Submitted:
16 August 2024
Posted:
19 August 2024
Read the latest preprint version here
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by cognitive impairment and memory loss. Oxidative stress (OS) has emerged as a central element in the pathophysiology of AD, linking various pathological features including amyloid-β (Aβ) plaque formation, tau hyperphosphorylation, and synaptic dysfunction. This review comprehensively examines the role of OS in AD, focusing on the mechanisms of reactive oxygen species (ROS) production, mitochondrial dysfunction, and their impact on neuronal integrity. Additionally, the review highlights recent advances in antioxidant therapy. Elevated ROS levels in the aging brain exacerbate oxidative damage to lipids, proteins, and DNA, contributing to neuronal atrophy and synaptic loss. Mitochondrial dysfunction further amplifies OS, disrupting cellular energy metabolism and promoting neurodegeneration. Despite the strong association between OS and AD, antioxidant therapies have shown inconsistent clinical outcomes. Emerging strategies targeting offer promising avenues for therapeutic intervention. This review highlights the need for a multifaceted approach in understanding and mitigating OS-related damage in AD, aiming to pave the way for more effective treatments and improved patient outcomes.
Keywords:
Alzheimer's disease
; Oxidative stress
; Mitochondrial dysfunction
; Antioxidant
; Reactive oxygen species.
1. Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that significantly impacts the elderly population and primarily manifests as cognitive impairment, memory loss, and compromised daily activities [1]. Globally, AD stands as the foremost cause of dementia accounting for 60-80% of all cases, and is the sixth leading cause of death among Americans aged 65 and older [2,3]. Approximately 6.7 million Americans currently live with AD with worldwide cases exceeding 55 million- a number projected to nearly triple by 2050 [3,4]. This escalating prevalence, along with an aging global demographic, underscores the formidable challenge AD presents to healthcare systems worldwide [3].
AD is marked by two primary neuropathological hallmarks: the extracellular deposition of amyloid-β (Aβ) peptides forming diffuse and neuritic plaques and the intracellular accumulation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau proteins [5,6]. These core features are widespread across the brain, inducing neuronal atrophy and synaptic loss, ultimately leading to neurodegeneration [6,7]. While these hallmarks are fundamental to understanding AD, they are part of a more complex pathogenesis. The development of AD also involves a broader spectrum of pathophysiological changes, including neuroinflammation, blood-brain barrier (BBB) dysfunction, mitochondrial dysfunction, and oxidative stress (OS) [8,9,10]. Multiple hypotheses have been proposed to explain these diverse aspects of AD, encompassing the amyloidogenic cascade, tauopathy, neurovascular dysfunction, and the impacts of OS and neuroinflammation [9,11]. Each of these hypotheses is integrated through various mechanistic relationships, suggesting a multifaceted and interconnected approach is crucial for fully understanding and addressing the progression of AD, as shown in Figure 1 [12]. Building upon these core hypotheses, OS emerges as a central element in the pathophysiology of AD, particularly as it bridges several core hypotheses including amyloid cascade, tau protein, inflammation, and metal ions [13,14]. As the brain ages, its metabolic balance tilts towards a pro-oxidative state, exacerbating the imbalance between the generation of reactive oxygen species (ROS) such as superoxide radicals (O2•−)and hydrogen peroxide (H2O2), and the body's antioxidant defenses [13,14,15]. This imbalance leads to significant tissue damage, particularly in the presence of metal ions like iron and copper, which catalyze harmful redox reactions [16,17].
The brain's high oxygen demand which accounts for 20% of the total body consumption is coupled with its abundance of peroxidation-sensitive lipids and cerebrospinal fluid that poorly binds iron hence heightening its vulnerability to oxidative damage [18]. Additionally, the brain's modest antioxidant defenses can cause higher susceptibility to OS and neurodegeneration [19]. This environment can lead to neuronal damage through possible mechanisms such as increased intracellular calcium, excitotoxicity, and the breakdown of cellular components including lipids, proteins, and DNA [20,21]. The neurotoxicity of Aβ peptides and hyperphosphorylated tau proteins synergistically lead to the accumulation of neurofibrillary tangles, synaptic loss, and cholinergic denervation, which are well-documented and exacerbated by OS [10,22,23].
Furthermore, mitochondrial dysfunction is a significant factor in AD's progression, evidenced by the reduced number and impaired functionality of mitochondria, largely due to defects in the electron transport chain (ETC) enzymes [24,25]. This mitochondrial impairment fosters ROS accumulation and further OS, disrupting calcium homeostasis and signal transduction, ultimately contributing to synaptic loss and neurodegeneration [26,27]. Extensive research, including studies by Butterfield’s group, has documented extensive oxidative damage in AD brains, linking it to the marked accumulation of Aβ and neurofibrillary tangles [24,28,29]. The complex interplay between ROS/RNS and the cellular antioxidant defenses underscores the importance of understanding these reactive species' sources, regulation, and effects. This knowledge is crucial for developing therapeutic strategies to mitigate OS-related damage in various diseases, including AD [30]. This body of work underscores the critical role of redox-mediated mechanisms in both the pathogenesis and progression of AD [31].
Recent therapeutic advances have been particularly noteworthy in light of this growing health concern. In particular, the U.S. Food and Drug Administration (FDA) has granted accelerated approval of Aducanumab and Lecanemab, reflecting the general urgency for therapeutic strategies for AD [32]. These monoclonal antibodies target amyloid-beta peptides implicated in AD pathology and are designed to modify disease progression, contrasting with traditional symptomatic treatments such as acetylcholinesterase inhibitors and N-methyl-D-aspartate antagonists like memantine [33]. The emergence of these therapies highlights a shift towards more targeted approaches that address the underlying mechanisms of AD, reflecting the ongoing need for a comprehensive and multidimensional strategy in combating this pervasive disease.
Figure 1.
This figure illustrates various pathophysiological mechanisms involved in Alzheimer's disease.
Figure 1.
This figure illustrates various pathophysiological mechanisms involved in Alzheimer's disease.
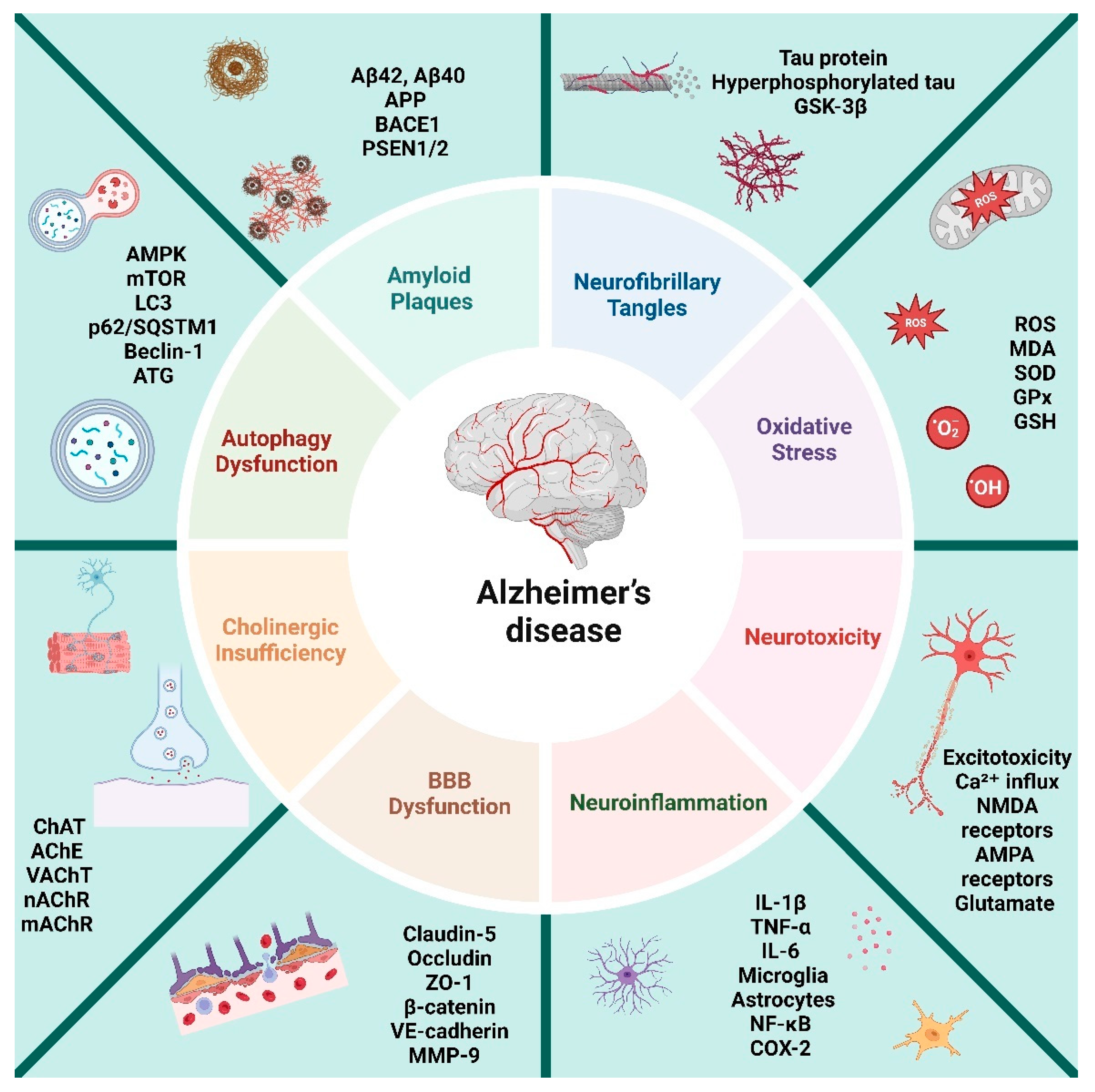
Despite mounting evidence implicating OS as a pivotal factor in the development and progression of AD. However, the exact mechanisms and interactions between OS and AD pathology remain unclear. Additionally, clinical trials utilizing antioxidant therapies have yielded inconsistent and often disappointing results. This review aims to provide a comprehensive exploration of the intricate roles played by OS and mitochondrial dysfunction in AD pathophysiology. We will delve into the complex interactions between oxidative damage and key pathological features of AD, such as amyloid-beta plaques, tau hyperphosphorylation, and synaptic dysfunction. Furthermore, we will assess how mitochondrial dysfunction exacerbates disease progression and contributes to neurodegeneration. By synthesizing current research findings, this review seeks to enhance our understanding of the mechanisms underlying AD and the dual role that ROS play in both neuronal health and disease. In addition, we will highlight recent advancements in antioxidant therapies and innovative strategies targeting oxidative stress, offering promising avenues for the development of more effective treatments for AD.
2. Method
In preparing this review, we systematically searched the PubMed and Google Scholar databases for articles published from 1994 to 2024. This search strategy incorporated key terms, such as oxidative stress, mitochondrial dysfunction, and Alzheimer’s disease, to capture studies that specifically addressed these factors in AD. The inclusion criteria targeted research articles that provided insights into the impact of oxidative stress and mitochondrial dysfunction on AD’s pathological markers or progression, whether through in vitro, in vivo, or clinical investigations. Articles focusing on non-neurological aspects of oxidative stress or mitochondrial dysfunction were excluded from the review to maintain a clear focus on AD. Our aim was to present a balanced overview to ensure a broad representation of the current research landscape.
3. Oxidative Stress
OS results from an imbalance between the production of species ROS and reactive nitrogen species (RNS), and the cell's ability to detoxify these reactive intermediates or repair the resulting damage [34,35]. This imbalance, often linked to mitochondrial dysfunction, can lead to significant damage to cellular components including lipids, proteins, polysaccharides, and DNA [36]. Elevated ROS levels can cause free radical damage to cell membranes and DNA, adversely affecting cellular function and survival. This damage can create a self-perpetuating cycle, as OS can destroy biomolecules and further increase ROS production [37]. Therefore, maintaining adequate levels of antioxidants and antioxidant enzyme activity is crucial to mitigate the harmful effects of OS [38].
