Submitted:
18 July 2024
Posted:
19 July 2024
You are already at the latest version
Abstract
The analysis of chemical warfare agents and their degradation products has become increasingly important for public and military safety in recent years. In the article, we drew attention to the possibilities of the HPLC-ICP-MS analytical technique, which has been relatively rarely used for identifying and quantitatively analyzing highly toxic and hazardous substances in environmental samples. This technique enables the identification and determination of minimal amounts (ppt range) of chemicals in various matrices. This fact is important for the analysis of chemical warfare agents and other highly harmful compounds, even small amounts of which can have serious consequences for living organisms. The article critically analyzes the results of scientific research on the identification and quantitative analysis of extremely toxic organophosphorus, organosulfur and organoarsenic substances, their derivatives and degradation products using high-performance liquid chromatography (HPLC) coupled with inductively coupled plasma mass spectrometry (ICP-MS).
Keywords:
HPLC-ICP-MS
; chemical warfare agents
; OPCW
; verification
Highlights
- Coupling HPLC with ICP-MS is a powerful tool to determine trace amounts of organophosphorus, organosulfur and organoarsenic substances, their derivatives and degradation products in environmental samples.
- HPLC-ICP-MS can be successfully used as a complementary to GC-MS and HPLC-MS techniques to conduct forensic analysis of complex environmental matrices containing CWA and other toxic substances.
1. Introduction
The ICP-MS technique was designed in the early 1980s. Houk et. al. [1] and Gray [2] published the first scientific research reports on the ICP-MS. Inductively coupled plasma was initially used for ionization in the optical emission spectroscopy (OES) technique. ICP-OES allowed for analyzing most metals and some non-metals at the ppm to ppb level. One of the disadvantages of this detection was the measurement windows that allowed the determination of only a few selected elements during a single-run analysis. One of the most significant advantages of the ICP technique was the addition of a mass spectrometry unit.
This connection allowed the sensitivity to be increased by three orders of magnitude compared to ICP-OES: from ppb to ppt. It also made it possible to increase the number of determined elements in a single run analysis from several to even several dozen. Inductively coupled argon plasma, used in the ICP-OES technique, with a quadrupole mass detector could be combined with the chromatographic techniques (HPLC-ICP-MS or GC-ICP-MS). The HPLC-ICP-MS technique is not the first-choice technique for the analysis of CWA, but it is a promising technique that can be used to verify the identification of toxic substances. [3]
The analysis of chemical warfare agents, and their environmental degradation products, using gas or liquid chromatography in conjunction with mass spectrometry, has been reviewed extensively.[4,5,6,7,8,9,10,11,12] Relative to HPLC-ICP-MS these methods are burdened with certain disadvantages. In the case of gas chromatography, CWA degradation products often require a time-consuming derivatization process at the sample preparation stage. Additionally, the complexity of the matrix complicates the analysis. For HPLC analyses, using atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI) to ionize the sample may deleteriously affect the mobile phase components and sample matrix components. In addition, the use of the APCI and ESI ionization modes is highly dependent on the analyzed compounds. It, therefore, sometimes requires expensive radiolabeled analogs, which increases the analysis cost. Strong ionization, used in the ICP technique, is virtually independent of the matrix type. This feature enables the analysis of matrices of samples considered too complicated for mass spectrometric analysis using ESI or APCI. An essential advantage of ICP-MS is also the lack of complicated sample preparation procedures.
