
Submitted:
10 October 2024
Posted:
14 October 2024
You are already at the latest version
Abstract
Runs of homozygosity (ROH) are contiguous homozygous segments occurring throughout the genome. ROH's number and size heavily depend upon shared parental ancestry, particularly relevant in the context of parental consanguinity. Homozygosity mapping, a valuable technique for the identification of genes associated with human genetic diseases, is based on the presence of ROH. Next-generation sequencing (NGS) improved this process by allowing simultaneous homozygosity mapping and detection of disease-causing variants. In this work, we present the methodology to automate the creation of personalized multigene panels based on whole-exome sequencing (WES) data using ROH (identified by combining two different algorithms: ROHMMCLI and HomozygosityMapper) and/or Human Phenotype Ontology (HPO) terms, and its integration in a Django Web application. The new resources’ applicability and impact on diagnostics are demonstrated through the genetic characterization of two siblings affected by a recessive disease. Resorting to a sample of 3,941 patients generated in this work, we provide an extensive analysis of ROH at a genomic scale for the first time in the Portuguese population. This work also describes the development of a new classification approach based on ROH features. In summary, this research advances ROH analysis from WES data, emphasizing its diagnostic potential and significance in population genetics’ characterization.
Keywords:
Regions of homozygosity
; bioinformatics model
; variant prioritization
; whole-exome sequencing
; consanguinity
; multigene panels
; recessive diseases
1. Introduction
Runs of homozygosity (ROHs) are continuous segments of the genome identical in both copies of a chromosome pair (alleles) ranging from tens of kilobases to megabases [1]. Homozygosity refers to having two identical alleles of a gene inherited from each parent [2], and due to common ancestry between the parents or due to identity-by-descent (IBD), it is called autozygosity [3]. ROHs can arise from consanguineous marriages (estimated to affect ~10% of people worldwide) [4], inbreeding [5,6], or founder effect [7], increasing the risk of recessive diseases in the offspring. ROHs may also be “runs of hemizygosity”, when there is a deletion in one copy of a chromosome, leading to a loss of heterozygosity [8].
ROH patterns reflect the level of kinship and autozygosity, both reduced by people’s mobility and globalization [9]. Short ROH are characteristic of admixed populations resembling ancient parental relatedness, whereas longer ROH reflect higher consanguinity levels and recent parental relatedness [10,11,12,13]. ROH patterns reflect population and demographic history [14,15], including differences in consanguinity and number of ROHs between ethnic subgroups [10,16,17,18,19,20,21]. Understanding these patterns in diverse populations is essential for assessing disease risk and identifying disease-causing genetic variants, particularly in admixture isolates [11,22,23,24].
As consanguinity increases, so does the number and size of ROHs, raising the risk of autosomal recessive (AR) diseases. ROH analysis increases the diagnostic rate of recessive diseases, especially in consanguineous families, for finding candidate genes [25,26,27,28,29], disease-causing homozygous variants [30], and corroborating the historical context of communities [31]. Furthermore, it is crucial for identifying candidate genes for specific recessive diseases [32,33,34,35,36], even in non-consanguineous families [37]. Biodemographic and genetic studies provide insights into population structure and its link to diseases, by exploring the human genome's significance in population history and consanguinity practices [14,38].
Homozygosity mapping (ROH detection) aids gene discovery by assuming individuals with AR diseases likely have homozygous markers surrounding the disease locus, searching for and identifying regions harboring the affected gene. If other relatives also have the disease, the strategy includes identifying ROHs exclusive to affected individuals within the family [39]. It was first applied in 1987 by Lander and Botstein in consanguineous families affected by a recessive disease using restriction fragment length polymorphisms (RFLPs) [40]. Homozygosity mapping evolved to utilize Single Nucleotide Polymorphism (SNP) array data, and with the advent of next-generation sequencing (NGS), software tools were designed to accommodate this sequencing data as input [39].
NGS introduction enables simultaneous homozygosity mapping and variant detection, generating vast data volumes surpassing previous technologies in speed and cost-effectiveness. Since 454 sequencing by Roche, NGS has evolved through second-generation (short-read) and third/fourth-generation (long-read) technologies [41]. Second-generation sequencing generates short DNA fragments (100-600 bp), with Illumina being widely used for genetic testing [41,42]. Third/fourth-generation sequencing achieves reads of over 10 kb, effectively detecting genome-wide repeats and structural variants, suitable for diagnostic and clinical applications [41,43]. The two main technologies are provided by Pacific Biosciences (PacBio) [44], and Oxford Nanopore (ONT) [45].
NGS applications include single-gene, targeted multigene panel, whole-exome sequencing (WES), whole-genome sequencing (WGS), and transcriptome (RNA sequencing), all effective for genetic testing [46]. WES, which targets protein-coding exons, where ~85% of the known Mendelian disease variants occur, has become a mainstream approach due to its cost-effectiveness and simplified data management [47,48,49]. WES can be done individually or in trio (enhanced variant identification) [50]. Its limitations include sensitivity to GC-rich regions, reliance on Sanger sequencing to confirm low-quality variants, challenges with variants of uncertain significance (VUS), shared homology between genomic regions (segmental duplications/ pseudogenes and failure to genotype highly repetitive regions completely and especially when large repeats expansions [2,51,52].
Re-analyzing genomic data enhances diagnostic rates by uncovering novel gene-disease associations, improving bioinformatics techniques for CNV detection and variant calling, incorporating consanguinity assessment (ROH filter) to narrow down the list of candidate variants, and integrating the Human Phenotype Ontology (HPO) [51] terms. HPO terms describe human phenotypic information in a standardized way (used for supporting clinical diagnostics and genetic research). Estimating consanguinity through ROH analysis allows for an unbiased determination of parental or ancestral consanguinity, overcoming the limitations of self-reports or inferences based on family context [52].
Homozygosity mapping tools, both adapted and new, emerged [39], enhancing diagnostic rates by combining WES data and ROH analysis [53,54]. The software can be based on sliding-window or hidden Markov model (HMM) algorithms [39]. Sliding-window algorithms, originally designed for SNP array data analysis, move a fixed-size window along the chromosome to find stretches of consecutive homozygous SNPs [39]. PLINK [39] is widely used on its own [16,55,56,57,58,59,60,61,62,63,64,65], as a complementary analysis [66], or integrated into other algorithms [67]. Other software followed, such as Obelisc [68], GERMLINE [39], EX-HOM (EXome-HOMozygosity) [69], and HomozygosityMapper (HM) [70]. PLINK, GERMLINE and HomozygosityMapper (HM) were subsequently adapted for WES data [39,71]. Other software created includes HOMWES [72], GARLIC [73], HomSI [39,74], and Automap [75].
Hidden Markov models (HMM) represent observed data as outputs generated by hidden states, modelled as a Markov chain [76]. In ROH detection, HMMs estimate the likelihood of a genotype (observation) being homozygous or heterozygous (hidden states) [77]. The software tools available are H3M2 [77,78]; IBDSeq and GIBDLD [78]; BEAGLE [79]; ROHMM and BCFtools/RoH [77,80]; and Python packages FILTUS and hapROH [81,82,83].
The accuracy of these tools can be influenced by many factors, such as the choice of algorithm used, sample sequencing depth and coverage, SNPs density and sequence quality, the need for phased data, loss of short and medium-sized ROHs, and false positives [39,82]. These factors should be considered when selecting the appropriate software for a project [39,83].
This work presents new bioinformatics approaches to address the creation of personalized multigene panels based on WES data using ROH and/or HPO terms, integrated into a Django Web application. Its impact on diagnostics is illustrated by the genetic characterization of two siblings affected by a recessive disease. Analysis of ROH at a genomic scale in a representative sample of 3,941 patients advances ROH analysis using WES data, highlighting its diagnostic potential and significance in population genetics.
2. Materials and Methods
The dataset used in this work consisted of WES samples from patients who performed genetics tests at the Center for Predictive and Preventive Genetics (CGPP), Portugal.
2.1. Creation of Personalized Multigene Panels Based on ROH
Multigene panels based on the patient’s ROH focus on the analysis of regions of the genome more likely to contain recessive disease-causing variants. By targeting genes within these ROH, the panels are more likely to identify relevant genetic variants, particularly in consanguinity context or shared ancestry.
The samples used for the creation of these panels were analyzed using two Homozygosity Mapping algorithms, HomozygosityMapper (HM), which uses a sliding-window algorithm, and ROHMMCLI, which uses a hidden Markov model (HMM) algorithm. Each patient has a pseudo-anonymized ID without any personal information.
Both algorithms output data in different formats: HM outputs a raw data text file with chromosome, position and score, while ROHMMCLI outputs a BED file. To generate the Uniform Resource Locator (URL), a connection to the HM database is initiated and the project number (project_no) is retrieved using the patient ID. With the URL generated, the data is collected and saved in a BED File. Then, the HM and ROHMMCLI BED files are merged using a shell script. This script takes the patient ID and the current date as inputs and is divided into four Linux commands, as follows:
- Clean up the ROHMMCLI BED file to contain only chromosome, start and end positions.
- Merge the HM and the cleaned ROHMMCLI BED files, using bedtools merge with option -d of 1000000 bp, the maximum distance between ROHs to be merged.
- Use bedtools intersect to find overlaps between the merged BED file and the coding sequence coordinates BED file, producing another BED file with the list of gene coordinates found within ROHs.
- Create a text file with a list of gene Entrez IDs present in the identified ROHs.
The process of obtaining the gene list is outlined in Figure 1.
The file containing all coding sequence coordinates was generated using two in-house developed tools (gtf2tsv.py and tsv2bed.py). For this work, the file used was a GTF file named GCF_000001405.25_GRCh37.p13_genomic.gff.gz, representing the RefSeq annotations release version 105.20220307 of the human genome build GRCh37, and the “feature” column was filtered for “CDS” (available at: https://ftp.ncbi.nlm.nih.gov/genomes/all/annotation_releases/9606/105.20220307/GCF_000001405.25_GRCh37.p13/) (Figure 2).
The final step to generate multigene panels consists of comparing the genes’ list with the coverage for the representative transcript of each gene, described in Figure 3. Genes are divided into three lists based on the percentage of horizontal coverage at 20x: white (≥0.9), grey (0.1-0.9), and black (≤0.1). Only the white and grey genes are included in the multigene panel. Another list, containing the genes that were not assorted to any of the previous lists, is generated.
Copy Number Variations (CNVs), more specifically heterozygous deletions, can mimic ROH, given that the single nucleotide variants (SNVs) encompassed by the deletion cannot be heterozygous (and are in fact hemizygous). To incorporate such a possible impact in the multigene panel creation, the following steps were implemented:
- Find the CNV results for the sample in analysis and filter by CNVs with a span above 500,000 bp and that are ‘Heterozygous Deletion’, resulting in a BED file with CNV’s genomic coordinates.
- Filter by non-empty files, meaning files that contain CNVs.
- The shell script uses bedtools jaccard tool to calculate the Jaccard index for each CNV that intersects an ROH, using the merged ROH results and CNV BED files.
The Jaccard index (equation 1) is a single statistic that reflects the similarity of the two BED files based on the intersections between them, where a value of 0.0 indicates no overlap and 1.0 represents complete overlap.
Where the intersection is the difference between the end of the ROH and the start of the CNV, the ROH length is the difference between the end and start coordinates of the ROH and the CNV length is the difference between the end and start coordinates of the CNV.
2.2. Creation of Personalized Multigene Panels Based on HPO Terms
Multigene panels based on HPO terms ensure that gene selection is targeted for the patient’s specific phenotype. These panels are designed to ensure that only the genes possibly associated with the phenotype are analyzed, and therefore the most likely to harbor disease-causing variants responsible for the patient’s phenotype, increasing the accuracy and relevance of genetic testing.
A Python script that takes as input an HPO term was created to establish the connection to the HPO API and retrieve the list of genes’ Entrez IDs associated with the HPO identifier (https://hpo.jax.org/api/hpo/term/{hpoId}/genes, version 1.7.13, accessed May 2023). By parsing the JSON file retrieved by the HPO API, we were able to get the gene entrez ID and gene symbol. Then the white, grey and black lists for the gene panels were created, as previously described in Figure 3.
2.3. Creation of Personalized Multigene Panels Based on ROH and HPO Terms
The integration of ROH and HPO term analysis may offer an even more personalized approach. This method focuses on the individual's ROH and examines whether the genes associated with the patient's specific HPO terms are located within these regions.
A new script was generated based on the previously described script for creating multigene panels. To obtain a BED file with the coordinates of the genes from the HPO terms’ gene list, the command-line tool tsv2bed.py was used. The merged BED file results containing the ROHs and the HPO terms genes’ BED file are merged to get a final list of the genes from the HPO terms within the ROHs. The corresponding flowchart is presented in Figure 4.
From the list of genes obtained, the process of generating the white, grey and black lists for the gene panels is the same as previously described in Figure 3.
2.4. Django Web Application Development
The process of personalized multigene panel creation based on ROH, HPO and both, simultaneously, was made using Python 3 and shell scripting. To deploy a more user-friendly interface, a Django web application was developed. The design/style of all HTML pages for this application (app) was made using Cascading Style Sheets (CSS) for visual consistency.
The home page of this app contains a title (“Personalized multigene panels”), a small description of the app, and three buttons: “HPO term-based panels”, “ROH based panels” and “HPO term and ROH-based panels”. Each button is linked to a different HTML file, with different HTML forms to submit the input data.
For the “HPO term-based panels” button, the form handles multiple HPO terms at the same time. For the “ROH-based panels” button, the form contains two different types of input, a text area to fit the multiple input lines, and a drop-down list of the possible HM threshold options.
For the “HPO term and ROH-based panels” button, the form is a combination of the ones from the previously described HTML files. The only difference is that the text area only allows the analysis of one sample at a time with multiple HPO terms.
2.5. Establishing the First Portuguese ROH Characterization on a Genomic Scale
To establish the first Portuguese ROH characterization on a genomic scale, the dataset initially consisted of over 12,000 WES samples. Since there were municipalities that were over-represented, normalization and down-sampling processes were automated. The final number of samples was 3,941 WES samples (detailed process in Supplementary File S1).
The process of establishing the first Portuguese ROH characterization started with the assessment of the ROH levels through the genome-wide autozygosity measure from ROH (), calculated using equation 2.
Here, is the total length of all of an individual’s ROHs above a specified minimum length and is the length of the autosomal genome covered by WES, after removing the centromeres (which are excluded to prevent overestimating autozygosity).
For each sample, three values were calculated, using three ROH minimum size thresholds to calculate : 0.5, 1.5 and 5 Mb.
The calculation of involved using the Integrative Genomics Viewer (IGV) to determine the genomic coordinates of the first and last genes on the p and q arms of each chromosome. These coordinates were used to calculate the size of each chromosomal arm using Equation 3:
The sum of both chromosomal arms’ sizes corresponds to the size of the autosome without the centromeres. value is the total size of all autosomes, calculated as 2638.813981 Mb.
The patients’ address information was obtained from the internal database, consisting of patients’ ID, postcode, municipality and district names. All patients’ VCF files were previously processed by HM and ROHMMCLI and both results were merged. Homozygosity mapping data was organized into standardized CSV (*.csv) files containing the detailed patient ROH profile (chromosome, ROH’s start and end position, ROH length (bp), ROH length (Mb), and ROH length/chromosome length), and the general patient’s profile (Number of ROH, Number of ROH > 1 Mb and Sum ROH (Mb)).
To calculate the FROH, the information needed was combined into separate CSV (*.csv) files:
- One containing ROH > 0.5 Mb;
- One containing ROH > 1.5 Mb;
- One containing ROH > 5 Mb.
Then, the was calculated per patient, grouping the ROHs, from each CSV (*.csv) file and summing its total per patient.
The value of FROH was calculated for each patient using the corresponding value, for each minimum ROH size, resulting in an CSV (*.csv) file containing the patients’ ID and FROH. Portugal comprises 18 districts and 2 Autonomous Regions (Açores and Madeira), divided into a total of 308 municipalities. With this information, and the address information, patients were grouped by municipality, and the mean FROH was calculated (per municipality) and used to create Maps of the Portugal Mailand and Autonomous Regions (Açores and Madeira) per municipality.
There were two types of maps, the classical and the interactive ones, created for each FROH mean calculated using minimum ROH’s size of 0.5, 1.5 and 5 Mb.
To create the maps several data were necessary. The geographical data, at the municipality level (shapefile) was obtained from dados.gov (https://dados.gov.pt/en/datasets/concelhos-de-portugal/, accessed June 6, 2023), the Portuguese Public Administration's open data portal. Then we established the connection to the internal SQL database to get the municipality and respective districts’ association, the CSV (*.csv) files per municipality and a CSV (*.csv) file containing the number of people per municipality and respective ratio, so that only the municipalities with representativity were used for the maps. For the creation of the maps, since we were dealing with geospatial data, we used the GeoPandas package. The maps created were stored as PNG (*.png) files.
