
Submitted:
07 August 2024
Posted:
08 August 2024
You are already at the latest version
Abstract
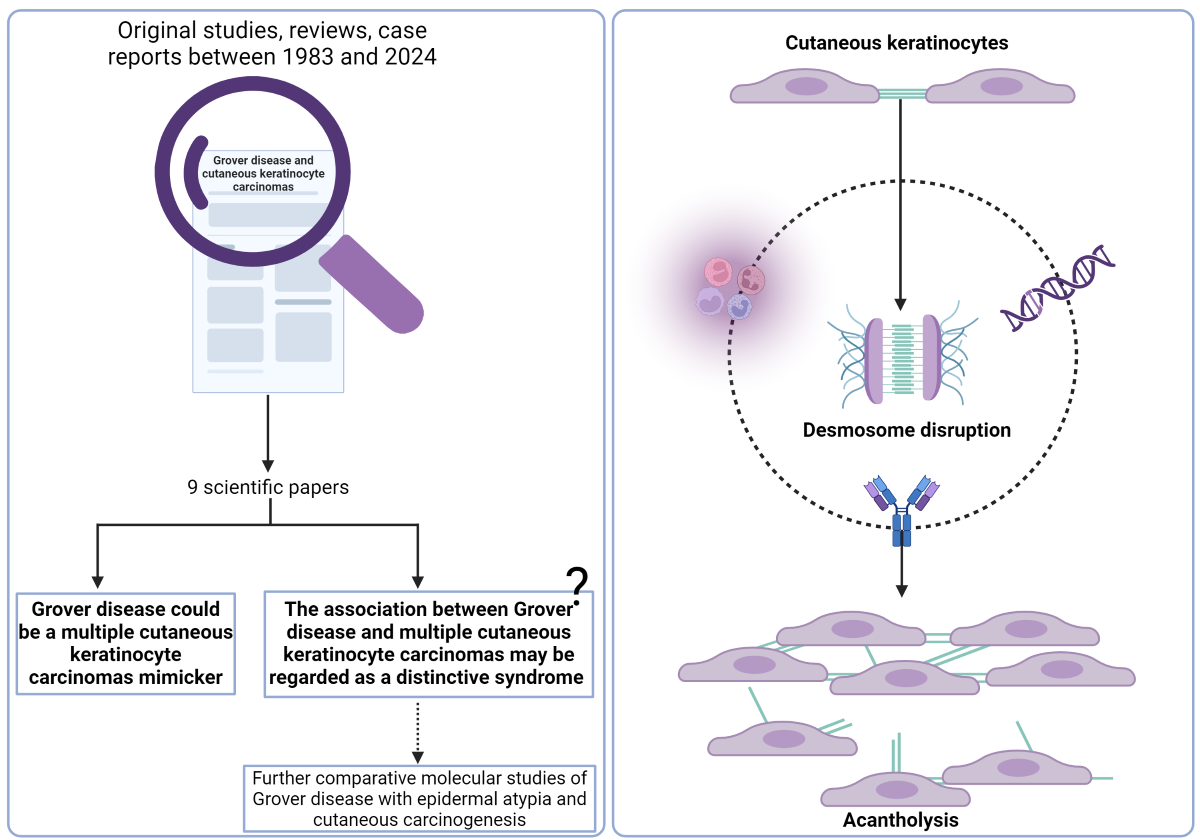
(1) Background: better mechanistic understanding of desmosome disruption and acantholysis in Grover disease (GD) may improve management of this disease. Recent molecular studies highlighted promising pathways to be explored, by directly comparing GD and selected features of associated skin diseases. The association between GD and cutaneous keratinocyte carcinomas, the most prevalent non-melanoma skin cancers (NMSC), is not completely characterized. (2) Objective: To review the medical literature regarding GD-associated cutaneous keratinocyte cancers, focusing on molecular features, pathophysiological mechanisms and disease associations, to help guide future research and patient management. (3) Result: GD has been associated with a variety of skin conditions, but its association with skin cancers has been rarely reported. Between 1983 and 2024 only 9 scientific papers presented data supporting this association. Interestingly, we found that GD may mimic multiple NMSCs, as few authors reported GD cases misdiagnosed as multiple cutaneous squamous cell carcinomas for more than 4 years, or the presence of superficial basal cell carcinoma–like areas associated with focal acantholysis. (4) Conclusions: (a) GD may be an imitator of multiple NMSCs and (b) the relationship between GD and NMSCs may reveal promising pathways for the mechanistic understanding of desmosome disruption and acantholysis in GD and may even lead to its reclassification as a distinctive syndrome.
Keywords:
cutaneous keratinocyte cancers
; non-melanoma
; basal cell carcinomas
; cutaneous squamous cell carcinomas
; Grover disease
; transient acantholytic dermatosis
1. Introduction
Grover disease (GD), also known as transient acantholytic dermatosis, is an acquired papular dermatosis first mentioned in the medical literature in 1970 by Ralph Grover [1]. It is a relatively common disease, but the true incidence is likely underestimated due to its similarity to several other conditions [2]. GD predominantly affects Caucasian middle-aged males, with a notable male-to-female disparity ranging from 3:1 to 7:1 [3]. While diagnosis primarily relies on clinical presentation, histopathological examination serves as crucial support. The hallmark of GD is acantholysis (Figure 1), a phenomenon where intercellular connections such as desmosomes are lost and epidermal cells detach from one another, leading to characteristic lesions [4]. In normal skin, the intercellular space of desmosomes includes the glycoproteins desmogleins and desmocollins, which in conjunction with the cytoplasmic counterparts plakoglobin and plakophilin suport intercellular attachment. Numerous diseases have been reported to be associated with GD, with many of them being oncologic in nature [5,6,7]. A study involving 72 patients with GD revealed concomitant skin cancer in 12% of subjects [6]. However, current evidence is insufficient to conclude a definitive correlation between skin cancers and GD.
