Submitted:
16 September 2024
Posted:
17 September 2024
You are already at the latest version
Abstract
No one can dismiss the fundamental role played by water in several important biochemical processes, including the folding of globular proteins. The so-called hydrophobic effect is the theoretical construction to rationalize how water molecules stabilize the folded state. However, over the years, analyses have been published that lead to the conclusion that water destabilizes the folded state. The aim of the present work is to state that the gain in translational entropy of water molecules (due to the decrease in water accessible surface area associated with folding) is the driving force of protein folding.
Keywords:
water
; globular proteins
; folding
; hydrophobic effect
1. Introduction
According to the famous and influential review article published by Walter Kauzmann in 1959 [1], the hydrophobic effect must play a major role in the folding of polypeptide chains and in the conformational stability of the folded state in water and aqueous solutions. The idea is that nonpolar side chains, to avoid the unfavorable contact with water molecules, cluster together to produce a nonpolar core, resembling an organic liquid (i.e., the oil drop model). The Kauzmann’s explanation was based on the values of the Gibbs free energy change of transfer for hydrocarbon species from an organic liquid to water, that proved to be largely positive and entropy-dominated [2]. The entropy dominance was attributed to the special ability of water molecules to form ordered 3D structures to host nonpolar species, which cannot be involved in H-bonds. The Kauzmann’s classic explanation ascribes water the fundamental role to overcome the large conformational entropy loss associated with the internal rotation degrees of freedom of the polypeptide chain, and to pass from the huge ensemble of denatured and swollen conformations to the few ones associated with the folded state. The hydrophobic effect is usually described as a water-mediated attraction. The Kauzmann’s classic explanation was widely accepted and is still considered valid [3,4,5]. Although there are several problems with it (as it will be clarified below), the main idea that water is the engine of protein folding is correct. There, it was a surprise to read the title of an article recently published in Protein Science [6]: “Water-mediated interactions destabilize proteins”. This title is the simple consequence of an erroneous division of the relevant thermodynamic quantities and points out the need to define a correct statistical mechanical model for the folding-unfolding transition of polypeptide chains in water. It is important to recognize that also in the past a similar situation occurred [7,8,9,10,11]. Measured macroscopic thermodynamic quantities are path-independent state functions and cannot provide, by themselves, any molecular level description. To overcome this limitation, a specific path must be defined. However, the same physical process could be described by different paths, each one leading to a different set of thermodynamic quantities. Therefore, the selection of a proper statistical mechanical model is necessary to define a path whose steps are physically meaningful [12,13,14]. For instance, it was rapidly recognized on analyzing the protein structures solved by means of X-ray crystallography, that their average volume packing density is around 0.72 [15,16] (i.e., the volume packing density is given by the ratio of the volume occupied by all the atoms constituting the protein to the volume of the envelope containing the whole protein molecule in the folded state). The latter value implies that the folded state resembles a solid, not an organic liquid (the volume packing density of the hexagonal close packing structure is 0.74, whereas that of common organic liquids is around 0.5 [16]). This picture is supported by the measured values of the adiabatic compressibility of several globular proteins dissolved in aqueous solutions [17,18]. In fact, Schrödinger considered globular proteins as “aperiodic crystals” [19], while Liquori proposed to consider them as “crystal molecules” [20]. Thus, the search for an organic liquid mimicking the interior of globular proteins has been un-successful because the premise (i.e., the oil drop model) is not correct [21,22,23].
2. General Analysis
The present treatment starts from the words written by Sumi and Imamura (the authors of the Protein Science article [6]) in the first sentence of their Abstract: “Proteins are folded to avoid exposure of the nonpolar groups to water because water-mediated interactions between nonpolar groups are a promising factor in the thermodynamic stabilities of protein”. Accepting literally this sentence, one should conclude that all the nonpolar groups of a polypeptide chain are buried in the interior of the folded state. This is absolutely not true because proteins are hetero-polymers, and the chain connectivity renders impossible such a complete segregation: all the nonpolar moieties inside the globular structure to avoid the contact with water, and all the polar moieties outside, on the surface of the globule, to make contacts with water molecules (consider that also the backbone consists of both nonpolar groups and the strongly polar peptide groups). Such a situation holds for micelles, not for globular proteins. Chothia and colleagues, by performing detailed analyses of protein 3D structures [24,25], pointed out that the average composition is the following: (a) buried surface, (62 ± 1)% nonpolar, (31 ± 2)% polar, and (7 ± 2)% charged; (b) accessible surface, (56 ± 4)% nonpolar, (26 ± 4)% polar, and (18 ± 5)% charged. These results have recently been verified and confirmed [26]. Buried and accessible surfaces are similar in chemical nature, and more than fifty percent of the accessible surface of folded structures is made by nonpolar groups and more than a third of buried surface is made by the sum of polar and charged groups. These numbers are the simple consequence of the hetero-polymeric nature of globular proteins and underscore that group additivity approaches cannot properly describe quantitatively the conformational stability of such fine-tuned macromolecules [27]. In other words, a globular protein is not the sum of a certain number of chemically different side chains, because the latter are covalently linked to the polypeptide chain and are not independent of each other [28]. Group additivity cannot hold for globular proteins. Thus, the hydration thermodynamics of the folded and unfolded states cannot be obtained as the sum of group contributions (even when the latter are weighted for their water accessible surface area, WASA [15]). The folded state must be treated as a unique object (and the same must hold for the unfolded state, using an object of different shape). The question becomes: how detailed does the object need to be in order to reliably describe the folded state? The answer to this question depends on the process to describe, in this case the interaction of water molecules with the folded and unfolded states. It is necessary to adopt precise definitions to derive meaningful quantities. The folding-unfolding transition can be fruitfully analyzed by means of the following thermodynamic cycle, reported in Figure 1, that consists of the following steps: unfolding in water and unfolding in the ideal gas phase, hydration of the folded state and hydration of the unfolded state.