The interactive maps were created using the explore method on a Geodata Frame and were saved as HTML files.
2.6. Consanguinity Classification Approach
The set of samples used to build the clustering model was meticulously chosen from the internal database based on the patients’ information concerning consanguinity. A total of 9,160 WES samples were collected for the analysis. This included 9,020 (98,5%) individuals with “unknown” consanguinity, 84 (0.92%) known non-consanguineous samples, and 56 (0.61%) known consanguineous samples. Of the 56 consanguineous samples, 34 (60.7%) were stringent (i.e. parents were first-degree cousins). Each sample was submitted to HM to find homozygous blocks of the exome and the raw data was provided in a text file containing the chromosome, position and their corresponding homozygosity scores from HM.
2.6.1. Feature Extraction
For each sample we generated features pertaining to the descriptive statistics of the ROH embedded within each chromosome. For the purposes of this analysis, we considered ROH to be at least 2 consecutive chromosome positions with homozygosity scores greater than or equal to 64 (i.e., 80% of the highest observed score, 80, as defined by [71]). Specifically, the following features were generated for each sample with respect to each chromosome x:
- Count_x: the number of ROH in chromosome x.
- Sum_x: the sum of ROH sizes in chromosome x.
- Min_x: the minimum ROH size in chromosome x.
- Max_x: the maximum of ROH size in chromosome x.
- Mean_x: the mean of ROH in chromosome x.
- STD_x: the standard deviation of ROH size in chromosome x.
To make these features more concrete, we provide an illustrative example. Suppose an individual possesses three ROHs within chromosome 1 (x = 1), with sizes 3, 7, and 4 Mb. The following features are extracted from chromosome 1: Count_1 = 3; Sum_1 = 14; Min_1 = 3; Max_1 = 7; Mean_1 = 4.67; and STD_1 = 1.6997. This feature extraction process is then repeated for the individual’s remaining chromosomes.
Following feature extraction, to test the predictive quality of various feature sets in our experiments, we created three separate representations of the data, dictated by the following sets of features:
- Tier 0: includes "Count_x" and "Sum_x" features only;
- Tier 1: includes "Count_x," "Sum_x," "Min_x," and "Max_x" features only;
- Tier 2: includes "Count_x," "Sum_x," "Min_x," "Max_x," "Mean_x," and "STD_x" features.
2.6.2. Outlier Detection
We formulate the task of consanguinity classification as an outlier detection problem. For our experiments, we randomly selected 50% of all labeled data points (70 total data points) to be reserved for testing. Using the remaining 50% of labeled data points for validation and 100% of unlabeled data points for training, we proceeded to establish the semi-supervised outlier detection pipeline. First, we projected the data into a low-dimensional (2-D) space using classical multidimensional scaling (MDS) [84], which is a manifold learning approach that aims to preserve pairwise Euclidean distances between points from the high-dimensional representation in the low-dimensional data representation. Following dimensionality reduction, we then fit an elliptic envelope [85] to the data with “unknown” consanguinity labels, validating the optimal contamination hyperparameter (i.e., the proportion of the data estimated to be outliers) using the remaining 50% of the labeled samples. Given the imbalanced class distribution, the F1-score was used to both optimize the contamination hyperparameter and evaluate the model on the reserved test set.
3. Results
The results obtained are presented in this section. Figure 5 contains an overview of the results obtained.
3.1. Personalized Multigene Panels
Personalized multigene panels streamline diagnostics by narrowing down the number of genes analyzed, leading to a more targeted and efficient diagnosis. In cases of suspected AR diseases, or when the patient's consanguinity status is known, ROH analysis can help in the selection of the appropriate multigene panel. These panels can also be further tailored to the patient's specific phenotype by using HPO terms. Integrating this process into a web application provides a user-friendly interface, making it accessible to other professionals within the genetic testing center.
During the process of creating the web application for personalized multigene panels based on ROH, HPO and a combination of both, several tests were conducted.
For testing the personalized multigene panels based on ROH, several randomly selected patients were used and the identified genes’ coordinates were checked to ensure they fell within the identified ROHs.
The testing process of the creation of personalized multigene panels based on HPO terms was divided into three parts:
- The creation of 15 multigene panels based on a single HPO term: HP:0001627 (Abnormal heart morphology), HP:0001047 (Atopic dermatitis), HP:0005584 (Renal cell carcinoma), HP:0001789 (Hydrops fetalis), HP:0011842 (Abnormal skeletal morphology), HP:0000846 (Adrenal insufficiency), HP:0003155 (Elevated circulating alkaline phosphatase concentration), HP:0000548 (Cone/cone-rod dystrophy), HP:0011510 (Drusen), HP:0000365 (Hearing impairment), HP:0000925 (Abnormality of the vertebral column), HP:0001949 (Neoplasm of the gastrointestinal tract), HP:0007373 (Motor neuron atrophy), HP:0006530 (Abnormal pulmonary interstitial morphology), HP:0012211 (Abnormal renal physiology), HP:0001733 (Pancreatitis), HP:0000556 (Retinal dystrophy);
- The creation of 3 multigene panels based on multiple HPO terms: HP:0000077 (Abnormality of the kidney), HP:0100243 (Leiomyosarcoma) and HP:0100522 (Thymoma); HP:0100574 (Biliary tract neoplasm) and HP:0003003 (Colon cancer); and HP:0003198 (Myopathy) and HP:0003473 (Fatigable weakness);
- The creation of 5 personalized multigene panels based on a single HPO previously manually prepared and curated: HP:0000126 (Hydronephrosis), HP:0001250 (Seizure), HP:0010566 (Hamartoma), HP:0012091 (Abnormality of pancreas physiology term) and HP:0012114 (Endometrial carcinoma), and comparison with the obtained results.
3.1.1. Output Obtained for Each Multigene Panel
The output for the multigene panels based on ROH, for each input line, is a CSV (*.csv) file with the gene symbols that belong to each of the lists (white, grey, black). If CNVs’ results are available for the sample being analyzed, a BED file with the CNVs’ genomic coordinates is retrieved, as well as a text file with the Jaccard index of the overlap.
The output for the multigene panels based on HPO terms, for each input line, is a CSV (*.csv) file with the gene symbols that belong to each of the lists (white, grey, black). The output provided by the different multigene panels depends on the number of samples and on the number of HPO terms.
As for personalized multigene panels, simultaneously based on ROHs and HPO terms, the output is a CSV (*.csv) file with the gene symbols from the HPO term(s) in analysis within the ROHs identified in the sample being analyzed. If CNVs’ results are available for the sample in analysis, a BED file with the CNVs is retrieved, as well as a text file with the Jaccard index.
3.1.2. Application of New Bioinformatic Resources in a Clinical Case
The clinical case presented consists of two siblings with a phenotype of epilepsy, myoclonus and dystonia with onset during infancy, daughters of a consanguineous couple (Figure 6). Even after conducting several genetic tests, including an analysis of the entire WES data, both remained genetically undiagnosed for several years.
The HPO term used was HP:0001336 (Myoclonus). Myoclonus is characterized by involuntary random muscular contractions happening at rest, due to a stimulus or during voluntary movements. Figure 7 presents the HTML forms filled with the HPO term used and the path to the VCF file of one of the sisters to create the personalized multigene panel.
The CSV (*.csv) output file contains the “Summary”, “White list”, “Grey list” and “Black list”. According to the HPO API, HP:0001336 (Myoclonus) comprises 360 genes (accessed September 25, 2023). The results obtained consisted of 11 genes listed for sister II:1 and 3 genes listed for sister II:2. All genes identified in both sisters were from the white list, no genes were in the grey and black lists. The white lists’ results from the sisters’ selected genes, resulting from the intersection of the genes within the identified ROH and those associated with the HPO term used, are presented in Table 1. From the 360 genes associated with HP:0001336, 3 genes were commonly shared between the 2 sisters: CSTB, SIK1 and SLC32A1.
Assuming an AR inheritance and the strong correlation between the phenotype associated with defects in CSTB gene and the patients’ phenotypes, variant data was further inspected. Integrative Genomics Viewer (IGV), a visualization tool, was used in the genomic data analysis. The VCF files, BAM files and respective index files were loaded to IGV, as well as the all_cds.bed file as reference. The analysis was conducted using the Human reference genome version GRCh37. The visualization results for the CSTB gene are depicted in Figure 8. No disease-causing variants were identified in the genomic regions covered by WES data.
Considering that the mutational spectrum associated with disease-causing variants in the CSTB gene includes the expansion of a repetitive region [86], the 5’UTR region where the dodecamer repeat CCC-CGC-CCC-GCG is located was visually inspected (Figure 9). As it is demonstrated no reads are aligned in this region in both patients, whereas these are present in the control sample. This is compatible with a large expansion of the dodecamer repeat in both gene’s alleles. A biallelic expansion within the pathogenic range (≥30 repeats) was indeed confirmed by targeted conventional approaches (fragment analysis and long-range PCR), making this variant diagnostic for the disease.
3.2. First Portuguese ROH Characterization on a Genomic Scale
After the down-sampling process, the 3,941 samples were submitted to ROH analysis to contribute to the portrayal of the first Portuguese landscape of ROH at a genomic level. The lack of data regarding ROH distribution in Portugal can be filled in with the study presented in this section, being of great interest for genetic testing and population genetics.
3.2.1. Distribution of ROHs per Length in Portugal
We began by analyzing the distribution of ROHs larger than 0.5 Mb, identifying a total of 19,407 ROHs. For an overview of the results, Figure 10 depicts the distribution of these ROHs across different length intervals in Mb.
3.2.2. Maps of Portugal and Respective Data for FROH > 0.5, 1.5 and 5 Mb
The results from the genome-wide autozygosity measure from ROH (FROH) are presented in this section. The FROH mean values per municipality, with ROHs of size greater than 0.5, 1.5 and 5 Mb, are presented in Supplementary File S2, and the details for the interactive maps, designed for more detailed and interactive navigation, are presented in Supplementary File S3.
Here we present the resulting classical maps of the representative Portuguese sample of 3,941 patients for the ROH characterization on a genomic scale. In
Figure 11 the classical map of Portugal presents the geographical distribution of the mean FROH, for ROHs with size greater than 0.5 Mb (FROH > 0.5 Mb), per municipality. The mean value of FROH > 0.5 Mb is 0.004.
The municipality of Alter do Chão from Portalegre district is the municipality with the highest value of FROH > 0.5 Mb (0.088). The lowest value of FROH > 0.5 Mb (0.0004) is from Manteigas’ municipality (Guarda district). The municipality of Machico has the highest value of FROH > 0.5 Mb (0.025) of the Autonomous Region of Madeira. In the Autonomous Region of Açores the values of FROH > 0.5 Mb are not very high, with the highest one being 0.026 in Vila do Porto municipality.
The mean FROH > 0.5 Mb intervals and the corresponding number of individuals per interval is presented in Table 2. From the 3,941 samples, 3,800 are represented in the map. The first interval, from 0.000 to 0.004, contains the highest number of samples (3,104). There is a drastic drop in the number of samples that within the second interval (from 0.004 to 0.006) and the number decreases until the last interval (from 0.034 to 0.088).
In Figure 12 the classical map of Portugal for the FROH mean for ROHs with size higher than 1.5 Mb (FROH > 1.5 Mb), per municipality, is presented. The mean value of FROH > 1.5 Mb is 0.003.
The municipality of Alter do Chão from Portalegre district is still the municipality with the highest value of FROH > 1.5 Mb, with a value of 0.085, which is lower than the 0.088 from the previous FROH > 0.5 Mb. The lowest value of FROH > 1.5 Mb (0.0001) is from Felgueiras municipality from the district of Porto. The municipality of Machico still contains the highest value of FROH > 1.5 Mb (0.024) in the Autonomous Region of Madeira. The municipality of Vila do Porto with a mean FROH of 0.024 is the municipality with the highest value from the Autonomous Region of Açores.
Table 3 presents the corresponding number of samples that fit within each FROH > 1.5 Mb interval. The total number of samples with FROH > 1.5 Mb is considerably low, 2,110 out of the 3,941. The behavior of the distribution of samples within each interval is similar to the one observed in Table 2. The first interval, from 0.000 to 0.003, contains the highest number of samples (1,430). There is a drastic drop in the number of people with FROH belonging to the interval from 0.003 to 0.005 and the number decreases until the last interval (from 0.033 to 0.085).
In Figure 13 the classical map of Portugal for the FROH mean for ROHs with a size higher than 5 Mb (FROH > 5 Mb), per municipality, is presented. The mean value of FROH > 5 Mb is 0.002. The decreasing tendency of the mean FROH value is expected, since with each minimum threshold of the ROHs size, the number of people with the mean FROH value equal to zero increases.
The municipality of Alter do Chão from Portalegre district is still the municipality with the highest value of mean FROH > 5 Mb. The value of mean FROH is 0.074, lower than the 0.085 observed in when FROH > 1.5 Mb. The lowest value of FROH > 5 Mb (0.0001) is from Santo Tirso from the district of Porto. The municipality of Machico is still the one with the highest value of mean FROH > 5 Mb (0.017) in the Autonomous Region of Madeira, but also showing a decreasing tendency, because the number of people with ROHs > 5 Mb is lower. The municipality of Vila do Porto with a mean FROH of 0.02 is the municipality with the highest value in the Autonomous Region of Açores.
Table 4 presents the corresponding number of samples that fit within each FROH > 5 Mb interval. The total number of samples with FROH > 5 Mb is even lower than before, 758 out of the 3,941. Contrary to the previous tables (Table 2 and Table 3), the first interval, in this case from 0.000 to 0.002, has the lowest number of samples (38) out of all the intervals. The interval with the highest number of people is from 0.002 to 0.004 with 318 people, then the distribution tendency is similar to the previous tables, showing a decrease up to the last interval.
The FROH mean distribution throughout Portugal is heterogeneous, as seen in the maps (Figure 11, Figure 12 and Figure 13). The number of municipalities with no sample data is 16. Overall, the ROH minimum size thresholds applied to calculate the FROH mean caused the non-representativeness of some municipalities. This effect is explained by the scarce number of individuals with ROHs of size greater than 1.5 Mb. Initially, there were 3,941 samples, after applying the first threshold (only considering the ROHs with size above 0.5 Mb) the sample size dropped to 3,800, then to 2,110 with the 1.5 Mb threshold and finally to 758 with the 5 Mb threshold.
3.2.3. Comparison with Other Studies
To compare our results, we used the reference values from a similar study developed using an insular population from the Orkney Isles in northern Scotland [87].
Table 5 contains the mean of all FROH calculated for all samples, the mean of the means per municipality and also the previously mentioned reference values for FROH [87].
The mean FROH values are lower than the mean FROH of the mean FROH values per municipality for all ROH thresholds. When comparing the Portuguese mean FROH values with the FROH reference values, the mean FROH for FROH > 0.5 Mb is 0.0042, which is inferior to 0.0315. The Portuguese mean value for FROH > 1.5 Mb is 0.0033 and for FROH > 5 Mb is 0.0020, both are above the reference values presented in [87], 0.0021 and 0.0001.
To compare the results, we used a study examining the prevalence of consanguineous marriages in Portugal between 1980 and 1986 [88]. The map from this study was colorized to align with the FROH mean for ROHs larger than 0.5, 1.5, and 5 Mb, and is presented in Figure 14 [88]. According to this figure, the Autonomous Region of Madeira exhibits the highest number of consanguineous marriages, closely followed by the Autonomous Region of the Açores. This observation can be attributed to the isolation of island populations, due to limited population mobility during the 1980s. However, with the advent of improved transportation infrastructure, population movement to and from the islands has become more accessible. In Portugal Mainland, the district with the highest incidence of consanguineous marriages is Bragança [88]. Furthermore, the top five districts (as shown in Table 6) with the highest number of consanguineous marriages, listed in descending order, are Madeira, Açores, Bragança, Viseu, and Vila Real.
Our findings, as presented in Table 6, reveal that the top five districts with the highest FROH mean, considering thresholds of 0.5, 1.5, and 5 Mb, remain consistent. The ranking from highest to lowest FROH mean value for 0.5 and 1.5 Mb thresholds is the following: Portalegre, Viseu, Bragança, Madeira, and Vila Real. Meanwhile, the ranking for the 5 Mb threshold is Portalegre, Bragança, Viseu, Madeira, and Vila Real.
The demographic origins of ROH [89] explored in our samples, are presented in Supplementary File S4.
3.3. Consanguinity Classification Results
The results of the outlier detection consanguinity classification approach can be found in Table 7. Meanwhile, visualizations of the low-dimensional data, separated by training and validation vs. testing datasets are shown in composite Figure 15.
