
Submitted:
21 July 2024
Posted:
22 July 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
This study aimed (1) to map the immunogenic epitopes (B and T cells) within the major BCoV structural proteins. These epitopes are believed to induce a robust im-mune response through the interaction with major histocompatibility complex (MHC class II) molecules (2) to design some novel BCoV multi-epitope-based vac-cines. To achieve these goals, we applied several integrated In-silico prediction computational tools to map these epitopes within the major BCoV structural pro-teins. The predicted epitopes were linked to some immunostimulants such as Toll-like receptors-2 (TLR2) and TLR-4. The tertiary structure of each epitope was modeled through the Alpha fold-2 colab machine learning tools. The stability and purity of each epitope were assessed using the Ramachandran plot and the Z Score values. Each multiepitope-based vaccine candidate conjugated with the Chlorotox-in B toxin as an adjuvant. We designed the vaccine constructs using various expres-sion vectors. We also predicted the affinity binding of these vaccines with the target protein using molecular docking. Our designed multiepitope vaccine candidates per each BCoV structural protein showed high antigenicity, immunogenicity, non-allergic, non-toxic, and high water solubility. Further studies are highly en-couraged to validate the efficacy of these novel BCoV vaccines in the natural host.
Keywords:
AI
; In silico predction
; BCoV
; multiepitopes
; vacciens
; adjuvant
; T Cell
; B Cell
; MHC-I
; MHC-II
; TLR2
; TLR4
1. Introduction
BCoV is one of the significant causes of enteritis in young calves; it may also be responsible for many respiratory outbreaks in young calves [1,2]. BCoV participates in the development of bovine respiratory disease complex in association with other bacterial pathogens [1,3]. BCoV infection in young calves leads to diarrhea, marked reduction in the body weight gain, dehydration, and death if it is superimposed with other viral pathogens, particularly Rotavirus and other bacterial pathogens such as Mannheimia haemolytica and some other parasitic diseases, particularly cryptosporidium [4]. BCoV belongs to the Betacoronavirus along with other essential coronaviruses affecting humans, such as the severe acute respiratory syndrome-2 (SARS-CoV-2), the Middle East Respiratory Syndrome coronavirus (MERS-CoV), and the human coronavirus-OC43 (HCoV-OC43). The viral genome is a single-strand positive sense RNA about 31 kb in length. The viral genome is flanked by two untranslated regions at both ends. The 5' two-thirds of the genome contains Gene-1, composed of two overlapping open reading frames (ORF1a and ORF1b) with ribosomal frameshifting sites in between. Gene-1 is further processed into 16 non-structural proteins (NSPs; NSp1-NSP16). The 3' end of the genome encompasses the major structural proteins (S, HE, M, and N). The BCoV genome is organized as follows (5'UTR-Gene-1, S, HE, M, E, N-UTR-3'). Unfortunately, a limited number of BCoV vaccines are commercially available in the market. This BCoV vaccine is based mainly on old technology that uses the live attention approach. This vaccine did not offer good immunity for the cattle population over several years. Other major disadvantages of the live attenuated-based coronavirus vaccines are the reversion to virulence and the possibility of recombination of some field isolates. Another common challenge in the coronavirus vaccinology is the continuous change of the genetic makeup of coronaviruses over time through mutations, recombination, and new host adaptations. These factors contributed to the vaccination failure of conventional vaccines. There is an urgent need to develop some novel BCoV vaccines that match the currently circulating field strain of this virus and offer a good immune response in the vaccinated cattle population. The most scientific vaccine design and development approach is to map the immunogenic epitopes across the viral genome. There are several generations of vaccines, including the (recombinant, DNA, and subunit-based vaccines. The major obstacle facing these new vaccines is the lack of a good delivery system for these vaccines to the target cells. Several nanoparticles have been recently used to enhance the delivery of the vaccines to the target cells. Several multiepitope-based vaccines have been recently designed and tested for many viral diseases affecting humans, particularly SARS-CoV-2 [5,6,7,8,9,10,11] and various species of animals and birds [12,13,14,15,16,17].
The main goal of this research was the development of some novel multiepitopes nanoparticle-based vaccines for Bovine coronavirus (BCoV). We used a combination of some in-silico prediction tools and some other computational tools, which provide a robust framework for the accelerated identification and evaluation of vaccine candidates. Initially, it begins with predicting and analyzing B-cell and T-cell epitopes with high antigenicity and strong binding affinities to MHC class II molecules, essential for eliciting potent immune responses. Followed by predicting the antigenicity, non-toxicity, and non-allergic nature through VaxiJen pro, Toxinpred, Allertop 2.0 and mapping the epitopes via the immune epitope database (IEDB), immune simulation with Immsim, structural modeling using AlphaFold2 colab. This multidisciplinary approach not only enhances the efficiency of vaccine development process but also offers a strategic pathway for generation of targeted immunogenic responses against BCoV. The finding from this study present promising epitope candidate that merit further experimental validation, potentially leading to the development of a next generation BCoV vaccine with improved efficacy and safety profiles. Further, it anticipates the linear B-cell epitopes, which are critical for inducing a humoral immunological response, which in turn primes B cells to produce antibodies. The HTLs recognize foreign antigens and activate B- and cytotoxic T-cells, which in turn cause the immune system to eradicate pathogenic microorganisms. The identification and evaluation of the helper T-cell epitopes binding to MHC class II molecules are important same as that of CTL epitopes and B-cell epitopes, as it also plays a vital role in generating antibodies like B cell and macrophages in turn eliminating the viral infection. In the immune response against viral infections, peptides play a pivotal role in activating helper T cells, which in turn stimulate B cells to differentiate into plasma cells, producing antibodies. However, when neutralizing antibodies alone is insufficient for complete viral eradication, the immune system relies on CD8+ cytotoxic T cells and CD4+ helper T cells. These T-cell-mediated responses are crucial for combating viral infections within host cells, with MHC-peptide complexes serving as critical components akin to antigen-antibody interactions. To neutralize the antibody response and improvise when exposed to toxins and infections, it is crucial to forecast the epitopes in both B-cell and T-cell.
This cutting-edge immunoinformatic method was used to build a vaccination that targets several epitopes. As a potential medication candidate, it should have the following essential characteristics: (i) it should have overlapped B-cell, CTL, and HTL epitopes; (ii) it should be water soluble, immunogenic, antigenic, non-toxic, and non-allergic. The Final vaccine will be created using linkers between adjuvants and epitopes based on these criteria [18]. Finding B-cell epitopes is necessary for the planning and creation of vaccines based on epitopes. Followed by the T-cell epitope prediction is also much more important since these molecules (MHC-I and MHC-II) are potential sources of elevated immune system activity. The need for Prediction and inclusion of B-cell epitopes is necessary in developing vaccine construct, as it involves the derivation of humoral immune response and, they could be easily recognized by the B-cell receptors or secreted antibodies. The action of these epitopes in the vaccine construct is more important as it triggers the efficient immune response and passes the signal to the cytotoxic T-cells to produce an increased number of antibodies to perform its action against the viral genome [19,20]. The Prediction of Cytotoxic T-cell epitopes binding to MHC class I molecules is important and plays a key role in cellular immunity and elimination of virus infection.
The adjuvants, PADRE (Pan HLA-DR reactive epitope), and the Universal T-helper epitopes are the next crucial characters in the vaccination constructions. The latter is considered a simple carrier epitope, useful in making synthetic or recombinant vaccines, and is known to enhance the response when coupled with adjuvants. While the former one, by augmenting the immune response, adjuvants empower vaccines to elicit higher levels of antibody production, thereby facilitating a decrease in the antigen dosage required for optimal immunization outcomes [21,22]. For example, the adjuvants such as cholera toxin subunit-B (CTB) (PDB ID 1XTC), 50S ribosomal protein L7/L12 (NCBI ID: P9WHE3), and so on., Additionally, the linkers can give flexibility and stabilize globular conformation while preserving the vaccine construct's structural integrity. The docking analysis was conducted between the vaccine of all the structural proteins (HE, S, E, M and HE) of BCoV and Toll-like receptors (TLR2 and TLR4). As the toll-like receptors have the potential to initiate an immune response with the production of inflammatory cytokines when they detect the conservative pathogens associate molecules on the range of microbes such as bacteria virus [23]. With referred to the previous research work, several studies show the impact of Toll like receptors in enhancing the immune response of SARS-CoV. The molecular docking analysis was performed with the help of Biovia Discovery Studio, and the details of the interaction between the amino acids were analyzed using the PDBSum computational tool.
2. Materials and Methods:
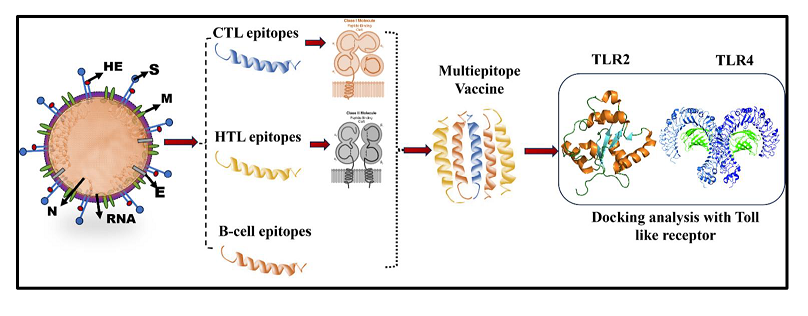
Figure 1 demonstrates the methodology of predicting the epitopes from the desired protein sequence (S spike, N nucleocapsid, M membrane, E envelop, and HE Hemagglutinin-Esterase protein) followed by the analysis of its characteristic features and checking the interaction with the Toll like receptors (TLR2 and TLR4) [24] to enhance its immune response with the binding energy value from molecular docking analysis.The immune simulation is used to confirm the type of binding contact and its attributes through their immunogenic properties [12].
