
Submitted:
09 January 2025
Posted:
10 January 2025
You are already at the latest version
Abstract
Cardiac muscle cells are specialised rod-shaped cells with a single nucleus that are connected to each other by intercalated discs. They are extremely important for the functioning of the heart, as they are responsible for the propagation and effectiveness of contraction. Mitochondria within these cells are vital for adenosine triphosphate (ATP) synthesis, redox balance, calcium homeostasis, and lipid synthesis. Heat shock protein 60 (Hsp60), a mitochondrial chaperonin, plays a crucial role in protein folding. The expression of Hsp60 increases when the heart is under stress, such as during a heart attack or heart failure. Hsp60 is located not only inside the cell, but has also been localised in small and large vesicles, in the cell membrane and freely in the cell culture medium or in the blood. And it is precisely in the latter form that this chaperonin could play a decisive role as an immunomodulator. In this review, we have brought together various studies to discuss and highlight the distribution of Hsp60 in cells, its association with apoptosis and its action as a damage-associated molecular pattern (DAMP) that interacts with Toll-like receptors to modulate immune responses. The link to heart disease arises from a connection to heart-specific microRNAs whose expression can be affected by dysregulation of Hsp60. In summary, we hypothesise that Hsp60 may also have an immunomodulatory effect under stress conditions.
Keywords:
Heart
; diseases
; heat shock proteins
; Hsp60
; cardiac cells
1. Introduction
The process of decomposition of glucose by oxidative phosphorylation (OXPHOS) is performed to produce a substantial quantity of adenosine triphosphate (ATP) which is a duty for mitochondria and manifests its capacity. The mitochondrial electron transport chain (ETC), which is made up of organic molecules and protein complexes, facilitates OXPHOS-dependent ATP production by transferring electrons to molecular oxygen and establishing the electrochemical gradient required for ATP synthase activity [1].
Along with producing energy and serves as the building blocks of production for other cellular components, mitochondria has a significant role in signaling organelles, governing crucial biological activities like cell division, growth and advancement, and death in addition to maintaining the redox and calcium (Ca2+) balance. Thus, mitochondria play a crucial role in both physiological and pathological processes such as inflammation, cancer, and tissue repair [2,3,4,5]. In order to produce ATP from a variety of accessible substrates and respond to external stimuli, mitochondria must convert various energy sources into a steady flow of energy. This substrate flexibility allows for quick variations in cardiac workload, such as those seen during exercise, as well as changes in the environment, including the amount of oxygen and nutrients in the blood. Since the heart muscle requires large amounts of energy to ensure effective contraction and ion transport, fatty acids are its favored substrates, providing 60–90% of the required energy [6].
In addition to being necessary for the differentiation and maturation of cardiomyocytes, mitochondria also undergo maturation. In the developing heart, they gain bulk, undergo structural development, and become functionally specialised [7]. The expression of heat-shock protein 60 (Hsp60) is strongly associated with heat stress, but its expression has also been described in non-stressed cells of the heart, mainly in the cytoplasm of cardiomyocytes [8]. The induction and upregulation of Hsp60 in the myocardium was also found in response to high temperature, as demonstrated by enzyme-linked immunosorbent assay (ELISA). This indicates the importance of Hsp60 for the protection of myocardial cells [9].
The aim of this review is to explore and explain the complex relationships between the composition and structure of cardiac muscle cells and their functions, the role of mitochondria and their function in heart disease, and the role of Hsp60 in cardiac health and disease, which is becoming increasingly important.
2. Heat Shock Proteins, Chaperones and Chaperonins
Heat-shock proteins (Hsps) are ubiquitous proteins expressed by prokaryotes and eukaryotes and are essential for life. Their sequence is highly conserved and they belong to a larger family of proteins responsible for proteostasis [10]. They have a molecular range of 10 to 100 kDa and were first discovered and described in the salivary gland of flies. Based on their molecular weight, they are categorized into small and large heat shock proteins (Hsps), for example, Hsp40, 60, 70, and 90. [11]. Stresses that can cause the expression of Hsps are starvation, heat, cold, and hypoxia [12,13].
Hsps are known as molecular chaperones because they control the folding of cell proteins with the help of conformational changes triggered by the binding of ATP. A misfolding caused by a malfunction of the Hsps leads to death [14,15].
Chaperones also prevent the aggregation of proteins, facilitate proper folding, and regulate secretion in vivo [14,16,17,18,19]. The presence of molecular chaperones is crucial for the conversion of protein chains into folded proteins [21].
Hsp60 is known as chaperonin due to its molecular weight of 60 kDa. It is one of the historically best-conserved proteins in both eukaryotic and prokaryotic cells. The genomic sequence is highly conserved and it is expressed in all cells of a living organ [21]. The first discovered localization of Hsp60 was within the mitochondria, where it regulates the homeostasis of mitochondrial proteins [22,23].
The double ring structure of Hsp60 in eukaryotes depends on the cofactor Hsp10. In the absence of ATP and Hsp10, Hsp60 is a single heptamer ring. Double ring structures develop between the single rings in an ATP-dependent manner, and Hsp60/Hsp10 forms complexes that resemble footballs [24,25]. The integrity of the mitochondrial respiratory chain depends on mitochondrial Hsp60.
Nearly half of the proteins in the mitochondrial matrix interact with the Hsp60 and HSP10 chaperonins, which both serve also in the folding of these proteins. Even 19 interactors, which exhibit significant cellular abundance, make for more than sixty percent of the mass of theproteins that co-immunoprecipitate with these chaperonins. This means that the impaired folding of a wider range of mitochondrial matrix proteins may be the cause of several disorders, like neuronal ones. The impairment may be caused by DNA mutations of interacting proteins or in the same chaperonins [26].
A wide range of mitochondrial proteins that are involved in several metabolic processes, including the three main routes for energy production—the respiratory chain, the cycle of tricarboxylic acid, and fatty acid oxidation— constitute the Hsp60 interactome [26]. In addition to Hsp60, other chaperones like the Hsp70 chaperone homolog, HspA9, and the matrix protease LNOP1, may interact with the mitochondrial proteins representing a higher order of complexes made up of unfolded or partially folded proteins and two or more protein quality control components [26]. On the other side, a proteomic analysis of the LONP1 interactome has previously identified both Hsp60 and HspA9 as LNOP1 interactors [27].
It is interesting to note that Hsp60 is also found in the bloodstream and in extramitochondrial compartments such as the cytosol, the plasma membrane and the extracellular space [28]. Hsp60 regulates the activity of other cellular processes, such as cell division, apoptosis, migration and immunological responses, depending on the different localisation of the protein [29]. Expression of Hsp60 is abundant in cardiac tissue and has been observed in many subcellular compartments within cardiomyocytes, such as the cytoplasm, extracellular space and mitochondria [30,31,32,33,34,35]. Most of the Hsp60 in cardiac cells is located in the mitochondria, whereas the cytoplasm contains only 20–40% of the protein [31].
The reduced release of mitochondrial cytochrome c, the impaired caspase-3 activity, the increased ATP recovery and the increased activities of mitochondrial complexes III and IV are all related to the protective role of overexpressed Hsp60 and Hsp10 [21].
