Submitted:
22 July 2024
Posted:
23 July 2024
You are already at the latest version
Abstract
Analysis of tRNA sequence structural features from forty mammals proves neither isoacceptors nor isodecoders are degenerate, and model organisms are inadequate representatives for multiple aspects of the translational process. It is proposed tRNA participate in protein construction beyond mere transport of amino acids to ribosomes: specifically, to facilitate amino acid modification and promote development of protein folding characteristics.
Keywords:
mammals
; tRNA
; comparative genomics
; model organisms
Introduction
In writing research paper Discussion sections, many geneticists possess an unfortunate tendency to communicate as if they were physicists, firmly believing investigations reveal universally applicable facts regarding gene structure and function upon extrapolation of data from less compositionally complex organisms. This error was enshrined into scientific and economic dogma once the National Institutes of Health in the United States developed a group of model organisms into whose research most government dollars pour (Bellen et al. 2021). When researching genomic characteristics of mammals, there is nothing sacrosanct about the genetic traits of Bacteria (Escherichia coli), Amoebae (Dictyostelium discoideum), Fungi (Saccharomyces cerevisiae), Invertebrates (Caenorhabditis elegans, Drosophila melanogaster), On the other hand, Plants (Arabidopsis thaliana) and Vertebrates (Danio rerio, Xenopus laevis) would seem within the realm of legitimacy. When researchers assert discoveries in model species are applicable to all Bacteria, Eukarya, Archaea—no representatives of the latter appear on the NIH list—they misjudge.
Genetics as science is the epitome of individualism. Saccharomyces cerevisiae is the most studied model organism for Eukarya, and reference strain S288c is often used. However, in a collection of 1011 variations, 123 of 1072 genes (11.5%) defined as essential for S288c activity as a living entity are absent in those other 1010 strains (Peter et al. 2018). As a second example, tRNA gene copy number variation ascertained in two families of three persons each revealed both children’s genomes exhibited distinctions from their parents, indicating inheritance is not always controlling (Iben and Maraia 2014). Taxonomic nomenclature recognizes species level or higher organizational units (subspecies descriptors are relatively rare), yet no pair of creatures, human or otherwise, are fungible commodities: supposedly identical twins become distinguishable when epigenetic factors emanating from surroundings are factored in.
Model organisms model themselves; with respect to others, they obscure meaningful information more than they reveal. In most circumstances, genetic features may be deemed attributes of others of their species. In certain situations, those characteristics might possibly be extended to genus level. Gene traits infrequently pertain per se to families, clades or other levels of biological complexity. Absolute claims of genetic (or genomic) applicability beyond individuals is improper unless analogous inquiries into other taxonomic units have been extensively explored among multiple candidates. Escherichia coli no more represents Bacteria (Ankeny and Leonelli 2011) than Joe Biden represents American presidents or Heads of State worldwide.
Allegedly, there are thirty-three ribosomal proteins encoded in all three domains of life, and each domain supplements this collection with sets of other proteins exclusively used by its members (Kisly and Tamm 2023). The idea of a universal core underlies advocacy of a naming procedure for codifying them, written in a paper with twenty-five authors, each a highly respected biologist (Ban et al. 2014). Universal is an absolutist term allowing no exceptions. Do these twenty-five believe no organism—unicellular or multicellular; inhabiting land, sea, air—has functionally adapted to its surroundings by evolutionarily discarding one or more of these thirty-three proteins, or merged two or more into a unified whole, such that it no longer possesses a 1:1 match with the thirty-three denominated universal core?
Force equals mass times acceleration has earned its title as a Universal Law of Motion; F = ma is valid throughout the expanding universe over its 13.7-billion-year history. There is no realistic possibility for it to fail in particular spatial locations at specific times under precisely determined conditions. The same cannot be remotely construed an analogous interpretation for a universal core of ribosomal proteins.
It is true the bulk of US research dollars is delegated to those select organisms on the NIH list. Therefore, it is understandable that scientists focus on them in their investigations (Dietrich et al. 2014). The problem is inductive inference beyond conservative propriety. Saccharomyces cerevisiae prefers A/U at tRNA wobble position 34, but mammals prefer C/G (Zhou et al. 2016). Mammalian ribosomes are larger than those of yeast; have more rRNA extension segments and more connections to eukarya specific ribosomal proteins (Chandramouli et al. 2008).
Pinkard et al. (2020) proclaimed 4-box codons possessing C in third position do not basepair with tRNA possessing G34 in mammals; they insisted it is always I34, producing noncanonical basepair C3/I34, obtained from A34 by deamination at a pre-tRNA stage in the nucleus (Torres et al. 2015). The originators of this doctrine (Grosjean et al. 2010) grounded their assertion on fifty genomes (60% bacterial, 26% archaeal, 14% eukaryl). Of the latter, one is mammalian and the study included no other vertebrates. Their insistence was absolute: “Without exception, tRNA harboring anticodon A34NN never coexists with tRNA harboring anticodon G34NN in any amino acid family boxes.” Their confidence was misplaced. In the present investigation, tRNAAla(GGC) is encoded in seven of forty mammals: tRNAGly(GCC) is found in all forty; tRNALeu(GAG) in one; tRNAPro(GGG) in two; tRNASer(GGA) in five; tRNAThr(GGU) in six; tRNAVal(GAC) in nine. In the context of 4-codon box isoacceptors, only tRNAArg(GCG) satisfies their belief by being absent from the genomes of these animals.
Criticism is not the goal. The solution is not to forego the use of model organisms; it is to always be conscious that data accumulated apply with reasonable certainty only to a studied individual. Simple prokaryotic strains of a single species exhibit divergent genomic properties: seven Methanosarcina mazei strains possessing a combined 387 tRNA contained sixty-three unique strings among them, thirty-five (56%) found in all seven and eleven (17%) in single strains (Laibelman 2022). It is imperative that comparisons of structure and function for genes not be subject to speculation unless the objects of theory are closely related phylogenetically on other grounds.
One measure of genetic separation between species is estimated divergence time. Logically, the longer spans of time passage since entities became genetically unable to interbreed, the larger the number of differences should be found when comparing genomic sequences in detail. This is a truism not only on the scale of single nucleotides, but cumulatively on the protein level these genes encode and the functions their gene products evoke. Geological timescales use many nomenclatural schemes, with each division measured in hundreds of millions (or billions) of years having a numerical range likewise spread over several million years. In determining species divergence times, recourse is typically made both to fossil records and molecular clocks.
The reliability of the fossil record in yielding an accurate calendar for species divergence times is completely dependent on their discovery: missing links in continuity invariably leads to guesswork. The actual existence of artifacts cannot be ascertained theoretically; even if real, geographic accessibility plus large doses of both skill and luck in recovering them sufficiently intact to undergo chemical analysis by radioactive carbon isotope dating is an ever-present challenge. Use of molecular clocks has its own set of problematics arising from severe methodological diversity generating variable timescale approximations, signifying independent sets of genes should be employed (Wang et al. 1999). However, casual inspection of the literature reveals numerous phylogenetic maps at species level discordant with each other because the molecules used as their basis vary enormously (Smith et al. 2015).
The importance of assigning divergence times becomes heightened when temporal gaps between model organisms are considered. Based on the molecular clock technique, and using Homo sapiens as the point of reference, model organism divergence times in units of millions of years (mya), according to TimeTree (http://www.timetree.org) are:

The greater the divergence time of a model from an anthropocentric perspective, the less useful it is as an indicator of human genetics and biochemistry. Since the most common models employed in this discipline are Saccharomyces cerevisiae and Escherichia coli, we should not be willing to take their data as universally valid.
The issue has been explored at length because this paper’s content bears iconoclastic overtones. The paper on tRNA sequences in Archaea initiated an attack on conventional orthodoxy surrounding the structure of the Standard Genetic Code (Laibelman 2022). Here, tRNA sequences in Eukarya, specifically mammals, extends that challenge in directions factual and hypothetical, frequently contrary to consensus views established almost exclusively on model organism research. The goals are: (i) to explore the myriad features presented by mammalian eukaryl tRNA; (ii) demonstrate how their impact on translation differs significantly from aspects uncovered in model organism study; (iii) propose ideas, and raise questions, in need of experimental investigation.
The analyses performed here utilize mature processed tRNA sequences from forty mammalian species symbolized by single specimens from each. The fact that these creatures are inherently individual embodiments, and cannot even be declared representative of their species with unimpeachable certainty, much less for any larger descriptive taxonomic index, circumscribes all conclusions , , , except those disproving prior claims of universality by express counterexample. These forty were chosen from a larger list whose codon usage data appears in the HiveCuts database (https://hive.biochemistry.gwu.edu/cuts/; updated April 2022; accessed November 2022) because they have their full catalog of tRNA sequences stored in the Genomic tRNA database (http://gtrnadb.ucsc.edu; updated June 2021; accessed December 2022).
To unify divergence time estimates so as not to confuse the issue by citing diverse sources and techniques of measurement, results are taken solely from the TimeTree website (http://www.timetree.org). TimeTree is updated periodically to reflect growth in documentation, with the latest update (TimeTree 5) released in 2022 (Kumar et al. 2022). The authors specified this version contains 137,306 species, or 41% more than the previous edition published five years earlier (Kumar et al. 2017). The TimeTree of Life contains a single timeline referenced to Homo Sapiens; however, species are mostly clustered into larger groups for clarity. This feature detracts from its utility because umbrella-covered divergence times cannot accurately depict values for closely connected organisms. This criticism is especially appropriate in the present case where the selected forty mammals combined had a last universal common ancestor just 180 million years ago.
One more general comment merits mention. There are two methodological assumptions serving to ground comparative genomics research: (i) analyses rely on accurate sequencing by researchers without introducing errors during result tabulation; (ii) collections of sequences retained in public databases are reliable expressions of original data without instituting errors during receipt, storage, or release. Published sequence information is rarely independently replicated; hence, conclusions drawn by those other than the initial investigators presume the information is a priori correct. These epistemological conditions are here in play with respect to information accessed from GtRNAdb (Chan and Lowe 2009).
Materials and Methods
Transfer RNA sequences for forty (40) mammals using conventional Genus species nomenclature were downloaded from GtRNAdb (http://gtrnadb.ucsc.edu; release 19, June 2021; accessed December 2022). Animals were selected from a larger compilation of mammalian codon usage data established by the HiveCuts database (https://hive.biochemistry.gwu.edu/cuts/; updated April 2022; accessed November 2022). The larger HiveCuts set comprised 145 species, but only these forty were found in GtRNAdb. The remaining 105 either lack tRNA sequencing or, for unknown reasons, are not contained within GtRNAdb. No further efforts were made to obtain their data from other sources. Table 1 presents the animals studied along with general background information from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov; accessed November 2021).
Only tRNA labeled mature sequences were used, meaning all known introns were removed as well as leader sequences. Sequences from each organism were presorted in GtRNAdb: first by reference amino acid using their common three-letter short form code, and second by anticodon triplet per amino acid. Sequence length expressed in basepairs (bp) from encoded DNA was replaced by a designation of nt (nucleotides) to reflect that these are single stranded RNA molecules. No other changes were made. These forty mammals contained 19,776 tRNA sequences written horizontally with 5’ end on the extreme left and 3’ end on the extreme right of each row in a spreadsheet, with columns arranged alphabetically by animal genus. Sequences were organized by a tripartite descriptor composed of corresponding amino acid + anticodon triplet + nt length.
The first order of business was to remove exact duplicates from each genome in order to provide a set of unique sequences for every animal. To accomplish this winnowing process, each batch of tRNA strings were separately subjected to the sort command of the spreadsheet. This function arranged them alphabetically (A/C/G/U) starting from the extreme left of each string. Visual inspection of successive lines enabled detection of adjacent pairs exhibiting 100% nucleotide-for-nucleotide matches across the entire length of the two sequences. All but one of these exact matches were discarded, and the anticodon triplet highlighted in bold red font. Proceeding through 19,776 sequences reduced to 12,578 (63.6%) the number of unique sequences differing by as little as one nucleotide between any two strings.
Some species contained suppressor tRNA (sup (NNN)) and/or sequences whose anticodon triplet were not determined by scientists performing the original sequencing (Undet (NNN)). These were held separate from tRNA expressing traditionally translatable amino acids. For forty mammals, they numbered 299 (295 unique), meaning 12,283 unique sequences from 19,477 (63.1%) total for relevant amino acid-associated anticodon triplet-containing strings.
With unique sequences by mammal per tripartite descriptor on hand, three types of matchings were performed, although the same methodology was employed for all: (1) internal comparison of strings for each animal; (2) external comparison of strings across all mammals; (3) external comparison of strings between these forty Eukarya and 186 Archaea, or selected other model organisms. Making extensive use of cell background color options for the second and third matchings to facilitate distinguishing between species, the appropriate aggregation of sequences were subjected to the spreadsheet sort command. Again, only 100% nucleotide-for-nucleotide match across the entire string length was an acceptable criterion. In the first comparison, the anticodon triplet was excluded from the determination of perfect match precisely to see if all other nucleotides were identical; comparison inclusive of anticodon triplet was previously performed in acquisition of unique sequences by removal of identical copies. For the second and third sets of comparison, anticodon triplets had to be exact replicas. After each test, sequences fulfilling the 100% criterion were transferred to separate spreadsheet tables for permanent reference, while those failing to meet the guideline were discarded from that comparison.
In no instance was any sophisticated alignment algorithm utilized. At no time was any statistical package invoked because all that was needed as data accumulating from this study was simple counting of sequences fulfilling demand expectations and consequent percentage computations.
Results
Table 1 provides a list of forty mammals studied. It includes scientific and common names, whole genome size (in Gbp), number of protein coding DNA sequences (CDS), and a three-letter code so their names need not be written in full every time. This code consists of a Capitalized first letter matching the first letter of the genus followed by two lower case letters indicating the first two letters of the species. In three cases, this is insufficient to distinguish among them, so a second letter for genus is used: Mamu for Macaca mulatta, Mimu for Microcebus murinus, Mumu for Mus musculus. Gbp and CDS values were obtained from the National Center for Biotechnology Information website (https://www.ncbi.nlm.nih.gov; accessed November 2021). These numbers may be outdated, but are sufficient for background, playing no role in the analyses undertaken.
Table S1 provides phylogenetic divergence in a grid of pairwise species separation times for easy reference, and recorded in units of million years ago (mya). It is symmetrical along the diagonal (upper left → lower right), so only half is displayed for clarity. When examining Table S1, search for the cell intersecting the appropriate column/row combination for those species of interest. Complete scientific names are given in the first column, but each row is headed by the abbreviation code. Oan is an unanticipated outlier, as it diverged from the thirty-nine other species 180 mya.
Many results uncovered by studying tRNA sequences from Archaea (Laibelman 2022) are here reified either in their entirety or slightly modified. Additional outcomes emerge only upon exploring the much larger numbers of strings necessarily incorporated into the more complex biochemistry innate to multicellular Eukarya in comparison with unicellular Archaea. Isoacceptors refers to sets of mRNA codon triplets translated into one amino acid according to the notion of codon degeneracy (redundancy). Since mRNA codons always act in conjunction with aminoacylated tRNA in a 1:1 relation, isoacceptors will likewise refer to relevant anticodon triplet tRNA. Isodecoder is employed when tRNA with an identical anticodon triplet encode nucleotide differences elsewhere when comparing entire sequences (Goodenbour and Pan 2006). In the Archaea paper, they are called unique sequences (unique strings): 186 organisms cumulatively encode 8869 sequences (4658 unique, 52.5%) for an average of ~48 strings per genome. Here, forty mammalian Eukarya generate 19,776 (12,578 unique, 63.6%), averaging ~494 per species.
Nonetheless, the most essential findings from that prior research are confirmed. Above all, it can no longer be doubted that, as forcefully advocated, The Standard Genetic Code Lacks Redundancy for Amino Acid Codons. This assessment constitutes a major departure from current dogma and demands a rethinking, and revision, of several core beliefs about genetics as revealed through genomics. Like the Archaea, mammalian Eukarya tRNA sequences display diversity in characteristics for every isoacceptor regardless of associated amino acid. Although the Standard Genetic Code interpretation suggests equality among codon triplets for specific residues after translation, this image offers a fundamentally incomplete and inaccurate picture. It is crucial to look at information provided by aminoacylated tRNA sequences bound to codon triplets. The range of properties exhibited by different anticodon triplets associated with single amino acids offers sufficient evidence to remove any semblance of degeneracy.
As in Archaea, the lengths of encoded tRNA strings in animals vary greatly per isoacceptor. The preferred length in number of nucleotides (nt) is defined as the largest number of total sequences for that anticodon triplet expressed in each genome. Glycine, for example, displays as preferred lengths:
- 39 sequences tRNAGly(ACC) → 73 nt (2×), 74 nt (1×)
- 388 sequences tRNAGly(CCC) → 71 nt (36×), 73 nt (4×)
- 540 sequences tRNAGly(GCC) → 71 nt (40×)
- 435 sequences tRNAGly(UCC) → 71 nt (2×), 72 nt (36×), 73 nt (2×)
The full range of expressed string lengths for glycine-associated tRNA covers 64−78 nt. Table S2 proves this is not an isolated cherry-picked example; length variance is typically found for 62 anticodon triplets (standard sixty-one plus UCA for selenocysteine). In general, there is a narrower range of tRNA lengths in these Eukarya than in Archaea, in part because forty of the former are tabulated as opposed to 186 of the latter. Preferred length for glycine isoacceptors is the same for tRNAGly(CCC), but different for tRNAGly(GCC) and tRNAGly(UCC); tRNAGly(ACC) has just one representative among these Archaea.
Table S2 offers a breakdown by animal and isoacceptor, with preferred length shown in bold red font. Table S3 summarizes preferred length distributions by isoacceptor; ties in length are listed as paired values; for example, 72/76 is a column heading indicating one species (Mlu according to Table S2) for which these lengths tie as most times found in tRNAPro(UGG). Only tRNAAla(AGC) and tRNAThr(CGU) display variability in preferred length, with the former invoking a species split between 72 nt (nineteen genomes) and 73 nt (twenty genomes), plus a 72/73 nt tie (one genome). For threonine isoacceptor, there is a wide spread favored: 72 nt (10×), 73 nt (2×), 74 nt (6×), 72/74 nt (17×), 72/73/74 nt (4×), 72/73/74/75 nt (1×). A meaning for length variability is presented in the Discussion. It cannot be presumed inconsequential, and the array illustrated by glycine-connected anticodons suffices to break degeneracy when tRNA are bound at the ribosomal A site with their correlate mRNA codon triplets.
What Goodenbour & Pan (2006) denominated as isodecoders applies to a greater extent than they focused on in their paper. They looked at eleven Eukarya including Cfa, Hsa, Mumu, Ptr, Rno. Their definition of isodecoders removed the most abundant differently sequenced string from the count; hence, their number of isodecoders per isoacceptor is relative to that string most often encoded by identical sequences. Neither in the Archaea survey nor here is this distinction made: the definition of unique tRNA equals the Goodenbour & Pan number plus one, representing an absolute value. Henceforth, isodecoders will be used interchangeably with unique.
