Submitted:
13 May 2025
Posted:
13 May 2025
You are already at the latest version
Abstract
Myalgic Encephalomyelitis (ME), sometimes known as Chronic Fatigue Syndrome (CFS), is a complex illness marked by fatigue, post-exertional malaise, cognitive dysfunction, autonomic instability, and immune abnormalities. This paper proposes a unifying hypothesis centred on dysfunction of lecithin–cholesterol acyltransferase (LCAT) and deficiency of phosphatidylcholine (PC), leading to altered membrane lipid composition, excess arachidonic acid release, and chronic inflammation. These changes may promote dysregulated production of prostaglandins and leukotrienes, imbalance in PPAR-γ activity, and disruptions in sex hormone metabolism, potentially contributing to conditions like polycystic ovary syndrome (PCOS), endometriosis and mast cell activation syndrome (MCAS). We propose that different combinations of LCAT dysfunction and PC deficiency result in distinct physiological subtypes. These include a neuronal insulin-hypersensitive subtype with reduced norepinephrine transporter (NET) expression and high extracellular norepinephrine; a high membrane arachidonic acid subtype with preserved PC but increased NET expression and neuronal insulin resistance; and a third group characterised by both elevated norepinephrine and downregulated β₂-adrenergic receptors. Each subtype presents with a different pattern of autonomic, metabolic, and immune dysregulation. Additional variation may arise from differences in acetylcholine synthesis, norepinephrine production capacity, and feedback mechanisms within the adrenergic system. This framework helps explain the heterogeneity of ME and offers a biologically coherent basis for further investigation into its pathophysiology.
Keywords:
Introduction
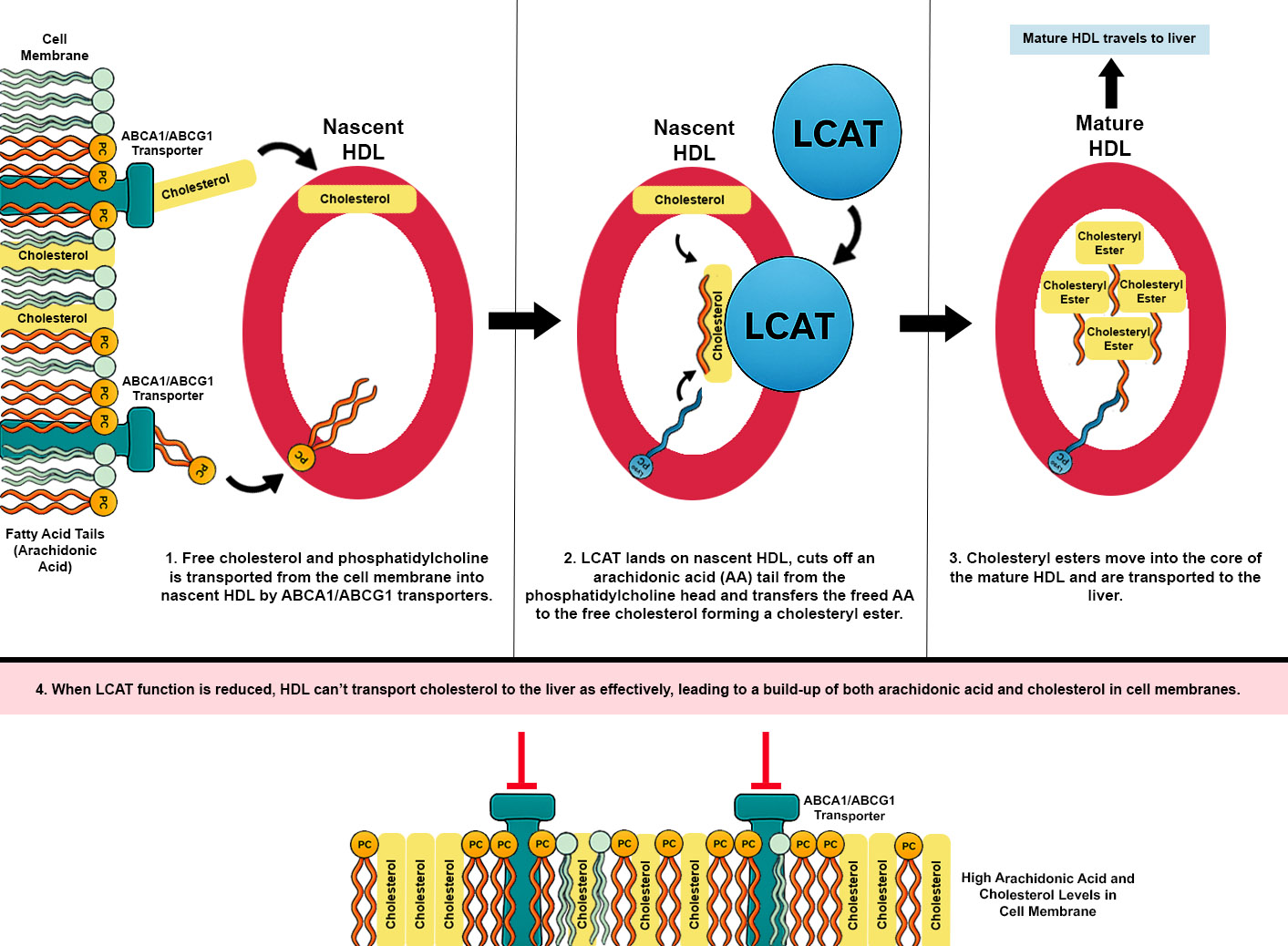
Lecithin–Cholesterol Acyltransferase Dysfunction and Cholesterol Accumulation
Phosphatidylcholine Deficiency and Membrane Integrity
- (i)
- Impaired synthesis – for example, genetic variants or functional defects in enzymes like PEMT (phosphatidylethanolamine N-methyltransferase) could limit endogenous PC production. The PEMT enzyme uses S-adenosylmethionine (SAMe) as a methyl donor to convert phosphatidylethanolamine (PE) to PC. Therefore, genetic variants in enzymes involved in methyl group metabolism—such as MTHFR and BHMT, which are important for SAMe synthesis—may indirectly impair PC synthesis by limiting PEMT activity;
- (ii)
