
Submitted:
13 September 2024
Posted:
14 September 2024
You are already at the latest version
Abstract
To date, the percentage composition of the lung microbiome in bronchopulmonary cancer has not been summarized. Existing studies on identifying the lung microbiome in bronchopulmonary cancer through 16S rRNA sequencing have shown variable results regarding the abundance of bacterial taxa.
Objectives: To identify the predominant bacteria in the lung microbiome of bronchopulmonary cancer from samples collected through bronchoalveolar lavage and to assess their potential role in the diagnosis of bronchopulmonary cancers.
Data Sources: A systematic review of English articles using MEDLINE, Embase, and Web of Science. Search terms included lung microbiome, lung cancer, bronchoalveolar lavage.
Study Selection: Randomized clinical trials that investigated the lung microbiome in bronchopulmonary cancer with samples collected via bronchoalveolar lavage.
Data Extraction: Independent extraction of articles using predefined data fields, including study quality indicators.
Data Synthesis: Nine studies met the inclusion criteria, focusing on those that utilized a percentage expression of the microbiome at the phylum or genus level. There was noted heterogeneity between studies, both in terms of phylum and genus, with a relatively constant percentage of the Firmicutes phylum and the genera Streptococcus and Veillonella being mentioned. Significant differences were also observed regarding the inclusion criteria for study participants, the method of sample collection, and data processing.
Conclusions: To date, there is no consistent percentage pattern at the phylum or genus level in bronchopulmonary cancer, with the predominance of a phylum or genus varying across different patient cohorts, resulting in non-overlapping outcomes.
Keywords:
lung microbiome
; lung cancer
1. Introduction
Lung cancer stands as the second most frequently diagnosed cancer and held the grim distinction of being the primary cause of cancer-related deaths in 2020. It accounted for about one in 10 (11.4%) of all diagnosed cancers and a staggering one in 5 (18.0%) of all cancer-related deaths. Notably, lung cancer is the foremost contributor to both cancer morbidity and mortality among men. In contrast, among women, it ranks third in terms of incidence, following breast and colorectal cancer, but rises to the second position for mortality, surpassed only by breast cancer.[1]
The human microbiota encompasses 10–100 trillion symbiotic microbial cells hosted by each individual, predominantly bacteria located in the gut. Clarifying the definition of the human microbiome has been challenging due to confusion surrounding terminology. For instance, "microbiota" is frequently used to refer to the microbial taxa associated with humans, while "microbiome" is used interchangeably but specifically denotes the catalog of these microbes and their genes. Initially, "metagenomics" described the shotgun analysis of total DNA. However, its current usage is expanding to include investigations of marker genes like the 16S rRNA gene.What sets today apart is not just the capacity to observe evident distinctions, but rather the capability to employ advanced molecular techniques to comprehend the reasons behind these differences. This enables us to gain insights into why variations exist and understand the mechanisms through which transformations from one state to another can be influenced. Scientists can now produce millions of sequences per sample, enabling the examination of variations in microbial communities across body sites and among individuals. [2]
The composition of the adult microbiome is shaped by a combination of factors such as host genetics, diet, environmental influences, and external factors like antibiotic usage, which has the potential to disrupt the balance in the gut. Any significant alteration in microbial composition can adversely impact the human host, potentially playing a role in the initiation of various conditions, including cancer. In contrast to the microbe-friendly conditions found in the gut, oral cavity, and upper airways, the lower airway seems to present an inhospitable environment, characterized by limited nutrients and high oxygen stress, creating challenges for microbial survival. Drawing on metaphors of adapted island models, one could liken the lower airway to a desert in terms of microbial colonization and replication. While it was traditionally believed to be sterile, emerging evidence now reveals a diverse array of microbial species in the lungs of healthy individuals. These species are predominantly anaerobes, including gram-negative Prevotella and Veillonella spp, as well as gram-positive Coprococcus and Dorea spp. [3]
Similar to any mucosal surface in the human body, the host immune system is not oblivious to microbial exposures. In fact, the microbial products present in the lower airways result from a delicate equilibrium between the influx of microbes and the removal of microbes through clearance mechanisms. [4]
The microbial composition and biomass in the upper and lower respiratory tracts differ significantly. In the upper respiratory tract, which includes the nasal cavity, paranasal sinuses, pharynx, and supraglottic portion of the larynx, bacteria are prevalent. Additionally, there are topographical variations in microbial composition within the upper respiratory tract. For instance, the dominant taxa in the nasal cavity and nasopharynx consist of Moraxella, Staphylococcus, Corynebacterium, Haemophilus, and Streptococcus species. In contrast, the oropharynx shows a high abundance of Prevotella, Veillonella, Streptococcus, Leptotrichia, Rothia, Neisseria, and Haemophilus species.[5]On the other hand, the lower respiratory tract, which includes the trachea and lungs, maintains a relatively low microbial biomass. It plays a major role in lower airway mucosal immunology. This low biomass is upheld by rapid microbial clearance through various physiological mechanisms. These mechanisms facilitate the lower respiratory tract in performing its crucial function: the exchange of oxygen and carbon dioxide. [3,4]
Lung bacteria, as measured by quantification of bacterial DNA, are roughly 100-fold lowerin concentration than oral bacteria Healthy lungs are subject to constant exposure to oropharyngeal bacteria via subclinical aspiration, confirmed both radiographically and ecologically in healthy, asymptomatic volunteers. [6]
In healthy lungs, the microbial biomass is significantly lower, ranging from to bacteria per gram of tissue[7], compared to the much higher microbial density in the lower gastrointestinal (GI) tract, which typically ranges from to bacteria per gram of tissue. [8] Despite the shared embryological origin of the GI tract and lungs as mucosa-lined luminal organs, their micro-anatomical features exhibit notable differences. This divergence in microbial abundance and anatomical characteristics underscores the distinct environments and functions of these organs within the body. The composition of the lung microbiome is thought to be influenced by the equilibrium of three key factors: the influx of microbes into the airways, the removal of microbes from the airways, and the relative reproduction rates of community members present in the airways. The latter is determined by the regional growth conditions, including factors such as pH, oxygen levels, and nutrient availability.[9]
