROS content in the yeast cells exposed to oxidative stress, the H2DCF-DA (2',7'-dichlorofluorescin diacetate, Sigma Aldrich) assay was performed. Yeast cells were grown in the liquid YPD medium under the conditions described above and exposed to oxidative stress. After 1 or 2 hours of incubation in oxidative stress, the culture was centrifuged (6000 rpm, 10 min at 4 °C), washed twice in PBS buffer (pH 7.4), diluted, counted and 1.5 x 107 cells were incubated for 30 min in the dark with 10 µM H2DCF-DA reagent. Then the cells were centrifuged, washed and transferred to the black 96-well plate (Perkin Elmer). The fluorescence of the cells was measured directly using a Synergy HT plate reader (BIO-TEK). The excitation and emission wavelengths for fluorescein derivative are as follows: λex=485 nm and λem=520 nm, respectively. The quantification of the data was performed with the KC4 software (BIO-TEK).
3.2.4. γ-Toxin Preparation
Recombinant γ-toxin (from Kluyveromyces lactis) was overexpressed in BL21(DE3) bacteria containing the pET28-smt3 plasmid encoding the 10x His-Smt3-Ulp1-γ-toxin fusion protein. Bacteria were cultured in 2 liters of LB medium inoculated with 1% inoculum from the overnight culture and grown at 37 °C for approximately 4 hrs, until reached logarithmic growth phase, OD600=0.4-0.6. Then, the culture was cooled to 4 °C by incubation on ice, and 0.4 mM IPTG with 2% ethanol was added to induce protein overexpression. The culture was maintained at 17 °C with shaking (200 rpm) for the next 16 hrs. The culture was centrifuged (4000 rpm, 30 min, 4 °C), the pellet was washed with water to remove the residual medium, and frozen at -70 °C. For protein isolation, the bacterial pellet was suspended in buffer containing 100 mM Tris-HCl pH 8.8, 500 mM NaCl, 10% glycerol supplemented with Complete Protease Inhibitor, EDTA free (Roche) and sonicated. After completion of this phase, insoluble material was removed by centrifugation (17 000 rpm, 30 min, 4 °C). The supernatant was applied to the Nickel affinity resin (Ni-NTA agarose, Thermo Fisher Scientific) equilibrated with the same buffer. The resin was washed with a buffer containing 50 mM Tris-HCl, pH 8.0, 250 mM NaCl, 10% glycerol, and the pure protein was eluted with buffer containing 50 mM Tris-HCl, pH 8.0, 250 mM NaCl, 10% glycerol, 500 mM imidazole. All preparation steps were carried out at 4 °C. The protein was dialyzed to 50 mM Tris-HCl pH 8.0, 100 mM NaCl, concentrated, frozen in liquid nitrogen and stored at -80 °C until use. Results of γ-toxin purification were placed in the Supplementary Material, Figure S5.
3.2.5. γ-toxin tRNA digestion
The reaction was performed according to the protocol previously described by Lentini et al [41]. Total cellular RNA samples (5 µg) isolated from yeast treated with H2O2 were incubated with γ-toxin (~14 µg /reaction) in 10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT buffer for 15-30 min at 30 °C. The resulting reaction products were analyzed by Northern blot using the tRNAGlu-specific DNA probe and RT-PCR with tRNAGlu-specific primers. The sequence of the probe and primers used can be found in the Supplementary Material, Table S4.
3.2.6. Northern Blot (NB) Analysis
The reaction mixtures after γ-toxin digestion (15 µL) were mixed with NB loading buffer (90% formamide with a mixture of tracking dyes: 0.01% bromophenol blue, 0.01% xylenocyanol), denatured for 2 minutes at 95 °C and loaded onto a 15% denaturing polyacrylamide gel for electrophoresis. After electrophoresis, the RNA was electro-transferred from the gel onto a Hybond-Ny+ membrane (Amersham Bioscience, Little Chalfont, UK). The membrane was placed in a tube and incubated with ULTRAhyb™ Ultrasensitive Hybridization Buffer (Thermo Fisher Scientific) at 42 °C for 1 hour. Then the 32P-labeled synthetic DNA probe complementary to yeast tRNAGlu or control 25S rRNA was added and incubated overnight at 42 °C with slow rotation. The sequences of probes are as follows: S. cerevisiae tRNAGlu probe: 5'-ATAGCCGTTACACTATATCGGA-3' and positive control probe for S. cerevisiae 25S rRNA 5'-GATTCTCACCCTCTATGACG-3'. After overnight incubation, unbound probes were washed out twice for 10 minutes (SSPE 2x, 0.1% SDS buffer), the membranes were air-dried, placed in a cassette with X-ray film and exposed to radiation for several hours at room temperature in the dark.
3.2.7. qRT-PCR Analysis
Real-time qRT-PCR reactions were performed with total cellular RNA from S. cerevisiae that was (i) not treated with γ-toxin, [γ-toxin (-)], or (ii) after γ-toxin hydrolysis, [γ-toxin (+)]. The RNA (1 µg), the tRNAGlu-specific primers and the components of the LightCycler® RNA Amplification SYBR Green I kit (Roche) were mixed according to the manufacturer's protocol. The reaction was performed on the Light Cycler instrument (Roche) in capillaries in 10 μl solution. The primers used in the experiment were specific for S. cerevisiae (sc) tRNAGlu: sc-tRNAGlu Fw: 5'-TCCGATATAGTGTAACGGCTAT-3'; sc-tRNAGlu Rv: 5'- CTCCGATACGGGGAGTCG -3'; and specific for the control S. cerevisiae 25S rRNA: 25S rRNA Fw 5'-GAAATCTGGTACCTTCGGTG-3'; 25S rRNA Rv 5'-GATTCTCACCCTCTATGACG-3'. The following program was used for the reaction: Step1. Reverse transcription: 55 °C 10 min, reverse transcriptase denaturation: 95 °C 30s; Step2. PCR (45 cycles): 95 °C 10s; 60 °C 10s; 72 °C 10s; Step3. Tm of the PCR products analysis according to the Light Cycler instruction.
3.2.8. Yeast Total RNA Preparation and Pull-Down of tRNAGlu
Yeast samples (e.g. 2 g) were suspended in 0.5 mL sterile water and then in 5 mL TRI Reagent (Thermo Fisher Scientific) with 2 g glass beads (0.5 mm) (Sigma Aldrich). Yeast suspension was vortexed vigorously to efficiently perform cell lysis. Total cellular RNA was prepared according to the TRI Reagent protocol by extraction with chloroform and precipitation of the RNA with isopropanol. The resulting pellet was washed with 70% ethanol and air dried. The RNA concentration was determined spectrophotometrically by measuring the absorbance at 260 nm.
The tRNAGlu was isolated from total cellular RNA by the pull-down method with the specific biotinylated probe complementary to the anticodon loop domain (5'-biotin-CCGGTCTCCACGGTGAAAGCGTGATGTGATAG-3') bound to the Streptavidin-agarose Resin (Pierce, Thermo Fisher Sci.). Approximately 500 μL of the streptavidin-agarose slurry was added to the sterile Eppendorf tube, washed three times with (i) RNase-free deionized water, three times with (ii) 100 mM sodium phosphate buffer pH 7.4 containing 125 mM NaCl, and then incubated with 15 nmol of the biotinylated DNA probe for 1 hour at room temperature with vigorous shaking (~2000 rpm). After incubation, the agarose beads were washed several times (at least six times) with 100 mM sodium phosphate buffer/125 mM NaCl to remove the unbound oligonucleotide. The washing step was monitored spectrophotometrically (Nanodrop, BIO-TEK). Approximately 500 mg of the total RNA solution was added to the agarose beads, incubated at 80 °C for 5 minutes, cooled and incubated overnight with vigorous shaking (~2000 rpm) at 4 °C. The next day, the resin was washed at least six times with 100 mM sodium phosphate buffer, pH 7.4, until the absorbance of the wash solution reached 0. The bound tRNAGlu was eluted by addition of 200 μl deionized water, incubation at 80 °C for 2 min and centrifugation (3000 rpm, 3 min). The elution step was repeated at least six times. The solution with eluted tRNAGlu was lyophilized using the vacuum SpeedVac concentrator (Savant). The amount of isolated tRNAGlu was determined spectrophotometrically at λ=260 nm. The samples were stored at -80 °C until further use.
3.3. Protocols for Human Cells
3.3.1. Human Cells Culture
Human cells: HeLa, K562, MOLT-4, A375, A549 were cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, FBS, (Gibco, Thermo Fisher Scientific) and antibiotics: Penicillin-Streptomycin (Gibco, Thermo Fisher Scientific); HEK293 and modified HEK293, A431, U-87 MG, MCF-7 were cultured in DMEM medium (Gibco, Thermo Fisher Scientific) supplemented with 10 % FBS (Gibco, Thermo Fisher Scientific) and the antibiotics penicillin and streptomycin (Gibco, Thermo Fisher Scientific). The complete list of cell lines, their origin and culture conditions can be found in the Supplementary Material, Table S2.
3.3.2. Cytotoxicity Assay
The cytotoxic effect of the oxidizing agents (H2O2, NaAsO2 or NaClO) on the viability of the tested cells was determined using the MTT assay. The cell suspension of 7x 103 cells per well was seeded in the 96-well plate in 200 µL of the appropriate medium, Table S2. After 24 hours of incubation at 37 °C in the 5% CO2 atmosphere, the culture medium was replaced with fresh medium containing the respective reagent at the following concentrations: 0, 5, 10, 25, 50, 100, 200 µM (H2O2, NaAsO2) or 0, 1, 5, 10, 20, 50, 100 µM (NaClO) and cell culture under oxidative stress conditions was continued for the next 1 or 2 hours. Untreated cells were used as positive control (100% viability) and medium without cells was used as background. After the incubation, 25 µL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma Aldrich) solution in PBS (5 mg/mL) was added to each well and the cells were incubated at 37 °C for another 2 hours. Finally, 95 μL of hot lysis buffer (20% SDS, 50% aqueous dimethylformamide, pH 4.5) was added to each well and the plates were incubated overnight at 37 °C. The absorbance of the lysed cells was measured at 570 nm and 630 nm using the FLUOstar Omega plate reader (Biogenet) and ΔAbs=Abs570-Abs630 was calculated. The results were presented as a dependence of the percentage of live cells on the concentration of oxidizing agent and the IC50 coefficient was determined for each cell line.