3. Structure of the Cardiac Muscle Cell and Role of Mitochondria
The cardiac muscle cell shares similar characteristics with the skeletal muscle fiber cell in both structure and function. The cardiac muscle cell has a single nucleus, is rod-shaped, and is connected to other cells through intercalated discs, which serve as crucial structural connections. These protein discs enhance the effectiveness of cardiac contraction by aligning the cells with each other, thus creating a three-dimensional mesh of myocytes within a supporting matrix of fibrous tissue in the cardiac wall. The cardiac cell contains myofibrils that are structurally like those in the skeletal muscle cell, though the proteins, despite having similar structures, have different amino acid sequences, allowing antibodies to distinguish between cardiac and skeletal proteins. The names of the sarcomere proteins are the same, the structure observed under the electron microscope is similar, and mitochondria are abundant in both cell types.
Mitochondria in cardiomyocytes are in a space parallel to the myofibrils. Cardiomyocytes contain about 5,000 to 8,000 mitochondria per cell [36]. More than 95% of the ATP in the myocardium is synthesized by mitochondria. Additionally, mitochondria play important roles in regulating redox status, calcium homeostasis, and lipid synthesis [37].
Mitochondria transform fatty acids taken from the bloodstream into ATP, providing 60-90% of the myocardium's energy supply. They also counteract the accumulation of reactive oxygen species (ROS) through detoxification proteins and enzymes, such as the mitochondrial antioxidant manganese superoxide dismutase (MnSOD) [38]. Many biochemical processes and pathways in mitochondria require high levels of Ca2+ and other ions like Na+, H+, and K+. Therefore, the mitochondrial membranes are rich in ion channels selective for these ions, which help preserve mitochondrial membrane potential [39].
Mitochondria also play a crucial role in lipid homeostasis. While most lipids are synthesized in the endoplasmic reticulum (ER), some components are synthesized in the inner mitochondrial membrane (IMM). One of the most important components of the IMM is cardiolipin, an abundant phospholipid first isolated from animal hearts, which constitutes about 20% of the total lipid composition of the IMM [40].
Given the abundance of mitochondria in cardiomyocytes and their important roles in energy supply and lipid homeostasis, it is not surprising that mitochondrial dysfunction is strongly linked to the development of cardiomyopathy and an increased risk of heart failure [41].
4. Hsp60 Expression and Localization in the Healthy Heart Tissue
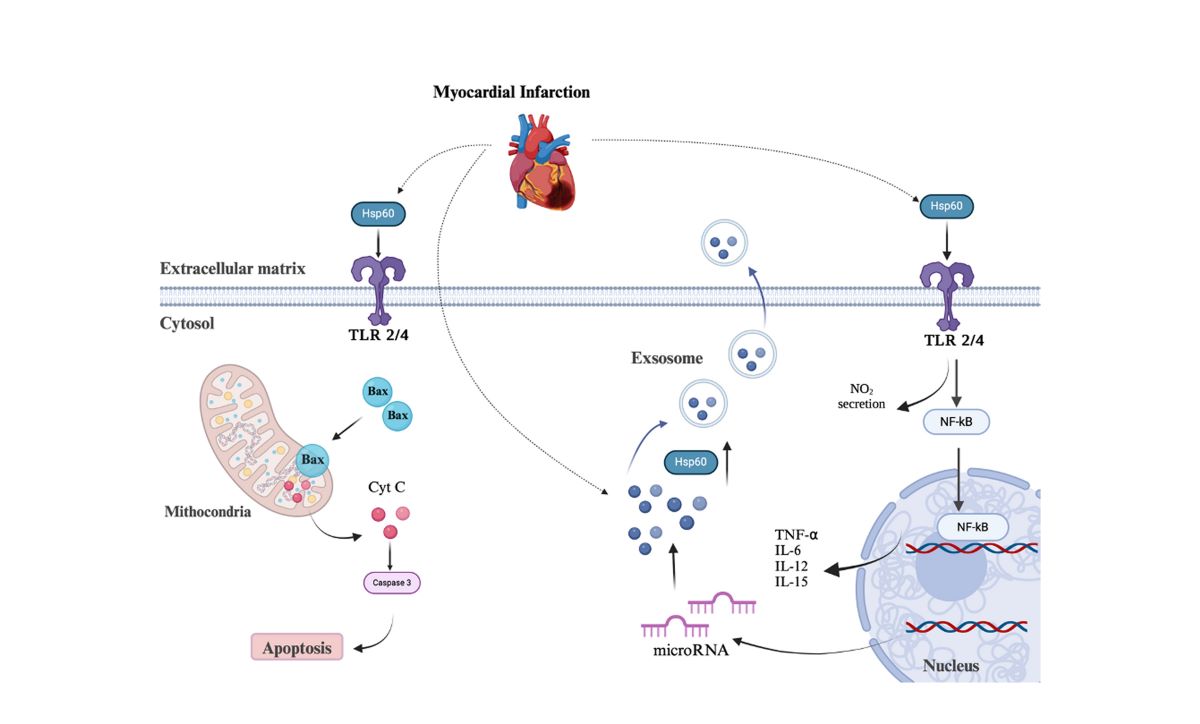
The localization of heat shock proteins in cardiac tissue, such as Hsp60 and the associated Hsp10, is similar to their localization in other tissues (Figure 1). Hsp60 has been found inside mitochondria, in the cytoplasm of cardiac cells [42], on the outer membrane, and within exosomes [43]—small vesicles released by cardiomyocytes—and finally in the blood [44]. In its cytosolic localization, Hsp60 is frequently associated with Bcl-2-associated X protein (Bax) and apoptosis. The expression levels of Hsp60 are typically so low that researchers have used heat shock to detect the localization of this protein. For example, in studies involving rats exposed to 42±1°C for 0 (control), 20, 80, and 100 minutes, researchers detected high levels of both Hsp60 and Hsp10 after 100 minutes, exhibiting a punctate distribution typical of mitochondrial localization [45].
The mechanism through which Hsp60 is released into the extracellular space, either inside extracellular vesicles or freely in the interstitial space, is not well understood and is widely debated. Hsp60 may be released as a free protein because of cell lysis, but transport mediated by exocytosis cannot be excluded. Recently, we demonstrated the release of Hsp60 inside small and large vesicles by immortalized muscle cell lines (C2C12 mouse myoblasts). We also showed that induced expression of high levels of Hsp60 can enhance its localization inside extracellular vesicles [46]. Immunolocalization studies using transmission electron microscopy revealed either a sub-membrane or an intramembrane localization. In both cases, overexpression of Hsp60 increased its presence in extracellular vesicles, with higher levels inside large vesicles compared to small extracellular vesicles. Although we did not use cardiomyocytes, we hypothesize that overexpression of Hsp60 using the same expression plasmid would induce a similar effect in isolated cardiomyocytes, leading to the induced and controlled release of Hsp60. The induced release of the protein inside extracellular vesicles or freely in the medium may explain the role of extracellular Hsp60 as an immune system modulator.
Hsp60 may play both pro-inflammatory and anti-inflammatory roles depending on its interactions with cell-surface receptors, including Toll-like receptors (TLRs). It may also bind to other proteins during an immune response to assist in their presentation to lymphocytes [47].
Human Hsp60, acting as a damage-associated molecular pattern (DAMP), elicits a rapid release of nitric oxide (NO), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, IL-12, and IL-15 from macrophages. It also can upregulate costimulatory molecules of major histocompatibility complex class I (MHC-I) and II (MHC-II), Cluster of Differentiation 86 (CD86), and CD40, promoting the maturation of dendritic cells (DCs) and enhancing the antigen-presenting capacity of antigen-presenting cells (APCs) [48].
