Submitted:
23 July 2024
Posted:
24 July 2024
You are already at the latest version
Abstract
Epidermodysplasia verruciformis (EV) is a rare genodermatosis caused by β-human papillomaviruses (HPV) in immunodeficient patients. EV is characterized by flat warts and pityriasis-like lesions and might be isolated or syndromic, associated with some other infectious manifestations. We report here three patients from 2 independent families with syndromic EV for 2 of them. By whole exome sequencing, we found that patients carry new homozygous variants in STK4, both leading to a premature stop codon. STK4 deficiency causes a combined immunodeficiency characterized by a broad infectious susceptibility to bacteria, viruses and fungi. Auto-immune manifestations were also reported. Deep immunophenotyping revealed multiple cytopenia in the three affected patients, in particular deep CD4+ T cells deficiency. We report here the fourth and the fifth cases of syndromic EV due to STK4 deficiency.
Keywords:
Epidermodysplasia verruciformis
; human papillomavirus
; STK4 deficiency
; CD4+ T cells
1. Introduction
Epidermodysplasia verruciformis (EV) is a genodermatosis characterized by ppityriasis versicolor and flat wart-like lesions. EV is a rare disease, with around 500 cases reported so far, and is the consequence of β-papillomavirus (β-HPV) infection [1, 2]. Patients with EV, after decades, have a high risk of developing actinic keratosis and non-melanoma skin cancer (NMSC), particularly cutaneous squamous cell carcinoma (cSCC), cSCC in situ (Bowen’s disease) and, to a lesser extent basal cell carcinoma (BCC). NMSC typically occurs in lesions exposed to the sun [3]. EV can be isolated or syndromic, including additional clinical features, mainly infectious [3]. Since the forties, it has been hypothesized that EV might also be a genetic disease [4]. In 2002, EVER1 and EVER2 have been identified as the two first genes underlying isolated EV [5]. More recently, CIB1 deficiency was reported in patients with isolated EV and as a new partner of EVER1 and EVER2 [6]. EVER1, EVER2 and CIB1 deficiencies are believed to result in a keratinocyte intrinsic deficiency, underlying exquisite susceptibility to β-HPVs [6]. In contrast, syndromic EV have been associated with T cell deficiencies, sharing CD4+ T cell lymphopenia [7,8]. These etiologies of atypical EV include loss-of-function (LOF) mutations of RHOH [9], STK4 [10,11,12], CORO1A [13], FLT3LG [14], TRAC [15], DCLRE1C (encoding the Artemis protein) [16], DOCK8 [17,18], RASGRP1 [19], LCK [20], TPP2 [21] and ITK [22]. Here we report 2 novel cases of STK4 deficiency associated with atypical EV.
2. Clinical Reports
2.1. Clinical Phenotypes and Genotypes
2.1.1. Family 1
From birth to 10 years old, the proband (P1) clinical history was unremarkable. He received all childhood vaccines (BCG, MMR, diphteria, tetanus, poliovirus, and HBV) without any complications. However, at the age of 10, he presented recurrent apyretic diarrhea episodes, with no pathogen identified. At the age of 12 years old, P1 growth curve presented a negative deviation from the norm. He had no cardiac clinical symptoms and no electrocardiogram or echocardiographic abnormalities.
P1 was also admitted to pediatrics consultation for generalized flat warts and pityriasis versicolor-like skin lesions, that progressed from the age of 6 years old (Figure 1A). The lesions were initially localized on the face and then generalized to the neck, trunk, arms and the back.
The histology analysis showed hyperkeratosis and parakeratosis, mild acanthosis, and the presence of koïlocytes, keratinocytes with pale-stained cytoplasm in the upper epidermis associated with high levels of intranuclear viral replication (Figure 1B). All together these observations suggested a diagnosis of epidermodysplasia verruciformis. The genotyping of HPV by PCR confirmed the diagnosis, with the identification of HPV5 in a punch biopsy of a lesion from his right arm.
His 16 years old sister (P2) did not develop EV, or any HPV-related skin disease but she presented recurrent low respiratory tract infections since the age of 3 years old with no pathogen identified. She also received all childhood vaccines (BCG, MMR, diphteria, tetanus, poliovirus, and HBV) without any complication and her growth curve presented a deviation from the norm too.
No secondary immunodeficiency was noted in both sibling and no auto-immune manifestation was reported so far. In term of treatment, both siblings have been treated with intravenous immunoglobulins substitution, and antibiotic prophylaxis against opportunistic infections (Trimethoprim/Sulfamethoxazole). In addition, P1 had a treatment with imiquimod application on the skin and skin protection from UV radiation and the skin lesions improved.
2.1.2. Family 2
An 8 years old girl (P3) from consanguineous marriage parents, presented in pediatric consultation for profuse pityriasis versicolor-like skin lesions the upper and lower limbs, in the trunk and at the face (Figure 2A). These lesions have been evolving since the age of 3 years old. Skin histology of a lesion was typical of EV lesions (Figure 2B).
By PCR on a lesion from the right forearm of the patient, we identified HPV8 confirming the EV diagnosis. She had also recurrent respiratory infections and diarrhea, with stature weight repercussion, from the age of 3 years old. She didn’t have secondary immunodeficiency (HIV, diabetes, immune suppressor treatment) or any familial history of primary immunodeficiency or similar lesions. She received all childhood vaccines (BCG, MMR, diphteria, tetanus, poliovirus, and HBV) without any complications. She had no cardiac clinical symptoms and no electrocardiogram or echocardiographic abnormalities. No auto-immune symptom was reported so far.
In term of treatment, she has been treated with intravenous immunoglobulins substitution, and antibiotic prophylaxis against opportunistic infections (Trimethoprim/Sulfamethoxazole). In addition, P3 had a treatment with imiquimod application on the skin and skin protection from UV radiation and the clinical conditions improved.