3.3.3. Intracellular ROS Level Determination
The intracellular ROS content was determined using the DCFDA (2',7'-dichlorofluorescin diacetate) Cellular ROS Detection Assay Kit (Abcam) according to the manufacturer’s protocol after the cells had been exposed to the oxidizing mixture (H2O2, NaAsO2 or NaClO).
Adherent cells were grown in RPMI-1640 or DMEM medium supplemented with 10% FBS and antibiotics as required. Cells were harvested and the suspension of 25 x 103 cells/well was seeded into clear bottom black 96-well microplate (PerkinElmer). Cells were allowed to grow overnight, then washed with PBS buffer and incubated with 25 µM DCFDA solution at 37 °C, 5% CO2 in the dark. After 45 minutes of incubation, cells were washed in PBS buffer and treated with oxidants (0, 5, 10, 25, 50, 100, 200 µM H2O2; 0, 5, 10, 25, 50, 100, 200 µM NaAsO2; or 0, 1, 5, 10, 20, 50, 100 µM NaClO) in 100 µL/well RPMI-1640 or DMEM colorless medium containing 10% FBS and antibiotics. The fluorescence of the cells was measured after 0, 1, 2 and 4 hours of incubation using the FLUOstar Omega plate reader (Biogenet) at Ex/Em= 485/520 nm in endpoint mode in the presence of oxidizing compounds in the medium.
Suspension cells were grown in RPMI-1640 medium supplemented with 10% FBS and antibiotics. Then cells were harvested by centrifugation, washed in PBS buffer and stained with 20 µM DCFDA solution, 1 x 106 cells/mL, 37 °C, in the dark. After incubation for 30 minutes, cells were washed in PBS, suspended in colorless RPMI-1640 medium containing 10% FBS and antibiotics to 1 x 105 per well, seeded in clear bottom black 96-well microplates (PerkinElmer) and exposed to the appropriate oxidant used as described above. The fluorescence of the cells was measured after 0, 1, 2 and 4 hours using the FLUOstar Omega plate reader (Biogenet) at Ex/Em= 485/520 nm in endpoint mode in the presence of the oxidizing compounds in the medium.
3.3.4. Isolation of Total Cellular RNA and the Total Cellular tRNA Fraction
The human cells (3x 106) were grown in the culture flask (25 cm2) in 5 mL of a suitable medium containing 10% FBS and antibiotics for 24 hours. Then the cells were exposed to the oxidizing agent used at a concentration determined by the IC50 for each cell line, as described above. After 2 hours of incubation of the cells in the oxidizing environment, the cells were washed with PBS buffer, harvested and lysed in 1 mL of TRI Reagent (Thermo Fisher Sci.). Total cellular RNA was prepared according to the TRI Reagent protocol. The resulting total RNA preparation was stored at -80 °C until use. The RNA concentration was determined spectrophotometrically by measuring the absorbance at 260 nm.
Further steps to isolate the total cellular tRNA fraction followed the procedure described by Su et al [36]. In brief, total RNA samples were denatured by incubation at 80 °C for 2 minutes and the total cellular tRNA fraction was isolated using an Agilent SEC-3, 300Å HPLC column, 150 mm length × 7.8 mm inner diameter (Perlan Technologies) on an FPLC AKTA purifier: Box-900, pH/C-900, UV-900, P-900, Frac 920 system (GE) in the aqueous 100 mM ammonium acetate phase, flow rate of 0.5 mL/min. The resulting tRNA fraction was precipitated in ice-cold ethanol (2.5 volumes) in the presence of 3 M sodium acetate, pH 5.2, (0.1 volumes) and stored at -80 °C until use.
3.3.5. tRNA Hydrolysis to Nucleosides
The tRNA hydrolysis was performed according to the described protocols [36,45]. In brief, the tRNA sample (~10 µg) was hydrolyzed with a combination of two nucleases, Benzonase (endonuclease from Serratia marcescens, Sigma-Aldrich), 20 units per reaction, in 50 mM Tris-HCl pH 8.0, 1 mM MgCl2, 4 hrs at 37 °C, Phosphodiesterase I from Crotalus adamanteus venom (Sigma-Aldrich), 0.8 units per reaction, in 50 mM Tris-HCl, 20 mM MgCl2, 16 hrs at 37 °C and finally Alkaline phosphatase (EURX, Poland), 10 units per reaction, in manufacturer's buffer (1M diethananolamine, 10 mM, p-nitrophenylophosphate, 0.25 mM MgCl2, pH 9.8), 1 h at 37 °C. The sample after hydrolysis was filtered with a 10 000 MW cut-off spin filter (Amicon) and dried in a vacuum centrifuge (Savant).
3.4. CRISPR/Cas9 Genome Editing Experiments
3.4.1. Transfection
HEK293 cells were seeded in 6-well plate, 3x 105 cells per well, in high glucose DMEM medium supplemented with 10% FBS and antibiotics. After 24 hours incubation at 37 °C. 5% CO2, when a cell confluence of about 60% was reached. transfection of the sgRNA-Cas9 complex was performed according to the instruction of Lipofectamine “CRISPRMAX”. Two mixtures were prepared for one well of the 6-well plate. Tube 1: TrueCut Cas9 v2 (6.25 µg) was mixed with sgRNA (1.2 µg) and Cas9 Plus Reagent (12.5 µL) in OPTI MEM I medium (125 µL). Tube 2: Lipofectamine “CRISPRMAX” (7.5 µL) was diluted in OPTI MEM I medium (125 µL). The solution from Tube 1 was immediately added to Tube 2, the mixture was incubated for 10 minutes at room temperature and the formed complex was added to the cells. HEK293 cells were incubated at 37 °C, 5% CO2 for the next 3 days. After incubation, cells from each well were counted, diluted to the concentration of approximately 1 cell per 100 µL of medium, and aliquoted into 96-well plates (three 96-well plates per one sgRNA applied, for a total of 9 plates for one gene) to obtain single clones. The rest of the cells were washed with PBS and stored at -70 °C for further testing.
3.4.2. Western Blot
The proteins contained in the 20-40 µg cell lysate were separated by electrophoresis in 10-15% SDS-PAGE and electro-transferred from the gel to Immobilon-P, PVDF membrane (Sigma Aldrich) in Rapid Transfer Buffer (VWR International). The PVDF membrane was blocked by incubation in 5% non-fat milk in TBS buffer (20 mM Tris-HCl pH 7.4, 0.9% NaCl) and then incubated with primary antibodies: rabbit anti Cat (1:1000) or mouse anti SOD1 (1:1000) or rabbit anti SOD2 (1:1000) or as reference mouse anti GAPDH (1:1000) (Cell Signaling), overnight at 4 °C. The next day, the membranes were washed 3 times for 10 minutes each in TBST buffer (TBS with 0.1% Tween 20) and incubated with secondary antibodies against rabbit or mouse conjugated with alkaline phosphatase, dilution 1:4000 (Sigma-Aldrich) for 2 hours at room temperature. The membranes were then washed 3 times for 10 minutes each in TBST buffer and incubated for 5 minutes at room temperature with a chemiluminescent substrate for alkaline phosphatase (DuoLuX® Chemiluminescent and Fluorescent Substrate, Alkaline Phosphatase, Vector Lab., Biokom). The protein bands were visualized with the gel visualization system (Uvitec).
3.4.3. T7 Endonuclease I Based Mutation Detection
Genomic DNA was isolated from modified HEK293 cells using the Genomic Mini AX Tissue Spin kit (A&A Biotechnology) according to the manufacturer’s protocol. The PCR primers were designed for the detection of mutations in the human genes Cat, SOD1 or SOD2 caused by CRISPR/Cas9 genome editing system. The primer sequences for the three target genes are listed in Table S3. The PCR reaction was performed under the conditions described in the T7 endonuclease 1-based mutation detection protocol in the EnGen Mutation Detection kit (NewEngland Biolabs). The PCR mixture contained: 200 ng of genomic DNA, 12.5 µL of Q5 Hot Start High-Fidelity (2x) master mix, 0.5 µM of appropriate forward and reverse primers, and nuclease-free water up to 25 µL. Thermocycling was performed under the following conditions: 1) initial denaturation, one cycle: 98 °C, 30 s; 2) 35 cycles: denaturation 98 °C, 5 s, annealing 62 °C, 10 s, extension 72 °C, 20 s, and 3) the final extension 72 °C, 2 minutes. A small amount of PCR product (e.g. 5 µL) was analyzed on an agarose gel to check the amplification of a single product of correct size compared to the DNA ladder size.
Heteroduplex formation: the mixture of 5 µL of PCR product with 2 µL of NEB buffer2 (10x) and 12 µL of ddH2O was denatured at 95 °C for 5 minutes and then cooled to room temperature for 20 minutes. Then 1 µL of EnGen T7 endonuclease was added and the heteroduplex digestion reaction was incubated at 37 °C for 15 minutes. After digestion, 1 µL of Proteinase K was added to the mixture and incubated at 37 °C for 5 minutes to inactivate the T7 endonuclease-1. The products of heteroduplex digestion were analyzed on the agarose gel.