The F1-scores associated with our outlier detection model remained in the range 0.9412 - 0.9615 across all feature set tiers of the held-out testing data, which was comparable to the performance range of 0.9310 - 0.9655 on the validation set, indicating successful generalization of each model to unseen data points. The inclusion of additional descriptive statistic features (i.e., Min, Max, Mean, STD) provided only marginal predictive benefit on the held-out test set, as evidenced by an F1-score increase of only 0.0203, demonstrating that the “Count_x” (i.e., the number of ROH in each chromosome) and “Sum_x” (i.e., the sum of ROH lengths in each chromosome) features provided the majority of the predictive power with respect to consanguinity classification in this framework. Moreover, the optimal contamination hyperparameters were observed in the range 0.0786 - 0.1190, which may inform the true proportion of the population who may be labeled consanguineous.
4. Discussion
Clinical diagnostics is evolving towards more personalized approaches, demanding the development of new genetic tests and the adaptation of existing ones. There are platforms created to support virtual gene panel curation, such as Genomics England PanelApp [90]. It is a database for storing virtual gene panel information while gathering community feedback, helping to build a consensus on the evidence needed to establish a gene-disease association.
With this work, we automated the process of creating personalized multigene panels based on different scenarios. Generically, all panels described in this work are less time-consuming to create, releasing professionals to other tasks to increase the number of tests carried out, and help narrow down the number of genes being analyzed. Since all the described tests are based on WES data, physicians can request a reanalysis of patient data without the need for additional sample sequencing, thereby conserving resources. In terms of storage, one sequencing per patient and a more personalized approach means less data being analyzed and consequently less allocated space.
Multigene panels based only on the specific ROH identified in a patient narrow the analysis to genes located within the identified ROH, thereby increasing the likelihood of detecting recessive disease-causing variants in those genes. Since the CNV assessment was also included in these panels, the diagnostic technician analyzing the case knows where the CNVs overlap with ROH, eliminating this confounding factor from ROH analysis.
The multigene panels based only on HPO terms take into consideration the possible phenotype or phenotypes that the patient presents. Resorting to a publicly available database, HPO, the panel is built only with the genes that are within the specified terms.
The use of HPO terms and ROH overlap results, simultaneously, is an even more personalized approach, by narrowing-down the list of genes to create personalized multigene panels. In this case, only the genes associated with the HPO term(s) in analysis are taken into account and their presence is checked in the patient’s ROHs. This allows a higher level of personalization, proven to be useful by the clinical case of the two sisters previously presented.
After having the gene list from the two sisters’ case, the common genes between the two were analyzed and visualized on IGV. The results were interpreted using Online Mendelian Inheritance in Man (OMIM) database to find the phenotype associated with the genes. According to OMIM, the CSTB gene is associated with the phenotype “Epilepsy, progressive myoclonic 1A (Unverricht and Lundborg)” (gene MIM number 601145 and phenotype MIM number 254800) and the mode of inheritance is AR. This result is in accordance with the analysis done, since the ROHs are used to target diseases with AR modes of inheritance. The gene SIK1, on the other hand, is associated with the phenotype “Developmental and epileptic encephalopathy 30”, with an autosomal dominant (AD) mode of inheritance. The gene SLC32A1 does not currently have an associated phenotype, but the gene encodes an amino acid transporter that loads the Gamma-aminobutyric acid (GABA) and glycine to synaptic vesicles. Even though there were no variants present in these genes, the diagnosis of Epilepsy was only possible through this multigene panel approach, through the visualization of the biallelic expansion on the CSTB gene.
Knowing our cohort background in terms of ROH is also important. In our study we showed the ROH distribution in Portugal (Figure 10), where most ROHs (9,358 ROHs) are within the size range of 0.5 to 1.0 Mb, followed by a decreasing tendency as the length of the ROHs increases until 4.0 Mb. Then, there is a small peak of 708 ROHs in the interval from 4.0 to 5.0 Mb, followed by a decreasing number of ROHs until the interval from 10.0 and 15.0 Mb, and another decreasing tendency in the next intervals. The minimum value is 0.5 Mb, the maximum value is 72.42 Mb and the mean value is 2.29 Mb. This is typical of a more ancient parental relatedness of the overall population.
Using the same sample with the 3,941 exomes, and by calculating the FROH per individual using the thresholds 0.5, 1.5 and 5 Mb, it was possible to build three maps using the mean value for each municipality. Consequently, we found that the top five districts exhibiting higher FROH were Portalegre, Viseu, Bragança, Madeira, and Vila Real. Furthermore, another notable finding from this study was the striking similarity between the patterns of admixed and consanguinity demographics observed in the Portuguese population when examining the Number of ROHs versus Sum of ROHs, available at Supplementary File S4.
The overall mean value of FROH for the thresholds 0.5, 1.5 and 5 Mb decreased as the minimum threshold increased. There are less samples with ROHs above a certain minimum length, with a smaller number of ROHs but with bigger sizes, per individual. The municipality of Alter do Chão from Portalegre district is the municipality with the highest value of mean FROH for all the presented minimum ROH size thresholds (0.5, 1.5 and 5 Mb), which might indicate more consanguinity.
The disparities observed in the data can derive from various factors, one significant contributor being the sample sizes utilized. In our study, we analyzed a sample of 3,941 individuals, with 3,800 exhibiting a FROH distinct from zero. In contrast, the comparative study only included 49 individuals. Notably, we compared populations from diverse geographic regions: an insular population from the Orkney Isles in northern Scotland[87] with a comprehensive Portuguese population encompassing individuals from Portugal Mainland as well as the Autonomous Regions of Madeira and the Açores. Another differing factor was the way Lauto was calculated, since the reference study referred using the length of the autosomal genome covered by SNPs in an array, excluding the centromeres, and in our study, we used the length of the autosomal genome covered by WES, excluding the centromeres.
According to the results from the study shown in Figure 14 [88], the Autonomous Region of Madeira exhibits the highest number of consanguineous marriages, closely followed by the Autonomous Region of the Açores. This observation can be attributed to the isolation of island populations, due to limited population mobility during the 1980s. However, improved transportation infrastructure has since increased population mobility to and from the islands. In Portugal Mainland, the district with the highest incidence of consanguineous marriages is Bragança [88]. Furthermore, the top five districts (as shown in Table 6) with the highest number of consanguineous marriages, listed in descending order, are Madeira, the Açores, Bragança, Viseu, and Vila Real.
Our findings, revealed that the top five districts with the highest FROH mean remain consistent, considering thresholds of 0.5, 1.5, and 5 Mb. The ranking from highest to lowest FROH mean value for 0.5 and 1.5 Mb thresholds is the following: Portalegre, Viseu, Bragança, Madeira, and Vila Real. Meanwhile, the ranking for the 5 Mb threshold is Portalegre, Bragança, Viseu, Madeira, and Vila Real.
Portalegre stands out with the highest FROH mean values across all three thresholds in our data, despite having fewer consanguineous marriages compared to other districts. This might be due to our sample including fewer individuals from Portalegre, which may suggest that those from this region who were referred for genetic testing were more likely to have been screened due to consanguinity. Surprisingly, the results show low FROH values across all three thresholds for the Autonomous Region of the Açores, which are not in accordance with the reference data on consanguineous marriages. This is possibly due to insufficient sample localization. Porto, the district with the lowest number of consanguineous marriages, also shows the lowest FROH mean across all thresholds. Additionally, we were unable to acquire information regarding the country of origin or birth of individuals included in the sample, this parameter was not used as an exclusion criterion. Moving forward, we should take this into consideration because certain countries present higher levels of consanguinity due to religious and cultural practices. We must also acknowledge the potential presence of samples from immigrants residing in our country, which could introduce biases into the data.
The presence of an admixed pattern denotes our country’s history, whilst the consanguineous pattern is a result of the marriages between cousins, leading to an increase in the sum of ROHs.
Having a model to predict patients consanguinity based on ROH features is useful in clinical centers. They can be used for the genetic test decision process and for assessing the risk of recessive diseases, knowing that the presence of consanguinity increases the risk of having recessive genetic diseases. Tier 0 (count and sum of ROH) of the model presented provided the majority of the predictive power for consanguinity classification.
Transitioning from WES to WGS might open some doors in terms of genetic testing, by adding insights into the non-coding regions of the genome. This will be of great interest particularly for undiagnosed patients and accelerate the diagnosis.
With this work, we demonstrated the applicability and utility of the newly developed resources and their impact on diagnostics, by solving the genetic etiology of a rare recessive disease. The representative sample of 3,941 WES used in this work, allowed us to provide the extensive analysis of ROH on a genomic scale for the first time ever in the Portuguese population. In summary, this research advances ROH analysis using WES data, highlighting its diagnostic potential and significance in population genetics’ characterization.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.Org, Supplementary File S1: WES Representative Sample of the Portuguese Population; Supplementary File S2: FROH for ROH of size greater than 0.5, 1.5 and 5 Mb, per municipality; Supplementary File S3: Interactive Maps; Supplementary File S4: Demographic origins of ROH in Portugal.
Author Contributions
Conceptualization, P.F.S., J.O.; methodology, S.V., M.R., F.A., J.S., N.M., P.F.S, J.O.; software, S.V., M.R., J.S., F.A. D.S.; writing—original draft preparation, S.V.; writing—critical review, J.O.; writing—review and editing, P.F.S., M.R. F.A., J.S., N.M., J.P.F.; supervision, P.F.S., J.O.; project administration, J.P.F., J.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki, the generation of the WES sample representative of the Portuguese Population was carried out within the scope of a project approved by the Institutional Ethics Committee of i3s (protocol code 10/CECRI/2023 and date of approval: 28-09-2023).
Informed Consent Statement
The project’s work involves the analysis of human sequencing genetic data. All patients involved in this study were part of the diagnostic process by their referring medical doctor. The clinician obtains the patient’s written informed consent (or from legal guardians if minors) to specifically participate in the genetic research or pilot genetic studies. Samples containing human cells/tissues are stored in CGPP biobank. All data used is fully anonymized and kept according to the authorization of CNPD (Portuguese Data Protection Authority) to CGPP. Data is stored in an encrypted database on a dedicated server at the IBMC/i3S data center, accessible only through the internal network by authorized personnel under confidentiality agreements. Data is fully anonymized and handled in an aggregated manner. Any personally identifiable information (including personal identifiers) will be removed, making re-identification virtually impossible. This approach adheres to the principle of confidentiality and respects the privacy rights of the individuals whose data is being analyzed. We also had a Data Protection Impact Assessment (DPIA) carried out with the active collaboration of the Data Protection Officer of our institution.
Data Availability Statement
The pseudocode used in this work will be available for consultation in the MSc dissertation, which will be accessible in November 2024 (http://hdl.handle.net/10773/39751). Please note that no public repository is currently available.
Acknowledgments
The authors express their gratitude for the initial conditions provided by Jorge Sequeiros (founder of CGPP and former UnIGENe research group PI) and the assistance of Victor Mendes (CGPP laboratory database manager).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Magi, A., Tattini, L., Palombo, F., Benelli, M., Gialluisi, A., Giusti, B., Abbate, R., Seri, M., Gensini, G. F. ranco, Romeo, G., & Pippucci, T. (2014). H3M2: detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics (Oxford, England), 30(20), 2852–2859. https://doi.org/10.1093/bioinformatics/btu401. [CrossRef]