2.1.3. Genotype
The consanguinity of parents and the clinical history of the 3 patients suggested an inborn error of immunity. To test this hypothesis, we performed genetic investigations in P1 and P3 by combining a deep sequencing array (using a custom-designed Illumina SNP array) with the 407 genes involved in inborn errors of immunity [23] and also whole exome sequencing. In both P1 and P3, we identified 2 predicted LOF homozygous variants in STK4, an essential splice site (c.1305+1G>A) in P1 and a premature stop codon (c.750G>A, p.W250*) in P3. We did not find any other candidate variants in other genes related syndromic EV. The familial segregation confirmed that P1, P2 and P3 were homozygous for their respective allele and their parents and healthy sibling were heterozygous (Figure 3A,B), suggesting that STK4 was the disease-causing gene in these 2 families.
2.2. Immunological Phenotype
We then performed immunological analyses in both clinical and research laboratories. In clinical laboratory, P1 and P3 had a profound lymphopenia, with a decreased T, B and NK cell counts as compared with normal range. In addition, P3 had also neutropenia. In contrast, P2, P1’s sister, had a normal count on T, B and NK, with the exception of a mild CD4+ T cells lymphopenia. The HLA-DR expression was found normal on CD19+ and CD14+ cells in both siblings. In contrast, the immunoglobulin levels were normal in P1, and P2, with a slight hypogammaglobulinemia observed P2. As for P3, there was a slight hypergammglobulinemia and Hyper Ig E (Table 1 and Table 2).
A deeper immunophenotyping has been done using CyTOF method (Figure 4). The proportion of NK, CD3+, Treg and gamma-delta T cells in P1, P2 and P3, are in the normal range of healthy donors (Figure 4A). However, we observed a strong reduction of CD4+ T cells, proportion in T lymphocytes, an absence of MAIT cells, and a higher proportion of CD8+ T cells, leading to an inverted CD4/CD8 ratio. In addition, the proportion of recent thymic emigrant cell, as well as naïve CD4 and CD8 T cells, are strongly decreased. In contrast, memory CD4 and CD8 subsets are in the normal range, or even increased in term of proportion (Figure 4B). The B cell compartment is also showing some major differences, such as a decrease of memory B cells and an increase of ABC subset (Figure 4C). Altogether, these immunological results are similar to the ones previously reported in patients with STK4 deficiency.
3. Discussion
Human autosomal recessive (AR) STK4 deficiency was first reported in 2012 in 7 patients with progressive T cell deficiency, and a broad range of infectious susceptibilities [24,25]. Since then, 29 additional cases of STK4 deficiency have been reported. All variants identified are loss-of-function [10,11,12,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. The clinical phenotype related to STK4 deficiency is characterized by recurrent pulmonary bacterial infections, recurrent skin infections, including HPV warts (9 out 36) [10,12,25,26,38] including 3 with syndromic EV [10,11,12] and mucocutaneous candidiasis, but also chronic EBV infections. Auto-immune manifestations were reported in 13 cases [11,12,24,30,31,32,37,38]. The immunological phenotype of patients with STK4 deficiency is characterized by a profound CD4 lymphopenia due to a decreased proliferation, increased susceptibility to apoptosis and dysregulation of the transcription factor Forkhead box protein O1 (FOXO1) and its downstream targets in T cells. Leukocytes also show defective adhesion and chemotaxis [10,22,25,38,39].
We report here three children from two independent families with rare variants in STK4 deficiency and a broad clinical phenotype, including syndromic EV for 2 of them. Respiratory infections were noted in P2 and P3, as reported in many cases (ref of Cagdas here). P1 had recurrent diarrhea with no pathogens identified. Gastroenteritis was already reported in patients with STK4 deficiency [38]. A negative deviation from the norm of growth curve was noted in the 3 patients reported here, as in previously reported before [34].
In term of immunologic phenotype, cytopenia is a common feature in STK4 deficiency, and we indeed observed it in the 3 patients reported here [38]. Furthermore, neutropenia is secondary to infections, autoimmunity, immunomodulatory agents, and chemotherapy [30,40]. In P3, neutropenia was developed during the infectious episodes. Allergic manifestations are also reported in STK4 deficiency (Asthma, atopic dermatitis) [38]. Despite P3 having high hyper IgE, none of the patients reported here developed any allergic symptoms for now.
Autoimmune diseases were also associated with STK4 deficiency. The presence of autoantibodies, such as antinuclear and anticardiolipin antibodies, has been described in STK4 deficiency [11,12,24,30,31,33,37,38]. The autoimmunity in STK4 deficiency may also be due to the defective regulation of development and function of regulatory T cells through modulation of FOXO1/FOXO3. In our patients, no autoimmune manifestation was reported so far.
Some patients with STK4 deficiency are particularly prone to EBV-driven infections and EBV-induced lymphoproliferation [38], this was absent in our patients.
To conclude, we describe here 3 new clinical phenotypes related to STK4 deficiency. We treated our patients with IVIG, antibacterial prophylactic agents associated with imiquimod application on the skin and skin protection from UV radiation for EV patients. Although HSCT is the curative treatment in most CIDs, the survival rate after HSCT is about 50% in STK4 deficiency [36] and further studies are needed to recommend HSCT as a safe therapy for patients with STK4 deficiency.