A better mechanistic understanding of desmosome disruption and acantholysis would open the door for targeted molecular therapies and improved disease management. Recent molecular studies highlighted novel promising pathways to be explored, by directly comparing GD and selected features of associated skin diseases. However, the association between GD and cutaneous keratinocyte carcinomas, the most prevalent non-melanoma skin cancers (NMSC), is not completely characterized.
Here, we present insights into GD-associated keratinocyte carcinomas, focusing on molecular features, pathophysiological mechanisms and significance of the association for improved management of patients in the future.
2. Methods
To obtain a comprehensive view of the association between Grover Disease (GD) and cutaneous keratinocyte carcinomas, we searched PubMed articles including the terms „Grover disease” OR „Grovers’s disease” OR „transient acantholytic dermatosis” AND „skin cancer” OR „keratinocyte carcinomas” OR „non-melanoma” OR „basal cell carcinoma” OR „squamous cell carcinoma” OR „actinic damage”. Between 1983 and 2023 eight articles were published on the topic (original studies, reviews and case reports and case series), and additionally one in press case report presented relevant data regarding this association.
3. Results
Table 1 summarizes core information within the 9 scientific papers that we found to contain data supporting the link between GD and NMSCs.
In a study group consisting of 72 patients with GD, the authors reported findings related to: hematoxylin-eosin-stained biopsy specimens; immunohistochemistry (IHC) for eight biopsy specimens stained for BRST-2, low-molecular-weight keratin CAM 5.2 and CD44; direct immunofluorescence; and indirect immunofluorescence used to detect major basic protein (MBP) [6]. Clinicopathologic correlations were furthermore discussed, and the comorbidities summarized. In the study group, the authors identified all patterns of acantholysis recognized in the literature at that time (1999), with a predominance of pemphigus vulgaris-like patterns. A prevalent histological finding was the perivascular lymphocytic infiltrate, sometimes admixed with eosinophils, neutrophils and plasma cells. Indirect immunofluorescence revealed large variation in eosinophil degranulation degree, staining one of the eosinophil granule mediators – MBP. Direct immunofluorescence detected changes considered nonspecific and IHC staining for markers of sweat glands was positive in 25% cases for CD44 and negative for BRST-2 and CAM 5.2. Of particular interest for our review is the consistent association between GD and cutaneous malignancies: 17% of patients with GD were diagnosed with concomitant skin cancer and another 15% had concurrent dermatitis. Details about skin cancer subtype or cells responsible for malignant proliferation at this level are not available from this study [6].
In another study, 33 cases of GD were analyzed with a focus on identification and interpretation of epidermal atypia present in GD cases. The research team described the presence of epithelial buds, nuclear pleomorphism, dyskeratosis or altered granular layer of the skin in histologically confirmed cases of GD [8]. Surprisingly, 64% of cases associated varying degrees of keratinocytic atypia and the authors reported for the first time the presence of superficial basal cell carcinoma-like areas associated with focal acantholysis. The authors highlighted that dysmaturative pattern is not an uncommon phenomenon in GD and anticipated the challenge prominent squamous atypia may represent in order to differentiate between GD with epidermal dysmaturation pattern and cutaneous keratinocyte cancers or pre-malignant tumors such as actinic keratosis, Bowen disease or basal cell carcinoma (BCC). The authors suggested that a useful clue for differentiating them is the confinement of the cleft to the basal layer in the acantholytic actinic keratosis in contrast with the cleft that affects all layers of epidermis in dysmaturative GD [8].
A few years later, a report of a 69-year-old man with a 4-year history of erythemato-papular rash on his trunk and extremities confirmed that some biopsies of GD could be mistaken for malignant epithelial lesions [7]. Multiple biopsies at the early stage of the eruption led to the diagnosis of multiple cutaneous squamous cell carcinomas (SCC), which was followed by appropriate surgical treatment. After these four years of disease, the authors reconsidered the case from multiple perspectives: the medical history of the patient and patient’s family were unremarkable; the eruption had a chronic course, worsening under exposure to heat and sweating; symptoms and systemic findings were absent; skin cancer screening was ambiguous; physical examination and dermoscopy revealed focally eroded papules, yellow crypts with white halos in a radiating pattern, all of these findings prompting a new biopsy. The main microscopic features included acanthosis, focal acantholytic dyskeratosis, parakeratosis and a perivascular lymphohistiocytic infiltrate. Correlating the medical history, clinical, dermoscopic and microscopic characteristics of the case, extensive GD was diagnosed. The systemic and local treatment with retinoids and corticosteroids improved the disease course significantly [7].
Regarding the extensive or atypical form of GD, a systematic review presented morphological features and associations of disease. The extensive form of GD was characterized by the distribution of the eruption on the trunk plus at least one additional non-truncal area of the body. When analyzing 72 cases of extensive or atypical GD, the authors found it is much more frequently associated with a malignancy (in 61% of patients) compared with typical GD. Regarding the association of extensive GD with skin neoplasia, half of the nonhematologic cancers reported (12 cases) were metastatic melanoma [5]. Particularly interesting, even if NMSCs were not numerically reported in the review, two of the cited studies presented 10 male patients aged 38-63 years treated in various London and Paris hospitals between 1969 and 1993 with GD and multiple non-melanoma skin cancers (NMSCs) and/or actinic damage: BCCs, SCCs, actinic keratosis and Bowen disease. The authors have proposed that the association between GD and multiple NMSCs would probably best be regarded as a distinctive syndrome [9,10].
