Submitted:
27 September 2024
Posted:
29 September 2024
You are already at the latest version
Abstract
Introduction: microRNA-181a (miR-181a) is a crucial post-transcriptional regulator of many mRNA transcripts and ncRNAs, influencing cell proliferation, cancer cell stemness, apoptosis, and immune responses. Its abnormal expression has been characterized in numerous cancers, making it a significant genomic vulnerability and biomarker in cancer research. Areas Covered: Here, we summarize miR-181a’s correlation with poor patient outcomes across numerous cancers, and the mechanisms governing miR-181a’s activity and processing. We comprehensively describe miR-181a’s involvement in multiple regulatory cancer signaling pathways, cellular processes, and the tumor microenvironment. We also discuss current therapeutic approaches to targeting miR-181a, highlighting their limitations and future potential. Expert Opinion: miR-181a is a clinically relevant pan-cancer biomarker with potential as a therapeutic target in cancer. Its regulatory control of tumorigenic signaling pathways and immune responses positions it as a promising candidate for more personalized treatments. The success of miR-181a as a target relies on the development of specific therapeutics platforms. Future research on miR-181a's role in the tumor microenvironment and the RNA binding proteins that regulate its stability will help uncover new techniques to targeting miR-181a. Further research into miR-181a serum levels in patients undergoing therapy will help to better stratify patients and enhance therapeutic success.
Keywords:
biomarker
; cancer
; miR-181a
; microRNA
; microRNA therapeutics
; microRNA processing
; microRNA regulation
; signaling pathways
; signal transduction
Article Highlights
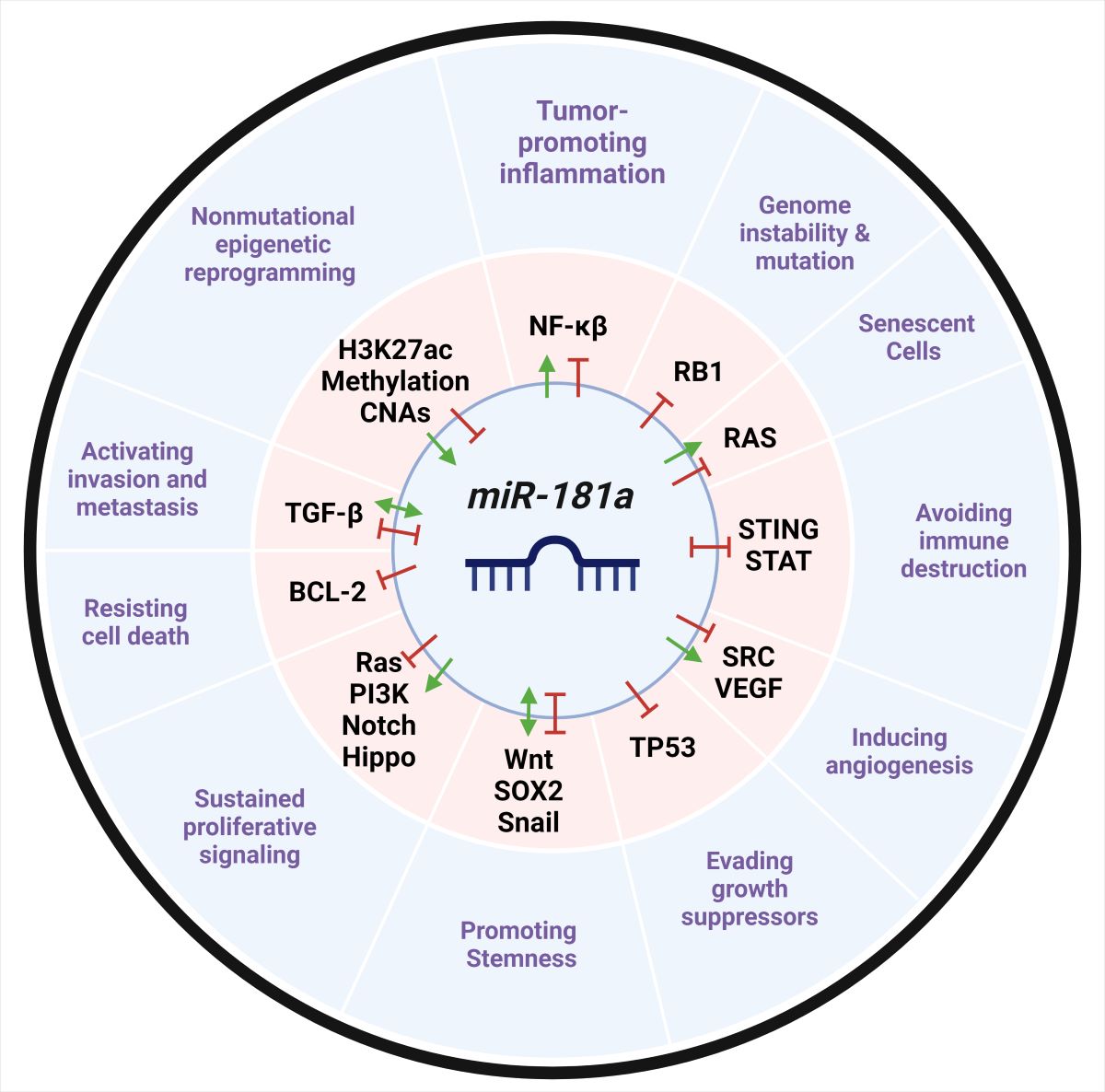
- Regulatory Role: microRNA-181a can function as an oncogene or a tumor suppressor by post-transcriptionally regulating many mRNA transcripts and non-coding RNAs involved in cancer.
- Aberrant Expression: microRNA-181a is aberrantly expressed in a large majority of cancers contributing to tumor progression, increased proliferation, immune suppression, and apoptosis.
- Clinical Relevance: miR-181a is a pan-cancer biomarker with altered expression profiles that can be detected in the serum of patients.
- Therapeutic Potential: Pre-clinical studies suggest that targeting miR-181a in vivo can inhibit cancer progression and that knockout of miR-181a is not toxic and presents a potentially favorable safety profile. Thus, miR-181a serves as an ideal therapeutic target.
- Immune Microenvironment: miR-181a plays a major role in immune cell development, particularly NK and T-cells, and can influence the tumor microenvironment.
- Next Steps: Development of a therapeutic platform for targeting miR-181a via nanoparticles, natural products, small molecules, or repurposed drugs is presents a polyvalent therapeutic approach to inhibiting cancer progression.
1. Introduction
Oncogenesis is a result of dysfunctions in cellular machinery. Tumor suppressive redundancies, feedback loops, extrinsic immune surveillance, programmed cell death, and other mechanisms exist to guard against transformation. When these mechanisms fail, neoplasia and carcinogenesis result as competitive advantages that overcome these defenses, permitting cell survival, proliferation, and metastasis. Countless happenstances can result in transformation; hence, due to their wide breadth of regulatory reach, microRNAs (miRNA) have emerged as potent, corruptible mediators of oncogenesis.
miRNAs are evolutionarily conserved non-coding RNAs of about 21-25 nucleotides in length that primarily mediate post-transcriptional repression of messenger RNA (mRNA). This targeted repression of mRNA networks is conducted via homology between the miRNA’s seed sequence and the 3’ untranslated region (3’UTR) of the mRNA. miRNA families share a common mRNA recognition seed sequence. These family members are often derived from a single phylogenic ancestor and can be found in clusters in the genome. miRNAs are localized intragenically or, more frequently, intergenically. Intragenic miRNAs, or miRtrons, exist within the intronic regions of protein-coding genes [1,2]. Intragenic miRNAs can sometimes have their own promoters within the transcriptional region of a protein-coding gene, in addition to being regulated by the host gene’s promoter [3,4]. In contrast, intergenic miRNAs have their own promoters. They are found as either independent transcription units or clusters. Clusters include two or more miRNAs transcribed from adjacent miRNA genes within ten kilobases (kb) of each other. These clusters are transcribed in the same orientation and are not separated by a transcriptional unit or miRNA in the opposite direction [5]. Thus, intergenic miRNAs can be transcribed as individual or polycistronic miRNAs [6,7,8].
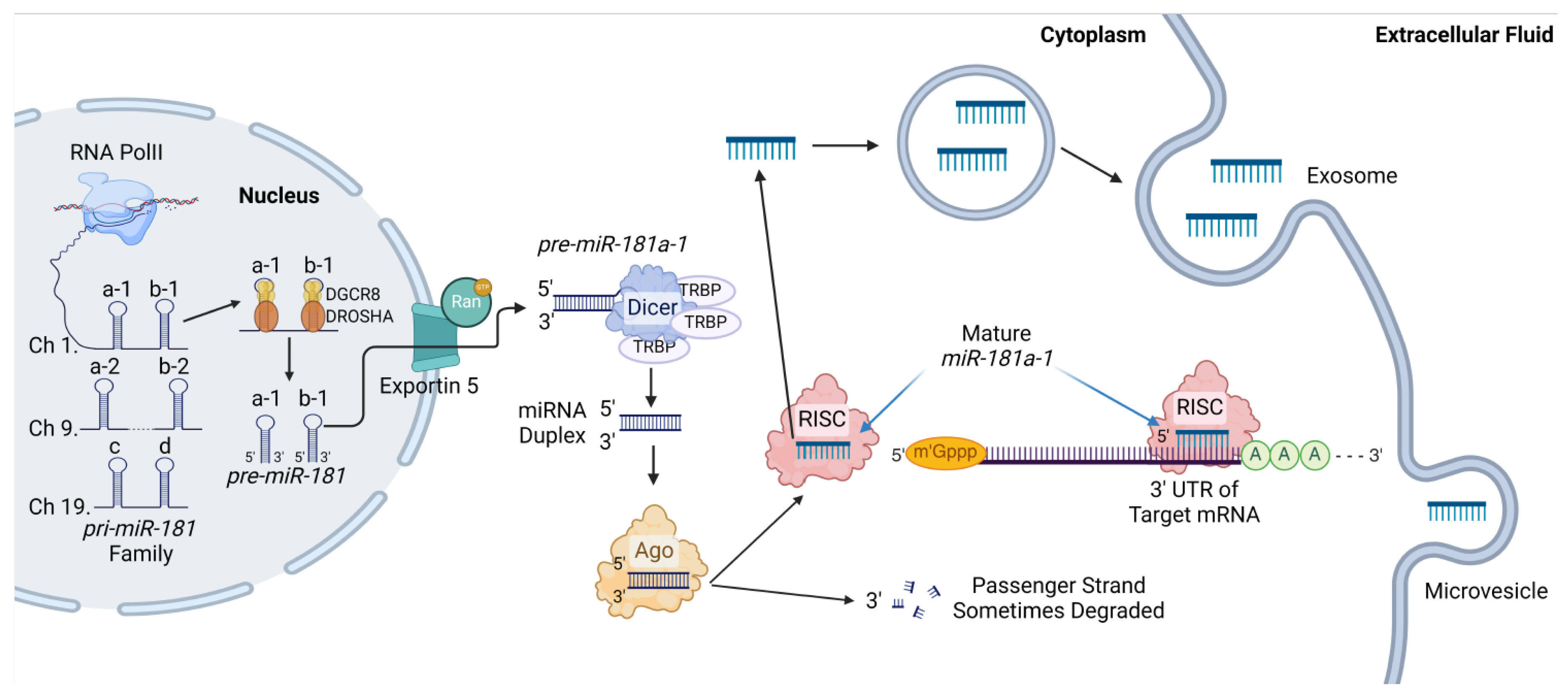
In canonical miRNA biogenesis (Figure 1), miRNA coding regions are detected by RNA polymerase II (PolII) and transcribed into large primary microRNAs (pri-miRNA) in the nucleus. There, the pri-miRNA is processed by DROSHA and DGC8 into a pre-miRNA transcript (~60-70 nt) before being shunted to the cytoplasm by Exportin 5 and Ran-GTP. Once in the cytoplasm, DICER and TRBP cleave the pre-miRNA generating a double-stranded duplex containing the mature miRNA and the complementary passenger strand. The RNA duplex is then loaded onto Argonaut (Ago), where the mature strand interacts with Ago, and the passenger strand is expelled. The Ago-miRNA unit is here-on named the RNA-induced silencing complex (RISC). RISC interacts with the target mRNA’s 3’UTR via nucleotide complementarity, facilitating miRNA-mediated mRNA silencing [6,9]. In the context of cancer, these miRNAs can target oncogenic and tumor-suppressive mRNA transcripts. Thus, dysregulation of miRNAs can lead to oncogenic progression. This review will highlight the dysregulation and cancer promoting role of a particular miRNA, miR-181a. Abnormal expression of miR-181a can lead to disruption in multiple signaling pathways, providing cancer cells with the functional characteristics and hallmarks of cancer central to carcinogenesis [10].
Figure 1.
The miR-181a biogenesis pathway following canonical miRNA biogenesis. miR-181 is transcribed by RNA Polll, generating one of three primary transcripts. miR-181a and miR-181b can be transcribed from chromosome 1 or 9. miR-181c and miR-181d are transcribed from chromosome 19. Following miR-181a-1 synthesis, its primary miR-181a/b-1 transcript is cleaved by the Drosha-DGCR8 microprocessor complex into individual pre-miR181a/b transcripts and exported into the cytoplasm via Exportin-5-Ran-GTP. In the cytoplasm, DICER-TRBP cleaves the hairpin structure to the mature miR-181a length. The mature strand is then loaded onto argonaut (Ago), and the passenger strand is expelled and sometimes degraded. To facilitate mRNA regulation, the Ago-mature miRNA unit, now termed RNA-induced silencing complex (RISC), is guided to the 3’UTR on target mRNA sequences and facilitates translational repression. Alternatively, mature miR-181a can be exported out of the cell in exosomes and microvesicles. Figure generated using BioRender.
Figure 1.
The miR-181a biogenesis pathway following canonical miRNA biogenesis. miR-181 is transcribed by RNA Polll, generating one of three primary transcripts. miR-181a and miR-181b can be transcribed from chromosome 1 or 9. miR-181c and miR-181d are transcribed from chromosome 19. Following miR-181a-1 synthesis, its primary miR-181a/b-1 transcript is cleaved by the Drosha-DGCR8 microprocessor complex into individual pre-miR181a/b transcripts and exported into the cytoplasm via Exportin-5-Ran-GTP. In the cytoplasm, DICER-TRBP cleaves the hairpin structure to the mature miR-181a length. The mature strand is then loaded onto argonaut (Ago), and the passenger strand is expelled and sometimes degraded. To facilitate mRNA regulation, the Ago-mature miRNA unit, now termed RNA-induced silencing complex (RISC), is guided to the 3’UTR on target mRNA sequences and facilitates translational repression. Alternatively, mature miR-181a can be exported out of the cell in exosomes and microvesicles. Figure generated using BioRender.
2. miR-181a Overview
2.1. miR-181 Family
First discovered in 2003 by three independent groups, the microRNA-181 (miR-181) family consists of four mature miRNA members (miR-181a/b/c/d) from six different loci in the genome and remains well conserved among vertebrates (Figure 1) (Table 1) [11,12,13]. miR-181a-1 and miR-181a-2 are located on chromosomes 1 and 9, respectively, and ultimately produce the same mature miR-181a. miR-181b-1 and miR-181b-2 are located on chromosomes 1 and 9, respectively, and ultimately produce the same miR-181b. On chromosome 1, miR-181a-1 and miR-181b-1 are 61 nucleotides (nt) apart and are encoded by the MIR181A1B1 gene. Both miR-181a-1 and miR-181b-1 are co-transcribed through their own promoter [14]. The minimal regulatory region for miR-181a-1 and miR-181b-1 on chromosome 1 has been mapped to the 615 nt upstream of miR-181a-1 on Chr 1q31.3. In contrast, miR-181a-2 and miR-181b-2 are encoded by MIR181A2B2 on chromosome 9 and are spaced 1158 nt apart. Distinctly, miR-181a-2 and miR-181b-2 are miRtrons of the NR6A1 gene, which is transcribed antisense to MIR181A2B2 [14]. Since miR-181a and miR-181b are polycistronic miRNAs, they are co-transcribed within the same pri-miRNA transcript. Although their mature 5p transcripts are identical across chromosomes 1 and 9, their mature 3p sequences are not. Furthermore, their pri-miRNAs and pre-miRNAs encoded by MIR181A1B1 and MIR181A2B2 are unique [14,15]. The other family members, miR-181c and miR-181d are located on chromosome 19 in an uncharacterized sequence and are spaced 66 nt apart.
All four mature 5p members of the miR-181 family share the same seed sequence from the second to the eighth nucleotide: “ACAUUCA.” microRNA recognition sequences (MRE) are complementary binding sequences in target mRNAs that use nucleotide complementarity to interact with the seed sequence of miRNAs and facilitate mRNA repression and silencing. These MREs are commonly located in the 3’UTR; however, it is noteworthy that a large portion of miR-181a MREs exist in coding regions and in 5’UTRs [16]. Although only one miR-181a MRE is required to confer repression, increased frequencies of miR-181a MREs in mRNA transcripts lead to increased target repression [16].
2.2. Genomic Variations of miR-181a
Investigation into the potential consequences of miRNA’s genetic variation is still developing. Although more than 30 single-nucleotide variations (SNVs) of miR-181a/b within chromosome 1 are known to exist, there is limited literature describing their roles in miR-181a’s activity.
One single-nucleotide polymorphism (SNP), rs322931 (C>T), has recently garnered attention. The minor allele of rs322931 is associated with lower miR-181a expression in the brain and the blood [17]. Variations in rs322931 are also associated with the miR-181 family’s role in regulating inflammation across various cell types, particularly the colon. Patients carrying the SNP rs322931 have exhibited aggravated inflammatory responses. For instance, in peripheral blood samples of Chron’s disease patients, those carrying rs322931 had increased inflammatory factors, including TNF-α, IL-1β, IL-6, CRP, SSA, AAT, AAG, and HPT [18]. Additionally, rs322931 is associated with a heightened predisposition to systemic lupus erythematosus, a disease characterized by chronic inflammation and immunologic abnormalities [19]. Moreover, higher expression levels of the rs322931 SNP correlates with increased happiness and significant risk for ischemic stroke [17,20,21,22].
In CRC patients, a SNP in MIR181A1 with a minor allele frequency >5%, rs12039395 G>T, was evaluated. In these patients, the G-allele was significantly more prevalent than the T-allele; however, this group was the first to identify that miR-181a-5p is overexpressed in both cancerous and normal tissue samples carrying the G-allele over the T-allele. Given that rs12039395 is found 1.31 Kb upstream of pri-miR-181a-1, primary structures containing the G-allele have a significantly different predicted loop formation with higher minimum free energy requirements than the T-allele. This suggests a potentially less complex structure with greater accessibility for protein modulators that contribute to its elevated mature expression [23]. Another, SNP rs7550394, residing in LINC01221 genomically adjacent to miR-181a/b-1, has also been associated with changes in miR-181a expression levels and pathogenicity [17]. Further exploration of the incidence of these SNPs could elucidate the molecular underpinnings contributing to changes in miR-181a processing, while also identifying increased associations between normal and pathological states and their response to therapeutic options in cancer and other diseases.
Aberrations in the seed sequence of a miRNA can cause striking differences in the function, number, and types of target sequences it regulates post-transcriptionally. Mutations in the first two nucleotides of miR-181’s seed sequence (AC), or the fifth and sixth nucleotides (UC), can abolish the miRNA’s activity, while mutations in the third and fourth nucleotides (AU), do not impair its function [24]. Chira et al. conducted a bioinformatic analysis examining the base composition in the seed region of mature miR-181 genes and concluded that an A>G substitution in the 4th nt would result in the most significant change in the number of predicted targets, suggesting that miR-181’s target activity is heavily influenced by binding at this specific purine [25].
The impact of changes in the nucleotide sequences outside of the seed region on the structural conformation of pri- and pre-miR-181a is crucial for investigating the interactions of its processing and binding partners. icSHAPE (in vivo click selective 2-hydroxyl acylation and profiling experiment) is a technique used to predict the secondary structure of RNA in cells by measuring nucleotide flexibility at each base [26]. icSHAPE-MaP combined with RNA-immunoprecipitation studies (RIP) suggest that the secondary structure of pre-miR-181a-1 matches the predicted structure found in miRBase, apart from the last few nucleotides on the 3’ end [27]. Although the wild type structure of pre-miR-181a is known, identification of mutant pri- and pre-miR-181a structures could be beneficial in elucidating structural components key to their function. Through ectopic expression of miR-181a-1 mutants, Liu et al. note that mutations in the nt 8-9 (AA), 10-11 (CG), 14-15 (GU), 20-21 (GA), or 22-23 (GU) can reduce its activity, while mutations in nt 16-17 (CG) can increase its activity. Moreover, mutations in the pre-miR-181a loop, particularly nt 26-27 (GG), 30-31 (UU), and 32-33 (CA), resulted in reduced downstream miR-181a activity [24]. Chira et al. also examined how base substitutions can impact the secondary structure of miR-181 family members. For instance, in the case of an A>G substitution in the 4th nt in miR-181a, this new secondary structure conformation resulted in a more extended 5’ arm by one residue and a free U residue at the 3’ end, creating a clamp-like structure [25]. Interestingly, this substitution leads to a reduced class of target genes, each associated with pro-oncogenic roles in human cancers, including Tripartite Motif Containing 5 (TRIM5) and CDK2AP1. This novel study highlights the potential for miRNAs to undergo “reprogramming” for a niche group of targeted interactions [25].
2.3. Canonical and Noncanonical miR-181a Processing
Canonical biogenesis of miRNAs includes the processing of pri-miRNAs by DROSHA/DGCR8, trafficking of pre-miRNAs into the cytoplasm by Exportin 5, and pre-miRNA to mature miRNA processing by DICER (Figure 1). However, some miRNAs are produced in noncanonical DICER-independent pathways. Exploration of miRNA processing specific to the miR-181 family remains in its infancy but suggests miR-181a could be processed via canonical and noncanonical mechanisms. In DICER-deficient murine sarcoma models, expression of all miR-181 family members was reduced but not eliminated, demonstrating the existence of a DICER-independent processing pathway used by the miR-181 family [28]. These findings were validated in human HCT116 DICER-/- cells where miR-181a-5p was expressed, although at reduced levels compared to cells with wt DICER [29]. These findings are mostly supported by the Huntsman group observing that miR-181a-5p was still expressed in hemizygous DICER+/fl-D1693N induced murine oviductal mutants and their corresponding tumors. However, in this model miR-181a-1-3p and miR-181a-2-3p were lowly expressed, suggesting that miR-181a-5p processing could be controlled independent of DICER while miR-181a-3p processing largely requires DICER [30].
Outside of DICER, other miRNA mediators have been shown to process miR-181a through canonical and noncanonical mechanisms. In HCT116 DROSHA-/- cells, miR-181a-5p levels were greatly reduced, suggesting that this miRNA is heavily processed by DROSHA [83]. These findings were further affirmed by Wang et al. demonstrating a direct interaction of pri-miR-181a and DROSHA in a breast cancer context [29]. In XPO5-/- HCT116 cells, miR-181a-5p levels were not changed, suggesting that pre-miR-181a may not be exported into the cytoplasm alone by Exportin 5, and there is another key transporter of miR-181a that remains to be identified [29]. Further investigation of miR-181a’s processing would elucidate critical machinery involved in its expression and could point to targetable interactions in miR-181a dysregulated diseases.
Upon mature miR-181a synthesis, little is known about this microRNA’s targeting mechanisms. More than 1,300 conserved target sites within transcripts have been predicted for miR-181a-5p using the TargetScan [31] software and 635 targets in miRTarBase [32]. However, further analysis of the miR-181a/b targetome has captured 2,995 target transcripts using an Ago2-miR-181a-mRNA chimeric enhanced UV crosslinking and chimeric eCLIP immunoprecipitation procedure [16]. This study identified new targets not previously predicted by target identification software. The authors propose that these targets contain a noncanonical MRE that the miR-181a seed sequence binds to. Canonically, the seed sequence, ACAUUC, binds to a complementary MRE found in mRNA, UGUAAG. However, an alternative seed match sequence, UGUUAG, was identified in nearly half of the targets [16]. This A-to-U seed match variant demonstrates that absolute matching of seed sequences is not required for miR-181a targeting and suggests that the targetome of miR-181a could be much larger than anticipated.
Targeting of miR-181a to these MREs is dependent on miR-181a-RISC interaction [16]. Once mature miR-181a interacts with the 3’UTR of its targets, mRNA-mediated gene regulation occurs via RNA destabilization rather than translational inhibition. To our knowledge, this is the first time miR-181a's gene silencing mechanism has been characterized [16]. Further elucidation miR-181a’s mechanism of posttranscriptional gene expression regulation could elucidate miR-181a–mRNA networks that are dysregulated and contribute to disease progression.