3.5. Analysis of tRNA Derived Nucleosides by LC-MS/MS
All described LC-MS/MS analyses were performed in the LabExperts laboratory (Lodz, Poland). The LC-MS/MS analysis of nucleoside mixture derived from yeast tRNA was carried out on QTRAP 6500+ mass spectrometer (Sciex, USA), coupled with Exion LC System (Sciex, USA). The LC-MS/MS analysis of nucleoside mixture derived from human tRNA was carried out on ZenoTOF 7600 mass spectrometer (Sciex, USA), coupled with ExionAC LC System (Sciex, USA). Chromatographic separation was conducted on an InfinityLab Poroshell 120 PFP, 2.1 x 100 mm, 2.7 µm, narrow bore LC column (Agilent, USA) thermostated at 50 °C. The major chromatography parameters were as follows: injection volume, 30 μL; constant flow, 0.5 mL/min; the mobile phase combining solvent A: 0.1% formic acid in water (LC-MS grade) and solvent B: 0.1% formic acid in acetonitrile (ACN) (LC-MS grade), total analysis time 7 min. The gradient started with 0.5 min of pre-injection conditioning, followed by gradient separation starting from 2% B increasing to 5% B after 1 min. followed by further increase to 60% B after 4 min and left at 60% B till 5 min. Initial conditions were restored from 5.1 till 7 min. of the run. A summary of the applied gradient conditions is provided in the Supplementary Table S5. The development of the method included optimization of the ion source parameters, selection of fragment ions and optimization of the MRMhr parameter scanning. An electrospray ion source (ESI) was used. Detailed parameters are presented in the Supplementary Table S5. The MS/MS detection was made in positive ionization scheduled multiple reaction-monitoring (sMRM) mode. The optimized ESI ions source parameters were as follows: CUR: 25; IS: 5000 V; TEMP: 400 °C; GS1: 50 and GS2: 40. The quantitation of the monitored compounds was performed on the basis of standard curves prepared for synthetic nucleoside standards within a range of concentrations from 0.01 nM to 1 µM.
4. Conclusions
Aerobic organisms, including humans, use oxygen to produce energy in the mitochondrial respiratory chain, in which the oxygen molecule undergoes a four-electron reduction and the energy released during this process is used to generate ATP. The electron flow through the respiratory chain is not completely dense, some electrons "escape" and reduce the oxygen via a one-electron process. The result of this reduction is a superoxide anion radical (O2•¬), which is converted into hydrogen peroxide (H2O2) and other oxygen species. It has been estimated that about 1-4% of the oxygen consumed by mitochondria undergoes one-electron reduction and this is the main source of O2•¬ radical in aerobic cells [46–48]. Reactive oxygen species (ROS) can also be generated in the body by external physical factors such as ionizing radiation, ultraviolet radiation, and ultrasound, as well as by some intracellular enzymatic reactions catalyzed by oxidases, reductases, dehydrogenases, and so on. Aerobic cells have developed a defense system against the destructive effects of ROS, consisting of low molecule antioxidants, antioxidant and repair enzymes. In addition to the undoubtedly positive functions that ROS fulfill in the body: e.g. as components of signal transduction pathways, in cell cycle progression, differentiation, cell development and apoptosis, however, when the generation of ROS exceeds their degradation by antioxidant enzymes a state of oxidative stress arises in the cell. Excessive ROS concentrations cause oxidative damage to proteins, nucleic acids, polysaccharides and lipids and lead to cell dysfunction and cell death. ROS react non-specifically with cellular components, modifying and damaging them. Oxidatively damaged biomolecules impair cellular functions and contribute to the pathology of a variety of disorders.
One of the molecules that fulfills an irreplaceable function in the cell and is susceptible to damage by ROS is transfer nucleic acid. Understanding the mechanisms of damage of modified nucleosides that occur in natural cellular tRNAs and the consequences of this damage is essential for expanding knowledge of the regulation of gene expression at the codon-anticodon interaction level. We focused our interest on the eukaryotic (yeast or human) tRNA containing 5-substituted-2-thiouridine (R5S2U) in the wobble position of the anticodon. Under conditions of external or internal oxidative stress caused by the addition of the oxidizing reagent to the medium during culture, R5S2U is desulfurized. Using the highly sensitive and selective technique LC-MS/MS, we analyzed the nucleosides derived from the cellular tRNAGlu or low-weight (< 200-nt) RNA fraction and detected not only the modifications mcm5S2U or cm5S2U, but also the desulfuration products mcm5H2U, cm5H2U, mcm5U and cm5U and confirmed our hypothesis that the 5-substituted S2U-containing tRNAs can be damaged in growing cells exposed to oxidative stress. The amount and type of R5S2U desulfuration products depended on the type of cells examined (yeast or human), the type of oxidation reagent (H2O2, NaAsO2 or NaClO), its concentration in the culture medium and the exposure time. The observed changes in the cellular content of R5S2U and the formation of its desulfuration products, R5H2U and R5U, were quite subtle and differed markedly from the reactions performed in vitro, in the absence of cells, where the S2U desulfuration efficiency of 100% was easily achieved. This effect can be explained by the activity of the antioxidant system in the cells. Disruption of this system, e.g. by inhibiting the expression of genes coding for antioxidant enzymes, leads to a significant sensitization of the cells to oxidative stress and to a stronger effect of R5S2U-tRNA desulfuration. The process of R5S2U-tRNA desulfuration triggered by prolonged oxidative stress in living cells can impair the function of transfer RNAs and alter the translation of genetic information.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1 Yeast cell lines used in the studies, media and culture conditions; Table S2 Human cell lines used in the studies, media and culture conditions; Table S3 Sequences of sgRNAs targeting the human SOD1, SOD2 and Cat genes, PCR primers designed to detect the introduced mutations; Table S4 Sequences of PCR primer and Northern blot probes used in γ-toxin assay for yeast tRNAGlu analyse; Table S5 Modification of HEK293 cells. Efficiency of CRISPR/Cas9 gene editing; Table S6 The list of nucleoside standards used in studies, molecular formula and UV characteristics; Table S7 LC-MS/MS analysis conditions (A) LC gradient, (B) TOF MS scanning parameters, (C) TOF MS/MS scanning parameters; Figure S1 T7 Endonuclease assay for selected clones of modified HEK293 with damaged SOD2 or Cat genes selected for genomic DNA sequencing; Figure S2 The result of sequencing of genomic DNA isolated from HEK293 cells with the damaged SOD2 or Cat genes. Comparison of the sequences obtained from the modified clones (upper strand) with the original gene sequence (lower strand); Figure S3. The results of the spotting test to investigate the viability of yeast cells exposed to oxidizing agents for 1 or 2 hours; Figure S4. Determination of the ROS level in yeast cells after incubation with NaClO., the 2,7 dichloro-dihydrofluorescein diacetate assay; Figure S5 Visualization of γ-toxin (~27 kDa) protein in SDS-PAGE; Figure S6. MTT assay for cancer cells exposed to the oxidative reagents NaAsO2 or NaClO. Evaluation of cell viability as a function of reagent concentration; Figure S7. ROS generation in the cancer cells exposed to H2O2, NaAsO2 or NaClO determined by 2,7 dichloro-dihydrofluorescein diacetate assay; Figure S8. The natural intracellular ROS level existing in cancer cells, evaluated by 2,7-dichloro-dihydrofluorescein-diacetate assay; Figure S9 Structural formula of the nucleoside standards used in the LC-MS/MS (MRMhr) studies; Figure S10 The extracted ions chromatogram (XIC) of the mixture of ten nucleoside standards used in the mass studies; Figure S11 Standard curves prepared for nucleoside standards; Figure S12 Identification of mcm5S2U desulfuration products in yeast tRNAGlu by LC-MS/MS, yeast exposed to NaAsO2 or NaClO during culture.
Author Contributions
M.S. project design and management, funding acquisition, design of experiments, cell culture, tRNA isolation and hydrolysis, manuscript writing and editing; R.S. LC-MS/MS analyses; A.D. synthesis of nucleoside standards; K.K-G. cell viability and ROS determination experiments; P.S. gamma-toxin purification; P.B. supervision on behalf of the project consortium Partner; B.N. project conceptualization, manuscript review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre in Poland, Project Number UMO-2016/23/B/NZ1/02316] to M.S.
Data Availability Statement
All relevant data are within the paper and Supplementary Materials.
Acknowledgments
Authors thank Prof. Elzbieta Sochacka (Lodz University of Technology, Poland) for her achievements in the chemistry of sulfur-modified nucleosides and scientific inspiration. Authors thank Prof M. Forte for kindly provided S. cerevisiae strain M3 and Dr Andonis Karachitos and Dr Martyna Baranek (Adam Mickiewicz University, Poznan, Poland) for S. cerevisiae M3Δsod1 strain, that were used in the published studies.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambow;itz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods. 2015, 12, 835-837. [CrossRef]
- Yoluc, Y.; van de Logt, E.; Kellner-Kaiser, S. The Stress-Dependent Dynamics of Saccharomyces cerevisiae tRNA and rRNA Modification Profiles. Genes (Basel). 2021, 28, 1344. [CrossRef]
- Yoluç, Y.; Ammann, G.; Barraud, P.; Jora, M.; Limbach, PA. Motorin, Y.; Marchand, V.; Tisné, C.; Borland, K.; Kellner, S. Instrumental analysis of RNA modifications. Crit Rev Biochem Mol Biol. 2021, 56, 178-204. [CrossRef]
- Sarin, L.P.; Kienast, S.D.; Leufken, J.; Ross, R.L; Dziergowska A.; Debiec, K.; Sochacka, E.; Limbach, P.A.; Fufezan, C.; Drexler, H.C.A.; Leidel, S.A. Nano LC-MS using capillary columns enables accurate quantification of modified ribonucleosides at low femtomol levels. RNA 2018, 24, 1403-1417. [CrossRef]
- Heiss, M.; Hagelskamp, F.; Marchand, V.; Motorin, Y.; Kellner, S. Cell culture NAIL-MS allows insight into human tRNA and rRNA modification dynamics in vivo. Nat Commun. 2021, 12, 89. [CrossRef]
- Amalric, A.; Bastide, A.; Attina, A.; Choquet, A.; Vialaret, J.; Lehmann, S.; David, A.; Hirtz, C. Quantifying RNA modifications by mass spectrometry: a novel source of biomarkers in oncology. Crit Rev Clin Lab Sci. 2022, 59, 1-18. [CrossRef]
- de Crécy-Lagard, V.; Boccaletto, P.; Manglebrg, C.G.; Sharma, P.; Lowe, T.M.; Leidel, S.A.; Bujnicki, J.M. Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res. 2019, 47, 2143-2159. [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; Avşar, G.; Romitelli, A.; Pir, P.; Dassi, E.; Conticello, S.G.; Aguilo, F.; Bujnicki, J.M. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231-D235. [CrossRef]
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. 2011, The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 39, D195-201. [CrossRef]
- McCown, P.J.; Ruszkowska, A.; Kunkler, C.N., Breger, K.; Hulewicz, J.P.; Wang, M.C.; Springer, N.A.; Brown, J.A. Naturally occurring modified ribonucleosides. Wiley Interdiscip Rev RNA. 2020, 11, e1595. [CrossRef]
- Machnicka, M.A.; Olchowik, A.; Grosjean, H.; Bujnicki, J.M. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014, 11, 1619-1629. [CrossRef]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395-404. [CrossRef]
- Grosjean, H.; de Crecy-Lagard, V.; Marck, C. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS Lett. 2010, 584, 252–264.