Patients with lung cancer are at a heightened risk for microbial infections. The impact of repeated microbial exposure on reshaping the lung's immune system has been gaining recognition. Furthermore, the roles of pathogens in lung disease are being rigorously investigated, highlighting the importance of understanding how these interactions may influence disease outcomes. Inflammation resulting from microbial infections is increasingly understood to play a role in the development and progression of cancer. [10]
RNA extracted from bacteria is a valuable tool for detecting and identifying microbes in various specimens, regardless of growth conditions. The 16S rRNA gene, with its distinct structure, is a major universal target for bacterial identification. Its nine variable regions and conserved regions allow the design of nearly universal primers, making it widely used in diagnostic departments. However, due to ongoing advancements, there is variability in DNA extraction and analysis processes, and no consensus on a gold standard practice has been established. [6]
It is important to name certain terms in order to better understand the study of the microbiome.Dysbiosis, characterized by a deviation from a normal microbial composition, is linked to various adverse biological phenomena, at times leading to clinical consequences. In the context of the lung, dysbiosis can exert a substantial influence on the initiation and advancement of respiratory diseases, underscoring the clinical imperative to comprehend the biology of the lung microbiome. In the last decade, there has been a swift proliferation of research employing culture-independent genomic techniques to characterize the microbial environment in the lung. [4]
Various higher-level metrics are frequently employed to characterize the microbiome within a sample. While these metrics may not offer insights into alterations in the abundance of individual taxa, they enable a more comprehensive assessment of overall changes or distinctions in the microbial composition. Examples of such measures include alpha and beta diversity.Within a single sample, diverse metrics collectively known as alpha diversity are employed to estimate diversity. These metrics encompass aspects such as richness (the number of different taxa) or distribution (evenness) within a microbial sample, aiming to capture a blend of both properties. Alpha diversity serves as a metric for microbiome diversity within an individual sample, whereas beta diversity gauges the similarity or dissimilarity between two communities. Like alpha diversity, numerous indices exist, each capturing distinct facets of community heterogeneity. These indices differ in how they account for variation in rare species, whether they consider presence/absence alone or incorporate abundance, and how they interpret shared absence. One widely adopted measure, Bray-Curtis dissimilarity, is popular for its consideration of both the size (overall abundance per sample) and shape (abundance of each taxon) of communities (Bray, 1957). [4]
Beta diversity is a term used to quantify how much a community's membership or structure differs between two samples. A recent examination of taxon-based assessments of beta diversity revealed that certain metrics, like Canberra and Gower distances, possess enhanced capabilities to distinguish clusters. In contrast, other metrics, such as chi-squared and Pearson correlation distances, are better suited for uncovering the impacts of environmental gradients on communities. [11]
Various pipelines are available for the analysis of microbial community data, including mothur, WATERS (Workflow for the Alignment, Taxonomy, and Ecology of the Microbial Environment), the RDP (Ribosomal Database Project) pyrosequencing tools, and QIIME (Quantitative Insights Into Microbial Ecology, pronounced “chime”). [12,13,14,15]QIIME serves as a platform for analyzing high-throughput sequencing data, allowing users to import raw sequence data and easily generate measures of inter- and intra-sample diversity. Ensuring consistency in identifying Operational Taxonomic Units (OTUs) and establishing unanimously accepted measures of diversity within and between samples is vital for comparing results across studies. However, the concept of OTUs is becoming progressively challenging as sequence data accumulate, and explicitly phylogenetic approaches gain popularity. [4]
To enhance analyses that depend on a restricted set of taxonomic names, 16S rRNA sequences can be grouped into operational taxonomic units (OTUs) at the 97% similarity level (3% difference). This degree of sequence-based clustering is commonly acknowledged for distinguishing bacterial organisms below the genus level. However, it's important to note that assuming this level of clustering consistently defines microbial species or strains would be inaccurate. [16]
LEfSe (Linear discriminant analysis Effect Size) is a tool designed for high-dimensional biomarker discovery, focusing on identifying features such as genes, pathways, or taxa that distinguish between two or more biological conditions. It integrates statistical significance, biological relevance, and effect size to pinpoint features that are not only differentially abundant but also biologically meaningful. By combining standard statistical tests with criteria for biological consistency and relevance, LEfSe effectively identifies the elements most likely to account for differences between studied groups. [17]
2. Material and Methods
The systematic review was conducted to answer the question: “What is the composition of the lung microbiome in bronchopulmonary cancer?” The study was planned, conducted, and reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).[18]
2.1. Search Strategy
We performed a systematic literature search using PubMed (Medline), Cochrane, and Web of Science databases to identify relevant studies published up to 10 November 2023.
The following keywords were used: (("lung"[MeSH Terms] OR "lung"[All Fields]) AND ("microbiota"[MeSH Terms] OR "microbiota"[All Fields] OR "microbiome"[All Fields]) AND ("lung neoplasms"[MeSH Terms] OR ("lung"[All Fields] AND "neoplasms"[All Fields]) OR "lung neoplasms"[All Fields] OR ("lung"[All Fields] AND "cancer"[All Fields]) OR "lung cancer"[All Fields]) AND ("bronchoalveolar lavage"[MeSH Terms] OR ("bronchoalveolar"[All Fields] AND "lavage"[All Fields]) OR "bronchoalveolar lavage"[All Fields])) AND (medline)).
2.2. Study Selection and Eligibility Criteria
Randomized clinical trials that analyzed the lung microbiome in bronchopulmonary cancer were included. The studies had to employ bronchoalveolar lavage as the method of sample collection, and the data processing technique must have been 16S rRNA sequencing. These studies should have expressed their results as percentages, especially at the phyla and genera levels. Articles written in English were considered. The study selection ranged from the year 2007 until 10 November 2023. Studies that evaluated the microbiome through sputum examination or postoperative assessments were excluded. Patients of any age, with any smoking status, either with a prior confirmed diagnosis of cancer before sample collection or with a suspicion of cancer confirmed later, were included.