ΔGd = ΔGconf + ΔG●(U) - ΔG●(F)
Considering that hydration consists of cavity creation and switching on solute-water energetic attractions, Equation (1) becomes:
ΔGd = ΔE(intra) - T⋅ΔSconf + ΔGc(U) + Ea(U-water) - ΔGc(F) - Ea(F-water)
That can be rearranged to:
ΔGd = [ΔGc(U) - ΔGc(F)] - T⋅ΔSconf + [Ea(U-water) - Ea(F-water) + ΔE(intra)]
Equation (3) has the merit of grouping together the terms due to (a) the translational entropy of water molecules, (b) the conformational entropy of the chain, (c) the protein-water and intra-protein energetic attractions. See text for details.
Following the statistical mechanical analysis by Arieh Ben-Naim [29], hydration must refer to the transfer of a solute molecule from a fixed position in the gas phase to a fixed position in water. In all liquids, the molecules are very close-by, and it is necessary to create, at a fixed position, a void space, a cavity, whose size is appropriate to host the solute molecule [30,31,32] (i.e., cavity creation may appear an un-necessary construct; actually, it is the simplest theoretical manner to account for the basic fact that each molecule has its own body, and so two molecules cannot occupy the same space at the same time). In any liquid, cavity creation is associated with a Gibbs free energy cost, ΔGc, whose magnitude increases upon raising the liquid number density [33]. To create a cavity at a fixed position while keeping constant temperature and pressure, the liquid volume has to increase by the van der Waals volume, VvdW, of the cavity. There is a further and fundamental consequence. The centers of liquid molecules should at most lie on the solvent accessible surface area of the cavity (i.e., cavity WASA if the liquid is water), if the latter must be void (look with care to Figure 2) [34].
The cavity presence leads to a solvent-excluded volume effect which affects all the liquid molecules, since they are in continuous translational motion (note that the equipartition theorem holds for the kinetic energy of the molecules in real liquids [35]). The solvent-excluded volume effect is operative in any liquid, its magnitude is particularly large in water due to its high number density [33], and does not depend on the polar or nonpolar chemical nature of the solute molecule (i.e., it can be considered a geometric effect). A statistical mechanical theory of hydration, based on these ideas, works well in rationalizing both the poor solubility of hydrocarbons and the good solubility of alcohols in water [36], and the large negative entropy change occurring in both cases [37]. Cavity creation always leads to a decrease in the translational entropy of liquid molecules for the drop in the accessible configurational space. This holds in any liquids and is exaggerated in water due to its high number density which arises from the small size of water molecules [33]. Such a general mechanism for the entropy reduction associated with hydration has nothing to do with the claimed increase in tetrahedral structural order for the formation of something resembling icebergs or clathrates [37]. The existence of such ordered structures is not possible in a liquid phase; indeed, they have never been observed [38,39,40]. This is a weak point of the Kauzmann’s classic explanation. It is true that the hydration entropy loss is due to water molecules, not to an increase in their tetrahedral order, but to a loss in their translational entropy.
A further point needs clarification. The above reasoning indicates that, keeping fixed temperature and pressure, ΔGc must scale with the cavity WASA and so a dependence on cavity shape has to emerge [33,34]. Indeed, fixing the cavity VvdW and changing the cavity shape from a sphere to a family of thinner and longer prolate spherocylinders, the ΔGc magnitude increases [34,41]. This was verified by means of both classic scaled particle theory, SPT, calculations [41,42], and computer simulations in detailed water models [43,44,45]. Such a result has a direct consequence for the conformational stability of globular proteins. Experimental measurements have firmly established that: (a) the difference in molecular volume between the folded and unfolded states is so small to be neglected [46,47,48]; (b) there is a large WASA increase associated with unfolding [15,24]. The latter indicates that there is a large increase in the magnitude of the solvent-excluded volume effect associated with protein unfolding. Water molecules lose much more translational entropy when the protein is unfolded than when the protein is folded [49]. Of course, looking at the inverse process, one can state that the folding of polypeptide chains leads to a large gain in translational entropy of water molecules. The latter is the folding driving force, and water molecules are the engine of the process. It has been shown that, since the WASA change upon unfolding plays the main role, simple geometric objects, such as a sphere modeling the folded state and a prolate spherocylinder modeling the unfolded state (the two objects have the same VvdW, but very different WASA; see Figure 2), can lead to reliable values for the Gibbs free energy decrease caused by the decrease in the magnitude of the solvent-excluded volume effect upon the folding of the polypeptide chain [50,51]. The magnitude of the latter can be large enough to overcome the loss in conformational entropy of the polypeptide chain, ensuring the thermodynamic stability of the folded state. It is useful to provide a numerical example. A polypeptide chain of 76 residues (corresponding to the protein ubiquitin) has VvdW = 7790 Å3 (i.e., the average VvdW of a residue in native structures is 102.5 Å3 [52]); the corresponding folded state is a sphere of 12.3 Å radius. A series of prolate spherocylinders, all having the above VvdW, can describe conformations belonging to the unfolded ensemble, or better, can be used to “model” the folding process. The geometric features of these objects are listed in Table 1, together with the classic SPT-ΔGc values, calculated at 20 °C and 1 atm, in a hard sphere fluid having the experimental density of water and particle diameter σ(H2O) = 2.8 Å (i.e., the distance corresponding to the first maximum of the oxygen-oxygen radial distribution function of water at room temperature[53]).