- Agris, P.F.; Eruysal, E.R.; Narendran, A.; Väre, V.Y.P.; Vangaveti, S.; Ranganathan, S.V. Celebrating wobble decoding: Half a century and still much is new. RNA Biol. 2018, 15, 537-553. [CrossRef]
- Agris, P.F.; Narendran, A.; Sarachan, K.; Väre, V.Y.P.; Eruysal, E. The Importance of Being Modified: The Role of RNA Modifications in Translational Fidelity. Enzymes, 2017, 41, 1-50. [CrossRef]
- Lorenz, C.; Lünse, C.E.; Mörl, M. tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules, 2017, 7, 35. [CrossRef]
- Chanfreau, G.F. Impact of RNA Modifications and RNA-Modifying Enzymes on Eukaryotic Ribonucleases. Enzymes, 2017, 41, 299-329. [CrossRef]
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic Carbon-Sulfur Bond Formation in Natural Product Biosynthesis. Chem Rev. 2017, 117, 5521-5577. [CrossRef]
- Zheng, Y.Y; Wu, Y.; Begley, T.J.; Sheng, J. Sulfur modification in natural RNA and therapeutic oligonucleotides. RSC Chem Biol. 2021, 2, 990-1003. [CrossRef]
- El Yacoubi, B.; Bailly, M.; de Crécy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet. 2012, 46, 69-95. [CrossRef]
- Tomikawa, C.; Ohira, T.; Inoue, Y.; Kawamura, T.; Yamagishi, A.; Suzuki, T.; Hori, H. Distinct tRNA modifications in the thermo-acidophilic archaeon, Thermoplasma acidophilum. FEBS Lett. 2013, 587, 3575–3580.
- Björk, G.R.; Hagervall, T.G. Transfer RNA modification: Presence, synthesis, and function. EcoSal Plus 2014, 6.
- Durant, P.C.; Bajji, A.C.; Sundaram, M.; Kumar, R.K.; Davis, D.R. Structural effects of hypermodified nucleosides in the Escherichia coli and human tRNALys anticodon loop: the effect of nucleosides s2U, mcm5U, mcm5s2U, mnm5s2U, t6A, and ms2t6A. Biochemistry. 2005, 44, 8078-8089. [CrossRef]
- Nawrot, B.; Sierant, M.; Szczupak, P. Selenium modified bacterial tRNAs. In: Sugimoto, N. (eds) Handbook of Chemical Biology of Nucleic Acids. Springer, Singapore. 2023. [CrossRef]
- Shigi, N. Biosynthesis and functions of sulfur modifications in tRNA. Front Genet. 2014, 5, 67. [CrossRef]
- Madore, E.; Florentz, C.; Giegé, R.; Sekine, S.; Yokoyama, S.; Lapointe, J. Effect of modified nucleotides on Escherichia coli tRNAGlu structure and on its aminoacylation by glutamyl-tRNA synthetase. Predominant and distinct roles of the mnm5 and s2 modifications of U34. Eur J Biochem. 1999, 266, 1128-1135. [CrossRef]
- Rodriguez-Hernandez, A.; Spears, J.L.; Gaston, K.W.; Limbach, P.A.; Gamper, H.; Hou, Y.M.; Kaiser, R.; Agris, P.F.; Perona, J.J. Structural and mechanistic basis for enhanced translational efficiency by 2-thiouridine at the tRNA anticodon wobble position. J Mol Biol. 2013, 425, 3888-3906. [CrossRef]
- Urbonavicius, J.; Stahl, G.; Durand, J.M.; Ben Salem, S.N.; Qian, Q.; Farabaugh, P.J.; Björk, G.R. Transfer RNA modifications that alter +1 frameshifting in general fail to affect -1 frameshifting. RNA, 2003, 9, 760-768. [CrossRef]
- Brégeon, D.; Colot, V.; Radman, M.; Taddei, F. Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev., 2001, 15, 2295-2306. [CrossRef]
- Corsaro, A.; Pistara, V. Conversion of the Thiocarbonyl Group into the Carbonyl Group. Tetrahedron 1998, 54:15027-15062.
- Sochacka, E.; Kraszewska, K.; Sochacki, M.; Sobczak, M.; Janicka, M.; Nawrot, B. The 2-thiouridine unit in the RNA strand is desulfured predominantly to 4-pyrimidinone nucleoside under in vitro oxidative stress conditions. Chem Commun (Camb). 2011, 47, 4914-4916. [CrossRef]
- Sochacka, E.; Bartos, P.; Kraszewska, K., Nawrot, B. Desulfuration of 2-thiouridine with hydrogen peroxide in the physiological pH range 6.6-7.6 is pH-dependent and results in two distinct products. Bioorg Med Chem Lett. 2013, 23, 5803-5805. [CrossRef]
- Chwialkowska, A., Wielgus, E.; Leszczynska, G.; Sobczak, M.; Mikolajczyk, B.; Sochacka, E.; Nawrot B. An efficient approach for conversion of 5-substituted 2-thiouridines built in RNA oligomers into corresponding desulfured 4-pyrimidinone products. Bioorg Med Chem Lett. 2015, 25, 3100-3104. [CrossRef]
- Bartos, P.; Ebenryter-Olbinska, K.; Sochacka, E.; Nawrot, B. The influence of the C5 substituent on the 2-thiouridine desulfuration pathway and the conformational analysis of the resulting 4-pyrimidinone products. Bioorg Med Chem. 2015, 23, 5587-5594. [CrossRef]
- Sochacka, E.; Szczepanowski, R.H.; Cypryk, M.; Sobczak, M.; Janicka, M.; Kraszewska, K.; Bartos, P.; Chwialkowska; A.; Nawrot B. 2-Thiouracil deprived of thiocarbonyl function preferentially base pairs with guanine rather than adenine in RNA and DNA duplexes. Nucleic Acids Res. 2015, 43, 2499-2512. [CrossRef]
- Su, D.; Chan, C.T.; Gu, C.; Lim, K.S.; Chionh, Y.H.; McBee, M.E.; Russell, B.S.; Babu, I.R.; Begley, T.J.; Dedon, P.C. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat Protoc. 2014 9, 828-841. [CrossRef]
- Blachly-Dyson, E.; Peng, S.; Colombini, M.; Forte, M. Selectivity changes in site-directed mutants of the VDAC ion channel: structural implications. Science 1990, 247, 1233-1236. [CrossRef]
- Blachly-Dyson, E.; Peng, S.Z.; Colombini, M.; Forte, M. Probing the structure of the mitochondrial channel, VDAC, by site-directed mutagenesis: a progress report. J Bioenerg Biomembr 1989, 21, 471-483. [CrossRef]
- Robbins, D.; Zhao, Y. Manganese superoxide dismutase in cancer prevention. Antioxid Redox Signal. 2014, 20, 1628-1645. [CrossRef]
- Scibior-Bentkowska, D.; Czeczot, H. Cancer cells and oxidative stress. Postepy Hig Med Dosw, 2009; 63, 58-72. e-ISSN 1732-2693.
- Lentini, J.M.; Ramos, J.; Fu, D. Monitoring the 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) modification in eukaryotic tRNAs via the γ-toxin endonuclease. RNA. 2018 24, 749-758. [CrossRef]
- Vorbruggen, .H.; Strehlke, P. Nucleosidsynthesen, VII. Eine einfache Synthese von 2-Thiopyrimidin-nucleosiden. Chem.Ber. 1973, 106, 3039-3061. [CrossRef]
- Fu, Y.; Dai, Q.; Zhang, W,; Ren, J.; Pan, T.; He, C. The AlkB domain of mammalian ABH8 catalyzes hydroxylation of 5-methoxycarbonylmethyluridine at the wobble position of tRNA. Angew Chem I,nt Ed Engl. 2010, 49, 8885-8888. [CrossRef]
- Kwolek-Mirek M, Zadrag-Tecza R. Comparison of methods used for assessing the via-bility and vitality of yeast cells. FEMS Yeast Res. 2014 Nov;14(7):1068-79. [CrossRef]
- Szczupak, P.; Sierant, M.; Wielgus, E.; Radzikowska-Cieciura, E.; Kulik, K.; Krakowiak, A.; Kuwerska, P.; Leszczynska, G.; Nawrot, B. Escherichia coli tRNA 2-Selenouridine Synthase (SelU): Elucidation of Substrate Specificity to Understand the Role of S-Geranyl-tRNA in the Conversion of 2-Thio- into 2-Selenouridines in Bacterial tRNA. Cells 2022, 11, 1522. [CrossRef]
- Boveris, A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984, 105, 429-435. [CrossRef]
- Cadenas, E., Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000, 29, 222-230. [CrossRef]
- Nohl, H. Generation of superoxide radicals as byproduct of cellular respiration. Ann Biol Clin (Paris). 1994, 52, 199-204.
Scheme 1.