At the molecular level, OS involves various molecules and free radicals derived from molecular oxygen [39]. Free radicals are chemical species with an unpaired electron in their outer shell, making them highly reactive [40]. Molecular oxygen in its ground state is a bi-radical with two unpaired electrons sharing the same spin, making it relatively unreactive [34]. However, when one of these electrons is excited, it changes its spin, allowing the oxygen molecule to react readily with other electron pairs, particularly double bonds. This reaction produces singlet oxygen, a highly potent oxidant [30].
It is important to note that ROS must be present at low levels and cannot be eliminated, as they play a vital role in normal cellular functions including inducing plasticity changes in response to various cellular changes. However, excessive ROS levels are detrimental to cells [37,38].
The reduction of oxygen by one electron generates relatively stable intermediates, leading to the formation of an O2•− [34,41]. This anion is a precursor to most ROS and a key player in OS chain reactions. Antioxidants can partially reduce O2•− to form a hydroxyl radical (OH•), one of the most potent oxidants [39,41]. This process is catalyzed by reduced transition metals, which can be re-reduced by O2•− hence further perpetuating the cycle [35,41]. O2•− can also react with other radicals such as nitric oxide (NO•) and form peroxynitrite (ONOO−), an extremely potent oxidant driving reactive RNS [30,42].
ROS and RNS play dual roles in intracellular signaling, cell proliferation, and survival [43,44]. However, uncontrolled increases in their steady-state concentrations can lead to free radical-mediated chain reactions targeting critical cellular components [44,45]. In vivo, O2•− is primarily produced by mitochondria and regulated by both enzymatic and non-enzymatic processes [46,47]. The ETC leaks electrons to oxygen making it a significant source of O2•− in many tissues [45]. Key enzymatic sources of O2•− include Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidases in various cell membranes, cytochrome P450 enzymes, and H2O2-dependent oxygenases [39,48]. Another source is the conversion of xanthine dehydrogenase to xanthine oxidase [49]. Non-enzymatic production occurs via direct oxygen transfer by reduced coenzymes or prosthetic groups like flavin or iron-sulfur clusters or by xenobiotics after enzymatic reduction [50,51].
Mitochondria also employ several mechanisms to mitigate ROS and regulate the steady-state concentration of O2•− [52]. Superoxide dismutase (SOD) enzymes dismutate O2•− to H2O2, which is further reduced to water [51,52]. Mitochondrial SOD (MnSOD or SOD2) eliminates O2•− in the matrix or inner membrane, while copper-zinc SOD (SOD1) functions in the cytoplasm [53]. Cytochrome c in the intermembrane space reduces superoxide anion, regenerating oxygen [54]. Glutathione peroxidase (GPx) and catalase decompose O2•− and hydroxyl radicals (OH•). Additionally, ubiquinol (QH2) acts as a reducing agent, detoxifying various peroxides [55]. Mitochondria also possess DNA repair enzymes to correct oxidative damage, maintaining genetic integrity [30].
Apart from direct ROS generation, glial cells such as astrocytes and microglia, alongside redox-active metal ions, like copper and iron, can also contribute to cerebral levels of ROS/RNS [56]. This increased level can alter DNA, RNA, lipids, and proteins, potentially exacerbating the production of ROS/RNS molecules [42,57]. Consequently, the resulting DNA oxidation can affect gene transcription and replication, while the RNA oxidation primarily results in strand breaks and ribosomal dysfunction [57,58]. Lipid peroxidation, particularly that of unsaturated fatty acids, produces compounds like isoprostanes, malondialdehyde (MDA), and 4-hydroxynonenal (HNE), which impair cellular membrane integration and protein functions [59,60]. The resulting oxidative damage to mitochondrial components can exacerbate ROS production, damage mitochondrial DNA, and trigger apoptosis through mechanisms involving the redox-sensitive protein Thioredoxin-1 (Trx-1) and Apoptosis Signal-regulating Kinase 1 (ASK-1), as well as other redox proteins and the p53 pathway, which activates proapoptotic genes [61,62].
Figure 2.
This figure illustrates the relationship between mitochondrial dysfunction, OS, and AD. At the core of cellular metabolism, mitochondria generate energy through the electron transport chain (ETC), a series of protein complexes embedded in the inner mitochondrial membrane. The ETC facilitates the transfer of electrons from NADH and FADH2 through complexes I to IV, culminating in the reduction of oxygen to water and the synthesis of ATP via ATP synthase. During this process, ROS are generated as by-products. Excessive ROS production, as indicated in the figure, leads to OS, damaging cellular components such as lipids, proteins, and DNA. This oxidative damage is particularly detrimental to neuronal cells, contributing to the pathogenesis of AD. The schematic highlights how metabolic intermediates like glucose and pyruvate fuel the tricarboxylic acid (TCA) cycle, producing substrates for the ETC. However, dysregulation in these pathways exacerbates ROS production, ultimately promoting neurodegenerative processes associated with AD, as depicted by the brain image showing areas affected by the disease.
Figure 2.
This figure illustrates the relationship between mitochondrial dysfunction, OS, and AD. At the core of cellular metabolism, mitochondria generate energy through the electron transport chain (ETC), a series of protein complexes embedded in the inner mitochondrial membrane. The ETC facilitates the transfer of electrons from NADH and FADH2 through complexes I to IV, culminating in the reduction of oxygen to water and the synthesis of ATP via ATP synthase. During this process, ROS are generated as by-products. Excessive ROS production, as indicated in the figure, leads to OS, damaging cellular components such as lipids, proteins, and DNA. This oxidative damage is particularly detrimental to neuronal cells, contributing to the pathogenesis of AD. The schematic highlights how metabolic intermediates like glucose and pyruvate fuel the tricarboxylic acid (TCA) cycle, producing substrates for the ETC. However, dysregulation in these pathways exacerbates ROS production, ultimately promoting neurodegenerative processes associated with AD, as depicted by the brain image showing areas affected by the disease.
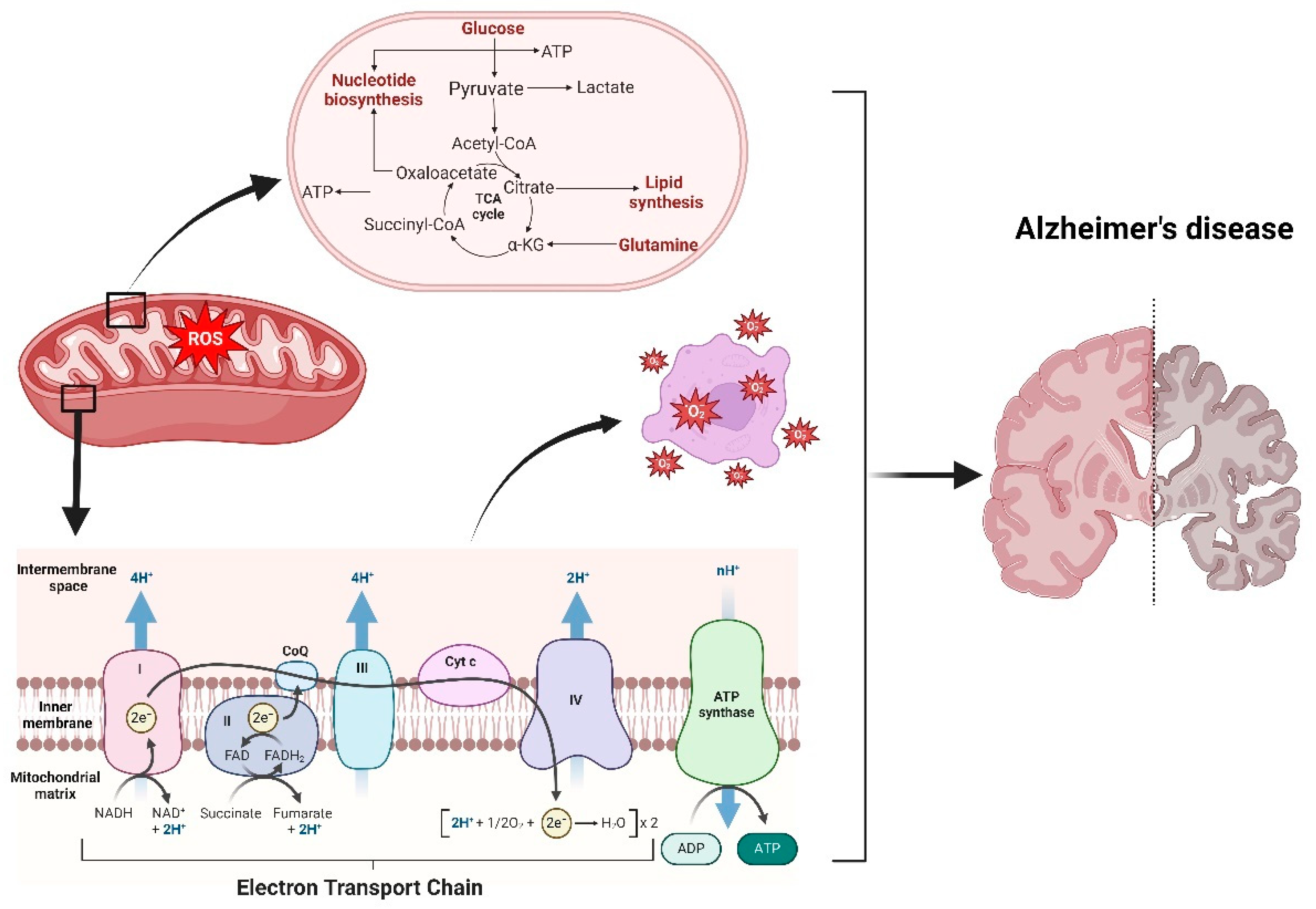
The complex interplay between ROS and RNS with cellular antioxidant defenses is crucial for understanding the sources, regulation, and mechanisms of action of these reactive species [30]. The accompanying figure illustrates how mitochondrial dysfunction leads to ROS production, highlighting the electron transport chain's role and its implications in OS and AD as shown in Figure 2.
4. Oxidative Stress in Alzheimer's Disease
The brain is highly susceptible to ROS-induced OS due to its significant energy demand, high oxygen requirement, and mitochondrial activity [14]. Neuronal cells contain high levels of lipids and iron but have fewer antioxidant enzymes compared to other tissues, making the brain particularly vulnerable to oxidative damage [63]. Both ROS and RNS can produce damaging reactive intermediates [64]. The OS burden increases with aging and is associated with decreased antioxidant defenses and neurogenesis [27,64].
In AD, extensive oxidative damage is observed and it’s closely linked to the abnormal accumulation of Aβ and NFTs [65]. Biometals such as iron, zinc, and copper play critical roles in Aβ aggregation and neurodegeneration, with copper exacerbating OS as a potent mediator of OH• found in elevated levels in amyloid plaques [16]. Elevated levels of oxidative damage markers are found in individuals with symptoms of preclinical Alzheimer's disease (PCAD), amnestic mild cognitive impairment (aMCI), and AD [39].
Markers of protein oxidation, such as protein carbonyls (PCs), are elevated in AD-affected brain regions rich in Aβ-peptide senile plaques [39]. Furthermore, lipid peroxidation markers, including protein-conjugated HNE, F2-isoprostanes, and F4-isoprostanes, are increased in patients diagnosed with AD, aMCI, and particularly in the hippocampus of patients with PCAD. [66]. Elevated 3-nitrotyrosine (3-NT) levels indicate damage by ONOO− and the biomarker 8-hydroxy-deoxyguanosine (8-OHdG) reflects oxidative damage to both nuclear and mitochondrial DNA [67,68]. RNA oxidation is also significant in AD reflected by oxidized, glycated, and nitrated proteins found within neuritic plaques and NFTs [69].