It is worth noting that the GC-ICP-MS technique allows for qualitative and quantitative analysis of volatile toxic organic compounds without the need to analyze standards, thanks to the possibility of blocking the retention time and calibration independent of the chemical compound. [13]
Following the contemporary security requirements for identifying CWA and other very toxic substances, four levels of identification are distinguished: first identification level – presumptive; second identification level – field confirmatory; third identification level – theatre validation and fourth identification level – field definitive. The fourth level of identification requires the employment of multiple state-of-the-art, independent, established protocols and technologies by scientific experts in internationally recognized laboratories to determine the unambiguous identity of a chemical hazard with the highest level of confidence and degree of certainty necessary to support strategic-level decisions. [14] We believe chromatographic analytical techniques coupled with ICP-MS perfectly supplement the most used qualitative and quantitative analytical methods. The unambiguous identification of degradation products related to CWAs represents a cornerstone in the instrumental analysis plan established by the Organization for the Prohibition of Chemical Weapons (OPCW) and exercised by OPCW-certified laboratories around the globe during yearly proficiency tests (PTs). [15] The high polarity often associated with these degradation products (i.e., alcohols, amines, phosphonic and sulfonic acids) make their detection by GC-MS particularly difficult because of their low volatility. As most of the preliminary line of analytical methods involve GC-MS and related hyphenated methods, derivatization of these dialkylethanolamines is a necessary step during the execution of these analyses. Derivatization of analytes accomplishes their conversion into more volatile species (i.e., lessened polarity) that exhibit different chromatographic profiles (i.e., peak shape and different retention times) from the original, unmodified analyte.
This review describes the possibilities of the ICP-MS technique coupled mainly with high-performance liquid chromatography (HPLC) in the analysis of environmental samples potentially containing CWA, their derivatives, and products of their degradation, as well as other highly toxic substances.
2. HPLC-ICP-MS in the Analysis of Organophosphorus Compounds
2.1. The importance of the analysis of organophosphorus nerve agents, their hydrolysis products, and metabolites
Organophosphorus nerve agents (OPNA) are the most toxic and deadliest compounds. These electrophilic compounds react with the nucleophilic serine residue in the active site of acetylcholinesterase (AChE), the enzyme responsible for the neurotransmitter acetylcholine (ACh) breakdown. By blocking AChE, organophosphate chemical warfare agents paralyze the nervous system [16]. Although nerve agents were used before World War II they have found continuous use in modern times [17,18]. The first recorded attack occurred on March 15, 1988, when Saddam Hussein's troops used sarin in Halabja in the Iran-Iraq War [19,20]. About 5,000 Kurdish citizens died then. One of the more shocking to the world was the terrorist attack by the Aum Shinrikyo (Supreme Truth) sect on the Tokyo subway on March 20, 1995 [21,22]. Fourteen people were killed then, but if the amount of sarin used had been distributed differently, the death toll could have been much higher. There is undeniable evidence that sarin was used on a large scale in Syria between 2013 and 2018 [23]. Kim Jong-Nam, the half-brother of Kim Jong-Un, the leader of North Korea, died of poisoning with the VX compound. The incident took place at Kuala Lumpur Airport in February 2017 [24,25]. The attacks with newly synthetized nerve agents took place in Salisbury in 2018 [26,27]. The detected substance was a compound with code A-234, belonging to a group of compounds known as Novichok.
Sergei Skripal and his daughter, Yulia, were poisoned first with this compound, followed three months later by two British citizens, Charlie Rowley and Dawn Sturgess [28].
2.2. Hydrolysis of Organophosphorus Based Nerve Agents
To illustrate the structure and behavior in environmental conditions, Figure 1 shows the possible pathways of the tabun (GA) and VX hydrolysis. The product of GA degradation is ethyl phosphoric acid (EPA), while the product of VX hydrolysis is methyl phosphonic acid (MPA). The VX hydrolysis leads through several intermediates (EMPA, EA 2192, EMPTA) [29]. Degradation reactions of other OPNA, leading to the final product, which is MPA, are presented in Figure 2. It should be borne in mind that the degradation products of organophosphorus CWA are low volatile and highly polar. However, OPNAs’ degradation products (OPNA DP) have almost equal toxicity than the initial CWA, so their analysis may be an excellent alternative to researching parent compounds. Moreover, the presence of specific degradation products of chemical warfare agents in the environment indicates, with high probability, the use of a toxic agent.