In Figure 3 the SPT-ΔGc values are reported as a function of the cavity WASA (panel A), and of the cavity cylindrical length (panel B).
The first plot confirms the dependence on the cavity shape and WASA; the second one confirms the large Gibbs free energy decrease associated with the chain collapse, and resembles a side of the funnel that should guide protein folding [54,55]. The ΔGc decrease upon folding has a magnitude larger than that associated with the conformational entropy loss; the latter can roughly be estimated by considering an equal and temperature-independent contribution of 19 J K-1molres-1 for all the residues, as suggested by the results of large-scale computer simulations [56]. In the present case, T⋅Nres⋅ΔSconf = 423.3 kJ mol-1, at 20 °C and 1 atm, proves to be smaller than the ΔGc difference between the sphere and the penultimate spherocylinder listed in the Table 1, amounting to 522.4 kJ mol-1. For the same couple of cavities, perfoming the classic SPT calculations in carbon tetrachloride, using its experimental density at 20 °C and 1 atm and the particle diameter σ(CCl4) = 5.37 Å [50], ΔΔGc = 755.5 – 434.7 = 320.8 kJ mol-1; the latter value is smaller than the T⋅Nres⋅ΔSconf contribution reported above. This clarifies that the magnitude of the solvent-excluded volume effect in CCl4 would be not enough to overwhelm the loss in conformational entropy of the polypeptide chain. The large magnitude of the solvent-excluded volume effect is one of the reasons which render water “special” for protein folding [50], together with the tetrahedral, 2-donor and 2-acceptor, H-bonding capabilities.
To conclude, it is necessary to account for a last contribution to the folding process [49,50,51]: the direct energetic attractions between water molecules and the folded state and those between water molecules and the unfolded state; and finally, the difference in intramolecular energetic attractions between the folded and unfolded states (see the caption of Figure 1). The calculation of this energetic contribution is a formidable task. Nevertheless, several evidence indicate that, in water, the sum of the three terms should be close to zero [51]. For instance, the large decrease in the number of intermolecular protein-water H-bonds upon folding is almost entirely compensated by the intra-protein H-bonds existing in the secondary structure elements of the folded state. According to George Rose [57,58,59], a H-bond satisfaction principle holds: a peptide group must be involved in H-bonds with water molecules or with other protein groups; otherwise, the folded state would not be stable. A similar situation holds for the other weak attractive energetic interactions. It is not correct to look at the contribution of only the intra-protein energetic attractions, that favor the folded state for sure; it is necessary to look at the overall balance of the energetic attractions, those with water molecules and the intra-protein interactions in the two states.
It is important to underscore that other scientists have developed theoretical and computational approaches whose outcomes confirm that water is the engine of protein folding, and the main contribution is due to the gain in translational entropy of water molecules, caused by the decrease in the solvent-excluded volume associated with folding [60,61,62,63,64,65].
3. Conclusions
The approaches calculating the hydration thermodynamics by means of group additivity contributions cannot account for the solvent-excluded volume effect caused in water (and in all liquids) by the presence of the folded state and of the unfolded state, respectively, of a globular protein. It should be clear that the solvent-excluded volume effect is non-additive by definition. In fact, the decrease in translational entropy of liquid molecules produced by a large object with a given shape cannot be obtained by summing the effects caused by a certain number of small and quasi-spherical groups, whose total van der Waals volume corresponds to that of the large object. For instance, a sphere with radius rc = 12.3 Å and VvdW = 7790 Å3 can be considered the sum of 10 smaller spheres, each having VvdW = 7790/10 = 779 Å3, rc = 5.71 Å and WASA = 635 Å2. The classic SPT-ΔGc to create such a spherical cavity in water amounts to 166 kJ mol-1, at 20 °C and 1 atm; it is evident that 10 times 166 kJ mol-1 is not equal to the classic SPT-ΔGc to create in water the spherical cavity with rc = 12.3 Å; the same is true also for the WASA values (look at the numbers listed in Table 1). A similar situation emerges by considering small spheres each having VvdW = 7790/20 = 389.5 Å3, rc = 4.53 Å and WASA = 442 Å2. These numerical examples should help in convincing readers that additivity approaches fail in recognizing the role played by water molecules, arriving at the wrong conclusion that water destabilizes the folded state of globular proteins. In contrast, it is necessary to recognize the need to consider the solvent-excluded volume effect associated with the presence of a solute molecule in water, its relationship with the translational entropy of water molecules, and the decrease in its magnitude when a polypeptide chain folds.