It was mentioned that of 8869 archaeal tRNA sequences, 4658 are unique (52.5%), and of 19,776 eukaryl tRNA, 12,578 are unique (63.6%). A higher uniqueness percentage for Eukarya than Archaea is not surprising. If sequence variations in tRNA strings exist, there should be a greater number (larger percentage) in structurally and functionally more complex organisms when nucleotide differences in any position are meaningful. Table 2 displays raw totals and percentages: (i) with all anticodon triplets considered; (ii) with suppressor genes and undetermined anticodons (tRNAUndet(NNN)) removed. The second condition ensures proper amino acid translatable sequences may be investigated separately. Totals in Table 2 show removal of suppressor and unassignable genes has minimal impact: uniqueness percentage declines from 63.6% to 63.1% because those sequences removed from the tally are almost always unique relative to each other.
Table 2 also contains the number of isoacceptors for each organism under condition (ii). These forty creatures possess numbers of isoacceptors from 46 in Mlu to 60 in Vpa. Bta has next to most isoacceptors at 54, revealing Vpa as an outsider in this regard, a status which will prove significant. Using condition (ii) data, normalized uniqueness percentages range from 42.1% for Mdo to 85.4% for Vpa. In addition to Mdo, Sha (42.9%) and Dor (47.6%) are on the conservative side of 50%. Vpa is not as much of an outlier in isodecoders compared to isoacceptors; Ttr encodes 80.7% uniqueness, and there are six animals above 70%.
Although informative, Table 2 does not speak to the main issue at hand, which is proving a lack of degeneracy among isoacceptors. Table 3 accomplishes this task by expressing uniqueness percentages as a function of anticodon triplet. It shows sixteen are encoded by relatively few strings: one for tRNALeu(GAG) in Laf up to sixty-five for tRNASec(UCA) present in every animal but Mlu. Their unique-to-total sequence percentages equal 81−100%. Suppressor sequences—twenty-four tRNAsup(CUA), eighty-nine tRNAsup(UCA), forty-one tRNAsup(UUA)—present uniqueness levels of 100%, 99%, 100%, respectively. Isoacceptor strings register uniqueness percentages from tRNALeu(UAG) (97.5%) to tRNAHis(GUG) (24.8%) based on 147−1673 sequences.
For any given amino acid—whether associated with 2-box, 3-box, 4-box, or 6-box isoacceptor—percentages are not the same except for lysine-affiliated tRNA, where tRNALys(CUU) equals 63.1%, while tRNALys(UUU) comes in at 63.0%. Alanine is fairly close at 82.0%, 83.3%, 100%, 80.9% for anticodon triplets AGC, CGC, GGC, UGC, respectively. Other 4-box amino acids are not a match for each other; for example, valine has 61.3%, 55.6%, 91.7%, 86.1% for anticodons AAC, CAC, GAC, UAC, respectively. Arginine, leucine, serine 6-box isoacceptors are unalike whether structured as 6-box or 2-box plus 4-box units; they appear more like valine than like alanine. Aside from lysine, the 2-box groupings are grossly different, with glutamic acid illustrative: tRNAGlu(CUC) is 42.3% and tRNAGlu(UUC) is 71.5%. Table S4 unites Table 2 and Table 3. Demonstrably large variances per isoacceptor constitute unimpeachable evidence for tRNA inequivalence. Given a 1:1 correspondence with mRNA codons, a lack of redundancy among the latter is a necessary conclusion.
A comparison of archaeal to eukaryl tRNA uniqueness percentage per isoacceptor indicates six matches out of sixty-three possible pairs: standard sixty-one for twenty amino acids plus selenocysteine plus initiator methionine distinct from elongator methionine. Matches are arbitrarily defined as ±5% for each isoacceptor pair. Matched (Archaea %, Eukarya %) are: tRNAAsn(GUU) (46.9, 45.6); tRNAAsp(GUC) (39.6, 40.3); tRNACys(GCA) (55.2, 54.0); tRNAPro(UGG) (53.5, 51.0); tRNASer(GCU) (63.2, 59.6); tRNAVal(CAC) (58.1, 55.6). Only for aspartic acid are these mammals less conservative than relatively simple unicellular prokaryotes, with the remaining five isoacceptor pairings contrary to the overall domain relationship under condition (ii) of 52.3% vs. 63.1%. As a consequence of inequality between isoacceptors as well as among isodecoders, they might not translate the same amino acid. Both assertions are addressed in the Discussion section.
Table S5 offers a compilation of unique sequences for every animal sorted by species, anticodon triplet, associated length. Aside from a reference for research, it permits additional string features related to skewness and translational ambiguity found for Archaea to have demonstrated similar applicability in mammals. Think of each 1D tRNA sequence as a barcode not only distinguishing apples from oranges via isoacceptors, but also segregating fuji apples from gala apples by isodecoders. In this metaphor, total copy numbers equate to ratios for each type of apple. When arranged linearly left to right (5’ → 3’), the amino acid attachment point is on the extreme right. Stacking same-length sequences means anticodon triplets sometimes fail to possess perfect vertical alignment. Instead, inserted nucleotides left of the triplet induce rightward skew; inserted nucleotides to the right cause leftward skew; deleted nucleotides left of the triplet induce leftward skew; deleted nucleotides to the right cause rightward skew. As shown in Table S5, skewness is rare, occurring mostly when there exists distinct 5’- and/or 3’ -terminal nucleotide triplets within isodecoders. Bta provides examples of skewness.
Ala (AGC) 72 nt
GGCGGUAUAGCUCAGUGGUAGAGCACAUGCUUAGCAUGCAUGAGACCCUGGGUUCAAUCCCCAGUACUGCCA
GGGGAUGUAGCUCAGUGGUAGAGCGCAUGCUUAGCAUGCAUGAGGUCCCGGGUUCGAUCCCCAGCAUCUCCA
GGGGGUAUAGCUCAGUGGCAGAGCACAUGCUUAGCAUGCACGAGACCCUGGGUUCAAUCCCCAGUAUCUCCA
GGGGGUAUAGCUCAGUGGUAGAGCGCAUGCUUAGCAUGCAUGAGGCCCUGGGUUCAAUCCCCAGUACCUCCA
GGGGGUAUAGCUCAGUGGUAGAGUGCGUGCUUAGCAUGUAUGAGGUCCUGAGUUCAAUCCCCAGUACCUCCA
GGGGGUGUAGCUCAGUGGUAGAGCGCGUGCUUAGCAUGCACGAGGCCCCGGGUUCAAUCCCCGGCACCUCCA
GGGGGUGUAGCUCAGUGGUAGAGCGCGUGCUUAGCAUGCACGAGGCCCUGGGUUCAAUCCCCAGCACCUCCA
GGGGGUGUAGCUCAGUGGUAGAGCGCGUGCUUAGCAUGUACGAGGUCCCGGGUUCAAUCCCCGGCACCUCCA
GGGGGUGUAGCUCAGUGGUAGAGUGUAUGCUUAGCAUGCACGAGGUGCCAGGUUCAAAUCCUGGCACUUCCA
UCCCUGGCAGUCCAGUGGUUAGGACUUGGCACCAGCACUGCCAGGGCCCAGGUUCGAUCCUUGGUUGGGGAA
Ala (AGC) 73 nt
GGGGAAUUAGCUCAAAUGGUAGAGCGCUCGCUUAGCAUGUGAGAGGUAGCGGGAUCGAUGCCCGCAUUCUCCA
GGGGAAUUAGCUCAAGUGGUAGAGCGCUCGCUUAGCAUGUGAGAGGUAGUGGGAUCGAUGCCCACAUUCUCCA
GGGGAAUUAGCUCAAGUGGUAGAGCGCUCGCUUAGCAUGUGAGAGGUAGUGGGAUCGAUGCCCGCAUUCUCCA
GGGGAAUUAGCUCAAGUGGUAGAGCGCUUGCUUAGCAUGUGAGAGGUAGUGGGAUCGAUGCCCACAUUCUCCA
GGGGGAUUAGCUCAAAUGGUAGAGCGCUCGCUUAGCAUGCGAGAGGUAGCGGGAUCGAUGCCCGCAUCCUCCA
GGGGGAUUAGCUCAAAUGGUAGAGCGCUCGCUUAGCAUGCGAGAGGUAGUGGGAUCGAUGCCCAUAUCCUCCA
GGGGGUGUAGCUCA GUGGUAGAGCGCGUGCUUAGCAUGCACGAGGCCCCGGGUUUCAAUCCCCGGCACCUCCA
Table S5 also provides a visual demonstration of anticodon triplet ambiguity by highlighting it in bold red font. Ambiguity results if adjacent nucleotides extend the triplet region, and the pattern occurs in three formats: (i) letter replication beyond XXX; (ii) palindromic repeats XYXYX; (iii) triplet duplication XYXXYX, XYYXYY, XXYXXY, XYZXYZ. Four ambiguous anticodons appear with frequencies above 50%: tRNAArg(CCU) as CCUCCU (63.5%); tRNAGlu(CUC) as CUCUC (86.8%); tRNALys(UUU) as UUUU (97.4%); tRNAVal(CAC) as CACAC (73.3%). Skewness and ambiguity might appear together, emphasizing sequence individuality: CUCUC extends in the 5’ direction, while CACAC extends towards a 3’ terminus. In addition to these four, ambiguity occurs rarely, and usually as single events within an animal genome:

In sum, only tRNA connected to alanine, aspartic acid, cysteine, glutamine, leucine, methionine, selenocysteine, tryptophan are free of translational ambiguity. Apparent symmetry leading to ambiguity may be broken by nucleotide modification occurring post-transcriptionally. These alterations are not considered due to an insufficiency of knowledge for these species. Although vast amounts of research on the topic exists, mammal-relevant work focuses on eukaryotic model organisms such as Saccharomyces cerevisiae (Berg and Brandl 2021; Dannfald et al. 2021). As stated in the Introduction, it is not presumed changes apply automatically to other Eukarya after one billion years divergence time. Data collected on nucleotide modifications might be significant in the present context; that it must be, without more general proof of applicability in mammals, is a viewpoint subject to legitimate criticism.
Upon collecting unique sequences in Table S5, an unmistakable pattern pertaining to initial (position 1) nucleotide identity and isoacceptor, independent of length, was observed:
- Adenosine for tRNAiMet(CAU) and tRNALeu(UAA)
- Cytidine for tRNATyr(AUA) and tRNATyr(GUA)
- Uridine for tRNAAsp(AUC), tRNAAsp(GUC), tRNAGlu(CUC), tRNAGlu(UUC)
- Guanosine for all other tRNA (fifty-five, counting initiator and elongator methionines separately plus selenocysteine)
Theoretically, first position mutations are conceivable for isodecoders from each isoacceptor in every mammal. In practice, no acceptable mutations were encoded for tRNAiMet(CAU); if they occurred in the past, they were quickly excised from genomes, consistent with its special role in translation. Although there seems no easily discernible justification for an eight plus fifty-five isoacceptor division, eighteen animals followed the pattern with six or fewer total isodecoder deviations. Table S6 presents a matrix of initial position codon mutation numbers with respect to each amino acid. Of 12,283 unique sequences for amino acid-related tRNA, 1236 violations (10%) were discovered. This is not a product of neutral drift because particular tRNA are especially susceptible to alteration at position 1, with sequences for alanine, arginine, glutamic acid, glycine, lysine, tryptophan affected most (69% of pattern deviations), whereas asparagine, histidine, isoleucine, methionine, phenylalanine, proline, selenocysteine, threonine, tyrosine are impacted least (7.5%).
Animals most susceptible to pattern violation are Bac, Bta, Oar, Ttr, a group itself connected. Bac and Ttr from order Cetacea diverged 34 million years ago; Bta and Oar from order Artiodactyla separated 24.6 mya; Bac/Ttr had their last common ancestor with Bta/Oar 58 mya (Table S1). These four species comprise the closest land-based and sea-based living creatures (Foote et al. 2015). A review of sequences (Table S5) in context with the matrix (Table S6) indicates this group is responsible for establishing uridine as rival to guanosine starting nucleotide for twenty-six isoacceptors (fourteen amino acids). Uridine is normally preferred for only acidic amino acids, but these four species altered (infected?) isoacceptors for every amino acid except histidine, leucine, proline, selenocysteine, tyrosine. The mutated strings possess starting 5’ and ending 3’ triplets identifiable as carryovers from aspartic and glutamic acid sequences. In other words, acidic amino acid tRNA are especially prone to undergo mutation at the anticodon triplet, illustrated by conversions tRNAAsp(AUC) → tRNAAla(AGC); tRNAAsp(GUC) → tRNAAsn(GUU); tRNAGlu(CUC) → tRNALys(CUU); tRNAGlu(UUC) → tRNAGln(UUG).
In addition to encoded tRNA strings whose anticodons were deemed unassignable (145 total, 142 unique) by scientists performing the sequencing analyses, a small number definitively assigned to specific isoacceptors still had nucleobases whose identity was uncertain. It is now possible to suggest resolution of these instances with high probability based on correlation with other sequences in those animals. Table S7 lists recommended corrections along with justification. Certainty is impossible, unless sequencings are independently repeated, since nucleobase alteration not conforming to precedent is an option. There is, however, one exception to impossibility of certainty: in Ggo, there exists an unusual 98 nt tRNAPro(AGG) encoded sequence with a 20 nt section of unknown bases:
GGCUCGUUGGUCUAGGGGUAUGNNNNNNNNNNNNNNNNNNNNGGUAUGAUUCUCGCUUAGGGUGCGAGAGGUCCCGGGUUCAAAUCCCGGACGAGCCC
Regardless of identities for those missing twenty bases, removal leaves a 78 nt string possessing a six nt overlap (underlined bases) producing a mature 72 nt tRNAPro(AGG) precisely matching nucleotide-for-nucleotide a sequence existing in this animal’s genome (Table S5). The remaining six examples from Table S7 contain consecutive nucleobase strings of 1−4 unknowns (i.e., N → NNNN).
Having determined unique tRNA sequences within each genome, they were examined to evaluate whether identical patterns existed excluding the anticodon triplet, which had to be distinct from each other since perfect matches throughout the entire length had been removed. Table S8 provides a complete list of all successful pairings and Table S9 arranges that information by the number of mammals encoding each internal sequence match. From the data it becomes obvious specific isoacceptors more frequently possess a match inside the genome primarily associated with alanine, glutamine, glutamic acid, glycine, leucine, proline, serine, valine, though not all appear in every genome. Aspartic acid, histidine, isoleucine, lysine, phenylalanine each merited inclusion once; arginine and tryptophan twice; threonine in four genomes; cysteine in five. Asparagine, methionine, selenocysteine, tyrosine are absent. Of more significance than recitation of amino acids are the sets of anticodon triplets grouped. An in-depth analysis of meaning is forthcoming in the Discussion, but a preview has been mentioned with respect to uridine mutations for position 1.
This internal matching exercise also permitted an analysis of the tRNA in each genome labeled as Undet (NNN) by initial researchers (according to GtRNAdb). From Table S4, five species have more than two unassignable tRNA: Oar (four), Bta (eleven), Bac (seventeen), Ttr (twenty-one), Vpa (seventy-six). The first four have been acknowledged to be phylogenetically related, and outlier status exhibited by Vpa has been noted in a different context. Executing a spreadsheet sort command allowed nearest neighbor strings to be determined. Plausible assignment of anticodon triplet could often be made, with the results in Table S10 segregated as probable (109), possible (sixteen), still unassignable (seventeen). Strings called probable differed from a neighbor string used as reference by 0−9 nucleotides as an arbitrary cutoff; those denoted possible deviated from their reference by 10−14 nucleotides.
Assigned tRNA contained either an exact match to the reference anticodon or a mutation in one base. Strings still regarded as unassignable either had no close matches to any nearest neighbor, or the best alignment instituted a gap in the anticodon triplet preventing evaluation. Table S10 is organized by mammal, anticodon assignment, tRNAUndet(NNN) plus nearest neighbor used as the basis for assignment. With its relatively enormous number of undetermined tRNA, Vpa commands special attention. Of its seventy-six unknowns, the evaluation process led to sixty-four in the probable group along with three possibles and nine still uncertain. For a species with 833 unique tRNAAla((AGC), 63% of tRNAUndet(NNN) used one of these as its reference string.
The next matching activity involving unique tRNA sequences compared them in all forty animals. Extensive usage of cell background colors facilitated distinguishing species. Unique sequences, omitting suppressor and original undeterminable strings, were subjected to the spreadsheet sort command. Again, 100% nucleotide-for-nucleotide pairwise match was the only acceptable criterion. Successful matches per isoacceptor with associated length were tabulated; the minimum number of inclusive genomes was two and the maximum forty. Table S11 reports 631 amino acid-translatable tRNA sequences were found. As shown in Table 2 arranged by animal, 5617 matched out of 12,283 unique sequences (45.7%), with a larger percentage of matches incurred by smaller genomes. In a separate test, two tRNAsup(UCA) and one tRNAUndet(NNN) aligned perfectly, deriving from cetaceans Bac and Ttr in all three cases (also shown in Table S11).
If the classic dogma held true, every isoacceptor would have at least one isodecoder matched in all forty species, corresponding to unmodified versions of twenty classical amino acids; it is not expected, nor does it occur, that every animal encodes tRNASec(UCA). Just twenty-nine sequences appear with 100% nucleotide match in all forty species (Table 5 and Table S11); 42.9% connected two animals, with every value from 2−40 genomes represented (Table 5), Of crucial importance, this set of twenty-nine isodecoders covers at least one isoacceptor for each canonical amino acid, with two for arginine, glycine, isoleucine, leucine, lysine, proline, serine, tryptophan, valine. The tryptophan sequences reference single isoacceptor tRNATrp(CCA), and there is a single nucleotide difference between them. Methionine’s match exists only for the initiator version; the highest interspecies acceptance for elongator methionine sequences is thirty-eight species (absent in Mdo, Sha). The import cannot be overstated: sixty-one isoacceptors do not find exact sequence matches in every mammal, substantiating the fact that 1:1 codon/anticodon complexes are not degenerate.
The ten tRNA sequences present in thirty-nine animals were studied to assess how deviant, based upon number of nucleobase differences, was the genome of the absent species. Results are detailed in Table S12. They involve tRNA sequences related to two alanine, two arginine, one glycine, one leucine, one lysine, one proline, one serine, one threonine. More than three mismatched nucleotides was arbitrarily defined as a not close match; two alanine (Sar, Vpa), glycine (Oan), serine (Ocu), threonine (Vpa) strings satisfied this negative condition, whereas the lysine sequence (Eeu) was borderline with three variants. Both tRNAArg(UCU) (Bac, Dno) altered a single nucleobase generated by two nearest neighbors each, and this was also found for two sequences from Ame for tRNAPro(UGG) plus a single neighbor from Mlu for tRNALeu(CAG). The arginine pair reference the same isoacceptor, and tRNALeu(CAG) was already covered in the collection of twenty-nine sequences. If the four amino acid associated strings are accepted into the consensus set, addition of two expands those covered in all forty mammals to thirty isoacceptors, which is barely halfway to sixty-one isoacceptors in the Standard Genetic Code.