2.3. The Use of the HPLC-ICP-MS Technique for the Analysis of Organophosphorus Compounds
The HPLC-ICP-MS technique, due to the achievement of extraordinary sensitivity at ppt concentration and excellent separation of elements, is beneficial in analyzing trace amounts of OPNA and their degradation products. Initially, this technique concerning the analysis of organophosphorus compounds was not perfect due to the high potential of the first ionization of 31P+ ion, which is 10.5 eV, as well as the interference caused by the presence of 14N16O1H+ and 15N16O+ ions (m/z 31).[31] However, using a particular system in the ICP-MS apparatus, part of the collision cell, the so-called octopole reaction system (ORS), with helium as the collision gas, eliminated interference and reduced the background signal.
The first work on the analysis of degradation products of OPNA using gas chromatography combined with the ICP-MS technique was published by Richardson and Caruso.[31] Their research optimized and validated a method that allows the separation and detection of seven organophosphorus compounds: EMPA, IMPA, EDPA, IBMPA, PMPA, MPA, and CMPA in less than 10 minutes, with a detection limit of less than 50 pg/mL. This method has been successfully implemented for river water and soil samples. Thanks to the technique of gaseous sample injection used in gas chromatography, interferences of atmospheric origin in the form of signals from 14N16O1H+ and 15N16O+ ions were less numerous than in the case of analyzes conducted using liquid chromatography. That fact results in a lower background level for the m/z 31 signal. The achieved background level was so low that using a collision reaction cell to improve this parameter in the research was unnecessary. Due to the best stability of the obtained products, TBDMS was used for the derivatization of the tested compounds. Derivatization reaction conditions were optimized and ran at 80°C for 45 min. The derivatization process, necessary to obtain more volatile compounds for gas chromatography analysis, increases the sample preparation time. Earlier work of this team [34] optimized the analysis method of organophosphorus chemical warfare agents using the ICP-MS technique combined with liquid chromatography. The work results show that the use of gas chromatography, in this case, reduces the detection limit by 64 to 89 percent while maintaining the correct separation of compounds. The exact results obtained for both studies are shown in Table 1. However, it should be noted that despite the better sensitivity of the results obtained, the analysis is burdened with the extension of time for sample preparation procedures (caused by, e.g., derivatization).
Analysis of OPNA DP is challenging in food samples because some products contain significant amounts of phosphoric acid (V), used in the food industry under the designation E338. Phosphates can also be found in food of plant origin because it is used to produce fertilizers in agriculture. Kubachka and coworkers published a study, in which they describe the method of OPNA DP analysis in food samples. [35] The matrix for the study were samples of products available in stores, such as bottled water, fruit juice, infant formula, isotonic drink, and lettuce. Three methods of separation of compounds were investigated in the research. One using the Dionex - IonPac AS7 column with a pre-column, and two methods using the Hamilton PRP-X100 column. Some parameters of the separation techniques used in the study are summarized in Table 2. [35]
The technique of analytes separation using the Dionex column was used to identify VX and sarin degradation products: EMPA and IMPA in the presence of MPA and H3PO4 using LC-ICP-MS. It allowed a satisfactory baseline to be achieved. The separation and identification of all these compounds took less than 5 min. Separation techniques using the Hamilton PRP-X100 column extended the analysis time. By increasing the number of analytes (OPNA DP), the peaks of IPHEP with MPA, and IBHMP with H3PO4 overlapped. Further research allowed the separation of ten OPNA degradation products within 25 min. The technique, combining the PRP-X100 column with the ammonium carbonate solution elution system, was previously successfully implemented in analyzing OPNA with a structure like MPA and a pKa value like H3PO4. The analysis was based not only on ICP ionization but also on electrospray ionization (ESI). To carry out analyses identifying 31P+ ions, the ICP-MS system equipped with a collision-reaction cell with helium as the reaction gas was used. To maintain the stability of the signal from the m/z 31 ion and, at the same time, reduce the noise level, the helium flow was optimized to 2 mL/min. Electrospray ionization analysis was performed in positive mode. The implemented technique is compatible with LC-ICP-MS and ESI-LC/MS analyses if the buffer concentration is kept relatively low, resulting in good separation of seven OPNA DP in less than 30 min. The compatibility of the techniques allows for identifying and verifying the structure of OPNA DP derivatives. In addition, the studies showed that a similar level of the LOD characterized both methods, but the ICP-MS technique is much more sensitive than the electrospray ionization technique.