2.4. RNA Binding Protein Interactions with miR-181a
RNA binding proteins (RBP) are pivotal in governing RNA processing through splicing factors, post-transcriptional regulators, and stabilizers. However, the specific RNA binding partners associated with miR-181a remain relatively unidentified. One study postulated that an RBP belonging to the heterogenous nuclear ribonucleoprotein class, Synaptotagmin-binding Cytoplasmic RNA-Interacting Protein (SYNCRIP), plays a role in miR-181a processing. Knockdown of SYNCRIP reduced precursor and mature miR-181a levels and restored mesenchymal to epithelial transitioning (MET) via the control of TGF-β, a known target pathway of miR-181a [33]. While this study suggests SYNCRIP as a potential RBP of miR-181a, their direct interaction remains unclear.
Lin-28 is a famous RNA binding protein that regulates developmental processes and directly regulates let-7. It has been shown in pan-cancer models that miR-181a directly targets the 3’UTR of Lin28, an inhibitor of let-7 maturation [34]. Given that let-7 dysregulation has been shown to play a crucial role in carcinogenesis, miR-181a mediated reduction in mature let-7 expression could have greater implications on tumor formation, which remains to be explored.
2.5. miR-181a’s Classical Role
miR-181’s regulatory network encompasses key mediators of stemness and cell fate to control development in various tissue types [35,36]. Properly orchestrated vasculogenesis and angiogenesis are critical from early embryonic development through adulthood [37,38]. The nature of these processes requires tight spatial and temporal regulation and coherence of various biological processes [38]. The miR-181 family can execute the multi-pathway regulation required to coordinate endothelial differentiation and angiogenesis through direct targeting of key mediators such as PROX1, ERK, MMP-2, MMP-9, PDGFRA, and more [37,39]. Furthermore, miR-181a/b are indispensable for controlling differentiation. For example, in retinal development, proper axonal guidance and cell-fate specification by facilitating a process that allows the TGF-β pathway to tune MAPK signaling [40]. Here, TGF-β increases mature miR-181a expression via SMAD2/3 mediated processing which in turn targets ERK2 mRNA for degradation. Knockdown of miR-181a in vitro and in vivo ablates this crosstalk, resulting in developmental defects of the visual system. Paradoxically, miR-181a promotes differentiation in adipocytes and osteoblasts through negative regulation of TGF-β signaling, indicating tissue-specific regulatory activity [41,42,43]. In these cell types, miR-181a can promote differentiation by blunting the TGF-β pathway by directly targeting TGF-βR1 [44]. Furthermore, in osteogenesis, miR-181a/b promote differentiation by targeting PTEN, thus enhancing PI3K signaling [41,45].
miR-181a has also emerged as a critical regulator of T-cell development [46]. Appropriate T-cell maturation is predicated on clonal deletion of self-recognizing thymocytes. This inextricable feature for avoiding autoimmunity is carefully orchestrated within the thymus, wherein even weakly affinitive self-peptides can induce negative selection. High expression of miR-181a has been shown to govern T-cell receptor (TCR) signaling making early thymocytes more sensitive to cognate antigens. This results in a more rigorous negative selection during development only to be downregulated in differentiated T-cells, thus increasing the activation threshold as a protective measure against autoimmunity [46,47]. In this way, miR-181a primes TCR signaling intermediates by governing the expression of multiple negative regulators. Li et al. demonstrated that miR-181a acts as a rheostat through repression of key phosphatases that negatively regulate the TCR signaling cascade [47]. miR-181a primes TCR signaling intermediate LCK uniquely through multi-target repression of phosphatases meant to restrain the pathway such as PTPN22, SHP-1, and DUSP5/6 (via ERK dephosphorylation) [47]. miR-181a has also been implicated as a key mediator of γδ T-cell maturation. High intrathymic miR-181a expression in immature γδ T-cells restricts differentiation by targeting MAP3K2 and Notch2 [48]. Importantly, miR-181a expression declines in T-cells over time, suggesting that miR-181a plays a key role in age-related adaptive immunity [46]. This decline in expression is thought to be controlled by transcription factor YY1, binds to the enhancer region of MIR181A1B1. As the expression of YY1 decreases with age, so does pri-miR-181a [46].
Beyond lymphocytic cells, miR-181a’s role in myeloid cells has also been recently described. For example, CD34+ hematopoietic progenitor cells require miR-181a to differentiate into mature CD56+ NK cells. At each stage of NK cell development, miR-181a expression steadily increases, emphasizing its role in NK cell development. Further, miR-181a is essential for CD56+ NK cells to produce IFN-γ but does not affect natural cytotoxicity receptors on the surface of NK cells [49,50]. Interestingly, miR-181a-5p expression levels decrease in CD27+ NK cells as animals age [50,51]. In M1, M2, and M3 acute myeloid leukemia (AML) patients, miR-181a is elevated suggesting that it plays a prominent role in leukemogenesis. The authors go on to show that miR-181a decreases during granulocytic and macrophage differentiation [52].
Taken together, these studies highlight miR-181a’s role in regulating cellular plasticity and differentiation in numerous cellular contexts. It is precisely these characteristics of miR-181a’s regulome that make its dysregulation a potent and pervasive mediator of oncogenesis. In this review, we describe the current literature surrounding miR-181a as a highly relevant miRNA in human cancer, discussing both the critical signaling pathways it regulates and is modulated by.
3. miR-181a in a Cancer Context
miR-181a plays a pivotal role in cancer, either as a tumor suppressor or an oncomiR. This duality is due to context-specific expression levels or genomic modifications, underscoring its use as a biomarker in clinical settings. The expression level of miR-181a in cells concerning its ability to enhance, stabilize, or inhibit target mRNA functions is critical in defining its role in cancer.
miR-181a can function as a tumor suppressor and upon dysregulation is downregulated in a subset of cancers, including, leukemias, gliomas, melanomas, oral squamous cell carcinomas (OSCC), non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), cervical cancer, and retinoblastomas (RB) [53,54,55,56,57,58,59,60,61,62,63,64,65]. In RBs, miR-181a targets NRAS through 3’UTR binding, thereby regulating RAS-induced oncogenic signals [61]. In colorectal cancer (CRC), miR-181a represses PLAG1/IGF2 signaling, subduing proliferative signals and sensitizing CRC cells to 5-fluoruracil chemotherapy treatment [66]. miR-181a-2-3p also targets HIF-2α, suppressing cancer stem cell (CSC) phenotypes [67]. In the ABC subgroup of diffuse large B cell lymphoma (DLBCL), miR-181a suppresses constitutive activity of NF-Kβ signals, controlling cellular proliferation and mediating cell death [68]. Collectively, it is evident that increased miR-181a expression levels in these cases are crucial in defining the anti-oncogenic role of miR-181a compared to normal cells.
Despite evidence supporting miR-181a’s tumor suppressive roles, significantly more studies suggest its oncogenic functions in tumor initiation and progression. Higher expression levels of miR-181a are associated with poor prognosis and increased the risk of disease progression in patients diagnosed with breast cancer (BC) [69,70,71,72,73,74,75], high-grade serous cancer (HGSC) [76,77,78,79], endometrial cancer [80], CRC [81,82,83,84], gastric cancer (GC) [60,85,86,87], NSCLC [88], brain stem gliomas [89], AML [52,90,91], acute lymphoblastic leukemia (ALL) [55,92,93], chronic lymphocytic leukemia (CLL) [94], pancreatic cancer (PaCa) [95], hepatocellular carcinoma (HCC) [96], or thyroid cancer [97]. In HGSC, the most aggressive subtype of ovarian cancer (OC), miR-181a has been shown to induce stem-like properties and drive tumor progression and recurrence [76,79,98,99,100,101]. miR-181a is also enriched in the serum and circulating tumor cells of BC, GC, and OC patients, further emphasizing its role as a putative biomarker [73,75,79,102,103]. In BC, miR-181a expression positively correlates with tumor aggressiveness and with primary grade 3 tumors, showing higher miR-181a expression levels than grade 1 or 2 [104]. The expression levels of miR-181a also increase during the progression from non-neoplasia to dysplasia in inflammatory bowel disease-associated CRC [105]. Moreover, miR-181a promotes transformation, tumor growth, and suppresses immune stimulation in several cancers leading to a pro-tumorigenic environment [71,81,98,106]. These hallmark capabilities acquired upon high miR-181a expression underscoring this miRNA’s role as an oncogenic driver
Mechanistically, both overexpression and under-expression of miR-181a are associated with aberrant activation of several cellular pathways involved in tumorigenesis, including TGF-β, STAT, WNT, PTEN/AKT, and MAPK. Regulation of miR-181a is critical for maintaining homeostasis, and its dysregulation is concomitant with cancer pathogenesis. Here, we will provide a concise overview of numerous crucial signaling pathways that contribute to cancer and involve miR-181a. Understanding these dynamics is essential in discovering novel therapeutic modalities of targeting miR-181a in these oncomiR-driven diseases.
4. Genomic and Epigenetic Changes at the miR-181a Loci in Cancer
The diversity seen in miR-181a expression levels across cancer lineages can be tied to changes in ploidy number due to genomic instability and epigenetic alterations at the miR-181a loci on chromosome 1 & 9. In head and neck cancer and ovarian cancer, where miR-181a overexpression is established as a known driver of oncogenesis; there are notable increased copy number gains at this locus [107,108,109]. In contrast, miR-181a copy number losses are notable in lymphoid cancer lineages [110]. In this cancers, miR-181a can function as a tumor suppressor and is known to be downregulated [55]. Ultimately, miR-181a copy number alterations could confer cancer progression, tumor heterogeneity, and drive resistance.
4.1. Acetylation Marks
Increased chromatin accessibility, marked by elevated H3K27ac marks, is a recognized driver of disease pathogenesis, particularly in cancer. For instance, in HGSC where miR-181a is oncogenic, H3K27ac modifications at the miR-181a promoter are elevated and are further amplified as cells become platinum-resistant [100]. Our group further showed that treatment with bromodomain and extra-terminal motif (BET) inhibitors, which target acetyl-lysine groups on open chromatin and suppress transcription, strongly reduce miR-181a expression. However, one study challenged this concept showing that, treatment with TSA, a histone deacetylase inhibitor (HDACi) which improves chromatic accessibility, has been shown to further elevate miR-181a expression in ALL where miR-181a is overexpressed [111]. Conversely, in contexts where miR-181a is tumor suppressive, including some CRC cases, butyrate supplementation, another HDACi, has been demonstrated to synergize with miR-181a, enhancing anti-proliferative effects [112].
4.2. Methylation Marks
The Polycomb group of proteins Repressive Complex 1 and 2 (PRC1 and PRC2) are epigenetic regulators that establish gene silencing. PRC2 mediates histone methyltransferase activity on histone H3 lysine 27. Trimethylation of this residue (H3K27me3) induces chromatin compaction and has been shown to recruit PRC1 to aid in the repression of target gene sites [113,114]. Dysregulation of these epigenetic writers is well characterized in cancer.
Interestingly, in BC and prostate cancer (PrCa) where miR-181a can be tumor suppressive, PRC2 H3K27me3 deposition at the miR-181a and miR-181b loci represses their expression enabling cancer progression. The histone methyltransferase subunit of PRC2, EZH2, was shown to play a significant role in this transferase activity, where inhibition of EZH2 increased miR-181a expression and vise versa. Furthermore, in a normal cell context, miR-181a would be able to directly target BMI1 and RING2, E3 ubiquitin-protein ligase subunits of PRC1 that modulate monoubiquitylation of histone H2A lysine 119, thereby preventing the transcriptional repression of target genes. However, inhibition of miR-181a expression by aberrantly expressed EZH2 prevents BMI1 and RING2 inhibition, leading to cancer progression [115]. The role of EZH2 in regulating miR-181a has also been implicated in astrocytic and GBM tumors [116].
The EpimiR database has shown that H3K27me3 reduces miR-181a/b expression [117]. Interestingly, miR-181a expression is upregulated upon DNA methyltransferase inhibitor (DNMTi) treatment in melanoma and CRC cell lines [118,119,120]. Further, hypermethylation of CpG islands in CRC reduced miR-181a expression and this effect was reversed upon DNMTi [66]. The role epigenetic regulation plays on miR-181a accessibility, expression, and activity remains relatively unexplored, and further investigation could point to new therapeutic avenues for controlling this oncomiR.
5. Post-Transcriptional Modifications on miR-181a in Cancer
Post-transcriptional modifications, such as RNA methylation via S-adenosylmethionine, can control the temporal binding of microRNAs to their target mRNAs. Recent findings indicate that the miR-181a transcript is frequently methylated in PaCa [121]. Further exploration of miR-181a methylation levels in serum samples could provide a unique diagnostic strategy for PaCa patients.
6. Competing Endogenous RNAs with miR-181a in Cancer
Just like miRNAs, long non-coding RNAs (lncRNAs) and circular RNAs (cirRNAs) also harbor distinct MREs that allow them to regulate the post-transcriptional activity of miRNAs. This ability to function as competitive inhibitors of miRNA activity characterizes this group of non-coding RNAs as competing endogenous RNAs (ceRNAs). In some cancer contexts including, CRC, NSCLC, BC, PrCa, endometrial, multiple myelomas, and colon adenocarcinoma (COAD), miR-181a functions as a tumor suppressor. In these cancers, ceRNAs like ANRIL, SNHG7, LUCAT1, CCAT1, LINC01232, and lncRNA-MALAT1 complementarily bind to and sequester miR-181a through a process known as miRNA sponging, which ultimately promotes oncogenesis [122,123,124,125,126,127,128,129]. Some of these ceRNAs against miR-181a, such as lncRNA-MALAT1, have even been implicated in secondary complications—like cirrhosis—associated with their primary cancers, such as in chatliver cancer [130]. In other cases, lncRNAs such as muscleblind-like 1 antisense RNA 1 (lncRNA-MBNL1-AS1) impede PrCa progression by sequestering miR-181a-5p, thereby abrogating miR-181a’s repression of PTEN and activation of the PI3K pathway [131]. Similarly, in glioma models, lncRNA-CASC2 directly binds to miR-181a in the RISC complex, thereby preventing miR-181a’s ability to function as an oncomiR—this sponging of miR-181a reduces tumor growth by upregulating PTEN and increasing sensitivity to temozolomide (TMZ) chemotherapy. However, overexpression of miR-181a can abrogate these effects by inhibiting lncRNA-CASC2 [132]. Alternatively, miR-181a can be upregulated by lncRNAs like HOTAIR and RELA in papillary thyroid cancer (PTC) [133].
7. Signaling Pathways Modulating miR-181a Expression
miR-181a has been shown to regulate numerous signaling pathways that will be discussed later. Conversely, many of these same pathways contain negative and positive feedback loops that control expression of miR-181a. Here, we briefly summarize the direct regulators of miR-181a expression and activity in both tumor suppressing and tumor promoting roles.
7.1. TGF-β Signaling
Canonical transforming growth factor-β (TGF-β) signaling through the SMAD pathway is responsible for regulating the expression of hundreds of genes, particularly in developmental signaling pathways. Stimulation of this pathway suppresses early-stage tumor growth but promotes transformation, epithelial to mesenchymal transition (EMT), and metastasis in later stages of disease. miR-181a has been shown to both be regulated by TGF-β and modulate downstream TGF-β signaling – the latter of which will be discussed later in this review (section 8.1). Notably, TGF-β upregulates miR-181a expression [134,135]. The Lutz group mapped the transcriptional start sites of miR-181a and identified that the MIR181A2B2 promoter was strongly transactivated by SMAD3 and SMAD4 transcription factors, suggesting that TGF-β signaling increases miR-181a expression [14].
TGF-β upregulates miR-181a more strongly in metastatic BC than nonmetastatic BC, promoting metastasis, micrometastatic outgrowth, and increased BC lethality [136,137]. This TGF-β dependent increase in mature miR-181a expression occurs in a time and dose-dependent manner. However, it is noteworthy that TGF-β supplementation also dose dependently decreases both pri-miR-181a and pre-miR-181a transcripts. Mechanistically, the authors suggest that TGF-β induces the binding of SMAD2/3 and DROSHA to the pri-miR-181a-1 transcript, leading to increased processing of the pri- transcripts into mature transcripts [137]. Likewise, miR-181a gene expression was found to be dependent on both TGF-β and activin in multiple cancer cell lines. Specifically in BC, the upregulation of miR-181a through activin and TGF-β is necessary for cell migration [138]. These findings contrast with Zhang et al.’s conclusion that miR-181a is suppressed by activin in mouse granulosa cells [139] (Figure 2).
Figure 2.
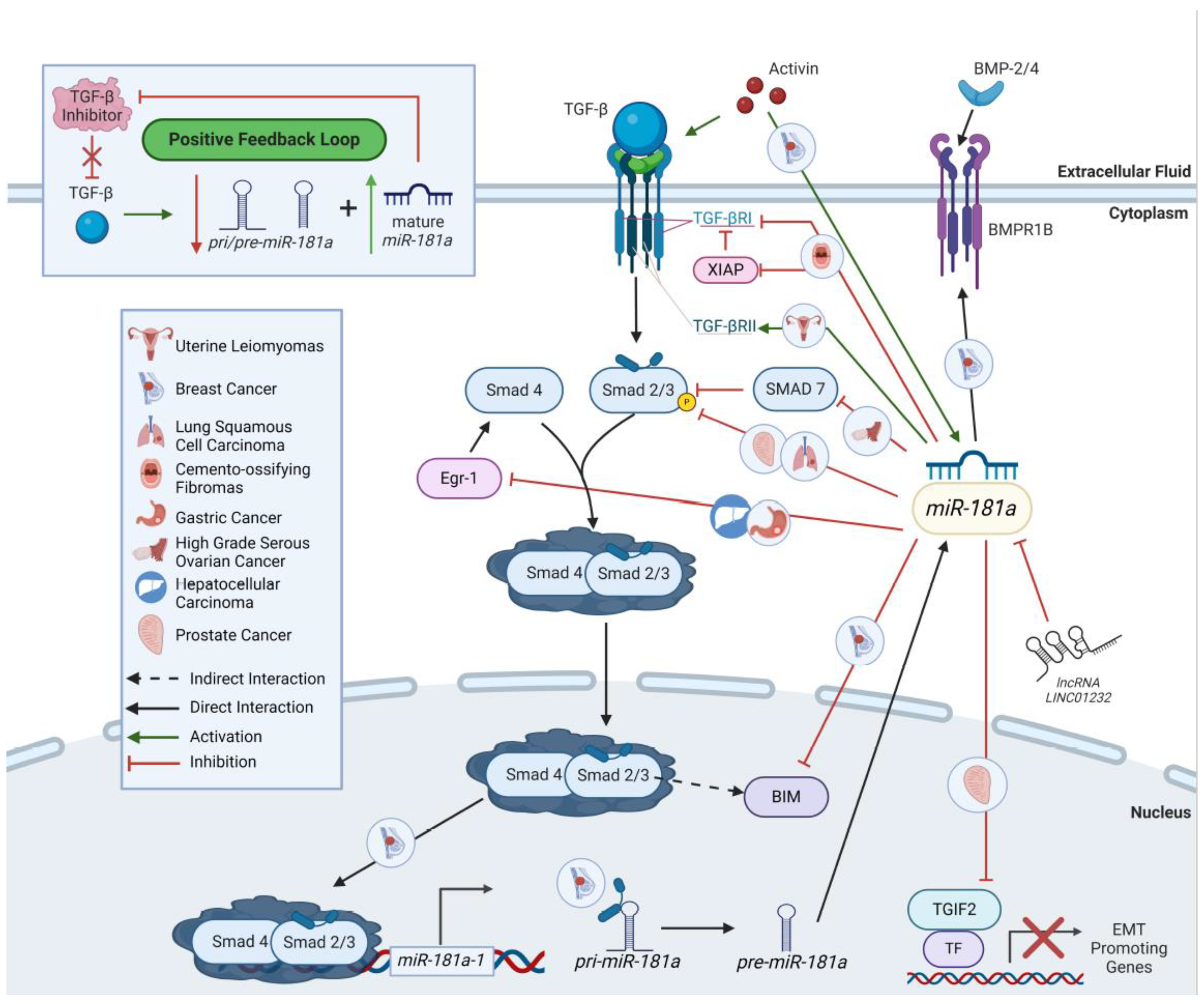
The role of miR-181a in the regulation of TGF-β signaling across cancer types. miR-181a can promote and TGF-β signaling by upregulating TGF-βRI, inhibiting XIAP, an inhibitor of TGF-βRII, and directly inhibiting SMAD7, an inhibitor of TGF-β signaling. Increased TGF-β signaling also leads to SMAD2/3/4 transactivation of miR-181a transcription, SMAD2/3 complexing with pri-miR-181a, and increased processing of pri-/pre-miR181a, elevating mature miR-181a levels. Activin treatment also promotes TGF-β signaling and miR-181a. Alternatively, miR-181a can inhibit TGF-β signaling by targeting SMAD2, Egr-1, TGF-βRI, BIM and TGIF2 directly. Alternatively, SMAD2 expression is also affected by LINC01232, which can sponge miR-181a, facilitating the increase of SMAD2. miR-181a is predicted to interact with BMPR1B, a crucial regulator of SMAD signaling. Figure generated using BioRender.
Figure 2.
The role of miR-181a in the regulation of TGF-β signaling across cancer types. miR-181a can promote and TGF-β signaling by upregulating TGF-βRI, inhibiting XIAP, an inhibitor of TGF-βRII, and directly inhibiting SMAD7, an inhibitor of TGF-β signaling. Increased TGF-β signaling also leads to SMAD2/3/4 transactivation of miR-181a transcription, SMAD2/3 complexing with pri-miR-181a, and increased processing of pri-/pre-miR181a, elevating mature miR-181a levels. Activin treatment also promotes TGF-β signaling and miR-181a. Alternatively, miR-181a can inhibit TGF-β signaling by targeting SMAD2, Egr-1, TGF-βRI, BIM and TGIF2 directly. Alternatively, SMAD2 expression is also affected by LINC01232, which can sponge miR-181a, facilitating the increase of SMAD2. miR-181a is predicted to interact with BMPR1B, a crucial regulator of SMAD signaling. Figure generated using BioRender.
7.2. Wnt Signaling
Canonical Wnt/β-catenin signaling is crucial in the regulation of cell fate. Evidence suggests that Wnt signaling can transcriptionally activate miR-181a in HCC [140]. Functionally, seven putative β-catenin/TCF4 binding sites have been identified in the promoter region of miR-181a-2 [140]. Further studies have demonstrated a direct interaction of the TCF4/β-catenin complex and the TCF/LEF complex in the promoter region of miR-181a-2/miR-181b-2 [140]. Importantly, these findings suggest the existence of a positive feedback loop between miR-181a and Wnt signaling. Further supporting this conclusion, miR-181a regulates the expression of NLK (Nemo-like kinase) and SFRP4, both inhibitors of Wnt/β-catenin signaling, ultimately stimulating further downstream activity [101,141,142] (Figure 3).
Figure 3.
