Submitted:
22 October 2024
Posted:
24 October 2024
You are already at the latest version
Abstract
This review introduces the development and applications of novel plant growth promotor ‘SOMRE’ and animal growth promoter ‘VED’ to increase food production, stop world hunger, greening deserts, stop global warming, and restore the green ‘Earth’. A concrete method to use ‘SOMRE’ and ‘VED’ as ‘the earth medicine’ is proposed. Part 1 describes ‘SOMRE’ helping the growth of vegetables, grains, flowers, herbs, endangered and native plants in the Gobi Desert. It increased plant-based food and achieved greening a part of the Gobi Desert. Part 2 explains ‘VED’, increasing the production of cashmere wool in goats, dramatically improving their reproductivity and their meat production. In humans, it demonstrated to eliminate the itchiness, skin spots, and acne scars, improved wound healing and ED, etc. It has the potential to be used as a treatment for dementia and osteoporosis. Part 3 is the experimental part. Following the described procedures, readers can get hold of ‘SOMRE’, ‘VED’ and related compounds which are a treasure trove of pharmacologically active substances and new ideas.
Keywords:
Medicine for the earth
; SOMRE
; VED
; Plant food
; animal food
; green desert
; α2-blocker
; pharmacologically active substances
; restore the green Earth
1. Part 1: Introduction
In the near future, the total population of the earth will over 8 billion. Especially, in India and Africa, food production and supply are stagnant due to various factors, and starvation deaths occur one after another. A United Nations report released in July 2022 predicted that nearly 670 million people would face hunger in 2030. Hunger-stricken regions are further exacerbated by human causes such as climate change, wars, civil strife, population increase and imbalanced logistics disruptions. Food loss is extremely serious in developed countries. Triggered by the war in Ukraine, food security issues have surfaced, with food producing countries hoarding food and restricting exports to food needing countries because of their own priority. This is an important issue for the present Japan, which has a low food self-sufficiency rate. In Japan the expansion of arable land is limited. However, we should not rely on crickets or genetically engineered food substitutes until we can guarantee their safety. We determined to develop safe technologies that increase food production by utilizing plants, animals, and seafood inherited from our ancestors as they are. And we have aimed new technologies with safe low-molecular-weight organic compounds that will double the amount of food production.
The total amount of water on Earth has not changed since prehistoric times. Ironically, human ingenuity/intellectual activity is destroying and transforming nature. Extremely uneven distribution of water, depletion of groundwater, floods, droughts, heavy snowfall, climate change due to desertification of land and sea, reduction of arable land, uneven westerly winds and ocean currents have been caused and changed from the original nature. Furthermore, they create and spread harmful pesticides and chemicals that had not existed in the natural world. Human beings are destroying the environment in which they live by devastating the lush land and sea with their own hands. It is slowly killing the ‘Earth’, like cancer cells trying to live and eventually killing their hosts. In order to save the ‘Earth’, we need to stop desertification and return the world's deserts to green areas.
The challenge is to stop climate change by greening land and sea deserts, restoring lush sea beds, forests and grasslands. Restore clean soil, water, and air to increase food production. The authors consider these difficult problems can be solved by developing new safe and harmless technologies that allow plants to perform their functions as they are. We believe there is no need to resort to GMOs, artificial meat, or insect diets. Food can be broadly classified into vegetable, animal, and marine foods, and there is a demand for the development of new technologies that can be applied to increase food production for all of them.
In his childhood, the author experienced food shortage in Japan after the World War 2, and also saw many starvation deaths with his own child eyes. With a dream to prevent this tragedy from happening in Japan, India, Africa, and the rest of the world, he chose a laboratory, researching plant growth hormones, during his university and graduate school days. In the laboratory, the structure determination study of the diterpene, enmein (1) contained in Isodon trichocarpus Kudo had been continued. The natural product was obtained in a relatively large amount. The author selected 1 as a starting material and studied its structural transformation into the gibberellin skeleton (Scheme 1), plant growth hormone, with the aim of synthesizing a new plant growth promoter. The research required new reactions which achieve the internuclear inert methyl group attacking method,[2] formation of six membered B–ring of gibberellin skeleton, and subsequent B–ring contraction to five membered–ring. These problems were extremely difficult to solve and required five and a half years of effort. Finally, he was able to establish a synthetic route to gibberellin A15 (2)[3] and also determine the absolute configuration of 1. It was also found that the plant growth–promoting action of total synthetic (dl)-gibberellin A15 achieved by Nagata et al. [4] is half that [5] of our natural 2.
In spite of the success, the author could not satisfy [6] because a substance with a complex chemical structure such as gibberellin is inevitably expensive due to the long synthetic process of 22 steps and the low overall yield. It was clear that the method was not suitable for large-scale synthesis and used for the purpose of increasing food production to save the world from hunger.
Since then, he had noticed indole-3-acetic acid (IAA) which was well known plant growth hormone with simple structure and studied IAA derivatives.[7] He could produce various new IAA derivatives but he could not satisfy them from the point of plant-, animal-, and marine-derived food production-enhancing agents that meet the conditions of being highly safe, having a simple synthetic route, using versatile materials, and being able to mass-synthesize.
In the process, he focused on tryptophan, essential amino acid. Tryptophan is a heterocyclic compound with a unique indole skeleton. Many metabolites and derivatives have been isolated and structurally determined as alkaloids and natural products, contributing to the world as bioactive substances and pharmaceuticals. Therefore, the author became interested in the metabolic process of tryptophan and IAA in the human/plant body and came up to the imaginary 1-hydroxyindole hypothesis (Scheme 2).[6]
Those around us considered the author's own hypothesis to be ridiculous. At that time, only electrophilic substitution reactions were known in indole chemistry.[8] While the author's hypothesis is that tryptophan is oxidized in the body to form 1-hydroxy- (A) or 1-hydroperoxytryptophan derivatives (B), as shown in Scheme 2.[6] He then imagined that the hydroxy group at the 1-position would be converted to a leaving group appropriately, resulting in an unprecedented nucleophilic substitution reaction.[6,8] By applying this hypothesis to the metabolic process of tryptophan, he was able to imagine a series of fantasy metabolites[6] that could be produced in plants and animals. He had a hunch that there would be a ‘medicine’ candidate [6,9,10,11,12] among these unknown compounds, for concrete and actionable ‘global solutions’ to the food crisis, end hunger, climate change. [6,9,10,11,12]
The author was worried whether the 1-hydroxyindole compound, a completely imaginary compound that has never been isolated as a natural product, could actually exist in the real world. It was a challenge with an uncertain future. In fact, it took about 40 years of research to overcome the difficult problems of realizing a general synthetic method for 1-hydroxyindoles and opened the door to unprecedented 1-hydroxyindole chemistry. Fortunately, we were able to develop ‘SOMRE #1’ (details in section 8) and ‘VED #1’ (details in section 18) as shown in Figure 1, [6,9,10,11,12] which are safe low-molecular-weight organic compounds based on 1-hydroxyindole chemistry.
‘SOMRE #1’ is an indole derivative and is used as a dilute aqueous solution of 1 ppm to 1 ppb. By simply soaking the seeds or roots of plants, vegetables, grains, trees, in the aqueous solution for a certain period of time (usually about one hour), the length and thickness of the roots and stems elongate at least 1.5—3 fold. It can also be sprayed on the leaves. In the basic experiments in Japan, we were able to increase the crop yield of almost all vegetables by at least 1.3 time. [6,9,10,11,12]
In 2008, in the Gobi Desert in China, we proposed a method of immersing the seeds of desert indigenous plants in an aqueous solution of ‘SOMRE #1’ and then spraying them from an airplane over the desert surface to make it green all at once.[13] With the cooperation of the NPO in Kanazawa, ‘The Society for Wrapping the World's Deserts in Greenery’,[14] we repeated preliminary experiments over four years[14] and obtained results that exceeded the expectations[13,14,15] of Chinese researchers. With the consent, permission, and cooperation of the local Chinese government who witnessed these results, we implemented a method of aerial spraying of ‘SOMRE-treated seeds’ from an airplane over the Gobi Desert,[16] during dry season of 2010, resulting in the successful partial greening of the desert.
On the other hand, in India, with the cooperation of Mr. Awano and Dr. Mishra's laboratory at KIIT university, we have achieved increased production of rice and vegetables. [17,18] The results were published in an international journal published in India.[19]
In addition, we introduced ‘SOMRE’ and ‘VED’ as new technologies to young master's students participating in the Abe Initiative,[20] who will lead the future of Africa. Using these technologies, we are proposing and collaborating to start businesses to alleviate hunger and improve the environment in their home countries and greening Sahara Desert [20,21] by increasing plant and animal food production.[21] On the other hand, we demonstrated that ‘SOMRE’ is effective in growing and preserving ‘Himekomatsu’ (Japanese pine),[22] one of the endangered plants. In addition, in order to protect the endangered Japanese traditional technique of lacquer culture, we could develop efficient lacquer seedling production technology using ‘SOMRE’ and made a presentation at the Japan Society for Bioscience, Biotechnology and Agrochemistry in March 2022.[23]
On December 4, 2017, we received the 2017 Environment Minister's Award for Global Warming Prevention Activities for our global contribution activities through the greening of the Gobi Desert in China using ‘SOMRE’ and volunteer activities to increase food production in India. [17,18,19]
As stated above, the author could build the possibility of increasing the production and yield of major grains such as rice, soybeans, wheat, potatoes, corn and more using ‘SOMRE’. [6,9,10,11,12] There would be a prospect of a sufficient supply of plant food and feed for animals, pets, and livestock.
In parallel with the above-mentioned plant food research, we have also focused our efforts on developing production enhancers for animal foods and discovered ‘VED’. [6,9,10,11,12] ‘VED’ is also an indole derivative. It is α2 receptor blockade and peripheral vasodilator. It improves blood circulation, [24,25] and enhances growth and reproductive ability of animals. [26,27] A four-year demonstration experiment in the Gobi Desert in China using cashmere goats proved that goats can produce many offsprings and increase the production of animal food.[27] In addition, the increase in cashmere wool production was outstanding, [25,28] bringing high incomes to the nomads. In humans, ‘VED’ is a product targeted at middle–aged and elderly people[29,30] that improves forgetfulness (dementia), myocardial infarction/cerebral infarction,[31] anti–aging, sexual dysfunction (ED) improvement,[32] Alzheimer's disease, Parkinson's disease.[33,34] Hair growth,[35] inhibition of platelet aggregation,[36] and promotion of metabolism have demonstrated the potential to be a comprehensive QOL (quality of life) improving medicine.[37,38]
Specifically, through 12 activities in the Gobi Desert, we have achieved our goal of reducing the number of cashmere goats, which is one of the causes of desertification. [25,39] We raised goats employing pasture and feed harvested at ‘SOMRE’, and succeeded in increasing cashmere wool production [39] and breeding experiments. [25,26,27] In addition, we started to increase the production of animal foods (cows, pigs, goats). On the other hand, in the examination of safety of ‘VED’ with fish,[40] we found it is not toxic.[40] They grew large and healthy. In addition, with the aim of improving human QOL in an aging society, we created cosmetics containing ‘VED’ and saved many patients from the suffering from itch [41] of atopic dermatitis. It has also been proven to be useful for the disappearance of skin spots [37] due to aging. Furthermore, we were able to discover a tribromo-melatonin and tribromo-tryptophan derivative (detail in section 20-11) that can be easily obtained as a promising therapeutic medicine candidate [42] for osteoporosis [43] which afflicts the elderly, through the development of 1-hydroxyindole chemistry.
In this review, we will introduce actual examples of ‘SOMRE’ and ‘VED’ that will help increase food production to eliminate hunger from the world. In addition, we will introduce the success of partial greening the Gobi Desert based on our proposal of a concrete and practical way to green the desert. By applying this method to the world's deserts, we can restore green ‘Earth’ and stop climate change. We will also explain how VED helps improve a person's QOL, and how it works in conjunction with ‘SOMRE’ to become a ‘medicine for the earth’.
2. Greenhouse Gases and Climate Change. What Causes Global Warming?
Humans have challenged nature (including Antarctic and Arctic), opened waterways, constructed dams, and eroded mountains, and have been happy with the joy of winning. But unexpected changes in ocean currents, meandering westerly winds, and other unforeseen changes on a global scale have occurred, leading to climate change and global warming. Now, the earth is suffering from a fever. The symptoms of climate change have intensified, with desertification of land and sea, loss of forests, loss of glaciers, droughts, floods, crop failures, changes and declines in marine and fisheries resources, global food crises, starvation, and extinction of species. There are many urgent problems that need to be solved for organisms living on the earth.
The First Working Group of the United Nations Intergovernmental Panel on Climate Change [44] has concluded that CO2, a greenhouse gas, is the cause and culprit. It has become a common understanding in the world. Based on the idea that if we remove CO2, global fevers should be alleviated and healed, intellectuals from around the world gathered together and held numerous COP meetings,[44] transcending political and economic systems to reduce CO2. We are narrowing down our wisdom. Japan has set a goal of reducing greenhouse gas (CO2) emissions by 46% by 2030,[45] but developing the technology to achieve this goal is an extremely difficult task.
We should notice that besides the alleged culprit, CO2, there are many worse greenhouse gases. For example, methane (CH4), which is the main component of natural gas and is generated by the action of microorganisms from organic matter in the soil of lakes and marshes, waste matter, generated from methane hydrate in frozen soil and deep sea. Nitrogen oxide (N2O) is another example, generated by burning fuel. The United Nations subcommittee has identified CO2 as the culprit and enemy, and has set completely irrelevant goals and policies to control its generation, leading global political, economic, and environmental activities in the wrong direction.
The reason is obvious if we pay attention to the global warming coefficient [44] that causes the greenhouse effect. That is, according to the IPCC Fourth Report,[44] when the global warming potential of CO2 is 1.0, that of CH4 is 25 times, and that of N2O is as high as 298 times. Furthermore, that of hydrofluorocarbons (HFCs), which did not exist before the Industrial Revolution, is 1,430 times that of CO2. Sulfur hexafluoride (SF6), produced for electrical insulation, is 22,800 times. Perfluorocarbons (PFCs) and nitrogen trifluoride (NF3), generated from semiconductor manufacturing processes, is 7,390 and 17,200 times, respectively. Even if we reduce CO2 emissions, if we do not reduce or eliminate these powerful greenhouse gases, we will not be able to cure the disease of the earth.
Unfortunately, HFCs, SF6, PFCs, NF3, PFOS, PFAS, etc. are extremely stable chemicals and are difficult to decompose by chemical reactions. Even if it is decomposed by applying high-energy radiation, harmful fluorine atoms are generated. We think the only way is to react with Ca to convert it into safe minerals such as CaF2 (fluorite). But who could devise an economical technology to recover and concentrate these potent greenhouse gases, which have diffused into the atmosphere in dilute concentrations, into gaseous or liquid feedstocks that can be subjected to chemical reactions? These essential technologies are not yet available to mankind.
CO2 is unique among greenhouse gases. Even if it exists in the atmosphere at a dilute concentration of about 420 ppm (as of 2022), it can be eliminated without the need for collection, accumulation, or concentration. Plants can employ CO2 as useful resources as they are and contribute to the maintenance of life on earth. In other words, plants form a green world environment in forests and in sea beds, use sunlight as an energy source, react CO2 with water, and produce forests, wood, various organic compounds that can be used as medicines, proteins, carbohydrates. They convert CO2 into lipids, proteins, sugars, create the bodies of our animals, and also convert into various foods. In the chemical reaction process, oxygen gas essential for respiration is also produced, and oxygen that is decreasing by 4 ppm per year is produced and replenished.
As long as plants exist, CO2 is not harmful but rather a beneficial substance. CH4 can also be converted to CO2 by burning, so it can be positioned as a food resource as well, but there remains the problem of how to concentrate and burn the dilute CH4 gas released into the atmosphere. Massive emissions of non-CO2 greenhouse gases are rapidly accelerating the anomaly of global warming. Is there no choice but to sit back and accept the worst-case scenario of global warming, in which almost all life forms die suddenly at the same time that the earth emits a high temperature?
We have a solution to take. Humans should immediately stop producing, using, and emitting greenhouse gases other than CO2, even if they give up convenience, comfort, and cultural life. Concentration of CO2 has increased by only 100 ppm in about 200 years since the industrial revolution due to human activity. Therefore, as a specific “cure for the earth” as mentioned above, it is only necessary to green the whole earth and ask plants, which are precious living organisms, to demonstrate their abilities in food production and environmental greening. It is the only solution to take. Even if we give up on human convenience, comfort, and cultural life, we should immediately stop producing, using, and emitting greenhouse gases other than CO2.
3. Medicine of the Earth: Cure for the World’s Food Crisis and Global Warming
Since the birth of mankind, the plants that created the global environment as “indigenous life forms” have been cut down, burned, and eradicated. Mankind transformed the lush Sahara area into the world's largest barren desert. Following a similar process, the world's second and fourth largest deserts, the Taklamakan and Gobi Deserts, were born. The Aral Sea is disappearing and turning into a desert due to mis-planned use of river water. Cultivating the earth's surface all over the world and sprinkling a large amount of groundwater on cultivated land using a center–pivot method has repeatedly caused depletion of groundwater and salt accumulation in the soil, giving birth to deserts and barren land. Currently, one-fourth of the earth's total land area is barren land or desert.
At the bottom of the sea, overexploitation of seaweed and seagrass and destruction of fishing reefs are repeated, promoting desertification of the sea and depriving fish and shellfish of their right to live. Like the screams of livestock in a slaughterhouse, the earth and the sea bed are filled with the death screams of living plants, losing the green clothes. The “naked earth” is crying.
The authors have developed ‘SOMRE’ and ‘VED’ as a one-of-a-kind medicines for curing the Earth. [6,9,10,11,12] The final forecast scenario will dramatically improve on time if we act on now. It is a technology that greens the vast land and sea desert and returns the green clothes to the “naked earth”. It is a technology that makes all life forms happy, including humans.
4. Expectation from Greening the Desert
Although it would take several decades, the effect would be immeasurable if the entire desert, which covers quarter of the earth's surface area, could be greened. [9,10,11,12] 1) First, plants in wide area absorb a large amount of CO2, so climate change will gradually stop. 2) Plants are almost the only living organisms that produce oxygen, which is essential for many living organisms including humans. The oxygen concentration in the earth's atmosphere also returns to its original state. Oxygen becomes ozone through a photoreaction in the stratosphere, and the ozone hole is repaired, restoring a bio-friendly environment. 3) The surface of the desert is covered with plants, and yellow sand from the Taklamakan and Gobi Deserts stops. 4) The surface of the earth will be greened, the temperature will drop, and clouds will be brought in, and rain will return to the desert. 5) Rain falls on high mountains in desert areas, snow returns to the mountain tops, and underground water revives. 6) Since the vast desert receives a huge amount of rain, torrential rains and sea level rise will stop, and no country will sink. 7) The desert will become cultivated land and become a base for food such as grains, vegetables and fruits. Various plant species grow and endangered species are revived. We will also be able to provide clothing and housing materials. 8) Restoration of animal habitats and restoration of biodiversity. However, these wonderful privileges are actually things that the earth has had since before the birth of mankind.
5. Mechanisms Behind Desertification: Ideas for Realizing Greening [11]
How could we make the desert green with plants? To get a hint, let's consider the mechanisms of desertification and greening. Each desert has its own personality. As a model of a general desert, the Gobi Desert in the Arashan League of Inner Mongolia in China is considered as an example. [10,11,12] In the Gobi Desert, groundwater exists about a hundred meter below the ground surface (Figure 2). [10,11,12]
Water oozes toward the ground surface and rises due to capillary action and then evaporation. Thus, humidity gradient is formed. On the surface, moisture content is 0%. The amount of moisture increases with depth, and a 100% moisture is present about hundred meter’s underground.
As a result, even if the surface temperature of the desert is as high as 50 °C or higher, we can feel moisture if we dig 30 to 50 cm below the ground surface. In fact, in the Gobi Desert, a stratum with a depth of between 30 and 50 cm contains about 1% (weight %) of moisture.
Desert plants that flourished in the past dropped their seeds to the surface (buried seeds) in order to leave offspring. Even in a natural environment, in Gobi Desert, with an annual poor rainfall of 100 to 200 mm or less, there is a rainy season of about three months. Buried seeds wait for the rainy season, and when the rain arrives, they come into contact with water and begin to germinate and start rooting all at once. [10,11,12]
If the root length can grow into a depth between 30 and 50 cm in the sand layer during the three months of the rainy season, they have a chance to survive due to the presence of trace amounts of water. Short-rooted plants with a root length below 30 cm or less would die in the following severe dry season because of the water shortage. Alternatively, cashmere goats raised by nomads will dig up the roots and eat them. Even if plants are lucky enough to survive, they will freeze to death in the next severe winter of –30 °C. Thus, many buried seeds die and disappear without producing offspring. In this way, desertification progresses without even a single plant left.
During the rainy season, if the seeds can elongate roots to a moist sand layer at a depth of 30–50 cm, the individual plant can survive. The long roots endure the dry season, resist feeding damage by goats, and do not freeze to death in cold winter. The next year in the spring, the roots that have grown sufficiently sprout and the individual continues to live. When the rainy season comes around again, the roots and trunks become longer and thicker, and the plants grow more. Finally, the frequency of rainfall would increase and desert would be covered with green plant. [10,11,12]
Based on the above consideration, we concluded that whether or not we can green the desert depends on whether we can develop a new root growth promoter that can grow roots two to three times longer (more than 30–50 cm) during the short rainy season than before.[11]
6. Philosophy for Greening Desert: Conception of ‘Phantom Substances’, ‘SOMRE’ and ‘VED’
Based on the mechanism of desertification considered in the previous section 5, the following two points are the key for the success of desert greening. [6,11] 1) Whether is it possible to create a cashmere hair thickening agent and breeding medicine that can maintain the nomadic life even if the number of cashmere goats is reduced? 2) Is it possible to create a new safe root/stem elongation agent that doubles or more the root/stem growth/elongation ability inherent in plants?
As for the first point: Although goats cause feeding damage, it is not possible to prohibit the breeding of cashmere goats, which are the source of income and means of living for nomads. If it were possible to develop a technology that would allow goats to grow twice as fast as before and produce more cashmere, even if they ate the same amount of pasture and fodder, it would also be possible to improve the reproductive capacity of goats, produce more young goats, and increase meat production.[25,26,27] If the technology could be developed, nomads will be able to earn the same or even more cash income even if they reduce the number of goats they raise. As a result, it becomes possible to reduce the number of grazing goats that cause feeding damage, and to stop desertification and protect grasslands. For this required technology we could develop ‘VED’, so please refer to section 18.
As for the second point: We have developed ‘SOMRE’, a plant root and stem elongator. By repeatedly using ‘SOMRE’ and ‘VED’ in the desert, the greening of the desert will progress and the nomads will be able to live happily. Furthermore, the rain that seeps into the ground accumulates over many years and becomes groundwater, and eventually the groundwater level rises. Thus, the desert will be finally covered with green.
In desert greening we believe that the following four views [10,11] are also important points to be considered.
3) We should not destroy the local natural vegetation; we do not bring in plants that grow in other countries because they are resistant to drought and heat. Even if greening is successful, the original environment and ecosystem of the land must not be destroyed.
4) We should rely only on the rain in the rainy season to plant trees in a natural way to revive and regenerate the old environment. An artificial irrigation method that pumps up groundwater should not be adopted. Deserts that have artificially depleted their groundwater are helpless for thousands of years until the groundwater is replenished. Excessive application of groundwater to the surface of desert causes salt accumulation. There are many examples of mankind's past failures in turning fertile land into barren land in this way. Collecting water from surrounding air powered by solar cell is the recommended technique.
5) We do not use polymeric water-retaining materials or water-absorbing agents. Even biodegradable macromolecules (polymers) become monomers and metabolites during the reaction process until they are degraded by soil bacteria and become CO2, producing acrylic acid, acetic acid, and formic acid, and so on. There is a high risk that the soil will become acidic and become barren, where plants cannot grow.
6) We do not use soil conditioners or extra chemicals. Constituents change the composition ratio of the soil. As a result, the microbial environment of the soil changes and cannot be returned to its original environment. In addition, abnormally proliferating pathogenic microorganisms will be mixed with or attached to yellow sand, and will rain down all over the world along with the above-mentioned chemical substances.
By the way, the author often encountered the phenomenon that after the seeds germinated, the roots did not develop well and the seeds gradually withered. From these experiences, we came to think that it is important to grow healthy roots first in order to grow plants. Dreaming of alleviating world hunger, greening deserts with poor rainfall, and covering the earth with green trees, the desert needs plant’s roots three times longer (more than 30–50 cm) than normal (20 cm).[12,13] At the same time, if there is a “phantom substance or technology” that can bring out the rooting power of buried seeds left by old plants and revive them, it will be possible to solve the food crisis and help endangered species. We thought that the technology would be “a medicine for the earth” that would revive life, ensure biodiversity, cover the entire planet with greenery, and stop global warming.
In fact, in a growth test from seeds of local indigenous plants in the dry Gobi Desert with a low annual rainfall of 100 to 250 mm, were carried out.[11] The control plants sprouted, while in the SOMRE-treated plants buds were hidden just below the surface of the hot ground. We have had many similar experiences in the initial growth. However, with the passage of time, the control plants withered due to the lack of water supply, whereas the SOMRE-treated seeds grew vigorously and the buds grew steadily, eventually surpassing the growth of the control plants and taking root. From these facts, it seemed that the SOMRE-treated plants patiently waited for the roots to grow, and only when the supply of water and minerals from the roots was established, they would shift to the growth stage of stems and leaves. In other words, the author was able to obtain confirmation that the germination and rooting processes were not concurrent.
7. Naming of ‘SOMRE’ and ‘VED’ [10,11]
The chemical substance name of our novel plant root/stem growth stimulant is 1H-2-bromoindole-3-carbaldehyde (Scheme 4, SOMRE #1, 12). Since the name is long, a simpler name was needed. It was named “ソムレ(甦群列)” in Japanese with the hope that plants that have lost their vigor in a severe water shortage environment will revive in groups and rows just by being sprayed. Luckily, the author's name in English is SOMEI, and the function is root elongation. Therefore, by taking the SOM of the inventor's name, taking the r and e from the root elongation that expresses biological activity, and combining them, it became SOMRE. And the Japanese and English pronunciations are the same. Later, we learned that the pronunciation of SOMRE in Chinese became “粗如来”, which means “medicine of the god”.
The chemical substance name for VED is Nb-nonanoyltryptamine (details in section 18). As a simple name, it was named based on its function. That is an α2-blocker, has a peripheral vasodilator, and is useful for erectile dysfunction (ED) treatment. We combined the initials of each word, vasodilator, erectile dysfunction, and named it VED.
8. Synthesis of a Group of ‘SOMRE’ Compounds [11]
8.1. Responsibility of Synthetic Chemists [6]
The author is a synthetic chemist and has been discussing what a synthetic chemist should be.[6] In other words, synthetic chemist should not simply target and synthesize the structurally and pharmacologically attractive natural products. They are the intellectual properties of the person who discovered and determined the chemical structure of them. We should be grateful to those who have presented us with targets worthy of synthesis. During his target synthetic process, he can create new reactions and new compounds related to the target.
In addition, he should always research whether the new compounds he has created has new physical properties, functions, or pharmacological effects. As time permits, he should continue to improve targeted natural product synthetic methods, and further develop short step, efficient, and economical synthetic method. His such efforts should be highly appreciated. For anyone who fails to make this effort will be condemning his own created new compound to death.[6] Suppose later that someone discovers new physical properties, functions, or pharmacological actions in the compound he created. At that time, he should be grateful, but should not claim priority [6] intellectual property rights over time. He should be ashamed of having been an executioner. Suppose that his novel compound obtained as a target compound or a synthetic intermediate is an excellent antidementia or osteoporosis medicine. It is unlikely that other synthetic chemists, who make a point of not imitating others, will make the compound again. Therefore, mankind will forever lose the chance to obtain the cure. The responsibility as the originator of new compounds is significant. However, in the field of synthesis of natural products with complex structural formulas, based on the development of novel reactions, the subject of total synthesis will continue to be a highly regarded art.
The author also proposed the absolute evaluation indicators [6] such as originality rate (OR), intellectual property factor (IPF), and application potential factor (APF) that can compare and evaluate the superiority and inferiority beyond history, instead of evaluating the results by the number of citations (citation index). If you are interested, please refer to our literature.[6] Keeping in mind the above claim and to stop the global desertification, the synthesis of SOMRE must be an original synthetic method, short process, and the yield of each process is good. In addition, a large amount of SOMRE should be synthesized at the lowest possible cost.
8.2. Creation of General Synthetic Method for 1-Hydroxyindole [11]
To achieve the claims mentioned in the section 8-1, the author came up with the idea of using indole compounds (C, Scheme 3) and 2,3-dihydroindole compounds (indolines, D), which are components of coal tar that are discarded in large quantities in the iron industry. Coke, which is necessary to reduce iron ore to produce iron metal, is produced by heating coal. During this process, coal tar is produced as a coke by-product. Therefore, C and D are ideal inexpensive raw materials.
Using them as starting materials, if it is possible to develop new uses as “a medicine for the earth” that will increase food production, save world hunger, and restore the green earth, it will open the way for effective utilization of coal resources.
In order to test the 1-hydroxyindole hypothesis,[6] we must create a method to synthesize 1-hydroxyindoles (E, Scheme 3), which has an unprecedented structural formula as a natural product.[46] After much effort, we succeeded in discovering the general synthetic method for imaginary 1-hydroxyindole compounds from indole (4) as shown in Scheme 4.[46] The first step is the reduction of indole with an appropriate reagent (Et3SiH or NaBH3CN) to 2,3-dihydroindole (indoline, 3). In the second step, 3 was oxidized with 30% hydrogen peroxide or urea hydrogen peroxide addition compound in the presence of such catalyst as sodium tungstate (Na2WO4・2H2O) or 2Na2O・P2O5・12WO4・18H2O, to give a quite unstable 1-hydroxyindole (5). Subsequent methylation successfully afforded stable 1-methoxyindole (6). So, anyone can apply this two-step method to any indole or bioactive indole compounds to create the corresponding unprecedented 1-hydroxy- and 1-methoxyindole derivatives. The group of 1-hydroxyindole compounds is a treasure trove of new biologically active compounds as shown in section 20.
8.3. Synthesis of SOMRE Compounds [11]
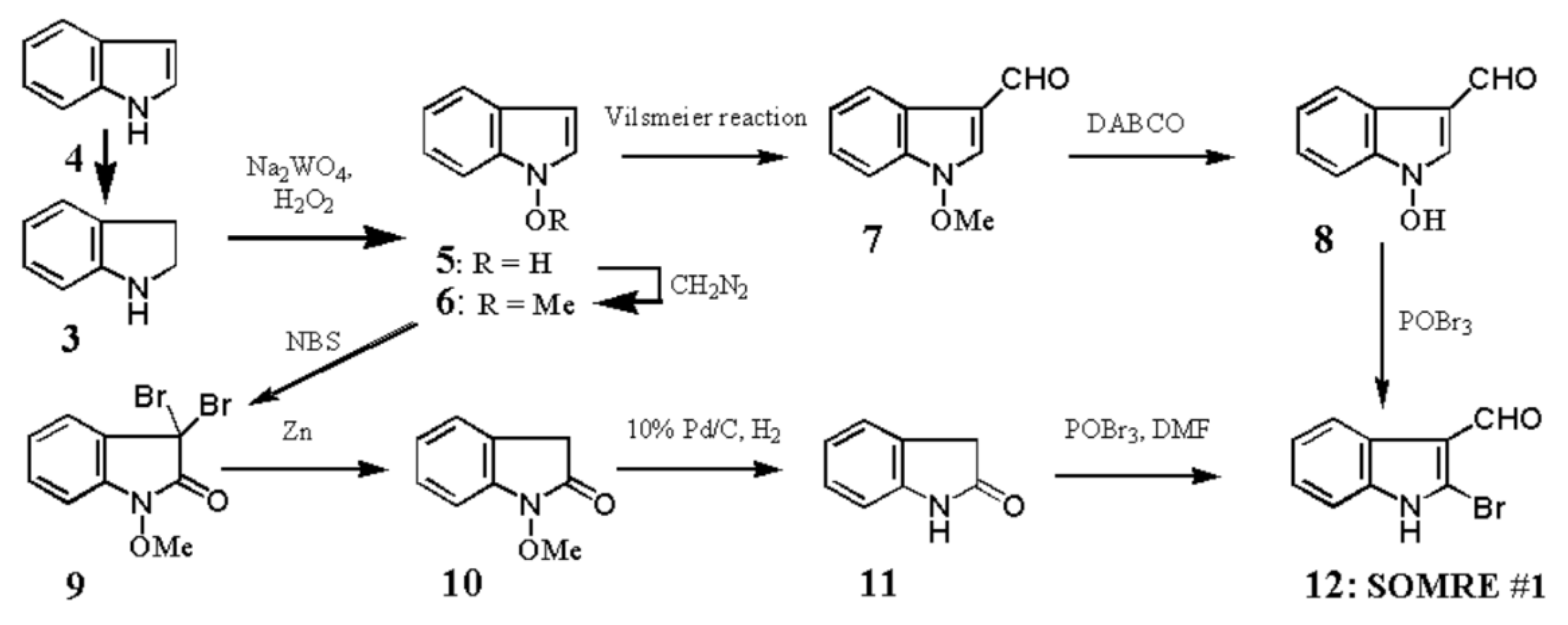
1-Hydroxyindoles (E) has an acidic hydroxy group. Therefore 1-hydroxy group can easily be methylated with CH2N2 or dimethyl sulfate/K2CO3 to give 1-methoxyindole (6). Either by transforming the 1-hydroxy group to 1-methoxygroup or introduction of electron withdrawing group at the 3-position, the stability of the resulted compounds is highly elevated. Based on this stability, few derivatives had been isolated as natural product. Starting from 4, we succeeded in creating SOMRE #1 (12) by the two routes shown in Scheme 4.[11]
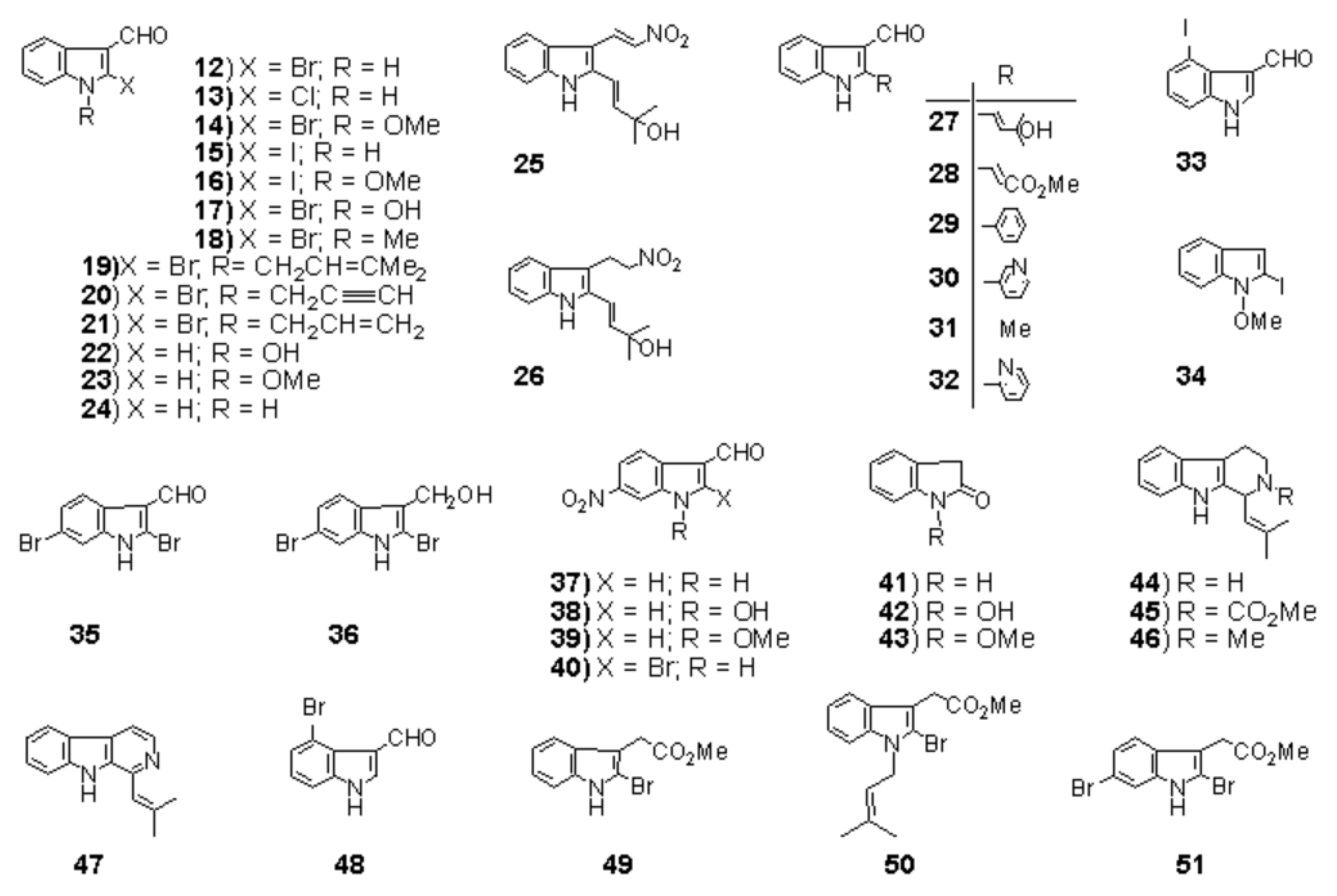
The first step is the Vilsmeier reaction of 6 to produce 91% yield of 1-methoxyindole-3-calbaldehyde (7), which is a phytoalexin of radish [47] and it has insecticidal and bactericidal action. Demethylation of 7 with DABCO afforded 1-hydroxyindole-3-carbaldehyde (8) in a quantitative yield. Treatment of 8 with POBr3 produced SOMRE #1 (12) in 33% yield. In the second route, 6 was converted to 9 in 59% yield by the reaction with NBS. The following zinc treatment produced 1-methoxy-2-oxindole (10) in 91% yield. Subsequent reduction with Pd/C with hydrogen produced 2-oxindole (11) in 95% yield. 11 was then led to 12 in 77% yield by the reaction with POBr3. Each step mentioned above proceeded in good yield. Using compound 12 as a starting material, we were able to synthesize a variety of 2-substituted indole-3-carbaldehydes [11] and related compounds, 12—51, by combining Heck, Still, and other known reactions. Among the 40 compounds shown in Figure 3, each had exhibited its own pharmacological effects in plant growth tests, including stem lengthening or shortening and root growth promotion or inhibition. So, these group compounds were named as the SOMRE family and assigned SOMRE numbers to each member. For example, the compound 27 is SOMRE #16, 25 is SOMRE #14.
Figure 3.
SOMRE family compounds.
Scheme 4.
Synthesis of SOMRE #1.
8.4. About SOMRE #1 (12)
Depending on the type of plant, the compound having the strongest effect of elongating and promoting roots and stems may be a different substance among the 40 SOMRE compounds. Among them, SOMRE #1 (12) has the simplest structure, is easy to synthesize, and is economical. As expected, it exhibits the effect of promoting root and stem elongation in all plants examined so far. We understand, therefore, it is wise to continue the study with 12 as the optimal candidate for the ‘Earth Medicine’. In this review, we would like to summarize and report our research results on 12.
8.5. Safety of SOMRE #1[10,11,12]
- SOMRE #1 is used at concentrations range from 1.0 to 0.001 ppm, and even from 1.0 ppb to 0.001 ppb. Therefore, safety was examined at the highest concentration used, i.e., 1.0 ppm aqueous solution.
- Regarding fish safety:[40] ‘medaka’, a species of Japanese fish, which is known to be sensitive to toxic substances, was employed. A plurality of medaka fish were grown in an aqueous solution of SOMRE #1 (1.0 ppm). Even after 2 years and 5 months, they are growing steadily. Comparing to the beginning, their physical conditions are longer and their waists are thicker. There is no bad effect on the growth of ‘medaka’. As a result, the safety was judged to be extremely high.
- Animal (rabbits and rats) safety: seeds treated in SOMRE #1 (1.0 ppm) aqueous solution have grown plants and are helping to green the Gobi Desert in China. At the time of germination, young seedlings, stems, and roots, were good food for wild rabbits and mice. Grown plants from seeds suffered from their ingestion damage, and the survival rate dropped to 87.6%.[14] However, it was far better results than the highest survival rate (78.3%) conducted under Chinese method by their expert’s experiments.[14] Under the conditions, no dead rabbits or rats were found around. Therefore, the safety was judged to be extremely high.
- Regarding animal (goat) safety: in the Gobi Desert, SOMRE #1 (1.0 ppm) was mixed in food and fed to cashmere goats for total four years. The goats grew up healthy and produced more cashmere wool than usual. In addition, the reproductive effect was enhanced and many off springs were produced. As a result, the safety was judged to be extremely high.
- Ame's test: negative. No mutagenicity.
- Safety for humans
a) Skin: in the Gobi Desert greening project, volunteer 15 men and women carried out the task of soaking the seeds in the SOMRE #1 water solution for 4 hours. Both hands were kept immersed in the SOMRE #1 (1.0 ppm) aqueous solution during this operation. Since there was no extra water in the desert, we went home without being able to wash our hands and resumed our normal life. One month later, none of the workers felt any abnormality in their skin.
b) Body: in the Gobi Desert afforestation project, working in the sand dunes makes us thirsty, and we needed drinking water. We didn’t carry extra water with us except for SOMRE #1 (1.0 ppm) aqueous solution, so we had to drank 500 mL of SOMRE #1 solution on hand for 3 days, but there was no change in our physical conditions. At home, we drank 100 mL of SOMRE #1 solution every day for 1 year, but we did not feel any troubles and bad physical effect. Based on these results, the toxicity to humans is considered to be extremely low.
9. Plant Food Production with SOMRE #1
9.1. General Procedure for Using SOMRE #1[6,11]
Four types of SOMRE aqueous solutions with different concentrations and dechlorinated water for the control were prepared, thus using boiled and dechlorinated tap water, the SOMRE #1 (12) crystals were dissolved to make an aqueous solution with a concentration of 1.0 ppm. Then, dilute 10-fold, 100-fold, and 1,000-fold to make SOMRE aqueous solutions with concentrations of 0.1, 0.01, and 0.001 ppm, and these five types of solutions were prepared as one set. [6,11]
On the other hand, five different containers of the same volume are prepared. A plant to be studied is selected, and its seeds or root parts of seedlings are put into five kinds of containers in equal numbers. Each of the 5 kinds of solutions prepared in each container is added in such an amount that the seed or root part of seedling is immersed as shown in Figure 4 (only 4 containers are shown). Usually after soaking for 1 h, the seeds or seedlings are taken out and sown or planted in the field while the seeds or roots are still wet. After that, just grow normally without using fertilizers, fungicides, or insecticides. [6,11]
The duration of soaking in the SOMRE aqueous solution varies depending on the hardness of the seed husk. Cruciferous seeds cannot be soaked because their shells are thin and when they get wet, the shells peel off and the seeds break. For that reason, after sowing the seeds in the furrows, each seed is thoroughly sprayed with each solution using a sprayer. After an hour, sprinkle soil and water as usual. In such cases of acorns and jatropha whose seed coat is wrapped in a hard shell; it is better to soak them in each solution for 3 to 7 days. [6,11] SOMRE #1, root and stem elongation agent for plants, was applied to almost all plants. Some plants which showed increased harvests and growth promotion are summarized in section 22-2, Figure 117[10,11]
If we can increase the yield of vegetables whose roots are edible, such as taro, sweet potato, potato, and cassava, we will be able to solve the global food shortage. If the roots and stems become strong, the yield of grains such as rice, wheat, amaranthus, millet, and soybeans can be expected to increase, and Japan's food self–sufficiency rate could be increased. If the yields of sugar cane and corn increase, it will be possible to secure food and fodder, and supply the remainder for bioethanol.
Such ‘dream’ expands further. Increased yields of herbal medicines and herbs would be possible (section 10). It can also be applied to slope planting, growing windbreak forests and sand protection forests. Strengthen the roots of broadleaf trees to revitalize the forest, and extend the roots of conifers such as cedar and cypress. If so, it will be possible to prevent disasters such as fallen trees and landslides in cedar forests, apply it to planting on slopes, and grow windbreak forests and sand protection forests.
Applications for increasing fruit yield, lawn cultivation, gardening, landscaping, increasing seed production, improving the germination rate of seeds, improving the production rate of seedlings, increasing the success rate of garden plants and cuttings, etc. are also conceivable.
We think that it can also be used to produce fragrances and perfume oils. Production of biodiesel oil by increasing the production of sesame, soybean, and jatropha cultivation (section 10-3) would be possible.
9.2. Detailed Application to Edible Plants
9.2.1. The Monocotyledonous and Dicotyledonous Plant [11]
The SOMRE group of compounds are usually effective at 1.0—0.001 ppm. As shown in Table 1 at a concentration of 3 ppm, SOMRE #1 elongated the roots of the monocotyledonous rice plant to about 1.7 times (168%, 79.5 mm) the length of the control 46.8 mm (100 %). In addition, SOMRE #14 at a concentration of 3 ppm elongated the roots of cucumber, a dicotyledonous plant, about twice (190%, 22.9 mm) as much as the control (12.1 mm, 100%). Furthermore, we tested plants such as daikon radish (Brassicaceae), eggplant (Solanaceae), lisianthus (Gentianaceae), onion (Liliaceae), buckwheat (Polygonaceae), burdock (Asteraceae), and carrot (Apiaceae). As expected, SOMRE exerted a remarkable elongation effect on each root.
9.2.2. Tomato[11]
Tomato seeds were grown in a field according to the general procedure of section 9–1. The average number of tomatoes per tomato stick was 10.6 in the control and 20.0 in the SOMRE 1.0 ppm treatment as shown in Table 2.
The average weight of one ripe tomato was 114 g for control and 125 g for that of SOMRE treatment. When the total tomato yield of the control was taken as 100%, SOMRE treatment increased the corresponding yield by 188%. The tomato taste was sweeter and more delicious than the control. Figure 5 is a photo of tomatoes in the early stages of growth. Right side three plants are grown from seeds treated with 1.0 ppm concentration of SOMRE #1 solution, and left side three plants are control. The results of average height, girth of plant, number of fruits, and weight of fruit at the harvest time are summarized in Table 3, respectively. Figure 6 is the crop of tomato, where the left eight tomatoes are control, while the right eight ones are SOMRE treated. As is clear from these results, SOMRE treated plants grew higher and thicker than the control and the harvest increased.
Next year to clarify the reason, tomato seeds were treated with SOMRE 0.1 ppm solution. The SOMRE treated roots near the surface of the ground are highly stretched and dense, and SOMRE treated roots were three times as long (207 cm) as the control roots, which grew only 69 cm long (Figure 7). Consequently, the long roots can absorb a lot of nutrients and minerals from the ground. As a result, the tomato yield increased 2.1 times. As for the taste, not only was it sweeter, but it was remarkably delicious as compared with the control. People, who said they hated tomatoes, were happy to eat them.
It was clear that this was the result of the growth of healthy roots and increased absorption of nutrients and minerals from the ground. Through these basic researches, we discovered that (1) there is a specific SOMRE compound that is most compatible with each plant, even if it is a plantbelonging to the same family, depending on the type of variety, and (2) making roots and stems thicker and longer. It turns out that there is an appropriate concentration for each. Furthermore, we noticed in the control seeds, the growth of the root length and stems varied from individual to individual.
On the other hand, the SOMRE-treated seeds grew almost the same longer length. This suggests the possibility that rooting instructions by SOMRE, that is activation of genes controlling growth, occur almost simultaneously inside seeds. The SOMRE group of compounds would serve as a clue to develop a new academic field on plant growth.
9.2.3. Tamba Black Soybeans
In Japan, soybeans are an important source of protein as foods such as miso, soy sauce, natto, and tofu. Prepare 5 petri dishes and put 5 Tamba black soybean seeds in each petri dish.
Water (control) and aqueous solutions of SOMRE 1.0, 0.1, 0.01 and 0.001 ppm were added in an amount sufficient to soak the seeds, and left at a temperature of 15.0 — 19.5 °C from April 19 to April 22. Figure 8 shows the state of rooting, and Table 4 shows the results of measurement of root length.
The experiment was repeated three times and similar results were obtained with good reproducibility. Therefore, we found that the necessary treatment to increase the yield of tamba black soybeans was to soak the seeds in a 0.1 ppm SOMRE aqueous solution. Indeed, by soaking the seeds in a 0.1 ppm SOMRE aqueous solution for 1 hour before sowing, we were able to obtain an expected yield increase (140%).
9.2.4. Edamame
What about edamame? A seed rooting test was performed according to the general procedure of 9-1. Fifteen seeds were used and the elongated root length was measured (Table 5). At SOMRE concentrations of 0.01 and 0.001 ppm, the rooting rate was inferior to that of the control by 10-20%, but at 1.0 and 0.1 ppm, it was 100%. Taking the total root length of the control (417 cm) as 100%, it was 495 cm (119%) in the case of SOMRE 1.0.
It was found that the SOMRE 1.0 ppm aqueous solution is suitable for the purpose of increasing the yield, unlike the case of Tamba black soybeans. Consequently, as shown in Table 6, the crop of edamame increased by 2.4 times higher than the control. Even for the similar beans it was found that the optimal SOMRE concentration for increasing yields changes when cultivar changes.
9.2.5. Turnip (Hakurei)
Using water and SOMRE 1.0 ppm aqueous solution, two sets of 10 turnip seeds were made on April 3, treated according to the general procedure of 9–1, and then planted in the field. After cultivating, it was harvested on May 22 (Figure 9).
The respective total leaf and root (bulb) weight were measured (Table 7). The total control weight was 1,900 g and the total root (bulb) weight was 1,365 g. Assuming each to be 100%, when SOMRE 1.0 ppm was used, the total weight was 2,634 g and the total weight of roots was 1,925 g, increasing yields by 139% and 141%, respectively. Not only is the bulb large, but when we eat it, it is sweeter and has a deep taste, which is really delicious.
We found that fruits grown in SOMRE result in delicious across the board. Later, when increasing the yield of rice (paddy rice) in India, the sugar content was measured in section 14. It was found that the use of SOMRE resulted in higher sugar content and improved quality than the control.
9.2.6. Garlic
We purchased white 6–sided garlic cloves (from Aomori), separated them into individual pieces on October 11, 2016, and then added four pieces each to water (control) and SOMRE concentrations of 1.0, 0.1, 0.01, and 0.001 ppm aqueous solutions. The base portion of each piece was left to soak. The roots were growing smoothly, and on October 17, splendid rooting was observed. The rooting rate was 75% for the control and 75, 75, 75 and100% for 1.0, 0.1, 0.01 and 0.001 ppm, respectively.
After taking photos, the plants were transplanted to the field, and the SOMRE 1.0 and 0.001 ppm treated groups grew better than the control as shown in Figure 10. Harvested on June 2, 2017, as the stem part withered. The weight of the combined root and stem parts and the weight of the cut bulb parts were measured (Table 8、Figure 11). The total weight of the control garlic bulb was 137.9 g. Taking this as 100%, SOMRE 1.0, 0.1, 0.01 and 0.001 ppm were 154.9 g (112%), 99.5 g (72%), 118 g (86%) and 167.6 g (122%) respectively.
After taking photos, the plants were transplanted to the field, and the SOMRE 1.0 and 0.001 ppm treated groups grew better than the control as shown in Figure 10. Harvested on June 2, 2017, as the stem part withered. The weight of the combined root and stem parts and the weight of the cut bulb parts were measured (Table 8、Figure 11). The total weight of the control garlic bulb was 137.9 g. Taking this as 100%, SOMRE 1.0, 0.1, 0.01 and 0.001 ppm were 154.9 g (112%), 99.5 g (72%), 118 g (86%) and 167.6 g (122%) respectively.
As a result, garlic production could be increased by 122% by treating with 0.001 ppm SOMRE aqueous solution.
9.2.7. Sweet Potato[12]
From the viewpoint of increasing food production, we examined the sweet potato, which saved the Kyoho famine in Japanese history. Sweet potato ‘Beniaka’ vines were immersed in SOMRE 1.0 ppm solution for 1 h, and planted in a field. Half a year later, they are harvested. The SOMRE treatment increased weight yield by 250% compared to the control, as shown in Figure 12. In the case of ‘Silk sweet’, the control yielded 2,088 g, while the SOMRE 1.0 ppm yielded 3,660 g, increase of 175%.
9.2.8. Potato
Next, we examined the potato that saved the Irish famine in history. Potatoes are stems rather than roots, but SOMRE has a yield–increasing effect regardless of roots or stems.
The results of ‘Kitaakari’ were shown in Figure 13 and Table 9. The Figure and Table show the weight distribution map of each harvested potatoes in a bar graph. The horizontal axis divides the weight of one piece into 25g intervals. The vertical axis is the total weight of potatoes present in each segment. The gray bar graph is the control result and the reddish–brown bar graph is the result of 0.1 ppm SOMRE. The rightmost segment shows that there are 4 potatoes weighing 325g to 350g less and the total weight is 659 g. From this graph, it can be seen that the weight distribution of individual potato changes from left to right, i.e., from small to large, when the SOMRE concentration is increased from 0.001 to 0.1 ppm. The total yield was 1,891 g (100%) for the control and 3,101 g for SOMRE 0.1 ppm, an increase of 164%.
For ‘Andes Red’, similar results were obtained. Comparing the total yield, the control yielded 1,400 g, while the use of 0.1 ppm SOMRE yielded 2,575 g, which was 184% higher than that of the control, an increase of about two times.
‘Awakening of the Incas’ resulted 140% increase in yield compared to the control when SOMRE 0.1 ppm was used. The result of ‘Danshaku’ is shown in Figure 14 and 15. The largest control potato, purchased from green grocery has weight 113 g, and major and minor diameter are 7.5 and 6.5 cm, respectively. While 0.1 ppm SOMRE treated potatoes are almost the same, having weight 510 g, major and minor diameters are 13 and 11 cm, respectively.
In the case of potatoes, as a result of investigations with many cultivars such as ‘Kitaakari’, ‘Andes red’, ‘Awakening of the Incas’, and ‘Danshaku’, it was found that using 0.1 ppm of SOMRE gave the maximum yield.
9.2.9. Taro, Green Onion, and Moroheiya
9.2.10. Kohlrabi
Next, we examined kohlrabi, a vegetable that is related to cabbage and broccoli. In November, 5 groups of 6 seeds each were immersed in water (control), 1.0, 0.1, 0.01, and 0.001 ppm SOMRE solution. Each group had a good germination rate and grew well. Figure 17 is a photo of the leaves and bulbs. Figure 18 shows the state in which the leaf part and the bulb part are separated. Based on these results, the weights of the six kohlrabi bulbs in each group are summarized in the Table 11.
For the control, the total weight was 710.4g. The results for SOMRE 0.001, 0.01, 0.1, and 1.0 ppm treatments were 1,015 g, 1,062 g, 1,127 g, and 1,775 g, respectively.
Given that the control yield is 100%, the yields for the SOMRE treatments are 143%, 150%, 159%, and 250%. With the SOMRE 1.0 ppm treatment gave the highest yield.
9.2.11. Upland Rice
How about upland rice which is one of the staple foods in Japan. Seeds of Upland Rice ‘Norin #1’ (‘Mochiokabo’) were divided into groups of 29 grains each, and the seedlings were grown by immersing them separately in a water, SOMRE 1.0, 0.1, 0.01, and 0.001 ppm solutions for one day and night.
A group of 29 grown seedlings was planted in groups as shown in Figure 19. Figure 20 shows how each of them is growing. Figure 21 is a photo at the time of harvest. The rice ears of each group are shown in Figure 22. The left end is the control group. SOMRE 0.001, 0.01, 0.1, 1.0 ppm treated groups were placed sequentially on the right side.
Figure 23 is a photo taken by spreading the ears of rice one by one to see how the rice is attached. From these photographs, it can be seen that compared with the control, SOMRE treatment increases yield regardless of concentration. Figure 24 is a comparison photo of the amount of paddy actually harvested. Figure 25 is a photo comparing the amount of brown rice obtained after threshing. From these experiments, we confirmed that SOMRE treatment increases yield regardless of its concentration.
The results are summarized in Table 12. Te total weight of paddy and brown rice after threshing in the control was 57.1 g and 42.5 g, respectively.
When the amount of brown rice in the control was 100%, those of 0.1 and 1.0 ppm were the respective yields of 375% and 238%, showing a significant increases in yield.
In order to clear the cause of this yield–increasing effect, we investigated the number of branches, that is the number of tillers, and found that the control had 79 branches, while 0.001 ppm had 91 branches, and 0.01, 0.1, and 1.0 ppm had 111, 143, and 162 branches, respectively.
SOMRE has a tillering action12 as well as a rooting and elongation action for branched roots, and promotes the thickening and growth of the roots of the branches. It was found that the growth was moderately controlled and the size of flowers and fruits also
increased, and the yield increased. This result is the reason behind the increase in rice production in India, which will be discussed in section 14.
9.2.12. Wheat
What about wheat, one of the staple foods? Rooting test was carried out from October 20th to December 1st using 20 seeds of wheat obtained from Matsudo City Hall. The rooting rate was 80% in the control, whereas SOMRE improved to 95% regardless of concentration. An intermediate course of the growth was observed and visually the SOMRE–treated barley showed better growth and stiffer berries than the control.
They grew well as expected and we were looking forward to the harvest. One morning, a large number of sparrows gathered in the normally quiet field, making a lot of noise. We didn't pay attention to anything. But in the afternoon, we were shocked when we saw the field to see how the wheat was growing. The wheat just before the harvest had been eaten across the board.
We chased away the sparrows and collected the remaining wheat berries, but sadly we were unable to compare the yields. It was fortunate to confirm that the grain size of the wheat was clearly larger by the SOMRE treatment than the control as shown in Figure 26.
9.2.13. Corn
A rooting test of corn (‘pure white’) was carried out according to the general procedure of 9–1. All concentrations of SOMRE resulted in longer roots than the control. SOMRE 0.01 and 0.001 ppm concentrations were the longest (Figure 27).
The relationship between corn variety and harvest depending on SOMRE concentration is shown in Table 13. When ‘pure white’, ‘gold rush’, and ‘honey bantam’ seeds were treated with SOMRE at concentrations of 1.0 ppb, 0.1 ppm, and 0.001 ppm, the yields were 2 times, 1.5 times, and 2 times higher than the control, respectively.
To our surprise, as a new discovery, we found that even lower concentrations of SOMRE, 0.1 and 0.01 ppb had a good rooting rate and long root growth as shown in Table 13. So far, we have had no studies conducted at concentrations of less than 0.001 ppm for other plants. In the near future, a new research question has been raised that we must reexamine the SOMRE effects on the roots/stems of each plant at ppb concentrations.
9.2.14. Radish (‘Sakurajima’)
According to the general procedure of 9–1, a root growth test of common radish ‘Okra’ was carried out. Figure 28 is a photo after harvesting. The weight of the harvested control was 2.3 kg (100%) while that of the radish treated with 0.1 ppm SOMRE increased to 3.1 kg (134%).
On the other hand, seeds of ‘Sakurajima radish,’ a cultivar that grows larger than common radishes, were treated with 1.0 ppm SOMRE solution. It grew into a larger radish as shown in Figure 29 with a weight of 7.05 Kg (190%) compared to 3.7 kg (100%) of the harvested control.
Normally, when vegetables grow large, they are no longer edible for humans, and become fodder for livestock. This idea was overturned by SOMRE. The taste was delicious and filled the bellies of many people.
10. Herbs
10.1.1. Basil
Herbs have been used for herbal medicines, fragrances, and flavorings of food and drinks since ancient times. From June 19 to June 24, using 15 seeds purchased from ‘Daiso Sangyo’, control (water), using 5 kinds of solutions of SOMRE aqueous concentration of 1.0, 0.1, 0.01, 0.001 ppm, rooting tests were performed at 22.5—24.5°C.
The rooting rate in the control was 73%, which was not much different from the case of using each SOMRE solution, but it was high in the SOMRE aqueous concentrations of 1.0 and 0.001 ppm, 80% and 100%, respectively, as shown in Table 14. Further comparing the root length, when the total root length of the control (242 cm) was taken as 100%, it was 288 and 358 cm at SOMRE concentrations of 1.0 and 0.001 ppm, respectively, which were 119% and 148%. Reflecting this result, even after transplanting to the field, the subsequent growth was smooth, and a high harvest of basil was obtained.
10.1.2. Coriander
Cilantro seeds were purchased from Tohoku Co., Ltd. Rooting test at 26.0—33.0°C from August 14th to August 23rd, using 15 seeds each, control (water), 5 types of SOMRE solutions with concentrations of 1.0, 0.1, 0.01 and 0.001 ppm, were carried out as shown in Table 15.
Seeds were with low rooting rate, only 13% in control. The use of each SOMRE solution improved them. It was higher at SOMRE aqueous solution concentrations of 1.0 and 0.01 ppm, and the rooting rate was 47% and 27%, respectively. Comparing the root lengths, when the total root length of the control (98 cm) was taken as 100%, those at SOMRE concentrations of 1.0 and 0.01 ppm were 451 cm (460%) and 219 cm (223%), respectively. By employing SOMRE solution, when it was transplanted to the field, it grew well after that, and we achieved a higher harvest of coriander than the control.
10.2. Application to Herbal Medicine
10.2.1. Licorice
Licorice has been used as a medicine in the East and the West since prehistoric times. In Chinese herbal medicine, it is added as an ingredient in about 70% of them.
It is popular medicine such as ‘Shakuyakukanzoto’, ‘Yokukansan’, ‘Kakkonto’, ‘Saireito’, ‘Hochuekkito’, ‘Ninjinyoeito’, and ‘Shoseiryuto’, and they are known for their antitussive, expectorant, stomachic, anti-inflammatory, antispasmodic, and analgesic effects. The medicinal ingredient is glycyrrhizic acid, which is said to be 150 to 200 times sweeter than sugar. It is useful as a sweetener, cosmetics, and medicine, and its demand is increasing all over the world. The roots, which are used for medicinal purposes, grow deep underground and take several years to grow and be harvested. Because the plant digs deep underground, there are concerns about environmental destruction, and there are concerns that resources will be depleted due to overhunting, and the world is eagerly awaiting the development of an effective cultivation method.
Figure 30.
Seeds, shaved off outer hard shells.

Figure 31.
The results of the average rooting rate.
Figure 32.
Average length of grown roots.
Figure 33.
Immersing roots to SOMRE solution.

Figure 34.
Digging the ditch for plantingtreated roots.

Figure 35.
Harvest of licorice.

Figure 36.
Results, control, and SOMRE.

Given the above background, it became an easy target for SOMRE's deployment. First, we examined the problem of seed germination. The outer shells of the 20 hard–shelled seeds in each group were partially shaved off with a cutter to allow SOMRE solution to penetrate (Figure 30). Then, they were immersed in water (control) and SOMRE solutions with concentrations of 1.0, 0.1, 0.01, and 0.001 ppm at 27.5°C to 33°C for 1 h. Thereafter, they were sown in “verdenite soil” and the growth was observed from September 6th to 13 rd. Figure 31 summarizes the results of the average rooting rate, and Figure 32 summarizes the results of the average length of grown roots.
The rooting rate for the control was 80%, whereas it was 100% for SOMRE 1.0 and 0.001 ppm concentrations, respectively. The average root length of the control was 100 mm, while at SOMRE 1.0 and 0.001 ppm concentrations it was 140 and 120 mm, respectively. In this way, SOMRE was found to be useful for efficiently producing licorice seedlings from seeds.
Given the above background, it became an easy target for SOMRE's deployment. First, we examined the problem of seed germination. The outer shells of the 20 hard–shelled seeds in each group were partially shaved off with a cutter to allow SOMRE solution to penetrate (Figure 30). Then, they were immersed in water (control) and SOMRE solutions with concentrations of 1.0, 0.1, 0.01, and 0.001 ppm at 27.5°C to 33°C for 1 h. Thereafter, they were sown in “verdenite soil” and the growth was observed from September 6th to 13 rd. Figure 31 summarizes the results of the average rooting rate, and Figure 32 summarizes the results of the average length of grown roots.
The rooting rate for the control was 80%, whereas it was 100% for SOMRE 1.0 and 0.001 ppm concentrations, respectively. The average root length of the control was 100 mm, while at SOMRE 1.0 and 0.001 ppm concentrations it was 140 and 120 mm, respectively. In this way, SOMRE was found to be useful for efficiently producing licorice seedlings from seeds.
10.2.2. Ginseng
Cultivation of ginseng, a traditional Chinese herbal medicine, is generally considered difficult to grow. Although it is highly popular and in high demand among middle–aged and older people who are health conscious. If the yield increases, it will be useful for social contribution.
Since improvement of the rooting rate is required for growth, we decided to examine the effect of SOMRE #1. The seeds were obtained from China with the help of Dr. Shuichi Takahashi. From December 2014 to January 2015, a rooting test was carried out for 21 days at a temperature of 9–16.0°C using 10 seeds each as shown in Figure 37 and Figure 38.
The rooting rate of the control was as low as 40%. While with the use of SOMRE solution, although the values fluctuated depending on the concentration, they increased, reaching 90% and 80% at SOMRE solution concentrations of 1.0 and 0.001 ppm, respectively. Furthermore, when the root length was compared, when the control total root length (42 cm) was taken as 100%, the SOMRE concentrations of 1.0 and 0.001 ppm were 108 (257%) and 108 cm (257%), respectively (Table 16).
Based on the above results, even in the case of seeds such as ginseng that are expensive and usually have a low germination rate, the use of the SOMRE aqueous solution can improve the rooting rate and make the roots grow longer and stronger. We believe SOMRE is the best of choice for increasing ginseng production
10.2.3. Maca
Maca is a cruciferous plant native to the Andes Mountains, and is rich in nutrients. It has been passed down as a ‘food of life’ to people living in the Andes Mountains. It is said to have been a food for the privileged class. In modern times, the NASA of the United States adopted it as food for astronauts, and it suddenly became popular worldwide. It is highly popular and in high demand among middle–aged and older people who are health conscious. If the yield increases, it will be useful for social contribution.
We decided to examine the effect of SOMRE #1. The seeds were obtained from Seed company ‘Premo’. A rooting test was carried out for one week at a temperature of 18.0–21.0°C using 5 seeds each as shown in Table 17. When the root length was compared, when the control total root length (55 cm) was taken as 100%, the SOMRE concentrations of 0.001 and 0.01 ppm were 64 (116%) and 73 cm (133%), respectively (Table 17). After transplanting these seedlings to planter, they grew steadily (Figure 39). However, unfortunately, a stray cat or dog, maybe, used the planter as a litter box, and spilled almost all soil from the planter, and the Maca died.
However, since it has been found that SOMRE is beneficial for the growth of maca, we hope that the use of a 0.001 ppm SOMRE solution will lead to increased maca production.
10.3. Jatropha
We next examined jatropha, a non-edible plant that grows even in poor soil. If the shortcoming of low rooting rate can be improved, the oil collected from the fruit will be useful as a biodiesel fuel.
Figure 40 and Figure 41 show the results of a rooting test conducted from June 14 to 18 using 5 seeds for each of the 2 cultivars, ‘Indonesia 12’ and ‘Brazil 1’, using water and an aqueous solution of SOMRE. In ‘Indonesia 12’ the control showed a rooting rate of 62%, while the SOMRE 1.0 and 0.1 ppm solutions had superior rooting rates of 87% and 100%, respectively (Figure 40). ‘Brazil 1’ had a particularly low rooting rate, only 25% in the control (Figure 41). On the other hand, when SOMRE 1.0 ppm solution was used, a good germination rate of 87% was achieved.
Figure 42 shows the growth of the rooted seedlings of the two varieties in the planter on the 13 days after they were transferred to the planter. They are growing well and, 21 days after they are transferred to the garden (Figure 43). After 88 days, it grew to the height of the author's waist (Figure 44) and flower buds began to grow from the tips (Figure 45). In Japan we have a winter season. And in January it snowed and the tree withered (Figure 46 and Figure 47).
We got the impression that a good harvest would be fully expected in tropical and subtropical regions such as Africa.
Figure 40.
Rooting test of Indonesia 12.