Recently, it has been suggested that Hsp60 may act as a ligand for TLR 2 and 4, modulating the immune response and inducing the release of TNF-α, IL-6, and IL-8 [49]. Activation of TLR-4 and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway in injured cardiac tissue leads to apoptosis and impaired contractility of the cardiac tissue [50].
5. Mitochondrial Dysfunction in Cardiac Diseases: The Role of HSP60 Release and Its Protective Function
Mitochondrial dysfunction plays a crucial role in various cardiac disorders, such as heart failure and myocardial infarction. Impaired function leads to energy imbalance, increased oxidative stress, and cardiomyocyte death. HSP60 is vital for the proper folding of mitochondrial proteins, and essential for maintaining mitochondrial health. In cardiac cells, HSP60 is primarily found in the mitochondria, with about 20-40% located in the cytoplasm [51]. HSP60 and HSP10 create a single-loop structure, forming an ATP-dependent chaperone system in the mitochondria. The role of HSP60 in this system is crucial for protein folding and preventing the accumulation of misfolded proteins [52].
In pathological conditions, HSP60 can be released into the extracellular space, where it acts as a DAMP. This interaction with TLRs on immune cells can lead to a systemic inflammatory response. In response to mitochondrial stresses, such as oxidative stress and ischaemia/reperfusion (I/R) injury, the unfolded mitochondrial protein response (UPRmt) is activated. It is hypothesised that the UPRmt may be associated with damaging events in the heart. For example, studies have demonstrated that the localisation of HSP60, a mitochondrial chaperone molecule, on the cell surface activated the innate immune system, inducing the release of TNF-α. This was observed in a coronary artery ligation model in rats with heart failure, where increased myocyte apoptosis was also noted [53].
Recent studies show that mitochondrial HSP60 helps protect heart cells. When this gene was deleted in adult heart cells, it led to major changes in how mitochondrial complexes work, a drop in the mitochondrial membrane potential, and an increase in ROS. This resulted in dilated cardiomyopathy, heart failure, and death in mice. An analysis of mitochondria without HSP60 found that about 20% of the proteins in them depend on HSP60 to fold correctly [51].
6. Hsp60 and Cardiac Diseases
Cardiac diseases include unstable angina, heart attack, heart failure, arrhythmia (abnormal heart rhythms), valve disease, high blood pressure, congenital heart conditions, and inherited heart conditions. While there are no published papers on the expression and role of Hsp60 in unstable angina, there are a few studies on the role of this protein in heart attacks, heart failure and myocardial infarction (MI).
Figure 2 is showing the expression levels of Hsp60 in cardiac diseases.
Hsp60 has been identified as one of the markers of MI, as demonstrated in both human and animal models. Numerous studies have been published showing the overexpression and release of Hsp60 in the blood stream of animals and patients with signs of heart failure or coronary heart disease. For example, in a study on a population of Chinese individuals, it was shown that Hsp60 levels increase both on the first day and seven days after MI [54]. While in animal studies, intracellular overexpression and release of Hsp60 into the interstitial space of cardiac tissue were observed in C57BL/6 mice with induced MI [55]. The increase in Hsp60 was related to apoptosis.
However, recent studies have identified Hsp60 as a ligand for TLR-2 [56,57] and TLR-4 [58,59], capable of modulating the immune response when released into the extracellular space. The expression of Hsp60 is not merely a reaction to a hypoxic environment but is finely regulated by cardiac-related micro-RNAs. Researchers demonstrated the overexpression of miR-1 after the induction of MI and the simultaneous downregulation of Hsp60 [60]. Carvedilol, a nonselective β-adrenergic receptor (β-AR) antagonist, was shown to reduce miR-1 expression and restore Hsp60 levels in response to tissue damage [61]. The negative regulation of miR-1 on Hsp60 expression was previously demonstrated in a 2012 study on a mouse model of cardiac ischemia-reperfusion injury.
The decreased expression of miR-1 after carvedilol therapy in the MI heart model used in this work suggests a special relationship between carvedilol and miR-1, which may be the mechanism behind the relationship between miR-1 and carvedilol in MI [56]. Huet al. discovered that carvedilol prevents cardiomyocytes from apoptosis by inhibiting the production of miR-1 in cardiomyocytes. This cardioprotective effect was associated with increased Hsp60 expression in cardiomyocytes during ischaemia. Carvedilol-responsive miRNAs are increasingly recognised [56].
In atrial fibrillation (AF), a common arrhythmia associated with congestive heart failure and cardiovascular disease [62,63,64], there is a documented depletion of Hsps. AF-induced proteostasis derailment and consequent electropathological remodeling may be caused by abnormalities in the heat shock response [65,66,67]. Heat shock proteins for the mitochondria, Hsp60 and Hsp10, were discovered to be overexpressed in atrial tissue from atrial fibrillation (AF) patients in two more experiments [68,69]. According to some research, variations in Hsp60 expression are linked to varying levels of atrial myolysis at various phases of AF [70].
Increased blood levels of Hsp60 can cause reactions in several organs and distant cells, such as endothelial cells and other components of blood vessel walls and heart muscle [52]. Hsp60 is a known risk factor due to its strong correlation with the development and progression of atherosclerosis. However, ample data indicate that Hsp60 is also important in the later stages of cardiac illness development [52].
Hsp60 plays a role in the development of atherosclerosis and heart failure. In patients suffering from heart failure, it has been observed that Hsp60 migrates to the surface of the heart muscle before being released into the bloodstream [71]. Elevated levels of Hsp60 were found to be correlated with increased apoptosis and worsening heart failure [72].
This pathological condition may have been accelerated by high concentrations of extracellular Hsp60, which has been shown to induce cardiomyocyte death [31,73]. The work of Tian, Jing et al. provides concrete evidence that extracellular Hsp60 stimulates TLR4 and myeloid differentiation factor 88 (MyD88) in cardiomyocytes, but not TLR2 or transcription factor (Trif). They showed that the expression of TLR2 and TLR4 is induced by extracellular Hsp60, regardless of whether it is endogenous (produced by the ischaemic myocardium) or exogenous. The release of Hsp60 from cardiomyocytes during myocardial ischaemia increases the production of inflammatory cytokines via the TLR4-MyD88-p38/NF-κB pathway and increases the expression of TLR2 and TLR4 via the TLR4-MyD88-JNK/NF-κB pathway [74]. In the same vein, Hsp60 requires MyD88, c-Jun NH2-terminal kinase (JNK) and NF-κB to increase the expression of TLR2 and TLR4. Cytokine generation by Hsp60–TLR4 was dependent on the activation of NF-κB, p38 and MyD88. TLR4 signalling via TIR domain-containing adaptor-inducing interferon-β (TRIF) and MyD88 [75].