Author Contributions
Conceptualization, G.G.; methodology, M.C. and G.G.; validation, M.C. and G.G.; writing—original draft preparation, M.C. and G.G.; writing—review and editing, M.C. and G.G.; supervision, G.G.; project administration, G.G.; funding acquisition, G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Università degli Studi del Sannio, FRA 2023.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. In Advances in Protein Chemistry; Elsevier, 1959; Volume 14, pp. 1–63. ISBN 978-0-12-034214-3.
- Dill, K.A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Blokzijl, W.; Engberts, J.B.F.N. Hydrophobic Effects. Opinions and Facts. Angewandte Chemie International Edition in English 1993, 32, 1545–1579. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Ball, P. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Imamura, H. Water-mediated Interactions Destabilize Proteins. Protein Science 2021, 30, 2132–2143. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.P.; Privalov, P.L.; Gill, S.J. Common Features of Protein Unfolding and Dissolution of Hydrophobic Compounds. Science 1990, 247, 559–561. [Google Scholar] [CrossRef]
- Herzfeld, J. Understanding Hydrophobic Behavior. Science 1991, 253, 88. [Google Scholar] [CrossRef]
- Muller, N. Does Hydrophobic Hydration Destabilize Protein Native Structures? Trends in Biochemical Sciences 1992, 17, 459–463. [Google Scholar] [CrossRef]
- Oobatake, M.; Ooi, T. Hydration and Heat Stability Effects on Protein Unfolding. Progress in Biophysics and Molecular Biology 1993, 59, 237–284. [Google Scholar] [CrossRef] [PubMed]
- Makhatadze, G.I.; Privalov, P.L. Energetics of Protein Structure. In Advances in Protein Chemistry; Elsevier, 1995; Vol. 47, pp. 307–425 ISBN 978-0-12-034247-1.
- Smith, P.E.; Van Gunsteren, W.F. When Are Free Energy Components Meaningful? J. Phys. Chem. 1994, 98, 13735–13740. [Google Scholar] [CrossRef]
- Mark, A.E.; Van Gunsteren, W.F. Decomposition of the Free Energy of a System in Terms of Specific Interactions. Journal of Molecular Biology 1994, 240, 167–176. [Google Scholar] [CrossRef]
- Boresch, S.; Karplus, M. The Meaning of Component Analysis: Decomposition of the Free Energy in Terms of Specific Interactions. Journal of Molecular Biology 1995, 254, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The Interpretation of Protein Structures: Estimation of Static Accessibility. Journal of Molecular Biology 1971, 55, 379-IN4. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Lim, W.A. An Analysis of Packing in the Protein Folding Problem. Quart. Rev. Biophys. 1993, 26, 423–498. [Google Scholar] [CrossRef] [PubMed]
- Gekko, Kunihiko.; Noguchi, Hajime. Compressibility of Globular Proteins in Water at 25.Degree.C. J. Phys. Chem. 1979, 83, 2706–2714. [CrossRef]
- Kharakoz, D.P. Protein Compressibility, Dynamics, and Pressure. Biophysical Journal 2000, 79, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, E.; Penrose, R. What Is Life?: With Mind and Matter and Autobiographical Sketches; 1st ed.; Cambridge University Press, 1992; ISBN 978-0-521-42708-1.
- Liquori, A.M. The Stereochemical Code and the Logic of a Protein Molecule. Quart. Rev. Biophys. 1969, 2, 65–92. [Google Scholar] [CrossRef]
- Nozaki, Y.; Tanford, C. The Solubility of Amino Acids and Two Glycine Peptides in Aqueous Ethanol and Dioxane Solutions. Journal of Biological Chemistry 1971, 246, 2211–2217. [Google Scholar] [CrossRef]
- Damodaran, S.; Song, K.B. The Role of Solvent Polarity in the Free Energy of Transfer of Amino Acid Side Chains from Water to Organic Solvents. Journal of Biological Chemistry 1986, 261, 7220–7222. [Google Scholar] [CrossRef]
- Andrew Karplus, P. Hydrophobicity Regained. Protein Science 1997, 6, 1302–1307. [Google Scholar] [CrossRef]
- Miller, S.; Janin, J.; Lesk, A.M.; Chothia, C. Interior and Surface of Monomeric Proteins. Journal of Molecular Biology 1987, 196, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Harpaz, Y.; Gerstein, M.; Chothia, C. Volume Changes on Protein Folding. Structure 1994, 2, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Merlino, A.; Pontillo, N.; Graziano, G. A Driving Force for Polypeptide and Protein Collapse. Phys. Chem. Chem. Phys. 2017, 19, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A. Additivity Principles in Biochemistry. Journal of Biological Chemistry 1997, 272, 701–704. [Google Scholar] [CrossRef]
- Dill, K.A. Polymer Principles and Protein Folding. Protein Science 1999, 8, 1166–1180. [Google Scholar] [CrossRef] [PubMed]
- Ben-Naim, A. Solvation Thermodynamics; Springer US: Boston, MA, 1987; ISBN 978-1-4757-6552-6. [Google Scholar]
- Lee, B. A Procedure for Calculating Thermodynamic Functions of Cavity Formation from the Pure Solvent Simulation Data. The Journal of Chemical Physics 1985, 83, 2421–2425. [Google Scholar] [CrossRef]
- Pohorille, A.; Pratt, L.R. Cavities in Molecular Liquids and the Theory of Hydrophobic Solubilities. J. Am. Chem. Soc. 1990, 112, 5066–5074. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Graziano, G. Scaled Particle Theory Study of the Length Scale Dependence of Cavity Thermodynamics in Different Liquids. J. Phys. Chem. B 2006, 110, 11421–11426. [Google Scholar] [CrossRef]
- Graziano, G. Dimerization Thermodynamics of Large Hydrophobic Plates: A Scaled Particle Theory Study. J. Phys. Chem. B 2009, 113, 11232–11239. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; Harper’s chemistry series; Harper & Row: New York, 1976; ISBN 978-0-06-044366-5.