Figure 41.
Rooting test of Brazil 1.

Figure 42.
Growth of the rooted seedling.

Figure 43.
Growing in the garden.

Figure 44.
Height of the growing plant.

Figure 45.
Flower bud.

Figure 46.
Snow fell on the plants.

Figure 47.
Withered plants.

10.4. Mushrooms and Bacteria
10.4.1. Fungi and Mushrooms
Could SOMRE help grow mushroom fungi? At the beginning of July, a cloud ear cultivation block was purchased from Mori Sangyo Co., Ltd., and cultivation was attempted by spraying water as a control and a 1.0 ppm aqueous solution as SOMRE #1 according to the cultivation guidelines. In about 10 days, the primordia of the fungus grew little by little, and eventually grew larger day by day. As compared to the control, the fungus grows larger due to the action of the SOMRE (Figure 48: shot from the side. Figure 49: shot from the above).
We found that SOMRE exerts an effect on the growth of Auricularia fungus. As a future goal, we hope that promoting the growth of other fungi, such as actinomycetes, which are used in the production of antibiotics, could contribute to the cheap production of medicines.
10.4.2. Effect on Plant Pathogens
In a previous report[12], in a ‘bok choy’ growing experiment, the leaves of the control group were attacked by insects and bacteria and had holes in the leaves, whereas the leaves of the SOMRE–treated group remained beautiful as shown in Figure 50.[12] First we considered that this phenomenon is due to SOMRE functioning as a bactericide and insecticide. Therefore, just in case, we examined the action of SOMRE #1 on a few plant pathogens[12] (conducted by Prof. Suzuki, 2007).
As a result, even if SOMRE aqueous solution with a high concentration of 1,000 ppm was used, it did not show any effect against gray mold, rice blast, cruciferous plant soft rot, brown streak of rice, and leaf blight of rice. Antibacterial tests against Bacillus subtilis (NBRC 3134) and Escherichia coli (NBRC 14237) showed no effect at all. On the other hand, rice sheath blight had a weak inhibitory effect, and seedling take–off fungus had an inhibitory effect. Based on these results, we confirmed that SOMRE at a concentration of 1.0 ppm or less had no antibacterial or bactericidal action.
It is surmised again that the effect seen in ‘bok choy’ is the result of an increase in the amount of phytoalexin produced by the plant itself, which has thick roots and has grown healthily.
11. Development of the SOMRE #1 kit
1.1.1. SOMRE #1 Kit
When SOMRE #1 is actually applied to agriculture, it is often difficult to decide which concentration of SOMRE solution should be used for which crops. If we grow crops one by one in the field and see the results, we will miss the planting time. As a simple method to overcome this problem, we developed a SOMRE Kit that allows to find the appropriate SOMRE in just one week (Figure 51).
Figure 51.
SOMRE #1 kit.
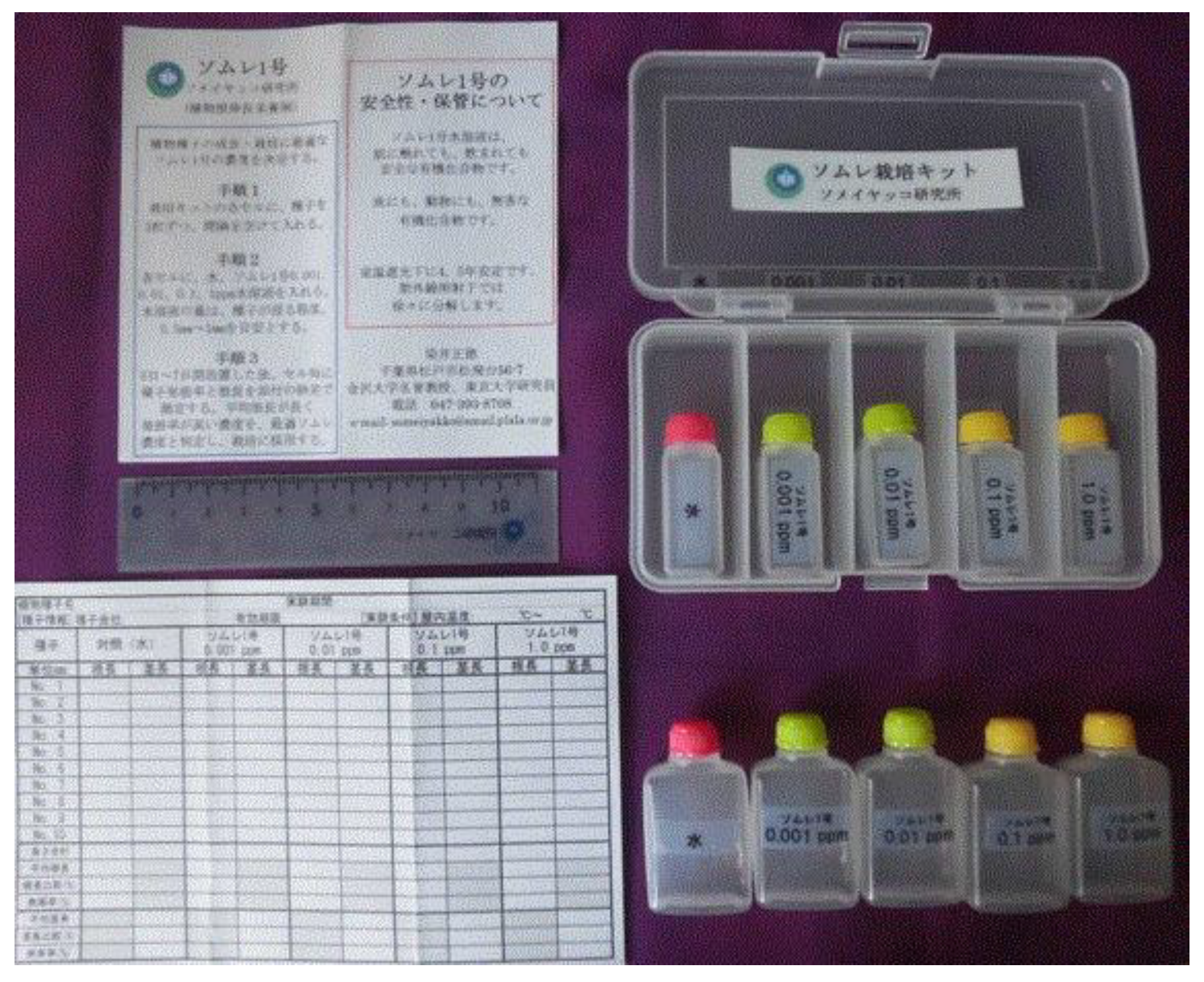
11.2. How to use the SOMRE #1 kit
A set of 5 types of liquid (water, 1.0, 0.1, 0.01 and 0.001 ppm SOMRE solutions) and a lidded container having five transparent compartments for observing rooting was prepared. The set is lightweight and can be carried anywhere. Three to five seeds, depending on their size of the plant under consideration, are placed in each chamber according to the accompanying instructions.
Then, add the same amount of each of the 5 types of liquid so that the seeds are soaked, and cover with a lid. All we have to do is the simple steps stated above and leave it for one week. Results are usually seen within a week.
Then fill in the necessary items such as the start date and time, temperature in the attached table. One can observe the progress of rooting. The rooting state of the seeds can be observed every day, and when the rooting is remarkable, the root length is measured after 4 or 5 days and recorded in the table. Good results can be obtained by soaking the seeds in a concentration of SOMRE solution that gives the longest roots of the selected plant, and then start farming process.
11.3. Examples
Examples of root growth of okra, corn, and burdock are shown below.
11.3.1. Okra
Figure 52 shows the state of rooting of okra, and the result is shown in Table 18. The rooting rate was 33% in the control but increased to 100% in SOMRE 0.001 ppm. The total root length was 14 cm in the control, while 113 cm in SOMRE 0.001 ppm, which was 8.07 times longer than the control. Treatment of okra seeds with 0.001 ppm solution of SOMRE resulted in the 150% improved yields.
11.3.2. Corn (‘Gold Rush’)
The rooting rate was 25% in the control, while it increased to 75% in SOMRE 0.01 ppm. The total root length was 15 cm in the control, but 113 cm in 0.01 ppm SOMRE, which was 7.53 times longer. Therefore, treatment of corn seeds with 0.01 ppm SOMRE solution can achieve improved yields.
11.3.3. Burdock (‘Tegaru’)
In the case of ‘tegaru burdock’, the rooting rate of the control was 83%, and it was almost the same at each concentration of SOMRE.
Results are shown in Figure 54 and Table 20. The total root length was 62 cm in the control, while 108 cm in 1.0 ppm SOMRE, which was 1.7 times longer than the control. The total root length was 62 cm in the control, while 108 cm in SOMRE 1.0 ppm, which was 1.7 times longer than the control. It can be expected that an improved yield can be achieved by treating the seeds of burdock root with SOMRE 1.0 ppm solution.
In the actual field, we were able to obtain the harvest expected with this kit.
11.4. Recommendation to Use SOMRE Kit
Even if we explain how to use SOMRE and show slides, there are many people who are skeptical and unable to make a decision to employ SOMRE #1. However, with the SOMRE kit, you can see the difference in root growth with your own eyes within just one week. When the results were actually applied to the field, researchers and farmers can feel at ease to be able to obtain the expected harvest with this kit.
12. With ‘SOMRE’ and ‘VED’ in Hand, Let's Go to the Gobi Desert
About 50 years ago, when ‘SOMRE’ and ‘VED’ were first created, we have actively made presentations at academic conferences and exhibitions. We applied for scientific research grants with the ‘dream’ of greening the desert, increasing food production, and preventing global climate change. But at the time, no one paid attention to us. During these days, we were able to create various ‘SOMRE’ and ‘VED’ compounds, so we wished to check whether they would work as expected as ‘Medicine for the Earth’ in the real desert.
In January 2005, we had Hokkoku Shinbun publish an article in order to obtain collaborators for the above ‘dream’. Luckily, we came across an NPO in Kanazawa, the Society for Wrapping the World's Deserts in Greenery. And as a comrade who challenges the common ‘dream’, the author was able to get the opportunity to work on the greening of the Gobi Desert in the Inner Mongolia Autonomous Region of China, which we had been longing for.
By the way, there is no word for the Gobi Desert in the Inner Mongolia Autonomous Region. The Gobi Desert is a coined word by the Japanese that refers to the aggregation of three deserts, the Batakirin, the Maous, and the Tenger Desert. Therefore, in this review, we refer to the Tenger Desert when describing the location precisely. We will also use the term Gobi Desert, which is familiar in a broad sense.
12.1. Seed Rooting Experiments Using Desert Indigenous Plants [11,12]
Which SOMRE compound is compatible with each of the seeds of the many plants that grow naturally in the Tenger Desert (Figure 55). What concentration is appropriate? Investigations were made using SOMRE #1 to #22. Among them, the seeds of ‘sand jujubes (砂棗)’ and ‘flower buds (花棒)’, which grow as trees, were selected as plants that could prevent the occurrence of sandstorms and stop the movement of fluid dunes.
As for the preliminary study, we prepared 1, 3, and 10 ppm aqueous solutions of SOMRE #1, 1.0 and 2.0 ppm aqueous solutions of indole acetic acid (IAA) for comparison, and control water, respectively. Seeds of ‘sand jujube’ were selected and soaked for 30 min. A circular desert experimental site with a diameter of about 5 m was divided into 6 sections, and grooves with a depth of about 5 cm were dug in each section, and seeds pretreated by the above solutions were sown. Since it was the first experiment and was also in the dry and water deficient season, the plants were grown by watering as needed for 73 days from August 2.[10,11,12]
Rats ate the saplings that had grown in all plots. The IAA plots were also damaged, and the 1 ppm plot wiped out. However, we found that there was no significant difference in the growth between the IAA 1.0 and 2.0 ppm plots, and the growth rate was lower than those in the SOMRE treated plots.
Rodents are adept at recognizing and avoiding harmful plants. Subsequent experiments also confirmed the safety of the SOMRE compound group, based on the fact that rats preferred to eat SOMRE–treated plants over IAA–treated plants. The results of this initial experiment are shown in Table 21. It was found that SOMRE #1–treated plants grew longer and stronger roots than the control or IAA–treated plants.
12.2. Rooting Experiments under Natural Conditions Using Seeds, Seedlings, and Trees of Native Plants in the Desert [10,11,12]
12.2.1. Only with Rain Fall as Water Source, under Natural Conditions
In the experiment from the next year, we decided to avoid artificial watering, aiming for the rainy season from May, and to grow the plants only with scarce rainfall under natural conditions.
A new native plants experimental site was prepared, and water (control), 1 ppm aqueous solutions of SOMRE #1 and #4 were prepared for ‘sand jujube’. The root length that rooted and grew during the short rainy season averaged 19 cm in the control plots. In the section treated with SOMRE #1, the roots were longer and deeper, so it was difficult to dig out, and the tip part was cut off in the middle. Even so, it was elongated to 53 cm, about 2.5 times as long. It was a pity, so we dug up a few and used them as specimens (Figure 56).
On the other hand, in the section treated with SOMRE #4, the roots had grown even longer, so digging was abandoned because it extends deep underground. When the seeds were treated with SOMRE, the roots reached a sandy layer with moisture about 30 to 50 cm below the surface of desert, where was an essential condition for survival in the Gobi Desert as illustrated in Section 5. The roots stretched leisurely into the deeper sand layer. On the other hand, during the harsh winter when the temperature dropped to –30 °C, the seedlings grown from the seeds treated with the SOMRE solution did not freeze to death because the roots of the grown seedlings were deep. While the control group almost froze to death in the desert. In the spring of the following year, it sprouted and grew vigorously again.
From these experiments, we confirmed that as long as seeds were treated with SOMRE #1 solution, even under natural conditions with little rainfall, individuals could take root throughout the year and grow into young trees.
12.2.2. Seedlings Treated with SOMRE Solution [10,11,12]
In April 2007, roots of 2,700 flower–buds (‘hana–bou’) seedlings were immersed in a 1.0 ppm aqueous solution of SOMRE #1 for 30 min. Two seedlings were paired about 30 cm apart from each other and the seedlings were planted on a semi–permanent sand dune in the Tenger Desert with a pair spacing of about 1.0 m (Figure 57).[48] Meanwhile, a group of Professors from Ningxia University, faculty of agriculture, carried out afforestation in parallel with us for comparison, employing their best afforestation method.
They carried out evaluation two months later.[49] We received the results in Japan. The survival rate of the China’s method was 78.3%, while the result of our SOMRE method was 87.9%. This result was widely reported in the Arashan newspaper (Figure 58).[48] In May 2008, 1 year and 2 months after planting, the plants endured the dry season and harsh winter. They grew up under the natural conditions. We were able to demonstrate that the use of the SOMRE aqueous solution dramatically improved the survival rate under the harsh natural conditions of the desert. The seedling height of ‘flower buds’, which was 30 to 50 cm at the time of planting, has grown to 1.0 to 1.3 m, and the tallest tree has grown to 1.8 m, making it a thick tree. In contrast, the number of trees that survived the Chinese conventional method was small, and the height of the few remaining trees were about 70 cm at the highest. Fortunately, the desert has changed to green area (Figure 59) and rabbits, insects, rats, and other creatures shave returned (Figure 60). The ecosystem is recovered quickly.
From these experiments, we confirmed that even in the case of seedlings, as long as the roots were treated with SOMRE #1 solution, they would take root throughout the year under the natural conditions of the Tenger Desert and would surely grow into trees.
12.2.3. Trees Treated with SOMRE Solution
In Japan, when transplanting trees, they are usually planted in a root–wrapped state. If the roots are left unwrapped, they will not be protected and will dry out, reducing the percentage of roots taking. Chinese people in the desert didn't care. In addition, to our surprise, the Chinese method of planting trees was to cut the root lengths to the same with scissors (Figure 61). The author thought they have no knowledge about plants, so they were doing this for the sake of efficiency in their work and for ease of manipulation. They had cut off the growing point at the tip of the root. All the seedlings were treated like this. The Communist Party was watching our every move behind us, and our experts' opinions were ignored. And we had to follow their amateur methods.
Figure 61.
Trim the roots to the same length.

Figure 62.
Soaking ‘hiba’s root in SOMRE solution.

Figure 63.
Soaking roots while praying for growth.

Figure 64.
After planting ‘hiba’ tree.

Also, the roots of the ‘hiba’ tree were cut into chunks while they were still thick. In May 2007, 1.0 ppm aqueous solution of SOMRE #1 was poured to the ‘hiba’ roots for 30 minutes as shown in Figure 62 through 64. Since they were expensive trees, we were allowed only three trees to be treated with SOMRE. We then planted our three trees together with their 97 ‘hiba’ trees as control following the Chinese
method. In March 2008, 10 months later, all the control trees withered and died during the summer and winter. While under the same conditions, our three SOMRE–treated trees took root and survived.
From these facts, we found that even trees can survive and grow under the natural conditions of the Tenger desert as long as their roots are treated with SOMRE #1 solution.
13. Greening the Gobi Desert
13.1. Preliminary Experiment of Air Seeding [11]
It is impossible to dig trenches and pits, scatter seeds in them, and plant seedlings and trees in vast desert, much less on a global scale, because of labor shortages and economic problems. We believe that the only way to make the desert green area is to use airplanes (or drones) to spread SOMRE–treated seeds over a big desert.
As a preliminary experiment, at the end of May 2007, we attempted the world's first trial. On the day before rain was predicted by the weather forecast, the seeds of ‘flower buds’ soaked in a 1.0 ppm aqueous solution of SOMRE #1 (Figure 65). Then they were thrown in the air on the surface of a typical fluidized dune located at the interface between semi–fixed and fluidized dunes in the Tenger Desert, while walking[50] as shown in Figure 66 and Figure 67. They were left under natural conditions, where several millimeters of rain had fallen. Two months later, in early August, we had an impressive result. Thirty seeds (0.1%) out of 30,000 scattered seeds had grown into seedlings.
13.2. World’s First Air Scattering of SOMRE Treated Seeds by Airplane [16]
Encouraged by the preliminary test results described in section 13–1, we convinced that attempt to sow seeds, pretreated with a 1.0 ppm aqueous solution of SOMRE #1, to air spray into a fluidized dune is not a dream story, but to become real story.
In air spray, development of a technology to reliably embed seeds in the sand of the target area is needed. At the same time, in order to increase the probability of growing into young plants, it is conceivable to make the seeds into clay balls, give them an appropriate weight, and scatter them in the air. Based on this idea, in mid–May 2008, 25,700 mixed seeds of ‘flower–buds’ and ‘jujubes’ treated with SOMRE 1.0 ppm aqueous solution were made into clay balls (Figure 68), and the grains were spread over the surface of the fluidized dune with the cooperation of volunteers. We threw them into the air while walking.
In October 2008, an on–site inspection was conducted with officials from the local government of Arashan. Figure 69 shows a survey of the number of young trees that have germinated and grown in the Gobi Desert by throwing out seeds while walking. And we confirmed that 127 seedlings had taken root and were growing well as shown inside the red circle in Figure 69. Chinese researcher said that at this plant size, the roots are grown over 2.5 meters long and deep.
These results mean that the preliminary experiment of sowing SOMRE soaked seeds from an airplane would be successful and that the greening of fluidized sand dunes (desert), which was impossible with the conventional method, is proved to be no longer a pipe dream.
With the cooperation of the local government, who witnessed these results firsthand, we carried out the world's first aerial spraying of SOMRE–soaked seeds on the Gobi Desert on June 20, 2010. Figure 70 was the soaking seeds into SOMRE solution. Figure 71 was the photo in filtering seeds through the bamboo colander. In Figure 72 the treated seeds were packed in bags for loading onto an airplane.
However, from our point of view, it was a reckless challenge to carry out the project while ignoring the weather forecast. Figure 73 shows SOMRE–treated seeds are loaded onto an airplane, and Figure 74
shows seeds being dispersed from airplane.[16] Later, we have received the reports from the Chinese collaborators that good results have been achieved.
There are still many issues to be investigated in the near future, such as the soaking time of seeds suitable for rooting, the particle size of clay balls, the ratio of clay and sand, the hardness of balls, how many days before rain should be sown, etc.
However, we believe that we will be able to find improved conditions and techniques, soon.
14. Expansion to India [17,18]
14.1. Rice Harvest; Indian Agriculture [19]
In India, environmental deterioration due to desertification and food shortages are rapidly progressing. It is reported that food crisis and hunger are severe problems in India and Africa. Continuing from the previous report,[11] with the cooperation of Mr. Ryoji Awano and Dr. S. Mishra of KIIT University, in order to solve these problems, we considered expanding SOMRE's agriculture technique to Odisha, India.
First, we spent one year to examine in detail whether or not SOMRE #1 has an impact on Indian soil. On the other hand, the effects of SOMRE on major Indian vegetables were examined in parallel. As a result, we found that the root elongation effect of the main Indian vegetables was significantly better than that of the control, as well as the Japanese vegetables, without any negative impact on the Indian soil environment.
Since 2015 with the cooperation of the Indian local government and agricultural researchers concerned, Dr. S. Mishra and Awano conducted four–year basic experiments on rice harvesting.[18] The results are shown in Figure 75 and Table 22. Rice yield in India is usually 5,100 kg/1 ha.[19] However, when the paddy was treated with 1.0 and 0.1 ppm SOMRE aqueous solution and grown, the yield was 6,511 Kg and 6,833 Kg/1 ha, respectively.
Assuming that the yield of the control was taken as 100%, the yields of 1.0 and 0.1 ppm were 127% and 134%, respectively, and achieved high yield increases. The sugar content is as high as 0.5% in the rice that uses 0.1 ppm, and the quality is also improved, making it possible to produce delicious rice.
14.2. Harvest Other Than Rice; Indian Agriculture [19]
Other Indian crops[19] such as black gram, sunflower and amaranthus also showed high yield increases of 113%, 122% and 126% respectively. The results are shown in Table 23. In the case of okra, the result is as low as 104%. However, when 0.001 ppm SOMRE solution was used in Japan, a 150% increase in yield was obtained. We suggest that the experiment should be done again in India using 0.001 ppm SOMRE solution.
With SOMRE, India would be able to improve food production and save hunger. Furthermore, we were able to show the possibility of being useful for arid land greening and environmental improvement.
14.3. Further Expansion to India
In order to save India from starvation, it is necessary to build a system to increase food production. Together with our collaborator Mr. Ryoji Awano, we are actively working to promote SOMRE. The current situation is described in the India–Japan Online Agri Mission, 2021 hosted by JETRO and IJCCI, ‘Japanese Technology’ x ‘Indian Market’, ‘India Online Presentation Challenge by Small and Medium Enterprises, Biostimulant (New Model Agricultural materials). SOMRE will change Indian agriculture’, and we gave a presentation on May 26, 2021 to many people involved in agriculture in India, including Mr. S. Janakiraman, Karnataka branch manager of the India–Japan Chamber of Commerce and Industry. The contents can be seen in the video from the URL described in the literature column.[51]
15. Deployment to Africa
15-1. Nerica Rice [12]
Nerica rice was developed with the aim of improving the food situation in Africa by crossing high–yielding Asian rice with pollen from African rice, which is resistant to drought and pests.
In contrast to the 120–140 days of conventional Asian rice, the cultivation period is shortened by about 30–50 days, and the cultivation period of Nerica is 90–100 days. Improving its harvest is a powerful means of relieving the food crisis. However, the first drawback point is that the initial growth rate is slow, and the second point is that it does not have a competitive advantage over the weeds seen in African rice. SOMRE has the characteristics of elongating roots, defeating weeds, and speeding up the growth rate, so there is a possibility that these two drawback problems can be improved.
Therefore, seeds were purchased from the Japan Burkina Faso Friendship Association, and a rooting test was carried out at a temperature of 13—19 °C from April 12 to April 21 using 20 grains of each seed.
At a low SOMRE concentration of 0.001 ppm, the rooting rate was 70%, lower than the control's 85%, but at higher concentrations of 0.01, 0.1, and 1.0 ppm, it was equal to or better than 85, 80, and 90%, respectively, as shown in Figure 76 and Figure 77. In terms of total root length, at a low concentration of 0.001 ppm, 105 cm is shorter than the control's 155 cm. However, at 0.01, 0.1, and 1.0 ppm, it is 169, 176, and 278 cm, respectively, and becomes longer as the concentration increases (Table 24). Assuming that of the control is 100%, it has grown to 109, 121, and 170%, and an increase in harvest is expected. The time required for growth is also shortened, and the growth competition with weeds could sufficiently win. Based on these results, we expect that SOMRE will contribute to improving the food situation in Africa by promoting the growth of NERICA rice and increasing the yield, just as India has achieved increased rice yields.
15.2. Baobab and Jatropa
The baobab is a tree called the ‘tree of life’ that lives in the harsh environment of Africa where other plants cannot grow. The roots look like they are standing upside down and stretching toward the sky. With a lifespan that can reach 5,000 to 6,000 years, a single tree can store 4,500 liters of water, making it a valuable plant that can provide life–sustaining water for people and animals in Africa. The bark and fruit are used as food, and are used as a panacea for diarrhea, fever, lumbago, trauma, recovery from fatigue, and head ache. The fibers of the bark are used to make ropes and clothing, and the trunks are hollowed out to provide shelter and storage for supplies.
Baobab seeds were purchased and tested for rooting. It did not germinate in water (control). While 1.0 ppm of SOMRE #1 accelerated the germination rate (Figure 78). On June 11, 2015, the germinated seeds were potted, and started to grow. On July 25, the seedlings grew to about 20 cm (Figure 79). The growth rate was fast (Figure 80), and 5 months later, on November 9, the tree height reached 100.5 cm (Figure 81). If it grew in this speed, it would become a large tree.
However, in December, winter begins in Japan. The temperature drops and it snows. Tropical plants could not survive in an outdoor environment without a greenhouse, and gradually withered. We expect that SOMRE would help it grow vigorously and fast under the tropic conditions.
15.3. Continued Expansion into Africa in Anticipation of the Times
A plant called Myrothamnus, which grows wild in the Sahara Desert, dries out and withers during the dry season. But once it rains and comes into contact with water, it reverts to green leaves in a few hours. It is called the “resurrection tree” because it has a mysterious vitality that allows it to survive in the dry desert environment. We believe SOMRE #1 would help this tree grow flourish.
15.4. Continued Expansion into Africa in Anticipation of Future [20,21,22]
In March 2016, “the ABE Initiative Masters and Internships Program”, Africa Business Networking Fair 2016 was held in Tokyo.[20] A large number of young master's students from all over Africa have participated in the program, hoping to contribute to the development of their home countries by acquiring the excellent Japanese technology. We joined together with Japanese leading companies and introdued about the potential of ‘SOMRE #1’ and ‘VED #1’, those are new technologies to realize increased food production, restore the devastated environment and deserts, and stop the expansion of the Sahara Desert, including the succesive achievements of greening activities in the Gobi Desert in China. It can change desert a green environment.The center–pivot method of groundwater irrigation, which is especially used in arid regions, is a poor technology that, in the long run, depletes groundwater, causes salt accumulation, and promotes desertification. We explained accumulating failures caused by human, such as the depletion of the Ogallala aquifer in the central part of the United States, the Gobi Desert in China, salt accumulation in cultivated land in India, the expansion of the Sahara Desert, and desertification in Europe.
As the author mentioned in the introduction, the total volume of water on Earth has remained constant and localized since prehistoric times. If ‘SOMRE #1’ is applied to weed seeds under natural conditions, the roots will grow and the desert surface will be covered with greenery with just one rainfall. Technology is being developed to collect water from the atmosphere using solar power even in the desert. Combined with ‘SOMRE’, water supply is possible, and grasslands can be restored and maintained. Desert surface temperatures could be reduced. Clouds will eventually gather, bring rain, and make the desert green. It will be possible to produce and increase the yield of crops and vegetables. We can restore the natural environment and turn the desert into a lush food base.
Figure 82 and Figure 83 show our booth at the ABE Initiative Master's Program and Internship held in March 2016.[20] It shows many students are gathering. Bright young minds were eager to bring back the ‘SOMRE’ and ‘VED’ technologies to their home countries and contribute to their country's environment and food supply.[21] Mr. Osward from Malawi and Mr. Koffi from Côte d'Ivoire, who were particularly active, held meetings at the International Center in ‘Shinanomachi’, as shown in Figure 84. Both of them brought ‘SOMRE’ back to their home countries when they returned and began concrete preliminary tests for increasing food production and greening the environment.
On April 23, 2021, in order to save the food crisis in Africa, we participated in Malawi's third event AFRI–CONVERSE 2021 #3, held for companies participating in JICA international student internships sponsored by JICA / UNDP. Mr. Osward presented the start-up plan using ‘SOMRE’ and the preliminary test results,[21] and he was able to obtain support from the Malawi government and JICA Malawi, and he decided to conduct a field test from 2022. We are looking forward to seeing his results.
16. Application of SOMRE to flowers and bulbs[22]
16.1. Hyacinth [22]
As a representative of flowers and bulbs, the results of hyacinth are introduced. 20 days after the bulbs were immersed in water (control), SOMRE solutions, beautiful flowers bloomed.
What is interesting is the number of flower buds. In water treatment, 21 flowers bloomed. On the other hand, the number of flowers increased to 15, 26, and 24 when 0.001, 0.01, and 0.1 ppm SOMRE solutions were used, and the maximum number of flowers, 34, resulted at 1.0 ppm (Table 25). As the concentration of SOMRE solution increased, the root length and density also increased, and growth was promoted. The most beautiful flower was produced by the action of SOMRE 1.0 ppm treatment (Figure 85 and Figure 86). Figure 85 is the photo of grown hyacinth. Figure 86 is the photo from left to right: control, 0.001, 0.01, 0.1, 1.0 ppm SOMRE treated flower.
16.2. Polygi and Calendula [22]
The overall growth state of Polygi and the state of each flower at each concentration of control and SOMRE are shown in Figure 87. The total number of flower buds on the 4 control plants was 80. On the other hand, the numbers of 4 flower buds grown at SOMRE concentrations of 0.001, 0.01, 0.1, and 1.0 ppm were 161, 130, 218, and 259, respectively. If the control value is 100%, those of SOMRE concentrations of 0.001, 0.01, 0.1, 1.0 ppm were 201, 163, 273, and 324%, respectively. As a result, when growing polygi, we can obtain many beautiful flowers by using a SOMRE concentration of 1.0 ppm.
In the case of Calendula, the condition of each flower at each concentration of control and SOMRE is shown in Figure 88. The number of petals of the control flower was 1,408. On the other hand, the number of petals of flowers grown at SOMRE concentrations of 0.001, 0.01, 0.1, and 1.0 ppm was 1,542, 1,928, 1,995, and 4,724, respectively. If the control value is 100%, SOMRE concentrations of 0.001, 0.01, 0.1, and 1.0 ppm were 110, 137, 142, and 336%.
In addition, the control flower was 4- or 5-fold flower, and when the SOMRE concentration is 0.001, 0.01, 0.1, 1.0 ppm, 6–7 fold, 7–8 fold, 7–8 fold, and 9–10-fold flower as shown in Figure 88. It turned out that using SOMRE, the number of flower seeds was more than three of the control. Consequently, we can use SOMRE for making beautiful Calendula flowers bloom.
16.3. Crocuses; ‘Pickwick’, ‘Jeanne d’arc’, ‘Flower Records’ [22]
We studied three types of crocuses, such as ‘Pickwick’, ‘Jeanne d’Arc’, and ‘Flower Records’. Thus, five bulbs of each plant are immersed in water and SOMRE 0.1 ppm aqueous solution for 1 h and grow in a planter. Control of ‘Pickwic’ and ‘Jeanne d’Arc’ did not germinate, but 4 ‘Flower records’ grew and flowered. Among the SOMRE–treated bulbs, one ‘Pickwic’, four ‘Jeanne d’Arc’, and all five ‘Flower records’ grew and flowered as shown in Figure 89. In conclusion, SOMRE has the potential to fill the world with beautiful flowers.
17. Protecting Endangered Plants [22]
17.1. Cultivation of ‘Himekomatsu’ by SOMRE #1[22]
In Chiba Prefecture, the number of ‘Himekomatsu’ trees has decreased to only 80, and it is designated as the prefecture's most important protected species as an endangered plant, and efforts are being made to protect and restore it. We were certified by the Chiba Biodiversity Center as supporters for the conservation of them and received a 38 cm tall poor sapling on February 8, 2016 as shown in Figure 90.
The roots of the seedling were immersed in a 1.0 ppm aqueous solution of SOMRE #1 for 1 h, potted, and then planted in a field. After that, the seedlings regained their vigor and grew vigorously, reaching a height of 84 cm (Figure 91) in 2017 and 139 cm in 2018.
The report compiled by the Chiba Prefectural Biodiversity Center on the growth status of the ‘Himekomatsu’ conservation supporters in each region says that since 2016 the number of dead trees has been increasing with the passage of time. Under these circumstances, the ‘Himekomatsu’ entrusted to us reached a height of 165 cm in 2019, over 210 cm in height as of October 2020, and in 2023, as you can see in the Figure 92. The tree is 370cm tall, twice the height of a person, and is growing vigorously.
We believe that SOMRE treatment will also help to regenerate other endangered plants.
17.2. Traditional Japanese Culture[23]
17.2.1. Lacquer Culture[23]
Lacquer is used as a surface coating film for national treasures, important cultural properties, architecture (shrines, temples), fixtures (jubako, bowls, chopsticks, trays), works of art, ornaments, etc., and its robustness, heat resistance, and water resistance. Not only does it protect the interior covered with lacquer, but its rich luster, color tone, and brilliance are the cornerstones of Japanese culture. Kyoto's Rokuonji Golden Pavilion, Hiraizumi's Chusonji Golden Pavilion, a national treasure, and other buildings with lacquered walls and floors covered with gold leaf are world–class in beauty. It is the mission of the Japanese to protect this important traditional Japanese culture and the techniques of lacquer and gold leaf crafts.
Lacquer sap (urushi) is a collection of sap that is secreted from the trunks of mature lacquer trees that have grown for 10 to 15 years after being planted. About 200 mL of sap can be obtained from one tree, and the tree is then cut down to complete its life cycle (Figure 93 and Figure 94). This is a very cruel method called ‘Koroshikaki’.
Japanese lacquer becomes more durable as it ages, and its color and gloss also improve. The demand for Japanese lacquer, which is required for repairing traditional precious national treasures and various cultural properties, is increasing with the passage of time. The annual production volume in 2009 was about 1.4 tons, which is far from the demand (about 69 tons).
In order to make up for this shortage, a large amount of Chinese lacquer has been imported in recent years, and Chinese lacquer is used for 98% of the demand. However, because the chemical composition of the lacquer is different from that of Japanese lacquer, Chinese lacquer will crack after two years. Supply measures and solutions for Japanese lacquer are required to maintain Japan's ancient traditions and culture.
Currently, lacquer production areas in Japan are mainly ‘Johoji’ in Iwate Prefecture. Lacquer seeds from ‘Johoji’ are used to produce many lacquer seedlings. For this reason, we decided to use lacquer seeds from ‘Johoji’ (Figure 95). We also tried branches of a lacquer tree that had been fallen and abandoned after tapping lacquer were cut, as branch materials for lacquer cuttings in a lacquer plantation in ‘Ninohe City’, Iwate Prefecture. Furthermore, after tapping the lacquer, the roots of the lacquer trees that had been fallen and abandoned were cut and used as materials for the roots of the lacquer roots.
It is already known that certain wavelengths of light promote plant growth. Therefore, we compared the rooting method of lacquer seeds, which is usually performed, and the method of treating with SOMRE #1 aqueous solution. Rooting and germination experiments were carried out under light irradiation. As a result, we discovered a new technology that only adding SOMRE treatment to the usual way can improve the germination rate of sumac seeds, improve the success rate of cuttings, and regenerate from roots.
This means that we succeeded in developing a new technology to regenerate the life of the lacquer tree as a new life without losing the life of the lacquer tree. Since we can fully expect to produce and increase the production of urushi, we are currently conducting further investigations. Since we are considering a patent application, we will refrain from describing the detail.
17.2.2. Japanese Paper ‘Washi’ Production[23]
Beautiful to the touch, durable and long–lasting, handmade Japanese paper is one of the arts and crafts mainly protected and supported by the three prefectures of the Hokuriku region (Ishikawa, Toyama, and Fukui). Invitations, guarantee paper, fusuma, calligraphy paper, business card holders, accessory cases, etc., have supported our daily lives and formed the traditional culture of Japan.
In recent years, as lifestyles have changed, the demand for Japanese paper has decreased, and this has led to a decline in the production of raw materials such plant as ‘kozo’, ‘mitsumata’, and ‘ganpi’. It takes 1 year for ‘kozo’, 3 years for ‘mitsumata’, and 15 to 20 years for ‘ganpi’ to grow enough to be harvested as a raw material for paper. It is necessary to plant and nurture trees systematically. ‘Neri’, which is indispensable for making handmade Japanese paper, is a slime produced by the roots of the annual ‘tororo–aoi’ plant. The production of ‘kozo’, ‘mitsumata’, ‘ganpi’, and ‘tororo–aoi’ is facing the problems of securing people to inherit the techniques and securing livelihoods due to the drop in demand, in addition to the problem of an aging population. Omitama City, Ibaraki Prefecture is currently the center of production of ‘tororo–aoi’.
We hope that SOMRE can solve this problem as well. SOMRE increases the rate of seed rooting and accelerates plant growth. If we can shorten the growth period from 10 years to 5 years, we can improve productivity, protect traditional culture, and contribute to further development. Based on this policy, we would like to examine whether ‘SOMRE’ works as expected for the production of ‘kozo’, ‘mitsumata’, ‘gampi’, and ‘tororo–aoi’.
We believe that SOMRE could not only help maintain, pass on, and develop new Japanese traditional culture, but also fill the Earth with plants and return it to a rich, green environment.
18. VED Compounds
18.1. Conception and Development of VED Compounds
The author has been suffering for a long time from the ‘itchiness’ of atopic dermatitis that did not improve after seeing a dermatologist. He wanted to create new medicine that had never been isolated as a natural product and cure his illness. The mechanism by which itching occurs is still unknown, but from his many years of experience, he felt it is related to blood circulation and active oxygen scavenger. As explained in Section 8-2, he developed his own 1-hydroxyindole hypothesis[6] into tryptophan and IAA with the aim of developing root and stem promoters for plants. Therefore, it was natural for him to turn his attention to yohimbine (54, Figure 96).
The alkaloid 54 is a Chinese herbal medicine and well known with its peripheral vasodilator as an α2 receptor blocker, and is prescribed for erectile dysfunction (ED).[52] The author conceived the idea to apply the chemistry of 1-hydroxyindole to synthesize a fantasy compound, 1-hydroxyyohimbine (55b), expecting for development of medicine for saving his own itchiness.
18.2. 1-Hydroxyyohimbine[52]
Yohimbine skeleton loses its action in response to a subtle change in the structure, for example introduction of a methyl group to the 1 position (55a) eliminate α2 blocking action. We created 1-hydroxyyohimbine[52] (55b), the N–OH form, which is the expected product after functioning as active oxygen scavenger. And then we found the α2 blocker action of 55b was maintained without diminishing. Additionally, the strong toxicity of yohimbine was reduced. As a result, we could discover its possibility to become a new ED treatment medicine.
Based on the author's intuition on the relationship between vasodilation, itching, and skin diseases, we searched for a substance with a simpler structure, improving blood circulation and active oxygen scavenger. During exploring for creating a non–toxic and safe α2 blocker having peripheral vasodilator action, improving blood circulation, promoting growth and reproductive ability of animals, and producing offspring, for animal food production, we have analyzed the chemical structure of yohimbine, comparing the chemical structures of compounds produced in the imaginary metabolic process of tryptophan, we paid attention to the structure shown in the dotted parenthesis of 56, namely tryptamine derivatives 57.
18.3. VED Compounds: Synthesis of VED #1 (Nb-Nonanoyl Tryptamine) and 1–Hydroxy-Nb-acyl Tryptamines[53]
Many physiologically active substances such as hallucinogens, neurotransmitters, and various alkaloids are known to be tryptamine derivatives.[8] Tryptamine itself exists in the body as a trace biogenic amine, but it is an unstable substance that is extremely difficult to handle. When isolated, it forms colorless crystals, but is oxidized by oxygen in the air, quickly turning into a black, sticky liquid. Due to this instability, its pharmacological tests could not be carried out until now. The author thought this is the reason why the potential hidden in this precious treasure, tryptamine, could not be discovered.
In order to make it possible to freely handle this unstable and easily oxidizable organic molecule, the author came up with the idea of creating 1-hydroxyindole derivative, which is presumed to be produced as a result of functioning as an antioxidant. Or adding an acyl group to Nb to make it resistant to oxidation. We set up a research policy to synthesize Nb-acyl-1-hydroxytryptamine, which is expected to be formed by oxidation in the body and to investigate its physical properties and pharmacological actions.
As mentioned earlier, the 1-hydroxyindole derivative is an imaginary compound that has never been isolated as a natural product. Therefore, it was not known whether the target compound could exist in the real world. Therefore, it took several decades for us to create a general synthetic method for 1-hydroxyindoles.[6] We were lucky to create our own general synthetic method (Scheme 3),[54] which is simple and easy to oxidize 2,3-dihydroindoles with a mild oxidizing agent, H2O2. As a result, it became possible to convert arbitrary indole compounds to the corresponding new 1-hydroxyindole derivatives.[54] Consequently, it became possible to synthesize any desired 1-hydroxy-Nb-acyltryptamines by the method shown in Scheme 5.[6,54]
First, tryptamine was reacted with various fatty acids (56) using the mixed acid anhydride method to synthesize Nb-acyl tryptamines (57) in good yield. All compounds of 57 were stable crystals. Subsequent reduction with triethyl silane led to the corresponding 2,3-dihydroindoles (58). Furthermore, when 58 was reacted with hydrogen peroxide using sodium tungstate as a catalyst, the imaginary new group of Nb-acyl-1-hydroxyindole derivatives (59a-g) were successfully obtained as stable crystals.[54]
All these compounds showed α2 receptor inhibitory effect.[55] The effect became stronger as the chain length of the Nb side chain became longer. When the number of Nb side chain was 9 (59d), it was the strongest.[55] Its strength was 79.0% of that of yohimbine as 100%
We also investigated the strength of the compound group 57a-g in which the 1-substituent is hydrogen, two steps before the synthesis of 59a-g. The α2 receptor inhibitory effect was equivalent to that of compound groups 59a-g (Table 26).[55] This is the reason why we named 57d as VED #1, taking the acronyms of Vasodilator and Erectile Disfunction of pharmacological actions. Regarding the safety of VED #1, the Ames test, human skin allergy test, and toxicity test were conducted by public institutions, and we could confirm that there is no toxicity.[55]
When VED #1 was made into a skin care cream and applied it to the author's itchy skin, he surprised that the itchiness disappeared almost instantly[41] (section 20-1). Despite seeing dermatologists for many years, there was no effective treatment, especially at night he could not stand the “itch” and continued to be sleep deprived. Desperately wanting to escape from suffering, every time a new compound was created, he applied it to the itchy areas as a trial. And finally, the moment to apply VED #1 to his skin came. The itch disappeared as soon as he applied it, and the pain of the wound being caused by scratching decreased,[37,38] and the wound healed in a week.
19. VED #1 Experiment Using Cashmere Goats at Gobi Desert
We selected two dairy farms run by nomads in the Arashan region of Inner Mongolia, China, and carried out experiment twelve times from July 2005 to May 2008 for the purpose of cashmere hair growth,[25] breeding,[27] and increasing dairy farm production at two independent grazing lands of cashmere goats in the Gobi Desert.[26,27,28] The experiments were repeated and the reproducibility was confirmed.
19.1. Cashmere Hair GROWTH experiment[24,25]
We purchased a total of 8 cashmere goats from two ranches, 6 and 2 goats, respectively, run by two nomads, respectively. They were divided into the first set consisting of 3 VED–treated animals and untreated 3 control animals, and the second set consisting of 1 VED–treated animal and 1 untreated control animal. That is, a total of two sets were prepared and bred under the same conditions for one year.
Despite being given the same amount of feed, the goats in the VED group grew faster, healthier and bigger. As a result, as shown in Table 27, the amount of cashmere hair increased by 1.2 to 1.7 times (Figure 98) the weight of the control group (Figure 97). We observed the hair of the control group was white like cotton, whereas the hair of the VED group was silky white and shiny, indicating high quality.
19.2. Breeding by VED, Animal Food Production[26,27]
Although the locals have tried various tricks and efforts to breed the cashmere goats, there was still the problem of a low percentage of female goats becoming pregnant. In our breeding experiments, two male goats were placed into 90 female goats, and mating was allowed freely as much as possible. In the control group 20–30 kids were born. Interestingly, in the group where 2 males were fed 1.0 mg VED once daily in the diet, all 90 females became pregnant and gave birth 90 kids.
According to the farmer's observations, the two goats go into heat when they take VED, and their penises lengthen and they begin to act. Figure 99 shows a part of the 90 baby goats. It is noteworthy that survival rate of the kid goats was 100%. Over the second to fourth years, re–experiments were conducted to confirm reproducibility. The VED–taking goats were living happy lives as seed goats, and the expected results were obtained.
Due to the age of the farmer, the number of goats to be cared for was reduced to 80 in the fourth year, but during the four years, a total of 350 (90+90+90+80) goats were successfully born from a total of 350 female goats.
As for nomads, the production of cashmere wool, the sale of young goats, and a sufficient supply of meat became possible, and the author will never forget the happy faces of the ranchers.
19.3. Increasing Animal Food Production and to Livestock and Poultry[26,27,28,29,30]
Many other applications are possible due to the success with cashmere goats. Animal hair production, which is popular with some people, is being banned in the spirit of animal welfare because it is produced by killing animals.
We believe VED could be used for shearing annually to produce expensive animal hair such as sheep, angora rabbits, picuna, etc. VED technology could be applied to increase the production of livestock such as cattle, pigs, and chickens, as well as poultry, and would be of great help in increasing the production of animal foods.[29,30] We believe that it will also be useful for the breeding of rare species of animals and birds that live in zoos. In China, the author wanted ‘panda’ to breed, but they wouldn't let us experiment. I think it will be possible to apply it to endangered species of animals. We believe that it will also help to maintain and restore biodiversity.
19.4. Hair Thickening, Improvement of ed, and Rejuvenation (Anti-Aging)[33,34,35]
We considered that VED is also effective in treating ED in humans.[29,30] After confirming the results shown in section 19-2, the interpreter, a Chinese man in his 40s, voluntarily tried for medicating his ED. VED played wonderful role, and his body was full of energy, became active, and his child was born soon.[30]
Based on the epoch–making results of increasing the cashmere hair production in the Gobi Desert, a VED hair tonic for thinning hair in humans was created and tested, and we found to be as effective in humans as it is in goats. In a mouse back hair test, VED (59d and 61d) was slightly weaker than ‘Minoxidil’ for hair growth.[35] However, in a mouse beard hair extension test, we found it to be strongly more effective than ‘Minoxidil’.[35]
In the case of humans, we found it is useful for improving thinning hair regardless of age or sex. In particular, a girl diagnosed with alopecia areata regained her bushy hair after only half a year of VED spray. A bald male exchange student from Africa tried it (Figure 100). After 10 months use, the hair returned as shown in Figure 101. He was very grateful to us.
20. VED Is a Treasure Trove of Various Pharmacological Actions
We have found that Nb-acyl-1-hydroxy- and -tryptamines are novel and structurally simple α2-blockers[53,55] for the treatment of erectile dysfunction. Furthermore, they have potent inhibitory activities on platelet aggregation.[36] Daikon and wasabi phytoalexins are weak fungicidal alkaloids[47,82] having stabilized 1-methoxyindole structure. Quite recently, we found that 1-hydroxy-Nb-nonanoyltryptamine had potent hair growth action[35] Judging from these facts: we hope that the chemistry of 1-hydroxyindole is a treasure field where a lot of new biologically active compounds are buried under the ground to be dug up.
Among the Nb-acyl tryptamines, Nb-nonanoyl tryptamine (59d) had the strongest α2 blocker action, and was used to investigate various pharmacological tests as much as possible. Consequently, the secret veils of 59d have been revealed one after another, and then we have found that VED #1 (59d) is a treasure trove of various pharmacological actions as follows.
20.1. Instant Disappearance of ‘Itching’[41]
Many people suffering from the itching of atopic dermatitis have been saved by VED #1 cream thus far. One of the voices of joy: my child's atopic dermatitis didn't heal even with the medicine prescribed by the dermatologist, and he cried at night and couldn't sleep. I felt sorry for him and was having a hard time every day. When I applied your VED cream, the itching subsided with just one application and she fell asleep. It's like magic and I'm surprised. Thank you.
20.2. Skin Spots, Wrinkles, Dullness, Rough Skin, Pimples Treatment[32,37]
Due to peripheral vasodilation, the supply of nutrients, minerals, etc. to the basal cells of the skin becomes sufficient, promoting proliferation and increasing the rate of metabolism. As a result, the senescent cells that form blemishes and the cells that form pimples are pushed out of the body by new cells and discharged. Figure 102 is a progress photo of a 67-year-old girl using the cream, with aged spots and wrinkles disappearing (Figure 103).
Even men and women in their 80s having many dark spots, wrinkles, pimples, and so on, on their faces, hands, and body, said their troubles fade after a year use of our cream, and they feel younger both mentally and physically. The author also shares their joy.
20.3. Improvement of Acne and Acne Scars[32,37]
VED cream is highly effective for treating acne scars. In December 2021, a 16-year-old junior high school boy was prescribed the medicine ‘Duac’ by a doctor at a dermatology clinic, and he got worse day by day, as shown in Figure 104. He became depressed and could no longer go to school. Her mother found us on the internet and started using VED cream. At first the boy didn't trust anything other than ‘doctor's medicine’. However, symptoms did not improve and her mother repeatedly recommended our cream, he gave it try and immediately experienced the ‘difference’.
He stopped using the doctor's medication quickly and switched to using ‘VED cream’. Then it started to get better and better (Figure 105), and as of June 10, 2022, his beautiful youthful skin was back (Figure 106), and rescued from depression.
20.4. Treatment of Wounds and Cuts, Elimination of Pain[32,38]
One of the voices of joy: for about 40 years, steroid treatment and other attempts were made. However, atopic dermatitis did not improve at all. But I applied your VED cream. I could get rid of the itch, so I didn't have to scratch it. The scars have healed and my skin has improved. I am very grateful to you. My doctor is present here.
His doctor’s opinion: The effect is great, so I will employ it for my patients for a long time. I would also like to use it for my patients suffering from interurticaria. The pain disappear and the wound will heal faster. VED cream healed scratches quickly and could be applied to cut and abraded skin.
Not only children, but even 80–year–olds can heal their wounds in about two weeks as shown in Figure 107 and Figure 108. Pain subsides from the moment of application. Figure 108 is the photo taken two weeks after applying the cream.
20.5. Platelet Aggregation Inhibitory Action and Dementia Suppression[36]
Almost all 1-hydroxyindole derivatives (60—65) showed potent inhibitory effects in the platelet aggregation test of arachidonic acid using rabbit platelet–rich blood. Its strength was one order higher than that of ‘cilostazol’ (Table 28).[36]
‘Cilostazol’ (66), which has an inhibitory effect on platelet aggregation, is under development as a substance that inhibits the progression of dementia.[56] The 1-hydroxyindole derivatives are also expected to be developed in the future as candidate medicines for the treatment of the disease.
20.6. Treatment of Frostbite, Poor Blood Circulation, Hemorrhoids, and Burns[37]
A 70–year–old man suffered frostbite on the heels of both his feet during the extreme cold. His blood circulation was poor, and his skin was purple and bloated and edematous (Figure 109). Since VED has a peripheral vasodilating effect, it was applied to the affected area as a spray. The initial pain disappeared immediately after spray. One month later, the formation of a separated layer was observed, (Figure 110) and a week later, the local part was separated like a thin film and peeled off and the lesion was completely cured after 3 months.
People often lose their fingers and toes due to poor blood circulation due to diabetes. We believe that the use of VED would realize the possibility of restoring blood circulation and healing.
Similarly, burnt skin regenerates quickly as the pain disappears. The blisters will gradually subside, the liquid from the blisters will be absorbed, and the skin will peel off and heal quickly. This is because of its metabolism speeds up effect.
20.7. Arthralgia, Muscle Pain, Improvement of Arrhythmia, Cramps[37]
After hard sports, farm work, and physical labor, muscle pain and joint pain remain in the legs and lower back. It took longer for the pain to disappear with age, and after reaching the age of 60, muscle pain remained for 4 to 7 days after work, and it was always difficult to start a new work. However, after taking VED, muscle soreness appears within the day and feel as if he became younger. Almost 100% of muscle soreness disappeared in one day, whether taken VED before or after exercise. We have thus succeeded in developing a technique that is healthier and more effective than drinking alcohol to deal with the pain.
On the other hand, the company president in his 50s, who was prescribed nitroglycerin for arrhythmia, often interrupted meeting. He listened to the explanation of VED #1 and voluntarily took 1.0 mg of VED. He recovered quickly and led the meeting for three straight hours without troubles. Five years have passed since then, but he uses VED habitually and is not under the care of a doctor.
A 75-year-old man had sudden leg cramps when waking up,[37] and a 70-year-old man had leg cramps during playing baseball as a catcher (as shown in Figure 111). The muscles in their legs were cramped and tense, and the pain was so severe that they couldn't move. At this time, when they applied VED cream to the skin of the stiffened muscle, the pain began to disappear in just a few minutes. Over time, the pain disappeared and they were able to move legs freely.
It is thought that the pain disappears due to the dilation of peripheral blood vessels distributed near the skin surface. We think conventional medical knowledge of the etiology of cramps appears to be erroneous.
20.8. Regarding Suppression of Apoptosis and Coexistence with Cancer[33,34]
ROS17/2.8 cells, exposed to various concentrations of VED #1, were cultured under starvation conditions, and the number of surviving cells was measured, and chromatin condensation was observed by DAPI staining. We found that VED inhibited starvation–induced cell death and chromatin condensation. Furthermore, we found an inhibitory effect on apoptosis. This effect was found to induce the expression of BcI-2 mRNA and suppress the expression levels of Bax, Bak and Bad mRNAs. These facts indicate that VED is useful for diseases caused by a decrease in normal cells caused by neurodegeneration such as Alzheimer's disease, Parkinson's disease and cerebral ischemia.
The author’s sister–in–law was diagnosed with terminal pancreatic cancer and was given one month to live. With no effective anti-cancer drugs, she was left to die. Since VED #1 also activates protein synthesis and immunity enhancement, she voluntarily took it in the hope that it would activate immune cells and prolong the time coexisting with cancer. She regained her appetite and gradually regained her energy, giving her and her family a bright hope. Seven months later, she was still cheerful and was able to chat with her caregivers.
Occasionally, when relatives are not present, visiting doctor met her, and he recommended her to get a ‘corona vaccine’, and he gave it with her consent. After the vaccination, her condition suddenly changed and she died two days later. She had high hopes for VED and was happy that her life expectancy was extended by half a year.
20.9. Dementia, Cerebral Hemorrhage, Cerebral Infarction, Myocardial Infarction[24,31,36]
In May 2018, the 76-year-old author suddenly became paralyzed on the right side of the body. It was during the consecutive holidays. He went to a nearby hospital, but there was no neurosurgery. His family could search for a suitable hospital three days later. Driven by his daughter, he was taken to the hospital in their own car.
MRI and CT scans revealed a hemorrhage in his left brain, and a spherical black shadow containing 5 mL of blood. The doctor told that there is no ‘cure medicine’ at present, so he decided to continue observation at the doctor's discretion.
The author considered that he would be forced to live in bed until his death. By the way, he has already developed VED #1. It has a platelet aggregation inhibitory[36] and peripheral vasodilating effects, so he believed it would be sure to dissolve clumps of hardened blood clots and help absorb and expel blood clots. He self–administered 10.0 mg of VED daily in the hope that it would remove the clots from the brain. Meanwhile, he started rehab lightly on his own.
First, he could not walk, could not write or speak, and his memory was shaky. After taking VED, the effect gradually began to appear, and after three months, he was able to walk indoors, albeit clumsily. Half a year later, he was able to walk almost stably, and his language was restored. He was surprised by the amazing effects of VED, and he secretly hopes that it will be of use to many people. And six years later, he is walking around without any inconvenience. Diagnosis by CT and MRI examinations revealed that bleeding clots had almost disappeared, as his expectation.
An 89-year-old woman with dementia used VED–containing cosmetics, and she was pleased to be seen that her skin became clearer with less spots. During that time, her family noticed that her dementia was becoming less severe. With her family's wishes, taking VED showed gradual improvement, and she surprised her family by showing her willingness to go out and work in the fields. A year later she passed away at the end of her life. The author regretted that we should have known each other sooner.
VED improves cerebral blood flow, improves blood circulation, suppresses platelet aggregation, makes blood smoother, and has a vasodilating effect, so it may be a therapeutic medicine for myocardial infarction and arrhythmia. This is a subject of near future investigation as a medicine that would be useful in an aging society.
20.10. A panacea Solving Skin/Body Troubles
There are people in the world who are suffering from various skin and body problems. Cosmetics and medicines have been developed for each trouble, and patients have to buy corresponding each product. If we could develop a single substance with multiple functions, it will become an ideal substance (almighty substance).
We have completed patent acquisition of VED that can deal with almost all skin troubles and related body troubles, backed by evidences and patents concerning to sections 20-1 to 20-11. In other words, we were able to create the ideal (almighty) substance that we were aiming at. In this way, we have provided ‘Ritayakko’ products (skin care cream, hair nourishing agent, and ‘pinpingenki supplement’ (nutritional supplement for healthy longevity)) that use VED #1 as the main ingredient, and have saved many people around so far.
20.11. Potential Drug for Osteoporosis[42,43]
Considering the bone tissue of osteoporosis patients, osteoblasts and osteoclasts coexist and interact closely with each other. The usual evaluation method for developing osteoporosis drugs, either osteoblasts or osteoclasts are cultured alone. A test compound is added to each system to determine whether it exhibits a growth or inhibitory effect. Therefore, the known evaluation method is not suitable for developing new medicines.
We applied the assay test using gills of goldfish developed by Suzuki and Hattori.[57,58] An excellent feature of the method is that it is a system in which osteoblasts and osteoclasts coexist within the gills and interact with each other. Therefore, it can be said this is the most excellent assay method for discovering drugs for treating osteoporosis.
The test compounds were synthesized as shown in Scheme 6. The reaction of Nb-acetyl-5-methoxy- tryptophan methyl ester (67) with bromine afforded 2,4,6-tribromo derivatives (68) exclusively together with small amount of 2,6-dibromo- and 2,4,7-tribromoderivatives. Further allylation, propargylation, and benzylation of 68 afforded high yields of the corresponding 1-allyl- (69a) , 1-propargyl- (69b), and 1-benzyl- (69c) tryptophan derivatives.
The same series of reactions were successfully applied to melatonin (70), which was obtained by treating 60 with 50% (MeOH)2·BF3 in good yield. Bromination of 70 afforded excellent yield of 2,4,6-tribromo- melatonin (71). Subsequent allylation, propargylation, and benzylation afforded high yields of the corresponding 1-allyl- (72a) , 1-propargyl- (72b), and 1-benzyl- (72c) 2,4,6-tribromomelatonin derivatives.
We subjected 1-substituted 2,4,6-tribromo- derivatives (69a-c) and 72a-c to the gills assay test. Happily, we found that 69c has the strong effect of activating osteoblasts. The compound 72c was also discovered to strongly promote the activation of osteoblasts and suppressing osteoclasts.
The test compounds were synthesized as shown in Scheme 6. In the acute toxicity test of 1-benzyl-2,4,6-tribromomelatonin (69c), according to the OECD (Europe) guidelines, 2 g per 1 kg of rat body weight was administered, and after 2 weeks of breeding, toxicity was not observed. Furthermore, no toxicity was observed in a mutagenicity test in which rats were administered 2 g per kg of body weight and evaluated after feeding for 2 weeks according to the OECD guidelines. Addition of S-9 mix (rat liver homogenate) did not change.[57,58] Therefore, we were able to discover compounds 69c and 72c as potential therapeutic candidate compounds.[42,43,57,58]
21. Proposals of a Concrete and Actionable ‘Cure for the Earth’[10,11,12]
21.1. Aerial Spraying of SOMRE–Treated Seeds onto the Desert
The author proposed a concrete ‘Earth Cure’, shown in Figure 112, to both green the desert and to prevent generation of yellow sand for stopping global warming based on the scientific grounds described above, at the 7th International Bio Forum held at Tokyo Big Sight on July 2, 2008.
Figure 112 is a schematic diagram of the Gobi Desert. It shows the progress of desertification toward the right–side direction. The desert shows a transition from bare fluidized dunes to semi–fixed dunes, and then to green areas. As a ‘healing method for the earth’, first select the time before the rainy season. Then, plant SOMRE–treated trees in a wide long belt, at a location about few km from the front of the sand dunes. Next, seedlings whose roots were soaked in SOMRE solution are planted in the same manner on the green side. Similarly on the further green side, SOMRE–treated seeds are sown. It is a method of counterattacking the advance of the moving sand dunes by arranging the walls of plants in three stages. In addition, the grass square ‘草方格’, which is the wisdom of our predecessors, is also adopted and placed here and there in the flowing sand dunes. For each grass square, trees, saplings and seeds should be treated with SOMRE solution.[10,11,12] The seeds treated with SOMRE solution should be sown by air–plane (Figure 73 and Figure 74), drone, or tractor. Employing the method, a single person can handle vast desert surfaces in one day.[16]
Seeds of grass and weeds should also be treated with SOMRE liquid and air-scattered. VED #1 is applied to goats and other livestock while growing pasture. Raise goats and livestock with less grass than before, and promote the greening of the desert more reliably while maintaining the current standard of living for nomads.[16]
21.2. To the Deserts of the World[10,11,12]
We believe that it is also possible to apply SOMRE to the world's deserts, each of which has its own personality. Kalahari watermelon is a valuable plant in the Kalahari Desert of Africa. This watermelon has no sugar, so it can be stored well. Therefore, as the only source of water, it is stored for a long time after harvesting and is even used for the first bath. We found that the SOMRE #1 aqueous solution showed remarkable effects in improving the germination rate of the watermelon seeds, promoting rooting, and much harvest.
Among various SOMRE compounds, in just a few weeks, using SOMRE kit, we can find the right SOMRE product that are compatible with plants that grow naturally in various deserts around the world. If we are willing to use the selected SOMRE, even now, mankind can deploy the Earth medicine described in Section 5 to deserts around the world to restore the green Earth filled with plants.
22. In conclusion, ‘SOMRE’ and ‘VED’ Save the Earth beyond the SDGs
22.1. Realization of an Eco-Friendly World with ‘SOMRE’ and ‘VED’
A boyhood dream of covering the Earth with plants, transforming deserts into lush forests, transforming them into food bases to save food crises and prevent climate change comes true. The author was finally able to acquire the new technologies ‘SOMRE’ and ‘VED’ necessary for that realization, just before he reached retirement age. We must stop the earth's land surface area will become barren deserts. In a SOMRE world, there is no need to pump up groundwater and water the desert. Using only natural rainfall as a water resource, the vast desert can be greened at once by spraying SOMRE–treated seeds from an airplane (or drone) onto the desert on the days before and after the rain is expected according to the weather forecast. Plants grow by extending their roots and stems deep into the ground, weeds grow abundantly, and grasslands regenerate.[59]
When the surface of the desert is covered with greenery and the surface temperature drops, clouds are brought in and rain return, and climate change slows down. Deserts form vast lush CO2 absorption zones. Plants release oxygen while converting CO2 into sugar, protein, and food. The desert becomes a food base.
Treating roots and stems with SOMRE eliminates the need to use pesticides, allowing rice, vegetables, soybeans, wheat, potatoes, and corn to grow healthily and agricultural production to thrive. When the rain returns, the green environment will be ready, and forests and lakes will be revived. Fruit trees, baobabs, jatropha, rubber trees, palms and mangroves grow. CO2 is absorbed at the bottom of the sea, seaweed grows vigorously, and desertification of the sea stops. Fish and shellfish grow and become a rich food base. Land and sea biodiversity will be secured, global warming and climate change will be controlled, and the global environment will be improved. Cool air, soil, and clean water fill the ground and underground. Plants in forests and grasslands serve as fodder for livestock and animals, and also provide habitats for animals and birds. Dairy farming also flourishes. Through the cooperation of a huge number of plants, CO2 will be converted into food, yields will increase, and hunger will disappear from the world. On the other hand, in the world where VED is used, plants grown in the SOMRE world are used as fodder, and many kinds of animals, birds, and insects grow and procreate. Since VED is an animal drug, pets, cows, pigs, and livestock grow up healthy. The dairy industry develops without the use of growth promoters or hormones, and without polluting the soil. Human and animal wastes are a source of nutrients and fertilizers for plants, and grow a rich green earth. Humans, animals, and fishes and shellfishes maintain their health with ‘VED’ with sufficient food and feed in a cool atmosphere and green environment. In addition, soybeans and corn mixed with VED are used as feed for breeding, increasing animal food production.
The harmful effects of ultraviolet rays on terrestrial organisms are protected by the ozone layer generated from the oxygen produced by plants. VED protects animal and human skin from cancer–causing spots. The body can be protected from aging and maintain health due to the effect of improving QOL due to the effect of promoting metabolism. As shown in Figure 114, ‘SOMRE’ and ‘VED’ create a green earth, ensure a virtuous cycle of resources, and establish a peaceful world. It is the only concrete and viable technology solution that can be used to restore and protect the Earth's ecosystems.
If humans, only with a desire to seek profit, do not enter the peaceful world described above, and do not artificially destroy the natural environment or take selfish actions, war, food loss, and starvation due to food depletion will disappear. A peaceful and safe world of SOMRE and VED can be realized. The author hopes that all life forms that coexist on the planet Earth will live out their lives in harmony.[60]
In India, a Nobel Prize–winning researcher at Monsanto, once called the ‘Green Revolution,’ turned vast farmlands into barren land due to soil contamination and salt accumulation. Even recently, they developed ‘glyphosate’, an ingredient in the herbicide ‘Roundup’, which is suspected to be carcinogenic, and is polluting farmlands around the world. More than 40,000 lawsuits have been filed for damages. Scientists should reflect on the foolishness of being dominated by the desire to seek selfish profit for the sake of temporary profit. Don't kill the Earth itself.
In India, SOMRE has demonstrated its ability to increase yields of rice and Indian vegetables. The authors started efforts to gather promising young people from African countries such as Malawi, Cote d'Ivoire, Zimbabwe, and Namibia who participated in the ‘Abe Initiative’. And also began to form a team for developing Africa's agriculture and livestock, relieving hunger in African countries, and improving the natural environment. Ultimately, the authors would like to realize the greening of the Sahara Desert with the help of native plants. As a basic experiment, ‘SOMRE’ has successfully grown baobabs and Kalahari watermelons. If we can increase the yield of plants whose roots and stems are edible, such as sweet potato, taro, cassava, maize, and millet, it will be possible to save the people currently suffering from hunger. Without relying on other countries for food, which is important for food security, each country can produce and secure the necessary amount, and solve the food shortage in Africa. While securing food and fodder, the rest can be supplied for bioethanol. ‘SOMRE and VED’ can make the competition between humans who buy farmland meaningless. World hunger and natural environment can be protected.
Since prehistoric times, plants and animals have coexisted with humans on Earth, respecting each other's lives. ‘SOMRE’ and ‘VED’ will maintain their relationship and will not break. We do not manipulate the genes of plants, animals, or fish. We will use them as they are now. It is a safe and environmentally friendly technology that increases food production and supply. It is possible to stably produce and supply seeds, achieving high food yields and high profitability.
The authors have demonstrated the fact that ‘SOMRE’ and ‘VED’ can increase the current food yield by at least 110–150%. The authors believe that if the increase in food production resulting from the greening of deserts is added to this, the Earth can comfortably support a population of 8.8 to 12 billion people. We don't think there is any need to create new organisms or new strains after genetic manipulation or alternative foods or insect foods with unknown safety and hazards.
Humans are part of nature. Harmony with nature should be pursued in the spirit of harmony in the mountains, rivers, plants, and all things as Japanese culture says. All living things can live a peaceful and healthy life. The chemistry of ‘SOMRE’ and ‘VED’ is ‘hope’ to save the entire planet, the Earth, which has green plants and the earth, clean water and life forms wrapped in the atmosphere, beyond the SDGs.
22.2. Summary of Some Plants, Which Increased Crop with ‘SOMRE’
Figure 115 summarizes the plants that have been examined so far and have been found to increase yield and promote growth using SOMRE #1. Vegetables/crops, flowers/herbs, herbal medicines, and cuttings of various mature trees grow well.
Other endangered plants and native plants of the Gobi Desert also grow well. All of the plants described here increased their crop yields by at least 110–150% over the control, and the dryland/desert native plants were significantly growing two to three times more than the control.
22.3. Space Age Considerations
Solar panel based on silicon will have serious disposal problems in the near future. In terms of disposal issues, establishment of rapid silicon recycling technology, and power generation efficiency, we should switch to ‘perovskite’ solar cells. These would be a favorable energy generation technologies compatible with the ‘SOMRE’ and ‘VED’ world described in section 22-1.
The world can be realized in the spaceships sailing through space. On new planets, it can also be deployed to construct dome like space colony or station, which has healthy and rich green environments.
The authors sincerely hope that outer space will not be polluted by rockets and spaceships waste that were made by humans.
Part 3 is an experimental section that allows readers to actually synthesize chemicals stated in this review such as SOMREs, VEDs, 1-hydroxyindoles, 1-hydroxyyohimbines, and so on. By following these methods, readers could synthesize the corresponding new 1-hydroxyindole compounds from any indole compound, and thus discover new pharmacologically active substances.
23. Experimental
Melting points were determined on a Yanagimoto micro melting point apparatus and are uncorrected. Infrared (IR) spectra with a Shimadzu IR-420, a Shimadzu IR-460, and a Horiba FT-720 spectrophotometers, UV spectra with a Shimadzu UV 2400 PC spectrophotometer, and 1H-NMR spectra with JEOL FX-100, JEOL EX 270, and JEOL GSX 500 spectrometers, with tetramethyl silane as an internal standard. Mass spectra (MS) were recorded on a Hitachi M-80 or JEOL SX-102A spectrometer. Preparative thin-layer chromatography (p-TLC) was performed on Merck Kiesel-gel GF254 (Type 60) (SiO2) or Merck Aluminum Oxide GF254 (Type 60/E) (Al2O3). Column chromatography was performed on silica gel (SiO2, 100—200 mesh, from Kanto Chemical Co. Inc.) or activated alumina (Al2O3, 300 meshes, from Wako Pure Chemical Industries, Ltd.) throughout the present study.
The solution of diazomethane (CH2N2) in diethyl ether (Et2O) was prepared as follows: a solution of potassium hydroxide (KOH) (5.50 g, 98.0 mmol) in H2O (8.0 mL) was placed in a 500 mL round bottom flask and cooled in an ice bath. The 95% EtOH (25 mL), Et2O (60.0 mL), and p-tolylsulfonylmethylnitrosoamide (21.5 g, 100 mmol) were added and the whole was slowly distilled to give the Et2O solution including about 3 g of CH2N2.
23.1. Synthesis of SOMRE and Related Derivatives Based on the 1-Hydroxyindole Chemistry