Author Contributions
Conceptualization, A.E.K.; Investigation, A.E.K., H.O, F.M., J.E.B., L.L, T.L.V. and R.C.; Resources, A.E.K., H.O, and A.A.B.; Funding acquisition, E.J. and J.-L.C.; writing—original draft preparation, A.E.K.; writing—review and editing, H,O., F.M, J.E.B., T.L.V., R.C., H.K, V.B, E.J., J.-L.C. and A.A.B.; supervision, J.-L.C. and A.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was founded by Laboratory of Clinical Immunology—Inflammation and Allergy (LICIA), Faculty of Medicine and Pharmacy, Hassan II University, Casablanca, Morocco and The Laboratory of Human Genetics of Infectious Diseases is supported by the Howard Hughes Medical Institute, the Rockefeller University, the St. Giles Foundation, the National Institutes of Health (NIH) (R01AI143810), the National Center for Advancing Translational Sciences (NCATS), NIH Clinical and Translational Science Award (CTSA) program (UL1TR001866), the program “Investissement d’Avenir” launched by the French Government and implemented by the Agence Nationale de la Recherche (ANR) (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the Square Foundation, Institut National de la Santé et de la Recherche Médicale (INSERM), and the University of Paris Cité. T.L.V. is funding by the MD-PhD program of Imagine Institute.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Ibn Rochd university hospital of Casablanca (protocol code 9/22 and date of approval: 22 April 2022).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Upon request.
Acknowledgments
The authors wish to thank the patients and their families for their approval and support of the study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Orth, G. Host defenses against human papillomaviruses: lessons from epidermodysplasia verruciformis. Curr Top Microbiol Immunol. 2008, 321, 59–83. [Google Scholar] [PubMed]
- Przybyszewska, J.; Zlotogorski, A.; Ramot, Y. Re-evaluation of epidermodysplasia verruciformis: Reconciling more than 90 years of debate. J. Am. Acad. Dermatol. 2017, 76, 1161–1175. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.J.; Imahorn, E.; Itin, P.; Uitto, J.; Orth, G.; Jouanguy, E.; Casanova, J.-L.; Burger, B. Epidermodysplasia Verruciformis: Inborn Errors of Immunity to Human Beta-Papillomaviruses. Front. Microbiol. 2018, 9, 1222. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Bahru, J.; Loo, D.S.C.; C, H.T.; Chin, K.N.; Tan, K.K. Familial Epidermodysplasia Verruciformis of Lewandowsky and Lutz. Arch. Dermatol. 1972, 105, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ramoz, N.; Rueda, L.-A.; Bouadjar, B.; Montoya, L.-S.; Orth, G.; Favre, M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 2002, 32, 579–581. [Google Scholar] [CrossRef] [PubMed]
- de Jong SJ, Créquer A, Matos I, Hum D, Gunasekharan V, Lorenzo L, et al. The human CIB1–EVER1–EVER2 complex governs keratinocyte-intrinsic immunity to β-papillomaviruses. Journal of Experimental Medicine. 1 août 2018;215(9):2289-310.
- Béziat, V.; Casanova, J.-L.; Jouanguy, E. Human genetic and immunological dissection of papillomavirus-driven diseases: new insights into their pathogenesis. Curr. Opin. Virol. 2021, 51, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lazarczyk, M.; Dalard, C.; Hayder, M.; Dupre, L.; Pignolet, B.; Majewski, S.; Vuillier, F.; Favre, M.; Liblau, R.S. EVER Proteins, Key Elements of the Natural Anti-Human Papillomavirus Barrier, Are Regulated upon T-Cell Activation. PLOS ONE 2012, 7, e39995. [Google Scholar] [CrossRef] [PubMed]
- Crequer, A.; Troeger, A.; Patin, E.; Ma, C.S.; Picard, C.; Pedergnana, V.; Fieschi, C.; Lim, A.; Abhyankar, A.; Gineau, L.; et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J. Clin. Investig. 2012, 122, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Crequer, A.; Picard, C.; Patin, E.; D’amico, A.; Abhyankar, A.; Munzer, M.; Debré, M.; Zhang, S.-Y.; de Saint-Basile, G.; Fischer, A.; et al. Inherited MST1 Deficiency Underlies Susceptibility to EV-HPV Infections. PLOS ONE 2012, 7, e44010. [Google Scholar] [CrossRef]
- Sharafian, S.; Ziaee, V.; Shahrooei, M.; Ahadi, M.; Parvaneh, N. A Novel STK4 Mutation Presenting with Juvenile Idiopathic Arthritis and Epidermodysplasia Verruciformis. J. Clin. Immunol. 2019, 39, 11–14. [Google Scholar] [CrossRef]
- Gutierrez-Marin, P.A.; Castano-Jaramillo, L.M.; Velez-Tirado, N.; Villamil-Osorio, M.; Patiño, E.; Reina, M.F.; Hernandez, M.T. STK4 deficiency and epidermodysplasia verruciformis-like lesions: A case report. Pediatr. Dermatol. 2023, 41, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Stray-Pedersen, A.; Jouanguy, E.; Crequer, A.; Bertuch, A.A.; Brown, B.S.; Jhangiani, S.N.; Muzny, D.M.; Gambin, T.; Sorte, H.; Sasa, G.; et al. Compound Heterozygous CORO1A Mutations in Siblings with a Mucocutaneous-Immunodeficiency Syndrome of Epidermodysplasia Verruciformis-HPV, Molluscum Contagiosum and Granulomatous Tuberculoid Leprosy. J. Clin. Immunol. 2014, 34, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Momenilandi M, Lévy R, Sobrino S, et al. FLT3L governs the development of partially overlapping hematopoietic lineages in humans and mice. Cell. 2024;187(11):2817-2837.