- Oliveira, J., Pereira, R., Santos, R., & Sousa, M. (2018). Evaluating runs of homozygosity in exome sequencing data - Utility in disease inheritance model selection and variant filtering. Communications in Computer and Information Science, 881, 268–288. https://doi.org/10.1007/978-3-319-94806-5_15. [CrossRef]
- Peripolli, E., Munari, D. P., Silva, M. V. G. B., Lima, A. L. F., Irgang, R., & Baldi, F. (2017). Runs of homozygosity: current knowledge and applications in livestock. In Animal Genetics (Vol. 48, Issue 3, pp. 255–271). Blackwell Publishing Ltd. https://doi.org/10.1111/age.12526. [CrossRef]
- Oniya, O., Neves, K., Ahmed, B., & Konje, J. C. (2019). A review of the reproductive consequences of consanguinity. European Journal of Obstetrics and Gynecology and Reproductive Biology, 232, 87–96. https://doi.org/10.1016/j.ejogrb.2018.10.042. [CrossRef]
- Marchi, N., Mennecier, P., Georges, M., Lafosse, S., Hegay, T., Dorzhu, C., Chichlo, B., Ségurel, L., & Heyer, E. (2018). Close inbreeding and low genetic diversity in Inner Asian human populations despite geographical exogamy. Scientific Reports, 8(1), 1–10. https://doi.org/10.1038/s41598-018-27047-3. [CrossRef]
- Yengo, L., Wray, N. R., & Visscher, P. M. (2019). Extreme inbreeding in a European ancestry sample from the contemporary UK population. Nature Communications, 10(1). https://doi.org/10.1038/s41467-019-11724-6. [CrossRef]
- Slatkin, M. (2004). A Population-Genetic Test of Founder Effects and Implications for Ashkenazi Jewish Diseases. In Am. J. Hum. Genet (Vol. 75). https://doi.org/10.1086/423146. [CrossRef]
- Dong, J.-T. (2001). Chromosomal deletions and tumor suppressor genes in prostate cancer. In Cancer and Metastasis Reviews (Vol. 20). https://doi.org/10.1023/A:1015575125780. [CrossRef]
- Nalls, M. A., Simon-Sanchez, J., Gibbs, J. R., Paisan-Ruiz, C., Bras, J. T., Tanaka, T., Matarin, M., Scholz, S., Weitz, C., Harris, T. B., Ferrucci, L., Hardy, J., & Singleton, A. B. (2009). Measures of autozygosity in decline: Globalization, urbanization, and its implications for medical genetics. PLoS Genetics, 5(3). https://doi.org/10.1371/journal.pgen.1000415. [CrossRef]
- Ceballos, F. C., Hazelhurst, S., & Ramsay, M. (2019). Runs of homozygosity in sub-Saharan African populations provide insights into complex demographic histories. Human Genetics, 138(10), 1123–1142. https://doi.org/10.1007/s00439-019-02045-1. [CrossRef]
- Lemes, R. B., Nunes, K., Carnavalli, J. E. P., Kimura, L., Mingroni-Netto, R. C., Meyer, D., & Otto, P. A. (2018). Inbreeding estimates in human populations: Applying new approaches to an admixed Brazilian isolate. PLoS ONE, 13(4). https://doi.org/10.1371/journal.pone.0196360. [CrossRef]
- Ben Halim, N., Nagara, M., Regnault, B., Hsouna, S., Lasram, K., Kefi, R., Azaiez, H., Khemira, L., Saidane, R., Ammar, S. Ben, Besbes, G., Weil, D., Petit, C., Abdelhak, S., & Romdhane, L. (2015). Estimation of Recent and Ancient Inbreeding in a Small Endogamous Tunisian Community Through Genomic Runs of Homozygosity. Annals of Human Genetics, 79(6), 402–417. https://doi.org/10.1111/ahg.12131. [CrossRef]
- Kang, J. T. L., Goldberg, A., Edge, M. D., Behar, D. M., & Rosenberg, N. A. (2017). Consanguinity Rates Predict Long Runs of Homozygosity in Jewish Populations. Human Heredity, 82(3–4), 87–102. https://doi.org/10.1159/000478897. [CrossRef]
- Pemberton, T. J., Absher, D., Feldman, M. W., Myers, R. M., Rosenberg, N. A., & Li, J. Z. (2012). Genomic patterns of homozygosity in worldwide human populations. American Journal of Human Genetics, 91(2), 275–292. https://doi.org/10.1016/j.ajhg.2012.06.014. [CrossRef]
- Kirin, M., Mcquillan, R., Franklin, C. S., Campbell, H., & Mckeigue, P. M. (2010). Genomic Runs of Homozygosity Record Population History and Consanguinity. PLoS ONE, 5(11), 13996. https://doi.org/10.1371/journal.pone.0013996. [CrossRef]
- Hunter-Zinck, H., Musharoff, S., Salit, J., Al-Ali, K. A., Chouchane, L., Gohar, A., Matthews, R., Butler, M. W., Fuller, J., Hackett, N. R., Crystal, R. G., & Clark, A. G. (2010). Population genetic structure of the people of Qatar. American Journal of Human Genetics, 87(1), 17–25. https://doi.org/10.1016/j.ajhg.2010.05.018. [CrossRef]
- Mezzavilla, M., Cocca, M., Maisano Delser, P., Badii, R., Abbaszadeh, F., Hadi, K. A., Giorgia, G., & Gasparini, P. (2022). Ancestry-related distribution of Runs of homozygosity and functional variants in Qatari population. BMC Genomic Data, 23(1). https://doi.org/10.1186/s12863-022-01087-1. [CrossRef]
- Scott, E. M., Halees, A., Itan, Y., Spencer, E. G., He, Y., Azab, M. A., Gabriel, S. B., Belkadi, A., Boisson, B., Abel, L., Clark, A. G., Rahim, S. A., Abdel-Hadi, S., Abdel-Salam, G., Abdel-Salam, E., Abdou, M., Abhytankar, A., Adimi, P., Ahmad, J., … Zhang, S. Y. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nature Genetics, 48(9), 1071. https://doi.org/10.1038/NG.3592. [CrossRef]
- Yang, X., Al-Bustan, S., Feng, Q., Guo, W., Ma, Z., Marafie, M., Jacob, S., Al-Mulla, F., & Xu, S. (2014). The influence of admixture and consanguinity on population genetic diversity in Middle East. Journal of Human Genetics, 59(11), 615–622. https://doi.org/10.1038/jhg.2014.81. [CrossRef]
- Ceballos, F. C., Gürün, K., Altınışık, N. E., Gemici, H. C., Karamurat, C., Koptekin, D., Vural, K. B., Mapelli, I., Sağlıcan, E., Sürer, E., Erdal, Y. S., Götherström, A., Özer, F., Atakuman, Ç., & Somel, M. (2021). Human inbreeding has decreased in time through the Holocene. Current Biology, 31(17), 3925-3934.e8. https://doi.org/10.1016/j.cub.2021.06.027. [CrossRef]
- Ece Kars, M., Nazlı Bas, A., Emre Onat, O., Bilguvar, K., Choi, J., Itan, Y., Ça, C., Palvadeau, R., Casanova, J.-L., Cooper, D. N., Stenson, P. D., Yavuz, A., Bulus, H., Günel, M., Friedman, J. M., & Özçelik, T. (n.d.). The genetic structure of the Turkish population reveals high levels of variation and admixture. https://doi.org/10.1073/pnas.2026076118/-/DCSupplemental. [CrossRef]
- Binzer, S., Imrell, K., Binzer, M., Kyvik, K. O., Hillert, J., & Stenager, E. (2015). High inbreeding in the Faroe Islands does not appear to constitute a risk factor for multiple sclerosis. Multiple Sclerosis, 21(8), 996–1002. https://doi.org/10.1177/1352458514557305. [CrossRef]
- Karafet, T. M., Bulayeva, K. B., Bulayev, O. A., Gurgenova, F., Omarova, J., Yepiskoposyan, L., Savina, O. V., Veeramah, K. R., & Hammer, M. F. (2015). Extensive genome-wide autozygosity in the population isolates of Daghestan. European Journal of Human Genetics, 23(10), 1405–1412. https://doi.org/10.1038/ejhg.2014.299. [CrossRef]
- McLaughlin, R. L., Kenna, K. P., Vajda, A., Heverin, M., Byrne, S., Donaghy, C. G., Cronin, S., Bradley, D. G., & Hardiman, O. (2015). Homozygosity mapping in an Irish ALS case-control cohort describes local demographic phenomena and points towards potential recessive risk loci. Genomics, 105(4), 237–241. https://doi.org/10.1016/j.ygeno.2015.01.002. [CrossRef]
- Alabdullatif, M. A., Al Dhaibani, M. A., Khassawneh, M. Y., & El-Hattab, A. W. (2017). Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clinical Genetics, 91(4), 616–622. https://doi.org/10.1111/cge.12872. [CrossRef]
- Wang, J. C., Ross, L., Mahon, L. W., Owen, R., Hemmat, M., Wang, B. T., El Naggar, M., Kopita, K. A., Randolph, L. M., Chase, J. M., Aguilera, M. J. M., Siles, J. L., Church, J. A., Hauser, N., Shen, J. J., Jones, M. C., Wierenga, K. J., Jiang, Z., Haddadin, M., … Sahoo, T. (2015). Regions of homozygosity identified by oligonucleotide SNP arrays: Evaluating the incidence and clinical utility. European Journal of Human Genetics, 23(5), 663–671. https://doi.org/10.1038/ejhg.2014.153. [CrossRef]
- Prasad, A., Sdano, M. A., Vanzo, R. J., Mowery-Rushton, P. A., Serrano, M. A., Hensel, C. H., & Wassman, E. R. (2018). Clinical utility of exome sequencing in individuals with large homozygous regions detected by chromosomal microarray analysis. BMC Medical Genetics, 19(1). https://doi.org/10.1186/s12881-018-0555-3. [CrossRef]
- Hengel, H., Buchert, R., Sturm, M., Haack, T. B., Schelling, Y., Mahajnah, M., Sharkia, R., Azem, A., Balousha, G., Ghanem, Z., Falana, M., Balousha, O., Ayesh, S., Keimer, R., Deigendesch, W., Zaidan, J., Marzouqa, H., Bauer, P., & Schöls, L. (2020). First-line exome sequencing in Palestinian and Israeli Arabs with neurological disorders is efficient and facilitates disease gene discovery. European Journal of Human Genetics, 28(8), 1034–1043. https://doi.org/10.1038/s41431-020-0609-9. [CrossRef]
- Palombo, F., Graziano, C., Al Wardy, N., Nouri, N., Marconi, C., Magini, P., Severi, G., La Morgia, C., Cantalupo, G., Cordelli, D. M., Gangarossa, S., Al Kindi, M. N., Al Khabouri, M., Salehi, M., Giorgio, E., Brusco, A., Pisani, F., Romeo, G., Carelli, V., … Seri, M. (2020). Autozygosity-driven genetic diagnosis in consanguineous families from Italy and the Greater Middle East. Human Genetics, 139(11), 1429–1441. https://doi.org/10.1007/s00439-020-02187-7. [CrossRef]
- Knopp, C., Rudnik-Schöneborn, S., Eggermann, T., Bergmann, C., Begemann, M., Schoner, K., Zerres, K., & Ortiz Brüchle, N. (2015). Syndromic ciliopathies: From single gene to multi gene analysis by SNP arrays and next generation sequencing. Molecular and Cellular Probes, 29(5), 299–307. https://doi.org/10.1016/j.mcp.2015.05.008. [CrossRef]
- de Farias, A. A., Nunes, K., Lemes, R. B., Moura, R., Fernandes, G. R., Melo, U. S., Zatz, M., Kok, F., & Santos, S. (2018). Origin and age of the causative mutations in KLC2, IMPA1, MED25 and WNT7A unravelled through Brazilian admixed populations. Scientific Reports, 8(1). https://doi.org/10.1038/s41598-018-35022-1. [CrossRef]
- Wakil, S. M., Ramzan, K., Abuthuraya, R., Hagos, S., Al-Dossari, H., Al-Omar, R., Murad, H., Chedrawi, A., Al-Hassnan, Z. N., Finsterer, J., & Bohlega, S. (2014). Infantile-onset ascending hereditary spastic paraplegia with bulbar involvement due to the novel ALS2 mutation c.2761C>T. Gene, 536(1), 217–220. https://doi.org/10.1016/j.gene.2013.11.043. [CrossRef]
- Lobo-Prada, T., Sticht, H., Bogantes-Ledezma, S., Ekici, A., Uebe, S., Reis, A., & Leal, A. (2017). A homozygous mutation in GPT2 associated with nonsyndromic intellectual disability in a consanguineous family from costa rica. In JIMD Reports (Vol. 36, pp. 59–66). Springer. https://doi.org/10.1007/8904_2016_40. [CrossRef]
- Guo, T., Tan, Z. P., Chen, H. M., Zheng, D. yuan, liu, L., Huang, X. G., Chen, P., Luo, H., & Yang, Y. F. (2017). An effective combination of whole-exome sequencing and runs of homozygosity for the diagnosis of primary ciliary dyskinesia in consanguineous families. Scientific Reports, 7(1). https://doi.org/10.1038/s41598-017-08510-z. [CrossRef]
- Costa, P., Zanus, C., Faletra, F., Ventura, G., di Marzio, G. M., Cervesi, C., & Carrozzi, M. (2019). Epileptic encephalopathy with microcephaly in a patient with asparagine synthetase deficiency: a video-EEG report∗. Epileptic Disorders, 21(5), 466–470. https://doi.org/10.1684/epd.2019.1100. [CrossRef]
- Khan, R., Shabbir, R. M. K., Raza, I., Abdullah, U., Naeem, M. A., Ahmed, A., Malik, S., Hu, Z., & Xia, K. (2020). A founder RDH5 splice site mutation leads to retinitis punctata albescens in two inbred Pakistani kindreds. Ophthalmic Genetics, 41(1), 7–12. https://doi.org/10.1080/13816810.2019.1709124. [CrossRef]
- Yu, W., You, X., Wang, D., Dong, K., Su, J., Li, C., Liu, J., Zhang, Q., You, F., Wang, X., Huang, J., Qiao, B., & Duan, W. (2015). Microarray analysis unmasked two siblings with pure hereditary spastic paraplegia shared a run of homozygosity region on chromosome 3q28-q29. Journal of the Neurological Sciences, 359(1–2), 351–355. https://doi.org/10.1016/j.jns.2015.10.057. [CrossRef]
- Calderón, R., Hernández, C. L., García-Varela, G., Masciarelli, D., & Cuesta, P. (2018). Inbreeding in Southeastern Spain: The Impact of Geography and Demography on Marital Mobility and Marital Distance Patterns (1900–1969). Human Nature, 29(1), 45–64. https://doi.org/10.1007/s12110-017-9305-z. [CrossRef]
- Pippucci, T., Magi, A., Gialluisi, A., & Romeo, G. (2014). Detection of runs of homozygosity from whole exome sequencing data: State of the art and perspectives for clinical, population and epidemiological studies. Human Heredity, 77(1–4), 63–72. https://doi.org/10.1159/000362412. [CrossRef]
- Lander, E. S., & Botstein, D. (1987). Homozygosity Mapping: A Way to Map Human Recessive Traits with the DNA of Inbred Children. Science, 236(4808), 1567–1570. https://doi.org/10.1126/SCIENCE.2884728. [CrossRef]
- Hu, T., Chitnis, N., Monos, D., & Dinh, A. (2021). Next-generation sequencing technologies: An overview. Human Immunology, 82(11), 801–811. https://doi.org/10.1016/j.humimm.2021.02.012. [CrossRef]
- Pereira, R., Oliveira, J., & Sousa, M. (2020). Bioinformatics and computational tools for next-generation sequencing analysis in clinical genetics. In Journal of Clinical Medicine (Vol. 9, Issue 1). MDPI. https://doi.org/10.3390/jcm9010132. [CrossRef]
- Thompson, J. F., & Milos, P. M. (2011). The properties and applications of single-molecule DNA sequencing. Genome Biology, 12(2), 1–10. https://doi.org/10.1186/GB-2011-12-2-217/TABLES/1. [CrossRef]
- Rhoads, A., & Au, K. F. (2015). PacBio Sequencing and Its Applications. https://doi.org/10.1016/j.gpb.2015.08.002. [CrossRef]
- Zhang, L., Chen, F. X., Zeng, Z., Xu, M., Sun, F., Yang, L., Bi, X., Lin, Y., Gao, Y. J., Hao, H. X., Yi, W., Li, M., & Xie, Y. (2021). Advances in Metagenomics and Its Application in Environmental Microorganisms. Frontiers in Microbiology, 12, 766364. https://doi.org/10.3389/FMICB.2021.766364. [CrossRef]
- Qin, D. (2019). Next-generation sequencing and its clinical application. Cancer Biology and Medicine, 16(1), 4–10. https://doi.org/10.20892/j.issn.2095-3941.2018.0055. [CrossRef]