In light of the above observations, we included in this review the first report of BCCs associated with GD in a middle-aged female (in press in Proceedings of EADV Congress 2024) [11]. A 62-year-old Caucasian female presented to our clinic with a disseminated eruption of pruritic papules that had developed and progressed continuously over the last 8 years. Initially diagnosed and treated as submammary intertrigo, the eruption subsequently slowly extended to involve the abdomen, lumbar region, and anterior and posterior thorax. During this time, some lesions self-resolved, but others appeared in crops, waxing and waning spontaneously for years. In the same year as the eruption onset, the patient observed an atypical lesion on the right flank, which was excised and diagnosed as superficial pigmented BCCs. Over the next few years, she developed 7 additional BCCs at different locations: the first 3 were completely excised (on the forehead, right flank, and left calf), and 4 were treated with topical 5% imiquimod (on the lumbar region, right forearm, chest, and posterior thorax). All of the surgically excised BCCs were focally pigmented superficial subtypes. Histopathology showed: an apparently multifocal tumor proliferation in continuity with the overlying epidermis, represented by islands, nests, and anastomosing cords in the papillary dermis, composed of basaloid cells with an inverted nucleus-to-cytoplasm ratio and hyperchromatic nuclei with slightly irregular contours and relatively rare mitoses; palisading arrangement of tumor cells at the periphery; focal accumulation of melanin pigment within the cytoplasm; minimal desmoplastic reaction; maximum tumor thickness = 0.3 mm; peritumoral skin is slightly acanthotic, with areas of hyperortho- and parakeratosis, focal spongiosis, and moderate perivascular lymphomonocytic inflammatory infiltrate with rare interstitial eosinophils (eczematous aspect). The patient had no other personal history and no known family history of skin diseases or cancers. She reported occasional sun exposure during holidays.
During the physical examination, we observed Fitzpatrick type II skin phototype, multiple seborrheic keratoses, and a disseminated symmetrical eruption composed of erythematous papules with a whitish-scaling center, mainly on the trunk. Given the personal history of multiple BCCs and the peculiar clinical presentation with central hyperkeratotic papules, a biopsy was performed from an abdominal papule. Histopathological examination revealed: hyperparakeratosis and moderate acanthosis with unevenly elongated epidermal ridges; dyskeratosis with the formation of round and granular bodies; foci of acantholysis with intraepidermal cleavage; moderate lymphocytic inflammatory infiltrate, including eosinophils, vaguely distributed perivascularly and diffusely interstitially in the upper dermis, consistent with Grover's disease (GD), displaying Darier-like characteristics. Dermoscopic evaluation revealed erythematous papule with central stellar-like, yellowish keratotic plug. Based on the evolution (chronic course), clinical, dermoscopic, and histopathological exams, and the negative family history of hereditary diseases, GD with a Darier-like pattern was diagnosed.
To our knowledge, there are no reported cases documenting the coexistence of multiple BCCs with GD in a middle-aged female. Moreover, the predominance of GD in males makes this case exceptional. The clinical diagnosis was particularly challenging due to the atypical presentation of the papules, which diverged from the classic descriptions of GD, as keratotic lesions are rarely mentioned in the literature [3]. The difficulty in distinguishing GD from NMSCs, especially on the trunk, emphasizes the importance of comprehensive evaluation, including histopathology, to guide appropriate management. Given the well-established association between BCCs and immunosuppression [12] and considering some case reports of GD associated with non-skin cancers, our patient necessitated careful assessment to rule out any underlying malignancies.
Two other recently published articles addressed the coexistence of GD and bullous diseases in patients with aggressive cutaneous carcinomas [13,14]. Pinto-Pulido et all reported the case of a 90-year-old man with advanced earlobe SCC treated with a PD-1 inhibitor who developed a pruritic eroded eruption on the neck and abdomen after two months from therapy initiation, with the extension of the lesions on the back and legs during next month. Coexistence of GD and bullous pemphigoid (BP) was supported by histologic and direct immunofluorescence (DIF) features. Histology revealed parakeratotic hyperkeratosis, suprabasal acantholysis and dyskeratosis, while DIF showed strong linear C3 and IgG positivity along the basement membrane [13]. Another recent article reported association of GD with pemphigus foliaceus observed in the case of a 63-year-old woman with a recurrent BCC on the right lower eyelid with infiltration of the orbital floor and compromise of the rectus and inferior oblique muscles, treated with an antagonist of the smoothened receptor (SMO) that inhibits the Hedgehog signaling pathway and extensive surgery. A few months after the treatment, the patient developed an extensive erythemato-squamous eruption, involving the surgery scars, trunk and face. Multiple skin biopsies revealed superficial blister with acantholytic cells, “corps ronds” and grain formation, dyskeratosis. DIF showed intercellular IgG deposition. By correlating the microscopic and molecular results with clinical course of the disease, the authors considered the eruption is the expression of concurrent GD and pemphigus foliaceus [14].
4. Discussion
4.1. Grover Disease (GD) – from Clinical to Molecular Understanding
The prototypical presentation of GD manifests as a sudden onset of self-limited, pruritic papular or papulo-vesicular eruptions primarily localized on the trunk followed by extremities [3]. Other areas involved less frequently include the neck (21%), face/scalp (17%), axilla (4%), and mucosal membranes (1%). When the rash spreads to additional body parts beyond the trunk, it is categorized as extensive GD, while involvement of at least one non-truncal area characterizes atypical GD [5]. Despite its historical designation as “transient acantholytic dermatosis”, it encompasses three main clinical forms: transient eruptive, persistent pruritic, and chronic asymptomatic [2]. The duration has been correlated with age, as older individuals are more likely to have extensive, longer-lasting eruptions. "Grover disease" is the more commonly used term and may be more appropriate since the disease has demonstrated persistence and various morphologies beyond acantholysis [15].
