
Submitted:
11 August 2024
Posted:
14 August 2024
You are already at the latest version
Abstract
Steroidogenic factor 1 (SF-1) is a nuclear receptor that regulates steroidogenesis and reproductive development. NR5A1/SF-1 variants are associated with a broad spectrum of phenotypes across individuals with disorders of sex development (DSD). Oligogenic inheritance has been suggested as an explanation. However, testing the impact of specific gene variants involved in a network remains difficult. To confirm the hypothesis that NR5A1/SF-1-related DSD follow an oligogenic mode of inheritance, we investigated a constellation of gene variants identified in a 46,XY severely undervirilized individual. Candidate genes were revealed by whole exome sequencing, and pathogenicity was predicted by different in silico tools. We found variants in NR1H2 and INHA associated with steroidogenesis, sex development, and reproduction. Functional testing was conducted in cell models. Novel SF-1 and NR1H2 binding sites in the AR and INHA gene promoters were found. Transactivation studies showed that wild-type NR5A1/SF-1 regulates INHA and AR gene expression, while the NR5A1/SF-1 variant had decreased transcriptional ac-tivity. NR1H2 was found to regulate AR gene transcription; however, the NR1H2 variant showed normal activity. This study expands the NR5A1/SF-1 network of interacting partners while strengthening the hypothesis that the broad phenotype observed in 46,XY DSD individuals may be caused by oligogenic pathogenicity.
Keywords:
steroidogenic factor 1 (SF-1/NR5A1)
; inhibin
; androgen receptor
; differences of sex development (DSD)
; 46
; XY DSD
; hypospadias
; oligogenicity
1. Introduction
Steroidogenic factor 1 (NR5A1/SF-1) is a nuclear receptor and a master regulator of steroidogenesis and reproductive development. NR5A1/SF-1 controls several steps of gonadal and adrenal development [1, 2]. Therefore, disruption of NR5A1/SF-1 may lead to abnormalities in steroidogenic and reproductive tissues. Nr5a1/Sf-1 knockout mice have a sex reversal phenotype and adrenocortical insufficiency, while heterozygous Nr5a1/Sf-1 mice exhibit hypoplasia of the adrenal glands and testes [3, 4]. Human genetic variants in NR5A1/SF-1 may lead to disorders/differences of sex development (DSD) associated with a wide range of phenotypes, and very few NR5A1/SF-1 carriers show an adrenal phenotype; thus, mice models do not recapitulate the broad phenotype seen in humans [5-7]. NR5A1/SF-1 variants are mostly found in a heterozygous state and are scattered throughout the whole gene without any obvious hotspots [5-7]. To assess the pathogenicity of identified NR5A1/SF-1 variants, numerous in vitro studies showed mixed results concerning confirmation of disease-causing mechanism [6].
NR5A1/SF-1 has a wide network of interactions, including many transcription factors, co-modulators, posttranslational modulators, and signaling molecules [1]. Therefore, it was suggested that the broad phenotypes among patients with DSD may be explained by oligogenic inheritance, where multiple genetic variants together with NR5A1/SF-1 might contribute to a specific DSD phenotype of an individual [5, 6, 8-13]. Oligogenic causation has been reported for other endocrine disorders, for instance, congenital hypogonadotropic hypogonadism or congenital hypothyroidism [14-17]. In both, a synergistic or collaborative role of different gene variants was assumed probable [14-17]. Similarly, NR5A1/SF-1 variants, in combination with other variants in DSD-related genes, were identified in several individuals using next-generation sequencing (NGS) methods [6, 8, 9, 11-13, 18-21]. However, mechanistic confirmation of oligogenicity in DSD related to NR5A1/SF-1 is still missing.
Bioinformatic tools for testing combinatory variants are beneficial for identifying the potential oligogenicity but are scarce [9, 22-24]. Moreover, the contribution of the predicted variants needs to be confirmed experimentally by in vitro or ex vivo studies [25]. The activity of NR5A1/SF-1 as a transcription factor has classically been analyzed in cell models by testing its transactivation activity on promoter constructs of targeted genes and by nuclear translocation studies [6]. These studies have enhanced our understanding of the effect of NR5A1/SF-1 on specific target genes. Therefore, in this study, we investigated the oligogenic mechanism of action of genetic variants found in a 46,XY individual with a severe DSD phenotype carrying an NR5A1/SF-1 mutation using bioinformatic and in vitro, cell-based methods. We performed whole exome sequencing (WES) and comprehensive data analysis guided by the patient’s phenotype to identify candidate variants in additional genes, which were then investigated by transactivation studies in different cell models to elucidate their interaction with NR5A1/SF-1 and beyond.
2. Results
2.1. Phenotypic Characterization
The patient manifested at birth with a 46,XY DSD consisting of micropenis, scrotal hypospadias, bilateral cryptorchidism, and absence of Müllerian ducts (previously reported in [8, 26]). The patient had hypospadias repair at the age of 3.8 and 4.5 years and right and left orchidopexy at the age of 2.5 and 5.7 years, respectively. Adrenocorticotropic hormone (ACTH) stimulation test was performed at the age of 3 years and revealed a normal cortisol response. At 11 years of age, ultrasound showed testes in the scrotum (volume of 1 cm3 and 0.8 cm3). The patient had spontaneous puberty at the age of 11.8 years with normal testosterone (T) and luteinizing hormone (LH) levels, but elevated follicle-stimulating hormone (FSH) levels (38.2 mIU/ml) for Tanner stage. Normal ACTH and cortisol levels were confirmed. Testicular biopsy was taken at the age of 12.4 years, revealing seminiferous tubules devoid of germinal cells. An anthropometric evaluation at the age of 14.8 years indicated a weight of 57.9 kg (-0.28 SDS), height of 167.2 cm (-0.25 SDS), and BMI of 20.7 kg/m2 (-0.18 SDS). Growth velocity was 9.4 cm/year (3.45 SD). The patient had a testicular volume of 6 ml/8 ml, with Tanner stage 3 for pubic hair and genital status; breast development was B1. Further biochemical evaluation was performed at the age of 15 years, presented in Table 1. Family history revealed healthy parents and was unremarkable for DSD.
2.2. Genotypic Characterization
The index patient and the father carry a heterozygous c.58G>C; p.(Val20Leu) variant in the NR5A1 gene [26]. This variant was previously classified as pathogenic according to the ACMG criteria and most of the in silico tools (Table 2). Because of the discrepancy of phenotype between father and son, WES was performed. Variant analysis was conducted using a tailored algorithm to search for oligogenic etiology of DSD linked to NR5A1/SF-1 [8]. A single heterozygous variant c.675T>G; p.(Ser225Arg) in the INHA gene was found in the patient, but not in the healthy father; this variant was classified as a variant of uncertain significance (VUS) [8]. Recently, the aforementioned algorithm was updated [10], and the WES data of the index patient were reanalyzed. This reanalysis revealed four additional heterozygous variants in the patient in different genes NR1H2, TCF7L2, NIBAN1, and SCUBE2 (Table 2). The INHA variant was re-classified as benign (B) according to the ACMG criteria [27] and in silico tools (Table 2). Three of the newly identified candidate variants were classified as VUS, while in silico tools showed variable predictions. The variant in the TCF7L2 gene was classified as likely benign (LB) according to the ACMG and in silico tools (Table 2). Additionally, in ORVAL, the variants in the TCF7L2 c.1535C>G; p.(Pro512Arg), NIBAN1 c.929G>A; p.(Arg310His), and SCUBE2 c.692C>T; p.(Thr231Ile) genes were predicted to form a pathogenic oligogenic combination with the NR5A1 gene (Table 2).
To investigate the possible contribution of the newly identified variants to the DSD phenotype of the patient, we searched the literature for reported interactions between the different genes and NR5A1/SF-1 (Table 3). In addition, we searched for the phenotype associated with these variants in human and mice models (Table 3). Apart from the NR5A1/SF-1 gene, which is associated with a wide phenotypic spectrum of DSD [5, 6], we found that only two genes (NR1H2 and INHA) were involved in steroidogenesis, sex development, and/or reproduction. Therefore, the identified variants in the three other genes were excluded from further studies due to their different biological roles (see Table 3 for more details).
2.3. Characterization of the Identified Variants in NR5A1/SF-1, NR1H2 and INHA
We conducted a conservation comparison for the three variants NR5A1/SF-1 c.58G>C; p.(Val20Leu), c.515_516insCAA; p.(Arg171_Lys172insAsn) NR1H2/LXRβ and c.675T>G; p.(Ser225Arg) INHA, to predict the effect on protein function. The comparison of SF-1 and inhibin α similarity across species revealed that the variants and the surrounding regions are highly conserved (Figure S1). Similarly, the insertion of the asparagine amino acid in the LXRβ variant protein (encoded by the NR1H2 gene) may affect two conserved amino acids across different species (Figure S1).
2.4. In Vitro Functional Testing of Selected Variants
The pathogenicity of the c.58G>C; p.(Val20Leu) NR5A1/SF-1 variant was previously assessed by activation testing on three different promoter constructs of three steroid enzymes (e.g. -152_CYP11A1, -227_CYP17A1 and -301_HSD3B2) in HEK293T and NCI-H295R cells revealing impaired transcriptional activation on all three gene promoters (Figure 1) [26]. In pursuit of explaining the DSD phenotype of the patient in comparison with the healthy carrier father for NR5A1/SF-1 variant, two additional variants in INHA and NR1H2 genes, were functionally studied in vitro for their possible disease-causing effect.