Table 6 employs a standardized format for those twenty-nine strings in order to evaluate which, if any, nucleobases are invariant. This three-point scheme uniformly aligns 5’ terminus + anticodon triplet + 3’ terminus, inserting gap spaces where needed to compensate for unequal lengths of the natural strings (71−83 nt). By this technique, anticodon triplets are at positions 35−37, and no nucleotide modifications are entertained in consideration of conservation; 2D cloverleaf and nucleotide numbering system (Sprinzl et al. 1998) are inconsequential. Nine positions (bold font) express strict conservation: U8, G10, A14, G18, G63, U65, C66, A68, C71. These sequences supply a variety of lengths, 5’ beginning and 3’ ending nucleotide triplets:
- lengths → 71 nt (1×), 72 nt (12×), 73 nt (8×), 74 nt (4×), 82 nt (3×), 83 nt (1×)
- 5’ terminus → AGC (1×), CCU (1×), GAC (4×), GCA (1×), GCC (4×), GCG (1×), GCU (1×), GGC (4×), GGG (3×), GGU (2×), GUA (1×), GUC (2×), GUU (2×), UCC (2×)
- 3’ terminus → ACA (3×), ACG (2×), CCA (3×), CCC (2×), CCU (3×), CUA (1×), GAA (1×), GAG (1×), GCA (5×), GCG (2×), GGA (1×), UCA (2×), UCG (3×)
Table 7 permits tracing a relative phylogenetic relationship among these animals by determining the number of matches against a species chosen as arbitrary standard. Since we are homocentric creatures by nature, it is convenient to use Hsa as reference. Ideally, larger numbers of common isodecoder strings should imply closer phylogenetic relationships in a single ordering. Hsa has a representative in 164 of 631 matches (26.0%). In common with those 164 tRNA are 142 held by Ptr, followed by 128 for Ggo, 121 for Ppy, 117 for Nle and so on, with Oan last at sixty-one sequences. Comparing this order with TimeTree divergence times gives a near-perfect fit: less time divergence equals more matches. This is wholly to-be-expected, for it is the root rationale for constructing phylogenetic relationships in the first place.
These are raw data results, and it is prima facie reasonable to predict larger numbers of encoded tRNA would lead to a greater number commonly held because chances of agreement likely increase with opportunities. However, a string length of 73 nt generates 473 ≈ 8.9×1043 permutations; leucine and serine tRNA of 82 nt produce 482 ≈ 2.3×1049 distinct strings. Genome size differences are of order 130−1436 isodecoders, with reference Hsa having 254 (Table 2). On the scale of possibles, genome size variance for unique tRNA are insignificant in terms of increasing the likelihood of a perfect match.
Knowing there are 631 encoded sequences among 2−40 mammalian species producing perfect matches enables comparison of this collection with unique tRNA strings from other lifeforms in order to determine whether model organisms serve as proper representatives for all biological constructs related to translational processes. It is informative to proceed according to TimeTree divergence times:
- Fifty-two unique tRNA from Escherichia coli (strain K12) failed to find 100% matches, focusing specifically on respective tRNAiMet(CAU) sequences (Table S13a).
- 186 Archaea containing 4608 unique tRNA with 12,283 unique mammalian tRNA, including those 631 interspecies-matched sequences, culminated in finding not a single perfect agreement, in particular for respective tRNAiMet(CAU) sequences (Table S13a). Such failure implies either: (i) no horizontal gene transfer occurs; (ii) occurs, but transferred strings are later altered in one or more nucleobase positions once internalized by an organism; (iii) an insufficient number of eukaryl species were examined. This test does not dismiss horizontal gene transfer as a possibility between domains; it only draws a tentative inference with respect to this process occurring across sets of tRNA genes.
- Arabidopsis thaliana, Oryza sativa, Zea mays with 171, 193, 304 unique tRNA, respectively, failed to find 100% matches, in particular for respective tRNAiMet(CAU) sequences (Table S13a).
- Fifty-five unique tRNA from Saccharomyces cerevisiae (strain S288c) failed to find 100% matches, in particular for respective tRNAiMet(CAU) sequences (Table S13a).
- Caenorhabditis elegans and Drosophila melanogaster encoding 152 and eighty-four unique tRNA, respectively, produced four matches: tRNAGln(CUG), tRNAGln(UUG), tRNALys(CUU), tRNAPro(CGG), all from Drosophila melanogaster. A focus on respective tRNAiMet(CAU) sequences failed to find matches to the consensus mammalian version (Tables S13a and S13b). There were fourteen cases where variations appeared in no more than three nucleobases, with just two containing Caenorhabditis elegans strings. In no case did both invertebrates match a single mammalian tRNA (Table S13b). Among four perfect alignments, the anticodon triplets are related by single base mutation: CUG → UUG; CUG → CUU; CUG → CGG. To be clear, these four are not identical to each other throughout their length; they are separately the same as representative mammalian tRNA. The mutation pattern is reminiscent of radioactive isotopic decay whereby element A is converted to element B by gain or loss of a helium nucleus (α process) or to element C by gain or loss of an electron (β process).
- Alligator mississippiensis and Gallus gallus with 391 and 158 unique tRNA, respectively, displayed numerous perfect pairings related to all isoacceptors. Twenty-five of the twenty-nine strings encoded by all mammals were also found in the two nonmammalian vertebrates, as well as nine of those ten strings contained in all mammals but one (i.e., sequences with thirty-nine-member genomes). Among those twenty-five was the consensus tRNAiMet(CAU) sequence (Table S13c).
Discussion
Genetics theorists directly confront Sophie’s Choice on a regular basis: a cherished idea must die. Perhaps genomes from evolutionarily divergent species should be radically different due to: (i) natural selection for environmental adaptation; (ii) unpredictable random mutation; (iii) genetic drift leading to an accumulation of inaccuracies in transcription and/or translation occurring over time, Contrarily, one may expect genes to demonstrate high degrees of sequence similarity for analogous function despite influential factors pushing for change. Consequently, geneticists might be surprised by gene identicality, if adhering to the first view, or by variance, if advocates of the second perspective. They cannot have it both ways at the same time for a given thematic issue. Call it the central conflict in evolutionary theory: taxonomy is predicated on a supposition that kingdoms, phyla, families, species normatively establish separate pursuits in life for life; yet, despite aspirations towards individuality, encountering issues concerning predators and prey, sexual reproduction, attracting or avoiding effects of natural phenomena (gravity, electromagnetic radiation, weather) means they sometimes converge in discovering ways to survive and prosper.
Like proteins, nucleotide sequences display degrees of homology over their length. Unlike them—where components can be same (valine vs. valine), similar (leucine vs. valine), or different (arginine vs. valine)—nucleotide comparisons in two molecules face a binary option; even if modified, they are either identical or not. The notion of similar nucleotides is a fallacy, although some perceive A/G purines similar and C/U pyrimidines likewise. Why does similarity not apply? The pairs engage in different numbers and strengths of hydrogen bonds during complementary base pairing, affecting the energetics of interaction in nonequivalent ways: three hydrogen bonds in GC equal to 14.3 Kcal/mol, as opposed to two hydrogen bonds in AU worth 10.6 Kcal/mol (Halder et al. 2019).
All pieces of data reported in Results collectively lead to three theses about the roles played by tRNA in mammalian, and probably vertebral lifeforms in general, though only two are proposed as novel: (i) isodecoders provide a record of benign mutations; (ii) beyond transporting amino acids to ribosomes, tRNA participate in modifying them before and during translation; (iii) secondary structure development in growing proteins depends on tRNA sequences.
When tRNA from forty mammals display patterns of both identicality and uniqueness, it must be in consequence of different aspects pertaining to structure and function. Structural parameters regarding length, sequence, skewness, anticodon ambiguity all express unrelenting diversity. Nucleotide differences between isodecoders represent mutation history almost by definition, since hazardous or lethal alterations would probably have been exorcised from genomes if longevity or survival were at stake. Evidence for a historical interpretation should be, at minimum, a finding that phylogenetically proximate species undergo similar changes. Table 7 confirms this indirectly by verifying a correlation between animal divergence time and incidence of string identity relative to humans. Contrarily, if evolutionarily separated creatures jointly develop mutations absent in more closely-related organisms, then other reasons for their presence must be discovered while still respecting a doctrine of historical record: do they serve cognizable novel purposes?
Both sequence identicality and deviation require justification. The law of large numbers enters in force, and probability statistics work in both directions. A 4-base code of 73 nt generates 473 ≈ 8.9×1043 possible sequences. If two strings, each 73 nt, are perfect matches across their length, the odds of this happening are one in 42×73 ≈ 8.0×1087 or, in general, one in 4MN for M creatures possessing N identical nucleotides. Therefore, when twenty-nine tRNA strings display nucleotide-for-nucleotide matches across forty species diverging 6−180 mya, it can only be attributed to sharing the same function each time; the odds against coincidence are truly astronomical, although an exact accounting would necessitate factoring in different lengths among these twenty-nine.
Sequence deviation is far more probable because the odds of a single base change is one in three rather than one in four for complete randomness. If, in a reference mammal, nucleotide A is replaced by C in mammals 2 and 3, then it is chosen from set C/G/U in each, so the odds are one in nine, whereas starting from scratch means selection from set A/C/G/U. For N identical variations between two species from any reference sequence chosen from among 8.9×1043 options, the probability is one in 32N. and for M creatures the chances are one in 3MN. Longer leucine and serine affiliated tRNA make the odds much worse since baseline numbers of permutations rise exponentially. This is another strong argument against coincidence. The conclusion must be similarity in sequence—defined by number of nucleobase changes between any pair—need not, at a degree of variance possibly as small as a single alteration, result in identical purposes. On the other hand, large amounts of variation, however large is quantified, may yield the same outcome or exhibit differential effects. Probability alone is inadequate for assessment; as in real estate, location (of changes) means everything with respect to value.
A second justification for matching sequences altered in exactly the same way from a reference is convergent evolution. As a philosophical principle of adaptation, its invocation automatically concedes a purpose for tRNA sequence variation above and beyond historical record or sheer coincidence. The only battle remaining is to assign the correct purpose(s). Referral to Table S11 is again made: twenty-nine of 631 tRNA held in common by forty mammals, with 602 sequences matched by two to thirty-nine animals. That represents a lot of convergent evolution definitely against the odds. Some of those 602 differ from the twenty-nine by a single nucleotide. For example, tRNALeu(CAG), 83 nt long, where the upper is found in all mammals investigated, while the lower is present in all but Mlu. From the general formula, odds (M = 39, N = 1) for deviation from the top tRNA is one in 339 ≈ one in 4.1×1018.
GUCAGGAUGGCCGAGCGGUCUAAGGCGCUGCGUUCAGGUCGCAGUCUCCCCUGGAGGCGUGGGUUCGAAUCCCACUCCUGACA
GUCAGGAUGGCCGAGCGGUCUAAGGCGCUGCGUUCAGGUCGCAGUCUCCCCUGGAGGCGUGGGUUCGAAUCCCACUUCUGACA
Identicality in sequence causing identicality in function is transparently logical, providing a firm ground for rational thought. Quasi-identicality throughout a string length implies a concomitant range of functional activity from absolutely identical to relatively similar. If divergence time between species is sufficiently narrow, the continuum scale of in vivo action tends more towards clone-like outcomes. When nucleotide sequences are dissimilar enough, they, nonetheless, could evoke similar biochemical endpoints if organisms are proximate physiologically and inhabit analogous geographical niches; should conditional criteria be unmet, the balance tips towards otherness in action. Large-scale deviance in tRNA leading to altered function is the logical converse of the first postulate, but causally neither necessary nor sufficient for guaranteeing a result. It remains to answer a crucial question: how much difference is enough to make a difference?
Returning to tRNALeu(CAG), and bearing in mind the hypothesized additional functions for tRNA, is it conceivable the single nucleobase change is assignable to participation in leucine modification in the intervening period between transcription and translation, or to involvement in co-translational folding of the peptide chain? Absolutely. C/U structural variance could lead to different points of contact between tRNA and rRNA, or between tRNA and ribosomal proteins, because C4 in cytidine has an NH2 attached able to act as both hydrogen bond donor or acceptor, whereas uridine C4 is engaged in a double bond to O capable of hydrogen bond acceptor status only. In addition, the C−N single bond (1.37Å) is longer than a C=O double bond (1.27Å), so it extends farther into the ribosomal space (Heyrovska 2008). Nothing here constitutes proof; it is all conjecture, but plausible, deserving of confirmation or rejection.
The influence of sequence is paramount for any discussion of tRNA function beyond its known role as amino acid conveyor to ribosomes. The other structural parameters (length, skewness, anticodon ambiguity) are relevant to disclosing full tRNA participation in translation. Table S3 covering preferred length distributions reveals predominant tRNA lengths are 72−73 nt for isoacceptors besides those leucine or serine related (82 nt). Exceptions include: 71 nt (tRNAGly(CCC), tRNAGly(GCC)); 74 nt (tRNAAsn(GUU), tRNAIle(AAU), tRNAIle(UAU), tRNAThr(AGU)); 83 nt (tRNALeu(CAG), tRNALeu(UAA)); 84 nt (tRNALeu(CAA)); 87 nt (tRNASec(UCA)). Long strings beyond 87 nt are accommodated by flexible variable arms; lengths less than 71 nt present challenges to cloverleaf construction (Sprinzl et al. 1998).
Sequences beyond those 71−87 nt limits should be viable unless proven otherwise. Hamashima et al. speculated that atypically long tRNA sequences housed in that variable arm, excluding leucine and serine isoacceptors, may not be aminoacylated at all, but serve other functional roles in all branches of Eukarya (Hamashima et al. 2015). This hypothesis begs for experimental approval or disapproval. An exploration of novel variable armed tRNA from bacterial genomes show Escherichia coli aminoacylation activity in some strings, but these authors conceded other sequences could possess otherwise unspecified nonaminoacylation functions (Mukai et al. 2017).
In this set of mammalian tRNA, forty-three sequences are shorter than 71 nt and six longer than 87 nt. Regardless of whether length distortions from that preferred for a given isoacceptor are less than, within, or beyond this length range, either they are of in vivo significance, meaning affect functionality, or represent irrelevant extravagances. Using resource material unnecessarily requires energy probably better employed for other situations, so the second choice is a priori less plausible. Lynch & Marinov engaged in an exhaustive analysis of energetic costs for Bacteria and Eukarya in cells undergoing gene replication, transcription, translation (Lynch and Marinov 2015). For tRNA, only the first two processes are pertinent. Using their calculations: (i) during M phase of a cell cycle, growth and maintenance energetic costs for a cell once formed are Escherichia coli << Saccharomyces cerevisiae << Mus musculus; (ii) during S phase of a cell cycle, transcription of mRNA after production engages energetic costs such that Escherichia coli < Saccharomyces cerevisiae < Arabidopsis thaliana; (iii) in absolute values, transcription costs exceed replication costs by at least one order of magnitude.
It is reasonable to accept energetic costs of mRNA would be sufficiently close to those for tRNA (nucleobase synthesis, processing into mature form, modifications as necessary) to render them roughly interchangeable, and those for Arabidopsis thaliana to adequately serve as proxy for Mus musculus in that both represent structurally complex Eukarya. Consequently, transcription of nonpreferred tRNA lengths for no legitimate purpose is inefficient. It is more rational to believe they topologically alter relationships among nucleotides, affecting folding ability into the conventional 3D shape. Spatial adjustments may also change potential contact points with other translation active biomaterials, affecting properties such as thermodynamic binding stability, kinetic translation rate, decomposition frequency.
Using a baseline of 71 nt established by glycine-related isoacceptors, deletions and insertions can be obtained from: (i) isodecoders by removal or addition of at least one nucleotide; (ii) other isoacceptors by nucleotide polymorphism at the anticodon triplet plus removal or addition of one or more nucleotides; (iii) foreign sources through horizontal gene transfer (Keeling and Palmer 2008). They are illustrated, where underlining indicates nucleobase mutations, and compiled in Table S14.
- Type (i) from Bac; tRNAArg(ACG) of 84 nt
isodecoder test
GGGCCAGUGGCGCAA UGGAUAACGCGUCUGACUACGGAUCAGAAGAUUCUAGGUUCGACUCCUGGCUGGCUCG
GGGCCAGUGGCGCAAUGGAUAACGCGUUGAUAACGCGUCUGACUACGGAUCAGAAGAGUCUAGGUUCGACUCCUGGCUGGCUCG
Addition constitutes an 11-nucleotide repetition of UGGAUAACGCG. From Tables S2 and S3, 73 nt is preferred in all forty mammals. It is conceivable this was an inadvertent sequencing or recording error by the original researchers. In the Results section, correction of a 98 nt tRNAPro(AGG) from Ggo was demonstrated to fall into this category. Using a cutoff of three or fewer base alterations from the reference sequence as distinguishing probable from possible, Table S14 offers fifteen probable and four possible tRNA outside the 71−87 nt range as belonging to this category (not including two mentioned in the text).
- Type (ii) from Fca; tRNAGln(UUG) of 85 nt
isodecoder test
GGCCCCAUGGUGUAA U GGUU AGCACUCUGGACUUUGAAUC CAGCGAUC CGA GUUCAAA UC UCGG UGGGACCU
GCC UGG GUGGCUCAGUCGGUUGAGCG UCU GACUUUGGCUCAGGUCA CGAUCUCGCGGUCCGUGGGUUCGAGCCCCACGUCGGGCU
isoacceptor test
GCCUGGGUGGCUCAGUCGGUUGAGCGUCUGACUUUGGCUCAGGUCACGAUCUCGCGGUCCGUGGGUUCGAGCCCCACGUCGGGCU
GCCUGGGUGGCUCAGUCGGUUGAGCGUCUGACUUCAGCUCAGGUCACGAUCUCGCGGUCCGUGGGUUCGAGCCCCACGUCGGGCU
Isodecoders of preferred length 72 nt for tRNAGln(UUG) differ by many nucleobases even with gaps introduced in both sequences (one sample comparison shown), but an isoacceptor gives a perfect match. According to Table S2, preferred length for tRNAsup(UCA) is species-dependent, although 85 nt in Fca. As shown in Table S14, three tRNA are assigned to this type, not counting that shown here.
- Type (iii) from Ame; tRNAArg(UCU) of 74 nt
isodecoder test
GGCUCUGUGGCGCAAUGGA UAGCGCAUUGGACUUCUAAUUCAAAGGUUGUGGGUUCGAGUCCCACCAGAGUCG
GUCUCUGUGGCGCAAUGGACGAGCGCGCUGGACUUCUAAU CCAGAGGUUCCGGGUUCGAGUCCCGGCAGAGAUG
isoacceptor test
GUCUCUGUGGCGCAAUGGACGAGCGCGCUGGACUUCUAAUCCAGAGGUUCCGGGUUCGAGUCCCGGCAGAGAUG
GUCUCUGUGGCGCAAUGGGUUAGCGCGUUCGGCUGUUAACUGAAAGGUUGGUGGUUCGAGCCCACCCAGGGACG
A gap space was added to maximize alignment in the isodecoder test because the preferred length for tRNAArg(UCU) is 73 nt. So many mutations were found in these comparison tests that the most likely source is external due to gene transfer. The feasibility of tRNA gene movement was discounted between Archaea and Mammals at the close of the Results section, leaving Bacteria or organelles (mitochondria) as suspects. Table S14 indicates twenty-six strings could not be matched by any sequence encoded.
Anticodon ambiguity was described as an intrinsic feature of some isoacceptors signifying their nondegeneracy. Encoding frequency varies from always (UUUU) to usually (CCUCCU) to occasionally (UAUAU) to rarely (GGGG), and leads to two related phenomena: ribosomal pausing and frameshifting. Pausing occurs when translation momentarily halts while mRNA/tRNA strands struggle to gain the proper relationship ensuring correctly attached amino acid in the ribosomal A site is joined to the peptide present in the P site. Frameshifting is a consequence of incorrectly spaced complementary base pairing.