The consequences of oxidative and nitrosative damage include disrupted glucose metabolism, loss of ion gradients, impaired action potentials, and calcium imbalance [66]. Brain membrane phospholipids rich in polyunsaturated fatty acids are particularly susceptible to free radical attacks, leading to lipid peroxidation - a prominent feature in AD [16,70]. OS also affects protein function and can impair critical enzymes like GS and creatine kinase, which are reduced in AD pathology. This reduction is linked to altered glutamate concentrations, increased excitotoxicity, and decreased energy metabolism [71].
Due to the clear link between OS and AD, recent studies are attempting to establish reliable oxidative and nitrosative stress biomarkers for early AD diagnosis [56]. HNE is widely used as an indicator of OS and is increased in AD brains, particularly in the early stages [72]. Other indicators of oxidative and nitrosative stress in both early and late AD stages include PCs and protein nitration [73,74]. Observed increased levels of ROS and RNS are often accompanied by a decrease in antioxidant defenses in AD, leading to an increase in the total oxidative capacity [74].
5. Mitochondrial Dysfunction in Alzheimer's Disease
Mitochondria, the powerhouse of the cell, are pivotal in oxidative phosphorylation-driven energy production and generating adenosine triphosphate (ATP) [75]. This process makes them a significant source of reactive ROS due to ETC activity in the inner membrane [45]. A considerable amount of cellular H2O2 originates from mitochondria, which can quickly convert to superoxide (O2-) due to electron leakage during energy production [45,76].
Oxidative damage severely impacts proteins involved in mitochondrial ATP production and glycolysis [77]. Impaired cellular metabolism leads to increased ROS production and creates a feedback loop of OS and increased cellular damage [78]. Reduced ATP levels cause heightened mitochondrial activity, further escalating ROS production and electron leakage from the ETC [79]. This suboptimal mitochondrial functioning accompanied by increased ROS and decreased ATP production, is critical in AD pathogenesis [80].
Mitochondrial dysfunction is evident early in AD and affects various aspects of mitochondrial function including energy metabolism, calcium homeostasis, and the expression of mitochondrial and DNA (mtDNA) [81]. Glucose metabolism, closely linked to cognitive function, is an essential measure for monitoring AD progression [82]. In AD, energy metabolism genes such as those of the mitochondrial ETC, are decreased in the posterior cingulate cortex, a region affected early in the disease progression [83].
The reduced glucose metabolism in the AD brain is associated with decreased activity of mitochondrial ETC enzymes like pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase complex, and cytochrome oxidase [82,84]. This reduction correlates with clinical symptoms and is consistent with the presence of Aβ plaques. Furthermore, AD brains show higher levels of mtDNA oxidation and mutations compared to age-matched controls due to the proximity of mtDNA to ROS generation sites and the lack of protective histone proteins [85,86,87].
These mutations impact mitochondrial function and the number of mitochondria, which are lower in AD brains compared to controls [88]. Interestingly, mitochondrial size in AD brains is increased, which is attributed to impaired fusion and fission dynamics resulting from abnormal protein expression. Dihydrosphingosine phosphate lyase (DPL1) is a protein mainly expressed in the cytoplasm and is recruited to mitochondria during fission. DPL1 is increased in AD, and may also interact with Aβ and phosphorylated tau, contributing to AD pathophysiology [89]. Therefore, excessive mitochondrial fission may enhance oxidative and nitrosative stress in AD pathology.
Calcium, an essential cellular signaling messenger, is dysregulated in AD which consequently leads to neurons becoming more susceptible to neurodegeneration [90]. ROS produced from mitochondrial damage can disrupt calcium homeostasis and impair the endoplasmic reticulum's ability to buffer calcium which is detrimental to cell signaling and survival [91]. Increased levels of calcium-related enzymes in AD patients further highlight this proposed mechanism of cellular dysregulation [90].
Finally, mitochondria are crucial in apoptosis, with ROS activating caspases that induce pro-apoptotic proteins like Bcl-2-associated X protein (Bax) to translocate to the mitochondrial membrane [92]. This translocation results in the formation of the mitochondrial membrane permeability transition pore, releasing cytochrome c and triggering cell death [93]. Although there is some controversy regarding these processes, the role of mitochondrial dysfunction in AD is evident [94]. The intricate interplay between mitochondrial dysfunction and OS underscores the importance of early detection and potential therapeutic strategies aimed at restoring mitochondrial function and mitigating oxidative damage in AD [79].
6. Neurobiological Implications
6.1. Oxidative Stress and Aβ plaques
OS has several neurobiological implications that contribute to the pathogenesis of AD, including amyloid plaque formation, tau hyperphosphorylation, and synaptic dysfunction. The formation of Aβ plaques is intricately linked to OS, with Aβ peptides, particularly Aβ42, prone to aggregation and forming insoluble fibrils that deposit as plaques in the brain [95]. OS enhances the production and aggregation of Aβ [96]. ROS can induce the expression of amyloid precursor protein (APP) and influence the activity of secretases that process APP, favoring the production of amyloidogenic Aβ fragments [42]. Moreover, Aβ itself can generate ROS, creating a vicious cycle where OS promotes Aβ production, and Aβ, in turn, generates more ROS [96]. This self-propagating loop exacerbates oxidative damage and plaque formation, with the presence of biometals like iron, copper, and zinc in amyloid plaques further accelerating ROS production through Fenton reactions [96].
Further compounding this issue, Aβ has been shown to enhance ROS production and cause mitochondrial dysfunction, exacerbating OS [23,97]. Research indicates that brain regions with higher Aβ expression exhibit greater levels of protein oxidation and lipid peroxidation compared to the cerebellum, which is relatively low in Aβ [98]. Amyloid plaques predominantly contain Aβ with a methionine sulfoxide modification, suggesting that lipid peroxidation is an early event in neurodegeneration [99]. When incorporated into lipid bilayer membranes, Aβ induces OS, leading to lipid peroxidation and subsequent damage to nucleic acids and proteins [23,100]. Proteins are particularly susceptible to oxidative damage, which can result in irreversible structural modifications such as unfolding, aggregation, and the disassociation of subunits, ultimately leading to functional loss [101] .
In both AD patients and transgenic mice models, Aβ has been shown to interact with the mitochondrial enzyme Aβ-binding alcohol dehydrogenase (ABAD), causing mitochondrial dysfunction, increased ROS production, and eventual apoptosis [101,102]. Apoptosis, a primary mode of cell death in AD progression, is closely associated with elevated oxidative and nitrosative stress (OS&NS), as observed in AD models [103]. Treatment of AD fibroblasts with the Aβ peptide leads to the oxidation of anti-apoptotic proteins such as vimentin and heat shock protein 60 (HSP60). Similarly, neuroblastoma cells treated with Aβ (1-42) show increased oxidation of anti-apoptotic proteins glutaredoxin-1 (GRX-1) and TRX-1 [104] . Additionally, elevated levels of the pro-apoptotic protein p53 and its oxidized form have been detected in the AD brain, further contributing to increased apoptosis [105,106]. Overall, the induction of Aβ significantly contributes to the heightened oxidative status observed in AD.
6.2. Oxidative Stress and Tau Hyperphosphorylation:
Tau protein plays a critical role in stabilizing microtubules and ensuring smooth neuronal signal transmission [107]. Numerous studies have demonstrated that OS contributes to the hyperphosphorylation of tau proteins [108]. The peroxidation end product Carbonyl-4-HNE has been identified as a key factor in the accumulation of hyperphosphorylated tau [109]. However, the exact relationship between tau hyperphosphorylation and OS remains unclear. In animal studies, exposure to OS -inducing compounds has been shown to increase the activity of glycogen synthase kinase 3β (GSK3β), a Ser/Thr kinase that hyperphosphorylates tau proteins, thereby exacerbating disease pathology [109,110]. Besides GSK3β, OS also influences other kinases, promoting tau hyperphosphorylation [109]. Additional research has indicated that OS leads to a reduction of peptidyl prolyl cis-trans isomerase 1 (Pin1) in AD brains [110]. Pin1 is crucial for the dephosphorylation of tau proteins [108]. The decrease in antioxidant molecules like GSH and the use of buthionine sulfoximine further contribute to increased tau hyperphosphorylation under OS conditions [109]. Moreover, OS can directly induce tau hyperphosphorylation by affecting protein phosphatase 2A (PP2A) [109]. Under OS, GSK3β activity is heightened while PP2A activity is diminished, which in turn activates the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, leading to apoptosis [109,110].
Studies show that low doses of GSK-3β inhibitors protect neuron cells from OS-induced apoptosis, while higher doses can have the opposite effect [111]. Increased GSK-3β and the p25 activator of cyclin-dependent kinase 5 halt mitochondrial movement in neurons, but GSK-3β inhibition reverses axonal transport disruption caused by Tau overexpression [112,113]. ROS-mimicking mitochondrial OS promotes Tau phosphorylation by increasing GSK-3β activity [114].
Overall, OS, mitochondrial dysfunction, and Aβ involvement are interconnected with Tau pathology, with Tau pathology also inducing OS and mitochondrial damage, partly by modulating Aβ toxicity [52].
6.3. Oxidative Stress and Glutamatergic Signaling and Synaptic Dysfunction
Glial pathology and neurotransmitter system dysfunction are critical factors in various neurodegenerative diseases, including AD [115,116]. Astrocytes play essential roles such as regulating synaptic transmission, supplying nutrients to neurons, controlling vasodilation, maintaining BBB permeability, and responding to injury and immune challenges [117]. Glutamate, the primary excitatory neurotransmitter in the mammalian CNS, is integral to brain plasticity but excessive levels contribute to neurodegenerative diseases, brain trauma, seizures, and cerebral ischemic injury [118,119,120].
Glutamate activates ionotropic receptors (NMDAR, AMPA, and kainate) and metabotropic receptors [121]. Astrocytes are crucial for glutamate recycling, primarily through glutamate uptake mediated by glutamate aspartate transporter (GLAST or EAAT1) and glutamate transporter 1 (GLT-1 or EAAT2), with the enzyme GS converting glutamate to glutamine [122,123]. Proper regulation of glutamate is essential for neuronal survival; low levels compromise neuron viability, while excess leads to excitotoxicity, characterized by increased intracellular calcium through heightened NMDAR activity, impairing synaptic function and causing neuronal cell death [123,124]. In AD, impaired glutamate availability and NMDAR function exacerbate OS, resulting in increased neuronal damage and cell death [125].
Synapses, specialized neuronal regions for signaling, rely on precise mechanisms including neurotransmitter biosynthesis, delivery, synaptic vesicle formation, receptor binding, and neurotransmitter removal [126]. Calcium is critical in mediating synaptic transmission by triggering synaptic vesicle release through voltage-gated calcium channels [127]. Disruption in any step can severely impact synaptic function and cognitive abilities. In AD, progressive memory impairment is linked to inhibited long-term potentiation (LTP) and enhanced long-term depression (LTD) in the hippocampus, with synapse loss correlating strongly with cognitive impairment [128,129].
Extensive research indicates a direct relationship between OS and synaptic dysfunction in AD [125,130]. ROS, Aβ, and phosphorylated tau (pTau) independently and synergistically affect NMDAR activity, crucial for excitatory synaptic transmission and plasticity [131,132]. Aβ reduces surface NMDA receptors, triggers NMDA-mediated calcium influx, and induces excitotoxicity, exacerbating oxidative stress and impairing neuronal function [133,134]. Memantine targets non-synaptic NMDA receptors involved in excitotoxicity due to glutamate spillover [135,136]. Additionally, soluble Aβ species promote AMPA receptor internalization, affecting synaptic plasticity and causing synaptic dysfunction and dendritic spine loss [137].