The transcriptional regulation of INHA by NR5A1/SF-1 was tested by transfecting four INHA promoter constructs in steroidogenic adrenal NCI-H295R cells and Leydig MA-10 cells, which both express endogenous NR5A1/SF-1. Only the two longer constructs -520INHA and -2050INHA containing a consensus NR5A1/SF-1 binding site (5’-TCATGGCCA-3’ at -222/-214) were activated by SF-1, while the two constructs lacking the NR5A1/SF-1 and/or cAMP responsive element (5’-TGCGTCA-3’ at -205/-199) were not (Figure 2A and 2B). In order to confirm that this activation was specifically achieved by NR5A1/SF-1, the constructs were co-transfected with WT or variant c.58G>C; p.(Val20Leu) NR5A1/SF-1 in non-steroidogenic HEK293T cells that do not express NR5A1/SF-1. Similar results were found; only the constructs -520INHA and -2050INHA were activated by the WT NR5A1/SF-1 (Figure 2C). However, variant c.58G>C; p.(Val20Leu) NR5A1/SF-1 showed impaired activation (Figure 2C) of the INHA promoters. Overall, these results indicate that SF-1 is a transcriptional regulator of INHA expression.
As the role of INHA in sex development appears to be through the regulation of the hypothalamic-pituitary-gonadal (HPG) axis [36], we investigated the combined impact of NR5A1/SF-1, inhibin α, and activin A on GnRHR gene expression. WT SF-1 was found to activate the -2300GnRHR promoter construct harboring a NR5A1/SF-1 binding site at -142/-134, while mutant SF-1 showed significantly lower activation (Figure 2D). By contrast, the addition of activin A, or overexpression of WT INHA, in the absence or presence of NR5A1/SF-1, had no additional impact on -2300GnRHR promoter activation (data not shown). To assess the impact of inhibin α on the NR5A1 expression, we overexpressed INHA in adrenal NCI-H295R cells, which express endogenous NR5A1. However, neither WT nor mutant inhibin α had an effect on NR5A1 expression levels (data not shown).
To test the impact of the identified variants in the NR1H2 and NR5A1/SF-1 genes, the androgen receptor (AR) was chosen as a target. The AR was reported to regulate NR1H2/LXβ expression [55] and to interact with NR5A1/SF-1 as part of the transcriptional machinery modulating the expression of specific genes (e.g. LHβ) [56]. However, its regulation by these nuclear factors has not been reported so far. Therefore, we first tested whether the AR promoter is regulated by endogenous NR5A1/SF-1 (Figure 3A and 3B). The -3000AR promoter-reporter construct was significantly activated in steroidogenic MA-10 Leydig cells (Figure 3A); however, no activation was detected in the adrenal NCI-H295R cells (Figure 3B). The possible NR5A1/SF-1 binding sites in the AR promoter were searched manually and found 5’-TGACCTCT-3’ at -1705/-1698 and 5’-TGGCCTCC-3’ at -1412/-1404. Interestingly, the -3000AR construct was found differentially regulated by NR5A1/SF-1 overexpression in three different cell lines (Figure 3C-F). WT NR5A1/SF-1 significantly activated the -3000AR construct in HEK293T and MA-10 cells, but not in NCI-H295R cells (Figure 3C-E). Surprisingly, the mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 activated the AR construct in HEK293T and NCI-H295R cells (Figure 3C and 3D) more than in MA-10 Leydig cells (Figure 3E). Lastly, the AR was tested for its transcriptional regulation by LXRβ/RXRA in HEK293T cells. Both the WT and mutant c.515_516insCAA, p.(Arg171_Lys172insAsn) LXRβ/RXRA hetero-tetramers were able to significantly activate the -3000AR construct, but no significant difference was found for the variant (Figure 3F).
NR5A1/SF-1 variants are reported in 46,XY and 46,XX individuals presenting with variable severity of DSD ranging from healthy to opposite sex phenotypes. So far, genotype-phenotype correlation has not been found [5, 6]. Oligogenic inheritance could be a possible explanation for this broad phenotype, where multiple gene variants may contribute to a unique DSD phenotype of each individual [5, 6, 8, 9, 11, 26, 57]. In this study, we show that oligogenicity may explain the phenotype of a 46,XY DSD patient carrying a c.58G>C, p.(Val20Leu) NR5A1/SF-1 variant inherited from his healthy carrier father. By conducting WES analysis on both the father and the son, we identified five additional gene variants in the patient only. Four of these variants had not been previously reported. According to literature, only the INHA and NR1H2 genes are involved in steroidogenesis, sex development, and/or reproduction, while the other genes are either involved in diabetes or cancer.
NR5A1/SF-1 is a regulatory hub for numerous interacting partners [1]. Conducting functional assays, we were able to show that both INHA and NR1H2 are part of the NR5A1/SF-1 interaction network.
The INHA gene encodes the α subunit needed for the assembly of the dimeric glycoproteins termed A and B inhibins that suppress FSH secretion from the pituitary and play an important role in modulating the activin levels [36]. In addition, inhibins play a role in Sertoli and Leydig cell function, spermatogenesis, and sperm count [58]. The rat inhibin α subunit can be detected at a very early stage of testicular development following the formation of the testicular cord, and it is thought to play an important autocrine/paracrine role [59]. In mice, disruption of the Inha gene leads to the development of gonadal sex cord-stromal tumor and infertility [60]. In contrast, the human inhibin α subunit has been detected in the fetal testis only by 16 weeks of gestation following gonadal differentiation, specifically in interstitial and Sertoli cells; it contributes to normal testicular development [59]. Biallelic INHA variants were found associated with 46,XY DSD in humans [37, 39]. A homozygous 2 bp deletion c.208_209delAG, p.(R70Gfs*3) in the INHA gene was found in two brothers with hypospadias, hypergonadotropic hypogonadism, gynecomastia and azoospermia [37]. Still, the specific functional role of INHA in male sex development and reproduction is largely unknown.
In this study, we identified a regulatory NR5A1/SF-1 binding site in the human INHA gene promoter and showed that INHA is transcriptionally regulated by NR5A1/SF-1, whereas the NR5A1/SF-1 variant showed reduced activity on the INHA promoter. Investigating the specific contribution of the c.675T>G, p.(Ser225Arg) INHA variant to the phenotype of the patient was more challenging. The c.675T>G, p.(Ser225Arg) INHA variant affects a highly conserved amino acid (Figure S1) located in the αN pro-domain in the inhibin α precursor protein, which is further processed to obtain its mature and active form [61]. To date, very little information is available regarding the function of this region and its underlying regulatory mechanisms. However, it is predicted to contribute to the proper folding, processing, and export of inhibins (predominantly inhibin B) from Sertoli cells in the testis to the serum [61-63].
It has been previously reported that Nr5a1/Sf-1 can stimulate Gnrhr expression in mice and humans [64, 65]. Additionally, activin A was shown to enhance Gnrhr expression in mice; however, its role in regulating human GnRHR is unknown [66]. Therefore, we explored the potential collaborative activation of the human GnRHR gene by activin A and NR5A1/SF-1, and their inhibition by inhibin α. However, we did not observe any additional increase by activin A. Similarly, upon the addition of the WT inhibin α, GnRHR expression was not affected in the presence and absence of activin A. Therefore, the mechanistic proof of the contribution of the c.675T>G, p.(Ser225Arg) INHA variant to the phenotype found in our patient remains elusive.
Another variant identified in the patient was in the NR1H2 gene, which encodes the liver X receptor β (LXRβ), an important modulator of lipid and cholesterol homeostasis [67]. It forms an obligate heterodimer with the retinoid X receptor (RXR) to govern gene transcription by binding to specific LXR-responsive elements [67]. The Nr1h2 gene was found to be strongly expressed at 16.5 days postcoitum (dpc) in the mouse embryonic testis, specifically in Sertoli cells, where its expression persists into adulthood [68]. lxrβ-/- knockout mice present with excessive cholesterol accumulation in Sertoli cells and dysregulated spermatogenesis, while lxrαβ-/- mice present with a severe infertility phenotype [69]. Similarly, lower expression levels of NR1H2 were detected in the testis of infertile men with azoospermia [34, 35]. However, the specific function of NR1H2 in the human developing testis has not been elucidated.
Due to the fact that both NR1H2 and NR5A1/SF-1 play important roles in androgen homeostasis and male fertility [1, 32, 34, 35, 69-71], we tested their transcriptional activity on the AR promoter. Functional studies showed that NR5A1/SF-1 is a cell-specific transcriptional regulator of the AR in Leydig MA-10 cells but not in adrenal NCI-H295R cells. Overexpression of WT NR5A1/SF-1 enhanced the AR reporter activity, while the mutant NR5A1/SF-1 impaired transactivation. By contrast, transactivation studies of the NR5A1/SF-1 variant with the AR reporter in adrenal NCI-H295R and non-steroidogenic HEK293T cells revealed contradictory results suggesting that the specific background of the Leydig cell is necessary for showing the specific interplay.
AR activity is regulated by complex mechanisms [72]. It is influenced by various transcription factors and coregulators involved in multiple cellular pathways [72-74]. The most recent study showed that AR activity is modulated by the transcription factor disheveled-associated activator of morphogenesis 2 (DAAM2), a cytoskeletal regulator of formin and actin. In vitro studies of genital skin-derived fibroblasts (GSF) from patients with androgen insensitivity syndrome (AIS) type II and DAAM2 variants showed reduced dihydrotestosterone (DHT)-induced AR activity compared to WT GSF [73]. Moreover, the AR is epigenetically regulated; alterations in methylated CpG regions within the proximal AR promoter were found to inhibit AR transcription in GSF from several patients with AIS type II [74]. In our study, we show that the LXRβ/RXRA heterodimer is a transcription activator of the AR, strengthening NR1H2/LXRβ association with male fertility in line with previous reports [32, 34, 35, 69]. However, the c.515_516insCAA; p.(Arg171_Lys172insAsn) NR1H2/LXRβ VUS had similar transcriptional activity on the AR reporter as WT, thus its contribution to the DSD phenotype is in doubt.