According to existing studies, dermoscopy has emerged as a valuable tool in aiding the diagnosis of GD, also providing insights into disease subtypes [16]. Typically, dermoscopic examination reveals centrally tan to brown keratotic areas with a star-like or discrete roundish/linear appearance, often surrounded by a whitish halo against a background ranging from white to pink to tan [16,17,18]. It was shown that the Darier-like subtype corresponds with this pattern, whereas the spongiotic subtype is characterized by a yellowish-red background and white scales [16].
The characteristic histologic features of the disease include focal acantholysis and varying degrees of dyskeratosis [19]. The histology of GD can present challenges in diagnosis due to predominantly focal involvement and its varied appearances, displaying various proportions of different intraepidermal acantholytic patterns [20]. In addition to classical histopathological subtypes such as Darier-like, spongiotic, Hailey-Hailey-like, and pemphigus vulgaris/foliaceus-like [19], recent descriptions have included subtypes like dysmaturative, porokeratotic, vesicular, lichenoid, and pseudoherpetic, which may manifest in isolation or combination [2,21]. The dermal infiltrate typically consists of lymphocytes and eosinophils. Establishing a definitive diagnosis may require analyzing multiple biopsy specimens, as they often exhibit nonspecific features or closely resemble conditions such as folliculitis, actinic keratosis, drug eruptions, or insect bites [2].
From an electron transmission microscopy perspective, the tonofibrils in GD are poorly developed, with damage seeming to initiate proximal to their insertion in desmosomes [22]. While the structure of desmosomes in the spinous layer appears normal, their numbers vary across studies, ranging from decreased [23] to increased numbers [24]. The latter indicates a potential compensatory mechanism for defective desmosomal function [24]. Notably, half-split desmosomes, observed in diseases like pemphigus vulgaris/foliaceus or Darier's disease, are absent in GD [24,25]. A plausible explanation for acantholysis could be the impairment of desmosomal plaque proteins linking tonofilaments to desmosomal cadherins [24].
4.2. The Intriguing Puzzle of Grover’s Disease Pathophysiology
Despite its initial mention many years ago and the well-deciphered clinical features since then, the precise etiology and pathophysiology of GD remain unclear. Some triggers, such as sweating, heat, sunlight, ionizing radiation, and prolonged bed rest, have been noted in many cases [3,4]. Over time, numerous hypotheses have emerged regarding its pathogenesis, but some have been contradicted by subsequent studies, underscoring the ongoing challenges in understanding this enigmatic dermatological disorder (Figure 1).
One of the earliest pathogenic theories proposed for GD suggested that occlusion of damaged intraepidermal eccrine ducts led to the characteristic skin eruption. It was hypothesized that this occlusion caused leakage of molecules into the epidermis, triggering acantholysis. However, subsequent studies contradicted this hypothesis: (1) no histological/IHC evidence of a relationship between the acantholytic areas and the eccrine apparatus were identified- no leakage of sweat molecules such as carcinoembryonic antigen or epithelial mucins has been demonstrated [6,20,26], [27]; (2) GD tends to spare the acral surfaces, where a high number of eccrine glands exist [20].
Other authors suggest that GD may arise from alterations in skin integrity rather than direct epidermal destruction, such as significant xerosis [12,15]. Recently, Bui et al. demonstrated moderate-to-strong staining of claudin-4 in disrupted keratinocytes both surrounding and within the acantholytic and bullous areas in acantholytic disorders, including cases of GD [28]. In non-affected areas, keratinocytes located in the stratum granulosum, as well as those surrounding hair follicles and adnexal glands, exhibited a normal staining pattern characterized by weak-to-moderate expression in all cases included in this study. The distinctive staining pattern described by them displayed a tapering effect as it transitioned away from the disrupted regions [28]. Tight junctions (TJs), which are predominantly located in the granular cell layer of skin, are essential for maintaining the epidermal barrier, ensuring intercellular integrity and regulating paracellular permeability. In addition to their barrier function, TJs are involved in keratinocyte proliferation, differentiation, adhesion, and apoptosis [29]. Claudins, a diverse family of integral membrane proteins, are critical components of TJs and exhibit specific expression patterns across different cells and tissues, which are crucial for the selective permeability of each tissue. In particular, claudin-1 and claudin-4, which shows a distinct expression pattern in the stratum granulosum, regulated by the ∆Np63 transcription factor from the p53 family, are vital for skin barrier functions [30,31]. The knockdown of claudin-4 in mice results in severe transepidermal water loss and early death, underscoring its crucial role in maintaining an effective epidermal barrier. Moreover, Li et al. demonstrated that the significant upregulation of claudin-4 could act as a compensatory mechanism to maintain barrier function in affected skin regions, potentially counteracting the barrier disruption caused by the downregulation of claudin-1 in these disorders [32].
In the case of linear GD, there is a hypothesis suggesting that the disease's cause might reveal a previously hidden tolerance to abnormal clones, often keratinocytes, nestled within specific embryonic Blaschko lines [24]. Furthermore, a study demonstrating the loss of desmoplakin and plakoglobin in desmosomes in GD has led to a hypothesis that this disease may result from an impairment of the dynamics of desmoplakin or other desmosomal plaque proteins [24,33].