Cardiomyocytes are protected from apoptosis by intracellular Hsp60 in the cytosol and mitochondria [76], while translocation of Hsp60 to the plasma membrane has been associated with apoptosis [77]. During apoptosis, Hsp60 connects to a number of proteins involved in proapoptotic processes, such as the anti-apoptotic elements procaspase-3 [78,79,80], surviving [81], cyclophilin D [82], p53 [81] and Bcl-XL, Bcl-2 homologous antagonist/killer (Bak) and Bax [83]. Hsp60 belongs to the cell systems because it is an anti-apoptotic molecule that blocks both Bak and Bax. Once Bax levels bound to the mitochondrial membrane are increased, Hsp60 decreases [84]. Nevertheless, mitochondrial Hsp60 could induce apoptosis in response to stress within cells by directly binding to procaspase-3 and stimulating its conversion to caspase-3 [79]. A decrease in Bcl-2 and an increase in Bax expression have been associated with apoptosis induced by downregulated Hsp60 [56,76].
A study found that under stressful circumstances, adult cardiac myocytes release ubiquitinated Hsp60 inside exosomes [43], however there is no evidence that Hsp60 is then released by exosomes. Another study suggests that Hsp60 seems to remain unchanged in exosomes released under various conditions [85].
The heart's chambers have different protein compositions, particularly regarding the Hsp60 profile. Research indicates that the upper and lower sections of the left and right ventricles differ in protein composition. In one study, the upper left ventricle (LV) showed a significant increase in five proteins, including Hsp60. This finding has significant implications for diseases such as acute MI, as this region is most affected by ischemic injury. However, the base-apical protein profile of the right ventricle did not show such an increase in the levels of Hsp60 [86].
A study investigated the effects of recurrent exposure to high temperatures through quick hot water baths as a mean of reducing total blood pressure, minimizing heart remodeling, and enhancing mechanical performance in patients with hypertension-induced cardiac hypertrophy. Hsps, including Hsp90, Hsp70, and Hsp60, were measured in LV tissue samples due to their sensitivity to elevated temperatures, which lead to increased expression [87]. Certain research findings indicate a correlation between modifications in HSP60 expression and variances in atrial myolysis throughout different phases of atrial fibrillation [88]. Trandopril was shown to have benefits in left ventricular dysfunction following acute myocardial infarction in an animal experiment by preventing the reduction in mitochondrial activity, lowering reactive oxygen species formation, and altering HSP60 synthesis [89].
Cardiovascular diseases (CVDs) result from various stressors that damage the heart's structure and function by affecting cardiac tissues, especially the myocardium. A major factor in the progressive reduction of oxygen and nutrition supply to the myocardium is plaque accumulation in the coronary arteries. Furthermore, localized inflammation plays a significant role in ongoing detrimental processes, including resident cell release of cytokines that trigger inflammatory reactions and attract immune cells to affected areas. In these settings, recurrent tissue damage and the initiation of programmed cell death are common outcomes that lead to organ dysfunction if the initial stressor is not removed [90].
Researchers found that the absence of Hsp60 caused the heart chambers to enlarge, and the left ventricle performed less effectively in another study. Early mortality and the lung-to-body weight ratio both significantly increased concomitantly. According to their research, the loss of Hsp60 in mature cardiomyocytes leads to dilated cardiomyopathy (DCM), which in turn causes heart failure and deadly consequences [91]. In mice, Hsp60 is essential for maintaining normal cardiac function and structure. The absence of Hsp60 in mature cardiac muscle cells causes DCM and heart failure by disrupting the equilibrium of mitochondrial proteins and impairing mitochondrial function [91].
In a study by Knowlton et al., the levels of several Hsps in patients with ischemic cardiomyopathy, DCM, and a control group were examined. According to the study, DCM patients had twice the levels of Hsp27 and Hsp60 compared to the control group [92]. In another study, Niizeki T et al. discovered a relationship between Hsp60 levels and the prognosis and severity of congestive heart failure in 112 patients. Additionally, they observed that elevated Hsp60 levels were associated with an increased risk of progressive heart failure [93]. In this study, they claimed that individuals with chronic heart failure had significantly higher levels of Hsp60 than those in a control group. The study also found that, compared to the control group, patients with chronic heart failure had higher serum levels of Hsp60, with this increase being even more pronounced in patients with a higher New York Heart Association (NYHA) functional class. This increase was particularly notable for patients with severe chronic heart failure assigned to NYHA functional class IV [93].
In a study performed by Al-Zghoul, et al. [94], they measured the amounts of Hsp90, Hsp60, and heat shock factor-1 (HSF-1) mRNA in the tissues of the heart, brain, and muscle on days 12, 14, 16, and 18. Compared to the control group, thermal modification resulted in a significant increase in HSF-1, Hsp60, and Hsp90 expression. While peak expressions in the heart occurred on embryonic days (ED) 12 and 16, the highest levels of Hsp60 were found in muscle tissue on ED 12 and 18. The strongest expressions in the brain were observed on EDs 14 and 16 [94].
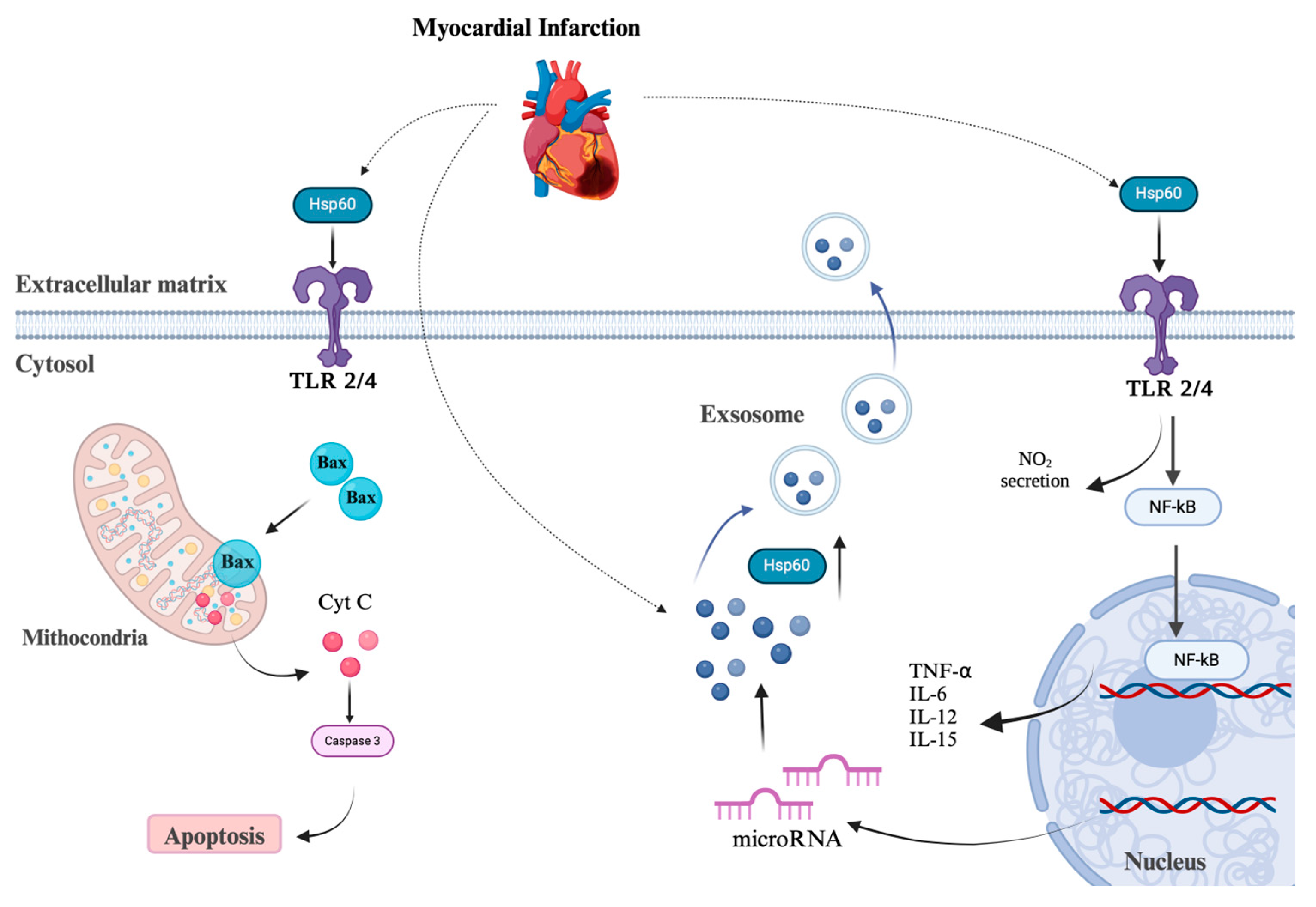
Hsp60 affects both innate and adaptive immune responses and is essential for immune system regulation. It induces apoptosis to promote cell death when its cellular location is disturbed in patients with cardiovascular diseases. On the other hand, exogenous Hsp60 stimulates the immune system, leading to a systemic inflammatory state marked by increased production of TNF-α and other inflammatory mediators, which further accelerates the progression of heart failure [52]. These hypotheses are shown in Figure 3.