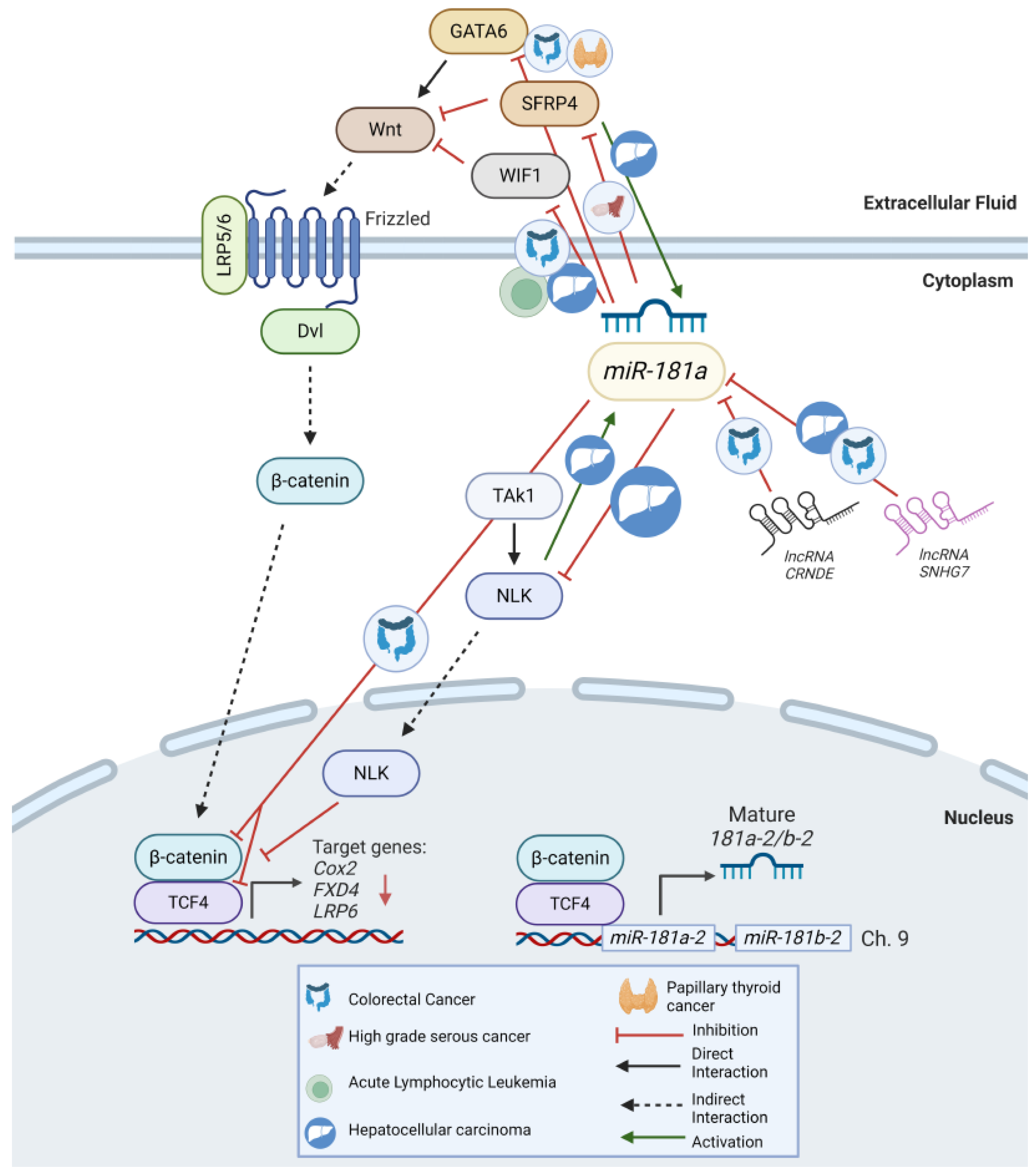
The role of miR-181a in the regulation of Wnt signaling across cancer types. In HGSC, miR-181a suppresses the Wnt antagonists SFRP4 and NLK. In ALL, HCC and CRC, miR-181a directly inhibits WIF1, altering cell growth and proliferation. In CRC alone, miR-181a overexpression reduced the Wnt target gene expression of COX2, FXD4, and LRP6. miR-181a can also function as a tumor suppressor in the Wnt/ β-catenin pathway. In CRC, miR-181a suppresses the Wnt signaling activators β-catenin and TCF4. Alternatively, β-catenin and TCF4 bind to the promoter region of MIR181A2B2 leading to increased miR-181a expression. miR-181a is also sponged by lncRNA-SNHG7, leading to GATA6 upregulation in HCC and PTC. Additionally, in HCC miR-181a is directly regulated by Wnt signaling through NLK and SFRP4. Figure generated using BioRender.
Figure 3.
The role of miR-181a in the regulation of Wnt signaling across cancer types. In HGSC, miR-181a suppresses the Wnt antagonists SFRP4 and NLK. In ALL, HCC and CRC, miR-181a directly inhibits WIF1, altering cell growth and proliferation. In CRC alone, miR-181a overexpression reduced the Wnt target gene expression of COX2, FXD4, and LRP6. miR-181a can also function as a tumor suppressor in the Wnt/ β-catenin pathway. In CRC, miR-181a suppresses the Wnt signaling activators β-catenin and TCF4. Alternatively, β-catenin and TCF4 bind to the promoter region of MIR181A2B2 leading to increased miR-181a expression. miR-181a is also sponged by lncRNA-SNHG7, leading to GATA6 upregulation in HCC and PTC. Additionally, in HCC miR-181a is directly regulated by Wnt signaling through NLK and SFRP4. Figure generated using BioRender.
7.3. STAT Signaling
The STAT (signal transducer and activators of transcription) family of proteins are well characterized mediators of apoptosis, cell signaling, cell proliferation, and differentiation. STAT1, a transcription factor activated by cytokines and growth factors, stimulates the transcription of genes crucial for cell viability. Activation of STAT1 can occur through Type-1 interferon (IFN) signaling, where IFN-α/β activates TYK2 and JAK1, leading to phosphorylation of STAT2. STAT2 then forms a heterodimer with STAT1, translocates to the nucleus, and activates IFN response elements for transcription. Alternatively, Type-2 interferon signaling activates STAT1 through IFN-γ receptor-mediated phosphorylation of JAK1 and JAK2, followed by STAT1 homodimer formation and nuclear translocation to initiate transcription of genes containing Gamma-Activated-Sequences (GAS) – particularly c-GAS, which activates STING-mediated signaling [143].
In tumorigenesis, STAT1 acts as a tumor suppressor by increasing major histocompatibility complex (MHC) expression and exhibiting anti-angiogenic properties [143]. In CRC, where high miR-181a expression correlates to poor patient prognosis, STAT1 inhibits CRC cell growth by down-regulating miR-181a. STAT1 directly inhibits miR-181a transcription by binding to the -890 base pair region of the miR-181a promoter. In this model, increased STAT1 promotes PTEN-mediated tumor suppression and reduces Akt activation. Taken together, STAT1 attenuates the increased cell viability and proliferative phenotypes concomitant with high miR-181a expression in CRC [144]. Notably, Zhu et al. subsequently demonstrated that miR-181a directly binds to the 3’UTR of STAT1, inhibiting its mRNA expression and suggesting a competitive dynamic between miR-181a and STAT1 [145]. In summary, the intricate interplay between STAT1 and miR-181a highlights their reciprocal regulation and significant impact on cancer cell maintenance and potential to evade immune activation.
7.4. SOX2
The sex determining region Y (SRY-type high mobility gene box) (SOX) gene family encodes transcription factors that are strongly involved in embryogenesis, gonad development, and differentiation. Aberrantly high SOX2 expression has been involved in tumorigenesis, and cancer stem cell maintenance [146,147]. In BC, SOX2 regulates miR-181a-5p expression given that knockdown reduced miR-181a-5p expression [148]. Interestingly, miR-181a directly targets the 3’ UTR of TUSC3, a tumor suppressor protein downstream of SOX2 that is dysregulated in ovarian and breast cancer [148,149,150,151]. Thus, cancer progression could be modulated through the SOX3-miR-181a-5p-TUSC3 axis [148].
7.5. HBx
In HCC, Hepatitis B Virus X protein (HBx) has been correlated with cancer development. Interestingly, HBx increases miR-181a promoter activity, engendering miR-181a induced proliferation and anti-apoptosis in hepatoma cells [152]. Limited research exists exploring this novel mechanism of HCC tumorigenesis; however, further in vivo studies elucidating this process may provide an effective therapeutic approach to targeting Hepatitis B Virus-induced HCC.
8. Signaling Pathways miR-181a Regulates
8.1. TGF-β
Dysregulation of the TGF-β signaling pathway has become synonymous with tumor progression across tumor lineages. The TGF-β signaling pathway has paradoxical roles in cell proliferation. In a pre-malignant state, it functions as a tumor suppressor by exerting cytostatic, pro-apoptotic, and tumor-suppressive effects. Upon loss of TGF-β’s tumor suppressive phenotypes, TGF-β signaling promotes oncogenic activity via cytoskeletal rearrangement, EMT, mobilization of cancer associated fibroblasts in the tumor microenvironment (TME), and stimulation of pro-metastatic cytokines [153].
Activation of this this pathway occurs upon TGF-β binding to and activates TGF-β receptors I and II. This receptor complex then phosphorylates and activates SMAD2/3, which subsequently engages with SMAD4 to translocate to the nucleus and interact with DNA-binding transcription factors to regulate transcription. SMAD7 can compete with SMAD2/3 to inhibit their activation outside the nucleus and DNA binding activity inside the nucleus. Numerous studies have implicated miR-181a’s interaction with these pathway mediators in both tumor suppressive and oncogenic contexts [154].
miR-181a activity in BC is highly controversial, with some groups suggesting miR-181a-5p is upregulated and acts as an oncomiR, while others suggest it is downregulated [74,122,155,156,157]. A bioinformatic analysis of three BC patient datasets revealed BMPR1B, a prominent modulator of TGF-β could be regulated by miR-181a [158]. Liu et al. showed that TGF-β-targeted DNA repair genes ATM and BRCA1 are direct targets of miR-181a and that this targeting can modulate tumor response to PARP inhibitors [159].
TGF-β-induced EMT is well documented in multiple other tumor types, causing enhanced invasion and metastasis [160]. Functionally, studies by Parikh et al. revealed miR-181a's involvement in downregulating a TGF-β inhibitor, SMAD7, to promote EMT signatures in HGSC. Re-expression of SMAD7 lacking a 3’UTR prevented miR-181a binding and, thus, rescued EMT phenotypes [76]. Similar lung squamous cell carcinoma (LUSC) studies show that miR-181a directly targets SMAD2. However, in LUSC, lncRNA, LINC01232, can sponge miR-181a, facilitating increased SMAD2 expression, TGF-β pathway stimulation, and enhanced proliferation, migration, and invasion [129].
In contrast, other studies in PrCa have identified a possible mechanism in which miR-181a inhibits SMAD function through transcriptional repressor TGIF2 favoring EMT transitions and inducing drug resistance mechanisms [161,162]. Additionally, Bi et al. demonstrated that miR-181a-5p is a negative regulator of early growth response factor 1 (Egr1), downregulating in TGF-β/Smad signaling [163]. miR-181a and TGF-β1 have also been shown to interact to promote peritoneal dissemination of GC [164]. In T-cell prolymphocytic leukemia (T-PLL), Erkeland et al. showed that miR-181a/b is highly expressed, correlates with poor survival, and significantly downregulates predicted target TGF-β3 [165].
miR-181a also targets various classes of TGF-β receptors across cancer types. High miR-181a-5p expression is correlated with increased TGF-βR2 and Insulin-like Growth Factor 2 mRNA Binding Protein 1 in advanced uterine leiomyomas [166]. miR-181a-5p is also upregulated in cemento-ossifying fibromas (COFs) and is correlated with increased XIAP expression, an inhibitor of cell-death caspases 3, 7, and 9 and TGF-βR1. Given that miR-181a directly targets the 3’UTR of XIAP and TGF-βR1, upregulation of miR-181a may lead to increased proliferative capacity [167] (Figure 2).
8.2. Wnt/β-Catenin
The Wnt/β-catenin signaling stands as one of the most evolutionarily well conserved paracrine and autocrine signaling pathways responsible for orchestrating cell fate determination and organogenesis during embryonic development. Mutations and dysregulation within this pathway consistently serve as critical drivers of recalcitrant cancer subtypes. Activation of Wnt signaling can occur through either the canonical (β-catenin dependent) or non-canonical (β-catenin independent) pathways. Canonical Wnt signaling stimulates the transcription of various transcription factors, extracellular matrix components, and cell adhesion proteins. In contrast, non-canonical Wnt signaling triggers other pathways, including the Wnt-dependent calcium or planar cell polarity pathway. Stimulation of canonical Wnt target genes augments tumor progression, fostering increased cancer cell initiation, persistence, invasion, and metastatic phenotypes.
Within the framework of Wnt signaling, miR-181a serves as a significant oncomiR across diverse cancer lineages by targeting WNT and its regulators [154]. As a result, miR-181a has garnered attention as an attractive target in Wnt/β-catenin-directed therapies. Recent research led by Nagaraj et al. showcased miR-181a suppression of the Wnt antagonist SFRP4, thereby promoting tumor initiation and stemness in HGSC [101]. Re-expression of SFRP4 in a miR-181a high cell line notably reduced β-catenin gene expression, leading to a robust decrease in tumor sphere formation ability. Furthermore, an inverse relationship between miR-181a and SFRP4 in primary and recurrent ovarian tumors underscores the significance of targeting this axis in HGSC treatment. In a similar study focusing on highly invasive HCC’s positive for epithelial cellular adhesion molecule (EpCAM) and alpha fetoprotein (AFP), Ji et al. demonstrated that miR-181a’s directly suppresses another Wnt antagonist, NLK, prolonging HCC stemness [142,168].
Evidence of constitutive Wnt/β-catenin activation has been reported in ALL, HCC, and CRC through the direct inhibition of WIF1 (Wnt inhibitory factor 1) by miR-181a-5p [55,81]. Thus, in ALL, blocking miR-181a activity significantly alters cell growth and proliferation by inducing cell cycle arrest at the G1/S phase [55]. Further, potentiation of Wnt signaling by miR-181a in CRC was evident through a TOPFlash assay, where treatment with a miR-181a mimetic reduced the expression of Wnt target genes COX2, FXD4, and LRP6 [112].
Other studies suggest that miR-181a functions as a tumor suppressor in the Wnt/β-catenin pathway. miR-181a can directly bind to the 3’UTR of β-catenin and TCF4, crucial activators of Wnt signaling. These studies additionally implicate lncRNA-CRNDE as an overexpressed sponge of miR-181a in CRC, promoting cell proliferation and chemoresistance [169]. Similarly in HCC, lncRNA-SNHG7 sponges miR-181a-5p leading to upregulation of GATA6, a crucial transcription regulator of intestinal cell phenotypic reprogramming [127]. Given that GATA6 has been shown to stimulate Wnt signaling [170], it is possible that the SNHG7/miR-181a/GATA6 axis could activate Wnt signaling in CRC. These results are corroborated by a new study in PTC where miR-181a promotes tumorigenesis by directly binding to GATA6 [142]. Combined, these studies suggest that miR-181a may exert repressive effects on Wnt signaling in some cancer subtypes (Figure 3).
8.3. Notch
The Notch gene is a transmembrane receptor initially discovered for its notched wing phenotype in Drosophila melanogaster. Years later, the Notch pathway became recognized as critically important in several fundamental cellular and developmental mechanisms, including regulation of cellular proliferation, stem cell differentiation, maintenance during embryogenesis, and apoptosis [171]. This signaling pathway is also highly oncogenic and is hyperactive in several cancers such as T-cell acute lymphoblastic leukemia (T-ALL), BC, and NSCLC [154]. Despite years of research, a Notch inhibitor has only recently been approved for the clinical treatment of desmoid tumors, highlighting the necessity to identify new targetable vulnerabilities in this pathway. Recent studies have implicated miR-181a modulation of the Notch pathway.
The Notch receptor, Notch2, is overexpressed in many cancers, including GBM. In this context, miR-181a is an oncosuppressor. Xiu et al. report that miR-181a downregulates Notch2, inhibiting the proliferation and differentiation of glioma cells [172]. Huang et al. support these findings, showing that increased miR-181a expression in GBM inhibits the stem-like properties of tumor-initiating glioblastoma cells via targeting the 3’UTR of Notch2 [173]. Furthermore, low miR-181a expression correlates with high Notch2 expression and poor patient prognosis, emphasizing the prognostic significance of miR-181a and its role as a tumor suppressor in Notch-driven GBMs.
Recognizing miR-181a’s crucial role in lymphocyte development and differentiation, it is not surprising that dysregulation of miR-181a causes perturbations in hematopoietic differentiation and stimulates oncogenesis through the Notch pathway. miR-181a and miR-181b are necessary for natural killer (NK) cell development and IFN-γ production. miR-181a downregulates NLK, a negative regulator of NK cell development, by directly targeting its 3’UTR, thus potentiating Notch proliferative signaling [49,142]. Additionally, miR-181a functionally targets the 3’UTR of Narp, a critical regulator of thymocyte development in the Notch pathway. In T-ALL, miR-181a acts as an oncogene, and loss of miR-181a/b promotes survival via potentiation of the intracellular domain of Notch1 (ICN1). Through loss of function analyses, Fragoso et al. demonstrated that deleting miR-181a-1/b-1 inhibits the development of notch oncogenes and tumor transformation [174]. These studies emphasize the significant onco-regulatory role of miR-181a on the Notch pathway and the treatment potential for targeting miR-181a in Notch-dependent cancers.
8.4. PTEN-PI3K-AKT-mTOR Network
The PI3K/AKT/mTOR (PAM) signaling pathway is a signal transduction network highly conserved across the Eukarya domain. This pathway is frequently activated in cancer and involves cell growth, survival, progression, and chemotherapy resistance [175]. The PAM signaling pathway has been recently linked to miR-181a expression in several cancer types, largely due to miR-181a’s inhibitory effect on the phosphatase and tensin homolog protein (PTEN). PTEN is a tumor suppressor in numerous cancers and a negative regulator of PI3K/AKT activity in normal cells [131].
In 2014, PTEN was identified as a direct target of miR-181a in CRC, and this interaction has since been confirmed in BC, NB, and HCC [21,152,176]. miR-181a knockdown effectively restores PTEN and E-cadherin expression and reduces the expression of EMT markers, resulting in inhibited cellular migration and invasion [96,152]. Reinforcing these findings in NB cells, PTEN overexpression reversed the effects of miR-181a upregulation, inducing cell death and increased ROS production [21]. Furthermore, in endometrial cancer, a study found that non-obese patients showed a correlation between increased miR-181a expression and decreased PTEN expression [80]. In CRC, miR-181a overexpression repressed PTEN and increased AKT activation, the direct downstream target of PTEN inactivation [144,176]. This increase in p-AKT induced a metabolic shift, increasing lactate secretion, glucose uptake, and glycolytic flux [176]. Together, these studies emphasize the regulatory interactions of PTEN and miR-181a to drive PAM-mediated cancer progression.
miR-181a has also been shown to regulate and interact with other modulators of PTEN activity. NDRG2, an N-myc downregulated gene, was identified as a PTEN binding protein, regulating the phosphatase activity of PTEN. In BC, miR-181a directly targets the 3’ UTR of NDRG2, demonstrating an inverse correlation between miR-181a and NDRG2 expression. miR-181a-5p suppresses NDRG2, resulting in PTEN dephosphorylation and increased phosphorylation of Akt, thus facilitating tumor progression through NDRG2-induced activation of PTEN/AKT signaling pathway [74]. Further, miR-181a and lncRNA-XIST (X-inactivated specific transcript) directly regulate each other; knockdown of miR-181a increases XIST expression and vice versa. Interestingly, overexpression of XIST abolished miR-181a’s inhibitory effects on PTEN [96]. In PrCa, lncRNA-MBNL1-AS1 inhibits tumor progression by sponging miR-181a-5p, thereby abrogating miR-181a’s repression of PTEN and downstream activation of the PAM pathway [131].
Without PTEN activation, downstream proliferative signaling proteins, such as PI3K, AKT, and mTOR, can also be regulated by miR-181a [177]. In CRC, increased miR-181a expression significantly decreases PIK3R3 expression, the regulatory subunit of PI3K, and a predicted target of miR-181a [112]. Knockdown of miR-181a in HCC reduces p-AKT and p-mTOR expression and subsequently markers of EMT signaling [96]. miR-181a and miR-181d also directly suppress PHLPP2 and INPP4B phosphatases, augmenting Akt phosphorylation, S-phase entry, and cell proliferation in estrogen receptor-positive BC [136,178]. In GC, Hao et al. found that miR-181a downregulates TCL1 family Akt coactivator A’s (TCL1A) expression and, therefore the activation of Akt family members. Interestingly, this miR-181a mediated inhibition of Akt activation increased c-MYC expression, further upregulating miR-181a, and establishing a miR-181a-5p-TCL1A-Akt/mTOR-c-MYC feedback loop. However, it is worth noting that miR-181a does not directly bind to the 3’UTR of TCLA1 or c-MYC, so the molecular mechanism of this interaction remains unknown [179].
miR-181a also modulates chemoresistance via the AKT/ERK pathway. In BC, miR-181a directly inhibits EPDR1 (Ependymin Related 1), a transmembrane protein that facilitates cell adhesion. EPDR1 inhibits PI3K/Akt activation, so increased expression of miR-181a in BC facilitates activation of PI3K/Akt signaling and reduces sensitivity to Epirubicin, a BC chemotherapeutic [180]. In NSCLC, Ping et al. show that miR-181a is upregulated in gefitinib-resistant NSCLC, and knockdown of miR-181a reduced p-AKT in gefitinib-resistant cells [181]. In T-cell leukemias and lymphomas, high miR-181a expression also correlates to increased p-AKT expression, cell proliferation, and acquired chemoresistance [182].
Contrary to these findings, some studies show that when miR-181a acts as a tumor suppressor, PAM signaling can be suppressed. For example, hormone replacement therapy, including norethisterone, correlates with an increase BC. In this cancer subtype miR-181a acts as a tumor suppressor. Here, Cai et al. report that exogenous miR-181a suppresses hormone-induced tumorigenesis through the deactivation of PAM pathway proteins including mTOR, EGRF, PTEN, and PGRMC1 [183]. In another BC and CRC example, the transmembrane protein MMP-14 (matrix metallopeptidase) phosphorylates EGFR, activates the MAPK and PI3K signaling pathways, and promotes oncogenic signaling [184]. Thus, upregulation of MMP-14 correlates with poor patient prognosis. miR-181a directly targets the 3’UTR of MMP-14, reducing PAM signaling, in vivo tumor invasion, angiogenesis, and migration [185]. These studies point out that miR-181a function as a tumor suppressor by inhibiting PAM signaling.
In summary, miR-181a plays a crucial role in regulating the PTEN-PAM signaling network in both oncogenic and tumor-suppressive contexts (Figure 4A). Further evaluation of the context-dependent mechanisms of miR-181a’s regulatory control of this pathway may elucidate how PTEN-PAM signaling promotes cancer progression and chemoresistance.
Figure 4.
(A) The role of miR-181a in regulating PI3K/PTEN signaling across cancer types. To upregulate this pathway, miR-181a directly targets of critical tumor suppressor PTEN in CRC, BC, NB, and HCC. In BC, miR-181a also directly targets NDRG2, resulting in PTEN dephosphorylation and increased Akt phosphorylation. To downregulate this pathway, miR-181a also interacts with several lncRNAs, such as the XIST, CASC2, and MBNL1-AS1. miR-181a also reduces PIK3R3 in CRC, PHLPP2 and INPP4B phosphatases in BC, EPDR1 in BC, and TCL1A expression in GC. miR-181a also directly targets MMP-14 in CML thereby preventing propagation of PAM signaling. (B) The role of miR-181a in the regulation of Ras signaling across cancer types. miR-181a can function as a tumor suppressor by directly regulating Ras isoforms, NRAS and KRAS, in AML, OSCC, and NSCLC. Further, miR-181a inhibits RalA leading to increased cell cycle arrest and apoptosis. Further, miR-181a supplementation has been shown to increase chemo-sensitivity when combined with BRAF inhibitors.As an oncomiR, miR-181a can inhibit RASSF1A thereby preventing the degradation of MDM2 leading to inhibition of P53. In HCC, miR-181a targets CDX2, promoting cell differentiation and enriched stem cell properties. miR-181a also modulates chemoresistance by targeting proteins GAS7 in NSCLC. In BC, miR-181a directly represses Bim, a pro-apoptotic factor that interacts with ERK, thereby promoting cancer progression. (C) The role of miR-181a in the regulation of P38/MAPK signaling across cancer types. miR-181a interacts with DUSP5/6 to enhance TCR signaling. Additionally, miR-181a can regulate p38 MAPK activation by targeting MKP-5 in HCC. Figure generated using BioRender. .
Figure 4.
(A) The role of miR-181a in regulating PI3K/PTEN signaling across cancer types. To upregulate this pathway, miR-181a directly targets of critical tumor suppressor PTEN in CRC, BC, NB, and HCC. In BC, miR-181a also directly targets NDRG2, resulting in PTEN dephosphorylation and increased Akt phosphorylation. To downregulate this pathway, miR-181a also interacts with several lncRNAs, such as the XIST, CASC2, and MBNL1-AS1. miR-181a also reduces PIK3R3 in CRC, PHLPP2 and INPP4B phosphatases in BC, EPDR1 in BC, and TCL1A expression in GC. miR-181a also directly targets MMP-14 in CML thereby preventing propagation of PAM signaling. (B) The role of miR-181a in the regulation of Ras signaling across cancer types. miR-181a can function as a tumor suppressor by directly regulating Ras isoforms, NRAS and KRAS, in AML, OSCC, and NSCLC. Further, miR-181a inhibits RalA leading to increased cell cycle arrest and apoptosis. Further, miR-181a supplementation has been shown to increase chemo-sensitivity when combined with BRAF inhibitors.As an oncomiR, miR-181a can inhibit RASSF1A thereby preventing the degradation of MDM2 leading to inhibition of P53. In HCC, miR-181a targets CDX2, promoting cell differentiation and enriched stem cell properties. miR-181a also modulates chemoresistance by targeting proteins GAS7 in NSCLC. In BC, miR-181a directly represses Bim, a pro-apoptotic factor that interacts with ERK, thereby promoting cancer progression. (C) The role of miR-181a in the regulation of P38/MAPK signaling across cancer types. miR-181a interacts with DUSP5/6 to enhance TCR signaling. Additionally, miR-181a can regulate p38 MAPK activation by targeting MKP-5 in HCC. Figure generated using BioRender. .