- Barbitoff, Y. A., Polev, D. E., Glotov, A. S., Serebryakova, E. A., Shcherbakova, I. V., Kiselev, A. M., Kostareva, A. A., Glotov, O. S., & Predeus, A. V. (2020). Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Scientific Reports, 10(1). https://doi.org/10.1038/s41598-020-59026-y. [CrossRef]
- Choi, M., Scholl, U. I., Ji, W., Liu, T., Tikhonova, I. R., Zumbo, P., Nayir, A., Bakkaloğlu, A., Ozen, S., Sanjad, S., Nelson-Williams, C., Farhi, A., Mane, S., & Lifton, R. P. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America, 106(45), 19096–19101. https://doi.org/10.1073/pnas.0910672106. [CrossRef]
- Bartha, Á., & Győrffy, B. (2019). Comprehensive outline of whole exome sequencing data analysis tools available in clinical oncology. In Cancers (Vol. 11, Issue 11). MDPI AG. https://doi.org/10.3390/cancers11111725. [CrossRef]
- Warman Chardon, J., Beaulieu, C., Hartley, T., Boycott, K. M., & Dyment, D. A. (2015). Axons to Exons: the Molecular Diagnosis of Rare Neurological Diseases by Next-Generation Sequencing. Current Neurology and Neuroscience Reports, 15(9), 1–8. https://doi.org/10.1007/S11910-015-0584-7/TABLES/2. [CrossRef]
- Gargano, M. A., Matentzoglu, N., Coleman, B., Addo-Lartey, E. B., Anagnostopoulos, A. V., Anderton, J., Avillach, P., Bagley, A. M., Bakštein, E., Balhoff, J. P., Baynam, G., Bello, S. M., Berk, M., Bertram, H., Bishop, S., Blau, H., Bodenstein, D. F., Botas, P., Boztug, K., … Robinson, P. N. (2024). The Human Phenotype Ontology in 2024: phenotypes around the world. Nucleic Acids Research, 52(D1), D1333–D1346. https://doi.org/10.1093/nar/gkad1005. [CrossRef]
- Bullich, G., Matalonga, L., Pujadas, M., Papakonstantinou, A., Piscia, D., Tonda, R., Artuch, R., Gallano, P., Garrabou, G., González, J. R., Grinberg, D., Guitart, M., Laurie, S., Lázaro, C., Luengo, C., Martí, R., Milà, M., Ovelleiro, D., Parra, G., … Vendrell, T. (2022). Systematic Collaborative Reanalysis of Genomic Data Improves Diagnostic Yield in Neurologic Rare Diseases. Journal of Molecular Diagnostics, 24(5), 529–542. https://doi.org/10.1016/j.jmoldx.2022.02.003. [CrossRef]
- Matalonga, L., Laurie, S., Papakonstantinou, A., Piscia, D., Mereu, E., Bullich, G., Thompson, R., Horvath, R., Pérez-Jurado, L., Riess, O., Gut, I., van Ommen, G. J., Lochmüller, H., Beltran, S., Renieri, A., Dursun, A., Matilla-Duenas, A., Cormand, B., Rivolta, C., … Sabater, M. (2020). Improved Diagnosis of Rare Disease Patients through Systematic Detection of Runs of Homozygosity. Journal of Molecular Diagnostics, 22(9), 1205–1215. https://doi.org/10.1016/j.jmoldx.2020.06.008. [CrossRef]
- Becker, J., Semler, O., Gilissen, C., Li, Y., Bolz, H. J., Giunta, C., Bergmann, C., Rohrbach, M., Koerber, F., Zimmermann, K., De Vries, P., Wirth, B., Schoenau, E., Wollnik, B., Veltman, J. A., Hoischen, A., & Netzer, C. (2011). Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. American Journal of Human Genetics, 88(3), 362–371. https://doi.org/10.1016/j.ajhg.2011.01.015. [CrossRef]
- Mezzavilla, M., Vozzi, D., Badii, R., Khalifa Alkowari, M., Abdulhadi, K., Girotto, G., & Gasparini, P. (2015). Increased rate of deleterious variants in long runs of homozygosity of an inbred population from Qatar. Human Heredity, 79(1), 14–19. https://doi.org/10.1159/000371387. [CrossRef]
- Yang, T. L., Guo, Y., Zhang, L. S., Tian, Q., Yan, H., Papasian, C. J., Recker, R. R., & Deng, H. W. (2010). Runs of homozygosity identify a recessive locus 12q21.31 for human adult height. Journal of Clinical Endocrinology and Metabolism, 95(8), 3777–3782. https://doi.org/10.1210/jc.2009-1715. [CrossRef]
- Wang, L. S., Hranilovic, D., Wang, K., Lindquist, I. E., Yurcaba, L., Petkovic, Z. B., Gidaya, N., Jernej, B., Hakonarson, H., & Bucan, M. (2010). Population-based study of genetic variation in individuals with autism spectrum disorders from Croatia. BMC Medical Genetics, 11(1), 134. https://doi.org/10.1186/1471-2350-11-134. [CrossRef]
- Gross, A., Tönjes, A., Kovacs, P., Veeramah, K. R., Ahnert, P., Roshyara, N. R., Gieger, C., Rueckert, I. M., Loeffler, M., Stoneking, M., Wichmann, H. E., Novembre, J., Stumvoll, M., & Scholz, M. (2011). Population-genetic comparison of the Sorbian isolate population in Germany with the German KORA population using genome-wide SNP arrays. BMC Genetics, 12. https://doi.org/10.1186/1471-2156-12-67. [CrossRef]
- Ghani, M., Sato, C., Lee, J. H., Reitz, C., Moreno, D., Mayeux, R., George-Hyslop, P. S., & Rogaeva, E. (2013). Evidence of recessive Alzheimer disease loci in a Caribbean Hispanic data set: genome-wide survey of runs of homozygosity. JAMA Neurology, 70(10), 1261–1267. https://doi.org/10.1001/JAMANEUROL.2013.3545. [CrossRef]
- Yang, T. L., Guo, Y., Zhang, J. G., Xu, C., Tian, Q., & Deng, H. W. (2015). Genome-wide Survey of Runs of Homozygosity Identifies Recessive Loci for Bone Mineral Density in Caucasian and Chinese Populations. Journal of Bone and Mineral Research : The Official Journal of the American Society for Bone and Mineral Research, 30(11), 2119–2126. https://doi.org/10.1002/JBMR.2558. [CrossRef]
- Ghani, M., Reitz, C., Cheng, R., Vardarajan, B. N., Jun, G., Sato, C., Naj, A., Rajbhandary, R., Wang, L. S., Valladares, O., Lin, C. F., Larson, E. B., Graff-Radford, N. R., Evans, D., De Jager, P. L., Crane, P. K., Buxbaum, J. D., Murrell, J. R., Raj, T., … Yu, L. (2015). Association of Long Runs of Homozygosity With Alzheimer Disease Among African American Individuals. JAMA Neurology, 72(11), 1313–1323. https://doi.org/10.1001/JAMANEUROL.2015.1700. [CrossRef]
- Bandrés-Ciga, S., Price, T. R., Barrero, F. J., Escamilla-Sevilla, F., Pelegrina, J., Arepalli, S., Hernández, D., Gutiérrez, B., Cervilla, J., Rivera, M., Rivera, A., Ding, J. hui, Vives, F., Nalls, M., Singleton, A., & Durán, R. (2016). Genome-wide assessment of Parkinson’s disease in a Southern Spanish population. Neurobiology of Aging, 45, 213.e3. https://doi.org/10.1016/J.NEUROBIOLAGING.2016.06.001. [CrossRef]
- Barbieri, C., Barquera, R., Arias, L., Sandoval, J. R., Acosta, O., Zurita, C., Aguilar-Campos, A., Tito-Álvarez, A. M., Serrano-Osuna, R., Gray, R. D., Mafessoni, F., Heggarty, P., Shimizu, K. K., Fujita, R., Stoneking, M., Pugach, I., & Fehren-Schmitz, L. (2019). The Current Genomic Landscape of Western South America: Andes, Amazonia, and Pacific Coast. Molecular Biology and Evolution, 36(12), 2698–2713. https://doi.org/10.1093/MOLBEV/MSZ174. [CrossRef]
- Font-Porterias, N., Caro-Consuegra, R., Lucas-Sánchez, M., Lopez, M., Giménez, A., Carballo-Mesa, A., Bosch, E., Calafell, F., Quintana-Murci, L., & Comas, D. (2021). The Counteracting Effects of Demography on Functional Genomic Variation: The Roma Paradigm. Molecular Biology and Evolution, 38(7), 2804–2817. https://doi.org/10.1093/MOLBEV/MSAB070. [CrossRef]
- Cruz, P. R. S. da, Ananina, G., Secolin, R., Gil-Da-Silva-Lopes, V. L., Lima, C. S. P., França, P. H. C. de, Donatti, A., Lourenço, G. J., Araujo, T. K. de, Simioni, M., Lopes-Cendes, I., Costa, F. F., & Melo, M. B. de. (2022). Demographic history differences between Hispanics and Brazilians imprint haplotype features. G3 (Bethesda, Md.), 12(7). https://doi.org/10.1093/G3JOURNAL/JKAC111. [CrossRef]
- Ruan, X., Kocher, J. P. A., Pommier, Y., Liu, H., & Reinhold, W. C. (2012). Mass homozygotes accumulation in the NCI-60 cancer cell lines as compared to HapMap Trios, and relation to fragile site location. PloS One, 7(2). https://doi.org/10.1371/JOURNAL.PONE.0031628. [CrossRef]
- Santoni, F. A., Makrythanasis, P., & Antonarakis, S. E. (2015). CATCHing putative causative variants in consanguineous families. BMC Bioinformatics, 16(1). https://doi.org/10.1186/S12859-015-0727-5. [CrossRef]
- Sonehara, K., & Okada, Y. (2020). Obelisc: an identical-by-descent mapping tool based on SNP streak. Bioinformatics, 36(24), 5567. https://doi.org/10.1093/BIOINFORMATICS/BTAA940. [CrossRef]
- Garone, C., Pippucci, T., Cordelli, D. M., Zuntini, R., Castegnaro, G., Marconi, C., Graziano, C., Marchiani, V., Verrotti, A., Seri, M., & Franzoni, E. (2011). FA2H-related disorders: A novel c.270+3A>T splice-site mutation leads to a complex neurodegenerative phenotype. Developmental Medicine and Child Neurology, 53(10), 958–961. https://doi.org/10.1111/j.1469-8749.2011.03993.x. [CrossRef]
- Seelow, D., & Schuelke, M. (2012). HomozygosityMapper2012-bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Research, 40(W1). https://doi.org/10.1093/nar/gks487. [CrossRef]
- Seelow, D., Schuelke, M., Hildebrandt, F., & Nürnberg, P. (2009). HomozygosityMapper - An interactive approach to homozygosity mapping. Nucleic Acids Research, 37(SUPPL. 2). https://doi.org/10.1093/nar/gkp369. [CrossRef]
- Kancheva, D., Atkinson, D., De Rijk, P., Zimon, M., Chamova, T., Mitev, V., Yaramis, A., Maria Fabrizi, G., Topaloglu, H., Tournev, I., Parma, Y., Battaloglu, E., Estrada-Cuzcano, A., & Jordanova, A. (2016). Novel mutations in genes causing hereditary spastic paraplegia and Charcot-Marie-Tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole-exome sequencing. Genetics in Medicine, 18(6), 600–607. https://doi.org/10.1038/GIM.2015.139. [CrossRef]
- Szpiech, Z. A., Blant, A., & Pemberton, T. J. (2017). GARLIC: Genomic Autozygosity Regions Likelihood-based Inference and Classification. Bioinformatics (Oxford, England), 33(13), 2059–2062. https://doi.org/10.1093/BIOINFORMATICS/BTX102. [CrossRef]
- Görmez, Z., Bakir-Gungor, B., & Saǧiroǧlu, M. Ş. (2014). HomSI: a homozygous stretch identifier from next-generation sequencing data. Bioinformatics, 30(3), 445–447. https://doi.org/10.1093/BIOINFORMATICS/BTT686. [CrossRef]
- Quinodoz, M., Peter, V. G., Bedoni, N., Bertrand, B. R., Cisarova, K., Salmaninejad, A., Sepahi, N., Rodrigues, R., Piran, M., Mojarrad, M., Pasdar, A., Asad, A. G., Sousa, A. B., Santos, L. C., Superti-Furga, A., & Rivolta, C. (n.d.). AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. https://doi.org/10.1038/s41467-020-20584-4. [CrossRef]
- Yoon, B.-J. (2009). Hidden Markov Models and their Applications in Biological Sequence Analysis. Current Genomics, 10(6), 402. https://doi.org/10.2174/138920209789177575. [CrossRef]
- Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., & Durbin, R. (2016). BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics, 32(11), 1749–1751. https://doi.org/10.1093/BIOINFORMATICS/BTW044. [CrossRef]
- Zhuang, Z., Gusev, A., Cho, J., & Pe’er, I. (2012). Detecting Identity by Descent and Homozygosity Mapping in Whole-Exome Sequencing Data. PLoS ONE, 7(10). https://doi.org/10.1371/journal.pone.0047618. [CrossRef]
- Browning, S. R., & Browning, B. L. (2010). High-Resolution Detection of Identity by Descent in Unrelated Individuals. American Journal of Human Genetics, 86(4), 526–539. https://doi.org/10.1016/j.ajhg.2010.02.021. [CrossRef]
- Çelik, G., & Tuncalı, T. (2022). ROHMM—A flexible hidden Markov model framework to detect runs of homozygosity from genotyping data. Human Mutation, 43(2), 158–168. https://doi.org/10.1002/HUMU.24316. [CrossRef]
- Vigeland, M. D., Gjøtterud, K. S., & Selmer, K. K. (2016). FILTUS: A desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics, 32(10), 1592–1594. https://doi.org/10.1093/BIOINFORMATICS/BTW046. [CrossRef]
- hapROH · PyPI. (n.d.). Retrieved March 27, 2023, from https://pypi.org/project/hapROH/.
- Ringbauer, H., Novembre, J., & Steinrücken, M. (2021). Parental relatedness through time revealed by runs of homozygosity in ancient DNA. Nature Communications, 12(1). https://doi.org/10.1038/S41467-021-25289-W. [CrossRef]
- Kruskal, J. B., & Hill, M. (1964). Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29, 1–27. https://doi.org/10.1007/BF0228956585. [CrossRef]
- Rousseeuw, P. J., & Van Driessen, K. (1999). A fast algorithm for the minimum covariance determinant estimator. Technometrics, 41(3), 212–223. https://doi.org/10.1080/00401706.1999.10485670. [CrossRef]
- Lalioti, M. D., Mirotsou, M., Buresi, C., Peitsch, M. C., Rossier, C., Ouazzani, R., Baldy-Moulinier, M., Bottani, A., Malafosse, A., & Antonarakis, S. E. (1997). Identification of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM1). American Journal of Human Genetics, 60(2), 342. /pmc/articles/PMC1712389/?report=abstract.
- McQuillan, R., Leutenegger, A. L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., Smolej-Narancic, N., Janicijevic, B., Polasek, O., Tenesa, A., MacLeod, A. K., Farrington, S. M., Rudan, P., Hayward, C., Vitart, V., Rudan, I., Wild, S. H., Dunlop, M. G., Wright, A. F., … Wilson, J. F. (2008). Runs of Homozygosity in European Populations. American Journal of Human Genetics, 83(3), 359. https://doi.org/10.1016/J.AJHG.2008.08.007. [CrossRef]
- Santos, H. G., Dias, J. A., Pimenta, Z. P., Homenagem Ao Professor, E., & Guignard, J. (n.d.). SUMÁRIO 41 INCIDÊNCIA DE CASAMENTOS CONSANGUINEOS NA POPULAÇÃO INCIDÊNCIA DE CASAMENTOS CONSANGUÍíNEOS NA POPULAÇÃO PORTUGUESA-1980-1986.
- Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M., & Wilson, J. F. (2018). Runs of homozygosity: Windows into population history and trait architecture. In Nature Reviews Genetics (Vol. 19, Issue 4, pp. 220–234). Nature Publishing Group. https://doi.org/10.1038/nrg.2017.109. [CrossRef]
- Martin, A. R., Williams, E., Foulger, R. E., Leigh, S., Daugherty, L. C., Niblock, O., Leong, I. U. S., Smith, K. R., Gerasimenko, O., Haraldsdottir, E., Thomas, E., Scott, R. H., Baple, E., Tucci, A., Brittain, H., de Burca, A., Ibañez, K., Kasperaviciute, D., Smedley, D., … McDonagh, E. M. (2019). PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. In Nature Genetics (Vol. 51, Issue 11, pp. 1560–1565). Nature Publishing Group. https://doi.org/10.1038/s41588-019-0528-2. [CrossRef]
Figure 1.
Flowchart representing the automation of the creation of multigene panels based on ROH (DB - Database, DF - Dataframe).
Figure 1.
Flowchart representing the automation of the creation of multigene panels based on ROH (DB - Database, DF - Dataframe).
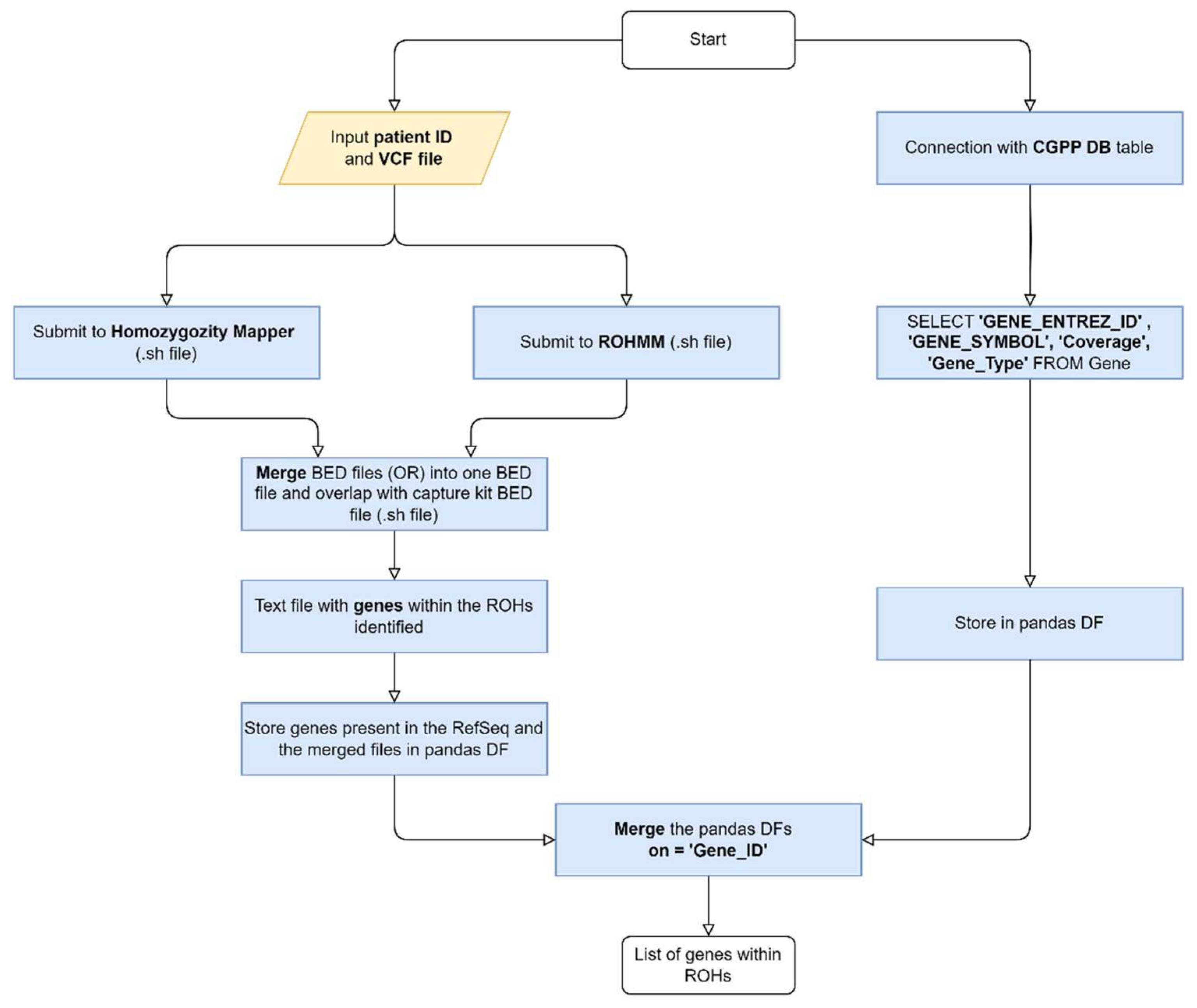
Figure 2.
Flowchart to obtain the reference BED file.