7. Conclusions
Heart muscle cells have only one nucleus and a basic structure similar to the skeletal muscle. Both striated muscles have a complex structure and their contraction is strictly dependent on the abundance and efficiency of their mitochondria. Energy, redox balance. Calcium homeostasis and lipid synthesis are important processes that take place inside mitochondria. They are so abundant in the cardiac cells that they appear as lines of mitochondria all around the myofibrils. The abundancy of mitochondria is important to preserve energy requirements and structural integrity of the heart tissue. As an outcome, mitochondria health is necessary to avoid cardiac disorders, while malfunction and dysfunction play an important role in commencing of cardiomyopathy and heart failure.
In the context of cardiac physiology and disease, the role of Hsp60 becomes more crucial. It has a wide variety of functions: from the role in the process of protein folding to setting and establishing immunological responses. The localization of Hsp60 varies inside the cell: known is its distribution inside mitochondria, in the cytoplasm, and inside extracellular vesicles [46].
The expression of this chaperonin seems to be closely related to heart failure, especially in response to cardiac stresses. Some of heart diseases like heart failure, myocardial infarction, and other cardiac disorders show an increase in the levels of Hsp60, suggesting a role of Hsp60 as a biomarker for stress and injury also in the heart.
Recently it has been highlighted a higher expression of Hsp60 in heart disorders, and its interactions with TLRs. This evidence let us suppose that this chaperone may act as a DAMP, activating a cascade of signals that can regulate inflammation or the synthesis and release of inflammatory interleukins such as IL-6. The importance of Hsp60 in heart homeostasis is pointed up in the fact that heart tissue specific micro-RNAs may influence its expression, highly linked to heart failure and cardiac remodeling.
Future studies need to focus on the therapeutic possibilities of influencing mitochondrial function and Hsp60 to alleviate heart disease. The exact pathway through which Hsp60 is released in the blood freely available or inside small vesicles needs to be unraveled. The mechanism through which the extracellular Hsp60 or extracellular vehicles bearing Hsp60, may bind to TLR2 and TLR4 must be elucidated. How Hsp60 may influence the synthesis and release of interleukins and if we can consider it as a DAMP must be studied.
In short, the link between mitochondrial activity, cardiomyocyte morphology and Hsp60 is described as a complex network necessary for the maintenance of cardiac health. This information and knowledge may lead to the development of a new clinical approach to cardiac interventions and the improvement of post-intervention therapies. By expanding our knowledge and understanding of the various functions and processes implied in this article, as well as the fundamental role that disruption in this network plays in heart disease, we may be able to effectively treat heart disorders.
Author Contributions
VS: AC and VDF equally contributed to writing and drawing of the paper.
Funding
This research received no external funding
Abbreviations
| Adenosine triphosphate | ATP |
| Oxidative phosphorylation | OXPHOS |
| Electron transport chain | ETC |
| Mitochondrial antioxidant manganese superoxide dismutase | MnSOD, SOD2 |
| Endoplasmic reticulum | ER |
| Inner mitochondrial membrane | IMM |
| Toll-like receptors | TLRs |
| Damage Associated Molecular Pattern | DAMP |
| Nitric oxide | NO |
| Interleukin | IL |
| Major histocompatibility complex | MHC |
| Antigen-presenting cells | APCs |
| Myocardial infarction | MI |
| Atrial fibrillation | AF |
| Left ventricle | LV |
| Acute myocardial infarction | AMI |
| Cardiovascular diseases | CVDs |
| Dilated cardiomyopathy | DCM |
| Chronic Heart Failure | CHF |
| Embryonic days | ED |
| unfolded mitochondrial protein response | UPRmt |
| ischaemia/reperfusion | I/R |
References
- Tzameli, I. The evolving role of mitochondria in metabolism. Trends in Endocrinology & Metabolism. 2012, 23, 417–419. [Google Scholar]
- Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nature cell biology. 2018, 20, 745–54. [Google Scholar] [CrossRef]
- Bock FJ, Tait SW. Mitochondria as multifaceted regulators of cell death. Nature reviews Molecular cell biology. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- de Souza Breda CN, Davanzo GG, Basso PJ, Câmara NOS, Moraes-Vieira PMM. Mitochondria as central hub of the immune system. Redox biology. 2019;26:101255.
- Papa L, Djedaini M, Hoffman R. Mitochondrial role in stemness and differentiation of hematopoietic stem cells. Stem cells international. 2019;2019(1):4067162.
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN, et al. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circulation research. 2016, 118, 1960–91. [Google Scholar] [CrossRef]
- Wang X, Zhang X, Wu D, Huang Z, Hou T, Jian C, et al. Mitochondrial flashes regulate ATP homeostasis in the heart. Elife. 2017;6:e23908.
- Duan Y, Tang H, Mitchell-Silbaugh K, Fang X, Han Z, Ouyang K. Heat Shock Protein 60 in Cardiovascular Physiology and Diseases. Front Mol Biosci. 2020;7:73.
- Yan J, Bao E, Yu J. Heat shock protein 60 expression in heart, liver and kidney of broilers exposed to high temperature. Research in Veterinary Science. 2009;86(3):533-8.
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324-32.
- Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress and Chaperones. 2009;14(1):105-11.
- Han D, Huang SS, Wang W-F, Deng D-F, Hung SS. Starvation reduces the heat shock protein responses in white sturgeon larvae. Environmental biology of fishes. 2012;93:333-42.
- Michaud MR, Teets NM, Peyton JT, Blobner BM, Denlinger DL. Heat shock response to hypoxia and its attenuation during recovery in the flesh fly, Sarcophaga crassipalpis. Journal of insect physiology. 2011;57(1):203-10.
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92(3):351-66.
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nature structural & molecular biology. 2009;16(6):574-81.