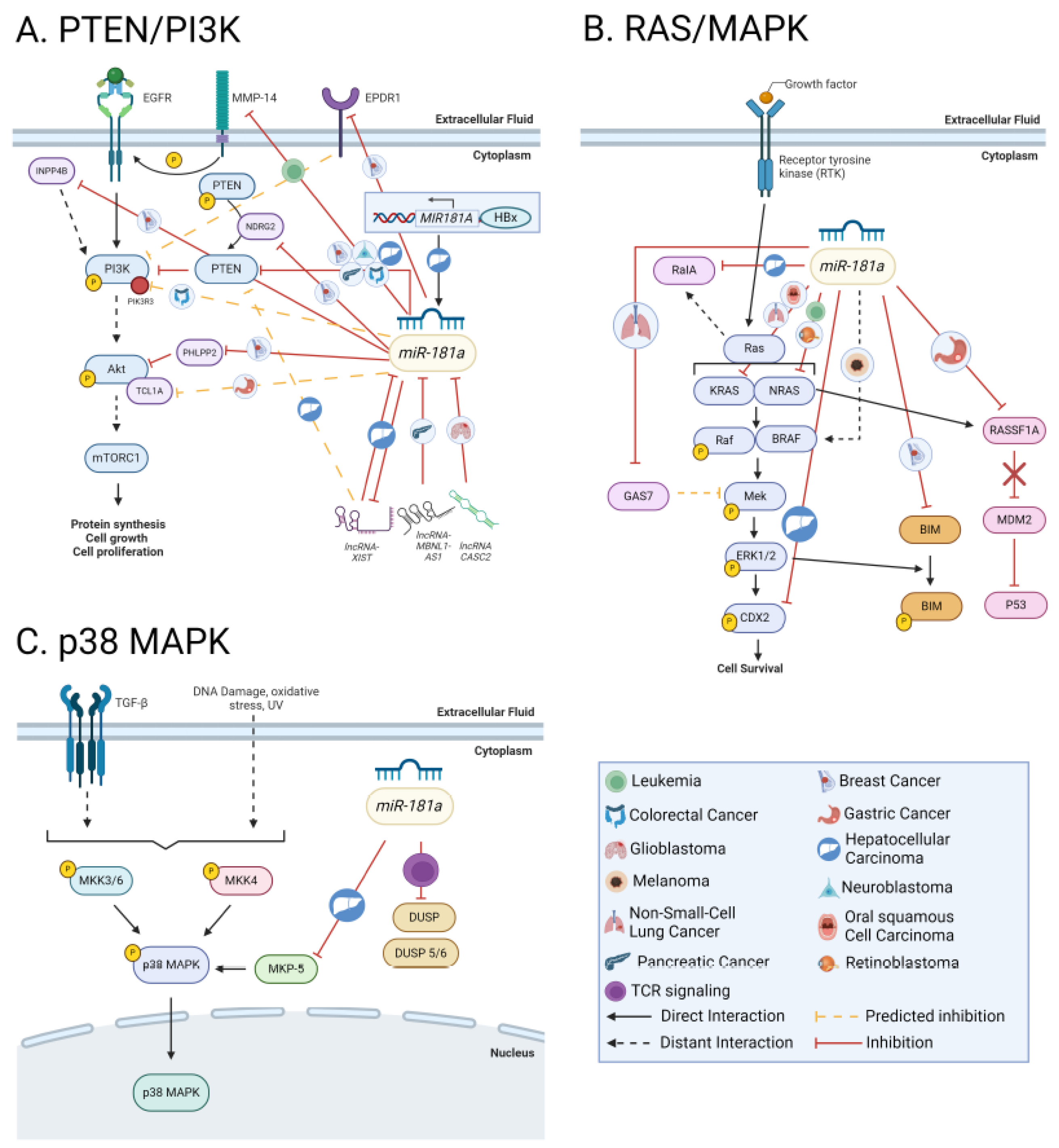
8.5. RAS/MAPK/ERK
The Ras family of proto-oncogenes is a group of small GTPases, including NRAS, HRAS, and KRAS. This family plays diverse roles in regulating cell division, differentiation, survival, metabolism, and programmed cell death. Downstream of these GTPase proteins are the canonical RAS/RAF1/MAPK and PI3K/AKT pathways, which drive gene expression, governing cell survival and cell cycle progression, respectively [186]. Dysregulation of these GTPases is well-characterized across various cancer types particularly, CRC and PaCa. A growing body of literature from the last decade suggests that miR-181a could significantly influence these pathways as either a tumor suppressor or an oncomiR.
In the Ras-MAPK-ERK pathway, miR-181a can function as a tumor suppressor. It directly regulates the Ras family by binding to the 3’ UTRs of NRAS, KRAS, and MAPK1, but not HRAS [44,60,187]. Huang et al. report that miR-181a downregulates NRAS, KRAS, and MAPK1 in AML, thereby decreasing cell proliferation [187]. In RB’s, miR-181a-5p’s 3’UTR inhibition of NRAS was shown to decrease proliferation, migration, and invasion while enhancing apoptosis in RB cells [61]. Similarly, miR-181a's regulation of KRAS in OSCC and NSCLC downregulates KRAS gene and protein expression, resulting in increased senescence and suppressed proliferation, migration, colony formation, and anchorage independent cell growth [60,188]. In CML, Gu et al. found that miR-181a directly targets the 3’UTR of RalA, a signaling molecule downstream of Ras, inducing G2 phase arrest and promoting apoptosis [189]. They later showed that miR-181a mimetic treatment combined with imatinib increases treatment sensitivity and reduces phosphorylation of P38 MAPK, SAPK/JNK, Akt, and Erk1/2 [106].
Downstream of Ras signaling, miR-181a can also enhance MAPK and ERK’s oncogenic activity through other mechanisms. Treatment with miR-181a mimics in BC models enhances the basal phosphorylation and activation of ERK, Akt, and c-JUN [136,190]. Additionally, miR-181a expression modulates caudal-type homeodomain transcription factor (CDX2), an intestine-specific transcription factor downstream of MAPK and ERK. In highly invasive EpCAM+ and AFP+ HCC, CDX2 is a major protein regulator of MAPK and ERK signaling. In this model, miR-181a promotes HCC stemness by targeting the 3’UTR of CDX2 and GATA6, promoting HCC cell differentiation and enriched stem cell properties [141,142,191]. In BC, miR-181a directly interacts with the 3’UTR of Bim, a pro-apoptotic factor that interacts with ERK, thereby repressing Bim translation and promoting tumor dissemination [136].
miR-181a has also been shown to modulate chemoresistance through aberrant RAS/MAPK/ERK signaling. Ping et al. noted that miR-181a-5p is upregulated in gefitinib-resistant NSCLC. Here, miR-181a drives chemoresistance by directly targeting GAS7 (growth arrest specific-7), increasing AKT and ERK activation, migration, and invasion [181]. In contrast, miR-181a expression can promote chemo-responsiveness. BRAF inhibitors, such as dabrafenib, are used in melanoma treatment but lack efficacy due to developed resistance. Barbato et al. found that miR-181a/b replacement therapy in melanoma cells with downregulated miR-181a/b increased dabrafenib sensitivity when combined with BRAF inhibitors. Clinically, this group saw increased chemo-responsiveness in patients with higher miR-181a [58].
In GC, where miR-181a functions as an oncogene, studies show that miR-181a-5p is a novel regulator of the Ras-MAPK pathway through its interaction with the Hippo pathway. miR-181a negatively regulates the highly conserved tumor suppressor, Ras-associated domain family member (RASSF6) by binding to its 3’UTR [86,192]. This prevents degradation of MDM2, thereby inhibiting P53 and ultimately inducing multiple proliferative phenotypes, including EMT, invasion, and metastasis. Inhibition of miR-181a reverses these phenotypes and prevents cell cycle transition. These findings highlight the various influential roles miR-181a has across the different dimensions of cancer-promoting signaling pathways (Figure 4B).
8.5.1. p38 MAPK
The p38 subgroup of MAP kinases serves as a center for signal transduction while playing a dual role in regulating cell death and survival. Mechanistically, p38 MAPK activation is mediated by dual phosphorylation of its Thr and Tyr residues in the TXY motif, differentiating it from ERK and JNK kinases. p38 is generally activated by environmental stress, inflammatory cytokines, and TGF-ꞵ signaling [193].
miR-181a interacts with the p38 MAPK pathway via its downstream dual specificity phosphatases (DUSPs). DUSPs are a class of genes and associated proteins that dephosphorylate Ser, Thr, and Tyr residues, ultimately regulating protein activity [194]. Specifically, DUSP5 and DUSP6 are controlled by miR-181a [47]. Okada et al. expanded on this work, suggesting that miR-181a’s regulation of DUSPs could be exploited as a promising cancer immunotherapy [195]. It is known that miR-181a augments the sensitivity of TCR-mediated T-cell responses and that DUSP5/6 negatively regulate TCR signaling. The authors suggest that miR-181a directly targets DUSP5 and DUSP6, which in turn upregulate TCR signaling and reduce the T-cell activation threshold. Leveraging this miR-181a/DUSP interaction could serve as a potential therapeutic angle to shift immune-cold environments to immune-hot microenvironments [195].
miR-181a can also regulate p38 MAPK activation by targeting the 3’UTR of MAPK phosphatase 5 (MKP-5) [196]. MKP-5 blocks the enzymatic activation of p38 [197]. However, in HCC cell lines exposed to carcinogens benzo[a]anthracene and benzo[k]fluoranthene, miR-181a is significantly upregulated, leading to MKP-5 suppression and continued p38 MAPK activation. Phenotypically, this drives migration [196]. Given these juxtaposing roles of miR-181a in driving p38 MAPK-mediated tumor suppression or, conversely, cancer migration, further characterization of miR-181a’s activity in the p38 MAPK pathway is needed (Figure 4C).
8.6. Snail
The Snail family of zinc-finger transcription factors consisting of SNAI1 (Snail), SNAI2 (Slug), and SNAIL3 (Smuc) plays a significant role in embryonic development [198]. Corruption of this crucial pathway illustrates how developmental pathways are repurposed to drive cancer progression. In the context of cancer, the Snail family promotes EMT and CSC maintenance [199]. Specifically, SNAI1 and SNAI2 stimulate tumor metastasis and immune suppression within the TME [200]. In BC, miR-181a, along with transmembrane bax inhibitor motif-containing 6 (TMBIM6), induces SNAI1 and SNAI2 expression by initiating upstream ERK activation [190]. In PrCa, migration and invasion inhibitory protein (MIIP) inhibits miR-181a-5p, resulting in reduced Snail and Twist expression and inhibited tumor growth [201]. Additionally, miR-181a is also implicated in the migration and invasion of salivary adenoid cystic carcinoma via regulation of the MAPK-Snai2 pathway. The Inhibition of miR-181a increases the expression of MAP2K1, MAPK1, and SNAI2, leading to enhanced metastatic potential [199]. In summary, miR-181a significantly influences the Snail family, particularly, SNAI1 and SNA2, to modulate carcinogenesis.
8.7. Hippo-YAP/TAZ
The canonical Hippo pathway is a kinase cascade that restricts the activity of two transcriptional activators, Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). Activated YAP and TAZ induce cytoplasmic retention and ubiquitin-mediated proteasomal degradation and prevent activation of the transcription factor TEAD. TEAD stimulates the transcription of various genes responsible for cell proliferation, migration, and survival [202]. The interaction between this pathway and several noncoding RNAs has been well-researched, as discussed in a recent review from Zhang et al [203].
Recent studies implicate miR-181a's involvement in the Hippo pathway. In PTC, exosomal miR-181a targets the 3’UTR of mixed lineage leukemia 3 (MLL3/KMT2C), a histone-lysine N-methyltransferase. These exosomes reduce the enhancer activity of disheveled binding antagonist of beta-catenin 2 (DACT2) in adjacent endothelial cells, thus activating the YAP/VEGF signaling pathway and promoting proliferation, migration, tumor formation, and capillary network formation [204].
In multiple myeloma, miR-181a-5p is downregulated. Re-expression of miR-181a in myeloma cells inhibits proliferation, adhesion, and promotes apoptosis. The authors show that this re-expression via a miR-181a mimic increases LATS1, p-YAP1, and reduces total YAP1 mRNA and protein expression. Another connection between the miR-181a and Hippo-YAP/TAZ pathway was identified in CML. Long-term exposure to the tyrosine kinase inhibitor, imatinib mesylate (IM), results in dysregulation of the Akt/Erk1/2 signaling pathway, miR-181a inhibition, decreased YAP levels, and increased sensitivity to IM treatment [205]. Although multiple studies demonstrate that elevated miR-181a levels are concomitant with activation of YAP/TAX, the molecular mechanism driving miR-181a’s stimulation of the Hippo pathway remains elusive [206].
8.8. JAK/STAT
The Janus kinase/signal transducers and activator of transcription (JAK/STAT) signaling pathway mediates cellular processes crucial for proliferation, differentiation, migration, apoptosis, immune cell development, and hematopoiesis [207]. Its activation is triggered by cytokines such as IL-6 and growth factors like EGF, which lead to the phosphorylation of intracellular JAKs and downstream STAT signaling.
Several studies have linked the post-translational regulation of this pathway to miRNAs, including miR-181a. In BC, miR-181a inhibits PIAS3 but not SOCS3, both of which are inhibitors of the JAK/STAT pathway [72]. Elevated levels of miR-181a in breast tumor exosomes can activate STAT signaling by targeting SOCS3, which favors the expansion of immature early myeloid-derived suppressor cells and suppresses T-cell immunity [72]. Conversely, other research in T-cell large granular lymphocytic leukemia (T-LGL) indicates that miR-181a targets the 3’UTR of both SOCS3 and DUSP6, an ERK signaling inhibitor [208]. Knockdown of miR-181a restores DUSP6 and SOCS3 expression and induces apoptosis [208]. In GBM models, Schafer et al. found that aberrantly high metalloprotease-disintegrin ADAM8 expression downregulates miR-181a via activation of STAT3 and MAPK [209]. In liver metastasis of CRC, miR-181a-5p enrich extracellular vesicles appear to activate hepatic stellate cells directly by targeting SOCS3 and activating IL6/STAT3 signaling. Overall, miR-181a influences tumor progression and immune stimulation in various cancers by regulating JAK/STAT signaling (Figure 5A).
Figure 5.
(A) The role of miR-181a in the regulation of JAK/STAT signaling across cancer types. miR-181a directly inhibits PIAS3 and SOCS3 in BC. Similarly, in T-LGL and CRLM, miR-181a seems to directly target DUSP6 and SOCS3. A competitive regulatory loop also exists where miR-181a directly inhibits STAT1 and STAT1 binds to the promoter of miR-181a preventing its transcription. (B) The role of miR-181a in the regulation of STING activation and signaling across cancer types. As we report, miR-181a regulates STING activation in high-grade serous cancer and in triple-negative breast cancer thus stimulating innate immune responses. Figure generated using BioRender.
Figure 5.
(A) The role of miR-181a in the regulation of JAK/STAT signaling across cancer types. miR-181a directly inhibits PIAS3 and SOCS3 in BC. Similarly, in T-LGL and CRLM, miR-181a seems to directly target DUSP6 and SOCS3. A competitive regulatory loop also exists where miR-181a directly inhibits STAT1 and STAT1 binds to the promoter of miR-181a preventing its transcription. (B) The role of miR-181a in the regulation of STING activation and signaling across cancer types. As we report, miR-181a regulates STING activation in high-grade serous cancer and in triple-negative breast cancer thus stimulating innate immune responses. Figure generated using BioRender.
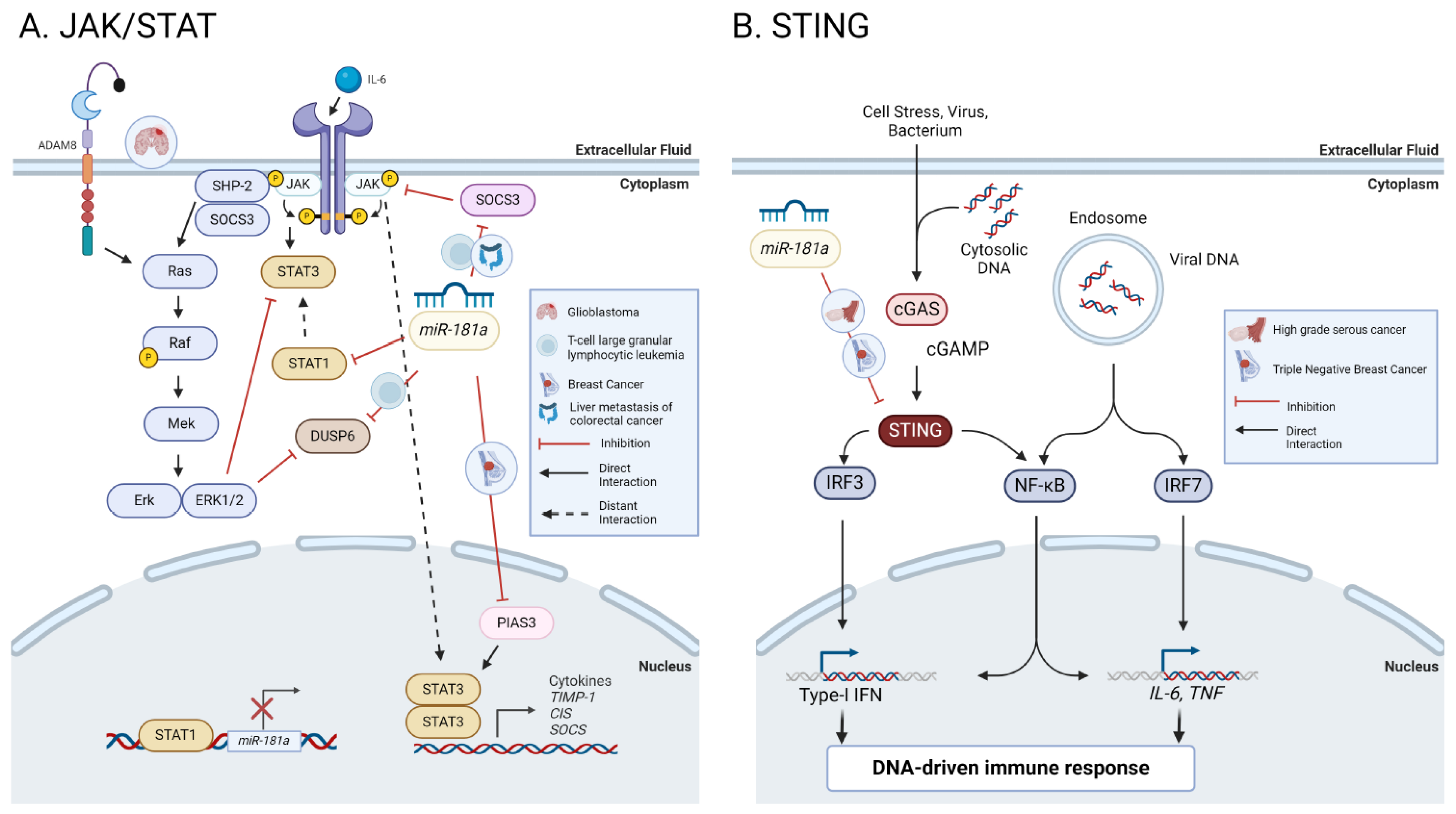
8.9. STING
The cGAS-STING (Cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING)) innate immune signaling pathway is a vital host defense network that produces Type-1 IFNs upon recognition of cytosolic DNA. Typically, this pathway is activated by viral DNA; however, high genomic instability and DNA damage in cancers can produce nucleic acids in the cytosol, stimulating this pathway in a tumor suppressive manner [210]. When cytosolic DNA binds to cGAS, it is converted to cGAMP which then binds to and activates STING. STING translocates to from the ER to the Golgi where it interacts with TBK1. TBK1 activates STING and the transcription factor interferon regulatory factor 3 (IRF3). Phosphorylated IRF3 promotes transcription of Type-I IFNs and may stimulate nuclear factor-κB (NF-κB) signaling [211].
Our group has recently shown that overexpression of miR-181a induces genomic instability, mitotic defects, and nuclear rupture in HGSC by directly inhibiting the tumor suppressor gene RB1 [98]. This increased miR-181a expression transforms fallopian tube secretory cells (FTSEC) and promotes early HGSC development through RB1 inhibition. Further, our group showed that STING targeting by miR-181a enabled cancer cells to bypass interferon-mediated cell death [98]. Supporting this work, a recent study found that miR-181a is a crucial regulator of the STING pathway in BRCA-mutated triple-negative breast cancer (TNBC) and OC. They observed that miR-181a overexpression in TNBC and OC downregulates the STING pathway and promotes resistance to the PARP inhibitor, Olaparib [71]. Altogether, this research suggests a novel connection between miR-181a and the STING pathway (Figure 5B).
8.9.1. NF-κβ
NF-κβ is a family of inducible transcription factors that regulate immune and inflammatory responses, and cell survival and growth. It comprises five structurally related members: NF-κβ1 (p50), NF-κβ2 (p52), RelA (p65), RelB, and c-Rel, which mediate target gene transcription [212]. NF-κβ signaling can be activated through Toll-like receptors (TLRs), interleukin-1 receptors (IL-1R), tumor necrosis factor receptors (TNFR), growth factors receptors (GFRs), and protein kinase C (PKC). Additionally, there is proposed crosstalk between STING and the NF-κβ signaling network [213]. STING, along with aIκβ kinase epsilon (IKKε), drives NF-κβ signaling by activating the IKK complex, which leads to a pro-inflammatory cytokine response [213].
In CRC, increased miR-181a expression is correlated with elevated IL-1β levels through activation of the NF-κβ pathway [214]. In this context, miR-181a directly suppresses PTEN, resulting in IL-1β induction and enhanced cell viability. These findings established an IL-1β/NF-κβ/miR-181a/PTEN pathway that may contribute to CRC progression [215]. In addition, another independent study in ABC-like subgroups of DLBCL demonstrated that miR-181a suppressed the constitutive activity of NF-κβ signals in vitro and in vivo, thus regulating cellular proliferation and survival [68]. Furthermore, in hypopharyngeal squamous cell carcinoma, a pathway reporter assay revealed that downregulation of miR-181a results in decreased NF-κβ levels [216]. Overall, miR-181a plays a crucial role in modulating NF-κβ signaling across multiple cancer lineages, influencing inflammation, cell survival, and tumor progression.
10. Immune Microenvironment
The tumor microenvironment (TME) is a complex ecosystem comprised of extracellular matrix components, stroma, and diverse immune cell populations, which regulate cancer progression. Recent research highlights miR-181a's involvement in the TME. In liver metastases of CRC, miR-181a-rich extracellular vesicles lead to the reprogramming of the TME and the formation of pre-metastatic niches through the IL6/STAT3 signaling pathway [217]. Extracellular vesicles from HNSCC cancer cell lines containing miR-181a, among other miRNAs can disrupt dendritic cell function by decreasing cytokine production [218].
miR-181a also interacts with the NK cells, T-cells, and other immune cells within the TME. For instance, Amado et al. showed that increased miR-181a-5p in murine CD4+ and CD8+ T-cells leads to decreased IFNγ production [219]. γδ T-cells are a subgroup of T-cells that have IFNγ-associated type 1 inflammatory and cytokine properties and also express a variety of critical immune determinants like NK cell receptor NK2GD. Studies by Gordino et al. demonstrate that miR-181a prevents γδ T-cells’ differentiation into TNF-α + effectors by regulating MAP3k2 and Notch2 signaling, inhibiting NK2GD functions in both differentiating γδ T-cells and circulating T-cells in PrCa [48]. Similarly, in MM, miR-181a overexpression decreases several inflammatory and adhesion factors including, IL-1, TNFα, IFNγ, and Muc-1, ICAM-1, VCAM-1 [206]. These studies revealed a novel role of miR-181a in regulating NK cell functions, which is particularly relevant as diverse strategies are currently being adopted to manipulate NK2GD to improve next-generation immunotherapies.
10.1. Immune Checkpoints
Immune checkpoints are a grouping of inhibitory pathways responsible for maintaining self-tolerance and modulating the duration and amplitude of immune responses [220]. One such immune checkpoint, programmed cell death (PD-1) and its ligands (PD-L1 and PD-L2), play a crucial role in maintaining peripheral tolerance [221]. These co-inhibitory proteins are expressed on the surface of antigen-stimulated T-cells and, when activated, can inhibit T-cell proliferation, survival, cytokine production, and other effector functions [222]. Cisplatin-induced miR-181a expression negatively regulates PD-L1 expression via the c-FOS subunit of the transcription factor AP-1. Since cisplatin induces negative feedback regulation of PD-L1 expression, increased miR-181a expression could lead to a downregulation of PD-L1 [223]. Similarly, miR-181a regulates PDL-1 expression through the INF-γ pathway [224,225]. The circular RNA circHMGB2 also promotes immunosuppression and resistance to anti-PD-1 therapy in lung adenocarcinomas and squamous cell carcinomas through the miR-181a-5p/CARM1 axis [226].