Table 30.
Preparation of 1-methxyindole (6) from 2,3-dihydroindole (3)[54]..
 |
1-Methoxyindole (6) from 2,3-dihydroindole (3)[54] — General method A (Table 30, Entry 1) :
A solution of Na2WO4·2H2O[70] (2.834 g, 8.42 mmol) in H2O (40.0 mL) was added to a solution of 3 (5.015 g, 42.1 mmol) in MeOH (375 mL). 30% H2O2 (47.657 g, 421 mmol) was added to the resultant solution at 0 °C with stirring. After stirring for 15 min at rt (16 °C), K2CO3 (20.456 g, 147 mmol) and a solution of Me2SO4 (7.972 g, 631 mmol) in MeOH (25.0 mL) were added to the reaction mixture. After stirring for 90 min at rt (16 °C), brine (330 mL) was added and the whole was extracted with CHCl3 (200 mL x 3). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a black oil, which was column-chromatographed on SiO2 with CHCl3–hexane (1:4, v/v) to give 6 (3.361 g, 15%). [12,61] 6: colorless oil. Mass and all spectral data are identical with those reported by Acheson et al.[62]
General method B (Table 1, Entry 3): A solution of Na2WO4·2H2O (13.2 mg, 0.04 mmol) in H2O (0.5 mL) was added to a solution of 3 (47.5 mg, 0.39 mmol) in MeOH (4.0 mL). 30% H2O2 (452.5 mg, 4.0 mmol) was added to the resultant solution at 0 °C with stirring. After stirring for 30 min at rt (17 °C), ethereal CH2N2 (excess) was added to the reaction mixture with stirring at rt until the starting material was not detected on tlc monitoring. Brine was added and the whole was extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave oil, which was purified by p-TLC on SiO2 with CH2Cl2–hexane (7:3, v/v) as a developing solvent. Extraction of a band having an Rf value of 0.92–0.79 with CH2Cl2 afforded 6 (29.6 mg, 50%).
Entry 6: In the same procedure for Entry 3, Na2WO4·2H2O (27.0 mg, 0.08 mmol), 3 (48.8 mg, 0.41 mmol), 30% H2O2 (464.9 mg, 4.10 mmol) were used. And the same work-up as Entry 3 afforded 6 (31.1 mg, 52%).
Entry 12: In the same procedure for Entry 3, 2Na2O·P2O5·12WO4·18H2O (23.8 mg, 0.007 mmol), 3 (50.3 mg, 0.42 mmol), 30% H2O2 (479.2 mg, 4.22 mmol) were used. And the same work-up as Entry 3 afforded 6 (35.9 mg, 58%).
General method C (Table 1, Entry 13): A solution of Na2WO4·2H2O (591.4 mg, 1.79 mmol) in H2O (10.0 mL) and urea·H2O2 compound (8.437 g, 89.64 mmol) were added to a solution of 3 (1.068 g, 8.96 mmol) in MeOH (100.0 mL) at 0 °C with stirring. After stirring at rt for 15 min, K2CO3 (22.300 g, 161.3 mmol) and then a solution of Me2SO4 (3.391 g, 26.9 mmol) in MeOH (10.0 mL) were added to the reaction mixture. After the same work-up as Entry 3, 6 (717.5 mg, 54%) was obtained.
Entry 14: m-Chloroperbenzoic acid (231.6 mg, 0.94 mmol), (n-Bu)4NHSO4 (7.1 mg, 0.02 mmol) and sat. aq. NaHCO3 (5.0 mL) were added to a solution of 3 (111.4 mg, 0.94 mmol) in acetone–CH2Cl2 (1:1, v/v, 5.0 mL) at 0 °C with stirring. After stirring for 5 min, brine was added. The whole was extracted with CH2Cl2 and ethereal CH2N2 (excess) was added to the extract. After stirring for 3 min, the solvent was evaporated under reduced pressure to leave oil, which was purified by column-chromatography on SiO2 to afford 6 (47.5 mg, 35%).
Entry 15: In the same procedure as Entry 14, solvent was changed to CH2Cl2 (5.0 mL) only, where m-chloroperbenzoic acid (223.2 mg, 0.90 mmol), (n-Bu)4NHSO4 (7.2 mg, 0.02 mmol), 3 (107.0 mg, 0.90 mmol), and sat. aq. NaHCO3 (5.0 mL) were used. After usual work-up, 6 (52.9 mg, 40%) was obtained.
Scheme 4.
Synthesis of SOMRE #1 (12).
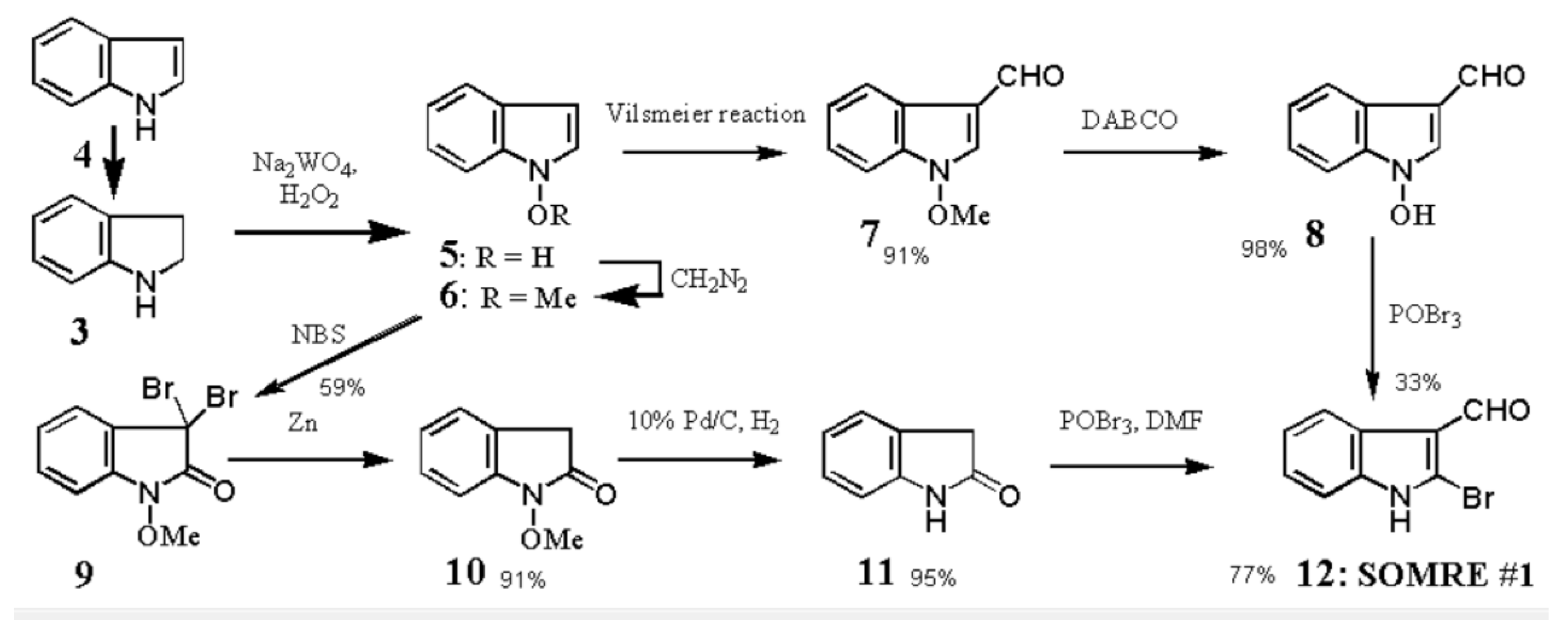
1-Methoxyindole-3-carbaldehyde (7)[63] from 1-methoxyindole (6) — POCl3 (8.4mL) was added to an ice cooled anhydrous DMF (31.0 mL) with stirring. A solution of 6 (12.341 g) in anhydrous DMF (10.0 mL) was added to the resultant viscous solution and stirring was continued at rt for 2 h. Then, crushed ice and 16% aq. NaOH (100 mL) were added to the reaction mixture and the whole was extracted with ether. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to give a crystalline solid. Recrystallization from ether-hexane afforded 7 (13.411 g, 91.3%) as colorless prisms. 7: mp 50.0–51.0 °C. IR (KBr): 2810,1660–1650, 1375, 1240 cm-1. 1H-NMR (CCl4) δ: 3.98 (3H, s), 6.81–7.31(3H, m, 7.52 (1H, s), 7.80–8.16 (1H,m), 9.57 (1H, s). MS m/z: 175 (M+): Anal. Calcd. for C10H9NO2·1/2H2O: C, 66.84; H, 5.33; N, 7.80. Found: C, 67.04; H, 5.11; N, 7.89.
3,3-Dibromo-1-methoxy-2-oxindole (9) from 1-methoxyindole (6) — NBS (3.637 g, 20.43 mmol) was added to a solution of 6 (1.001g, 6.81 mmol) in t-BuOH (70 mL) and the mixture was stirred at rt for 30 min. After evaporation of the solvent, H2O was added to the residue. The whole was extracted with benzene. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a yellow solid, which was column-chromatographed on SiO2 with CH2Cl2–hexane (2:1, v/v) to give 9 (1.281 g, 59%). 9: mp 73—75°C (pale yellow prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 1745 cm–1. 1H-NMR (CDCl3) δ: 4.06 (3H, s), 6.95 (1H, dd, J=7.7, 1.5 Hz), 7.15 (1H, br dt, J=1.5, 7.7 Hz), 7.34 (1H, br dt, J=1.5, 7.7 Hz), 7.56 (1H, dd, J=7.8, 1.5 Hz). MS m/z: 319, 321, and 323 (M+, 79Br and 81Br). Anal. Calcd for C9H7Br2NO2: C, 33.64; H, 2.18; N, 4.36. Found: C, 33.51; H, 2.09; N, 4.51.
1-Methoxy-2-oxindole (10) from 9 — Zink powder (103.2 mg, 1.6 mmol) was added to a solution of 9 (50.5 mg, 0.16 mmol) in AcOH (5 mL) and the mixture was stirred at rt for 1.5 h. Unreacted Zn was filtered off and washed with CH2Cl2–MeOH (95:5, v/v). H2O was added to the combined washing and the filtrate. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CH2Cl2 to give 10 (16.6 mg, 65%). Spectral data are identical with the authentic sample prepared according to our previous procedures.[73]
2-Oxindole (11) from 10 — A solution of 10 (155.3 mg, 0.95 mmol) in MeOH (10 mL) was hydrogenated in the presence of 10% Pd/C (50 mg) at rt and 1 atm for 1 h. Catalyst was filtered off and the filtrate was evaporated under reduced pressure to leave a crystalline solid, which was column-chromatographed on SiO2 with CH2Cl2 to give 11 (120.4 mg, 95%), whose physical data were identical with the commercially available sample.
1-Hydroxyindole-3-carbaldehyde (8) from 1-Methoxyindole-3-carbaldehyde (7) — i) Method A:
DABCO (386.3 mg, 3.45 mmol) was added to a solution of 7[3,17] (61.2 mg, 0.35 mmol) in DMF–H2O (3:1, v/v, 2 mL) and the mixture was heated at 100°C for 21 h with stirring. After addition of H2O, the whole was made acidic (pH 4) with 6% HCl and extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a brown solid, which was column-chromatographed on SiO2 with CHCl3–MeOH (97:3, v/v) to give 8 (55.0 mg, 98%). 8: mp 154—156 °C (decomp, pale yellow prisms, recrystallized from AcOEt-hexane). IR (KBr): 3107, 1616 (br), 1558 (br), 1516, 1309, 1238 cm-1. 1H-NMR (DMSO-d6) δ: 7.26 (1H, dt, J=1.3, 7.3 Hz), 7.34 (1H, dt, J=1.3, 7.3 Hz), 7.52 (1H, d, J=7.3 Hz), 8.11 (1H, d, J=7.3 Hz), 8.43 (1H, s), 9.84 (1H, s), 12.20 (1H, br s, disappeared on addition of D2O). Anal. Calcd for C9H7NO2: C, 67.07; H, 4.38; N, 8.69. Found: C, 66.86; H, 4.37; N, 8.66.
ii) Method B: KI (2.753 g, 16.6 mmol) was added to a solution of 7 (32.3 mg, 0.19 mmol) in DMF–H2O (3:1, v/v, 4 mL) and the mixture was heated at 160°C for 24 h with stirring. After the same work-up as described in the method B, unreacted 7 (10.5 mg, 33%), 8 (0.7 mg, 3%), and 8 (16.3 mg, 55%) were obtained in the order of elution.
SOMRE #1 (2-Bromoindole-3-carbaldehyde (12) from 2-Oxindole (11) — A solution of phosphorus tribromide (8.0 ml, 78.7 mmol) in anhydrous CHCl3 (20 mL) was added dropwise to anhydrous DMF (30 mL) at 0 °C within 10 min with stirring. The mixture became viscous and then turned to yellow white solid. Although magnetic stirring bar stopped stirring, a solution of 2-oxindole (11, 4.056 g, 30.5 mmol) in anhydrous CHCl3 (60 mL) was added to the yellow white solid and the whole was immersed in an ultra sound bath for 1 h at rt and allowed to stand for 10 h. Again, the whole was immersed in an ultra sound bath for 1 h at rt. With ice cooling, H2O (100 mL) was added to the reaction mixture and the yellow solid solved. The whole was then made slightly alkaline (pH 9) with 40% aqueous NaOH. After separation of organic layer, the water layer was extracted with CH2Cl2–MeOH (9:1, v/v, 4 times, total 400 mL). The combined organic layer and the extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave crystals. Repeated recrystallization from MeOH afforded 12. Total yield : 5.281 g (77%). 12: mp 209.0–210.5°C[1b] (colorless needles, , recrystallized from MeOH, lit.[67] mp 196—198°C). IR (KBr): 3100, 1645 cm-1. 1H-NMR (DMSO-d6) δ: 6.96–7.54 (3H, m), 7.88–-8.14 (1H, m), 9.82 (1H, s). MS m/z: 223 and 225 (M+, 79Br and 81Br). Anal. Calcd for C9H6BrNO: C, 48.25; H,2.70; N, 6.25. Found: C, 48.26; H, 2.70; N, 6.35.
SOMRE #1 (2-Bromoindole-3-carbaldehyde (12) from 1-Hydroxyindole-3-carbaldehyde (8) — A solution of 8 (27.6 mg, 0.17 mmol) in anhydrous THF (3 mL) was added to POBr3 (310.0 mg, 1.08 mmol) and the mixture was stirred at rt for 15 h. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give 12 (12.6 mg, 33%) and 24 (5.3 mg, 21%) in the order of elution.
Figure 3.
SOMRE family compounds 12-51.
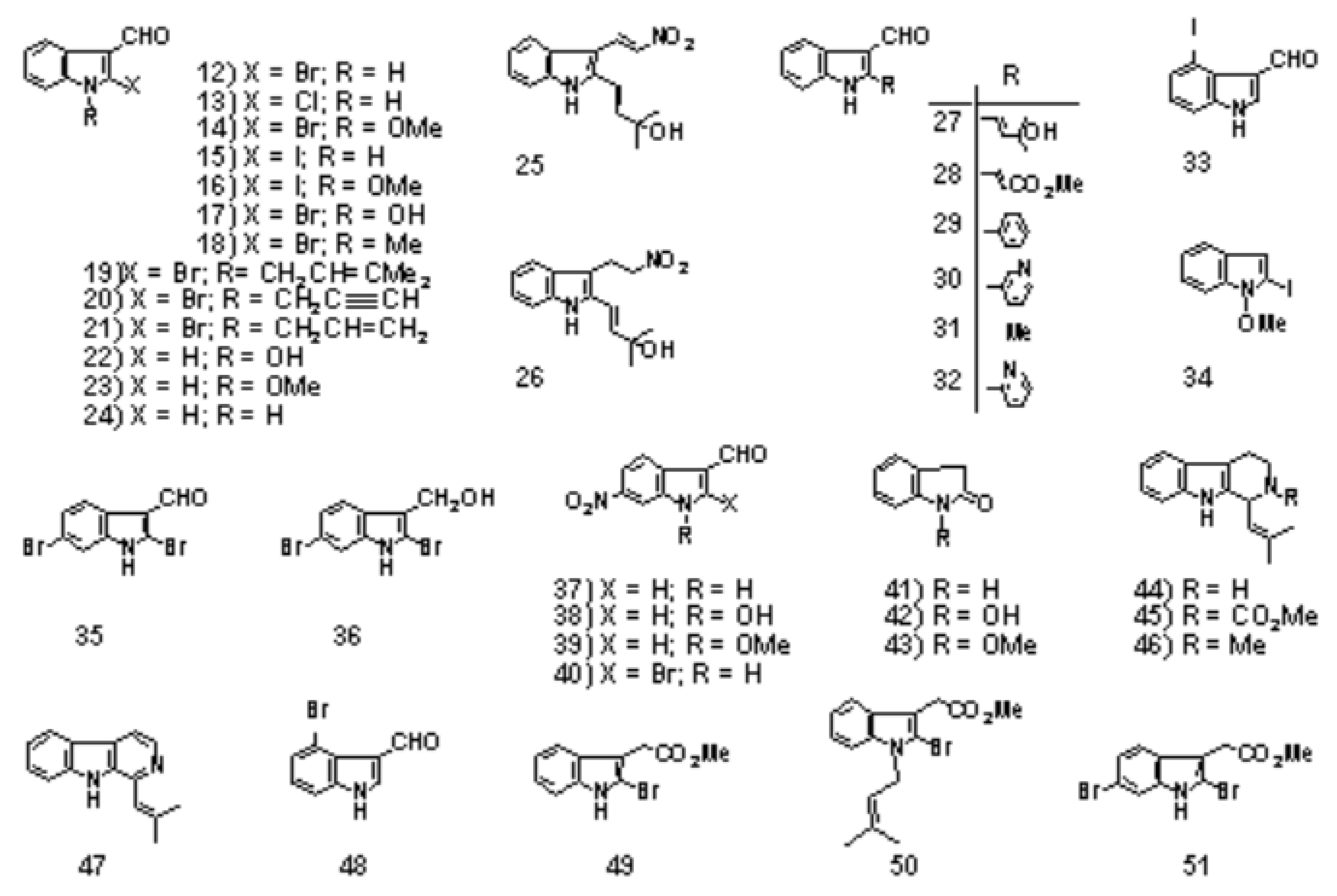
2-Chloroindole-3-carbaldehyde (13) from (8) — A solution of 8 (27.6 mg, 0.17 mmol) in anhydrous THF (3 mL) was added to POCl3 (45.9 mg, 0.3 mmol) and DMF (22.0 mg, 0.3 mmol), and the mixture was stirred at rt for 5.5 h. The whole was made basic with 8% aqueous NaOH and extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a solid, which was column-chromatographed on SiO2 with CH2Cl2 to give 13 (20.6 mg, 67%). Physical data were identical with those of the authentic sample.[68]
2-Bromo-1-methoxyindole-3-carbaldehyde (14) from 1-Methoxy-2-oxindole (10)[69] — Anhydrous DMF (9 mL) was added to a solution of POBr3 (0.2 mL) in anhydrous CHCl3 (6 mL) at 0°C and stirring was continued at rt for 15 min. To the resulting solution was added a solution of 10[70] (236.3 mg, 1.45 mmol) in DMF (5 mL) at 0°C and the mixture was stirred at rt for 12 h. The whole was made basic with 8% aqueous NaOH and extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a solid, which was column-chromatographed on SiO2 with AcOEt–hexane (1:3, v/v) to give 14 (307.5 mg, 84%). 14: mp 97—98°C (colorless needles, recrystallized from MeOH). IR (KBr): 1653 cm-1. 1H NMR (CDCl3) δ: 4.19 (3H, s), 7.31 (1H, ddd, J=7.6, 7.3, 1.2 Hz), 7.36 (1H, ddd, J=7.8, 7.3, 1.2 Hz), 7.45 (1H, dd, J=7.8, 1.2 Hz), 8.32 (1H, dd, J=7.6, 1.2 Hz), 9.98 (1H, s). MS m/z: 253 and 255 (M+, 79Br and 81Br). Anal. Calcd for C10H8BrNO: C, 47.27; H, 3.17; N, 5.51. Found: C, 47.02; H, 3.22; N, 5.33.
2-Iodoindole-3-carbaldehyde (15) from 12 — KI (1.144 g, 6.89 mmol) and CuI (659.5 mg, 3.46 mmol) were added to a solution of 12 (152.8 mg, 0.68 mmol) in DMF (15 mL) and the mixture was heated at 120°C for 48 h. After evaporation of the solvent under reduced pressure, H2O was added to the residue. The whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give an inseparable mixture (151.9 mg) of 12 and 15 in a ratio of 1:3.1 (1H-NMR analysis). The yields of 12 and 15 were calculated to be 29.8 mg (20%) and 122.1 mg (62%), respectively. To obtain 2 mg of pure 15, repeated HPLC and column-chromatography were required. 15: mp 224—226°C (colorless needles, recrystallized from MeOH). IR (KBr): 3138, 1639 cm–1. 1H-NMR (DMSO-d6) δ: 7.19 (1H, td, J=7.5, 1.2 Hz), 7.22 (1H, td, J=7.5, 1.2 Hz), 7.42 (1H, dd, J=7.5, 1.2 Hz), 8.09 (1H, dd, J=7.5, 1.2 Hz), 9.72 (1H, s), 12.81 (1H, br s). High-resolution MS m/z: Calcd for C9H6INO: 270.9494. Found: 270.9478.
2-Iodo-1-methoxyindole-3-carbaldehyde (16) from 2-Iodo-1-methoxyindole (34) — POCl3 (0.2 mL, 2.15 mmol) was added to DMF (2 mL, 25.8 mmol) at 0°C and the stirring was continued at rt for 15 min. To the solution was added a solution of 34 (137.1 mg, 0.50 mmol) in DMF (2 mL) at 0°C and the mixture was stirred at rt for 2 h. The whole was made basic with 8% aqueous NaOH and extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under the reduced pressure to leave a solid, which was column-chromatographed on SiO2 with AcOEt–hexane (1:5, v/v) to give 16 (105.6 mg, 70%). 16: mp 132—134°C (colorless needles, recrystallized from MeOH). IR (KBr): 1645 cm–1. 1H-NMR (CDCl3) δ: 4.18 (3H, s), 7.29 (1H, t, J=7.3 Hz), 7.33 (1H, t, J=7.3 Hz), 7.46 (1H, d, J=7.3 Hz), 8.33 (1H, d, J=7.3 Hz), 9.79 (1H, s). MS m/z: 301 (M+). Anal. Calcd for C10H8INO2: C, 39.89; H, 2.68; N, 4.65. Found: C, 40.33; H, 2.87; 4.47.
2,6-Dibromoindole-3-carbaldehyde (35) with/without 2-Bromo-1-hydroxyindole-3-carbaldehyde (17) from 2-Bromo-1-methoxyindole-3-carbaldehyde (14) — i) Method A: BBr3 (2 mL, 21.2 mmol) was added to a solution of 14 (50.0 mg, 0.20 mmol) in anhydrous CH2Cl2 (5 mL) at 0°C and the mixture was refluxed for 21 h with stirring. The mixture was poured into an ice water and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (95:5, v/v) to give 35 (36.3 mg, 61%). 35: mp 269—270°C (decomp., colorless prisms, recrystallized from MeOH). IR (KBr): 3082, 1631 cm-1. 1H NMR (CD3OD) δ: 7.35 (1H, dd, J=8.5, 1.8 Hz), 7.56 (1H, d, J=1.8 Hz), 8.04 (1H, d, J=8.5 Hz), 9.91 (1H, s). 1H NMR (CDCl3) δ: 7.41 (1H, dd, J=8.5, 1.2 Hz), 7.51 (1H, d, J=1.2 Hz), 8.17 (1H, d, J=8.5 Hz), 8.76 (1H, br s), 10.01 (1H, s). MS m/z: 301, 303, and 305 (M+ 79Br2, 79Br81Br, and 81Br2). Anal. Calcd for C9H5Br2NO: C, 35.68; H, 1.66; N, 4.62. Found: C, 35.64; H, 1.72; N, 4.57.
ii) Method B: BBr3 (4.0 mL, 42.3 mmol) was added to a solution of 14 (97.0 mg, 0.33 mmol) in anhydrous CH2Cl2 (10 mL) at 0°C and the mixture was stirred at rt for 24 h. After the same work-up as described in the Method A, the crude product was column-chromatographed on SiO2 with CHCl3 to give unreacted 14 (30.8 mg, 32%), 35 (18.2 mg, 16%), and 17 (12.7 mg, 14%) in the order of elution. 17: mp 192—194°C (decomp., colorless needles, recrystallized from AcOEt–hexane). IR (KBr): 1630 cm–1. 1H-NMR (CD3OD) δ: 7.27 (1H, dd, J=7.8, 7.3 Hz), 7.34 (1H, J=7.8, 7.3 Hz), 7.50 (1H, d, J=7.8 Hz), 8.16 (1H, d, J=7.8 Hz), 9.86 (1H, s). High-resolution MS m/z: Calcd for C9H679BrNO2: 238.9582. Found: 238.9579. Calcd for C9H681BrNO2: 240.9561. Found: 240.9556.
2-Bromo-1-methylindole-3-carbaldehyde (18) from 12 — A solution of MeI (969.2 mg, 6.83 mmol) in THF (10 mL) was added to a mixture of 12 (1.07 g, 4.75 mmol), Bu4NBr (306.9 mg, 0.952 mmol), and K2CO3 (3.55 g, 25.7 mmol) in THF (60 mL), and the mixture was stirred at rt for 5 h. After addition of brine, the whole was extracted with CH2Cl2–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CH2Cl2 to give 18 (1.11 g, 98%). 18: mp 118—118.5°C (colorless prisms, recrystallized from hexane). IR (KBr): 1638 cm–1. 1H-NMR (CDCl3) δ: 3.84 (3H, s), 7.14—7.36 (3H, m), 8.14—8.36 (1H, m), 9.98 (1H, s). MS m/z: 237 and 239 (M+, 79Br and 81Br). Anal. Calcd for C10H8BrNO: C, 50.45; H, 3.39; N, 5.88. Found: C, 50.44; H, 3.35; N, 6.09.
2-Bromo-1-(3-methyl-2-buten-1-yl)indole-3-carbaldehyde (19) from 12 — A solution of prenyl bromide (1.06 g, 7.17 mmol) in THF (10 mL) was added to a mixture of 12 (1.02 g, 4.54 mmol), Bu4NBr (300.8 mg, 0.933 mmol), and K2CO3 (3.57 g, 25.8 mmol) in THF (60 mL), and the mixture was stirred at rt for 6 h. After the same work-up as described in the preparation of 18, 1.22 g (92%) of 19 was obtained. 19: 78.5—79°C (colorless needles, recrystallized from hexane). IR (KBr): 1650 cm–1. 1H-NMR (CDCl3) δ: 1.75 (3H, d, J=1.2 Hz), 1.92 (3H, d, J=1.2 Hz), 4.83 (2H, d, J=7.0 Hz), 5.18 (1H, th, J=7.0, 1.2 Hz), 7.11—7.35 (3H, m), 8.15—8.39 (1H, m). 10.00 (1H, s). MS m/z: 291 and 293 (M+, 79Br and 81Br). Anal. Calcd for C14H14BrNO: C, 57.55; H, 4.83; N, 4.79. Found: C, 57.58; H, 4.84; N, 4.80.
2-Bromo-1-propargylindole-3-carbaldehyde (20) from 12 — A solution of propargyl bromide (826.6 mg, 6.95 mmol) in THF (10 mL) was added to a mixture of 12 (1.00 g, 4.50 mmol), Bu4NBr (280.1 mg, 0.88 mmol), and K2CO3 (3.19 g, 23.1 mmol) in THF (40 mL), and the mixture was stirred at rt for 22 h. After addition of brine, the whole was extracted with CH2Cl2–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a crystalline solid, which was recrystallized from MeOH to give 20 (1.01 g) as colorless flakes. The mother liquor was subjected to p-TLC on SiO2 with CH2Cl2–hexane (3:2, v/v) as a developing solvent. Extraction of the band having an Rf value of 0.35—0.43 with CH2Cl2–MeOH (95:5, v/v) gave 20 (60.9 mg). Total yield of 20 was 1.07 g (91%). 20: mp 151.5—152.5°C. IR (KBr): 1636 cm–1. 1H-NMR (CDCl3) δ: 2.37 (1H, t, J=2.5 Hz), 5.00 (2H, d, J=2.5 Hz), 7.14—7.54 (3H, m), 8.14—8.39 (1H, m), 10.00 (1H, s). MS m/z: 261 and 263 (M+, 79Br and 81Br). Anal. Calcd for C12H8BrNO: C, 54.98; H, 3.08; N, 5.34. Found: C, 54.82; H, 2.98; N, 5.47.
1-Allyl-2-bromoindole-3-carbaldehyde (21) from 12 — A solution of allyl bromide (108.0 mg, 0.89 mmol) in THF (2 mL) was added to a mixture of 12 (100.3 mg, 0.45 mmol), Bu4NBr (29.3 mg, 0.09 mmol), and K2CO3 (307.8 mg, 2.23 mmol) in THF (6 mL), and the mixture was stirred at rt for 1 h. After the same work-up as described in the preparation of 18, 115.3 mg (98%) of 21 was obtained. 21: mp 87—88°C (colorless prisms, recrystallized from CHCl3). IR (KBr): 1652 cm–1. 1H-NMR (CDCl3) δ: 4.90 (2H, ddd, J=5.0, 1.8, 1.2 Hz), 5.23 (1H, dt, J=17.0, 1.8 Hz), 5.26 (1H, dt, J=10.5, 1.2 Hz), 5.94 (1H, ddt, J=17.0, 10.5, 5.0 Hz), 7.26—7.33 (3H, m), 8.31—8.34 (1H, m), 10.06 (1H, s). MS m/z: 263 and 265 (M+, 79Br and 81Br). Anal. Calcd for C12H10BrNO·1/4H2O: C, 53.65; H, 3.94; N, 5.21. Found: C, 53.84; H, 3.73; N, 5.21.
(E,E)-2-Methyl-4-[3-(2-nitrovinyl)indol-2-yl]-3-buten-2-ol (25) from 27 — NH4OAc (736.1 mg, 9.50 mmol) was added to a solution of 27 (435.7 mg, 1.90 mmol) in MeNO2 (26 mL) and the mixture was heated at 90°C for 4 h with stirring. After cooling to rt, the resulting precipitates (16, 406.1 mg) were collected by filtration and washed with MeOH. The filtrate and washings were combined and H2O was added. The whole was extracted with CH2Cl2–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a crystalline solid, which was column-chromatographed on SiO2 with CH2Cl2–MeOH (95:5, v/v) to give 25 (75.7 mg). Total yield of 25 was 481.8mg (93%). 25: mp 246.5—247°C (decomp., red needles, recrystallized from MeOH). IR (KBr): 3250, 1578, 1360 cm–1. 1H NMR (pyridine-d5) δ: 1.55 (6H, s), 7.02 (1H, d, J=16.0 Hz), 7.26—7.59 (4H, m), 7.85—8.05 (1H, m), 8.52 (1H, d, J=13.2 Hz), 8.77 (1H, d, J=13.2 Hz). MS m/z: 272 (M+). Anal. Calcd for C15H16N2O3: C, 66.16; H, 5.92; N, 10.29. Found: C, 65.97; H, 5.89; N, 10.46.
(E)-2-Methyl-4-[3-(2-nitroethyl)indol-2-yl]-3-buten-2-ol (26) from 25 — NaBH4 (173.5 mg, 4.59 mmol) was added to a solution of 25 (206.0 mg, 0.76 mmol) in MeOH (30 mL) and the mixture was stirred at rt for 1 h. After addition of AcOEt, the whole was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give 26 (199.5 mg, 96%). 26: mp 122.5—123°C (colorless prisms, recrystallized from benzene). IR (KBr): 3520, 3310, 1554, 1380 cm–1. 1H-NMR (CD3OD) δ: 1.23 (6H, s), 3.48 (2H, t, J=7.5 Hz), 4.62 (2H, t, J=7.5 Hz), 6.31 (1H, d, J=16.0 Hz), 6.71 (1H, d, J=16.0 Hz), 6.83—7.52 (4H, m). MS m/z: 274 (M+). Anal. Calcd for C15H18N2O3: C, 65.67; H, 6.61; N, 10.21. Found: C, 65.89; H, 6.58; N, 10.23.
E-2-(3-Hydroxy-3-methyl-1-butenyl)indole-3-carbaldehyde (27) from 12 — General Procedure: A solution of 12 (405.4 mg, 1.81 mmol), (3-hydroxy-3-methyl-1-butenyl)tributyltin (1.00 g, 2.81 mmol), Pd(OAc)2 (42.3 mg, 0.19 mmol), and Bu4NCl (1.00 g, 3.61 mmol) in DMF (6 mL) was heated at 115—120°C for 3 h with stirring. After evaporation of the solvent under reduced pressure, brine was added, and the whole was extracted with AcOEt–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (2:1, v/v) to give 27 (362.1 mg, 87%). 27: mp 194—195°C (colorless prisms, recrystallized from MeOH). IR (KBr): 3165, 2975, 1614 cm–1. 1H-NMR (CD3OD) δ: 1.47 (6H, s), 6.75 (1H, d, J=16.0 Hz), 7.02—7.48 (3H, m), 7.20 (1H, d, J=16.0 Hz), 7.99—8.23 (1H, m), 10.18 (1H, s). MS m/z: 229 (M+). Anal. Calcd for C14H15NO2: C, 73.34; H, 6.59; N, 6.11. Found: C, 73.17; H, 6.60; N, 6.32.
Methyl (E)-3-(3-Formylindol-2-yl)acrylate (28) from 12 — In the general procedure for 27, 12 (102.2 mg, 0.46 mmol), 2-(methoxycarbonyl)vinyl tributyltin (254.2 mg, 0.68 mmol), Pd(OAc)2 (12.1 mg, 0.05 mmol), and Bu4NCl (248.6 mg, 0.90 mmol) were used. After the same work-up as described in the preparation of 27, 70.2 mg (67%) of 28 was obtained. 28: mp 261—262°C. IR (KBr): 3050, 1703, 1628 cm–1. 1H-NMR (pyridine-d5) δ: 3.72 (3H, s), 6.95 (1H, d, J=16.0 Hz), 7.25—7.49 (3H, m), 8.50 (1H, d, J=16.0 Hz), 8.59—8.77 (1H, m), 10.71 (1H, s). The NH proton signal was not observed. MS m/z: 229 (M+). Anal. Calcd for C13H11NO3: C, 68.11; H, 4.84; N, 6.11. Found: C, 67.89; H, 4.76; N, 6.04.
2-Phenylindole-3-carbaldehyde (29) from 12 — In the general procedure for 27, 12 (41.1 mg, 0.18 mmol), tetraphenyl tin (117.3 mg, 0.27 mmol), Pd(OAc)2 (4.3 mg, 0.02 mmol), and Bu4NCl (97.7 mg, 0.35 mmol) were used. After the same work-up and column-chromatography as described in the preparation of 27, 29 (27.7 mg, 68%) and 12 (3.9 mg, 10%) were obtained in the order of elution. 29: mp 260.5—263°C (colorless prisms, recrystallized from MeOH). IR (KBr): 3120, 1628 cm–1. 1H-NMR (pyridine-d5) δ: 7.18—7.90 (8H, m), 8.53—8.91 (1H, m), 10.25 (1H, s). MS m/z: 221 (M+). Anal. Calcd for C15H11NO: C, 81.43; H, 5.01; N, 6.33. Found: C, 81.51; H, 4.92; N, 6.35.
2-(3-Pyridyl)indole-3-carbaldehyde (30) from 12 — In the general procedure for 27, 12 (103.1 mg, 0.46 mmol), (3-pyridyl)trimethyl tin (221.0 mg, 0.91 mmol), Pd(OAc)2 (9.7 mg, 0.04 mmol), and Bu4NCl (261.3 mg, 0.94 mmol) were used. After the same work-up and column-chromatography as described in the preparation of 27, 30 (39.1 mg, 38%), 31 (10.9 mg, 15%), and indole-3-carbaldehyde (5.5 mg, 8%) were obtained in the order of elution. 30: mp 246—246.5°C (colorless needles, recrystallized from MeOH). IR (KBr): 3430, 3130, 1628 cm–1. 1H-NMR (pyridine-d5) δ: 7.28—7.70 (4H, m), 8.14 (1H, ddd, J=8.0, 2.2, 1.8 Hz), 8.75—8.96 (2H, m), 9.27 (1H, dd, J=2.2, 0.8 Hz), 10.41 (1H, s). MS m/z: 222 (M+). Anal. Calcd for C14H10N2O: C, 75.65; H, 4.54; N, 12.61. Found: C, 75.48; H, 4.77; N, 12.50.
2-Methylindole-3-carbaldehyde (31) from 12 — In the general procedure for 27, 12 (467.8 mg, 2.09 mmol), tetramethyl tin (562.2 mg, 3.12 mmol), Pd(OAc)2 (42.7 mg, 0.19 mmol), and Bu4NCl (1.34 g, 4.84 mmol) were used. After the same work-up and column-chromatography as described in the preparation of 27, 31 (130.0 mg, 39%) and indole-3-carbaldehyde (18.1 mg, 6%) were obtained in the order of elution. 31: mp 204—205°C (colorless needles, recrystallized from MeOH). IR (KBr): 3250, 1635 cm–1. 1H-NMR (CD3OD) δ: 2.63 (3H, s), 6.89—7.39 (3H, m), 7.79—8.12 (1H, m), 9.82 (1H, s). MS m/z: 159 (M+). Anal. Calcd for C10H9NO: C, 75.45; H, 5.70; N, 8.80. Found: C, 75.50; H, 5.62; N, 8.82.
2-(2-Pyridyl)indole-3-carbaldehyde (32) from 12 — In the general procedure for 27, 12 (100.5 mg, 0.45 mmol), (2-pyridyl)trimethyl tin (1.07 g, 4.43 mmol), Pd(OAc)2 (10.9 mg, 0.05 mmol), and Bu4NCl (246.1 mg, 0.89 mmol) were used. After the same work-up and column-chromatography as described in the preparation of 27, unreacted 12 (21.4 mg, 21%), 32 (5.0 mg, 5%), 31 (0.5 mg, 1%), and indole-3-carbaldehyde (2.4 mg, 4%) were obtained in the order of elution. 32: mp 225.5—226.5°C (colorless needles, recrystallized from MeOH). IR (KBr): 3060, 1619 cm–1. 1H-NMR (pyridine-d5) δ: 7.12—7.70 (4H, m), 7.75 (1H, dd, J=7.5, 2.0 Hz), 8.70 (1H, dt, J=8.0, 1.0 Hz), 8.65—8.78 (1H, m), 8.78—8.98 (1H, m), 11.04 (1H, s). MS m/z: 222 (M+). Anal. Calcd for C14H10N2O: C, 75.65; H, 4.54; N, 12.61. Found: C, 75.42; H, 4.38; N, 12.83.
Hydroxy-6-nitroindole-3-carbaldehyde (38) from 1-Methoxy-6-nitroindole-3-carbaldehyde (39) — i) Method A: The formation of 38 in 90% yield by the reaction of 39 with DABCO was reported in the preceding paper.
ii) Method B: A solution of KI (960.0 mg, 5.78 mmol) in H2O (1 mL) was added to a solution of 39 (32.8 mg, 0.15 mmol) in DMF (3 mL), and the mixture was heated at 120°C for 36 h with stirring. After evaporation of the solvent under reduced pressure, AcOEt was added to the residue. The same work-up of the organic layer as described in the Method A for 38, gave unreacted 39 (7.3 mg, 22%), 37 (1.1 mg, 4%), and 38 (17.9 mg, 58%) in the order of elution. 38 is identical with the authentic sample.72
2-Bromo-6-nitroindole-3-carbaldehyde (40) from 38 — POBr3 (529.4 mg, 1.85 mmol) was added to a solution of 38 (60.9 mg, 0.30 mmol) in anhydrous THF (4 mL) and the mixture was stirred at rt for 5 h. After evaporation of the solvent, H2O was added to the residue, and the whole was extracted with CHCl3–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give 40 (65.3 mg, 82%) and 37 (6.2 mg, 11%) in the order of elution. 40: mp 296—298°C (decomp., colorless prisms, recrystallized from AcOEt). IR (KBr): 1631 cm–1. 1H-NMR (DMSO-d6) δ: 8.12 (1H, dd, J=8.8, 1.6 Hz), 8.24 (1H, d, J=8.8 Hz), 8.27 (1H, d, J=1.6 Hz), 9.95 (1H, s). The proton at the 1-position did not appear. MS m/z: 268 and 270 (M+, 79Br and 81Br). Anal. Calcd for C9H5BrN2O3: C, 40.18; H, 1.87; N, 10.41. Found: C, 40.16; H, 1.93; N, 10.47.
2,6-Dibromoindol-3-ylmethanol (36) from 35 — NaBH4 (150.0 mg, 3.95 mmol) was added to a solution of 35 (65.2 mg, 0.22 mmol) in MeOH (10 mL), and the mixture was stirred at rt for 1 h. After evaporation of the solvent under reduced pressure, AcOEt was added to the residue. The whole was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (95:5, v/v) to give 36 (44.2 mg, 67%). 36: colorless oil. IR (film): 3398, 3213, 1614 cm–1. 1H-NMR (CDCl3) δ: 1.48 (1H, br s), 4.80 (2H, s), 7.26 (1H, dd, J=8.5, 1.7 Hz), 7.43 (1H, d, J=1.7 Hz), 7.55 (1H, d, J=8.5 Hz), 8.18 (1H, br s). High-resolution MS m/z: Calcd for C9H779Br2NO: 302.8895. Found: 302.8915. Calcd for C9H779Br81BrNO: 304.8874. Found: 304.8852. Calcd for C9H781Br2NO: 306.8854. Found: 306.8829.
1-(2-Methyl-1-propenyl)-1,2,3,4-tetrahydro-β-carboline (44) from 26 — A solution of 26 (174.9 mg, 0.64 mmol) in THF (19.5 mL) was added to a mixture of Zn powder (897.2 mg, 13.7 mmol) [washed with 6% HCl (4 mL)] in 6% HCl (6.5 mL) at 0°C and the mixture was refluxed for 5 min with stirring. Unreacted Zn was filtered off and the filtrate was evaporated under reduced pressure. The residue was made basic by adding 8% aqueous NaOH and the whole was extracted with CH2Cl2–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:2:0.2, v/v) to give 44 (22.5 mg, 50%). 44: mp 160—161°C (colorless prisms, recrystallized from CH2Cl2, lit.,[10] mp 158—159°C). IR (KBr): 3400, 1092 cm–1. 1H-NMR (CDCl3) δ: 1.82 (3H, d, J=1.2 Hz), 1.88 (3H, d, J=1.2 Hz), 2.65—2.89 (2H, m), 2.89—3.55 (2H, m), 4.83 (1H, br d, J=9.5 Hz), 5.26 (1H, dt, J=9.5, 1.2 Hz), 6.92—7.33 (3H, m), 7.33—7.53 (1H, m), 7.64 (1H, br s). MS m/z: 226 (M+). Anal. Calcd for C15H18N2·1/4H2O: C, 78.05; H, 8.07; N, 12.13. Found: C, 78.47; H, 8.09; N, 11.84.
2-Methoxycarbonyl-1-(2-methyl-1-propenyl)-1,2,3,4-tetrahydro-β-carboline (45) from 44 — A solution of ClCO2Me (38.6 mg, 0.41 mmol) in CH2Cl2 (1 mL) was added to a solution of 44 (54.0 mg, 0.24 mmol) and Et3N (80.0 mg, 0.79 mmol) in CH2Cl2 (3 mL) and the mixture was stirred at rt for 2 h. Saturated NaHCO3 was added and the whole was extracted with CH2Cl2–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was recrystallized from MeOH to give 45 (47.5 mg) as colorless prisms. The mother liquor was subjected to p-TLC on SiO2 with CH2Cl2–MeOH (95:5, v/v) as a developing solvent. Extraction from the band having an Rf value of 0.93—1.00 with CH2Cl2–MeOH (95:5, v/v) gave 45 (10.3 mg). Total yield of 45 was 58.7 mg (85%). 45: mp 186—189°C (lit.,[74] mp 180—181°C). IR (KBr): 3400, 3040, 1092 cm–1. 1H NMR (CDCl3) δ: 1.77 (3H, d, J=1.4 Hz), 1.98 (3H, d, J=1.4 Hz), 2.64—2.88 (2H, m), 2.88—3.37 (1H, m), 3.73 (3H, s), 4.39 (1H, d, J=12.5 Hz), 5.30 (1H, d, J=10.0 Hz), 5.88 (1H, d, J=10.0 Hz), 6.91—7.32 (3H, m), 7.36—7.52 (1H, m), 7.59 (1H, br s). High-resolution MS m/z: Calcd for C17H20N2O2: 284.1524. Found: 284.1526.
Borrerine (46) from 45 — LiAlH4 (98.6 mg, 2.60 mmol) was added to a solution of 45 (46.3 mg, 0.16 mmol) in THF (8 mL) at 0°C and the mixture was refluxed for 5 h with stirring. After addition of MeOH and 10% aqueous Rochelle salt under ice cooling, the whole was extracted with CH2Cl2–MeOH (95:5, v/v). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was subjected to p-TLC on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:5:0.5, v/v) as a developing solvent. Extraction from the band having an Rf value of 0.74—0.90 with CHCl3–MeOH–28% aq. NH3 (46:5:0.5, v/v) gave 46 (28.2 mg, 70%). 46: mp 105—106°C (colorless prisms, recrystallized from hexane, lit.[10] mp 102—103°C). IR (KBr): 3200 cm–1. 1H-NMR (CDCl3) δ: 1.86 (3H, d, J=1.2 Hz), 1.88 (3H, d, J=1.2 Hz), 2.48—3.29 (4H, m), 4.05 (1H, dt, J=9.5, 1.2 Hz), 5.18 (1H, br d, J=9.5 Hz), 6.92—7.34 (3H, m), 7.34—7.63 (2H, m). High-resolution MS m/z: Calcd for C16H20N2: 240.1625. Found: 240.1630.
Vulcanine (47) from 44 — A solution of t-BuOCl (42.4 mg, 0.39 mmol) in THF (1 mL) was added to a suspension of 44 (37.0 mg, 0.16 mmol) and powdered NaOH (32.1 mg, 0.80 mmol) in THF (4 mL) and the mixture was stirred at rt for 64 h. H2O was added and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was subjected to p-TLC on SiO2 with AcOEt–hexane (1:1, v/v) as a developing solvent. Extraction from the band having an Rf value of 0.47—0.65 with AcOEt gave 47 (18.5 mg, 51%). 47: pale yellow oil. IR (CHCl3): 1645, 1623, 1567, 1492, 1452, 1420, 1380, 1316, 1235 cm–1. 1H-NMR (CDCl3) δ: 2.00 (3H, d, J=1.2 Hz), 2.01 (3H, d, J=1.2 Hz), 6.60 (1H, t, J=1.2 Hz), 7.28 (1H, ddd, J=8.1, 6.4, 1.7 Hz), 7.51 (1H, ddd, J=8.1, 1.7, 1.0 Hz), 7.53 (1H, ddd, J=8.1, 6.4, 1.0 Hz), 7.82 (1H, d, J=5.4 Hz), 8.11 (1H, d, J=8.1 Hz), 8.46 (1H, d, J=5.4 Hz), 8.57 (1H, br s, disappeared on addition of D2O). 13C-NMR (CDCl3) δ: 20.5, 26.7, 111.9, 113.0, 119.0, 120.3, 121.6, 121.8, 128.8, 129.5, 134.0, 136.8, 140.7, 140.8, 143.8. UV λmax (MeOH) nm (log ε): 214 (4.36), 239 (4.49), 260 (sh, 4.23), 292 (4.14), 356 (3.79). MS m/z: 222 (M+), 207, 182, 103. High-resolution MS m/z: Calcd for C15H14N2: 222.1157. Found: 222.1154. 47·HCl: mp 189—192°C (yellow needles, recrystallized from Et2O–MeOH, lit.,[75] mp 103°C). IR (KBr): 3340, 1640, 1599, 1442, 761 cm–1. 1H-NMR (CD3OD) δ: 1.93 (3H, d, J=1.2 Hz), 2.22 (3H, d, J=1.2 Hz), 6.74 (1H, t, J=1.2 Hz), 7.47 (1H, ddd, J=8.1, 6.8, 1.0 Hz), 7.76 (1H, dt, J=8.3, 1.0 Hz), 7.80 (1H, ddd, J=8.3, 6.8, 1.0 Hz), 8.35 (1H, d, J=6.4 Hz), 8.41 (1H, dt, J=8.1, 1.0 Hz), 8.55 (1H, d, J=6.4 Hz). 13C-NMR (CD3OD) δ: 20.9, 26.6, 113.9, 114.2, 116.6, 121.5, 123.0, 124.1, 129.5, 133.1, 134.8, 135.2, 137.4, 145.4, 152.5. Anal. Calcd for C15H14N2·HCl·1/4H2O: C, 68.44; H, 5.93; N, 10.64. Found: C, 68.51; H, 5.83; N, 10.68.
Table 28.
Effects of 1-Hydroxyindoles on Arachidonic Acid Induced Platelet Aggregation in Rabbit PRP 60-66.
Table 28.
Effects of 1-Hydroxyindoles on Arachidonic Acid Induced Platelet Aggregation in Rabbit PRP 60-66.
 |
Scheme 6.
Synthesis of candidates of osteoporosis.
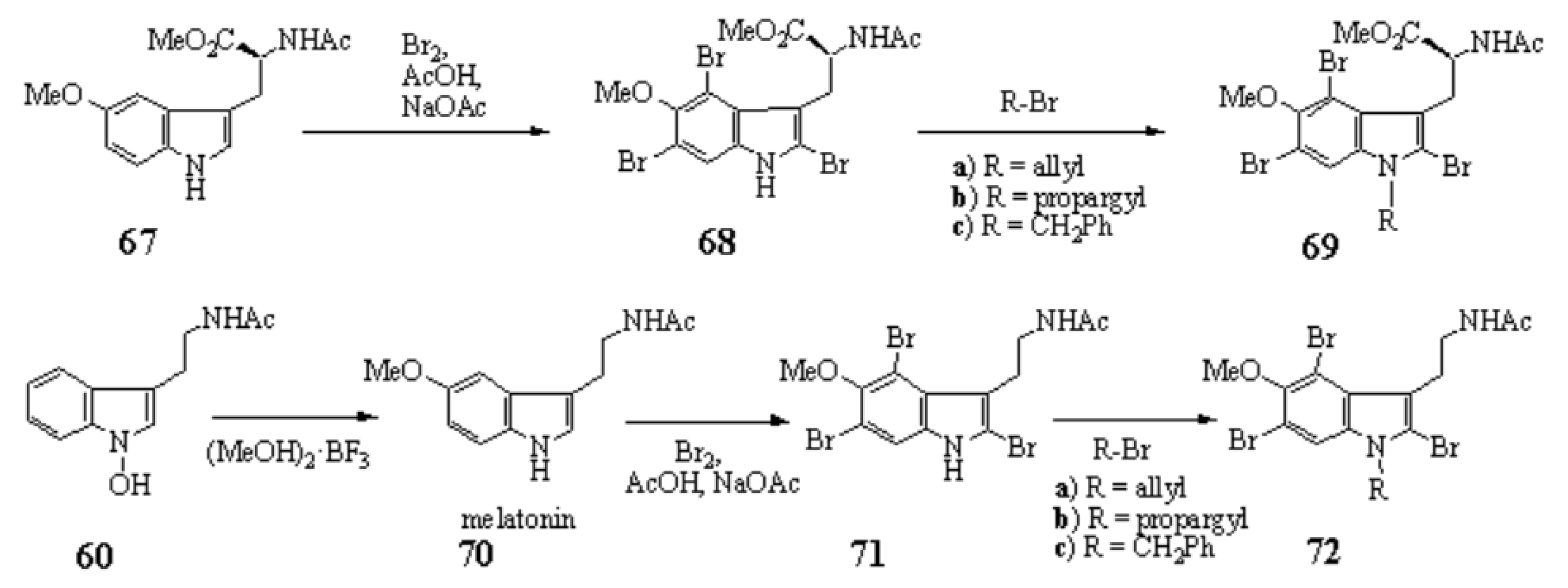
23.2. Tryptophan Derivatives:
Synthesis of (S)-(+)-N-acetyl-2,4,6-tribromo-5-methoxytryptophan methyl ester (68) from (S)-(+)-N-acetyl-5-methoxy tryptophan methyl ester (67) — To a solution of (S)-(+)-N-acetyl-5-methoxytryptophan methyl ester (67)[84] (56.6 mg, 0.20 mmol) in 4.5 mL of AcOH was added a solution of Br2 (1.0 mL, 0.59 mmol), separately prepared by dissolving 458.0 mg of Br2 and 41.8 mg of NaOAc in 5.0 mL of AcOH, and the mixture was stirred at room temperature for 30 min. After the addition of 10% aqueous Na2S2O2, the mixture was made alkaline by adding 40% aqueous NaOH under ice cooling and extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a yellow solid, which was column-chromatographed on SiO2 with CHCl3–MeOH (99.5:0.5, v/v) to give an inseparable 1:2 mixture (28.4 mg) of (S)-(+)-N-acetyl-2,6-dibromo- and (S)-(+)-N-acetyl-2,4,7-tribromo--5-methoxytryptophan methyl ester, and 68 (53.7 mg, 52%) in the order of elution. 68: mp 198–199°C (colorless granules, recrystallized from AcOEt). IR (KBr): 3307, 1730, 1647, 1556, 1300, 1232, 1028 cm-1. 1H-NMR (CDCl3) δ: 1.88 (3H, s), 3.29 (1H, dd, J = 9.8, 14.6 Hz), 3.58 (1H, dd, J = 5.2, 14.6 Hz), 3.77 (3H, s), 3.88 (3H, s), 5.01 (1H, ddd, J = 5.2, 8.6, 9.8 Hz, changed to dd, J = 5.2, 9.8 Hz on addition of D2O), 6.17 (1H, br d, J = 8.6 Hz, disappeared on addition of D2O), 7.37 (1H, s), 8.70 (1H, br s, disappeared on addition of D2O). MS m/z: 530 (M+), 528 (M+), 526 (M+), 524 (M+). Anal. Calcd for C15H15Br3N2O4: C, 34.19; H, 2.87; N, 5.32. Found: C, 34.24; H, 2.89; N, 5.18. Optical Rotation [α]D26 +14.8° (DMSO, c 0.200). [α]D27 +1.47° (MeOH, c 0.204). [α]D28 +4.4° (CHCl3, c 0.203).
Synthesis of (S)-(+)-N-acetyl-1-allyl-2,4,6-tribromo-5-methoxytryptophan methyl ester (69a) from 68 — To a solution of 68 (39.8 mg, 0.08 mmol) in N,N-dimethylformamide (DMF, 2.5 mL) was added K2CO3 (36.5 mg, 0.26 mmol) and allyl bromide (0.13 mL, d = 1.398, 1.51 mmol). After stirring at room temperature for 30 min, water was added to the reaction mixture. The whole was extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a yellow oil. Purification by column-chromatography on SiO2 with CHCl3 to give 69a (42.4 mg, 99%). 69a: mp 191–192°C (colorless needles, recrystallized from AcOEt). IR (KBr): 3303, 1732, 1645, 1547, 1228, 1016 cm-1. 1H-NMR (CDCl3) δ: 1.86 (3H, s), 3.34 (1H, dd, J = 9.8, 14.7 Hz), 3.62 (1H, dd, J = 5.4, 14.7 Hz), 3.74 (3H, s), 3.89 (3H, s), 4.75 (2H, m), 4.83 (1H, d, J = 17.1 Hz), 5.01 (1H, ddd, J = 5.4, 8.7, 9.8 Hz, changed to dd, J = 5.4, 9.8 Hz on addition of D2O), 5.19 (1H, d, J = 10.3 Hz), 5.86 (1H, tdd, J = 4.8, 10.3, 17.1 Hz), 6.12 (1H, br d, J = 8.7 Hz, disappeared on addition of D2O), 7.41 (1H, s). MS m/z: 570 (M+), 568 (M+), 566 (M+), 564 (M+). Anal. Calcd for C18H19Br3N2O4: C, 38.12; H, 3.38; N, 4.94. Found: C, 37.97; H, 3.43; N, 4.86. Optical Rotation [α]D26 +13.8° (CHCl3, c 0.203).
Synthesis of (S)-(+)-N-acetyl-2,4,6-tribromo-5-methoxy-1-propargyltryptophan methyl ester (69b) from 68 — To a solution of 68 (23.6 mg, 0.04 mmol) in DMF (2.0 mL) was added K2CO3 (21.6 mg, 0.16 mmol) and propargyl bromide (0.08 mL, d = 1.335, 0.9 mmol). After stirring at room temperature for 30 min, water was added to the reaction mixture. The whole was extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a pink oil. Purification by column-chromatography on SiO2 with CHCl3 to give 69b (23.9 mg, 94%). 69b: mp 284–285°C (decomp., measured with a sealed tube, colorless needles, recrystallized from CHCl3–MeOH). IR (KBr): 3284, 3224, 2114, 1724, 1647, 1552, 1230, 1016 cm-1. 1H-NMR (DMSO-d6) δ: 1.82 (3H, s), 3.28 (1H, dd, J = 7.1, 14.9 Hz), 3.33 (1H, dd, J = 8.7, 14.9 Hz), 3.33 (1H, t, J = 2.4 Hz), 3.48 (3H, s), 3.80 (3H, s), 4.47 (1H, ddd, J = 7.1, 7.1, 8.7 Hz, changed to dd, J = 7.1, 8.7 Hz on addition of D2O), 5.13 (2H, dt, J = 2.4, 4.2 Hz), 8.02 (1H, s), 8.44 (1H, br d, J = 7.1 Hz, disappeared on addition of D2O). MS m/z: 568 (M+), 566 (M+), 564 (M+), 562 (M+). Anal. Calcd for C18H17Br3N2O4: C, 38.26; H, 3.03; N, 4.96. Found: C, 38.11; H, 3.12; N, 4.83. Optical Rotation [α]D24 +7.7° (DMSO, c 0.202).
Synthesis of (S)-(+)-N-acetyl-1-benzyl-2,4,6-tribromo-5-methoxytryptophan methyl ester (69c) from 68 — To a solution of 68 (19.6 mg, 0.04 mmol) in DMF (1.5 mL) was added K2CO3 (18.0 mg, 0.13 mmol) and benzyl bromide (0.09 mL, d = 1.44, 0.7 mmol). After stirring at room temperature for 30 min, water was added to the reaction mixture. The whole was extracted with AcOEt–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a yellow oil. Preparative thin-layer chromatography was performed on SiO2 with CHCl3–MeOH (99:1, v/v) as a developing solvent. Extraction of the band having an Rf value of 0.18 to 0.29 with CHCl3–MeOH (95:5, v/v) afforded 69c (22.0 mg, 96%). 69c: mp 226–227°C (colorless needles, recrystallized from MeOH). IR (KBr): 3298, 1732, 1643, 1550, 1414, 1230, 1018 cm-1. 1H-NMR (DMSO-d6) δ: 1.80 (3H, s), 3.28 (1H, dd, J = 7.1, 14.4 Hz), 3.40 (1H, dd, J = 8.5, 14.7 Hz, appeared on addition of D2O), 3.47 (3H, s), 3.79 (3H, s), 4.55 (1H, ddd, J = 7.1, 7.1, 8.5 Hz, changed to dd, J = 7.1, 8.5 Hz on addition of D2O), 5.53 (2H, s), 6.96 (2H, d, J = 7.1 Hz), 7.25 (1H, t, J = 7.1 Hz), 7.31 (2H, t, J = 7.1 Hz), 7.90 (1H, s) 8.45 (1H, d, J = 7.1 Hz, disappeared on addition of D2O). MS m/z: 620 (M+), 618 (M+), 616 (M+), 614 (M+). Anal. Calcd for C22H21Br3N2O4: C, 42.82; H, 3.43; N, 4.54. Found: C, 42.69; H, 3.47; N, 4.56. Optical Rotation [α]D24 +8.3° (CHCl3, c 0.204)
- 1)
- Tryptamine derivatives
Melatonin (70) from Nb-acetyl-1-hydroxytryptamine (60) — 50% (MeOH)2·BF3 (15.0 mL) was added to a solution of 60 (150.0 mg, 0.688 mmol) in MeOH (10.0 mL) under ice cooling, and the mixture was refluxed for 30 min with stirring. After evaporation of the solvent, the whole was made neutral by adding 8% NaOH under ice cooling and extracted with CH2Cl2–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CH2Cl2–MeOH (98:2, v/v) to give 70 (121.6 mg, 72%). 70 was identical with commercially available authentic sample in every spectral data.
2,4,6-Tribromomelatonin (71) from melatonin (70) — A 0.56 M solution of Br2 in AcOH (containing 1 mmol of NaOAc, 3.30 mL, 1.85 mmol) was added to a solution of 70 (144.5 mg, 0.62 mmol) in AcOH (12 mL), and the mixture was stirred at rt for 1.5 h. After the addition of Na2S2O3 (1.0 mL) and H2O, the mixture was made basic with 40% NaOH under ice cooling and extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–MeOH (99:1, v/v) to give 71 (272.2 mg, 94%). 71: mp >300 °C (decomp., colorless powder, recrystallized from MeOH). IR (KBr): 3371, 3370, 1653, 1543, 1446, 1406, 1306, 1022 cm-1. 1H-NMR (CDCl3) δ: 1.78 (3H, s), 2.97 (2H, t, J=7.3 Hz), 3.25 (2H, q, J=7.3 Hz), 3.77 (3H, s), 7.52 (1H, s), 7.89 (1H, br t, J=5.6 Hz, disappeared on addition of D2O), 12.15 (1H, br t, J=7.3 Hz, disappeared on addition of D2O). Anal. Calcd for C13H13Br3N2O2: C, 33.29; H, 2.79; N, 5.97. Found: C, 33.27; H, 2.82; N, 5.85.
1-Allyl-2,4,6-tribromomelatonin (72a) from 2,4,6-tribromomelatonin (71) — General procedure. K2CO3 (31.1mg, 0.22 mmol) was added to a solution of 71 (30.2 mg, 0.064 mmol) in DMF (2.0 mL), and the mixture was stirred at rt for 1.5 h. To the resultant mixture, allyl bromide (0.11 mL, 1.28 mmol) was added and stirred at rt for 1.5 h. After the addition of H2O, the mixture was extracted with AcOEt–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt to give 72a (31.0 mg, 95%). 72a: mp 142–143 °C (colorless fine needles, recrystallized from AcOEt–hexane). IR (KBr): 3284, 1633, 1562, 1456, 1412, 1298, 1018 cm-1. 1H-NMR (CDCl3) δ: 1.93 (3H, s), 3.24 (2H, t, J=6.6 Hz), 3.58 (2H, q, J=6.6 Hz), 3.89 (3H, s), 4.76 (2H, dt, J=4.9, 1.7 Hz), 4.89 (1H, d, J=16.6 Hz), 5.20 (1H, d, J=10.3 Hz), 5.55 (1H, br t, disappeared on addition of D2O), 5.87 (1H, ddt, J=16.6, 10.3, 4.9 Hz), 7.4 (1H, s). Anal. Calcd for C16H17Br3N2O2: C, 37.75; H, 3.37; N, 5.50. Found: C, 37.75; H, 3.37; N, 5.42.
1-Propargyl-2,4,6-tribromomelatonin (72b) from 2,4,6-tribromomelatonin (71) — In the general procedure for the preparation of 72a, K2CO3 (31.9mg, 0.22 mmol), 71 (30.1 mg, 0.064mmol), and propargyl chloride (0.09 mL, 1.28 mmol) were used. After work–up, 31.6 mg (97%) of 72b was obtained. 72b: mp 199–200 °C (colorless fine needles, recrystallized from AcOEt–hexane). IR (KBr): 3286, 2117, 1628, 1558, 1456, 1435, 1410, 1294, 1018 cm-1. 1H-NMR (CDCl3) δ: 1.93 (3H, s), 2.34 (1H, t, J=2.4 Hz), 3.23 (2H, t, J=6.6 Hz), 3.58 (2H, q, J=6.6 Hz), 3.89 (3H, s), 4.91 (2H, d, J=2.4 Hz), 5.54 (1H, br t, J=6.6 Hz, disappeared on addition of D2O), 7.58 (1H, s). Anal. Calcd for C13H15Br3N2O2: C, 37.90; H, 2.98; N, 5.53. Found: C, 37.78; H, 3.00; N, 5.44.
1-Benzyl-2,4,6-tribromomelatonin (72c) from 2,4,6-tribromomelatonin (71) — In the general procedure for the preparation of 74a, K2CO3 (31.8mg, 0.30 mmol), 71 (40.1 mg, 0.086 mmol), and benzyl bromide (0.20 mL, 1.72 mmol) were used. After work–up, 40.3 mg (83%) of 72c was obtained. 72c: mp 218–219 °C (colorless fine needles, recrystallized from MeOH). IR (KBr): 3280, 1630, 1547, 1454, 1414, 1360, 1298, 1014 cm-1. 1H-NMR (CDCl3) δ: 1.91 (3H, s), 3.26 (2H, t, J=6.6 Hz), 3.61 (2H, td, J=12.7, 6.6 Hz), 3.88 (3H, s), 5.36 (2H, s), 5.54 (1H, br t, J=6.6 Hz, disappeared on addition of D2O), 7.01 (2H, d, J=6.6 Hz), 7.27-7.33 (3H, m), 7.39 (1H, s). Anal. Calcd for C20H19Br3N2O2: C, 42.97; H, 3.43; N, 5.01. Found: C, 42.76; H, 3.40; N, 4.86.
23-2. Synthesis of VED and related derivatives through 1-hydroxy tryptamines
Scheme 5 Synthesis of 1-hydroxy-Nb-acyltryptamines
Nb-Propionyl tryptamine (57a) from tryptamine — Et3N (1.89 mL, 13.6 mmol) and ClCO2Me (1.05 mL, 1.36 mmol) were added to a solution of propionic acid (913 mg, 12.3 mmol) in anhydrous CHCl3 (30 mL) and the mixture was stirred at 0 °C for 30 min. To the resulting mixture, tryptamine (2.17 g, 13.6 mmol) was added and the mixture was stirred at rt for 30 min. After addition of H2O the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a residue, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give 57a (2.51g, 94%). 57a: mp 88–89 °C (colorless fine needles, recrystallized from Et2O). IR (KBr): 3377, 1635, 1563, 1453, 1368, 1250 cm-1. 1H-NMR (CDCl3) δ: 1.11 (3H, t, J=7.0 Hz), 2.14 (2H, q, J=7.6 Hz), 2.98 (2H, dt, J=6.6, 0.7 Hz), 3.61 (2H, q, J=6.6 Hz), 5.50 (1H, br s), 7.04 (1H, d, J=2.2 Hz), 7.13 (1H, ddd, J=7.8, 7.1, 1.0 Hz), 7.21 (1H, ddd, J=7.8, 7.1, 1.2 Hz) 7.37 (1H, dt, J=7.8, 1.0 Hz), 7.61 (1H, ddd, J=7.8, 1.2, 0.7 Hz), 8.09 (1H, br s). MS m/z: 216 (M+). Anal. Calcd. For C13H16N2O·1/8H2O: C, 71.45; H, 7.50; N, 12.82. Found: C, 71.77; H, 7.34; N, 12.52.
Nb-Valeryl tryptamine (57b) from tryptamine — Et3N (1.57 mL, 11.3 mmol) and ClCO2Me (0.87 mL, 11.3 mmol) were added to a solution of valeric acid (1.05 g, 10.3 mmol) in anhydrous CHCl3 (30 mL) and the mixture was stirred at 0 °C for 30 min. To the resulting mixture, tryptamine (1.81 g, 11.3 mmol) was added and the mixture was stirred at rt for 30 min. After addition of H2O the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a residue, which was column-chromatographed on SiO2 with AcOEt–hexane (2:3, v/v) to give 57b (2.29g, 91%). 57b: mp 93–94 °C (colorless powder, recrystallized from AcOEt–hexane). IR (KBr): 3377, 3237, 2927, 1630, 1561, 1450 cm-1. 1H-NMR (CDCl3) δ: 0.88 (3H, t, J=7.3 Hz), 1.26–1.34 (2H, m), 1.53–1.60 (2H, m), 2.11 (2H, t, J=7.5 Hz), 2.98 (2H, t, J=6.6 Hz), 3.61 (2H, q, J=6.6 Hz), 5.56 (1H, br s), 7.04 (1H, s), 7.13 (1H, ddd, J=7.9, 7.0, 0.9 Hz), 7.22 (1H, ddd, J=7.9, 7.0, 0.9 Hz), 7.38 (1H, dt, J=7.9 Hz), 7.61 (1H, d, J=7.9 Hz), 8.09 (1H, br s). Anal. Calcd. For C15H20N2O: C, 73.73; H, 8.25; N, 11.47. Found: C, 73.48; H, 8.23; N, 11.42.
Nb-Heptanoyl tryptamine (57c) from tryptamine — Et3N (1.19 mL, 8.58 mmol) and ClCO2Me (0.66 mL, 8.58 mmol) were added to a solution of heptanoic acid (1.01 g, 7.80 mmol) in anhydrous CHCl3 (30 mL) and the mixture was stirred at 0 °C for 30 min. To the resulting mixture, tryptamine (1.37 g, 8.58 mmol) was added and the mixture was stirred at rt for 30 min. After addition of H2O the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a residue, which was column-chromatographed on SiO2 with CHCl3 to give 57c (2.02 g, 95%). 57c: mp 97–98 °C (colorless powder, recrystallized from AcOEt–hexane). IR (KBr): 3410, 1632, 1565, 1457, 1425 cm-1. 1H-NMR (CDCl3) δ: 0.87 (3H, t, J=7.3 Hz), 1.21–1.31 (6H, m), 1.57 (2H, quint, J=7.5 Hz), 2.10 (2H, t, J=7.5 Hz), 2.98 (2H, t, J=6.6 Hz), 3.61 (2H, q, J =6.6 Hz), 5.52 (1H, br s), 7.04 (1H, d, J=2.2 Hz), 7.13 (1H, ddd, J= 8.1, 7.0, 1.0 Hz), 7.22 (1H, ddd, J= 8.1, 7.0, 1.0 Hz, 7.38 (1H, dt, J=8.1, 1.0 Hz) 7.61 (1H, d, J=8.1 Hz), 8.08 (1H, br s). Anal. Calcd. For C17H24N2O: C, 74.96; H, 8.88; N, 10.29. Found: C, 74.80; H, 8.92; N, 10.26.
VED #1 [Nb-Nonanoyl tryptamine (57d)] from tryptamine — Et3N (0.99 mL, 7.09 mmol) and ClCO2Me (0.55 mL, 7.09 mmol) were added to a solution of nonanoic acid (1.02g, 6.45 mmol) in anhydrous CHCl3 (30 mL) and the mixture was stirred at 0 °C for 30 min. To the resulting mixture, tryptamine (1.14 g, 7.09 mmol) was added and the mixture was stirred at rt for 30 min. After addition of H2O the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a residue, which was column-chromatographed on SiO2 with AcOEt–hexane (1:2, v/v) to give 57d (1.78g, 93%). 57d: mp 101–102 °C (colorless fine needles, recrystallized from CHCl3–hexane). IR (CHCl3): 2950, 1652, 1506, 1165 cm-1. 1H-NMR (CDCl3) δ: 0.87 (3H, t, J=7.0 Hz), 1.22–1.31 (10H, m), 1.57 (2H, br quint, J=7.0 Hz), 2.10 (2H, t, J=7.6 Hz), 2.98 (2H, t, J=6.7 Hz), 3.61 (2H, q, J=6.7 Hz, collapsed to t , J=6.7 Hz on addition of D2O), 5.52 (1H, br s, disappeared on addition of D2O), 7.04 (1H, s), 7.13 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.21 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.38 (1H, d, J=8.1 Hz), 7.61 (1H, d, J=8.1 Hz), 8.09 (1H, br s, disappeared on addition of D2O). Anal. Calcd for C19H28N2O: C, 75.96; H, 9.39; N, 9.33. Found: C, 75.66; H, 9.49; N, 9.24.
2,3-Dihydro-Nb-propionyltryptamine (58a) from 57a — A mixture of 57a (1.02 g, 4.74 mmol) and Et3SiH (1.89 mL, 11.9 mmol) in TFA (20 mL) was stirred at rt for 30 min. After evaporation of the solvent, the residue was made alkaline with 8% NaOH and extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH–28%NH4H (46:1:0.1, v/v) to give 58a (1.01 g, 98%). 58a: yellow viscous oil. IR (film): 3315, 2970, 1635, 1606, 1547, 1486, 1461 cm-1. 1H-NMR (CDCl3) δ: 1.13 (3H, t, J=7.5 Hz), 1.78 (1H, dtd, J=13.6, 7.9, 6.0 Hz), 2.00 (1H, dddd, J=13.6, 7.9, 7.0, 5.0 Hz), 2.16 (2H, q, J=7.5 Hz), 2.82(1H, br s, disappeared on addition of D2O), 3.26–3.42 (4H, m), 3.72 (1H, t, J=8.8 Hz), 5.62 (1H, br s, disappeared on addition of D2O), 6.67 (1H, d, J=7.3 Hz), 6.75 (1H, td, J=7.3, 0.9 Hz), 7.05 (1H, br t, J=7.3 Hz), 7.10 (1H, d, J=7.3 Hz). HR–MS m/z: Calcd for C13H18N2O: 218.1419. Found: 218.1431.
2,3-Dihydro-Nb-valeryltryptamine (58b) from 57b — A mixture of 57b (102.1 mg, 0.42 mmol) and Et3SiH (0.17 mL, 1.05 mmol) in TFA (3.0 mL) was stirred at rt for 30 min. After evaporation of the solvent, the residue was made alkaline with 8% NaOH and extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH–28%NH4H (46:1:0.1, v/v) to give 58b (88.1 mg, 86%). 58b: yellow viscous oil. IR (film): 3290, 2930, 1640, 1605, 1552, 1484, 1461 cm-1. 1H-NMR (CDCl3) δ: 0.91 (3H, t, J= 7.5 Hz), 1.33 (2H, sext, J=7.5 Hz), 1.59 (2H, quint, J=7.5 Hz), 1.73 (1H, dtd, J=13.6, 8.0, 6.1 Hz), 1.99 (1H, dddd, J=13.6, 8.0, 7.0, 5.0 Hz), 2.13 (2H, t, J=7.5 Hz), 2.75 (1H, br s, disappeared on addition of D2O), 3.27–3.42 (4H, m), 3.72 (1H, t, J=8.6 Hz), 5.60 (1H, br s, disappeared on addition of D2O), 6.68 (1H, d, J=7.3 Hz), 6.75 (1H, td, J=7.3, 0.9 Hz), 7.05 (1H, br t, J=7.3 Hz), 7.11 (1H, d, J=7.3 Hz). HR–MS m/z: Calcd for C15H22N2O: 246.1732. Found: 246.1743.
Nb-Heptanoyl-2,3-dihydrotryptamine (58c) from 57c — A mixture of 57c (1.04 g, 3.82 mmol) and Et3SiH (1.52 mL, 9.54 mmol) in TFA (20 mL) was stirred at rt for 30 min. After evaporation of the solvent, the residue was made alkaline with 8% NaOH and extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (2:1, v/v) to give 58c (912.7 mg, 87%). 58c: pale yellow viscous oil. IR (film): 3310, 2960, 1634, 1606, 1544, 1484, 1461 cm-1. 1H-NMR (CDCl3) δ: 0.88 (3H, t, J= 7.5 Hz), 1.25–1.34 (6H, m), 1.60 (2H, quint, J=7.5 Hz), 1.78 (1H, dtd, J=13.6, 7.9, 5.9 Hz), 1.99 (1H, dddd, J=13.6, 7.9, 7.1, 5.1 Hz), 2.12 (2H, t, J=7.7 Hz), 2.68 (1H, br s, disappeared on addition of D2O), 3.27–3.42 (4H, m), 3.72 (1H, t, J=8.6 Hz), 5.60 (1H, br s, disappeared on addition of D2O), 6.68 (1H, d, J=7.5 Hz), 6.75 (1H, td, J=7.5, 1.1 Hz), 7.05 (1H, br t, J=7.5 Hz), 7.10 (1H, d, J=7.5 Hz). HR–MS m/z: Calcd for C17H26N2O: 274.2045. Found: 274.2057.
2,3-Dihydro-Nb-nonanoyltryptamine (58d) from 57d — A mixture of 57d (1.10 g, 3.65 mmol) and Et3SiH (1.45 mL, 9.10 mmol) in TFA (20 mL) was stirred at rt for 30 min. After evaporation of the solvent, the residue was made alkaline with 8% NaOH and extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give 58d (862.4 mg, 78%). 58d: mp 41–42.5 °C (colorless powder, recrystallized from AcOEt–hexane). IR (KBr): 3300, 2935, 2870, 1638, 1546, 1486, 1465 cm-1. 1H-NMR (DMSO-d6) δ: 0.84 (3H, t, J=7.0 Hz), 1.20–1.27 (10H, m), 1.45–1.57 (3H, m). 1.83 (1H, dtd, J=13.2, 7.6, 5.6 Hz), 2.04 (2H, t, J=7.5 Hz), 3.05 (1H, ddd, J=9.3, 8.1, 2.2 Hz), 3.09–3.16 (3H, m), 3.54 (1H, td, J=8.6, 1.7 Hz), 5.40 (1H, br s, disappeared on addition of D2O), 6.47 (1H, d, J=7.5 Hz), 6.52 (1H, td, J= 7.5, 0.7 Hz), 6.90 (1H, br t, J=7.5 Hz), 7.00 (1H, d, J=7.5 Hz), 7.80 (1H, br t, J= 6.1 Hz, disappeared on addition of D2O). Anal. Calcd for C19H30N2O: C, 75.45; H, 10.00; N, 9.26. Found: C, 75.25; H,10.16; N, 9.24.