The ICP-MS technique is also beneficial in many other studies contributing to broadening knowledge about organophosphorus compounds. For instance, Carletti et al. published exciting studies on the biochemical properties of phosphotriesterase (PTE) - an enzyme detoxing the body from harmful organophosphorus compounds, including agricultural pesticides and nerve and convulsant warfare agents (OPNA). [36] The active center of this enzyme consists of two zinc cations that another element can replace. The most effective but least stable variant of this enzyme contains Co2+ cations. Using CCT-ICP-MS (Collision-reaction Cell Technology-ICP-MS) and X-ray fluorescence spectrometry was confirmed that the Zn-PTE recombinant contains only Zn2+ cations, while when the Co-PTE recombinant contains Zn2+ and Co2+ cations in equilibrium amounts. For one of the research stages, a compound related to VX O,O-(3-chloro-4methyl-2oxo-2H-chromen-7-yl) ethyl methyl phosphonate was used. The hydrolysis activity of this VX analog under the influence of Zn-PTE was too slow to reach the average inhibitory concentration (IC50). However, it was adequate for Co-PTE. It has also been proven that an increase in the concentration of ZnCl2 in the reaction medium inhibits the Co-PTE enzyme. Studies indicate that the most significant benefits in decontaminating organophosphorus compounds are obtained by Co-PTE containing two Co2+ cations. However, such a compound is highly unstable, and its production on an industrial scale would be difficult and expensive.
3. ICP-MS in the Analysis of Organosulfur Compounds
3.1. The Importance of the Organosulfur Compounds Analysis
The ICP-MS technique is used to analyze not only OPNA but also necrotic chemical warfare agents and their derivatives and metabolites. Sulfur mustard (HD) is one of the best-known chemical warfare agents. So-called mustard gas was used for the first time during World War I near Ypres.[37] This agent was also used later, for example, in China in 1937-1945 or the Iran-Iraq war in 1983-1988.[38,39,40] Now, the greatest threat is posed by the remains of chemical weapons, which have been sunk, among others, in the Baltic Sea. At the bottom of this basin are about 15,000 tons of poisonous warfare agents, 63% of which is sulfur mustard, filling both barrels and artillery shells or bombs.[41,42,43]
3.2. Degradation Reactions of Organosulfur Compounds
Among the organosulfur compounds, the most dangerous from the point of view of use on the battlefield is bis(2-chloroethyl)sulfide (HD). It belongs to vesicant chemical warfare agents but also causes intense general poisoning effects.[44,45] Sulfur mustard is readily hydrolyzed in water, forming 2-((2-chloroethyl)thio)ethanol (known as half sulfur mustard). The major product of hydrolysis is thiodiglycol (TDG). Other products of sulfur mustard degradation may include 1,4-thioxane and 1,4-dithiane.[46,47] However, it should be remembered that the number of degradation products may include over 20 additional compounds. [47,48] The hydrolysis of sulfur mustard and the main degradation products are shown in Figure 3.
3.3. Use of ICP-MS Technique to Analyze Organosulfur Compounds
Many works describe the methods of analysis of organosulfur poison agents, e.g., using gas chromatography coupled with mass spectrometry and other techniques.[49,50,51] In the work of Kroening et al., five sulfur mustard hydrolysis products were analyzed using the HPLC-ICP-MS technique. [52] For comparison, HPLC-ESI-MS was used in a positive ion mode. The analyses were carried out using a single and tandem mass spectrometer (ESI-MS and ESI-MS/MS). That is the first work describing the analysis of sulfur mustard degradation products using HPLC-ICP-MS, enriched with a collision cell module. One of the significant advantages of that technique is the presence of a collision cell with collision gas – xenon allows to eliminate of the interference of the 32S+ channel, derived, e.g., from 16O16O+ and 14N18O+ ions. The optimal gas flow for these analyses was established at 0.25 mL/min. The most considerable multi-foggy interference was minimized by matching the appropriate voltage to the octopole and quadrupole. In this study, displacement chromatography was used to improve the analysis parameters. 2-mehyl-3-pentanol, 3-pentanol, and 2,2-dimethyl-3-pentanol were used as displacers. The results were verified using different column types and displacement coefficients. The considered methods were successfully applied to the analysis of river water samples. The selected parameters of the analysis are shown in Table 3.