2.1. Retrieval of the BCoV Structural Proteins Sequences
The protein sequences of different BCoV structural were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/). The unique ID of the BCoV structural proteins are (the Hemagglutinin-Esterase protein HE-3CL5 (PDB id) the spike glycoprotein S-P15777, the nucleocapsid protein N-P10525, the membrane proteins M-P69704, the envelope protein E-P15779, were obtained from the Uniport website (https://www.uniprot.org/uniprotkb/P15777/entry). We used the VaxiJen v2.0 and the ANTIGEN pro server to evaluate the protective features of the examined BCoV structural proteins using a threshold value of 0.4 as described elsewhere. Each BCoV was subjected to a rigorous analysis using various bioinformatic tools and several parameters, including the architecture of the major domains, the conservation, and the homology among various BCoV isolates. The comprehensive analysis of individual BCoV structural proteins sheds light on their genetic structures and evolutionary relationship with various BCoV isolates and other coronaviruses. We used the TMHMM online tools to identify the transmembrane helices ™ per each BCoV structural protein. We assigned a value (0-1) as a marker for the good TM helix as previously described [25].
2.2. Prediction and Mapping of B Cells and T Cells Epitopes within the Major Structural Proteins of the BCoV
The default parameters of the IEDB program ((http://tools.iedb.org/main/) were applied as described elsewhere [26] and used to predict the B cell epitopes within the major structural proteins of the BCoV (HE, S, E, M, and N). It was concluded that all the residues containing five amino acids or more and scores over the cutoff point of 0.5 are associated with an individual epitope. The predicted B-cell epitope sequences above were used to predict the CTLs and HTLs epitopes by using the Immune Epitopes Database (IEDB). The epitope ranking score is used to predict them; the lower the value, the better the binding affinity.
2.3. Evaluation of the Major Physicochemical Properties (Antigenicity, Allergenicity, Toxicity, Solubility, and Immunogenicity) to Design the BCoV Multi-Epitope-Based Vaccine Candidates
Briefly, to assess the safety and effectiveness of the vaccine candidates, the ideally designed vaccine must meet the following requirements: it must be antigenic, non-allergenic, non-toxic and water-soluble. Hence, the physicochemical properties of the designed muti-epitopes-based BCoV vaccines were assessed using various bioinformatics software's as previously described. The toxicity and antigenicity nature of the constructed vaccine were evaluated with the VaxiJen and Antigen servers as previously described for the SARS-CoV-2 vaccine candidates [27]. Additionally, the Aller Top and the MHC immunogenicity servers were used to evaluate the immunogenicity and the allergenicity of the designed BCoV vaccine candidates as described [28].
2.4. Designing of the Potential BCoV Multi-Epitope-Based Vaccines from the Major Structural Proteins and the Confirmation of Their Structural Arrangement
Both the NetMHCpan-4.1 and the NetMHCIIpan-4.0 online tools were used to predict the binding affinity of the BCoV antigen of interest with the cellular MHC- class-I and MHC-class-II as previously described [29]. The cholera toxin subunit B was selected as an adjuvant for the potential BCoV vaccines to enhance and sustain the immune response. This adjuvant is linked to the N-terminal domains of the designed vaccine candidates using some EAAAK linkers. We also used the AAY to link the universal T-helper epitope (PADRE or TpD) to the designed multi-epitope-based immunogens as previously described [12]. Moreover, the KK linkers were utilized to join the LBL epitopes with the CTL and HTL epitopes linked together by the AAY linkers, as described in (Figure 2).
2.5. Analysis of the Secondary and the Tertiary Structures of the Designed Multiepitopes BCoV Based Vaccine Candidates
To predict and validate the secondary and tertiary structures of a designed BCoV vaccine candidate, we used several sophisticated computational methods and online tools in this study. Initially we begin with predicting the secondary structure for the final vaccine construct through PSIPRED v4.0 server (http://bioinf.cs.ucl.ac.uk/psipred/), and then the homology models were made using the Biovia Discovery Studio (https://www.3ds.com/products/biovia/discovery-studio) and Alphafold2 colab (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb), and the assessments of the Z-score, the RMSD, and the Ramachandran plot analysis were used to validate the models as previously described [30].
2.6. Molecular Docking and Binding Affinities of the Designed Epitopes-Based Vaccine Candidates with the Cellular Immunoreceptors
The molecular docking can assess Protein-receptors complex binding analysis. These techniques emerged primarily to evaluate the binding affinity of the potential vaccination candidates with host immune receptors. In this scenario, both the Toll receptors like TLR2 and the TLR4 were used as host cell receptors, while the designed BCoV vaccine constructs served as ligands. These receptors function as regulatory receptors for viral antigens, and their complexes further initiate the cascades of several signaling pathways that mobilize immune cells to counteract viral infections [24]. The vaccine constructs were, therefore, docked to the TLR4 (PDB ID:4G8A) and the TLR2 (PDB ID:2Z7X). The Dassault Systems integrated Biovia Discovery studio server was used to evaluate this protein-protein docking validation procedure. Moreover, the PDBSum was utilized to get the graphical representation of the interactions between the vaccine candidates and the host immune receptors as previously described for other viruses such as SARS-COV-2 and Ebola virus vaccine designs [31].
2.7. Applications of the In-Silico Immune Simulation and Computational Immunology to Evaluate the Immunogenic Properties of the Designed Multiepitopes BCoV-Based Vaccine Candidates
The In-silico immune simulation analysis was conducted to analyze the immunogenic property and the interactions between the designed BCoV vaccine candidates with the viral proteins. Utilizing the CIMMSIM v10.1 server (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php?page=1), the machine learning platforms, the immune responses of the designed immunogens at several time intervals were analyzed as previously described [32]. The immune stimulation capabilities of the designed vaccine candidate in the simulation and in the production of the downstream immune response, such as the cytotoxic T-cells, the helper T-cells, the B cells, and the other immune cells also performed as previously described for the SARS-CoV-2 vaccine design [33]. We used the default parameters such as random seed:1234, simulated volume: 10, and the simulation steps 1000.
2.8. Application of a Combination of Codon Optimization and In-Silico Cloning for the Design of the Epitopes-Based Vaccine Constructs
To simulate the performance of the potential vaccine candidate into the selected expression system, either the prokaryotic or the eukaryotic systems, we applied the in-silico cloning approach as previously described [34]. Thus, to guarantee effective translation and enhanced protein synthesis, the codon corresponding to the vaccine candidate must be optimized in accordance with the target expression system. The Vector Builder codon optimization program (https://en.vectorbuilder.com/tool/codon-optimization.html) was used to optimize the vaccine construct's codon usage for the highest possible protein production in the E. coli K-12 strain, followed by the Snap gene tools (https://www.snapgene.com/), were used to create the cloning of chimeric vaccine construct of all the structural proteins of BCoV referring pET-28a (+) as the compatible plasmid vector.
3. Results and Discussion
3.1. Prediction of the Secondary Structures of the BCoV Major Structural Proteins
The Psipred and TMHMM 2.0 were used to predict the secondary structures and the presence of protein in the transmembrane complexes. These tools predict the location of specific amino acids of these proteins, whether the extracellular, the intracellular, or on the surface membrane. Our results showed that the secondary structure of all the structural proteins consists of the alpha helix (pink color), beta-strand (yellow), and the coil turns (grey), shown in the supplementary table as Table S1. As from the previous reference, the range (0 to 1) denotes the suitable transmembrane helices, which is also confirmed by the results presented in Figure 3a–e. The color codes denote (black: the transmembrane, green: (intracellular), and orange; outside the membrane (extracellular).
3.2. Prediction of B Cell and T Cell (MHC Class I and MHC Class II) Epitopes within the BCoV Major Structural Proteins
3.2.1. The B Cell Epitopes Prediction and Assessment
B cell epitopes are important components in many vaccines because they are responsible for triggering the humoral immune response in the immunized/infected hosts. They also potentiate the stimulation of the cytotoxic T cells. The linear/continuous B-cell epitopes were predicted for the major BCoV structural proteins with the help of the Immune Epitope Database (IEDB) computational tool using the default parameters. Each B cell predicted epitope consists of more than five amino acids and was constructed and shown in (Table 1). The list of these potential B cell epitopes was used as the input source for predicting the T cell epitopes, including the classes of MHC (Class I and Class II). The interpreted graphical results (Figure 4a–e) show that the green color peaks represent the presence of epitopes, and the yellow color peaks show the non-epitope sequences across the structural proteins of BCoV.
3.2.2. Prediction of the T Cell Epitopes within the BCoV Major Structural Proteins and Their Binding/Interaction with the MHC -I and the MHC-II Proteins.
3.2.2.1. Cytotoxic T-Lymphocyte Epitope Identification and Evaluation (CTL):
The Prediction of Cytotoxic T-cell epitopes binding to the MHC class I molecules plays a key role in cellular immunity and in the elimination of viral infections. The epitope prediction analysis started with the application of the identified B-cell epitope sequences as an input into the T-cell binding interaction to MHC class I am using the IEBD tool. The epitope predictor version 2023.09 of the NetMHCpan 4.1 EL was used for the Prediction of the alleles (BoLA 1-6, BoLA amani, BoLA T7, BoLA T5, BoLA T2c, BoLA T2b, BoLA JSP.1, BoLA T2a, BoLA HD6, BoLA gb1.7, BoLA D18.4, BoLA, and the AW10) using the default parameters. The predicted epitopes were sorted in a descend peptide predicted score. Confirmation of the epitope prediction was also applied using the IC50 predictions. Once the results were generated, the desired CTL epitopes were filtered initially based on the predicted score of less than or equal to 0.4. The best epitopes were screened, identified, and selected based on their immunogenicity, water solubility, non-allergic, antigenicity, and non-toxic properties.