2.3. Objectives
The primary objective is to identify the composition and ratio of the lung microbiome in bronchopulmonary cancer, with an assessment of its potential role in the diagnosis of bronchopulmonary cancer. Secondary objectives include evaluating methods of sample collection, processing, and assessing potential compositional differences based on these methods.
2.4. Data Extraction and Synthesis
Study characteristics (first author, country, number of patients, sample data, analytical method, main result) were extracted from the included articles and summarized in Table 1. Data extraction was performed by one author (L.S) and independently reviewed by an additional author.
From: Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372:n71. doi: 10.1136/bmj.n71

| Authors | Country | Inclusion Criteria | No* | What was compared | Sample | Metod of analysis | alpha diversity | Main results |
|---|---|---|---|---|---|---|---|---|
| Bingula R et al. (2020) | France | NSCLC eligible for surgical treatment; 18-80 yo; IMB < 29,9; no previous airway surgery or cancer treatment, no AB, Corticotherapy, Immunospressive drugs or pulmonary infections for at least the past 2 months | 15 | microbiota in saliva, BAL (obtained directly on excised lobe), non-malignant, peritumoural and tumour tissue |
the removed lung or lung lobe was placed in a sterile vessel and the tumour position was determined by palpation. First, a piece of non-malignant lung distal to the tumour (opposite side of the lobe) with an average size of 1 cm3 wa5s clamped 2 × 40 mL of sterile physiological saline into the bronchus; was retrieved (8–10 mL in total) |
Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3-V4. |
Shannon diversity index and Faith’s phylogenetic Diversity No differences in alpha diversity metrics were detected between four lung samples |
At phylum level: Firmicutes 45.7%; Bacteriodes 13.3%; Actinobacteria 11.9%; Proteobacteria 28%; Fusobacteria 0.23%; Cyanobacteria 0.16%; Acidobacteria0.11%; Other 0.07% At genus level: Pseudomonas 10.3%; Blautia 5.9%; Streptococcus 5.1%; Capnocytophaga 4.8%; Acinetobacter 2.9%; Prevotella 2.3% Propionibacterium 2.3%; Lactobacillus 2.1%; Sphingomonas 1.8%; Bacteroides 1.5%; Veillonella 1.4%; Other each <1% |
| Wang K et al. (2019) |
China | primary bronchogenic carcinoma-confirmed; no glucocorticoid or antibiotic treatment for at least 30 days before sample collection; |
47 | the difference in microbiota diversity in the oral cavity and fluid bronchoalveolar lavage (BALF) of patients with lung cancer and healthy controls |
local anesthesia, flexible fiberoptic bronchoscopy, subsegmental bronchus in the involved focal lobe 3x 50 mL of sterile normal saline were instilled, gently aspirated. Suction channel use was avoided until the tip of the bronscope extended beyond the carina; pooled and collected in a siliconized plastic bottle placed on ice | Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V4. QIAamp DNA Microbiome Kit |
Shannon and Simpson indexes Lung cancer patients had less lung and oral microbiota diversity than healthy controls |
At phylum level: FIRMICUTES 38.42%; FUSOBACTERIA 5.12%; SPIROCHAETES 0.11%; TENERICUTES 0.11%; SYNERGISTETES 0.03%; |
| Jang, H.J. et al. (2021) |
South Korea | pathologically diagnosed with non-small cell lung cancer (NSCLC) | 84 | the differences in the lung microbiomes of patients with lung cancer. | rinsed mouth twice with sterile saline; topical anesthesia (lidocaine) using a nebulizer; sedated with midazolam and fentanyl; When the bronchoscope reached the “involved” airway containing the lung mass or the lung nodule, the bronchi were washed with 30–50 mL sterile saline (0.9%); approximately 15 mL BAL fluid was acquired for sequencing analysis; samples were immediately stored at -70 °C in a freezer, and DNA extraction was performed within 24 h | Illumina HiSeq technology, performed 16S ribosomal rRNA targeted region V3-V4. FastDNA® SPIN Kit for Soil CleanPCR kit |
Shannon and Simpson the difference was not significant (p = 0.307 for Shannon; p = 0.540 for Simpson index). |
At phylum level: PDL-1>10%: Bacteroidetes 39.4%; Firmicutes 30.5%; Proteobacteria19.1%; Fusobacteria 6.4%; Acinetobacter 3.2% PDL-1<10% Bacteroidetes 39.4%; Proteobacteria 28.2%; Firmicutes 23.2%; Fusobacteria 5.1%; Acinetobacter 2.8% At genus level: PDL-1>10%: Prevotella; Streptococcus; Veillonella; Haemophilus; Neisseria; Porphyromonas; Fusobacterium; Megasphaera; Leptotrichia; Rothia; Escheichia; PDL-1<10%: Prevotella; Neisseria; Haemophilus; Veillonella; Streptococcus; Porphyromonas; Fusobacterium; Megasphaera; Leptotrichia; Rothia; Pseudomonas; |
| Zhuo M et al. (2020) | China | lung cancer - no one with cancer treatment | 50 | association of the microbiota with lung cancer | Bronchoendoscope, which avoided contamination of the upper respiratory tract or oral microbiota, was performed to obtain paired BALF samples in lung cancer patients (one from the cancerous lung, the other from the contralateral non-cancerous lung). All samples were immediately frozen and maintained at -80C until further DNA extraction |
Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3- V4 PowerSoil DNA Isolation Kit |
Shannon diversity index and Simpson diversity index Cancer lung was not significantly different from normal lung in a-diversity |
At phylum level: Affected lung: Proteobacteria: 34.2%; Firmicutes: 27.96%; Bacteroides: 21.46%; Actinobacteria: 5.79%; Fusobacteria: 5.39%; Cyanobacteria: 1.23%; Spirochaerae: 1.12%; TM7 (Saccharibacteria): 0.53%; Acidobacteria: 0.53%; Tenericutes: 0.5%; Others: 1.2% Normal lung: Proteobacteria: 32.95%; Bacteroides: 26.65%; Firmicutes: 26.46%; Fusobacteria: 5.02%; Actinobacteria: 4.39%; Spirochaerae: 0.97%; TM7 (Saccharibacteria): 0.65%; Cyanobacteria: 0.56%; Acidobacteria: 0.55%; Tenericutes: 0.32%; Others: 1.43%. At genus level: Affected lung: Streptococcus: 10.78%; Neisseria: 7.54%; Alloprevotella: 5.22%; Prevotella_7: 4.88%; Haemophilus: 4.8%; Veillonella: 4.25%; Fusobacterium: 4.14%; Prevotella: 3.93%; Ochrobactrum: 3.25%; Porphyromonas: 3.25%; Other: 47.95%. Normal lung: Streptococcus: 12.04%; Neisseria: 9.37%; Prevotella_7: 7.1%; Alloprevotella: 6.57%; Haemophilus: 5.65%; Prevotella: 5.28%; Porphyromonas: 4.78%; Veillonella: 4.53%; Fusobacterium: 3.96%; Stenotrophomonas: 3.86%; Other: 47.95% |
| Gomes S et al. (2019) |
Portugal | subjects undergoing bronchoscopy for evaluation of lung disease at three hospitals in Portugal | 49 | Microbiota in LC vs controler | Sample collection was targeted toward affected lung segments and done by bronchoscope wedging into subsegmental lung regions; was used only bronchoscope working channel washes, which were done twice with a minimum volume of 15 mL (0.9% saline solution) | V3-V4, V4-V6 regions of the 16S rRNA gene DNA Mini kit (Qiagen) |
Simpson and Shannon SCC cases were in average more diverse than ADC |