1-Hydroxy-Nb-propionyltryptamine (59a) from 58a — A solution of 30% H2O2 (1.11 g, 9.80 mmol) in MeOH (3.0 mL) was added to a solution of 58a (211.8 mg, 0.97 mmol) and Na2WO4·2H2O (64.1 mg, 0.19 mmol) in MeOH (7.0 mL) and H2O (1.0 mL) under ice cooling with stirring. Stirring was continued at rt for 15 min. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (99:1, v/v) to give 59a (150.2 mg, 67%). 59a: mp 132–133 °C (colorless fine prisms, recrystallized from CHCl3). IR (KBr): 3290, 3100, 2935, 1598, 1566, 1352 cm-1. 1H-NMR (DMSO-d6) δ: 0.99 (3H, t, J=7.6 Hz), 2.06 (2H, q, J=7.6 Hz), 2.79 (2H, t, J=7.3 Hz), 3.30 (2H, td, J=7.3, 6.1 Hz, collapsed to t, J=7.3 Hz, on addition of D2O), 6.98 (1H, dd, J=8.0, 7.3 Hz), 7.13 (1H,dd, J=8.0, 7.3 Hz), 7.24 (1H, s), 7.32 (1H, d, J=8.0 Hz), 7.53 (1H, d, J=8.0 Hz), 7.84 (1H, br t, J=6.1 Hz, disappeared on addition of D2O), 11.01 (1H, s, disappeared on addition of D2O). Anal. Calcd for C13H16N2O2: C, 67.22; H, 6.94; N, 12.06. Found: C, 66.94; H, 6.95; N, 12.02.
1-Hydroxy-Nb-valeryltryptamine (59b) from 58b — A solution of 30% H2O2 (2.37 g, 20.9 mmol) in MeOH (5.0 mL) was added to a solution of 58b (513.3 mg, 2.09 mmol) and Na2WO4·2H2O (138.0 mg, 0.42 mmol) in MeOH (20 mL) and H2O (2.5 mL) under ice cooling with stirring. Stirring was continued at rt for 15 min. After addition of H2O, the whole was extracted with CHCl3. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (1:1, v/v) to give 59b (331.2 mg, 61%). 59b: mp 114.5–115 °C (colorless powder, recrystallized from CHCl3). IR (CHCl3): 3125, 2922, 1649, 1513 cm-1. 1H-NMR (DMSO-d6) δ: 0.86 (3H, t, J=7.4 Hz), 1.25 (2H, sext, J=7.4 Hz), 1.47 (2H, quint, J=7.4 Hz), 2.05 (2H, t, J=7.4 Hz), 2.78 (2H, t, J=7.4 Hz), 3,30 (2H, td, J=7.4, 6.1 Hz, collapsed to t, J=7.4 Hz, on addition of D2O), 6.98 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.12 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.23(1H, s), 7.32 (1H, d, J=8.1 Hz), 7.53 (1H, d, J=8.1 Hz), 7.86 (1H, br t, J=6.1 Hz, disappeared on addition of D2O), 11.00 (1H, s, disappeared on addition of D2O). Anal. Calcd for C15H20N2O2: C, 69.20; H, 7.74; N, 10.76. Found: C, 69.17; H, 7.70; N, 10.68.
1-Hydroxy-Nb-heptanoyltryptamine (59c) from 58c — A solution of 30% H2O2 (461 4 mg, 4.07 mmol) in MeOH (1.0 mL) was added to a solution of 58c (111.3 mg, 0.41 mmol) and Na2WO4·2H2O (27.3 mg, 0.08 mmol) in MeOH (4.0 mL) and H2O (0.5 mL) under ice cooling with stirring. Stirring was continued at rt for 30 min. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–hexane (2:1, v/v) to give 59c (79.8 mg, 68%). 59c: mp 83–83.5 °C (colorless prisms, recrystallized from CHCl3–hexane). IR (KBr): 3280, 2930, 1601, 1555, 1435, 1358, 1241 cm-1. 1H-NMR (DMSO-d6) δ: 0.86 (3H, t, J=7.5 Hz), 1.21–1.29 (6H, m), 1.47 (2H, quint., J=7.5 Hz), 2.04 (2H, t, J=7.5 Hz), 2.78 (2H, t, J=7.5 Hz), 3.29 (2H, td, J=7.5, 6.1 Hz, collapsed to t, J=7.5 Hz, on addition of D2O), 6.98 (1H, ddd, J= 8.1, 7.1, 1.0 Hz), 7.12(1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.24 (1H, s), 7.32 (1H, dt, J=8.1, 1.0 Hz), 7.52 (1H, dt, J=8.1, 1.0 Hz), 7.86 (1H, br t, J=6.1 Hz, disappeared on addition of D2O), 11.00 (1H, s, disappeared on addition of D2O). Anal. Calcd for C17H24N2O2: C, 70.80; H, 8.39; N, 9.71. Found: C, 70.73; H, 8.40; N, 9.64.
1-Hydroxy-Nb-nonanyltryptamine (59d) from 58d — A solution of 30% H2O2 (451.3 mg, 3.98 mmol) in MeOH (1.0 mL) was added to a solution of 58d (119.3 mg, 0.40 mmol) and Na2WO4·2H2O (26.7 mg, 0.08 mmol) in MeOH (4.0 mL) and H2O (0.5 mL) under ice cooling with stirring. Stirring was continued at rt for 30 min. After addition of H2O, the whole was extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (99:1, v/v) to give 59d (75.8 mg, 61%). 59d: mp 82.5–83 °C (colorless powder, recrystallized from CHCl3–hexane). IR (CHCl3): 3155, 2915, 1648, 1510, 1457 cm-1. 1H-NMR (DMSO-d6) δ: 0.86 (3H, t, J=7.4 Hz), 1.15–1.30 (10H, m), 1.47 (2H, quint., J=7.4 Hz), 2.03 (2H, t, J=7.4 Hz), 2.78 (2H, t, J=7.4 Hz), 3.30 (2H, td, J=7.4, 6.1 Hz, collapsed to t, J=7.4 Hz, on addition of D2O), 6.98 (1H, ddd, J= 8.1, 7.1, 1.0 Hz),7.12(1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.24 (1H, s), 7.32 (1H, d, J=8.1 Hz), 7.52 (1H, d, J=8.1 Hz), 7.86 (1H, br t, J=6.1 Hz, disappeared on addition of D2O), 11.01 (1H, s, disappeared on addition of D2O). Anal. Calcd for C19H28N2O2: C, 72.11; H, 8.92; N, 8.85. Found: C, 72.09; H, 8.96; N, 8.85.
1-Allyloxyindole (73a) from (3) — Prepared according to the general method B for 6, where Na2WO4·2H2O (611.6 mg, 1.85 mmol) in H2O (10.0 mL) was added to a solution of 3 (1.108 g, 9.29 mmol) in MeOH (20.0 mL). 30% H2O2 (10.561 g, 101.2 mmol) in MeOH (20.0 mL) was added to the resultant solution at 0 °C with stirring. After stirring for 15 min at rt (20 °C), K2CO3 (3.86 g, 27.8 mmol) and allyl bromide (3.313 g, 27.4 mmol) were added and stirred at rt for 1.5 h. Brine was added and the whole was extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave oil, which was purified by column-chromatography on SiO2 with CH2Cl2–hexane (1:9, v/v) to give 73a (711.3 mg, 44%). 73a: colorless oil. IR (film): 3050, 1449, 1220, 740 cm-1. 1H-NMR (CDCl3) δ: 4.67 (2H, dt, J=6.6, 1.2 Hz), 5.21 (1H, m), 5.35 (1H, d, J=3.4 Hz), 5.86–6.25 (1H, m), 6.30 (1H, dd, J=3.5, 1.0 Hz), 6.94–7.61 (5H, m). High resolution MS m/z: Calcd for C11H11NO: 173.0782. Found: 173.0811.
Scheme 7 Synthesis of 1-hydroxyindole derivatives (1)
1-Benzoyloxyindole (73b) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (57.1 mg, 0.17 mmol) in H2O (1.0 mL), 3 (103.0 mg, 0.86 mmol) in MeOH (8.0 mL), and 30% H2O2 (981.2 mg, 8.66 mmol) in MeOH (2.0 mL) were used. The reaction mixture was extracted with benzene and benzene layer was dried over Na2SO4. After filtering off Na2SO4, K2CO3 (583.3 mg, 3.89 mmol) and benzoyl chloride (483.2 mg, 2.59 mmol) were added to the benzene solution and stirred at rt for 1.5 h. H2O was added and the whole was extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave oil, which was purified by column-chromatography on SiO2 with CH2Cl2–hexane (3:7, v/v) to give 73b (100.8 mg, 49%). 73b: mp 55.5–56.0 °C (lit.[64] mp 49–50 °C, pale brown needles, recrystallized from MeOH). IR (KBr): 1767, 1600, 1446, 1323, 1236, 1184, 1075, 1039, 1012, 1002, 754, 729, 700 cm-1. UV λmaxMeOH nm (log ε): 217 (4.50), 265 (3.94), 293 (3.60). 1H-NMR (CDCl3) δ: 6.53 (1H, d, J=3.7 Hz), 6.96–7.35 (4H, m), 7.35–7.82 (4H, m), 8.21 (2H, dd, J=8.2, 1.7 Hz). MS m/z: 237 (M+). Anal. Calcd for C15H11NO2: C, 75.94; H, 4.67; N, 5.90. Found: C, 75.85; H, 4.62; N, 5.84.
1-t-Butyldimethylsilyloxyindole (73c) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (75.9 mg, 0.23 mmol) in H2O (1.3 mL), 3 (136.9 mg, 1.15 mmol) in MeOH (10.0 mL), and 30% H2O2 (1.304 g, 11.5 mmol) in MeOH (3.0 mL) were used. Silylation was carried out according to the method for 73b with K2CO3 (715.5 mg, 5.18 mmol) and t-butyldimethylsilyl chloride (520.2 mg, 3.45 mmol). After usual work-up and purification, 73c (133.1 mg, 47%) was obtained. 73c: colorless oil. IR (film): 1472, 1436, 1266, 1074, 1035, 836, 787, 739 cm-1. 1H-NMR (CDCl3) δ: 0.23 (6H, s), 1.10 (9H, s), 6.31 (1H, d, J=3.4 Hz), 7.01 (1H, t, J=6.6 Hz), 7.07 (1H, d, J=3.4 Hz), 7.17 (1H, t, J=6.6 Hz), 7.31 (1H, d, J=6.6 Hz), 7.53 (1H, d, J=6.6 Hz). High resolution MS m/z: Calcd for C14H21NOSi: 247.1390. Found: 247.1376.
1-Methallyloxyindole (73d) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (26.7 mg, 0.08 mmol) in H2O (0.5 mL), 3 (48.0 mg, 0.40 mmol) in MeOH (4.0 mL), and 30% H2O2 (461.7 mg, 4.0 mmol) in MeOH (1.0 mL) were used. Methallylation was carried out with K2CO3 (253.3 mg, 1.80 mmol) and methallyl chloride (110.5 mg, 1.20 mmol). After usual work-up and purification, 73d (4.2 mg, 6%) was obtained. 73d: colorless oil. IR (film): 1653, 1450, 1323, 1222, 740 cm-1. 1H-NMR (CDCl3) δ: 1.97 (3H, t, J=1.2 Hz), 4.60 (2H, s), 5.03 (2H, m), 6.32 (1H, dd, J=3.4, 0.7 Hz), 7.00–7.63 (5H, m). High resolution MS m/z: Calcd for C12H13NO: 187.0972. Found: 187.0984.
1-Benzyloxyindole (73e) and 3-benzyl-1-benzyloxyindole (74a) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (60.3 mg, 0.18 mmol) in H2O (1.0 mL), 3 (108.7 mg, 0.91 mmol) in MeOH (8.0 mL), and 30% H2O2 (1.036 g, 9.13 mmol) in MeOH (2.0 mL) were used. Benzylation was carried out with K2CO3 (568.1 mg, 4.11 mmol) and benzyl bromide (483.2 mg, 2.74 mmol). After usual work-up and purification, 73e (96.0 mg, 47%) and 74a (14.7 mg, 5%) were obtained. 73e: colorless oil. IR (film): 1455, 1323, 1221, 1074, 1032, 756, 740, 697 cm-1. 1H-NMR (CDCl3) δ: 5.15 (2H, s), 6.23 (1H, d, J=3.4 Hz), 6.98 (1H, d, J=3.4 Hz), 6.98–7.22 (2H, m), 7.22–7.44 (6H, m), 7.44–7.60 (1H, m). High resolution MS m/z: Calcd for C15H13NO: 223.0996. Found: 223.0992. 74a: colorless oil. IR (KBr, film): 1494, 1450, 734, 695 cm–1. 1H-NMR (CDCl3) δ: 4.00 (2H, s), 5.12 (2H, s), 6.11 (1H, s), 6.85–7.11 (14H, m). High resolution MS m/z: Calcd for C22H15NO: 313.1465. Found: 313.1468.
1-Prenyloxyindole (73f) and 3-prenyl-1-prenyloxyindole (74b) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (61.9 mg, 0.19 mmol) in H2O (1.0 mL), 3 (111.2 mg, 0.93 mmol) in MeOH (8.0 mL), and 30% H2O2 (1.087 g, 10.1 mmol) in MeOH (2.0 mL) were used. Prenylation was carried out with K2CO3 (470.6 mg, 6.84 mmol) and prenyl bromide (380.2 mg, 0.51 mmol). After usual work-up and purification 73f (13.5 mg, 7%) and 74b (9.3 mg, 4%) were obtained. 73f: pale yellow oil. IR (film): 3050, 2980, 2930, 1670, 1450, 1324, 1222, 1075, 1030, 740 cm-1. 1H-NMR (CDCl3) δ: 1.48 (3H, s), 1.68 (3H, s), 4.54 (2H, d, J=7.9 Hz), 5.44 (1H, t, J=7.2 Hz), 6.21 (1H, dd, 3.6, 1.0 Hz), 6.88–7.50 (5H, m). High resolution MS m/z: Calcd for C13H15NO: 201.1153. Found: 201.1155. 74b: pale red oil. IR (film): 2980, 2920, 1666, 1612, 1447, 1375, 1088, 1008, 735 cm–1. 1H-NMR (CDCl3) δ: 1.58 (3H, s), 1.74 (3H, s), 1.76 (6H, s), 3.38 (2H, dd, J=7.0, 1.0 Hz), 4.60 (2H, d, J=7.5 Hz), 5.28–5.61 (2H, m), 6.94–7.56 (5H, m). High resolution MS m/z: Calcd for C18H23NO: 269.1753. Found: 269.1765.
1-Prenyloxyindole (73f) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (65.2 mg, 0.19 mmol) in H2O (1.0 mL), 3 (116.4 mg, 0.98 mmol) in MeOH (9.0 mL), and 30% H2O2 (1.143 g, 10.0 mmol) in MeOH (1.0 mL) were used. Prenylation was carried out with NEt3 (1.4 mL, 9.8 mmol), (n-Bu)4NBr (32.3 mg, 0.10 mmol), and prenyl bromide (1.339 g, 8.71 mmol). After usual work-up and purification,73f (35.7 mg, 18%) was obtained.
1-Prenyloxyindole (73f) from 73c — A solution of prenyl bromide (112.2 mg, 0.72 mmol) in anhydrous THF (1.0 mL), KOt-Bu (91.7 mg, 0.82 mmol), and a solution of (n-Bu)4NF·3H2O (120.6 mg, 0.34 mmol) in anhydrous THF (1.0 mL) were added to a solution of 73c (75.4 mg, 0.37 mmol) in anhydrous THF (1.0 mL) at rt. After usual work-up and purification, 73f (75.4 mg, 100%) was obtained.
1-Cinnamyloxyindole (73g) and 3-cinnamyl-1-cinnanyloxyindole (74c) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (57.9 mg, 0.17 mmol) in H2O (1.0 mL). 3 (104.1 mg, 0.87 mmol) in MeOH (8.0 mL), and 30% H2O2 (991.9 mg, 8.7 mmol) in MeOH (2.0 mL) were used. Cinnamylation was carried out with K2CO3 (544.6 mg, 3.91 mmol) and cinnamyl bromide (520.8 mg, 2.61 mmol). After usual work-up and purification, 73g (110.5 mg, 51%) and 74c (80.2 mg, 22%) were obtained. 73g: pale brown oil. IR (film): 3050, 3017, 2920, 1495, 1449, 1323, 1221, 1074, 1030, 965, 757, 738, 691 cm-1. 1H-NMR (CDCl3) δ: 4.81 (2H, d, J=6.0 Hz), 6.31 (1H, dd, J=3.5, 1.0 Hz), 6.25–6.72 (2H, m), 6.96–7.60 (10H, m). High resolution MS m/z: Calcd for C17H15NO: 249.1148. Found: 249.1150. 74c: pale yellow oil. IR (film): 3050, 3017, 2920, 1496, 1449, 964, 738, 692 cm–1. 1H-NMR (CDCl3) δ: 3.58 (2H, dd, J=5.3, 1.0 Hz), 4.77 (2H, d, J=5.5 Hz), 6.13–6.72 (4H, m), 6.93–7.61 (15H, m). High resolution MS m/z: Calcd for C26H23NO: 365.1800. Found: 365.1789.
1-Methoxymethoxyindole (73h) from 3— Prepared according to the general method C for 6, where Na2WO4·2H2O (63.7 mg, 0.19 mmol) in H2O (1.0 mL), 3 (115.1 mg, 0.96 mmol) in MeOH (10.0 mL), and urea·H2O2 compound (927.2 mg, 9.66 mmol) were used. Methoxymethylation was carried out with K2CO3 (2.403 g, 17.38 mmol), (n-Bu)4NBr (31.1 mg, 0.09 mmol), and methoxymethyl chloride (233.3 mg, 2.89 mmol) in benzene (1.0 mL). After usual work-up and purification, 73h (66.0 mg, 39%) was obtained. 73h: mp 27.0–27.5 °C (colorless prisms, recrystallized from hexane). IR (KBr): 2970, 1445, 1330, 1230, 1182, 1100, 1089, 1030, 920, 740 cm-1. 1H-NMR (CDCl3) δ: 3.66 (3H, s), 5.17 (2H, s), 6.37 (1H, d, J=3.5 Hz), 7.11 (1H, t, J=7.4 Hz), 7.20–7.26 (3H, m), 7.42 (1H, d, J=8.1 Hz), 7.58 (1H, d, J=7.9 Hz). MS m/z: 177 (M+). Anal. Calcd for C10H11NO2: C, 67.78; H, 6.26; N, 7.90. Found: C, 67.64; H, 6.33; N, 7.86.
1-Methoxymethoxyindole (73h) from 73c — A solution of methoxymethyl chloride (56.2 mg, 0.69 mmol) in anhydrous THF (1.0 mL), KOt-Bu (91.2 mg, 0.81 mmol), and a solution of (n-Bu)4NF·3H2O (111.1 mg, 0.35 mmol) in anhydrous THF (1.0 mL) were added to a solution of 73c (85.4 mg, 0.34 mmol) in anhydrous THF (1.0 mL) at rt. After usual work-up and purification, 73h (59.7 mg, 98%) was obtained.
1-(2-Methoxyethoxymethoxy)indole (73i) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (61.4 mg, 0.19 mmol) in H2O (1.0 mL), 3 (111.1 mg, 0.93 mmol) in MeOH (9.0 mL), and 30% H2O2 (1.073 g, 9.46 mmol) in MeOH (1.0 mL) were used. Methoxyethoxymethylation was carried out with NEt3 (2.6 mL, 18.6 mmol), (n-Bu)4NBr (29.9 mg, 0.09 mmol), and 2-methoxyethoxymethyl chloride (1.038 g, 8.35 mmol). After usual work-up and purification, 73i (23.2 mg, 11%) and indole (2.2 mg, 2%) were obtained. 73i: colorless oil. IR (KBr): 2920, 2890, 1450, 1320, 1220, 1105, 1030, 920, 875, 845, 760, 740 cm-1. 1H-NMR (CDCl3) δ: 3.41 (3H, s), 3.59–3.65 (2H, m), 3.93–3.99 (2H, m), 5.27 (2H, s), 6.36 (1H, dd, J=3.5, 1.0 Hz), 7.10 (1H, ddd, J=7.9, 6.9, 1.0 Hz), 7.22 (1H, ddd, J=8.3, 6.9, 1.0 Hz), 7.33 (1H, d, J=3.5 Hz), 7.42 (1H, dd, J=8.3, 1.0 Hz), 7.58 (1H, ddd, J=7.9, 1.0, 1.01 Hz). High resolution MS m/z: Calcd for C12H15NO3: 221.1050. Found: 221.1049.
1-(2-Methoxyethoxymethoxy)indole (73i) from 73c — A solution of 2-methoxyethoxymethyl chloride (98.8 mg, 0.79 mmol) in anhydrous THF (1.0 mL), KOt-Bu (97.2 mg, 0.86 mmol), and a solution of (n-Bu)4NF·3H2O (125.8 mg, 0.39 mmol) in anhydrous THF (1.0 mL) were added to a solution of 73c (94.9 mg, 0.38 mmol) in anhydrous THF (1.0 mL) at rt. After usual work-up and purification, 73i (83.3 mg, 98%) was obtained.
3-Tosyloxyindole (75) from 3 — Prepared according to the general method B for 6, where Na2WO4·2H2O (559.6 mg, 1.69 mmol) in H2O (10.0 mL), 3 (1.009 g, 8.48 mmol) in MeOH (80.0 mL), and 30% H2O2 (9.610 g, 84.8 mmol) in MeOH (20.0 mL) were used. Tosylation was carried out with K2CO3 (5.270 g, 38.2 mmol) and tosyl chloride (4.848 g, 25.5 mmol). After usual work-up and purification, 75 (252.2 mg, 10%) was obtained. 75: mp 112–114°C (colorless prisms, recrystallized from MeOH). IR (KBr): 3390, 3120, 1595, 1453, 1370, 1190, 1175, 1090, 1063, 843, 812, 742, 723, 657, 555, 545, 503 cm-1. 1H-NMR (CDCl3) δ: 2.41 (3H, s), 6.95–7.30 (8H, m), 7.75 (2H, d, J=8.3 Hz). MS m/z: 287 (M+). Anal. Calcd for C15H13NO3S: C, 62.70; H, 4.56; N, 4.87. Found: C, 62.70; H, 4.53; N, 4.72. 76 was not isolated because of its instant instability to change to 75.
1-Hydroxy-6-nitroindole (78a) from 77a — Prepared according to the general method B for 6, where Na2WO4·2H2O (18.8 mg, 0.06 mmol), 77a (101.3 mg, 0.57 mmol) in MeOH (10.0 mL), and 30% H2O2 (0.58 mL, 5.7 mmol) were used. After usual work-up and purification,78a (80.1 mg, 79%) was obtained. 78a: mp 153–155 °C (decomp., pale orange needles, recrystallized from CHCl3). IR (KBr): 3240, 1617, 1586, 1514, 1481, 1357, 1332, 1280, 1095, 1056, 863, 811, 751, 731 cm-1. UV λmaxMeOH nm (log ε): 264 (4.01), 321 (3.94), 361 (3.73). 1H-NMR (CDCl3–CD3OD. 95:5, v/v) δ: 6.43 (1H, d, J=3.3 Hz), 7.51 (1H, d, J=3.3 Hz), 7.61 (H, d, J=8.8 Hz), 7.95 (1H, dd, J=8.8, 2.1 Hz), 8.42 (1H, dd, J=2.1 Hz). MS m/z: 178 (M+). Anal. Calcd for C8H6N2O3: C, 53.94; H, 3.39; N, 15.72. Found: C, 54.03; H, 3.45; N, 15.73.
1-Hydroxy-5-nitroindole (78b) from 77b — Prepared according to the general method B, where Na2WO4·2H2O (81.6 mg, 0.25 mmol) in H2O (2.0 mL), 77b (202.9 mg, 1.23 mmol) in MeOH (20.0 mL), and 30% H2O2 (1.26 mL, 12.3 mmol) were used. After usual work-up and purification, 77b (52.7 mg, recovery, 26%), 5-nitroindole (9.4 mg, 5%), and 78b (91.7 mg, 42%) were obtained. The mixture of 78b and 5-nitroindoe was successfully separated by column chromatography on Al2O3 with benzene–EtOAc (10:1, v/v). 78b: mp 175–176 °C (decomp., brown needles, recrystallized from CHCl3). IR (KBr, film): 1613, 1584, 1508, 1358, 1339, 756, 720 cm-1. UV λmaxMeOH nm (log ε): 254 (4.07), 275 (4.20), 331 (3.81). 1H–NMR (CDCl3–CD3OD, 95:5, v/v) δ: 6.45 (H, d, J=3.4 Hz), 7.52 (1H, d, J=3.4 Hz), 7.60 (1H, d, J=8.9 Hz), 7.98 (1H, dd, J=8.8, 2.2 Hz), 8.38 (1H, br s). MS m/z: 178 (M+). Anal. Calcd for C8H6N2O3: C, 53.94; H, 3.39; N, 15.72. Found: C, 53.66; H, 3.37; N, 15.71.
1-Hydroxy-2-phenylindole (78d) from 2,3-dihydro-2-phenylindole (77d) — Prepared according to the general method B for 6, where Na2WO4·2H2O (17.4 mg, 0.053 mmol), 77d (51.5 mg, 0.26 mmol) in 4.0 mL of MeOH, and 30% H2O2 (299.4 mg, 2.64 mmol) in MeOH (1.0 mL) were used. The crude product was purified by p-TLC on SiO2 with CH2Cl2–MeOH (98:2, v/v) as a developing solvent to afford 78d (30.9 mg, 56%). 78d: mp 174.0–175.0 °C (decomp., pale yellow needles, recrystallized from CHCl3, lit.,[65] mp 175 °C). IR (KBr): 2400, 1625, 1370, 756, 740, 683 cm-1. 1H-NMR (10% CD3OD in CDCl3) δ: 6.52 (1H, s, C3–H, deuterated during measuring), 6.88–7.62 (7H, m), 7.68–7.92 (2H, m). MS m/z: 209 (M+). Identical with the authentic sample prepared from benzoin oxime.[66]
1-Methoxy-6-nitroindole (79a) from 2,3-dihydro-6-nitroindole (77a) — Prepared according to the general method B for 6, where Na2WO4·2H2O (9.0 mg, 0.027 mmol), 77a (44.8 mg, 0.27 mmol) in MeOH (3.0 mL), and 30% H2O2 (309.7 mg, 2.73 mmol) were used. After methylation, usual work-up, and purification, 79a (31.3 mg, 60%) and 6-nitroindole (3.8 mg, 9%) were obtained. 79a: mp 90.0–91.0 °C (yellow needles, recrystallized from MeOH). IR (KBr): 1613, 1584, 1508, 1358, 1339, 756, 720 cm-1. 1H-NMR (CDCl3) δ: 4.17 (3H, s), 6.45 (1H, dd, J=3.4 and 1.0 Hz), 7.52 (1H, d, J=3.4 Hz), 7.60 (1H, d, J=8.8 Hz), 7.98 (1H, dd, J=8.8, 2.2 Hz), 8.38 (1H, br d, J=2.2 Hz). MS m/z: 192 (M+). Anal. Calcd for C9H8N2O3: C, 56.25; H, 4.20; N, 14.58. Found: C, 56.21; H, 4.17; N, 14.73.
1-Methoxy-5-nitroindole (79b) from 2,3-dihydro-5-nitroindole (77b) — Prepared according to the general method B for 6, where Na2WO4·2H2O (16.4 mg, 0.05 mmol), 77b (40.8 mg, 0.29 mmol) in MeOH (3.0 mL), and 30% H2O2 (282.0 mg, 2.48 mmol) in MeOH (2.0 mL) were used. After methylation, usual work-up, and purification, 79b (23.2 mg, 49%), unreacted starting material (17.1 mg, 42%), and 5-nitroindole (1.1 mg, 3%) were obtained. 79b: mp 89.5–90.5 °C (yellow plates, recrystallized from MeOH). IR (KBr): 1615, 1580, 1512, 1324, 1066, 737 cm-1. 1H-NMR (CDCl3) δ: 4.14 (3H, s), 6.54 (1H, dd, J=3.6, 0.8 Hz), 7.38 (1H, d, J=3.6 Hz), 7.44 (1H, d, J=9.0 Hz), 8.12 (1H, dd, J=9.0, 2.1 Hz), 8.54 (1H, d, J=2.1 Hz). MS m/z: 192 (M+). Anal. Calcd for C9H8N2O3: C, 56.25; H, 4.20; N, 14.58. Found: C, 56.25; H, 4.17; N, 14.50.
7-Iodo-1-methoxyindole (79c) from 2,3-dihydro-7-iodoindole (77c) — Prepared according to the general method B for 6, where Na2WO4·2H2O (14.7 mg, 0.045 mmol), 79c (218.7 mg, 0.89 mmol) in MeOH (5.0 mL), and 30% H2O2 (303.6 mg, 2.68 mmol) in MeOH (4.0 mL) were used. After usual work-up and purification, 79c (62.6 mg, 26%), 7-iodoindole (9.9 mg, 5%), and unreacted starting material (117.5 mg, 54%) were obtained. 79c: mp 35.0–35.5 °C (colorless plates, recrystallized from hexane). IR (KBr): 1544, 1333, 1276, 1033, 948, 775 cm-1. 1H-NMR (CDCl3) δ: 4.08 (3H, s), 6.31 (1H, d, J=3.4 Hz), 6.82 (1H, t, J=7.6 Hz), 7.29 (1H, d, J=3.4 Hz), 7.54 (1H, dd, J=7.6, 1.0 Hz), 7.68 (1H, dd, J=7.6, 1.0 Hz). MS m/z: 273 (M+). Anal. Calcd for C9H8INO: C, 39.59; H, 2.95; N, 5.12. Found: C, 39.53; H, 2.99; N, 5.13.
1-Methoxy-2-phenylindole (79d) — a) From 2,3-dihydro-2-phenylindole (77d); prepared according to the general method B for 6, where Na2WO4·2H2O (17.7 mg, 0.054 mmol), 77d (52.3 mg, 0.27 mmol) in MeOH (4.0 mL), and 30% H2O2 (304.1 mg, 2.68 mmol) in MeOH (1.0 mL) were used. After usual work-up and purification, 79d (40.0 mg, 67%) and 2-phenylindole (3.9 mg, 8%) were obtained. 79d: mp 47.0–48.0 °C (lit.[15] mp 49–51 °C, pale yellow plates, recrystallized from MeOH). IR (KBr): 1597, 956, 760, 741 cm-1. 1H-NMR (CDCl3) δ: 3.73 (3H, s), 6.56 (1H, s), 7.00–7.66 (7H, m), 7.73–7.91 (2H, m). Anal. Calcd for C15H13NO: C, 80.69; H, 5.87; N, 6.27. Found: C, 80.77; H, 5.91; N, 6.04.
b) From 1-Hydroxy-2-phenylindole (78d): ethereal CH2N2 (excess) was added to a solution of 78d (30.9 mg, 0.15 mmol) in MeOH (3.0 mL) with stirring at rt until the starting material was not detected on tlc monitoring. The crude product was purified by p-TLC on SiO2 with EtOAc–hexane (1:4, v/v) as a developing solvent to afford 79d (28.3 mg, 86%).
Methyl 1-methoxyindole-2-carboxylate (79f) from 2,3-dihydroindole-2-carboxylic acid (77e) — Prepared according to the general method B, where Na2WO4·2H2O (20.8 mg, 0.063 mmol), 77e (51.5 mg, 0.31 mmol) in MeOH (4.0 mL), and 30% H2O2 (358.2 mg, 3.16 mmol) in MeOH (1.0 mL) were used. After methylation and usual work-up and purification, 4 (10.4 mg, 22%) and 79f (11.4 mg, 18%) were obtained. 79f: mp 40.5–41.5 °C (colorless plates, recrystallized from MeOH). IR (KBr): 1723, 1239, 1209, 1085, 738 cm-1. 1H-NMR (CDCl3) δ: 3.93 (3H, s), 4.19 (3H, s), 7.08 (1H, d, J=1.2 Hz), 7.02–7.54 (3H, m), 7.61 (1H, dt, J=7.9, 1.0 Hz). MS m/z: 205 (M+). Anal. Calcd for C11H11NO3: C, 64.38; H, 5.40; N, 6.83. Found: C, 64.19; H, 5.45; N, 7.00.
Scheme 8.
Synthesis of 1-hydroxyindole derivative (2)
3-Acetylaminomethyl-1-hydroxyindole (82b) from 3-acetylaminomethyl-2,3-dihydroindole (80) — Prepared according to the method for 6, where Na2WO4·2H2O (149.8 mg, 0.45 mmol) in H2O (4.5 mL), 80 (688.0 mg, 3.16 mmol) in MeOH (55.0 mL), and 30% H2O2 (2.574 g, 22.7 mmol) in MeOH (5.0 mL) were used. After usual work-up and purification, 82b (302.5 mg, 66%) was obtained. 82b: mp 132.5–133.0 °C (pale yellow prisms, recrystallized from CH2Cl2). IR (KBr): 3330, 2810, 1603, 1538, 1406, 1366, 1320, 1244, 1103, 1028, 1006, 743, 667, 570 cm-1. 1H-NMR (5% CD3OD–CDCl3) δ: 1.96 (3H, s), 4.49 (2H, s), 7.08 (1H, dd, J=7.7, 7.3 Hz), 7.18 (1H, s), 7.22 (1H, dd, J=8.1, 7.3 Hz), 7.45 (1H, d, J=8.1 Hz), 7.54 (1H, d, J=7.7 Hz). MS m/z: 204 (M+). Anal. Calcd for C11H12N2O2·1/8H2O: C, 63.99; H, 5.98; N, 13.57. Found: C, 64.15; H, 5.79; N, 13.60.
1-Hydroxy-Nb-trifluoroacetylindole-3-methanamine (82a) from 81 — Prepared according to the method for 82b, where Na2WO4·2H2O (148.7 mg, 0.45 mmol) in H2O (4.5 mL), 81 (551.2 mg, 2.26 mmol) in MeOH (50.0 mL), and 30% H2O2 (2.575 g, 22.6 mmol) in MeOH (5.0 mL) were used. After usual work-up, the crude product was column-chromatographed on SiO2 with EtOAc–hexane (3:1, v/v) to give 82a (414.5 mg, 71%). 82a: mp 123.5–124.5 °C (colorless needles, recrystallized from CH2Cl2). IR (KBr): 3390, 3305, 1694, 1566, 1541, 1353, 1252, 1206, 1178, 1169, 1151, 1103, 755, 747, 682 cm-1. 1H-NMR (CD3OD) δ: 4.59 (2H, s), 7.03 (1H, dd, J=8.0, 0.9 Hz), 7.17 (1H, dd, J=8.0, 0.9 Hz), 7.28 (1H, s), 7.38 (1H, ddd, J=8.0, 0.9, 0.7 Hz), 7.57 (1H, ddd, J=8.0, 0.9, 0.7 Hz). High resolution MS m/z: Calcd for C11H9F3N2O2: 258.0615. Found: 258.0617.
1-Methoxy-Nb-trifluoroacetylindole-3-methanamine (83a) from 2,3-dihydro-3-trifluoroacetyl- indole-3-methanamine (81) — Prepared according to the method for 80b, where Na2WO4·2H2O (14.8 mg, 0.04 mmol) in H2O (0.5 mL), 81 (54.1 mg, 0.22 mmol) in MeOH (6.0 mL), and 30% H2O2 (266.6 mg, 2.35 mmol) in MeOH (1.0 mL) were used. After methylation and work-up, the product was purified by column-chromatography on SiO2 with CHCl3–hexane (3:1, v/v) to give 83a (46.5 mg, 77%). 83a: mp 70.5–71.0 °C (colorless prisms, recrystallized from benzene–hexane). IR (KBr): 3290, 1715, 1695, 1561, 1208, 1180, 1159, 738, 722 cm-1. UV λmaxMeOH nm (log ε): 219 (4.57), 272 (3.73), 288 (3.72). 1H-NMR (CDCl3) δ: 4.10 (3H, s), 4.68 (2H, d, J=5.3 Hz), 6.43 (1H, br s), 7.18 (1H, t, J=7.8 Hz), 7.31 (1H, s), 7.31 (1H, t, J=7.8 Hz), 7.46 (1H, d, J=7.8 Hz), 7.57 (1H, d, J=7.8 Hz). High resolution MS m/z: Calcd for C12H11F3N2O2: 272.0772. Found: 272.0756.
1-Methoxy-Nb-acetylindole-3-methanamine (83b) from 3-acetylaminomethyl-2,3-dihydroindole (80) — Prepared according to the method for 82b, where Na2WO4·2H2O (22.8 mg, 0.07 mmol) in H2O (0.5 mL), 80 (64.9 mg, 0.34 mmol) in MeOH (6.0 mL), and 30% H2O2 (388.0 mg, 3.42 mmol) in MeOH (1.0 mL) were used. After methylation and work-up, the product was purified by column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (100:1:0.1, v/v) to give 83b (43.6 mg, 59%). 83b: mp 132.5–133.0 °C (colorless prisms, recrystallized from benzene). IR (KBr): 3200, 3040, 1623, 1533, 1450, 1245, 735 cm-1. UV λmaxMeOH nm (log ε): 222 (4.51), 273 (3.72), 288 (3.72). 1H-NMR (CDCl3) δ: 1.98 (3H, s), 4.07 (3H, s), 4.56 (2H, d, J=5.1 Hz), 5.65 (1H, br s), 7.14 (1H, t, J=8.2 Hz), 7.24 (1H, s), 7.28 (1H, t, J=8.2 Hz), 7.43 (1H, d, J=8.2 Hz), 7.60 (1H, d, J=8.2 Hz). MS m/z: 218 (M+). Anal. Calcd for C12H14N2O2: C, 66.04; H, 6.47; N, 12.84. Found: C, 65.91; H, 6.47; N, 12.71.
3-Acetylaminomethyl-1-methoxyindole (83b) from 82b — Ethereal CH2N2 (excess) was added to a solution of 82b (11.8 mg, 0.058 mmol) in MeOH (1.0 mL) and stirring was continued at rt for 30 min. After evaporation of the solvent under reduced pressure, the residue was column-chromatographed on SiO2 with CH2Cl2–MeOH (97:3, v/v) to give 81b (10.7 mg, 85%).
2,3-Dihydro-3-tosylaminomethylindole (85) from 3-tosylaminomethylindole (84) — 95% NaBH3CN (247.7 mg, 3.74 mmol) was added to a solution of 84 (201.8 mg, 0.67 mmol) in AcOH (10.0 mL) at rt and stirring was continued for 5 h. After adding H2O under ice cooling, the solvent was evaporated under reduced pressure. The residue was made alkaline by adding H2O and 8% NaOH. The whole was extracted with CH2Cl2–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a residue, which was column-chromatographed on SiO2 with CH2Cl2–MeOH (99:1, v/v) to give 84 (16.5 mg, recovery, 8%) and 85 (144.3 mg, 71%) in the order of elution. 85: colorless hard oil. IR (KBr): 3320, 3050, 2910, 1592, 1481, 1460, 1423, 1323, 1155, 1041, 755 cm-1. 1H-NMR (CD3OD) δ: 2.42 (3H, s), 2.92 (1H, dd, J=12.8, 5.3 Hz), 3.06 (1H, dd, J=12.8, 5.3 Hz), 3.27 (1H, dd, J=9.3, 5.7 Hz), 3.30–3.38 (1H, m), 3.51 (1H, dd, J=9.3, 8.6 Hz), 6.63 (1H, d, J=7.9 Hz), 6.65 (1H, td, J=7.3, 0.9 Hz), 6.98 (1H, t, J=7.9 Hz), 7.03 (1H, d, J=7.3 Hz), 7.37 (2H, m), 7.72 (2H, m). MS m/z: 302 (M+). Anal. Calcd for C16H18N2O2S: C, 63.55; H, 6.00; N, 9.26. Found: C, 63.45; H, 6.04; N, 9.16.
1-Hydroxy-3-tosylaminomethylindole (86) from 85 — Prepared according to the method for 82b, where Na2WO4·2H2O (60.6 mg, 0.18 mmol) in H2O (1.8 mL), 85 (277.4 mg, 0.92 mmol) in MeOH (13.0 mL), and 30% H2O2 (1.049 g, 9.26 mmol) in MeOH (5.0 mL) were used. After usual work-up, 86 (197.5 mg, 68%) was obtained. 86: mp 134.0–135.5 °C (colorless needles, recrystallized from CHCl3). IR (KBr): 3390, 3320, 1396, 1318, 1303, 1230, 1157, 1097, 1020, 817, 732 cm-1. 1H-NMR (CD3OD) δ: 2.39 (3H, s), 4.18 (2H, s), 6.94 (1H, ddd, J=8.0, 7.0, 1.0 Hz), 7.08 (1H, s), 7.11 (1H, ddd, J=8.0, 7.0, 1.0 Hz), 7.28 (2H, m), 7.29 (1H, dt, J=8.0, 1.0 Hz), 7.39 (1H, dt, J=8.0, 1.0 Hz), 7.68 (2H, m). MS m/z: 316 (M+). Anal. Calcd for C16H16N2O3S·1/4H2O: C, 59.89; H, 5.18; N, 8.73. Found: C, 59.91; H, 5.00; N, 8.71.
1-Methoxy-3-tosylaminomethylindole (87) from 86 — Ethereal CH2N2 (excess) was added to a solution of 86 (32.3 mg, 0.10 mmol) in MeOH (1.0 mL) and stirring was continued at rt for 30 min. After evaporation of the solvent under reduced pressure, the residue was column-chromatographed on SiO2 with CH2Cl2 to give 87 (32.2 mg, 96%). 87: mp 176.0–178.5 °C (colorless prisms, recrystallized from MeOH). IR (KBr): 3300, 1600, 1423, 1315, 1243, 1145, 1089, 1024, 952, 866, 812, 746, 681, 541 cm-1. 1H-NMR (5% CD3OD–CDCl3) δ: 2.43 (3H, s), 4.02 (3H, s), 4.25 (2H, s), 7.07 (1H, ddd, J=8.0, 7.1, 1.0 Hz), 7.10 (1H, s), 7.23 (1H, ddd, J=8.2, 7.1, 1.0 Hz), 7.28 (2H, m), 7.37 (1H, dt, J=8.2, 1.0 Hz), 7.41 (1H, dt, J=8.0, 1.0 Hz), 7.74 (2H, m). MS m/z: 330 (M+). Anal. Calcd for C17H18N2O3S: C, 61.80; H, 5.49; N, 8.48. Found: C, 61.71; H, 5.50; N, 8.41.
Methyl N-(2,3-dihydroindol-3-yl)methyl succinamate (90) and N-(2,3-Dihydroindol-3-yl)methyl- succinimide (89) from methyl N-(indol-3-yl)methyl succinamate (88) — Prepared according to the method for 85, where 95% NaBH3CN (66.0 mg, 1.00 mmol) and 88 (49.8 mg, 0.19 mmol) in AcOH (2.0 mL) were used. After usual work-up and purification, 89 (6.5 mg, 15%) and 90 (41.0 mg, 82%) were obtained. 90: mp 73.0–74.0 °C (colorless prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 3290, 1726, 1638, 1609, 1542, 1483, 1433, 1338, 1197, 1174, 745 cm-1. 1H-NMR (CD3OD) δ: 2.50 (2H, t, J=6.8 Hz), 2.61 (2H, t, J=6.8 Hz), 3.25 (1H, dd, J=9.3, 5.5 Hz), 3.28–3.32 (1H, m), 3.41–3.47 (2H, m), 3.56 (1H, t, J=9.3 Hz), 3.66 (3H, s), 6.68 (1H, d, J=7.6 Hz), 6.69 (1H, td, J=7.6, 1.0 Hz), 7.00 (1H, t, J=7.6 Hz), 7.14 (1H, d, J=7.6 Hz). MS m/z: 262 (M+). Anal. Calcd for C14H18N2O3: C, 64.10; H, 6.92; N, 10.68. Found: C, 64.11; H, 6.90; N, 10.69. 89: mp 94.5–96.0 °C (colorless prisms, recrystallized from MeOH). IR (KBr): 3370, 2990, 2820, 1778, 1689, 1608, 1405, 1351, 1247, 1124, 1151, 755 cm-1. 1H-NMR (CD3OD) δ: 2.71 (4H, s), 3.29 (1H, dd, J=9.5, 5.1 Hz), 3.48 (1H, dd, J=9.5, 8.4 Hz), 3.57 (1H, m), 3.64 (1H, dd, J=13.1, 8.7 Hz), 3.71 (1H, dd, J=13.1, 5.4 Hz), 6.66 (1H, t, J=7.8 Hz), 6.68 (1H, td, J=7.3, 1.0 Hz), 7.01 (1H, m), 7.08 (1H, d, J=7.3 Hz). MS m/z: 230 (M+). Anal. Calcd for C13H14N2O2: C, 67.81; H, 6.13; N, 12.17. Found: C, 67.77; H, 6.11; N, 12.08.
Methyl N-(1-hydroxyindol-3-yl)methyl succinamate (91a) from 90 — Prepared according to the method for 82b, where Na2WO4·2H2O (14.6 mg, 0.04 mmol) in H2O (0.4 mL), 90 (56.8 mg, 0.21 mmol) in MeOH (3.5 mL), and 30% H2O2 (249.9 mg, 2.21 mmol) in MeOH (1.0 mL) were used. After usual work-up and purification, 91a (37.5 mg, 63%) was obtained. 91a: mp 115.5–116.0 °C (colorless needles, recrystallized from EtOAc). IR (KBr): 3350, 3120, 2930, 1710, 1637, 1535, 1443, 1387, 1358, 1243, 1218, 1174, 735 cm-1. 1H-NMR (CD3OD) δ: 2.48 (2H, t, J=6.7 Hz), 2.62 (2H, t, J=6.7 Hz), 3.61 (3H, s), 4.48 (2H, s), 7.01 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.15 (1H, ddd, J=8.3, 7.1, 1.0 Hz), 7.24 (1H, s), 7.36 (1H, dt, J=8.3, 1.0 Hz), 7.54 (1H, dt, J=8.1, 1.0 Hz). MS m/z: 276 (M+). Anal. Calcd for C14H16N2O4: C, 60.86; H, 5.84; N, 10.14. Found: C, 60.72; H, 5.85; N, 10.12.
Methyl N-(1-methoxyindol-3-yl)methyl succinamate (91b) from 91a — Ethereal CH2N2 (excess) was added to a solution of 91a (25.5 mg, 0.09 mmol) in MeOH (1.0 mL) and stirring was continued at rt for 30 min. After evaporation of the solvent under reduced pressure, the residue was column-chromato- graphed on SiO2 with CH2Cl2–MeOH (99:1, v/v) to give 91b (25.5 mg, 95%). 91b: mp 75.0–76.5 °C (colorless prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 3280, 1730, 1634, 1537, 1445, 1349, 1319, 1201, 1136, 736 cm-1. 1H-NMR (CD3OD) δ: 2.49 (2H, t, J=7.1 Hz), 2.62 (2H, t, J=7.1 Hz), 3.61 (3H, s), 4.05 (3H, s), 4.48 (2H, s), 7.06 (1H, ddd, J=7.8, 7.1, 1.0 Hz), 7.20 (1H, ddd, J=8.3, 7.1, 1.0 Hz), 7.36 (1H, s), 7.39 (1H, dt, J=8.3, 1.0 Hz), 7.58 (1H, dt, J=7.8, 1.0 Hz). MS m/z: 290 (M+). Anal. Calcd for C15H18N2O4: C, 62.05; H, 6.25; N, 9.65. Found: C, 62.05; H, 6.33; N, 9.60.
2-Methylthiomethylindole (93) from gramine (92) — MeI (1.45 mL, 23.3 mmol) was added to a solution of 92 (399.3 mg, 2.30 mmol) in THF (23.0 mL) and stirred at rt for 1 h. The solvent was evaporated under reduced pressure to leave a residue, which was dissolved in MeOH (20.0 mL). To the resultant solution, 15% aqueous NaSMe (10.7 mL, 23.3 mmol) was added and stirred at rt for 15 h. After addition of H2O, the whole was extracted with CH2Cl2–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a residue, which was column-chromatographed on SiO2 with CHCl3–MeOH–28% aq. NH3 (100:20:2, v/v) to give 93 (325.7 mg, 80%) and 92 (65.7 mg, recovery, 17%) in the order of elution. 93: mp 91.0–92.5 °C (colorless prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 3310, 1645, 1555, 1456, 1421, 1354, 1253, 1097, 745, 639 cm-1. 1H-NMR (CD3OD) δ: 1.98 (3H, s), 3.89 (2H, s), 7.00 (1H, ddd, J=8.1, 7.2, 0.9 Hz), 7.09 (1H, ddd, J=8.2, 7.2, 1.1 Hz), 7.14 (1H, s), 7.32 (1H, dd, J=8.2, 0.9 Hz), 7.62 (1H, dd, J=8.1, 1.1 Hz). Anal. Calcd for C10H11NS: C, 67.79; H, 6.25; N, 7.88. Found: C, 67.76; H, 6.25; N, 7.90.
Scheme 9.
Synthesis of 1-hydroxyinole derivative (3)
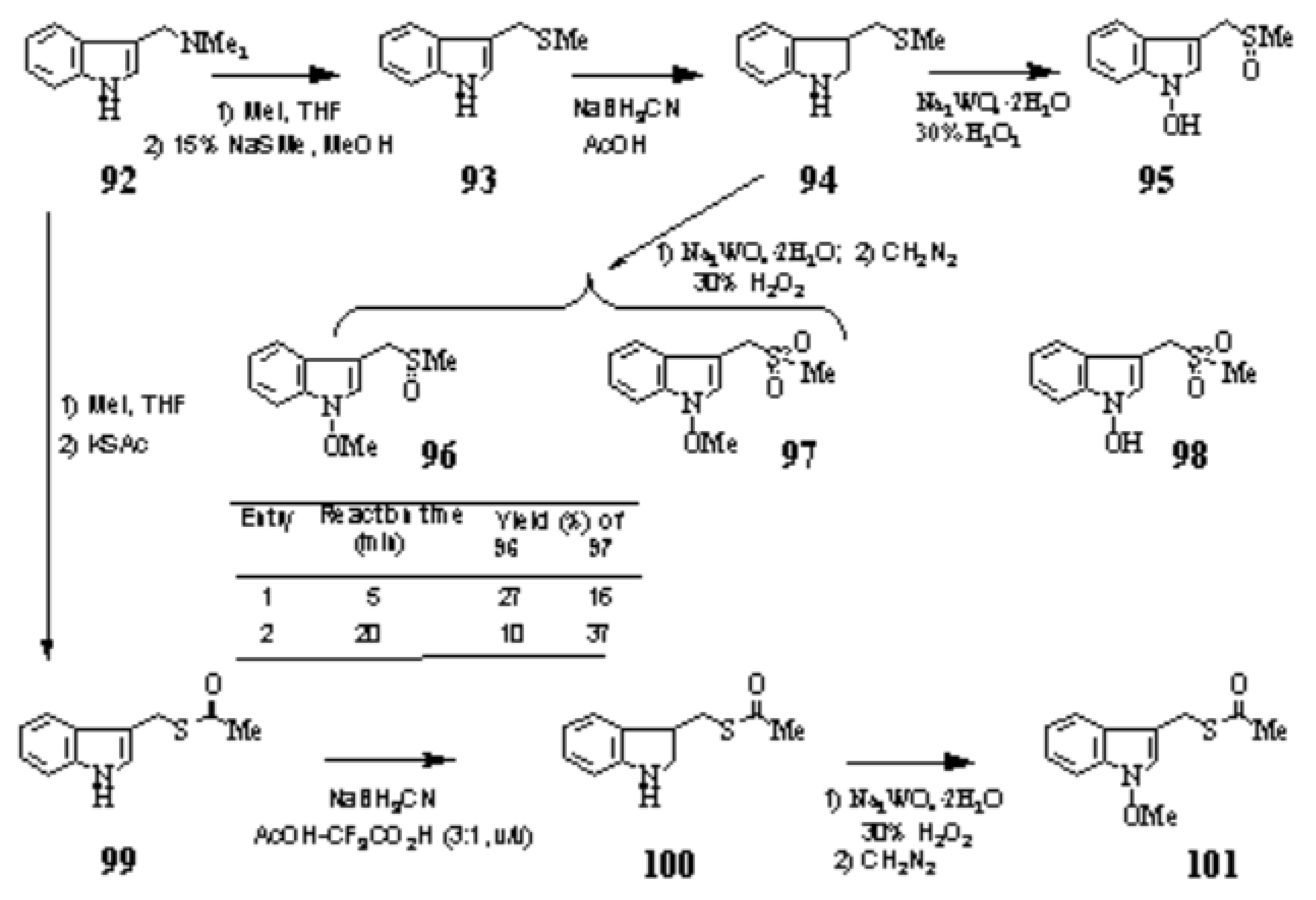
2,3-Dihydro-3-methylthiomethylindole (94) from 93 — Prepared according to the method for 85, where 95% NaBH3CN (355.5 mg, 5.37 mmol) and 93 (100.2 mg, 0.56 mmol) in AcOH (6.0 mL) were used. After usual work-up and purification, 93 (21.8 mg, recovery, 22%) and 94 (55.5 mg, 55%) were obtained. 94: colorless oil. IR (film): 3370, 2920, 1607, 1488, 1465, 1249, 747 cm-1. 1H-NMR (CDCl3) δ: 2.15 (3H, s), 2.68 (1H, dd, J=12.8, 9.4 Hz), 2.91 (1H, dd, J=12.8, 5.1 Hz), 3.44 (1H, dd, J=9.2, 6.2 Hz), 3.49–3.55 (1H, m), 3.75 (1H, t, J=9.2 Hz), 6.68 (1H, d, J=7.6 Hz), 6.75 (1H, t, J=7.6 Hz), 7.06 (1H, t, J=7.6 Hz), 7.17 (1H, d, J=7.6 Hz). MS m/z: 179 (M+). Anal. Calcd for C10H13NS: C, 66.99; H, 7.31; N, 7.81. Found: C, 67.10; H, 7.36; N, 7.93.
1-Hydroxy-3-methylsulfinylmethylindole (95) from 94 — Prepared according to the method for 82b, where Na2WO4·2H2O (47.5 mg, 0.14 mmol) in H2O (0.7 mL), 94 (128.9 mg, 0.72 mmol) in MeOH (6.0 mL), and 30% H2O2 (803.7 mg, 7.09 mmol) in MeOH (1.0 mL) were used. Then, a solution of Me2S (0.42 mL, 5.76 mmol) in MeOH (1.0 mL) was added to the reaction mixture. After usual work-up and purification, 95 (41.2 mg, 27%) was obtained. 95: mp 114.0–115.0 °C (pale orange prisms, recrystallized from EtOAc). IR (KBr): 2580, 1349, 1322, 1240, 1093, 1007, 947, 735 cm-1. 1H-NMR (CD3OD) δ: 2.53 (3H, s), 4.23 (1H, d, J=13.7 Hz), 4.30 (1H, d, J=13.7 Hz), 7.08 (1H, ddd, J=8.1, 7.0, 1.0 Hz), 7.20 (1H, ddd, J=8.1, 7.0, 1.0 Hz), 7.39 (1H, s), 7.42 (1H, d, J=8.1 Hz), 7.62 (1H, d, J=8.1 Hz). High resolution MS m/z: Calcd for C10H11NO2S: 209.0510. Found: 209.0508.
1-Methoxy-3-methylsulfinylmethylindole (96) and 1-methoxy-3-methylsulfonylmethylindole (97) from 94 — [Entry 1]: Prepared according to the method for 82b, where Na2WO4·2H2O (35.5 mg, 0.11 mmol) in H2O (0.5 mL), 94 (94.7 mg, 0.53 mmol) in MeOH (4.0 mL), and 30% H2O2 (600.3 mg, 5.30 mmol) in MeOH (1.0 mL) were used. After stirring at rt for 5 min, a solution of Me2S (0.31 mL, 4.23 mmol) in MeOH (1.0 mL) was added and stirred for 30 min. Ethereal CH2N2 (excess) was then added and stirred for 30 min. After usual work-up and purification by column-chromatography on SiO2 with CH2Cl2–MeOH (99:1, v/v), 97 (20.1 mg, 16%) and 96 (31.8 mg, 27%) were obtained. 96: mp 67.0–69.0 °C (colorless prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 3420, 1453, 1435, 1349, 1321, 1093, 1063, 1025, 965, 946, 747 cm-1. 1H-NMR (CD3OD) δ: 2.54 (3H, s), 4.11 (3H, s), 4.21 (1H, dd, J=13.8, 0.6 Hz), 4.30 (1H, dd, J=13.8, 0.6 Hz), 7.13 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.25 (1H, ddd, J=8.3, 7.1, 1.0 Hz), 7.45 (1H, dt, J=8.3, 1.0 Hz), 7.53 (1H, s), 7.66 (1H, dt, J=8.1, 1.0 Hz). MS m/z: 223 (M+). Anal. Calcd for C11H13NO2S·1/8H2O: C, 58.58; H, 5.81; N, 6.21. Found: C, 58.49; H, 5.84; N, 6.14. 97: mp 101.5–102.5 °C (colorless plates, recrystallized from CH2Cl2–hexane). IR (KBr): 3100, 2930, 1455, 1320, 1263, 1244, 1147, 1120, 968, 945, 747, 736 cm-1. 1H-NMR (CDCl3) δ: 2.75 (3H, s), 4.13 (3H, s), 4.41 (2H, s), 7.21 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.31 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.48 (1H, dt, J=8.1, 1.0 Hz), 7.48 (1H, s), 7.62 (1H, dt, J=8.1, 1.0 Hz). MS m/z: 239 (M+). Anal. Calcd for C11H13NO3S: C, 55.21; H, 5.48; N, 5.85. Found: C, 55.19; H, 5.47; N, 5.81.
3-Acetylthiomethylindole (99) from 92 — MeI (0.12 mL, 1.85 mmol) was added to a solution of 92 (32.2 mg, 0.19 mmol) in THF (2.0 mL) at rt and stirring was continued for 1 h. The solvent was evaporated under reduced pressure to leave a residue, which was dissolved in DMF–H2O (3:1, v/v, 2.0 mL). To the resultant solution, KSCOMe (31.7 mg, 0.28 mmol) was added and stirred at rt for 2 h. After usual work-up and purification by column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (100:20:2, v/v), 99 (29.8 mg, 79%) and 92 (6.9 mg, recovery, 21%) were obtained. 99: colorless oil. IR (film): 3350, 1676, 1454, 1419, 1352, 1339, 1136, 1116, 1095, 959, 740 cm-1. 1H-NMR (CDCl3) δ: 2.34 (3H, s), 4.35 (2H, d, J=0.7 Hz), 7.14 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.18 (1H, d, J=2.4 Hz), 7.21 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.35 (1H, dt, J=8.1, 1.0 Hz), 7.60 (1H, d, J=8.1 Hz), 8.03 (1H, br s). High resolution MS m/z: Calcd for C11H11NOS: 205.0561. Found: 205.0541.
3-Acetylthiomethyl-2,3-dihydroindole (100) from 99 — Prepared according to the method for 94, where 95% NaBH3CN (42.6 mg, 0.64 mmol) and 99 (26.2 mg, 0.13 mmol) in AcOH–CF3CO2H (3:1, v/v, 1.5 mL) were used. After usual work-up, 100 (17.8 mg, 67%) was obtained. 100: colorless oil. IR (film): 3370, 1692, 1611, 1487, 1465, 1252, 1138, 957, 748 cm-1. 1H-NMR (CDCl3) δ: 2.36 (3H, s), 3.10 (1H, dd, J=13.6, 8.3 Hz), 3.28 (1H, dd, J=13.6, 5.4 Hz), 3.30 (1H, dd, J=9.1, 6.1 Hz), 3.48–3.53 (1H, m), 3.68 (1H, t, J=9.1 Hz), 6.65 (1H, d, J=7.5 Hz), 6.73 (1H, td, J=7.5, 1.0 Hz), 7.06 (1H, t, J=7.5 Hz), 7.19 (1H, d, J=7.5 Hz). High resolution MS m/z: Calcd for C11H13NOS: 207.0718. Found: 207.0763.
3-Acetylthiomethyl-1-methoxyindole (101) from 99 — Crude 100, prepared with 95% NaBH3CN (60.6 mg, 0.92 mmol) and 99 (37.9 mg, 0.18 mmol) in AcOH–CF3CO2H (3:1, v/v, 2.0 mL), was dissolved in MeOH (1.5 mL). To the resultant solution, a solution of Na2WO4·2H2O (12.3 mg, 0.04 mmol) in H2O (0.2 mL) and then a solution of 30% H2O2 (207.8 mg, 1.83 mmol) in MeOH (0.5 mL) were added under ice cooling and stirred at rt for 20 min. Ethereal CH2N2 (excess) was added to the mixture and stirring was continued at rt for 1 h. After usual work-up and purification, 101 (6.6 mg, 15%) was obtained. 101: colorless oil. IR (film): 2940, 1689, 1452, 1354, 1232, 1133, 1097, 1031, 954, 758, 737 cm-1. 1H-NMR (CDCl3) δ: 2.34 (3H, s), 4.06 (3H, s), 4.29 (2H, d, J=0.7 Hz), 7.13 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.24–7.27 (2H, m), 7.43 (1H, dt, J=8.1, 1.0 Hz), 7.57 (1H, dt, J=8.1, 1.0 Hz). High resolution MS m/z: Calcd for C12H13NO2S: 235.0667. Found: 235.0685.
Scheme 10.
Synthesis of 1-hydroxyindoles (4).
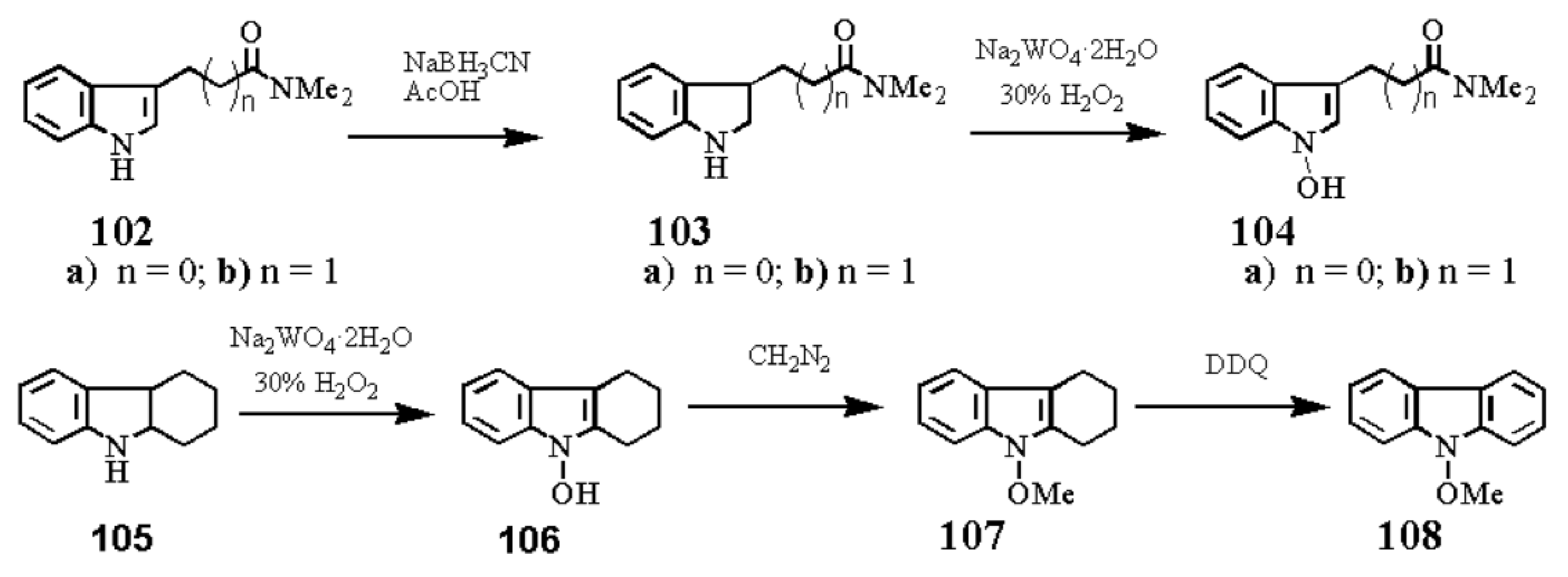
2,3-Dihydro-N,N-dimethylindole-3-acetamide (103a) from N,N-dimethylindole-3-acetamide (102a) — Prepared according to the method for 94, where 95% NaBH3CN (394.5 mg, 5.96 mmol) and 102a (241.1 mg, 1.20 mmol) in AcOH (10.0 mL) were used. After work-up, 103a (236.6 mg, 97%) was obtained.103a: colorless oil. IR (film): 3310, 2930, 1630, 1488, 1465, 1407, 1322, 1254, 1143, 748 cm-1. 1H-NMR (CDCl3) δ: 2.58 (1H, dd, J=16.0, 8.9 Hz), 2.73 (1H, dd, J=16.0, 4.8 Hz), 2.94 (3H, s), 2.97 (3H, s), 3.23–3.26 (1H, m), 3.78–3.86 (2H, m), 6.65 (1H, d, J=7,7 Hz), 6.71 (1H, td, J=7.3, 1.0 Hz), 7.04 (1H, d, J=7.7 Hz), 7.10 (1H, d, J=7.3 Hz). High resolution MS m/z: Calcd for C12H16N2O: 204.1263. Found: 204.1255.
2,3-Dihydro-N,N-dimethylindole-3-propionamide (103b) from N,N-dimethylindole-3-propionamide (102b) — Prepared according to the method for 94, where 95% NaBH3CN (303.5 mg, 4.59 mmol) and 102b (201.4 mg, 0.93 mmol) in AcOH (10.0 mL) were used. After usual work-up and purification, 103b (199.8 mg, 98%) was obtained. 103b: colorless oil. IR (film): 3290, 2920, 1628, 1486, 1459, 1399, 1247, 1143, 747 cm-1. 1H-NMR (CDCl3) δ: 1.88–1.96 (1H, m), 2.11–2.18 (1H, m), 2.33–2.45 (2H, m), 2.95 (3H, s), 2.99 (3H, s), 3.24 (1H, dd, J=8.8, 6.4 Hz), 3.33–3.39 (1H, m), 3.70 (1H, t, J=8.8 Hz), 6.64 (1H, d, J=7.6 Hz), 6.72 (1H, td, J=7.6, 1.0 Hz), 7.03 (1H, d, J=7.6 Hz), 7.11 (1H, d, J=7.6 Hz). High resolution MS m/z: Calcd for C13H18N2O: 218.1419. Found: 218.1427.
N,N-Dimethyl-1-hydroxyindole-3-acetamide (104a) from 103a — Prepared according to the method for 82b where Na2WO4·2H2O (131.6 mg, 0.40 mmol) in H2O (4.0 mL), 103a (407.4 mg, 2.00 mmol) in MeOH (3.0 mL), and 30% H2O2 (2.214 g, 19.5 mmol) in MeOH (5.0 mL) were used. After usual work-up, 104a (321.4 mg, 74%) was obtained. 104a: mp 146.0–147.0 °C (colorless prisms, recrystallized from CHCl3–hexane). IR (KBr): 2600, 1590, 1405, 1316, 1216, 1086, 758, 741 cm-1. 1H-NMR (CDCl3) δ: 2.97 (3H, s), 2.97 (3H, s), 3.61 (2H, s), 6.52 (1H, s), 6.98 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.16 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.23 (1H, d, J=8.1 Hz), 7.43 (1H, d, J=8.1 Hz), 10.72 (1H, s, D2O exchange). Anal. Calcd for C12H14N2O2: C, 66.04; H, 6.47; N, 12.84. Found: C, 65.74; H, 6.36; N, 12.69.
N,N-Dimethyl-1-hydroxyindole-3-propionamide (104b) from 103b — Prepared according to the method for 82b, where Na2WO4·2H2O (208.4 mg, 0.63 mmol) in H2O (6.0 mL), 103b (688.0 mg, 3.16 mmol) in MeOH (55.0 mL), and 30% H2O2 (3.543 g, 31.3 mmol) in MeOH (5.0 mL) were used. After usual work-up and purification, 104b (483.9 mg, 66%) was obtained. 104b: mp 144.0–145.0 °C (colorless prisms, recrystallized from CHCl3–hexane). IR (KBr): 2760, 1598, 1402, 1310, 1140, 1026, 736 cm-1. 1H-NMR (DMSO-d6) δ: 2.63 (2H, dd, J=8.2, 7.2 Hz), 2.82 (3H, s), 2.88 (2H, dd, J=8.2, 7.2 Hz), 2.93 (3H, s), 6.97 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.12 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.23 (1H, s), 7.31 (1H, d, J=8.1 Hz), 7.51 (1H, d, J=8.1 Hz), 10.98 (1H, s, D2O exchange). Anal. Calcd for C13H16N2O2·1/4H2O: C, 65.94; H, 7.02; N, 11.83. Found: C, 66.14; H, 6.85; N, 11.80.
9-Hydroxy-1,2,3,4-tetrahydrocarbazole (106) from 1,2,3,4,4a,9a-hexahydrocarbazole (105) — Prepared according to the method for 82b, where Na2WO4·2H2O (28.7 mg, 0.087 mmol) in H2O (1.0 mL), 105 (71.8 mg, 0.41 mmol) in MeOH (10.0 mL), and 30% H2O2 (0.45 mL, 3.92 mmol) were used. After usual work-up and purification, 105 (13.3 mg, 18%) and 106 (50.1 mg, 65%) were obtained. 106: yellow oil. IR (film): 3061, 2931, 2857, 1458, 1238, 1178, 740 cm-1. 1H-NMR (CD3OD) δ: 1.73–1.84 (4H, m), 2.55–2.57 (2H, m), 2.64–2.66 (2H, m), 6.83 (1H, t, J=7.9 Hz), 6.94 (1H, t, J=7.9 Hz), 7.18 (1H, d, J=7.9 Hz), 7.23 (1H, d, J=7.9 Hz). High resolution MS m/z: Calcd for C12H13NO: 187.0997. Found: 187.1001.
9-Methoxy-1,2,3,4-tetrahydrocarbazole (107) from 1,2,3,4,4a,9a-hexahydrocarbazole (105) — Prepared according to the method for 82b, where Na2WO4·2H2O (19.3 mg, 0.028 mmol), 105 (50.6 mg, 0.29 mmol) in MeOH (4.0 mL), and 30% H2O2 (331.6 mg, 2.92 mmol) in MeOH (1.0 mL) were used. After methylation with CH2N2 and work-up, product purification was carried out by p-TLC on SiO2 with CH2Cl2–hexane (7:3, v/v) as a developing solvent to afford 107 (32.2 mg, 55%). 107: colorless oil. IR (KBr): 2942, 2842, 1459, 1443, 1230, 1046, 735 cm-1. 1H-NMR (CDCl3) δ: 1.68–2.08 (4H, m), 2.52–2.92 (4H, br m), 3.99 (3H, s), 6.88–7.48 (4H, m). High resolution MS m/z: Calcd for C13H15NO: 201.1152. Found: 201.1134.
9-Methoxy-1,2,3,4-tetrahydrocarbazole (107) from 105 — K2CO3 (173.3 mg, 1.25 mmol) and Me2SO4 (0.053 mL, 0.56 mmol) were added to a solution of 105 (67.0 mg, 0.33 mmol) in acetone (10.0 mL) and the mixture was stirred at rt for 2 h. After usual work-up and purification, 107 (54.8 mg, 70%) was obtained.
9-Methoxycarbazole (108) from 107 — Dichlorodicyanoquinone (469.4 mg, 2.38 mmol) was added to a solution of 107 (188. 9 mg, 0.94 mmol) in benzene (30.0 mL) and stirred at rt (14 °C) for 3 h. Precipitates were filtered off through silica gel and washed with CH2Cl2. Washings and filtrates were combined and evaporated under reduced pressure to leave a crystalline solid, which was column-chromatographed on SiO2 with hexane–EtOAc (9:1, v/v) as an eluent to afford 108 (121.1 mg, 65%). 108: mp 40.0–41.0 °C (colorless needles, recrystallized from MeOH). IR (KBr): 1601, 1450, 1320, 1233, 1052, 946 cm-1. 1H-NMR (CDCl3) δ: 4.12 (3H, s), 7.19 (1H, dd, J=7.3, 2.7 Hz), 7.25 (1H, dd, J=7.3, 2.7 Hz), 7.34–7.58 (4H, m), 8.02 (2H, dt, J=7.3, 1.0 Hz). MS m/z: 197 (M+). Anal. Calcd for C13H11NO: C, 79.17; H, 5.62; N, 7.10. Found: C, 79.36; H, 5.55; N, 7.21.
A mixture of diastereoisomers, 4-nitro-1,2,2a,3,4,5-hexahydrobenz[cd]indole (110) from 4-nitro- 1,3,4,5-tetrahydrobenz[cd]indole (109) — Prepared according to the method for 94, where 95% NaBH3CN (60.8 mg, 0.97 mmol) and 109 (35.9 mg, 0.18 mmol) in AcOH–CF3CO2H (3:2, v/v, 2.0 mL) were used. After usual work-up, crude 110 was subjected to p-TLC on SiO2 with CH2Cl2–hexane (3:1, v/v) as a developing solvent. Extraction of the band having an Rf value of 0.39–0.14 with CH2Cl2–MeOH (95:5, v/v) afforded pure 110 (34.4 mg, 95%). Although 1H-NMR analysis of 110 showed 2:1 mixture of diastereoisomers, further separation was not examined.
1-Hydroxy-4-nitro-1,3,4,5-tetrahydrobenz[cd]indole (111) from a diastereoisomer’s mixture (110) — Prepared according to general method C for 82b, where Na2WO4·2H2O (10.5 mg, 0.03 mmol) in H2O (0.2 mL), urea·H2O2 (138.6 mg, 1.47 mmol), and diastereoisomer’s mixture, 110 (30.2 mg, 0.15 mmol), in MeOH (2.0 mL) were used. The reaction mixture was adjusted to pH 4 by adding 0.6% HCl and extracted with CH2Cl2–MeOH (95:5, v/v). After usual work-up and purification, 111 (16.9 mg, 52%) was obtained. 111: mp 134–134.5 ℃ (colorless prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 3427, 3112, 2971, 1604, 1530, 1442, 1419, 1349, 1149, 1001, 848, 769, 751 cm-1. 1H-NMR (CDCl3) δ: 3.50 (2H, br s), 3.52 (1H, dd, J=15.6, 4.4 Hz), 3.61 (1H, dd, J=15.6, 9.3 Hz), 4.98 (1H, ddd, J=13.7, 9.3, 4.4 Hz), 6.79 (1H, br s, D2O exchange), 6.89 (1H, br s), 7.01 (1H, br s), 7.21 (1H, dd, J=7.8, 7.3 Hz). High resolution MS m/z: Calcd for C11H10N2O3: 218.0690. Found: 218.0692.
Scheme 11.
Synthesis of 1-hydroxyinoles (5).
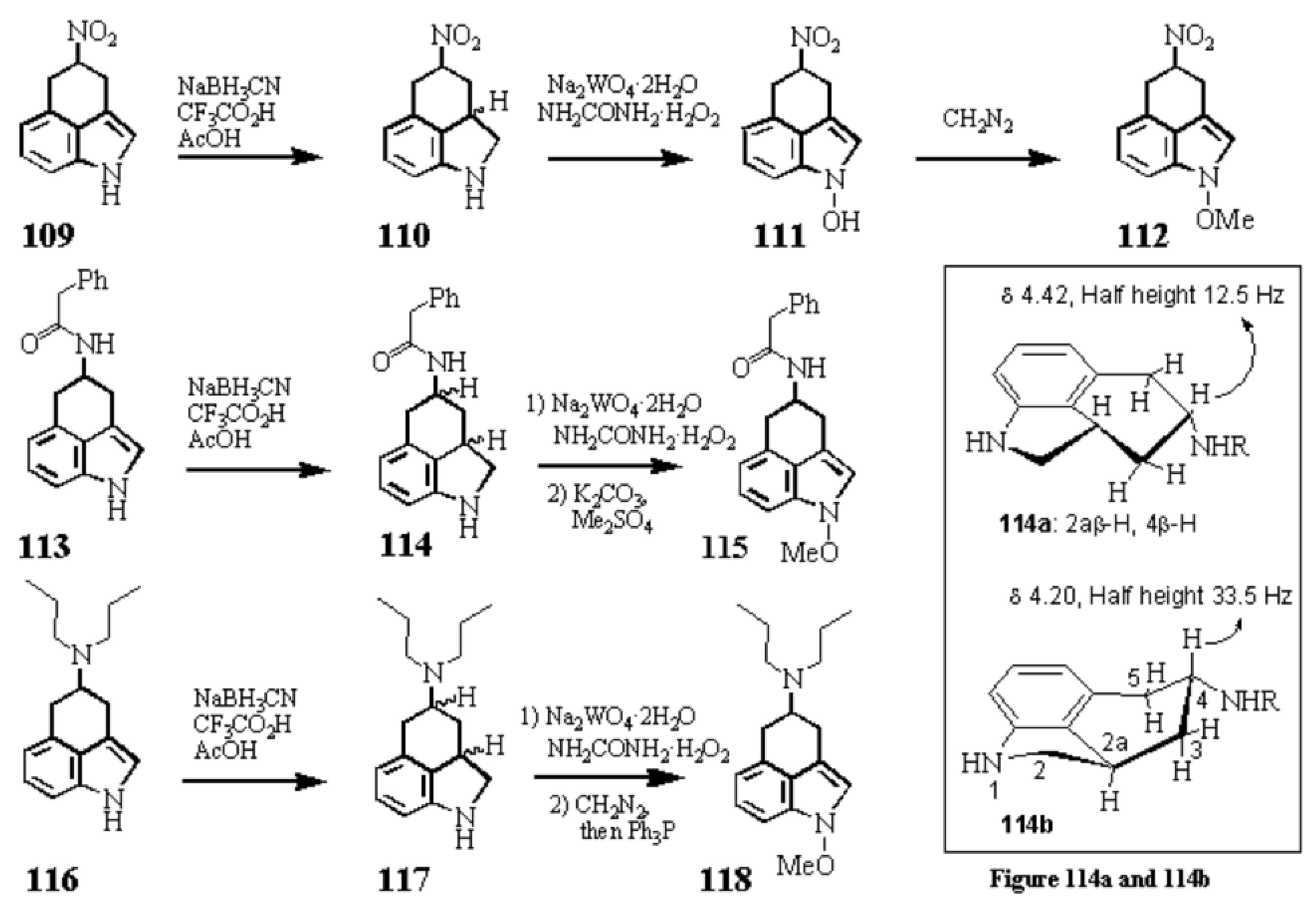
1-Methoxy-4-nitro-1,3,4,5-tetrahydrobenz[cd]indole (112) from 111 — Diazomethane-ether solution (excess) was added to a solution of 111 (7.5 mg, 0.04 mmol) in MeOH (1.0 mL) at rt with stirring for 0.5 h. After evaporation of solvent under reduced pressure, the residue was purified by p-TLC on SiO2 with CH2Cl2–hexane (1:1, v/v) as a developing solvent. Extraction of the band having an Rf value of 0.50–0.31 with CH2Cl2–MeOH (95:5, v/v) afforded 112 (5.1 mg, 64%). 112: pale brown oil. IR (KBr): 3450, 2941, 1605, 1521, 1440, 1561, 1540, 983, 761, 747 cm-1. 1H-NMR (CDCl3) δ: 3.48 (2H, dd, J=17.3, 1.0 Hz), 3.54 (1H, dd, J=15.6, 4.6 Hz), 3.61 (1H, dd, J=15.6, 9.3 Hz), 4.07 (3H, s), 4.99 (1H, ddt, J=9.3, 7.3, 4.6 Hz), 6.90 (1H, d, J J=7.3 Hz), 7.02 (1H, s), 7.20 (1H, dd, J=7.8, 7.3 Hz), 7.25 (1H, d, J=7.8 Hz). High resolution MS m/z: Calcd for C12H12N2O3: 232.0847. Found: 232.0893.
A mixture of diastereoisomers, 4-(N-phenylacetylamino)-1,2,2aβ,3,4β,5-hexahydrobenz[cd]indole (114a) and 4-(N-phenylacetylamino)-1,2,2aα,3,4β,5-hexahydrobenz[cd]indole (114b) from 4-(N-phenylacetylamino)-1,3,4,5-tetrahydrobenz[cd]indole (113) — Prepared according to the method for 94, where 95%NaBH3CN (46.0 mg, 0.73 mmol) and 113 (40.1 mg, 0.14 mmol) in AcOH–CF3CO2H (4:1, v/v, 2.0 mL) were used. After work-up and purification, 114a (17.0 mg, 47%) and 114b (16.7 mg, 41%) were obtained. 114a: colorless oil. IR (KBr) : 3261, 3037, 2910, 2843, 1637, 1603, 1532, 1491, 1452, 1333, 1247, 1233, 761, 718, 692 cm-1. 1H-NMR (CD3OD) δ: 1.45 (1H, dt, J=12.8, 3.2 Hz), 2.29 (1H, dt, J=12.8, 4.6 Hz), 2.70 (1H, d, J=18.3 Hz), 2.96 (1H, dd, J=18.3, 6.4 Hz), 3.00 (1H, dd, J=11.9, 8.3 Hz), 3.10–3.19 (1H, m), 3.46 (1H, d, J=14.2 Hz), 3.50 (1H, d, J=14.2 Hz), 3.57 (1H, dd, J=8.3, 7.3 Hz), 4.39–4.46 (1H, m), 6.51 (2H, t, J=7.3 Hz), 6.94 (1H, dd, J=8.3, 7.3 Hz), 7.19–7.24 (1H, m), 7.27 (2H, s), 7.28 (2H, s). High resolution MS m/z: Calcd for C19H20N2O: 292.1575. Found: 292.1578. 114b: mp 161–162℃ (colorless prisms, recrystallized from EtOAc–hexane). IR (KBr): 3233, 3054, 2928, 2883, 1637, 1551, 1452, 1280, 1243, 762, 727, 691 cm-1. 1H-NMR (CD3OD) δ: 1.39 (1H, q, J=11.9 Hz), 2.24 (1H, dt, J=11.9, 3.7 Hz), 2.48 (1H, dd, J=16.5, 11.9 Hz), 2.99 (1H, dd, J=11.9, 8.3 Hz), 3.06 (1H, dd, J=16.5, 6.4 Hz), 3.12–3.22 (1H, m), 3.51 (2H, s), 3.58 (1H, t, J=8.3 Hz), 4.20 (1H, ddt, J=11.9, 6.4, 3.7 Hz), 6.49 (2H, d, J=7.3 Hz), 6.91 (1H, dd, J=8.3, 7.3 Hz), 7.20– 7.26 (1H, m), 7.30 (2H, s), 7.31 (2H,s). MS m/z: 292 (M+). Anal. Calcd for C19H20N2O: C, 78.05; H, 6.90; N, 9.58. Found: C, 77.91; H, 6.92; N, 9.42.