3.2.2.2. Prediction of the Putative T Helper-Cell Epitopes (HTL) within the Major BCoV Structural Proteins:
The helper T-cell epitopes binding to the MHC class II molecules are as important as that of the CTL epitopes and B-cell epitopes. They play a key role in the stimulation of antibody production by B cells. It also activates macrophages to help eliminate viral infections. We started by filtering the epitopes based on the percentile rank of less than or equal to 4 using the IEBD online tools. We used the recommended epitope predictor version 2023.09 of the NetMHCpan 4.1 EL for the alleles of HLA DRB 1-5 (due to the lack of identified cattle alleles, we used similar parameters used for the Prediction of the Human alleles as previously described). The best epitopes were screened, identified, and selected, as discussed above. The supplementary table Table S2 shows the filtered best epitopes of both CTL and HTL binding to MHC class I and II molecules to be used in the design of the downstream vaccine candidate.
3.3. Homology Modeling and Epitope Visualization of the Potential Vaccine Candidates Based on the Identified Epitopes from the Major BCoV Structural Proteins.
3.3.1. Designing of the Potential Vaccine Constructs Based on the Identified Epitopes in the Major Structural Proteins of the BCoV and Their Conjugation with the Adjuvants.
Our criteria for the potential ideal vaccine constructs are (i) the overlapping between the CTL, the HTL, and the B-cell epitopes, (ii) should have the characteristic nature of being antigenic, non-allergic, non-toxic, immunogenic, and water-soluble, (iii) should have a higher affinity towards the alleles. Based on these criteria, we designed several multiepitope subunit vaccine candidates for the major structural proteins of BCoV. We used the top-ranked (B-cell, T cell (MHC class I and II)) epitopes and then combined each vaccine candidate with the adjuvants and linkers. Previous research used the universal T-helper epitope PADRE (Pan HLA-DR reactive epitope) to enhance the immune response. The Cholera Toxin B (CTB) adjuvant is the other interesting contender that enhances the immune response by activating both the innate and adaptive immune systems.
The main action of CTB in conjugated vaccines is binding to the ganglioside GM1 receptors on the epithelial cell surface. Thus, the CTB adjuvants could be a powerful tool to enhance the efficacy of vaccines by promoting systemic immune responses. They are attached to the N-terminal of the constructed vaccine through the linker EAAK.
The next step in the candidate vaccine design was to conjugate the (CTL, HTL, and linear B cell epitopes) as a multiepitope vaccine with the adjuvant and PADRE, the linkers such as AAY for CTL, GPGPG for HTL, and KK for B-cell epitopes were utilized as shown in Table 2.
3.3.2. Prediction of the 3D Structures of the Designed Multi-Epitope-Based Vaccines for the Major Structural Proteins of BCoV
The Prediction of the tertiary structure of the BCoV candidate vaccines was made using machine learning tools, particularly the Alphafold2 colab. Both the Ramachandran plot and Z-score were also used to validate the structural quality of the developed model, and the results are displayed in supplementary Figure S1a–e. The Z-score of the predicted models was used to select the best model, and the presence of residues in the most advantageous parts of the model validates its highest quality and, consequently, the stability and dependability of the antigen design. When used in tandem, Ramachandran plots and AlphaFold homology modeling greatly improve the accuracy and dependability of vaccine design, guaranteeing the production of stable and successful vaccine candidates.
3.3.3. Visualization of the Multiepitope (B-Cell, CTL, and HTL of T-Cell) of the Major BCoV Structural Proteins
Figure 5.
(a-e). Results of the Visualization of the B cell (green color) and T cell (MHC I (purple color) and MHC II (red color) class) with the predicted epitopes (a) S, (b) N, (c) M, (d) E and (e) HE generated by the Dassault system discovery studio tools.
Figure 5.
(a-e). Results of the Visualization of the B cell (green color) and T cell (MHC I (purple color) and MHC II (red color) class) with the predicted epitopes (a) S, (b) N, (c) M, (d) E and (e) HE generated by the Dassault system discovery studio tools.

3.4. Results of the Molecular Docking of the Designed BCoV Vaccine Constructs with the Immunoreceptors (TLR2 and TLR4).
The results of the docking analysis for the designed vaccine candidates based on the BCoV major structural proteins and the Toll-like receptors (TLR2 and TLR4) are shown in Figure 6. Previous studies showed the impacts of Toll-like receptors in enhancing the immune response of coronaviruses, particularly the SARS-CoV. The molecular docking analysis was performed using the Biovia Discovery Studio, and the details about the interaction between the amino acids were analyzed by the PDBSum computational tool. The results are displayed in supplementary Figure S2a for the TLR2 and in Figure S2b,c for the TLR4.
3.5. In-Silico Immune Simulation
Computational tools such as C- C-Immsim are widely used to predict the immune response and identify effective vaccine candidates. In these models, we used these tools to predict the immune response of the designed vaccine candidates through the interaction between the BCoV antigens, B-cell, T-cell, and cytokines. The simulation was performed using the antigen sequence data from the major BCoV structural proteins (HE, S, M, E, N). The simulation steps were set to 100, while the simulation volume was set to 10. From the supplementary Figure S3 (1-5), it is prominent to understand that the vaccine construct of all the structural proteins shows a the promising trend of enhanced primary and secondary immune responses against BCoV antigens expressed in our designed vaccine constructs.
3.6. The Codon Optimization and In-Silico Cloning of the BCoV Multi-Epitope-Based Vaccines Based on the Individual Structural Proteins:
We used the codons matching each of the vaccine candidates; we used the prokaryotic expression system (E. Coli) to express the target multiepitope-based vaccine candidates described above for the major BCoV structural proteins; results are shown in the supplementary Figure S4a–e. We used the vector builder software (https://en.vectorbuilder.com/) to optimize the multiepitope-based vaccine candidates before their insertion into the prokaryotic expression vector. The recombinant plasmids were created by computationally inserting the modified codon-optimized sequences into the pET-28a (+) vector using the snap Gene program, as shown in the supplementary Figure S5a–e.
4. Discussion
BCoV continues to pose a great risk to the two sections of the cattle industry (dairy and beef). Despite the intensive application of some commercial BCoV vaccines, the virus continues to circulate among most cattle populations across the globe, including the USA. The current BCoV vaccine mainly depends on the live attenuation approach, and the vaccination is usually done through the mucosal surfaces in the respiratory tracts. The commercial BCoV vaccines were not tested in pregnant animals [35,36]. There are major factors that hamper the success of most coronaviruses, including BCoV. First, the poor-proof reading capabilities of CoVs. Second, there are possibilities for recombination among various strains/isolates of CoVs. Taking into consideration the dynamic changes of CoVs, including BCoV on the genomic levels, the side effects of the live attenuated vaccines, particularly reversion to virulence, there is a high demand for the design of some novel vaccines that could be upgradable to match the field circulating strains of BCoV at any time. The applications of AI, simulation, and molecular docking paved the way for the development of novel vaccines not only for BCoV but for other emerging and re-emerging viral diseases. The immuno-informatic approaches using some advanced computational tools could be used to predict and design the multiepitope-based vaccine targeting key structural proteins of BCoV. The usage of this bioinformatics analytical tool to predict the characteristic features of the constructed vaccines, such as antigenicity, allergenicity, and structural validation, alongside molecular docking and in-silico immune simulation, leads to the development of a vaccine capable of eliciting strong cellular and humoral immune responses, paving the way the development of some novel BCoV vaccines to protect cattle against the field viral infection [37].
However, it gathers information from immunology, proteomics, and genomes to provide a thorough understanding of pathogen-host interactions and make identifying new targets easier. Furthermore, it facilitates customized treatment by customizing vaccines to each recipient's DNA profile, improving both efficacy and safety. As we all know, the members of BCoV contain the following structural proteins such as Spike protein, the genome which is responsible for selection and entry into the target cells, and could be the best candidate for a therapeutic targeted approach, which will pave the way to kill deadly pathogens. Other proteins, such as nucleocapsid protein, are involved in RNA packaging, envelope protein in virus assembly, membrane protein in maintaining viral structure, and HE protein is involved in viral entry and immune evasion. We adapted several consecutive steps to develop novel BCoV vaccines based on the multiepitope-conjugated approach [38,39]. First, we conducted a multiple sequence alignment of several BCoV isolates using the Mebus strain (accession no: U00735.2) as a reference strain. We predicted the linear B-cell epitopes within the major BCoV structural proteins. Second, we used the predicted B-cell epitopes as input sequences to predict the T-cell epitopes and the binding affinity score for both MHC I and MHC II class molecules. Third, it is essential to analyze the predicted epitopes' characteristic features as the vaccine construct's major backbone. The following computational tools have been used to analyze the complexity, structural properties, and characteristic nature of these proteins as well as the epitopes predicted from these protein structures. Tools such as VaxiJen 2.0 and Aller top were used to study the antigenic properties and allergic nature of the candidates. This could be visualized through the specific range which is from 0.4 to 0.5, indicating the sequence as antigenic, and from this analysis, we confirmed that the selected protein sequences could be the best candidate to be utilized as antigenic mortal [40,41]. We used the Allertop software tools to check and confirm the allergenicity of each vaccine candidate. Similarly, we tested the potential toxicity of each vaccine candidate using the Toxinpred server tools. Once the epitopes meet the assigned requirements to be vaccine candidates, the epitopes, adjuvant, and linkers are built for the major BCoV structural proteins [42,43].