- Feldman DE, Frydman J. Protein folding in vivo: the importance of molecular chaperones. Current opinion in structural biology. 2000;10(1):26-33.
- Braig, K. Chaperonins. Current opinion in structural biology. 1998;8(2):159-65.
- Fink, AL. Chaperone-mediated protein folding. Physiological reviews. 1999;79(2):425-49.
- Agashe VR, Hartl F-U, editors. Roles of molecular chaperones in cytoplasmic protein folding. Seminars in cell & developmental biology; 2000: Elsevier.
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295(5561):1852-8.
- Duan Y, Tang H, Mitchell-Silbaugh K, Fang X, Han Z, Ouyang K. Heat shock protein 60 in cardiovascular physiology and diseases. Frontiers in molecular biosciences. 2020;7:73.
- Cheng MY, Hartl F-U, Martin J, Pollock RA, Kalousek F, Neuper W, et al. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337(6208):620-5.
- Ostermann J, Horwich AL, Neupert W, Hartl F-U. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature. 1989;341(6238):125-30.
- Ishida R, Okamoto T, Motojima F, Kubota H, Takahashi H, Tanabe M, et al. Physicochemical properties of the mammalian molecular chaperone HSP60. International Journal of Molecular Sciences. 2018;19(2):489.
- Gomez-Llorente Y, Jebara F, Patra M, Malik R, Nisemblat S, Chomsky-Hecht O, et al. Structural basis for active single and double ring complexes in human mitochondrial Hsp60-Hsp10 chaperonin. Nature Communications. 2020;11(1):1916.
- Bie AS, Cömert C, Körner R, Corydon TJ, Palmfeldt J, Hipp MS, et al. An inventory of interactors of the human HSP60/HSP10 chaperonin in the mitochondrial matrix space. Cell Stress Chaperones. 2020;25(3):407-16.
- Kao TY, Chiu YC, Fang WC, Cheng CW, Kuo CY, Juan HF, et al. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015;6(2):e1642.
- Meng Q, Li BX, Xiao X. Toward developing chemical modulators of Hsp60 as potential therapeutics. Frontiers in molecular biosciences. 2018;5:35.
- Henderson B, Fares MA, Lund PA. Chaperonin 60: a paradoxical, evolutionarily conserved protein family with multiple moonlighting functions. Biological Reviews. 2013;88(4):955-87.
- Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. American Journal of Physiology-Heart and Circulatory Physiology. 2007;292(6):H3052-H6.
- Lin L, Kim S-C, Wang Y, Gupta S, Davis B, Simon SI, et al. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. American Journal of Physiology-Heart and Circulatory Physiology. 2007;293(4):H2238-H47.
- Pockley AG, Wu R, Lemne C, Kiessling R, de Faire U, Frostegård J. Circulating heat shock protein 60 is associated with early cardiovascular disease. Hypertension. 2000;36(2):303-7.
- Lewthwaite J, Owen N, Coates A, Henderson B, Steptoe A. Circulating human heat shock protein 60 in the plasma of British civil servants: relationship to physiological and psychosocial stress. Circulation. 2002;106(2):196-201.
- Giannessi D, Colotti C, Maltinti M, Del Ry S, Prontera C, Turchi S, et al. Circulating heat shock proteins and inflammatory markers in patients with idiopathic left ventricular dysfunction: their relationships with myocardial and microvascular impairment. Cell stress & chaperones. 2007;12(3):265.
- Blasi C, Kim E, Knowlton AA. Improved metabolic control in diabetes, HSP60, and proinflammatory mediators. Autoimmune Diseases. 2012;2012(1):346501.
- Wang X, Zhang X, Wu D, Huang Z, Hou T, Jian C, et al. Mitochondrial flashes regulate ATP homeostasis in the heart. Elife. 2017;6.
- Nguyen BY, Ruiz-Velasco A, Bui T, Collins L, Wang X, Liu W. Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Br J Pharmacol. 2019;176(22):4302-18.
- Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94(2):329-54.
- Singh, H. Mitochondrial ion channels in cardiac function. Am J Physiol Cell Physiol. 2021;321(5):C812-c25.
- Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells. 2019;8(7).
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, 2nd, Kitsis RN, et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ Res. 2016;118(12):1960-91.
- Giannessi D, Colotti C, Maltinti M, Del Ry S, Prontera C, Turchi S, et al. Circulating heat shock proteins and inflammatory markers in patients with idiopathic left ventricular dysfunction: their relationships with myocardial and microvascular impairment. Cell Stress Chaperones. 2007;12(3):265-74.
- Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol. 2007;292(6):H3052-6.
- Pockley AG, Wu R, Lemne C, Kiessling R, de Faire U, Frostegård J. Circulating heat shock protein 60 is associated with early cardiovascular disease. Hypertension. 2000;36(2):303-7.
- Cheng Y, Sun J, Chen H, Adam A, Tang S, Kemper N, et al. Expression and location of HSP60 and HSP10 in the heart tissue of heat-stressed rats. Exp Ther Med. 2016;12(4):2759-65.
- Di Felice V, Barone R, Trovato E, D'Amico D, Macaluso F, Campanella C, et al. Physiactisome: A New Nanovesicle Drug Containing Heat Shock Protein 60 for Treating Muscle Wasting and Cachexia. Cells. 2022;11(9).
- Osterloh A, Meier-Stiegen F, Veit A, Fleischer B, von Bonin A, Breloer M. Lipopolysaccharide-free heat shock protein 60 activates T cells. J Biol Chem. 2004;279(46):47906-11.
- Pockley AG, Muthana M, Calderwood SK. The dual immunoregulatory roles of stress proteins. Trends Biochem Sci. 2008;33(2):71-9.
- Swaroop S, Sengupta N, Suryawanshi AR, Adlakha YK, Basu A. HSP60 plays a regulatory role in IL-1β-induced microglial inflammation via TLR4-p38 MAPK axis. J Neuroinflammation. 2016;13:27.
- Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72(3):384-93.
- Duan Y, Tang H, Mitchell-silbaugh K, Fang X, Han Z, Ouyang K. Heat Shock Protein 60 in Cardiovascular Physiology and Diseases. Frontiers in Molecular Biosciences. 2020;7.
- Krishnan-Sivadoss I, Mijares-Rojas IA, Villarreal-Leal RA, Torre-Amione G, Knowlton AA, Guerrero-Beltrán CE. Heat shock protein 60 and cardiovascular diseases: An intricate love-hate story. Med Res Rev. 2021;41(1):29-71.
- Rocca C, Soda T, De Francesco EM, Fiorillo M, Moccia F, Viglietto G, et al. Mitochondrial dysfunction at the crossroad of cardiovascular diseases and cancer. Journal of Translational Medicine. 2023;21(1):635.
- Zhang X, He M, Cheng L, Chen Y, Zhou L, Zeng H, et al. Elevated heat shock protein 60 levels are associated with higher risk of coronary heart disease in Chinese. Circulation. 2008;118(25):2687-93.
- Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu Z, et al. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018;9(7):769.
- Hu Y, Chen X, Li X, Li Z, Diao H, Liu L, et al. MicroRNA-1 downregulation induced by carvedilol protects cardiomyocytes against apoptosis by targeting heat shock protein 60. Molecular medicine reports. 2019;19(5):3527-36.