- Graziano, G. Contrasting the Hydration Thermodynamics of Methane and Methanol. Phys. Chem. Chem. Phys. 2019, 21, 21418–21430. [Google Scholar] [CrossRef] [PubMed]
- Bologna, A.; Graziano, G. Remarks on the Hydration Entropy of Polar and Nonpolar Species. Journal of Molecular Liquids 2023, 391, 123437. [Google Scholar] [CrossRef]
- Soper, A.K.; Finney, J.L. Hydration of Methanol in Aqueous Solution. Phys. Rev. Lett. 1993, 71, 4346–4349. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, P.; Aldiwan, N.; Soper, A.K.; Creek, J.L.; Koh, C.A. Decreased Structure on Dissolving Methane in Water. Chemical Physics Letters 2005, 415, 89–93. [Google Scholar] [CrossRef]
- Graziano, G. Structural Order in the Hydration Shell of Nonpolar Groups versus That in Bulk Water. ChemPhysChem 2024, 25, e202400102. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. The Gibbs Energy Cost of Cavity Creation Depends on Geometry. Journal of Molecular Liquids 2015, 211, 1047–1051. [Google Scholar] [CrossRef]
- Pierotti, R.A. A Scaled Particle Theory of Aqueous and Nonaqueous Solutions. Chem. Rev. 1976, 76, 717–726. [Google Scholar] [CrossRef]
- Wallqvist, A.; Berne, B.J. Molecular Dynamics Study of the Dependence of Water Solvation Free Energy on Solute Curvature and Surface Area. J. Phys. Chem. 1995, 99, 2885–2892. [Google Scholar] [CrossRef]
- Patel, A.J.; Varilly, P.; Chandler, D.; Garde, S. Quantifying Density Fluctuations in Volumes of All Shapes and Sizes Using Indirect Umbrella Sampling. J Stat Phys 2011, 145, 265–275. [Google Scholar] [CrossRef]
- Sosso, G.C.; Caravati, S.; Rotskoff, G.; Vaikuntanathan, S.; Hassanali, A. On the Role of Nonspherical Cavities in Short Length-Scale Density Fluctuations in Water. J. Phys. Chem. A 2017, 121, 370–380. [Google Scholar] [CrossRef]
- Royer, C.A. Revisiting Volume Changes in Pressure-Induced Protein Unfolding. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 2002, 1595, 201–209. [Google Scholar] [CrossRef]
- Chalikian, T.V. Volumetric Properties of Proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 207–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Makhatadze, G.I. Molecular Determinant of the Effects of Hydrostatic Pressure on Protein Folding Stability. Nat Commun 2017, 8, 14561. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. On the Molecular Origin of Cold Denaturation of Globular Proteins. Phys. Chem. Chem. Phys. 2010, 12, 14245. [Google Scholar] [CrossRef]
- Graziano, G. On the Mechanism of Cold Denaturation. Phys. Chem. Chem. Phys. 2014, 16, 21755–21767. [Google Scholar] [CrossRef]
- Pica, A.; Graziano, G. Shedding Light on the Extra Thermal Stability of Thermophilic Proteins. Biopolymers 2016, 105, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Liquori, A.M.; Sadun, C. Close Packing of Amino Acid Residues in Globular Proteins: Specific Volume and Site Binding of Water Molecules. International Journal of Biological Macromolecules 1981, 3, 56–59. [Google Scholar] [CrossRef]
- Head-Gordon, T.; Hura, G. Water Structure from Scattering Experiments and Simulation. Chem. Rev. 2002, 102, 2651–2670. [Google Scholar] [CrossRef]