At phylum level: Proteobacteria 38.7%; Firmicutes 25.4%; Actinobacteria 16.5%; Bacteroidetes 13.3%; Spirochaetes 2.2%; Fusobacteria 2.1%; TM7 0.7%; OD1 0.5%; SR1 0.3%; Tenericutes 0.2%; Synergistetes 0.1%; Others 0.0%; At genus level: Haemophilus 29.5%; Streptococcus 10.9%; Corynebacterium 8.2%; Actinomyces 7.4%; Prevotella 5.8%; Veillonella 5.0%; Neisseria 3.6%; Selenomonas 2.8%; Parvimonas 2.4%; Porphyromonas 2.4%; Aggregatibacter 2.1%; Treponema 2.1%; Fusobacterium 2.1%; Propionibacterium 2.0%; Bulleidia 1.9%; Peptostreptococcus 1.2%; Pseudomonas 1.1%; Granulicatella 0.9%; Oribacterium 0.9%; Actinobacillus 0.8%; Bifidobacterium 0.6%; Campylobacter 0.5%; Sphingobacterium 0.5%; Staphylococcus 0.5%; Sphaerochaeta 0.5%; Filifactor 0.4%; Leptotrichia 0.4%; Scardovia 0.3%; Stenotrophomonas 0.3%; Moraxella 0.3%; Capnocytophaga 0.3%; Rothia 0.2%; Lactobacillus 0.2%; Megasphaera 0.2%; Morganella 0.2%; Acholeplasma 0.2%; Flavobacterium 0.1%; Catonella 0.1%; Aerococcus 0.1%; Cupriavidus 0.1%; TG5 0.1%; Sphingomonas 0.1%; Phenylobacterium 0.1%; Pedobacter 0.1%; Dialister 0.1%; Others 0.1% |
| Seixas S et al (2021) |
Portugal | did not include in the non-LC group any subject with a primary diagnosis of COPD or ILD. No healthy controls were collected. For second goal, was selected three homogenous patient groups with a single CLD diagnosis (controlled for other comorbidities) |
49 | LC vs other lung disease | Sample collection targeted affected lung segments BALF samples had a minimum volume of 15 mL (0.9% saline solution) and were initially stored by pulmonologists at − 20 to 4 °C according to the facilities available at the participating hospitals. Samples were then transported on ice to research centers where they were stored at − 80 °C until needed |
Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V4 DNA Mini kit (Qiagen) |
Shannon, ACE, Simpson, Fisher and Phylogenetic (Faith’s) diversity indices Alpha-diversity indices did not vary significantly between LC and non-LC groups |
At phylum level: Firmicutes 47.11%; Proteobacteria 31.35%; Bacteroidetes 15.52%; Actinobacteria 2.80%; At genus level: Escherichia/Shigella 8.80 %; Bacillus 7.66%; Streptococcus 7.45%; Salmonella 7.40%; Staphylococcus 7.27 %; Lactobacillus 6.41 %; Prevotella 6.09%; Veillonella 6.00 %; Pseudomonas 3.56%; Haemophilus 3.21 %; Others (each <1%) |
| Lee SH et al. (2016) |
South Korea | Patients who were admitted for evaluation of lung masses were prospectively enrolled in this study at a 2500-bed tertiary uni-versity medical centre in Seoul, South Korea between May and September 2015. Excluded: less than 20 years of age, pregnant, or had undergone any procedure other than bronchoscopy to evaluate the lung mass. |
20 | characterized and compared the microbiomes of patients with lung cancer and those with benign mass-like lesions. | topical anaesthesia (lido-caine) by nebulizer and then were sedated with midazolam and fentanyl.. BAL was performed following a standardized protocol on the opposite side of the lung mass, and 10 mL of BALF was acquired from each patients using about 30 ml sterile 0.9% saline. If a patient had a lung mass on the right upper lobe, BAL was performed on the left upper lobe | Illumina HiSeq technology, performed 16S ribosomal rRNA targeted region V1-V3 |
Chao1 estimation and Shannon more complex diversity with higher abundance and α-diversity |
At phylum level: Bacteroidetes: 39.5%; Firmicutes: 29.7%; Proteobacteria: 22.8%; Fusobacteria: 4.5%; Actinobacteria: 2.1%; Spirochaetes: 0.4%; TM7: 0.5%; SR1: 0.3%; Tenericutes: 0.1% At genus level: Prevotella: 30.8%; Neisseria: 13.8%; Veillonella: 11.4%; Streptococcus: 10.9%; Haemophilus: 7.2%; Alloprevotella: 6.1%; Fusobacterium: 2.2%. Megasphaera: 2.2%; Porphyromonas: 2.0%; Leptotrichia: 1.8%; Campylobacter: 1.1%; Actinomyces: 0.8%. |
| Liu B et al. (2022) | China | patients with LC were recruited in the Zibo Municipal Hospital. The exclusion criteria included the uses of antibiotics, corticoids, probiotics, prebiotics or immunosuppressive drugs in the past 3 months; hypertension; diabetes; previous airway surgery; preoperative radiotherapy and chemotherapy; and atomization treatment |
7 | excavate the features of the lung microbiota and metabolites in patients and verify potential biomarkers for lung cancer diagnosis. |
Sterile saline samples of bilateral lungs were obtained by bronchoscopy in patients with LC. Paired samples of bronchoalveolar lavage fluid (BALF) included the one from the cancerous lobe and the other from the contralateral noncancerous lobe. |
Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3-V4 FastDNA Spin Kit (MP Biomedicals, Shanghai, China) |
Shannon, Chao, ace Lower abundance in alpha diversity |
At phylum level: Proteobacteria 45.05%; Firmicutes 28.31%; Bacteroidota 14.89%; Actinobacteriota 7.15%; Fusobacteriota 2.41%; Patescibacteria 1.25%; others 0.94%; At genus level: Pseudomonas 35.14%; Streptococcus 14.34%; Prevotella 9.55%; Neisseria 6.81%; Veillonella 4.85%; Actinomyces 4.6%; Granulicatella 3.53 %; Alloprevotella 3.25%; Leptotrichia 1.27 %; Fusobacterium 1.13 %; Porphyromonas 1.12 %; Haemophilus 1.07 %; Rhodococcus 0.91 %; Klebsiella 0.05 %; Lactobacillus 0.12 %; Bacillus 0.11 %; others 12.15 %; |
| Jang, H.J. et al. (2023) |
South Korea | patients who were pathologically diagnosed with NSCLC |
84 | the histological type-based differences in the lung microbiomes of patients with lung cancer. |
topical anesthesia (lidocaine) via nebulizer; sedation with midazolam and fentanyl when the bronchoscope arrived in the “involved” airway containing lung masses or lung nodules, the bronchi were flushed with 30 to 50 mL of sterile saline (0.9%). Approximately 15 mL of BAL fluid samples were obtained from each patient for sequencing analysis. BAL fluid samples were immediately placed at –70°C in a freezer, and DNA extraction was conducted within 24 hours |
Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3-V4 |
Shannon and Simpson α -diversity was different between the two types of lung cancer. |
At phylum level: ADK Bacteroidetes 40.8%; Proteobacteria 24.9%; Firmicutes 24.1%; Fusobacteria 6.0%; Actinobacteria 2.8% SCC Bacteroidetes 35.0%; Firmicutes 29.3%; Proteobacteria 27.8%; Fusobacteria 3.8%; Actinobacteria 3.3%; |
* Number of participants with cancer and BAL procedure; NSCLC: Non-Small Cell Lung Cancer; BMI: body mass index; AB: antibiotics; BALF: broncho-alveolar lavage fluid; ILD: interstitial lung disease; COPD: chronic onbstructive pulmonary disease; CLD: chronic lung disease; LC: lung cancer; ADK: adenocarcinoma; SCC: Squamous cell carcinoma.
3. Results
3.1. Literature Search
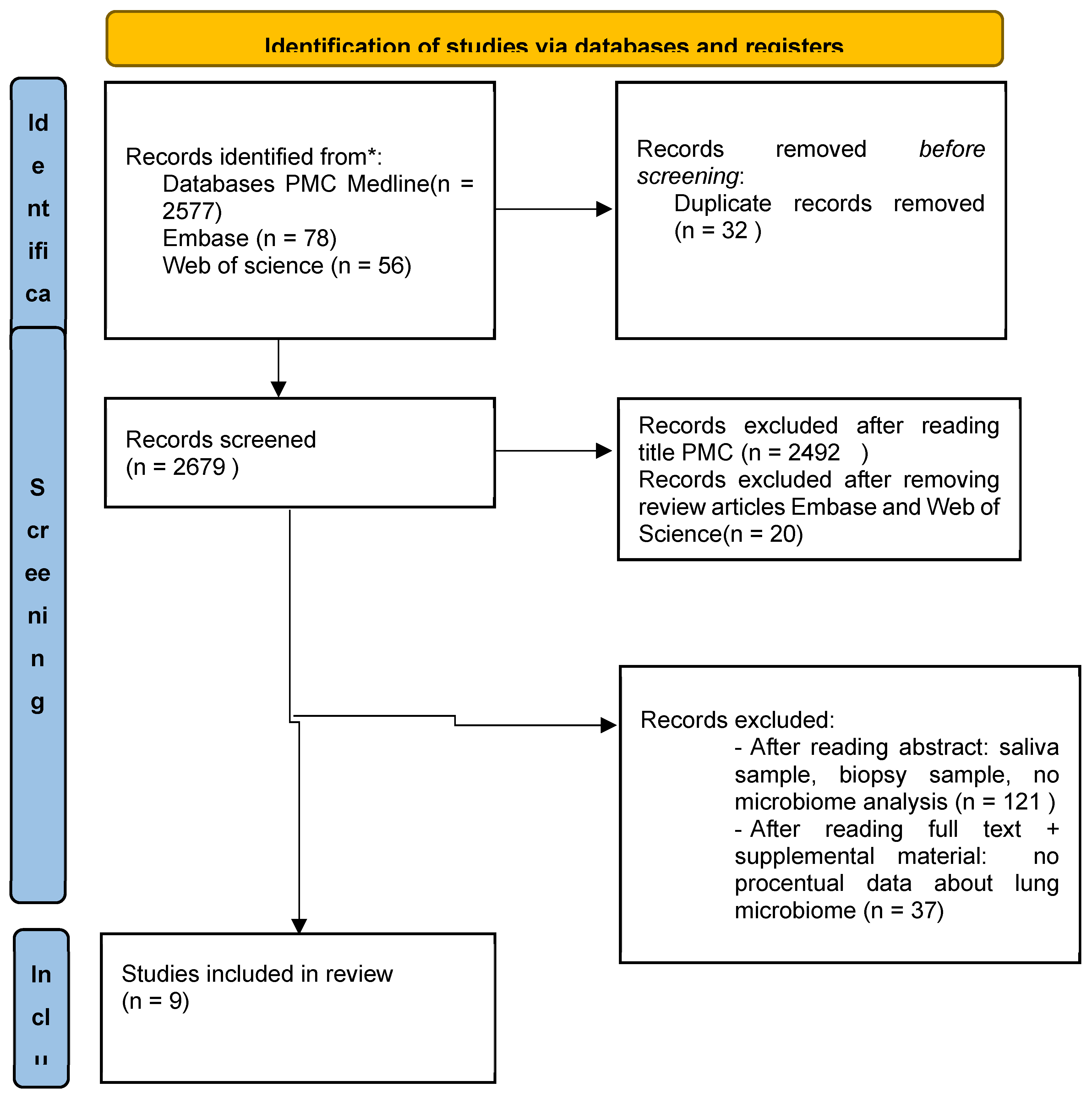
Out of 2711 studies identified in the search, 32 were excluded after removing duplicates, and 20 were excluded after reviewing the reviews (Web of Science and Embase). Additionally, 2492 studies from PMC were excluded after title screening, leaving 167 articles. After abstract review, 121 studies were excluded for not being relevant to the question addressed in the systematic review, leaving 37 studies. Following a full-text and supplemental materials review of the remaining 37 articles, 28 were excluded for not providing percentage data about the microbiome. Overall, 9 studies were selected for inclusion in the systematic review.