The oxidative desulfuration takes place with the formation of two products: mcm5H2U (5-methylcarboxymethyl-4-pirymidinone riboside) and mcm5U (5-methylcarboxymethyl uridine).
Scheme 1.
The oxidative desulfuration takes place with the formation of two products: mcm5H2U (5-methylcarboxymethyl-4-pirymidinone riboside) and mcm5U (5-methylcarboxymethyl uridine).
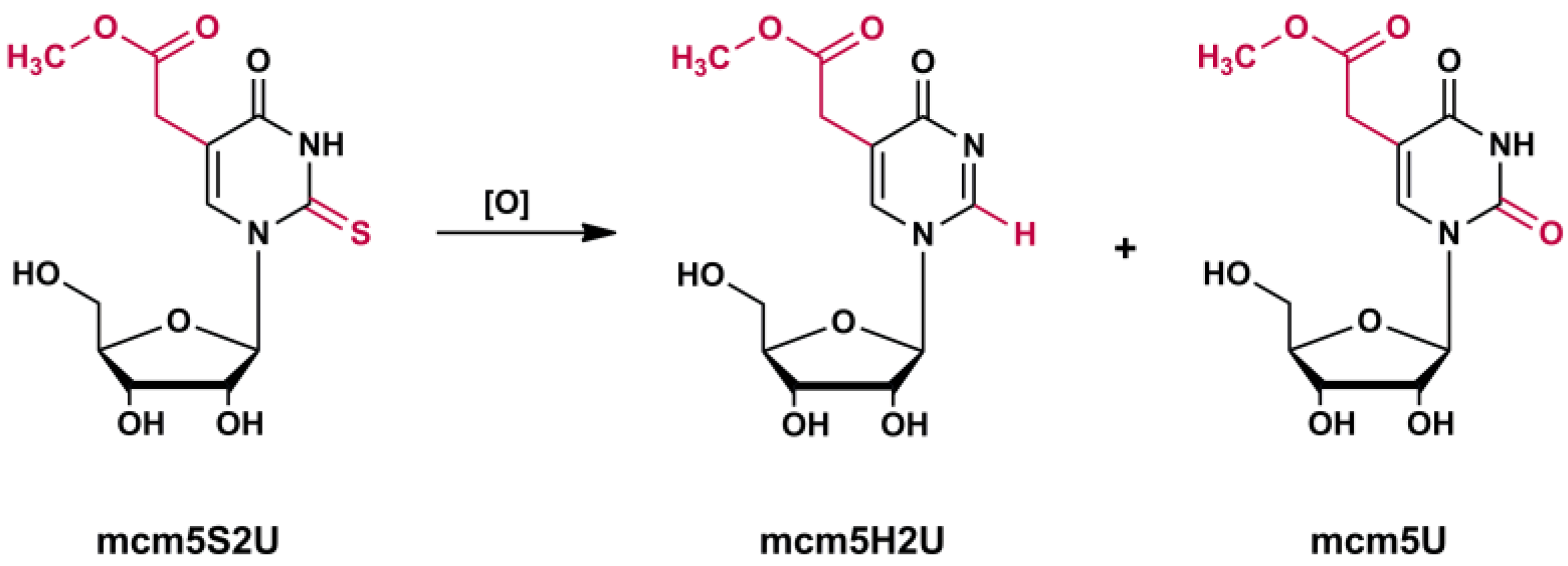
Figure 1.
Example of Western blot analysis of HEK293ΔCat, HEKΔSOD1 and HEKΔSOD2 clones. Selected clones 1 and 2 with mutated Cat gene show no expression of Cat and correct expression of the other genes (SOD1, SOD2 and GAPDH reference gene. Selected clones 3 and 4 with mutated SOD2 gene show no expression of SOD2 gene and correct expression of other genes (SOD1, Cat and GAPDH reference gene). Clones 5 and 6 with mutated SOD1 gene show expression of all studied genes: SOD1, SOD2, Cat and GAPDH.
Figure 1.
Example of Western blot analysis of HEK293ΔCat, HEKΔSOD1 and HEKΔSOD2 clones. Selected clones 1 and 2 with mutated Cat gene show no expression of Cat and correct expression of the other genes (SOD1, SOD2 and GAPDH reference gene. Selected clones 3 and 4 with mutated SOD2 gene show no expression of SOD2 gene and correct expression of other genes (SOD1, Cat and GAPDH reference gene). Clones 5 and 6 with mutated SOD1 gene show expression of all studied genes: SOD1, SOD2, Cat and GAPDH.
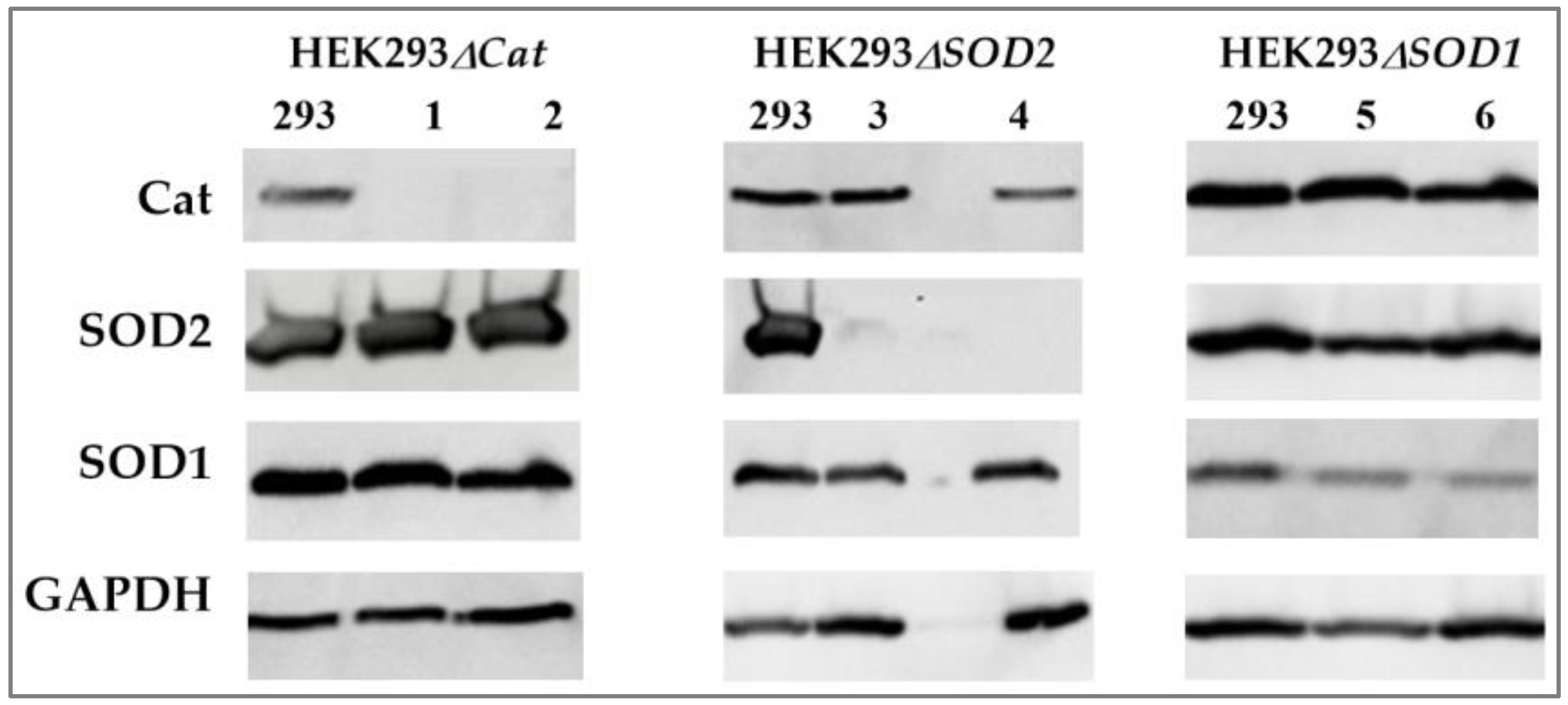
Figure 2.
Evaluation of the viability of yeast grown under oxidative stress by H2O2, results of the spotting test. The red line indicates the H2O2 concentration at which the yeast retains >50% viability.
Figure 2.
Evaluation of the viability of yeast grown under oxidative stress by H2O2, results of the spotting test. The red line indicates the H2O2 concentration at which the yeast retains >50% viability.