11. Cell cycle & DNA Repair Regulation
11.1. TP53
The tumor suppressor p53 is activated in response to DNA damage and hypoxia. Upon activation, it induces cell cycle arrest and apoptosis. TP53 is widely mutated and dysregulated across cancer lineages. The interaction of TP53 and miR-181a has been well summarized by the Baradaran group [154]. More recent studies have shown that miR-181a is significantly downregulated in CLL patients with attenuated TP53 compared to patients with wild-type TP53 [94] . Upregulation of miR-181a has also been linked to p53 through the modulation of its transcriptional targets, Bax and Bcl-2 in CRC [227]. Knarr et al. report that the upregulation of miR-181a in HGSC is part of an early mechanism contributing to tumor development. We found that small regions of histologically normal FTSECs carrying a TP53 mutation also have miR-181a upregulation [98]. Additionally, in NSCLC, Rezaei et al. report that the overexpression of TP53 upregulates miR-181a. This correlation suggests that miR-181a could be involved in G1 phase cell cycle regulation via the p53 pathway [154].
11.2. G1/S Regulation
Cell cycle control mechanisms and their role in cancer have been well-characterized and exploited in cancer treatments. More recently, miR-181a has been connected to several cell cycle regulatory pathways, including the G1/S control. In NSCLC, miR-181a is downregulated and plays a tumor-suppressive role by regulating CDK1 expression and downregulating Cyclin D1 and Cyclin B1 [228]. Additionally, miR-181a-5p targets E2F7 in NSCLC, another important cell cycle modulator of Cyclin B1 and Cyclin D1 [124].
mi-181a has also been linked to the RB1 pathway, which drives the progression from the G1 to the S phase of the cell cycle. In thyroid cancer and HGSC, where miR-181a is significantly upregulated, miR-181a directly represses RB1, promoting unregulated cell cycle progression and preventing apoptosis [97,98]. Further, miR-181a targeting of RB1 in FTSEC promotes transformation and increased genomic instability [98]. In uveal melanoma, miR-181a is overexpressed and targets the 3’UTR of CTDSPL, inhibiting its translation. This inhibition leads to increased phosphorylation of RB1 and accumulation of the downstream cell cycle effector E2F1, ultimately negatively regulating cell cycle progression [229]. A similar relationship exists in multiple myeloma, where miR-181a is upregulated in tumor tissue. When miR-181a was silenced in vitro, there was a significant decrease in S-phase cells, a decrease in proliferative and invasive tendencies, and a return of apoptotic function [230]. Collectively these studies indicate miR-181a's oncogenic role in cell cycle regulation, but more research is needed to understand the mechanisms behind this phenotype.
Furthermore, researchers found that in AML where miR-181a upregulated, miR-181a overexpression drives a decreased ratio of G1-phase cells and an increased ratio of S-phase cells. To assess this mechanism, the authors found that miR-181a directly targets Ataxia-telangiectasia mutated (ATM), which plays an important role in DNA damage control [231]. miR-181a also promotes G1/S transition and cell proliferation in clear cell renal cell carcinoma by directly binding and suppressing the antitumor protein KLF6 [232]. Corroborating this finding, when miR-181a was inhibited in ALL cells, a G1/S cell cycle arrest was observed. Together these findings suggest that miR-181a is involved in inhibiting the G1/S phase regulation and cell cycle promotion across cancer types.
11.3. G2/M Regulation
In addition to miR-181a's role in G1/S cell cycle regulation, research across several cancer types suggests that miR-181a also regulates G2/M cell cycle progression. In non-malignant FTSECs, overexpression of miR-181a, showed an increased in the G2/M subpopulation suggesting increased proliferation [98]. In BC and GC, miR-181a targeting of ATM was shown to regulate DNA repair and cell cycle regulation at the G2M checkpoint [104,233]. In GC, miR-181a inhibition of ATM led to increased proliferation and inhibited apoptosis [233]. In BC, increased levels of miR-181a also decrease ATM and BRAC1 functions in tumors while also blocking G2/M transition [104].
12. Apoptosis
Apoptosis is a naturally occurring process by which a cell experiences programmed death. This can include both extrinsic and intrinsic pathways, both of which are regulated by miR-181a.
12.1. BCL-2
BCL-2 (B-cell lymphoma 2) is an outer mitochondrial membrane protein that regulates cell death by blocking apoptosis. BCL-2 can inhibit pro-apoptotic signals and promote chemoresistance when overexpressed in cancer cells. BCL-2 expression is also linked to miR-181a activity in several cancers. In CLL patient samples, treatment with a miR-181a mimetic reduces the expression of multiple apoptotic genes, including BCL-2, MCL-2, XIAP, and TCL-1 by directly binding to the 3’UTR of BCL-2, MCL-1, and XIAP [94,234]. Treatment with a miR-181a mimic in CLL models was found to sensitize cells to fludarabine treatment and induce apoptosis [94]. Furthermore, treatment with a miR-181a mimic in daunorubicin (DNR) resistant CLL lines sensitized them to DNR via direct inhibition of BCL-2 [234]. These findings suggest that miR-181a plays an tumor suppressive important role in cancer pathogenesis and sensitivity to apoptosis-inducing agents through its action on BCL-2.
miR-181a has also been linked to chemosensitivity in BC by targeting BCL-2 [235]. Additional work confirms miR-181a's direct inhibition of BCL-2 in BC, inducing apoptosis [94]. Majzoub et al. screened the antitumor activity of different thiosemicarbazone derivatives in TNBC. They found that combined Cisplatin + thiosemicarbazone compound 4 treatment increased miR-181a-5p expression, resulting in the downregulation of BCL-2 [236]. Interestingly, treatment with a miR-181a mimic in low-invasion BC cells resistant to Adriamycin increased chemosensitivity in a BCL-2-dependent manner, juxtaposing previous studies [235]. Taken together, these studies demonstrate how the efficacy of many BC chemotherapeutic agents relies upon increased miR-181a expression to facilitate apoptosis in a BCL-2-dependent manner.
Transient miR-181a overexpression in GBM cells downregulates BCL-2 and sensitizes glioma cells to radiation treatment. This may indicate that miR-181a could modulate radiosensitivity and be a molecular driver of cellular instability through BCL-2 signaling in GBM models [214]. In melanoma research, miR-181a has been linked to lncRNA-LHFPL3-AS1-long's regulation of CSC apoptosis. Zhang et al. found that miR-181a-5p is sponged by LHFPL3-AS1-long, thereby preventing miR-181a-5p's degradation of BCL-2 and restoring apoptosis [237]. In B-cell lymphoma pathogenesis, Kozoski et al. identified a less direct relationship between miR-181a and BCL-2. They previously found that miR-181a directly targets BCL-2; however, upon completing a rescue experiment in which they transfected a plasmid lacking the 3’UTR of BCL-2, BCL-2 did not alleviate the effect of miR-181a on cell proliferation or viability. Thus, further investigation is needed to draw conclusions between miR-181a and BCL-2 in this context[68].
12.2. Other Apoptotic Pathways
miR-181a is linked to various apoptotic pathways across several cancers. In leukemia research, Zhou et al. investigated the effect of miR-181a overexpression on cell apoptosis and DNA damage post-chemotherapy. They observed that miR-181a overexpression increased several DNA damage markers, such as caspase-3 and γH2Ax, and enhanced Poly [ADP-ribose] polymerase 1 (PARP1) acetylation by downregulating histone deacetylase SIRT1, suggesting a role in repairing DNA damage and PARP1-mediated cell death [238].
In acute promyelocytic leukemia, PML/RARα oncogenic fusion protein upregulates miR-181a, increasing colony formation and resistance to apoptosis. Given that RASSF1A is downregulated in this cancer, it was hypothesized that miR-181a interacts with RASSF1A. They show that miR-181a binds to the 3'UTR of RASSF1A, downregulating its expression and inhibiting cell proliferation and apoptosis [239]. Similarly, in GC, miR-181a drives carcinogenesis through direct interaction with RASSF1A, promoting cell proliferation and reducing apoptosis [239,240].
Other studies suggest that miR-181a-5p promotes apoptosis in BC via Bax and Caspase-9 [241]. In GBM, miR-181a increases sub-G1 cell cycle arrest, inducing apoptosis by regulating several genes, including, caspase-9, Bcl-2, and SIRT1 [53]. miR-181a also directly targets PDCD4, a known marker of programmed cell death often lost in BC cases [242,243]. In vitro studies by Gu et al. demonstrate that miR-181a suppresses BC cell growth, viability, and has a pro-apoptotic role [241]. Elevated miR-181a expression in cryogenically normal acute myeloid leukemia (CA-AML) is also correlated with increased apoptosis, decreased cell viability, delayed leukemogenesis, favorable outcomes, and improved overall survival by directly targeting the 3’UTR of the homeobox gene PBX3, a transcription factor that drives poor prognosis in CA-AML [244]. A similar relationship is seen in cervical cancer, where miR-181a-5p overexpression inhibits apoptosis by targeting inositol polyphosphate-5-phosphatase A (INPP5A) [245]. Alternatively, in lung adenocarcinoma, Wang et al. found that miR-181a in exosomes derived from M1 macrophages promotes apoptosis via the miR-181a-5p/ETS1/STK16 axis [246].
13. Angiogenesis
Angiogenesis is a tightly regulated process by which new blood vessels are formed from pre-existing microvasculature. Pro-angiogenic factors like vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and epidermal growth factor (EGF) (along with their receptors VEGFR, FGFR, and EGFR) modulate hypoxia and normoxia in tumors [247]. The enzyme Src, linked to VEGF-induced angiogenesis with malignant activation, is present across many cancers, including BC, CRC, PaCa, OC, HNSCC, lung, bladder, neuronal tumors, chronic myeloid leukemia (CML), and MM. miR-181a regulates VEGF-induced angiogenesis via Src activation [248]. Studies have shown that miR-181a directly targets the 3’UTR of Src-related tumor suppressor factor SRC kinase signaling inhibitor 1 (SRCIN1) in CRC. Inhibition of SRCIN1 by miR-181a activates Src, triggering VEGF secretion and leading to angiogenesis [222]. Furthermore, in BC, Taylor et al. found that increased miR-181a expression enhances Src phosphorylation and activation [136]. miR-181a is also involved in angiogenesis in chondrosarcoma and clear cell renal cell carcinoma. When miR-181a expression is increased, mRNA and protein expression levels of VEGF are upregulated, contributing to increased tumor microvasculature [249].
Transitioning to matrix metalloproteinases (MMPs), miR-181a MMPs, which mediate angiogenesis through modulation of anti- and pro-angiogenetic factors. In BC, miR-181a directly targets MMP-14, decreasing cell migration, invasion, and angiogenesis [161]. miR-181a was also found to target MMP-14 in supportive study in human fibrosarcoma and BC models [162]. In CRC, miR-181a directly binds to reversion-inducing cysteine-rich protein with Kazal motifs (RECK), driving MMP-9 upregulation and phosphorylation of SMAD2/3 [163]. In summary, miR-181a plays a critical role in angiogenesis across various cancers by modulating VEGF signaling, Src activation, and the expression of MMPs, thereby influencing tumor growth and progression.
14. Hormonal Signaling
In BC, miR-181a-5p plays a role in both ERα and ERβ downregulation [69,70]. This is unsurprising given that the 3’UTR of estrogen receptor 1 (ESR1) contains a binding site for the miR-181a seed sequence [178]. miR-181a also directly targets the 3’UTR of progesterone receptor (PGR) [250]. Gilam et al. validate this finding and show that miR-181a negatively regulates PGR in BC [251]. Further, Maillot et al. suggest that pri-miR-181a/b are primary targets of ER-mediated transcriptional activity. Consequently, estradiol reduces miR-181a/b expression, while anti-estrogen treatment increases miR-181a expression. Although miR-181a’s effects on estradiol and BC progression are not fully explored, it is known that miR-181a can abrogate the estradiol-dependent increase in cell proliferation via downregulation of PR [250]. miR-181a also reduces PGRMC1 expression, thereby attenuating progestin-induced cell growth in BC [241].
15. miR-181a as a Non-Toxic Targetable oncomiR
As emerging studies highlight miR-181a's role as an oncogenic driver in particular contexts and explore ways to inhibit its activity, it is important to note that the knockout of this gene is not lethal or toxic to the host, making it a promising therapeutic target. For example, in an HCC model, knockout of miR-181a did not change the number of hematopoietic or endothelial cells, suggesting a non-toxic effect on rapidly dividing cells, despite miR-181a’s role in development [252]. These findings suggest a favorable safety profile for miR-181a targeting. In another study, Paterson et al. developed a miR-181a knockout C57BL/6J mouse model and found minor phenotypic effects, such as elevated blood pressure, increased mean arteriole blood pressure, and increased salt sensitivity. However, there was no difference in food or water intake, mouse weight, and only slightly larger spleens in the miR-181a knockout group compared to the control [253]. Another group showed that germline miR-181a1/b1 or miR-181a2/b2 knockout did not cause gross phenotypic abnormalities in growth, development, or survival [254]. These studies establish proof of concept, suggesting that miR-181a inhibitory treatments could be safe and potentially have no significant toxicities.
Given miR-181a’s abundance and role in T-cell development activation, and function, it was imperative to investigate the effects on T-cells following miR-181a deletion [255]. Kim et al. reviewed miR-181a regulatory pathways in T-cell differentiation and aging, noting that miR-181a/b modulates T-cell tolerance by controlling T-cell selection and peripheral T-cell function, indicating a correlation between miR-181a/b and the active immune system [46]. They showed that miR-181a-1/b-1 murine germline deletion increases in the intrinsic reactivity of naïve T-cells to self-antigens without causing spontaneous autoimmunity [256]. The Krueger lab found that knockout of miR-181a is not detrimental to T-cell function [257]. Although whole-body murine knockout of miR-181a results in impaired T-regulatory cell development, total frequency, and diversity in T-regulatory cells in the periphery are not changed due to homeostatic expansion [257]. These findings were corroborated by Fragoso et al., who showed that germline knockout of miR-181a1/b1 did not change the cellularity of hematopoietic organs, including bone marrow, spleen, thymus, and peripheral blood [174]. Additionally, the Krueger lab showed show that whole body miR-181a/b knockout does not result in a substantial compensatory takeover by other miRNAs in the thymus, suggesting that cellular functions remain stable in the absence of miR-181a and further supporting the positive developing safety profile of miR-181a targeting [16]. In work on allogeneic hematopoietic stem cell transplantation therapy, Sang et al. found that miR-181a affects the differentiation of naive CD41+ T-cells by targeting IFN-γ production [224]. Further research showed that miR-181a/b-1 deficient mice display aberrant double positive thymocyte development and a complete block in early invariant natural killer T-cell development, resulting in impaired T-cell receptor signaling [254,258]. This study suggests that loss of miR-181a could compromise TCR signaling and the immune system; however, as stated earlier, an abundance of studies suggest that T-cell function is not harmed despite impaired development. Future studies clarifying how loss of miR-181a affects immune function in the context of cancer will be critical to understanding the regulatory role of this miRNA. Collectively, these studies establish miR-181a as a promising therapeutic target with a potentially favorable safety profile. The non-toxic effects observed across various knockout models and organs, combined with miR-181a’s crucial role in lymphocyte regulation, underscore the potential for developing miR-181a-targeted therapies.
16. Genetically Engineered miR-181a Murine Models
In more recent studies, genetically engineered mouse models (GEMMs) have been created to highlight the role of miR-181a across cancers. Valencia et al. created GEMMs of KRAS-driven lung and pancreatic cancers. After identifying miR-181a/b as a miRNA cluster upregulated by oncogenic KRAS, they used several miR-181a-KRAS GEMMs to evaluate the loss of function of miR-181a/b. They demonstrated the key role of miR-181a/b in tumorigenesis, and further defined the need for miR-181a targeted therapies in KRAS-mutant cancers [259]. Similarly, in HCC, a miR-181a-altered GEMM was created to elucidate miR-181a expression in combination with TGF-β treatment to combat hepatocyte EMT. Here, miR-181a was significantly upregulated in response to TGF-β treatment, and aberrantly expressed in HCC and chronic liver disease [134]. These studies underscore the importance of miR-181a in cancer development. Continued development of GEMMs is crucial for further contextually characterization miR-181a as an oncomiR in pan-cancer models.
17. Nanoparticles as a Delivery Platform for miR-181a
Despite increasing knowledge of miR-181a, and the use of complementary oligos, sponges, and mimics to ascertain the relevance of its genetic manipulation in disease-based contexts, hurdles pertaining to the delivery of antagomiRs and mimics still exist and limit the clinical application of microRNA-based therapeutics. AntagomiRs are synthetic oligos that complementarily bind and sequester miRNAs, thereby preventing them from interacting with MREs and target mRNA networks. Nanoparticles (NP) have been employed for several decades to deliver miRNAs and antagomiRs across various cancer types, providing an efficient and safe mechanism for miRNA-driven diseases.
When miR-181a functions as a tumor suppressor, lower levels of miR-181a are associated with poorer outcomes in cancer. Treatment with miR-181a mimetics can counteract these cancer-driving phenotypes in vitro but not in vivo. In AML, Huang et al. successfully delivered miR-181a mimics to AML blasts via transferrin-targeted anionic lipid-based lipopolyplex nanoparticles [260]. This increased miR-181a expression, downregulated known targets KRAS, NRAS, and MAPK, reduced proliferation, impaired colony formation, and increased sensitivity to cytarabine treatment [187]. This miR-181a NP delivery model in murine AML represents a clinically relevant miRNA delivery platform resulting in improved survival outcomes. In RB research, miR-181a NP delivery has also shown success in rat xenograft models. Co-delivery of a miR-181a mimic along with the chemotherapeutic agent melphalan via lipid NP significantly decreased immunogenicity, and improved chemosensitivity and cytotoxicity [261]. miR-181a is also downregulated in GBM clinical samples. Using a hyaluronan-decorated lipid nanoparticle (HA-LNP) to deliver miR-181a to GBM cells in vivo and in vitro, they found that this treatment platform induced cell death and delayed tumor growth [262]. miR-181a has also been used as a radiosensitizer by inducing DNA damage post-radiotherapy for rectal cancer. Successful loading of miR-181a into a MnO2 @ZIF-8-polyethylene glycol nanocomplex prevented miRNA degradation, reduced tumor proliferation, enhanced radiosensitivity, and improved local TME hypoxia [263]. These studies illustrate the promise of miR-181a mimetic treatment as a novel therapeutic approach for cancers where miR-181a acts as a tumor suppressor.
Other highly aggressive cancers are driven by miR-181a overexpression and lack effective systemic treatment options for reducing miR-181a expression. Recently, anti-miR-181a oligos loaded into Janus-based nanotubes and delivered into chondrosarcoma xenograft models, reduced miR-181a expression, restored RGS16, a known target of miR-181a, and improved survival outcomes [264]. Future studies confirming the use of anti-miR-181a oligos to systemically or organ specifically knockdown miR-181a in other cancer models would point to an easy and safe therapeutic delivery platform to be exploited by clinics.
18. Pharmacological Inhibition of miR-181a
18.1. Epigenetic Drugs Controlling miR-181a Activity
In our previous work, we developed a functional endogenous miR-181a biosensor for high-throughput therapeutic screens. Using this sensor, we sorted for OVCAR3 cells highly expressing miR-181a and screened >3,000 bioactive compounds. Notably, 50% of the hit compounds that reduced miR-181a activity were bromodomain and extra-terminal motif (BET) inhibitors, indicating epigenetic regulation of miR-181a by the BET protein family [100]. Another study demonstrated that MC3324, a hybrid lysine-specific histone demethylase inhibitor, reduced miR-181a-5p expression and estrogen receptor alpha (ERα) protein levels in BC [70]. The authors observed an inverse correlation between miR-181a and ERα, with MC3324 treatment reversing this correlation, inducing miR-181a-5p, and reducing ERα expression [70]. Together, these findings suggest that epigenetic modulation could be a therapeutic strategy for controlling miR-181a expression and oncomiR function. Further research on epigenetic regulators and their correlation with miR-181a expression in tissue specific contexts is needed to determine if these mechanisms could enhance treatment sensitivity.
18.2. Natural Products
In addition to synthetically derived miR-181a therapeutic options, several natural products have been shown to regulate the miR-181a family. For example, the fingerroot compound pinostrobin reduces miR-181b-5p expression and induces ATM-mediated apoptosis in acute leukemia [265]. Previous studies have implicated pinostrobin’s anticancer effects in BC, GBM, OC, and leukemias. Future research might reveal that pinostrobin treatment has similar beneficial effects across miR-181a-driven cancer types. Similarly, all-trans-retinoic acid (ATRA), a naturally occurring derivative of vitamin A (retinal), downregulates miR-181a/b in acute promyelocytic leukemia. Mechanistically, miR-181a/b targets the 3’ UTR of the ATRA-regulated tumor suppressive gene RASSF1A, which attenuates ATRA-induced granulocytotic differentiation through Cyclin D1 regulation [239]. New research points to an interaction between miR-181a and the Chinese medicinal herb astragaloside IV (AS-IV). AS-IV has been studied in NSCLC anlotinib resistance. AS-IV treatment decreases miR-181a-3p expression and activates endoplasmic reticulum-associated degradation (ERAD) and unfolded protein response (UPR) pathways, thereby attenuating anlotinib resistance. Thus, these authors suggest a promising new NSCLC therapy combining AS-IV treatment with inhibition of the miR-181a-3p/UPR-ERAD axis [266]. Curcumin, a common anti-inflammatory additive in skin care products and compound found in turmeric. Interestingly, curcumin has been shown to induce cisplatin sensitivity by increasing ATG5 expression, a known target of miR-181a-3p, in OC [267,268].
Resveratrol (RV) is an active polyphenol substance found in knotweed and grapes. It is a phytoestrogen known to bind to ERs and is known for its immunoregulatory and antibacterial effects, as well as the alleviation of menopausal symptoms. In cancer, RV has known antitumor properties, including induction of apoptosis in BC and inhibition of migration and invasion in PaCa [269,270]. Although this mechanism has not been explored extensively in cancer, one study in myocardial fibrosis showed that RV treatment reduces miR-181a expression and inhibits TGF-β-mediated hyperproliferation [271]. Given miR-181a’s well characterized role in TGF-β-mediated tumor suppression and oncogenic progression, further studies examining if RV treatment could reduce miR-181a driven invasion and metastasis would offer an innovative and accessible treatment option for miR-181a driven cancers. In contrast, another study showed that RV treatment increased miR-181a expression, thereby repressing the TRAF6/TAK1 inflammatory signaling pathway in osteoclasts [272]. Both of these studies illustrate the potential of RV as an indirect targeting agent for miR-181a; however, further characterization of its mechanism is crucial to determining its use and efficacy.
18.3. Drug Repurposing
Given the difficulty of developing targeted small moelcules, particularly for miRNAs, some studies have found success in repurposing drugs for targeting miR-181a. Perftoran, a perfluorodecalin emulsion, initially developed as an oxygen-carrying plasma additive for acute blood-loss anemia, has recently been investigated for its effect on several oncomiRs, including miR-181a [273]. One study analyzed its impact on the photosensitizing agent indocyanine green (ICG) used in photodynamic therapy (PDT) for lung cancer. They found that Perftoran treatment suppresses miR-181a and inhibits ICG/PDT-hypoxia [274]. Another study found that vincristine and prednisone treatment in naïve, but not recurrent, leukemia patients induced pro-apoptotic gene expression and suppressed exosomal miR-181a expression [92]. These findings highlight the potential of repurposing existing drugs to efficiently target miR-181a driven cancers and offer a promising avenue of developing new therapeutic strategies in miRNA-driven diseases.
19. miR-181a Governs Cancer Therapeutic Efficacy
19.1. miR-181a Modulates Radiosensitivity
Radiotherapy is a component of many cancers’ standard of care; however, like any treatment, its efficacy is limited by radio-resistance. Dysregulation of essential genes modulating pro-apoptotic signaling pathways has been postulated to drive this therapeutic resistance, but identification of these genetic drivers still remains a challenge. In GBM models, miR-181a was found to be reduced by >2.6 fold upon irradiation. Upon miR-181a overexpression, GBM cells were sensitized to radiation therapy [214]. Similarly, in cervical cancer models, miR-181a was found to be upregulated in radio-resistant samples compared to radio-sensitive samples [275]. Mechanistically, the authors’ postulate that miR-181a's direct inhibition of PRKCD’s, a pro-apoptotic kinase, inhibits irradiation-induced apoptosis and decreases G2/M block promoting insensitivity to radiation. miR-181a’s inhibition of PRKCD was further validated by Su et al. [52]. Combined, these studies suggest that miR-181a may be a good biomarker for identifying sensitivity to radiotherapy.
19.2. miR-181a Contributes to Chemotherapy Resistance
The interaction of miR-181a-5p with platinum-based therapeutics and other chemotherapeutic agents has been well characterized. miR-181a expression has been correlated to increased chemotherapy resistance across several cancer types. Specifically, in leukemia research, Li et al. found that miR-181a could play a role in developing resistance to the chemotherapeutic agent DNR. Treatment with a miR-181a mimic and DNR significantly reduced cell survival and increased BCL-2 expression, suggesting that miR-181a could sensitize leukemia cells in a BCL-2-dependent manner [234]. Similarly, in chemoresistant cervical squamous cell carcinoma, miR-181a enhances chemoresistance in vitro and in vivo by directly targeting PRKCD, an apoptotic target gene of miR-181a [276]. miR-181a expression levels are also elevated across several other cisplatin-treated models, including NSCLC, T-cell leukemia/lymphoma, and OC [76,182,223]. In contrast, in melanomas, increased miR-181a expression has been linked to reduced signaling down the chemoresistant pathway TFAM, leading to a restoration of chemosensitivity [58] . These findings highlight the complex role of miR-181a in modulating chemotherapy resistance and sensitivity.