- Excessive breakdown – chronic inflammation or allergic activation could elevate phospholipase A₂ (PLA₂) activity, which cleaves PC and releases arachidonic acid; and
- (iii)
- Transport defects – the distribution of PC to peripheral tissues might be hampered by liver dysfunction or lipoprotein abnormalities.
Lecithin–Cholesterol Acyltransferase Variants, Cholesterol Dysregulation, and Red Blood Cell Deformability
Arachidonic Acid, Eicosanoids and Immune Activation
Peroxisome Proliferator-Activated Receptor Gamma Activation and Hormone Imbalance
Pyruvate Dehydrogenase Complex Inhibition
Histamine Exacerbation of Dysregulation
Endometriosis
Neuronal vs. Non-Neuronal Insulin Sensitivity
Insulin’s Role in Regulating Norepinephrine Transporters
β₂-Adrenergic Receptor & Overtraining Syndrome
Noradrenergic Neuronal Insulin Resistance and Increased Norepinephrine Uptake
Insulin Signaling and Dopamine Transporter Regulation in the Striatum
Noradrenergic Subtypes of ME (Low vs. High Norepinephrine)
Subtype 1: Low Norepinephrine (Hypoadrenergic)
Subtype 2: High Norepinephrine (Hyperadrenergic)
Subtype 3: High Norepinephrine with β₂-Adrenergic Receptor Downregulation
Acetylcholine Synthesis
Renin–Angiotensin–Aldosterone System (RAAS) and Vasopressin
Glymphatic Clearance and Unrefreshing Sleep
SARS-CoV-2 Infection as a Trigger for ME
- Mast Cell Activation and PLA2 Surge: COVID-19, especially severe cases, is characterised by a massive inflammatory response. Mast cells are often activated in this process, releasing histamine and tryptase. These mediators can directly activate phospholipase A₂ (particularly secretory PLA2), leading to increased breakdown of membrane phospholipids and a flood of arachidonic acid. Indeed, extremely high levels of sPLA2-IIA have been documented in patients after SARS-CoV-2 infection [27] . In Long COVID, it has been observed that patients frequently meet criteria for mast cell activation syndrome (MCAS)[28]. This sequence aligns precisely with the initial steps of our proposed model, in which mast cell activation drives PLA₂ activity, leading to the release of AA, subsequent production of prostaglandins and leukotrienes, and ultimately resulting in inflammation and tissue damage. In a post-COVID patient predisposed to PC deficiency, this acute PLA2 spike could drastically deplete PC pools. Over months, insufficient PC could persist, impairing membrane integrity and LCAT function, thus lingering as a chronic issue even after the virus is gone.