Some studies have proposed an immunomediated pathogenesis of GD [4,34]. However, it remains unclear whether the reported antibodies against desmogleins are the cause of GD or if they appear as a consequence of GD, representing an immune response to the desintegrated desmosomes due to acantholysis [34]. The most recent studies have revealed intriguing findings regarding GD. The transcriptomic profile of GD, Hailey-Hailey disease, and Darier disease exhibits a significant overlap, indicating an increase in keratinocyte differentiation and a decrease in cell adhesion and actin organization [35]. The downregulation of the latter was suggested to occur as a result of decreased serum response factor/ myocardin-related transcription factor (SRF/MRTF) activity. Importantly, these characteristics were not observed in other inflammatory disorders such as psoriasis and atopic dermatitis [35].
Because Grover disease has been reported after checkpoint (cytotoxic T-lymphocyte-associated protein 4- CTLA4) inhibitor therapies, and patients receiving these therapies have been found to have higher serum levels of interleukin-4 (IL-4), it was hypothesized that the upregulation of this interleukin plays an important role in the pathogenesis of GD [36]. This theory is further supported by the successful treatment of refractory cases of Grover disease with Dupilumab [36,37,38]. Dupilumab is a human monoclonal IgG4 antibody that targets and inhibits IL-4 and interleukin-13 (IL-13) signaling by specifically binding to the IL-4Rα subunit common to both IL-4 and IL-13 receptor complexes [39]. The positive response to this therapy indicates that type-2 inflammation may be involved in the pathogenesis of GD. Given that age-related changes in the immune system can lead to a pro-inflammatory state with dysregulation of the Th1/Th2 balance towards a Th2-inflammation profile, this type-2 inflammation may also help explain the increased incidence of GD with age [37].
Another recent study, which performed genetic analysis on 15 archival tissues from patients with GD, found that 80% were associated with somatic damaging single-nucleotide variants (SNVs) in ATP2A2. Notably, UV light-induced mutagenesis may have contributed to the development of the lesions, because all variants were C>T or G>A substitutions. This finding underlines the role of somatic mutations in aquired diseases [40]. SERCA2, a sarco/endoplasmic reticulum Ca2+-adenosine triphosphate (ATP)ase pump encoded by the ATP2A2 gene, plays a crucial role in maintaining homeostatic cytoplasmic Ca2+ levels. Defects in ATP2A2 associated with Darier disease result in both acantholysis and apoptosis, which are partly due to alteration in the synthesis, trafficking, and folding of desmosomal proteins, as well as abnormal cytokeratin expression [41]. Unlike GD, Darier disease is an autosomal dominant condition that manifests earlier in life. Additionally, it has been hypothesized that some cases of GD might actually be mosaic forms of Darier disease [40].
To date, there have been no mechanistic studies of GD, nor have there been studies utilizing GD patient-derived keratinocytes, resulting in limited understanding of desmosomal dysfunction in this disease. Furthermore, this knowledge gap is compounded by the absence of an animal model closely resembling the symptomatology of the human disease [35].
4.3. Grover Disease (GD) Association with Cutaneous Keratinocyte Carcinomas
The association between GD and NMSCs is far from being completely elucidated, but there are sufficient scientific evidence to support the relevance of continuing studies on this topic.
One the one hand, there are several authors who have emphasized the challenge to differentiate between GD with keratinocyte atypia in early stages and NMSCs. In this regard, Aljarbou and colleagues examined biopsy specimens from GD cases, focusing on atypical histopathological changes, revealing in over 60% of cases actinic keratosis–like changes, nuclear pleomorphism, dyskeratosis in all layers of epidermis and an altered granular layer [8]. A few years later, Kotzerke and team confirmed that GD could be a multiple NMSCs mimicker: the authors reconsidered the diagnosis of multiple cutaneous SCCs after 4 years of monitoring the case of a 69-year-old man who had multiple biopsies [7]. Davis et al. reported in a retrospective study conducted at Mayo Clinic Rochester that 12 (16.6%) of 72 cases of GD had skin cancer as concurrent condition [6]. Details about the clinical or microscopic features of the 12 cutaneous malignancies associated with GD were missing and would have been valuable, as the above authors raised awareness recently regarding the challenge to differentiate between GD with epidermal atypia and cutaneous keratinocyte cancer [7,8].
On the other hand, several papers highlighted the link between actinic damage, medical history of NMSCs and GD (Table 1). We elected to include our own case of multiple BCCs associated with GD (in press to EADV Congress 2024) [11]. This case is of a 62-year-old female who developed 8 basal cell carcinomas and persistent GD during an almost ten-year period. To our knowledge, this is the first association between Darier-type GD and multiple BCCs in a famale patient with no significant condition in her past medical history. Few authors have previously described coexistence of GD and multiple BCCs in a case series of male patients [9,10]. A number of authors presented the coexistence of GD and single cutaneous carcinoma, rather associated with different potential etiopathogenic factors of GD [13,14]. Notable from the literature is the case of a 63-year-old woman with a recurrent BCC on her right lower eyelid, treated with vismodegib and large exenteration. Three months after surgery, the patient was diagnosed with concurrent GD and pemphigus foliaceus sustained by clinical, histological, and immunofluorescence examinations [14]. Another case report presented a 90-year-old man treated with pembrolizumab for advanced earlobe SCC, who simultaneously developed GD and bullous pemphigoid two months after initiating pembrolizumab [13].
5. Conclusions
We reviewed the medical literature regarding GD-associated cutaneous keratinocyte cancers, focusing on molecular features, pathophysiological mechanisms and disease associations, to help guide future research and patient management. GD has been associated with a variety of skin conditions, but its association with skin cancers has been rarely reported. Between 1983 and 2024 only 8 articles presented data supporting this association.