20. Concluding Remarks
Mounting evidence has demonstrated miR-181a’s critical role in cancer progression. Aberrant expression of miR-181a can cause dysregulation across numerous signaling pathways, enabling cancer cells to acquire the functional capabilities and hallmarks of cancer needed for tumorigenesis (Figure 6) [10]. Mechanistically, miR-181a targets multiple mRNA transcripts that are included in all the hallmarks of cancer [10]. Further, it is regulated by numerous ncRNAs, transcription factors, and post-transcriptional modulators that drive these cancer-relevant pathways. In some cases, the restoration of miR-181a expression can reinstate tumor suppressive phenotypes; however, in many cancer lineages, miR-181a overexpression can drive oncogenesis and chemotherapy resistance. Overall, these studies clearly define miR-181a as a promising, predictive biomarker with relevant clinical applications. Although therapeutic approaches to targeting miRNAs in general, and miR-181a specifically, remain in the early stages, current studies show promising options for targeting miR-181a using nanoparticles, repurposed drugs, and natural products. Given miR-181a’s promising safety profile, further development of miR-181a targeted therapies and small molecules could significantly impact oncology treatment options and increase sensitivity to current chemotherapies.
Figure 6.
Schematic depicting miR-181a’s oncogenic and tumor suppressive function across numerous signaling pathways in cancer. miR-181a dysregulation can enable cancer cells to acquire hallmark capabilities required by this neoplastic disease [10]. miR-181a can modulate the function of several key molecular pathways, including RB1, Ras, BCL-2, and SRC/VEGF, leading to genomic instability, senescent cells, apoptotic dysfunction, angiogenesis, and an evasion of growth suppressors. miR-181a dysregulation can also lead to the overactivation of other pathways and proteins such as the activation of Wnt, SOX2, TGF-β, and Ras/PI3K/Notch across several cancer types. This overactivation can promote stemness, activate invasion and metastasis, and increase proliferation capabilities, respectively. miR-181a dysfunction also impacts cancer epigenetics, causing nonmutational epigenetic reprogramming though the overactivation of H3K27ac methylation CNAs. Additionally, miR-181a overexpression can impact immune regulation through the inhibition of NF-κβ, STAT, and STING signaling. Figure generated using BioRender.
Figure 6.
Schematic depicting miR-181a’s oncogenic and tumor suppressive function across numerous signaling pathways in cancer. miR-181a dysregulation can enable cancer cells to acquire hallmark capabilities required by this neoplastic disease [10]. miR-181a can modulate the function of several key molecular pathways, including RB1, Ras, BCL-2, and SRC/VEGF, leading to genomic instability, senescent cells, apoptotic dysfunction, angiogenesis, and an evasion of growth suppressors. miR-181a dysregulation can also lead to the overactivation of other pathways and proteins such as the activation of Wnt, SOX2, TGF-β, and Ras/PI3K/Notch across several cancer types. This overactivation can promote stemness, activate invasion and metastasis, and increase proliferation capabilities, respectively. miR-181a dysfunction also impacts cancer epigenetics, causing nonmutational epigenetic reprogramming though the overactivation of H3K27ac methylation CNAs. Additionally, miR-181a overexpression can impact immune regulation through the inhibition of NF-κβ, STAT, and STING signaling. Figure generated using BioRender.
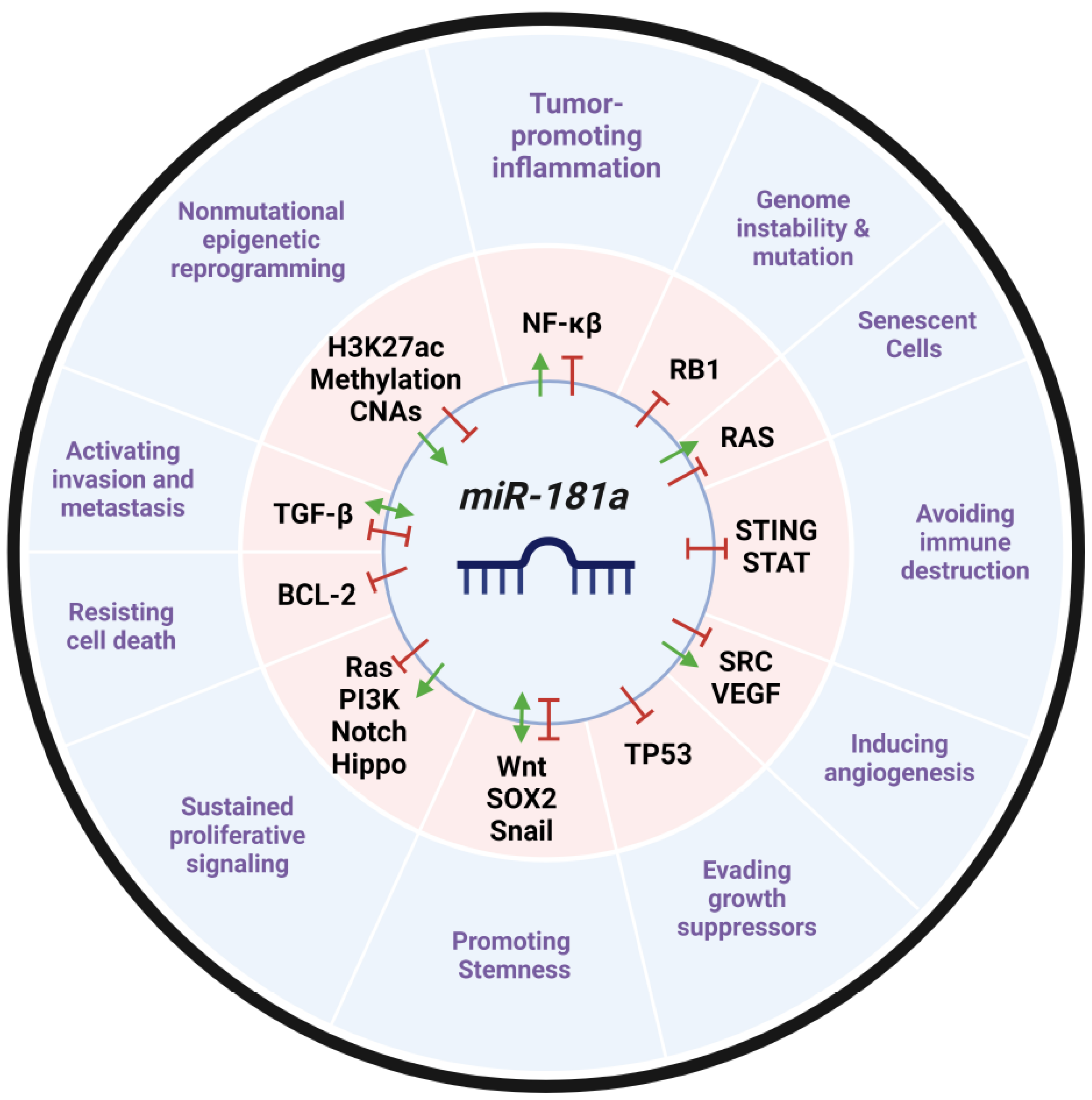
21. Expert Opinion
miR-181a is a novel and exciting therapeutic target that can be applied to various cancer types. It’s ability to act on multiple cancer driving pathways including cell growth, developmental, angiogenic, apoptotic, and immune stimulatory pathways emphasize its multi-faceted regulatory control mechanisms. Slight variation in miR-181a’s expression levels can significantly alter its wide breadth of regulatory reach on numerous target mRNA networks and disrupt the balance required for cancer cell maintenance and homeostasis. This altered expression profile is easily measurable and characterized in many cancers underscoring the diagnostic potential of miR-181a as a biomarker and its targetable potential. Given that most precision cancer treatments focus only on a single pathway, there is a demonstrated clinical need to develop polyvalent therapeutic approaches that can act on many molecular targets. Tailoring treatments to be directed towards miR-181a would offer a unique and central therapeutic approach that can achieve these goals while inhibiting tumor growth, metastasis, recurrence, and stimulating the immune system.
Indeed, the development of RNA therapeutic approaches is exploding and has the potential to revolutionize precision medicine. Since the first Phase I antagomir clinical trial in 2010 (ClinicalTrials.gov Identifier: NCT02031133), more than 22 Phase I, II, and III miRNA based clinical trials have been conducted and we expect to see miR-181a therapeutic approaches joining the list in the next 10 years [277]. However, the development of these miRNA-based treatments is not without its barriers. The implementation of miR-181a therapy in cancer requires the development of the appropriate therapeutics’ platform. Such systems including nanoparticles, natural products, and repurposed drugs have been preclinically explored but not thoroughly evaluated for their clinical potential despite mounting evidence suggesting miR-181a is an ideal molecular target with a promising safety profile and the power to reduce tumor formation in vivo. Future studies should home in on developing robust and clinically relevant miR-181a therapeutic platforms.
One of the key advantages of miR-181a as a genetic vulnerability in cancer is that its aberrant expression has been widely characterized in normal and tumor patient samples. Furthermore, accumulating evidence validating these expression characteristics in serum samples collected from patients demonstrates the diagnostic relevance and the clinical accessibility of using miR-181a as a patient biomarker. Diagnostically, measuring miR-181a levels prior to treatment could allow clinicians to infer how patients will respond to treatment and what their prognosis will be. Future pre-clinical studies measuring miR-181a serum samples in normal, neo-adjuvant, adjuvant, and recurrent patients can enable clinicians to further stratify patients and their ability to respond to chemotherapeutics and future miR-181a targeted therapies.
Developing a miR-181a inhibitory therapeutic involves determining the appropriate target. Instead of targeting the pri- or pre-miR-181a structure, some studies have tried to target its biogenesis mediators and stabilizers. Investigators have identified that miR-181a expression can be modulated through epigenetic drugs; however, as with many therapies in this class, off-target effects including altered expression of non-target genes and lack of precision can lead to complications and a lack of predictability. Furthermore, very few studies have examined the RNA-binding proteins that directly interact with miR-181a to either stabilize it or facilitate its synthesis, processing, and degradation. Future biochemical studies are crucial to clarify the protein modulators of miR-181a and therefore identify alternative therapeutics angles to modulating miR-181a expression and activity.
MiR-181a’s function in cancer cells is crucial to their survival. However, mechanistically, cancer cells require a team of supporting cells in their microenvironment to propagate and metastasize. Given miR-181a’s crucial role in lymphocyte and myeloid cell development, it is conceivable that miR-181a reprogramming of the immune system in the tumor microenvironment is occurring in a pro-tumorigenic fashion. Future studies characterizing miR-181a’s immune-modulatory role as an oncogene in these stromal cells will be crucial in determining miR-181a’s mechanism in cancer progression and further stratifying patients and their predicted therapeutic responses. Additionally, this information can inform future miR-181a therapeutic development, administration, and dosing when designing these therapeutic agents.
Overall, the challenges of miR-181a future investigations are two-fold: 1) further characterization of miR-181a’s mechanism of action and interacting partners and 2) targeted miR-181a therapeutic development. More than 1,400 papers have been published discussing miR-181a since 2008 and we expect to see these avenues more thoroughly explored in the next 5 years with strong pre-clinical therapies rising to clinical investigation.
Funding
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 1841052 (GM) and by the National Cancer Institute of the National Institutes of Health under Award Number R01CA197780 (AD). Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation.
Acknowledgments
The authors would like to thank BioRender for all schematic generation.
Disclosure Statement
The authors report there are no competing interests to declare. The authors report no conflicts of interest.
Abbreviations
| Abbreviation | Definition |
| AFP | Alpha fetoprotein |
| Ago | Argonaut |
| Akt | Protein kinase B |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| AS-IV | astragaloside IV |
| ATM | Ataxia telangiectasia mutated protein kinase |
| ATRA | all-trans-retinoic acid |
| BC | Breast cancer |
| BCL-2 | B-cell leukemia/lymphoma 2 protein |
| BET | bromodomain and extra-terminal motif |
| CA-AML | Cryogenically normal acute myeloid leukemia |
| CDX2 | Caudal-type homeodomain transcription factor |
| ceRNAs | Competing endogenous RNAs |
| cGAS | Cyclic GMP AMP Synthase |
| cirRNAs | Circular RNAs |
| CLL | Chronic lymphocytic leukemia |
| CML | Chronic myelogenous leukemia |
| COAD | Colon adenocarcinoma |
| COFs | Cemento-ossifying fibromas |
| CRC | Colorectal cancer |
| CSC | Cancer stem cell |
| DACT2 | Disheveled binding antagonist of beta-catenin 2 |
| DLBCL | Diffuse Large B-Cell Leukemia |
| DNMTi | DNA methyltransferase inhibitor |
| EGF | Epidermal growth factor |
| Egr1 | Early growth response factor 1 |
| EMT | Epithelial to mesenchymal transition |
| EOlC | Epithelial ovarian cancer |
| EpCAM | epithelial cellular adhesion molecule |
| EPDR1 | Ependymin Related 1 proteinn |
| ER | Estrogen receptor |
| ERAD | endoplasmic reticulum-associated degradation |
| ERK2 | ERK mRNA |
| ESR1 | Estrogen receptor 1 |
| FGF | Fibroblast growth factor |
| FTSEC | Fallopian tube secretory cells |
| GAS7 | Growth arrest specific-7 |
| GC | Gastric cancer |
| GEMM | Genetically engineered mouse model |
| HCC | Hepatocellular carcinoma |
| HDACi | Histone deacetylase inhibitor |
| HGSC | High-grade serous cancer |
| IFN | Interferon |
| IGF2BP1 | Insulin Like Growth Factor2 mRNA Binding Protein 1 |
| IKKε | IKK-inducible kinase |
| IM | imatinib mesylate |
| INPP5A | Inositol Polyphosphate-5-Phosphatase A |
| IRF3 | Interferon regulatory factor 3 |
| JAK | Janus kinase |
| kb | kilobases |
| lncRNAs | Long non-coding RNAs |
| LUSC | Lung squamous cell carcinoma |
| MET | Mesenchymal to epithelial transition |
| MHC | Major histone compatibility complex |
| MIIP | Migration and invasion inhibitory protein |
| miR-181 | microRNA 181 |
| miRNA | microRNA |
| MKP-5 | MAPK phosphatase 5 |
| MMP | Matrix metallopronteinase |
| MRE | microRNA recognition element |
| mRNA | Messenger RNA |
| NB | Neuroblastoma |
| NDRG2 | N-myc downstream regulated gene 2 |
| NF-κβ | Nuclear factor kappa B |
| NK | Natural Killer Cells |
| NLK | Nemo-like kinase |
| NP | Nanoparticle |
| NSCLC | Non-small cell lung cancer |
| nt | Nucleotides |
| PaCa | Pancreatic cancer |
| PAM | PI3K-Akt-mTOR signaling pathway |
| PARP1 | Poly[ADP-ribose] polymerase 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PGR | Progesterone Receptor |
| PI3K | phosphoinositide-3-kinase |
| PN | Pinostrobin |
| PolII | RNA polymerase II |
| PrCa | Prostate caner |
| pri-miRNA | Primary microRNA |
| PTC | Papillary thyroid cancer |
| RBP | RNA binding proteins |
| RISC | RNA-induced silencing complex |
| RTK | Receptor tyrosine kinase |
| RV | Resveratrol |
| SNP | Single nucleotide polymorphism |
| SNV | Single nucleotide variation |
| SRCIN1 | Src kinase signaling inhibitor 1 |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of interferon gene |
| SYNCRIP | Synaptotagmin-binding Cytoplasmic RNA-Interacting Protein |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TCGA | The Cancer Genome Atlas |
| TCR | T-cell receptor |
| TFs | Transcription factors |
| TGF-β | Canonical transforming growth factor-β |
| TKI | Tyrosine kinase inhibitor |
| T-LGL | T-cell large granular lymphocyte |
| TMBIM6 | Transmembrane Bax Inhibitor Motif-containing 6 |
| TME | Tumor microenvironment |
| TNBC | Triple negative breast cancer |
| T-PLL | T-cell prolymphocytic leukemia |
| TRIM5 | Tripartite Motif Containing 5 |
| UPR | Unfolded protein response |
| VEGF | Vascular endothelial growth factor |
| WIF1 | Wnt inhibitory factor 1 |
| XIST | X-inactivated specific transcript |
| YAP | Yes-associated protein |
References
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol Biol. 2017, 1509, 1–10. [Google Scholar] [PubMed]
- Okamura, K.; Hagen, J.W.; Duan, H.; et al. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007, 130, 89–100. [Google Scholar] [CrossRef]
- Pepe, F.; Pagotto, S.; Soliman, S.; et al. Regulation of miR-483-3p by the O-linked N-acetylglucosamine transferase links chemosensitivity to glucose metabolism in liver cancer cells. Oncogenesis. 2017, 6, e328. [Google Scholar] [CrossRef] [PubMed]
- Veronese, A.; Visone, R.; Consiglio, J.; et al. Mutated beta-catenin evades a microRNA-dependent regulatory loop. Proc Natl Acad Sci U S A. 2011, 108, 4840–5. [Google Scholar] [CrossRef] [PubMed]
- Bell-Hensley, A.; Das, S.; McAlinden, A. The miR-181 family: Wide-ranging pathophysiological effects on cell fate and function. J Cell Physiol. 2023, 238, 698–713. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116, 281–97. [Google Scholar] [CrossRef]
- Kamanu, T.K.; Radovanovic, A.; Archer, J.A.; et al. Exploration of miRNA families for hypotheses generation. Sci Rep. 2013, 3, 2940. [Google Scholar] [CrossRef]
- Olena, A.F.; Patton, J.G. Genomic organization of microRNAs. J Cell Physiol. 2010, 222, 540–5. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; De Giorgis, M.; et al. microRNAs Biogenesis, Functions and Role in Tumor Angiogenesis. Front Oncol. 2020, 10, 581007. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; et al. Vertebrate microRNA genes. Science. 2003, 299, 1540. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; et al. New microRNAs from mouse and human. RNA. 2003, 9, 175–9. [Google Scholar] [CrossRef] [PubMed]
- Dostie, J.; Mourelatos, Z.; Yang, M.; et al. Numerous microRNPs in neuronal cells containing novel microRNAs. RNA. 2003, 9, 180–6. [Google Scholar] [CrossRef]
- Presnell, S.R.; Al-Attar, A.; Cichocki, F.; et al. Human natural killer cell microRNA: differential expression of MIR181A1B1 and MIR181A2B2 genes encoding identical mature microRNAs. Genes Immun. 2015, 16, 89–98. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Verheyden, N.A.; Klostermann, M.; Brüggemann, M.; et al. A high-resolution map of functional miR-181 response elements in the thymus reveals the role of coding sequence targeting and an alternative seed match. Nucleic Acids Res. 2024 May 23.
- Wingo, A.P.; Almli, L.M.; Stevens, J.S.; et al. Genome-wide association study of positive emotion identifies a genetic variant and a role for microRNAs. Mol Psychiatry. 2017, 22, 774–783. [Google Scholar] [CrossRef]
- Zhong, C.; Yao, Q.; Han, J.; et al. SNP rs322931 (C>T) in miR-181b and rs7158663 (G>A) in MEG3 aggravate the inflammatory response of anal abscess in patients with Crohn's disease. Aging (Albany NY). 2022, 14, 3313–3324. [Google Scholar] [CrossRef] [PubMed]
- He, Y.L.; Yang, J.; Zeng, Z.N.; et al. Interaction of miR-181b and. Biomed Res Int. 2020, 2020, 4757065. [Google Scholar]
- Han, X.; Zheng, Z.; Wang, C.; et al. Association between MEG3/miR-181b polymorphisms and risk of ischemic stroke. Lipids Health Dis. 2018, 17, 292. [Google Scholar] [CrossRef]
- Li, S.; Zhu, P.; Wang, Y.; et al. miR-181a targets PTEN to mediate the neuronal injury caused by oxygen-glucose deprivation and reoxygenation. Metab Brain Dis. 2023, 38, 2077–2091. [Google Scholar] [CrossRef]
- Lancaster, T.M.; Ihssen, N.; Brindley, L.M.; et al. Further support for association between GWAS variant for positive emotion and reward systems. Transl Psychiatry. 2017, 7, e1018. [Google Scholar] [CrossRef] [PubMed]
- El-Korany, W.A.; Zahran, W.E.; Alm El-Din, M.A.; et al. Rs12039395 Variant Influences the Expression of hsa-miR-181a-5p and PTEN Toward Colorectal Cancer Risk. Dig Dis Sci. 2024 Jun 28.
- Liu, G.; Min, H.; Yue, S.; et al. Pre-miRNA loop nucleotides control the distinct activities of mir-181a-1 and mir-181c in early T cell development. PLoS One. 2008, 3, e3592. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Ciocan, C.; Bica, C.; et al. Artificial miRNAs derived from miR-181 family members have potential in cancer therapy due to an altered spectrum of target mRNAs. FEBS Lett. 2023, 597, 1989–2005. [Google Scholar] [CrossRef]
- Flynn, R.A.; Zhang, Q.C.; Spitale, R.C.; et al. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat Protoc. 2016, 11, 273–90. [Google Scholar] [CrossRef]
- Luo, Q.J.; Zhang, J.; Li, P.; et al. RNA structure probing reveals the structural basis of Dicer binding and cleavage. Nat Commun. 2021, 12, 3397. [Google Scholar] [CrossRef]
- Ravi, A.; Gurtan, A.M.; Kumar, M.S.; et al. Proliferation and tumorigenesis of a murine sarcoma cell line in the absence of DICER1. Cancer Cell. 2012, 21, 848–55. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci U S A. 2016, 113, E1881–9. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.Y.; Ta, M.; et al. Biallelic Dicer1 Mutations in the Gynecologic Tract of Mice Drive Lineage-Specific Development of DICER1 Syndrome-Associated Cancer. Cancer Res. 2023, 83, 3517–3528. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; et al. The biochemical basis of microRNA targeting efficacy. Science. 2019, 366. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lin, Y.C.; Cui, S.; et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Riccioni, V.; Trionfetti, F.; Montaldo, C.; et al. SYNCRIP Modulates the Epithelial-Mesenchymal Transition in Hepatocytes and HCC Cells. Int J Mol Sci. 2022, 23. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Gao, L.; et al. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death Differ. 2012, 19, 378–86. [Google Scholar] [CrossRef] [PubMed]
- Kunze-Schumacher, H.; Krueger, A. The Role of MicroRNAs in Development and Function of Regulatory T Cells - Lessons for a Better Understanding of MicroRNA Biology. Front Immunol. 2020, 11, 2185. [Google Scholar] [CrossRef]
- Indrieri, A.; Carrella, S.; Carotenuto, P.; et al. The Pervasive Role of the miR-181 Family in Development, Neurodegeneration, and Cancer. Int J Mol Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Passos Gibson, V.; Hardy, P. The Role of MiR-181 Family Members in Endothelial Cell Dysfunction and Tumor Angiogenesis. Cells. 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Patel-Hett, S.; D'Amore, P.A. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011, 55, 353–63. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, J.; Michael, M.Z.; Harvey, N.L. Prox1 expression is negatively regulated by miR-181 in endothelial cells. Blood. 2010, 116, 2395–401. [Google Scholar] [CrossRef] [PubMed]
- Carrella, S.; Barbato, S.; D'Agostino, Y.; et al. TGF-β Controls miR-181/ERK Regulatory Network during Retinal Axon Specification and Growth. PLoS One. 2015, 10, e0144129. [Google Scholar] [CrossRef]
- Bhushan, R.; Grünhagen, J.; Becker, J.; et al. miR-181a promotes osteoblastic differentiation through repression of TGF-β signaling molecules. Int J Biochem Cell Biol. 2013, 45, 696–705. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, Y.; Xu, M.Q.; et al. miR-181a regulate porcine preadipocyte differentiation by targeting TGFBR1. Gene. 2019, 681, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Knarr, M.; Nagaraj, A.B.; Kwiatkowski, L.J.; et al. miR-181a modulates circadian rhythm in immortalized bone marrow and adipose derived stromal cells and promotes differentiation through the regulation of PER3. Sci Rep. 2019, 9, 307. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, D.; Xu, L.; Zhang, L.; et al. MiR-181a-5p regulates 3T3-L1 cell adipogenesis by targeting Smad7 and Tcf7l2. Acta Biochim Biophys Sin (Shanghai). 2016, 48, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, J.; Tycksen, E.; et al. MicroRNA-181a/b-1 over-expression enhances osteogenesis by modulating PTEN/PI3K/AKT signaling and mitochondrial metabolism. Bone. 2019, 123, 92–102. [Google Scholar] [CrossRef]
- Kim, C.; Ye, Z.; Weyand, C.M.; et al. miR-181a-regulated pathways in T-cell differentiation and aging. Immun Ageing. 2021, 18, 28. [Google Scholar] [CrossRef]
- Li, Q.J.; Chau, J.; Ebert, P.J.; et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007, 129, 147–61. [Google Scholar] [CrossRef] [PubMed]
- Gordino, G.; Costa-Pereira, S.; Corredeira, P.; et al. MicroRNA-181a restricts human γδ T cell differentiation by targeting Map3k2 and Notch2. EMBO Rep. 2022, 23, e52234. [Google Scholar] [CrossRef]
- Cichocki, F.; Felices, M.; McCullar, V.; et al. Cutting edge: microRNA-181 promotes human NK cell development by regulating Notch signaling. J Immunol. 2011, 187, 6171–5. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, S.; Li, X.; et al. Declined miR-181a-5p expression is associated with impaired natural killer cell development and function with aging. Aging Cell. 2021, 20, e13353. [Google Scholar] [CrossRef]
- Rady, M.; Watzl, C.; Claus, M.; et al. Altered expression of miR-181a and miR-146a does not change the expression of surface NCRs in human NK cells. Sci Rep. 2017, 7, 41381. [Google Scholar] [CrossRef]
- Su, R.; Lin, H.S.; Zhang, X.H.; et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene. 2015, 34, 3226–39. [Google Scholar] [CrossRef]
- Rezaei, T.; Hejazi, M.; Mansoori, B.; et al. microRNA-181a mediates the chemo-sensitivity of glioblastoma to carmustine and regulates cell proliferation, migration, and apoptosis. Eur J Pharmacol. 2020, 888, 173483. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cheng, Z.; Zhang, J.; et al. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008, 1236, 185–93. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Li, J.; Yun, X.; et al. miR-181a-5p, an inducer of Wnt-signaling, facilitates cell proliferation in acute lymphoblastic leukemia. Oncol Rep. 2017, 37, 1469–1476. [Google Scholar] [CrossRef]
- Simiene, J.; Dabkeviciene, D.; Stanciute, D.; et al. Potential of miR-181a-5p and miR-630 as clinical biomarkers in NSCLC. BMC Cancer. 2023, 23, 857. [Google Scholar] [CrossRef]
- Papadaki, C.; Monastirioti, A.; Rounis, K.; et al. Circulating MicroRNAs Regulating DNA Damage Response and Responsiveness to Cisplatin in the Prognosis of Patients with Non-Small Cell Lung Cancer Treated with First-Line Platinum Chemotherapy. Cancers (Basel). 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Barbato, A.; Iuliano, A.; Volpe, M.; et al. Integrated Genomics Identifies miR-181/TFAM Pathway as a Critical Driver of Drug Resistance in Melanoma. Int J Mol Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Guo, S.; Tong, S.; et al. Identification of Keratinocyte Differentiation-Involved Genes for Metastatic Melanoma by Gene Expression Profiles. Comput Math Methods Med. 2021, 2021, 9652768. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Bae, S.D.; Hong, H.S.; et al. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem Biophys Res Commun. 2011, 404, 896–902. [Google Scholar] [CrossRef]
- Ouyang, M.; Liu, G.; Xiong, C.; et al. microRNA-181a-5p impedes the proliferation, migration, and invasion of retinoblastoma cells by targeting the NRAS proto-oncogene. Clinics (Sao Paulo). 2022, 77, 100026. [Google Scholar] [CrossRef]
- Xu, P.; Zhou, D.; Yan, G.; et al. Correlation of miR-181a and three HOXA genes as useful biomarkers in acute myeloid leukemia. Int J Lab Hematol. 2020, 42, 16–22. [Google Scholar] [CrossRef]
- Shadbad, M.A.; Baradaran, B. hsa-miR-181a-5p inhibits glioblastoma development via the MAPK pathway: in-silico and in-vitro study. Oncology Research.