- LCAT and Cholesterol Transport in Inflammation: As mentioned, acute inflammation suppresses LCAT and HDL formation. SARS-CoV-2 infection often leads to low HDL cholesterol and dysfunctional cholesterol transport, which has been proposed as part of the immunopathology (HDL carries important immunomodulatory molecules too)[29]. If LCAT activity drops during the illness and doesn’t fully recover, the patient might be left with a pro-inflammatory lipid profile.
- Hyperinsulinemia and Insulin Resistance Post-Infection: Long COVID patients, even those who were not diabetic, have shown a propensity to develop insulin resistance or new-onset diabetes [30]. If SARS-CoV-2 leads to a long period of high glucose and insulin after infection, this could increase SNS activation in ME patients with neuronal insulin hypersensitivity.
Diagnostic and Therapeutic Implications
- Lipid Metabolism Markers: Testing for LCAT functionality could involve measuring the ratio of esterified to free cholesterol in plasma or a direct LCAT activity assay. A low cholesterol esterification percentage with normal cholesterol levels might indicate LCAT impairment. Additionally, assessing HDL particle numbers and sizes (via NMR or electrophoresis) could reveal if HDL is low or dysfunctional. For phosphatidylcholine, one could measure PC and phosphatidylethanolamine in red blood cell membranes (e.g., through lipidomics). A low PC:PE ratio in cell membranes would support the PC deficiency aspect. Low plasmalogen levels may also indirectly suggest phospholipid metabolic issues. High plasma arachidonic acid or a high omega-6:omega-3 ratio would be another red flag; this can be measured in blood fatty acid profiles. In a research setting, one might even check for genetic variants in PEMT, MTHFR, or ALOX5 that could contribute to the lipid profile.
- Catecholamine and Autonomic Testing: Patients can be stratified by autonomic subtypes using tests like standing plasma norepinephrine levels (to detect hyperadrenergic POTS, defined by NE > 600 pg/mL when upright), heart rate and blood pressure response to tilt-table testing, and quantitative sudomotor or pupillary response tests (which can reveal sympathetic nerve activity). Low DHPG (a NE metabolite) in the presence of high NE would support the NET dysfunction idea.
- PPAR-γ and Inflammatory Profiling: In patients suspected to have reduced PPAR-γ activity, genetic testing for common polymorphisms such as PPARG Pro12Ala may provide initial support. Given PPAR-γ’s regulatory role in lipid metabolism, immune modulation, and hormonal balance, indirect biomarkers may offer further diagnostic value. For example, elevated transforming growth factor beta 2 (TGF-β2) may serve as a surrogate indicator of reduced PPAR-γ signaling, as PPAR-γ normally suppresses TGF-β transcription. Additionally, hormonal profiles showing elevated estradiol may reflect increased activity of the enzyme CYP19A1, which is normally inhibited by PPAR-γ. Together, the combination of elevated TGF-β2 and high estradiol may help identify a PPAR-γ–deficient ME subtype. One could also measure adiponectin (a hormone increased by PPAR-γ activation) – low adiponectin might indicate PPAR-γ is not active.