Fourth, the secondary structures, including alpha helix, beta strands, and coil, were predicted using PSIPRED computational tools. The obtained data demonstrated the designed vaccine candidates showed a globular, flexible, stable, and stable conformation due to the existence of beta strands, coil turn, and an alpha helix. Fifth, we checked the stability of the designed vaccines using the Ramachandran plot and Zscore. Sixth, the tertiary structure of the designed BCoV vaccines was modeled by the AlphaFold colab2 machine learning algorithms tool. Seventh, the multiepitope-based vaccines were visualized using the Biovia discovery studio, Dassault system. This alignment step is critical as it determines the precise locations of each epitope within the 3D conformation of the designed candidate protein, allowing for precise structural analysis. Each epitope was assigned a color code to differentiate it from the rest and for easy identification and downstream analysis [44]. In turn, the amino acids present in the particular region were highlighted to provide a detailed view of the structural context. The representation of the surface was also generated to visualize the accessibility of the epitopes on the protein surface, which is essential for their recognition by the immune system. This will help identify and represent the different categories of the epitopes within the multiepitope vaccine construct, enhancing their potential efficacy. Eighth, the enhanced immune response of our designed BCoV vaccine candidates was tested with Toll-like receptors through binding interaction analytical testing, i.e., docking analytical method using the Z-docker. The protein data bank provided both the TLR-2 and TLR-4 receptors with the PDB ID 2Z7X and PDB ID PDB 4G8A. Both the toll-like receptors crystal structure was determined by the X-ray diffraction with a high resolution of 2.1 Ǻ. Our docking simulation results showed the firm binding affinities between the designed vaccine epitopes and the conjugated TLRs, facilitating effective immune recognition and the initiation of the robust immune response. The molecular docking is a crucial step in the vaccine development. It ensures the specific binding of the designed vaccine epitopes and the target host immune receptors. Below, we discuss the molecular docking results of the designed BCoV multiepitope vaccines based on the major viral structural proteins [45]. The Z score identifies several high-affinity bindings poses in the molecular docking results of the designed BCoV-HE vaccines. Our results showed that the best binding pose after refinement has an energy value of -8.3 kcal/mol. Their interaction with these receptors may help the BCoV to evade the host's immune responses through various mechanisms: (1) altering the signaling pathways (2) suppressing the immune response (3) potentiating the inflammation and tissue damage, the binding of the designed multiepitope vaccine with the TLRs might reduce the receptor availability on host cells, contributing to the viral immune evasion.
The binding analyses of the BCoV-S glycoprotein-designed vaccine candidates with the Toll-like receptors (TLR2 and TLR4) were performed as described above. The PDBsum results showed the interaction between the BCoV/S (chain A) and the TLR2 (chain B). We noticed the presence of 7 hydrogen bonds, three salt bridges, and 343 disulfide bonds. These bonds are called covalent bonds, which ensure stability, structural integrity, and functionality of the generated 3D structural proteins. These disulfide bonds link the different parts of the designed vaccine candidates' polypeptide chain to maintain the protein's folded conformation under various environmental conditions [46]. The BCoV candidate vaccine-TLR2 interaction residues are (Lys505-Sev261, Cys539-Thr247, Ile573-Asp250, Asn550-Lys252, Gln 554-Gly256, Phe325- and Ala274). The surface interaction between the spike protein and the TLR4 is explained, and it is further differentiated into two chain modes, as the TLR4 is a dimer molecule. The interaction with the other dimers was listed as (Ala 572-Arg86, Ser570-Arg86, Val32-Trp101, His68-Tgr145, Lys57-Tyr69, Gln91- and Gln73). Along with this, a few non-bonded contacts were formed.
The BCoV/E protein is a small, integral membrane protein that plays important roles in molecular pathogenesis, assembly, and release. It is also involved in the modulation of the host's immune response against BCoV infection. The docking analysis revealed that the E protein of BCoV interacts strongly with both the TLR2 and the TLR4. The best binding pose shows a binding energy of -7.9 kcal/mol and -8.4 kcal/mol. The details about most interaction sites with a disulfide bond, hydrogen bond, salt bridges, and non-contact bond for both TLRs. The most important key residues involved in this binding/interaction were (Asp144, Glu173, and Arg63 for TLR2 and Asp60, Arg87, and Lys165 for TLR4). The docking interaction between the BCoV/M protein and the Toll-like receptors (TLR2 and TLR4) was assessed. The docking simulation was conducted, and the resultant product provides detailed insights into the binding interactions [47,48]. The most favorable binding poses exhibit a binding energy of -8.7 kcal/mol. These results indicate that the BCoV/M protein also has a strong binding interaction with the TLRs.
In confirmation of the docking results, the PDBSum analysis showed the interaction of the active sites between the BCoV/M/TLR2/TLR4. The most prominent active sites for the TLR2 are (Asp208, Glu554, and Lys527) which create significant di-sulfide bonds with the BCoV/M protein. Similarly, for the TLR4, the interactions involved (Asp55, Arg98, and Phe101) show the important hydrogen bond interactions. Along with that, disulfide bonds and non-bonded contacts were formed. Thus, based on the results of the binding energy value and active interaction site with hydrogen and disulfide bond, it proves that the BCoV/M protein interaction with the TLRs was efficiently engaged and potentially triggers a robust immune response against the designed BCoV/M vaccine candidate.
We applied the analysis conducted above for various BCoV proteins to the designed BCoV/N candidate vaccine construct with the TLR2 and TLR4. The docking analysis using the ZDOCK showed a strong binding interaction between the designed BCoV/N vaccine constructs and the TLR2 and TLR4. The best pose shows a binding energy of -9.2 kcal/mol. This strong affinity suggests effective engagement of TLRs, which is crucial for robust immune responses. The key residues in this interaction are (Asp54, Glu76, and Arg98 ) on the TLR, forming stable hydrogen bonds and hydrophobic interactions with the designed BCoV vaccine constructs. This highlights the potential efficacy of our designed BCoV vaccine candidates [49,50]. The active interaction site from the PDBSum analysis for both toll-like receptors and the BCoV/N protein possess, for TLR2 there are 248 disulfide bonds and 13 hydrogen bonds between the chain A (N protein) and chain B (TLR2), whereas, for TLR4, there are 266 disulfide bonds altogether were presented in the chains from A, B and C and seven hydrogen bonds altogether. The active sites (Pro346-Arg302, Gly349-Glu241, Leu353-Glu215, Gln354-Ile211, Asp344-Gly245) on the TLR2 and the BCoV/N. Regarding the TLR4, the sites were chain A-chain B (Asn352-Met557), Chain B-Chain C (Asn409-Lys435), and the chain A-chain C (Arg343-Lys541, Glu42-Ser391, Asn393-Leu43).
The In-silico immune simulations using C-Immsim provided critical insights about the potential immune responses elicited by the designed BCoV vaccine constructs using various structural proteins. The simulations revealed robust activation of T-cell populations, including cytotoxic T cells and helper T cells, crucial for both cellular and humoral immunity. This comprehensive analysis demonstrated that the BCoV vaccine constructs in this study will induce strong humoral and cell-mediated immunity that might play important roles in protecting cattle against BCoV infection.
Based on the supplementary data, a high level of IgM, IgG1, and IgG2 antibodies and the other immune cells are expected at the time of the administration of the candidate vaccines. It also predicted the progression of the magnitude of the immune response with the progression of the time after administering these candidate vaccines (primary immune response).
In-silico cloning techniques and codon optimization were used to improve the expression and effectiveness of the candidate vaccines in the prokaryotic expression system. Our approach ensured that the vaccination candidates had reliable protein synthesis and effective translation using the optimized codons and utilizing computational tools such as vector builders and snap Gene [51]. The optimized sequence of the designed BCoV vaccine candidates was inserted into an expression vector for the target protein expression. The improved sequence was integrated, and the recombinant BCoV vaccines were cloned using the compatible plasmid vector pET-28a (+). This work aimed to create a viable cloning plan for the various BCoV multiepitope-based vaccine candidates. This method expedited the creation of vaccines and established a strong basis for upcoming clinical trials and experimental validations [52]. Based on the presented data above, we believe our designed BCoV multiepitope-based vaccine candidates will be strong and will be used for the next generation of BCoV vaccines.
Conclusions
We mapped the main epitopes within the major structural proteins of BCoV. We used these epitopes to design several BCoV multi-epitope-based vaccines that show high antigenicity immunogenicity and are non-toxic and non-allergic. These vaccine candidates are linked with strong immunostimulants (TLR2 and TLR4) as well as the Chlora toxin B adjuvant to ensure a sustained immune response for a long time in the vaccinated cattle against BCoV. The combination of AI tools, including epitope prediction, immune stimulation, and docking, provides a novel approach to vaccine development and reduces the time required for vaccine development and approval.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: The Ramachandran plot analysis of the designed BCoV vaccine constructs for (a) HE, (b) S, (c) E, (d) M, and (e) N analyzed by the Biovia discovery studio, Dassault system. Figure S2 (a) Mapping the positions of the binding of the BCoV candidate vaccine construct with the TLR2 for (a) HE, (b) S, (c) E, (d) M, and (e) N analyzed by the PDBSum online tool. Figure S2 (b) Mapping the positions of the binding of the BCoV candidate vaccine construct with the TLR4 for (a) HE, (b) S, (c) E, (d) M, and (e) N analyzed by the PDBSum online tool. Figure S2 (c) Mapping the positions of the binding of the BCoV candidate vaccine construct with the TLR4 for (d) M and (e) N analyzed by the PDBSum online tools. Figure S3 (1-5) Immune simulation analysis of (1) Hemagglutinin-Esterase protein (HE), (2) spike protein (S), (3) Envelop protein (E), (4) membrane protein (M) and (5) Nucleocapsid protein (N) using the C-ImmSim computational tool, (a-c) B-cell populations (IgM, IgG1, IgG2) in response to the designed BCoV candidate vaccines, B-cell populations in different states (active, internalized, and anergic), helper T-cell count in the resting and active states, B -cell populations )IgG and IgM) reached up to 7.2 cells per mm3, helper T-cell population, (d- h) cytotoxic T-cell count, natural killer cells throughout the 35 days, which reached a level of up to a maximum of 370 cells per mm3, dendritic cell population in the active and resting states, T-cytotoxic cell populations in different states, resting and active, activity in terms of counts and activation states of cytotoxic T cells, helper T cells and macrophages respectively, (i - m) elevation of cytokines and interleukin cells, (n) Elevation of Immunoglobulins at different concentrations of antigens. Figure S4 (a-e) Maps of the designed vaccine candidate constructs using the Codon optimization for the BCoV major structural proteins of (a) S, (b) N, (c) M, (d) E (e) and HE using the Vector builder computational tool analysis. Figure S5 (a-e) The in-silico cloning design of the expression constructs of the vaccine candidates based on the selected epitopes within the BCoV major structural proteins (a) (S), (b) N, (c) M, (d) (E) (e) HE proteins using the Snap gene computational tool analysis. Table S1: Results of the secondary structure prediction of the BCoV structural proteins a) HE, (b) S, (c) E, (d) M, and (e) N using the Protein Structure Prediction Server (PSIPRED). Table S2: Summary of the information of the shortlisted epitopes used for the design of the BCoV candidate vaccine constructs based on the major BCoV structural proteins (HE, S, E, M, N) binding with MHC class I and class II molecules.