1-Methoxy-4-(N-phenylacetylamino)-1,3,4,5-tetrahydrobenz[cd]indole (115) from a mixture of diastereoisomers, 4-(N-phenylacetylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indoles (114a, 114b) — Prepared according to general method C for 6, where Na2WO4·2H2O (6.6 mg, 0.02 mmol) in H2O (0.2 mL), urea·H2O2 (95.2 mg, 1.01 mmol), and diastereoisomer’s mixture, 114a and 114b (29.1 mg, 0.10 mmol) in MeOH (2.0 mL) were used. To the reaction mixture, K2CO3 (247.1 mg, 1.79 mmol) and Me2SO4 (80.0 mg, 0.64 mmol) were added and stirred at rt for 1.5 h. After usual work-up and purification, 115 (12.8 mg, 40%) was obtained. 115: mp 138–139 ℃ (colorless prisms, recrystallized from CH2Cl2–hexane). IR (KBr): 3301, 3053, 2943, 1635, 1543, 1493, 1442, 1342, 985, 752, 731 cm-1. 1H-NMR (CDCl3) δ: 2.74 (1H, dd, J=15.6, 5.9 Hz), 2.90 (1H, dd, J=15.6, 5.9 Hz), 3.01 (1H, dd, J=15.6, 3.7 Hz), 3.11 (1H, dd, J=15.6, 3.7 Hz), 3.43 (2H, s), 4.05 (3H, s), 4.61–4.69 (1H, m), 5.44 (1H, br d, J=8.3 Hz), 6.79 (1H, d, J=7.3 Hz), 6.89 (1H, s), 7.03 (1H, dd, J=7.3, 1.8 Hz), 7.13–7.22 (2H, m). MS m/z: 320 (M+). Anal. Calcd for C20H20N2O2: C, 74.98; H, 6.29; N, 8.74. Found: C, 74.70; H, 6.20; N, 8.31.
A mixture of diastereoisomers, 4-N,N-di(n-propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole (117), from 4-N,N-di(n-propylamino)-1,3,4,5-tetrahydrobenz[cd]indole (116) — Prepared according to the method for 94, where 95% NaBH3CN (27.2 mg, 0.43 mmol) and 116 (22.0 mg, 0.09 mmol) in AcOH–CF3CO2H (2:1, v/v, 1.5 mL) were used. After usual work-up, the reaction residue was subjected to p-TLC on SiO2 with CHCl3–MeOH–aq. 30% NH3–hexane (92:10:1:1, v/v) as a developing solvent. Extraction of the band having an Rf value of 0.53–0.24 with CHCl3–MeOH–aq. 30% NH3 (46:5:0.5, v/v) afforded 117 (19.0 mg, 86%). Although 1H-NMR analysis of 117 showed 6:1 mixture of diastereoisomers, further separation was not examined.
4-N,N-Di(n-propylamino)-1-methoxy-1,3,4,5-tetrahydrobenz[cd]indole (118) from a diastereoiso- mer’s mixture (117) — Prepared according to the method for 6, where Na2WO4·2H2O (8.1 mg, 0.02 mmol) in H2O (0.2 mL), urea·H2O2 (116.8 mg, 1.24 mmol), and diastereoisomer’s mixture, 117 (30.7 mg, 0.12 mmol) in MeOH (2.0 mL) were used. Then, the reaction mixture was treated with ethereal CH2N2 (excess), followed by addition of PPh3 (319.5 mg, 1.26 mmol) under ice cooling, and the whole was stirred at rt for 20 min. After usual work-up, products were separated by p-TLC on Al2O3 with EtOAc–hexane (1:14, v/v) to give 118 (15.8 mg, 46%) and 117 (4.2 mg, 14%). 118: pale brown oil. IR (KBr): 2950, 1606, 1460, 1441, 1375, 1152, 1071, 984, 747 cm-1. 1H-NMR (pyridine-d5) δ: 0.90 (6H, t, J=7.3 Hz), 1.42 (4H, sex, J=7.3 Hz), 2.47 (4H, t, J J=7.3 Hz), 2.73 (1H, ddd, J=15.1, 12.0, 1.5 Hz), 2.92 (1H, dd, J=15.1, 4.1 Hz), 2.95 (1H, d, J=12.0 Hz), 3.02 (1H, dd, J=15.1, 4.1 Hz), 3.20–3.27 (1H, m), 3.97 (3H, s), 6.98 (1H, d, J=7.8 Hz), 7.16 (1H, d, J=1.5 Hz), 7.29 (1H, dd, J=7.8, 6.8 Hz), 7.36 (1H, d, J=7.8 Hz). High resolution MS m/z: Calcd for C18H26N2O: 286.2043. Found: 286.2044.
Scheme 12.
Synthesis of 1-hydroxyinoles (6).
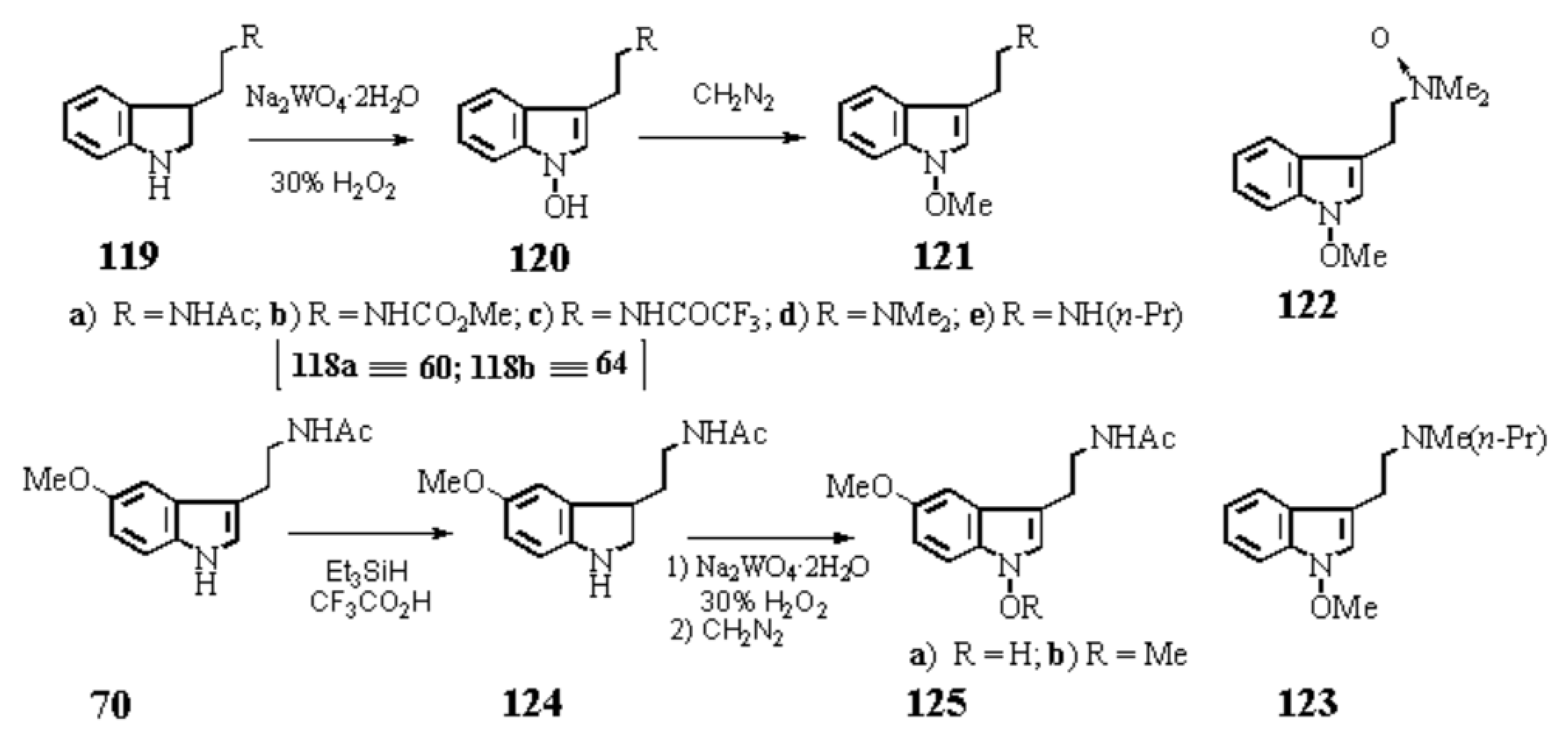
1-Hydroxy-Nb-acetyltryptamine (120a) from Nb-acetyl-2,3-dihydrotryptamine (119a)— Prepared according to the general method for 6, where Na2WO4·2H2O (66.4 mg, 0.20 mmol), 119a (205.2 mg, 1.00 mmol) in MeOH (20.0 mL), and 30% H2O2 (1.0 mL, 10.0 mmol) were used. After usual work-up and purification, 120a (121.5 mg, 55%) was obtained. 120a: mp 138.0–139.0 °C (colorless prisms, recrystallized from EtOAc). IR (KBr): 3250, 3105, 1619, 1602, 1580, 743 cm-1. UV λmaxMeOH nm (log ε): 225 (4.52), 281 (3.62), 295 (3.66). 1H-NMR (CD3OD) δ: 1.89 (3H, s), 2.89 (2H, t, J=7.3 Hz), 3.43 (2H, t, J=7.3 Hz), 6.99 (1H, dd, J=8.3, 8.3 Hz), 7.10 (1H, s), 7.12 (1H, dd, J=8.3, 8.3 Hz), 7.34 (1H, d, J=8.3 Hz), 7.52 (1H, d, J=8.3 Hz), 7.46 (1H, d, J=8.3 Hz). MS m/z: 218 (M+). Anal. Calcd for C12H14N2O2: C, 66.04; H, 6.47; N, 12.84. Found: C, 66.02; H, 6.53; N, 12.77.
1-Hydroxy-Nb-methoxycarbonyltryptamine (120b) from Nb-methoxycarbonyl-2,3-dihydro- tryptamine (119b) — Prepared according to the general method for 6, where Na2WO4·2H2O (56.1 mg, 0.17 mmol) in H2O (1.8 mL), 119b (185.9 mg, 0.85 mmol) in MeOH (18.0 mL), and 30% H2O2 (0.86 mL, 8.42 mmol) were used. After usual work-up and purification, 120b (131.5 mg, 67%) was obtained. 120b: mp 114.0–115.0 °C (colorless needles, recrystallized from CH2Cl2–hexane). IR (KBr): 3380, 3190, 1698, 1533, 1267, 983, 751 cm-1. UV λmaxMeOH nm (log ε): 225 (4.53), 295 (3.66). 1H-NMR (CD3OD) δ: 2.89 (2H, t, J=7.5 Hz), 3.36 (2H, t, J=7.9 Hz), 3.61 (3H, s), 6.99 (1H, t, J=7.9 Hz), 7.09 (1H, s), 7.13 (1H, t, J=7.9 Hz), 7.34 (1H, d, J=7.9 Hz), 7.53 (1H, d, J=7.9 Hz). MS m/z: 234 (M+). Anal. Calcd for C12H14N2O3: C, 61.53; H, 6.02; N, 11.96. Found: C, 61.40; H, 6.02; N, 11.90.
1-Hydroxy-Nb-trifluoroacetyltryptamine (120c) from Nb-trifluoroacetyl-2,3-dihydrotryptamine (119c) — Prepared according to the general method for 6, where Na2WO4·2H2O (57.8 mg, 0.18 mmol) in H2O (2.2 mL), 119c (218.6 mg, 0.85 mmol) in MeOH (20.0 mL), and 30% H2O2 (984.0 mg, 8.68 mmol) in MeOH (2.0 mL) were used. After usual work-up, the crude product was purified by column-chromatography on SiO2 with CH2Cl2–MeOH (99:1, v/v) to give 120c (165.3 mg, 72%). 120c: colorless oil. IR (film): 3310, 2935, 1721, 1698, 1566, 1553, 1451, 1354, 1205, 1098, 1008, 741 cm-1. 1H-NMR (5% CD3OD in CDCl3) δ: 2.99 (2H, t, J=6.6 Hz), 3.62 (2H, q, J=6.6 Hz), 7.07 (1H, s), 7.08 (1H, t, J=8.0 Hz), 7.22 (1H, t, J=8.0 Hz), 7.38 (1H, br s), 7.44 (1H, d, J=8.0 Hz), 7.53 (1H, d, J=8.0 Hz). High resolution MS m/z: Calcd for C12H11F3N2O2: 272.0772. Found: 272.0779.
Nb,Nb-Dimethyl-1-hydroxytryptamine (120d) from Nb,Nb-dimethyl-2,3-dihydrotryptamine (119d)— Prepared according to the general method for 6, where Na2WO4·2H2O (132.5 mg, 0.40 mmol) in H2O (4.0 mL), 119d (378.9 mg, 1.99 mmol) in MeOH (40.0 mL), and 30% H2O2 (2.0 mL, 19.6 mmol) in MeOH (40.0 mL) were used. After usual work-up, the crude product was purified by column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:5:0.5, v/v) to give 120d (70.5 mg, 55%). 120d: mp 179.5–180.0 °C (colorless needles, recrystallized from MeOH–H2O). IR (KBr): 2415, 1470, 1447, 1320, 1226, 838, 737 cm-1. UV λmaxMeOH nm (log ε): 223 (4.48), 292 (3.62). 1H-NMR (CD3OD) δ: 2.35 (6H, s), 2.64–2.68 (2H, m), 2.89–2.93 (2H, m), 6.99 (1H, dt, J=0.9 and 8.1 Hz), 7.09 (1H, s), 7.13 (1H, dt, J=0.9, 8.1 Hz), 7.34 (1H, dt, J=8.1, 0.9 Hz), 7.50 (1H, dt, J=8.1, 0.9 Hz). MS m/z: 204 (M+). Anal. Calcd for C12H16N2O: C, 70.56; H, 7.90; N, 13.71. Found: C, 70.35; H, 8.04; N, 13.66.
1-Hydroxy-Nb-n-propyltryptamine (120e) from Nb-n-propyl-2,3-dihydrotryptamine (119e) — Prepared according to the general method for 6, where Na2WO4·2H2O (56.5 mg, 0.17 mmol) in H2O (1.8 mL), 119e (173.4 mg, 0.85 mmol) in MeOH (18.0 mL), and 30% H2O2 (0.85 mL, 8.42 mmol) were used. After usual work-up, the crude product was purified by column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:5:0.5, v/v) to give 120e (96.3 mg, 52%). 120e: mp 147.0–148.0 °C (colorless needles, recrystallized from MeOH). IR (KBr): 2960, 2840, 1970, 1515, 1447, 1343, 1322, 1221, 1089, 785, 734 cm-1. 1H-NMR (CD3OD) δ: 0.90 (3H, t, J=7.3 Hz), 1.52 (2H, sext, J=7.3 Hz), 2.63 (2H, m), 2.94 (4H, m), 6.96 (1H, dd, J=8.0, 1.1 Hz), 7.09 (1H, s), 7.11 (1H, dd, J=8.0, 1.1 Hz), 7.37 (1H, ddd, J=8.0, 1.1, 0.7 Hz), 7.50 (1H, ddd, J=8.0, 1.1, 0.7 Hz). MS m/z: 218 (M+). Anal. Calcd for C13H18N2O·1/8H2O: C, 70.80; H, 8.34; N, 12.70. Found: C, 70.91; H, 8.29; N, 12.70.
1-Methoxy-Nb-acetyltryptamine (121a) from 120a — Ethereal CH2N2 (excess) was added to a solution of 120a (51.6 mg, 0.23 mmol) and stirred at rt for 1 h. After evaporation of the solvent under reduced pressure, the residue was column-chromatographed on SiO2 with CH2Cl2–MeOH (99:1, v/v) to give 121a (46.7 mg, 85%). 121a: colorless oil. IR (film): 3280, 3075, 1650, 1551, 1451, 740 cm-1. 1H-NMR (CDCl3) δ: 1.93 (3H, s), 2.93 (2H, t, J=6.6 Hz), 3.56 (2H, dt, J=5.9, 6.6 Hz), 4.06 (3H, s), 5.66 (1H, br s, D2O exchange), 7.11 (1H, s), 7.12 (1H, br d, J=8.3 Hz), 7.26 (1H, br d, J=8.3 Hz), 7.42 (1H, d, J=8.2 Hz), 7.56 (1H, d, J=8.3 Hz). High resolution MS m/z: Calcd for C13H16N2O2: 232.1210. Found: 232.1214.
1-Methoxy-Nb-methoxycarbonyltryptamine (121b) from 120b — Ethereal CH2N2 (excess) was added to a solution of 120b (39.1 mg, 0.18 mmol) and stirred at rt for 1 h. After usual work-up and purification, 121b (34.3 mg, 83%) was obtained. 121b: colorless oil. IR (film): 3320, 2930, 1705, 1525, 1452, 1254, 737 cm-1. 1H-NMR (CDCl3) δ: 2.93 (2H, t, J=6.6 Hz), 3.49 (2H, q, J=6.6 Hz), 3.66 (3H, s), 4.06 (3H, s), 4.76 (1H, br s), 7.10 (1H, s), 7.12 (1H, dt, J=1.1, 8.0 Hz), 7.25 (1H, dt, J=1.1, 8.0 Hz), 7.42 (1H, d, J=8.0 Hz), 7.57 (1H, d, J=8.0 Hz). High resolution MS m/z: Calcd for C13H16N2O3: 248.1160. Found: 248.1163.
1-Methoxy-Nb-trifluoroacetyltryptamine (121c) from 120c — Ethereal CH2N2 (excess) was added to a solution of 120c (32.7 mg, 0.12 mmol) and stirred at rt for 1 h. After usual work-up, 121c (26.8 mg, 78%) was obtained. 121c: mp 70.5–71.0 °C (colorless prisms, recrystallized from benzene–hexane). IR (KBr): 3270, 1732, 1702, 1567, 1454, 1215, 1186, 1148, 735 cm-1. UV λmaxMeOH nm (log ε): 223 (4.53), 276 (3.68), 290 (3.69). 1H-NMR (CDCl3) δ: 3.02 (2H, t, J=6.7 Hz), 3.67 (2H, q, J=6.7 Hz), 4.07 (3H, s), 6.35 (1H, br s), 7.12 (1H, s), 7.14 (1H, t, J=8.0 Hz), 7.28 (1H, t, J=8.0 Hz), 7.44 (1H, d, J=8.0 Hz), 7.56 (1H, d, J=8.0 Hz). High resolution MS m/z: Calcd for C13H13F3N2O3: 286.0928. Found: 286.0877.
Lespedamine (Nb,Nb-Dimethyl-1-methoxytryptamine, 121d) from 120d — Ethereal CH2N2 (excess) was added to a solution of 120d (13.1 mg, 0.064 mmol) in MeOH (5.0 mL) with stirring at rt until the starting material was not detected on tlc monitoring. After usual work-up and purification, 121d (8.0 mg, 57%) was obtained. 121d: colorless oil. IR (film): 2930, 2855, 2820, 2770, 1460, 1093, 1051, 1034, 1007, 953 cm-1 (lit.[18] 1459 cm-1). 1H-NMR (CDCl3) δ: 2.37 (6H, s), 2.65 (2H, t, J=8.0 Hz), 2.93 (2H, t, J=8.0 Hz), 4.05 (3H, s), 7.10 (1H, dt, J=0.9, 7.8 Hz), 7.10 (1H, s), 7.23 (1H, dt, J=0.9, 7.8 Hz), 7.40 (1H, dd, J=7.8, 0.9 Hz), 7.57 (1H, dd, J=7.8, 0.9 Hz) (lit.7b,18 1H-NMR (CCl4) δ: 2.19 (6H, s), 2.32–2.96 (4H, m), 3.92 (3H, s), 6.62–7.45 (5H, m)).
Lespedamine (121d) and lespedamine-Nb-oxide (122) from 2,3-dihydro-Nb,Nb-dimethyltryptamine (119d) — Prepared according to the general method for 6, where Na2WO4·2H2O (15.1mg, 0.04 mmol) in H2O (0.5 mL), 119d (43.9 mg, 0.23 mmol) in MeOH (5 mL), and 30% H2O2 (0.24 mL, 2.35 mmol) were used. After methylation and work-up, the product was purified by column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:2:0.2, v/v) to give 121d (13.1 mg, 26%) and 122 (17.0 mg, 31%) in the order of elution. 122: colorless oil. IR (film): 3420, 1644, 1453, 954, 742 cm-1. 1H-NMR (CDCl3) δ: 3.31 (6H, s), 3.37–3.40 (2H, m), 3.57–3.60 (2H, m), 4.06 (3H, s), 7.13 (1H, dt, J=1.1, 7.9 Hz), 7.18 (1H, s), 7.25 (1H, dt, J=1.1, 7.9 Hz), 7.42 (1H, ddd, J=7.9, 1.1, 0.9 Hz), 7.59 (1H, ddd, J=7.9, 1.1, 0.9 Hz). MS m/z: 234 (M+). Anal. Calcd for C13H18N2O2·MeOH: C, 63.13; H, 8.33; N, 10.52. Found: C, 63.24; H, 8.14; N, 10.74. High resolution MS m/z: Calcd for C13H18N2O2: 234.1368. Found: 234.1370.
1-Methoxy-Nb-n-propyltryptamine (121e) and 1-methoxy-Nb-methyl-Nb-n-propyltryptamine (121) from Nb-n-propyl-2,3-dihydrotryptamine (119e)— Prepared according to the general method for 6, where Na2WO4·2H2O (13.9 mg, 0.04 mmol) in H2O (0.4 mL), 119e (41.9 mg, 0.21 mmol) in MeOH (4.0 mL), and 30% H2O2 (0.21 mL, 2.06 mmol) were used. After methylation and work-up, the products were purified by column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:2:0.2, v/v) to give 123 (4.6 mg, 9%) and 121e (23.3 mg, 49%) in the order of elution. 121e: pale yellow oil. IR (film): 2960, 2930, 2875, 2820, 1451, 1094, 738 cm-1. 1H-NMR (CDCl3) δ: 0.90 (3H, t, J=7.5 Hz), 1.53 (2H, sext, J=7.5 Hz), 2.63 (2H, t, J=7.5 Hz), 2.97 (4H, m), 4.05 (3H, s), 7.10 (1H, dt, J=1.1 and 8.0 Hz), 7.11 (1H, s), 7.24 (1H, dt, J=1.1, 8.0 Hz), 7.41 (1H, dt, J=8.0, 1.1 Hz), 7.59 (1H, dt, J=8.0, 1.1 Hz). High resolution MS m/z: Calcd for C14H20N2O: 232.1575. Found: 232.1575. 123: colorless oil. IR (film): 2955, 2940, 2870, 2780, 1450, 1098, 1010, 956, 736 cm–1. 1H-NMR (CDCl3) δ: 0.93 (3H, t, J=7.3 Hz), 1.58 (2H, br sext, J=7.3 Hz), 2.40 (3H, s), 2.47 (2H, br t, J=7.3 Hz), 2.75 (2H, br t, J=7.5 Hz), 2.95 (2H, br t, J=7.5 Hz), 4.05 (3H, s), 7.11 (1H, dt, J=0.9, 8.1 Hz), 7.11 (1H, s), 7.24 (1H, dt, J=0.9, 8.1 Hz), 7.40 (1H, d, J=8.1 Hz), 7.57 (1H, d, J=8.1 Hz). High resolution MS m/z: Calcd for C15H22N2O: 246.1731. Found: 246.1734.
2,3-Dihydromelatonin (124) from melatonin (70) — A solution of 70 (1.01 g, 4.35 mmol) in CF3CO2H (20.0 mL) was added to Et3SiH (0.85 mL, 5.32 mmol) and stirred at 58 °C for 1 h. After usual work-up and purification, by column-chromatography on SiO2 with CHCl3–MeOH (97:3, v/v), 124 (847.7 mg, 83%) was obtained. 124: mp 83–84 °C (colorless prisms, recrystallized from EtOAc–hexane). IR (KBr): 3550, 1645, 1550, 1495, 1360, 1220, 1110, 1025, 890, 795, 740, 690, 585, 535 cm-1. 1H-NMR (CDCl3) δ: 1.73–1.79 (1H, m), 1.94–2.03 (1H, m), 1.95 (3H, s), 3.22–3.43 (4H, m), 3.70 (1H, br t, J=8.8 Hz), 3.75 (3H, s), 5.71 (1H, br s), 6.60 (1H, d, J=8.5 Hz), 6.62 (1H, dd, J=8.5, 2.20 Hz), 6.73 (1H, d, J=2.20 Hz). Anal. Calcd for C13H18N2O2: C, 66.64; H, 7.74; N, 11.96. Found: C, 66.47; H, 7.80; N, 11.91.
1-Hydroxymelatonin (125a) from 2,3-dihydromelatonin (124) — Prepared according to the general method B for 6, where Na2WO4·2H2O, (107.1 mg, 0.325 mmol) in H2O (3.8 mL), 124 (379.5 mg, 1.62 mmol) in MeOH (38.0 mL), and 30% H2O2 (1.8 mL, 15.9 mmol) were used. After usual work-up and purification by column-chromatographed on SiO2 with EtOAc, 125a (234.9 mg, 58%) was obtained. 125a: mp 113–114 °C (colorless prisms recrystallized from CHCl3–hexane. IR (KBr): 3600, 3200, 2900, 2850, 1610, 1560, 1480, 1360, 1280, 1260, 1215, 1170, 1095, 1035, 995, 950, 900, 823, 795, 760, 600 cm-1. 1H-NMR (CD3OD) δ: 1.91 (3H, s), 2.86 (2H, t, J=7.3 Hz), 3.42 (2H, t, J=7.3 Hz), 3.82 (3H, s), 6.80 (1H, dd, J=8.8, 2.4 Hz), 7.03 (1H, d, J=2.4 Hz), 7.07 (1H, s), 7.23 (1H, d, J=8.8 Hz). MS m/z: 248 (M+). Anal. Calcd for C13H16N2O3·1/8H2O: C, 62.32; H, 6.54; N, 11.18. Found: C, 62.20; H, 6.40; N, 11.01.
1-Methoxymelatonin (125b) from 1-hydroxymelatonin (125a) — Excess CH2N2 in Et2O was added to a solution of 125a (40.2 mg, 0.16 mmol) in MeOH (5.0 mL) at rt and stirred for 15 min. Evaporation of the solvent under reduced pressure afforded an oil, which was column-chromatographed on SiO2 with AcOEt to give 125b (39.1 mg,75.1%). 125b: pale yellow oil. UV λmaxMeOH nm: 306, 277. IR (film): 3280, 2930, 1643, 1553, 1480, 1440, 1220, 1090 cm-1. 1H-NMR (5% CD3OD in CDCl3) δ: 1.93 (3H, s), 2.87 (2H, t, J=6.8 Hz), 3.53 (2H, t, J=6.8 Hz), 3.85 (3H, s), 4.04 (3H, s), 6.91 (1H, dd, J=8.9, 2.3 Hz), 7.00 (1H, d, J=2.3 Hz), 7.08 (1H, s), 7.30 (1H, dd, J=8.9, 2.3 Hz). MS m/z: 262 (M+). High resolution MS m/z: Calcd for C14H18N2O3: 262.1317. Found: 262.1331.
(dl)-2-Acetoamino-3-(1-hydroxyindol-3-yl)propanol ((dl)-127) from (dl)-2-acetoamino-3-(2,3- dihydroindol-3-yl)propanol ((dl)-126) — Prepared according to the general method B for 6, where Na2WO4·2H2O (44.6 mg, 0.14 mmol), (dl)-126 (158.2 mg, 0.68 mmol) in MeOH (16 mL), and 30% H2O2 (0.69 mL, 6.76 mmol) were used. After usual work-up and purification, (dl)-127 (40.8 mg, 30%) was obtained. (dl)-127: colorless unstable oil. IR (film): 3265, 3110, 1629, 1547, 738 cm-1. 1H-NMR (CD3OD) δ: 1.88 (3H, s), 2.80 (1H, dd, J=14.5, 7.5 Hz), 3.00 (1H, dd, J=14.5, 6.5 Hz), 3.53 (2H, d, J=5.1 Hz), 4.14 (1H, m), 6.96 (1H, dd, J=7.6, 7.1 Hz), 7.10 (1H, s), 7.12 (1H, dd, J=7.6, 6.8 Hz), 7.32 (1H, d, J=7.1 Hz), 7.57 (1H, d, J=6.8 Hz). High resolution MS m/z: Calcd for C13H16N2O3: 248.1159. Found: 248.1146.
Scheme 13.
Synthesis of 1-hydroxyinole derivative (7).
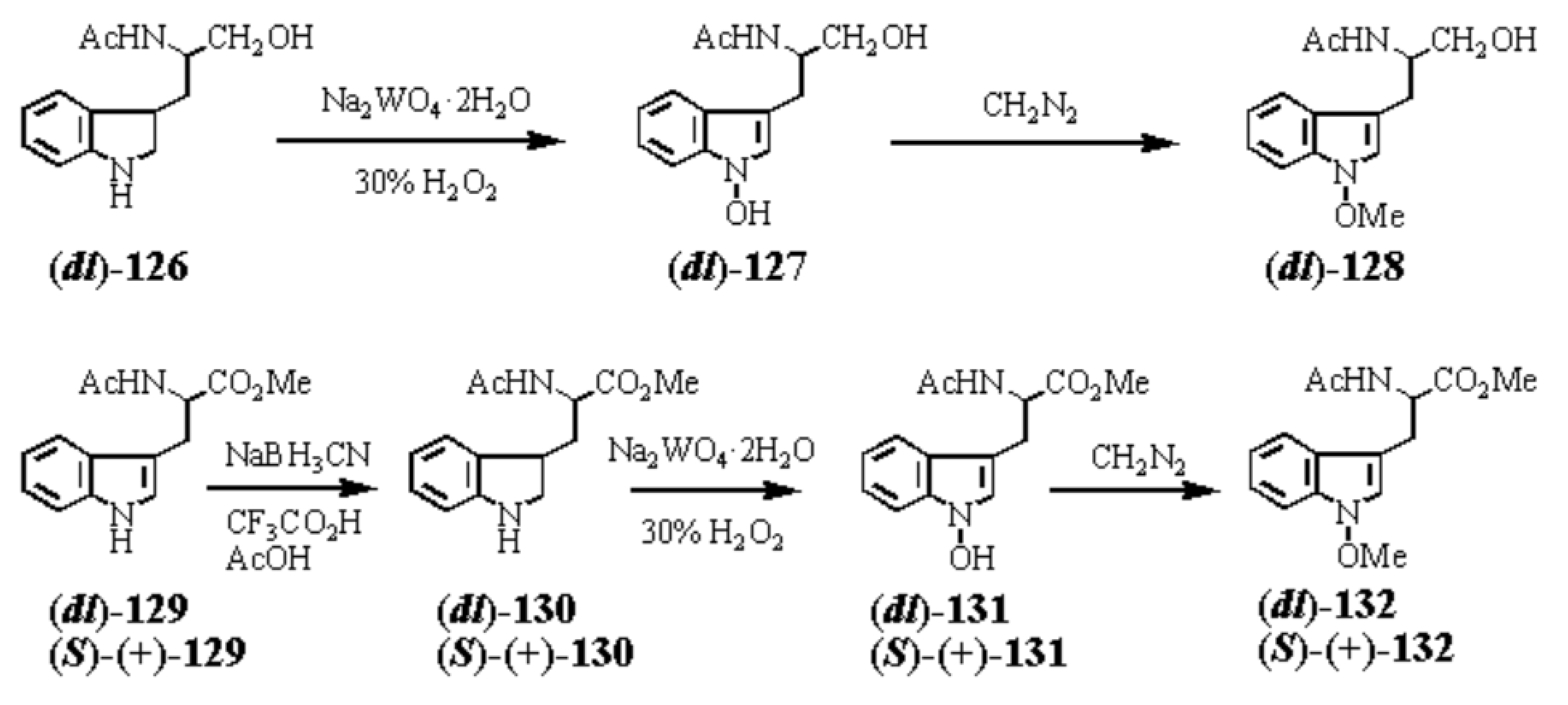
(dl)-2-Acetoamino-3-(1-methoxyindol-3-yl)propanol ((dl)-128) from (dl)-127) — Ethereal CH2N2 (excess) was added to a solution of (dl)-127 (32.8 mg, 0.13 mmol) in MeOH (3.0 mL) and stirring was continued at rt for 10 min. After usual work-up and purification by column-chromatography on SiO2 with CH2Cl2–MeOH (95:5, v/v), (dl)-128 (26.7 mg, 77%) was obtained. (dl)-128: mp 117–118°C (colorless prisms, recrystallized from EtOAc). IR (KBr): 3275, 3180, 3090, 1630, 1584, 1440, 1081, 1052, 757, 737 cm-1. UV λmaxMeOH nm (log ε): 223 (4.43), 278 (3.63), 291 (3.65). 1H-NMR (CDCl3) δ: 1.96 (3H, s), 2.54 (1H, br s, D2O exchange), 2.97 (2H, d, J=6.3 Hz), 3.63 (2H, dd, J=5.6, 3.9 Hz), 4.04 (3H, s), 4.08–4.38 (1H, m), 5.88 (1H, d, J=7 Hz), 7.08 (1H, dd, J=7.1, 6.8 Hz), 7.12 (1H, s), 7.23 (1H, dd, J=7.3, 6.8 Hz), 7.40 (1H, d, J=7.1 Hz), 7.60 (1H, d, J=7.3 Hz). MS m/z: 262 (M+). Anal. Calcd for C14H18N2O2: C, 64.11; H, 6.92; N, 10.68. Found: C, 63.89; H, 7.24; N, 10.54.
(S)-(+)-Nb-Acetyl-1-hydroxytryptophan methyl ester ((S)-(+)-131) from (S)-(+)-Nb-acetyl-2,3-di- hydrotryptophan methyl ester ((S)-(+)-130) — Prepared according to the general method B for 6, where Na2WO4·2H2O (40.2 mg, 0.12 mmol), (S)-(+)-130 (159.5 mg, 0.61 mmol) in MeOH (15.0 mL), and 30% H2O2 (0.62 mL, 6.09 mmol) were used. After usual work-up, the product was purified by p-TLC on SiO2 with CH2Cl2–MeOH (98:2, v/v) to give (S)-(+)-131 (89.7 mg, 53%). (S)-(+)-131: mp 116.0–117.0 °C (colorless prisms, recrystallized from MeOH–H2O). [α]D24 +11.8° (c=0.102, MeOH). IR (KBr): 3370, 3240, 1733, 1655, 1534, 745 cm-1. UV λmaxMeOH nm (log ε): 224 (4.53), 282 (3.64), 293 (3.66). 1H-NMR (5% CD3OD in CDCl3) δ: 1.90 (3H, s), 3.19 (1H, dd, J=15.0, 5.8 Hz), 3.27 (1H, dd, J=15.0, 5.2 Hz), 3.71 (3H, s), 4.86 (1H, dd, J=5.8, 5.2 Hz), 7.01 (1H, s), 7.06 (1H, t, J=8.3 Hz), 7.19 (1H, t, J=8.3 Hz), 7.42 (1H, d, J=8.3 Hz), 7.45 (1H, d, J=8.3 Hz). MS m/z: 276 (M+). Anal. Calcd for C14H16N2O4: C, 60.86; H, 5.84; N, 10.14. Found: C, 60.85; H, 5.88; N, 10.14.
(dl)-Nb-Acetyl-1-hydroxytryptophan methyl ester ((dl)-131) from (dl)-130 — Prepared under the same reaction conditions as described in the procedure for (S)-(+)-131. Yield was 73%. (dl)-131: mp 153.0–154.0 °C (decomp., colorless prisms, recrystallized from MeOH). IR (KBr): 3259, 3125, 1739, 1640, 1547, 727 cm-1. UV λmaxMeOH nm (log ε): 224 (4.55), 282 (3.65), 294 (3.68). 1H-NMR (CD3OD) δ: 1.92 (3H, s), 3.06 (1H, dd, J=13.9, 7.6 Hz), 3.28 (1H, dd, J=13.9, 5.9 Hz), 3.65 (3H, s), 4.66 (1H, dd, J=7.6, 5.9 Hz), 6.97 (1H, ddd, J=7.1, 6.8, 1.5 Hz), 7.09 (1H, s), 7.12 (1H, ddd, J=7.6, 6.8, 1.5 Hz), 7.32 (1H, dm, J=7.1 Hz), 7.47 (1H, dm, J=7.6 Hz). MS m/z: 276 (M+). Anal. Calcd for C14H16N2O4: C, 60.86; H, 5.84; N, 10.14. Found: C, 60.78; H, 5.92; N, 10.09.
(S)-(+)-Nb-Acetyl-1-methoxytryptophan methyl ester ((S)-(+)-132) from (S)-(+)-131 — Ethereal CH2N2 (excess) was added to a solution of (S)-(+)-131 (46.8 mg, 0.17 mmol) in MeOH (2.0 mL) and stirring was continued at rt for 15 min. After work-up and purification by column-chromatography on SiO2 with CH2Cl2–MeOH (98:2, v/v), (S)-(+)-132 (38.3 mg, 78%) was obtained. (S)-(+)-132: colorless oil. [α]D20 +16.8° (c=0.107, MeOH). IR (film): 3270, 1741, 1658, 1540, 736 cm-1. UV λmaxMeOH nm (log ε): 223 (4.47), 276 (3.66), 289 (3.68). 1H-NMR (CDCl3) δ: 1.97 (3H, s), 3.25 (1H, dd, J=14.6, 4.9 Hz), 3.31 (1H, dd, J=14.6, 5.4 Hz), 3.70 (3H, s), 4.05 (3H, s), 4.93 (1H, ddd, J=7.8, 5.4, 4.9 Hz), 6.03 (1H, d, J=7.8 Hz), 7.04 (1H, s), 7.11 (1H, dd, J=8.3, 7.8 Hz), 7.24 (1H, t, J=8.3 Hz), 7.40 (1H, d, J=8.3 Hz), 7.49 (1H, d, J=7.8 Hz). High resolution MS m/z: Calcd for C15H18N2O4: 290.1266. Found: 290.1296.
(dl)-Nb-Acetyl-1-methoxytryptophan methyl ester ((dl)-132) from (dl)-131 — Ethereal CH2N2 (excess) was added to a solution of (dl)-131 (40.1 mg, 0.15 mmol) in MeOH (2.0 mL) and stirring was continued at rt for 30 min. After usual work-up, (dl)-132 (35.1 mg, 83%) was obtained. (dl)-132: mp 95–96°C (colorless plates, recrystallized from MeOH-H2O). IR (KBr): 3235, 1737, 1657, 1545, 1443, 1376, 1313, 1241, 1210, 1173, 747, 741 cm-1. UV λmaxMeOH nm (log ε): 224 (4.50), 277 (3.70), 290 (3.17). 1H-NMR (CDCl3) δ: 1.97 (3H, s), 3.26 (1H, dd, J=15.1, 4.9 Hz), 3.31 (1H, dd, J=15.1, 5.4 Hz), 3.71 (3H, s), 4.05 (3H, s), 4.93 (1H, ddd, J=7.8, 5.4, 4.9 Hz), 5.88 (1H, d, J=7.8 Hz, D2O exchange), 7.04 (1H, s), 7.11 (1H, br d, J=8.3 Hz), 7.24 (1H, br d, J=8.3 Hz), 7.41 (1H, d, J=8.3 Hz), 7.49 (1H, d, J=8.3 Hz). MS m/z: 290 (M+). Anal. Calcd for C15H18N2O4: C, 62.06; H, 6.25; N, 9.65. Found: C, 62.04; H, 6.37; N, 9.52.
23.3. 1-Hydroxyyohimbine Derivatives, a Group of Potential New Medicine Candidates
23.3.1. Synthesis of 1-Hydroxyyohimbine Derivatives
2β,7β- (133) and 2α,7α-Dihydroyohimbine (134) from Yohimbine (52·Free) — General Procedure: NaBH3CN (610.7 mg, 9.72 mmol) was added to a solution of 52·Free (1056.4 mg, 2.98 mmol) in CF3COOH (20.0 mL) at 0°C. The mixture was stirred at rt for 3 h. After evaporation of the solvent, the whole was made alkaline with initially aq. 8% and then 0.8% NaOH under ice cooling, and extracted with CHCl3. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with AcOEt–CHCl3–MeOH–28% aq. NH3 (51.5:46:5:0.5, v/v) to give 134 (843.3 mg, 79%) and 133 (189.1 mg, 18%) in the order of elution. 133: mp 191—193.5°C (pale yellow needles, recrystallized from AcOEt–hexane). IR (KBr): 3502, 1722, 1608, 754 cm-1. 1H-NMR (CDCl3) δ: 1.05 (1H, q, J=11.5 Hz), 1.30—1.53 (5H, m), 1.60—1.78 (4H, m), 1.90—1.96 (1H, m), 2.06 (1H, dt, J=3.9, 11.5 Hz), 2.15—2.27 (2H, m), 2.25 (1H, dd, J=11.5, 2.0 Hz), 2.60 (1H, dt, J=11.5, 3.4 Hz), 2.71 (1H, dd, J=11.5, 2.7 Hz), 3.00 (1H, br s, disappeared on addition of D2O), 3.31 (1H, t, J=8.1 Hz), 3.42 (1H, br s), 3.78 (3H, s), 4.16 (1H, br s), 6.65 (1H, d, J=7.8 Hz), 6.77 (1H, dd, J=7.6, 7.3 Hz), 7.02—7.06 (2H, m). High-resolution MS m/z: calcd for C21H28N2O3: 356.2100, found 356.2096. [α]29D –69.9° (c=0.33, MeOH). 134: mp 190—193°C (colorless fine needles, recrystallized from AcOEt–hexane). IR (KBr): 3471, 2906, 1707, 1020 cm-1. 1H-NMR (CDCl3) δ: 1.36—1.61 (7H, m), 1.68 (1H, br s, disappeared on addition of D2O), 1.71—1.77 (1H, m), 1.83—2.06 (4H, m), 2.18 (1H, dt, J=11.5, 2.7 Hz), 2.30 (1H, dd, J=11.5, 2.2 Hz), 2.77 (1H, ddd, J=11.5, 3.4, 3.2 Hz), 2.83 (1H, dd, J=11.5, 2.2 Hz), 2.93 (1H, dt, J=6.6, 2.7 Hz), 3.10 (1H, s, disappeared on addition of D2O), 3.57 (1H, dd, J=6.6, 2.7 Hz), 3.76 (3H, s), 4.19 (1H, br s), 6.68 (1H, dd, J=7.8, 1.0 Hz), 6.72 (1H, ddd, J=7.6, 7.3, 1.0 Hz), 7.01 (1H, ddd, J=7.8, 7.6, 1.0 Hz), 7.08 (1H, dd, J=7.3, 1.0 Hz). High-resolution MS m/z: calcd for C21H28N2O3: 356.2100, found: 356.2111. Anal. Calcd for C21H28N2O3·1/8H2O: C, 70.31; H, 7.94; N, 7.81. Found: C, 70.30; H, 7.93; N, 7.78. [α]25D +90.64° (c=0.20, CHCl3).
Scheme 14.
Synthesis of new yohimbine derivatives (1).
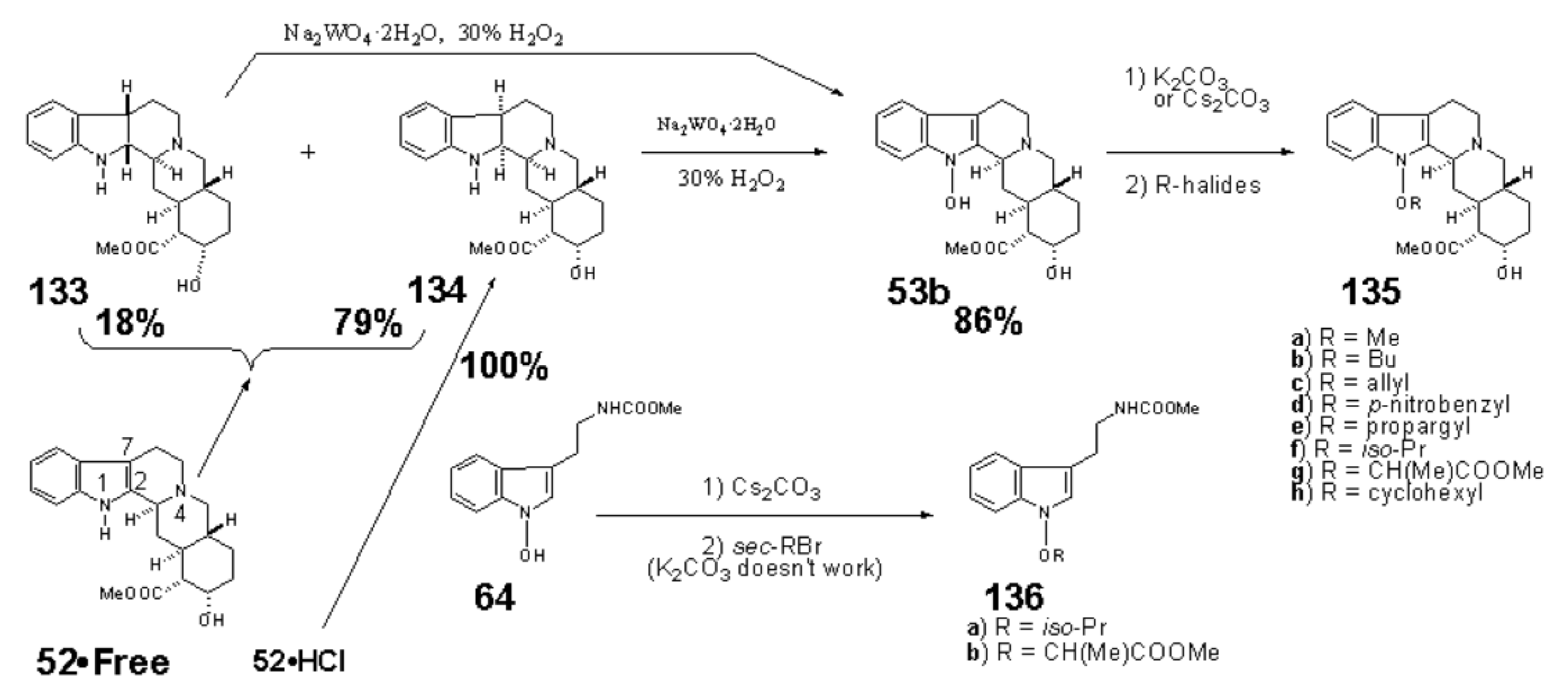
2α,7α-Dihydroyohimbine (134) from Yohimbine hydrochloride (52·HCl) — According to the general procedure, NaBH3CN (36.4 mg, 0.55 mmol), 52·HCl (107.6mg, 0.28 mmol), and CF3COOH (2.0 mL) were used. After column-chromatography on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:3:0.3, v/v), 134 (98.0 mg, 100%) was obtained.
Table 29.
Effect of yohimbine derivative on the contraction induced by clonidine. in rat thoracic aort.
Table 29.
Effect of yohimbine derivative on the contraction induced by clonidine. in rat thoracic aort.
 |
1-Hydroxyyohimbine (53b) from Yohimbine hydrochloride (52·HCl) — According to the general procedure, NaBH3CN (85.5 mg, 1.3 mmol), 52·HCl (101.0mg, 0.26 mmol), and CF3COOH (2.0 mL) were used. The resultant oil, obtained after general procedure, was dissolved in MeOH (9.0 mL). A solution of Na2WO4·2H2O (17.0 mg, 0.05 mmol) in H2O (1.0 mL) and 30% H2O2 (0.59 mL, 5.2 mmol) were added to the solution. The mixture was stirred at 0°C for 1 h. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a solid, which was column-chromatographed on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:5:0.5, v/v) to give 53b (82.1 mg, 86%). 53b: mp 224—226°C (decomp., colorless fine needles, recrystallized from MeOH). IR (KBr): 3505, 2945, 1711, 751 cm-1. 1H-NMR (CD3OD) δ: 1.19 (1H, q, J=11.5 Hz), 1.33—1.39 (1H, m), 1.43—1.57 (2H, m), 1.65 (1H, br t, J=13.4 Hz), 1.91 (1H, dq, J=13.4, 2.7 Hz), 1.99 (1H, dq, J=2.7, 11.5 Hz), 2.31 (1H, br d, J=11.5 Hz), 2.40 (1H, t, J=11.5 Hz), 2.63—2.76 (2H, m), 2.88—2.98 (3H, m), 3.10—3.15 (1H, m), 3.62 (1H, d, J=11.5 Hz), 3.73 (3H, s), 4.22 (1H, q, J=2.7 Hz), 6.98 (1H, t, J=7.6 Hz), 7.09 (1H, t, J=7.6 Hz), 7.29 (1H, d, J=7.6 Hz), 7.37 (1H, d, J=7.6 Hz). MS m/z: 370 (M+), 354 (M+–O), 353 (M+–OH). Anal. Calcd for C21H26N2O4: C, 68.09; H, 7.07; N, 7.56. Found: C, 67.97; H, 7.13; N, 7.60. [α]30D +7.75° (c=0.20, DMF).
1-Hydroxyyohimbine (53b) from 2β,7β-Dihydroyohimbine (133) — A solution of Na2WO4·2H2O (6.8 mg, 0.03 mmol) in H2O (0.3 mL) and 30% H2O2 (0.10 mL, 0.80 mmol) were added to a solution of 133 (28.2 mg, 0.80 mmol) in MeOH (3.0 mL). The mixture was stirred at rt for 2 h. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave a solid, which was column-chromatographed on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:5:0.5, v/v) to give 53b (12.7mg, 43%).
1-Methoxyyohimbine (135a) from 53b — An excess amount of ethereal CH2N2 was added to a solution of 53b (52.6 mg, 0.14 mmol) in MeOH (20.0 mL) and the whole was stirred at 0°C for 1 h. The solution was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:3:0.3, v/v) to give 135a (42.2 mg, 77%). 135a: mp 201—203°C (decomp., colorless prisms, recrystallized from acetone. Lit.[76] mp 198—201°C). IR (KBr): 3145, 1737, 743 cm-1. 1H-NMR (CDCl3) δ: 1.36—1.45 (2H, m), 1.49—1.62 (3H, m), 1.97—2.09 (2H, m), 2.32—2.39 (2H, m), 2.46 (1H, ddd, J=12.7, 3.2, 2.9 Hz), 2.63 (1H, dt, J=4.2, 11.2 Hz), 2.66—2.71 (1H, m), 2.89—2.98 (2H, m), 3.03—3.08 (1H, m), 3.36 (1H, br s, disappeared on addition of D2O), 3.49 (1H, br d, J=11.2 Hz), 3.77 (3H, s), 3.89 (3H, s), 4.21 (1H, br s), 7.09 (1H, ddd, J=7.8, 7.1, 1.0 Hz), 7.19 (1H, ddd, J=8.1, 7.1, 1.0 Hz), 7.34 (1H, dd, J=8.1, 1.0 Hz), 7.44 (1H, dd, J=7.8, 1.0 Hz). MS m/z: 384 (M+), 353 (M+–OMe). Anal. Calcd for C22H28N2O4: C, 68.72; H, 7.34; N, 7.29. Found: C, 68.65; H, 7.35; N, 7.23. [α]29D +20.54° (c=0.20, CHCl3).
1-n-Butyloxyyohimbine (135b) from 53b — According to the general procedure for 135c, K2CO3 (56.7 mg, 0.41 mmol), n-butyl iodide (30.8 mg, 0.17 mmol), and 53b (50.5 mg, 0.14 mmol) were used. After column-chromatography, 135b (57.8 mg, 99%) was obtained. 135b: mp 126—128.5°C (decomp., colorless fine needles, recrystallized from hexane). IR (KBr): 3458, 2920, 1739, 1151, 737 cm-1. 1H-NMR (CDCl3) δ: 1.34—1.43 (2H, m), 1.48—1.64 (3H, m), 1.97—2.06 (2H, m), 2.34 (1H, dd, J=11.5, 2.2 Hz), 2.35 (1H, t, J=11.5 Hz), 2.55 (1H, dt, J=12.9, 2.9 Hz), 2.62 (1H, dt, J=4.2, 10.7 Hz), 2.66—2.72 (1H, m), 2.90—2.98 (1H, m), 2.96 (1H, dd, J=11.5, 2.9 Hz), 3.05 (1H, ddd, J=11.5, 5.6, 2.2 Hz), 3.30 (1H, s, disappeared on addition of D2O), 3.51 (1H, dd, J=11.5, 2.2 Hz), 3.75 (3H, s), 4.20 (1H, d, J=1.2 Hz), 4.49 (1H, dddd, J=11.0, 6.6, 1.2, 1.0 Hz), 4.55 (1H, dddd, J=11.0, 6.1, 1.2, 1.0 Hz), 5.39 (1H, ddd, J=10.7, 1.2, 1.0 Hz), 5.44 (1H, dq, J=17.1, 1.2 Hz), 6.05 (1H, dddd, J=17.1, 10.7, 6.6, 6.1 Hz), 7.08 (1H, ddd, J=8.1, 7.8, 1.0 Hz), 7.18 (1H, dt, J=1.0, 8.1 Hz), 7.34 (1H, ddd, J=8.1, 1.0, 0.7 Hz), 7.43 (1H, br d, J=7.8 Hz). MS m/z: 410 (M+), 353 (M+–OCH2CH=CH2). Anal. Calcd for C24H30N2O4: C, 70.22; H, 7.37; N, 6.82. Found: C, 70.13; H, 7.50; N, 6.57. [α]30D +18.4° (c=0.21, CHCl3).
1-Allyloxyyohimbine (135c) from 53b — General procedure: K2CO3 (59.2 mg, 0.43 mmol) and a solution of allyl bromide (24.7 mL, 0.3 mmol) in DMF (1.0 mL) were successively added to a solution of 53b (52.8 mg, 0.14 mmol) in DMF (4.0 mL) and the whole was stirred at rt for 30 min. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (95:5, v/v) to give 135c (54.6 mg, 93%). 135c: mp 150—152°C (decomp., colorless fine needles, recrystallized from hexane). IR (KBr): 3464, 2935, 1738, 1151, 737 cm-1. 1H-NMR (CDCl3) δ: 1.01 (3H, t, J=7.3 Hz), 1.34—1.43 (2H, m), 1.48—1.63 (5H, m), 1.65—1.79 (2H, m), 1.97—2.07 (2H, m), 2.34 (1H, dd, J=11.2, 2.0 Hz), 2.36 (1H, t, J=11.2 Hz), 2.53 (1H, dt, J=12.9, 2.9 Hz), 2.63 (1H, dt, J=4.2, 11.2 Hz), 2.66—2.72 (1H, m), 2.90—2.98 (1H, m), 2.96 (1H, dd, J=11.2, 2.9 Hz), 3.05 (1H, ddd, J=11.2, 5.6, 2.0 Hz), 3.29 (1H, s, disappeared on addition of D2O), 3.48 (1H, d, J=11.2 Hz), 3.76 (3H, s), 3.98 (1H, dt, J=6.6, 8.5 Hz), 4.06 (1H, dt, J=6.6, 8.5 Hz), 4.20 (1H, br s), 7.07 (1H, ddd, J=8.1, 7.8, 1.0 Hz), 7.17 (1H, dt, J=1.0, 8.1 Hz), 7.31 (1H, dd, J=8.1, 1.0 Hz), 7.43 (1H, dd, J=7.8, 1.0 Hz). MS m/z: 426 (M+), 353 (M+–On-Bu). Anal. Calcd for C25H34N2O4: C, 70.39; H, 8.03; N, 6.57. Found: C, 70.26; H, 8.12; N, 6.48. [α]32D +21.46° (c=0.21, CHCl3).
1-p-Nitrobenzyloxyyohimbine (135d) from 1-Hydroxyyohimbine (53b) — According to the general procedure for 135c, K2CO3 (56.8 mg, 0.41 mmol), p-nitrobenzyl bromide (35.4 mg, 0.16 mmol), and 53b (50.3 mg, 0.14 mmol) were used. After column-chromatography, 135d (61.7 mg, 90%) was obtained. 135d: mp 148—149°C (decomp., yellow fine needles, recrystallized from AcOEt–hexane). IR (KBr): 3430, 2924, 1734 and 1703 (collapsed to 1710 in CHCl3), 1523, 1348, 739 cm-1. 1H-NMR (CDCl3) δ: 1.33—1.43 (2H, m), 1.50—1.61 (3H, m), 1.94—2.03 (2H, m), 2.28 (1H, br t, J=11.0 Hz), 2.33 (1H, dd, J=11.7, 2.2 Hz), 2.54 (1H, dt, J=12.7, 2.9 Hz), 2.57 (1H, dt, J=4.2, 11.0 Hz), 2.66—2.72 (1H, m), 2.89—2.98 (2H, m), 3.01—3.07 (1H, m), 3.04 (1H, s, disappeared on addition of D2O), 3.23 (1H, br d, J=11.0 Hz), 3.59 (3H, s), 4.21 (1H, br s), 5.02 (1H, d, J=10.6 Hz), 5.07 (1H, d, J=10.6 Hz), 7.12 (1H, ddd, J=8.1, 7.8, 1.0 Hz), 7.21 (1H, dt, J=1.0, 8.1 Hz), 7.32 (1H, dd, J=8.1, 1.0 Hz), 7.46 (1H, dd, J=7.8, 1.0 Hz), 7.59—7.62 (2H, A2 part of A2B2), 8.29—8.33 (2H, B2 part of A2B2). Anal. Calcd for C28H31N3O6: C, 66.52; H, 6.18; N, 8.31. Found: C, 66.40; H, 6.22; N, 8.18. [α]31D +48.77° (c=0.20, CHCl3).
1-Propargyloxyyohimbine (135e) from 53b — According to the general procedure for 135c, K2CO3 (223.6 mg, 1.62 mmol), propargyl bromide (70.8 mg, 0.60 mmol), and 53b (200.1 mg, 0.54 mmol) were used. After column-chromatography with AcOEt–hexane (1:1, v/v), 135e (57.8 mg, 99%) was obtained. 135e: mp 158—161°C (decomp., colorless fine needles, recrystallized from AcOEt–hexane). IR (KBr): 3565, 1720, 1265, 742 cm-1. 1H-NMR (CDCl3) δ: 1.35—1.43 (2H, m), 1.51—1.63 (3H, m), 1.97—2.07 (2H, m), 2.34 (1H, dd, J=11.5, 2.2 Hz), 2.36 (1H, d, J=11.2 Hz), 2.55 (1H, dt, J=12.7, 3.4 Hz), 2.60-2.71 (3H, m), 2.89—2.96 (1H, m), 2.96 (1H, dd, J=11.2, 3.4 Hz), 3.05 (1H, ddd, J=11.2, 6.1, 1.7 Hz), 3.46 (1H, br s, disappeared on addition of D2O), 3.58 (1H, dd, J=11.2, 1.7 Hz), 3.79 (3H, s), 4.21 (1H, d, J=1.2 Hz), 4.63 (1H, dd, J=15.1, 2.4 Hz), 4.71 (1H, dd, J=15.1, 2.4 Hz), 7.09 (1H, dt, J=1.0, 7.8 Hz), 7.19 (1H, dt, J=1.0, 7.8 Hz), 7.38 (1H, d, J=7.8 Hz), 7.43 (1H, d, J=7.8 Hz). MS m/z: 408 (M+). Anal. Calcd for C24H28N2O4·1/2H2O: C, 69.04; H, 7.00; N, 6.71. Found: C, 68.99; H, 6.82; N, 6.56. [α]24D +96.19° (c=0.21, MeOH).
1-Isopropyloxyyohimbine (135f) from 53b — a) Cs2CO3 Method: According to the general procedure for 135c, 53b (29.4 mg, 0.08 mmol), Cs2CO3 (28.6 mg, 0.09 mmol), and isopropyl bromide (29.3 mg, 0.24 mmol) were used. After column-chromatography with CHCl3–MeOH–28% aq. NH3 (46:1:0.1, v/v), 135f (32.3 mg, 99%) was obtained. 135f: pale brown viscous oil. IR (film): 3456, 2922, 1734 (br), 750 cm-1. 1H-NMR (CDCl3) δ: 1.29 (3H, d, J=6.1 Hz), 1.31 (3H, d, J=6.1 Hz), 1.41—1.64 (4H, m), 1.95—2.07 (2H, m), 2.38 (1H, dd, J=11.6, 2.0 Hz), 2.64—2.77 (3H, m), 2.97—3.07 (2H, br s), 3.07—3.17 (2H, br s), 3.55—3.65 (1H, br), 3.76 (3H, s), 4.23 (1H, br s), 4.50 (1H, sep, J=6.1 Hz), 4.72 (2H, br s, disappeared on addition of D2O), 7.02 (1H, t, J=7.3 Hz), 7.19 (1H, t, J= 7.3 Hz), 7.30 (1H, d, J=7.9 Hz), 7.43 (1H, d, J=7.3 Hz). MS m/z: 412 (M+), 353 (M+–OCHMe2). High-resolution MS m/z: calcd for C24H32N2O4: 412.2362, found: 412.2366. [α]25D +16.45° (c=0.15, CHCl3).
b) K2CO3 Method: According to the general procedure for 135c, K2CO3 (18.9 mg, 0.14 mmol), isopropyl bromide (19.8 mg, 0.16 mmol), and 53b (16.8 mg, 0.05 mmol) were used. After work-up, 135f (13.3 mg, 71%) was obtained.
(dl)-1-(1-Methoxycarbonyl)ethoxyyohimbine (135g) from 53b — According to the general procedure for 135c, 53b (30.3 mg, 0.08 mmol), Cs2CO3 (29.4 mg, 0.09 mmol), and (dl)-methyl-2-bromopropionate (41.0 mg, 0.25 mmol) were used. After column-chromatography with AcOEt, 135g (37.0 mg, 99%) was obtained. 135g: pale brown viscous oil. IR (film): 3509, 2923, 1739, 1710, 752 cm-1. 1H-NMR (CDCl3) δ: 1.30—1.45 (2H, m), 1.52—1.61 (3H, m), 1.55 (3H, d, J=6.8 Hz), 1.95—2.07 (2H, m), 2.34 (1H, dd, J=11.6, 2.0 Hz), 2.37—2.44 (1H, m), 2.52 (1H, dt, J=13.2, 2.7 Hz), 2.61—2.71 (2H, m), 2.86—2.95 (1H, m), 2.98 (1H, dd, J=11.6, 2.7 Hz), 3.01—3.08 (1H, m), 3.16 (1H, br s, disappeared on addition of D2O), 3.53 (1H, d, J=10.0 Hz), 3.66 (3H, s), 3.77 (3H, s), 4.20 (1H, br s), 4.66 (1H, q, J=6.8 Hz), 7.06 (1H, t, J=7.8 Hz), 7.16 (1H, t, J=7.8 Hz), 7.39 (1H, d, J=7.8 Hz), 7.44 (1H, d, J=7.8 Hz). High-resolution MS m/z: calcd for C25H32N2O6: 456.2260, found: 456.2263. [α]26D –22.11° (c=0.015, CHCl3).
1-Cyclohexyloxyyohimbine (135h) from 53b — According to the general procedure for 135c, 53b, (30.0 mg, 0.08 mmol), and Cs2CO3 (29.1 mg, 0.09 mmol) were used. The reaction time with cyclohexyl bromide (40.7 mg, 0.25 mmol) was prolonged to 130 min. After column-chromatography with CHCl3–MeOH–28% aq. NH3 (46:0.5:0.05, v/v), 135h (4.0 mg, 11%) and unreacted 53b (20 mg, 67%) were obtained. 135h: pale brown viscous oil. IR (film): 3446, 2927, 1738 (br), 739 cm-1. 1H-NMR (CD3OD) δ: 1.25—1.41 (8H, m), 1.46—1.71 (5H, m), 1.77—2.08 (6H, m), 2.35 (1H, dd, J=11.7, 2.7 Hz), 2.45 (1H, br t, J=11.0 Hz), 2.68—2.79 (2H, m), 2.90—3.02 (3H, m), 3.09—3.18 (1H, m), 3.73 (3H, s), 4.12—4.19 (1H, m), 4.20—4.25 (1H, m), 7.01 (1H, t, J=7.8 Hz), 7.13 (1H, t, J=8.3 Hz), 7.30 (1H, d, J=8.3 Hz), 7.39 (1H, d, J=7.8 Hz). High-resolution MS (EI) m/z: calcd for C27H34N2O4: 452.2675, found: 452.2677. [α]25D +8.46° (c=0.14, CHCl3).
N-Methoxycarbonyl-1-isopropyloxytryptamine (136a) from 1-Hydroxy-N-methoxy- carbonyltryptamine (64) — General procedure: 1-Hydroxy-N-methoxycarbonyltryptamine (64, 50.9 mg, 0.22 mmol) was added to a solution of Cs2CO3 (83.9 mg, 0.23 mmol) in MeOH (2.0 mL) and the whole was stirred at rt for 20 min. To the resultant residue obtained after evaporation of the solvent under reduced pressure, a solution of isopropyl bromide (160.0 mg, 1.32 mmol) in DMF (3.0 mL) was added and the whole was stirred at rt for 1 h. After addition of H2O, the whole was extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 136a (49.7 mg, 83%). 136a: colorless oil. IR (film): 1710 (br), 740 cm-1. 1H-NMR (CDCl3) δ: 1.36 (3H, d, J=6.2 Hz), 1.37 (3H, d, J=6.2 Hz), 2.93 (2H, t, J=6.6 Hz), 3.49 (2H, br q, J=6.6 Hz, collapsed to t on addition of D2O), 3.66 (3H, s), 4.52 (1H, sep, J=6.2 Hz), 4.74 (1H, br s, disappeared on addition of D2O), 7.06 (1H, s), 7.09 (1H, dt, J=0.8, 7.7 Hz), 7.22 (1H, dt, J=0.8, 7.7 Hz), 7.38 (1H, dd, J=7.7, 0.8 Hz), 7.55 (1H, dd, J=7.7, 0.8 Hz). High-resolution MS m/z: calcd for C15H20N2O3: 276.1482, found: 276.1474.
(dl)-N-Methoxycarbonyl-1-(1-methoxycarbonyl)ethoxy tryptamine (136b) from 64 — According to the general procedure for 136a, 64 (49.1 mg, 0.21 mmol), Cs2CO3 (76.0 mg, 0.23 mmol), and (dl)-methyl 2-bromopropionate (212.8 mg, 1.3 mmol) were used. After column-chromatography with AcOEt–hexane (1:2, v/v), 136b (64.9 mg, 97%) was obtained. 136b: colorless oil. IR (film): 3405, 1749, 1716 (br), 742 cm-1. 1H-NMR (CDCl3) δ: 1.66 (3H, d, J=7.0 Hz), 2.89 (2H, t, J=6.6 Hz), 3.47 (2H, br q, J=6.6 Hz, collapsed to t on addition of D2O), 3.66 (3H, s), 3.77 (3H, s), 4.73 (1H, br s, disappeared on addition of D2O), 4.85 (1H, q, J=7.0 Hz), 7.12 (1H, dt, J=0.7, 7.6 Hz), 7.21 (1H, s), 7.24 (1H, dt, J=0.7, 7.6 Hz), 7.40 (1H, d, J=7.6 Hz), 7.54 (1H, d, J=7.6 Hz). High-resolution MS m/z: calcd for C16H20N2O5: 320.1372, found: 320.1371.
Scheme 15.
New yohimbine derivatives (2).
7α-Acetoxy- (137a) and 7α,17α-dacetoxy-7H-yohimbine (137b) from 1-hydroxyyohimbine (53b) — 98% NaOAc (93.6 mg, 1.1 mmol) was added to a solution of 53b (207.1 mg, 0.56 mmol) in Ac2O (10.0 mL) at rt and stirred at 65 °C for 1 h. After evaporation of the solvent and adding H2O, the whole was made alkaline with 0.8% NaOH under ice cooling. The whole was extracted with CHCl3 and the extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (97:3, v/v) to give 137a (163.4 mg, 66%) and 137b (21.4 mg, 8%). 137a:Lit. [81] mp 120—122 °C (yellow powder, recrystallized from Et2O–hexane). IR (KBr): 1747, 1726, 1597, 1203, 1146, 1111, 1078, 1018, 775, 758 cm-1. 1H-NMR (CDCl3) δ: 1.36—1.63 (5H, m), 1.78—2.09 (4H, m), 2.06 (3H, s), 2.18 (1H, t, J=11.0 Hz), 2.37 (1H, dd, J=11.0, 2.0 Hz), 2.61—2.77 (3H, m), 2.91 (1H, dd, J=11.0, 3.4 Hz), 2.94 (1H, dd, J=11.0, 2.7 Hz), 3.15 (1H, s, disappeared on addition of D2O), 3.76 (3H, s), 4.18 (1H, s), 7.20 (1H, dd, J=7.6, 7.3 Hz), 7.37 (1H, dd, J=7.6, 7.3 Hz), 7.38 (1H, d, J=7.6 Hz), 7.61 (1H, d, J=7.6 Hz). MS m/z: 412 (M+). Anal. Calcd for C23H28N2O5·3/2H2O: C, 62.85; H, 7.11; N, 6.37. Found: C, 62.86; H, 7.28; N, 6.18. [α]23D +189° (c=0.221, CHCl3). 137b: mp 190—191 °C (pale yellow prisms, recrystallized from Et2O–hexane). IR (KBr): 1736, 1597, 1369, 1254, 1147, 1024, 754 cm-1. 1H-NMR (CDCl3) δ: 1.38—1.52 (4H, m), 1.59—1.68 (1H, m), 1.69 (1H. q, J=12.0 Hz), 1.90—2.03 (2H, m), 2.05 (3H, s), 2.06 (3H, s), 2.22 (1H, t, J=10.5 Hz), 2.30 (1H, dt, J=12.9, 3.0 Hz), 2.42 (1H, dd, J=11.5, 2.4 Hz), 2.64—2.78 (3H, m), 2.93 (1H, dd, J=11.0, 3.0 Hz), 3.06 (1H, dd, J=11.0, 2.4 Hz), 3.64 (3H, s), 5.41 (1H, dd, J=5.4, 2.4 Hz), 7.19 (1H, ddd, J=7.6, 7.3, 1.0 Hz), 7.36 (1H, ddd, J=7.8, 7.6. 1.0 Hz), 7.39 (1H, dd, J=7.3, 1.0 Hz), 7.59 (1H, dd, J=7.8, 1.0 Hz). Anal. Calcd for C25H30N2O6: C, 66.06; H, 6.65; N, 6.16. Found: C, 65.99; H, 6.78; N, 6.06. [α]30D +188° (c=0.201, CHCl3).
(2α,7α)- (141) and (2β,7β)-26-Nitro-benzofurano[2,3-n]yohimbine (142) from 53b — A solution of K2CO3 (374.2 mg, 2.71 mmol) and p-bromonitrobenzene (329.2 mg, 1.63 mmol) in DMF (3.0 mL) was added to a solution of 53b (500.5 mg, 1.35 mmol) in DMF (12.0 mL) and the whole was stirred at rt for 22 h. After addition of H2O, the whole was extracted with EtOAc. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with EtOAc–hexane (1:1, v/v) to give 141 (358.0 mg, 53%) and 142 (89.6 mg, 13%). 141: mp 275—276 °C (decomp., yellow powder, recrystallized from EtOAc–hexane). IR (film): 3545, 3392, 2360, 1596, 1506, 1387, 1327 cm-1. 1H-NMR (CDCl3) δ: 1.30—1.77 (5H, m), 1.84—2.17 (6H, m), 2.20—2.32 (1H, m), 2.35 (1H, dd, J=12.5, 2.5 Hz), 2.60—2.97 (3H, m), 3.07 (1H, brs, disappeared on addition of D2O), 3.83 (3H, s), 4.20 (1H, brs), 5.21 (1H, brs, disappeared on addition of D2O), 6.77 (1H, d, J=9.2 Hz), 6.82 (1H, dd, J=8.3, 1.7 Hz), 6.82 (1H, t, J=7.5 Hz), 7.01 (1H, dd, J=8.3, 1.7 Hz), 7.10 (1H, ddd, J=8.3, 8.3, 1.2 Hz), 8.12 (1H, dd, J=8.8, 2.5 Hz), 8.30 (1H, d, J=2.4 Hz). 13C-NMR (CDCl3) δ: 175.62 (C), 164.64 (C), 145.02 (C), 142.25 (C), 132.98 (C), 132.43 (C), 128.47 (CH), 126.02 (CH), 122.47 (CH), 121.04 (CH), 118.91 (CH), 111.93 (C), 110.68 (CH), 109.94 (CH), 66.52 (CH), 64.73 (CH), 61.42 (CH2), 55.11 (C), 52.25 (CH3), 52.03 (CH), 51.44 (CH3), 40.09 (CH), 36.14 (CH), 31.73 (CH2), 31.06 (CH2), 29.74 (CH2), 23.03 (CH2). MS m/z: 491 (M+). Anal. Calcd for C27H29N3O6·1/4H2O: C, 65.38; H, 5.99; N, 8.47. Found: C, 65.41; H, 5.89; N, 8.54. [α]23D +310° (c=0.221, MeOH). 142: yellow viscous oil. IR (film): 3508, 3352, 1728, 1518, 1338 cm-1. 1H-NMR (CDCl3) δ: 1.23—1.62 (4H, m), 1.74—1.92 (4H, m), 1.92—2.00 (2H, m), 2.06 (1H, ddd, J=11.7, 11.7, 2.9 Hz), 2.33 (1H, dd, J=10.7, 2.4 Hz), 2.38 (1H, dd, J=10.9, 2.1 Hz), 2.59 (1H, dd, J=14.2, 3.4 Hz), 2.72 (1H, ddd, J=11.7, 4.2, 4.2 Hz), 2.81 (1H, brs, disappeared on addition of D2O), 2.84 (1H, dd, J=11.2, 2.7 Hz), 3.83 (3H, s), 4.23 (1H, brs), 4.71 (1H, brs, disappeared on addition of D2O), 6.63 (1H, d, J=7.8 Hz), 6.90 (1H, t, J=7.6 Hz), 6.95 (1H, d, J=8.8 Hz), 7.13 (1H, td, J=7.7, 1.2 Hz), 7.39 (1H, d, J=7.1 Hz), 7.94 (1H, d, J=2.4 Hz), 8.09 (1H, d, J=8.8, 2.4 Hz). 13C-NMR (CDCl3) δ: 175.62 (C), 162.15 (C), 147.60 (C), 142.87 (C), 136.19 (C), 129.03 (CH), 128.39 (C), 125.57 (CH), 122.57 (CH), 122.55 (CH), 120.44 (CH), 119.41 (CH), 110.92 (CH), 110.21 (C), 109.68 (CH), 66.82 (CH), 66.27 (CH), 61.63 (CH2), 54.82 (C), 52.30 (CH), 52.04 (CH3), 51.20 (CH3), 39.95 (CH), 36.24 (CH), 31.78 (CH2), 31.29 (CH2), 29.40 (CH2), 23.04 (CH2). HR–MS m/z: Calcd for C27H29N3O6: 491.2057. Found: 491.2058. [α]23D +59.0° (c=0.370, MeOH).
Scheme 16 New yohimbine derivatives (3)
7α-(2-Methoxy-5-nitrophenyl)-7H-yohimbine (143) from 141 — Excess CH2N2 in Et2O was added to a solution of 141 (165.0 mg, 0.34 mmol) in MeOH (5 mL) and the whole was stirred at rt for 30 min. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 143 (142.8 mg, 84%) and unreacted 141 (26.7 mg, 16%). 143: mp 171.5—172 °C (yellow prisms, recrystallized from CHCl3–hexane). IR (KBr): 3392, 1734, 1589, 1344, 1269 cm-1. 1H-NMR (CDCl3) δ: 1.33—1.85 (5H, m), 1.87—2.04 (2H, m), 2.05—2.32 (2H, m), 2.41—2.71 (3H, m, 1H disappeared on addition of D2O), 2.87—3.24 (4H, m), 3.55 (3H, brs), 3.77 (3H, s), 4.67 (1H,, brs), 6.86 (1H, d, J=10.0 Hz), 7.05 (1H, d, J=8.3 Hz), 7.15 (1H, t, J=8.3 Hz), 7.35 (1H, ddd, J=8.3, 8.3, 1.7 Hz), 7.73 (1H, d, J=8.3 Hz), 8.22 (1H, dd, J=10.0, 3.3 Hz), 8.51 (1H, brs). 13C-NMR (CDCl3) δ: 184.96 (C), 175.18 (C), 162.15 (C), 155.02 (C), 142.95 (C), 141.78 (C), 128.12 (CH), 125.54 (CH), 125.09 (CH), 123.85 (CH), 121.76 (CH), 120.95 (CH), 111.37 (CH), 67.06 (CH), 61.40 (C), 61.26 (CH2), 59.09 (C), 55.85 (CH3), 52.26 (CH3), 51.80 (CH), 51.13 (CH2), 50.66 (CH), 40.11 (CH), 36.59 (CH2), 35.99 (CH), 31.45 (CH2), 31.41 (CH2), 23.06 (CH2). HR–MS m/z: Calcd for C28H31N3O6: 505.2213. Found: 505.2206. [α]29D +289° (c=0.141, DMF).
7α-(5-Amino-2-methoxyphenyl)-7H-yohimbine (144) from 143 — Tin powder (75.6 mg, 0.64 mmol) was added to a solution of 143 (30.9 mg, 0.06 mmol) in MeOH–8% HCl (3:1, v/v, 6.0 mL) and the whole was stirred at rt for 40 min. The resulting solution was made alkaline with 6% NaOH under ice cooling, and the whole was extracted with CHCl3–MeOH–28% aq. NH3 (46:3:0.3, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH–28% aq. NH3 (46:2:0.2, v/v) to give 144 (26.7 mg, 90%). 144: mp 262—264 °C (decomp., yellow prisms, recrystallized from EtOAc–MeOH). IR (KBr): 3452, 3336, 3234, 2933, 1745, 1500, 1236, 1144, 1109, 1018 cm-1. 1H-NMR (DMSO-d6) δ: 1.04 (1H, t, J=13.1 Hz), 1.61—1.43 (3H, m), 1.52—1.65 (2H, m), 1.70—1.79 (2H, m), 1.88 (1H, t, J=10.4 Hz), 2.15 (1H, brd, J=12.7 Hz), 2.25—2.33 (2H, m), 2.38—2.54 (1H, m), 2.65 (1H brd, J=11.5 Hz), 2.76 (1H, dd, J=10.9, 3.1 Hz), 2.83—3.11 [1H, m, on addition of D2O, it changed to 2.89 (1H, brd, J=10.3 Hz)], 3.27 (3H, s), 3.64 (3H, s), 4.12 (1H, s), 4.20 (1H, brd, J=4.4 Hz, disappeared on addition of D2O), 4.55 (2H, brs, disappeared on addition of D2O), 6.48 (1H, dd, J=8.5, 2.7 Hz), 6.63 (1H, d, J=8.5 Hz), 6.84 (1H, brs), 7.06 (1H, t, J=7.4 Hz), 7.14 (1H, d, J=7.4 Hz), 7.23 (1H, t, J=7.4 Hz), 7.49 (1H, d, J=7.4 Hz). MS m/z: 475 (M+). Anal. Calcd for C28H33N3O4·1/2H2O: C, 69.40; H, 7.07; N, 8.67. Found: C, 69.60; H, 7.10; N, 8.60. [α]30D +255° (c=0.210, MeOH).
17α-Acetoxy- (145b) and 7α-(5-acetylamino-2-methoxyphenyl)-7H-yohimbine (145a) from 144 — [Entry 1] Ac2O (1.0 mL) was added to a solution of 144 (33.5 mg, 0.07 mmol) in pyridine (2.0 mL) and the whole was stirred at rt for 40 min. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH–30% aq. NH3 (46:3:0.3, v/v) to give 145a (30.3 mg, 80%) and 145b (2.6 mg, 7%).
[Entry 2] Ac2O (1.0 mL) was added to a solution of 144 (28.4 mg, 0.06 mmol) in pyridine (2.0 mL) and the whole was stirred at rt for 2 days. The same work-up and purification as Entry 1 afforded 145b (31.8 mg, 94%). 145a: mp 180—182 °C (colorless powder, recrystallized from EtOAc–hexane). IR (KBr): 3434, 2927, 1731, 1668, 1608, 1548, 1500, 1244, 1146, 1024 cm-1. 1H-NMR (DMSO-d6) δ: 1.09 (1H, td, J=3.3, 13.2 Hz), 1.17—1.41 (3H, m), 1.50—1.58 (1H, m), 1.62 (1H, q, J=11.9 Hz), 1.70—1.78 (2H, m), 1.84 (1H, t, J=10.8 Hz), 2.03 (3H, s), 2.14—2.19 (1H, m), 2.26—2.30 (2H, m), 2.43—2.53 (1H, m), 2.69—2.64 (1H, m), 2.76 (1H, dd, J=10.8, 3.5 Hz), 2.83 (1H, brd, J=10.8 Hz), 3.42 (3H, s), 3.64 (3H, s), 4.10—4.14 (1H, m), 4.21 (1H, d, J=4.6 Hz, disappeared on addition of D2O), 6.84 (1H, d, J=8.8 Hz), 7.07 (1H, td, J=1.2, 7.5 Hz), 7.13 (1H, brd, J=7.5 Hz), 7.25 (1H, td, J=1.2, 7.5 Hz), 7.46 (1H, dd, J=7.8, 2.3 Hz), 7.51 (1H, brd, J=7.5 Hz), 7.72 (1H, brs), 9.56 (1H, s, disappeared on addition of D2O). MS m/z: 517 (M+). Anal. Calcd for C30H35N3O5·H2O: C, 67.27; H, 6.96; N, 7.84. Found: C, 67.29; H, 6.92; N, 7.54. [α]28D +249° (c=0.411, MeOH). 145b: mp 218—220 °C (orange prisms, recrystallized from EtOAc–hexane). IR (KBr): 3430, 2927, 1739, 1664, 1610, 1500, 1244, 1025 cm-1. 1H-NMR (DMSO-d6) δ: 1.05—1.14 (1H, m), 1.19—1.45 (3H, m), 1.63—1.75 (3H, m), 1.79—1.86 (1H, m), 1.87—1.93 (1H, m), 1.94 (3H, s), 2.03 (3H, s), 2.06—2.12 (1H, m), 2.30 (1H, t, J=12.9 Hz), 2.38—2.55 (2H, m), 2.69 (1H, d, J=11.7 Hz), 2.80 (1H, d, J=8.3 Hz), 2.82—3.15 (1H, m), 3.40 (3H, s), 3.63 (3H, s), 5.28 (1H, brd, J=2.7 Hz), 6.84 (1H, d, J=8.8 Hz), 7.08 (1H, t, J=7.7 Hz), 7.13 (1H, brd, J=7.7 Hz), 7.25 (1H, t, J=7.7 Hz), 7.47 (1H, dd, J=8.8, 2.1 Hz), 7.52 (1H, d, J=7.7 Hz), 7.72 (1H, brs), 9.58 (1H, brs, disappeared on addition of D2O). MS m/z: 559 (M+). Anal. Calcd for C32H37N3O6·1/2H2O: C, 67.59; H, 6.74; N, 7.39. Found: C, 67.38; H, 6.57; N, 7.42. [α]24D +215° (c=0.204, CHCl3).
7α-(5-Benzoylamino-2-methoxyphenyl)-7H-yohimbine (146) from 144 — Benzoyl chloride (0.02 mL, 0.16 mmol) was added to a solution of 144 (37.0 mg, 0.08 mmol) in pyridine (5.0 mL) and the mixture was stirred at rt for 1 h. After adding H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (95:5, v/v) to give 146 (38.8 mg, 83%). 146: mp 191—193 °C (decomp., colorless prisms, recrystallized from CHCl3–hexane). IR (KBr): 3435, 2927, 1732, 1649, 1500, 1111, 1026 cm-1. 1H-NMR (DMSO-d6) δ: 1.13 (1H, td, J=13.2, 3.4 Hz), 1.19—1.59 (4H, m), 1.66 (1H, q, J=11.8 Hz), 1.73—1.92 (2H, m), 2.19 (1H, d, J=12.7 Hz), 2.25—2.37 (2H, m), 2.39—2.55 (1H, m), 2.70 (1H, brd, J=11.8 Hz), 2.78 (1H, dd, J=10.9, 3.1 Hz), 2.84—3.00 (1H, m), 3.14 (1H, brd, J=13.7 Hz), 3.44 (3H, brs), 3.65 (3H, s), 4.09 (1H, brs, disappeared on addition of D2O), 4.13 (1H, brs), 6.90 (1H, d, J=8.8 Hz), 7.08 (1H, td, J=7.5, 1.1 Hz), 7.17 (1H, brd, J=7.5 Hz), 7.25 (1H, td, J=7.5, 1.1 Hz), 7.48—7.58 (4H, m), 7.68 (1H, dd, J=8.8, 2.4 Hz), 7.90—8.00 (3H, m), 9.86 (1H, brs, disappeared on addition of D2O). MS m/z: 579 (M+). Anal. Calcd for C35H37N3O5·H2O: C, 70.33; H, 6.58; N, 7.03. Found: C, 70.30; H, 6.36; N, 7.16. [α]28D +239° (c=0.232, MeOH).
7α-(5-Benzenesulfonylamino-2-methoxyphenyl)-7H-yohimbine (147) from 144 — Benzene sulfonyl chloride (0.03 mL, 0.24 mmol) was added to a solution of 144 (54.8 mg, 0.12 mmol) in pyridine (6.0 mL) and the mixture was stirred at rt for 20 min. After adding H2O, the whole was extracted with CHCl3–MeOH (97:3, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (95:5, v/v) to give 147 (68.0 mg, 95%). 147: mp 277—279 °C (decomp., colorless prisms, recrystallized from MeOH). IR (KBr): 3388, 2924, 1738, 1500, 1460, 1441, 1163 cm-1. 1H-NMR (DMSO-d6) δ: 1.03 (1H, td, J=13.4, 3.2 Hz), 1.18—1.32 (2H, m), 1.40 (1H, qd, J=12.5, 3.2 Hz), 1.50—1.64 (2H, m), 1.70—1.82 (2H, m), 2.01 (1H, t, J=11.8 Hz), 2.13 (1H, td, J=12.5, 3.2 Hz), 2.26 (1H, dd, J=11.8, 2.4 Hz), 2.45—2.52 (1H, m), 2.56 (1H, d, J=11.8 Hz), 2.67 (1H, d, J=11.0 Hz), 2.73 (1H, dd, J=11.0, 3.2 Hz), 2.80—3.00 (1H, m), 3.41 (3H, s), 3.65 (3H, s), 4.13 (2H, brs, disappeared 1H, on addition of D2O), 6.80 (1H, d, J=8.5 Hz), 6.92 (1H, brd, J=7.6 Hz), 7.00 (1H, dd, J=8.5, 2.4 Hz), 7.04 (1H, t, J=7.6 Hz), 7.13 (1H, brs), 7.23 (1H, td, J=7.6, 1.2 Hz), 7.48 (1H, t, J=7.6 Hz), 7.53 (2H, t, J=7.6 Hz), 7.60 (1H, tt, J=7.6, 1.7 Hz), 7.73 (2H, dd, J=7.6, 1.7 Hz), 9.55 (1H, brs, disappeared on addition of D2O). MS m/z: 615 (M+). Anal. Calcd for C34H37N3O6S·1/2H2O: C, 65.84; H, 6.09; N, 6.77. Found: C, 65.91; H, 6.12; N, 6.75. [α]25D +223° (c=0.200, MeOH).