- Mast Cell and Eicosanoid Markers: For patients suspected of mast cell/eicosanoid involvement, one can test serum or urine histamine, prostaglandin D2, and leukotriene E4. Elevations in these would confirm active mast cell/eicosanoid pathways. High arachidonic acid plus high leukotriene E4 in urine, for instance, would strongly point to the AA-leukotriene axis being upregulated. Those patients might benefit most from leukotriene inhibitors.
- Phosphatidylcholine Restoration: If PC deficiency is identified, a logical step is to replenish PC. PC supplements that are made from sunflower oil may be preferable as they are likely to contain less AA. Another approach is to provide choline (as alpha-GPC or citicoline) to drive endogenous PC synthesis; however, in those with PEMT issues, that might not be sufficient. By restoring PC, we not only repair membranes but also give LCAT its substrate back – potentially improving HDL function and cholesterol clearance. However, increasing PC levels may increase membrane AA levels.
- LCAT Augmentation: Currently, recombinant human LCAT therapy is an experimental treatment mainly for familial LCAT deficiency. A recombinant LCAT (called rhLCAT or ACP-501) has been shown to raise HDL cholesterol and enhance reverse cholesterol transport in animal models. If our hypothesis is correct, one could envision using rhLCAT in a subset of ME patients with proven LCAT dysfunction, to help clear excess cholesterol and reduce immune activation.
- Targeting Arachidonic Acid Pathways: For those with signs of AA/eicosanoid excess (e.g., high prostaglandins, pain, MCAS), omega-3 fatty acid supplementation (fish oil) is a foundational approach. Omega-3 (EPA/DHA) competes with AA in cell membranes and can reduce the substrate available for inflammatory eicosanoids, while also yielding anti-inflammatory resolvins. Dietarily, reducing omega-6 intake (from vegetable oils, etc.) and increasing omega-3 could shift the balance.
- COX-2 Inhibitors (like celecoxib or even over-the-counter NSAIDs if tolerated) can cut down prostaglandin production and may alleviate symptoms like joint/muscle pain and headaches. More specifically, leukotriene inhibitors could be game-changing for a subset: the leukotriene receptor antagonist montelukast (commonly used for asthma) might help not only asthma-like symptoms but also brain inflammation. Another option is a 5-LOX inhibitor, which would directly reduce leukotriene synthesis – this could benefit those with MCAS and severe inflammatory pain.
- PPAR-γ Agonism: If an ME patient has evidence of the PPAR-γ impaired profile (high inflammation, insulin resistance, perhaps PCOS), a PPAR-γ agonist could address multiple problems at once. The thiazolidinedione drugs pioglitazone and rosiglitazone are potent PPAR-γ activators used in diabetes. An alternative is using nutraceutical PPAR-γ agonists: compounds like resveratrol and omega-3 EPA have mild PPAR-γ activating properties. Another intriguing candidate is NRF2 activators (like sulforaphane) because NRF2 and PPAR-γ pathways intersect in controlling inflammation and metabolism. It is not clear if these treatments would be effective if the patient has genetic variants of PPAR-γ.
- Targeting PDH Inhibition and Restoring Metabolic Flexibility: In individuals with impaired PDH activity—potentially driven by PPAR-β/δ–mediated upregulation of PDK4, reduced PGC-1α expression, or cholesterol-induced mitochondrial dysfunction—therapeutic strategies should aim to reactivate PDH while supporting mitochondrial function and improving membrane lipid balance. Sodium dichloroacetate (DCA) is a direct inhibitor of PDK enzymes and can restore PDH activity by preventing phosphorylation-dependent inactivation. Cofactor support with thiamine (vitamin B1), alpha-lipoic acid, magnesium, and NAD+ precursors (e.g., nicotinamide riboside or niacin) may further enhance PDH complex function. PPAR-γ agonists such as pioglitazone could offer additional benefit by improving insulin sensitivity, suppressing inflammatory signaling, and indirectly modulating PDK expression, although they do not directly counter PPAR-β/δ activity. In cases where PPAR-β/δ activation is driven by high arachidonic acid or eicosanoid load, dietary reduction of omega-6 fats, PLA2 inhibition, or supplementation with omega-3 fatty acids (EPA/DHA) may reduce the stimulus for PDK4 upregulation. Where PGC-1α function is compromised—e.g., due to PPARGC1A rs8192678 variants—AMPK activators such as AICAR, metformin, or mild exercise mimetics may be considered to enhance mitochondrial gene expression. Finally, phosphatidylcholine or plasmalogen supplementation may improve membrane fluidity and reduce cholesterol-induced mitochondrial stress, helping to restore a metabolic environment more favorable for glucose oxidation.