Two key points emerged from reviewing the medical literature regarding GD-associated keratinocyte carcinomas: GD may be an imitator of multiple NMSCs; the accurate association between GD and NMSCs may reveal promising pathways for mechanistic understanding of desmosomes disruption and acantholysis in GD and may be regarded as a distinctive syndrome. We suggest that the newly identified association between GD and multiple NMSCs may offer new insights through further comprehensive and comparative molecular studies of GD with epidermal atypia and cutaneous carcinogenesis. Our analysis also represents a meaningful support for clinicians to consider the differential diagnosis of GD in patients with multiple NMSCs when no risk factor is identified and a pruritic, erythematous papular, or papulovesicular rash is observed at any time during the patient monitoring.
Author Contributions
R Nedelcu, A Dobre, G Turcu, and D Ion made substantial contributions to the conception of the study, the interpretation of data and have drafted the article. R Andrei, A Brinzea, F Pantelimon, M Niculescu and A Dobritoiu made substantial contributions to the acquisition and analysis of data and substantively revised the manuscript. R Radu, G Zaharia, E Balasescu, I Hulea and M Gherghiceanu made substantial contributions to the the acquisition of data and substantively revised the manuscript. Every author has approved the submitted version (and version substantially edited by journal staff that involves the author’s contribution to the study) and agrees to be personally accountable for the his own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and documented in the literature.
Funding
Publication of this paper was supported by the University of Medicine and Pharmacy Carol Davila, through the institutional program Publish not Perish.
Informed Consent Statement
Informed consent was obtained from the patient presented in this study.
Acknowledgments
Graphical Abstract Image was created with BioRender.com. The authors would like to thank dr. Emanuel Fertig for the valuable review of the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Grover RW. Transient Acantholytic Dermatosis. Arch Dermatol 1970;101:426–34. [CrossRef]
- Fernández-Figueras MT, Puig L, Cannata P, et al. Grover disease: a reappraisal of histopathological diagnostic criteria in 120 cases. Am J Dermatopathol 2010;32:541–9. [CrossRef]
- Bellinato F, Maurelli M, Gisondi P, Girolomoni G. Clinical features and treatments of transient acantholytic dermatosis (Grover’s disease): a systematic review. JDDG - Journal of the German Society of Dermatology 2020;18:826–33.
- Haniff S, Butler ME, Abou-Jaoude EA, Lenahan ML. An Unusual Trigger of Grover’s Disease (GD). Cureus 2023.
- Gantz M, Butler D, Goldberg M, Ryu J, McCalmont T, Shinkai K. Atypical features and systemic associations in extensive cases of Grover disease: A systematic review. J Am Acad Dermatol 2017;77:952-957.e1. [CrossRef]
- Davis MDP, Dinneen AM, Landa N, Gibson LE. Grover’s disease: clinicopathologic review of 72 cases. Mayo Clin Proc 1999;74:229–34. [CrossRef]
- Kotzerke M, Mitri F, Enk A, Toberer F, Haenssle H. A Case of Extensive Grover’s Disease in a Patient with a History of Multiple Non-Melanoma Skin Cancers. Case Rep Dermatol 2021;13:553–7.
- Aljarbou OZ, Asgari M, Al-Saidi N, Silloca-Cabana E, Alathamneh M, Sangueza O. Grover Disease With Epidermal Dysmaturation Pattern: a Common Histopathologic Finding. Am J Dermatopathol 2018; 0:1-5. [CrossRef]
- Fawcett HA, Miller JA. Persistent acantholytic dermatosis related to actinic damage. British Journal of Dermatology 1983; 109: 349-354. [CrossRef]
- Mokni M, Aractingi S, Grossman R, Verola O, Letessier S, Civatte J, Dubertret L. Persistent acantholytic dermatosis: sex-related differences in clinical presentation? Acta Derm Venereol. 1993;73(1):69-71. [CrossRef]
- Dobre A, Nedelcu R, Turcu G, Andrei R, Brinzea A, Balasescu E, Hulea I, Pantelimon F, Ion D. A rare case of Grover disease associated with multiple basal cell carcinomas. In Proceedings of European Academy of Dermatology & Venereology Congress, Amsterdam, Netherlands, in press 25-28 September 2024.
- Casanova JM, Pujol RM, Taberner R, Egido R, Fernandez E, Alomar A. Grover’s disease in patients with chronic renal failure receiving hemodialysis: clinicopathologic review of 4 cases. J Am Acad Dermatol 1999;41:1029–33. [CrossRef]
- Pinto-Pulido EL, Polo-Rodríguez I, González-Cañete M, Medina-Expósito I, Vélez-Velázquez MD, Medina-Montalvo S. Association of bullous pemphigoid and Grover disease induced by immune checkpoint therapy. An Bras Dermatol 2024;13:S0365-0596(24)00122-3. [CrossRef]
- Magdaleno-Tapial J, Valenzuela-Oñate C, Martínez-Doménech A, García-Legaz-Martínez M, Carballeira-Braña A, Sánchez-Carazo JL, Pérez-Ferriols A, Alegre-de Miquel V. Coexistence of Pemphigus Foliaceus and Grover Disease After a Radical Surgery for Basal Cell Carcinoma. Am J Dermatopathol 2019;41:744–746. [CrossRef]
- Aldana PC, Khachemoune A. Grover disease: review of subtypes with a focus on management options. Int J Dermatol 2020;59:543–50. [CrossRef]
- Errichetti E, De Francesco V, Pegolo E, Stinco G. Dermoscopy of Grover’s disease: Variability according to histological subtype. J Dermatol 2016;43:937–9. [CrossRef]
- Giacomel J, Zalaudek I, Argenziano G. Dermatoscopy of Grover’s disease and solitary acantholytic dyskeratoma shows a brown, star-like pattern. Australasian Journal of Dermatology 2012;53:315–6. [CrossRef]
- Specchio F, Argenziano G, Tiodorovic-Zivkovic D, et al. Dermoscopic clues to diagnose acantholytic dyskeratosis. Dermatol Pract Concept 2015;5:59. [CrossRef]
- Chalet M, Grover R, Ackerman AB. Transient Acantholytic Dermatosis: A Reevaluation. Arch Dermatol 1977;113:431–5. [CrossRef]
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med 2009;133:1490–4. [CrossRef]
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature. J Cutan Pathol 2017;44:486–9. [CrossRef]
- Grover RW. Transient Acantholytic Dermatosis: Electron Microscope Study. Arch Dermatol 1971;104:26–37. [CrossRef]
- Mota A, Correia TM, Lopes JM, Guimarães JM. Successful treatment of Grover’s disease with calcipotriol. European Journal of Dermatology 1998;8:33–5.