Author Contributions
Conceptualization, MGH. and MC.; methodology, MGH, ND, YMK, AS, RNE.; software, ND, YMK, AS, RNE.; validation, ND, YMK and AS.; formal analysis, ND, YMK and AS.; investigation, MGH, MC, RNE.; resources, MGH, MC, RNE ; data curation, MGH, ND, YMK, AS, RNE..; writing—original draft preparation, ND, MGH.; writing—review and editing, MGH, ND, YMK, AS, RNE, MC.; Visualization, MGH, ND, YMK, AS, RNE, MC.; supervision, MGH.; project administration, MGH, RNE, MC.; funding acquisition, MGH, RNE, MC. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Saif, L.J. Bovine respiratory coronavirus. Vet. Clin. North. Am. Food Anim. Pract. 2010, 26, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, B.; Sun, D. Advances in Bovine Coronavirus Epidemiology. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Blakebrough-Hall, C.; Hick, P.; Mahony, T.J.; Gonzalez, L.A. Factors associated with bovine respiratory disease case fatality in feedlot cattle. J. Anim. Sci. 2022, 100. [Google Scholar] [CrossRef] [PubMed]
- Thanthrige-Don, N.; Lung, O.; Furukawa-Stoffer, T.; Buchanan, C.; Joseph, T.; Godson, D.L.; Gilleard, J.; Alexander, T.; Ambagala, A. A novel multiplex PCR-electronic microarray assay for rapid and simultaneous detection of bovine respiratory and enteric pathogens. J. Virol. Methods 2018, 261, 51–62. [Google Scholar] [CrossRef]
- Abdelmageed, M.I.; Abdelmoneim, A.H.; Mustafa, M.I.; Elfadol, N.M.; Murshed, N.S.; Shantier, S.W.; Makhawi, A.M. Design of a Multiepitope-Based Peptide Vaccine against the E Protein of Human COVID-19: An Immunoinformatics Approach. Biomed. Res. Int. 2020, 2020, 2683286. [Google Scholar] [CrossRef] [PubMed]
- Arwansyah, A.; Arif, A.R.; Kade, A.; Taiyeb, M.; Ramli, I.; Santoso, T.; Ningsih, P.; Natsir, H.; Tahril, T.; Uday Kumar, K. Molecular modelling on multiepitope-based vaccine against SARS-CoV-2 using immunoinformatics, molecular docking, and molecular dynamics simulation. SAR QSAR Environ. Res. 2022, 33, 649–675. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kancharla, S.; Kolli, P.; Jena, M. Reverse vaccinology approach towards the in-silico multiepitope vaccine development against SARS-CoV-2. F1000Res 2021, 10, 44. [Google Scholar] [CrossRef]
- Pandey, A.; Madan, R.; Singh, S. Immunology to Immunotherapeutics of SARS-CoV-2: Identification of Immunogenic Epitopes for Vaccine Development. Curr. Microbiol. 2022, 79, 306. [Google Scholar] [CrossRef]
- Tahir Ul Qamar, M.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS One 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Umitaibatin, R.; Harisna, A.H.; Jauhar, M.M.; Syaifie, P.H.; Arda, A.G.; Nugroho, D.W.; Ramadhan, D.; Mardliyati, E.; Shalannanda, W.; Anshori, I. Immunoinformatics Study: Multi-Epitope Based Vaccine Design from SARS-CoV-2 Spike Glycoprotein. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Waqas, M.; Haider, A.; Rehman, A.; Qasim, M.; Umar, A.; Sufyan, M.; Akram, H.N.; Mir, A.; Razzaq, R.; Rasool, D.; et al. Immunoinformatics and Molecular Docking Studies Predicted Potential Multiepitope-Based Peptide Vaccine and Novel Compounds against Novel SARS-CoV-2 through Virtual Screening. Biomed. Res. Int. 2021, 2021, 1596834. [Google Scholar] [CrossRef] [PubMed]
- Elshafei, S.O.; Mahmoud, N.A.; Almofti, Y.A. Immunoinformatics, molecular docking and dynamics simulation approaches unveil a multi epitope-based potent peptide vaccine candidate against avian leukosis virus. Sci. Rep. 2024, 14, 2870. [Google Scholar] [CrossRef] [PubMed]
- Ghafoor, D.; Zeb, A.; Ali, S.S.; Ali, M.; Akbar, F.; Ud Din, Z.; Ur Rehman, S.; Suleman, M.; Khan, W. Immunoinformatic based designing of potential immunogenic novel mRNA and peptide-based prophylactic vaccines against H5N1 and H7N9 avian influenza viruses. J. Biomol. Struct. Dyn. 2024, 42, 3641–3658. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.M.; Tong, G.Z.; Wang, Y.F.; Qiu, H.J. [Multi-epitope DNA vaccines against avian influenza in chickens]. Sheng Wu Gong. Cheng Xue Bao 2003, 19, 623–627. [Google Scholar] [PubMed]
- Tataje-Lavanda, L.; Malaga, E.; Verastegui, M.; Mayta Huatuco, E.; Icochea, E.; Fernandez-Diaz, M.; Zimic, M. Identification and evaluation in-vitro of conserved peptides with high affinity to MHC-I as potential protective epitopes for Newcastle disease virus vaccines. BMC Vet. Res. 2023, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, H.N.; Lu, D.; Zhang, Y.F.; Wang, T.; Kang, R.M. The immunoreactivity of a chimeric multi-epitope DNA vaccine against IBV in chickens. Biochem. Biophys. Res. Commun. 2008, 377, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ma, X.; Wang, F.; Li, H.; Xiao, Y.; Zhao, X. Design and construction of a chimeric multi-epitope gene as an epitope-vaccine strategy against ALV-J. Protein Expr. Purif. 2015, 106, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sanami, S.; Nazarian, S.; Ahmad, S.; Raeisi, E.; Tahir Ul Qamar, M.; Tahmasebian, S.; Pazoki-Toroudi, H.; Fazeli, M.; Ghatreh Samani, M. In silico design and immunoinformatics analysis of a universal multi-epitope vaccine against monkeypox virus. PLoS One 2023, 18, e0286224. [Google Scholar] [CrossRef] [PubMed]
- Cia, G.; Pucci, F.; Rooman, M. Critical review of conformational B-cell epitope prediction methods. Brief. Bioinform. 2023, 24. [Google Scholar] [CrossRef]
- Jimenez-Vasquez, V.; Calvay-Sanchez, K.D.; Zarate-Sulca, Y.; Mendoza-Mujica, G. In-silico identification of linear B-cell epitopes in specific proteins of Bartonella bacilliformis for the serological diagnosis of Carrion's disease. PLoS Negl. Trop. Dis. 2023, 17, e0011321. [Google Scholar] [CrossRef]
- Abdulla, F.; Nain, Z.; Hossain, M.M.; Syed, S.B.; Ahmed Khan, M.S.; Adhikari, U.K. A comprehensive screening of the whole proteome of hantavirus and designing a multi-epitope subunit vaccine for cross-protection against hantavirus: Structural vaccinology and immunoinformatics study. Microb. Pathog. 2021, 150, 104705. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Akter, S.; Rubayet Ul Alam, A.S.M.; Siddique, M.A.; Saha, O.; Rahaman, M.M.; Sultana, M.; Crandall, K.A.; et al. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2, the etiologic agent of COVID-19 pandemic: an in silico approach. PeerJ 2020, 8, e9572. [Google Scholar] [CrossRef]
- Saravanan, D.; Mohan, M. Immunoinformatics-driven approach for development of potential multi-epitope vaccine against the secreted protein FlaC of Campylobacter jejuni. J. Biomol. Struct. Dyn. 1080. [Google Scholar] [CrossRef]
- Naz, A.; Shahid, F.; Butt, T.T.; Awan, F.M.; Ali, A.; Malik, A. Designing Multi-Epitope Vaccines to Combat Emerging Coronavirus Disease 2019 (COVID-19) by Employing Immuno-Informatics Approach. Front. Immunol. 2020, 11, 1663. [Google Scholar] [CrossRef] [PubMed]
- Pumchan, A.; Proespraiwong, P.; Sawatdichaikul, O.; Phurahong, T.; Hirono, I.; Unajak, S. Computational design of novel chimeric multiepitope vaccine against bacterial and viral disease in tilapia (Oreochromis sp.). Sci. Rep. 2024, 14, 14048. [Google Scholar] [CrossRef] [PubMed]
- Fleri, W.; Vaughan, K.; Salimi, N.; Vita, R.; Peters, B.; Sette, A. The Immune Epitope Database: How Data Are Entered and Retrieved. J. Immunol. Res. 2017, 2017, 5974574. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Jain, A.; Verma, S.K. Prediction of Epitope based Peptides for Vaccine Development from Complete Proteome of Novel Corona Virus (SARS-COV-2) Using Immunoinformatics. Int. J. Pept. Res. Ther. 2021, 27, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, X.; Fei, C. A Highly Effective System for Predicting MHC-II Epitopes With Immunogenicity. Front. Oncol. 2022, 12, 888556. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Hooft, R.W.; Sander, C.; Vriend, G. Objectively judging the quality of a protein structure from a Ramachandran plot. Comput. Appl. Biosci. 1997, 13, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.; Amini-Khoei, H.; Tahmasebian, S.; Ghatrehsamani, M.; Ghatreh Samani, K.; Edalatpanah, Y.; Rostampur, S.; Salehi, M.; Ghasemi-Dehnoo, M.; Azadegan-Dehkordi, F.; et al. Designing a novel multi-epitope vaccine against Ebola virus using reverse vaccinology approach. Sci. Rep. 2022, 12, 7757. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sood, D.; Chandra, R. Design and optimization of a subunit vaccine targeting COVID-19 molecular shreds using an immunoinformatics framework. RSC Adv. 2020, 10, 35856–35872. [Google Scholar] [CrossRef]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef] [PubMed]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar] [CrossRef] [PubMed]
- Sevinc Temizkan, S.; Alkan, F. Bovine coronavirus infections in Turkey: molecular analysis of the full-length spike gene sequences of viruses from digestive and respiratory infections. Arch. Virol. 2021, 166, 2461–2468. [Google Scholar] [CrossRef]
- Dashti, F.; Raisi, A.; Pourali, G.; Razavi, Z.S.; Ravaei, F.; Sadri Nahand, J.; Kourkinejad-Gharaei, F.; Mirazimi, S.M.A.; Zamani, J.; Tarrahimofrad, H.; et al. A computational approach to design a multiepitope vaccine against H5N1 virus. Virol. J. 2024, 21, 67. [Google Scholar] [CrossRef]
- Gupta, Y.; Savytskyi, O.V.; Coban, M.; Venugopal, A.; Pleqi, V.; Weber, C.A.; Chitale, R.; Durvasula, R.; Hopkins, C.; Kempaiah, P.; et al. Protein structure-based in-silico approaches to drug discovery: Guide to COVID-19 therapeutics. Mol. Aspects Med. 2023, 91, 101151. [Google Scholar] [CrossRef]
- M.Y. Khan, N.D., A. M.Y. Khan, N.D., A.U. Shah, R.N. ElAlaoui, M. Cherkaoui, M.G. Hemida, Identification of Some Potential Cellular Receptors and Host Enzymes That Could Potentially Refine the Bovine Coronavirus (BCoV) Replication and Tissue Tropism. A Molecular Docking Study, (2024).