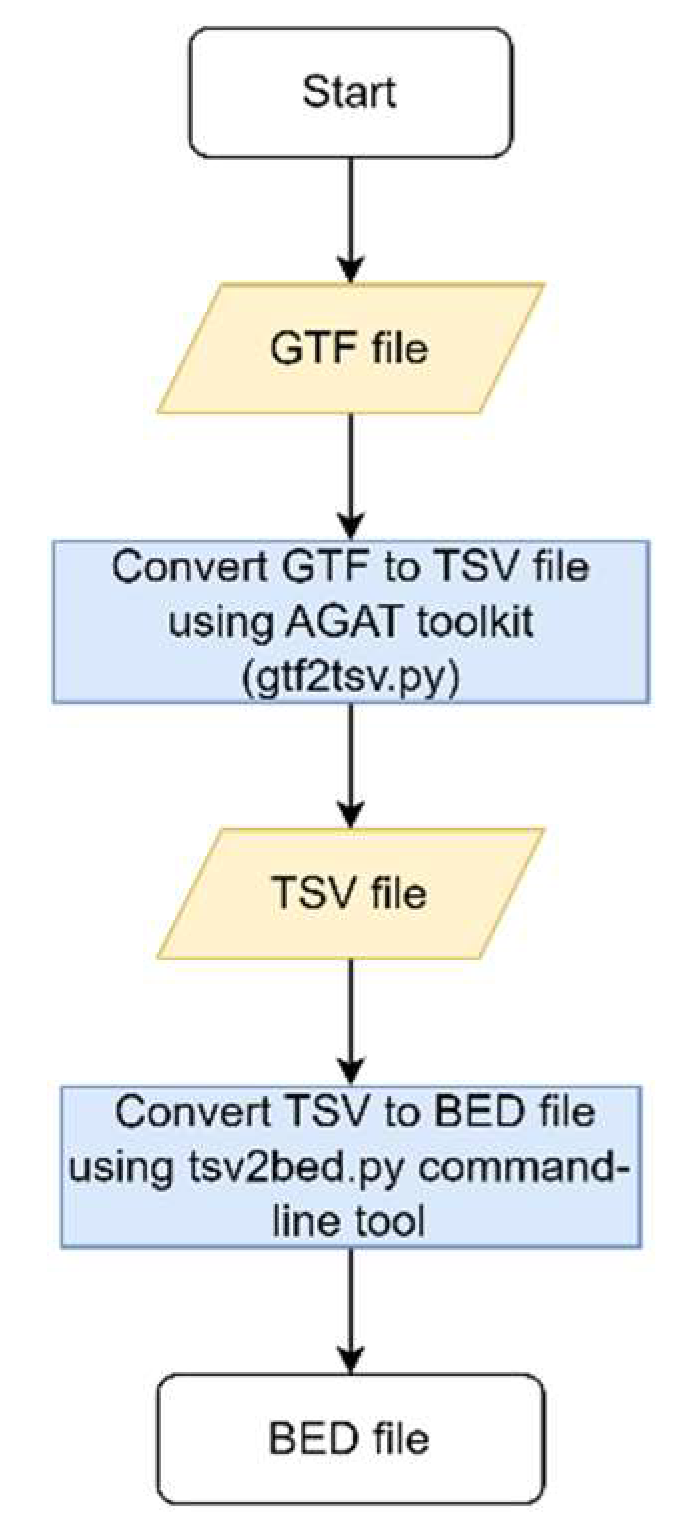
Figure 3.
The flowchart of the multigene panels lists: white, grey and black.
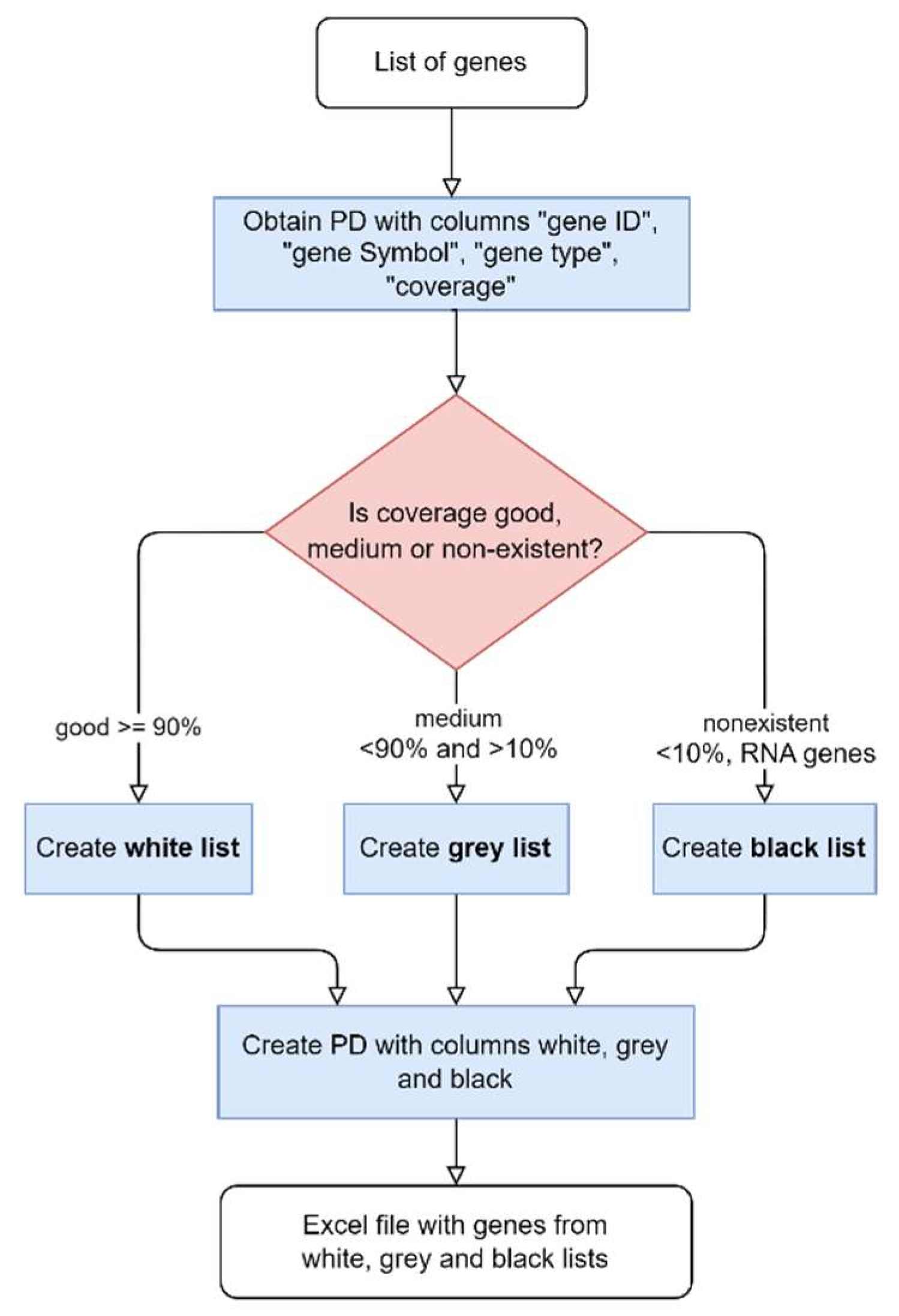
Figure 4.
Flowchart of the ROH and HPO multigene panels automation.

Figure 5.
Overview of the results regarding processes of generating the multigene panel application in a case study, the first Portuguese ROH characterization, and the clustering model.
Figure 5.
Overview of the results regarding processes of generating the multigene panel application in a case study, the first Portuguese ROH characterization, and the clustering model.
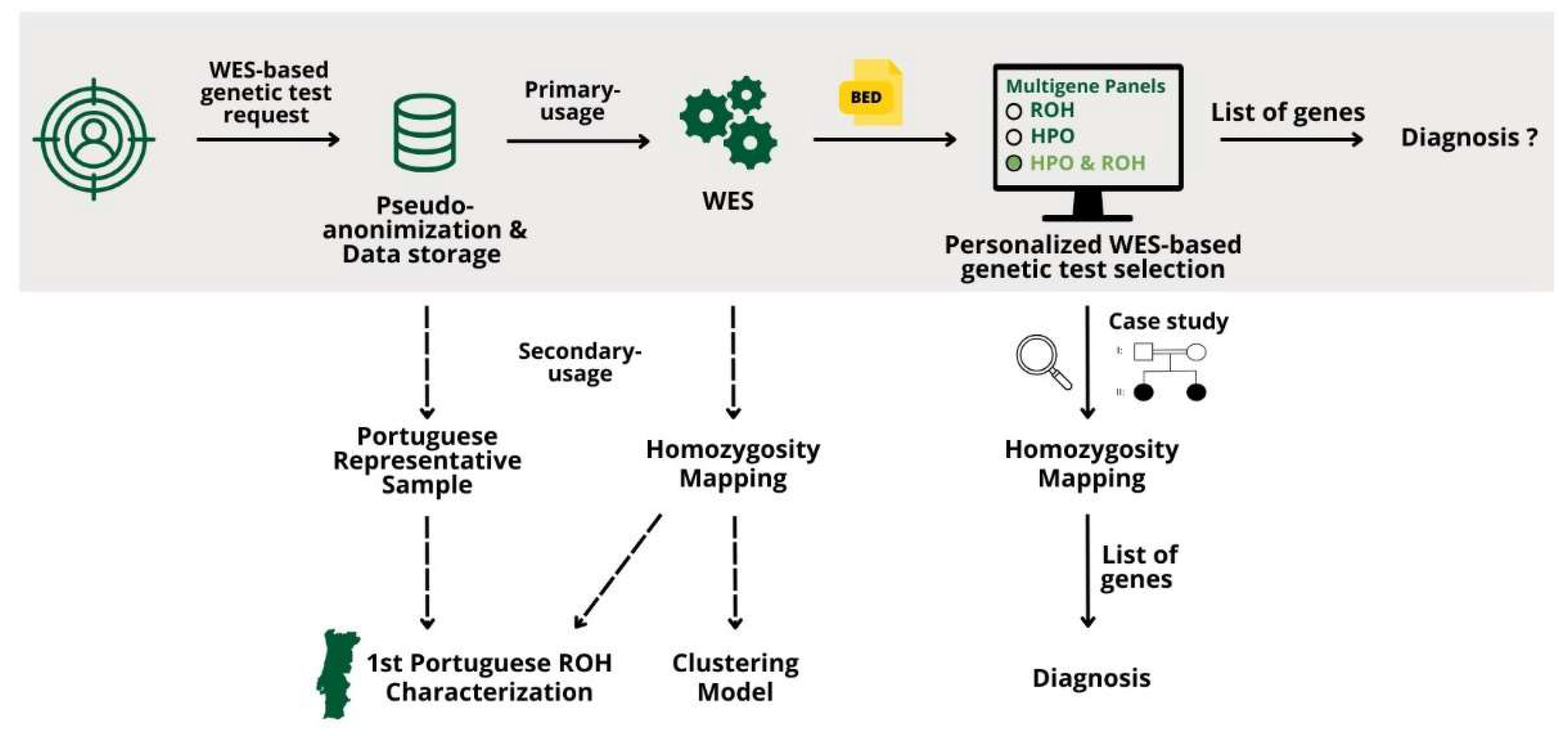
Figure 6.
Pedigree depicting two affected sisters, daughters of a consanguineous couple.
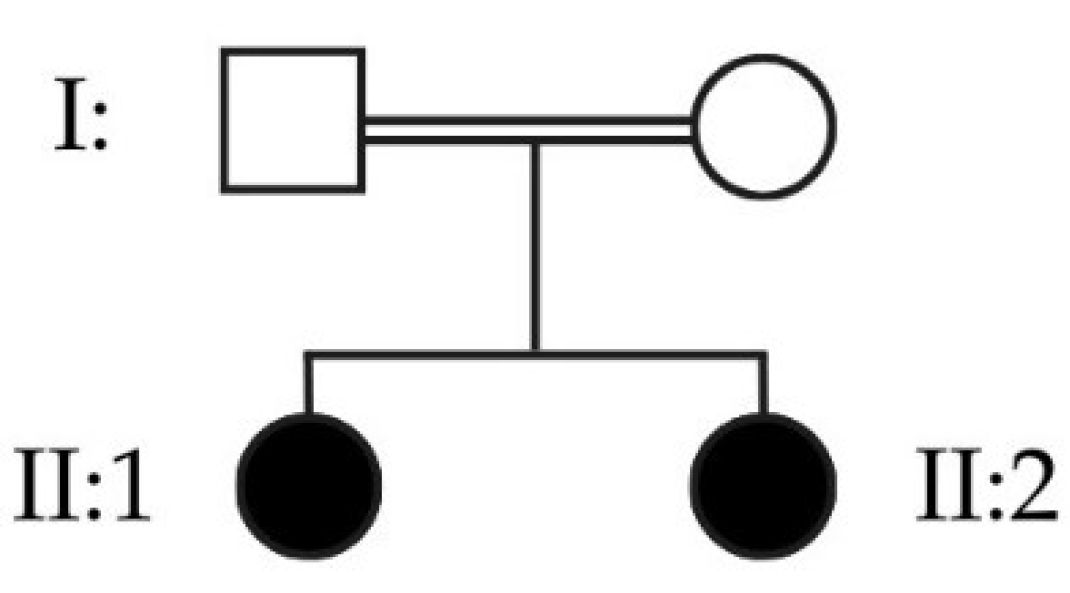
Figure 7.
Example of an input for the personalized multigene panels based on HPO term and ROH.

Figure 8.
IGV visualization of the reads mapped to the CSTB gene in both sisters (II:1 and II:2).
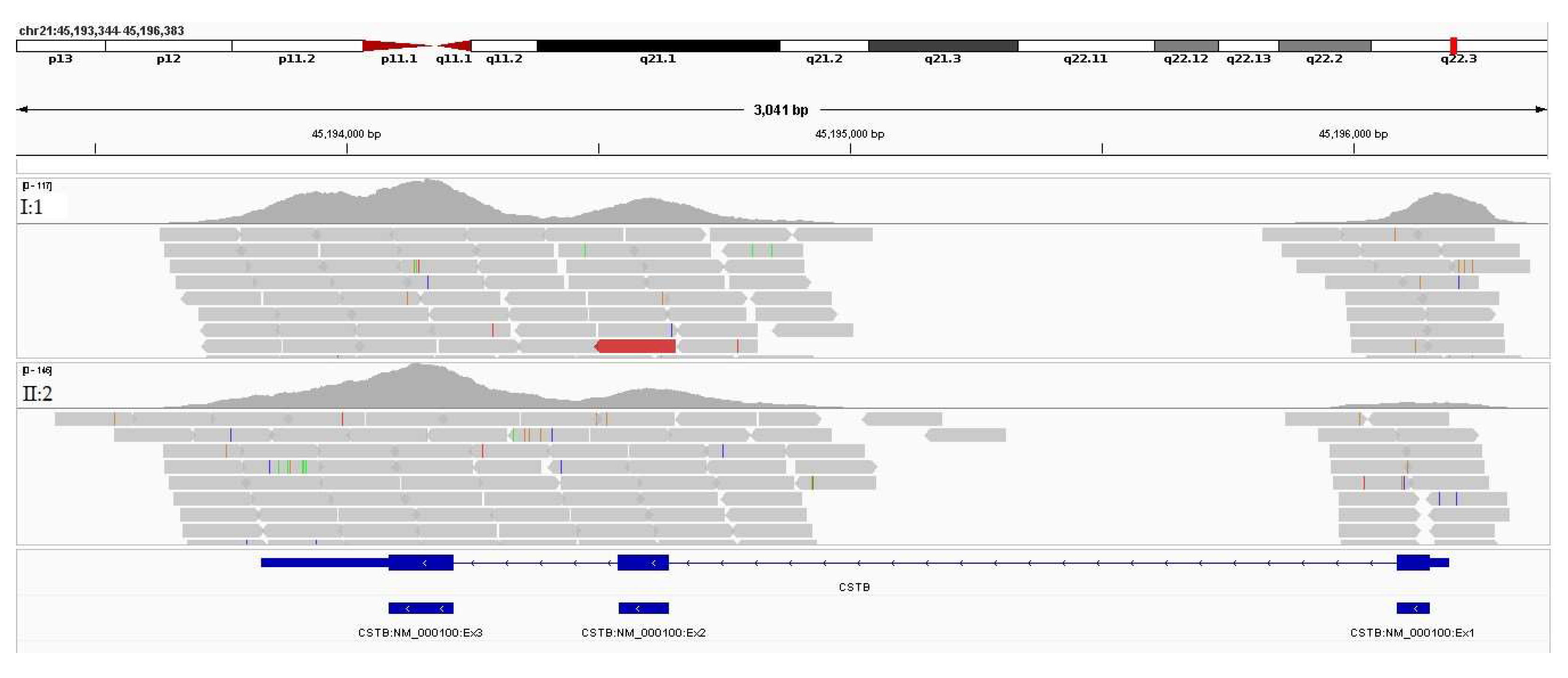
Figure 9.
BAM visualization depicting the region of the dodecamer repeat expansion in a control I, and in both sisters (II:1 and II:2). No reads are aligned in this region in both patients, suggesting that a possible expansion is biallelic (present in both CSTB alleles).
Figure 9.
BAM visualization depicting the region of the dodecamer repeat expansion in a control I, and in both sisters (II:1 and II:2). No reads are aligned in this region in both patients, suggesting that a possible expansion is biallelic (present in both CSTB alleles).