- Rawat, A.; Singh, A.; Dobbs, K.; Pala, F.; Delmonte, O.M.; Vignesh, P.; Jindal, A.K.; Gupta, A.; Suri, D.; Kaur, A.; et al. Skewed TCR Alpha, but not Beta, Gene Rearrangements and Lymphoma Associated with a Pathogenic TRAC Variant. J. Clin. Immunol. 2021, 41, 1395–1399. [Google Scholar] [CrossRef]
- Tahiat, A.; Badran, Y.R.; Chou, J.; Cangemi, B.; Lefranc, G.; Labgaa, Z.-M.; Oussalam, S.; Kaddouri-Slimani, A.; Belarbi, A.; Bendissari-Bouzid, K.; et al. Epidermodysplasia verruciformis as a manifestation of ARTEMIS deficiency in a young adult. J. Allergy Clin. Immunol. 2016, 139, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Sanal, O.; Jing, H.; Ozgur, T.; Ayvaz, D.; Strauss-Albee, D.M.; Ersoy-Evans, S.; Tezcan, I.; Turkkani, G.; Matthews, H.F.; Haliloglu, G.; et al. Additional Diverse Findings Expand the Clinical Presentation of DOCK8 Deficiency. J. Clin. Immunol. 2012, 32, 698–708. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, G.; Mo, X.; Wang, B.; Wu, F.; Chen, J.; Luo, H.; Zhu, L.; Xu, M.; Zhou, Q.; et al. A novel homozygous DOCK8 mutation associated with unusual coexistence of gross molluscum contagiosum and epidermodysplasia verruciformis in a DOCK8 deficiency patient. J. Eur. Acad. Dermatol. Venereol. 2017, 31, e504–e505. [Google Scholar] [CrossRef]
- Platt, C.D.; Fried, A.J.; Hoyos-Bachiloglu, R.; Usmani, G.N.; Schmidt, B.; Whangbo, J.; Chiarle, R.; Chou, J.; Geha, R.S. Combined immunodeficiency with EBV positive B cell lymphoma and epidermodysplasia verruciformis due to a novel homozygous mutation in RASGRPClin. Immunol. 2017, 183, 142–144. [Google Scholar] [CrossRef]
- Li SL, Duo LN, Wang HJ, Dai W, Zhou EYH, Xu YN, et al. Identification of LCK mutation in a family with atypical epidermodysplasia verruciformis with T-cell defects and virus-induced squamous cell carcinoma. Br J Dermatol. déc 2016;175(6):1204-9.
- Stepensky, P.; Rensing-Ehl, A.; Gather, R.; Revel-Vilk, S.; Fischer, U.; Nabhani, S.; Beier, F.; Brümmendorf, T.H.; Fuchs, S.; Zenke, S.; et al. Early-onset Evans syndrome, immunodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiency. Blood 2015, 125, 753–761. [Google Scholar] [CrossRef]
- Youssefian, L.; Vahidnezhad, H.; Yousefi, M.; Saeidian, A.H.; Azizpour, A.; Touati, A.; Nikbakht, N.; Hesari, K.K.; Adib-Sereshki, M.M.; Zeinali, S.; et al. Inherited Interleukin 2–Inducible T-Cell (ITK) Kinase Deficiency in Siblings With Epidermodysplasia Verruciformis and Hodgkin Lymphoma. Clin. Infect. Dis. 2019, 68, 1938–1941. [Google Scholar] [CrossRef]
- Invitae Primary Immunodeficiency Panel | Test catalog | Invitae [Internet]. [cité 3 juill 2023]. Disponible sur: https://www.invitae.com/en/providers/test-catalog/test-08100.
- Nehme, N.T.; Schmid, J.P.; Debeurme, F.; André-Schmutz, I.; Lim, A.; Nitschke, P.; Rieux-Laucat, F.; Lutz, P.; Picard, C.; Mahlaoui, N.; et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 2012, 119, 3458–3468. [Google Scholar] [CrossRef]
- Abdollahpour, H.; Appaswamy, G.; Kotlarz, D.; Diestelhorst, J.; Beier, R.; Schäffer, A.A.; Gertz, E.M.; Schambach, A.; Kreipe, H.H.; Pfeifer, D.; et al. The phenotype of human STK4 deficiency. Blood 2012, 119, 3450–3457. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, S.E.; Al-Mousawi, A.; Assing, K.; Hartling, U.; Grosen, D.; Fisker, N.; Nielsen, C.; Jakobsen, M.A.; Mogensen, T.H. STK4 Deficiency Impairs Innate Immunity and Interferon Production Through Negative Regulation of TBK1-IRF3 Signaling. J. Clin. Immunol. 2021, 41, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Radwan, N.; El-Owaidy, R.; El-Sayed, Z.A.; Abdel-Baky, A.; El-Haddad, A.; Rashad, H.; Khorshed, E.N.; Platt, C.D.; Wallace, J.G.; Chou, J.; et al. A Case of STK4 Deficiency with Complications Evoking Mycobacterial Infection. J. Clin. Immunol. 2020, 40, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, F.; Klein, C.; Poorpooneh, M.; Sherkat, R.; Khoshnevisan, R. A case report of sinusoidal diffuse large B-cell lymphoma in a STK4 deficient patient. Medicine 2020, 99, e18601. [Google Scholar] [CrossRef] [PubMed]
- Abolnezhadian F, Iranparast S, Ahmadpour F. Identical Twins with a Mutation in the STK4 Gene Showing Clinical Manifestations of the Mutation at Different Ages: A Case Report. Iran J Immunol. 2020;17(4):333-340.
- Halacli, S.O.; Ayvaz, D.C.; Sun-Tan, C.; Erman, B.; Uz, E.; Yilmaz, D.Y.; Ozgul, K.; Tezcan, I.; Sanal, O. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: A novel mutation. Clin. Immunol. 2015, 161, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Schipp, C.; Schlütermann, D.; Hönscheid, A.; Nabhani, S.; Höll, J.; Oommen, P.T.; Ginzel, S.; Fleckenstein, B.; Stork, B.; Borkhardt, A.; et al. EBV Negative Lymphoma and Autoimmune Lymphoproliferative Syndrome Like Phenotype Extend the Clinical Spectrum of Primary Immunodeficiency Caused by STK4 Deficiency. Front. Immunol. 2018, 9, 2400. [Google Scholar] [CrossRef] [PubMed]
- Sherkat R, Sabri MR, Dehghan B, Bigdelian H, Reisi N, Afsharmoghadam N, et al. EBV lymphoproliferative-associated disease and primary cardiac T-cell lymphoma in a STK4 deficient patient: A case report. Medicine (Baltimore). déc 2017;96(48):e8852.