OS significantly impacts synaptic integrity and function, especially in synaptic membranes rich in polyunsaturated fatty acids, which are highly susceptible to lipid peroxidation [138,139]. Aβ oligomers are particularly toxic to synapses, inducing OS by interacting with synaptic receptors and disrupting calcium homeostasis[140,141]. Excessive glutamate resulting from impaired astrocytic glutamate transporters overstimulates NMDA receptors, leading to a large influx of calcium into neurons [142]. Elevated intracellular calcium levels activate detrimental cellular events, including calcium-dependent enzymes like calpains, phospholipases, and nitric oxide synthase (NOS), which increase ROS production [142]. This overwhelms cellular antioxidant defenses, causing further oxidative damage to lipids, proteins, and DNA, disrupting membrane integrity, enzyme activity, and structural proteins [143]. Mitochondrial dysfunction exacerbates ROS production, reducing ATP production and releasing pro-apoptotic factors [142]. This disruption hampers neurotransmitter release and reuptake, impairing synaptic plasticity crucial for learning and memory [144]. Chronic inflammation further exacerbates synaptic damage through pro-inflammatory cytokines and additional ROS, contributing to synaptic loss and cognitive decline [142]. This complex interplay of factors underpins the synaptic dysfunction observed in AD, highlighting the critical roles of neurotransmitter systems, OS, and neuronal integrity in disease progression.
7. Oxidative Stress Impact on Cellular Functions
The impact of OS on cellular function is profound and multifaceted. ROS can oxidize essential biomolecules such as lipids, proteins, and nucleic acids, leading to significant impairment of cellular structures and functions [145]. Lipid peroxidation, for instance, damages cell membranes, affecting their fluidity and permeability [146]. This damage compromises the integrity and function of the cell membrane, leading to cellular instability. Similarly, protein oxidation can alter enzyme activities and disrupt protein-protein interactions, thereby interfering with critical cellular processes [147]. Oxidative damage to DNA is another severe consequence, resulting in mutations and impaired gene expression, which further exacerbate cellular dysfunction [148].
7.1. Protein Oxidation:
In AD, ROS mediates protein oxidation, introducing hydroxyl groups or generating protein-based carbonyls through the oxidation of amino acid residues such as lysine, arginine, proline, and threonine [149]. This process also involves the cleavage of peptide bonds via the α-amidation pathway or the oxidation of glutamyl residues [39]. ROS can also react with lipids, DNA, and sugars, producing reactive carbonyl derivatives and aldehydes that further react with proteins to form protein-bound carbonyls [39,149]. Measurement of protein carbonylation is a reliable indicator of oxidative damage linked to various OS conditions, aging, physiological disorders, and AD [150]. Functional changes in AD, such as decreased glucose metabolism in the parietal-temporal association cortices, are observed through fluorodeoxyglucose (FDG)-positron emission tomography (PET) analysis [151].
7.2. Lipid Oxidation:
Lipid peroxidation, an essential process in OS, results in the formation of several aldehyde by-products including MDA, HNE, and acrolein [152]. Among these, HNE and MDA are the most abundant, while acrolein is highly reactive [153].
Elevated levels of HNE-histidine and glutathione-HNE adducts have been observed in AD brains [154]. Proteomic analysis has shown a significant increase in protein-bound HNE in these brains [153]. F2-isoprostanes (F2-IsoPs) and neuroprostanes are also significantly elevated in patients with MCI and late-stage AD [155]. Increased acrolein levels are found in the hippocampus and temporal cortex of AD patients, regions where OS is exceptionally high [156]. Due to its high reactivity, acrolein is both a marker of lipid peroxidation and an initiator of OS, forming adducts with proteins, lipids, and nucleic acids [153].
Lipid peroxidation occurs not only in the brains of MCI patients but also in those with preclinical AD, suggesting that oxidative damage may play an early role in the disease pathogenesis [153]. Aβ induces lipid peroxidation of membranes, correlating strongly with the presence of antioxidant enzymes, amyloid plaques, and NFTs in AD brains [39]. Breakdown products of OS such as HNE, acrolein, MDA, and F2-isoprostanes, are more prevalent in AD brains compared to age-matched controls. Particularly, HNE can modify proteins, inhibiting neuronal glucose and glutamate transporters, Na-K ATPases, and disrupting intracellular calcium signaling, ultimately triggering apoptotic mechanisms [39,157,158,159,160].
The first evidence of Aβ-induced lipid peroxidation was demonstrated using electron paramagnetic resonance (EPR) methods [161]. Aβ addition to synaptosomes led to a rapid loss of the EPR signal, indicating lipid-centered free radical formation [14,161]. Subsequent studies using a murine model of mutant presenilin-1 (PS-1) supported these findings, showing oxidative damage in the brain [162]. A proposed mechanism involves the Aβ42 peptide, which in its oligomeric form can integrate into the lipid bilayer of neuronal membranes, adopting an α-helical structure. [23,158,163]. This conformation contributes to oxidative damage and neurotoxicity [158]. The interaction between specific amino acid residues within the peptide, such as the sulfur atom of methionine and the oxygen atom of isoleucine, facilitates one-electron oxidation, generating free radicals that propagate lipid peroxidation [164]. This process results in the formation of lipid peroxyl radicals and lipid hydroperoxides which combinedly contribute to the OS observed in AD [152,165].
In summary, lipid oxidation is a crucial factor in the OS associated with AD. The by-products of lipid peroxidation, particularly HNE, MDA, and acrolein, play significant roles in the disease's progression by modifying proteins and disrupting cellular functions, ultimately leading to neuronal damage and death.
7.3. DNA Oxidation:
OS significantly impacts DNA integrity in AD, contributing to its pathogenesis and disease progression [39,166]. ROS, particularly OH•, can cause various forms of DNA damage, including strand breaks, DNA-DNA and DNA-protein cross-linking, and the formation of oxidized base adducts [167,168]. These modifications can lead to mutations and altered protein synthesis [168,169].
DNA bases are highly susceptible to damage caused by OS, resulting in hydroxylation, protein carbonylation, and nitration [39]. One of the most prominent markers of oxidative DNA damage is 8-hydroxylamine (8-OHG), which forms when ROS reacts with guanine [170]. Due to its low oxidation potential, guanine is readily oxidized and forms several by-products including 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) and 7,8-dihydro-8-oxoguanine (8-OHG) [171,172]. ROS attack on DNA leads to more than 20 oxidized base adducts [173]. The most common oxidative DNA damage includes single-strand breaks (SSBs) and double-strand breaks (DSBs), with DSBs being more toxic and capable of altering gene transcription [174]. SSBs occur due to the breakdown of the DNA sugar-phosphate backbone following ROS oxidation, while DSBs can result in changes in the transcription of gene promoters near break sites [173,174].
The capacity to repair DNA damage is compromised in AD [175]. Studies indicate a decline in the efficiency of the base excision repair (BER) pathway and other DNA repair mechanisms [175,176]. This impairment is evidenced by reduced recruitment of repair proteins such as p53-binding protein 1 (53BP1) to sites of damage and lower levels of critical proteins involved in double-strain break (DSB) repair, including DNA PKcs and the MRN complex [177,178]. Elevated levels of γH2AX, a marker of DSBs, have been found in the neurons and astrocytes of AD patients, indicating persistent DNA damage [179].
OS significantly impacts DNA integrity in AD, leading to various forms of DNA damage, including strand breaks and oxidized base adducts. The compromised DNA repair capacity in AD exacerbates disease progression and highlights the need for targeted therapeutic interventions.
8. Biometals and Alzheimer’s disease Pathogenesis
8.1. Copper, Selenium and Zinc:
Copper (Cu2+) is an essential micronutrient and redox-active metal that plays a crucial role in AD [180]. It is required for the function of SOD1, a vital cellular antioxidant [181]. However, the interaction of Cu2+ with Aβ forms H2O2, which contributes to the production of OH• and exacerbates AD pathogenesis [182]. Zinc, while providing structural support to SOD1, in excess, promotes the hyperphosphorylation of tau, a protein crucial for microtubule stability [183]. Both Cu2+ and zinc can enhance the production of ROS, and damage proteins, DNA, and lipids [184,185]. Studies in mice have shown that chelating these metals from Aβ aggregates increases Aβ solubilization and decreases deposits, suggesting potential therapeutic strategies [184,186].
Selenium has a dual role in AD, acting both as an antioxidant and a pro-oxidant depending on its concentration [187]. High selenium levels can increase free radical formation by oxidizing sulfhydryl groups [187,188]. Conversely, selenium deficiency impairs GPx activity, leading to increased OS production [187]. As an antioxidant, selenium has the ability to help mitigate oxidative damage in AD.
8.2. Magnesium, Calcium, and Iron:
Magnesium and calcium also influence AD through their interactions with APP and presenilins [189,190]. Presenilins, which form the catalytic subunit of the γ-secretase complex, regulate zinc and Cu2+ uptake and are involved in cleaving APP [191]. Mutations in presenilin genes (PS1 and PS2) lead to the downregulation of calcium channels and mitochondrial transport proteins, disrupting calcium homeostasis and contributing to AD pathology [192]. Both magnesium and calcium stabilize γ-secretase and enhance its activity which reduces Aβ production [193].
Iron accumulation in the brain is associated with Aβ aggregation, inflammation, and OS [194,195]. Increased iron deposition exacerbates OS, which promotes further Aβ deposition in a proposed vicious cycle [196]. Aβ alters redox-inactive ferric iron (Fe3+) to redox-active ferrous iron (Fe2+), catalyzing the production of toxic free radicals via Fenton reactions [197,198]. Elevated iron levels have been observed in individuals with MCI, suggesting early involvement in AD [199]. APP contains an iron response element (IRE) that regulates its translation under iron-rich conditions, linking increased iron levels to higher APP production and toxic aggregate formation [200]. Additionally, APP interacts with ferroportin to facilitate iron export, and hepcidin regulates iron levels by controlling ferroportin activity [201]. Dysregulation of these pathways can contribute to AD pathology [198]. Disruption in iron homeostasis also influences tau protein aggregation through induction of tau aggregation by Fe3+, while reducing Fe2+ levels can reverse this process. [198]. Proteins involved in iron metabolism, such as divalent metal transporter 1 (DMT1) and ferroportin 1 (FPN1), affect brain iron load and AD progression [202].
Understanding the complex roles of these metals in AD provides insights into potential therapeutic targets for mitigating OS, protein aggregation, and neurodegeneration in AD pathology.
9. Antioxidant Deficiency in Alzheimer's Disease
RNS and ROS are typically regulated by a balance of endogenous and dietary antioxidants presence, such as glutathione (GSH), and antioxidant enzymes, including glutaredoxins, catalase, Trx-1, GPx, glutathione reductase (GR), and SOD [203]. These antioxidant systems are distributed across various subcellular compartments and often work in complementary ways to neutralize ROS and RNS [43].
In AD, these antioxidant defenses become insufficient due to increased ROS production, which can result from the downregulation or loss of function of antioxidant enzymes [78]. This insufficiency of antioxidative mechanisms can tilt the cellular reprogramming to a pro-oxidative state, which can contribute to additional OS [204]. GSH is widely expressed throughout the body and is crucial in reducing OS [205]. Increased levels of H2O2 induce GPx activity, which reduces H2O2 by oxidizing GSH into glutathione disulfide (GSSG) [206]. The GSH/GSSG ratio is an essential marker of OS. This ratio is often lowered in erythrocytes in AD patients and is likely due to increased GPx activity despite some studies reporting mixed results [207,208].
The activity of SOD1, a key antioxidant enzyme, is lower in the frontal lobes of AD patients suggesting that regional variations in central antioxidant enzyme activity may be significant in the pathogenesis of AD [209,210]. When compared to healthy controls, systemic SOD and GPx activities were found to be lower not only in AD patients but also in individuals with mild cognitive impairment (MCI), a precursor stage to AD [211,212]. This supports the notion that increased systemic OS is an early indicator of AD progression [213].