- For the Hypoadrenergic Subtype: the goal is to increase sympathetic tone or compensate for it. One potential medication that could help is midodrine, an alpha-1 agonist that raises standing blood pressure and can reduce orthostatic symptoms. Fludrocortisone, a mineralocorticoid, helps kidneys retain salt and water to expand blood volume (useful if aldosterone is low). Low-dose psychostimulants (like methylphenidate or modafinil) have been used off-label in ME to combat fatigue and might particularly help those with low central NE by promoting release of monoamines. Care must be taken not to overshoot and cause anxiety or insomnia.
- For the Hyperadrenergic Subtype: here the aim is to blunt the excessive NE effects. Beta-blockers (such as propranolol, bisoprolol or even low-dose atenolol) can be very helpful in POTS/hyperadrenergic patients to reduce heart rate; patients often report they feel more calm and can stand longer with a beta blocker. Ivabradine, which lowers heart rate by a different mechanism, is another POTS treatment that could be used if beta blockers are not tolerated. Clonidine or guanfacine (central α2 agonists) can reduce central sympathetic output. Correction of underlying PC deficiency to correct NET regulation would be the ideal treatment.
- For the Desensitised β2 Receptor Subtype: This is a more difficult to treat subtype– simply blocking NE might not help because receptors are already unresponsive, and increasing NE (with a drug like midodrine) might not work either because the receptors aren’t listening well. The ideal treatment is to correct the underlying dysfunction of NETs to lower extracellular norepinephrine levels. If low NET expression is due to PC deficiency, then supplementing PC should help correct extracellular NE levels. However, β2 receptors can take months to upregulate their surface expression back to normal, meaning that PC supplementation may have to be maintained for at least 3 months to achieve symptom improvement.
- Mast Cell Stabilisation and Antihistamines: Regardless of subtype, if mast cell activation is part of the picture (histamine symptoms, etc.), treatments to stabilise mast cells or block their mediators can dramatically improve quality of life. This includes H1 antihistamines (such as cetirizine, fexofenadine) and H2 blockers (famotidine) in combination, cromolyn sodium (oral or nebulized) to stabilise gut and lung mast cells, and supplements like quercetin which is a natural mast cell stabiliser. Ketotifen, a prescription H1 blocker that also stabilises mast cells, is sometimes used in severe cases. Reducing mast cell activation can also indirectly preserve PC levels (by reducing PLA2 activation) and improve sleep in ME patients. Some patients follow a low-histamine diet to minimise triggers.
- Hormonal Therapies: If a patient has clear hormone abnormalities like PCOS or thyroid dysfunction, standard treatments for those should be incorporated. For PCOS, beyond metformin and pioglitazone (addressing insulin resistance), ensuring adequate estrogen/progesterone balance via oral contraceptives or cyclic progesterone might help energy and mood. Spironolactone, which is used in PCOS for its anti-androgen and aldosterone-blocking effects, could also be beneficial in ME patients with PPAR-γ dysfunction by reducing aldosterone’s pro-inflammatory effects and helping with potassium retention.