- Al-Saud, B.; Alajlan, H.; Sabar, H.; Anwar, S.; Alruwaili, H.; Al-Hussain, T.; Alamri, N.; Alazami, A.M. STK4 Deficiency in a Patient with Immune Complex Glomerulonephritis, Salt-Losing Tubulopathy, and Castleman’s-Like Disease. J. Clin. Immunol. 2019, 39, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Guennoun, A.; Bougarn, S.; Khan, T.; Mackeh, R.; Rahman, M.; Al-Ali, F.; Ata, M.; Aamer, W.; Prosser, D.; Habib, T.; et al. A Novel STK4 Mutation Impairs T Cell Immunity Through Dysregulation of Cytokine-Induced Adhesion and Chemotaxis Genes. J. Clin. Immunol. 2021, 41, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Saglam, A.; Cagdas, D.; Aydin, B.; Keles, S.; Reisli, I.; Arslankoz, S.; Katipoglu, K.; Uner, A. STK4 deficiency and EBV-associated lymphoproliferative disorders, emphasis on histomorphology, and review of literature. Virchows Arch. 2022, 480, 393–401. [Google Scholar] [CrossRef]
- Vignesh, P.; Rawat, A.; Kumrah, R.; Singh, A.; Gummadi, A.; Sharma, M.; Kaur, A.; Nameirakpam, J.; Jindal, A.; Suri, D.; et al. Clinical, Immunological, and Molecular Features of Severe Combined Immune Deficiency: A Multi-Institutional Experience From India. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Uygun, V.; Keleş, S.; Daloğlu, H.; Öztürkmen, S.; Yalçın, K.; Karasu, G.; Yeşilipek, A. Hematopoietic stem cell transplantation in serine/threonine kinase 4 (STK4) deficiency: Report of two cases and literature review. Pediatr. Transplant. 2022, 27, e14439. [Google Scholar] [CrossRef] [PubMed]
- Cagdas, D.; Halacli, S.O.; Tan, C.; Esenboga, S.; Karaatmaca, B.; Cetinkaya, P.G.; Balcı-Hayta, B.; Ayhan, A.; Uner, A.; Orhan, D.; et al. Diversity in Serine/Threonine Protein Kinase-4 Deficiency and Review of the Literature. J. Allergy Clin. Immunol. Pr. 2021, 9, 3752–3766. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.S.; Willet, J.D.; Griffin, H.R.; Morgan, N.V.; O’boyle, G.; Arkwright, P.D.; Hughes, S.M.; Abinun, M.; Tee, L.J.; Barge, D.; et al. Defective Leukocyte Adhesion and Chemotaxis Contributes to Combined Immunodeficiency in Humans with Autosomal Recessive MST1 Deficiency. J. Clin. Immunol. 2016, 36, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Dotta, L.; Badolato, R. Primary immunodeficiencies appearing as combined lymphopenia, neutropenia, and monocytopenia. Immunol. Lett. 2014, 161, 222–225. [Google Scholar] [CrossRef]
Figure 1.
A. Pictures of the lesions on the face and the back of P1. B. Histological study of Punch skin biopsy showed hyperkeratosis and parakeratosis, mild acanthosis, and the presence of koilocytes, keratinocytes with pale-stained cytoplasm in the upper epidermis associated with high levels of intranuclear viral replication. The cytoplasm of the affected cells stains pale blue which is pathognomonic of epidermodysplasia verruciformis lesions without any sign of malignancy (Arrows).
Figure 1.
A. Pictures of the lesions on the face and the back of P1. B. Histological study of Punch skin biopsy showed hyperkeratosis and parakeratosis, mild acanthosis, and the presence of koilocytes, keratinocytes with pale-stained cytoplasm in the upper epidermis associated with high levels of intranuclear viral replication. The cytoplasm of the affected cells stains pale blue which is pathognomonic of epidermodysplasia verruciformis lesions without any sign of malignancy (Arrows).