3.2. Characteristics of the Included Studies
3.2.1. Studies Objective
One study compared the microbiota in saliva, BAL (obtained directly from the excised lobe), non-malignant, peritumoral, and tumor tissue from 18 NSCLC patients eligible for surgical treatment. Bronchoalveolar lavage was performed on 15 patients. [19] Another study, which conducted BAL on 47 patients, compared the differences in microbiota diversity in the oral cavity and bronchoalveolar lavage fluid (BALF) of patients with lung cancer and healthy subjects. [20]
Zeng W et al. (2022) compared the differences in composition and gene expression in the microbiota between benign lung disease and non-small cell lung cancer. [21] A study including 84 patients compared the differences in the lung microbiomes of patients with lung cancer al. [22] Another study investigated the association of the microbiota with lung cancer. [23] In Portugal, two studies were conducted, each including 49 lung cancer patients who underwent BAL; one compared the microbiota in lung cancer vs. controls [24], and the other compared the microbiota in lung cancer vs. other lung diseases. [25] In South Korea, a study including 20 lung cancer patients who underwent
BAL characterized and compared the microbiomes of patients with lung cancer and those with benign mass-like lesions. [26]
Liu B et al. (2022) explored the features of the lung microbiota and metabolites in patients and verified potential biomarkers for lung cancer diagnosis. [27] In a study with 84 patients, the histological type-based differences in the lung microbiomes of patients with lung cancer were compared [28]. The V3-V4 regions of the 16S rRNA were most frequently used for the identification and classification of microorganisms, likely due to their specificity and variability [19,22,23,27,28]. In one study, the V1-V3 regions of the 16S rRNA were used [26], while in two other studies the V4 region was used [20,25]. Gomes et al used the V3-4 and V4-V6 regions.
3.2.2. Inclusion/Exclusion Criteria
Inclusion and exclusion criteria varied among the studies, with differences likely attributable to the specific objectives of each study. Jang, H.J. et al. (2021) and (2023) focused on patients pathologically diagnosed with NSCLC between 2018 and 2020 [19,20]. Zhuo M et al. (2020) included lung cancer patients who had not received cancer treatment. [21] Gomes S et al. (2020) enrolled subjects undergoing bronchoscopy for lung disease evaluation at three Portuguese hospitals. [22] Seixas S et al (2021) selected lung cancer patients, excluding those primarily diagnosed with COPD or ILD from the non-LC group, and did not collect healthy control samples. [23] Wang K et al. (2019) included patients with primary bronchogenic carcinoma, confirmed and without glucocorticoid or antibiotic treatment for at least 30 days prior to sample collection. [24] Lee SH et al. (2016) considered patients admitted for lung mass evaluation between May and September 2015, excluding individuals under 20 years of age, pregnant women, or those who had undergone procedures other than bronchoscopy for lung mass evaluation. [25] Bingula R et al. (2020) included NSCLC patients eligible for surgical treatment, aged 18-80, BMI < 29.9, with no previous airway surgery or cancer treatment, no recent antibiotherapy, corticotherapy, immunosuppressive drugs, or pulmonary infections in the past two months. [26] Liu B et al. (2022) incorporated patients with lung cancer, excluding those who had used antibiotics, corticoids, probiotics, prebiotics, or immunosuppressive drugs in the past three months, as well as patients with hypertension, diabetes, previous airway surgery, preoperative radiotherapy and chemotherapy, and atomization treatment. [27]
3.2.3. Bronchoalveolar Lavage Sample Collection
The techniques used for sample collection varied across all studies. Jang HJ et al.(2021) (2023) employed topical anesthesia (lidocaine) via a nebulizer, followed by administration of midazolam and fentanyl. In the affected segment, the bronchi were flushed with 30 to 50 mL of sterile saline (0.9%), yielding approximately 15 mL of bronchoalveolar lavage (BAL) fluid. These samples were immediately frozen at -70 °C and DNA extraction was conducted within 24 hours. [19,20] Zhuo M et al. (2020) provided limited details on their collection method: using a transbronchoendoscope to avoid contamination, they obtained paired BALF samples from lung cancer patients - one from the cancerous lung and the other from the contralateral non-cancerous lung, with samples immediately frozen at -80C for subsequent DNA extraction. [21] Gomes S. et al.(2019) focused on affected lung segments, using bronchoscope wedging into subsegmental regions and collecting samples via bronchoscope working channel washes, done twice with a minimum of 15 mL, 0.9% saline solution. [22] Seixas S et al (2021) targeted affected lung segments, with BALF samples of at least 15 mL, 0.9% saline solution. Samples were initially stored at temperatures ranging from -20 to 4 °C and later transferred to -80 °C. [23] Wang K et al. (2019) used local anesthesia and flexible fiberoptic bronchoscopy to collect samples from subsegmental bronchi. Three aliquots of 50 mL sterile saline were instilled and gently aspirated, with the samples collected in a siliconized plastic bottle and stored on ice. [24] Lee SH et al. (2016) applied topical anesthesia (lidocaine) via nebulizer, followed by sedation, to perform bronchoalveolar lavage (BAL). The procedure was standardized to collect 10 mL of BALF using about 30 ml sterile saline from the lung opposite the mass. [25] Bingula R et al. (2020) extracted samples from removed lungs or lobes. Non-malignant lung tissue was identified, and 2 × 40 mL of sterile saline was instilled into the bronchus, retrieving 8–10 mL in total. [26] Lastly, Liu B et al. (2022) obtained sterile saline samples of bilateral lungs by bronchoscopy in lung cancer (LC) patients, collecting paired samples of BALF from both the cancerous and contralateral noncancerous lobes. [27]
3.2.4. Study Conclusions
Bingula R et al. (2020) confirmed that the pulmonary and oral microbiomes differ in both taxonomy and diversity, and that the tumor's location in the upper or lower lobes can influence the microbiome. The microbiome was not compared based on the type or stage of cancer. The number of patients was limited. [26]