10. Recent Advances in Alzheimer's Disease Therapeutics Targeting Oxidative Stress
Currently, the FDA-approved drugs for treating AD include galantamine, donepezil, memantine, rivastigmine, and the combination drug Namzaric (donepezil and memantine). These medications, however, provide only symptomatic relief without halting disease progression or altering its outcomes [214]. Tacrine was the first drug approved for AD treatment, but it was later withdrawn due to hepatotoxicity [110,215]. Acetylcholinesterase inhibitors are beneficial in almost all stages of dementia, yet their efficacy in mild cognitive impairment and prodromal AD remains unproven [215]. Memantine is effective for moderate to severe AD but does not significantly mitigate cognitive decline [215].
Recently, anti-amyloid-β antibodies, such as aducanumab, lecanemab, and gantenerumab, have garnered attention for AD treatment [32]. These monoclonal IgG1 antibodies target aggregated forms of Aβ, and growing clinical evidence supports the beneficial role of Aβ immunotherapy in improving AD conditions [216]. Emerging therapeutic developments include naturally occurring polyphenolic compounds that act as antioxidants and confer neuroprotection in AD [217]. Polyphenols either reduce the production of ROS or enhance antioxidant release [218]. These compounds can cross the BBB and promote neuroprotection [219]. For instance, α-lipoic acid, a polyphenolic compound, acts as a free radical scavenger and mitigates H2O2 or iron-induced pathologies by inhibiting ferroptosis. It reduces the iron required for converting H2O2 into OH• via the Fenton reaction by forming chelates with iron. Additionally, it decreases brain calcium content and calpain activity, thereby preventing neuronal cell death [220].
Polyphenols exhibit a broad range of biological activities against several human diseases, including AD [218,221]. They also potentially modulate gut dysbiosis [222]. Green tea polyphenols, rich in (-)-epigallocatechin-3-gallate (EGCG), scavenge free radicals, chelate metal ions, and inhibit the nuclear translocation of NF-кB, alleviating OS and protecting against various AD-promoting factors [223]. ROS have been found to disrupt the BBB by activating several signaling pathways, leading to tight junction activation, adherent junction modification, mitochondrial membrane pore activation, and cytoskeletal disorganization. This disruption results in BBB dysfunction and further exacerbates other pathological conditions, including neuroinflammation, progressing to AD. Naturally occurring polyphenols such as stilbenes, flavanones, isoflavones, and phenolic acids act as antioxidants, mitigating BBB dysfunction associated with increased OS [224]. Thus, polyphenols are considered potential therapeutic molecules for treating OS-induced AD.
Additionally, targeting mitochondria to reduce the production of free radicals (FRs) is a significant strategy in ameliorating OS-induced AD. Antioxidants like coenzyme Q10, MitoQ, dimebon, and α-lipoic acid are potent in alleviating mitochondrial dysfunction and associated oxidative damage, thereby reducing cognitive decline in AD patients [225]. ROS activates protein kinase C and the Mitogen-Activated Protein Kinase (MAPK) pathway, triggering the release of inflammatory cytokines and chemokines. These inflammatory cells synthesize FRs, further stimulating other inflammatory mediators. Consequently, anti-inflammatory drugs can be employed to reduce OS-induced damage and prevent neurodegeneration. Tumor Necrosis Factor-alpha (TNF-α) is a major stimulator of cytokines and other inflammatory mediators, leading to the abnormal cleavage of amyloid precursor protein (APP). TNF-α also stimulates the Nuclear Factor kappa B (NFκB) pathway, resulting in the production of Aβ. Inhibiting TNF-α in AD patients has been found to mitigate cognitive defects [226]. TNF-α inhibitors, such as etanercept, can reduce TNF-α-induced neuronal damage. Drugs targeting OS-induced AD are currently undergoing clinical trials, offering hope for more effective treatments.
Carvedilol, a β-blocker, is currently in phase IV clinical trials with 29 participants enrolled. This study, designed as a randomized, triple-blind, and parallel assignment, has shown that Carvedilol inhibits apoptosis, reduces ROS levels, and mitigates Aβ toxicity. By regulating Interleukin-1 beta (IL-1β) expression, Carvedilol promotes neuronal growth and survival. Furthermore, it activates the Nrf2/ARE pathway, increasing the levels of heme oxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase-1 (NQO-1) in HT22 cells, thereby alleviating OS [227,228]. Similarly, Donepezil is being evaluated in phase III trials for its therapeutic potential in AD patients through cognitive function assessments. This double-blind, randomized, parallel assignment study has demonstrated that Donepezil suppresses NF-κB in murine macrophages and promotes remyelination of neurons in a cuprizone-induced mouse model. As a piperidine derivative, Donepezil reversibly inhibits acetylcholinesterase, thereby improving cholinergic transmission by increasing acetylcholine levels. Additionally, Donepezil activates AMPK and other downstream pathways to mitigate OS [229,230,231]. Following this, Memantine is in phase III clinical trials with a randomized quadruple masking and factorial assignment study design involving 613 participants. Memantine blocks NMDA receptors, crucial for brain function, and has been shown to mitigate OS via Brain-Derived Neurotrophic Factor / Tropomyosin receptor kinase B (BDNF/TrkB) signaling in HUVECs. It also activates the Nrf2 pathway in SHSY5Y cells, reducing OS directly or indirectly [231,232,233]
In a related effort, Melatonin is undergoing phase III trials aimed at treating AD. As a pineal hormone, Melatonin inhibits β and γ secretase enzymes while increasing α secretase activity, thus reducing amyloidogenesis. It is highly efficient in early-stage AD for neuroprotection. Melatonin scavenges ROS and activates the Nrf2/HO-1 pathway, elevating antioxidant levels such as catalase, superoxide dismutase, and glutathione peroxidase [231,234,235]. On a similar note, Pramipexole is in phase II trials with an open-label, single-group assignment design involving 20 participants. Preclinical studies in APPswe/PS1dE9 mice models have shown that Pramipexole, a dopamine agonist, exhibits neuroprotective effects by scavenging free radicals and mitigating OS in mitochondria. It activates the Nrf2/HO-1 pathway, increases IL-10 generation, and improves cognitive functions. Additionally, Pramipexole activates the CREB pathway, reducing over-expressed RCAN1 levels [236,237,238,239].
Moreover, Resveratrol is in phase II trials to evaluate its effectiveness in preventing AD progression. This randomized, single-centered study with quadruple masking and parallel group assignment includes 119 participants. Preclinical studies in Tg19959 and APP/PS1 transgenic mice models have shown that Resveratrol reduces tau pathology by activating AMPK and reducing OS. It also inhibits the Phosphoinositide 3-Kinase / Protein Kinase B (PI3K/AKT) pathway and stimulates PP2A activation, promoting tau dephosphorylation and neuronal survival [240,241,242]. Adding to the list, Etanercept is in phase I clinical trials with an open-label, crossover assignment model involving 12 participants. Studies in AD mouse models have demonstrated that Etanercept decreases TNF-α levels in the brain, reducing ROS production via NADPH oxidase and improving cognitive function. As an anti-TNF-α drug, Etanercept reduces OS, evidenced by decreased malondialdehyde levels and increased antioxidant levels such as superoxide dismutase and glutathione peroxidase [243,244,245,246,247].
Similarly, Epigallocatechin-gallate (EGCG) is being evaluated in phase III trials for its benefits in early-stage AD. With 200 participants, this randomized, double-blind, crossover assignment study has shown that EGCG, which contains hydroxyl groups, neutralizes FRs, modulates pro-apoptotic proteins Bax and Bad, and regulates mitochondrial permeability. It also activates the Nrf2/ARE pathway, alleviating OS and providing neuroprotection [248,249]. Lastly, Genistein, in a phase II trial with a randomized, parallel assignment, and quadruple masking study design, involves 27 participants. Experiments on streptozotocin-induced rat models have shown that Genistein induces autophagy and promotes Aβ clearance. It hinders kinases such as cyclic Adenosine Monophosphate (cAMP)-dependent protein kinase, protein kinase C, and phosphorylase kinase. Genistein also promotes neuronal survival by attenuating OS via the Phosphoinositide 3-Kinase / Protein Kinase B (Akt) / Nuclear Factor Erythroid 2-Related Factor 2 / Kelch-like ECH-Associated Protein 1 (PI3K/Akt/Nrf2/Keap1) pathway and activates the cAMP/CREB-BDNF-TrkB signaling pathway, enhancing cAMP levels and CREB and TrkB phosphorylation [250,251,252].
11. Conclusions
Based on the comprehensive review of the literature on OS and AD, it is evident that OS plays a pivotal role in the pathogenesis and progression of AD. This review highlights the multifaceted mechanisms through which ROS and RNS contribute to neuronal damage, including lipid peroxidation, protein oxidation, and DNA damage. Mitochondrial dysfunction emerges as a critical factor, exacerbating ROS production and disrupting cellular energy metabolism, further contributing to the neurodegenerative processes observed in AD. The interplay between OS and key pathological features of AD, such as Aβ aggregation, tau hyperphosphorylation, and synaptic dysfunction, underscores the complexity of the disease. Aβ peptides, particularly Aβ42, are prone to aggregation, forming insoluble fibrils that deposit as plaques in the brain, a process enhanced by OS. Furthermore, OS-induced mitochondrial dysfunction leads to increased ROS production, creating a vicious cycle that perpetuates neuronal damage and cognitive decline.
Despite the strong evidence linking OS to AD, clinical trials with antioxidant therapies have yielded inconsistent results, highlighting the need for a more nuanced understanding of the disease's pathophysiology. Emerging therapeutic strategies, including the use of polyphenolic compounds and targeted mitochondrial antioxidants, show promise in mitigating OS-related damage. Additionally, recent advances in immunotherapy, such as the development of monoclonal antibodies targeting Aβ, represent a significant shift towards disease-modifying treatments.
Future research should focus on combination therapies that target multiple pathways implicated in AD, including OS, mitochondrial dysfunction, and neuroinflammation. Promoting a healthy lifestyle with a focus on antioxidant-rich diets, regular physical activity, and cognitive engagement may also play a role in delaying the onset and progression of AD.
In conclusion, while significant progress has been made in understanding the role of OS in AD, further studies are needed to translate these insights into effective therapeutic interventions. By addressing the intricate mechanisms underlying OS and its interplay with other pathological features of AD, there is hope for developing treatments that can slow or even halt the progression of this debilitating disease.
Author Contributions
All authors contributed to this manuscript’s conceptualization, writing, editing, and reviewing. All authors have read and agreed to the published version of the manuscript.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat Rev Dis Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Association, A.s. 2019 Alzheimer's disease facts and figures. Alzheimer's & dementia 2019, 15, 321–387. [Google Scholar]
- Association, A.s. What is Alzheimer’s Disease? Available online:. Available online: https://www.alz.org/alzheimers-dementia/what-is-alzheimers#:~:text=Alzheimer's%20disease%20accounts%20for%2060%2D80%25%20of%20dementia%20cases.&text=Alzheimer's%20is%20not%20a%20normal,affects%20a%20person%20under%2065. (accessed on 14 April 2024).
- Organization, W.H. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on).