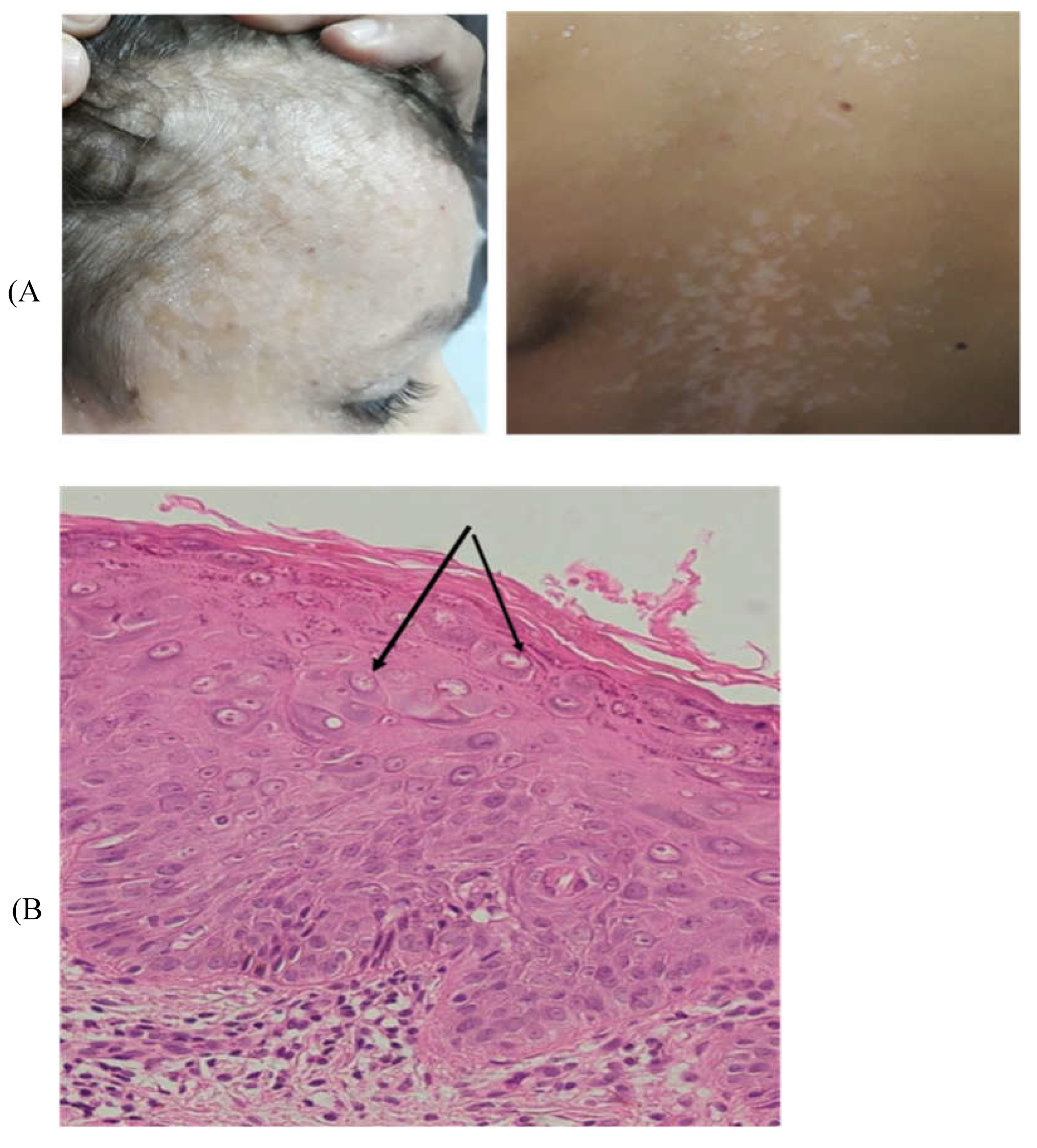
Figure 2.
A. Pictures of the lesions on the face and the hand of P3. B. Histological study of Punch skin biopsy showed hyperkeratosis and parakeratosis, mild acanthosis, and the presence of koilocytes, keratinocytes with pale-stained cytoplasm in the upper epidermis associated with high levels of intranuclear viral replication. The cytoplasm of the affected cells stains pale blue which is pathognomonic of epidermodysplasia verruciformis lesions without any sign of malignancy (Arrows).
Figure 2.
A. Pictures of the lesions on the face and the hand of P3. B. Histological study of Punch skin biopsy showed hyperkeratosis and parakeratosis, mild acanthosis, and the presence of koilocytes, keratinocytes with pale-stained cytoplasm in the upper epidermis associated with high levels of intranuclear viral replication. The cytoplasm of the affected cells stains pale blue which is pathognomonic of epidermodysplasia verruciformis lesions without any sign of malignancy (Arrows).
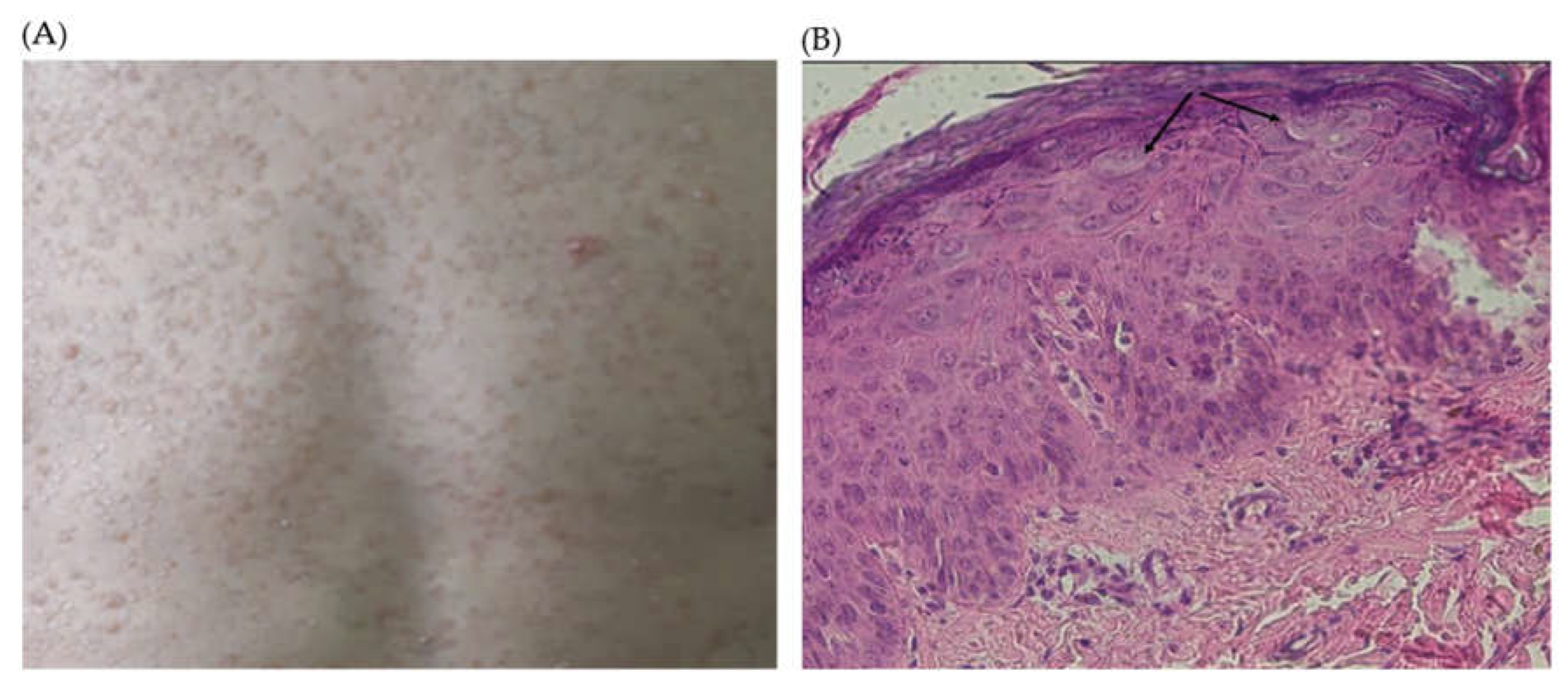
Figure 3.