An anticodon ambiguity consecutively extended one nucleotide leading to NaNaNaNa results in a corresponding ± 1 frameshift depending on whether the extra identical residue is located towards the 5’ or 3’ end. If ambiguity is palindromic of form NaNbNaNbNa then a ± 2 frameshift eventuates. A repeat of type NaNbNcNaNbNc (or permutations involving six nucleobases) does not technically create frameshifts since it is a multiple of three. Instead, it forces an aminoacylated tRNA to adopt a conformation contrary to that produced when the correct triplet is aligned. The question becomes whether shifted nucleotide triplets can still establish appropriate interactions with rRNA nucleotides of the large and small subunits (Harish and Caetano-Anollés 2012) or with neighboring ribosomal proteins (Wilson and Doudna Cate 2012).
Cognizing connections between anticodon ambiguity and ribosomal pausing permits an alternate explanation for data accumulated by Charneski & Hurst. Translation rate along Saccharomyces cerevisiae ribosomes decreases if positively charged arginine, histidine, lysine are found in (near) the Peptide Exit Tunnel because of, they speculated, electrostatic attraction to negative charges on phosphates in rRNA. Acknowledging histidine displayed weaker effects than the other two, differences were ascribed to pKa at physiological pH (Carneski and Hurst 2013).
Arginine codon AGG complements tRNAArg(CCU) encoded as CCUCCU 63.5% of the time among forty mammals and also present in the yeast genome as a unique tRNA string in a single occurrence. Repetition makes it difficult for mRNA to establish a correct pairing properly oriented to ensure other nucleotide interactions, slowing down processing. Similarly, mRNA codon AAA for lysine is partnered with tRNALys(UUU) always encoded as UUUU or UUUUU among studied mammals and as UUUU in the fungus. Again, searching for the right nucleotide positions induces temporary ribosomal pausing. Finally, histidine’s reduced effect is explained by matching mRNA codon CAC with tRNAHis(GUG). The yeast and mammal strings are almost palindromic: GUUGUG rather than GUGUG; in consequence, there is some decline in translation speed, but only a fraction caused by ambiguous anticodons in arginine and lysine, as observed.
Earlier, mention was made of a theoretical mutation process by which isoacceptors of both related and unrelated aminoacylated tRNA could be connected; for example, tRNATrp(CCA) → tRNACys(GCA) → tRNAGly(GCC) → tRNAAsp(GUC). String matching tests performed on the tRNA component for each animal’s genome gave results (Tables S8 and S9) supporting this thesis. Aside from some sort of directed process of mutation, the best alternative would be accumulated random errors during transcription (neutral drift). If alignments, discounting differences in anticodon triplet, between otherwise unrelated tRNA happened rarely, an error theory would be viable given a transcription mistake frequency of 10−4 or lower (Rozov et al. 2016). On that scale, a mammal in S phase of its cell cycle would need 27.4 years to accomplish one base change in the anticodon triplet: one mutation every 10,000 cell cycles, each cycle occurring once in twenty-four hours (for Hsa; Cooper and Adams 2022).
The problem is Tables S8 and S9 demonstrate the same alteration occurs in up to forty genomes. If all nucleotides outside the anticodon triplet remain perfectly matched, as the data insists they must be, the law of large numbers militates against fortuitous coincidence, even if anticodon mutations need not take place within a single 27.4 year period for every animal, but could extend over millions of years. In that time, other nucleobases would also be susceptible to the same neutral drift process, causing loss of alignment in other portions of tRNA molecules. Systemic mutation is a preferred explanation for these observations coupled with the notion that resultant sequence matched alignments are purposeful, so are maintained during evolution once they occur in each genome.
Mutations involve purine/purine, purine/pyrimidine, pyrimidine/pyrimidine for isoacceptor pairs, and may occur in any anticodon position, but mostly at the wobble base. Occasionally, a trio of strings are participants: tRNAPhe(GAA), tRNASer(GGA), tRNATrp(CCA) of 73 nt in Oar is an example; notice the length for the serine isoacceptor is unusual, meaning it must derive from phenylalanine tRNA via single nucleotide polymorphism. Existence of internally matched sequences does not typically reveal from which direction alteration occurred: tRNAPro(AGG) and tRNAPro(CGG) are perfectly aligned, except for anticodon triplet, in all forty animals, and tRNAPro(UGG) is a match for all but Ame. A forty-for-forty outcome also applies to the combination tRNAVal(AAC) and tRNAVal(CAC), with tRNAVal(UAC) a match in the Bta genome.
Although directionality is not ordinarily obvious, exceptions for which they could be elucidated have been cited already. In this respect, Vpa is a major contributor to unraveling the mysteries of systemic mutation. It transcribes 1436 unique tRNA, more than twice that in Laf (694), its closest rival (Table 2). Of these, 833 (58.0%) are tRNAAla(AGC) (Table S4) of every length 71−80 nt. This abundance does not appear for other isoacceptors in the genome, so its existence cannot be a consequence of a whole genome duplication event (Meyer and Schartl 1999). Another method to account for excessive accumulation is single gene multiplication. Supposedly, most duplicates are silenced after several million years (Lynch and Conery 2000). It is unknown whether the majority of these genes have been rendered nonfunctional in this manner.
Still another route justifying tRNA imbalance of this magnitude would require an exclusive gene transcription event occurring several times, but not shared by other tRNA genes, as if it were caught in a transcription loop repeat cycle. Reiterative transcription refers to a loop, but usually constitutes a process of repetitive single nucleotide addition, mainly A or U, to 3’ termini (Turnbough Jr. 2011). A distinction between gene duplication and gene reiterative transcription is clear-cut: in the first, a gene is duplicated X times, with each replicant transcribed once, leading to X copies; in the second, a single gene is transcribed X times. As envisioned, gene reiterative transcription would be analogous to chemical polymerization, as in conversion of ethylene to polyethylene. Of course, questions arise immediately: (i) what would be the genetic equivalent of a polymer initiation reagent? (ii) what would be the genetic equivalent of a polymer termination reagent?
A biochemically obvious reason why a species should contain this many isodecoder genes for a single isoacceptor is missing, regardless of the process by which it came to be. Could its natural habitat necessitate this response as a survival mechanism? Perhaps the surplus is intended to compensate for the animal being prone to deleterious, even lethal, mutations. The most recent sequencing of the Vpa genome acknowledged the presence of more recognizable protein coding sequences than is found in camel, cow, sheep (Richardson et al. 2019). However, this would fail to explain excessiveness of a single isoacceptor. Whatever the purpose might be, matched sequences within the genome reveal unusual agreements (Tables S8 and S9) absent in other mammals. Undoubtedly, mutation direction proceeded as AGC → GGC, AGC → AUC, AGC → AGG, AGC → AAC, respectively.
- 73 nt: tRNAAla(AGC) with tRNAAla(GGC)
- 75 nt: tRNAAla(AGC) with tRNAAla(GGC) (two pairs)
- 73 nt: tRNAAla(AGC) with tRNAAsp(AUC)
- 73 nt: tRNAAla(AGC) with tRNAPro(AGG)
- 73 nt: tRNAAla(AGC) with tRNAVal(AAC) (three pairs)
Of all tRNA isoacceptors, two stand out as unambiguously special because they possess activities irreplaceable by others: (i) tRNAiMet(CAU) is a key part of the Start codon/anticodon mRNA/tRNA couple; (ii) tRNACys(GCA) , and in some genomes tRNACys(ACA), because its attached cysteine is transformable into a dimer whose disulfide bonds have substantial effect on secondary protein structure. For tRNAiMet(CAU), a single isodecoder sequence is contained in all mammals, confirming its structural demand for special binding affinity with eukaryotic initiation factors (Table S11). Transfer RNACys(GCA) is encoded the most times, and represents the most unique sequences, for these animals, excluding tRNAAla(AGC) whose totals are inflated by Vpa (Table S4). As diametric opposite of initiator methionine tRNA singularity, cysteine tRNA multiplicity also suggests precise order and location of individual nucleotides are responsible for providing information; in this case, on which pair of cysteines engage in S−S bond formation.
Unsurprisingly, the consensus initiator methionine sequence lacks a duplicate in organism types other than vertebrates. Bacteria, Archaea, plants, fungi, invertebrates fail match tests (Table S13a), which supports the repetitive claim throughout this paper portraying them as poor models from which to draw conclusions about features concerning translational processes in mammals. Either tRNAiMet(CAU) strings matter or do not. If number and location of nucleotide changes lack significance, then no variable position can participate in rate critical interactions among tRNA, initiation factors, GTP within ternary complexes commencing translation. If relevant, they probably cause rate and/or thermodynamic stability differences during translation onset. Possible also, and not a mutually exclusive option, is that initiation factors for taxonomic groups (vertebrates, invertebrates, fungi, plants, prokaryotes) are structurally diverse in order to compensate for alterations in tRNA sequences (Lütcke et al. 1987; Gu et al. 2010). If no universal eukaryotic initiation factors exist, demarcation occurred around 500 million years ago during vertebrate and invertebrate separation. Employing nonvertebrates as models for features characteristic of translation related to vertebrate tRNA is like using tricycles to model jet airplanes because both have wheels.
Final protein conformations are frequently intimately connected to disulfide bond formation; 29% of all expressed proteins in Eukarya are said to contain cysteine-cysteine linkages (Narayan 2021). In addition, cysteine sulfur is readily oxidizable to sulfenic acid (RSOH), sulfinic acid (RSO2H), sulfonic acid (RSO3H), where R = HO2CCH(NH2)CH2− (remainder of amino acid). A survey of X-ray crystal structures in the Protein Data Bank turned up 1171 sulfenic acids, 469 sulfinic acids, 382 sulfonic acids (Ruiz et al. 2022). The authors issued a caveat: X-ray irradiation may promote oxidation, so molecules found might not all be biologically relevant. This tally does not include a plethora of derivatives, such as thioesters and thioamides (Poole 2015). The two most prominent oxidants are hydrogen peroxide and glutathione (Schulte et al. 2020), both abundantly present in cells. A diversity of tRNACys(GCA) sequences, and occasionally tRNACys(ACA), encoded in these animals should now be readily comprehensible.
It is hypothesized isoacceptors and their constituent isodecoders might not facilitate translation of the same amino acid. This proposition is contrary to conventional wisdom. How might nondegeneracy be justified as prima facie plausible beyond the structural impediments to redundancy already articulated? Again, tRNA sequence transcription errors are estimated to appear at a frequency of 10−5 to 10−4 (one event in 10,000−100,000 molecules) along with a similar misacylation rate (Rozov et al. 2016). Given a typical protein of 300−500 residues, the probability of introducing incorrect amino acids by either route is low.
Among twenty-nine tRNA perfect nucleotide match sequences appearing in all forty mammals (Table 5) is exactly one for tRNACys(GCA) despite more isodecoders (forty-three) held in common between two or more animals compared to every other isoacceptor. Of this total, six others are encoded in over twenty genomes and thirty-six are found in 2−6 species (Table S11). It is suggested the consensus string is used for unmodified cysteine not undergoing disulfide bond formation in any protein, while the remainder contain precise information signifying which pair of cysteines join together, or are converted into another type of modification for employment in select instances. At the other end of the spectrum is lysine, which appears twice among these twenty-nine: one for each isoacceptor and possessing the same length (73 nt). What can it mean in practical terms to claim codons AAG and AAA (synonymous with tRNALys(CUU) and tRNALys(UUU) respectively) fail to translate into the same lysine? Consensus sequences are shown with underlined residues denoting differences:
GCCCGGCUAGCUCAGUCGGUAGAGCAUGAGACUCUUAAUCUCAGGGUCGUGGGUUCGAGCCCCACGUUGGGCG
GCCCGGAUAGCUCAGUCGGUAGAGCAUCAGACUUUUAAUCUGAGGGUCCAGGGUUCAAGUCCCUGUUCGGGCG
Ideally, one would consider these as constituting a no frills option translating the canonical amino acid structure absent post-translational modifications. Notice: (i) identical beginning 5’ and ending 3’ sets of nucleotides containing the amino acid attachment stem; (ii) eleven changes (15%) in isolated clusters of 1−3 nucleobases; (iii) these mutations are of various types distributed unevenly throughout the strings, where some alterations are Watson-Crick complementary (CG, AU), some purine/pyrimidine (AC, GU), some purine/purine (AG), some pyrimidine/pyrimidine (CU).
The questions are obvious. If both strings translate the no frills option faithfully, then what do the diversity of mutations signify? If both strings translate the no frills option faithfully, then why do all forty animals encode both sequences, yet not also encode other isodecoders with such unanimity? No shortage exists (Table S11): there are fifteen other CUU versions shared by at least two mammals, and twenty-four more for UUU. Aside from one CUU isodecoder, none of the fourteen remaining are encoded in greater than seven species. Likewise, twenty-four UUU isodecoders are, except for two, not found in more than six animals. The variety of in vivo relevant modified lysines is extensive (Wang and Cole 2020), and it is at least plausible some may form while still in the ribosomal environment.
There still remains the binary option: either differences in sequence matter or do not. Can it be alleged variation is insignificant? Yes, but this postulate is contrary to an image of a refined evolutionary process where errors in transcription and/or translation—stages characterized as having a large number of moving parts engaged in a series of multiple steps—are very rare, as has been stipulated. A fundamental paradigm of Darwinian Evolution is: nature does everything for good reasons. Almost by definition, lethal or detrimental mutations are typically removed from genomes. Retained diversity in isoacceptor tRNA coding must serve meaningful purposes, must be important by design.
If this hypothesis is correct, remaining isodecoders shared nonunanimously indicate mutations in tRNA sequence are either (i) benign, producing the same amino acid or (ii) induce translation of amino acids in modified forms. Retention as a historical record of nucleotide mutation is reasonable. The second choice suggests isoacceptors and their subordinate isodecoders are not redundantly referencing the same amino acid. Where is the dividing line? How many nucleobase changes (mutations, not modifications) are sufficient to result in subsequent amino acid structural conversion?
It cannot be stated definitively due to a paucity of information; hence, a compelling incentive for experimental testing exists. Theoretically, single nucleotide mutation could lead to amino acid alterations ranging from simple (methylation) to complex (glycosylation) to esoteric (L → D stereoisomer change; Ehmsen et al. 2013). On the other hand, single nucleobase changes might be minor alterations due to transcriptional mistakes, rare as they appear to be, leading over time to neutral drift emendations. When the same sets of mutations are observed in phenotypically and phylogenetically divergent species, then base switching cannot be due to simple transcriptional laxity. It is reasonable to theorize that, if positioned in a crucial location, one nucleotide might signify assistance in producing a modified amino acid after aminoacylation has occurred. By argument extension, multiple bases could likewise assist in establishing structural changes eventually resulting in functional adjustments. A nonexhaustive catalog of conceivable transformations, besides those already mentioned, includes acetylation, hydroxylation, phosphorylation, ubiquinylation, methionine oxidation.
If it is still insisted there is no innate difficulty in affixing no frills labels to both tRNALys(CUU) and tRNALys(UUU), asserting they illustrate degenerate isoacceptors, what about those thirty-nine other strings unevenly distributed among species? It would demand a convoluted reasoning process to maintain a belief in unmitigated redundancy pertaining to all lysine isodecoders. If, however, it is conceded there is a two-fold division between the unanimous two and the remainder—in physics, this operation would be called a symmetry break—then a basis for distinction can only lie in admitting sequence is the decisive factor. In that case, there must be additional divisions, other symmetry breaking operations, involving thirty-nine strings: species ABCDE possess one sequence in common, but only ADF share another isodecoder.
It would appear there is a means to circumvent this reasoning chain. Instances of evolution-based intentional diversity among isodecoders appeared as early as 2006 when Dittmar et al. suggested they permitted translational control of product proteins beyond that presented through the dictates of mRNA sequences alone (Dittmar et al. 2006). This paper’s title announced Tissue-specific Differences in Human Transfer RNA Expression. By this account, functional differences between isodecoders—where this term appeared for the first time—indicated anatomical locations of operation, but made no mention of a lack of isoacceptor redundancy. To these authors. all tRNALys(CUU) and tRNALys(UUU) encoded the no frills option (canonically structured lysine), but each isodecoder had predominant expression and utilization in diverse organs. This proposition was later confirmed in a mouse study: eighty-six of 210 (41%) isodecoder tRNA showed tissue-dependent expression variance among cortex, cerebellum, medulla oblongata, spinal cord, heart, liver, tibialis (Pinkard et al. 2020).
This sophisticated explanation of functional distinctions does not circumvent the possibility here imagined: not all lysines, to continue the example, are canonical structures because chemical adjustments contribute substantively to those tissue-specific expressions. The explanation offered by Dittmar et al. and Pinkard et al. is not mutually exclusive to the current suggestion. According to a database of amino acid modifications (dbPTM; Li et al. 2022), cysteine and lysine undergo more change types in vivo than other amino acids. Perhaps Geslain & Pan were unwittingly alluding to amino acid structural alterations when positing that different tRNA sequences led to functional distinctions in aminoacylation efficiency as well as ribosomal binding during translation (Geslain and Pan 2010).
Source dbPTM is specifically denominated post-translational modifications, but no justification exists for the view they must be so constrained. It is now commonplace to explore co-translational folding when it was once believed all protein secondary and tertiary conformational structural changes transpired after a molecule was assembled. Nonetheless, post-translational folding is still mostly true (Ellgaard et al. 2016): folding time for a fifty kilodalton protein takes 15−30× as long as synthesis (30−60 min vs. 2 min).
Analogously, pre- or co-translational amino acid modifications could be viable developmental stages. Pre means during or after tRNA aminoacylation, whereby nucleotides assist in formulating compositionally altered amino acids; co signifies once canonical aminoacylated tRNA are engaged in association with the A site of the ribosome.
Mechanistically, the pre version of amino acid modification begins with synthetases responsible for initial aminoacylation. After transcription in the nucleus, tRNA are exported to the cytoplasm for first order amino acid binding catalyzed by synthetases. Amino acids, bound or unbound, might be structurally altered while in the cytoplasm prior to movement towards ribosomes and initiation of translation because the medium contains molecules able to serve as reagents and catalysts (Luby-Phelps 1999) assisted by nucleotides in tRNA acting as interaction partners to bind those needed. Also possible is aminoacylated tRNA being returned (cytoplasm → nucleus) for adjustments necessitated by encounters with stresses of diverse types (Avcilar-Kucukgoze and Kashina 2020).
If no structural changes are warranted prior to beginning protein construction, then the alternate pathway for amino acid modification (co version) is available. Once the tRNA is base paired with mRNA and the ternary complex formed with cofactors and GTP, there is room to access amino acid sidechains by modifying reagents present near the ribosome because the attachment point is orthogonal to the codon/anticodon interaction position. As in the pre version, specific nucleotides would be involved, only this time by connecting with rRNA or ribosomal proteins acting as catalysts for the conversion. Although no evidence points to an in vivo function for ribosomal proteins beyond stabilization of rRNA comprising ribosomal subunits (Draper and Reynaldo 1999), tRNA might develop transitory associations through: (i) positively charged amino acids in nearby proteins ionically bound to negatively charged/phosphate within tRNA; (ii) protein hydrophilic amino acids or rRNA nucleotides hydrogen bonding to ribose 2’-OH in tRNA; (iii) direct hydrogen bond formation between hydrophilic amino acids and tRNA nucleotides; (iv) aromatic ring pi bond stacking between aromatic amino acids and nucleotide bases.