Proper reporting of oligogenic variant combinations requires thorough genetic testing and functional evidence of pathogenicity of the causal variants [25, 75]. Advancement of NGS methods (WES, whole genome sequencing) has enhanced the yield of identifying possible genetic causes of DSD profoundly [24], and this is especially true for gene variants that occur in combination with NR5A1/SF-1 variants. In fact, more than 70 different gene variants have been reported in association with NR5A1/SF-1 variants in individuals with DSD (Table S1) [6, 8, 10-13, 57, 76]. We performed WES analysis in individuals with DSD and NR5A1/SF-1 variants as part of the SF1next study [5] and found several additional novel gene variants (unpublished data), suggesting digenic or oligogenic causation for the disease. To confirm the oligogenic disease mechanism of a DSD can be difficult as often appropriate experimental models are missing for modelling multiple genetic hits and/or assessing smaller effect size that can been only shown in combination. In our 46,XY DSD index patient, five gene variants were identified, of which only the NR5A1/SF-1 variant was also found in the healthy father. While the variants in TCF7L2, NIBAN1 and SCUBE2 were deemed irrelevant for the observed phenotype, the NR1H2 and INHA genes were found to be interacting partners of NR5A1/SF-1 and may therefore contribute towards the DSD phenotype. Future studies using patient-derived biomaterials may help in assessing oligogenic mechanisms. Cellular reprogramming of induced pluripotent stem cells (iPSC) carrying the specific, individual’s genetic background may inform on variants’ effect on steroidogenesis and sex development. Recently, in vitro systems for the differentiation of iPSCs towards gonadal progenitors and Sertoli-like cells have been established [77, 78]. Rescue experiments in iPSCs originating from a 46,XY DSD patient with an NR5A1/SF-1 variation showed the disease mechanism on sex determination [77]. Unfortunately, even these promising models have limitations, including the availability of patients’ biological materials and variability and difficulty of obtaining robust maturation of fully functional iPSC-derived somatic cells (e.g. Sertoli- and Leydig-like cells). Therefore, even these experiments may not fully recapitulate the phenotype when used for disease modeling. Moreover, the challenge to rescue multiple combined variants and assess their effect on the overall phenotype remains.
In conclusion, the use of NGS methods for achieving a molecular diagnosis in individuals with a DSD yielded a multitude of gene variants possibly associated with NR5A1/SF-1 variants and DSD. Studying the genetic profile of a 46,XY DSD patient through WES, we found five additional candidates and provided novel functional data showing that INHA, NR1H2/LXRβ, AR, and NR5A1/SF-1 interact. Although these studies support the hypothesis of oligogenic DSD, the final proof of the effect of the single variants involved remains a challenge.
4. Materials and Methods
4.1. Participants
The patient and his father included in this work were part of two previous genetic studies [8, 26] and the SF1next study [5].
4.2. In Silico Analyses and Variant Classification
The DNA of the index patient and the father were sequenced by WES (Novogene, UK) and analyzed with an in-house specific data filtering algorithm for gene variants related to DSD and/or NR5A1/SF-1 [8, 10]. We predicted the possible effect of identified genetic variants on the structure and function of the protein using Polyphen-2, (Polymorphism Phenotyping v2, http://genetics.bwh.harvard.edu/pph2/), Panther (Protein ANalysis THrough Evolutionary Relationships, http://www.pantherdb.org/tools/csnpScore.do), SNPs and Go (https://snps-and-go.biocomp.unibo.it/snps-and-go/), CADD (Combined Annotation Dependent Depletion, https://cadd.gs.washington.edu/) and the calibrated scores given by VarSome [27] for Revel (Rare Exome Variant Ensemble Learner), SIFT (Scale-invariant feature transform), Provean (Protein Variation Effect Analyzer), Mutation taster and M-CAP (Mendelian Clinically Applicable Pathogenicity). Variants were classified for pathogenicity according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) using VarSome [27]. We explored the possible pathogenicity of multiple variants’ combinatory effect using ORVAL (Oligogenic Resource for Variant AnaLysis) [79].
4.3. Plasmids
The human HA-tagged wild-type (WT) and the variant c.58G>C cDNA of NR5A1/SF-1 (NM_004959.5) in pcDNA3, empty control vector pcDNA3, and Renilla-TK (pRL-TK) were all available from previous work [26]. The human NR1H2 cDNA (NM_007121.5) in pCMV3-C-HA and RXRA cDNA (NM_002957.5) in pCMV3 vector were purchased (Sino Biologicals Inc, Eschborn, Germany). The human NR1H2 cDNA was used as a template to generate the NR1H2 variant expression vector by PCR-based site-directed mutagenesis using the following primers, forward (5’-CGGAAGAAGAAGATTCGGAACAAACAGCAGCAGGAG-3’), reverse (5’-CTCCTGCTGCTGTTTGTTCCGAATCTTCTTCTTCCG-3’), and the QuickChange protocol by Stratagene (Agilent Technologies Inc., Santa Clara, CA, USA).
4.4. Cloning
The 5’-untranslated region constructs of the different genes were produced by PCR using control human DNA extracted from blood leukocytes using the DNA isolation kit of Qiagen (QIAGEN, Aarhus, Denmark). The different forward primers used for PCR were as follows: -2056_INHA (5’-AGAGAGGGTACCTTGAGCACGAAGCCGCC-3’), -520_INHA (5’-AGA GAGGGTACCCTGAGGGGTGATGCACTTTGTC-3’), -213_INHA (5’-GAGGGTACCCA GACATCTGCGTCAGAGATAGGAG-3’), -198_INHA (5’-AGAGGGTACCGAGATAGGA GGTCTCAATGCCACG-3’) all with the KpnI restriction site included, while the following reverse primer including the XhoI restriction site was used in the four PCR reactions, (5’-GAGAGACTCGAGAGAACAAGTTCCCGGGCCAG-3’). For the generation of the -220_GnRHR construct, the forward primer, including the KpnI restriction site (5’-AGAGGTACCGGCCTGCTCTGTTTTAGCACT-3’) and the reverse primer, including the XhoI restriction site (5’-GAGCTCGAGATTTTCCCAGGACAGAGCTTCAAG-3’) were used. For the generation of the -3000_AR construct, the forward primer, including the HindIII restriction site (5’-AGAGAAGCTTTAAACTTTGGAGTCTTTCAGACCCAG-3’), and the reverse primer, including the XhoI restriction site (5’-GAGACTCGAGCCTTGAG CTTGGCTGAATCTTCC-3’) were used in the PCR reaction. All PCR products were digested with the indicated restriction enzymes and subcloned into the corresponding site in the pGL3 basic vector (Promega). The constructs were confirmed by direct sequencing. The -2300_GnRHR promoter construct in pGL3 was custom-made by Genscript (Rijswijk, Netherlands).
4.5. In Vitro Testing of Transactivation Activity by Dual Luciferase Assay
Non-steroidogenic, human embryonic kidney HEK293T cells (ATCC CRL-1573), steroidogenic NCI-H295R adrenal cells (ATCC CRL-2128), and mouse Leydig MA-10 cells (ATCC CRL-3050) were cultured as previously described [26, 80]. For all promoter activity experiments, cells were cultured on 12-well plates. For the INHA promoter activity experiments, NCI-H295R and MA-10 steroidogenic cells were transiently transfected with 950 ng from the different promoter luciferase reporter constructs: -2050INHA_pGL3, -520INHA_pGL3, -213INHA_pGL3 or -198INHA_pGL3; whereas HEK293T cells were transiently transfected with 200 ng WT or mutant NR5A1/SF-1 expression vectors, 800 ng of the different promoter luciferase reporter construct -2050INHA_pGL3, -520INHA_pGL3, -213INHA_pGL3 or -198INHA_pGL3 separately. For the GnRHR promoter activity experiment, HEK293T cells were transiently co-transfected with 200 ng WT or mutant NR5A1/SF-1 expression vectors and 800 ng of the promoter luciferase reporter constructs -2300GnRHR_pGL3 or -220GnRHR_pGL3. For the AR promoter activity experiments, MA-10 or NCI-H295R cells were transiently transfected with 950 ng of the -3000AR_pGL3 promoter luciferase reporter construct, while for the AR promoter experiments with overexpressed NR5A1/SF-1 or NR1H2/LXRβ, the cell lines were transiently transfected with 600 ng of the -3000AR_pGL3 promoter, and 200 ng WT or mutant of NR5A1/SF-1 expression vector (in the three cell lines), or with NR1H2/LXRβ and RXRA expression vectors, respectively. Lastly, 50 ng of the pRL-TK vector was used as an internal control in all transfection experiments. All transfections were carried out with Lipofectamine 2000TM (Invitrogen, Glasgow, UK) in Opti-MEM (1X)-reduced serum medium (Gibco, Thermo Fisher Scientific, US). Forty-eight hours after transfection, cells were washed with PBS, lysed, and assayed for luciferase activity with a dual-luciferase assay using a microplate Luminometer reader (Fluoroskan Ascent® FL & Fluoroskan Ascent®, Thermo Fisher). Specific Firefly luciferase readings were standardized against Renilla luciferase control readings. Experiments were repeated three to five times in duplicates and data were summarized giving the mean ± standard error of the mean (SEM). Statistical significance was examined by the Student’s t-test (GraphPad Prism, GraphPad Software, Boston, MA, USA). Significance was assumed with a p-value of less than 0.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Protein localization and conservation across species of the three variants NR5A1/SF-1 p.(V20L), NR1H2/LXRβ p.(R171_K172insN) and INHA/inhibin α p.(S225R). Table S1: Reported combined variants in NR5A1 and associated genes.
Author Contributions
All authors read and approved the final version of the manuscript. R.NE. Methodology and experimental investigations, data analysis, creation of tables and figures, manuscript writing, reviewing, and proofreading. C.K. Genetic data analysis. Manuscript reviewing and proofreading. I.MdLP. Genetic methodology and genetic data analysis. Manuscript reviewing and proofreading. K.S.S Manuscript reviewing and proofreading. F.M. Providing of clinical and genetic data of the patient. Manuscript reviewing and proofreading. N.CT. Providing of clinical and genetic data of the patient. Manuscript reviewing and proofreading. C.E.F. Study PI. Grant holder. Data analysis. Creation of tables and figures. Manuscript writing, reviewing, and proofreading. Responsible for the content and the decision to submit the manuscript. Corresponding author.