Wang K et al. (2019) showed that both in the lung and oral levels, patients with lung cancer have less lung and oral microbiota diversity than healthy controls, and the composition of the microbiome is different in patients with lung cancer compared to healthy subjects. Pseudomonas was enriched in patients with adenocarcinoma and small cell lung cancer, while Veillonella and Corynebacterium were abundant in BAL in patients with squamous carcinoma. Lactobacillus was enriched in patients with small cell lung cancer. Rothia was observed to be significantly different in the adenocarcinoma group. Treponema and clinical lung cancer markers, including SCCA, CA125, CK-19, CA-199, and CEA, were observed in the BALF samples. [24]
In the study comparing the microbiome in patients with low PD-L1 (<10%) versus high PD-L1 (≥10%) group by Jang, H.J. et al. (2021), it was highlighted that the abundances of Neisseria and Veillonella differed significantly in relation to PD-L1 expression levels and immunotherapy responses. There was also no significant difference in alpha and beta diversity, with Haemophilus being dominant in the immunotherapy non-responder group. [19]
Zhuo M et al. (2020) showed that there is a difference between the microbiome of the cancerous lung compared to the healthy lung in the same patient, and the genera Spiroplasma and Weissella were significantly enriched in the cancerous lung. The top three dominant phyla, classes, orders, and families were the same in both the healthy and cancerous lungs, as well as the top dominant genera. However, the third at the genera level was Alloprevotella in the affected lung and Prevotella in the healthy lung. There were no significant differences in terms of alpha diversity and overall composition of the microbiome. They found a greater abundance of phylum Tenericutes, as well as its class Mollicutes and its genus Spiroplasma. [21]
Gomes S et al. (2019) found that scuamos cell carcinoma cases were on average more diverse than adenocarcinoma, a result that can be related to a heavier smoking load in these patients. [22]
Seixas S et al. (2021) showed that COPD, ILD, and LC varied not only in microbial composition and evenness, but also in the proportions of Prevotella and Haemophilus. Regarding alpha or beta diversity, no significant differences were found between the non-cancer and lung cancer groups, possibly related to the heterogeneity of the cancer types, with only 34.7% of adenocarcinoma and 10.5% of scuamos cell carcinoma subtypes. In the cancer group, Streptococcus was significantly increased compared to the non-lung cancer group, and Prevotella was increased in the lung cancer group compared to the ILD group. [23]
Lee SH et al. (2016) compared the microbiomes of patients with lung cancer and those with benign mass-like lesions. In patients with lung cancer, an increased presence at the phyla level of Firmicutes (p=0.037) and TM7 (p=0.035) was observed compared to benign tumors. Also, at the genera level, a relative abundance was noted for Veillonella (p=0.003) and Megasphaera (p=0.022), suggesting a potential role in cancer for Veillonella and Megasphaera. Furthermore, a more complex diversity with higher abundance in α-diversity was noted in patients with cancer. [25]
The microbiome composition was different in patients with LC compared to controls, with a decrease in alpha diversity and abundance in Streptococcus, Prevotella, Veillonella, and Haemophilus in the study conducted by Liu B et al. (2022). Fusobacterium was also increased in patients with LC compared to controls. [27]
Jang HJ et al. (2023) in the study comparing the microbiome based on the histological type of lung cancer highlighted a significant difference between the two groups in terms of α- and β-diversities (p=0.004 for Chao1, p=0.001 for Simpson index, and p=0.011 for PERMANOVA), being significantly more diverse in patients with adenocarcinoma compared to squamous carcinoma. There was also a significant difference in stages I, II, and IIIA compared to stage IIIB and IV in terms of alpha diversity in patients with NSCLC. In patients with squamous carcinoma, Actinomyces graevenitzii was dominant. In patients with adenocarcinoma, populations of Haemophilus parainfluenza, Neisseria subflava, Porphyromonas endodontalis, and Fusobacterium nucleatum were significantly more abundant compared to squamous carcinoma. [20]
3.3. Proportional Distribution of Microbial Phyla and Genera in Lung Cancer
In three of the twelve samples, the dominant phylum is Firmicutes. Moreover, in all twelve samples, the percentage of Firmicutes exceeds 20%, ranging from 23.2% (with a 95% confidence interval between 0.14173216 and 0.32226784) to 47.11% (with a 95% confidence interval between 0.331336623 and 0.610863). [19,23]. In five of the samples, the dominant phylum was Bacteroidetes, with the highest percentage being 40.8% (95% confidence interval between 0.302900807 and 0.513099193) [20], while in the rest, it was below 40%, with some studies reporting as low as 13.3%. [11,16] Proteobacteria was the dominant phylum in four samples, with the highest percentage at 45.05% (95% confidence interval between 0.081921229 and 0.819078771), and the lowest at 19.1% (95% confidence interval between 0.106938063 and 0.275061937). [14,19]
At the phylum level in the study led by Zeng W et al., Firmicutes, Bacteroidetes, and Fusobacteriota were identified as being highly abundant in the lung cancer group. [28]
In healthy, non-smoking individuals, the Bacteroidetes phylum predominates, accounting for over 60% of the microbiome composition.[29,30,31]. Conversely, in smokers, the dominant phylum is Actinobacteria, exceeding 40%, followed by Proteobacteria and Bacteroidetes. [29] Advanced stages of Chronic Obstructive Pulmonary Disease (COPD) are characterized by a shift from the Bacteroidetes phylum towards Proteobacteria, and sometimes towards Firmicutes. [32] In sarcoidosis, the dominant phyla are Actinobacteria and Proteobacteria, while in Interstitial Lung Disease (ILD), Proteobacteria prevails. [33]Proteobacteria, identified as one of the predominant bacterial phyla in individuals with asthma. [34]
Regarding genus diversity, there is considerable variability observed. In some batches, Pseudomonas was found to have the highest percentage, 10.3% [26] and 35.14% [27], whereas in other batches, the percentage was significantly lower. [19] Streptococcus had a substantial presence across all batches, ranking within the top five genera in each. The presence of Veillonella, suspected in several studies of playing a role in bronchopulmonary cancer, varied between 1.4% [26] and 11.4% [25], being over 4% in most batches, except for Bingula et al. The presence of Haemophilus, known for its role in COPD, showed wide variability across batches, from less than 1% [26], 1.07% [27] to 29.5%. [22] A relatively constant presence in the "top" ranks was the genus Neisseria.