- Dill, K.A.; Chan, H.S. From Levinthal to Pathways to Funnels. Nat Struct Mol Biol 1997, 4, 10–19. [Google Scholar] [CrossRef]
- Wolynes, P.G. Evolution, Energy Landscapes and the Paradoxes of Protein Folding. Biochimie 2015, 119, 218–230. [Google Scholar] [CrossRef]
- Baxa, M.C.; Haddadian, E.J.; Jumper, J.M.; Freed, K.F.; Sosnick, T.R. Loss of Conformational Entropy in Protein Folding Calculated Using Realistic Ensembles and Its Implications for NMR-Based Calculations. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 15396–15401. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Porter, L.L.; Rose, G.D. Counting Peptide-water Hydrogen Bonds in Unfolded Proteins. Protein Science 2011, 20, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.D. Reframing the Protein Folding Problem: Entropy as Organizer. Biochemistry 2021, 60, 3753–3761. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.D. Protein Folding - Seeing Is Deceiving. Protein Science 2021, 30, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Harano, Y.; Kinoshita, M. Translational-Entropy Gain of Solvent upon Protein Folding. Biophysical Journal 2005, 89, 2701–2710. [Google Scholar] [CrossRef]
- Kinoshita, M. A New Theoretical Approach to Biological Self-Assembly. Biophys Rev 2013, 5, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M. Importance of Translational Entropy of Water in Biological Self-Assembly Processes like Protein Folding. IJMS 2009, 10, 1064–1080. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Harano, Y. Does Water Drive Protein Folding? Chemical Physics Letters 2013, 581, 85–90. [Google Scholar] [CrossRef]
- Kamo, F.; Ishizuka, R.; Matubayasi, N. Correlation Analysis for Heat Denaturation of Trp-cage Miniprotein with Explicit Solvent. Protein Science 2016, 25, 56–66. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Yamamori, Y.; Matubayasi, N. Probabilistic Analysis for Identifying the Driving Force of Protein Folding. The Journal of Chemical Physics 2018, 148, 125101. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the thermodynamic cycle describing the folding-unfolding transition of a protein. ΔG●(F) and ΔG●(U) are the Gibbs free energy changes associated with the hydration of the folded and unfolded states, respectively. ΔGd and ΔGconf are the Gibbs free energy changes for the unfolding of the protein in water and in the gas phase, respectively. According to the thermodynamic cycle, the denaturation Gibbs free energy change is given by:.
Figure 1.
Schematic representation of the thermodynamic cycle describing the folding-unfolding transition of a protein. ΔG●(F) and ΔG●(U) are the Gibbs free energy changes associated with the hydration of the folded and unfolded states, respectively. ΔGd and ΔGconf are the Gibbs free energy changes for the unfolding of the protein in water and in the gas phase, respectively. According to the thermodynamic cycle, the denaturation Gibbs free energy change is given by:.
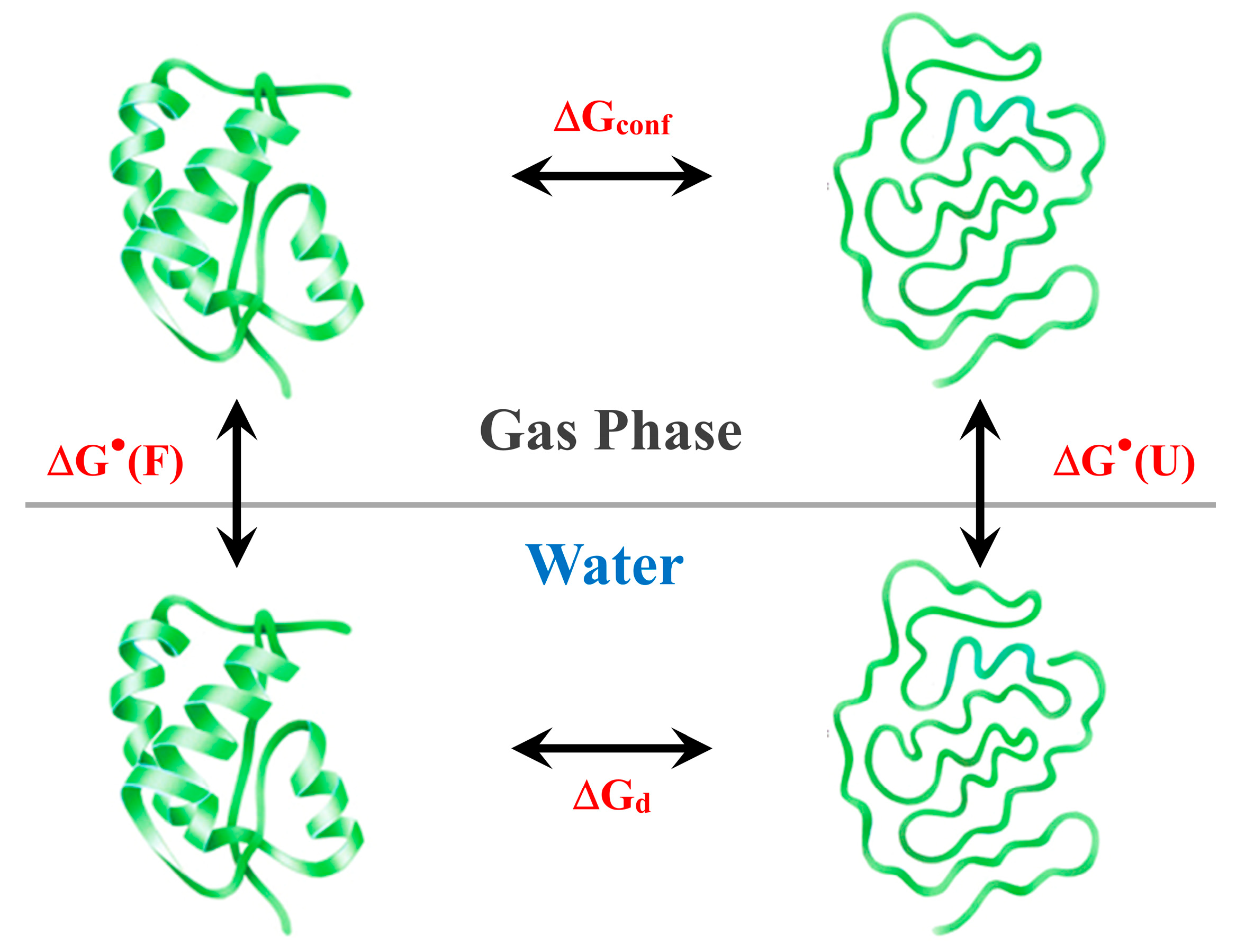
Figure 2.