Funding
The study was supported by a project grant of the Swiss National Science Foundation (320030-197725). I.MdLP. is supported by a Postdoctoral Fellowship Grant from the Education Department of Basque Government (Spain).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of the Hospital Infantil La Fe, Valencia, Spain and the Ethics Committee of Canton Bern, Switzerland (BASEC 2016-01210).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data were collected in a project-specific REDCap database governed by the Clinical Trials Unit (CTU) at University of Bern, Switzerland. Genetic data are also stored on servers of the University of Bern. These data can also be accessed upon reasonable request according ethical and informed consent.
Acknowledgments
We thank the patient and the family for providing their medical data and trust towards this research study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Schimmer, B.P.; White, P.C. , Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol 2010, 24, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Ikeda, Y.; Luo, X.; Caron, K.M.; Weber, T.J.; Swain, A.; Schimmer, B.P.; Parker, K.L. , Steroidogenic factor 1 plays multiple roles in endocrine development and function. Recent Prog Horm Res 1997, 52, 167–182, discussion 182–184. [Google Scholar] [PubMed]
- Luo, X.; Ikeda, Y.; Parker, K.L. , A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994, 77, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Bland, M.L.; Jamieson, C.A.; Akana, S.F.; Bornstein, S.R.; Eisenhofer, G.; Dallman, M.F.; Ingraham, H.A. , Haploinsufficiency of steroidogenic factor-1 in mice disrupts adrenal development leading to an impaired stress response. Proc Natl Acad Sci U S A 2000, 97, 14488–14493. [Google Scholar] [CrossRef] [PubMed]
- Kouri, C.; Sommer, G.; Martinez de Lapiscina, I.; Elzenaty, R.N.; Tack, L.J.W.; Cools, M.; Ahmed, S.F.; Fluck, C.E.; group, S.F. n. s. , Clinical and genetic characteristics of a large international cohort of individuals with rare NR5A1/SF-1 variants of sex development. EBioMedicine 2024, 99, 104941. [Google Scholar] [CrossRef] [PubMed]
- Naamneh Elzenaty, R.; Martinez de Lapiscina, I.; Kouri, C.; Sauter, K.S.; Sommer, G.; Castano, L.; Fluck, C.E.; group, S.F. n. s. , Characterization of 35 novel NR5A1/SF-1 variants identified in individuals with atypical sexual development: The SF1next study. J Clin Endocrinol Metab 2024. [Google Scholar] [CrossRef] [PubMed]
- Fabbri-Scallet, H.; de Sousa, L.M.; Maciel-Guerra, A.T.; Guerra-Junior, G.; de Mello, M.P. , Mutation update for the NR5A1 gene involved in DSD and infertility. Hum Mutat 2020, 41, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Camats, N.; Fernandez-Cancio, M.; Audi, L.; Schaller, A.; Fluck, C.E. , Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: an oligogenic origin? Eur J Hum Genet 2018, 26, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Camats, N.; Fluck, C.E.; Audi, L. , Oligogenic Origin of Differences of Sex Development in Humans. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Martinez de Lapiscina, I.; Kouri, C.; Aurrekoetxea, J.; Sanchez, M.; Naamneh Elzenaty, R.; Sauter, K.S.; Camats, N.; Grau, G.; Rica, I.; Rodriguez, A.; Vela, A.; Cortazar, A.; Alonso-Cerezo, M.C.; Bahillo, P.; Bertholt, L.; Esteva, I.; Castano, L.; Fluck, C.E. Genetic reanalysis of patients with a difference of sex development carrying the NR5A1/SF-1 variant p.Gly146Ala has discovered other likely disease-causing variations. PLoS One 2023, 18, e0287515. [Google Scholar] [CrossRef]
- Martinez de LaPiscina, I.; Mahmoud, R.A.; Sauter, K.S.; Esteva, I.; Alonso, M.; Costa, I.; Rial-Rodriguez, J.M.; Rodriguez-Estevez, A.; Vela, A.; Castano, L.; Fluck, C.E. , Variants of STAR, AMH and ZFPM2/FOG2 May Contribute towards the Broad Phenotype Observed in 46,XY DSD Patients with Heterozygous Variants of NR5A1. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Zidoune, H.; Ladjouze, A.; Chellat-Rezgoune, D.; Boukri, A.; Dib, S.A.; Nouri, N.; Tebibel, M.; Sifi, K.; Abadi, N.; Satta, D.; Benelmadani, Y.; Bignon-Topalovic, J.; El-Zaiat-Munsch, M.; Bashamboo, A.; McElreavey, K. , Novel Genomic Variants, Atypical Phenotypes and Evidence of a Digenic/Oligogenic Contribution to Disorders/Differences of Sex Development in a Large North African Cohort. Front Genet 2022, 13, 900574. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Wang, N.; Zhu, H.; Han, B.; Sun, F.; Yao, H.; Zhang, Q.; Zhu, W.; Cheng, T.; Cheng, K.; Liu, Y.; Zhao, S.; Song, H.; Qiao, J. , Next-generation sequencing reveals genetic landscape in 46, XY disorders of sexual development patients with variable phenotypes. Hum Genet 2018, 137, 265–277. [Google Scholar] [CrossRef]
- Gach, A.; Pinkier, I.; Wysocka, U.; Salacinska, K.; Salachna, D.; Szarras-Czapnik, M.; Pietrzyk, A.; Sakowicz, A.; Nykel, A.; Rutkowska, L.; Rybak-Krzyszkowska, M.; Socha, M.; Jamsheer, A.; Jakubowski, L. , New findings in oligogenic inheritance of congenital hypogonadotropic hypogonadism. Arch Med Sci 2022, 18, 353–364. [Google Scholar] [CrossRef]
- de Filippis, T.; Gelmini, G.; Paraboschi, E.; Vigone, M.C.; Di Frenna, M.; Marelli, F.; Bonomi, M.; Cassio, A.; Larizza, D.; Moro, M.; Radetti, G.; Salerno, M.; Ardissino, D.; Weber, G.; Gentilini, D.; Guizzardi, F.; Duga, S.; Persani, L. , A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet 2017, 26, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Petit, I.; Edouard, T.; Jacques, V.; Bournez, M.; Cartault, A.; Grunenwald, S.; Savagner, F. , Next-Generation Sequencing Analysis Reveals Frequent Familial Origin and Oligogenism in Congenital Hypothyroidism With Dyshormonogenesis. Front Endocrinol (Lausanne) 2021, 12, 657913. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.A.; Quinton, R.; Hall, J.E.; Gusella, J.F.; Seminara, S.B.; Crowley, W.F., Jr.; Pitteloud, N. , Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A 2010, 107, 15140–15144. [Google Scholar] [CrossRef]
- Hughes, L.A.; McKay-Bounford, K.; Webb, E.A.; Dasani, P.; Clokie, S.; Chandran, H.; McCarthy, L.; Mohamed, Z.; Kirk, J.M.W.; Krone, N.P.; Allen, S.; Cole, T.R.P. , Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD). Endocr Connect 2019, 8, 100–110. [Google Scholar] [CrossRef]
- Werner, R.; Monig, I.; Lunstedt, R.; Wunsch, L.; Thorns, C.; Reiz, B.; Krause, A.; Schwab, K.O.; Binder, G.; Holterhus, P.M.; Hiort, O. , New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PLoS One 2017, 12, e0176720. [Google Scholar] [CrossRef]
- Mazen, I.; Abdel-Hamid, M.; Mekkawy, M.; Bignon-Topalovic, J.; Boudjenah, R.; El Gammal, M.; Essawi, M.; Bashamboo, A.; McElreavey, K. , Identification of NR5A1 Mutations and Possible Digenic Inheritance in 46,XY Gonadal Dysgenesis. Sex Dev 2016, 10, 147–151. [Google Scholar] [CrossRef]
- de Oliveira, F.R.; Mazzola, T.N.; de Mello, M.P.; Francese-Santos, A.P.; Lemos-Marini, S.H.V.; Maciel-Guerra, A.T.; Hiort, O.; Werner, R.; Guerra-Junior, G.; Fabbri-Scallet, H. , DHX37 and NR5A1 Variants Identified in Patients with 46,XY Partial Gonadal Dysgenesis. Life (Basel) 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Audi, L.; Ahmed, S.F.; Krone, N.; Cools, M.; McElreavey, K.; Holterhus, P.M.; Greenfield, A.; Bashamboo, A.; Hiort, O.; Wudy, S.A.; McGowan, R.; The, E.U.C.A. , GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 'DSDnet'. Eur J Endocrinol 2018, 179, R197–R206. [Google Scholar] [CrossRef]