7α-(5-Mesylamino-2-methoxyphenyl)-7H-yohimbine (148) from 144 — MsCl (0.014 mL, 0.18 mmol) was added to a solution of 144 (42.3 mg, 0.09 mmol) in pyridine (5.0 mL) and the whole was stirred at rt for 40 min. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with EtOAc–MeOH (93:7, v/v) to give 148 (48.1 mg, 95%). 148: mp 183.0—185.0 °C (colorless fine prisms, recrystallized from MeOH). IR (KBr): 3438, 1732, 1498, 1327, 1146 cm-1. 1H-NMR (DMSO-d6) δ: 1.12 (1H, td, J=13.3, 3.4 Hz), 1.18—1.34 (2H, m), 1.37 (1H, qd, J=12.3, 3.4 Hz), 1.50—1.58 (1H, m), 1.65 (1H, q, J=11. 5 Hz), 1.71—1.80 (2H, m), 1.83—1.91 (1H, m), 2.16 (1H, d, J=12.3Hz), 2.28 (1H, dd, J=11.5, 2.0 Hz), 2.43—2.52 (1H, m), 2.65—2.71 (1H, m), 2.77 (1H, brd, J=11.5 Hz), 2.80—3.00 [1H, m, on addition of D2O, it changed to 2.80—2.90 (1H, m)], 2.80—3.00 [3H, m, changed to 2.94 (3H, s) on addition of D2O], 3.11 (1H, brd, J=13.3 Hz), 3.44 (3H, s), 3.65 (3H, s), 4.08 (1H, brs, disappeared on addition of D2O), 4.13 (1H, brs), 6.89 (1H, d, J=8.9 Hz), 7.07 (1H, t, J=7.2 Hz), 7.13 (1H, d, J=7.2 Hz), 7.14 (1H, dd, J=8.9, 2.7 Hz), 7.25 (1H, td, J=7.2, 1.2 Hz), 7.42 (1H, brs), 7.52 (1H, d, J=7.2 Hz), 9.05 (1H, brs, disappeared on addition of D2O). 13C-NMR (CDCl3) δ: 186.12 (C), 175.25 (C), 155.61 (C), 154.80 (C), 143.70 (C), 129.97 (C), 128.19 (C), 127.80 (CH), 125.36 (CH), 123.68 (CH), 123.10 (CH), 121.98 (CH), 120.74 (CH), 112.92 (CH), 67.15 (CH), 61.52 (CH), 61.35 (CH2), 59.22 (C), 55.59 (CH3), 52.27 (CH), 51.86 (CH3), 51.34 (CH2), 40.08 (CH), 39.00 (CH3), 36.89 (CH2), 36.04 (CH), 31.50 (CH2), 31.40 (CH2), 23.08 (CH2). Anal. Calcd for C29H35N3O6S·3/4H2O: C, 61.41; H, 6.49; N, 7.41. Found: C, 61.48; H, 6.39; N, 7.49. [α]25D +204° (c=0.250, CHCl3).
(2α,7α)- (150) and (2β,7β)-24,26-Dinitrobenzofurano[2,3-n]yohimbine (151) from 53b — A solution of K2CO3 (76.7 mg, 0.55 mmol) and 2,4-dinitrofluorobenzene (62.8 mg, 0.34 mmol) in DMF (1.0 mL) was added to a solution of 53b (102.8 mg, 0.28 mmol) in DMF (4.0 mL) and the whole was stirred at –8 °C for 70 min. After addition of H2O, the whole was extracted with EtOAc. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (99:1, v/v) to give 150 (106.0 mg, 71%) and 151 (35.2 mg, 24%). 150: yellow viscous oil. IR (film): 3481, 3286, 2934, 1701, 1622, 1606, 1535, 1523, 1473, 1437, 1375, 910 cm-1. 1H-NMR (CDCl3) δ: 1.34—1.39 (1H, m), 1.44—1.54 (4H, m), 1.88—1.91 (5H, m), 2.15 (1H, dt, J=12.4, 2.4 Hz), 2.22 (1H, dd, J=11.5, 2.4 Hz), 2.33 (1H, dd, J=11.5, 1.7 Hz), 2.67—2.70 (1H, m), 2.75—2.78 (1H, m), 2.87 (1H, dd, J=11.5, 1.7 Hz), 3.11 (1H, disappeared on addition of D2O), 3.84 (3H, s), 4.22 (1H, d, J=0.7 Hz), 5.41 (1H, s, disappeared on addition of D2O), 6.84 (1H, dd, J=8.0, 0.7 Hz), 6.85 (1H, td, J=8.0, 0.7 Hz), 6.99 (1H, dd, J=8.0, 0.7 Hz), 7.15 (1H, dd, J=8.0, 0.7 Hz), 8.48 (1H, d, J=2.2 Hz), 8.89 (1H, d, J=2.2 Hz). 13C-NMR (CDCl3) δ: 175.17 (C), 158.84 (C), 144.70 (C), 141.06 (C), 138.25 (C), 131.69 (C), 131.63 (C), 129.17 (CH), 122.59 (CH), 122.35 (CH), 121.76 (CH), 121.58 (CH), 116.78 (C), 115.15 (CH), 66.50 (CH), 64.76 (CH), 61.39 (CH2), 54.86 (C), 52.34 (CH3), 52.17 (CH), 51.44 (CH2), 40.08 (CH), 36.07 (CH), 32.01 (CH2), 31.06 (CH2), 29.73 (CH2), 22.97 (CH2). HR–MS m/z: Calcd for C27H28N4O8: 536.1907. Found: 536.1903. [α]28D +258° (c=0.106, MeOH). 151: yellow viscous oil. IR (film): 3556, 3381, 2935, 1716, 1619, 1608, 1535, 1336, 738 cm-1. 1H-NMR (CDCl3) δ: 1.25—1.60 (5H, m), 1.81—2.00 (5H, m), 2.16—2.22 (2H, m), 2.39—2.50 (2H, m), 2.64—2.67 (1H, m), 2.69 (1H, brs, disappeared on addition of D2O), 2.77—2.81 (2H, m), 3.82 (3H, s), 5.14 (1H, s, disappeared on addition of D2O), 6.67 (1H, d, J=7.8 Hz), 6.89 (1H, td, J=7.8, 1.0 Hz), 7.14 (1H, td, J=7.8, 1.0 Hz), 7.31 (1H, d, J=7.8 Hz), 8.21 (1H, dd, J=2.4, 1.5 Hz), 8.85 (1H, dd, J=2.4, 1.5 Hz). 13C-NMR (CDCl3) δ: 175.44 (C), 157.82 (C), 147.04 (C), 141.32 (C), 141.03 (C), 131.82 (C), 129.58 (CH), 128.37 (C), 122.78 (CH), 122.59 (CH), 121.30 (CH), 120.77 (CH), 114.19 (C), 109.92 (CH), 66.94 (CH), 64.13 (CH), 61.40 (CH2), 54.80 (C), 52.06 (CH3), 52.04 (CH), 50.00 (CH2), 39.38 (CH), 35.70 (CH), 31.25 (CH2), 30.22 (CH2), 29.07 (CH2), 22.89 (CH2). HR–MS m/z: Calcd for C27H28N4O8: 536.1907. Found: 536.1902. [α]28D +121° (c=0.101, MeOH).
(2α,7α)-1-Acetyl- (152) and -17-Acetoxy-24,26-dinitrobenzofurano[2,3-n]yohimbine (153) from 150 — Ac2O (1.0 mL) was added to a solution of 150 (40.4 mg, 0.08 mmol) in pyridine (2.0 mL) and the whole was stirred at 60 °C for 4 h. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with EtOAc–hexane (2:1, v/v) to give 153 (26.1 mg, 59%) and 152 (2 mg, 4%). 153: mp 200—201 °C (decomp., yellow powder, recrystallized from EtOAc–hexane). IR (KBr): 3423, 1749, 1728, 1599, 1572, 1543, 1373, 1355 cm-1. 1H-NMR (CDCl3) δ: 1.23—1.58 (5H, m), 1.63 (1H, m), 1.85—2.07 (4H, m), 2.02 (3H, s), 2.26—2.33 (1H, m), 2.36—2.45 (2H, m), 2.65—2.81 (2H, m), 2.90 (1H, dd, J=11.1, 3.1 Hz), 3.71 (3H, s), 5.42 (1H, brs, disappeared on addition of D2O), 5.43 (1H, d, J=2.4 Hz), 6.83 (1H, d, J=7.6 Hz), 6.85 (1H, t, J=7.6 Hz), 6.99 (1H, d, J=7.6 Hz), 7.15 (1H, t, J=7.6 Hz), 8.49 (1H, d, J=2.3 Hz), 8.90 (1H, d, J=2.3 Hz). 13C-NMR (CDCl3) δ: 171.65 (C), 169.99 (C), 158.87 (C), 144.76 (C), 141.02 (C), 138.31 (C), 131.71 (C), 131.52 (C), 129.22 (CH), 122.51 (CH), 122.33 (CH), 121.68 (CH), 121.53 (CH), 116.77 (C), 111.14 (CH), 69.26 (CH), 64.79 (CH), 61.25 (CH2), 54.83 (C), 52.01 (CH3), 51.42 (CH2), 51.36 (CH), 39.45 (CH), 36.16 (CH), 31.96 (CH2), 29.70 (CH2), 29.60 (CH2), 23.68 (CH2), 20.94 (CH3). MS m/z: 578 (M+). Anal. Calcd for C29H30N4O9·1/2H2O: C, 59.28; H, 5.32; N, 9.54. Found: C, 59.19; H, 5.33; N, 9.23. [α]24D +148° (c=0.220, CHCl3). 153: yellow viscous oil. IR (film): 1743, 1673, 1622, 1533, 1373, 1338 cm-1. 1H-NMR (CDCl3) δ: 1.02 (1H, dd, J=24.7, 12.7 Hz), 1.20—1.36 (2H, m), 1.37—1.47 (1H, m), 1.49—1.66 (2H, m), 1.94—2.03 (2H, m), 2.06 (3H, s), 2.21—2.27 (2H, m), 2.56—2.69 (2H, m), 2.67 (3H, s), 2.74—2.89 (1H, m), 2.83—2.91 (2H, m), 3.63 (3H, s), 4.25 (1H, dd, J=12.8, 2.1 Hz), 5.37 (1H, d, J=2.7 Hz), 7.19 (1H, t, J=7.6 Hz), 7.29 (1H, t, J=7.6 Hz), 7.42 (1H, d, J=7.8 Hz), 8.14 (1H, d, J=7.8 Hz), 8.18 (1H, d, J=2.3 Hz), 8.91 (1H, d, J=2.3 Hz). 13C-NMR (CDCl3) δ: 171.30 (C), 170.36 (C), 169.96 (C), 155.86 (C), 143.10 (C), 142.45 (C), 140.51 (C), 132.36 (C), 130.05 (CH), 129.00 (C), 125.00 (CH), 123.03 (CH), 121.57 (CH), 121.56 (CH), 117.45 (CH), 111.74 (C), 69.12 (CH), 62.77 (CH), 60.35 (CH2), 54.33 (C), 51.90 (CH3), 50.70 (CH), 41.10 (CH2), 38.71 (CH), 33.30 (CH), 32.34 (CH2), 29.74 (CH2), 25.51 (CH2), 24.52 (CH3), 23.11 (CH2), 21.02 (CH3). HR–MS m/z: Calcd for C31H32N4O10: 620.2118. Found: 620.2124. [α]31D –38.2° (c=0.192, CHCl3).
Scheme 17.
Synthesis of new yohimbine derivatives (4).
7α-(2-Methoxy-3,5-dinitrophenyl)-7H-yohimbine (154) from 150 — Excess CH2N2 in Et2O was added to a solution of 150 (5.0 mg, 0.008 mmol) in MeOH (0.5 mL) and the whole was stirred at rt for 30 min. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (95:5, v/v) to give 154 (5.0 mg, 98%). 154: yellow oil. IR (KBr): 2927, 1736, 1595, 1541, 1456, 1342, 1267, 1207 cm-1. 1H-NMR (CDCl3) δ: 1.13—1.36 (5H, m), 1.51—1.63 (2H, m), 1.72—1.76 (2H, m), 1.91 (1H, d, J=10.5 Hz), 2.12 (1H, d, J=10.8 Hz), 2.30 (1H, dd, J=10.5, 2.4 Hz), 2.33 (1H, t, J=10.0 Hz), 2.76 (2H, dd, J=10.5, 2.4 Hz), 2.84 (1H, brd, J=10.0 Hz), 3.02 (3H, brs), 3.62 (3H, s), 4.10 (1H, brt, J=2.4 Hz), 4.55 (1H, d, J=2.8 Hz, disappeared on addition of D2O), 7.18 (1H, t, J=7.6 Hz), 7.24 (1H, d, J=7.6 Hz), 7.36 (1H, t, J=7.6 Hz), 7.64 (1H, d, J=7.6 Hz), 8.75 (1H, d, J=2.4 Hz), 8.92 (1H, brs). 13C-NMR (CD3OD) δ: 186.15 C), 175.24 (C), 157.33 (C), 155.44 (C), 144.48 (C), 144.38 (C), 144.33 (C), 137.81 (C), 130.01 (CH), 128.51 (CH), 127.69 (CH), 123.72 (CH), 122.37 (CH), 121.82 (CH), 68.52 (CH), 62.92 (CH), 62.65 (CH3), 62.04 (CH2), 60.94 (C), 53.94 (CH), 52.09 (CH3), 51.85 (CH2), 41.22 (CH), 37.71 (CH2), 36.54 (CH), 33.40 (CH2), 32.56 (CH2), 24.18 (CH2). HR–MS m/z: Calcd for C28H30N4O8: 550.2063. Found: 550.2059. [α]26D +258° (c=0.101, MeOH).
(S)-(4aα,9aα)- (159) and (S)-(4aβ,9aβ)-3β-Methoxycarbonyl-11,13-dinitro-1,2,3,4-tetrahydro-9H- benzofurano[2,3-m]-β-carboline (160) from 155[77] — A solution of K2CO3 (168.5 mg, 1.2 mmol) and 2,4-dinitrofluorobenzene (84.6 mg, 0.45 mmol) in DMF (3.0 mL) was added to a solution of 155 (100.0 mg, 0.41 mmol) in DMF (7.0 mL) and the whole was stirred at 0 °C for 30 min. After addition of H2O, the whole was extracted with EtOAc. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with EtOAc–hexane–Et2O (1:1:1, v/v) to give 159 (77.5 mg, 46%), 160 (58.0 mg, 35%), and unreacted 155 (7.1 mg, 7%). 159: mp 158.0—160.0 °C (yellow needles, recrystallized from Et2O–hexane). IR (KBr): 3392, 1736, 1620, 1608, 1533, 1473, 1437, 1336 cm-1. 1H-NMR (CDCl3) δ: 2.02 (1H, dd, J=14.4, 11.4 Hz), 2.04 (1H, brs, disappeared on addition of D2O), 3.02 (1H, dd, J=14.4, 4.7 Hz), 3.23 (1H, d, J=14.2 Hz), 3.38 (1H, dd, J=11.4, 4.4 Hz), 3.69 (1H, d, J=14.2 Hz), 3.71 (3H, s), 5.41 (1H, brs, disappeared on addition of D2O), 6.80 (1H, d, J=7.8 Hz), 6.84 (1H, t, J=7.8 Hz), 7.12 (1H, d, J=7.8 Hz), 7.16 (1H, t, J=7.8 Hz), 8.51 (1H, dd, J=2.0, 1.0 Hz), 8.90 (1H, dd, J=2.0, 1.0 Hz). 13C-NMR (CDCl3) δ: 172.66 (C), 159.04 (C), 145.78 (C), 141.04 (C), 138.00 (C), 131.25 (C), 130.33 (C), 129.61 (CH), 122.78 (CH), 122.71 (CH), 121.88 (CH), 121.24 (CH), 113.33 (C), 110.54 (CH), 54.69 (C), 52.93 (CH), 52.47 (CH3), 48.65 (CH2), 34.50 (CH2). Anal. Calcd for C19H16N4O7·1/8H2O: C, 55.03; H, 3.95; N, 13.41. Found: C, 55.13; H, 3.94; N, 13.19. [α]28D +206° (c=0.111, MeOH). 160: yellow viscose oil. IR (film): 3402, 1732, 1618, 1608, 1533, 1473, 1433, 1333, 1255, 744 cm-1. 1H-NMR (CDCl3) δ: 2.21 (1H, brs, disappeared on addition of D2O), 2.55 (1H, dd, J=14.6, 6.6 Hz), 2.83 (1H, dd, J=14.6, 6.1 Hz), 3.39 (1H, d, J=14.4 Hz), 3.57 (1H, d, J=14.4 Hz), 3.60 (3H, s), 3.69 (1H, t, J=6.4 Hz), 5.17 (1H, s, disappeared on addition of D2O), 6.75 (1H, d, J=8.1 Hz), 6.89 (1H, td, J=8.1, 0.7 Hz), 7.17 (1H, td, J=8.1, 0.7 Hz), 7.24 (1H, d, J=8.1 Hz), 8.36 (1H, d, J=2.2 Hz), 8.87 (1H, d, J=2.2 Hz). 13C-NMR (CDCl3) δ: 173.22 (C), 158.70 (C), 146.70 (C), 141.03 (C), 138.80 (C), 131.45 (C), 129.82 (CH), 129.24 (C), 123.57 (CH), 122.78 (CH), 121.82 (CH), 121.07 (CH), 113.09 (C), 110.14 (CH), 54.49 (C), 52.39 (CH3), 51.51 (CH), 46.57 (CH2), 32.30 (CH2). HR–MS m/z: Calcd for C19H16N4O7: 412.1019. Found: 412.1024. [α]27D –171° (c=0.100, MeOH).
(S)-(4aα,9aα)-3β-Methoxycarbonyl-Nb-Methyl- (162), -Na-methyl- (161), and -Na,Nb-dimethyl- 11,13-dinitro-1,2,3,4-tetrahydro-9H-benzofurano[2,3-m]-β-carboline (163) from 159 — Excess CH2N2 in Et2O was added to a solution of 159 (33.0 mg, 0.08 mmol) in MeOH (1.5 mL) and the whole was stirred at rt for 145 min. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 162 (5.4 mg, 16%), 161 (9.2 mg, 27%), 163 (6.4 mg, 18%), and unreacted 159 (5.4 mg, 16%). 162: pale viscose oil. IR (film): 3380, 1743, 1621, 1610, 1373, 1336 cm-1. 1H-NMR (DMSO-d6:D2O=5:1, v/v) δ: 2.26 (1H, dd, J=15.0, 11.7 Hz), 2.37 (3H, s), 2.86 (1H, dd, J=15.0, 5.0 Hz), 3.02 (1H, d, J=14.2 Hz), 3.11 (1H, dd, J=11.7, 5.0 Hz), 3.70 (3H, s), 3.70 (1H, m), 4.52—4.81 (1H, brs, disappeared on addition of D2O), 6.77 (1H, d, J=8.3 Hz), 6.85 (1H, t, J=8.3 Hz), 7.13 (1H, d, J=8.3 Hz), 7,16 (1H, t, J=8.3 Hz), 8.44 (1H, d, J=2.5 Hz), 8.89 (1H, d, J=2.5 Hz). 13C-NMR (CDCl3) δ: 172.30 (C), 159.35 (C), 146.16 (C), 140.95 (C), 138.36 (C), 130.91 (C), 129.68 (CH), 129.63 (C), 122.86 (CH), 122.36 (CH), 121.71 (CH), 121.07 (CH), 114.78 (C), 110.50 (CH), 59.85 (CH), 55.79 (CH2), 54.19 (C), 52.04 (CH3), 42.21 (CH3), 33.77 (CH2). HR–MS (FAB+) m/z: Calcd for C20H18N4O7: 426.1175. Found: 427.1257. [α]31D +154° (c=0.234, CHCl3). 161: yellow oil. IR (film): 2952, 1736, 1620, 1606, 1489, 1435, 1338, 1246, 756 cm-1. 1H-NMR (CDCl3) δ: 2.01 (1H, brs, disappeared on addition of D2O), 2.02 (1H, dd, J=14.4, 11.2 Hz), 2.99 (1H, dd, J=14.4, 5.4 Hz), 3.14 (3H, s), 3.24 (1H, d, J=14.4 Hz), 3.42 (1H, dd, J=11.2, 5.4 Hz), 3.71 (3H, s), 3.75 (1H, d, J=14.4 Hz), 6.67 (1H, d, J=7.8 Hz), 6.83 (1H, d, J=7.8 Hz), 7.10 (1H, d, J=7.8 Hz), 7.21 (1H, t, J=7.8 Hz), 8.47 (1H, d, J=2.4 Hz), 8.87 (1H, d, J=2.4 Hz). 13C-NMR (CDCl3) δ: 172.98 (C), 159.40 (C), 148.41 (C), 140.81 (C), 138.45 (C), 131.17 (C), 129.85 (C), 129.71 (CH), 122.36 (CH), 122.35 (CH), 121.71 (CH), 120.35 (CH), 116.24 (C), 108.17 (CH), 54.26 (C), 52.60 (CH), 52.41 (CH3), 46.12 (CH2), 33.57 (CH2), 28.55 (CH3). HR–MS m/z: Calcd for C20H18N4O7: 426.1175. Found: 426.1165. [α]26D +42.9° (c=0.107, MeOH). 163: yellow oil. IR (film): 2952, 1736, 1620, 1608, 1533, 1489, 1435, 1335, 1255, 1007, 744, 594 cm-1. 1H-NMR (CDCl3) δ: 2.14 (1H, dd, J=14.7, 11.0 Hz), 2.36 (3H, s), 2.88 (1H, dd, J=14.7, 6.1 Hz), 3.10 (1H, d, J=14.0 Hz), 3.12 (3H, s), 3.21 (1H, dd, J=11.0, 6.1 Hz), 3.70 (3H, s), 3.77 (1H, d, J=14.0 Hz), 6.61 (1H, d, J=7.8 Hz), 6.79 (1H, t, J=7.8 Hz), 7.10 (1H, d, J=7.8 Hz), 7.19 (1H, t, J=7.8 Hz), 8.39 (1H, dd, J=2.2, 0.7 Hz), 8.85 (1H, dd, J=2.2, 0.7 Hz). 13C-NMR (CDCl3) δ: 172.50 (C), 159.95 (C), 148.56 (C), 140.60 (C), 138.88(C), 130.61(C), 129.68 (CH), 129.36 (C), 122.54 (CH), 121.75 (CH), 121.43 (CH), 119.95 (CH), 117.92 (C), 107.61 (CH), 58.98 (CH), 53.64 (C), 53.56 (CH2), 51.83 (CH3), 41.71 (CH3), 33.04 (CH2), 28.53 (CH3). HR–MS m/z: Calcd for C21H20N4O7: 440.1332. Found: 440.1334. [α]27D +18.8° (c=0.110, MeOH).
Scheme 18.
Synthesis of new β-carboline derivatives (1).
162 from 159 — Me2SO4 (12.3 mg, 0.09 mmol) in MeOH (0.5 mL) was added to a solution of 159 (11.1 mg, 0.027 mmol) in MeOH (0.5 mL) and K2CO3 (13.4 mg, 13.0 mmol) at 0°C and the mixture was stirred at rt for 19 h. After addition of H2O, the whole was extracted with EtOAc. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (97:3, v/v) to give 162 (6.3 mg, 55%) and unreacted 159 (1.9 mg, 17%).
(S)-(4aα,9aα)-Nb-Acetyl- (164a) and -Na,Nb-diacetyl-3β-methoxycarbonyl-11,13-dinitro-1,2,3,4- tetrahydro-9H-benzofurano[2,3-m]-β-carboline (164b) from 159 — Ac2O (1.0 mL) was added to a solution of 159 (40.0 mg, 0.09 mmol) in pyridine (2.0 mL) and the whole was stirred at rt for 3 h. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 164a (30.8 mg, 59%) and 164b (14.6 mg, 30%). 164a: mp 198—200 °C (yellow prisms, recrystallized from EtOAc). IR (KBr): 3292, 1647, 1620, 1610, 1533, 1336 cm-1. 1H-NMR (CDCl3) δ: 2.19 (3H, s), 2.24 (1H, dd, J=14.8, 12.7 Hz), 3.14 (1H, dd, J=12.6, 6.7 Hz), 3.72 (3H, s), 3.88 (1H, d, J=15.1 Hz), 4.42 (1H, dd, J=12.6, 6.7 Hz), 4.53 (1H, d, J=15.1 Hz), 5.27 (1H, s, disappeared on addition of D2O), 6.74 (1H, d, J=8.1 Hz), 6.89 (1H, t, J=7.6 Hz), 7.18 (1H, t, J=7.8 Hz), 7.25 (1H, d, J=8.1 Hz), 8.46 (1H, d, J=2.2 Hz), 8.86 (1H, d, J=2.2 Hz). 13C-NMR (CDCl3) δ: 170.97 (C), 170.84 (C), 158.50 (C), 146.28 (C), 141.53 (C), 136.52 (C), 130.89 (C), 130.18 (CH), 128.16 (C), 123.36 (CH), 122.85 (CH), 122.17 (CH), 121.25 (CH), 113.31 (C), 110.26 (CH), 55.88 (C), 52.63 (CH3), 50.98 (CH), 48.31 (CH2), 31.74 (CH2), 20.98 (CH3). Anal. Calcd for C21H18N4O8·1/2EtOAc: C, 55.42; H, 4.45; N, 11.24. Found: C,55.22; H, 4.45; N, 11.03. [α]31D +63.1° (c=0.194, MeOH). 164b: yellow viscous oil. IR (film): 1747, 1685, 1655, 1614, 1541, 1340 cm-1. 1H-NMR (CDCl3) δ: 2.27 (3H, s), 2.28 (1H, dd, J=14.9, 12.9 Hz), 2.68 (3H, s), 3.16 (1H, dd, J=15.0, 6.7 Hz), 3.72 (3H, s), 3.95 (1H, d, J=15.4 Hz), 4.40 (1H, dd, J=12.9, 6.6 Hz), 5.26 (1H, brs), 7.19 (1H, t, J=7.6 Hz), 7.26 (1H, d, J=7.6 Hz), 7.35 (1H, t, J=7.9 Hz), 7.39 (1H, d, J=7.6 Hz), 8.49 (1H, d, J=2.2 Hz), 8.90(1H, d, J=2.2 Hz). 13C-NMR (CDCl3) δ: 170.89 (C), 170.66 (C), 169.04 (C), 157.24 (C), 142.52 (C), 141.49 (C), 135.21 (C), 131.63 (C), 130.43 (CH), 130.31 (C), 125.60 (CH), 123.17 (CH), 122.78 (CH), 122.56 (CH), 116.76 (CH), 110.67 (C), 56.16 (C), 52.59(CH3), 50.69 (CH), 47.74 (CH2), 32.66 (CH2), 25.44 (CH3), 20.83 (CH3). HR–MS m/z: Calcd for C23H20N4O9: 496.1231. Found: 496.1232. [α]31D +89.3° (c=0.273, CHCl3).
Scheme 19.
Synthesis of new β-carboline derivatives (2).
164b from 164a — Ac2O (0.5 mL) was added to a solution of 164a (10.0 mg, 0.02 mmol) in pyridine (1.0 mL) and the whole was refluxed for 3 h. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 164b (7.7 mg, 71%).
164a from 164b — Sat. NaHCO3 (1.0 mL) was added to a solution of 164b (16.6 mg, 0.03 mmol) in MeOH (4.0 mL) and the whole was stirred at rt for 100 min. After addition of H2O, the whole was extracted with CHCl3. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 164a (14.7 mg, 97%).
(S)-(4aα,9aα)-Nb-Chloroacetyl- (165a) and -Na,Nb-bis(chloroacetyl)-3β-methoxycarbonyl-11,13-di- nitro-1,2,3,4-tetrahydro-9H-benzofurano[2,3-m]-β-carboline (165b) from 159 — Chloroacetyl chloride (0.012 mL, 0.18 mmol) and Et3N (0.15 mL) was added to a solution of 159 (60.8 mg, 0.15 mmol) in CHCl3 (1.5 mL) and the whole was stirred at rt for 20 min. The solvent was evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3 to give 163a (17.2 mg, 24%), 165b (32.8 mg, 39%), and unreacted 159 (4.3 mg, 7%). 165a: mp 198—200 °C (yellow prisms, recrystallized from EtOAc). IR (KBr): 3382, 1749, 1729, 1668, 1654, 1619, 1610, 1373, 1336 cm-1. 1H-NMR (CDCl3) δ: 2.31 (1H, dd, J=14.9, 12.6 Hz), 3.17 (1H, dd, J=14.9, 6.5 Hz), 3.73 (3H, s), 3.95 (1H, d, J=15.1 Hz), 4.11 (1H, d, J=12.7 Hz), 4.29 (1H, d, J=12.7 Hz), 4.42 (1H, dd, J=12.6, 6.5 Hz), 4.61 (1H, d, J=15.1 Hz), 5.40 (1H, brs, disappeared on addition of D2O), 6.75 (1H, d, J=7.8 Hz), 6.89 (1H, t, J=7.8 Hz), 7.18 (1H, t, J=7.8 Hz), 7.24—7.28 (1H, m), 8.48 (1H, d, J=2.3 Hz), 8.85 (1H, d, J=2.3 Hz). 13C-NMR (CDCl3) δ: 170.21 (C), 166.82 (C), 158.16 (C), 146.09 (C), 141.75 (C), 136.20 (C), 131.17 (C), 130.33 (CH), 127.97 (C), 123.30 (CH), 122.85 (CH), 122.26 (CH), 121.50 (CH), 112.59 (C), 110.40 (CH), 55.88 (C), 52.81 (CH), 51.61 (CH3), 48.13 (CH2), 40.17 (CH2), 31.60 (CH2). HR–MS m/z: Calcd for C21H17N4O8Cl: 488.0734, 490.0705. Found: 488.0756, 490.0707. [α]28D +135° (c=0.141, DMF). 165b: mp 223—224 °C (pale yellow prisms, recrystallized from EtOAc). IR (KBr): 1747, 1735, 1662, 1541, 1373, 1340 cm-1. 1H-NMR (CDCl3) δ: 2.30 (1H, dd, J=15.3, 12.8 Hz), 3.13 (1H, dd, J=15.3, 6.1 Hz), 3.63 (3H, s), 3.98 (1H, d, J=15.9 Hz), 4.14 (1H, d, J=12.8 Hz), 4.23 (1H, d, J=13.1 Hz), 4.27 (1H, dd, J=12.8, 6.1 Hz), 4.42 (1H, d, J=12.8 Hz), 4.64 (1H, d, J=13.1 Hz), 5.26 (1H, d, J=15.9 Hz), 7.14—7.22 (1H, m), 7.31 (1H, t, J=7.8 Hz), 7.39 (1H, t, J=7.8 Hz), 7.76 1H, brd, J=7.8 Hz), 8.46 (1H, d, J=1.8 Hz), 8.81 (1H, d, J=1.8 Hz). 13C-NMR (CDCl3) δ: 170.90 (C), 167.15 (C), 165.56 (C), 156.52 (C), 142.97 (C), 140.39 (C), 134.65 (C), 132.04 (C), 130.77 (CH), 130.47 (C), 126.51 (CH), 123.47 (CH), 122.94 (CH), 122.74 (CH), 117.07 (CH), 109.90 (C), 56.20 (C), 52.79 (CH3), 51.59 (CH), 47.01 (CH2), 42.55 (CH2), 40.23 (CH2), 32.73 (CH3). Anal. Calcd for C23H18N4O9Cl2: C, 48.87; H, 3.21; N, 9.91. Found: C, 48.87; H, 3.29; N, 9.87. [α]30D +122° (c=0.200, DMF).
165b from 165a — Chloroacetyl chloride (54.8 mg, 0.49 mmol) and Et3N (0.1 mL) was added to a solution of 165a (11.7 mg, 0.02 mmol) in CHCl3 (2.0 mL) and the whole was stirred at rt for 20 min. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (99:1, v/v) to give 165b (17.9 mg, 88%).
165a from 165b — Sat. NaHCO3 (0.5 mL) was added to a solution of 165b (10.4 mg, 0.02 mmol) in MeOH (2.0 mL) and the whole was stirred at rt for 30 min. After addition of H2O, the whole was extracted with CHCl3–MeOH (95:5, v/v). The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was column-chromatographed on SiO2 with CHCl3–MeOH (99:1, v/v) to give 165a (7.5 mg, 83%).
In summary, we demonstrated further examples of rearrangement reactions based on 1-hydroxyindole chemistry and succeeded in the production of thus far unknown 7β- and 7α-heteroaryl yohimbines, and 4aα- and 4aβ-heteroaryl-1,2,3,4-tetrahydro-β-carboline derivatives. We hope these novel compounds reported in this text would become a new member of our group of biologically active compounds that we have created so far.[10]
23.3.2. Yohimbine α2-Blocker Test Method
Animals: Male Wistar rats were used in the present study. Animals were housed under controlled conditions (21–22ºC, relative humidity 50±5%). Food and water were freely available to all animals. This study was performed according to the Guideline for the Care and Use of Laboratory Animals of Toho University School of Pharmaceutical Sciences (which is accredited by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan), and the protocol of this study was approved by the Institutional Animal Care and Use Committee.
Preparation of rat thoracic aortic rings: Rats were killed by cervical dislocation and exsanguinated from the common carotid arteries. A section of the thoracic aorta between aortic arch and diaphragm was carefully removed and immersed in oxygenated Krebs-HEPES solution of the following composition (in mM): NaCl, 126.9; KCl, 5.9; CaCl2, 2.36; MgCl2, 1.18; HEPES, 10.03 and glucose, 11.8 (pH=7.4). The aorta was cleaned of loosely adhering fat and connective tissues and cut into ring segments about 2 mm in length. In this series of experiments, the endothelium was not removed.
Measurement of tension changes: The aortic tissue was then mounted using stainless steel hooks (outer diameter, 200 µm) under the resting tension of 2.0 g in a 5 mL organ bath (UC-5, UFER Medical Instrument, Kyoto, Japan) containing normal Tyrode’s solution (mM): NaCl, 158.3; KCl, 4.0; NaHCO3, 10.0; NaH2PO4:, 0.42; CaCl2, 2.0; MgCl2, 1.05 mM, glucose, 5.6), which was continuously gassed with 95% O2–5% CO2 being kept at 37±1ºC (pH=7.4). Tension changes of the muscle preparation were isometrically recorded with a force-displacement transducer (T7-8-240; Orientec, Tokyo, Japan; TB-612T, Nihon Kohden, Tokyo, Japan) connected to a carrier amplifier (AP-600G/AP-621G, Nihon Kohden, Tokyo, Japan; Signal Conditioner: Model MSC-2, Labo Support, Suita-City, Japan). Vascular preparations were equilibrated for 90 min in normal Tyrode’s solution, which was exchanged every 20–30 min. Before starting assessment of yohimbine derivatives, aortic preparations were contracted with isotonic high-KCl (80 mM) Tyrode’s solution (mM: NaCl, 82.3; KCl, 80.0; CaCl2, 2.0; MgCl2, 1.05; NaH2PO4, 0.42; NaHCO3, 10.0 and glucose, 5.6), in order to confirm the muscle normal contractility. After washing out, experiments were started after a subsequent 30 min equilibration period.
Assessment of relaxant potencies of yohimbine derivatives: Aortic ring preparations were contracted with an α2-adrenoceptor (α2-AR) agonist clonidine (10-7–10-6 M) in the presence of an NO synthase inhibitor nitro-l-arginine methyl ester (l-NAME, 10-4 M). When the sustained contraction induced by clonidine reached a steady-state level, yohimbine derivatives (10-5 M) were applied to the bath solution. When the relaxant effects of yohimbine derivatives reached their maximum level, yohimbine at 10-5 M was applied. The steady-state tension level before application of each yohimbine derivative and the tension level corresponding to yohimbine-induced maximum relaxation were defined as 0% and 100% relaxation, respectively. Relaxant potency of tested yohimbine derivatives was expressed as percentage relaxation to the maximum response to 10-5 M yohimbine.
Drugs: The following drugs were used in the present study: clonidine hydrochloride (Sigma-Aldrich, St. Louis, MO, USA); yohimbine hydrochloride (Wako Pure Chemical Industries, Ltd., Osaka, Japan); NG-nitro-l-arginine methyl ester hydrochloride (l-NAME) (Dojindo Laboratories, Kumamoto, Japan). Yohimbine derivatives tested in this study were dissolved in pure dimethyl sulfoxide (DMSO) at 10-2 M. Final DMSO concentrations in the bath medium did not exceed 0.1%, which did not affect the vascular responses. Other drugs were dissolved/diluted in/with distilled water. All drugs are expressed in molar concentrations (mol/L, M) in bathing solution.
Statistics: Data are presented as means±S.E.M. and n refers to the number of experiments.
23.3.3. Candidates for Curing Osteoporosis
We subjected 1-substituted 2,4,6-tribromotryptophan derivatives (69a-c) and -melatonin derivatives (72a-c) to the gills assay test (synthesis of chemicals are reported in Scheme 6). Happily, we discovered that 69c as a strong activator for osteoblast and suppressor for osteoclast. The compound 72c was also discovered to be strong promotor of osteoblasts and suppressor for osteoclasts.
The test compounds were synthesized as shown in Scheme 6. In the acute toxicity test of 1-benzyl- 2,4,6-tribromomelatonin (69c), according to the OECD (Europe) guidelines, 2 g per 1 kg of rat body weight was administered, and after 2 weeks of breeding, toxicity was not observed. Furthermore, no toxicity was observed in a mutagenicity test in which rats were administered 2 g per kg of body weight and evaluated after feeding for 2 weeks according to the OECD guidelines. Addition of S-9 mix (rat liver homogenate) did not change.[57,58]
Therefore, we were able to discover compounds 69c and 72c as potential therapeutic candidate compounds.[42,43,57,58] Even now, no effective therapeutic agent for osteoporosis that has an osteoblast-activating action is known. Combined use of 69c and 72c with VED #1 can not only treat osteoporosis, fractures, arthralgia, and dental treatment in humans, but also improve the bone metabolism of shrimp, shellfish, goldfish, ‘Nishikigoi’, farming, poultry, birds, and animals. It opens up the possibility of treating osteoporosis caused by aging in humans and guaranteeing a healthy life.
As noted previously in the safety test of VED #1, ‘medaka’ a kind of fish grew for 2 years and 6 months without any problems, and the body length became more than doubled.[40] From these results, we believe that mixing VED with fish feed and breeding them will contribute to the improvement of production in the fishery and aquaculture industries.
It is predicted that by the 2030, the aging of the population will progress worldwide and become a social problem in countries around the world. In an aging society, the number of osteoporosis patients is rapidly increasing. We can provide society with these compounds that have the potential to cure osteoporosis.
23-4. Structure elucidation by X ray analysis – A single crystal (0.10 x 0.20 x 0.30 mm) of (dl)-131was obtained by recrystallization from MeOH. Measurements were made on a Rigaku AFC5R diffractometer with graphite monochromated Cu-Kα radiation (λ=1.54178 Å). Crystal data for (dl)-131: C14H16N2O4, M=276.29; triclinic, space group P1(_) (#2), a=8.163 (1)Å, b=12.086 (1)Å, c=8.0126 (9)Å, α=107.940 (8)°, β=109.560 (9)°, γ=73.161 (8)°, V=693.2 (1)Å3, Z=2, Dcalc=1.324 g/cm3, F(000)=292, and μ(CuKα)=7.77 cm–1. The structure was solved by direct methods using MITHRIL[78] The non-hydrogen atoms were refined anisotropically. The final cycle of full-matrix least-squares refinement was based on 2685 observed reflections (I>3.00σ (I), 2θ <120.1°) and 245 variable parameters. The final refinement converged with R=0.039 and Rw=0.047.
Contrary to our expectation, (dl)-131 and (S)-(+)-131 are stable crystalline compounds enough to X-ray crystallographic analysis. Figure 116 (H, I) is an ORTEP drawing of (dl)-131. Numbering of (dl)-131 is shown in J. It should be stressed that 1-hydroxy group is deviated from the indole plane by 15.2°. This is the reason why 1-hydroxytryptophan derivatives undergo nucleophilic substitution.
The following measurements were made on a Rigaku/MSC Mercury diffractometer with graphite monochromated Mo-Kα radiation (λ=0.71069 Å). All calculations were performed using the teXsan package.[79] The structure was solved by a direct method (SIR).[80] The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included but not refined.
Crystal data for 53b: C23H36N2O9S, M=516.61; orthorhombic; space group, P212121 (#19); a=8.738(3) Å, b=14.732(4) Å, c=19.428 (6) Å, V=2501 (1) Å3, Z=4, Dcalc= 1.372 g/cm3. The final R- and Rw-factors after full-matrix least-squares refinements were 0.038 and 0.049 for 3499 observed reflections [I>3.00σ (I)], respectively. Positional parameters and B(eq) for 53b are shown in Table 32. Crystallization solvent is CH3SO3H.
Figure 118.
ORTEP Drawing and Numbering of 135a.
Crystal data for 135a: C22H28N2O4, M=384.47; monoclinic; space group, P21 (#4); a=8.200(3) Å, b=12.873(4) Å, c=9.296 (4) Å, β=97.769(4)°, V=972.2(6) Å3, Z=2, Dcalc= 1.313 g/cm3. The final R- and Rw-factors after full-matrix least-squares refinements were 0.035 and 0.050 for 4169 observed reflections [I>3.00σ (I)], respectively. Positional parameters and B(eq) for 135a are shown in Table33. The N-O bond is deviated from the indole plane by12.7°.
Crystal data for 137b: C25H30N2O6, M=454.52; monoclinic; space group, P21 (#4); a=8.1888(6) Å, b=10.7944(9) Å, c=13.8942(9) Å, β=97.262(1)°, V=1218.3(2) Å3, Z=2, Dcalc= 1.239 g/cm3, F(000)=484 and μ(Cu–Kα)=6.92 cm-1. The final R- and Rw-factors after full-matrix least-squares refinements were 0.030 and 0.036 for 1632 observed reflections [I>3.00σ (I)], respectively. Positional parameters and B(eq) for 137b are shown in Table 34.
Figure 119.
ORTEP Drawing and Numbering of 137b.
Figure 120.
ORTEP Drawing and Numbering of 148.
Crystal data for 148: C23H36N2O9S, M=516.61; orthorhombic; space group, P212121 (#19); a=8.738(3) Å, b=14.732(4) Å, c=19.428(6) Å, V=2501(1) Å3, Z=4, Dcalc= 1.372 g/cm3. The final R- and Rw-factors after full-matrix least-squares refinements were 0.038 and 0.049 for 3499 observed reflections [I>3.00σ (I)], respectively. Positional parameters and B(eq) for 148 are shown in Table 35. Crystallization solvents are H2O, CH3OH, and CH3SO3H.
Crystal data for 164a: C21H18N4O8, M=542.50; tetragonal; space group, P43 (#78); a=14.441(2) Å, b=14.449(1) Å, c=12.626(2) Å, V=2633.2(7) Å3, Z=4, Dcalc=1.368 g/cm3. The final R- and Rw-factors after full-matrix least-squares refinements were 0.060 and 0.091 for 5781 observed reflections [I>3.00σ (I)], respectively. Positional parameters and B(eq) for 164a are shown in Table 36. Crystallization solvent is CH3CO2Et.
Figure 121.
ORTEP Drawing and Numbering of 164a.
24. Awards for Global Warming Prevention Activities and Others
On December 4th, 2017, the Ministry of the Environment gave us the Environment Minister's Award for our activities titled ‘Global deployment of concrete and feasible new technologies to solve global warming and restore the green earth’ (Figure 122).
Activities: Developed a new and safe organic compound "SOMRE" as a plant root elongation agent.
Reason for commendation: SOMRE–treated indigenous plant roots grew two to three times longer than usual, and the rooting rate was 87.6%, resulting in partial greening of the fluid dunes. The authors also conducted the world's first airborne SOMRE soaked seed dispersal in the Gobi Desert. Furthermore, they verified the increase in yield of Indian rice. Contributed to the prevention of global warming by vast CO2 absorption sources in drylands and desert greening, and by improving food yields.
Fortunately, the author was able to contribute to society in small way, so in March 2023, he was awarded the Order of the Sacred Treasure in the Spring Conferment of Decorations. Encouraged by this, he intends to make further efforts to realize his dream and mission until the end of life.
Acknowledgments
The author took the first step in researching ‘SOMRE and VED’ in Kanazawa, located in the center of Japan's Sea of Japan coast, with the big dream and mission of saving the ‘whole earth’ from hunger and desertification. Every year, he applied for the Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, but for this topic he only received the lowest grade, ‘C’, and was unable to receive any funding. However, he was able to continue this research because of the advantage of the Japanese university course system. Thank you from the bottom of his heart. He was able to advance research with the cooperation of many people mentioned in the text, staff members, graduate and undergraduate students of Kanazawa University. The author will continue to research ‘SOMRE and VED’ for the rest of his life in order to go beyond the 17 goals of the United Nations and SDGs and restore and save ‘the Earth’ to a healthy state. The devoted support and cooperation of his family is worthy of special mention.
References and Notes
- Professor Emeritus of Kanazawa University. SOMEIYAKKO Kenkyusho, director. e-mail: someiyakko@smail.plala.or.jp. 56-7 Matsuhidai, Matsudo, Chiba 270–2214, Japan.
- M. Somei, Advances in internuclear deactivated methyl group attack methods, J. Synth. Org. Chem. Jpn., 1972, 30, 354.
- M. Somei and T. Okamoto, A Novel Method for Attacking Non-activated C-H Bond and Its Application to the Synthesis of Gibberellin-A15 from Enmein, Chem. Pharm. Bull., 1970, 18, 2135. [CrossRef]
- W. Nagata et al., Stereocontrolled total synthesis of dl-gibberellin A15, J. Am. Chem. Soc. 1971, 93, 5740. [CrossRef]
- Y. Isogai, et al., Activity of synthetic gibberellin A15 and dl-gibberellin A15 isolactone, Phyto- chemistry, 1974, 13, 337. [CrossRef]
- M. Somei, ‘Imagination and Creation: 1-Hydroxyindole Chemistry and the dream Challenge,’ Yakugaku Zasshi, 2008, 128, 527. M. Somei, Chemistry, 2007, 62, 116. M. Somei, ‘Topics in Heterocyclic Chemistry; A Frontier in Indole Chemistry: 1-Hydroxyindoles, 1-Hydroxytryptamines, and 1-Hydroxytryptophans,’ Vol. 6, ed. by Shoji Eguchi, Springer-Verlag, Berlin, 2006, pp. 77-111. M. Somei, ‘Advances in Heterocyclic Chemistry: Recent Advances in the Chemistry of 1-Hydroxyindoles, 1-Hydroxytryptophans, and 1-Hydroxytryptamines,’ Vol. 82, ed. by A. R. Katritzky, Elsevier Science (USA), 2002, pp. 101-155. M. Somei, Heterocycles, 1999. 50, 1157. M. Somei, J. Synth. Org. Chem. Jpn., 1991, 49, 205. .
- M. Somei et al., Syntheses of 3-Indoleacetic Acid and 3-Indoleacetonitrile Having a Halogen Group and a Carbon Functional Group at the 4-Position, Chem. Pharm. Bull., 1985, 33, 3696.
- G.W. Gribble, Contemp. Org. Chem., 1994, 1, 145. U. Pindur et al., J. Heterocycl. Chem., 1988, 25, 1. R. J. Sundberg, ‘The Chemistry of Indoles’, Academic Press, New York, 1970.
- M. Somei, Greening the desert and combating global warming, JICA Hokuriku, 2008.
- M. Somei, the challenge for greening the desert to prevent yellow sand and stop global warming, Koryoiki kyoiku, 2008, 70, 4.
- M. Somei, Indole chemistry for combating yellow sand and desertification directed towards stopping global warming, Heterocycles, 2011, 82, 1007–1027. M. Somei, ibid., 2008, 75, 1021. [CrossRef]
- M. Somei et al., ibid., 2007, 73, 537. M. Somei, J. Synth. Org, Chem., 1991, 49, 205.
- M. Somei, ‘Japan Sea Research: SOMRE, a Growth Regulator ‘Medicine’ for the Earth to increase Food Production and to Stop Global Warming’, 2014, 45, 105. N. Suzuki, The 127th Annual Meeting of Pharmaceutical Society of Japan, Toyama, March 2007. M. Somei et al., JP Patent, application No. 2023–071608. M. Somei et al., JP Patent, application No. 2023–071608.
- Greening the Gobi Desert: The Effects of Plant Promoters, The Hokkoku Shimbun, 2005, Oct. 21.
- Japanese researcher succeeded in plant growing and greening Gobi Desert, Inner Mongolia Arashan Zuqi Newspaper, 2007, June 18.
- Magic solution helps green the desert with jujube seedlings, The Chunichi Shinbun, 2007, June 5.
- Seeding by airplane to green 3000 hectares, The Hokkoku Shinbun, 2010, July 2.
- H. Somei et al., Greening Gobi Desert and increasing food production in India, Abstr. Paper, JSBBA Annual Meeting, Kyoto, 2021, March 20, Lecture No. 3H04-11. .
- R. Awano et al., SOMRE #1 deployed to India for increasing food production and improving the environment, Abstr. Paper, JSBBA Annual Meeting, Tokyo, 2019, March 24, Lecture No. 1E6a-13.
- K. Naik, et al., Effect of a root growth promoter on selected crops grown in India, Plant Physiology Reports, 2020, 25(2), 284.
- Abe initiative, Master’s degree and internship program of the African business education initiative for youth, Networking Fair 2016, March 24.
- Abe initiative, Master’s degree and internship program of the African business education initiative for youth, Networking Fair 2023, March 14. Abe initiative, Master’s degree and internship program of the African business education initiative for youth, Networking Fair 2019, March 13.
- H. Somei et al., Abstr. Paper, JSBBA Annual Meeting, Tokyo, 2019, March 24, Lecture No. 1E6a-12.
- M. Somei, et al., Abstr. Paper, JSBBA Annual Meeting, Kyoto, 2022, March 18, Lecture No. 4I06-07.
- The Nikkei Shinbun, Development of vasodilator substances, 2004, Nov. 17.
- The Hokuriku Chunichi Shinbun, Growth promotor of Cashmere goats, 2006, Sept. 27.
- The Yukan Fuji, Substances that could lead to new ED treatments, 2016, Jan. 25,.
- The Yukan Fuji, Efficacy demonstrated by testing male goats in heat, 2016, Feb. 1.
- The Hokkoku Shinbun, Prevent desertification by growing goat hair and increase cashmere, 2006, June 5.
- M. Somei, H. Shigenobu, and Y. Tanaka, a2 receptor antagonists and vasodilators containing indole derivatives as active ingredients, JP Patent 3964417. .
- M. Somei, K. Shigenobu, and Y. Tanaka, Receptor blocker and vasodilator comprising indole derivative as active ingredient, US Patent 7,872,040 B2.
- M. Somei, 1-Hydroxyindole derivatives, JP Patent 3795093.
- M, Somei, The 1st Cosmetics Development, Academic Forum, An organic compound that instantly relieves itching and heals wounds, Osaka, 2020, September 9-11th.
- 33. Y. Mikami and M. Somei, Apoptosis inhibitors, JP Patent 5370957.
- 34. Y. Mikami and M. Somei, Method of inhibiting apoptosis, US Patent 8,258,174 B2.
- 35. M. Natui, et al., Stimulation of the hair growth by a2-blocker SST-VED, Akita J. Med., 2014, 41, 23–33.
- 36. M. Somei et al., Preparations of 1-hydroxyindole derivatives and their potent inhibitory activities on.
- platelet aggregation, Heterocycles, 1996, 43, 1855.
- 37. M. Somei, Compositions containing N-acyltryptamines, JP Patent 5380170.
- M. Somei, Compositions containing N-acyltryptamines, JP Patent 5705939.
- 38. M. Somei, Compositions containing N-acyltryptamines, JP Patent 5705940.
- 39. The Hokuriku Chunichi Shinbun, Curbing desertification and increasing cashmere production, 2006, Sept. 27.
- 40. M. Somei, unpublished results: A breeding experiment using ‘medaka fish’, which are sensitive to toxic substances. SOMRE #1 is usually used at a concentration of 1.0 to 0.001 ppm. When ‘medaka fish’ were raised using a 1.0 ppm aqueous solution of SOMRE #1, the highest concentration normally used, they survived for two years and two months and grew large and healthy. They were then released back into the wild.
- 41. M. Somei, et al., Atopic dermatitis treatment that reduces itching, JP Patent 7048051. M. Somei, et.
- al., Atopic dermatitis treatment that reduces itching, US Patent 11,065,229 B2.
- 42. M. Somei, N. Suzuki, Y. Hattori, et al., Indole derivatives and their uses, JP Patent 4014052.
- M. Somei, et al., Indole derivative and application thereof, US Patent 8,053,462 B2.
- The Hokuriku Chunichi Shinbun, New osteoporosis drug shines a light, 2004, April. 22.
- The Hokkoku Shinbun, A new drug for osteoporosis that builds bones and inhibits destruction, 2004, April. 22.
- 43. M. Somei et al., Tryptophan derivatives and application thereof, US Patent 7,659,304 B2. M. Somei et al., Tryptophan derivatives and their uses, JP Patent 5070050. M. Somei et al., Tryptophan derivatives and application thereof, EP 1,911,744 B1. M. Somei et al., Tryptophan derivatives and their uses, CN 702,124.
- 44. COP28, 28th United Nations Climate Change conference, 2023, November 30–December 12, (Dubai). IPCC AR6 WG1, March 13–20 (Switzerland), and references cited therein. IPCC AR4, Nov.12–17, 2007 (Spain), and references cited therein. IPCC Assessment, and References cited therein. COP 27, Nov. 6—20 (Egypt), and references cited therein.
- 45. Ministry of education, culture, sports, science, and technology (MEXT), Japan meteorological agency (JMA), Climate change in Japan, 2020, Dec. and references cited therein.
- 46. M. Somei, A Frontier in Indole Chemistry: 1-hydroxyindoles, 1-hydroxytryptamines, and 1-hydroxytryptophans, Topics in Heterocyclic Chemistry, 2006, 6, 77–111. M. Somei, Recent advances in the chemistry of 1-hydroxyindoles, 1-hydroxytryptophans and 1-hydroxytryptamines, Advances in Heterocyclic Chemistry, Vol. 82, ed. by A. R. Katritzky, Elsevier Science (USA), 2002, pp. 101–155. M. Somei, 1-Hydroxyindoles, Heterocycles, 1999, 50, 1157–1211. d) M. Somei, Chemistry of 1-hydroxyindole and its derivatives, J. Synth. Org. Chem. Jpn., 1991, 49, 205–217.
- 47. M. Takasugi, et al., Spirobrassinin, a Novel Sulfur-containing Phytoalexin from the Daikon.
- Rhaphanus sativus L. var. hortensis (Cruciferae), Bull. Chem. Soc. Jpn., 1988, 61, 285.
- 48. The Chunichi Shinbun, Magic solution helps green the Alashan Desert, 2007, June 5.
- 49. Inner Mongolia Arashan Zuqi Newspaper, In the Alashan Desert, Professor Somei's drug achieved.
- a survival rate of 87.6%, 9.3% higher than conventional methods, 2007, June 18.
- 50. The Hokkoku Shinbun, Seeds planted directly in the Gobi Desert take root, 2008, March 22.
- 51. India-Japan Online Agri Mission, 2021: Vol.1 - Organic Bio-stimulant SOMRE, URL 2021:.
- https://youtu.be/t4bGY9fANAA.
- 52. K. Yoshino et al., Rearrangement Reaction in 1-Hydroxyindole Chemistry: A Synthesis of Novel.
- 7-Substituted Yohimbine, and 4a-Substituted 1,2,3,4Tetrahydro-β-Carboline Derivatives, 2019,.
- Heterocycles, 98, 1384. M. Somei et al., Heterocycles, 2006, 69, 259. M. Somei et al., Heterocycles, 2001, 55, 1237–1240.
- 53. M. Somei et al., The ideal synthetic method aimed at the leads for an 2-blocker, an inhibitor of.
- platelet aggregation, and an anti-osteoporosis Agent, Heterocycles, 2006, 68, 1565.
- 54. T. Kawasaki et al., Simple synthetic method for 1-hydroxyindole and its application to 1-hy-.
- droxytryptophan derivatives, Heterocycles, 2015, 90, 1038.
- 55. K. Yamada et al., Synthesis of Nb-acyl tryptamines and their 1-hydroxytryptamine derivatives as new.
- 2-blockers, Heterocycles, 2009, 79, 635.
- 56. J.Y. Lee et al., Efficacy of cilostazol administration in Alzheimer’s disease patients with white matter lesions: a positron-emission tomography study, Neurotherapeutics, 2017, 14, 784. [CrossRef]
- 57. N. Suzuki, et al., Novel tryptophan derivatives as potentially effective therapeutic drugs to treat bone.
- diseases, Am. J. Life Sci., 2015, 3 (3-2), 31. A. Hattori et al., Melatonin research in space and anti-aging medicine. Anti-Aging Medicine, 2013, 9, 356–363. N. Suzuki et al., Novel effects of melatonin: Effects on bones and development of drugs for bone disease using its derivatives, Comparative Endocrinology, 2011, 37 (No. 143), 194—203. N. Suzuki, et al., Response of osteoblasts and osteoclasts in regenerating scales to gravity loading, Biological Sciences in Space, 2009, 23, 211. N. Suzuki, et al., Novel bromo melatonin derivatives as potentially effective drugs to treat bone diseases, J. Pineal Res., 2008, 45, 229. N. Suzuki, et al., Novel bromo melatonin derivatives suppress osteoclastic activity and increase osteoblastic activity: implications for the treatment of bone diseases, J. Pineal Res., 2008, 44, 326. The Hokuriku Chunichi Shinbun, New osteoporosis drug shines a light, 2004, April 22. The Hokkoku Shinbun, A miracle cure for osteoporosis, 2004, April 22.
- 58. Y. Mikami, et al., Current status of drug therapies for osteoporosis and the search for stem cells.
- adapted for bone regenerative medicine, Anat Sci Int., 2014, 89:1. Y.Mikami et al., A new synthetic.
- compound, SST-VEDI-1, inhibits osteoblast differentiation with a down-regulation of the osterix.
- expressions, J. Biochem, 2009, 145, 239. Y. Mikami et al., JP Patent 5370957. Y. Mikami et al., US.
- Patent 8,258,174 B2.
- 59. H. Somei et al., “SOMRE” realizes greening of Gobi Desert and increased food production in India,.
- Abstr. Paper, JSBBA Annual Meeting, Sendai, 2021, March 18.
- 60. M. Somei, 13th International Biotechnology Exhibition & Conference, Biotech., 2014, May 14-16.
- M. Somei, Eco-Products, 2013, Dec. 12-14. M. Somei, 12th International Biotechnology Exhibition & Conference, Bio tech., 2013, May 8-10.
- 61. M. Somei and T. Shoda, A facile route to 1-acetoxy- and 1-methoxyindoles, Heterocycles, 16, 1523–1525 (1981).
- 62. R. M. Acheson, “New Trends in Heterocyclic Chemistry,” ed. by R. B. Mitra, N. R. Ayyangar, V. N. Gogte, R. M. Acheson, and N. Cromwell, Elsevier Scientific Pub. Co. New York, 1979, pp. 1–33; R. M. Acheson, P. G. Hunt, D. M. Littlewood, B. A. Murrer, and H. E. Rosenberg, J. Chem. Soc., Perkin Trans. 1, 1978, 1117.
- 63. M. Somei, H. Ohnishi, and Y. Shoken, A Practical Synthesis of the 1-Methoxy Analog of an Ergot Alkaloid, (±)-1-Methoxy-6,7-secoagroclavine, Chem. Pharm. Bull., 34, 677 (1986).
- 64. M. Somei, K. Imai, F. Yamada, T. Hayashi, Y. Nakai, and K. Tokumura, 1-Hydroxytryptophan derivatives have sp3 hybrid nitrogen, The 34th Congress of Heterocyclic Chemistry, Kanazawa, Japan, 2004. R. M. Acheson, M. H. Benn, J. Jacyno, and J. D. Wallis, Pyramidal bonding about nitrogen in 1-benzoyloxyindole determined by X-ray diffraction, J. Chem. Soc., Perkin Trans. 2, 1983, 497.
- 65. J. H. M. Hill, D. P. Gilbert, and A. Feldsott, Benzoin oximes in sulfuric acid. cyclization and fragmentation, J. Org. Chem., 1975, 40, 3735. E. Fischer, Chem. Ber., 1896, 29, 2062. [CrossRef]
- 66. N. Sugiyama, M. Akutagawa, T. Gasha, Y. Saiga, and H. Yamamoto, Bull. Chem. Soc. Japan, 1967, 40, 347.
- 67. M. Somei, H. Sato, and C. Kaneko, A short step synthesis of Lespedamine, Heterocycles, 1983, 20,.
- 1797. H. Morimoto and N. Matsumoto, Natural product, 1-methoxy-N,N-dimethyltryptamine,.
- Liebigs Ann. Chem., 1966, 692, 194. H. Morimoto and H. Oshio, Isolation of 1-methoxy-N,N-dimethyltryptamine, Liebigs Ann. Chem., 1965, 682, 212.
- 68. M. Somei, H. Ohnishi, and Y. Shoken, A practical synthesis of the 1-methoxy analog of an ergot alkaloid, (±)-1-Methoxy-6,7-secoagroclavine, Chem. Pharm. Bull., 1986, 34, 677.
- 69. M. Somei, F. Yamada, T. Kurauchi, Y. Nagahama, M. Hasegawa, K. Yamada, S. Teranishi, H. Sato, and C. Kaneko, Simple syntheses of serotonin, N-methyl serotonin, bufotenine, 5-methoxy-N-methyltryptamine, bufobutanoic acid, N-(indol-3-yl)methyl-5-methoxy-N- methyltryptamine, and lespedamine based on 1-hydroxyindole chemistry, Chem. Pharm. Bull., 2001, 49, 87, and references cited therein.
- 70. Oxidation of amines by this reagent was reported: S. Murahashi and T. Shiota, Short-step synthesis of amino acids and N-hydroxyamino acids from amines, Tetrahedron Lett., 1987, 28, 6469-6472; S. Murahashi, Jpn. Kokai Tokkyo Koho, 1987, JP 62212363 [Chem. Abstr., 1988, 109, 22847c].
- 71. M. Somei, K. Kobayashi, K. Shimizu, and T. Kawasaki, A simple synthesis of a phytoalexin, methoxybrassinin, Heterocycles, 1992, 33, 77. M. Somei, T. Kawasaki, K. Shimizu, Y. Fukui, and T. Ohta, Syntheses of methyl 1-hydroxyindole-3-acetate, Nb-acetyl-1-hydroxytryptamine, (±)- and (S)-1-hydroxytryptophan Derivatives, Chem. Bull., 1991, 39, 1905. T. Kawasaki, A. Kodama, T. Nishida, K. Shimizu, and M. Somei, Preparation of 1-hydroxyindole derivatives and a new route to 2-substituted indoles, Heterocycles, 1991, 32, 221.
- 72. M. Somei and T. Kawasaki, A new and simple synthesis of 1-hydroxyindole derivatives, Heterocycles,1989, 29, 1251.
- 73. B. Wright Jr. and K. H. Collins, 1-Methoxy-2-oxindole, J. Am. Chem. Soc., 1956, 78, 221.
- 74. E. Yamanaka, N. Shibata, and S. Sakai, Synthesis of Borrerine, Heterocycles, 1984, 22, 271; M. Doe.
- de Maindreville, J. Levy, F. Tillequin, and M. Koch, Syntheses of Borrerine, J. Nat. Prod., 1983, 46, . [CrossRef]
- 310; J. L. Pousset, J. Kerharo, G. Maynart, X. Monseur, A. Cave, and R. Goutarel, New alkaloid.
- isolated from Borreria verticillata. Phytochemistry, 1973, 12, 2308–10.
- 75. T. Gozler, B. Gozler, A. Linden, and M. Hesse, Vulcanine, a β-carboline alkaloid from Haplophyllum.
- vulcanicum, Phytochemistry, 1996, 43, 1425–1426. [CrossRef]
- 76. H. Takayama, N. Seki, M. Kitajima, N. Aimi, H. Seki, and S. Sakai, Heterocycles, 1992, 33, 121.
- 77. M. Somei, K. Noguchi, R. Yamagami, Y. Kawada, K. Yamada, and F. Yamada, the ideal synthetic method aimed at the leads for an 2-blocker, an inhibitor of pPlatelet aggregation, and an anti-osteoporosis Agent, Heterocycles, 2000, 53, 7.
- 78. C. J. Gilmore, MITHRIL-an integrated direct-methods computer program, J. Appl. Cryst., 1984, 17, 42–46.
- 79, 79. teXsan: Crystal Structure Analysis Package, Molecular Structure Corporation, 1985 & 1999.
- 80. M. C. Burla, M. Camalli, M. Cascarano, C. Giacovazzo, A. Guagliardi, and G. Polidori, SIR92, a program for automatic solution of crystal structures by direct methods, J. Appl. Cryst., 1994, 27, 435. [CrossRef]
- 81. N. Finch, C. W. Gemenden, I. H-C. Hsu, and W. I. Taylor, Oxidative transformations of indole alkaloids. 3. pseudoindoxyls from yohimbinoid alkaloids and their conversion to "invert" alkaloids, J. Am. Chem. Soc., 1965, 87, 2229-2235. [CrossRef]
- 82. K. Yamada, S. Tomioka, N. Tanizawa, and M. Somei, Novel syntheses of 1-hydroxy-6-, -5-nitroindole-3-carbaldehyde, and their derivatives as daikon-phytoalexin analogs, Heterocycles, 2004, 63, 1601–1611. F. Yamada, K. Yamada, H. Takeda, and M. Somei, Simple syntheses of analogs of a wasabi phytoalexin, Heterocycles, 2001, 55, 2361–2368. M. S. C. Pedras and J. L. Sorensen, Phytoalexin accumulation and antifungal compounds from the crucifer wasabi, Phytochemistry, 1998, 49, 1959–1965. M. Takasugi, et al., Novel sulfur-containing phytoalexins from the chinese cabbage Brassica campestris L. ssp. pekinensis (Cruciferae), Bull. Chem. Soc. Jpn., 1988, 61, 285–289.
Scheme 1.
Transformation of enmein (1) to gibberellin A15 (2) .
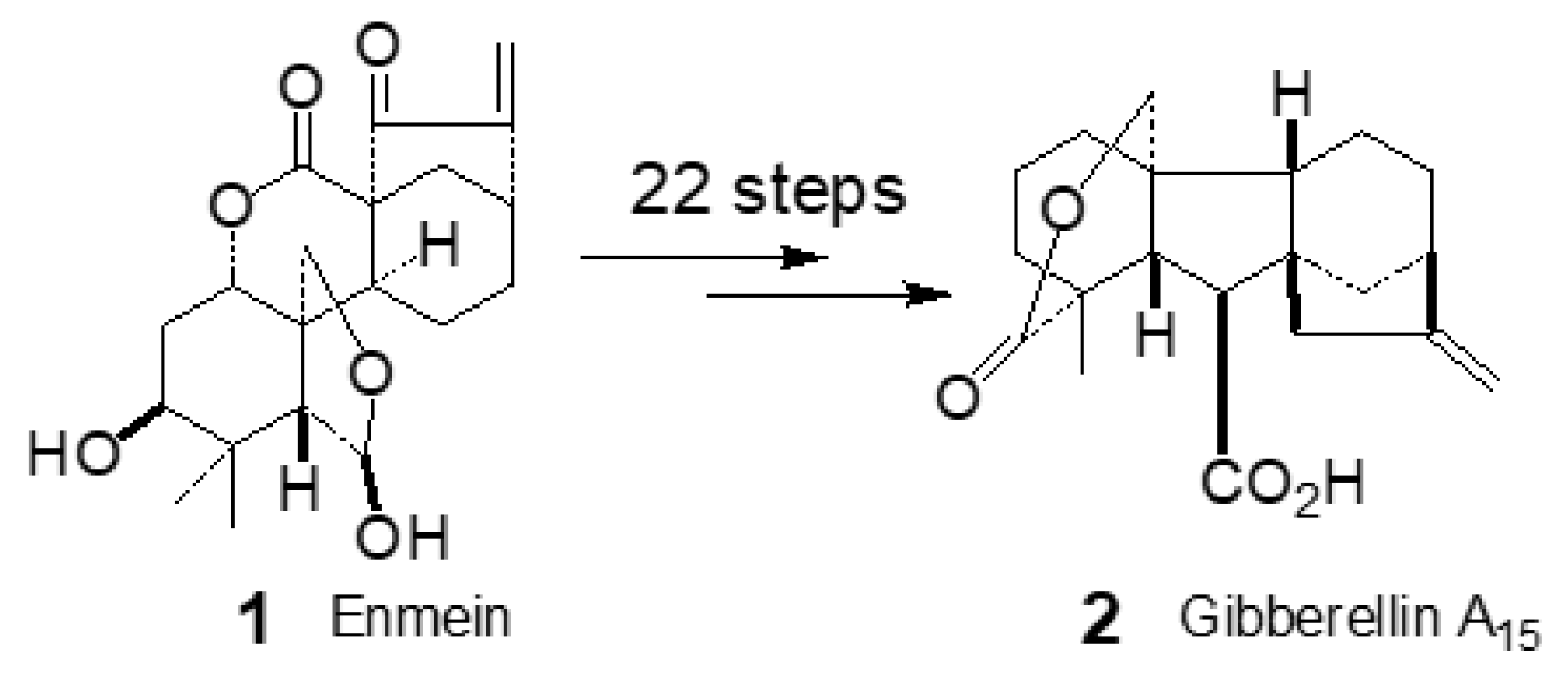
Scheme 2.
1-Hydroxyindole hypotheses, nucleophilic substitution.
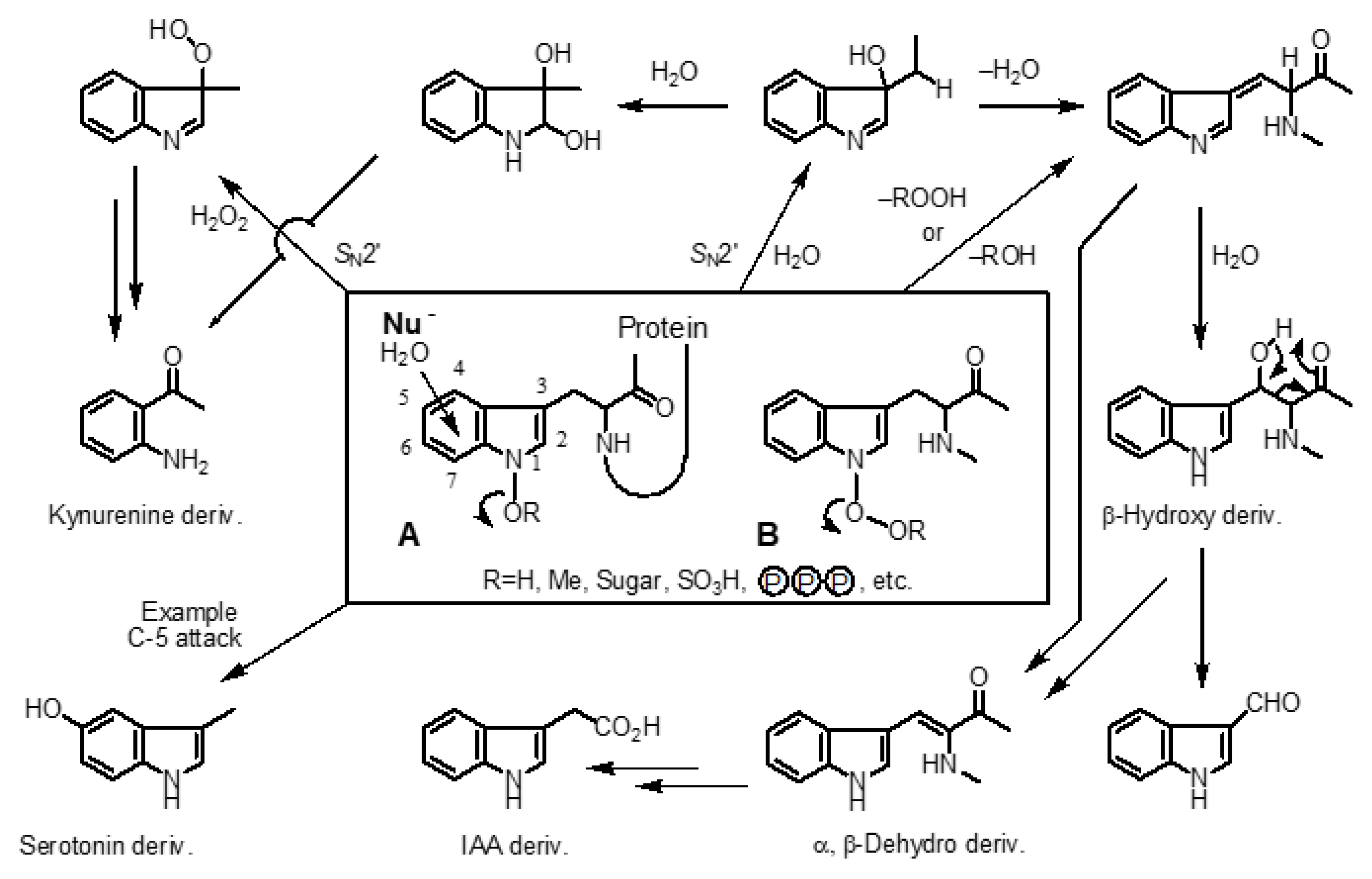
Figure 1.
SOMRE #1 and VED #1.