- Király, J.; Szabó, E.; Fodor, P.; et al. Expression of hsa-miRNA-15b, -99b, -181a and Their Relationship to Angiogenesis in Renal Cell Carcinoma. Biomedicines. 2024. [CrossRef]
- Zhou, W.Y.; Chen, J.C.; Jiao, T.T.; et al. MicroRNA-181 targets Yin Yang 1 expression and inhibits cervical cancer progression. Mol Med Rep. 2015, 11, 4541–6. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, X.; Wu, Z.; et al. DNA methylation-mediated repression of miR-181a/135a/302c expression promotes the microsatellite-unstable colorectal cancer development and 5-FU resistance via targeting PLAG1. J Genet Genomics. 2018, 45, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hallis, S.P.; Kim, S.K.; Lee, J.H.; et al. Association of NRF2 with HIF-2α-induced cancer stem cell phenotypes in chronic hypoxic condition. Redox Biol. 2023, 60, 102632. [Google Scholar] [CrossRef] [PubMed]
- Kozloski, G.A.; Jiang, X.; Bhatt, S.; et al. miR-181a negatively regulates NF-κB signaling and affects activated B-cell-like diffuse large B-cell lymphoma pathogenesis. Blood. 2016, 127, 2856–66. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.; Lamberti, J.; Saggese, P.; et al. Small Non-Coding RNA Profiling Identifies miR-181a-5p as a Mediator of Estrogen Receptor Beta-Induced Inhibition of Cholesterol Biosynthesis in Triple-Negative Breast Cancer. Cells. 2020, 9. [Google Scholar] [CrossRef]
- Benedetti, R.; Papulino, C.; Sgueglia, G.; et al. Regulatory Interplay between miR-181a-5p and Estrogen Receptor Signaling Cascade in Breast Cancer. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Bustos, M.A.; Yokoe, T.; Shoji, Y.; et al. MiR-181a targets STING to drive PARP inhibitor resistance in BRCA- mutated triple-negative breast cancer and ovarian cancer. Cell Biosci. 2023, 13, 200. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.; Zhang, R.; et al. Cancer exosome-derived miR-9 and miR-181a promote the development of early-stage MDSCs via interfering with SOCS3 and PIAS3 respectively in breast cancer. Oncogene. 2020, 39, 4681–4694. [Google Scholar] [CrossRef]
- Fouladi, H.; Ebrahimi, A.; Derakhshan, S.M.; et al. Over-expression of mir-181a-3p in serum of breast cancer patients as diagnostic biomarker. Mol Biol Rep. 2024, 51, 372. [Google Scholar] [CrossRef]
- Zhai, Z.; Mu, T.; Zhao, L.; et al. MiR-181a-5p facilitates proliferation, invasion, and glycolysis of breast cancer through NDRG2-mediated activation of PTEN/AKT pathway. Bioengineered. 2022, 13, 83–95. [Google Scholar] [CrossRef]
- Lu, C.; Lin, S.; Wen, Z.; et al. Testing the accuracy of a four serum microRNA panel for the detection of primary bladder cancer: a discovery and validation study. Biomarkers. 2024, 29, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Lee, C.; Joseph, P.; et al. microRNA-181a has a critical role in ovarian cancer progression through the regulation of the epithelial-mesenchymal transition. Nat Commun. 2014, 5, 2977. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Zannoni, G.F.; Beltrame, L.; et al. Identification of high-grade serous ovarian cancer miRNA species associated with survival and drug response in patients receiving neoadjuvant chemotherapy: a retrospective longitudinal analysis using matched tumor biopsies. Ann Oncol. 2016, 27, 625–34. [Google Scholar] [CrossRef] [PubMed]
- Panoutsopoulou, K.; Avgeris, M.; Magkou, P.; et al. miR-181a overexpression predicts the poor treatment response and early-progression of serous ovarian cancer patients. Int J Cancer. 2020, 147, 3560–3573. [Google Scholar] [CrossRef]
- Zuo, L.; Li, X.; Zhu, H.; et al. Expression of miR-181a in Circulating Tumor Cells of Ovarian Cancer and Its Clinical Application. ACS Omega. 2021, 6, 22011–22019. [Google Scholar] [CrossRef] [PubMed]
- Geletina, N.S.; Kobelev, V.S.; Babayants, E.V.; et al. PTEN negative correlates with miR-181a in tumour tissues of non-obese endometrial cancer patients. Gene. 2018, 655, 20–24. [Google Scholar] [CrossRef]
- Ji, D.; Chen, Z.; Li, M.; et al. MicroRNA-181a promotes tumor growth and liver metastasis in colorectal cancer by targeting the tumor suppressor WIF-1. Mol Cancer. 2014, 13, 86. [Google Scholar] [CrossRef]
- Zhao, S.; Mi, Y.; Zheng, B.; et al. Highly-metastatic colorectal cancer cell released miR-181a-5p-rich extracellular vesicles promote liver metastasis by activating hepatic stellate cells and remodelling the tumour microenvironment. J Extracell Vesicles. 2022, 11, e12186. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, J.; Handa, R.; Yamamoto, H.; et al. microRNA-181a is associated with poor prognosis of colorectal cancer. Oncol Rep. 2012, 28, 2221–6. [Google Scholar] [CrossRef]
- Pichler, M.; Winter, E.; Ress, A.L.; et al. miR-181a is associated with poor clinical outcome in patients with colorectal cancer treated with EGFR inhibitor. J Clin Pathol. 2014, 67, 198–203. [Google Scholar] [CrossRef]
- Li, J.; Xu, X.; Liu, C.; et al. miR-181a-2-3p Stimulates Gastric Cancer Progression. Front Bioeng Biotechnol. 2021, 9, 687915. [Google Scholar]
- Mi, Y.; Zhang, D.; Jiang, W.; et al. miR-181a-5p promotes the progression of gastric cancer via RASSF6-mediated MAPK signalling activation. Cancer Lett. 2017, 389, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, J. Ascites-derived hsa-miR-181a-5p serves as a prognostic marker for gastric cancer-associated malignant ascites. BMC Genomics. 2024, 25, 628. [Google Scholar] [CrossRef]
- Xue, W.X.; Zhang, M.Y. ; Rui Li, et al. Serum miR-1228-3p and miR-181a-5p as Noninvasive Biomarkers for Non-Small Cell Lung Cancer Diagnosis and Prognosis. Biomed Res Int. 2020, 2020, 9601876. [Google Scholar]
- Chen, X.; Dong, D.; Pan, C.; et al. Identification of Grade-associated MicroRNAs in Brainstem Gliomas Based on Microarray Data. J Cancer. 2018, 9, 4463–4476. [Google Scholar] [CrossRef] [PubMed]
- Qiang, P.; Pan, Q.; Fang, C.; et al. MicroRNA-181a-3p as a Diagnostic and Prognostic Biomarker for Acute Myeloid Leukemia. Mediterr J Hematol Infect Dis. 2020, 12, e2020012. [Google Scholar]
- Debernardi, S.; Skoulakis, S.; Molloy, G.; et al. MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia. 2007, 21, 912–6. [Google Scholar] [CrossRef]
- Haque, S.; Vaiselbuh, S.R. Vincristine and prednisone regulates cellular and exosomal miR-181a expression differently within the first time diagnosed and the relapsed leukemia B cells. Leuk Res Rep. 2020, 14, 100221. [Google Scholar] [CrossRef]
- Haque, S.; Vaiselbuh, S.R. Silencing of Exosomal miR-181a Reverses Pediatric Acute Lymphocytic Leukemia Cell Proliferation. Pharmaceuticals (Basel). 2020, 13. [Google Scholar] [CrossRef]
- Zhu, D.X.; Zhu, W.; Fang, C.; et al. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis. 2012, 33, 1294–301. [Google Scholar] [CrossRef]
- Lee, E.J.; Gusev, Y.; Jiang, J.; et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007, 120, 1046–54. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Chen, B.; Wang, X.; et al. Long non-coding RNA XIST regulates PTEN expression by sponging miR-181a and promotes hepatocellular carcinoma progression. BMC Cancer. 2017, 17, 248. [Google Scholar] [CrossRef]
- Le, F.; Luo, P.; Yang, Q.O.; et al. MiR-181a promotes growth of thyroid cancer cells by targeting tumor suppressor RB1. Eur Rev Med Pharmacol Sci. 2017, 21, 5638–5647. [Google Scholar]
- Knarr, M.; Avelar, R.A.; Sekhar, S.C.; et al. miR-181a initiates and perpetuates oncogenic transformation through the regulation of innate immune signaling. Nat Commun. 2020, 11, 3231. [Google Scholar] [CrossRef]
- Wiechert, A.; Saygin, C.; Thiagarajan, P.S.; et al. Cisplatin induces stemness in ovarian cancer. Oncotarget. 2016, 7, 30511–22. [Google Scholar] [CrossRef] [PubMed]
- Belur Nagaraj, A.; Joseph, P.; Ponting, E.; et al. A miRNA-Mediated Approach to Dissect the Complexity of Tumor-Initiating Cell Function and Identify miRNA-Targeting Drugs. Stem Cell Reports. 2019, 12, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Belur Nagaraj, A.; Knarr, M.; Sekhar, S.; et al. The miR-181a-SFRP4 Axis Regulates Wnt Activation to Drive Stemness and Platinum Resistance in Ovarian Cancer. Cancer Res. 2021, 81, 2044–2055. [Google Scholar] [CrossRef]
- Perepechaeva, M.L.; Studenikina, A.A.; Grishanova, A.Y.; et al. Serum miR-181a and miR-25 in patients with malignant and benign breast diseases. Biomed Khim. 2023, 69, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Ma, X.; Zhang, N.; et al. Targeting Oncogenic miR-181a-2-3p Inhibits Growth and Suppresses Cisplatin Resistance of Gastric Cancer. Cancer Manag Res. 2021, 13, 8599–8609. [Google Scholar] [CrossRef]
- Bisso, A.; Faleschini, M.; Zampa, F.; et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell Cycle. 2013, 12, 1679–87. [Google Scholar] [CrossRef]
- Kanaan, Z.; Rai, S.N.; Eichenberger, M.R.; et al. Differential microRNA expression tracks neoplastic progression in inflammatory bowel disease-associated colorectal cancer. Hum Mutat. 2012, 33, 551–60. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Feng, M.; Yin, Z.; et al. RalA, a GTPase targeted by miR-181a, promotes transformation and progression by activating the Ras-related signaling pathway in chronic myelogenous leukemia. Oncotarget. 2016, 7, 20561–73. [Google Scholar] [CrossRef]
- De Marco, C.; Zoppoli, P.; Rinaldo, N.; et al. Genome-wide analysis of copy number alterations led to the characterisation of PDCD10 as oncogene in ovarian cancer. Transl Oncol. 2021, 14, 101013. [Google Scholar] [CrossRef]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin Cancer Res. 2012, 18, 5806–15. [Google Scholar] [CrossRef]
- Dimova, I.; Orsetti, B.; Negre, V.; et al. Genomic markers for ovarian cancer at chromosomes 1, 8 and 17 revealed by array CGH analysis. Tumori. 2009, 95, 357–66. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Musto, P.; Di Martino, M.T.; et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin Cancer Res. 2013, 19, 3130–42. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Zhou, R.; et al. Gene silencing of MIR22 in acute lymphoblastic leukaemia involves histone modifications independent of promoter DNA methylation. Br J Haematol. 2010, 148, 69–79. [Google Scholar] [CrossRef]
- Ali, S.R.; Humphreys, K.J.; Simpson, K.J.; et al. Functional high-throughput screen identifies microRNAs that promote butyrate-induced death in colorectal cancer cells. Mol Ther Nucleic Acids. 2022, 30, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, X.; Zhao, M.; et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 2014, 5, 3127. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006, 441, 349–53. [Google Scholar] [CrossRef]
- Cao, Q.; Mani, R.S.; Ateeq, B.; et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011, 20, 187–99. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Purkait, S.; Takkar, S.; et al. Analysis of EZH2: micro-RNA network in low and high grade astrocytic tumors. Brain Tumor Pathol. 2016, 33, 117–28. [Google Scholar] [CrossRef] [PubMed]
- Morales, S.; Monzo, M.; Navarro, A. Epigenetic regulation mechanisms of microRNA expression. Biomol Concepts 2017, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008, 105, 13556–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, Y.L.; Zhang, P.; et al. SP1 mediates the link between methylation of the tumour suppressor miR-149 and outcome in colorectal cancer. J Pathol. 2013, 229, 12–24. [Google Scholar] [CrossRef]
- Dai, E.; Yu, X.; Zhang, Y.; et al. EpimiR: a database of curated mutual regulation between miRNAs and epigenetic modifications. Database (Oxford). 2014, 2014, bau023. [Google Scholar] [CrossRef]
- Konno, M.; Koseki, J.; Asai, A.; et al. Distinct methylation levels of mature microRNAs in gastrointestinal cancers. Nat Commun. 2019, 10, 3888. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, T.; Du, Y.; et al. LncRNA LUCAT1/miR-181a-5p axis promotes proliferation and invasion of breast cancer via targeting KLF6 and KLF15. BMC Mol Cell Biol. 2020, 21, 69. [Google Scholar] [CrossRef]
- Sun, C.; Shen, C.; Zhang, Y.; et al. LncRNA ANRIL negatively regulated chitooligosaccharide-induced radiosensitivity in colon cancer cells by sponging miR-181a-5p. Adv Clin Exp Med. 2021, 30, 55–65. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L. SNHG7 Contributes to the Progression of Non-Small-Cell Lung Cancer via the SNHG7/miR-181a-5p/E2F7 Axis. Cancer Manag Res. 2020, 12, 3211–3222. [Google Scholar] [CrossRef]
- Yuan, Y.; Long, Z. LncRNA LINC01232 Enhances Proliferation, Angiogenesis, Migration and Invasion of Colon Adenocarcinoma Cells by Downregulating miR-181a-5p. J Microbiol Biotechnol. 2023, 33, 398–409. [Google Scholar] [CrossRef]
- Shang, A.; Wang, W.; Gu, C.; et al. Long non-coding RNA CCAT1 promotes colorectal cancer progression by regulating miR-181a-5p expression. Aging (Albany NY). 2020, 12, 8301–8320. [Google Scholar] [CrossRef]
- Pan, D.; Chen, K.; Chen, P.; et al. Downregulation of LncRNA SNHG7 Sensitizes Colorectal Cancer Cells to Resist Anlotinib by Regulating miR-181a-5p/GATA6. Gastroenterol Res Pract. 2023, 2023, 6973723. [Google Scholar] [CrossRef]
- Yu, J.; Jiang, L.; Gao, Y.; et al. LncRNA CCAT1 negatively regulates miR-181a-5p to promote endometrial carcinoma cell proliferation and migration. Exp Ther Med. 2019, 17, 4259–4266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hua, M.; Zhang, N. LINC01232 promotes lung squamous cell carcinoma progression through modulating miR-181a-5p/SMAD2 axis. Am J Med Sci. 2023, 365, 386–395. [Google Scholar] [CrossRef]
- Khalaf, S.E.; Abdelfattah, S.N.; Hasona, N.A. Crosstalk Between Long Non-coding RNA MALAT1, miRNA-181a, and IL-17 in Cirrhotic Patients and Their Possible Correlation SIRT1/NF-Ƙβ Axis. Indian Journal of Clinical Biochemistry. 2024 2024/03/26.
- Ding, X.; Xu, X.; He, X.F.; et al. Muscleblind-like 1 antisense RNA 1 inhibits cell proliferation, invasion, and migration of prostate cancer by sponging miR-181a-5p and regulating PTEN/PI3K/AKT/mTOR signaling. Bioengineered. 2021, 12, 803–814. [Google Scholar] [CrossRef]
- Liao, Y.; Shen, L.; Zhao, H.; et al. LncRNA CASC2 Interacts With miR-181a to Modulate Glioma Growth and Resistance to TMZ Through PTEN Pathway. J Cell Biochem. 2017, 118, 1889–1899. [Google Scholar] [CrossRef]
- Lu, J.; Liu, X.; Cen, A.; et al. HYPOXIA induces lncRNA HOTAIR for recruiting RELA in papillary thyroid cancer cells to upregulate miR-181a and promote angiogenesis. J Endocrinol Invest. 2024 May 15.
- Brockhausen, J.; Tay, S.S.; Grzelak, C.A.; et al. miR-181a mediates TGF-β-induced hepatocyte EMT and is dysregulated in cirrhosis and hepatocellular cancer. Liver Int. 2015, 35, 240–53. [Google Scholar] [CrossRef]
- Wang, A.; Huang, Y.; Yang, X. TGF-β promotes proliferation and inhibits apoptosis of liver cancer Huh-7 cells by regulating MiR-182/CADM1. Tropical Journal of Pharmaceutical Research; 2023.
- Taylor, M.A.; Sossey-Alaoui, K.; Thompson, C.L.; et al. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013, 123, 150–63. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Y.; Tsuyada, A.; et al. Transforming growth factor-β regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene. 2011, 30, 1470–80. [Google Scholar] [CrossRef] [PubMed]
- Neel, J.C.; Lebrun, J.J. Activin and TGFβ regulate expression of the microRNA-181 family to promote cell migration and invasion in breast cancer cells. Cell Signal. 2013, 25, 1556–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, H.; Jiang, Y.; et al. MicroRNA-181a suppresses mouse granulosa cell proliferation by targeting activin receptor IIA. PLoS One. 2013, 8, e59667. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Yamashita, T.; Wang, X.W. Wnt/beta-catenin signaling activates microRNA-181 expression in hepatocellular carcinoma. Cell Biosci. 2011, 1, 4. [Google Scholar] [CrossRef]
- Ji, J.; Wang, X.W. New kids on the block: diagnostic and prognostic microRNAs in hepatocellular carcinoma. Cancer Biol Ther. 2009, 8, 1686–93. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Yamashita, T.; Budhu, A.; et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009, 50, 472–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lopez McDonald, M.C.; Kim, C.; et al. The complementary roles of STAT3 and STAT1 in cancer biology: insights into tumor pathogenesis and therapeutic strategies. Front Immunol. 2023, 14, 1265818. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.; Tan, F.; et al. STAT1 Inhibits MiR-181a Expression to Suppress Colorectal Cancer Cell Proliferation Through PTEN/Akt. J Cell Biochem. 2017, 118, 3435–3443. [Google Scholar] [CrossRef]
- Zhu, J.; Yao, K.; Guo, J.; et al. miR-181a and miR-150 regulate dendritic cell immune inflammatory responses and cardiomyocyte apoptosis via targeting JAK1-STAT1/c-Fos pathway. J Cell Mol Med. 2017, 21, 2884–2895. [Google Scholar] [CrossRef]
- Belotte, J.; Fletcher, N.M.; Alexis, M.; et al. Sox2 gene amplification significantly impacts overall survival in serous epithelial ovarian cancer. Reprod Sci. 2015, 22, 38–46. [Google Scholar] [CrossRef]
- Lu, Y.; Futtner, C.; Rock, J.R.; et al. Evidence that SOX2 overexpression is oncogenic in the lung. PLoS One. 2010, 5, e11022. [Google Scholar] [CrossRef]
- Liu, K.; Xie, F.; Gao, A.; et al. SOX2 regulates multiple malignant processes of breast cancer development through the SOX2/miR-181a-5p, miR-30e-5p/TUSC3 axis. Mol Cancer. 2017, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Vaňhara, P.; Horak, P.; Pils, D.; et al. Loss of the oligosaccharyl transferase subunit TUSC3 promotes proliferation and migration of ovarian cancer cells. Int J Oncol. 2013, 42, 1383–9. [Google Scholar] [CrossRef]
- Pils, D.; Horak, P.; Vanhara, P.; et al. Methylation status of TUSC3 is a prognostic factor in ovarian cancer. Cancer. 2013, 119, 946–54. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Edmiston, S.N.; Tse, C.K.; et al. Racial variation in breast tumor promoter methylation in the Carolina Breast Cancer Study. Cancer Epidemiol Biomarkers Prev. 2015, 24, 921–30. [Google Scholar] [CrossRef]
- Tian, Y.; Xiao, X.; Gong, X.; et al. HBx promotes cell proliferation by disturbing the cross-talk between miR-181a and PTEN. Sci Rep. 2017, 7, 40089. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFbeta in Cancer. Cell. 2008, 134, 215–30. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, T.; Amini, M.; Hashemi, Z.S.; et al. microRNA-181 serves as a dual-role regulator in the development of human cancers. Free Radic Biol Med. 2020, 152, 432–454. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Li, Y.; Ye, S.; et al. MicroRNA profiling implies new markers of chemoresistance of triple-negative breast cancer. PLoS One. 2014, 9, e96228. [Google Scholar] [CrossRef]
- Autenshlyus, A.I.; Perepechaeva, M.L.; Studenikina, A.A.; et al. Serum miR-181а and miR-25 Levels in Patients with Breast Cancer or Benign Breast Disease. Dokl Biochem Biophys. 2023, 512, 279–283. [Google Scholar] [CrossRef]
- Yang, C.; Tabatabaei, S.N.; Ruan, X.; et al. The Dual Regulatory Role of MiR-181a in Breast Cancer. Cell Physiol Biochem. 2017, 44, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Azadeh, M.; Salehzadeh, A.; Ghaedi, K.; et al. Integrated High-Throughput Bioinformatics (Microarray, RNA-Seq, and RNA Interaction) and qRT-PCR Investigation of. Adv Biomed Res. 2023, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhou, W.; Cheng, C.T.; et al. TGFβ induces "BRCAness" and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol Cancer Res. 2014, 12, 1597–609. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhou, X.M.; Li, Y.S.; et al. MicroRNA-181a promotes epithelial-mesenchymal transition in esophageal squamous cell carcinoma via the TGF-β/Smad pathway. Mol Med Rep. 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhiping, C.; Shijun, T.; Linhui, W.; et al. MiR-181a promotes epithelial to mesenchymal transition of prostate cancer cells by targeting TGIF2. Eur Rev Med Pharmacol Sci. 2017, 21, 4835–4843. [Google Scholar]
- Verma, S.; Pandey, M.; Shukla, G.C.; et al. Integrated analysis of miRNA landscape and cellular networking pathways in stage-specific prostate cancer. PLoS One. 2019, 14, e0224071. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.G.; Zheng, J.F.; Li, Q.; et al. MicroRNA-181a-5p suppresses cell proliferation by targeting Egr1 and inhibiting Egr1/TGF-β/Smad pathway in hepatocellular carcinoma. Int J Biochem Cell Biol. 2019, 106, 107–116. [Google Scholar] [CrossRef]
- Wang, C.; Ji, J.; Jin, Y.; et al. Tumor-mesothelium HOXA11-PDGF BB/TGF β1-miR-181a-5p-Egr1 feedforward amplifier circuity propels mesothelial fibrosis and peritoneal metastasis of gastric cancer. Oncogene. 2023 Nov 21.