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Molecular Neurodegeneration 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Molecular Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor perspectives in medicine 2011, 1, a006189. [Google Scholar]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial cell-mediated neuroinflammation in Alzheimer’s disease. International journal of molecular sciences 2022, 23, 10572. [Google Scholar]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. International journal of molecular sciences 2023, 24, 16288. [Google Scholar] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology & Experimental Neurology 2001, 60, 759–767. [Google Scholar]
- Mehta, R.I.; Mehta, R.I. The Vascular-Immune Hypothesis of Alzheimer's Disease. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Translational Neurodegeneration 2018, 7, 2. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.-Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing research reviews 2022, 77, 101619. [Google Scholar] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. Oxidative damage in neurodegeneration: Roles in the pathogenesis and progression of Alzheimer disease. Physiological Reviews 2024, 104, 103–197. [Google Scholar]
- Korovesis, D.; Rubio-Tomás, T.; Tavernarakis, N. Oxidative Stress in Age-Related Neurodegenerative Diseases: An Overview of Recent Tools and Findings. Antioxidants (Basel, Switzerland) 2023, 12. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer's disease. Biomed Rep 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: where are we now? Journal of neurochemistry 2006, 97, 1634–1658. [Google Scholar]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp Neurobiol 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Wang, H.; Patterson, C. Atherosclerosis: risks, mechanisms, and therapies; John Wiley & Sons, 2015. [Google Scholar]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Amartumur, S.; Nguyen, H.; Huynh, T.; Kim, T.S.; Woo, R.-S.; Oh, E.; Kim, K.K.; Lee, L.P.; Heo, C. Neuropathogenesis-on-chips for neurodegenerative diseases. Nature Communications 2024, 15, 2219. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid β-peptide. Trends in molecular medicine 2001, 7, 548–554. [Google Scholar]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Frontiers in aging neuroscience 2021, 13, 57. [Google Scholar]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Chakole, R.D.; Nemade, L.S.; Kale, N.K.; Borah, S.; Deokar, S.S. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis-an updated review. Mitochondrion 2023. [Google Scholar]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer's disease: Role in pathogenesis and novel therapeutic opportunities. British journal of pharmacology 2019, 176, 3489–3507. [Google Scholar] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-g.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2014, 1842, 1240–1247. [Google Scholar]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxidants & redox signaling 2013, 19, 823–835. [Google Scholar]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial dysfunction in Alzheimer’s disease: a biomarker of the future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer's disease. Biomedical reports 2016, 4, 519–522. [Google Scholar]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomedicine & Pharmacotherapy 2015, 74, 101–110. [Google Scholar]
- Food, U.; Administration, D. FDA grants accelerated approval for Alzheimer’s drug. FDA News Release 2021. [Google Scholar]
- Wang, Y. An insider's perspective on FDA approval of aducanumab. Alzheimers Dement (N Y) 2023, 9, e12382. [Google Scholar] [CrossRef]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxidative medicine and cellular longevity 2014, 2014. [Google Scholar]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-biological interactions 2006, 160, 1–40. [Google Scholar]
- Koopman, W.J.; Nijtmans, L.G.; Dieteren, C.E.; Roestenberg, P.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxidants & redox signaling 2010, 12, 1431–1470. [Google Scholar]
- Cheignon, C.m.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox biology 2018, 14, 450–464. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Current biology 2014, 24, R453–R462. [Google Scholar] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adh Migr 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005, 70, 200–214. [Google Scholar]
- Sheldon, R. Metal-catalyzed oxidations of organic compounds: mechanistic principles and synthetic methodology including biochemical processes; Elsevier, 2012. [Google Scholar]
- Doorn, J.A.; Petersen, D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chemico-biological interactions 2003, 143, 93–100. [Google Scholar]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med Cell Longev 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Lee, J.; Koo, N.; Min, D.B. Reactive oxygen species, aging, and antioxidative nutraceuticals. Comprehensive reviews in food science and food safety 2004, 3, 21–33. [Google Scholar]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J Physiol 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Therade-Matharan, S.; Laemmel, E.; Duranteau, J.; Vicaut, E. Reoxygenation after hypoxia and glucose depletion causes reactive oxygen species production by mitochondria in HUVEC. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2004, 287, R1037–R1043. [Google Scholar]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. The Journal of physiology 2003, 552, 335–344. [Google Scholar] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 1996, 382, 685–691. [Google Scholar]
- Kusano, T.; Nishino, T.; Okamoto, K.; Hille, R.; Nishino, T. The mechanism and significance of the conversion of xanthine dehydrogenase to xanthine oxidase in mammalian secretory gland cells. Redox Biol 2023, 59, 102573. [Google Scholar] [CrossRef]
- Corvo, M.L.; Marinho, H.S.; Marcelino, P.; Lopes, R.M.; Vale, C.A.; Marques, C.R.; Martins, L.C.; Laverman, P.; Storm, G.; Martins, M.B.A. Superoxide dismutase enzymosomes: Carrier capacity optimization, in vivo behaviour and therapeutic activity. Pharmaceutical research 2015, 32, 91–102. [Google Scholar]
- Fridovich, I. Superoxide radical and superoxide dismutases. Oxygen and Living Processes: An Interdisciplinary Approach, 1981; 250–272. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. Journal of Biological Chemistry 2001, 276, 38388–38393. [Google Scholar]
- Butler, J.; Koppenol, W.H.; Margoliash, E. Kinetics and mechanism of the reduction of ferricytochrome c by the superoxide anion. Journal of Biological Chemistry 1982, 257, 10747–10750. [Google Scholar] [PubMed]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Superoxide Anion Chemistry-Its Role at the Core of the Innate Immunity. International journal of molecular sciences 2023, 24. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.J.; Dubey, A.; Dawson, K.M.; Stutts, W.A.; Lal, H.; Sohal, R.S. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proceedings of the National Academy of Sciences 1996, 93, 4765–4769. [Google Scholar]
- Levine, R.L.; Williams, J.A.; Stadtman, E.P.; Shacter, E. [37] Carbonyl assays for determination of oxidatively modified proteins. In Methods in enzymology; Elsevier, 1994; Volume 233, pp. 346–357. [Google Scholar]
- Mustafa, S.A.; Karieb, S.S.; Davies, S.J.; Jha, A.N. Assessment of oxidative damage to DNA, transcriptional expression of key genes, lipid peroxidation and histopathological changes in carp Cyprinus carpio L. following exposure to chronic hypoxic and subsequent recovery in normoxic conditions. Mutagenesis 2015, 30, 107–116. [Google Scholar]
- Headlam, H.A.; Davies, M.J. Markers of protein oxidation: different oxidants give rise to variable yields of bound and released carbonyl products. Free Radical Biology and Medicine 2004, 36, 1175–1184. [Google Scholar] [PubMed]
- Nadeau, P.J.; Charette, S.J.; Toledano, M.B.; Landry, J. Disulfide bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-Jun NH2-terminal kinase activation and apoptosis. Molecular biology of the cell 2007, 18, 3903–3913. [Google Scholar] [PubMed]
- Yamamoto, H.; Ozaki, T.; Nakanishi, M.; Kikuchi, H.; Yoshida, K.; Horie, H.; Kuwano, H.; Nakagawara, A. Oxidative stress induces p53-dependent apoptosis in hepatoblastoma cell through its nuclear translocation. Genes to Cells 2007, 12, 461–471. [Google Scholar]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/RNS generation. Journal of biomedical science 2017, 24, 1–10. [Google Scholar]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Current neuropharmacology 2009, 7, 65–74. [Google Scholar]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev 2013, 2013, 316523. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch Toxicol 2015, 89, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal—A bioactive lipid peroxidation product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer's disease. Curr Alzheimer Res 2008, 5, 525–532. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer's Disease Pathology and Potential Therapies. Front Physiol 2020, 11, 598. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Bernard, L.P.; Sinha, C.; Ehrhart, J.; Nicklas, W.J. Excitotoxicity and oxidative stress during inhibition of energy metabolism. Dev Neurosci 1998, 20, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.A.; Xiong-Fister, S.; Markesbery, W.R.; Lovell, M.A. Elevated 4-hydroxyhexenal in Alzheimer's disease (AD) progression. Neurobiology of aging 2012, 33, 1034–1044. [Google Scholar]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radical Biology and Medicine 2017, 111, 253–261. [Google Scholar]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer's disease: role of lipid peroxidation in Alzheimer's disease pathogenesis. PROTEOMICS–Clinical Applications 2009, 3, 682–693. [Google Scholar]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 1997, 77, 731–758. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- Afzal, S.; Abdul Manap, A.S.; Attiq, A.; Albokhadaim, I.; Kandeel, M.; Alhojaily, S.M. From imbalance to impairment: the central role of reactive oxygen species in oxidative stress-induced disorders and therapeutic exploration. Front Pharmacol 2023, 14, 1269581. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer's Disease. Front Aging Neurosci 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer's Disease: A Biomarker of the Future? Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer's Disease. Front Neurosci 2020, 14, 530219. [Google Scholar] [CrossRef]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A 2008, 105, 4441–4446. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction - the beginning of the end in Alzheimer's disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimers Res Ther 2011, 3, 15. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; Trimmer, P.A.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; et al. Alzheimer's disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol 2000, 48, 148–155. [Google Scholar]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Gao, R.; Ma, S.L. Is Mitochondria DNA Variation a Biomarker for AD? Genes (Basel) 2022, 13. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer's disease. Neurosci Bull 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer's Disease: A Target for New Drug Development. J Alzheimers Dis Parkinsonism 2017, 7. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu Rev Genet 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol Neurodegener 2020, 15, 30. [Google Scholar] [CrossRef]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer's disease. Indian J Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer's Disease. Which Comes First: The Chicken or the Egg? Antioxidants (Basel, Switzerland) 2021, 10. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Reviews in the Neurosciences 2018, 29, 613–627. [Google Scholar]
- Butterfield, D.A. The 2013 discovery award from the society for free radical biology and medicine: Selected discoveries from the Butterfield Laboratory of oxidative stress and its sequelae in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free radical biology & medicine, 2014; 157. [Google Scholar]
- Boutte, A.M.; Woltjer, R.L.; Zimmerman, L.J.; Stamer, S.L.; Montine, K.S.; Manno, M.V.; Cimino, P.J.; Liebler, D.C.; Montine, T.J. Selectively increased oxidative modifications mapped to detergent-insoluble forms of Aβ and β-III tubulin in Alzheimer's disease. The FASEB journal 2006, 20, 1473–1483. [Google Scholar] [PubMed]
- Allan Butterfield, D. Amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free radical research 2002, 36, 1307–1313. [Google Scholar]