Amino acid acceptor stem and TΨC loop interact with the large subunit, in contrast to anticodon and D loop connections to the small subunit (Harish and Caetano-Anollés 2012). Transfer RNA contacts with ribosomal proteins while in the A site include uS12, uS13, uL16, plus uS9, uL5, uL16 when in the P site (Timsit et al. 2021). This list omits potential Eukarya-specific ribosomal protein participation. These agents would be likely, not necessarily sole, candidates for incipient catalysis of modified amino acid generation. Postulating points of contact between tRNA nucleotides and amino acids present in ribosomal proteins, or between rRNA expansion segments and tRNA, introduces meaning for finding isodecoders of different lengths related to the same anticodon triplet within each genome (Tables S2 and S3).
Length variance creates positional adjustments by altering topological relationships: tRNAAla(AGC) strings of 72 nt do not permit nucleobases to occupy the same space in its ribosomal environment as do nucleotides with 73 nt tRNAAla(AGC). Although the adjustment in nanometers is minor within the absolute confines of an L-shaped molecule, a one base movement relative to spatially immobilized amino acids in ribosomal proteins, or to the subunits themselves, could alter potential interactions allowing modifications within tRNA-bound residues. If 72 nt → 73 nt extensions might be significant, imagine the consequences for tRNAAla(AGC) strings whose length varies from 69 nt (Dor) to 80 nt (Vpa), a range different from those encoded by allegedly degenerate isoacceptors tRNAAla(CGC) and tRNAAla(UGC) (Table S2). The data shows the same argument is applicable to all other isoacceptors regardless of associated amino acid.
Transfer RNA: (i) of different lengths per isoacceptor cannot all refer to canonical amino acids without structural modifications; (ii) of a single length having matching isodecoder sequences, but not present in all species, cannot all refer to canonical amino acids without structural modifications; (iii) of a single length with matching isodecoder strings in all species could refer to canonical amino acids without structural modifications if it represents the only such set for that isoacceptor; (iv) of a single length with matching isodecoder sequences in all species and encoding the most identical copies probably refers to a canonical amino acid without structural modifications.
A second speculation regarding tRNA roles in translation concerns influencing folding patterns as proteins are constructed. A prime example has been declared: secondary structural organization through cysteine dimerization. If many cysteines are present, S−S bond construction could start before the entire molecule is synthesized to minimize likelihood of improper linkages distorting eventual molecular shapes and impeding intended activity. Cysteine dimerization has been found to take place as just described for bovine protein γB-crystallin. From its crystal structure, Cys18, Cys22, Cys78 are observed to be in close proximity, with four more cysteines present (Cys15, Cys32, Cys41, Cys109). Measuring translation speed for variable length partial primary sequences indicated folding took place within the Ribosomal Exit Tunnel amid S−S bond formation between Cys18 and Cys22, possibly through glutathione assistance (Schulte et al. 2020).
Tunnel dimensions are estimated at 100Å long plus 10−20Å wide (Wilson and Beckmann 2011). Approximately thirty residues are required to span the distance, and growing chains are visible when their lengths extend 60Å from a tRNA molecule situated in the P site. Given spatial restrictions, both disulfide bond-forming cysteines would need to be within that thirty residue limit if reaction were to take place before departure from the Exit Tunnel, as occurs in γB-crystallin, while still being subject to influence by tRNA. Interaction of cysteines with tRNA portions (nucleobase, ribose, phosphate), which need not be the same component for both, might be sufficient to bring them into close proximity enabling covalent bond construction.
Codons are known to vary in translation speed (Zouridis and Hatzimanikatis 2008; Buhr et al. 2016), and rate variance to depend more on precise isodecoder sequences than copy number counts (Zhou et al. 2016). Assumptions have been made that rate is inversely proportional to available co-translational folding time (O’Brien et al. 2014). Tunnel geometry offers sufficient space to initiate α-helix formation (Bhushan et al. 2010; Nilsson et al. 2015) as well as β-sheets (Marino et al. 2016). Protein folding could begin before reaching the Exit Tunnel, starting as early as when a segment arrives in the Peptidyl Transfer Center, if the needed amino acids are assembled and size of the PTC permits.
None of the research on co-translational folding within the Exit Tunnel makes mention of tRNA inclusion in the procedure, but has not been investigated, so is an open possibility awaiting study. As with proposed amino acid modification, it is conceived isodecoder tRNA in ribosomal A and P sites participate in folding efforts through interaction with ribosomal proteins and rRNA extension segments. Implicated tRNA must remain available before disappearing through the E site. If it could be evaluated, isodecoder residence time should correlate with situational chain folding events. Differential contact between tRNA and ribosomal proteins or rRNA explains why isodecoders do not vertically align even if a single length, justifying observations of skewness among strings, and certainly rationalizes sequence length differences.
Unicellular species display linear correlations between codon usage and tRNA abundance, but a connection between these variables is uncertain for Eukarya (Novoa et al. 2012). Most archaeal species encode one isodecoder per isoacceptor (Laibelman 2022) since multiple copies are pertinent to only 5.7% of total sequences; therefore, an inherent relationship between tRNA abundance and codon usage will be uncovered. A fundamental reason for the uncertainty within Eukarya is now capable of elucidation. There are two ways to assess copy number: (i) by reliance on exact multiple copies only; (ii) by including all isodecoders of variable length. The distinction is not solely with respect to number, but to assumptions underlying each method. Counting by (i) adheres to the rationale that like structure implies like function is an ironclad rule; following (ii) presumes functional degeneracy is maintained despite variable sequence structure and length.
Associated with the issue of codon interchangeability is the theme of codon usage bias. These two concepts are oppositional, for bias between isoacceptors implies absence of degeneracy: some aspect or feature endemic to these sequences must end the tie implied by a concept of redundancy. It was referred to earlier as a symmetry-breaking operation. The voluminous literature on codon usage bias absorbed into the HiveCuts database (https://hive.biochemistry.gwu.edu/cuts/) provides a distorted portrayal of features and significance by failing to accommodate agreement between mRNA activity and tRNA availability. Assessing statistical use of codons in isolation is like calculating the speed of a race car without taking into account identity and personal abilities of the driver: it can be done but is a flawed indicator of actual performance.
Codon usage bias intrinsically implies anticodon usage bias, an idea consistent with the notion of nondegeneracy exhibited through differential properties such as tRNA sequence and length. If preferred length for isoacceptors captures the set of isodecoders transcribed most often, optimal codon designations depend on usage for a particular assigned task, such as selective tissue distribution based on isodecoder. Colloquially, this is known as the right tool for the right job. As admitted by Hanson & Coller, difficulty in parsing effects of codon bias on translation efficiency derives from additional factors influencing codon usage (Hanson and Coller 2018).
Since the first introduction of the notion of isodecoders in 2006, the question arose as to whether mammalian tRNA variation revealed distinct functions differentially expressed, or reflected actual genetic redundancy, appearances notwithstanding (Pinkard et al. 2020). If the endorsed propositions are proven experimentally to have merit, then the first alternative applies without reservation. However, the case of Vpa raises real questions about scope because the isodecoder number for tRNAAla(AGC) strains credulity in conceptualizing nonduplicative functions for 833 unique sequences covering every length from 71−80 nt. There is certainly a limit, well below 833, how many ways alanine may be modified, or tRNA molecules affect protein conformation. Tissue expression variety cannot conceivably warrant forming a comparable magnitude of strings, because if that were the case, one would expect to see a similar augmentation of isodecoders for many isoacceptors in genomes of every animal. Since this isoacceptor alone suffers from such an extreme isodecoder imbalance in Vpa, it seems preferable to concede they record a history of evolutionarilyy benign mutations, with a majority illustrating redundancy in functionality, if not completely silenced.
Although tRNAAla(AGC) from Vpa is the greatest outlier in number of isodecoders per isoacceptor, it does not stand completely alone. Table S4 reveals twenty-eight isoacceptors encode over thirty unique isodecoders. Among them are five species with tRNALys(CUU) (Eeu 96, Cgr 43, Cfa 34, Fca 32, Mpu 32) plus four with tRNAGlu(UUC) (Tma 37, Ssc 36, Oar 33, Ttr 33). No evidence exists this pair’s possession of mirror-image anticodon triplets is anything other than coincidence, but one translating a basic amino acid and the other an acidic counterpart may be more than circumstantial if strings represent amino acids potentially modifiable. As with Vpa and alanine tRNA, the exorbitant tally for Eeu with lysine tRNA of multiple lengths also suggests most are probably in the benign mutation category, since selective tissue expression seems unable in principle to fully account for this isodecoder magnitude. The same can be said for tRNAGly(UCC) from Tma encoded as fifty-five unique strings in two lengths. Perhaps hypothesis of a transcriptional loop repeat cycle is not far-fetched, although lacking precedent and leading to questions: (i) how does a loop form mechanistically? (ii) how does a transcription event enter into, and exit from, a loop? (iii) what makes only some isoacceptors prone to gene reiterative transcription?
The most conservative conclusion is: unique isodecoders within isoacceptors create opportunities for: (i) enabling amino acid modification; (ii) affecting secondary structural features of growing proteins; (iii) targeting tissue expression selectively; (iv) recording a history of mutations with no tangible overt effects on functional diversity. The first three expand tRNA use beyond passive amino acid transporter: the biological equivalent of tugboats leading ships to port. The final response to the either/or queried by Pinkard et al. is both/and.
All emphasis so far has been on unique tRNA characteristics and their implications for aspects of translation. The flip side is to ponder the meaning of encoding exact duplicates in multiple copies. Table 2 reveals percentages of unique tRNA from 42.1% (Mdo) to 85.4% (Vpa), with a forty mammal average of 63.1%, not including suppressor and undetermined anticodon tRNA. Alternately stated, exact copy tRNA range from 14.6% to 57.9%, with a mean of 36.9%. Why so many identical copies? The statistic might be comprehensible if just a few cells in each tissue type were designated to produce tRNA capable of making proteins for that body part. However, since every cell in animal bodies is responsible for generating tRNA during a growth cycle S phase, massive perfect duplication—as few as 1−2 per isoacceptor per mammal, but as many as 15−20—is a mystery. Undoubtedly, repetition reduces risks from adverse chance mutation or potential misacylation leading to losses of protein product. However, isoacceptors do not generate similar numbers of identical sequences across-the-board, as Table 3 demonstrates in the aggregate, and Table S4 proves for individuals. Why do some isoacceptors (isodecoders) favor exact nucleotide matches in the form of cloned copies more than others within a given genome?
Mature mammalian tRNA possess an estimated half-life of around one hundred hours (Choe and Taylor 1972), or by a second report, thirty-six hours in resting cells and sixty hours during growth stages (Abelson et al. 1974). Protein synthesis occurs at an average of 2–5 peptide bonds per second, or roughly the speed for aminoacylated tRNA turnover at the A site (Pan 2018). Competitive nonspecific binding to ribosomal A site is rate limiting in each elongation cycle for every codon (Zouridis and Hatzimanikatis 2008). Binding affinity for isoacceptors in the A site varies with sequence and sidechain constitution of amino acid attached in Escherichia coli (Dale et al. 2009), which should be true for mammals, though not with the same numerical values given differences in sequences available.
If it takes approximately twenty-five seconds for a eukaryotic ribosome to synthesize a protein of 100 amino acids (Nilsson et al. 2015) and a typical tRNA t1/2 = 60 hr (taking the mid-value among three reported), then one molecule could be used for 4 AA/s × 3600 s/hr × 60 hr = 8.64×105 synthesis steps before 50% become unusable if the protein consisted of a single amino acid and the same isoacceptor (isodecoder) is circulated for every step. Since no such polypeptide exists, tRNA are used substantially fewer times before 50% are no longer functional. Multiple copy number variance among isoacceptors does correlate with usage in all domains (Santos and Del-Bem 2023).
Does it explain why some isodecoders demand more exact copies be produced? Yes, if isodecoder differences indicate unequal numbers and types of amino acid modifications because some alterations are needed to fulfill biochemical actions more frequently than others (phosphorylation vs. hydroxylation for example). Consequences of amino acid modification include adjustments to enzymatic catalysis kinetics, nonenzymatic protein thermodynamic stability, solubility, folding dynamics, cellular localization, thereby impacting signal transduction, gene expression, DNA repair, cell cycle control occurring in the nucleus, cytoplasm, Golgi apparatus, endoplasmic reticulum (Ramazi and Zahiri 2021).
This idea also justifies perceived correlation between high expression genes and tRNA abundance availability (Du et al. 2017). Sequence-dependent decay rates qualitatively explain differences in both number of exact multiple copies and which isodecoders are most often copied: each animal has different protein synthesis requirements for tissue-specific activity coupled to physiologically distinct homeostatic conditions affecting t1/2 in singular ways. Whether this entire construct affords a quantitative accounting for genome-specific tRNA encoding patterns demands experimentation on factors such as half-life as a function of sequence.
The simple fact tRNA have a half-life means they are intrinsically biochemically unstable in a host environment. Since five half-lives reduce effective concentration to ~3.1% of transcribed amounts in a normal cell, what makes diminished amounts of these molecules no longer functionally capable, and what happens to them upon reaching that state? Loss of tRNA does not harm an organism as long as cell cycle S phase time is shorter than t1/2 . To render them devoid of use implies they must become no longer able to either: (i) undergo aminoacylation; (ii) migrate to ribosomes; (iii) bind mRNA, translation factors and/or GTP. Efficiency suggests halting at the first stage is easiest because least complicated mechanistically.
Methodological clues were perhaps disclosed in a compendium of 111 post-transcriptional tRNA modifications (McCown et al. 2020) averaging thirteen per gene (Pinkard et al. 2020) in studied species from all three domains. Modifications can: (i) improve binding interactions with ribosomal subunits; (ii) reduce misreading during aminoacylation; (iii) enhance (or diminish) responses to environmental stress; (iv) lessen chance of frameshifting in the A site (Chan et al. 2010). They are found on ribose, nucleobase, or both at once.
According to McCown et al., ribose modifications are of two kinds: (i) methylation to produce 2’-OCH3, possibly with concurrent alteration on nucleobases; (ii) condensation of 2’-OH with C1-OH of ribose-5-phosphate, and accompanied by loss of a water molecule, to form a 2’-O-(1-ribosyl-5-phosphate) ether linkage. Type (ii) alterations have been observed only for purines in eukaryl tRNA at position 64 of the TΨC stem loop. Structural adjustments to nucleobases are more variable in location and type than on ribose, and subdivided into attachments to ring C or alterations on heteroatoms N and O. Conversions on carbon are typically small substituent adjustments such as methylation, thiomethylation, or changing of carbonyl (C=O) to thionyl (C=S), but more extensive transformations are known. Modifications to N or O are more diverse in composition, size, ring position. If sterically small (methylation), they often eliminate hydrogen bond capabilities. Some transformations occupy comparatively large spatial volumes, extending hydrogen bond opportunities, as either donor or acceptor, over greater distances than untouched purines or pyrimidines.
Applied to the problem of rendering tRNA incapable of aminoacylation suggests these modalities should be considered: (i) removal of the amino acid attachment nucleotide at the 3’ terminus, possibly via a phosphatase cleaving 3’-pN; (ii) blockage of terminal ribose 2’-OH and 3’-OH, denying synthetases the ability to catalyze reaction, via agents conducive to methylation, acetylation, 2’,3’-cyclic phosphodiester formation. The proposed avenues for inactivation are, at this time, hypothetical, but there are precedents for each route. Use of a phosphatase-like enzyme is accomplished during tRNA processing (Schwer et al. 2008). Formation of ribose 2’-OCH3 is a standard nucleotide modification as described, and there seems little reason to reject those same methyltransferases as enabling 3’-OCH3 synthesis. Twin acetylations can be performed using acetyl-CoA (Polevoda and Sherman 2002). Cyclic phosphate production is readily attainable (Yoshihisa 2014); enzymes catalyzing cyclic phosphodiester transformation on other kinds of substrates are known (Honda et al. 2016).
If the proposed approaches are shown to be valid, then the first question—what makes diminished amounts of these molecules no longer functionally capable—has been answered, though of course, many details still need development. As for the second query—what happens to them upon reaching that state—perhaps they are degraded into fragments upon reaching minimal concentration thresholds, and blocking their aminoacylation capabilities by an expressed route makes chemical destruction easier to clear them from the body. Fragments are known in various lengths, most commonly by dissection into two nearly equal pieces adjacent to the anticodon triplet. However, these are said to be produced by angiogenin as a response to environmental stresses (Keam & Hutvagner 2015), not fractures due to translational usage over a molecular lifetime. Still, angiogenin or analogs might participate if diminished tRNA capacity is perceived internally as a stressful situation onset.
Other tRF pieces are created post-transcriptionally before or after processing into mature, ready for aminoacylation, tRNA. In such cases, cuts are made in the D loop, TΨC loop, or past the 3’ terminus to generate fragments reportedly inducing primarily pathological physiological effects, at least in humans (Kumar et al. 2016). This, naturally, is additional motivation to remove them from the body. Although processing-related tRNA partition is not precisely on point, it does serve to establish possible mechanisms for destruction when tRNA have literally outlived their serviceable lifetime.
Should any of these pathways for disempowerment, degradation, elimination be demonstrable, they have the virtue of justifying encoding of multiple exact copies of isoacceptors to different extents in general, and explaining variable exact copy numbers for each isodecoder specifically. Precise sequences would determine translational usage frequency, kinetic half-life decay rates, mode of decomposition, ease of clearance from the body when inactivated.
The survey of archaeal tRNA (Laibelman 2022) offered an extensive analytical logical chain to justify the view that CCA 3’ termini are unnecessary despite numerous contrary assertions in print. It is well to remember the thesis of CCA as de rigeur derives from studies in the 1970s on aminoacylation of Escherichia coli. Given expressed reasons why unicellular organisms are inapplicable models for research on processes pertinent to multicellular mammals, a CCA debate provides fodder for this conclusion. Each of this particular bacterium’s fifty-two unique tRNA sequences (strain K12) terminate in CCA. In a literal sense, it is natural that aminoacylation demands this 3’ ending in order to be effective.
At a time when few genomes had been completely sequenced, it was common to read statements about rarity, or complete absence, of 3’-CCA in archaea and eukarya (Xiong et al. 2003), which seemed to explain why ATP:CTP nucleotidyltransferase was encoded, namely to force inclusion. By 2016, Ardell & Hou acknowledged forty-seven Archaea possessed 2140 tRNA sequences with encoded 3’-CCA in 650 (30.4%) of them (Ardell and Hou 2016). The aforementioned survey increased these totals substantially: 186 Archaea incorporated 3’-CCA 2031 times out of 4658 unique sequences (43.6%).