Therapeutic Strategies for Augmenting Lecithin–Cholesterol Acyltransferase Activity in ME/CFS & Long COVID
Recombinant LCAT Enzyme Therapy
Small Molecule LCAT Activators
Apolipoprotein A-I Mimetics
Clinical Implications
Conclusions
References
- Arron HE, Marsh BD, Kell DB, Khan MA, Jaeger BR, Pretorius E. Myalgic encephalomyelitis/chronic fatigue syndrome: the biology of a neglected disease. Front Immunol. 2024;15:1386607. [CrossRef]
- Maksoud R, Magawa C, Eaton-Fitch N, Thapaliya K, Marshall-Gradisnik S. Biomarkers for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): a systematic review. BMC Med. 2023;21:189. [CrossRef]
- Mandarano AH, Maya J, Giloteaux L, Peterson DL, Maynard M, Gottschalk CG, Hanson MR. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J Clin Invest. 2020;130(3):1491–1505. [CrossRef]
- Słomko J, Estévez-López F, Kujawski S, Zawadka-Kunikowska M, Tafil-Klawe M, Klawe JJ, et al. Autonomic phenotypes in chronic fatigue syndrome (CFS) are associated with illness severity: a cluster analysis. J Clin Med. 2020;9(8):2531. [CrossRef]
- Nguyen CB, Kumar S, Zucknick M, Kristensen VN, Gjerstad J, Nilsen H, Wyller VB. Associations between clinical symptoms, plasma norepinephrine, and deregulated immune gene networks in subgroups of adolescents with chronic fatigue syndrome. Brain Behav Immun. 2019;76:82–96. [CrossRef]
- Haase CL, Tybjærg-Hansen A, Qayyum AA, Schou J, Nordestgaard BG, Frikke-Schmidt R. LCAT, HDL cholesterol and ischemic cardiovascular disease: a Mendelian randomization study of HDL cholesterol in 54,500 individuals. J Clin Endocrinol Metab. 2012;97(2):E248–E256. [CrossRef]
- Kennedy G, Spence VA, McLaren M, Hill A, Underwood C, Belch JJF. Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med. 2005;39(5):584–589. [CrossRef]
- Muñoz Herrera OM, Zivkovic AM. Microglia and cholesterol handling: implications for Alzheimer’s disease. Biomedicines. 2022;10(12):3105. [CrossRef]
- van der Veen JN, Lingrell S, Gao X, Takawale A, Kassiri Z, Vance DE, Jacobs RL. Fenofibrate, but not ezetimibe, prevents fatty liver disease in mice lacking phosphatidylethanolamine N-methyltransferase. J Lipid Res. 2017;58(4):656–667. [CrossRef]
- Huang K, de Sá AGC, Thomas N, Phair RD, Gooley PR, Ascher DB, Armstrong CW. Discriminating myalgic encephalomyelitis/chronic fatigue syndrome and comorbid conditions using metabolomics in UK Biobank. Commun Med. 2024;4:248. [CrossRef]
- Suda T, Akamatsu A, Nakaya Y, Masuda Y, Desaki J. Alterations in erythrocyte membrane lipid and its fragility in a patient with familial lecithin:cholesterol acyltransferase (LCAT) deficiency. J Med Invest. 2002;49(3–4):147–55. (No DOI available).