- Dean, R.T.; FU, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochemical Journal 1997, 324, 1–18. [Google Scholar]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.-C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD enhances Aβ-induced cell stress via mitochondrial dysfunction. The FASEB Journal 2005, 19, 1–25. [Google Scholar]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxidative medicine and cellular longevity 2014, 2014, 427318. [Google Scholar]
- Akterin, S.; Cowburn, R.F.; Miranda-Vizuete, A.; Jiménez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in β-amyloid toxicity and Alzheimer's disease. Cell Death & Differentiation 2006, 13, 1454–1465. [Google Scholar]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. Journal of cellular and molecular medicine 2008, 12, 987–994. [Google Scholar] [PubMed]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Archives of biochemistry and biophysics 2008, 480, 85–94. [Google Scholar]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2012, 2, a006247. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid Med Cell Longev 2015, 2015, 151979. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer's Disease. Oxid Med Cell Longev 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer's disease: current knowledge of signaling pathways and therapeutics. Mol Biol Rep 2024, 51, 48. [Google Scholar] [CrossRef]
- Lee, K.Y.; Koh, S.H.; Noh, M.Y.; Park, K.W.; Lee, Y.J.; Kim, S.H. Glycogen synthase kinase-3beta activity plays very important roles in determining the fate of oxidative stress-inflicted neuronal cells. Brain Res 2007, 1129, 89–99. [Google Scholar] [CrossRef]
- Morel, M.; Authelet, M.; Dedecker, R.; Brion, J.P. Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 2010, 167, 1044–1056. [Google Scholar] [CrossRef]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Drummond, J.A.; Berg, S.; MacKay, D.; et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Salazar, A.; Bañuelos-Hernández, B.; Rodríguez-Leyva, I.; Chi-Ahumada, E.; Monreal-Escalante, E.; Jiménez-Capdeville, M.E.; Rosales-Mendoza, S. Oxidative Stress Modifies the Levels and Phosphorylation State of Tau Protein in Human Fibroblasts. Front Neurosci 2017, 11, 495. [Google Scholar] [CrossRef]
- Sanacora, G.; Rothman, D.L.; Mason, G.; Krystal, J.H. Clinical studies implementing glutamate neurotransmission in mood disorders. Annals of the New York Academy of Sciences 2003, 1003, 292–308. [Google Scholar]
- Krystal, J.H.; Tolin, D.F.; Sanacora, G.; Castner, S.A.; Williams, G.V.; Aikins, D.E.; Hoffman, R.E.; D'Souza, D.C. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug discovery today 2009, 14, 690–697. [Google Scholar] [PubMed]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [PubMed]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. New England Journal of Medicine 1994, 330, 613–622. [Google Scholar]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44, S14–23. [Google Scholar] [PubMed]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of disease: astrocytes in neurodegenerative disease. Nature clinical practice Neurology 2006, 2, 679–689. [Google Scholar] [PubMed]
- Kew, J.N.; Kemp, J.A. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 2005, 179, 4–29. [Google Scholar]
- Gjessing, L.; Gjesdahl, P.; Sjaastad, O. The free amino acids in human cerebrospinal fluid. 1972. [Google Scholar]
- Danbolt, N.C. Glutamate uptake. Progress in neurobiology 2001, 65, 1–105. [Google Scholar]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox biology 2015, 6, 260–271. [Google Scholar] [PubMed]
- Girouard, H.; Wang, G.; Gallo, E.F.; Anrather, J.; Zhou, P.; Pickel, V.M.; Iadecola, C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. Journal of Neuroscience 2009, 29, 2545–2552. [Google Scholar]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nature reviews neuroscience 2006, 7, 278–294. [Google Scholar]
- Szule, J.A.; Jung, J.H.; McMahan, U.J. The structure and function of ‘active zone material’at synapses. Philosophical Transactions of the Royal Society B: Biological Sciences 2015, 370, 20140189. [Google Scholar]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 1990, 27, 457–464. [Google Scholar]
- Jang, S.-S.; Chung, H.J. Emerging link between Alzheimer’s disease and homeostatic synaptic plasticity. Neural plasticity 2016, 2016, 7969272. [Google Scholar]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer's Disease. J Alzheimers Dis 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues in clinical neuroscience 2000, 2, 219–232. [Google Scholar]
- Frankland, P.W.; Bontempi, B. The organization of recent and remote memories. Nature reviews neuroscience 2005, 6, 119–130. [Google Scholar]
- Trushina, E.; McMurray, C. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci 2008, 31, 454–463. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr 2014, 54, 151–160. [Google Scholar] [CrossRef]
- Westra, M.; Gutierrez, Y.; MacGillavry, H.D. Contribution of Membrane Lipids to Postsynaptic Protein Organization. Front Synaptic Neurosci 2021, 13, 790773. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer's Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- McDaid, J.; Mustaly-Kalimi, S.; Stutzmann, G.E. Ca(2+) Dyshomeostasis Disrupts Neuronal and Synaptic Function in Alzheimer's Disease. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer's Disease. J Alzheimers Dis 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, D.; Kim, M.S.; Jeong, N.Y.; Jung, J. Roles of Gasotransmitters in Synaptic Plasticity and Neuropsychiatric Conditions. Neural Plast 2018, 2018, 1824713. [Google Scholar] [CrossRef] [PubMed]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem Sci 2016, 7, 489–498. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein oxidation - Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol 2021, 42, 101901. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Davies, M.J. The oxidative environment and protein damage. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 2005, 1703, 93–109. [Google Scholar]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparło, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging (Albany NY) 2018, 10, 868–901. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K. PET approaches for diagnosis of dementia. AJNR Am J Neuroradiol 2014, 35, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Singh, M.; Dang, T.N.; Arseneault, M.; Ramassamy, C. Role of by-products of lipid oxidation in Alzheimer's disease brain: a focus on acrolein. J Alzheimers Dis 2010, 21, 741–756. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochim Biophys Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef]
- Montine, T.J.; Peskind, E.R.; Quinn, J.F.; Wilson, A.M.; Montine, K.S.; Galasko, D. Increased cerebrospinal fluid F2-isoprostanes are associated with aging and latent Alzheimer's disease as identified by biomarkers. Neuromolecular Med 2011, 13, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.; Arseneault, M.; Murthy, V.; Ramassamy, C. Potential role of acrolein in neurodegeneration and in Alzheimer's disease. Curr Mol Pharmacol 2010, 3, 66–78. [Google Scholar] [PubMed]
- Ali, J.; Aziz, M.A.; Rashid, M.M.O.; Basher, M.A.; Islam, M.S. Propagation of age-related diseases due to the changes of lipid peroxide and antioxidant levels in elderly people: A narrative review. Health Sci Rep 2022, 5, e650. [Google Scholar] [CrossRef]
- Mark, R.J.; Lovell, M.A.; Markesbery, W.R.; Uchida, K.; Mattson, M.P. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. Journal of neurochemistry 1997, 68, 255–264. [Google Scholar] [PubMed]
- Selley, M.; Close, D.; Stern, S. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiology of aging 2002, 23, 383–388. [Google Scholar]
- Tamagno, E.; Robino, G.; Obbili, A.; Bardini, P.; Aragno, M.; Parola, M.; Danni, O. H2O2 and 4-hydroxynonenal mediate amyloid β-induced neuronal apoptosis by activating JNKs and p38MAPK. Experimental neurology 2003, 180, 144–155. [Google Scholar]
- Butterfield, D.A. Brain lipid peroxidation and alzheimer disease: Synergy between the Butterfield and Mattson laboratories. Ageing Res Rev 2020, 64, 101049. [Google Scholar] [CrossRef] [PubMed]
- Abdul, H.M.; Sultana, R.; St Clair, D.K.; Markesbery, W.R.; Butterfield, D.A. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med 2008, 45, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiology of aging 2002, 23, 655–664. [Google Scholar]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal 2013, 19, 823–835. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: mechanisms and analysis. Chemical reviews 2011, 111, 5944–5972. [Google Scholar]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic acids research 2007, 35, 7497–7504. [Google Scholar] [PubMed]
- Collins, A.R.; Dusinska, M.; Gedik, C.M.; Stĕtina, R. Oxidative damage to DNA: do we have a reliable biomarker? Environmental health perspectives 1996, 104, 465–469. [Google Scholar] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. Journal of neurochemistry 1998, 71, 2034–2040. [Google Scholar]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell calcium 2003, 34, 385–397. [Google Scholar]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. Journal of Neuroscience 1999, 19, 1959–1964. [Google Scholar]
- Lovell, M.A.; Gabbita, S.P.; Markesbery, W.R. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. Journal of neurochemistry 1999, 72, 771–776. [Google Scholar]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 1994, 36, 747–751. [Google Scholar]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J Cell Physiol 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Potlapalli, R.; Quan, H.; Chen, L.; Xie, Y.; Pouriyeh, S.; Sakib, N.; Liu, L.; Xie, Y. Exploring DNA Damage and Repair Mechanisms: A Review with Computational Insights. BioTech (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Alexandrov, N.; Alexandrov, V. Computational science research methods for science education at PG level. Procedia Computer Science 2015, 51, 1685–1693. [Google Scholar]
- Milanowska, K.; Rother, K.; Bujnicki, J.M. Databases and bioinformatics tools for the study of DNA repair. Molecular biology international 2011, 2011. [Google Scholar]
- Lei, T.; Du, S.; Peng, Z.; Chen, L. Multifaceted regulation and functions of 53BP1 in NHEJ-mediated DSB repair (Review). Int J Mol Med 2022, 50. [Google Scholar] [CrossRef]
- Zentout, S.; Smith, R.; Jacquier, M.; Huet, S. New methodologies to study DNA repair processes in space and time within living cells. Frontiers in Cell and Developmental Biology 2021, 9, 730998. [Google Scholar]
- Watt, N.T.; Whitehouse, I.J.; Hooper, N.M. The role of zinc in Alzheimer's disease. Int J Alzheimers Dis 2010, 2011, 971021. [Google Scholar] [CrossRef]
- Summers, K.L.; Schilling, K.M.; Roseman, G.; Markham, K.A.; Dolgova, N.V.; Kroll, T.; Sokaras, D.; Millhauser, G.L.; Pickering, I.J.; George, G.N. X-ray absorption spectroscopy investigations of copper (II) coordination in the human amyloid β peptide. Inorganic Chemistry 2019, 58, 6294–6311. [Google Scholar]
- Xiao, G.; Fan, Q.; Wang, X.; Zhou, B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proceedings of the National Academy of Sciences 2013, 110, 14995–15000. [Google Scholar]
- Pham, E.; Crews, L.; Ubhi, K.; Hansen, L.; Adame, A.; Cartier, A.; Salmon, D.; Galasko, D.; Michael, S.; Savas, J.N. Progressive accumulation of amyloid-β oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. The FEBS journal 2010, 277, 3051–3067. [Google Scholar] [PubMed]
- Mezentsev, Y.V.; Medvedev, A.E.; Kechko, O.I.; Makarov, A.A.; Ivanov, A.S.; Mantsyzov, A.B.; Kozin, S.A. Zinc-induced heterodimer formation between metal-binding domains of intact and naturally modified amyloid-beta species: implication to amyloid seeding in Alzheimer’s disease? Journal of Biomolecular Structure and Dynamics 2016, 34, 2317–2326. [Google Scholar]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of copper in the onset of Alzheimer’s disease compared to other metals. Frontiers in aging neuroscience 2018, 9, 446. [Google Scholar] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.-S. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar]
- Vaz, F.N.C.; Fermino, B.L.; Haskel, M.V.L.; Wouk, J.; de Freitas, G.B.L.; Fabbri, R.; Montagna, E.; Rocha, J.B.T.; Bonini, J.S. The Relationship Between Copper, Iron, and Selenium Levels and Alzheimer Disease. Biol Trace Elem Res 2018, 181, 185–191. [Google Scholar] [CrossRef]