In a study, conducted by Jun-Chieh J. Tsay in 2021, several Operational Taxonomic Units (OTUs) identified as belonging to the genera Veillonella, Prevotella, and Streptococcus were found to be enriched in samples from subjects with a worse prognosis.[35]
In other analysis of 13 patient samples with Non-Small Cell Lung Cancer (NSCLC), the genera Streptococcus, Vibrio, and Enterobacter were identified as the most prevalent. [10]. In the study conducted by Zeng W et al., the bacteria Prevotella, Veillonella, and Neisseria were found to be highly abundant in the group of patients with lung cancer. [28]
Regarding COPD, besides the increased presence of the genus Streptococcus, the significant presence of Haemophilus in high percentages was notable. Additionally, in smokers, the percentage of Haemophilus is significant. [31,36] In healthy, non-smoking subjects, the predominant genera are Prevotella, Veillonella, Actinomyces, and Streptococcus [29,36,37,38]. In patients with sarcoidosis, the genera Prevotella, Streptococcus, and Corynebacterium are significantly present [33,39], while in patients with IPF, the most significant genus is Streptococcus (30%), but Prevotella, Veillonella, or Staphylococcus are also noteworthy. [33,40]Pleural fluid from cases of malignant pleural effusion, both from Lung Cancer and Mesothelioma, was found to be enriched with bacteria typically considered to be commensals from oral and gut origins, including genera such as Rickettsiella, Ruminococcus, Enterococcus, and the order Lactobacillales. [41]
3.4. Patient Demographics and Tumor Histology in Selected Studies
Of the nine selected studies, six were conducted on Asian populations, 292 patients, and three on European populations, 113 participants, totaling 405 patients. There were 279 male participants and 133 female participants. In one study, three samples were excluded (2 BALF due to an insufficient number of reads (< 1000), and one sample did not specify the sex of the patients whose samples were eliminated. In another study, four samples were excluded because the samples failed to amplify using PCR, without specifying the sex of the patients whose samples were excluded. It should also be noted that likely two studies were conducted on the same patient cohort, totaling 84 patients (Jang HJ 2021 and 2023). Regarding tumor histology, lung cancer ADK accounted for 218 cases, squamous cell carcinoma for 91 cases, 1 large cell carcinoma, small cell lung cancer for 39 cases, carcinoid for 2 cases, NOS for 10 cases, 4 metastases, 2 from colorectal cancer, one from breast cancer, and 1 from renal cancer, and 47 cases were unknown. [19,20,21,22,23,24,25,26,27]
3.5. Alpha Diversity
Regarding alpha diversity, studies comparing lung cancer with controls found that lung cancer patients had less lung and oral microbiota diversity than healthy controls, lower abundance in alpha diversity compared with the non-lung cancer group. Among those with lung cancer and benign tumors, it was noted that cancer patients exhibited more complex diversity with higher abundance and α-diversity. Alpha-diversity indices did not vary significantly between LC and non-LC groups. [23,24,25,27]
The results regarding the microbiome diversity in patients with squamous carcinoma and adenocarcinoma differ between the study conducted by Jang HJ et al. and that by Gomes S et al. [20,22]
No significant difference was noted in terms of alpha diversity in patients with PD-L1 < 10% compared to those with PD-L1 >10% (p = 0.307 for Shannon; p = 0.540 for Simpson index). In lung cancer patients, no significant difference in alpha diversity was observed between cancerous and healthy lungs: Shannon (P = 0.871) and Simpson diversity index (P = 0.627). [19,21]
4. Discussion
There is considerable variability in the inclusion and exclusion criteria across the studies, indicating tailored patient selection strategies based on the unique objectives and parameters of each study. Most studies primarily included patients with lung cancer, with specific subgroups like NSCLC being a common focus. Common exclusion criteria included recent treatment with antibiotics, corticoids, or other specific medications, underlying conditions like hypertension or diabetes, and previous surgeries or therapies related to the airways. [24,26,27]Most studies conducted to date have involved a small cohort of patients, with varying inclusion/exclusion criteria.
The existence of standardized inclusion and exclusion criteria would significantly contribute to obtaining more efficiently comparable results in microbiome studies. It's crucial to consider the use of antibiotics, as they are known to influence the microbiome. The anti-inflammatory effect of corticosteroids, as well as the potential impact of other medications such as immunosuppressants or probiotics, should also be considered. [42] Developing a comprehensive list of medications that could affect the composition of the pulmonary microbiome, to be restricted prior to sample collection, would be beneficial. The specific duration for which these treatments should be ceased prior to the procedure is another critical aspect. Chronic pathologies that can influence the microbiome composition, such as bronchial asthma, COPD, and ILD, must be taken into account. [6,32,42]The pulmonary microbiome in these conditions has been extensively studied and continues to be a research focus. Additionally, smoking status, including current and former smokers, significantly affects the microbiome composition and should be factored into the study design. [29] These elements could serve as inclusion criteria for studies focusing on patients with these associated pathologies. Conversely, they might also be considered as exclusion criteria to eliminate factors that could further alter the microbiome, such as recent pulmonary infections or autoimmune diseases. [43,44]Age restrictions and health status (e.g., absence of pregnancy, specific BMI range) were also crucial criteria in patient selection, reflecting the need to control for variables that could influence study outcomes.
There is a notable diversity in the sample collection methods across different studies, reflecting tailored approaches based on specific study requirements and patient conditions. Despite variations, common practices include the use of topical anesthesia, saline washes, and immediate freezing of samples for DNA extraction.
For future uniformity of results, it might be beneficial to establish a standard protocol for performing bronchoscopy for lavage purposes. This protocol could include pre-procedural rinsing of the cavity with saline solution, local anesthesia with or without sedation, specification of the volume of saline to be introduced for lavage, and ensuring that the lavage is performed prior to any biopsy procedures, if a biopsy is necessary. Additionally, the collection of a “background sample” is crucial for accurate interpretation of results. [38] Furthermore, it might be valuable to standardize the methods for storing and processing these samples, including specific temperature control requirements and time frames for processing to minimize degradation and ensure sample integrity. [45]Uniform data recording and reporting protocols could also be implemented to facilitate more effective comparison and analysis across different studies.
All included studies use Illumina sequencing platforms (MiSeq or HiSeq) for sample analysis. This choice is probably due to the high accuracy and efficiency these platforms offer in DNA sequencing. [19,20,21,22,23,24,25,26,27]
For sample preparation, there are variations in DNA extraction methods and the kits used, such as the FastDNA Spin Kit, PowerSoil DNA Isolation Kit, or kits from Qiagen. This variety suggests that there is no universal standard for DNA extraction from BAL samples, with each laboratory adapting the method according to its specific resources and objectives. [19,21,22,23,25]
Also, different databases and sequence classification programs (such as QIIME, EzTaxon-e, GenBank, or EzBioCloud) are used for taxonomic analysis, indicating diversity in data interpretation and in the identification of microorganisms present in the samples.
There is growing interest and an increasing number of studies regarding the pulmonary microbiome in lung cancer, as it may serve as a potential biomarker for diagnosis, monitoring progression, and treatment response in bronchopulmonary cancer.Given that bronchoscopy is frequently utilized in the diagnosis of lung cancer, the collection of bronchoalveolar lavage fluid (BALF) alongside biopsy specimens could represent a promising strategy to enhance predictive capabilities. [46]
To date, a pattern indicating the percentage expression of the lung microbiome in bronchopulmonary cancer cannot be utilized.
The continuation of studies on the lung microbiome in bronchopulmonary cancer is necessary, but there is a need for well-defined, universally accepted inclusion/exclusion criteria, similar collection techniques, and proper storage, transport, and processing of samples. Additionally, the databases used, DNA extraction techniques, and kits must provide reproducible, consistent data.
Larger patient cohort studies are needed to explore the pulmonary microbiome in bronchopulmonary cancer in relation to race, populations, environment, cancer type, cancer stage, associated pathologies, and smoking status.
Furthermore, it is essential to consider the integration of pulmonary microbiome studies with other types of data, such as genomic and proteomic analyses, to enable a more comprehensive and multidimensional approach in understanding the complex interactions in bronchopulmonary cancer.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; et al. Global Cancer Statistics 2020 GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr Rev. 2013, 70, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Luo, J.L.; Ali, M.K.; Spiekerkoetter, E.; Nicolls, M.R. The Human Respiratory Microbiome: Current Understandings and Future Directions. Am J Respir Cell Mol Biol. 2023, 68, 245–255. [Google Scholar] [CrossRef]
- Natalini, J.G.; Singh, S.; Segal, L.N. The dynamic lung microbiome in health and disease. Nat Rev Microbiol. 2023, 21, 222–235. [Google Scholar] [CrossRef]
- de Steenhuijsen Piters, W.A.A.; Binkowska, J.; Bogaert, D. Early Life Microbiota and Respiratory Tract Infections. Cell Host Microbe. 2020, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.; Ege, M.J. The Lung Microbiome E vM, editor.: European Respiratory Society; 2019.