4. ICP-MS in the Analysis of Organoarsenic Compounds
4.1. Significance of the Organoarsenic Compounds Analysis
Organoarsenic CWA were used during World War I, constituting a kind of "replacement" for sulfur mustard. These were, among others, Lewisite, diphenylchloroarsine DPCA (Clark I or DA), diphenylcyanoarsine DPCNA (Clark II or DC), phenylodichloroarsine PDCA (Pfiffikus) or Adamsite. Many ammunitions containing these agents were sunk in the Baltic Sea [53] and found in Belgium, Germany, China, and Japan [54], present in soil [55] and groundwater [56]. During World War II, organoarsenic CWA were produced in Japan, e.g., on the Okuno Island in the Seto Inland Sea. After World War II, some produced substances were solidified in concrete and illegally buried, among others, in closed gravel pits. To this day, there are cases of poisoning, such as, for example, contaminating workers in the construction of roads in the cities of Samukawa and Hiratsuka, in Kanagawa prefecture, or the city of Kamisu, prefecture Ibraki.[57,58]
The analysis of organoarsenic compounds is significantly difficult, especially in industrialized areas. In such situations, special analysis plays a significant role in determining whether organic or inorganic arsenic compounds are in the sample. In the case of environmental analyses, in which the remains of potentially used CWA are sought, the high probability of finding several organoarsenic compounds, which are products of the distribution of output toxic substances. Biological samples are exposed to arsenobetaine (AB), naturally found in living organisms, especially marine organisms, and human urine.
4.2. Degradation Reactions of Organoarsenic Compounds
4.3. The Use of the ICP-MS Technique for the Analysis of Organoarsenic Compounds
The analysis of arsenic compounds in biological and environmental samples is critical due to the still not fully understood mechanism of metabolic transformations of these compounds in living organisms and redox reactions in the environment.