- Erkeland, S.J.; Stavast, C.J.; Schilperoord-Vermeulen, J.; et al. The miR-200c/141-ZEB2-TGFβ axis is aberrant in human T-cell prolymphocytic leukemia. Haematologica. 2022, 107, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kang, D.; Kwon, M.Y.; et al. MicroRNAs as potential indicators of the development and progression of uterine leiomyoma. PLoS One. 2022, 17, e0268793. [Google Scholar] [CrossRef]
- Pereira TDSF, Brito, J. A.R.; Guimarães, A.L.S.; et al. MicroRNA profiling reveals dysregulated microRNAs and their target gene regulatory networks in cemento-ossifying fibroma. J Oral Pathol Med. 2018, 47, 78–85.
- Meng, F.; Glaser, S.S.; Francis, H.; et al. Functional analysis of microRNAs in human hepatocellular cancer stem cells. J Cell Mol Med. 2012, 16, 160–73. [Google Scholar] [CrossRef]
- Han, P.; Li, J.W.; Zhang, B.M.; et al. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR-181a-5p-mediated regulation of Wnt/β-catenin signaling. Mol Cancer. 2017, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Z.; Fu, B.; et al. GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS One. 2011, 6, e22129. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009, 66, 1631–46. [Google Scholar] [CrossRef] [PubMed]
- Xiu, M.X.; Liu, Y.M. The role of oncogenic Notch2 signaling in cancer: a novel therapeutic target. Am J Cancer Res. 2019, 9, 837–854. [Google Scholar] [PubMed]
- Huang, S.X.; Zhao, Z.Y.; Weng, G.H.; et al. Upregulation of miR-181a suppresses the formation of glioblastoma stem cells by targeting the Notch2 oncogene and correlates with good prognosis in patients with glioblastoma multiforme. Biochem Biophys Res Commun. 2017, 486, 1129–1136. [Google Scholar] [CrossRef]
- Fragoso, R.; Mao, T.; Wang, S.; et al. Modulating the strength and threshold of NOTCH oncogenic signals by mir-181a-1/b-1. PLoS Genet. 2012, 8, e1002855. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023, 22, 138. [Google Scholar] [CrossRef]
- Wei, Z.; Cui, L.; Mei, Z.; et al. miR-181a mediates metabolic shift in colon cancer cells via the PTEN/AKT pathway. FEBS Lett. 2014, 588, 1773–9. [Google Scholar] [CrossRef]
- Xie, X.H.; Jiang, M.; Xiong, Z.A.; et al. Upregulation of miR-181a-5p and miR-125b-2-3p in the Maternal Circulation of Fetuses with Rh-Negative Hemolytic Disease of the Fetus and Newborn Could Be Related to Dysfunction of Placental Function. Dis Markers. 2022, 2022, 2594091. [Google Scholar] [CrossRef]
- Strotbek, M.; Schmid, S.; Sánchez-González, I.; et al. miR-181 elevates Akt signaling by co-targeting PHLPP2 and INPP4B phosphatases in luminal breast cancer. Int J Cancer. 2017, 140, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Mei, H.; Luo, Q.; et al. TCL1A acts as a tumour suppressor by modulating gastric cancer autophagy via miR-181a-5p-TCL1A-Akt/mTOR-c-MYC loop. Carcinogenesis. 2023, 44, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guan, Y.; Zheng, X.; et al. Hypoxia-induced miR-181a-5p up-regulation reduces epirubicin sensitivity in breast cancer cells through inhibiting EPDR1/TRPC1 to activate PI3K/AKT signaling pathway. BMC Cancer. 2024, 24, 167. [Google Scholar] [CrossRef]
- Ping, W.; Gao, Y.; Fan, X.; et al. MiR-181a contributes gefitinib resistance in non-small cell lung cancer cells by targeting GAS7. Biochem Biophys Res Commun. 2018, 495, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.X.; Zheng, Z.; Xue, W.; et al. MicroRNA181a Is Overexpressed in T-Cell Leukemia/Lymphoma and Related to Chemoresistance. Biomed Res Int. 2015, 2015, 197241. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Wang, Y.; Houda, T.; et al. MicroRNA-181a suppresses norethisterone-promoted tumorigenesis of breast epithelial MCF10A cells through the PGRMC1/EGFR-PI3K/Akt/mTOR signaling pathway. Transl Oncol. 2021, 14, 101068. [Google Scholar] [CrossRef]
- Zarrabi, K.; Dufour, A.; Li, J.; et al. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem. 2011, 286, 33167–77. [Google Scholar] [CrossRef]
- Li, Y.; Kuscu, C.; Banach, A.; et al. miR-181a-5p Inhibits Cancer Cell Migration and Angiogenesis via Downregulation of Matrix Metalloproteinase-14. Cancer Res. 2015, 75, 2674–85. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef]
- Huang, X.; Schwind, S.; Santhanam, R.; et al. Targeting the RAS/MAPK pathway with miR-181a in acute myeloid leukemia. Oncotarget. 2016, 7, 59273–59286. [Google Scholar] [CrossRef]
- Ma, Z.; Qiu, X.; Wang, D.; et al. MiR-181a-5p inhibits cell proliferation and migration by targeting Kras in non-small cell lung cancer A549 cells. Acta Biochim Biophys Sin (Shanghai). 2015, 47, 630–8. [Google Scholar] [CrossRef]
- Fei, J.; Li, Y.; Zhu, X.; et al. miR-181a post-transcriptionally downregulates oncogenic RalA and contributes to growth inhibition and apoptosis in chronic myelogenous leukemia (CML). PLoS One. 2012, 7, e32834. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Choi, H.Y.; Kwak, Y.; et al. TMBIM6-mediated miR-181a expression regulates breast cancer cell migration and invasion via the MAPK/ERK signaling pathway. J Cancer. 2023, 14, 554–572. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Zheng, J.Y.; Du, J.J.; et al. What is the relationship among microRNA-181, epithelial cell-adhesion molecule (EpCAM) and beta-catenin in hepatic cancer stem cells. Hepatology. 2009, 50, 2047–8. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Kapanova, G.; Kussainov, A.Z.; et al. Regulation of RASSF by non-coding RNAs in different cancers: RASSFs as masterminds of their own destiny as tumor suppressors and oncogenes. Noncoding RNA Res. 2022, 7, 123–131. [Google Scholar] [CrossRef] [PubMed]
- New, L.; Han, J. The p38 MAP kinase pathway and its biological function. Trends Cardiovasc Med. 1998, 8, 220–8. [Google Scholar] [CrossRef]
- Pulido, R.; Lang, R. Dual Specificity Phosphatases: From Molecular Mechanisms to Biological Function. Int J Mol Sci. 2019, 20. [Google Scholar] [CrossRef]
- Okada, H.; Kohanbash, G.; Lotze, M.T. MicroRNAs in immune regulation--opportunities for cancer immunotherapy. Int J Biochem Cell Biol. 2010, 42, 1256–61. [Google Scholar] [CrossRef]
- Song, M.K.; Park, Y.K.; Ryu, J.C. Polycyclic aromatic hydrocarbon (PAH)-mediated upregulation of hepatic microRNA-181 family promotes cancer cell migration by targeting MAPK phosphatase-5, regulating the activation of p38 MAPK. Toxicol Appl Pharmacol. 2013, 273, 130–9. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, A.; Smith, A.; Gillieron, C.; et al. MKP5, a new member of the MAP kinase phosphatase family, which selectively dephosphorylates stress-activated kinases. Oncogene. 1999, 18, 6981–8. [Google Scholar] [CrossRef]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005, 132, 3151–61. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhou, X.; Li, S.; et al. MicroRNA-181a suppresses salivary adenoid cystic carcinoma metastasis by targeting MAPK-Snai2 pathway. Biochim Biophys Acta. 2013, 1830, 5258–66. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Sui, X.; Weng, L.; et al. SNAIL1: Linking Tumor Metastasis to Immune Evasion. Front Immunol. 2021, 12, 724200. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Yan, F.; Ru, Y.; et al. MIIP inhibits EMT and cell invasion in prostate cancer through miR-181a/b-5p-KLF17 axis. Am J Cancer Res. 2020, 10, 630–647. [Google Scholar] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Ji, H.; et al. The interplay between noncoding RNA and YAP/TAZ signaling in cancers: molecular functions and mechanisms. J Exp Clin Cancer Res. 2022, 41, 202. [Google Scholar] [CrossRef]
- Wang, Y.; Cen, A.; Yang, Y.; et al. miR-181a, delivered by hypoxic PTC-secreted exosomes, inhibits DACT2 by downregulating MLL3, leading to YAP-VEGF-mediated angiogenesis. Mol Ther Nucleic Acids. 2021, 24, 610–621. [Google Scholar] [CrossRef]
- Chorzalska, A.; Kim, J.F.; Roder, K.; et al. Long-Term Exposure to Imatinib Mesylate Downregulates Hippo Pathway and Activates YAP in a Model of Chronic Myelogenous Leukemia. Stem Cells Dev. 2017, 26, 656–677. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, T.; Jia, Y.; et al. LncRNA MALAT1/miR-181a-5p affects the proliferation and adhesion of myeloma cells via regulation of Hippo-YAP signaling pathway. Cell Cycle. 2019, 18, 2509–2523. [Google Scholar] [CrossRef]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb Perspect Biol. 2012, 4. [Google Scholar] [CrossRef]
- Assmann JLJC, Leon, L. G.; Stavast, C.J.; et al. miR-181a is a novel player in the STAT3-mediated survival network of TCRαβ+ CD8+ T large granular lymphocyte leukemia. Leukemia. 2022, 36, 983–993.
- Schäfer, A.; Evers, L.; Meier, L.; et al. The Metalloprotease-Disintegrin ADAM8 Alters the Tumor Suppressor miR-181a-5p Expression Profile in Glioblastoma Thereby Contributing to Its Aggressiveness. Front Oncol. 2022, 12, 826273. [Google Scholar] [CrossRef] [PubMed]
- Khoo, L.T.; Chen, L.Y. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012, 5, ra20. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; et al. NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, X.; Wang, Z.; et al. NF-κB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep. 2023, 42, 112185. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhu, W.; Shi, D.; et al. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol Rep. 2010, 23, 997–1003. [Google Scholar] [PubMed]
- Hai Ping, P.; Feng Bo, T.; Li, L.; et al. IL-1β/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch Biochem Biophys. 2016, 604, 20–6. [Google Scholar] [CrossRef]
- Learn-Han N-SML, Sidik, S.; Rahman, S.; et al. miR-181a regulates multiple pathways in hypopharyngeal squamous cell carcinoma. African Journal of Biotechnology; 2014.
- Braicu, C.; Gulei, D.; Cojocneanu, R.; et al. miR-181a/b therapy in lung cancer: reality or myth? Mol Oncol. 2019, 13, 9–25. [Google Scholar] [CrossRef] [PubMed]
- de Paula Silva, E.; Marti, L.C.; Andreghetto, F.M.; et al. Extracellular vesicles cargo from head and neck cancer cell lines disrupt dendritic cells function and match plasma microRNAs. Sci Rep. 2021, 11, 18534. [Google Scholar] [CrossRef]
- Amado, T.; Amorim, A.; Enguita, F.J.; et al. MicroRNA-181a regulates IFN-γ expression in effector CD8. J Mol Med (Berl). 2020, 98, 309–320. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012, 12, 252–64. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef]
- Sun, W.; Wang, X.; Li, J.; et al. MicroRNA-181a promotes angiogenesis in colorectal cancer by targeting SRCIN1 to promote the SRC/VEGF signaling pathway. Cell Death Dis. 2018, 9, 438. [Google Scholar] [CrossRef]
- Chen, Y.; Song, W.; Gao, Y.; et al. Increased PD-L1 Expression in Acquired Cisplatin-Resistant Lung Cancer Cells via Mir-181a. Tohoku J Exp Med. 2022, 257, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.; Zhang, C.; Zhang, D.; et al. MicroRNA-181a, a potential diagnosis marker, alleviates acute graft versus host disease by regulating IFN-γ production. Am J Hematol. 2015, 90, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2019, 29, 3766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.X.; Gao, J.; Long, X.; et al. The circular RNA circHMGB2 drives immunosuppression and anti-PD-1 resistance in lung adenocarcinomas and squamous cell carcinomas via the miR-181a-5p/CARM1 axis. Mol Cancer. 2022, 21, 110. [Google Scholar] [CrossRef]
- . !!! INVALID CITATION !!! [102].
- Shi, Q.; Zhou, Z.; Ye, N.; et al. MiR-181a inhibits non-small cell lung cancer cell proliferation by targeting CDK1. Cancer Biomark. 2017, 20, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; He, X.; Li, F.; et al. The miR-181 family promotes cell cycle by targeting CTDSPL, a phosphatase-like tumor suppressor in uveal melanoma. J Exp Clin Cancer Res. 2018, 37, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yang, J.; Yuan, R.; et al. Effects of miR-181a on the biological function of multiple myeloma. Oncol Rep. 2019, 42, 291–300. [Google Scholar] [CrossRef]
- Liu, X.; Liao, W.; Peng, H.; et al. miR-181a promotes G1/S transition and cell proliferation in pediatric acute myeloid leukemia by targeting ATM. J Cancer Res Clin Oncol. 2016, 142, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Ma, X.; Li, H.; et al. Up-regulation of miR-181a in clear cell renal cell carcinoma is associated with lower KLF6 expression, enhanced cell proliferation, accelerated cell cycle transition, and diminished apoptosis. Urol Oncol. 2018, 36, 93.e23–93.e37. [Google Scholar] [CrossRef]
- Zhang, X.; Nie, Y.; Li, X.; et al. MicroRNA-181a functions as an oncomir in gastric cancer by targeting the tumour suppressor gene ATM. Pathol Oncol Res. 2014, 20, 381–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hui, L.; Xu, W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting BCL-2. Acta Biochim Biophys Sin (Shanghai). 2012, 44, 269–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, J.; Li, S.; et al. The function role of miR-181a in chemosensitivity to adriamycin by targeting Bcl-2 in low-invasive breast cancer cells. Cell Physiol Biochem. 2013, 32, 1225–37. [Google Scholar] [CrossRef] [PubMed]
- El Majzoub, R.; Fayyad-Kazan, M.; Nasr El Dine, A.; et al. A thiosemicarbazone derivative induces triple negative breast cancer cell apoptosis: possible role of miRNA-125a-5p and miRNA-181a-5p. Genes Genomics. 2019, 41, 1431–1443. [Google Scholar] [CrossRef]
- Zhang, S.; Wan, H.; Zhang, X. LncRNA LHFPL3-AS1 contributes to tumorigenesis of melanoma stem cells via the miR-181a-5p/BCL2 pathway. Cell Death Dis. 2020, 11, 950. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, P.; Zhou, X.; et al. MiR-181a enhances drug sensitivity of mixed lineage leukemia-rearranged acute myeloid leukemia by increasing poly(ADP-ribose) polymerase1 acetylation. Leuk Lymphoma. 2021, 62, 136–146. [Google Scholar] [CrossRef]
- Bräuer-Hartmann, D.; Hartmann, J.U.; Wurm, A.A.; et al. PML/RARα-Regulated miR-181a/b Cluster Targets the Tumor Suppressor RASSF1A in Acute Promyelocytic Leukemia. Cancer Res. 2015, 75, 3411–24. [Google Scholar] [CrossRef]
- Yu, J.; Qi, J.; Sun, X.; et al. MicroRNA-181a promotes cell proliferation and inhibits apoptosis in gastric cancer by targeting RASSF1A. Oncol Rep. 2018, 40, 1959–1970. [Google Scholar] [CrossRef]
- Gu, M.; Wang, L.; Yang, C.; et al. Micro-RNA-181a suppresses progestin-promoted breast cancer cell growth. Maturitas. 2018, 114, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Ota, D.; Mimori, K.; Yokobori, T.; et al. Identification of recurrence-related microRNAs in the bone marrow of breast cancer patients. Int J Oncol. 2011, 38, 955–62. [Google Scholar] [PubMed]
- Cmarik, J.L.; Min, H.; Hegamyer, G.; et al. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A. 1999, 96, 14037–42. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, H.; Li, Y.; et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012, 119, 2314–24. [Google Scholar] [CrossRef]
- Yang, M.; Zhai, X.; Ge, T.; et al. miR-181a-5p Promotes Proliferation and Invasion and Inhibits Apoptosis of Cervical Cancer Cells via Regulating Inositol Polyphosphate-5-Phosphatase A (INPP5A). Oncol Res. 2018, 26, 703–712. [Google Scholar] [CrossRef]
- Wang, X.; Huang, R.; Lu, Z.; et al. Exosomes from M1-polarized macrophages promote apoptosis in lung adenocarcinoma via the miR-181a-5p/ETS1/STK16 axis. Cancer Sci. 2022, 113, 986–1001. [Google Scholar] [CrossRef]
- Schenone, S.; Manetti, F.; Botta, M. SRC inhibitors and angiogenesis. Curr Pharm Des. 2007, 13, 2118–28. [Google Scholar] [CrossRef]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–58. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wei, L.; Chen, Q.; et al. MicroRNA regulates vascular endothelial growth factor expression in chondrosarcoma cells. Clin Orthop Relat Res. 2015, 473, 907–13. [Google Scholar] [CrossRef]
- Maillot, G.; Lacroix-Triki, M.; Pierredon, S.; et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009, 69, 8332–40. [Google Scholar] [CrossRef]
- Gilam, A.; Shai, A.; Ashkenazi, I.; et al. MicroRNA regulation of progesterone receptor in breast cancer. Oncotarget. 2017, 8, 25963–25976. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, Y.; Zhang, F.; et al. Liver-specific deletion of miR-181ab1 reduces liver tumour progression via upregulation of CBX7. Cell Mol Life Sci. 2022, 79, 443. [Google Scholar] [CrossRef] [PubMed]
- Paterson, M.R.; Jackson, K.L.; Dona, M.S.I.; et al. Deficiency of MicroRNA-181a Results in Transcriptome-Wide Cell-Specific Changes in the Kidney and Increases Blood Pressure. Hypertension. 2021, 78, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Williams, A.; Goff, L.A.; et al. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity. 2013, 38, 984–97. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Li, G.; Kim, C.; et al. Regulation of miR-181a expression in T cell aging. Nat Commun. 2018, 9, 3060. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, S.A.; Loh, C.; Wang, S.; et al. mir-181a-1/b-1 Modulates Tolerance through Opposing Activities in Selection and Peripheral T Cell Function. J Immunol. 2015, 195, 1470–9. [Google Scholar] [CrossRef]
- Łyszkiewicz, M.; Winter, S.J.; Witzlau, K.; et al. miR-181a/b-1 controls thymic selection of Treg cells and tunes their suppressive capacity. PLoS Biol. 2019, 17, e2006716. [Google Scholar] [CrossRef]
- Ziętara, N.; Łyszkiewicz, M.; Witzlau, K.; et al. Critical role for miR-181a/b-1 in agonist selection of invariant natural killer T cells. Proc Natl Acad Sci U S A. 2013, 110, 7407–12. [Google Scholar] [CrossRef]
- Valencia, K.; Erice, O.; Kostyrko, K.; et al. The Mir181ab1 cluster promotes KRAS-driven oncogenesis and progression in lung and pancreas. J Clin Invest. 2020, 130, 1879–1895. [Google Scholar] [CrossRef]
- Huang, X.; Schwind, S.; Yu, B.; et al. Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res. 2013, 19, 2355–67. [Google Scholar] [CrossRef]
- Tabatabaei, S.N.; Derbali, R.M.; Yang, C.; et al. Co-delivery of miR-181a and melphalan by lipid nanoparticles for treatment of seeded retinoblastoma. J Control Release. 2019, 298, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Passos Gibson, V.; Tahiri, H.; Yang, C.; et al. Hyaluronan decorated layer-by-layer assembled lipid nanoparticles for miR-181a delivery in glioblastoma treatment. Biomaterials. 2023, 302, 122341. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Yan, Q.; Li, Z.; et al. Multifunctional miR181a nanoparticles promote highly efficient radiotherapy for rectal cancer. Materials Today Advances 2022, 16, 100317. [Google Scholar] [CrossRef]
- Sun, X.; Chen, Y.; Yu, H.; et al. Anti-miRNA Oligonucleotide Therapy for Chondrosarcoma. Mol Cancer Ther. 2019, 18, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Norkaew, C.; Subkorn, P.; Chatupheeraphat, C.; et al. Pinostrobin, a fingerroot compound, regulates miR-181b-5p and induces acute leukemic cell apoptosis. Sci Rep. 2023, 13, 8084. [Google Scholar] [CrossRef]
- Wang, L.; Sun, T.; Yang, X.; et al. Astragaloside IV Overcomes Anlotinib Resistance in Non-small Cell Lung Cancer through miR-181a-3p/UPR-ERAD Axis. Curr Comput Aided Drug Des. 2024 Jan 17.
- Sharifi-Rad, J.; Rayess, Y.E.; Rizk, A.A.; et al. Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical, Biotechnological and Medicinal Applications. Front Pharmacol. 2020, 11, 01021. [Google Scholar] [CrossRef]
- Ravindran, F.; Mhatre, A.; Koroth, J.; et al. Curcumin modulates cell type-specific miRNA networks to induce cytotoxicity in ovarian cancer cells. Life Sci. 2023, 334, 122224. [Google Scholar] [CrossRef] [PubMed]
- Dewangan, J.; Tandon, D.; Srivastava, S.; et al. Novel combination of salinomycin and resveratrol synergistically enhances the anti-proliferative and pro-apoptotic effects on human breast cancer cells. Apoptosis. 2017, 22, 1246–1259. [Google Scholar] [CrossRef]
- Li, W.; Ma, J.; Ma, Q.; et al. Resveratrol inhibits the epithelial-mesenchymal transition of pancreatic cancer cells via suppression of the PI-3K/Akt/NF-κB pathway. Curr Med Chem. 2013, 20, 4185–4194. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Y. ; Ong'achwa MJ, et al. Resveratrol Inhibits the TGF-. Biomed Res Int. 2018, 2018, 8730593. [Google Scholar]
- Xue, H.Y.; Liu, M.W.; Yang, G. Resveratrol suppresses lipopolysaccharide-mediated activation of osteoclast precursor RAW 264. 7 cells by increasing miR-181a-5p expression. Int J Immunopathol Pharmacol. 2023, 37, 3946320231154995. [Google Scholar] [PubMed]
- Latson, G.W. Perftoran: History, Clinical Trials, and Pathway Forward. In: Liu, H.; Kaye, A.D.; Jahr, J.S.; editors. Blood Substitutes and Oxygen Biotherapeutics. Cham: Springer International Publishing; 2022. p. 361-367.
- Gamal-Eldeen, A.M.; Alrehaili, A.A.; Alharthi, A.; et al. Effect of Combined Perftoran and Indocyanine Green-Photodynamic Therapy on HypoxamiRs and OncomiRs in Lung Cancer Cells. Front Pharmacol. 2022, 13, 844104. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Liang, L.; Yang, J.M.; et al. MiR-181a confers resistance of cervical cancer to radiation therapy through targeting the pro-apoptotic PRKCD gene. Oncogene. 2013, 32, 3019–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ke, G.; Han, D.; et al. MicroRNA-181a enhances the chemoresistance of human cervical squamous cell carcinoma to cisplatin by targeting PRKCD. Exp Cell Res. 2014, 320, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Seyhan, A.A. Trials and Tribulations of MicroRNA Therapeutics. Int J Mol Sci. 2024, 25. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