- Alhomaidah, D.; McGowan, R.; Ahmed, S.F. , The current state of diagnostic genetics for conditions affecting sex development. Clin Genet 2017, 91, 157–162. [Google Scholar] [CrossRef]
- Delot, E.C.; Vilain, E. , Towards improved genetic diagnosis of human differences of sex development. Nat Rev Genet 2021, 22, 588–602. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Gravel, B.; Nachtegael, C.; De Baere, E.; Loeys, B.; Vikkula, M.; Smits, G.; Lenaerts, T. , Toward reporting standards for the pathogenicity of variant combinations involved in multilocus/oligogenic diseases. HGG Adv 2023, 4, 100165. [Google Scholar] [CrossRef]
- Camats, N.; Pandey, A.V.; Fernandez-Cancio, M.; Andaluz, P.; Janner, M.; Toran, N.; Moreno, F.; Bereket, A.; Akcay, T.; Garcia-Garcia, E.; Munoz, M.T.; Gracia, R.; Nistal, M.; Castano, L.; Mullis, P.E.; Carrascosa, A.; Audi, L.; Fluck, C.E. , Ten novel mutations in the NR5A1 gene cause disordered sex development in 46,XY and ovarian insufficiency in 46,XX individuals. J Clin Endocrinol Metab 2012, 97, E1294–E1306. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. , VarSome: the human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Achermann, J.C. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev 2008, 2, 200–209. [Google Scholar] [CrossRef]
- Bashamboo, A.; McElreavey, K. , Human sex-determination and disorders of sex-development (DSD). Semin Cell Dev Biol 2015, 45, 77–83. [Google Scholar] [CrossRef]
- Budefeld, T.; Tobet, S.A.; Majdic, G. , Altered position of cell bodies and fibers in the ventromedial region in SF-1 knockout mice. Exp Neurol 2011, 232, 176–184. [Google Scholar] [CrossRef]
- Zhao, C.; Dahlman-Wright, K. , Liver X receptor in cholesterol metabolism. J Endocrinol 2010, 204, 233–240. [Google Scholar] [CrossRef]
- Robertson, K.M.; Schuster, G.U.; Steffensen, K.R.; Hovatta, O.; Meaney, S.; Hultenby, K.; Johansson, L.C.; Svechnikov, K.; Soder, O.; Gustafsson, J.A. , The liver X receptor-beta is essential for maintaining cholesterol homeostasis in the testis. Endocrinology 2005, 146, 2519–2530. [Google Scholar] [CrossRef]
- Nilsson, M.; Stulnig, T.M.; Lin, C.Y.; Yeo, A.L.; Nowotny, P.; Liu, E.T.; Steffensen, K.R. , Liver X receptors regulate adrenal steroidogenesis and hypothalamic-pituitary-adrenal feedback. Mol Endocrinol 2007, 21, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Rondanino, C.; Ouchchane, L.; Chauffour, C.; Marceau, G.; Dechelotte, P.; Sion, B.; Pons-Rejraji, H.; Janny, L.; Volle, D.H.; Lobaccaro, J.M.; Brugnon, F. , Levels of liver X receptors in testicular biopsies of patients with azoospermia. Fertil Steril 2014, 102, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, S.; Williamson, C.; Bevan, C.L. , Liver X Receptors and Male (In)fertility. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Barakat, B.; Itman, C.; Mendis, S.H.; Loveland, K.L. Activins and inhibins in mammalian testis development: new models, new insights. Mol Cell Endocrinol, 2012; 359, 66–77. [Google Scholar]
- Arslan Ates, E.; Eltan, M.; Sahin, B.; Gurpinar Tosun, B.; Seven Menevse, T.; Geckinli, B.B.; Greenfield, A.; Turan, S.; Bereket, A.; Guran, T. , Homozygosity for a novel INHA mutation in two male siblings with hypospadias, primary hypogonadism, and high-normal testicular volume. Eur J Endocrinol 2022, 186, K25–K31. [Google Scholar] [CrossRef] [PubMed]
- Sundblad, V.; Chiauzzi, V.A.; Andreone, L.; Campo, S.; Charreau, E.H.; Dain, L. , Controversial role of inhibin alpha-subunit gene in the aetiology of premature ovarian failure. Hum Reprod 2006, 21, 1154–1160. [Google Scholar] [CrossRef]
- Huhtaniemi, I. , The first report on homozygous INHA inactivation in humans. Eur J Endocrinol 2022, 187, C1–C2. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Mather, J.P.; Krummen, L.; Lu, H.; Bradley, A. , Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci U S A 1994, 91, 8817–8821. [Google Scholar] [CrossRef]
- Ito, M.; Park, Y.; Weck, J.; Mayo, K.E.; Jameson, J.L. , Synergistic activation of the inhibin alpha-promoter by steroidogenic factor-1 and cyclic adenosine 3',5'-monophosphate. Mol Endocrinol 2000, 14, 66–81. [Google Scholar]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. , Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 1998, 19, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Nateri, A.S.; Spencer-Dene, B.; Behrens, A. , Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 2005, 437, 281–285. [Google Scholar] [CrossRef]
- Lili, L.N.; Farkas, A.E.; Gerner-Smidt, C.; Overgaard, C.E.; Moreno, C.S.; Parkos, C.A.; Capaldo, C.T.; Nusrat, A. , Claudin-based barrier differentiation in the colonic epithelial crypt niche involves Hopx/Klf4 and Tcf7l2/Hnf4-alpha cascades. Tissue Barriers 2016, 4, e1214038. [Google Scholar] [CrossRef]
- Angus-Hill, M.L.; Elbert, K.M.; Hidalgo, J.; Capecchi, M.R. , T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A 2011, 108, 4914–4919. [Google Scholar] [CrossRef]
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.; Jose, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; Voshol, P.; Dor, Y.; Cuppen, E.; Fillat, C.; Clevers, H. , Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607. [Google Scholar] [CrossRef]
- Helgason, A.; Palsson, S.; Thorleifsson, G.; Grant, S.F.; Emilsson, V.; Gunnarsdottir, S.; Adeyemo, A.; Chen, Y.; Chen, G.; Reynisdottir, I.; Benediktsson, R.; Hinney, A.; Hansen, T.; Andersen, G.; Borch-Johnsen, K.; Jorgensen, T.; Schafer, H.; Faruque, M.; Doumatey, A.; Zhou, J.; Wilensky, R.L.; Reilly, M.P.; Rader, D.J.; Bagger, Y.; Christiansen, C.; Sigurdsson, G.; Hebebrand, J.; Pedersen, O.; Thorsteinsdottir, U.; Gulcher, J.R.; Kong, A.; Rotimi, C.; Stefansson, K. , Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 2007, 39, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Freathy, R.M.; Weedon, M.N.; Bennett, A.; Hypponen, E.; Relton, C.L.; Knight, B.; Shields, B.; Parnell, K.S.; Groves, C.J.; Ring, S.M.; Pembrey, M.E.; Ben-Shlomo, Y.; Strachan, D.P.; Power, C.; Jarvelin, M.R.; McCarthy, M.I.; Davey Smith, G.; Hattersley, A.T.; Frayling, T.M. , Type 2 diabetes TCF7L2 risk genotypes alter birth weight: a study of 24,053 individuals. Am J Hum Genet 2007, 80, 1150–1161. [Google Scholar] [CrossRef]
- Gummow, B.M.; Winnay, J.N.; Hammer, G.D. , Convergence of Wnt signaling and steroidogenic factor-1 (SF-1) on transcription of the rat inhibin alpha gene. J Biol Chem 2003, 278, 26572–26579. [Google Scholar] [CrossRef]
- Diana, P.; Carvalheira, G.M.G. , NIBAN1, Exploring its Roles in Cell Survival Under Stress Context. Front Cell Dev Biol 2022, 10, 867003. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.D.; Kobayashi, T.; Abe, M.; Tada, N.; Adachi, H.; Shiota, A.; Totsuka, Y.; Hino, O. , The endoplasmic reticulum stress-inducible protein Niban regulates eIF2alpha and S6K1/4E-BP1 phosphorylation. Biochem Biophys Res Commun 2007, 360, 181–187. [Google Scholar] [CrossRef]