Figure 2.
Mechanism of desertification and greening.
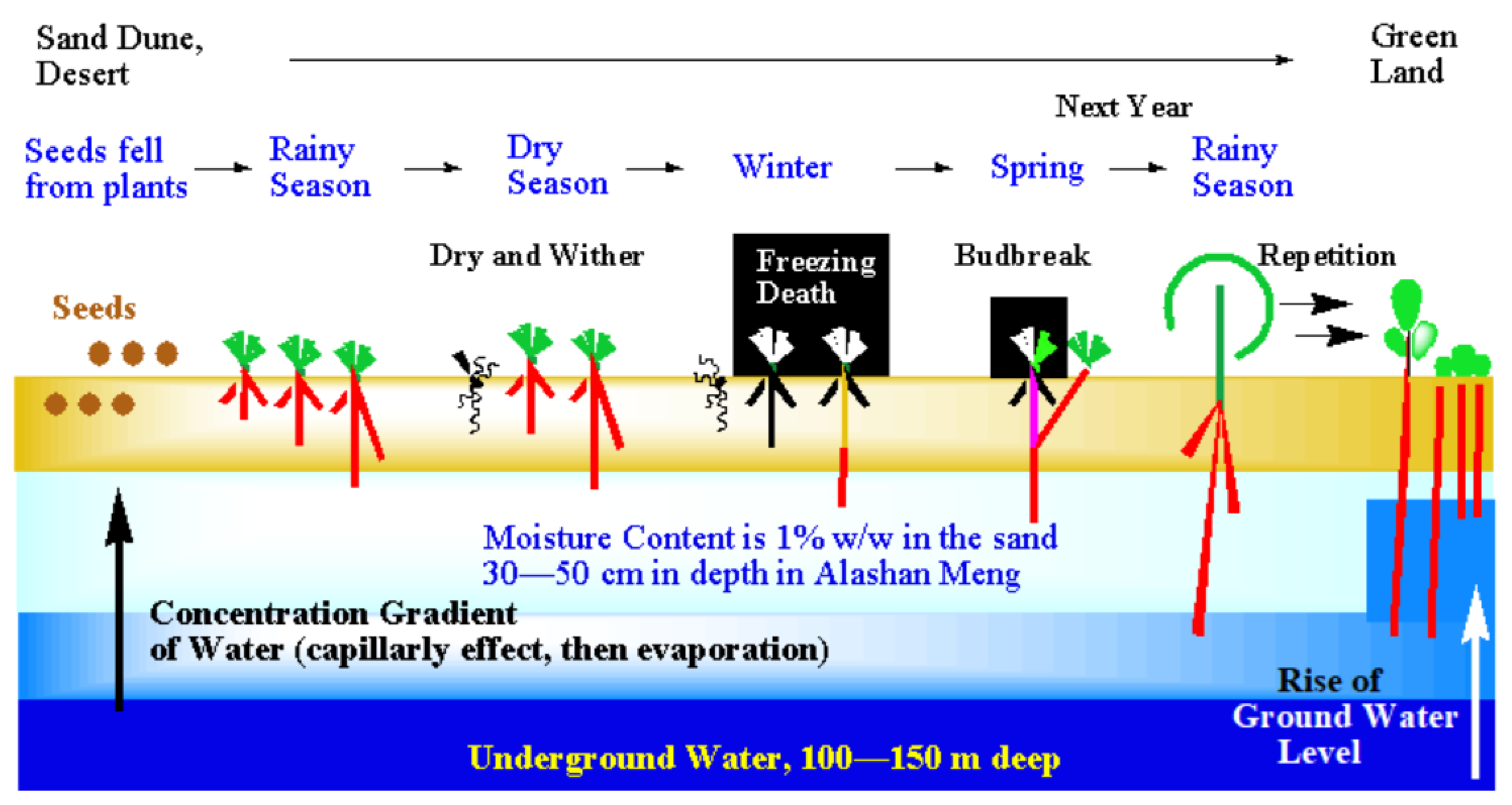
Scheme 3.
General synthetic method for 1-hydroxyindole.
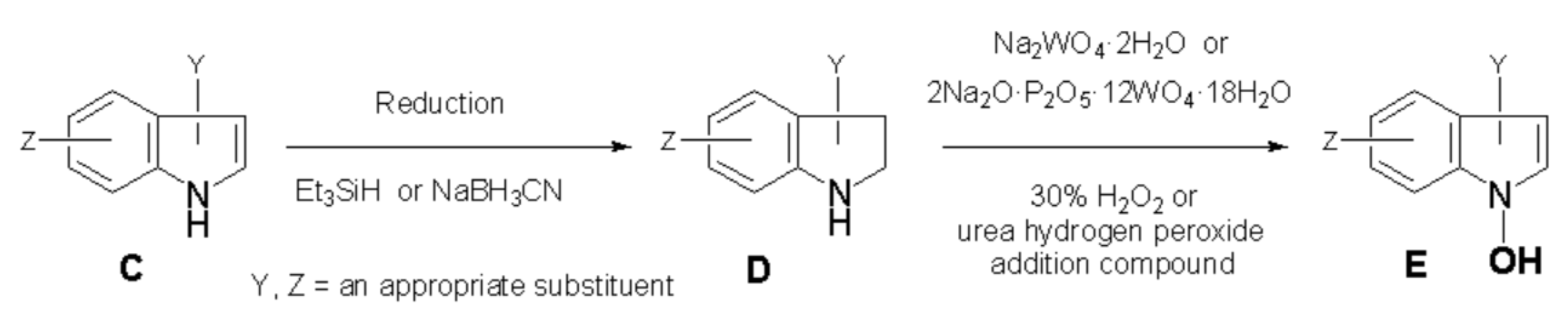
Figure 4.
How to immerse seeds in SOMRE solution.
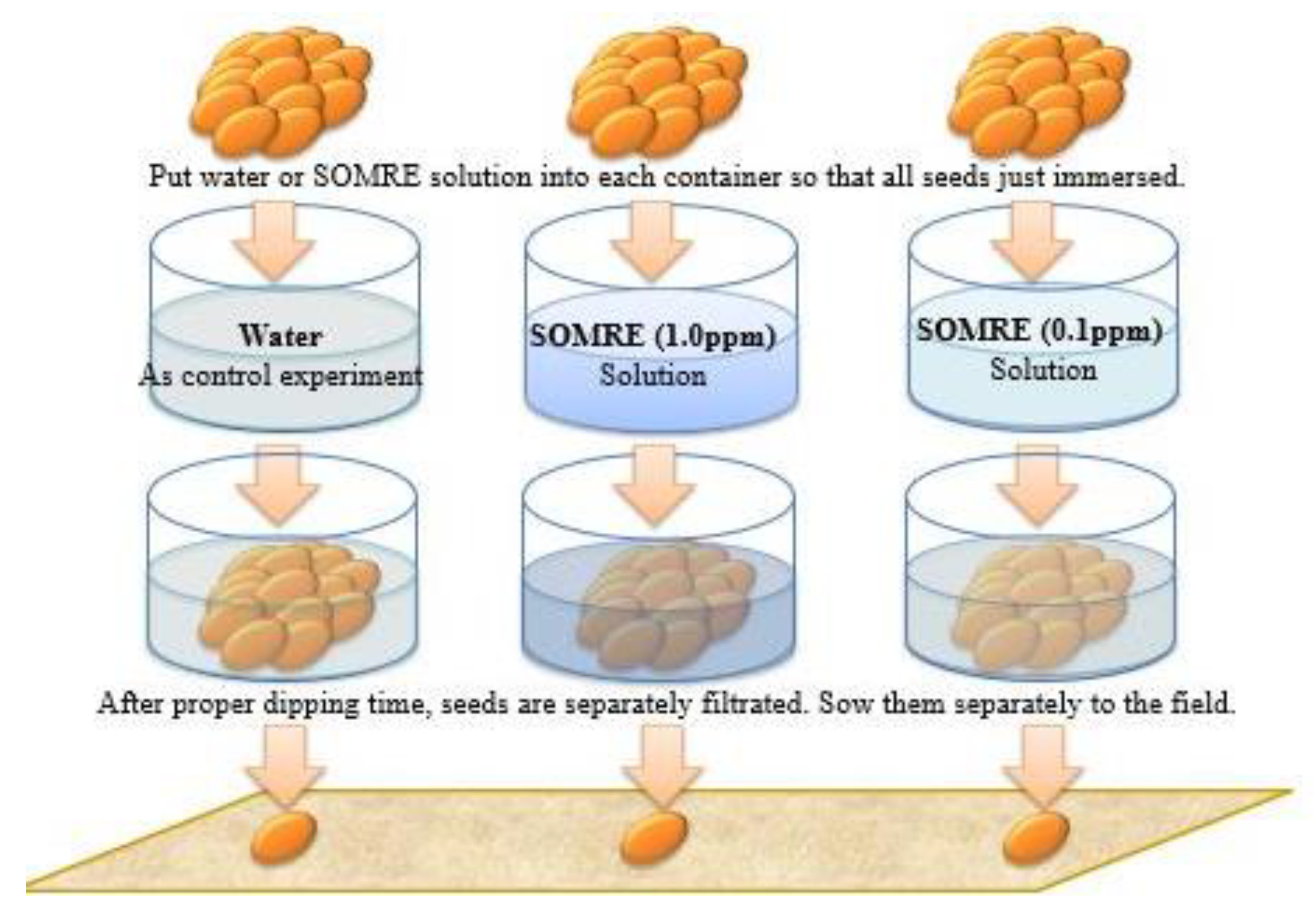
Figure 5.
Growing tomato plant.