- Kaur, B.; Karnwal, A.; Bansal, A.; Malik, T. An Immunoinformatic-Based In Silico Identification on the Creation of a Multiepitope-Based Vaccination Against the Nipah Virus. Biomed. Res. Int. 2024, 2024, 4066641. [Google Scholar] [CrossRef] [PubMed]
- Fathollahi, M.; Motamedi, H.; Hossainpour, H.; Abiri, R.; Shahlaei, M.; Moradi, S.; Dashtbin, S.; Moradi, J.; Alvandi, A. Designing a novel multi-epitopes pan-vaccine against SARS-CoV-2 and seasonal influenza: in silico and immunoinformatics approach. J. Biomol. Struct. Dyn. 1080. [Google Scholar] [CrossRef]
- Sami, S.A.; Marma, K.K.S.; Mahmud, S.; Khan, M.A.N.; Albogami, S.; El-Shehawi, A.M.; Rakib, A.; Chakraborty, A.; Mohiuddin, M.; Dhama, K.; et al. Designing of a Multi-epitope Vaccine against the Structural Proteins of Marburg Virus Exploiting the Immunoinformatics Approach. ACS Omega 2021, 6, 32043–32071. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Molaei, A.; Fard, N.A. Multi-epitopevaccines, from design to expression; an in silico approach. Hum. Immunol. 2024, 85, 110804. [Google Scholar] [CrossRef]
- Liang, S.; Zhang, S.; Bao, Y.; Zhang, Y.; Liu, X.; Yao, H.; Liu, G. Combined Immunoinformatics to Design and Evaluate a Multi-Epitope Vaccine Candidate against Streptococcus suis Infection. Vaccines (Basel) 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Kornbluth, R.S.; Stone, G.W. Immunostimulatory combinations: designing the next generation of vaccine adjuvants. J. Leukoc. Biol. 2006, 80, 1084–1102. [Google Scholar] [CrossRef] [PubMed]
- Hopp, M.T.; Holze, J.; Lauber, F.; Holtkamp, L.; Rathod, D.C.; Miteva, M.A.; Prestes, E.B.; Geyer, M.; Manoury, B.; Merle, N.S.; et al. Insights into the molecular basis and mechanism of heme-triggered TLR4 signalling: The role of heme-binding motifs in TLR4 and MD2. Immunology 2024, 171, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.J.I.; Hossain, M.F.; Neef, J.; Zwack, E.E.; Tsai, C.M.; Raafat, D.; Fechtner, K.; Herzog, L.; Kohler, T.P.; Schluter, R.; et al. TLR4 sensing of IsdB of Staphylococcus aureus induces a proinflammatory cytokine response via the NLRP3-caspase-1 inflammasome cascade. mBio 2024, 15, e0022523. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Wu, K.H.; Wu, H.P. Unraveling the Complexities of Toll-like Receptors: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Oladipo, E.K.; Ojo, T.O.; Olufemi, S.E.; Irewolede, B.A.; Adediran, D.A.; Abiala, A.G.; Hezekiah, O.S.; Idowu, A.F.; Oladeji, Y.G.; Ikuomola, M.O.; et al. Proteome based analysis of circulating SARS-CoV-2 variants: approach to a universal vaccine candidate. Genes. Genomics 2023, 45, 1489–1508. [Google Scholar] [CrossRef] [PubMed]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope Prediction by Novel Immunoinformatics Approach: A State-of-the-art Review. Int. J. Pept. Res. Ther. 2020, 26, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Yurina, V.; Adianingsih, O.R. Predicting epitopes for vaccine development using bioinformatics tools. Ther. Adv. Vaccines Immunother. 2022, 10, 25151355221100218. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Akhtar, N.; Sharma, N.R.; Kaushik, V.; Borkotoky, S. MERS virus spike protein HTL-epitopes selection and multi-epitope vaccine design using computational biology. J. Biomol. Struct. Dyn. 2023, 41, 12464–12479. [Google Scholar] [CrossRef]
Figure 1.
The proposed model workflow for the predicting of the linear B cell and T cell epitopes and the docking analysis of the binding interactions of these potential vaccines constructs with the TLR Complex.
Figure 1.
The proposed model workflow for the predicting of the linear B cell and T cell epitopes and the docking analysis of the binding interactions of these potential vaccines constructs with the TLR Complex.
Figure 2.
Schematic model of the design of the BCoV (B cells and T cells) multi-epitopes-based vaccine using the PADRE as linkers and the Cholera toxin subunit B as an adjuvant.
Figure 2.
Schematic model of the design of the BCoV (B cells and T cells) multi-epitopes-based vaccine using the PADRE as linkers and the Cholera toxin subunit B as an adjuvant.
Figure 3.
Identification of the transmembrane helices among the BCoV major structural proteins (a) HE, (b) S, (c) E, (d) M, and (e) N protein using the TMHMM 2.0 computational tools.
Figure 3.
Identification of the transmembrane helices among the BCoV major structural proteins (a) HE, (b) S, (c) E, (d) M, and (e) N protein using the TMHMM 2.0 computational tools.
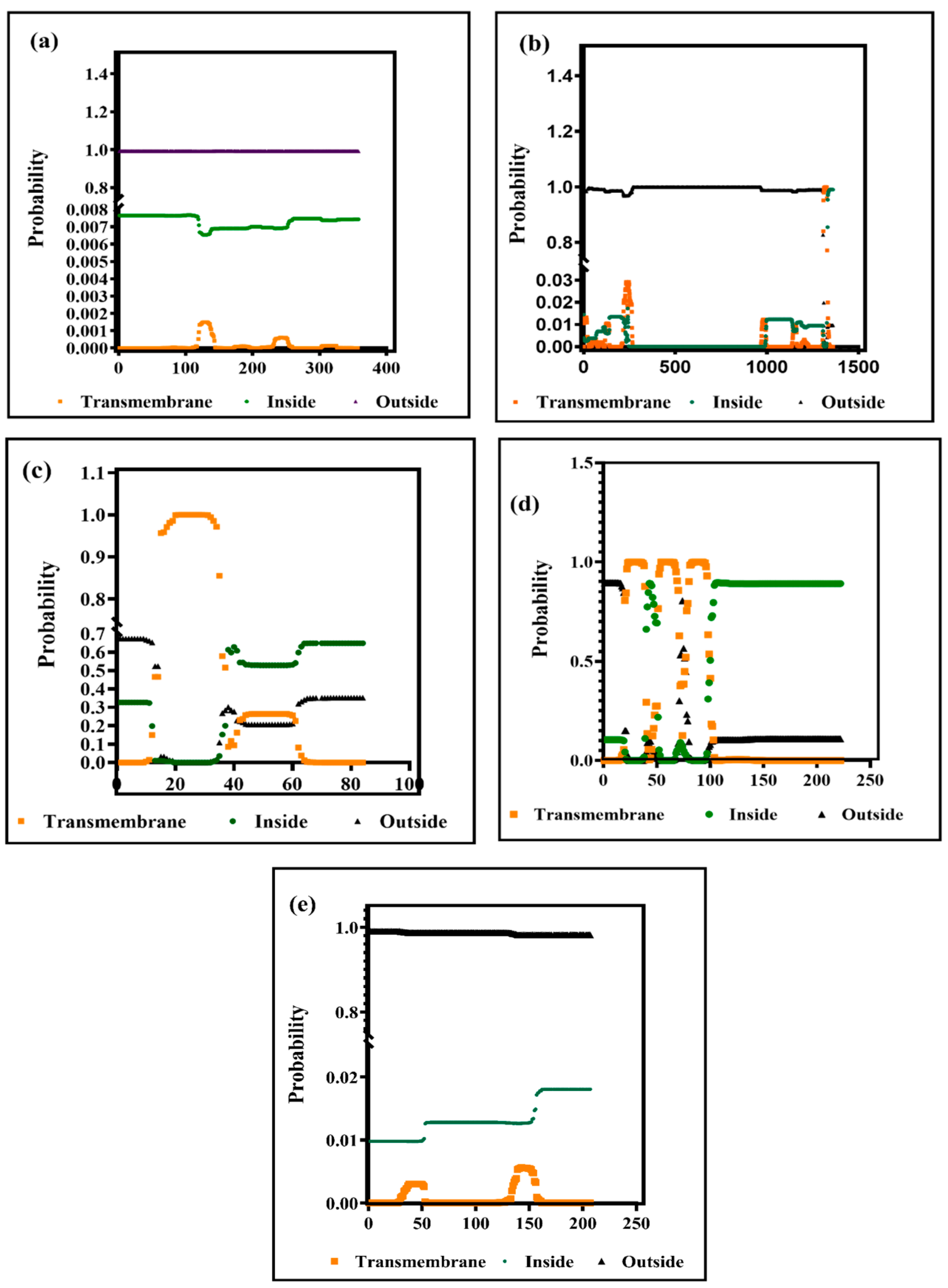
Figure 4.