- Jiang, K.; Xie, C.; Li, Z.; Zeng, H.; Zhao, Y.; Shi, Z. Selenium Intake and its Interaction with Iron Intake Are Associated with Cognitive Functions in Chinese Adults: A Longitudinal Study. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Ye, S.; Wang, T.T.; Cai, B.; Wang, Y.; Li, J.; Zhan, J.X.; Shen, G.M. Genistein protects hippocampal neurons against injury by regulating calcium/calmodulin dependent protein kinase IV protein levels in Alzheimer's disease model rats. Neural Regen Res 2017, 12, 1479–1484. [Google Scholar] [CrossRef]
- Oikawa, N.; Walter, J. Presenilins and γ-Secretase in Membrane Proteostasis. Cells 2019, 8. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer's disease-linked presenilin mutants and intracellular Ca(2+) handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Hoke, D.E.; Chua, Y.J.; Li, Q.X.; Culvenor, J.G.; Masters, C.; White, A.R.; Evin, G. Effect of Metal Chelators on γ-Secretase Indicates That Calcium and Magnesium Ions Facilitate Cleavage of Alzheimer Amyloid Precursor Substrate. Int J Alzheimers Dis 2010, 2011, 950932. [Google Scholar] [CrossRef]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim Biophys Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Baruch-Suchodolsky, R.; Fischer, B. Soluble amyloid beta1-28-copper(I)/copper(II)/Iron(II) complexes are potent antioxidants in cell-free systems. Biochemistry 2008, 47, 7796–7806. [Google Scholar] [CrossRef]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer's disease peptide β-amyloid (1-42). J R Soc Interface 2014, 11, 20140165. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.; Brooks, J.; Lermyte, F.; O'Connor, P.B.; Sadler, P.J.; Dobson, J.; Collingwood, J.F.; Telling, N.D. Iron stored in ferritin is chemically reduced in the presence of aggregating Aβ(1-42). Sci Rep 2020, 10, 10332. [Google Scholar] [CrossRef]
- Smith, M.A.; Zhu, X.; Tabaton, M.; Liu, G.; McKeel, D.W., Jr.; Cohen, M.L.; Wang, X.; Siedlak, S.L.; Dwyer, B.E.; Hayashi, T.; et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 2010, 19, 363–372. [Google Scholar] [CrossRef]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer's Disease: From Pathogenesis to Therapeutic Implications. Front Neurosci 2018, 12, 632. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol Neurodegener 2017, 12, 75. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- Dong, X.H.; Gao, W.J.; Kong, W.N.; Xie, H.L.; Peng, Y.; Shao, T.M.; Yu, W.G.; Chai, X.Q. Neuroprotective effect of the active components of three Chinese herbs on brain iron load in a mouse model of Alzheimer's disease. Exp Ther Med 2015, 9, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.; de Oliveira, J.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid Med Cell Longev 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: the interplay. Biomed Res Int 2014, 2014, 761264. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione "Redox Homeostasis" and Its Relation to Cardiovascular Disease. Oxid Med Cell Longev 2019, 2019, 5028181. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, M.A.; Mendoza-Núñez, V.M. Oxidative Stress Indexes for Diagnosis of Health or Disease in Humans. Oxid Med Cell Longev 2019, 2019, 4128152. [Google Scholar] [CrossRef]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [PubMed]
- Olufunmilayo, E.O.; Gerke-Duncan, M.B.; Holsinger, R.M.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants (Basel, Switzerland) 2023, 12. [Google Scholar] [CrossRef]
- Mao, P. Oxidative Stress and Its Clinical Applications in Dementia. J Neurodegener Dis 2013, 2013, 319898. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Herrmann, N.; Chen, J.J.; Saleem, M.; Oh, P.I.; Andreazza, A.C.; Kiss, A.; Lanctôt, K.L. Glutathione Peroxidase Activity Is Altered in Vascular Cognitive Impairment-No Dementia and Is a Potential Marker for Verbal Memory Performance. J Alzheimers Dis 2021, 79, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Studart, A.N.; Nitrini, R. Subjective cognitive decline: The first clinical manifestation of Alzheimer's disease? Dement Neuropsychol 2016, 10, 170–177. [Google Scholar] [CrossRef]
- Barthold, D.; Joyce, G.; Ferido, P.; Drabo, E.F.; Marcum, Z.A.; Gray, S.L.; Zissimopoulos, J. Pharmaceutical Treatment for Alzheimer's Disease and Related Dementias: Utilization and Disparities. J Alzheimers Dis 2020, 76, 579–589. [Google Scholar] [CrossRef]
- Joe, E.; Ringman, J.M. Cognitive symptoms of Alzheimer's disease: clinical management and prevention. Bmj 2019, 367, l6217. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab - Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer's Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef]
- Arias-Sánchez, R.A.; Torner, L.; Fenton Navarro, B. Polyphenols and Neurodegenerative Diseases: Potential Effects and Mechanisms of Neuroprotection. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Kaddoumi, A. Extra-Virgin Olive Oil in Alzheimer's Disease: A Comprehensive Review of Cellular, Animal, and Clinical Studies. International journal of molecular sciences 2024, 25. [Google Scholar] [CrossRef]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in alzheimer's disease: A review on emergent natural polyphenolic therapeutics. Complement Ther Med 2020, 49, 102294. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; Guo, C. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol 2018, 14, 535–548. [Google Scholar] [CrossRef]
- Rathod, N.B.; Elabed, N.; Punia, S.; Ozogul, F.; Kim, S.K.; Rocha, J.M. Recent Developments in Polyphenol Applications on Human Health: A Review with Current Knowledge. Plants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Bucciantini, M.; Leri, M.; Nardiello, P.; Casamenti, F.; Stefani, M. Olive Polyphenols: Antioxidant and Anti-Inflammatory Properties. Antioxidants (Basel, Switzerland) 2021, 10. [Google Scholar] [CrossRef]
- Afzal, O.; Dalhat, M.H.; Altamimi, A.S.A.; Rasool, R.; Alzarea, S.I.; Almalki, W.H.; Murtaza, B.N.; Iftikhar, S.; Nadeem, S.; Nadeem, M.S.; Kazmi, I. Green Tea Catechins Attenuate Neurodegenerative Diseases and Cognitive Deficits. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Kim, J.H.; Ju, I.G.; Kim, N.; Huh, E.; Son, S.R.; Hong, J.P.; Choi, Y.; Jang, D.S.; Oh, M.S. Yomogin, Isolated from Artemisia iwayomogi, Inhibits Neuroinflammation Stimulated by Lipopolysaccharide via Regulating MAPK Pathway. Antioxidants (Basel, Switzerland) 2022, 12. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Shi, X.; Ma, C. An overview on therapeutics attenuating amyloid β level in Alzheimer's disease: targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am J Transl Res 2016, 8, 246–269. [Google Scholar] [PubMed]
- Ekert, J.O.; Gould, R.L.; Reynolds, G.; Howard, R.J. TNF alpha inhibitors in Alzheimer's disease: A systematic review. Int J Geriatr Psychiatry 2018, 33, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Kisby, B.; Jarrell, J.T.; Agar, M.E.; Cohen, D.S.; Rosin, E.R.; Cahill, C.M.; Rogers, J.T.; Huang, X. Alzheimer's Disease and Its Potential Alternative Therapeutics. J Alzheimers Dis Parkinsonism 2019, 9. [Google Scholar] [CrossRef]
- Ouyang, Y.; Chen, Z.; Tan, M.; Liu, A.; Chen, M.; Liu, J.; Pi, R.; Fang, J. Carvedilol, a third-generation β-blocker prevents oxidative stress-induced neuronal death and activates Nrf2/ARE pathway in HT22 cells. Biochem Biophys Res Commun 2013, 441, 917–922. [Google Scholar] [CrossRef]
- Cui, X.; Guo, Y.E.; Fang, J.H.; Shi, C.J.; Suo, N.; Zhang, R.; Xie, X. Donepezil, a drug for Alzheimer's disease, promotes oligodendrocyte generation and remyelination. Acta Pharmacol Sin 2019, 40, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Atef, M.M.; El-Sayed, N.M.; Ahmed, A.A.M.; Mostafa, Y.M. Donepezil improves neuropathy through activation of AMPK signalling pathway in streptozotocin-induced diabetic mice. Biochem Pharmacol 2019, 159, 1–10. [Google Scholar] [CrossRef]
- Khalaf, S.S.; Hafez, M.M.; Mehanna, E.T.; Mesbah, N.M.; Abo-Elmatty, D.M. Combined vildagliptin and memantine treatment downregulates expression of amyloid precursor protein, and total and phosphorylated tau in a rat model of combined Alzheimer's disease and type 2 diabetes. Naunyn Schmiedebergs Arch Pharmacol 2019, 392, 685–695. [Google Scholar] [CrossRef]
- Hao, Y.; Xiong, R.; Gong, X. Memantine, NMDA Receptor Antagonist, Attenuates ox-LDL-Induced Inflammation and Oxidative Stress via Activation of BDNF/TrkB Signaling Pathway in HUVECs. Inflammation 2021, 44, 659–670. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Caporaso, R.; Basagni, F.; Catanzaro, M.; Abu, I.F.; Fagiani, F.; Fusco, F.; Masuzzo, S.; Albani, D.; et al. Merging memantine and ferulic acid to probe connections between NMDA receptors, oxidative stress and amyloid-β peptide in Alzheimer's disease. Eur J Med Chem 2019, 180, 111–120. [Google Scholar] [CrossRef]
- Shukla, M.; Govitrapong, P.; Boontem, P.; Reiter, R.J.; Satayavivad, J. Mechanisms of Melatonin in Alleviating Alzheimer's Disease. Curr Neuropharmacol 2017, 15, 1010–1031. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.C.; Liu, X.C.; Yang, S.H.; Song, L.L.; Zhou, S.J.; Deng, S.L.; Tian, L.; Cheng, L.Y. Melatonin Inhibits Oxidative Stress and Apoptosis in Cryopreserved Ovarian Tissues via Nrf2/HO-1 Signaling Pathway. Front Mol Biosci 2020, 7, 163. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer's Disease? CNS Neurosci Ther 2019, 25, 5–13. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer's disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Salman, M.; Akram, M.; Shahrukh, M.; Ishrat, T.; Parvez, S. Effects of pramipexole on beta-amyloid(1-42) memory deficits and evaluation of oxidative stress and mitochondrial function markers in the hippocampus of Wistar rat. Neurotoxicology 2022, 92, 91–101. [Google Scholar] [CrossRef]
- Wang, J.; Jia, Y.; Li, G.; Wang, B.; Zhou, T.; Zhu, L.; Chen, T.; Chen, Y. The Dopamine Receptor D3 Regulates Lipopolysaccharide-Induced Depressive-Like Behavior in Mice. Int J Neuropsychopharmacol 2018, 21, 448–460. [Google Scholar] [CrossRef]
- Chen, J.Y.; Zhu, Q.; Zhang, S.; OuYang, D.; Lu, J.H. Resveratrol in experimental Alzheimer's disease models: A systematic review of preclinical studies. Pharmacol Res 2019, 150, 104476. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; He, J.; Pan, C.; Wang, J.; Ma, M.; Shi, X.; Xu, Z. Resveratrol Activates Autophagy via the AKT/mTOR Signaling Pathway to Improve Cognitive Dysfunction in Rats With Chronic Cerebral Hypoperfusion. Front Neurosci 2019, 13, 859. [Google Scholar] [CrossRef]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauß, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci Rep 2017, 7, 13753. [Google Scholar] [CrossRef] [PubMed]
- Detrait, E.R.; Danis, B.; Lamberty, Y.; Foerch, P. Peripheral administration of an anti-TNF-α receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-α levels and memory deficits in mice. Neurochem Int 2014, 72, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am J Pathol 2009, 175, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Ortí-Casañ, N.; Wu, Y.; Naudé, P.J.W.; De Deyn, P.P.; Zuhorn, I.S.; Eisel, U.L.M. Targeting TNFR2 as a Novel Therapeutic Strategy for Alzheimer's Disease. Front Neurosci 2019, 13, 49. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer's Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Yang, M.; Chen, J.; Zhao, J.; Meng, M. Etanercept attenuates myocardial ischemia/reperfusion injury by decreasing inflammation and oxidative stress. PloS one 2014, 9, e108024. [Google Scholar] [CrossRef]
- Khalatbary, A.R.; Khademi, E. The green tea polyphenolic catechin epigallocatechin gallate and neuroprotection. Nutr Neurosci 2020, 23, 281–294. [Google Scholar] [CrossRef]
- Han, J.; Wang, M.; Jing, X.; Shi, H.; Ren, M.; Lou, H. (-)-Epigallocatechin gallate protects against cerebral ischemia-induced oxidative stress via Nrf2/ARE signaling. Neurochem Res 2014, 39, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Pierzynowska, K.; Podlacha, M.; Gaffke, L.; Majkutewicz, I.; Mantej, J.; Węgrzyn, A.; Osiadły, M.; Myślińska, D.; Węgrzyn, G. Autophagy-dependent mechanism of genistein-mediated elimination of behavioral and biochemical defects in the rat model of sporadic Alzheimer's disease. Neuropharmacology 2019, 148, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Shanmuganathan, B.; Manayi, A.; Nabavi, S.F.; Nabavi, S.M. Molecular and Therapeutic Targets of Genistein in Alzheimer's Disease. Mol Neurobiol 2017, 54, 7028–7041. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yang, G.; He, Y.; Xu, H.; Fan, H.; An, J.; Zhang, L.; Zhang, R.; Cao, G.; Hao, D.; Yang, H. Involvement of α7nAChR in the Protective Effects of Genistein Against β-Amyloid-Induced Oxidative Stress in Neurons via a PI3K/Akt/Nrf2 Pathway-Related Mechanism. Cell Mol Neurobiol 2021, 41, 377–393. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