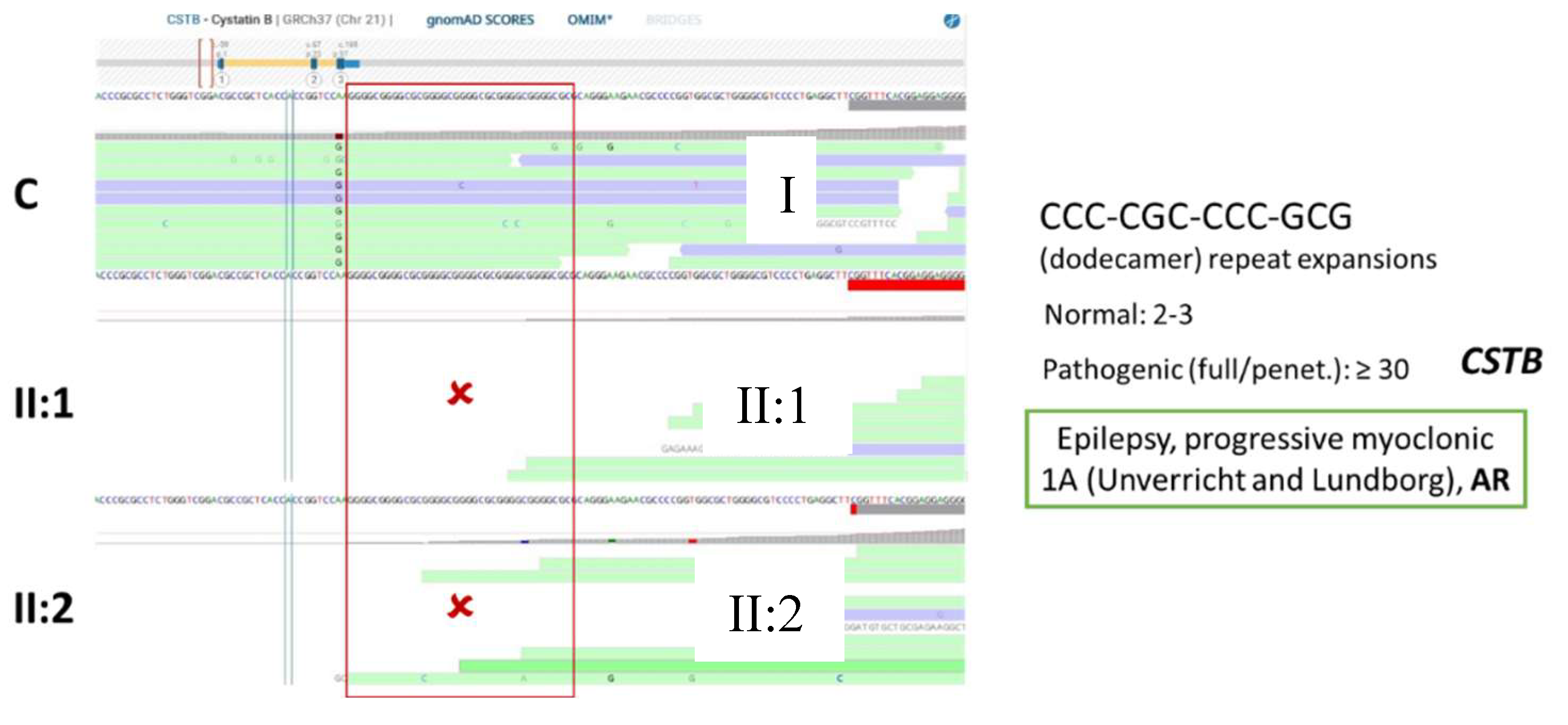
Figure 10.
Histogram depicting the distribution of ROH length above 0.5 Mb in a Portuguese cohort of 3,941 samples.
Figure 10.
Histogram depicting the distribution of ROH length above 0.5 Mb in a Portuguese cohort of 3,941 samples.
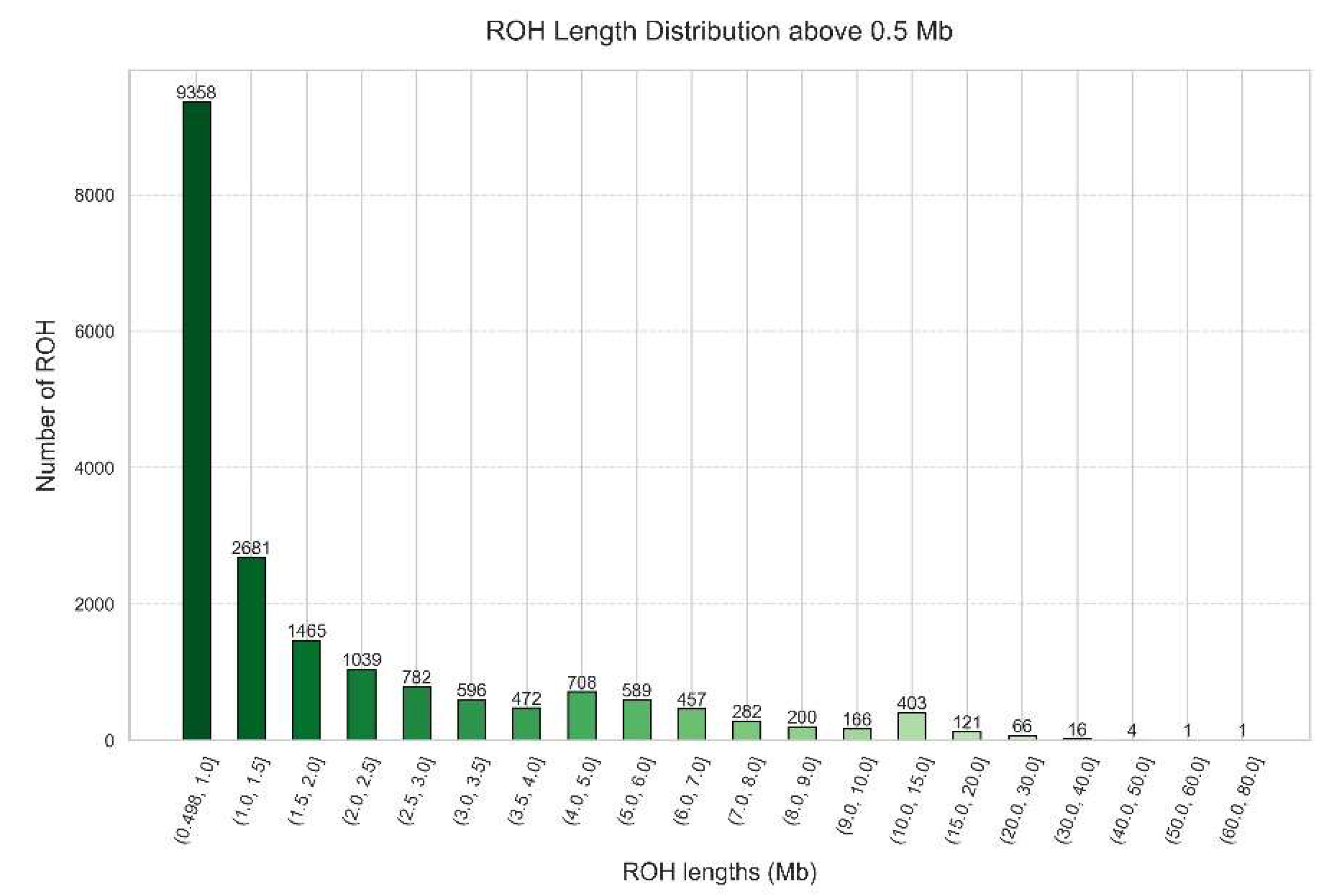
Figure 11.
Geographical distribution per municipality of FROH > 0.5 Mb in Portugal Mainland, Autonomous Region of Açores and Autonomous Region of Madeira.
Figure 11.
Geographical distribution per municipality of FROH > 0.5 Mb in Portugal Mainland, Autonomous Region of Açores and Autonomous Region of Madeira.
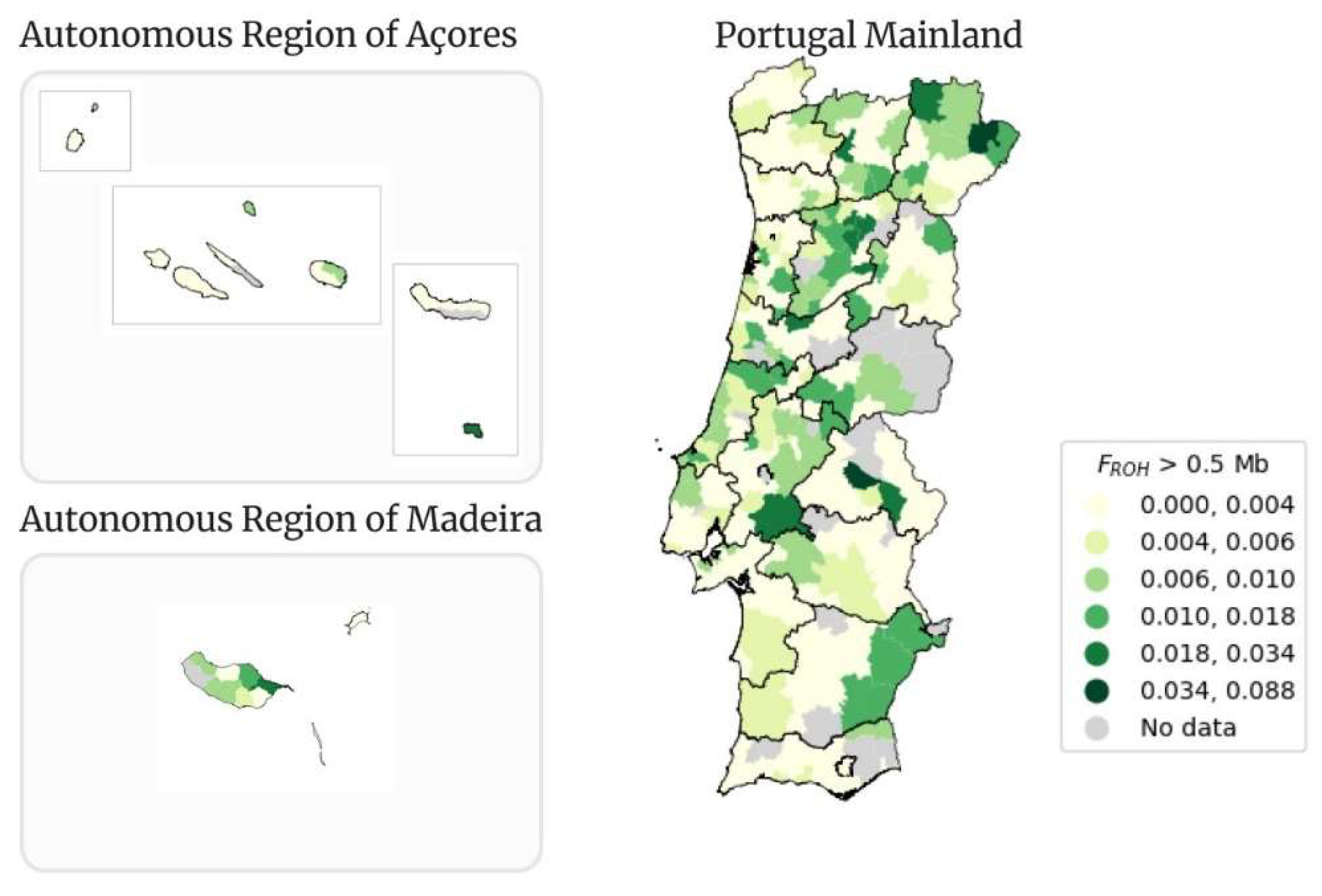
Figure 12.
Geographical distribution per municipality of FROH > 1.5 Mb in Portugal Mainland, Autonomous Region of Açores and Autonomous Region of Madeira.
Figure 12.
Geographical distribution per municipality of FROH > 1.5 Mb in Portugal Mainland, Autonomous Region of Açores and Autonomous Region of Madeira.
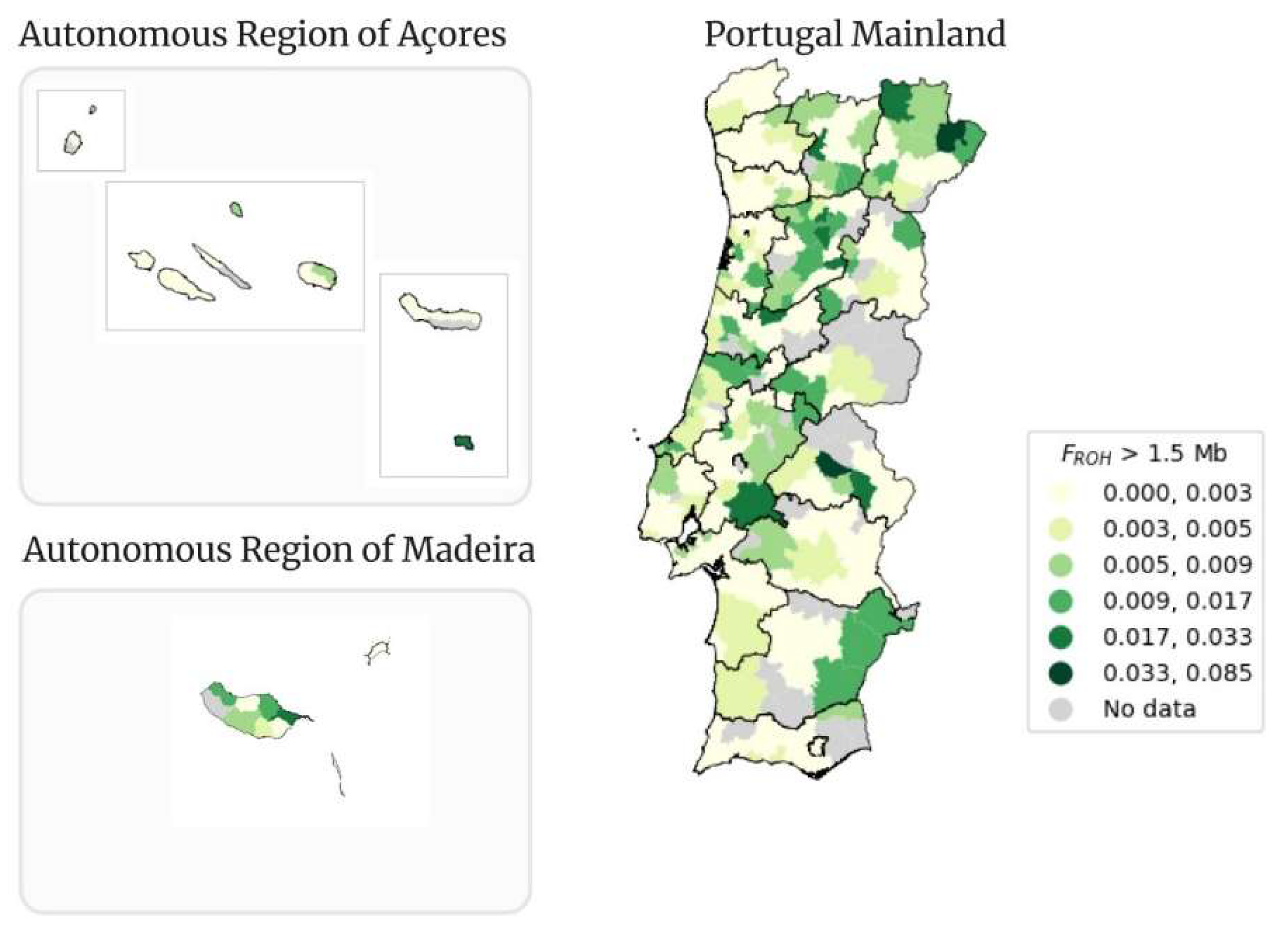
Figure 13.
Geographical distribution per municipality of FROH > 5 Mb in Portugal Mainland, Autonomous Region of Açores and Autonomous Region of Madeira.
Figure 13.
Geographical distribution per municipality of FROH > 5 Mb in Portugal Mainland, Autonomous Region of Açores and Autonomous Region of Madeira.