What does this recounting to do with mammalian tRNA? Forty mammals in possession of 12,283 unique tRNA strings (Table 2) encode 2670 (21.7%) with 3’-CCA (Table S15). Although inflated by Vpa (1069 CCA-containing sequences), removing them leaves one in seven (1601 of 11214, 14.3%). If these species yield 3’-CCA with sizable frequency, why should it be necessary for post-transcriptional addition to those tRNA that do not? Logic in demanding a dual pathway seems absent. Alternatively, 3’-CCA is not needed for aminoacylation in an absolute sense, and the function of nucleotidyltransferase is to repair defective tRNA. The enzyme has been proposed to serve exactly this function in Escherichia coli, where no need for automatic nucleotide addition exists, yet the enzyme is found in its DNA (Kim et al. 2009).
In Archaea, crystal structure evidence led to classifying nucleotidyltransferases from this domain as a Class I enzyme in contrast to Class II reserved for bacterial and eukaryl versions (Xiong et al. 2003). This is odd, since Bacteria are segregated from Archaea/Eukarya evolutionarily. Justification presumed gene transfer across Class II species, although absent experimental evidence. Even if a horizontal gene transfer thesis is correct, activity differences between bacterial and human types suffices to show they underwent structural and functional divergence since any theoretical time of transfer (Lizano et al. 2008). It can no longer be assumed humans, or any vertebrates, employ it to invariably add 3’-CCA.
All investigatory work on eukaryl tRNA with respect to aminoacylation began in the mid-1970s, and the role of nucleotidyltransferase is based on Saccharomyces cerevisiae. Could it be an indicator of direction for exploration in mammals? Absolutely. Could it serve as a sufficient symbol of structure and function? Absolutely not. With respect to necessity for 3’-CCA, the earliest work was that by Aebi et al. They claimed Eukarya did not encode it, so must be added. Essentiality was grounded on Escherichia coli data and assertions of it being a prerequisite for all domains. Although little evidence existed linking CCA nucleotidyltransferase to repair activity in eukaryotes at that time, it was presumed these molecules also fulfilled this role (Aebi et al. 1990).
A suggestion of ATP:CTP nucleotidyltransferase serving a repair function in Eukarya (Dupasquier et al. 2008) was made again by extrapolation without accompanying proof; their key phrase was “likely shared” with that assigned role in Escherichia coli. Bacterial and human versions display different kinetic binding ability: bacterial enzyme KM = 30 μM (CTP), 330 μM (ATP); human enzyme KM = 20 μM (CTP), 605 μM (ATP) (Lizano et al. 2008). Binding for ATP is 10−30× worse than CTP, bringing into question its utility, at concentrations used, for adenosine addition. Lizano et al. conceded the human form could exist as CC additive alone under certain conditions.
Bacterial, archaeal, fungal nucleotidyltransferase enzyme data are inadequate for elucidating fine details in mammals. Kinetic outcomes obtained in vitro are not precise descriptors for what occurs in vivo (Rudorf et al. 2014). Rate information depends on buffer composition, but cytosolic media contain agents excluded from formally designed laboratory experiments (Luby-Phelps 1999). Invasive explorations on living animals raise profound ethical issues, so simple organisms cannot be avoided, but interpretation of outcomes should not readily be extrapolated beyond their proper arena.
The same query regarding necessity for CCA addition to the 3’ terminus of every eukaryl tRNA could be asked about the demand for G−1 addition to the 5’ end for all eukaryl tRNAHis(GUG). If encoded in some entities (Bacteria and many Archaea), why not all needing it? Why an elaborate process of removal of a 5’ leading sequence from pre-tRNA followed by a multistep enzyme-catalyzed G−1 supplement to the residual string (Jackman and Phizicky 2006)? Is it possible G−1 is not critical for aminoacylation?
Abad et al. committed the same mistake almost universally made when transferring conditions in yeast to species evolutionarily divergent from it by over one billion years. They first allege conservation of existence of tRNAHis guanyltransferase (Thg 1), the enzyme catalyzing G−1 addition in Archaea, and then subject that datum to an inference of conservation of both sequence and function through the phrase “suggests similar mode of” (Abad et al. 2010). Yet, research on Methanopyrus kandleri, Methanosarcina acetivorans, Methanosarcina barkeri, Methanothermobacter thermautotrophicus archaeal Thg 1 led to recognition of distinctions in activity between archaeal and yeast enzyme forms; hence, “similar mode” and not same mode. Another instance: post-transcriptional addition of G−1 in Schizosaccharomyces pombe and Drosophila melanogaster becomes “by inference possibly in other eukaryotes as well” (Francklyn and Schimmel 1990). This ongoing diatribe by explicit quotation is not intended to single out individuals for criticism, but to demonstrate a pattern of logically impermissible thought.
Human Thg 1 has 52% sequence identity with the Saccharomyces cerevisiae enzyme (Hyde et al. 2010), whereas archaeal and yeast types average 15% identicality (Heinemann et al. 2012). Conceptually, homology is a sliding scale trait, and need not imply similar purpose unless there exists a high degree of it, especially for key residues. Does 15 % satisfy the standard? Does 52%? Discovery of a human Thg 1 analog neither demands a presence in other mammals nor an identical application, especially when the human version has been crystallized, but tRNAHis(GUG) not included (Heinemann et al. 2012), so a known binding site is missing. Further confounding its potential role in vertebrates is a lack of homology to other enzyme classes, and a mode of operation (3’ → 5’), based on bacterial and archaeal versions, directionally opposite generic nucleotide polymerases (5’ → 3’). Hyde et al. stipulated a human enzyme knockout test in vitro resulted in “cell cycle progression growth defects.” This observation does not bear directly on the role, if any, of G−1 on aminoacylation of tRNAHis(GUG) by its synthetase.
Even if tRNA guanyltransferase is discovered to have the claimed essential function in mammals as demonstrated for prokaryotes and fungi, the question is why tRNAHis(GUG) is unlike other isoacceptors in being the sole molecule demanding G−1 addition for aminoacylation? According to Himeno et al., the corresponding synthetase is the only enzyme missing a sequence consisting of amino acids H(I/L)GH. By inserting G nucleotide at the head of the amino acid acceptor stem, these researchers suggested extra base pair G−1/C73 enables the conformation of terminal CCA needed for aminoacylation (Himeno et al. 1989). Their work was confined to Escherichia coli, where 3’-CCA is omnipresent.
Tables S4 and S5 indicate Laf, Mpu, Oan, Vpa encode tRNAHis(AUG) and it is unknown whether this isoacceptor incorporates the same behaviors allocated to tRNAHis(GUG). If there is a single synthetase for both isoacceptors, they should display identical urgency for G−1 addition. Among forty mammals, tRNAHis(GUG) encodes 3’-GCA in every shared sequence (Table S11), but tRNAHis(AUG) contains 3’-UUA or UCA (Laf), UCG (Oan, Mpu). CCA (Vpa) (Table S5). A clear mutation pattern exists; bidirectional arrows signify uncertainty in cause and effect, which highlights a further urgency for an investigation of the isoacceptor on this unsettled topic:
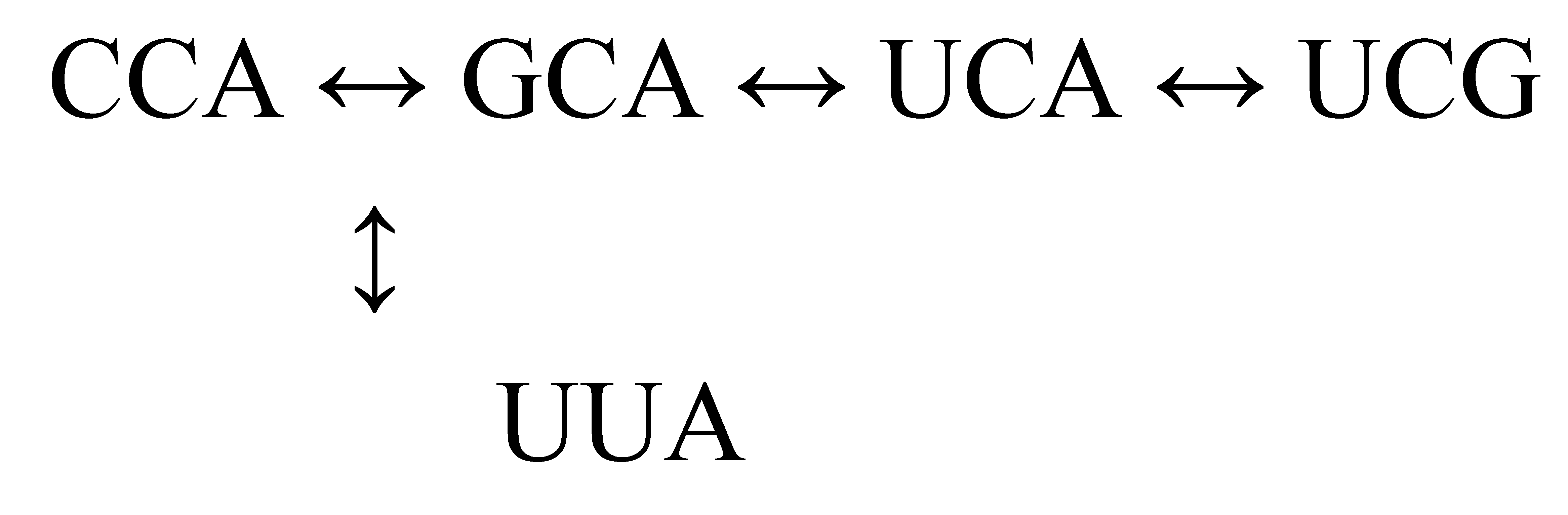
If mammals have no explicit need for 3’-CCA, then an urgency for G−1 is lessened considerably. Subsequent to the Himeno et al. publication, synthetases were divided into classes I a−c and II a−c, with histidine synthetase assigned to class IIa. Categorization was based on sequence and folding patterns. Only class I synthetases contain an H(I/L)GH signature (O’Donoghue and Luthey-Schulten 2003). The earlier supposition of histidine synthetase uniqueness as justification for G−1 insertion into associated tRNA cannot be maintained. At present, evidence for G−1 addition for mammals, and possibly vertebrates generally, is weak, and a solid justification altogether absent.
The close of the paper exploring archaeal tRNA offered a suggestion that certain themes needed review and revision in light of the research findings articulated in its pages and accompanying data tables (Laibelman 2022). That request is reiterated because this compilation of information and logical analyses raises numerous questions. There is an unfulfilled need for experimental studies designed to find evidence in support of, or rejecting, many enclosed speculations.
Mammals are not fungi is not intended to be comical, but taken seriously as warning. Geneticists must be responsibly conservative in oral and written communications, utilizing absolutes such as always, never, conserved, universal with extreme reluctance. There are some disciplines in which inductive logic is appropriate. In synthetic organic chemistry, inference is not merely useful, it is the foundation: because it can be inferred every primary alcohol is oxidizable in stages to aldehydes and then to carboxylic acids (RCH2OH → RCHO → RCO2H), transformations may be performed regardless of substrate, hence R represents every collection of aliphatic or aromatic carbon-based substitution patterns. Presuppositions of transferability for translational processes in the three domains as acceptable because all organisms contain ribosomes, mRNA, tRNA, relevant cofactors is akin to claiming cell cycle growth phases may be applied to Archaea, Bacteria, Eukarya without loss of generality. The fact is, phase times vary for each, and fine details become issues of life and death (Lynch and Marinov 2015).
Induction is not acceptable in genetics; the operative guideline must be it-takes-one-to-know-one. To research genetics of mammals, study mammals, or at least vertebrates. Legal, philosophical, technical constraints preclude in vivo work, so in vitro efforts on species such as Gallus gallus or Xenopus laevis will have to suffice. Still, that is scientifically preferable to, and results are more reliable than, any study on Saccharomyces cerevisiae. Fungi, or a bacterium like Escherichia coli, may be useful signposts for what might be worth exploring in other lifeforms, but they are not, and will never be, either necessary or sufficient if the goal is to gain knowledge about those other entities.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1. Pairwise Species Divergence Times. Table S2. tRNA Lengths. Table S3. Distribution of tRNA Preferred Lengths by Isoacceptor. Table S4. Ratio of Total to Unique tRNA Sequences by Isoacceptor. Table S5. Unique tRNA Sequences. Table S6. First Position Mutations in Isodecoders. Table S7. Potential Sequence Corrections. Table S8. Identical tRNA from Internal Matching. Table S9. Summary of Internal Matching tRNA Sequences. Table S10. Potential Undet (NNN) Assignments. Table S11. Interspecies Comparison of Unique tRNA Sequences. Table S12. Evaluation of Nucleotide Deviations. Table S13. tRNA Comparison among Mammals and Model Organisms. Table S14. Deletions and Insertions Match Tests. Table S15. tRNA with 3’-CCA by Isoacceptor.
References
- Abad, M.G.; Rao, B.S.; Jackman, J.E. Template-dependent 3 ′ –5 ′ nucleotide addition is a shared feature of tRNA His guanylyltransferase enzymes from multiple domains of life. Proc. Natl. Acad. Sci. 2009, 107, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Abelson, H.; Johnson, L.; Penman, S.; Green, H. Changes in RNA in relation to growth of the fibroblast: II. The lifetime of mRNA, rRNA, and tRNA in resting and growing cells. Cell 1974, 1, 161–165. [Google Scholar] [CrossRef]
- Aebi, M.; Kirchner, G.; Chen, J.Y.; Vijayraghavan, U.; Jacobson, A.; Martin, N.C.; Abelson, J. Isolation of a temperature-sensitive mutant with an altered tRNA nucleotidyltransferase and cloning of the gene encoding tRNA nucleotidyltransferase in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 16216–16220. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, R.A.; Leonelli, S. What’s so special about model organisms? Stud. Hist. Philos. Sci. Part A 2011, 42, 313–323. [Google Scholar] [CrossRef]
- Ardell, D.H.; Hou, Y.-M. Initiator tRNA genes template the 3′ CCA end at high frequencies in bacteria. BMC Genom. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Avcilar-Kucukgoze, I.; Kashina, A. Hijacking tRNAs From Translation: Regulatory Functions of tRNAs in Mammalian Cell Physiology. Front. Mol. Biosci. 2020, 7, 610617. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Beckmann, R.; Cate, J.H.D.; Dinman, J.D.; Dragon, F.; Ellis, S.R.; Lafontaine, D.L.J.; Lindahl, L.; Liljas, A.; Lipton, J.M.; et al. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Bellen, H.J.; Hubbard, E.J.A.; Lehmann, R.; Madhani, H.D.; Solnica-Krezel, L.; Southard-Smith, E.M. Model organism databases are in jeopardy. Development 2021, 148. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.D.; Brandl, C.J. Transfer RNAs: diversity in form and function. RNA Biol. 2020, 18, 316–339. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, S.; Gartmann, M.; Halic, M.; Armache, J.-P.; Jarasch, A.; Mielke, T.; Berninghausen, O.; Wilson, D.N.; Beckmann, R. α-Helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 2010, 17, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Buhr, F.; Jha, S.; Thommen, M.; Mittelstaet, J.; Kutz, F.; Schwalbe, H.; Rodnina, M.V.; Komar, A.A. Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol. Cell 2016, 61, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.T.Y.; Dyavaiah, M.; DeMott, M.S.; Taghizadeh, K.; Dedon, P.C.; Begley, T.J. A Quantitative Systems Approach Reveals Dynamic Control of tRNA Modifications during Cellular Stress. PLOS Genet. 2010, 6, e1001247. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2008, 37, D93–D97. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, P.; Topf, M.; Ménétret, J.-F.; Eswar, N.; Cannone, J.J.; Gutell, R.R.; Sali, A.; Akey, C.W. Structure of the Mammalian 80S Ribosome at 8.7 Å Resolution. Structure 2008, 16, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Charneski, C.A.; Hurst, L.D. Positively Charged Residues Are the Major Determinants of Ribosomal Velocity. PLOS Biol. 2013, 11, e1001508. [Google Scholar] [CrossRef] [PubMed]
- Choe, B.K.; Taylor, M.W. Kinetics of synthesis and characterization of transfer RNA precursors in mammalian cells. Biochim. et Biophys. Acta (BBA) - Nucleic Acids Protein Synth. 1972, 272, 275–287. [Google Scholar] [CrossRef]
- Cooper GM, Adams KW. 2022. The Cell: A Molecular Approach, 9th edition. Sunderland, MA: Sinauer Associates.