- Asahina A, Ishiko A, Saito I, Hasegawa K, Sawamura D, Nakano H. Grover’s Disease Following Multiple Bilateral Blaschko Lines: A Rare Clinical Presentation with Genetic and Electron Microscopic Analyses. Dermatology 2012;225:183–7. [CrossRef]
- Wang W, Amagai M, Ishiko A. Desmosome splitting is a primary ultrastructural change in the acantholysis of pemphigus. J Dermatol Sci 2009;54:59–61. [CrossRef]
- Hashimoto K, Moiin A, Chang MW, Tada J. Sudoriferous acrosyringeal acantholytic disease. J Cutan Pathol 1996;23:151–64. [CrossRef]
- Antley CM, Carrington PR, Mrak RE, Smoller BR. Grover’s disease (transient acantholytic dermatosis): relationship of acanthoiysis to acrosyringia. J Cutan Pathol 1998;25:545–9. [CrossRef]
- Bui CM, Vuong HG, Le MK, et al. Claudin-4 Upregulation in Acantholytic and Autoimmune-Mediated Bullous Disorders. Dermatopathology (Basel). 2023;11(1):1-7. Published 2023 Dec 21. [CrossRef]
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004 Jun;286(6):C1213-28. [CrossRef] [PubMed]
- Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta. 2008 Mar;1778(3):588-600. [CrossRef] [PubMed]
- Kubo T, Sugimoto K, Kojima T, Sawada N, Sato N, Ichimiya S. Tight junction protein claudin-4 is modulated via ΔNp63 in human keratinocytes. Biochem Biophys Res Commun. 2014 Dec 12;455(3-4):205-11. [CrossRef] [PubMed]
- Li J, Li Q, Geng S. All-trans retinoic acid alters the expression of the tight junction proteins Claudin-1 and -4 and epidermal barrier function-associated genes in the epidermis. Int J Mol Med. 2019 Apr;43(4):1789-1805. Erratum in: Int J Mol Med. 2024 Sep;54(3):75. doi: 10.3892/ijmm.2024.5399. PMID: 30816426; PMCID: PMC6414175. [CrossRef]
- Hashimoto K, Fujiwara K, Tada J, Harada M, Setoyama M, Eto H. Desmosomal dissolution in Grover’s disease, Hailey-Hailey’s disease and Darier’s disease. J Cutan Pathol 1995;22:488–501. [CrossRef]
- Phillips C, Kalantari-Dehaghi M, Marchenko S, et al. Is Grover’s disease an autoimmune dermatosis? Exp Dermatol 2013;22:781–4. [CrossRef]
- Roth-Carter QR, Burks HE, Ren Z, et al. Transcriptional profiling of rare acantholytic disorders suggests common mechanisms of pathogenesis. JCI Insight 2023;8. [CrossRef]
- Shelton E, Doolittle C, Shinohara MM, Thompson JA, Moshiri AS. Can't handle the itch? Refractory immunotherapy-related transient acantholytic dermatosis: prompt resolution with dupilumab. JAAD Case Rep. 2022;22:31-33. Published 2022 Feb 10. [CrossRef]
- Barei F, Torretta S, Morini N, Ferrucci S. A case of Grover disease treated with Dupilumab: Just serendipity or a future perspective? Dermatol Ther. 2022 May;35(5):e15429. [CrossRef] [PubMed] [PubMed Central]
- Butler DC, Kollhoff A, Berger T. Treatment of Grover disease with Dupilumab. JAMA Dermatol. 2021;153(3):353-356. [CrossRef]
- D'Ippolito D, Pisano M. Dupilumab (Dupixent): An Interleukin-4 Receptor Antagonist for Atopic Dermatitis. P T. 2018 Sep;43(9):532-535. [PubMed] [PubMed Central]
- Seli D, Ellis KT, Goldust M, Shah K, Hu R, Zhou J, McNiff JM, Choate KA. Association of Somatic ATP2A2 Damaging Variants With Grover Disease. JAMA Dermatol. 2023 Jul 1;159(7):745-749. [CrossRef] [PubMed] [PubMed Central]
- Hobbs RP, Amargo EV, Somasundaram A, et al. The calcium ATPase SERCA2 regulates desmoplakin dynamics and intercellular adhesive strength through modulation of PKCα signaling. FASEB J. 2011;25(3):990-1001. [CrossRef]
Figure 1.
The molecular background of GD pathophysiology.