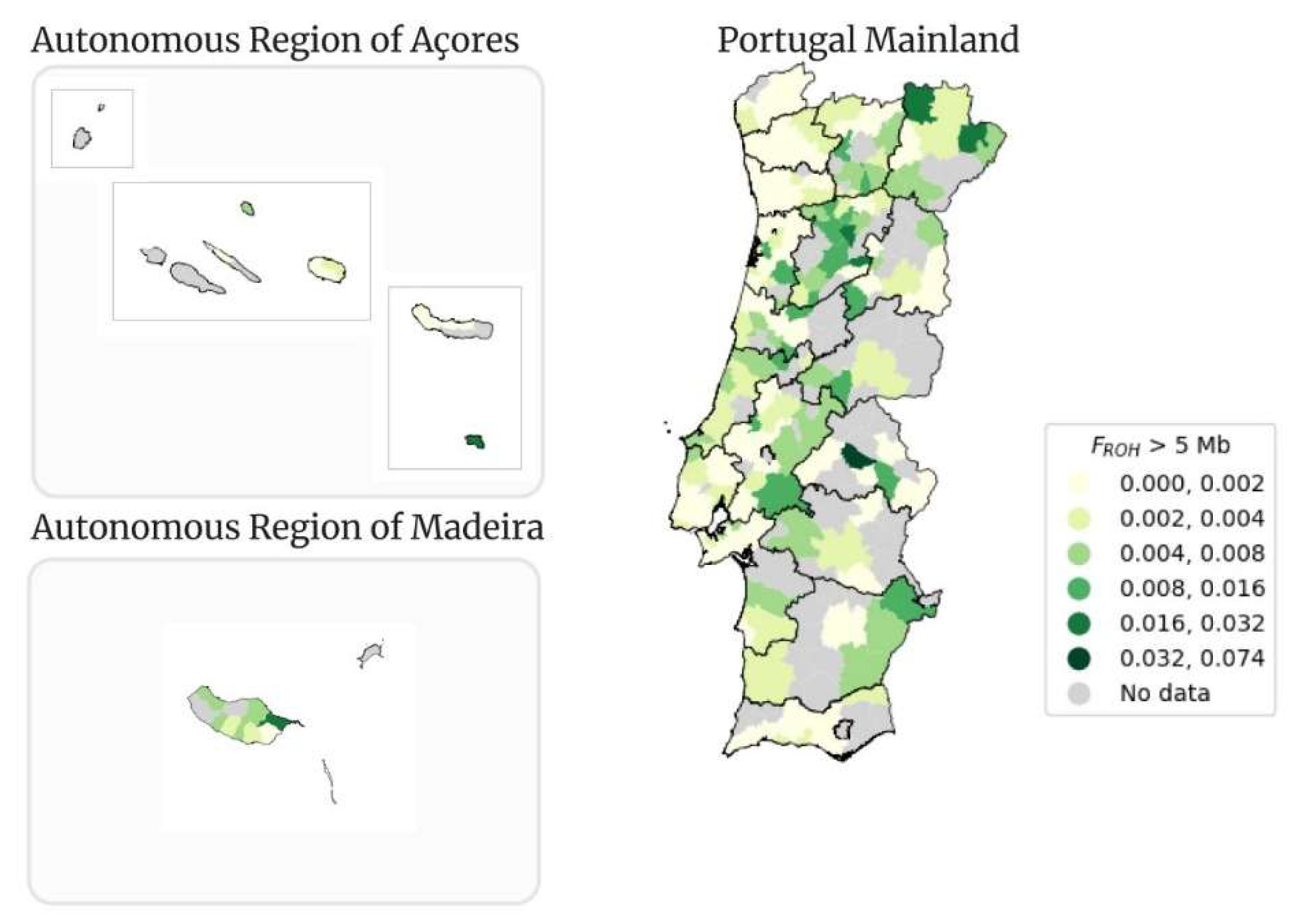
Figure 14.
Map of Portugal representing the consanguineous marriages between 1980 and 1986 (/100000) adapted from [88] (upper left) and the Portugal Mainland maps for the FROH calculated for ROHs of size above 0.5 Mb (upper right), 1.5 Mb (lower left) and 5 Mb (lower right).
Figure 14.
Map of Portugal representing the consanguineous marriages between 1980 and 1986 (/100000) adapted from [88] (upper left) and the Portugal Mainland maps for the FROH calculated for ROHs of size above 0.5 Mb (upper right), 1.5 Mb (lower left) and 5 Mb (lower right).
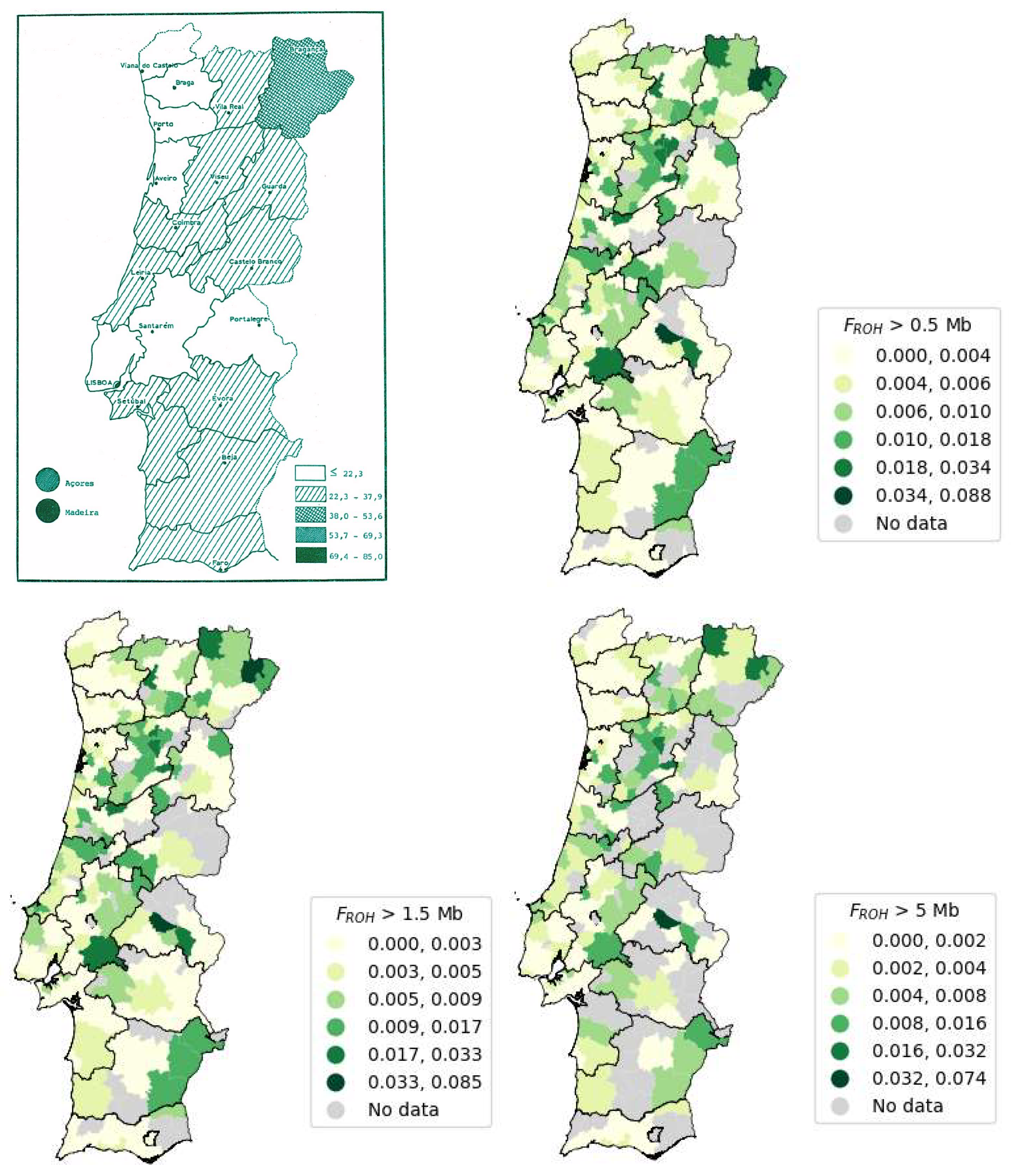
Figure 15.
Low-dimensional MDS representations of each “tier” dataset, where Tier 0: training and validation results (A) and testing results (B), Tier 1: training and validation results (C) and testing results (D); Tier 2: training and validation results (E) and testing results (F). Data points are colored according to their consanguinity labels: White “unknown” points do not possess a ground truth label; green “NCON” points represent non-consanguineous samples; red “CON” points represent consanguineous samples; and purple “CON_ST” represent stringent consanguineous points. The red dashed circles represent the elliptic envelope’s outlier decision boundary (i.e., points falling outside of the envelope are predicted to be consanguineous, either stringent or non-stringent).
Figure 15.
Low-dimensional MDS representations of each “tier” dataset, where Tier 0: training and validation results (A) and testing results (B), Tier 1: training and validation results (C) and testing results (D); Tier 2: training and validation results (E) and testing results (F). Data points are colored according to their consanguinity labels: White “unknown” points do not possess a ground truth label; green “NCON” points represent non-consanguineous samples; red “CON” points represent consanguineous samples; and purple “CON_ST” represent stringent consanguineous points. The red dashed circles represent the elliptic envelope’s outlier decision boundary (i.e., points falling outside of the envelope are predicted to be consanguineous, either stringent or non-stringent).
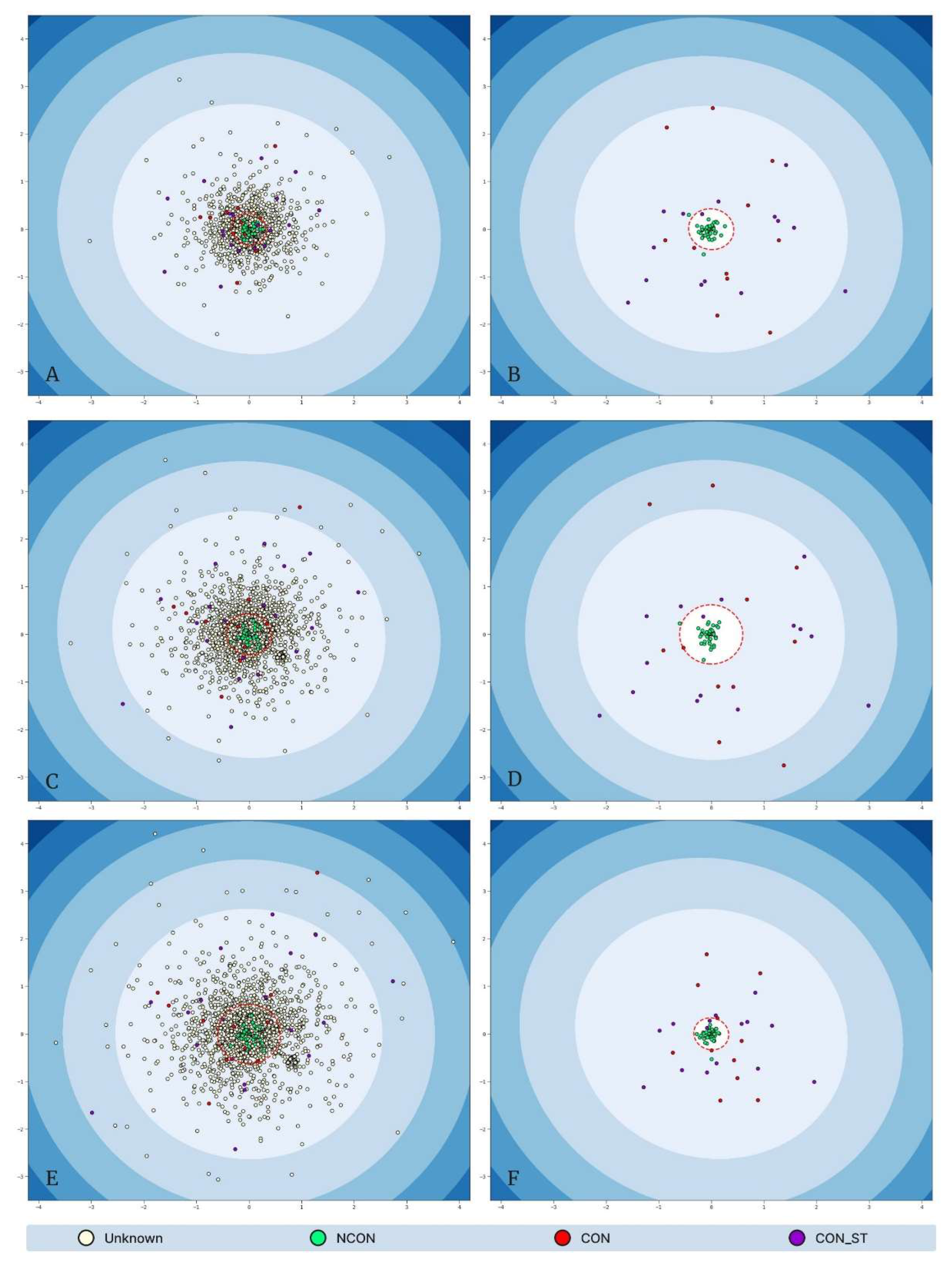

Table 1.
Resulting list of genes for each sister (II:1 and II:2).
| II:1 | II:2 |
|---|---|
| DHDDS | SIK1 |
| HMGCL | CSTB |
| MERC | SLC32A1 |
| SDHA | |
| SIK1 | |
| CSTB | |
| PIGV | |
| SLC25A19 | |
| SLC32A1 | |
| TERT | |
| TSEN54 |
Table 2.
Number of people with FROH > 0.5 Mb within each interval.
| FROH > 0.5 Mb Intervals | Number of samples |
|---|---|
| ]0.000, 0.004] | 3,104 |
| ]0.004, 0.008] | 210 |
| ]0.008, 0.010] | 156 |
| ]0.010, 0.018] | 107 |
| ]0.018, 0.034] | 124 |
| ]0.034, 0.088] | 99 |
Table 3.
Number of people with FROH > 1.5 Mb within each interval.
| FROH > 1.5 Mb Intervals | Number of samples |
|---|---|
| ]0.000, 0.003] | 1,430 |
| ]0.003, 0.005] | 196 |
| ]0.005, 0.009] | 162 |
| ]0.009, 0.017] | 107 |
| ]0.017, 0.033] | 126 |
| ]0.033, 0.085] | 89 |
Table 4.
Number of people with FROH > 5 Mb within each interval.
| FROH > 5 Mb Intervals | Number of samples |
|---|---|
| ]0.000, 0.002] | 38 |
| ]0.002, 0.004] | 318 |
| ]0.004, 0.008] | 146 |
| ]0.008, 0.016] | 113 |
| ]0.016, 0.032] | 91 |
| ]0.032, 0.074] | 52 |
Table 5.
FROH mean, FROH mean of means per municipality and comparative values for the different ROH size thresholds (0.5, 1.5 and 5 Mb).
Table 5.
FROH mean, FROH mean of means per municipality and comparative values for the different ROH size thresholds (0.5, 1.5 and 5 Mb).
| Mean FROH | Mean FROH of means per municipality | FROH comparative values [87] | |
|---|---|---|---|
| FROH > 0.5 Mb | 0.0042 | 0.0057 | 0.0315 |
| FROH > 1.5 Mb | 0.0033 | 0.0049 | 0.0021 |
| FROH > 5 Mb | 0.0020 | 0.0039 | 0.0001 |
Table 6.
FROH calculated for the 0.5, 1.5 and 5 Mb thresholds per district, and data from a study estimating the number of consanguineous marriages per district [88] .
Table 6.
FROH calculated for the 0.5, 1.5 and 5 Mb thresholds per district, and data from a study estimating the number of consanguineous marriages per district [88] .
| District | FROH > 0.5 Mb | FROH > 1.5 Mb | FROH > 5.0 Mb | Number of consanguineous marriages (10 000) [88] |
|---|---|---|---|---|
| Açores | 0.0046 | 0.0035 | 0.0023 | 78.7 |
| Aveiro | 0.0052 | 0.0041 | 0.0024 | 22.1 |
| Beja | 0.0048 | 0.0039 | 0.0022 | 22.8 |
| Braga | 0.0034 | 0.0025 | 0.0013 | 19.2 |
| Bragança | 0.0102 | 0.0090 | 0.0060 | 52.7 |
| Castelo Branco | 0.0066 | 0.0054 | 0.0034 | 19.9 |
| Coimbra | 0.0063 | 0.0053 | 0.0036 | 38.2 |
| Évora | 0.0039 | 0.0028 | 0.0016 | 34.5 |
| Faro | 0.0029 | 0.0019 | 0.0010 | 27.2 |
| Guarda | 0.0048 | 0.0038 | 0.0024 | 35.3 |
| Leiria | 0.0058 | 0.0047 | 0.0030 | 35.1 |
| Lisboa | 0.0039 | 0.0030 | 0.0017 | 20.2 |
| Madeira | 0.0077 | 0.0068 | 0.0041 | 133.6 |
| Portalegre | 0.0106 | 0.0092 | 0.0070 | 24.8 |
| Porto | 0.0026 | 0.0018 | 0.0010 | 14.4 |
| Santarém | 0.0056 | 0.0045 | 0.0032 | 27.6 |
| Setúbal | 0.0038 | 0.0029 | 0.0019 | 30.1 |
| Viana do Castelo | 0.0030 | 0.0021 | 0.0010 | 17.8 |
| Vila Real | 0.0070 | 0.0060 | 0.0037 | 38.3 |
| Viseu | 0.0105 | 0.0091 | 0.0059 | 38.7 |
Table 7.
Outlier detection F1-score results, separated by feature set tier.
| Dataset Tier (Feature Set) | Best Contamination Hyperparameter | Validation F1-Score | Test F1-Score |
|---|---|---|---|
| Tier 0 (Count_x, Sum_x) | 0.0786 | 0.9310 | 0.9412 |
| Tier 1 (Count_x, Sum_x, Min_x, Max_x) | 0.1190 | 0.9655 | 0.9434 |
| Tier 2 (Count_x, Sum_x, Min_x, Max_x, Mean_x, STD_x) | 0.1061 | 0.9474 | 0.9615 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