Schematic 2D representation of the van der Waals volume, VvdW, and the Water Accessible Surface Area, WASA, of a sphere and a prolate spherocylinder, the two objects chosen to represent the folded and the unfolded states, respectively, of a globular protein. The yellow sphere represents a water molecule that, rolling on the surface of the inner sphere or the inner spherocylinder, defines their WASA. The two objects must have the same VvdW to model the globular protein states; in the picture, they are not to scale.
Figure 2.
Schematic 2D representation of the van der Waals volume, VvdW, and the Water Accessible Surface Area, WASA, of a sphere and a prolate spherocylinder, the two objects chosen to represent the folded and the unfolded states, respectively, of a globular protein. The yellow sphere represents a water molecule that, rolling on the surface of the inner sphere or the inner spherocylinder, defines their WASA. The two objects must have the same VvdW to model the globular protein states; in the picture, they are not to scale.
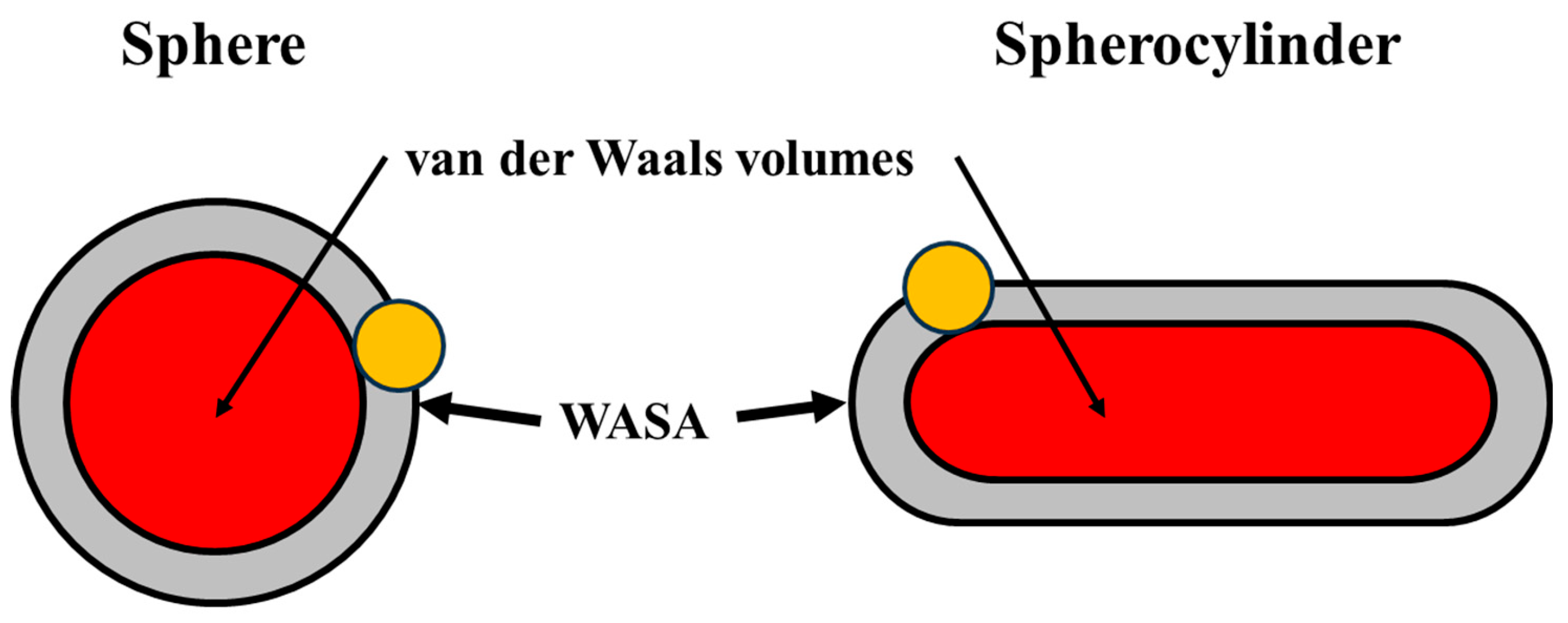
Figure 3.
Plots of the ΔGc values calculated by means of classic SPT relationships, at 20 °C and 1 atm, as a function of (A) WASA and (B) cylindrical length (l). Red squares indicate the calculated data points. Black curves were added as a guide for the eye.
Figure 3.
Plots of the ΔGc values calculated by means of classic SPT relationships, at 20 °C and 1 atm, as a function of (A) WASA and (B) cylindrical length (l). Red squares indicate the calculated data points. Black curves were added as a guide for the eye.