- Esmaeili, R.; Mohammadi, S.; Jafarbeik-Iravani, N.; Yadegari, F.; Olfatbakhsh, A.; Mazaheri, M.; Kaviani, A.; Rezaee, M.; Majidzadeh, A.K. , Expression of SCUBE2 and BCL2 Predicts Favorable Response in ERalpha Positive Breast Cancer. Arch Iran Med 2021, 24, 209–217. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chen, C.C.; Cheng, C.J.; Yang, R.B. , Domain and functional analysis of a novel breast tumor suppressor protein, SCUBE2. J Biol Chem 2011, 286, 27039–27047. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Roffler, S.R.; Yan, Y.T.; Yang, R.B. , Disruption of Scube2 Impairs Endochondral Bone Formation. J Bone Miner Res 2015, 30, 1255–1267. [Google Scholar] [CrossRef]
- Krycer, J.R.; Brown, A.J. , Cross-talk between the androgen receptor and the liver X receptor: implications for cholesterol homeostasis. J Biol Chem 2011, 286, 20637–20647. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.S.; Nilson, J.H. , AR suppresses transcription of the LHbeta subunit by interacting with steroidogenic factor-1. Mol Endocrinol 2001, 15, 1505–1516. [Google Scholar]
- Mazen, I.; Mekkawy, M.; Kamel, A.; Essawi, M.; Hassan, H.; Abdel-Hamid, M.; Amr, K.; Soliman, H.; El-Ruby, M.; Torky, A.; El Gammal, M.; Elaidy, A.; Bashamboo, A.; McElreavey, K. , Advances in genomic diagnosis of a large cohort of Egyptian patients with disorders of sex development. Am J Med Genet A 2021, 185, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, A.E.; De Kretser, D.M. , Inhibins in normal male physiology. Semin Reprod Med 2004, 22, 177–185. [Google Scholar] [CrossRef]
- Majdic, G.; McNeilly, A.S.; Sharpe, R.M.; Evans, L.R.; Groome, N.P.; Saunders, P.T. , Testicular expression of inhibin and activin subunits and follistatin in the rat and human fetus and neonate and during postnatal development in the rat. Endocrinology 1997, 138, 2136–2147. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Su, J.G.; Hsueh, A.J.; Bradley, A. , Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 1992, 360, 313–319. [Google Scholar] [CrossRef]
- Walton, K.L.; Makanji, Y.; Robertson, D.M.; Harrison, C.A. , The synthesis and secretion of inhibins. Vitam Horm 2011, 85, 149–184. [Google Scholar]
- Walton, K.L.; Kelly, E.K.; Johnson, K.E.; Robertson, D.M.; Stanton, P.G.; Harrison, C.A. , A Novel, More Efficient Approach to Generate Bioactive Inhibins. Endocrinology 2016, 157, 2799–2809. [Google Scholar] [CrossRef]
- Anawalt, B.D.; Bebb, R.A.; Matsumoto, A.M.; Groome, N.P.; Illingworth, P.J.; McNeilly, A.S.; Bremner, W.J. , Serum inhibin B levels reflect Sertoli cell function in normal men and men with testicular dysfunction. J Clin Endocrinol Metab 1996, 81, 3341–3345. [Google Scholar]
- Duval, D.L.; Nelson, S.E.; Clay, C.M. , A binding site for steroidogenic factor-1 is part of a complex enhancer that mediates expression of the murine gonadotropin-releasing hormone receptor gene. Biol Reprod 1997, 56, 160–168. [Google Scholar] [CrossRef]
- Ngan, E.S.; Cheng, P.K.; Leung, P.C.; Chow, B.K. , Steroidogenic factor-1 interacts with a gonadotrope-specific element within the first exon of the human gonadotropin-releasing hormone receptor gene to mediate gonadotrope-specific expression. Endocrinology 1999, 140, 2452–2462. [Google Scholar] [CrossRef]
- Fernandez-Vazquez, G.; Kaiser, U.B.; Albarracin, C.T.; Chin, W.W. , Transcriptional activation of the gonadotropin-releasing hormone receptor gene by activin A. Mol Endocrinol 1996, 10, 356–366. [Google Scholar]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. , Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front Immunol 2020, 11, 584303. [Google Scholar] [CrossRef] [PubMed]
- Annicotte, J.S.; Schoonjans, K.; Auwerx, J. , Expression of the liver X receptor alpha and beta in embryonic and adult mice. Anat Rec A Discov Mol Cell Evol Biol 2004, 277, 312–316. [Google Scholar] [CrossRef]
- Volle, D.H.; Mouzat, K.; Duggavathi, R.; Siddeek, B.; Dechelotte, P.; Sion, B.; Veyssiere, G.; Benahmed, M.; Lobaccaro, J.M. , Multiple roles of the nuclear receptors for oxysterols liver X receptor to maintain male fertility. Mol Endocrinol 2007, 21, 1014–1027. [Google Scholar] [CrossRef]
- Lee, J.H.; Gong, H.; Khadem, S.; Lu, Y.; Gao, X.; Li, S.; Zhang, J.; Xie, W. , Androgen deprivation by activating the liver X receptor. Endocrinology 2008, 149, 3778–3788. [Google Scholar]
- Naamneh Elzenaty, R.; du Toit, T.; Fluck, C.E. , Basics of androgen synthesis and action. Best Pract Res Clin Endocrinol Metab 2022, 36, 101665. [Google Scholar] [CrossRef]
- Hornig, N.C.; Holterhus, P.M. , Molecular basis of androgen insensitivity syndromes. Mol Cell Endocrinol 2021, 523, 111146. [Google Scholar] [CrossRef]
- Knerr, J.; Werner, R.; Schwan, C.; Wang, H.; Gebhardt, P.; Grotsch, H.; Caliebe, A.; Spielmann, M.; Holterhus, P.M.; Grosse, R.; Hornig, N.C. , Formin-mediated nuclear actin at androgen receptors promotes transcription. Nature 2023, 617, 616–622. [Google Scholar] [CrossRef]
- Hornig, N.C.; Rodens, P.; Dorr, H.; Hubner, N.C.; Kulle, A.E.; Schweikert, H.U.; Welzel, M.; Bens, S.; Hiort, O.; Werner, R.; Gonzalves, S.; Eckstein, A.K.; Cools, M.; Verrijn-Stuart, A.; Stunnenberg, H.G.; Siebert, R.; Ammerpohl, O.; Holterhus, P.M. , Epigenetic Repression of Androgen Receptor Transcription in Mutation-Negative Androgen Insensitivity Syndrome (AIS Type II). J Clin Endocrinol Metab 2018, 103, 4617–4627. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; Voelkerding, K.; Rehm, H.L.; Committee, A.L.Q.A. , Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Fabbri-Scallet, H.; Werner, R.; Guaragna, M.S.; de Andrade, J.G.R.; Maciel-Guerra, A.T.; Hornig, N.C.; Hiort, O.; Guerra-Junior, G.; de Mello, M.P. , Can Non-Coding NR5A1 Gene Variants Explain Phenotypes of Disorders of Sex Development? Sex Dev 2022, 16, 252–260. [Google Scholar] [CrossRef]
- Gonen, N.; Eozenou, C.; Mitter, R.; Elzaiat, M.; Stevant, I.; Aviram, R.; Bernardo, A.S.; Chervova, A.; Wankanit, S.; Frachon, E.; Commere, P.H.; Brailly-Tabard, S.; Valon, L.; Barrio Cano, L.; Levayer, R.; Mazen, I.; Gobaa, S.; Smith, J.C.; McElreavey, K.; Lovell-Badge, R.; Bashamboo, A. , In vitro cellular reprogramming to model gonad development and its disorders. Sci Adv 2023, 9, eabn9793. [Google Scholar] [CrossRef]
- Rodriguez Gutierrez, D.; Eid, W.; Biason-Lauber, A. , A Human Gonadal Cell Model From Induced Pluripotent Stem Cells. Front Genet 2018, 9, 498. [Google Scholar] [CrossRef] [PubMed]
- Renaux, A.; Papadimitriou, S.; Versbraegen, N.; Nachtegael, C.; Boutry, S.; Nowe, A.; Smits, G.; Lenaerts, T. , ORVAL: a novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res 2019, 47, W93–W98. [Google Scholar] [CrossRef]
- Udhane, S.S.; Pandey, A.V.; Hofer, G.; Mullis, P.E.; Fluck, C.E. , Retinoic acid receptor beta and angiopoietin-like protein 1 are involved in the regulation of human androgen biosynthesis. Sci Rep 2015, 5, 10132. [Google Scholar] [CrossRef]
Figure 1.
Previously reported transcriptional activity of the c.58G>C; p.(Val20Leu) NR5A1/SF-1 variant tested on three different steroidogenic promoter constructs in HEK293T and NCI-H295R cell lines [26].
Figure 1.
Previously reported transcriptional activity of the c.58G>C; p.(Val20Leu) NR5A1/SF-1 variant tested on three different steroidogenic promoter constructs in HEK293T and NCI-H295R cell lines [26].
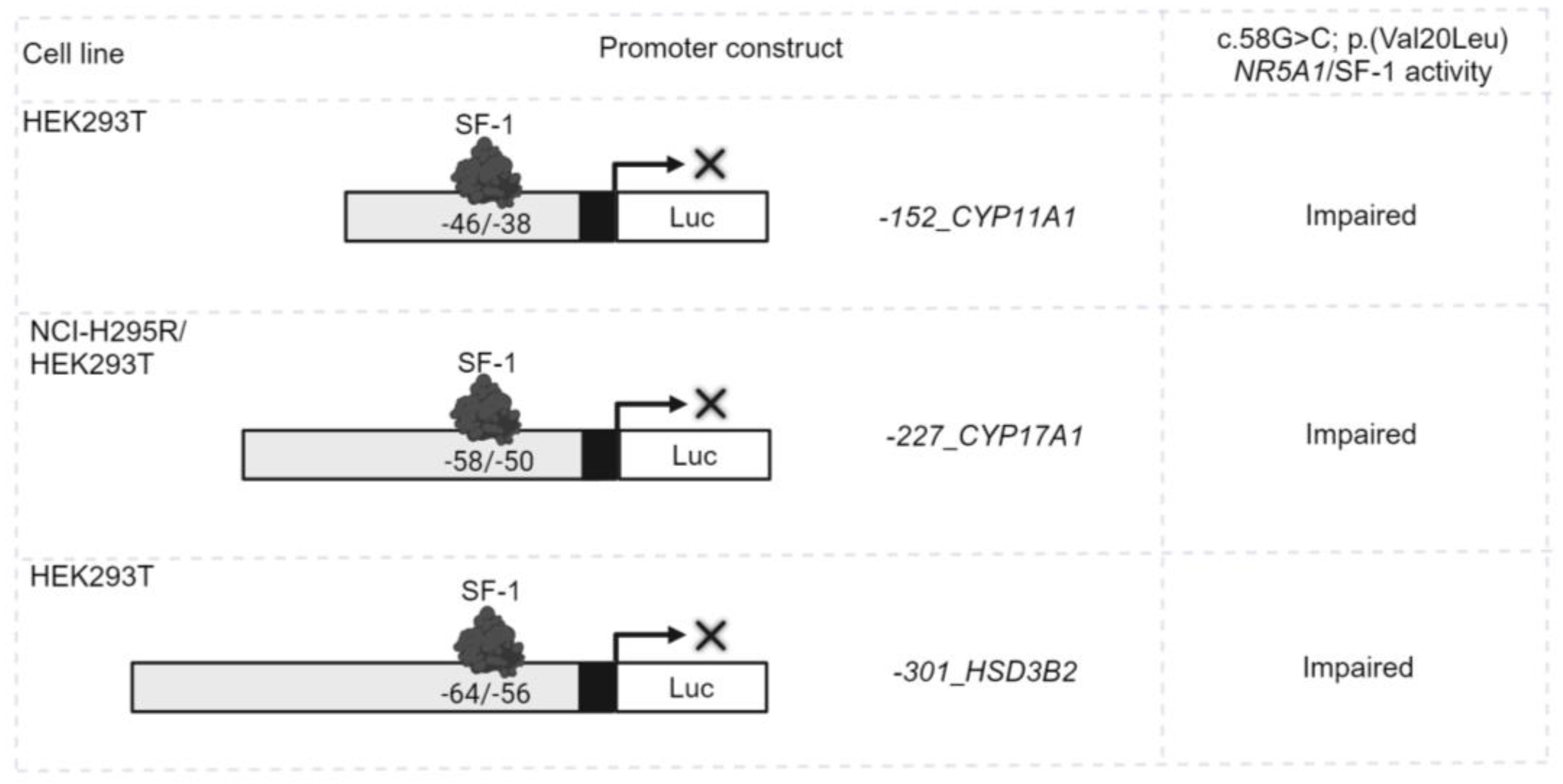
Figure 2.