- Cohen-Sfady M, Nussbaum G, Pevsner-Fischer M, Mor F, Carmi P, Zanin-Zhorov A, et al. Heat shock protein 60 activates B cells via the TLR4-MyD88 pathway. The Journal of Immunology. 2005;175(6):3594-602.
- Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. The Journal of clinical investigation. 2006;116(7):2022-32.
- Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. The Journal of Immunology. 2000;164(1):13-7.
- Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. The Journal of Immunology. 2000;164(2):558-61.
- Hu Y, Chen X, Li X, Li Z, Diao H, Liu L, et al. MicroRNA-1 downregulation induced by carvedilol protects cardiomyocytes against apoptosis by targeting heat shock protein 60. Mol Med Rep. 2019;19(5):3527-36.
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139(10):e56-e528.
- Farmakis D, Parissis J, Filippatos G. Acute heart failure: epidemiology, classification, and pathophysiology. The ESC textbook of intensive and acute cardiovascular care, 2nd ed Oxford. 2015:459-69.
- Komajda M, Anker SD, Cowie MR, Filippatos GS, Mengelle B, Ponikowski P, et al. Physicians' adherence to guideline-recommended medications in heart failure with reduced ejection fraction: data from the QUALIFY global survey. European journal of heart failure. 2016;18(5):514-22.
- Santhanakrishnan R, Wang N, Larson MG, Magnani JW, McManus DD, Lubitz SA, et al. Atrial fibrillation begets heart failure and vice versa: temporal associations and differences in preserved versus reduced ejection fraction. Circulation. 2016;133(5):484-92.
- Henning RH, Brundel BJ. Proteostasis in cardiac health and disease. Nature Reviews Cardiology. 2017;14(11):637-53.
- Brundel BJ, Shiroshita-Takeshita A, Qi X, Yeh Y-H, Chartier D, Van Gelder IC, et al. Induction of heat shock response protects the heart against atrial fibrillation. Circulation research. 2006;99(12):1394-402.
- Wiersma M, Meijering RA, Qi XY, Zhang D, Liu T, Hoogstra-Berends F, et al. Endoplasmic reticulum stress is associated with autophagy and cardiomyocyte remodeling in experimental and human atrial fibrillation. Journal of the American Heart Association. 2017;6(10):e006458.
- Kirmanoglou K, Hannekum A, Schäfler AE. Expression of mortalin in patients with chronic atrial fibrillation. Basic research in cardiology. 2004;99:404-8.
- Yang M, Tan H, Cheng L, He M, Wei Q, Tanguay RM, et al. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell stress & chaperones. 2007;12(2):142.
- Kim SC, Stice JP, Chen L, Jung JS, Gupta S, Wang Y, et al. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ Res. 2009;105(12):1186-95.
- Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, et al. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293(4):H2238-47.
- Kim S-C, Stice JP, Chen L, Jung JS, Gupta S, Wang Y, et al. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circulation research. 2009;105(12):1186-95.
- Tian J, Guo X, Liu X-M, Liu L, Weng Q-F, Dong S-J, et al. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovascular Research. 2013;98(3):391-401.
- Kawai T, Akira S, editors. TLR signaling. Seminars in immunology; 2007: Elsevier.
- Kirchhoff S, Gupta S, Knowlton A. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation. 2002;105(24):2899-904.
- Gupta S, Knowlton A. Cytosolic heat shock protein 60, hypoxia, and apoptosis. Circulation. 2002;106(21):2727-33.
- Xanthoudakis S, Roy S, Rasper D, Hennessey T, Aubin Y, Cassady R, et al. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. The EMBO journal. 1999.
- Chandra D, Choy G, Tang DG. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. Journal of Biological Chemistry. 2007;282(43):31289-301.
- Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of Jurkat cells. The EMBO journal. 1999.
- Ghosh JC, Dohi T, Kang BH, Altieri DC. Hsp60 regulation of tumor cell apoptosis. Journal of Biological Chemistry. 2008;283(8):5188-94.
- Ghosh JC, Siegelin MD, Dohi T, Altieri DC. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer research. 2010;70(22):8988-93.
- Gupta S, Knowlton A. HSP60, Bax, apoptosis and the heart. Journal of cellular and molecular medicine. 2005;9(1):51-8.
- Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proceedings of the National Academy of Sciences. 1998;95(9):4997-5002.
- Cappello F, Logozzi M, Campanella C, Bavisotto CC, Marcilla A, Properzi F, et al. Exosome levels in human body fluids: A tumor marker by themselves? Eur J Pharm Sci. 2017;96:93-8.
- Eckhardt A, Kulhava L, Miksik I, Pataridis S, Hlavackova M, Vasinova J, et al. Proteomic analysis of cardiac ventricles: baso-apical differences. Mol Cell Biochem. 2018;445(1-2):211-9.
- Oyama J, Maeda T, Sasaki M, Higuchi Y, Node K, Makino N. Repetitive hyperthermia attenuates progression of left ventricular hypertrophy and increases telomerase activity in hypertensive rats. Am J Physiol Heart Circ Physiol. 2012;302(10):H2092-101.
- Yang M, Tan H, Cheng L, He M, Wei Q, Tanguay RM, et al. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones. 2007;12(2):142-50.
- Toga W, Tanonaka K, Takeo S. Changes in Hsp60 level of the failing heart following acute myocardial infarction and the effect of long-term treatment with trandolapril. Biol Pharm Bull. 2007;30(1):105-10.
- Williams JW, Huang LH, Randolph GJ. Cytokine Circuits in Cardiovascular Disease. Immunity. 2019;50(4):941-54.
- Fan F, Duan Y, Yang F, Trexler C, Wang H, Huang L, et al. Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure. Cell Death Differ. 2020, 27, 587–600. [Google Scholar] [CrossRef]
- Knowlton AA, Kapadia S, Torre-Amione G, Durand JB, Bies R, Young J, et al. Differential expression of heat shock proteins in normal and failing human hearts. J Mol Cell Cardiol. 1998;30(4):811-8.
- Niizeki T, Takeishi Y, Watanabe T, Nitobe J, Miyashita T, Miyamoto T, et al. Relation of serum heat shock protein 60 level to severity and prognosis in chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2008;102(5):606-10.
- Al-Zghoul MB, Ismail ZB, Dalab AE, Al-Ramadan A, Althnaian TA, Al-Ramadan SY, et al. Hsp90, Hsp60 and HSF-1 genes expression in muscle, heart and brain of thermally manipulated broiler chicken. Res Vet Sci. 2015;99:105-11.
Figure 1.
Localization and release of heat-shock protein 60 (Hsp60).