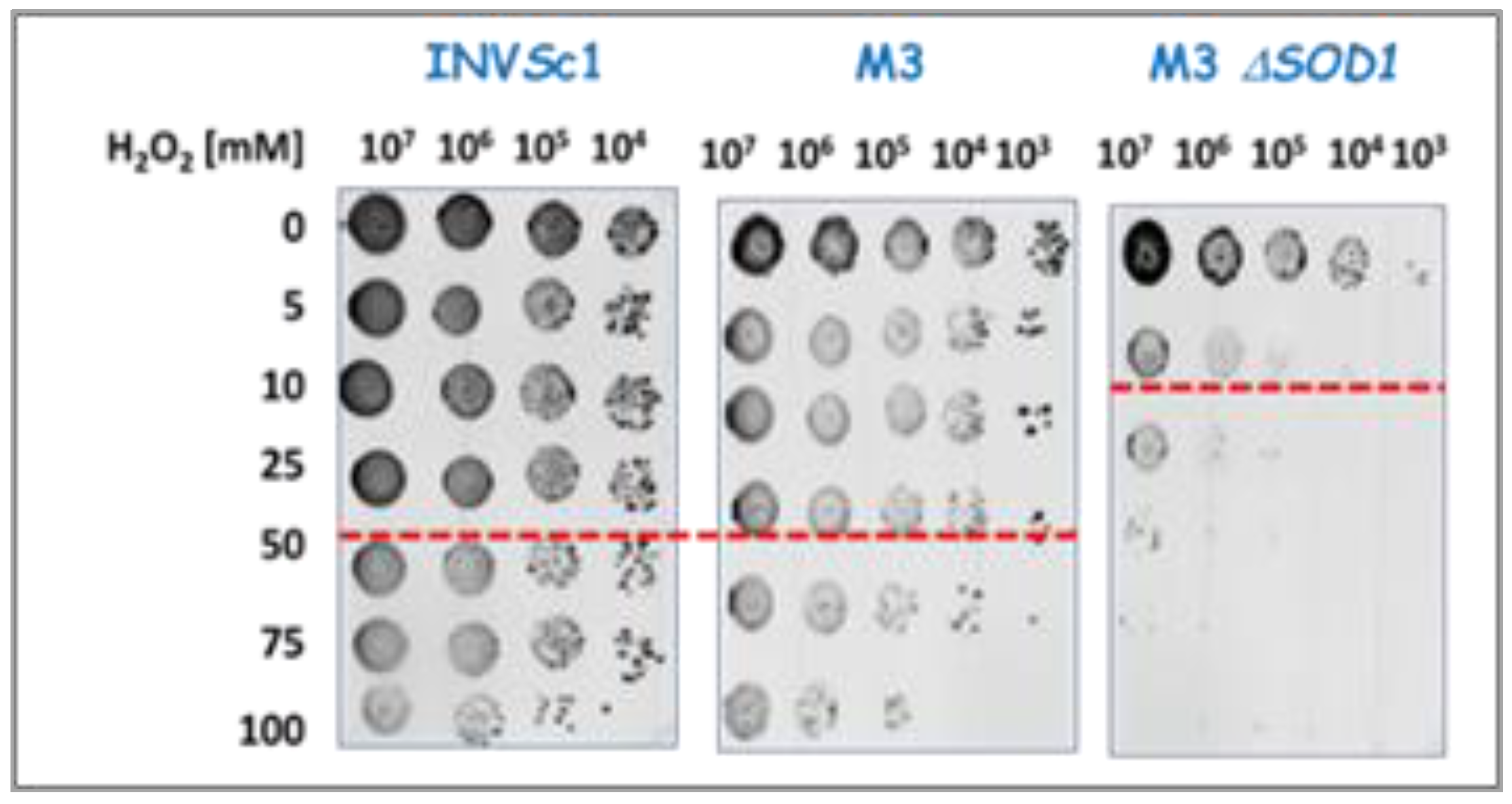
Figure 3.
Determination of the ROS content in yeast cells after incubation with H2O2 or NaAsO2. M3 sod1 (-) indicates the M3Δsod1 strain. The red and blue rectangles indicate the ROS content that forms under the influence of oxidizing agents at previously determined "safe concentrations” for the yeast strains M3 and M3Δsod1 respectively.
Figure 3.
Determination of the ROS content in yeast cells after incubation with H2O2 or NaAsO2. M3 sod1 (-) indicates the M3Δsod1 strain. The red and blue rectangles indicate the ROS content that forms under the influence of oxidizing agents at previously determined "safe concentrations” for the yeast strains M3 and M3Δsod1 respectively.
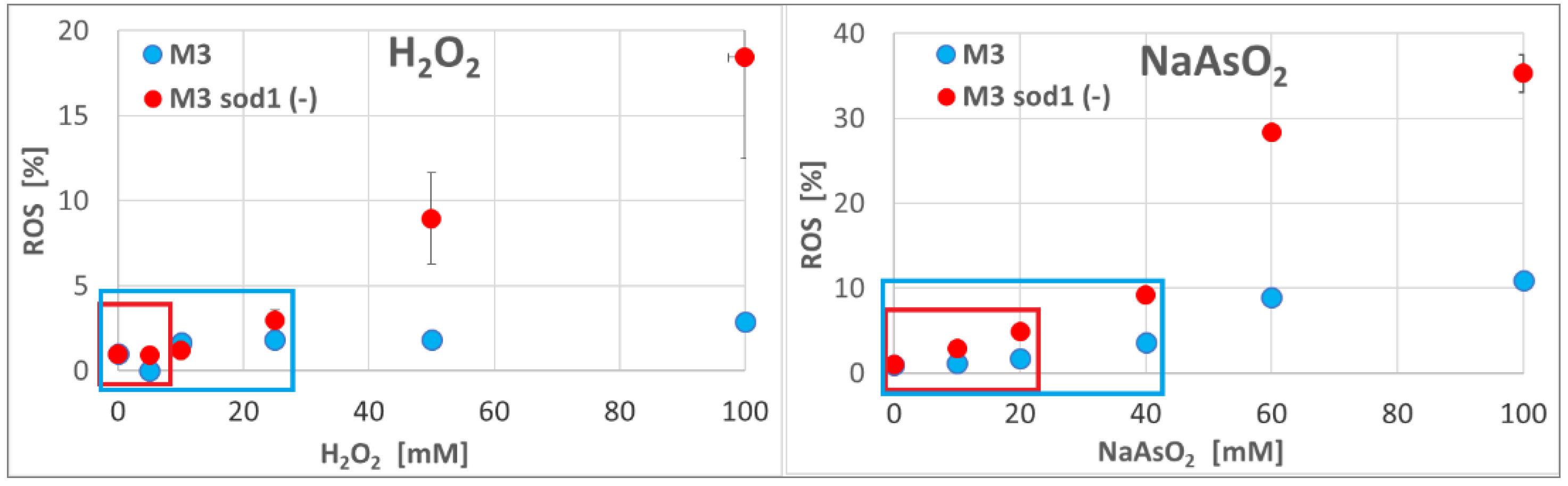
Figure 4.
Monitoring the reduction of mcm5S2U-tRNA in H2O2-treated yeast using the γ-toxin assay. (A) General overview of the assay, (B) Northern blot analysis of the tRNA hydrolysis products, (C) qRT-PCR analysis of the tRNA hydrolysis products, tRNA-specific primers recognize the full-length mcm5-tRNA.
Figure 4.
Monitoring the reduction of mcm5S2U-tRNA in H2O2-treated yeast using the γ-toxin assay. (A) General overview of the assay, (B) Northern blot analysis of the tRNA hydrolysis products, (C) qRT-PCR analysis of the tRNA hydrolysis products, tRNA-specific primers recognize the full-length mcm5-tRNA.
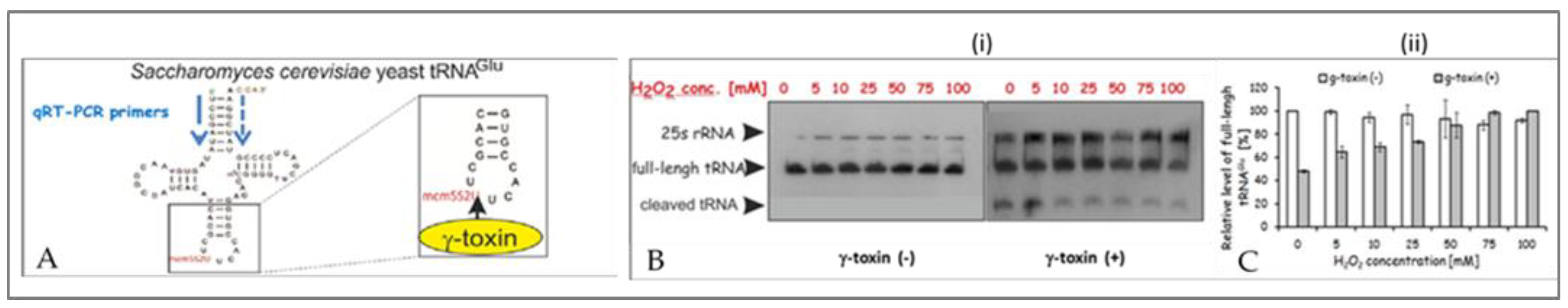
Figure 5.
Summary of the results of the MTT assay for human cancer cells cultured in the presence of H2O2.
Figure 5.
Summary of the results of the MTT assay for human cancer cells cultured in the presence of H2O2.
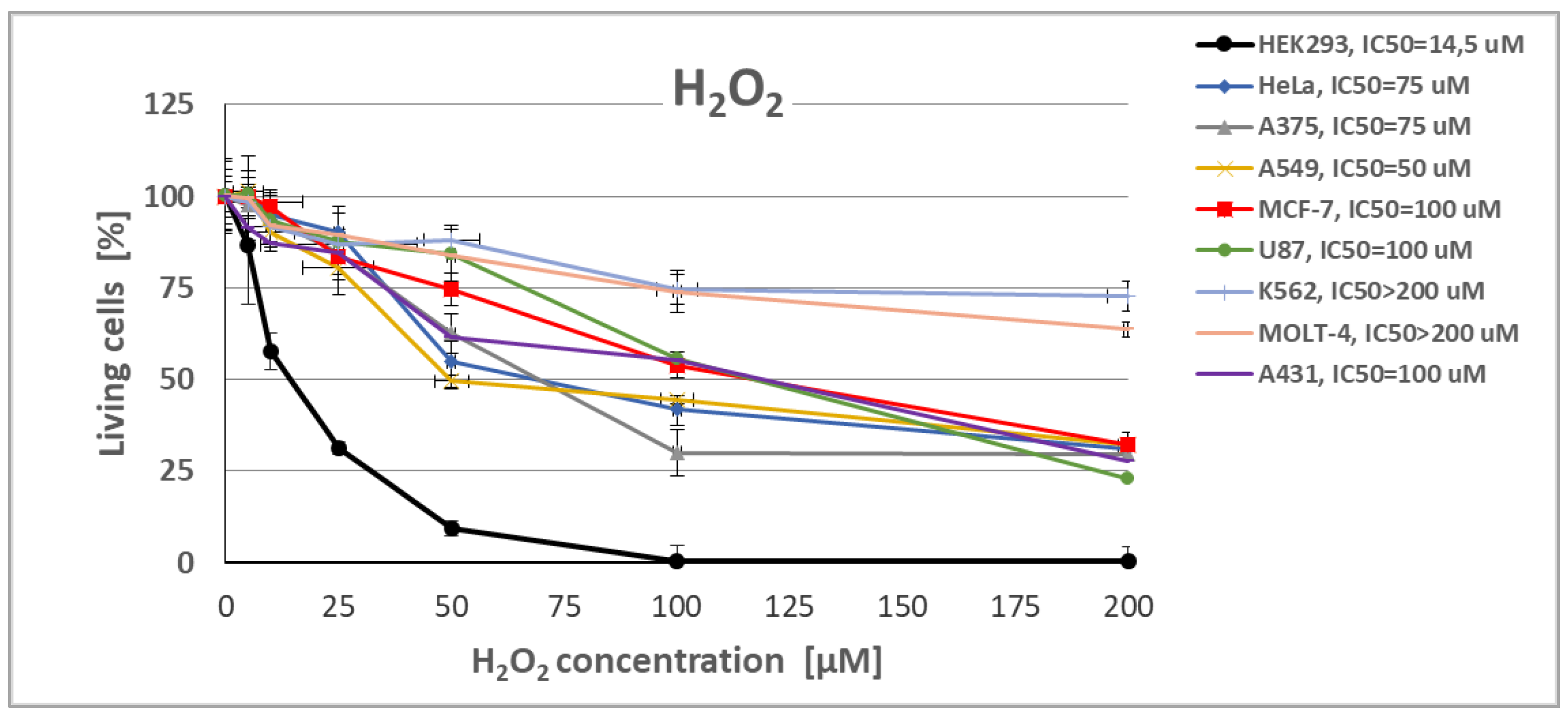
Figure 6.