NR5A1/SF-1 regulates the expression of genes crucial for the function of steroidogenic tissues and the hypothalamic-pituitary-gonadal axis. (A and B). Endogenous NR5A1/SF-1 transcriptional activity on different INHA promoter-reporter constructs in the steroidogenic cell lines: (A) adrenal NCI-H295R cells and (B) mouse Leydig MA-10 cells. Cells were transiently transfected only with the -198_INHA, -213_INHA, -520_INHA, -2050_INHA promoter luciferase reporter constructs. (C) The ability of the WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 to activate four different promoter-reporter constructs of the INHA gene was tested in the non-steroidogenic HEK293T cell line. The cells were transiently co-transfected with WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 and -198_INHA, -213_INHA, -520_INHA, -2050_INHA promoter luciferase reporter constructs. (D) The ability of the WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 to activate the two different promoter-reporter constructs of the GnRHR gene was tested in HEK293T cells. Cells were transiently co-transfected with WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 and -220_GnRHR, -2300_GnRHR promoter luciferase reporter constructs. In all experiments, the luciferase activity was measured with the Dual-Luciferase assay system (Promega). Results are shown as the mean ± standard error of the mean (SEM) of three to five independent experiments, all performed in duplicate. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001. RLU, relative light units.
Figure 2.
NR5A1/SF-1 regulates the expression of genes crucial for the function of steroidogenic tissues and the hypothalamic-pituitary-gonadal axis. (A and B). Endogenous NR5A1/SF-1 transcriptional activity on different INHA promoter-reporter constructs in the steroidogenic cell lines: (A) adrenal NCI-H295R cells and (B) mouse Leydig MA-10 cells. Cells were transiently transfected only with the -198_INHA, -213_INHA, -520_INHA, -2050_INHA promoter luciferase reporter constructs. (C) The ability of the WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 to activate four different promoter-reporter constructs of the INHA gene was tested in the non-steroidogenic HEK293T cell line. The cells were transiently co-transfected with WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 and -198_INHA, -213_INHA, -520_INHA, -2050_INHA promoter luciferase reporter constructs. (D) The ability of the WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 to activate the two different promoter-reporter constructs of the GnRHR gene was tested in HEK293T cells. Cells were transiently co-transfected with WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 and -220_GnRHR, -2300_GnRHR promoter luciferase reporter constructs. In all experiments, the luciferase activity was measured with the Dual-Luciferase assay system (Promega). Results are shown as the mean ± standard error of the mean (SEM) of three to five independent experiments, all performed in duplicate. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001. RLU, relative light units.
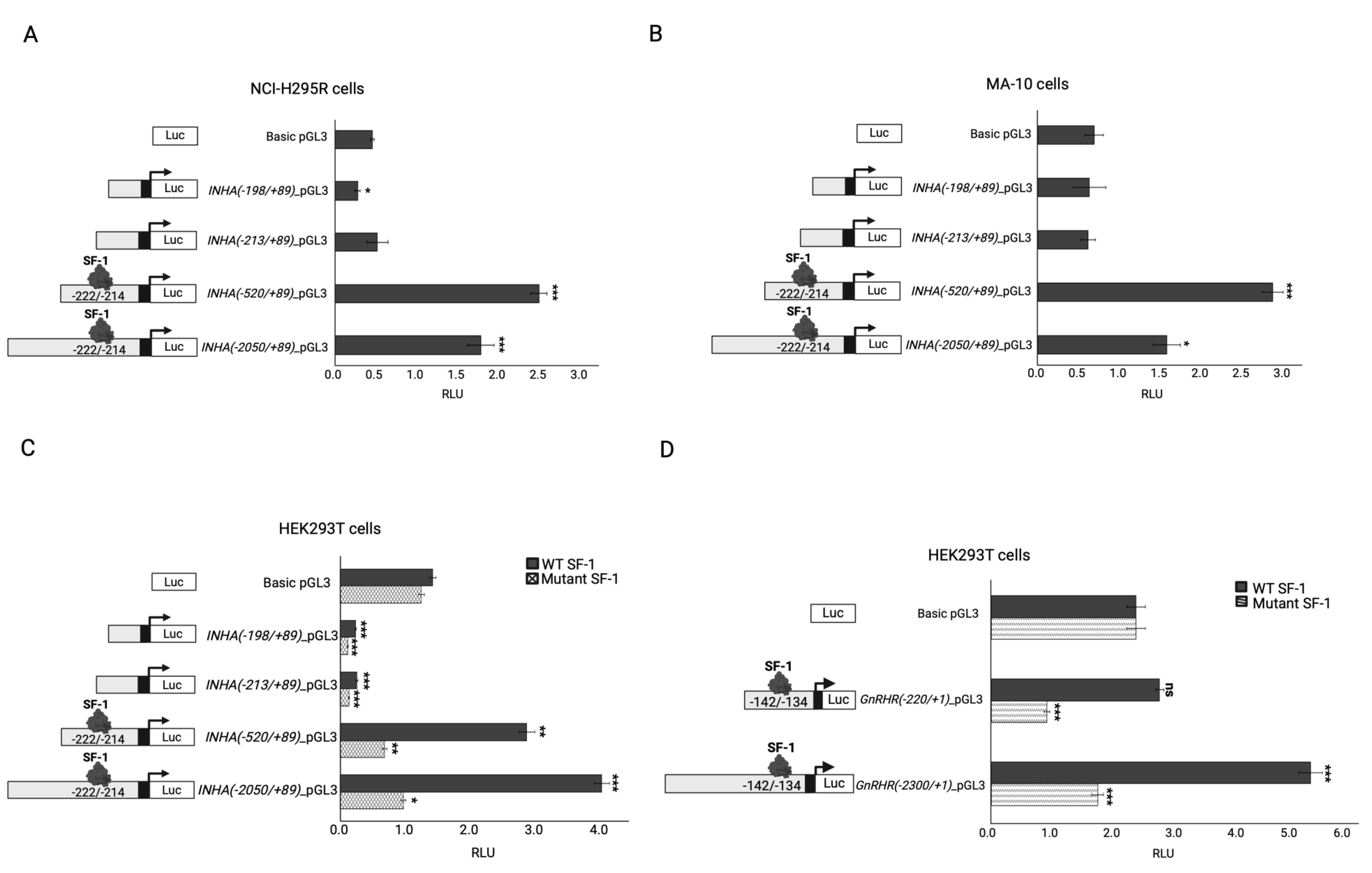
Figure 3.
The transcriptional regulation of the AR in different cell lines. (A-B) The AR promoter construct transcriptional regulation was investigated in the steroidogenic cell line MA-10 (A) and (B) NCI-H295R. Cells were transiently transfected only with the -3000_AR promoter luciferase reporter construct (C-D) The ability of the WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 to activate the AR promoter reporter construct was tested in (C) HEK293T, (D) NCI-H295R and E. MA-10 cells. The Cells were transiently co-transfected with WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 and -3000_AR promoter luciferase reporter construct. (F) The ability of the WT or mutant c.515_516insCAA; p.(Arg171_Lys172insAsn) NR1H2/LXRβ and WT RXRA hetero-tetramer to activate the AR promoter-reporter constructs was tested in HEK293T cells. Cells were transiently co-transfected with WT or mutant c.515_516insCAA; p.(Arg171_Lys172insAsn) NR1H2/LXRβ, WT RXRA, and the -3000_AR promoter luciferase reporter construct. In all experiments, the luciferase activity was measured with the Dual-Luciferase assay system (Promega). Results are shown as the mean ± standard error of the mean (SEM) of three to five independent experiments, all performed in duplicate. RLU, relative light units. Significance of the experimental group vs. the control group: *, p<0.05; **, p<0.01; ***, p<0.001. Significance between the experimental groups: #, p<0.05; ##, p<0.01.3. Discussion.
Figure 3.
The transcriptional regulation of the AR in different cell lines. (A-B) The AR promoter construct transcriptional regulation was investigated in the steroidogenic cell line MA-10 (A) and (B) NCI-H295R. Cells were transiently transfected only with the -3000_AR promoter luciferase reporter construct (C-D) The ability of the WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 to activate the AR promoter reporter construct was tested in (C) HEK293T, (D) NCI-H295R and E. MA-10 cells. The Cells were transiently co-transfected with WT or mutant c.58G>C; p.(Val20Leu) NR5A1/SF-1 and -3000_AR promoter luciferase reporter construct. (F) The ability of the WT or mutant c.515_516insCAA; p.(Arg171_Lys172insAsn) NR1H2/LXRβ and WT RXRA hetero-tetramer to activate the AR promoter-reporter constructs was tested in HEK293T cells. Cells were transiently co-transfected with WT or mutant c.515_516insCAA; p.(Arg171_Lys172insAsn) NR1H2/LXRβ, WT RXRA, and the -3000_AR promoter luciferase reporter construct. In all experiments, the luciferase activity was measured with the Dual-Luciferase assay system (Promega). Results are shown as the mean ± standard error of the mean (SEM) of three to five independent experiments, all performed in duplicate. RLU, relative light units. Significance of the experimental group vs. the control group: *, p<0.05; **, p<0.01; ***, p<0.001. Significance between the experimental groups: #, p<0.05; ##, p<0.01.3. Discussion.