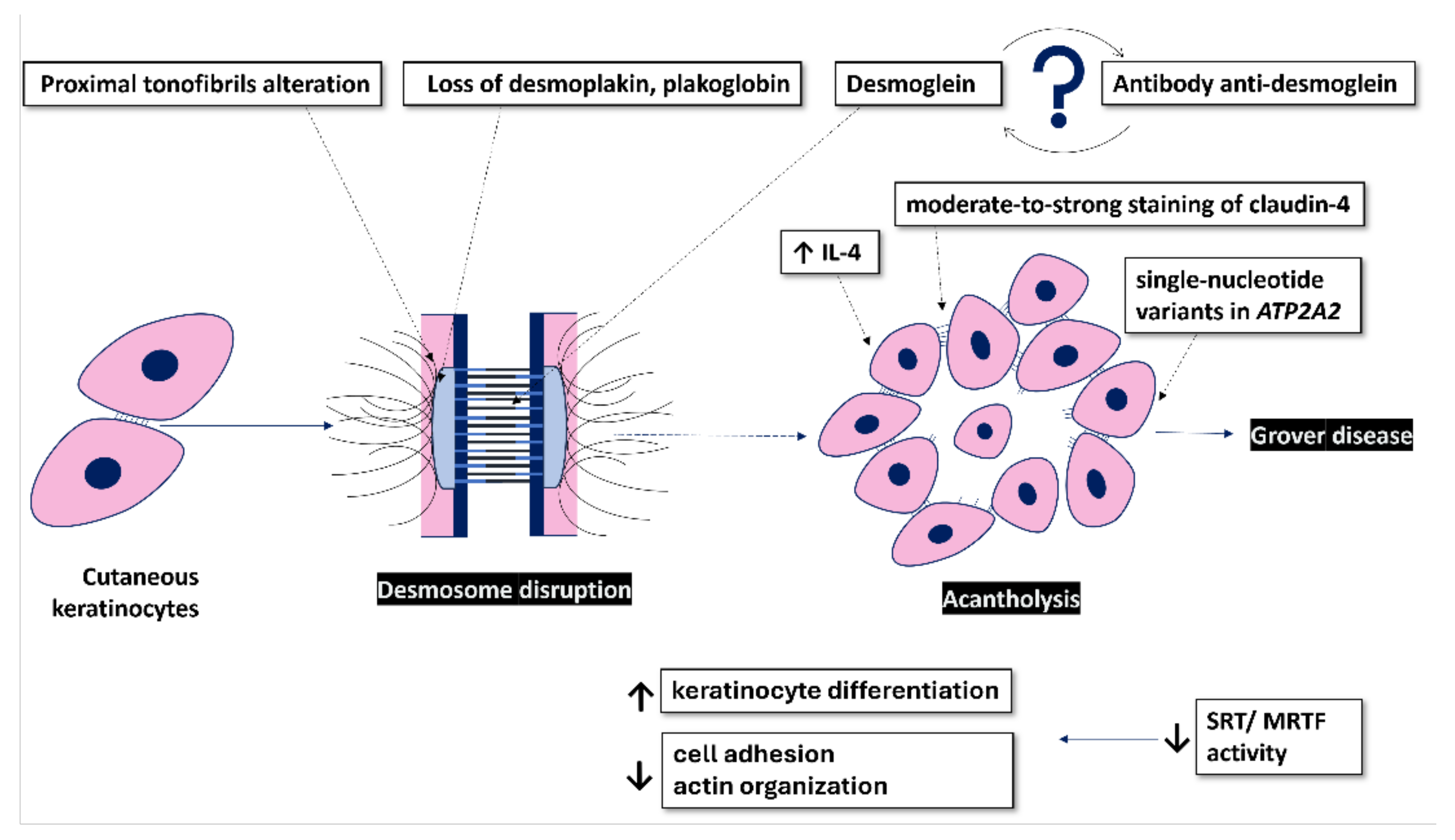

Table 1.
Summary of studies investigating Grover disease (GD) and cutaneous malignancies.
| Scheme . | Study type | Sample Size | Core Findings | Ref. |
|---|---|---|---|---|
| Davis et al. (1999) | Original article | 72 | 12 of GD patients (17%) associated skin cancer but details about skin cancer subtype were not available from this study | [6] |
| Aljarbou et al. (2018) | Original article | 33 | 20 cases (64%) of GD biopsies associated varying degrees of keratinocytic atypia that may represent a challenge to differentiate between GD with dysmaturative pattern and cutaneous keratinocyte cancers or pre-malignant tumors such as actinic keratosis (AKs), Bowen disease or basal cell carcinoma (BCC) | [8] |
| Kotzerke et al. (2021) | Case report | 1 | 69-year-old man misdiagnosed with multiple squamous cell carcinomas (SCCs), later identified as extensive GD after multiple biopsies and comprehensive analysis | [7] |
| Gantz et al. (2017) |
Systematic review | 72 | Extensive or atypical GD is associated with malignancy in 31 of cases (61%); half of non-hematologic cancers were metastatic melanoma (6 cases) | [5] |
| Fawcett HA & Miller JA (1983) |
Case series | 10 | 9 of 10 (90%) male patients associated at least one of the following: BCCs (6 patients), AKs (4 patients), SCCs including Bowen’s disease (2 patients), melanoma (1 patient) | [9] |
| Mokni M et al. (1993) |
Case report | 1 | 58 year-old man diagnosed with persistent acantholytic dermatosis associated with multiple AKs, SCCs, BCCs and Bowen’s disease | [10] |
| Dobre et al. (2024) | Case report | 1 | 62-year-old female with Darier-like pattern GD coexisted with 8 BCCs | [11] |
| Pinto-Pulido et al. (2023) | Case report | 1 | 90-year-old male developed GD and bullous pemphigoid after 2 months of PD-1 inhibitor treatment for invasive SCC | [13] |
| Magdaleno-Tapial et al. (2019) | Case report | 1 | Coexistence of GD with pemphigus foliaceus after invasive relapsing BCC treatment | [14] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