- Dale, T.; Fahlman, R.P.; Olejniczak, M.; Uhlenbeck, O.C. Specificity of the ribosomal A site for aminoacyl-tRNAs. Nucleic Acids Res. 2008, 37, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Dannfald, A.; Favory, J.-J.; Deragon, J.-M. Variations in transfer and ribosomal RNA epitranscriptomic status can adapt eukaryote translation to changing physiological and environmental conditions. RNA Biol. 2021, 18, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.R.; A Ankeny, R.; Chen, P.M. Publication Trends in Model Organism Research. Genetics 2014, 198, 787–794. [Google Scholar] [CrossRef]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue-Specific Differences in Human Transfer RNA Expression. PLOS Genet. 2006, 2, e221–2115. [Google Scholar] [CrossRef] [PubMed]
- Draper, D.E.; Reynaldo, L.P. RNA binding strategies of ribosomal proteins. Nucleic Acids Res. 1999, 27, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Du, M.-Z.; Wei, W.; Qin, L.; Liu, S.; Zhang, A.-Y.; Zhang, Y.; Zhou, H.; Guo, F.-B. Co-adaption of tRNA gene copy number and amino acid usage influences translation rates in three life domains. DNA Res. 2017, 24, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Dupasquier, M.; Kim, S.; Halkidis, K.; Gamper, H.; Hou, Y.-M. tRNA Integrity Is a Prerequisite for Rapid CCA Addition: Implication for Quality Control. J. Mol. Biol. 2008, 379, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, J.T.; Ma, T.M.; Sason, H.; Rosenberg, D.; Ogo, T.; Furuya, S.; Snyder, S.H.; Wolosker, H. D-Serine in Glia and Neurons Derives from 3-Phosphoglycerate Dehydrogenase. J. Neurosci. 2013, 33, 12464–12469. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; McCaul, N.; Chatsisvili, A.; Braakman, I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016, 17, 615–638. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.D.; Liu, Y.; Thomas, G.W.C.; Vinař, T.; Alföldi, J.; Deng, J.; Dugan, S.; E van Elk, C.; E Hunter, M.; Joshi, V.; et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015, 47, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.; Schimmel, P. Enzymatic aminoacylation of an eight-base-pair microhelix with histidine. Proc. Natl. Acad. Sci. USA 1990, 87, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Geslain, R.; Pan, T. Functional Analysis of Human tRNA Isodecoders. J. Mol. Biol. 2010, 396, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Goodenbour, J.M.; Pan, T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006, 34, 6137–6146. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; de Crécy-Lagard, V.; Marck, C. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS Lett. 2009, 584, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhou, T.; Wilke, C.O. A Universal Trend of Reduced mRNA Stability near the Translation-Initiation Site in Prokaryotes and Eukaryotes. PLOS Comput. Biol. 2010, 6, e1000664–e1000664. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.; Data, D.; Seelam, P.P.; Bhattacharyya, D.; Mitra, A. Estimating Strengths of Individual Hydrogen Bonds in RNA Base Pairs: Toward a Consensus between Different Computational Approaches. ACS Omega 2019, 4, 7354–7368. [Google Scholar] [CrossRef] [PubMed]
- Hamashima, K.; Tomita, M.; Kanai, A. Expansion of Noncanonical V-Arm-Containing tRNAs in Eukaryotes. Mol. Biol. Evol. 2015, 33, 530–540. [Google Scholar] [CrossRef]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2017, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Harish, A.; Caetano-Anollés, G. Ribosomal History Reveals Origins of Modern Protein Synthesis. PLOS ONE 2012, 7, e32776. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, I.U.; Nakamura, A.; O'Donoghue, P.; Eiler, D.; Söll, D. tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res. 2011, 40, 333–344. [Google Scholar] [CrossRef]
- Heyrovska, R. Structures of the Molecular Components in DNA and RNA with Bond Lengths Interpreted as Sums of Atomic Covalent Radii. Open Struct. Biol. J. 2008, 2, 1–7. [Google Scholar] [CrossRef]
- Himeno, H.; Hasegawa, T.; Ueda, T.; Watanabe, K.; Miura, K.-I.; Shimizu, M. Role of the extra G-C pair at the end of the acceptor stem of tRNAHbin aminoacylation. Nucleic Acids Res. 1989, 17, 7855–7863. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Morichika, K.; Kirino, Y. Selective amplification and sequencing of cyclic phosphate–containing RNAs by the cP-RNA-seq method. Nat. Protoc. 2016, 11, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Hyde, S.J.; Eckenroth, B.E.; Smith, B.A.; Eberley, W.A.; Heintz, N.H.; Jackman, J.E.; Doublié, S. tRNA His guanylyltransferase (THG1), a unique 3′-5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′-3′ DNA polymerases. Proc. Natl. Acad. Sci. 2010, 107, 20305–20310. [Google Scholar] [CrossRef] [PubMed]
- Iben, J.R.; Maraia, R.J. tRNA gene copy number variation in humans. Gene 2014, 536, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Phizicky, E.M. tRNAHis guanylyltransferase adds G−1 to the 5′ end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA 2006, 12, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.P.; Hutvagner, G. tRNA-Derived Fragments (tRFs): Emerging New Roles for an Ancient RNA in the Regulation of Gene Expression. Life 2015, 5, 1638–1651. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Palmer, J.D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Liu, C.; Halkidis, K.; Gamper, H.B.; Hou, Y.-M. Distinct kinetic determinants for the stepwise CCA addition to tRNA. RNA 2009, 15, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Kisly, I.; Tamm, T. Archaea/eukaryote-specific ribosomal proteins - guardians of a complex structure. Comput. Struct. Biotechnol. J. 2023, 21, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kuscu, C.; Dutta, A. Biogenesis and Function of Transfer RNA-Related Fragments (tRFs). Trends Biochem. Sci. 2016, 41, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Suleski, M.; Craig, J.M.; E Kasprowicz, A.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Laibelman, AM. The Standard Genetic Code Lacks Redundancy for Amino Acid Codons. BioRxiv 2022. [Google Scholar] [CrossRef]
- Li, Z.; Li, S.; Luo, M.; Jhong, J.-H.; Li, W.; Yao, L.; Pang, Y.; Wang, Z.; Wang, R.; Ma, R.; et al. dbPTM in 2022: an updated database for exploring regulatory networks and functional associations of protein post-translational modifications. Nucleic Acids Res. 2021, 50, D471–D479. [Google Scholar] [CrossRef]
- Lizano, E.; Scheibe, M.; Rammelt, C.; Betat, H.; Mörl, M. A comparative analysis of CCA-adding enzymes from human and E. coli: Differences in CCA addition and tRNA 3′-end repair. Biochimie 2008, 90, 762–772. [Google Scholar] [CrossRef]
- Luby-Phelps, K. Cytoarchitecture and Physical Properties of Cytoplasm: Volume, Viscosity, Diffusion, Intracellular Surface Area. Internat Rev Cytol. 1999, 192, 189–221. [Google Scholar] [CrossRef]
- Lütcke, H.; Chow, K.; Mickel, F.; Moss, K.; Kern, H.; Scheele, G. Selection of AUG initiation codons differs in plants and animals. EMBO J. 1987, 6, 43–48. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The Evolutionary Fate and Consequences of Duplicate Genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Marinov, G.K. The bioenergetic costs of a gene. Proc. Natl. Acad. Sci. 2015, 112, 15690–15695. [Google Scholar] [CrossRef]
- Marino, J.; von Heijne, G.; Beckmann, R. Small protein domains fold inside the ribosome exit tunnel. FEBS Lett. 2016, 590, 655–660. [Google Scholar] [CrossRef]
- McCown, P.J.; Ruszkowska, A.; Kunkler, C.N.; Breger, K.; Hulewicz, J.P.; Wang, M.C.; Springer, N.A.; Brown, J.A. Naturally occurring modified ribonucleosides. Wiley Interdiscip. Rev. RNA 2020, 11, e1595–e1595. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Schartl, M. Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 1999, 11, 699–704. [Google Scholar] [CrossRef]
- Mukai, T.; Vargas-Rodriguez, O.; Englert, M.; Tripp, H.J.; Ivanova, N.N.; Rubin, E.M.; Kyrpides, N.C.; Söll, D. Transfer RNAs with novel cloverleaf structures. Nucleic Acids Res. 2016, 45, 2776–2785. [Google Scholar] [CrossRef]
- Narayan, M. Securing Native Disulfide Bonds in Disulfide-Coupled Protein Folding Reactions: The Role of Intrinsic and Extrinsic Elements vis-à-vis Protein Aggregation and Neurodegeneration. ACS Omega 2021, 6, 31404–31410. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.B.; Hedman, R.; Marino, J.; Wickles, S.; Bischoff, L.; Johansson, M.; Müller-Lucks, A.; Trovato, F.; Puglisi, J.D.; O’brien, E.P.; et al. Cotranslational Protein Folding inside the Ribosome Exit Tunnel. Cell Rep. 2015, 12, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Novoa, E.M.; Pavon-Eternod, M.; Pan, T.; de Pouplana, L.R. A Role for tRNA Modifications in Genome Structure and Codon Usage. Cell 2012, 149, 202–213. [Google Scholar] [CrossRef] [PubMed]
- O’brien, E.P.; Vendruscolo, M.; Dobson, C.M. Kinetic modelling indicates that fast-translating codons can coordinate cotranslational protein folding by avoiding misfolded intermediates. Nat. Commun. 2014, 5, 2988. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue P, Luthey-Schulten Z. 2003. On the Evolution of Structure in Aminoacyl-tRNA Synthetases. Microbiol Molec Biol Rev, 67: 550–73. [CrossRef]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Pinkard, O.; McFarland, S.; Sweet, T.; Coller, J. Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Polevoda, B.; Sherman, F. The diversity of acetylated proteins. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free. Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: resources, tools and prediction methods. Database 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.F.; Munyard, K.; Croft, L.J.; Allnutt, T.R.; Jackling, F.; Alshanbari, F.; Jevit, M.; Wright, G.A.; Cransberg, R.; Tibary, A.; et al. Chromosome-Level Alpaca Reference Genome VicPac3.1 Improves Genomic Insight Into the Biology of New World Camelids. Front. Genet. 2019, 10, 586. [Google Scholar] [CrossRef]
- Rozov, A.; Westhof, E.; Yusupov, M.; Yusupova, G. The ribosome prohibits the G•U wobble geometry at the first position of the codon–anticodon helix. Nucleic Acids Res. 2016, 44, 6434–6441. [Google Scholar] [CrossRef] [PubMed]
- Rudorf, S.; Thommen, M.; Rodnina, M.V.; Lipowsky, R. Deducing the Kinetics of Protein Synthesis In Vivo from the Transition Rates Measured In Vitro. PLOS Comput. Biol. 2014, 10, e1003909. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, D.G.; Sandoval-Perez, A.; Rangarajan, A.V.; Gunderson, E.L.; Jacobson, M.P. Cysteine Oxidation in Proteins: Structure, Biophysics, and Simulation. Biochemistry 2022, 61, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.B.; Del-Bem, L.-E. The Evolution of tRNA Copy Number and Repertoire in Cellular Life. Genes 2022, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Schulte, L.; Mao, J.; Reitz, J.; Sreeramulu, S.; Kudlinzki, D.; Hodirnau, V.-V.; Meier-Credo, J.; Saxena, K.; Buhr, F.; Langer, J.D.; et al. Cysteine oxidation and disulfide formation in the ribosomal exit tunnel. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Schwer, B.; Aronova, A.; Ramirez, A.; Braun, P.; Shuman, S. Mammalian 2′,3′ cyclic nucleotide phosphodiesterase (CNP) can function as a tRNA splicing enzyme in vivo. RNA 2007, 14, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Moore, M.J.; Brown, J.W.; Yang, Y. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 2015, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.; Horn, C.; Brown, M.; Ioudovitch, A.; Steinberg, S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Timsit, Y.; Sergeant-Perthuis, G.; Bennequin, D. Evolution of ribosomal protein network architectures. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Torres, A.G.; Piñeyro, D.; Rodríguez-Escribà, M.; Camacho, N.; Reina, O.; Saint-Léger, A.; Filonava, L.; Batlle, E.; de Pouplana, L.R. Inosine modifications in human tRNAs are incorporated at the precursor tRNA level. Nucleic Acids Res. 2015, 43, 5145–5157. [Google Scholar] [CrossRef] [PubMed]
- Turnbough, C.L. Regulation of gene expression by reiterative transcription. Curr. Opin. Microbiol. 2011, 14, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.-C.; Kumar, S.; Hedges, S.B. Divergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proc. R. Soc. B: Biol. Sci. 1999, 266, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Cole, P.A. The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem. Biol. 2020, 27, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N.; Beckmann, R. The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr. Opin. Struct. Biol. 2011, 21, 274–282. [Google Scholar] [CrossRef]
- Wilson, D.N.; Cate, J.H.D. The Structure and Function of the Eukaryotic Ribosome. Cold Spring Harb. Perspect. Biol. 2012, 4, a011536–a011536. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Li, F.; Wang, J.; Weiner, A.M.; Steitz, T.A. Crystal Structures of an Archaeal Class I CCA-Adding Enzyme and Its Nucleotide Complexes. Mol. Cell 2003, 12, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Yoshihisa, T. Handling tRNA introns, archaeal way and eukaryotic way. Front. Genet. 2014, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.-H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. 2016, 113, E6117–E6125. [Google Scholar] [CrossRef] [PubMed]
- Zouridis, H.; Hatzimanikatis, V. Effects of Codon Distributions and tRNA Competition on Protein Translation. Biophys. J. 2008, 95, 1018–1033. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Mammals Included in Study.
| Abbreviation | Mammal | Common Name | Genome (Gbp) | CDS |
|---|---|---|---|---|
| Ame | Ailuropoda melanoleuca | giant panda | 2.44408 | 55,366 |
| Bac | Balaenoptera acutorostrata scammoni | minke whale | 2.43169 | 37,625 |
| Bta | Bos taurus | cow | 2.71585 | 63,696 |
| Cja | Callithrix jacchus | marmoset | 2.89820 | 85,133 |
| Cfa | Canis familiaris | dog | 2.39860 | 66,768 |
| Cpo | Cavia porcellus | guinea pig | 2.72322 | 37,349 |
| Csa | Chlorocebus sabaeus | green monkey | 2.93783 | 61,846 |
| Cgr | Cricetulus griseus | Chinese hamster | 2.39979 | 37,534 |
| Dno | Dasypus novemcinctus | nine-banded armadillo | 3.63152 | 38,178 |
| Dor | Dipodomys ordii | kangaroo rat | 2.23637 | 29,351 |
| Ete | Echinops telfairi | tenrec | 2.94712 | 24,821 |
| Eca | Equus caballus | horse | 2.50697 | 60,900 |
| Eeu | Erinaceus europaeus | Western European hedgehog | 2.71572 | 29,382 |
| Fca | Felis catus | domesticated cat | 2.42575 | 71,463 |
| Ggo | Gorilla gorilla gorilla | western lowland gorilla | 3.04487 | 51,654 |
| Hgl | Heterocephalus glaber | mole rat | 2.61820 | 60,814 |
| Hsa | Homo sapiens | human | 3.27209 | 123,378 |
| Laf | Loxodonta africana | African savannah elephant | 3.19674 | 41,267 |
| Mamu | Macaca mulatta mulatta | Indian rhesus macaque | 2.97133 | 67,977 |
| Mimu | Microcebus murinus | mouse lemur | 2.48741 | 59,023 |
| Mdo | Monodelphis domestica | opossum | 3.59844 | 49,112 |
| Mumu | Mus musculus | house mouse (balb c/) | 2.72822 | 92,499 |
| Mpa | Mus pahari | shrew mouse | 2.47501 | 42,226 |
| Mpu | Mustela putorius furo | ferret | 2.41088 | 48,107 |
| Mlu | Myotis lucifugus | brown bat | 2.03458 | 43,106 |
| Nle | Nomascus leucogenys | northern white-cheeked gibbon | 2.84398 | 51,330 |
| Opr | Ochotona princeps | pika | 2.23149 | 29,701 |
| Oan | Ornithorhynchus anatinus | platypus | 1.85928 | 38,747 |
| Ocu | Oryctolagus cuniculus | rabbit | 2.73746 | 38,463 |
| Oga | Otolemur garnettii | galago | 2.51972 | 32,556 |
| Oar | Ovis aries | sheep | 2.62815 | 60,628 |
| Ptr | Pan troglodytes | chimpanzee | 3.05040 | 80,811 |
| Ppy | Pongo pygmaeus | Sumatran orangutan | 3.06505 | 49,721 |
| Rno | Rattus norvegicus | Norway rat (RGSC) | 2.64792 | 74,754 |
| Sha | Sarcophilus harrisii | Tasmanian devil | 3.08667 | 45,893 |
| Sar | Sorex araneus | European shrew | 2.42316 | 23,427 |
| Ssc | Sus scrofa | pig | 2.50191 | 63,577 |
| Tma | Trichechus manatus latirostri | West Indian manatee | 3.10381 | 36,406 |
| Ttr | Tursiops truncatus | bottlenose dolphin | 2.37852 | 55,739 |
| Vpa | Vicugna pacos | alpaca | 2.11962 | 44,159 |
Table 2.
Summary of tRNA Data.
| All Anticodons | AA Anticodons (≠ sup, Undet) | 22 AA Anticodons (≠ sup, Undet) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mammals | Total | Unique | % Unique | Isoacceptors | Total | Unique | % Unique | Matched | % Matched |
| Ailuropoda melanoleuca | 432 | 256 | 59.3% | 48 | 428 | 252 | 58.9% | 152 | 60.3% |
| Balaenoptera acutorostrata scammoni | 720 | 555 | 77.1% | 50 | 660 | 496 | 75.2% | 164 | 33.1% |
| Bos taurus | 837 | 542 | 64.8% | 54 | 811 | 516 | 63.6% | 185 | 35.9% |
| Callithrix jacchus | 340 | 201 | 59.1% | 48 | 339 | 200 | 59.0% | 131 | 65.5% |
| Canis familiaris | 384 | 247 | 64.3% | 48 | 380 | 243 | 63.9% | 135 | 55.6% |
| Cavia porcellus | 350 | 202 | 57.7% | 47 | 348 | 200 | 57.5% | 121 | 60.5% |
| Chlorocebus sabaeus | 396 | 242 | 61.1% | 51 | 396 | 242 | 61.1% | 160 | 66.1% |
| Cricetulus griseus | 427 | 238 | 55.7% | 48 | 425 | 236 | 55.5% | 128 | 54.2% |
| Dasypus novemcinctus | 539 | 356 | 66.0% | 47 | 538 | 355 | 66.0% | 140 | 39.4% |
| Dipodomys ordii | 401 | 191 | 47.6% | 48 | 401 | 191 | 47.6% | 135 | 70.7% |
| Echinops telfairi | 290 | 186 | 64.1% | 47 | 289 | 185 | 64.0% | 117 | 63.2% |
| Equus caballus | 427 | 227 | 53.2% | 49 | 426 | 226 | 53.1% | 149 | 65.9% |
| Erinaceus europaeus | 447 | 282 | 63.1% | 47 | 445 | 280 | 62.9% | 130 | 46.4% |
| Felis catus | 600 | 336 | 56.0% | 49 | 585 | 321 | 54.9% | 151 | 47.0% |
| Gorilla gorilla gorilla | 394 | 239 | 60.7% | 50 | 394 | 239 | 60.7% | 158 | 66.1% |
| Heterocephalus glaber | 294 | 206 | 70.1% | 50 | 294 | 206 | 70.1% | 113 | 54.9% |
| Homo sapiens | 419 | 254 | 60.6% | 49 | 419 | 254 | 60.6% | 164 | 64.6% |
| Loxodonta africana | 962 | 700 | 72.8% | 53 | 956 | 694 | 72.6% | 172 | 24.8% |
| Macaca mulatta | 583 | 336 | 57.6% | 49 | 583 | 336 | 57.6% | 161 | 47.9% |
| Microcebus murinus | 328 | 178 | 54.3% | 49 | 328 | 178 | 54.3% | 131 | 73.6% |
| Monodelphis domestica | 464 | 196 | 42.2% | 48 | 463 | 195 | 42.1% | 117 | 60.0% |
| Mus musculus | 384 | 201 | 52.3% | 49 | 382 | 199 | 52.1% | 130 | 65.3% |
| Mus pahari | 346 | 178 | 51.4% | 48 | 346 | 178 | 51.4% | 123 | 69.1% |
| Mustela putorius furo | 380 | 247 | 65.0% | 49 | 378 | 245 | 64.8% | 144 | 58.8% |
| Myotis lucifugus | 240 | 130 | 54.2% | 46 | 240 | 130 | 54.2% | 102 | 78.5% |
| Nomascus leucogenys | 365 | 236 | 64.7% | 50 | 363 | 234 | 64.5% | 148 | 63.2% |
| Ochotona princeps | 323 | 168 | 52.0% | 47 | 323 | 168 | 52.0% | 119 | 70.8% |
| Ornithorhynchus anatinus | 699 | 432 | 61.8% | 53 | 695 | 428 | 61.6% | 109 | 25.5% |
| Oryctolagus cuniculus | 483 | 254 | 52.6% | 49 | 482 | 253 | 52.5% | 137 | 54.2% |
| Otolemur garnettii | 320 | 189 | 59.1% | 49 | 320 | 189 | 59.1% | 124 | 65.6% |
| Ovis aries | 774 | 532 | 68.7% | 52 | 762 | 520 | 68.2% | 179 | 34.4% |
| Pan troglodytes | 436 | 254 | 58.3% | 52 | 436 | 254 | 58.3% | 176 | 69.3% |
| Pongo pygmaeus | 411 | 254 | 61.8% | 50 | 411 | 254 | 61.8% | 159 | 62.6% |
| Rattus norvegicus | 386 | 203 | 52.6% | 47 | 386 | 203 | 52.6% | 124 | 61.1% |
| Sarcophilus harrisii | 389 | 168 | 43.2% | 47 | 387 | 166 | 42.9% | 109 | 65.7% |
| Sorex araneus | 333 | 185 | 55.6% | 47 | 331 | 183 | 55.3% | 119 | 65.0% |
| Sus scrofa | 495 | 273 | 55.2% | 50 | 493 | 271 | 55.0% | 155 | 57.2% |
| Trichechus manatus latirostris | 593 | 477 | 80.4% | 52 | 582 | 466 | 80.1% | 136 | 29.2% |
| Tursiops truncatus | 625 | 513 | 82.1% | 52 | 571 | 461 | 80.7% | 163 | 35.4% |
| Vicugna pacos | 1760 | 1514 | 86.0% | 60 | 1681 | 1436 | 85.4% | 147 | 10.2% |
| Totals | 19776 | 12578 | 63.6% | 19477 | 12283 | 63.1% | 5617 | 45.7% | |
Table 3.