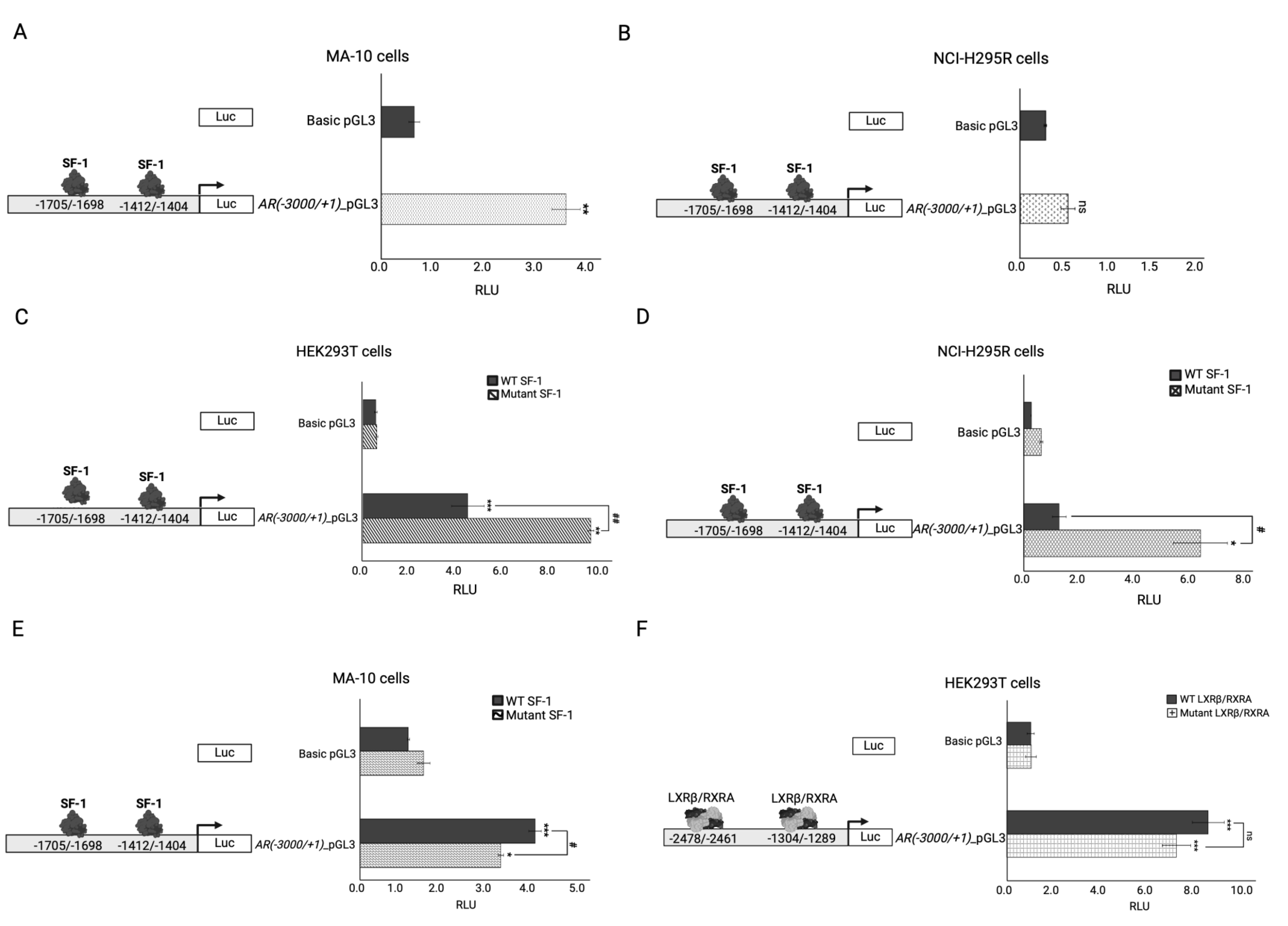

Table 1.
Biochemical characterization of the index patient at 15 years of age.
| Hormones/Markers | Biochemical value | Range | Units |
|---|---|---|---|
| Sex hormones | |||
| FSH | 85.1 | 0.95 – 11.95 | mU/ml |
| LH | 20.3 | 0.57 – 12.07 | mU/ml |
| Prolactin | 29.1 | 3.46 – 19.4 | ng/ml |
| Testosterone | 4.15 | 1 – 12 | ng/ml |
| AMH | 5.18 | 27 – 1141 | pM |
| Adrenal function | |||
| ACTH | 53.7 | 9.0 – 40.0 | pg/ml |
| Cortisol | 179 | 30 – 210 | ng/ml |
| DHEA-S | 2243 | 166 – 2427 | ng/ml |
ACTH, adrenocorticotropic hormone; AMH, anti Müllerian hormone; DHEA-S, dehydroepiandrosterone sulfate; FSH, follicle-stimulating hormone; LH, luteinizing hormone.
Table 2.
Genetic characterization of the different gene variants identified in a complex DSD case.
| Gene name | Gene transcript | Variant | Chromosome position | Type/ Consequence |
ACMG classification (criteria) | SIFT | Polyphen | Mutation Taster | Panther | SNPs and Go | M-CAP | Mutation assessor | REVEL | Provean | ORVAL - VarCoPP score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NR5A1 | ENST00000373588.9 | c.58G>C; p.(Val20Leu) | 9:124503338 | SNV/missense | P | Unc | B | Unc | Prdam | Dis | P | Unc | P | Unc | - |
| NR1H2 | ENST00000253727.10 | c.515_516insCAA;p.(Arg171_Lys172insAsn) | 19:50378563 | Ins/In-frame insertion | VUS | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| INHA | ENST00000243786.3 | c.675T>G; p.(Ser225Arg) | 2:219575100 | SNV/missense | B | B | B | B | ND | Dis | Unc | Unc | B | B | 0.5825 |
| TCF7L2 | ENST00000355995.9 | c.1535C>G; p.(Pro512Arg) | 10:113165647 | SNV/missense | LB | B | B | Unc | Prben | Neu | Unc | B | B | B | 0.9825 |
| NIBAN1 | ENST00000367511.4 | c.929G>A; p.(Arg310His) | 1:184823223 | SNV/missense | VUS | Unc | Prdam | Unc | Prdam | Neu | B | Unc | B | Unc | 0.8500 |
| SCUBE2 | ENST00000649792.2 | c.692C>T; p.(Thr231Ile) | 11:9066765 | SNV/missense | VUS | Unc | Prdam | Unc | Prdam | Neu | B | Unc | Unc | P | 0.8450 |
ACMG, American College Medical Genetics; B, benign; Dis, disease-causing; DSD, disorders of sex development; Ins, insertion; LB, likely benign; ND, not defined; Neu, Neutral; P, pathogenic; Prben, probably benign; Prdam, probably damaging; SNV, single nucleotide variant; Unc, uncertain; VUS, variant of uncertain significance.
Table 3.
Relevant information on selected candidate genes from literature.
| Gene/Protein | Biological function | Phenotype associated with this gene in humans | The phenotype associated with this gene in mice models | In vitro studies (NR5A1 related) | A possible contribution of this gene to the DSD phenotype of the patient? |
|---|---|---|---|---|---|
| NR5A1/SF-1 |
|
NR5A1 homozygous and heterozygous variants are associated with disorders of sex development: including adrenal insufficiency and 46,XY gonadal dysgenesis, ambiguous genitalia, hypospadias, micropenis, spermatogenic failure with normal genitalia and primary ovarian insufficiency [28, 29]. | The majority of heterozygous NR5A1/SF-1 variants located in the DNA binding domain present with impaired functional activity on different human steroidogenic enzyme promoters. While variants located elsewhere in the SF-1 protein present with variable activity. Mostly, no genotype-phenotype correlation was found [6]. | Yes | |
| NR1H2/LXRβ | Plays an important role as a modulator of lipid homeostasis and inflammation throughout the human body [31]. | Diseases associated with NR1H2 include type 2 diabetes and male infertility (azoospermia) [31-35]. |
|
LXRβ is involved in the basal expression levels of CYP11A1, StAR, and NR5A1 in NCI-H295R adrenal cells [33]. | Yes |
| INHA/Inhibinα | Antagonizes activin signaling in the reproductive hypothalamic-pituitary gonadal axis [36, 37]. | Homozygous INHA variants are associated with decreased prenatal and postnatal testosterone production and infertility in males, and primary ovarian failure in women [37-39]. | INHA knockout mice develop mixed or incompletely differentiated gonadal stromal tumors and die from cachexia syndrome [37, 40]. | Rat inhibin alpha gene expression is regulated by the synergistic activity of Nr5a1 and cAMP [41]. | Yes |
| TCF7L2/TCF-4 |
|
TCF7L2 variants are associated with an increased risk of type 2 diabetes [46-48]. |
Tcf7l2 knockout causes neonatal death in mice [42]. Conditional inactivation of Tcf7l2 in the adult intestinal epithelium in mice causes impaired cell proliferation in the small intestines and colon [45]. |
Tcf-4 is involved in the rat inhibin alpha gene expression: Tcf-4 disrupts β-catenin’s ability to synergize with Sf-1 on the inhibin alpha promoter in a dose-dependent manner [49]. | Unlikely |
| NIBAN1/FAM129A | Plays an important role in apoptosis, preventing cell death and tumor progression under stress conditions [50, 51]. | NIBAN1 expression has been described in several tumor subtypes, including microcarcinomas, papillary and follicular carcinoma, prostate cancer, as well as in Hashimoto’s Thyroiditis [50]. | Niban1 -/- mice are viable and show no obvious phenotype or any phenotypic abnormalities [51]. | Not found | Unlikely |
| SCUBE2/SCUB2 | Plays an important role as a tumor suppressor in different types of cancer [52, 53]. |
SCUBE2 expression is reduced in endometrial, breast, and colorectal cancers [52]. | Scube2 (-/-) mice have defective endochondral bone formation and impaired Indian hedgehog-dependent chondrocyte-mediated chondrocyte differentiation and proliferation [54]. | Not found | Unlikely |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