Figure 6.
Harvested tomatoes.

Figure 7.
Tomato root length comparison.

Figure 8.
Germination, Tamba Black Soybeans.

Figure 9.
Harvested turnips.

Figure 10.
Growing garlic.

Figure 11.
Harvest of Garlic.

Figure 12.
Harvest of sweet potatoes.

Figure 13.
Weight distribution of potatoes, ‘Kitaakari’.
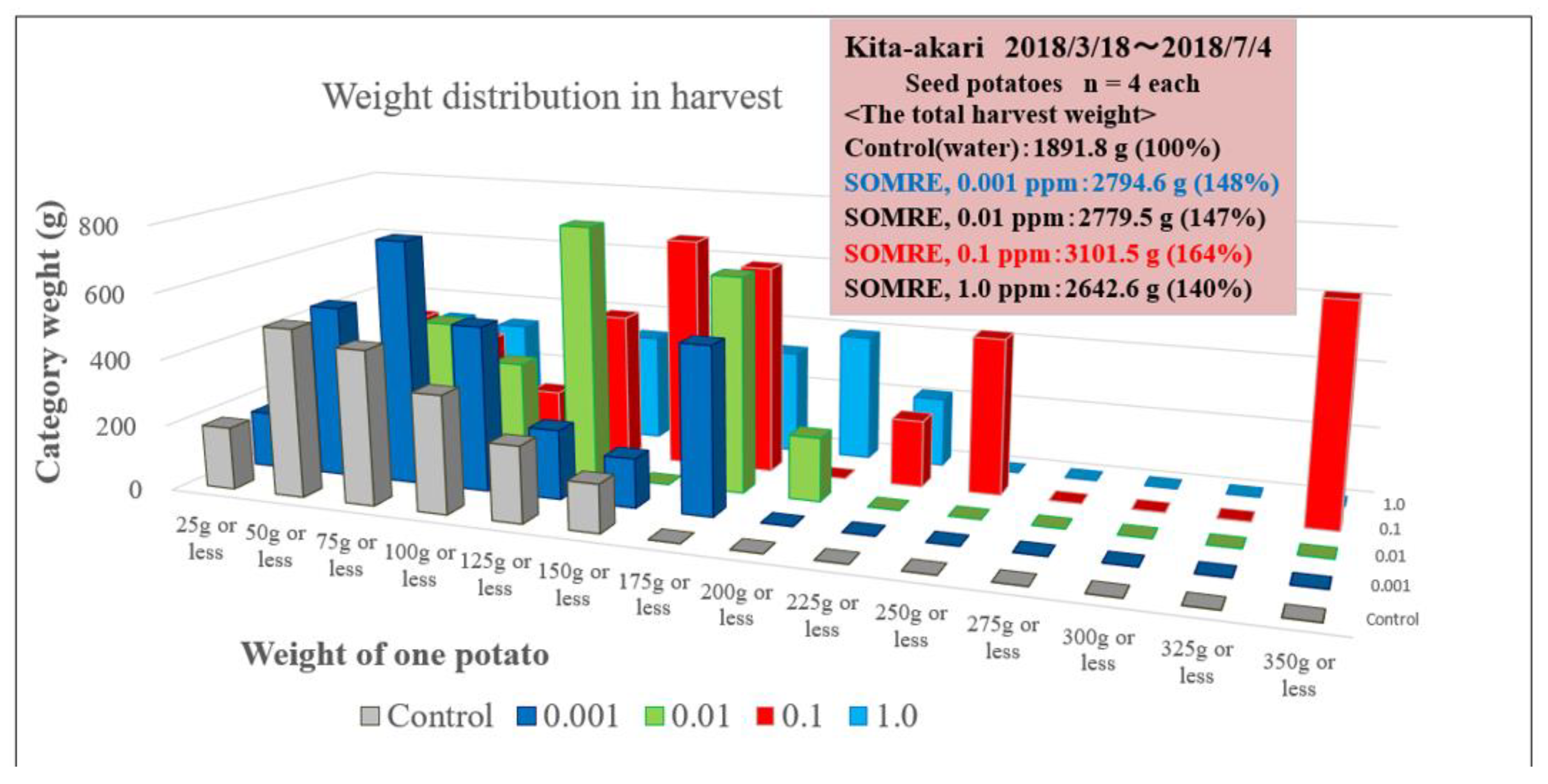
Figure 14.
Control potato, ‘Danshaku’, purchased from grocery.

Figure 15.
SOMRE 0.1 ppm treated Potato, ‘Danshaku’.
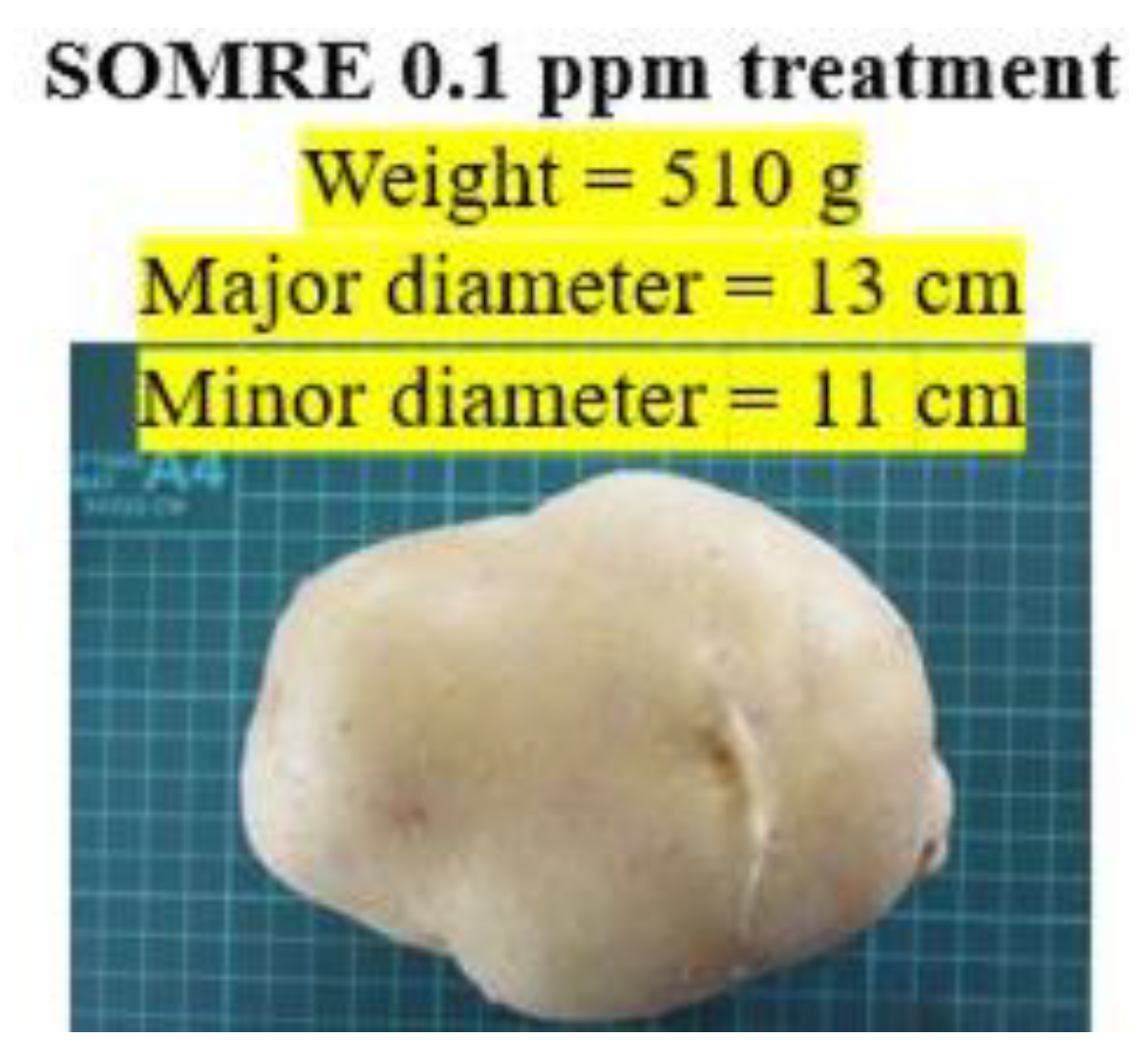
Figure 16.
Weight distribution of Taro.
Figure 17.
Leaves and bulbs of kohlrabi.

Figure 18.
Separeted leaves, bulbs of kohlrabi.

Figure 19.
Seedlings were planted.

Figure 20.
Growing state.

Figure 21.
Harvest.

Figure 22.
Rice ears of each group.

Figure 23.
Spreading each ear.

Figure 24.
Amount of harvested paddy.

Figure 25.
Brown rice after threshing.

Figure 26.
Grain size of the wheat.

Figure 27.
Rooting test of corn (‘pure white’) .

Figure 28.
Harvest of radish, ‘Okra’.

Figure 29.
Harvest of radish, ‘Sakurajima’.

Figure 37.
Rooting test of ginseng.
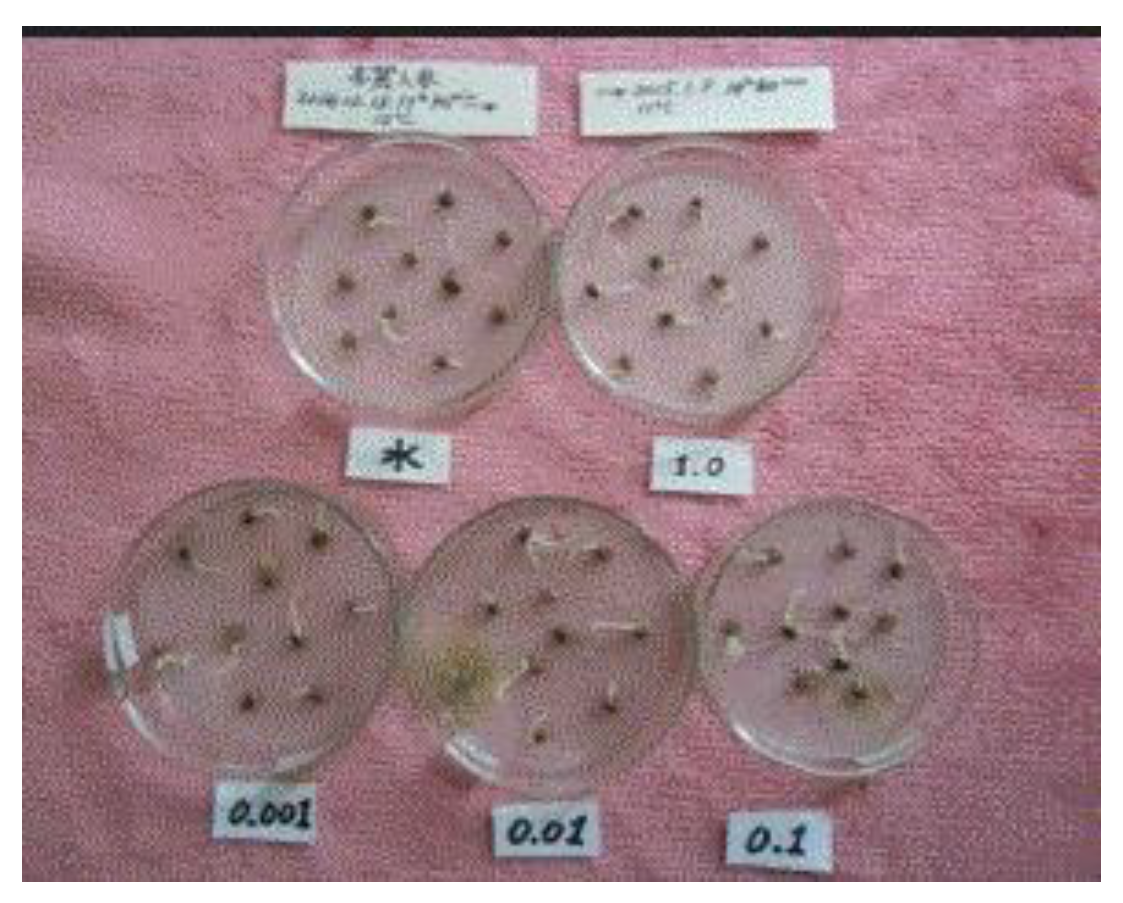
Figure 38.
Growing ginseng.

Figure 39.
Growing Maca.

Figure 48.
Growth of fungus, shot from the side.

Figure 49.
Growth of fungus, shot from the above.

Figure 50.
Leaves of growing ‘bok choy’.

Figure 52.
Rooting of okra.

Figure 53.
Rooting of corn.

Figure 54.
Rooting of burdock.

Figure 55.
Various seeds of desert native plants.

Figure 56.
Rooting test of sand jujube.

Figure 57.
Planting seedlings in the Gobi Desert.

Figure 58.
Arashan newspaper.

Figure 59.
Greening status a year and 7 months after planting.

Figure 60.
Rabbit returned.

Figure 65.
Soaking seeds in SOMRE solution.

Figure 66.
Throwing treated seeds to the desert.

Figure 67.
Throwing SOMRE treated seeds into the air.

Figure 68.
Making clay balls.

Figure 69.
Young trees growing in the desert.

Figure 70.
Soaking seeds into SOMRE solution.

Figure 71.
Filtering seeds through bamboo colander.

Figure 72.
Packing seeds to load onto airplane.

Figure 73.
Loading SOMRE treated seeds onto air plane onto the desert.

Figure 74.
Scattering SOMRE treated seeds.

Figure 75.
Rice harvesting in India.

Figure 76.
Rooting test of in the petri dish.

Figure 77.
Rooted seeds.

Figure 78.
Baobab germination on June 13.
Figure 79.
Baobab height, 20 cm on July 25.

Figure 80.
Height is 84 cm on Sept. 05.

Figure 81.
Height is 100.5 cm on Nov. 9.

Figure 82.
Our booth with students.

Figure 83.
African students.

Figure 84.
Active students.

Figure 85.
Comparison of hyacinth.

Figure 86.
Comparison of flower.

Figure 87.
Growth state of polygi.

Figure 88.
Growth state of calendula.

Figure 89.
Growth state of crocus.

Figure 90.
Poor sapling, ‘HIMEKOMATSU’.

Figure 91.
Growing vogorously.

Figure 92.
Height 370 cm, in 2023.

Figure 93.
Felled lacquer trees.

Figure 94.
Cut down lacquer trees.

Figure 95.
Lacquer seeds.

Figure 96.
Yohimbine and imaginary tryptamine derivatives for α2 blocker.
Scheme 5.
Synthesis of 1-Hydroxy-Nb-acyltryptamines.
Figure 97.
Hair length of control goat.

Figure 98.
Hair length of VED taking goat.

Figure 99.
A part of 90 baby goats, May 2007.

Figure 100.
young man, bald head.

Figure 101.
After 10 months use of VED spray.
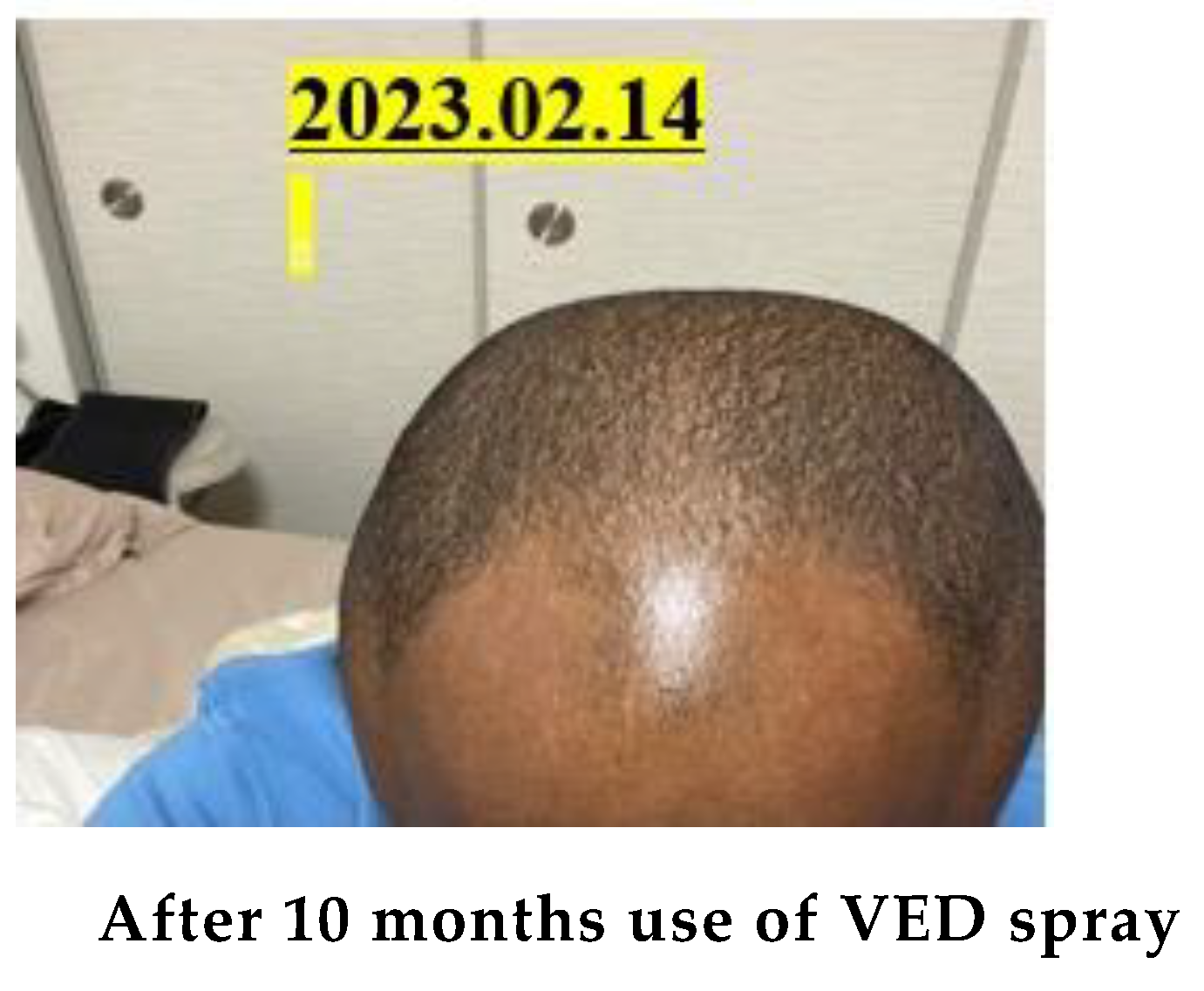
Figure 102.
67-year-old, aged spots and wrinkles .
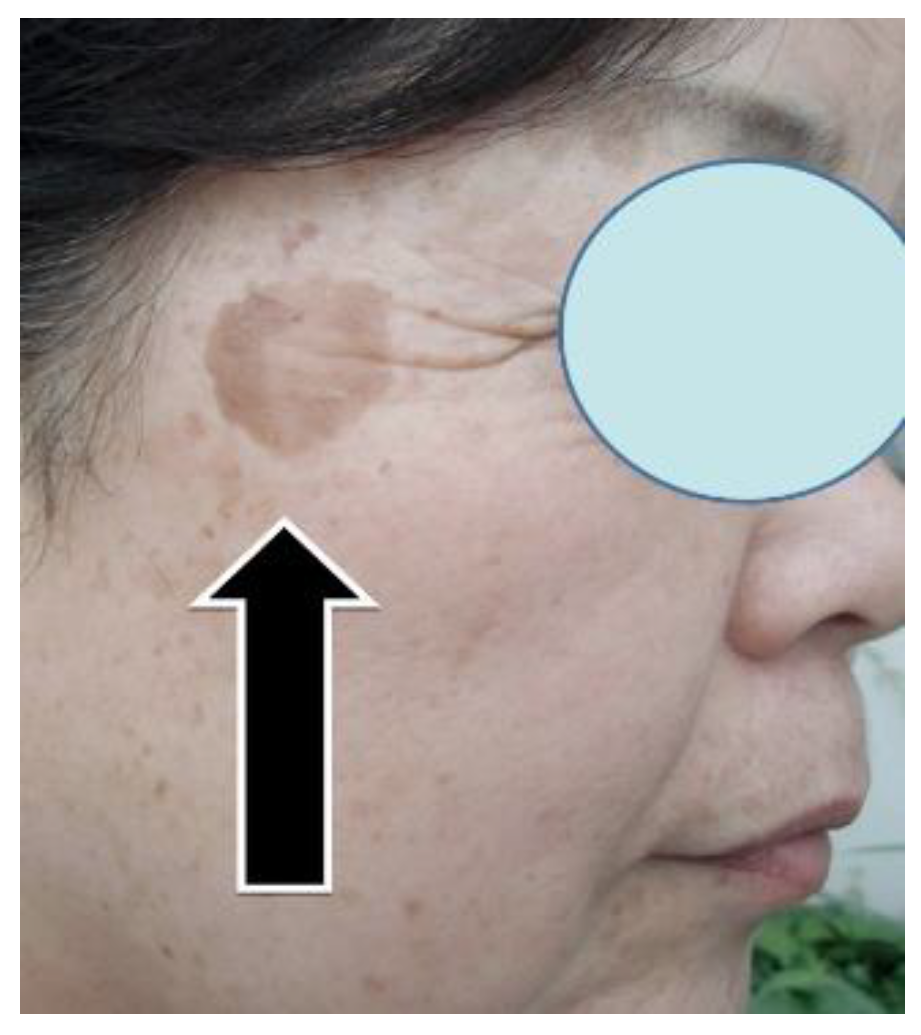
Figure 103.
Her spots and wrinkles disappeared.
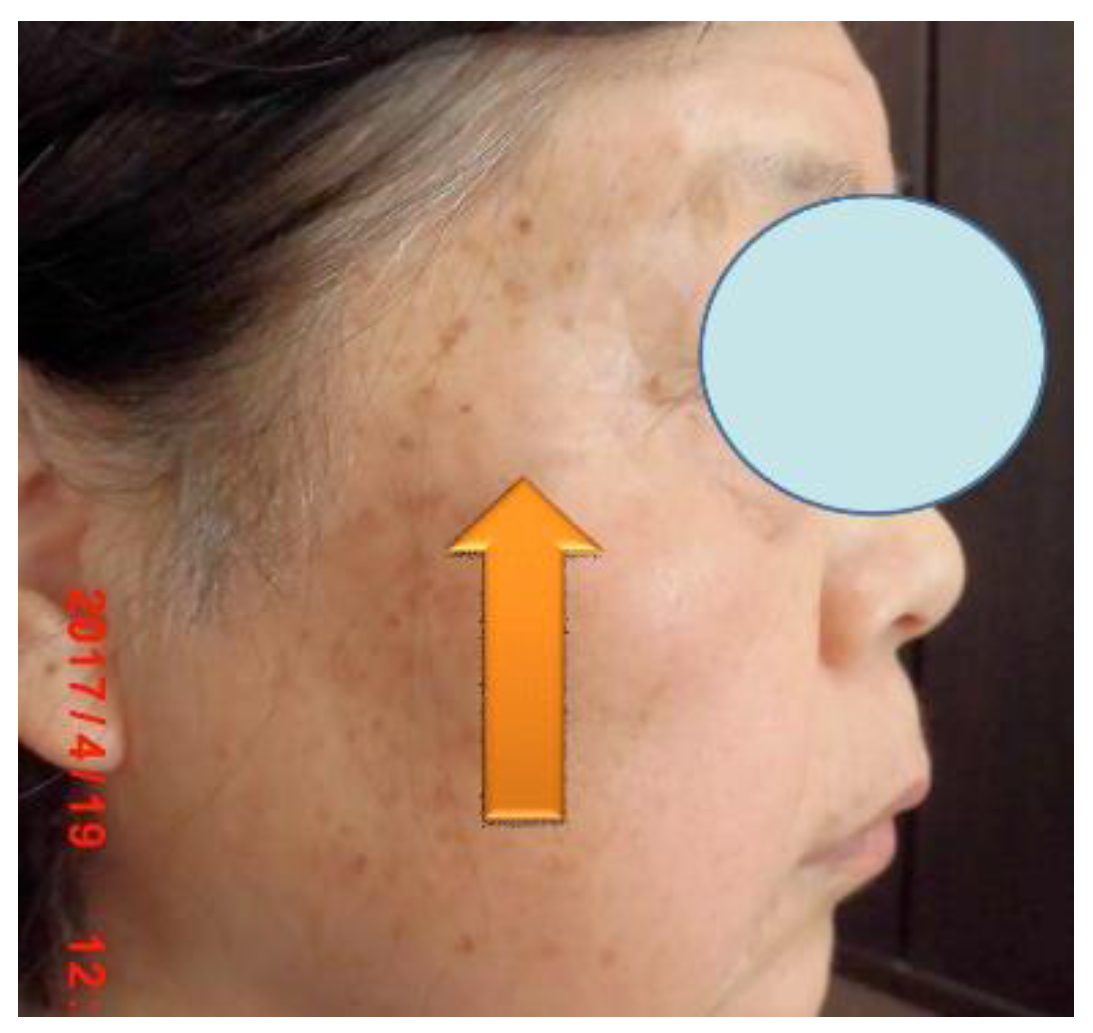
Figure 104.
Acne scars became worse due to medication prescribed by a doctor.
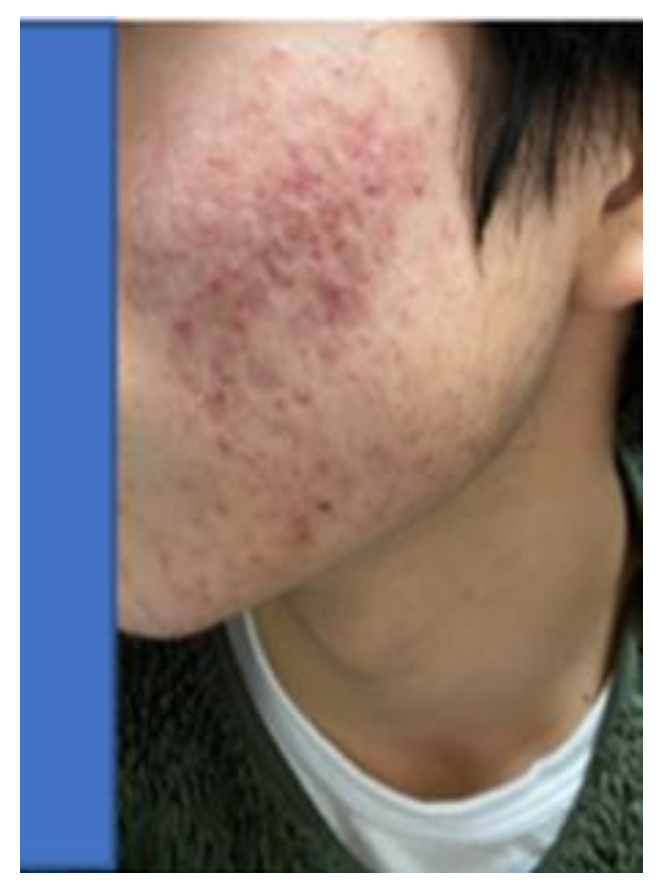
Figure 105.
VED cream usage history .
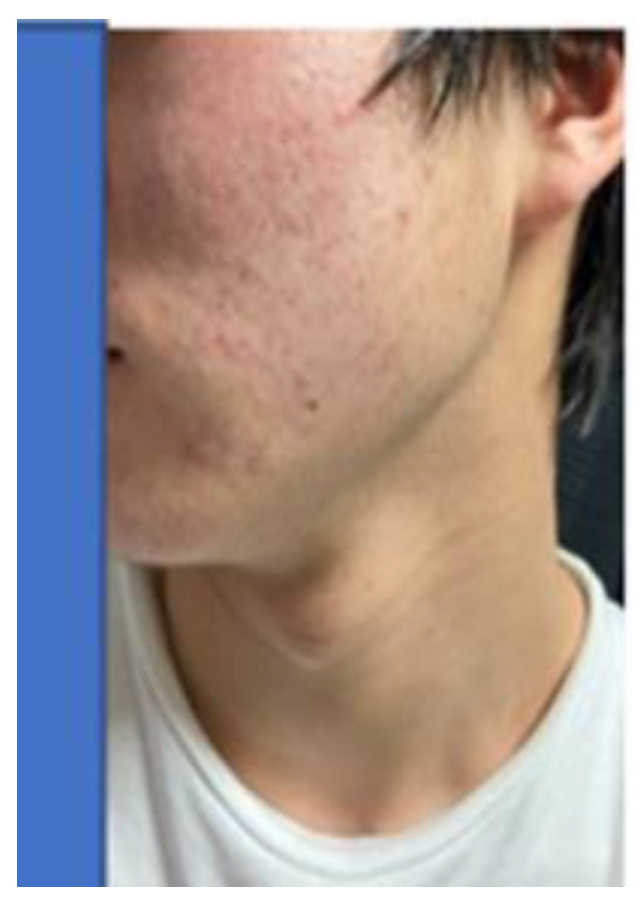
Figure 106.
Beautiful youthful skin was back, after 6 months.

Figure 107.
Wounds of 80–year–old man.
Figure 108.
Two week after applying VED cream.
Figure 109.
Edematous of 70–year–old man.
Figure 110.
Separated layer formed, a month later.
Figure 111.
Playing baseball as a catcher.

Scheme 6.
Synthesis of osteoporosis agents, tryptophan and melatonin derivatives.
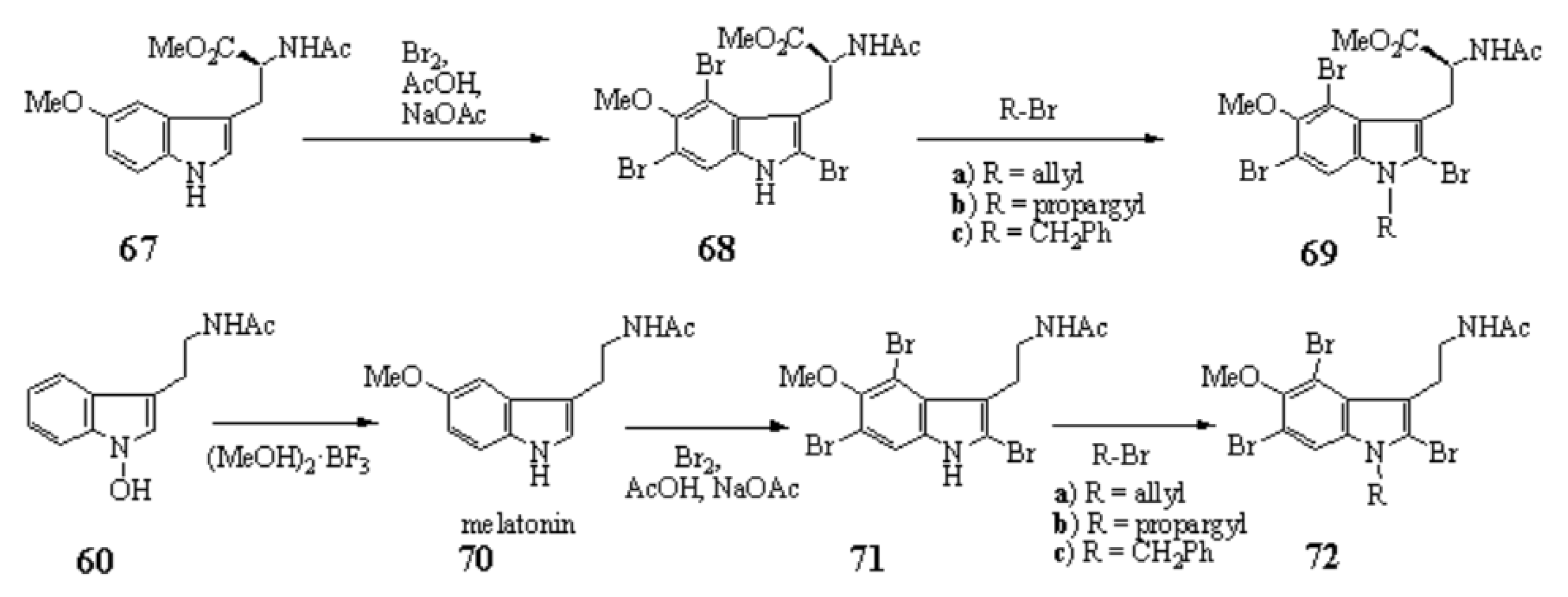
Figure 112.
Concrete ‘Earth Cure’ method.
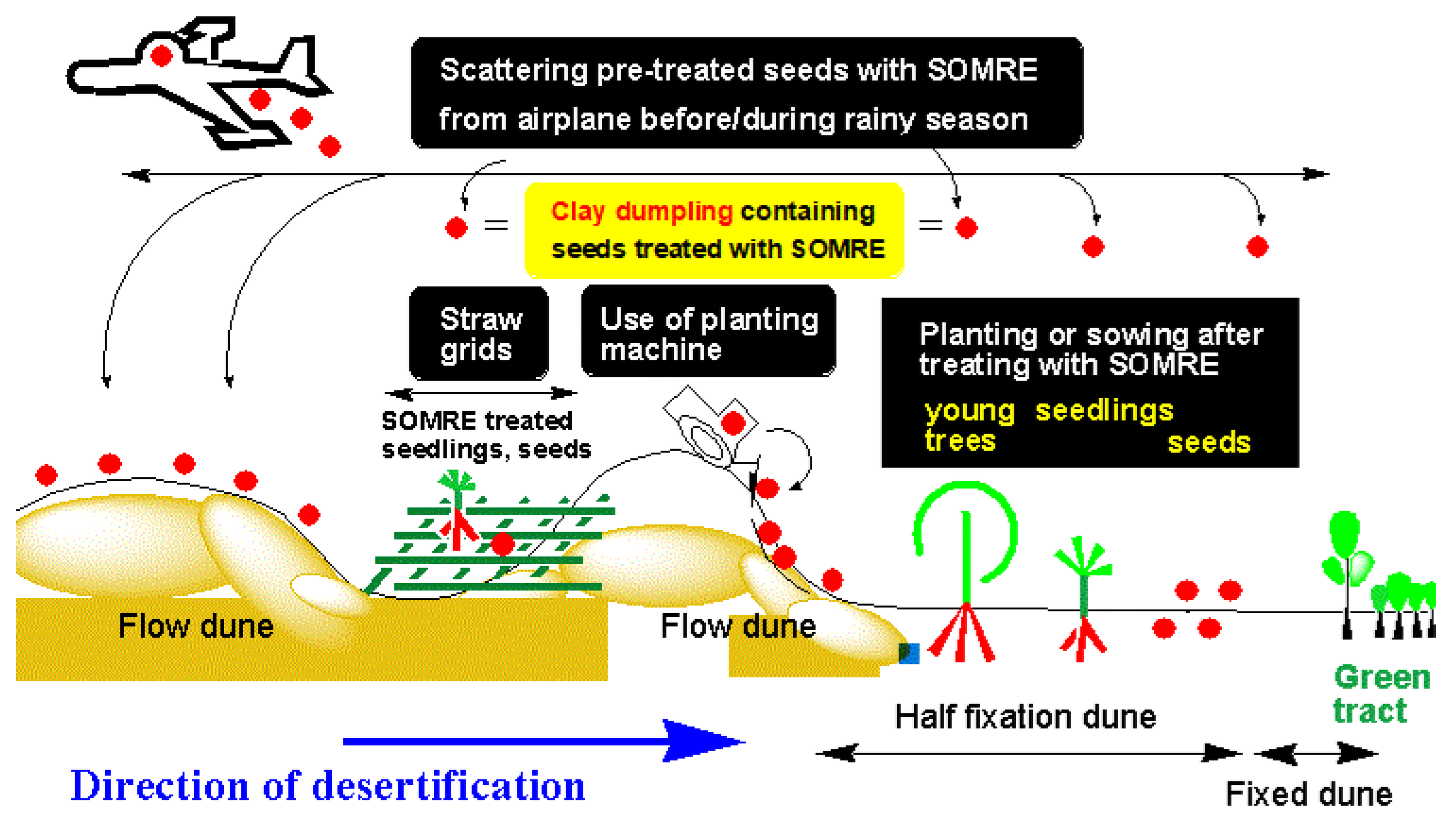
Figure 113.
Harvest of Kalahari watermelon; Far left; control. Right 4 fruits; SOMRE.

Figure 114.
Eco-friendly world with ‘SOMRE’ and ‘VED’.
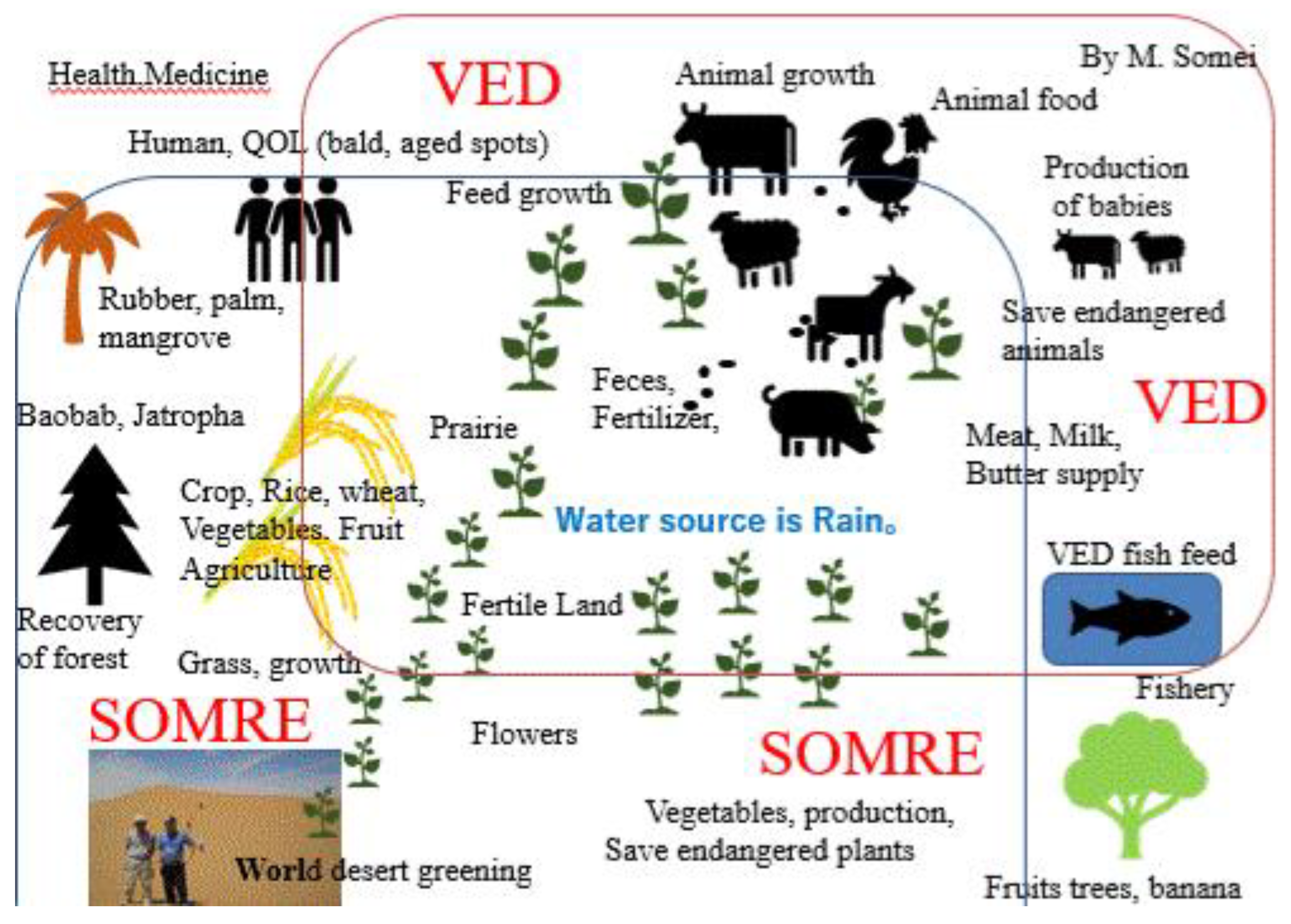
Figure 115.
Plants, which increased crop with SOMRE.

Figure 116.
Xray structure of (dl)-131: ORTEP drawing H, I. Numbering of (dl)-131: J.
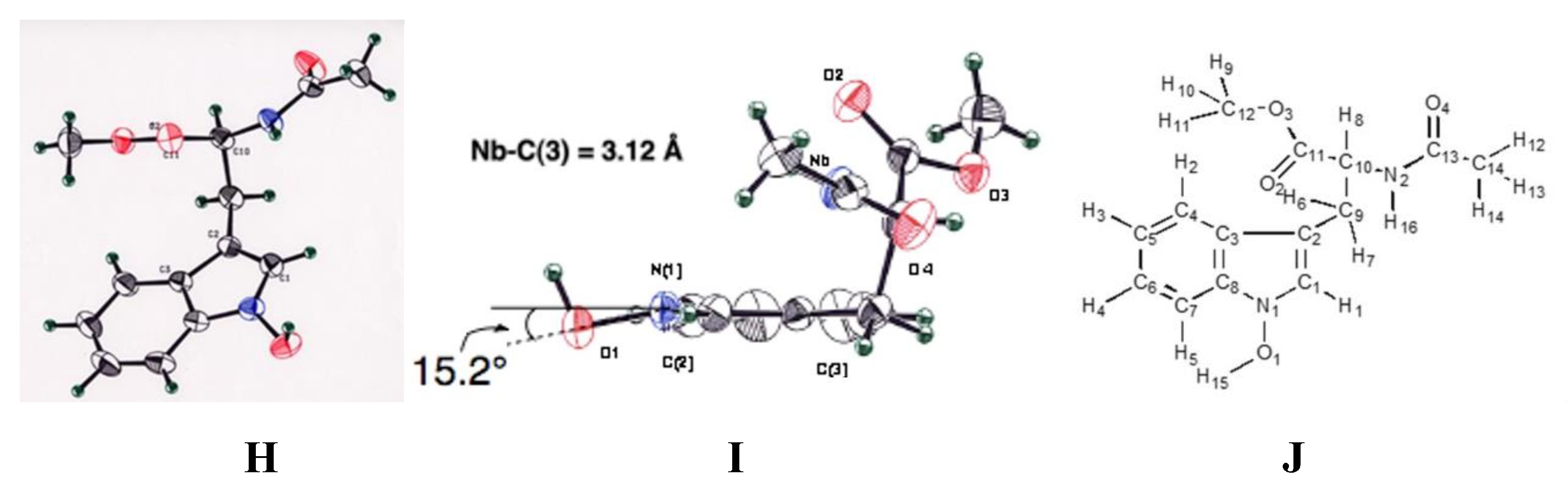
Figure 117.
ORTEP Drawing and Numbering of 53b.
Figure 122.
.

Table 1.
Average root length of rice and cucumber.
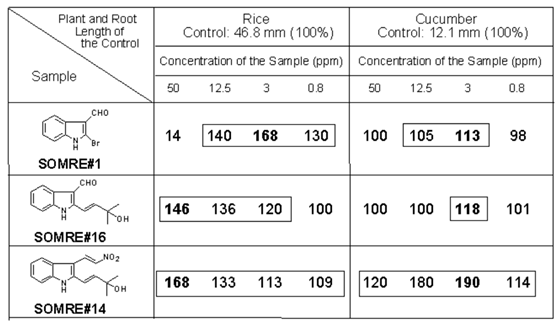 |
Table 2.
Crop of Tomato (solanaceae) per one stem.
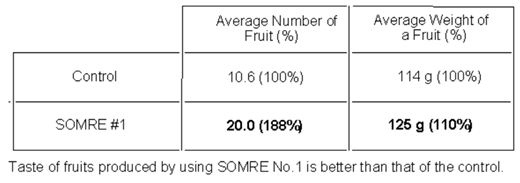 |
Table 3.
Average height, girth of tomato plant.
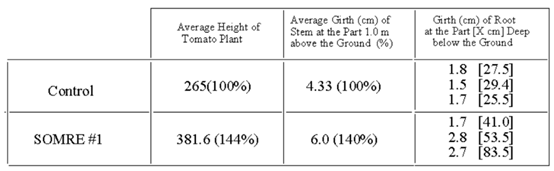 |
Table 4.
Root length of Tamba Black Soybeans.
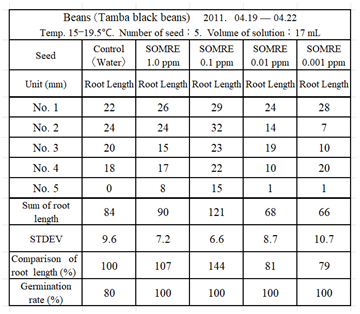 |
Table 5.
Root length of Edamame.
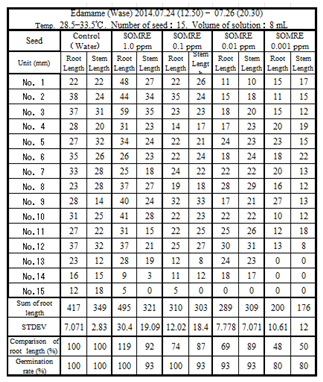 |
Table 6.
Crop of soybeans (Edamame) per one stem.
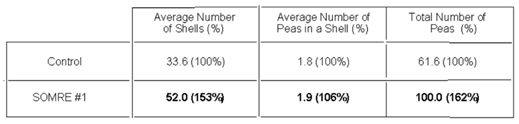 |
Table 7.
Leaf and root (bulb) weight of turnip.
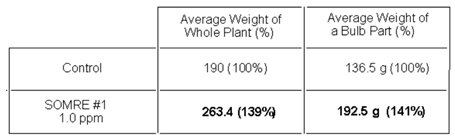 |
Table 8.
The weight of root and bulb part of garlic.
 |
Table 9.
Results of ‘Kitaakari’.
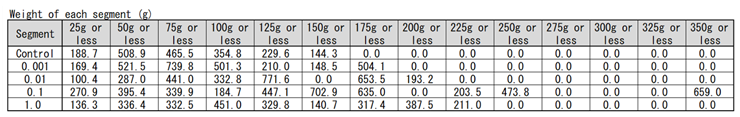 |
Table 10.
Results of Taro.
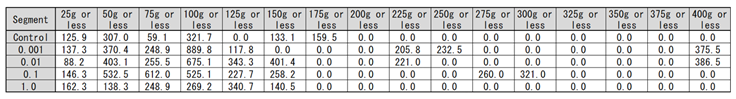 |
Table 11.
Results of Kohlrabi.
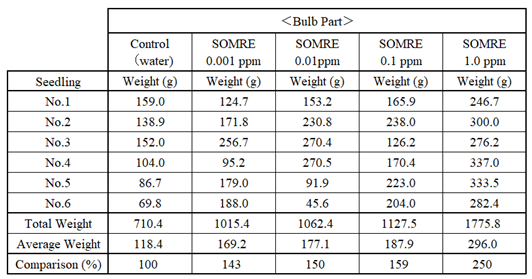 |
Table 12.
Weight of rice, comparing the amount of brown rice obtained after threshing.
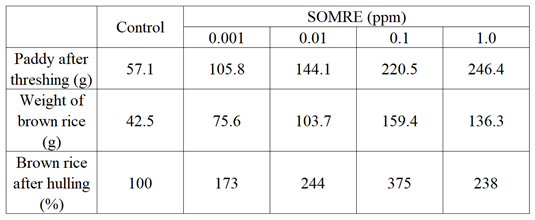 |
Table 13.
Optimal concentration for Corn.
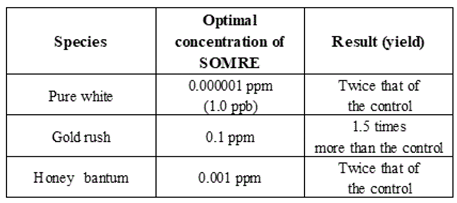 |
Table 14.
Results of basil.
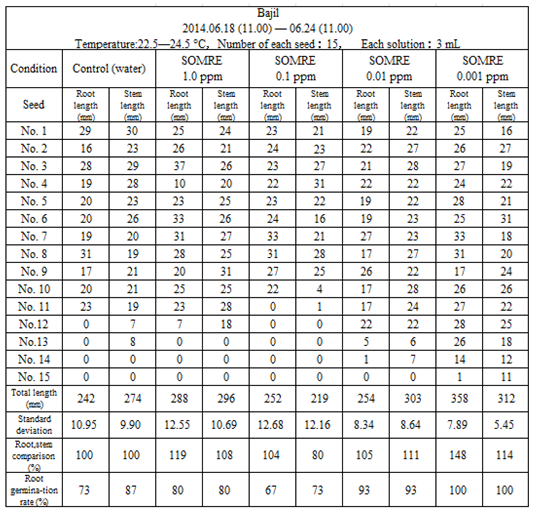 |
Table 15.
Results of Coriander.
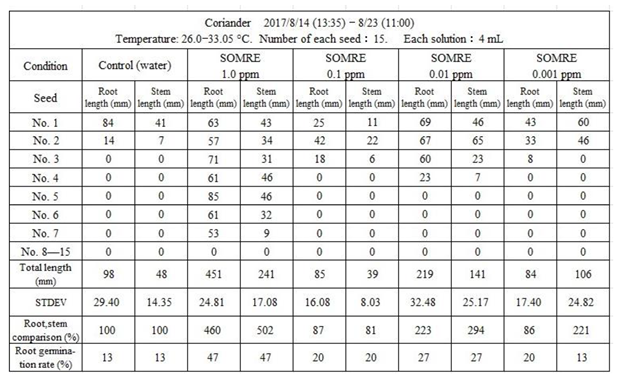 |
Table 16.
Results of Ginseng.
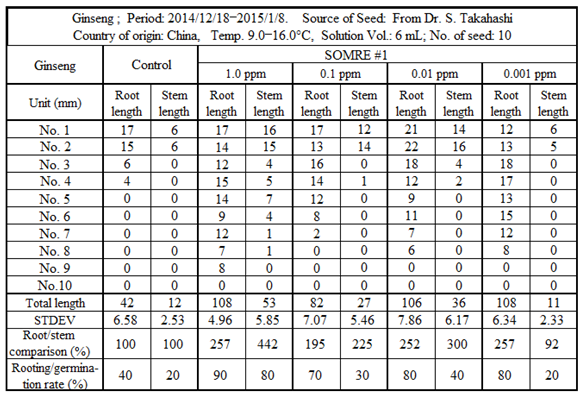 |
Table 17.
Results of Maca.
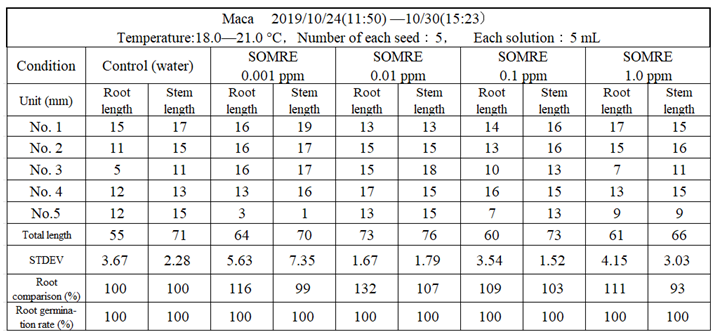 |
Table 18.
Results of okra.
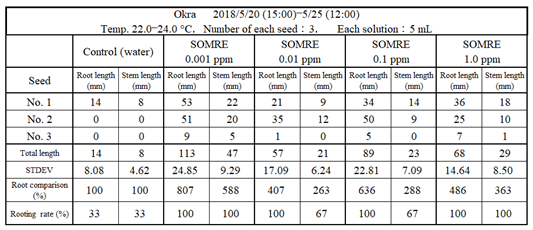 |
Table 19.
Results of corn.
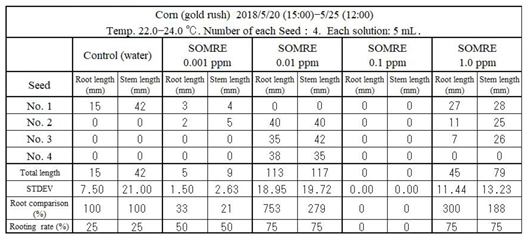 |
Table 20.
Results of burdock.
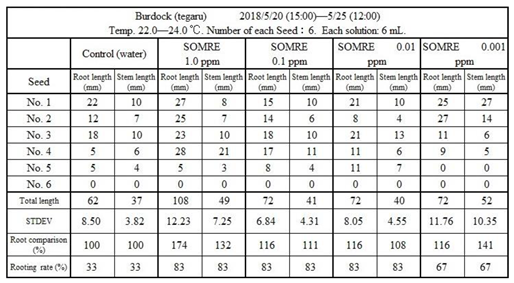 |
Table 21.
Rooting experiment of Indigenous plant in Gobi Desert.
 |
Table 22.
Results of Indian rice.
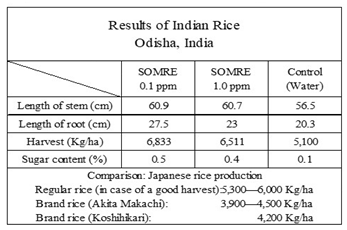 |
Table 23.
Harvest of other Indian crops.
 |
Table 24.
Results of Nerica rice.
 |
Table 25.
Results of hyacinth.
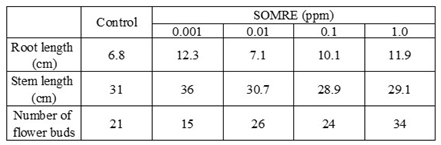 |
Table 26.
α2 Receptor inhibitory effect.
 |
Table 27.
Cashmere hair production in ranch I and II.
 |
Table 28.
Effects of 1-hydroxyindoles on arachidonic acid induced platelet aggregation. in rabbit PRP.
Table 28.
Effects of 1-hydroxyindoles on arachidonic acid induced platelet aggregation. in rabbit PRP.
 |
Table 31.
Positional Parameters and B (eq) for (dl)-131.
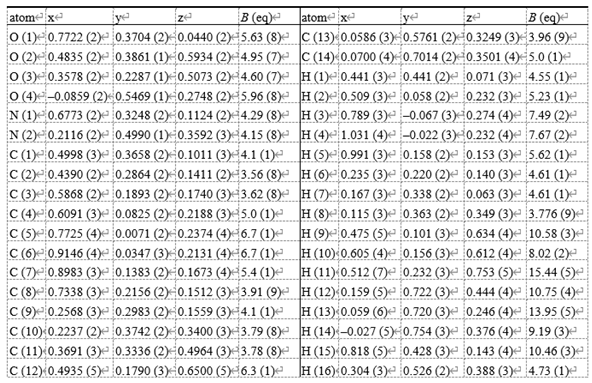 |
Table 32.
Positional Parameters and B (eq) for 53b.
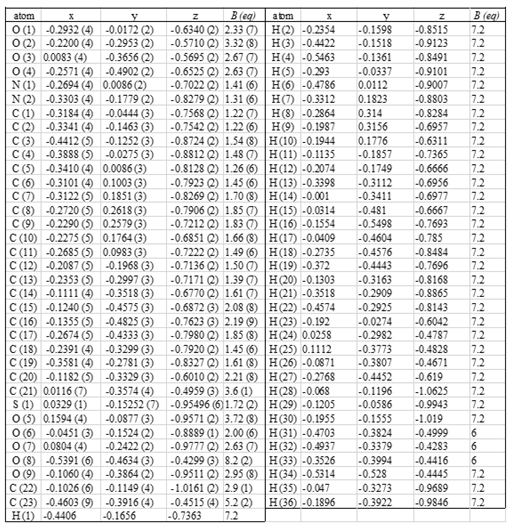 |
Table 33.
Positional Parameters and B (eq) for 135a.
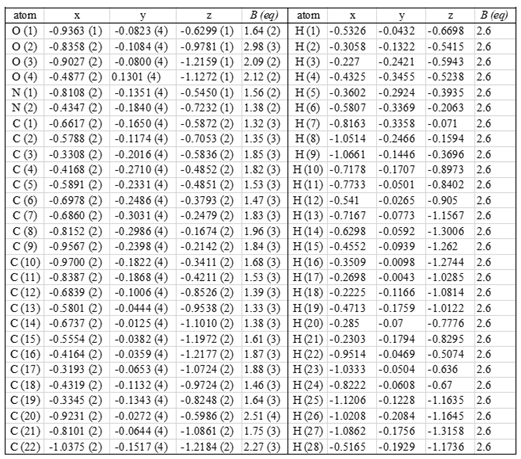 |
Table 34.
Positional Parameters and B (eq) for 137b.
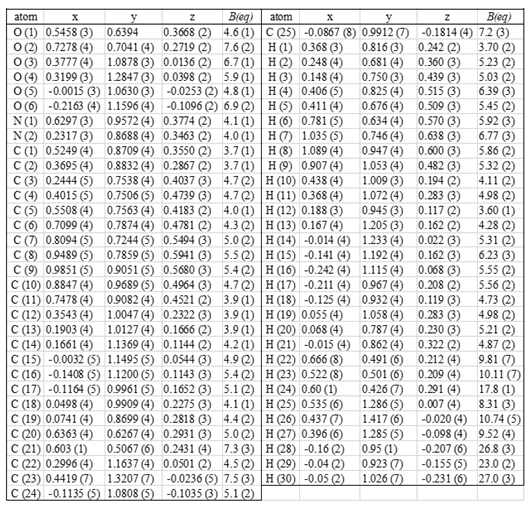 |
Table 35.
Positional Parameters and B (eq) for 148.
 |
Table 36.
Positional Parameters and B (eq) for 164a.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