Prediction of B-cell epitopes among the BCoV major structural proteins: (a) HE, (b) S, (c) E, (d) M, and (e) N.
Figure 4.
Prediction of B-cell epitopes among the BCoV major structural proteins: (a) HE, (b) S, (c) E, (d) M, and (e) N.
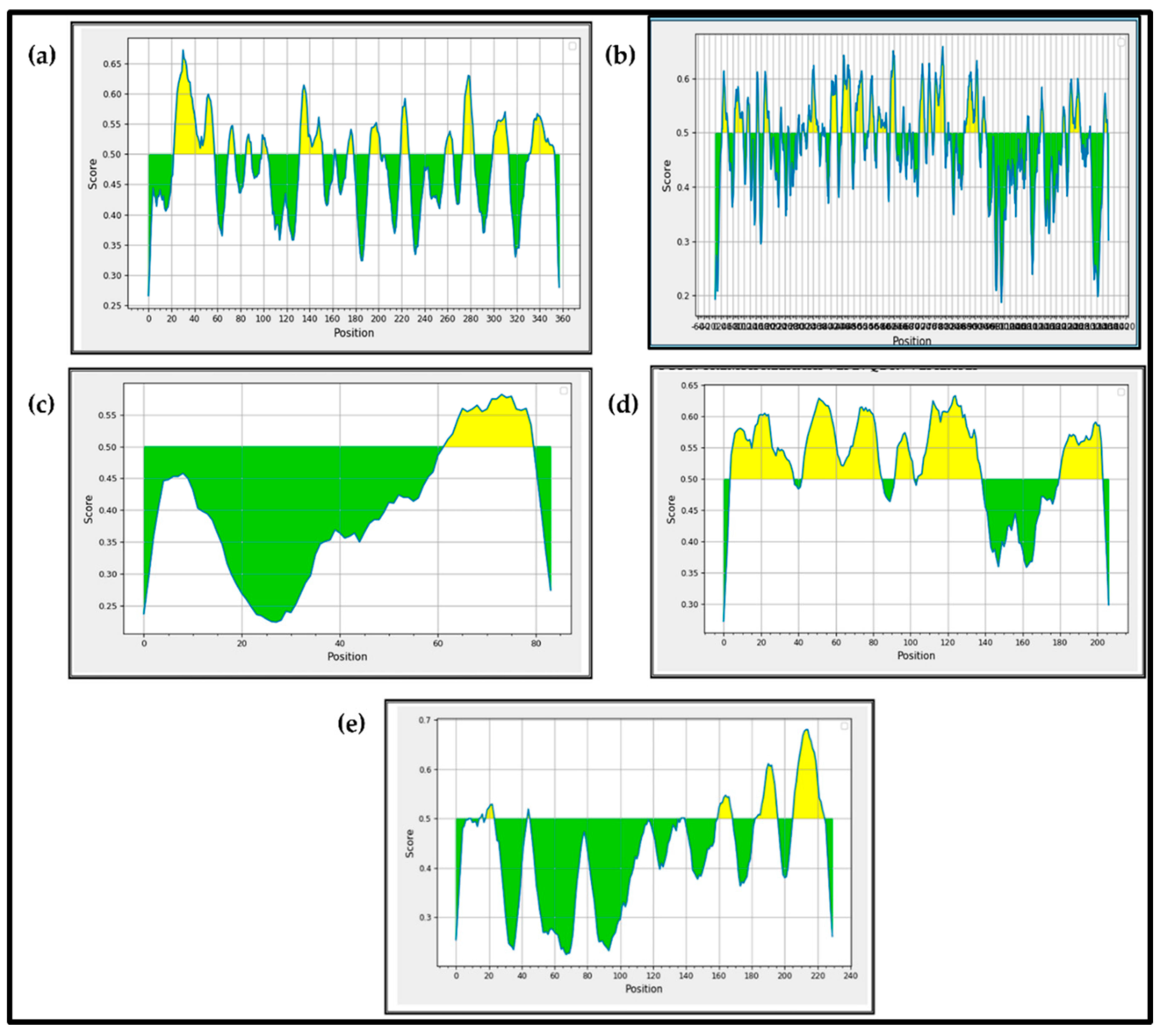
Figure 6.
The molecular docking analysis results of the designed vaccine candidates based on the individual structural proteins of the BCoV conjugated with the TLR2 and TLR4 using the Z-docker of the Biovia discovery studio, Dassault system.
Figure 6.
The molecular docking analysis results of the designed vaccine candidates based on the individual structural proteins of the BCoV conjugated with the TLR2 and TLR4 using the Z-docker of the Biovia discovery studio, Dassault system.
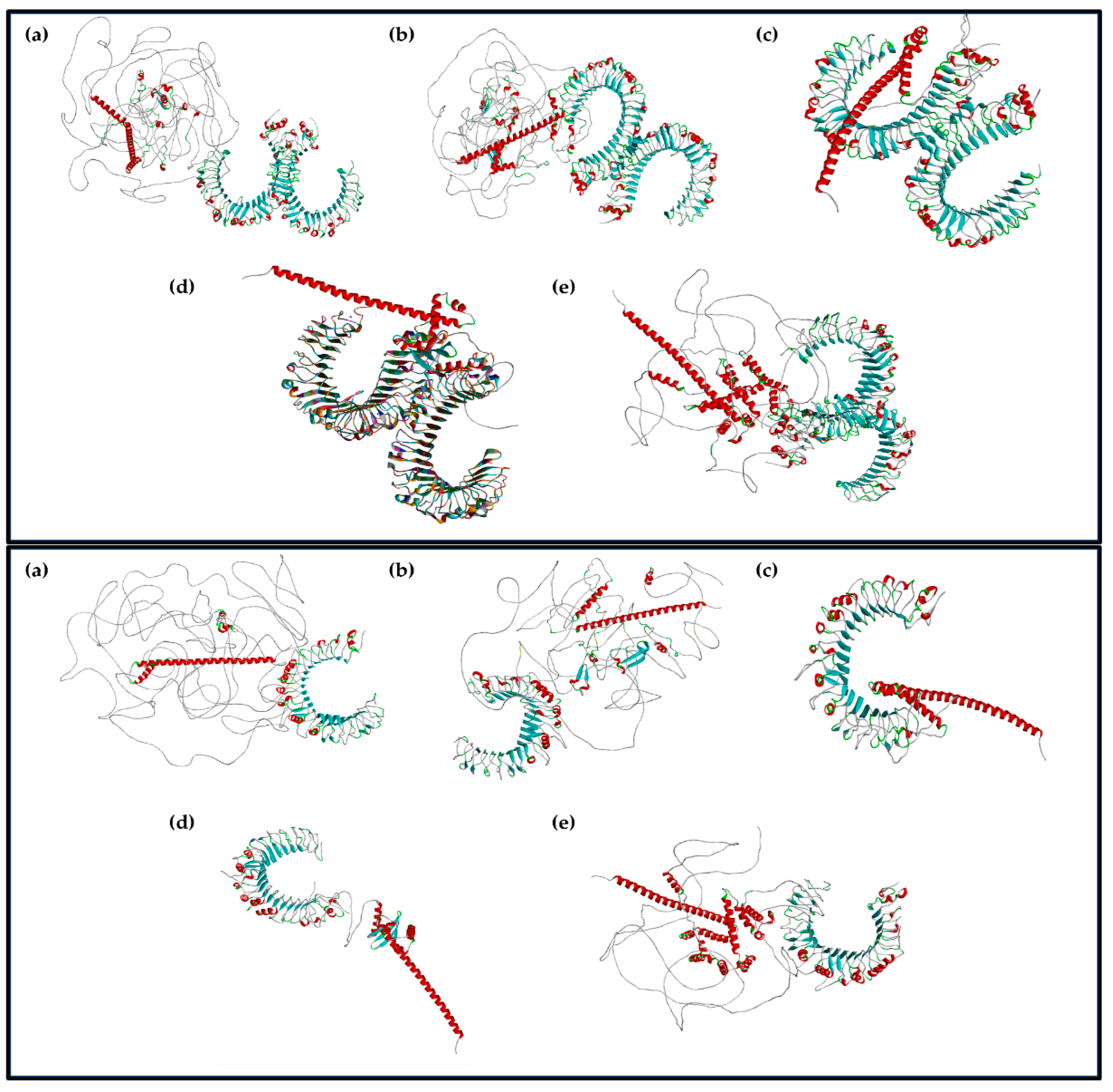

Table 1.