LC-MS/MS analysis, identification of R5S2U desulfuration products in tRNAGlu from Saccharomyces cerevisiae M3 and M3Δsod1 yeast exposed to H2O2 during culture. The LC-MS/MS experiments were repeated independently three times.
Figure 6.
LC-MS/MS analysis, identification of R5S2U desulfuration products in tRNAGlu from Saccharomyces cerevisiae M3 and M3Δsod1 yeast exposed to H2O2 during culture. The LC-MS/MS experiments were repeated independently three times.
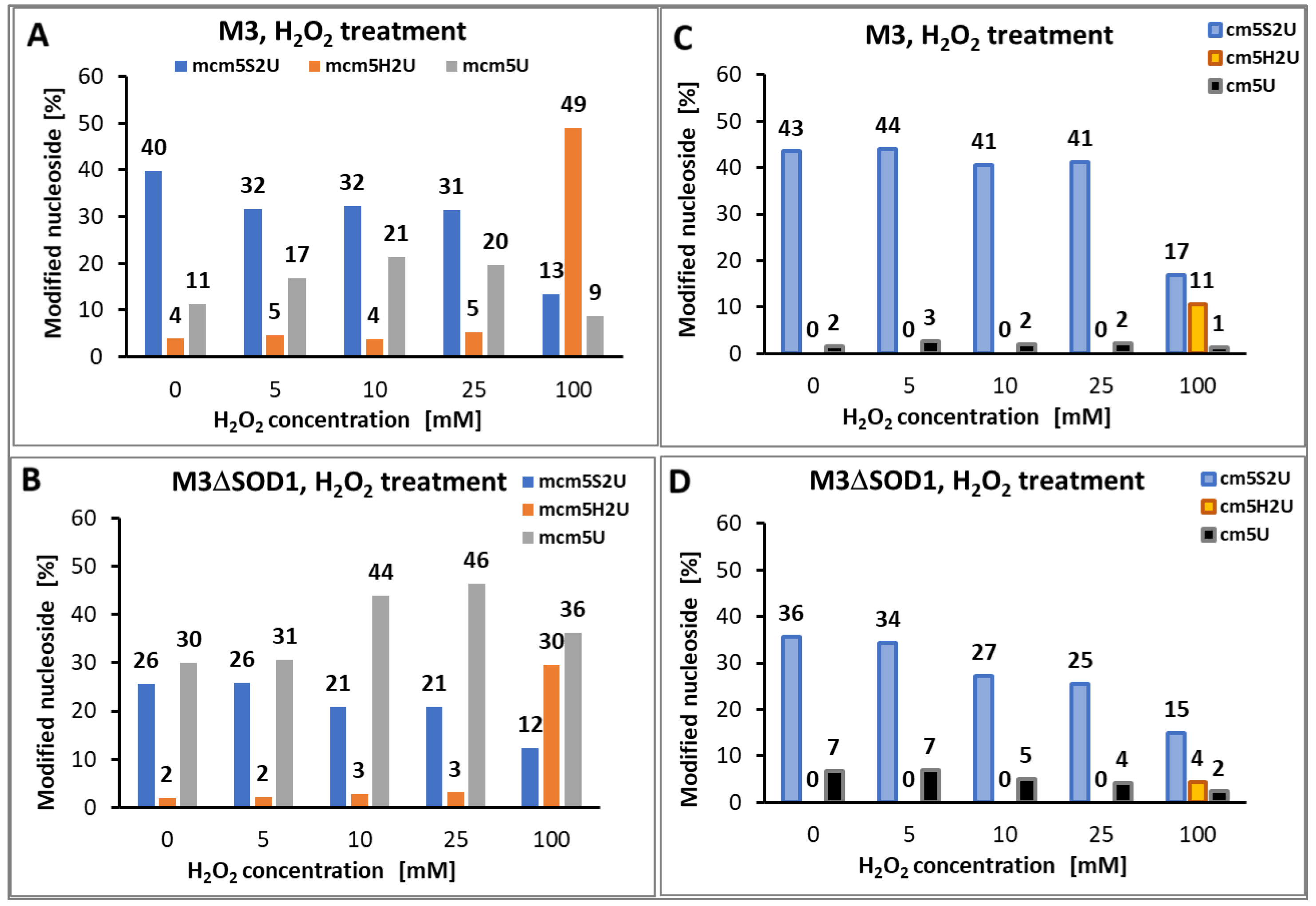
Figure 7.
LC-MS/MS analysis of the modified nucleosides in the low-weight RNA fraction (< 200 nt) in the cancer cells that were not treated with oxidizing reagents during culture. (A) mcm5S2U derivatives, (B) cm5S2U derivatives.
Figure 7.
LC-MS/MS analysis of the modified nucleosides in the low-weight RNA fraction (< 200 nt) in the cancer cells that were not treated with oxidizing reagents during culture. (A) mcm5S2U derivatives, (B) cm5S2U derivatives.
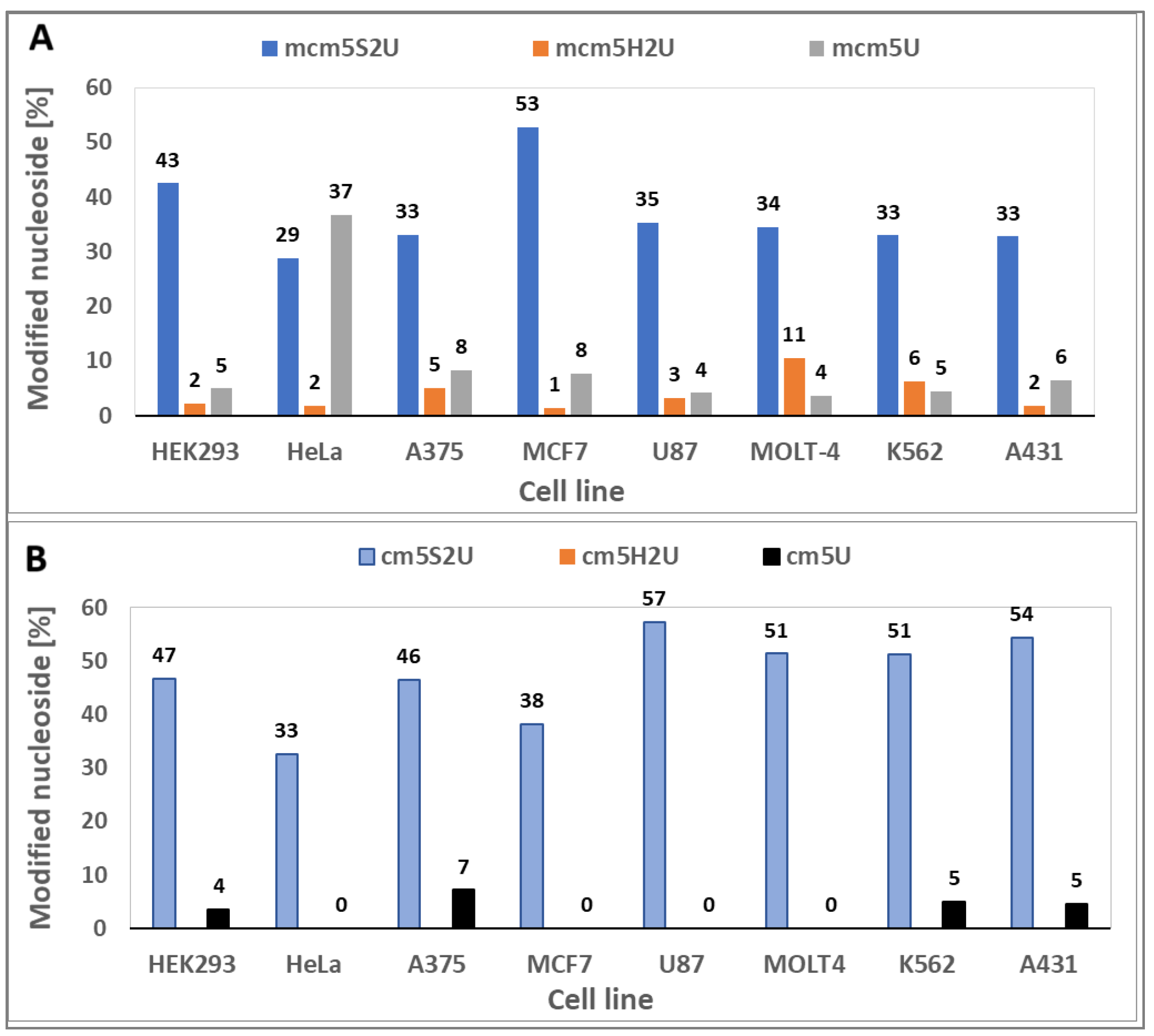
Figure 8.