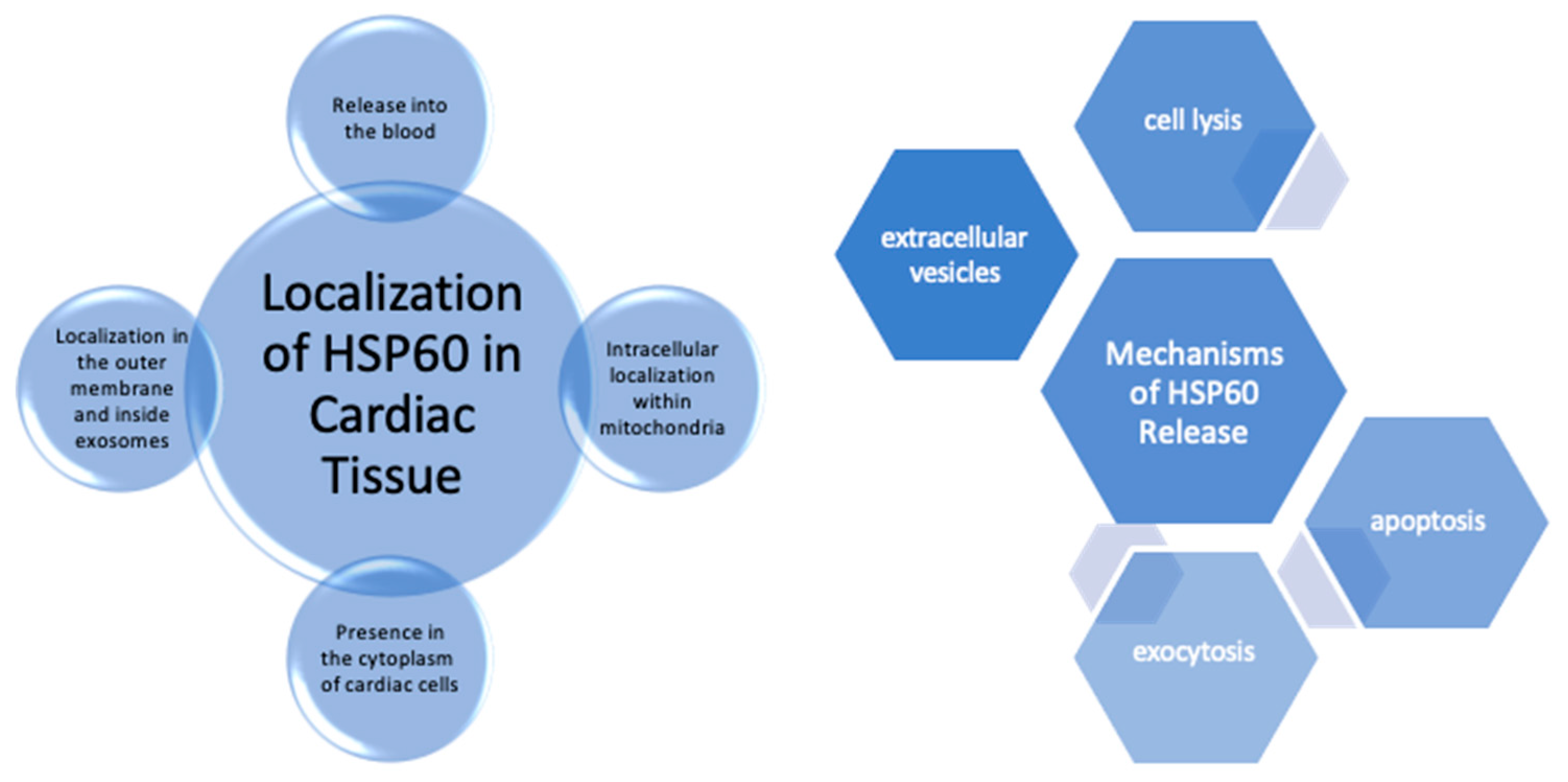
Figure 2.
Expression of HSP60 in Cardiac Disease.
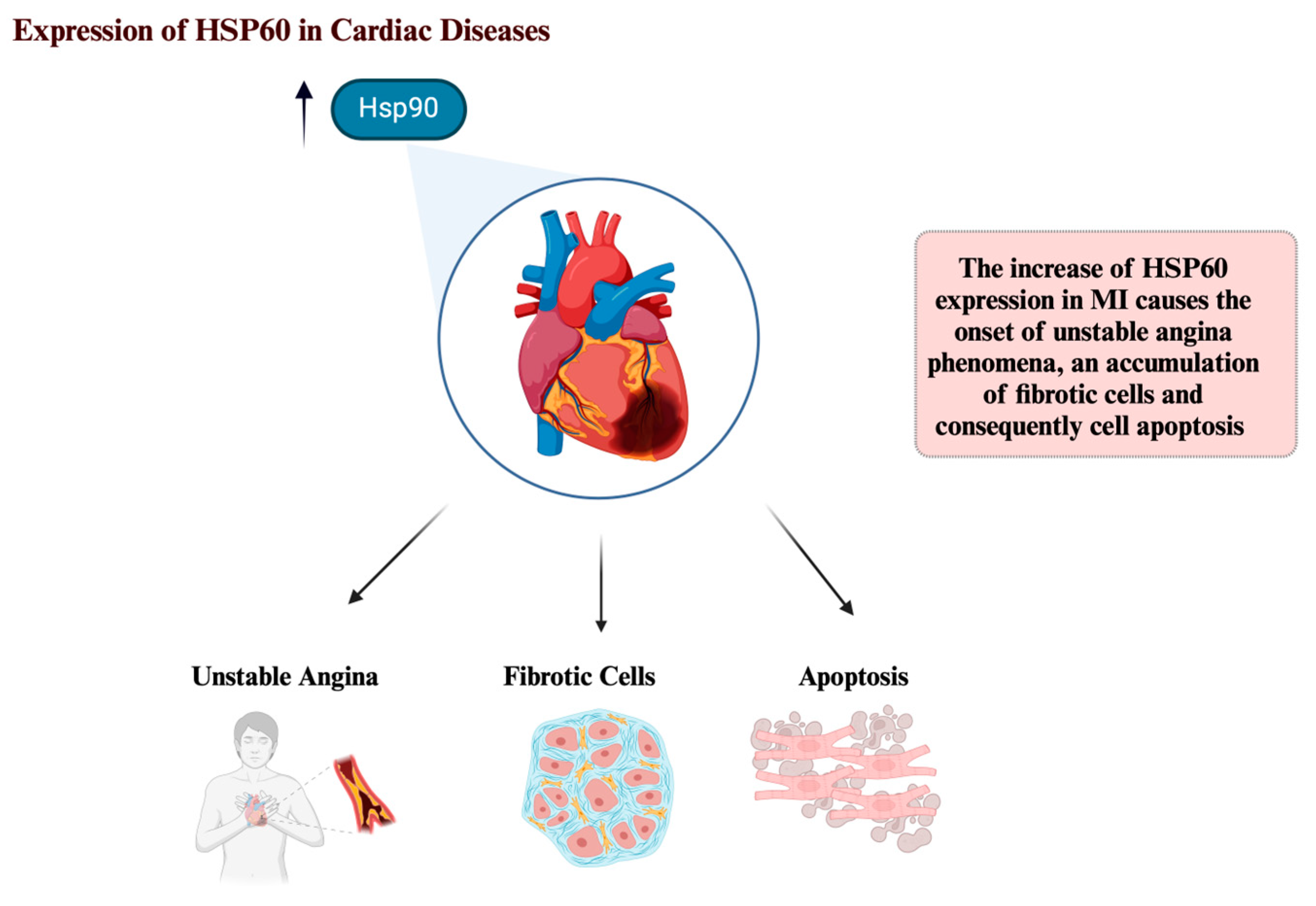
Figure 3.
This diagram is showing how heat-shock protein 60 (Hsp60) may play an important role in cardiovascular disease (CD) such as myocardial infarction (MI).
Figure 3.
This diagram is showing how heat-shock protein 60 (Hsp60) may play an important role in cardiovascular disease (CD) such as myocardial infarction (MI).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