- Niesor EJ, Nader E, Perez A, Lamour F, Benghozi R, Remaley A, et al. Red Blood Cell Membrane Cholesterol May Be a Key Regulator of Sickle Cell Disease Microvascular Complications. Membranes (Basel). 2022;12(11):1134. [CrossRef]
- Meurs I, Hoekstra M, van Wanrooij EJ, Hildebrand RB, Kuiper J, Kuipers F, et al. HDL cholesterol levels are an important factor for determining the lifespan of erythrocytes. Exp Hematol. 2005;33(11):1309–19. [CrossRef]
- Saha AK, Schmidt BR, Wilhelmy J, Nguyen V, Do J, Suja VC, et al. Red blood cell deformability is diminished in patients with Chronic Fatigue Syndrome. Clin Hemorheol Microcirc. 2019;71(1):113–116. [CrossRef]
- Thomas N, Gurvich C, Huang K, Gooley PR, Armstrong CW. The underlying sex differences in neuroendocrine adaptations relevant to Myalgic Encephalomyelitis Chronic Fatigue Syndrome. Front Neuroendocrinol. 2022;66:100995. [CrossRef]
- Fluge Ø, Mella O, Bruland O, Risa K, Dyrstad SE, Alme K, et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. 2016;1(21):e89376. [CrossRef]
- Robertson SD, Matthies HJG, Owens WA, Sathananthan V, Christianson NSB, Kennedy JP, et al. Insulin reveals Akt signaling as a novel regulator of norepinephrine transporter trafficking and norepinephrine homeostasis. J Neurosci. 2010;30(34):11305–16. [CrossRef]
- Nguyen CB, Kumar S, Zucknick M, Kristensen VN, Gjerstad J, Nilsen H, et al. Associations between clinical symptoms, plasma norepinephrine and deregulated immune gene networks in subgroups of adolescents with Chronic Fatigue Syndrome. Brain Behav Immun. 2019;76:82–96. [CrossRef]
- Walitt B, Singh K, LaMunion SR, Hallett M, Jacobson S, Chen K, et al. Deep phenotyping of post-infectious myalgic encephalomyelitis/chronic fatigue syndrome. Nat Commun. 2024;15:907. [CrossRef]
- Kavelaars A, Kuis W, Knook L, Sinnema G, Heijnen CJ. Disturbed neuroendocrine-immune interactions in chronic fatigue syndrome. J Clin Endocrinol Metab. 2000;85(2):692–6. [CrossRef]
- Goto T, Kikuchi S, Mori K, Nakayama T, Fukuta H, Seo Y, et al. Cardiac β-Adrenergic Receptor Downregulation, Evaluated by Cardiac PET, in Chronotropic Incompetence. J Nucl Med. 2021;62(7):996–998. [CrossRef]
- Fry AC, Schilling BK, Weiss LW, Chiu LZF. β2-Adrenergic Receptor Downregulation and Performance Decrements During High-Intensity Resistance Exercise Overtraining. J Appl Physiol. 2006;101(6):1664–72. [CrossRef]
- Lim EJ, Kang EB, Jang ES, Son CG. The Prospects of the Two-Day Cardiopulmonary Exercise Test (CPET) in ME/CFS Patients: A Meta-Analysis. J Clin Med. 2020;9(12):4040. [CrossRef]
- Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, et al. Phosphorylation of Cav1.2 on S1928 uncouples the L-type Ca2+ channel from the β2 adrenergic receptor. EMBO J. 2016;35(12):1330–45. [CrossRef]
- Figlewicz DP, Szot P, Chavez M, Woods SC, Veith RC. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994;644(2):331–4. [CrossRef]
- Dehlia A, Guthridge MA. The persistence of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) after SARS-CoV-2 infection: A systematic review and meta-analysis. J Infect. 2024;106297. [CrossRef]
- Kuypers FA, Rostad CA, Anderson EJ, Chahroudi A, Jaggi P, Wrammert J, et al. Secretory phospholipase As2 in SARS-CoV-2 infection and multisystem inflammatory syndrome in children (MIS-C). Exp Biol Med (Maywood). 2021;246(23):2543–52. [CrossRef]
- Sumantri S, Rengganis I. Immunological dysfunction and mast cell activation syndrome in long COVID. Asia Pac Allergy. 2023;13(1):50–53. [CrossRef]
- Al-Kuraishy HM, Hussien NR, Al-Niemi MS, Fahad EH, Al-Buhadily AK, Al-Gareeb AI, et al. SARS-CoV-2 induced HDL dysfunction may affect the host’s response to and recovery from COVID-19. Immun Inflamm Dis. 2023;11(5):e861. [CrossRef]
- Man DE, Andor M, Buda V, Kundnani NR, Duda-Seiman DM, Craciun LM, et al. Insulin Resistance in Long COVID-19 Syndrome. J Pers Med. 2024;14(9):911. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