Total and Unique tRNA Sequences by Isoacceptor.
| Isoacceptor | Total / Unique | % | Isoacceptor | Total / Unique | % |
|---|---|---|---|---|---|
| Ala (AGC) | 1673 / 1372 | 82.0% | Lys (CUU) | 883 / 557 | 63.1% |
| Ala (CGC) | 234 / 195 | 83.3% | Lys (UUU) | 616 / 388 | 63.0% |
| Ala (GGC) | 48 / 48 | 100% | |||
| Ala (UGC) | 477 / 386 | 80.9% | Met (CAU) | 381 / 294 | 77.2% |
| Arg (ACG) | 294 / 151 | 51.4% | iMet (CAU) | 393 / 103 | 26.2% |
| Arg (CCG) | 168 / 127 | 75.6% | |||
| Arg (CCU) | 349 / 315 | 90.3% | Phe (AAA) | 10 / 10 | 100% |
| Arg (GCG) | Phe (GAA) | 458 / 229 | 50.0% | ||
| Arg (UCG) | 265 / 232 | 87.5% | |||
| Arg (UCU) | 378 / 341 | 90.2% | Pro (AGG) | 400 / 115 | 28.8% |
| Pro (CGG) | 147 / 54 | 36.7% | |||
| Asn (AUU) | 38 / 38 | 100% | Pro (GGG) | 4 / 4 | 100% |
| Asn (GUU) | 656 / 299 | 45.6% | Pro (UGG) | 251 / 128 | 51.0% |
| Asp (AUC) | 56 / 54 | 96.4% | Sec (UCA) | 65 / 62 | 95.4% |
| Asp (GUC) | 561 / 226 | 40.3% | |||
| Ser (ACU) | 13 / 13 | 100% | |||
| Cys (ACA) | 29 / 28 | 96.6% | Ser (AGA) | 432 / 221 | 51.2% |
| Cys (GCA) | 1068 / 577 | 54.0% | Ser (CGA) | 152 / 144 | 94.7% |
| Ser (GCU) | 413 / 246 | 59.6% | |||
| Gln (CUG) | 467 / 278 | 59.5% | Ser (GGA) | 56 / 47 | 83.9% |
| Gln (UUG) | 241 / 185 | 76.8% | Ser (UGA) | 162 / 151 | 93.2% |
| Glu (CUC) | 395 / 167 | 42.3% | sup (CUA) | 24 / 24 | 100% |
| Glu (UUC) | 578 / 413 | 71.5% | sup (UCA) | 89 / 88 | 98.9% |
| sup (UUA) | 41 / 41 | 100% | |||
| Gly (ACC) | 39 / 39 | 100% | |||
| Gly (CCC) | 388 / 286 | 73.7% | Thr (AGU) | 432 / 325 | 75.2% |
| Gly (GCC) | 540 / 180 | 33.3% | Thr (CGU) | 192 / 187 | 97.4% |
| Gly (UCC) | 435 / 261 | 60.0% | Thr (GGU) | 9 / 9 | 100% |
| Thr (UGU) | 266 / 215 | 80.8% | |||
| His (AUG) | 7 / 7 | 100% | |||
| His (GUG) | 379 / 94 | 24.8% | Trp (CCA) | 446 / 345 | 77.4% |
| Ile (AAU) | 495 / 198 | 40.0% | Tyr (AUA) | 6 / 6 | 100% |
| Ile (GAU) | 36 / 29 | 80.6% | Tyr (GUA) | 485 / 260 | 53.6% |
| Ile (UAU) | 218 / 128 | 58.7% | |||
| Val (AAC) | 444 / 272 | 61.3% | |||
| Leu (AAG) | 276 / 123 | 44.6% | Val (CAC) | 437 / 243 | 55.6% |
| Leu (CAA) | 224 / 175 | 78.1% | Val (GAC) | 12 / 11 | 91.7% |
| Leu (CAG) | 305 / 148 | 48.5% | Val (UAC) | 273 / 235 | 86.1% |
| Leu (GAG) | 1 / 1 | 100% | |||
| Leu (UAA) | 163 / 154 | 94.5% | Undet (NNN) | 145 / 142 | 97.9% |
| Leu (UAG) | 158 / 154 | 97.5% |
Table 4.
Distribution of Total and Unique Sequences by Isoacceptor.
| Total Sequences | Unique Sequences | Total Sequences | Unique Sequences | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isoacceptor | 1−10 | 11−20 | 21−30 | > 30 | 1−10 | 11−20 | 21−30 | > 30 | Isoacceptor | 1−10 | 11−20 | 21−30 | > 30 | 1−10 | 11−20 | 21−30 | > 30 |
| Ala (AGC) | 2 | 24 | 7 | 7 | 11 | 25 | 3 | 1 | Lys (CUU) | 5 | 22 | 5 | 8 | 27 | 5 | 3 | 5 |
| Ala (CGC) | 37 | 3 | 37 | 3 | Lys (UUU) | 11 | 22 | 2 | 5 | 32 | 3 | 3 | 2 | ||||
| Ala (GGC) | 6 | 1 | 6 | 1 | |||||||||||||
| Ala (UGC) | 25 | 11 | 3 | 1 | 30 | 7 | 3 | Met (CAU) | 28 | 12 | 36 | 4 | |||||
| Arg (ACG) | 37 | 3 | 40 | iMet (CAU) | 28 | 10 | 2 | 40 | |||||||||
| Arg (CCG) | 39 | 1 | 39 | 1 | |||||||||||||
| Arg (CCU) | 30 | 7 | 2 | 1 | 30 | 7 | 2 | 1 | Phe (AAA) | 3 | 3 | ||||||
| Arg (GCG) | Phe (GAA) | 21 | 16 | 3 | 37 | 3 | |||||||||||
| Arg (UCG) | 36 | 3 | 1 | 37 | 2 | 1 | |||||||||||
| Arg (UCU) | 33 | 2 | 3 | 2 | 34 | 2 | 2 | 2 | Pro (AGG) | 30 | 8 | 1 | 1 | 38 | 1 | 1 | |
| Pro (CGG) | 40 | 40 | |||||||||||||||
| Asn (AUU) | 10 | 1 | 10 | 1 | Pro (GGG) | 2 | 2 | ||||||||||
| Asn (GUU) | 7 | 21 | 11 | 1 | 29 | 10 | 1 | Pro (UGG) | 38 | 2 | 40 | ||||||
| Asp (AUC) | 7 | 1 | 7 | 1 | Sec (UCA) | 38 | 1 | 38 | 1 | ||||||||
| Asp (GUC) | 11 | 23 | 6 | 37 | 1 | 2 | |||||||||||
| Ser (ACU) | 7 | 7 | |||||||||||||||
| Cys (ACA) | 13 | 13 | Ser (AGA) | 29 | 9 | 1 | 1 | 37 | 1 | 2 | |||||||
| Cys (GCA) | 2 | 5 | 21 | 12 | 8 | 29 | 3 | Ser (CGA) | 40 | 40 | |||||||
| Ser (GCU) | 27 | 12 | 1 | 36 | 3 | 1 | |||||||||||
| Gln (CUG) | 18 | 20 | 2 | 33 | 6 | 1 | Ser (GGA) | 3 | 1 | 1 | 3 | 1 | 1 | ||||
| Gln (UUG) | 36 | 4 | 37 | 3 | Ser (UGA) | 39 | 1 | 40 | |||||||||
| Glu (CUC) | 25 | 13 | 2 | 38 | 2 | sup (CUA) | 11 | 11 | |||||||||
| Glu (UUC) | 24 | 9 | 7 | 31 | 2 | 3 | 4 | sup (UCA) | 12 | 2 | 1 | 12 | 2 | 1 | |||
| sup (UUA) | 12 | 2 | 12 | 2 | |||||||||||||
| Gly (ACC) | 2 | 1 | 2 | 1 | |||||||||||||
| Gly (CCC) | 32 | 3 | 1 | 4 | 35 | 1 | 2 | 2 | Thr (AGU) | 29 | 10 | 1 | 37 | 2 | 1 | ||
| Gly (GCC) | 13 | 25 | 1 | 1 | 39 | 1 | Thr (CGU) | 38 | 2 | 39 | 1 | ||||||
| Gly (UCC) | 28 | 9 | 1 | 2 | 35 | 3 | 2 | Thr (GGU) | 6 | 6 | |||||||
| Thr (UGU) | 36 | 4 | 37 | 3 | |||||||||||||
| His (AUG) | 4 | 4 | |||||||||||||||
| His (GUG) | 28 | 11 | 1 | 40 | Trp (CCA) | 31 | 4 | 1 | 4 | 35 | 1 | 1 | 3 | ||||
| Ile (AAU) | 16 | 22 | 2 | 37 | 2 | 1 | Tyr (AUA) | 6 | 6 | ||||||||
| Ile (GAU) | 14 | 14 | Tyr (GUA) | 15 | 24 | 1 | 37 | 3 | |||||||||
| Ile (UAU) | 39 | 1 | 40 | ||||||||||||||
| Val (AAC) | 30 | 7 | 1 | 2 | 37 | 1 | 1 | 1 | |||||||||
| Leu (AAG) | 37 | 3 | 39 | 1 | Val (CAC) | 24 | 12 | 3 | 1 | 35 | 4 | 1 | |||||
| Leu (CAA) | 39 | 1 | 39 | 1 | Val (GAC) | 9 | 9 | ||||||||||
| Leu (CAG) | 34 | 6 | 39 | 1 | Val (UAC) | 35 | 4 | 1 | 35 | 4 | 1 | ||||||
| Leu (GAG) | 1 | 1 | |||||||||||||||
| Leu (UAA) | 38 | 2 | 40 | Undet (NNN) | 13 | 2 | 1 | 1 | 13 | 2 | 1 | 1 | |||||
| Leu (UAG) | 40 | 40 | |||||||||||||||
Table 5.
Interspecies Matched tRNA.
| Genomes | Matches | Genomes | Matches | Genomes | Matches | Genomes | Matches |
|---|---|---|---|---|---|---|---|
| 40 | 29 | 30 | 2 | 20 | 3 | 10 | 5 |
| 39 | 10 | 29 | 1 | 19 | 1 | 9 | 6 |
| 38 | 10 | 28 | 1 | 18 | 3 | 8 | 10 |
| 37 | 8 | 27 | 3 | 17 | 2 | 7 | 12 |
| 36 | 5 | 26 | 3 | 16 | 2 | 6 | 16 |
| 35 | 7 | 25 | 1 | 15 | 1 | 5 | 26 |
| 34 | 4 | 24 | 1 | 14 | 5 | 4 | 54 |
| 33 | 4 | 23 | 2 | 13 | 5 | 3 | 93 |
| 32 | 3 | 22 | 3 | 12 | 5 | 2 | 271 |
| 31 | 1 | 21 | 5 | 11 | 8 | Total | 631 |
Table 6.
Interspecies Aligned tRNA.
| Isoacceptor | Matches (40 Mammals only) — 3 Point Scheme |
|---|---|
| Ala (CGC) 72 nt | GGGGAUGUAGCUCAGUGGUAGA GCGCAU GCUUCGCAUGUAUGAGGCC CCGGGUUCGAUCCCCGGCAUCUCCA |
| Arg (ACG) 73 nt | GGGCCAGUGGCGCAAUGGAUAACGCGUCU GACUACGGAUCAGAAGAUU CCAGGUUCGACUCCUGGCUGGCUCG |
| Arg (CCG) 73 nt | GACCCAGUGGCCUAAUGGAUAAGGCAUCA GCCUCCGGAGCUGGGGAUU GUGGGUUCGAGUCCCAUCUGGGUCG |
| Asn (GUU) 74 nt | GUCUCUGUGGCGCAAUCGGUUAGCGCGUUCGGCUGUUAACCGAAAGGUU GGUGGUUCGAGCCCACCCAGGGACG |
| Asp (GUC) 72 nt | UCCUCGUUAGUAUAGUGGUGAGUAUCCC CGCCUGUCACGCGGGAGACC GG GGUUCGAUUCCCCGACGGGGAG |
| Cys (GCA) 72 nt | GGGGGUAUAGCUCAGUGGUAGAGCAUUU GACUGCAGAUCAAGAGGUC CCCG GUUCAAAUCCGGGUGCCCCCU |
| Gln (CUG) 72 nt | GGUUCCAUGGUGUAAUGGUUAGCACUCU GGACUCUGAAUCCAGCGAUC CGAGUUCAAAUCUCGGUGGAACCU |
| Glu (CUC) 72 nt | UCCCUGGUGGUCUAGUGGUUAGGAUUCG GCGCUCUCACCGCCGCGGCC CGGGUUCGAUUCCCGGUCAGGGAA |
| Gly (GCC) 71 nt | GCAUUGGUGGUUCAGUGGUAGAAUUCUC GCCUGCCACGCGGGAGGCC CGGGUUCGAUUCCCGGCCAAUGCA |
| Gly (UCC) 72 nt | GCGUUGGUGGUAUAGUGGUGAGCAUAGCU GCCUUCCAAGCAGUUGACC CGGGUUCGAUUCCCGGCCAACGCA |
| His (GUG) 72 nt | GCCGUGAUCGUAUAGUGGUUAGUACUCUG CGUUGUGGCCGCAGCAACC UCGGUUCGAAUCCGAGUCACGGCA |
| Ile (AAU) 74 nt | GGCCGGUUAGCUCAGUUGGUUAGAGCGUGGUGCUAAUAACGCCAAGGUC GCGGGUUCGAUCCCCGUACGGGCCA |
| Ile (UAU) 74 nt | GCUCCAGUGGCGCAAUCGGUUAGCGCGCGGUACUUAUAAUGCCGAGGUU GUGAGUUCGAGCCUCACCUGGAGCA |
| Leu (AAG) 82 nt | GGUAGCGUGGCCGAGCGGUCUAAGGCGCUGGAUUAAGGCUCCAGUCUCUUC GGGGGCGUGGGUUCGAAUCCCACCGCUGCCA |
| Leu (CAG) 83 nt | GUCAGGAUGGCCGAGCGGUCUAAGGCGCUGCGUUCAGGUCGCAGUCUCCCCUGGAGGCGUGGGUUCGAAUCCCACUCCUGACA |
| Lys (CUU) 73 nt | GCCCGGCUAGCUCAGUCGGUA GAGCAUGAGACUCUUAAUCUCAGGGUC GUGGGUUCGAGCCCCACGUUGGGCG |
| Lys (UUU) 73 nt | GCCCGGAUAGCUCAGUCGGUA GAGCAUCAGACUUUUAAUCUGAGGGUC CAGGGUUCAAGUCCCUGUUCGGGCG |
| iMet (CAU) 72 nt | AGCAGAGUGGCGCAG CGGAA GCGUGCUGGGCCCAUAACCCAGAGGUC GAUGGAUCGAAACCAUCCUCUGCUA |
| Phe (GAA) 73 nt | GCCGAAAUAGCUCAGUUGGGA GAGCGUUAGACUGAAGAUCUAAAGGUC CCUGGUUCGAUCCCGGGUUUCGGCA |
| Pro (AGG) 72 nt | GGCUCGUUGGUCUAGGGGUA UGAUUCUCGCUUAGGGUGCGAGAGGUC CCGGGUUCAAAUCCCGGACGAGCCC |
| Pro (CGG) 72 nt | GGCUCGUUGGUCUAGGGGUA UGAUUCUCGCUUCGGGUGCGAGAGGUC CCGGGUUCAAAUCCCGGACGAGCCC |
| Ser (AGA) 82 nt | GUAGUCGUGGCCGAGUGGUUAAGGCGA UGGACUAGAAAUCCAUUGGGGUCUCCCCGCGCAGGUUCGAAUCCUGCCGACUACG |
| Ser (GCU) 82 nt | GACGAGGUGGCCGAGUGGUUAAGGCGA UGGACUGCUAAUCCAUUGUGCUCUGCACGCGUGGGUUCGAAUCCCAUCCUCGUCG |
| Thr (AGU) 74 nt | GGCGCCGUGGCUUAGUUGGUUAAAGCGCCUGUCUAGUAAACA GGAGAUC CUGGGUUCGAAUCCCAGCGGUGCCU |
| Trp (CCA) 72 nt | GACCUCGUGGCGCAAC GGU AGCGCGUCUGACUCCAGAUCA GAAGGCU GCGUGUUCGAAUCACGUCGGGGUCA |
| Trp (CCA) 72 nt | GACCUCGUGGCGCAAC GGU AGCGCGUCUGACUCCAGAUCA GAAGGUU GCGUGUUCAAAUCACGUCGGGGUCA |
| Tyr (GUA) 73 nt | CCUUCGAUAGCUCAGCU GGU AGAGCGGAGGACUGUAGAUCCUUAGGUCGCU GGUUCGAUUCCGGCUCGAAGGA |
| Val (AAC) 73 nt | GUUUCCGUAGUGUAGU GGUUAUCACGUUCGCCUAACACGCGAAAGGUCCCC GGUUCGAAACCGGGCGGAAACA |
| Val (CAC) 73 nt | GUUUCCGUAGUGUAGU GGUUAUCACGUUCGCCUCACACGCGAAAGGUCCCC GGUUCGAAACCGGGCGGAAACA |
Table 7.
Unique tRNA Matches Relative to Hsa.
| Mammal | Matched | Divergence Time | Mammal | Matched | Divergence Time |
|---|---|---|---|---|---|
| Hsa | 164 | xx | Hsa | 164 | xx |
| Ptr | 142 | 6.4 mya | Ocu | 91 | 87 mya |
| Ggo | 128 | 8.6 mya | Opr | 90 | 87 mya |
| Ppy | 121 | 15.2 mya | Cpo | 89 | 87 mya |
| Nle | 117 | 19.5 mya | Cfa | 89 | 94 mya |
| Csa | 116 | 28.8 mya | Dno | 89 | 99 mya |
| Mamu | 111 | 28.8 mya | Ttr | 89 | 94 mya |
| Cja | 105 | 43 mya | Eeu | 88 | 94 mya |
| Bta | 100 | 94 mya | Tma | 88 | 99 mya |
| Oar | 100 | 94 mya | Mumu | 86 | 87 mya |
| Mimu | 99 | 74 mya | Sar | 84 | 94 mya |
| Ssc | 97 | 94 mya | Rno | 84 | 87 mya |
| Vpa | 97 | 94 mya | Hgl | 84 | 87 mya |
| Eca | 96 | 94 mya | Cgr | 84 | 87 mya |
| Fca | 96 | 94 mya | Mpa | 83 | 87 mya |
| Laf | 96 | 99 mya | Ete | 82 | 99 mya |
| Dor | 93 | 87 mya | Mlu | 81 | 94 mya |
| Bac | 92 | 94 mya | Mdo | 73 | 160 mya |
| Oga | 91 | 74 mya | Sha | 71 | 160 mya |
| Mpu | 91 | 94 mya | Oan | 61 | 180 mya |
| Ame | 91 | 94 mya |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