- Mathieu, E.; Escribano-Vazquez, U.; Descamps, D.; Cherbuy, C.; Langella, P.; et al. Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Frontiers. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet. 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Yagi, K.; Huffnagle, G.B.; Lukacs, N.W.; Asai, N. The Lung Microbiome during Health and Disease. Int J Mol Sci. 2021, 22, 10872. [Google Scholar] [CrossRef]
- Zheng, L.; Sun, R.; Zhu, Y.; Li, Z.; She, X.; et al. Lung microbiome alterations in NSCLC patients. Sci Rep. 2021, 11, 11736. [Google Scholar] [CrossRef]
- Kuczynski, J.; Liu, Z.; Lozupone, C.; McDonald, D.; Fierer, N.; et al. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nature methods. 2010, 7, 813–9. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Riddle, S.; McPhillips, T.; Ludäscher, B.; Eisen, J.A. Introducing W.A.T.E.R.S.: a Workflow for the Alignment, Taxonomy, and Ecology of Ribosomal Sequences. BMC Bioinformatics. 2010, 11, 317. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A Core Human Microbiome as Viewed through 16S rRNA Sequence Clusters. PLoS ONE. 2012, 7. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011, 12. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021, 372, n71. [Google Scholar] [CrossRef]
- Jang, H.J.; CJKKYSKYKSea. Relationship of the lung microbiome with PD-L1 expression and immunotherapy response in lung cancer. Respir Res. 2021, 22, 322. [Google Scholar] [CrossRef]
- Jang, H.J.; Lee, E.; Cho, Y.J.; Lee, S.H. Subtype-Based Microbial Analysis in Non-small Cell Lung Cancer. Tuberc Respir Dis. 2023, 86, 294–303. [Google Scholar] [CrossRef]
- Zhuo, M.; An, T.; Zhang, C.; Wang, Z. Characterization of Microbiota in Cancerous Lung and the Contralateral Non-Cancerous Lung Within Lung Cancer Patients. Front Oncol. 2020, 10, 1584. [Google Scholar] [CrossRef]
- Gomes, S.; Cavadas, B.; Ferreira, J.C.; Marques, P.I.; Monteiro, C.; Sucena, M.; et al. Profiling of lung microbiota discloses differences in adenocarcinoma and squamous cell carcinoma. Sci Rep. 2019, 9, 12838. [Google Scholar] [CrossRef] [PubMed]
- Seixas, S.; Kolbe, A.R.; Gomes, S.; Sucena, M.; Sousa, C.; Rodrigues, L.V.; et al. “Comparative analysis of the bronchoalveolar microbiome in Portuguese patients with different chronic lung disorders. Scientific reports. 2021, 11, 15042. [Google Scholar] [CrossRef]
- Wang, K.; Huang, Y.; Zhang, Z.; Liao, J.; Ding, Y.; Fang, X.; et al. A Preliminary Study of Microbiota Diversity in Saliva and Bronchoalveolar Lavage Fluid from Patients with Primary Bronchogenic Carcinoma. Med Sci Monit. 2019, 25, 2819–2834. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Sung, J.Y.; Yong, D.; Chun, J.; Kim, S.Y.; Song, J.H.; et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer. 2016, 102, 89–95. [Google Scholar] [CrossRef]
- Bingula, R.; Filaire, E.; Molnar, I.; Delmas, E.; Berthon, J.Y.; Vasson, M.P.; et al. Characterisation of microbiota in saliva, bronchoalveolar lavage fluid, non-malignant, peritumoural and tumour tissue in non-small cell lung cancer patients: a cross-sectional clinical trial. Respir Res. 2020, 21, 129. [Google Scholar] [CrossRef]
- Liu B, Li Y, Suo L, Zhang W, Cao H, Wang R et all. l. 2022;12:1058436. Published 2022 Nov 15. Characterizing microbiota and metabolomics analysis to identify candidate biomarkers in lung cancer. Front Oncol. 2022, 12, 1058436.
- Zeng, W.; Zhao, C.; Yu, M.; Chen, H.; Pan, Y.; Wang, Y.; et al. Alterations of lung microbiota in patients with non-small cell lung cancer. Bioengineered. 2022, 3, 6665–6677. [Google Scholar] [CrossRef]
- Liu, X.; Sun, W.; Ma, W.; Wang, H.; Xu, K.; et al. Smoking related environmental microbes affecting the pulmonary microbiome in Chinese population. Sci Total Environ. 2022, 829, 154652. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; Lisa McCloskey, L.; Beck, J.M.; et al. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Annals of the American Thoracic Society. 2015, 12, 821–830. [Google Scholar] [CrossRef]
- Karakasidis, E.; Kotsiou, O.S.; Gourgoulianis, K.I. Lung and Gut Microbiome in COPD. Journal of personalized medicine. 2023, 13, 804. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu Rev Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Shariff, M.; Chaturvedi, G.; Chaturvedi, G.; Sharma, A.; Goel, N.; et al. Comparative analysis of the alveolar microbiome in COPD, ECOPD, Sarcoidosis, and ILD patients to identify respiratory illnesses specific microbial signatures. Sci Rep. 2021, 11, 3963. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Nelson, C.E.; Brodie, E.L.; DeSantis, T.Z.; Baek, M.S.; et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. The Journal of allergy and clinical immunology. 2011, 127, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Jun-Chieh, J.T.; Benjamin GWu Sulaiman, I.; Gershner, K.; Schluger, R.; et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer discovery. 2021, 11, 293–307. [Google Scholar]
- Einarsson, G.G.; Comer, D.M.; McIlreavey, L.; Parkhill, J.; Ennis, M.; et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016, 71, 795–803. [Google Scholar] [CrossRef]
- Loverdos, K.; Bellos, G.; Kokolatou, L.; Vasileiadis, I.; Giamarellos, E.; et al. Lung Microbiome in Asthma: Current Perspectives. J Clin Med. 2019, 8, 1967. [Google Scholar] [CrossRef]
- Morris, A.; Beck, J.M.; Schloss, P.D.; Campbell, T.B.; Crotherset, K.; et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013, 187, 1067–1075. [Google Scholar] [CrossRef]
- Becker, A.; Vella, G.; Galata, V.; Rentz, K.; Beisswenger, C.; et al. The composition of the pulmonary microbiota in sarcoidosis – an observational study. Respir Res. 2019, 20. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russellet, K.E.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Kwok, B.; Wu, B.G.; Kocak, I.F.; Sulaiman, I.; Schluger, R.; et al. Pleural fluid microbiota as a biomarker for malignancy and prognosis. Sci Rep. 2023, 13, 2229. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019, 7, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Huffnagle, G.B. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. 2014, 2, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Joshua, V.; Artacho, A.; Abdollahi-Roodsaz, S.; Öckinger, J.; et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome. 2016, 4, 60. [Google Scholar] [CrossRef]
- Wiscovitch-Russo, R.; Singh, H.; Oldfield, L.M.; Fedulov, A.V.; Gonzalez-Juarbe, N. An optimized approach for processing of frozen lung and lavage samples for microbiome studies. PLoS One. 2022, 17, Apr. [Google Scholar] [CrossRef] [PubMed]
- Masuhiro, K.; Tamiya, M.; Fujimoto, K.; Koyama, S.; Naito, Y.; Osa, A.; et al. Bronchoalveolar lavage fluid reveals factors contributing to the efficacy of PD-1 blockade in lung cancer. JCI Insight. 2022, 7, e157915. [Google Scholar] [CrossRef]
- Doocey, C.M.; Finn, K.; Murphy, C.; Guinane, C.M. The impact of the human microbiome in tumorigenesis, cancer progression, and biotherapeutic development. BMC Microbiol. 2022, 22, 53. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