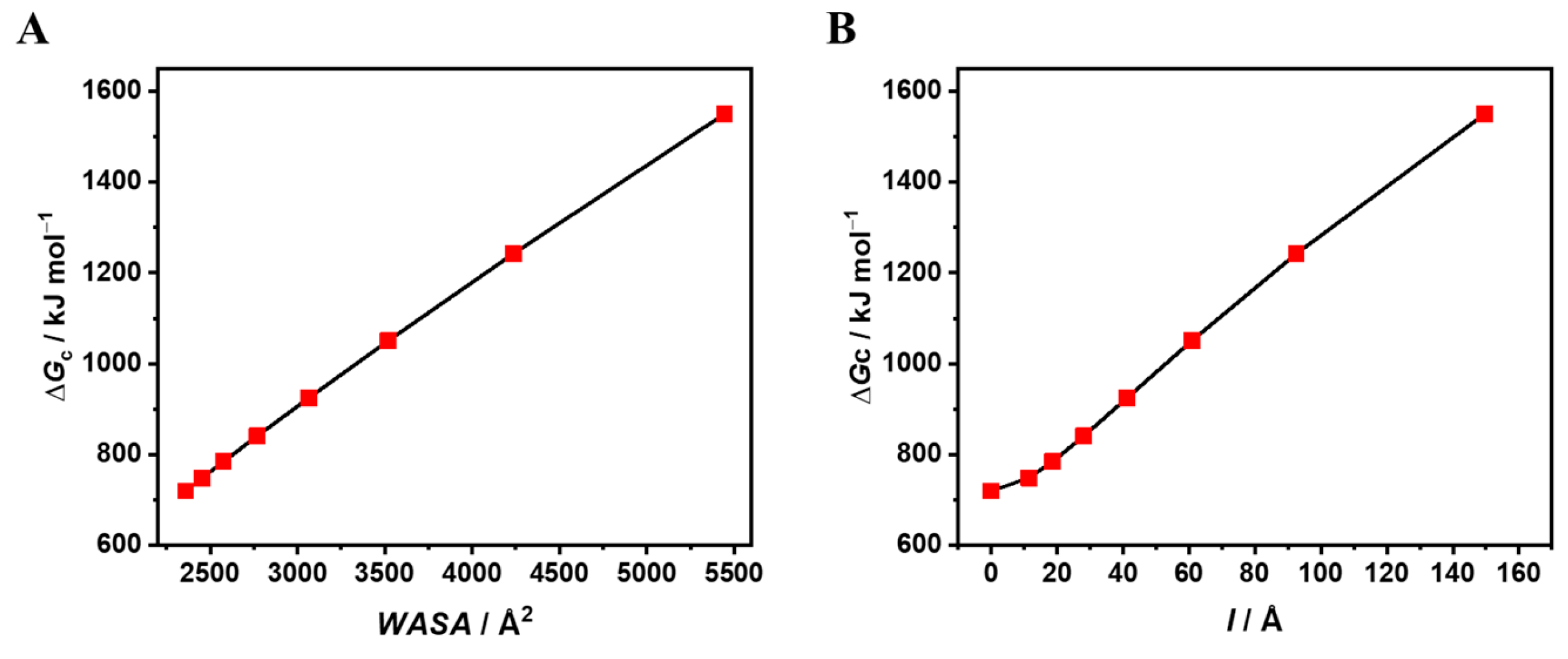

Table 1.
Classic SPT-ΔGc values to create prolate spherocylindrical cavities, at 20 °C and 1 atm, in a hard sphere fluid having the experimental density of water and particle diameter σ(H2O) = 2.8 Å. By fixing the cavity VvdW to that of a sphere of 12.3 Å radius, ΔGc values were calculated on increasing the cylindrical length-to-radius ratio (l/a). The values of the spherical cavity are reported in the first row.
Table 1.
Classic SPT-ΔGc values to create prolate spherocylindrical cavities, at 20 °C and 1 atm, in a hard sphere fluid having the experimental density of water and particle diameter σ(H2O) = 2.8 Å. By fixing the cavity VvdW to that of a sphere of 12.3 Å radius, ΔGc values were calculated on increasing the cylindrical length-to-radius ratio (l/a). The values of the spherical cavity are reported in the first row.
|
a Å |
l Å |
VvdW Å3 |
WASA Å2 |
ΔGc kJ mol-1 |
ΔGc/WASA J mol-1 Å-2 |
|---|---|---|---|---|---|
| 12.3 | / | 7790 | 2359 | 719.8 | 305.1 |
| 10.0 | 11.46 | 7790 | 2454 | 748.6 | 305.1 |
| 9.0 | 18.61 | 7790 | 2575 | 784.6 | 304.7 |
| 8.0 | 28.07 | 7790 | 2768 | 840.7 | 303.7 |
| 7.0 | 41.27 | 7790 | 3065 | 925.2 | 301.9 |
| 6.0 | 60.88 | 7790 | 3519 | 1050.9 | 298.6 |
| 5.0 | 92.51 | 7790 | 4235 | 1242.2 | 293.3 |
| 4.0 | 149.65 | 7790 | 5444 | 1550.1 | 284.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