LC-MS/MS analysis of the modified nucleosides in the low-weight RNA fraction (< 200 nt) in the cancer cells exposed to various oxidants (H2O2, NaAsO2, NaClO, (at concentrations below IC50) during culture (A) mcm5S2U derivatives, (B) cm5S2U derivatives.
Figure 8.
LC-MS/MS analysis of the modified nucleosides in the low-weight RNA fraction (< 200 nt) in the cancer cells exposed to various oxidants (H2O2, NaAsO2, NaClO, (at concentrations below IC50) during culture (A) mcm5S2U derivatives, (B) cm5S2U derivatives.
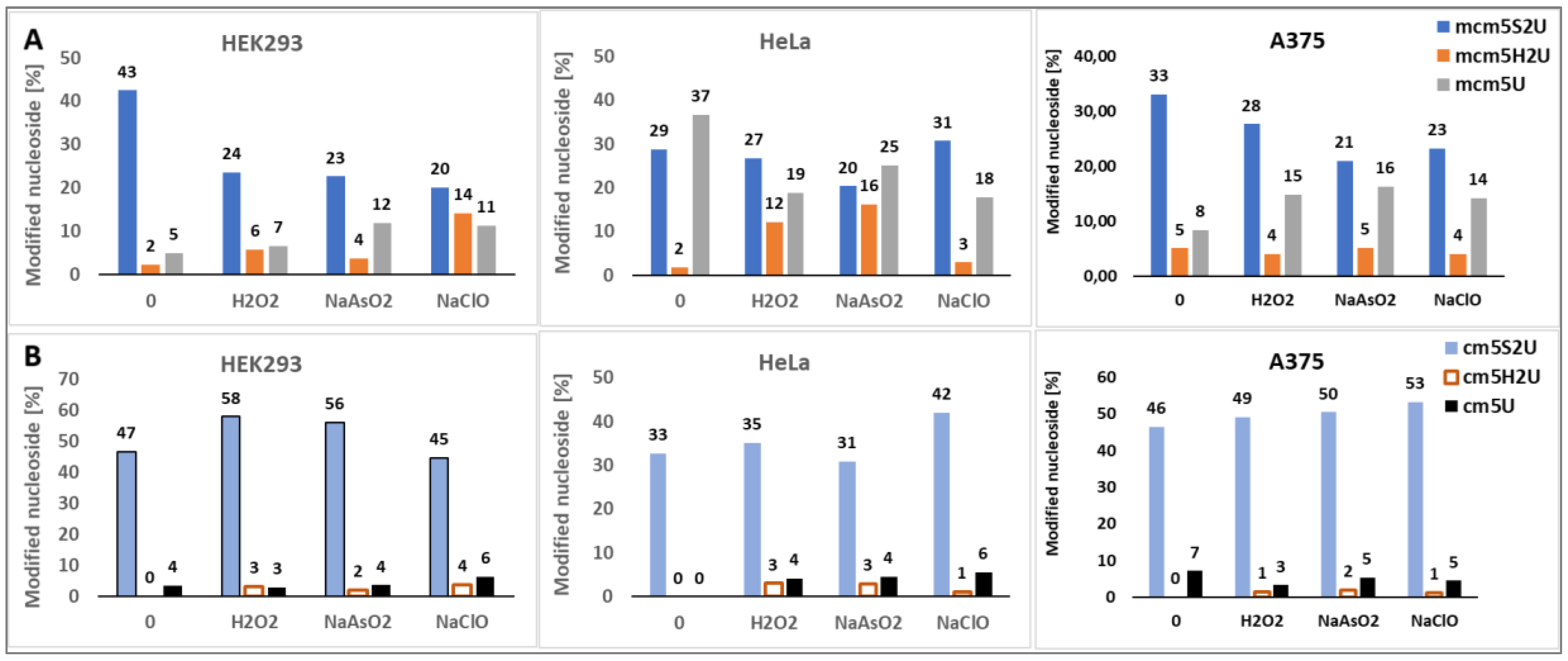
Figure 9.
LC-MS/MS analysis of the modified nucleosides in the low-weight RNA fraction (< 200 nt) in the HEK293 unmodified parent cells and HEK293 variant cells with damaged Cat or SOD2 gene. (A) HEK293, (B) HEK293ΔCat (3-1E1) clone, (C) HEK293ΔCat (2-2F7) clone, (D) HEK293ΔSOD2 (3-1D8) clone, (E) HEK293ΔSOD2 (1-1C6) clone, (F) HEK293ΔSOD2 (2-1H12) clone. Cells were exposed to H2O2 (0-50 µM) during growth.
Figure 9.
LC-MS/MS analysis of the modified nucleosides in the low-weight RNA fraction (< 200 nt) in the HEK293 unmodified parent cells and HEK293 variant cells with damaged Cat or SOD2 gene. (A) HEK293, (B) HEK293ΔCat (3-1E1) clone, (C) HEK293ΔCat (2-2F7) clone, (D) HEK293ΔSOD2 (3-1D8) clone, (E) HEK293ΔSOD2 (1-1C6) clone, (F) HEK293ΔSOD2 (2-1H12) clone. Cells were exposed to H2O2 (0-50 µM) during growth.
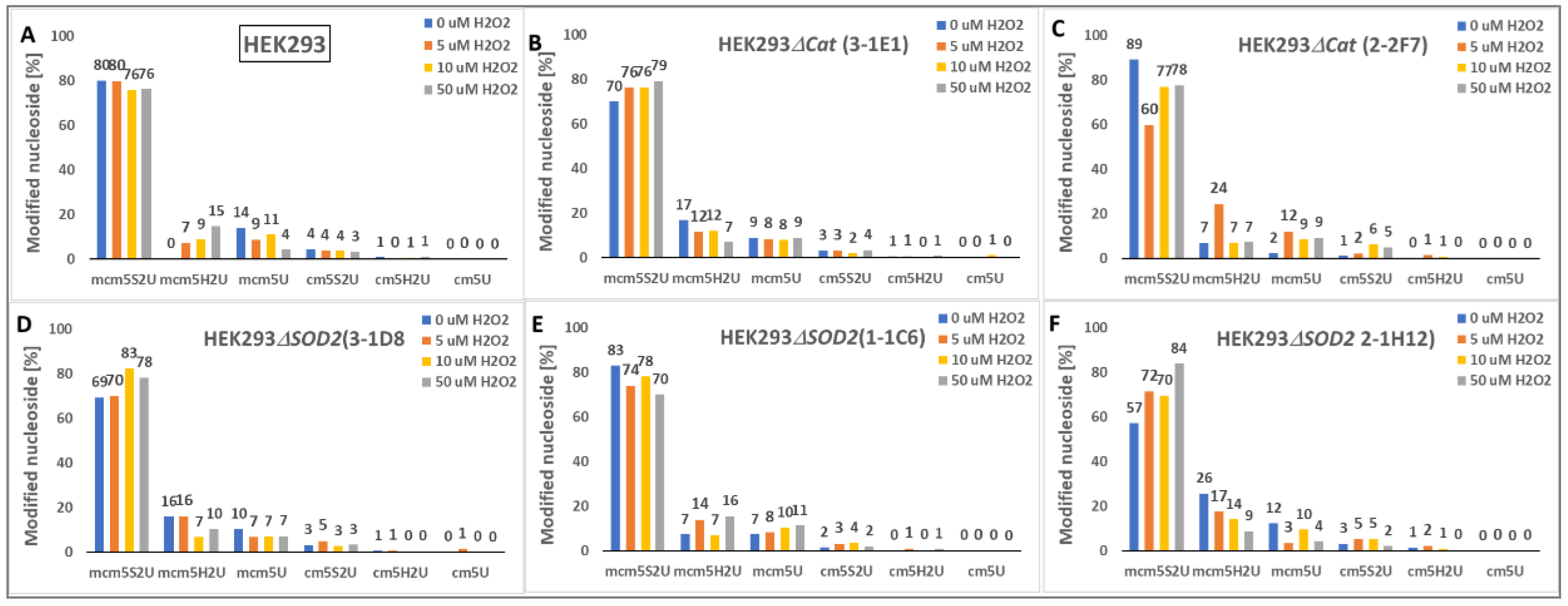

Table 1.
Evaluation of "safe concentrations" of oxidant reagents at which the viability of the yeast was >50%. Assessed on the basis of spotting test and the colony forming unit (CFU) assays.
Table 1.
Evaluation of "safe concentrations" of oxidant reagents at which the viability of the yeast was >50%. Assessed on the basis of spotting test and the colony forming unit (CFU) assays.
| Yeast strain | H2O2 [mM] |
NaAsO2 [mM] |
NaClO [mM] |
|---|---|---|---|
| INVSc1 | 50 | 100 | 10 |
| M3 | 25 | 40 | 7 |
| M3Δsod1 | 5 | 20 | 7 |
Table 2.
IC50 values determined with the MTT assay for human cancer cells cultured in the presence of oxidative reagents.
Table 2.
IC50 values determined with the MTT assay for human cancer cells cultured in the presence of oxidative reagents.
| Cell lines | H2O2 IC50 [µM] |
NaAsO2 IC50 [µM] |
NaClO IC50 [µM] |
|---|---|---|---|
| HEK293 | 14,5 | 200 | 100 |
| HEK293ΔSOD2 | 10 | nd | nd |
| HEK293ΔCat | 10 | nd | nd |
| HeLa | 50 | >200 | >100 |
| A375 | 70 | >200 | >100 |
| A549 | 50 | >200 | >200 |
| MCF-7 | 100 | 200 | 100 |
| U-87 MG | 100 | 200 | 100 |
| K562 | >200 | >200 | >100 |
| MOLT-4 | >200 | >200 | >100 |
| A431 | 100 | 200 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