Results of the B cell epitope sequences within the BCoV major structural proteins.
| Protein | B-cell epitope Sequence |
|---|---|
| HE | RSDCNHVVNTNPRNYSYMDLNPALCDSGKISSKAGNNYTGEGNFTPYSNDIWVYNGSAQSTALCKSGSLVLNFYLSGCFLSNTKYYDDTETITTGFLNKRKDFRWNNARQSDNMTNYVGVYDINHGDAQQGVFRYDNVSSVWPLYSYGRCP |
| S | VSINDVDTGAPSISTDIVDVTNYPTSGSTYRNMALKGTLLLSRLWFKPPFLSDTKVIKKGVMYSTTNLDNKLQHTICHPNLGNKRVELWHWDTGKSDFMSIAPSTGVYEDVYRRIPNLPDCNIEAWLNDKSVPSPLNWERKTFSNCNFGRKVDLQLGNLGYLQSFNYRIDTTVSVSRFNPSTWNRRFGFTEQFVFKPQPVGVFTHHDFCPCKLDGSLCVGNGPGIDAGYKNSGIGTCLCTPDPITSKSTGPYKCPQSGLAIKSDYCGGNPCTCQPQAFLGWSVDSCLQGHDVNSGTTCSTDLQKSNTDIATYYNSDYLTNKCNYVFNNTLSRQLQPINYFADNSTSSVVQTCDLTDYSTKRRSRRAITTGYRFTTFEPFTVNSVNDSLEPVGGDTTQLQVANSLMNGVTLSTKLKDGVNFNVDDINFSPVLGCLGSDCNKVSSRLSDVGFVEAYNNLEAQAQIDKSQSSRINFCGYYYPEPITGNKAPDVMLNISTPNLHDFKEELDQWFKNQTSVAPDLSLDYICDDYTGHQELVIKT |
| E | RGRQFYEFYNDVKPPVLD |
| M | IKFLKELGTGYSLSDTYKRGFLDKIGDTKVGNYRLPSTQKGSGMDTAL |
| N | SGPISPTNLEMFKPGVEELNPSKLLLLSYHQEGMILGSLELLSFKKERSLNLQRDKVCLLHQESQLLKLRGTGTDTTMATSVNCCHDFTILEQDRMPKTSMAPILTESSGSLVTRLMSIPRLLHAHPVEPLVQDRVVEPILATEP |
Table 2.
Structure and sequences of the designed multiepitope-based vaccine candidates based on the major BCoV structural proteins.
Table 2.
Structure and sequences of the designed multiepitope-based vaccine candidates based on the major BCoV structural proteins.
| Protein | Final Vaccine Construct |
|---|---|
| HE | MSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDELEAAAKAKFVAAWTLKAAAAAYALCDSGKISSKAAYAQSTALCKAAYFLNKRKDFAAYFRYDNVSSVAAYNKRKDFRWAAYNNARQSDNMAAYRQSDNMTNYAAYRYDNVSSVWAAYSGKISSKAAAYSSKAGNNYAAYTTGFLNKRKAAYVNTNPRNYAAYALCDSGKISSKAGNNGPGPARQSDNMTNYVGVYDGPGPGCDSGKISSKAGNNYTGPGPGDAQQGVFRYDNVSSVGPGPGDDTETITTGFLNKRKGPGPGDFRWNNARQSDNMTNGPGPGDINHGDAQQGVFRYDGPGPGDLNPALCDSGKISSKGPGPGDSGKISSKAGNNYTGGPGPGDTETITTGFLNKRKDGPGPGFLNKRKDFRWNNARQGPGPGFRWNNARQSDNMTNYGPGPGCFLSNTKYYDDTETGPGPGGFLNKRKDFRWNNARGPGPGGKISSKAGNNYTGEGGPGPGISSKAGNNYTGEGNFGPGPGITTGFLNKRKDFRWNGPGPGKDFRWNNARQSDNMTGPGPGKISSKAGNNYTGEGNGPGPGKRKDFRWNNARQSDNGPGPGLNKRKDFRWNNARQSGPGPGLNPALCDSGKISSKAGPGPGMDLNPALCDSGKISSGPGPGNNARQSDNMTNYVGVGPGPGNNYTGEGNFTPYSNDGPGPGNPALCDSGKISSKAGGPGPGPALCDSGKISSKAGNGPGPGPRNYSYMDLNPALCDGPGPGRKDFRWNNARQSDNMGPGPGRNYSYMDLNPALCDSGPGPGRSDCNHVVNTNPRNYGPGPGSSKAGNNYTGEGNFTGPGPGSYMDLNPALCDSGKIGPGPGTGFLNKRKDFRWNNAGPGPGTITTGFLNKRKDFRWGPGPGTTGFLNKRKDFRWNNGPGPGVYDINHGDAQQGVFRGPGPGYDDTETITTGFLNKRGPGPGYDINHGDAQQGVFRYGPGPGYMDLNPALCDSGKISGPGPGYSYMDLNPALCDSGKKKRSDCNHVVNTNPRNYSYMDLNPALCDSGKISSKAGNKKNYTGEGKKNFTPYKKSNDIWKKVYNGSAQSTALCKSGSLVLNKKFYLSGCKKFLSNTKYYDDKKTETITTGFKKLNKRKDFKKRWNNARQSDNMKKTNYVGVYDINHGDAKKQQGVFRYDNVSSVWPLYSYGRCP |
| S | MSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDELEAAAKAKFVAAWTLKAAAAAYDKSVPSPLNWAAYKSQSSRINFAAYLGNKRVELWAAYLNDKSVPSPLNWAAYLSDTKVIKKAAYNDKSVPSPLNWAAYRNMALKGTLLWAAYRRFGFTEQFAAYSQSSRINFAAYSAKSDFMSIAAYSTTNLDNKLQHAAYTNLDNKLQHAAYYRNMALKGTLAAYTSKSTGPYKAAYTTNLDNKLQHGPGPGSDVGFVEAYNNLEAQGPGPGDVGFVEAYNNLEAQAGPGPGFEPFTVNSVNDSLEPGPGPGTFEPFTVNSVNDSLEGPGPGPEPITGNKAPDVMLNGPGPGYYPEPITGNKAPDVMKKVSINDVDTGAPSISTDIVDVTNKKYPTSGSTYRNMALKGTLLLSRLWFKPPFLSDKKTKVIKKGVMYSKKTTNLDNKLQKKHTICHPNLGNKRVELWHWDTGKKGVVTKKKSSAKKKSDFMSKKIAPSTGVYEKKDVYRRIPNLPDCNIEAWLNDKSVPSPLNWERKTFSNCNFKKAKKFTCNKKIKKAAKKKGRKVDLQLGNLGYLQSFNYRIDTTKKVSVSRFNPSTWNRRFGFTEQFVFKPQPVGVFTHHDKKFCPCKLDGSLCVGNGPGIDAGYKNSGIGTKKQKKCLCTPDPITSKSTGPYKCPQKKSGLAIKSDYCGGNPCTCQPQAFLGWSVDSCLQGKKHDVNSGTTCSTDLQKSNTDIKKATYYNSKKDYLTNKKSKKKCNYVFNNTLSRQLQPINYFKKADNSTSSVVQTCDLTKKDYSTKRRSRRAITTGYRFTTFEPFTVNSVNDSLEPVGGKKTIGKKDTTQLQVANSLMNGVTLSTKLKDGVNFNVDDINFSPVLGCLGSDCNKVSSRKKLSDVGFVEAYNNKKLEAQAQIDKKKSQSSRINFCGKKYYYPEPITGNKKKAPDVMLNISTPNLHDFKEELDQWFKNQTSVAPDLSLDYIKKIGTKKCDDYTGHQELVIKT |
| E | MSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDELEAAAKAKFVAAWTLKAAAAAYYNDVKPPVLAAYRGRQFYEFYAAYYNDVKPPVLAAYRQFYEFYNDVAAYNDVKPPVLGPGPGQFYEFYNDVKPPVLDGPGPGRGRQFYEFYNDVKPPKKRGRQFYEFYNDVKPPVLD |
| M | MSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDELEAAAKAKFVAAWTLKAAAAAYTQKGSGMDTALAAYTGYSLSDTYKAAYRGFLDKIGDTKAAYYSLSDTYKAAYFLKELGTGYAAYKELGTGYSLAAYDTKVGNYRLAAYKGSGMDTALAAYYSLSDTYKRGPGPGIKFLKEKKLGTGYSLSDKKTYKRGFLDKKIGDTKKKVGNYRLPSTQKGSGMDTALKK |
| N | MSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDELEAAAKAKFVAAWTLKAAAAAYLQRDKVCLLAAYGSLELLSFKAAYSLNLQRDKVCLLAAYRSLNLQRDKAAYRSLNLQRDKVCLLAAYLLSFKKERSLAAYLQRDKVCLLHAAYLSFKKERSLAAYEELNPSKLLAAYSLELSFKKERSLAAYGMILGSLELAAYLSFKKERSLAAYHPVEPLVQDRVAAYVEELNPSKLAAYFKKERSLNLAAYGVEELNPSKLLAAYLSFKKERSLGPGPGAHPVEPLVQDRVVEPGPGPGHAHPVEPLVQDRVVEGPGPGLLSFKKERSLNLQRDGPGPGSLNLQRDKVCLLHQEGPGPGIPRLLHAHPVEPLVQGPGPGRSLNLQRDKVCLLHQGPGPGLHAHPVEPLVQDRVVGPGPGLLSFKKERSLNLQRDGPGPGHDFTILEQDRMPKTSGPGPGSLNLQRDKVCLLHQEGPGPGLLSFKKERSLNLQRDGPGPGCHDFTILEQDRMPKTGPGPGELLSFKKERSLNLQRKKSGPISPTNLEMFKPGVEELNPSKLLLLSYHQEGMKKILGSLELLSFKKERSLNLQRDKVCLLHQESQLLKLRGTGTDTTKKMATSVNCCHDKKFTILEQDRMPKTSMAPILTESSGSLVTRLMSIPRLKKLHAHPVEPLVQDRVVEPILATEP |
Cholera toxin subunit B / EAAAK / PADRE / AAY / CTL / AAY / HTL / GPGPG / B cell / KK (Adjuvant / linker/ PADRE / linker).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