Pedigrees and familial segregation done by Sanger sequencing of STK4 variants in family 1 (panel A) and family 2 (panel B).
Figure 3.
Pedigrees and familial segregation done by Sanger sequencing of STK4 variants in family 1 (panel A) and family 2 (panel B).
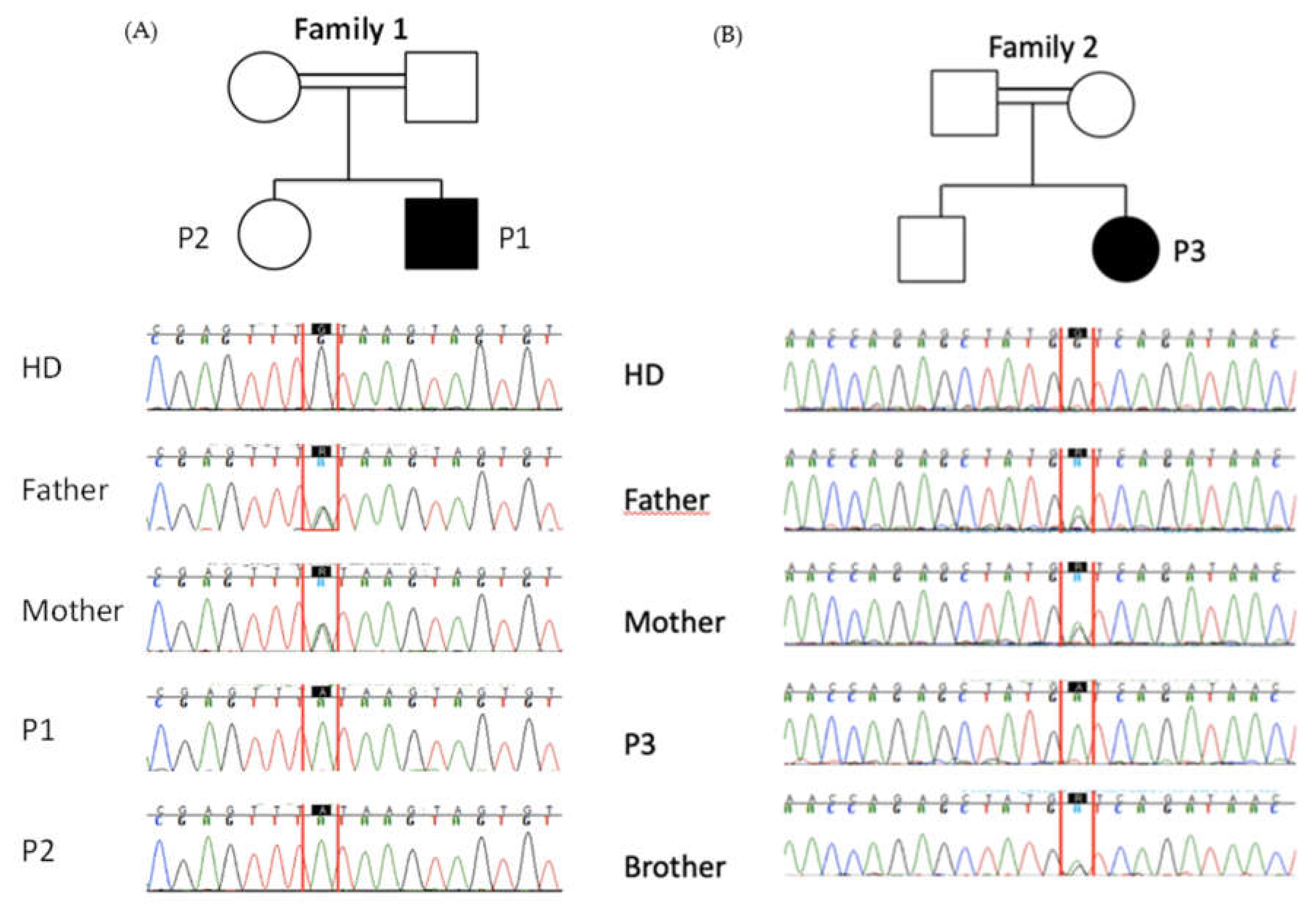
Figure 4.
CyTOF analysis on peripheral blood. A. Proportion of total and subsets of NK cells, of total T cells, total T cells, γδ T cells, iNKT and MAIT cells. B. Proportion of total and naïve, central memory, effector memory and EMRA CD4+ T cells, T-regulatory, recent thymic emigrant T cells and of total and naïve, central memory, effector memory and EMRA CD8+ T cell subsets. C. Proportion of total and memory, transitional, Age-associated B cells (ABC), plasmablasts and switched B cell subsets. Age-matched healthy donors (black), patients (red).
Figure 4.
CyTOF analysis on peripheral blood. A. Proportion of total and subsets of NK cells, of total T cells, total T cells, γδ T cells, iNKT and MAIT cells. B. Proportion of total and naïve, central memory, effector memory and EMRA CD4+ T cells, T-regulatory, recent thymic emigrant T cells and of total and naïve, central memory, effector memory and EMRA CD8+ T cell subsets. C. Proportion of total and memory, transitional, Age-associated B cells (ABC), plasmablasts and switched B cell subsets. Age-matched healthy donors (black), patients (red).
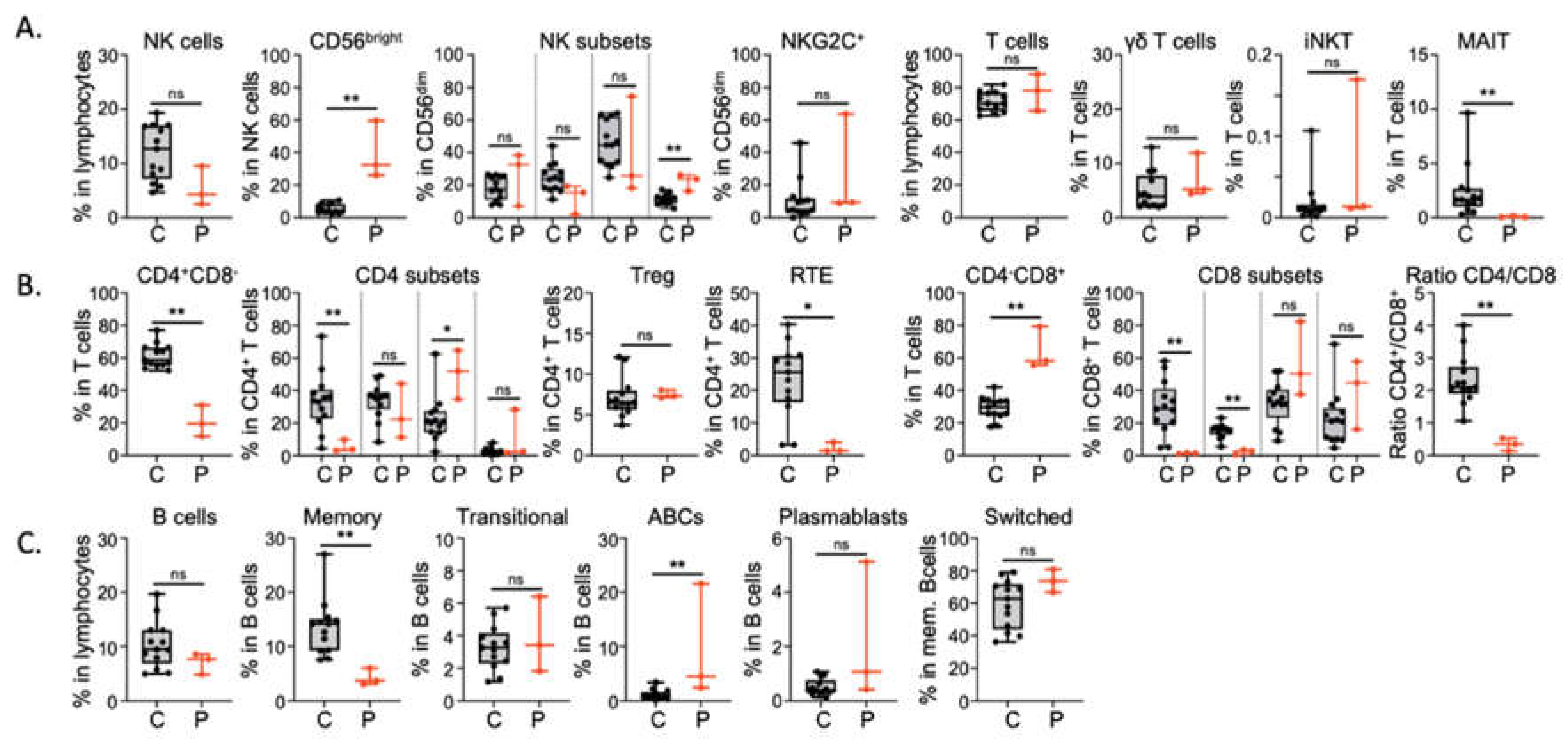

Table 1.
Lymphocyte subpopulations count.
| Lymphocyte subpopulations (N/mm3) |
P1 (12 years) |
P2 (8 years) |
P3 (6 years) |
Normal range (age matched) |
|---|---|---|---|---|
| T cells | ||||
| CD3+ | 771 | 2990 | 1489 | 1200-2600 |
| CD4+ | 253 | 470 | 243 | 650-1500 |
| CD8+ | 450 | 2330 | 1057 | 404-826 |
| B cells | ||||
| CD19+ | 128 | 430 | 131 | 270-860 |
| NK cells | ||||
| CD16+/CD56+ | 74 | 110 | 123 | 100-480 |
Table 2.
Immunoglobulin levels.
| Immunoglobulin levels (g/L) |
P1 (12 years) |
P2 (8 years) |
P3 (6 years) |
Normal range (age matched) |
|---|---|---|---|---|
| IgG | 11.62 | 5.17 | 17,3 | 6,10-16,16 |
| IgM | 0.49 | 0.46 | 2,09 | 0,22-2,40 |
| IgA | 1.99 | 3.21 | 2,17 | 0,84-4,99 |
| IgE (UI/mL) | 10 | 5.18 | 175,68 | <100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