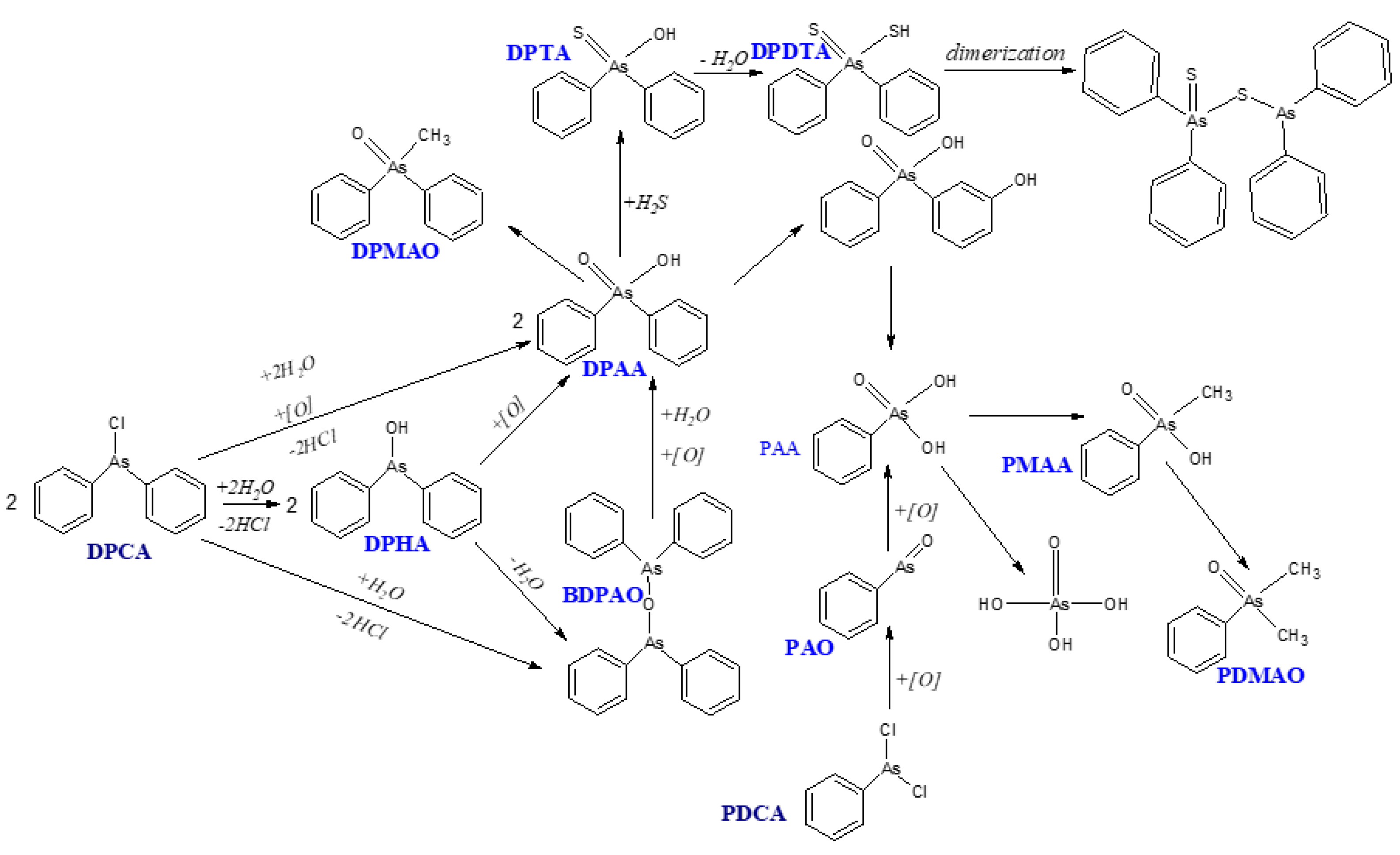
GC combined with mass spectrometry is an excellent tool for analyzing organoarsenic CWA. Unfortunately, low volatile arsenic compounds, such as DPAA, require prior derivatization with, e.g., 1-propanethiol or thioglycolic acid methyl ester. Derivatives have an unpleasant odor and are unstable at elevated temperatures, which may cause their thermal decomposition in GC’s injection port. That can cause problems with the accuracy and reliability of the analyzed results. For this reason, the analysis of organoarsenic compounds through HPLC-ICP-MS is an excellent alternative to GC-MS. ICP-MS also has some limitations, mainly for three reasons. These include simultaneous elution of compounds with identical m/z ratio during chromatographic separation, ionization variability in high-temperature plasma, which can generate signal attenuation or amplification, and inaccurate separation of arsenic ions from other ions in a mass spectrometer. Interference from other ions is the most significant problem. Suppose chlorine could be present in the test sample, which is especially the case in environmental samples. In that case, the ion 40Ar35Cl+ can be produced in the plasma with a m/z ratio 75, the same as the only stable isotope of arsenic, 75As+. The next, although much less likely, interferences may be caused by ions 150Nd2+ and 150Sm2+, which also have m/z 75. However, they are extremely rare in environmental samples. When using a single quadrupole for analysis, chlorine ions (ArCl+) formed in plasma can be eliminated using a collision cell module with helium. However, the best way is to use a double quadrupole, which removes interference from ArCl+, Nd2+ and Sm2+ ions. When ICP-QQQ is used, an ion with m/z 75 is isolated in the first quadrupole (Q1). It is then transported to an oxygen collision cell, where the 75As16O+ ion with m/z 91 is formed, which is isolated in the second quadrupole (Q2) and then transmitted to the MS detector. When monitoring an m/z 91 ion, there is no way m/z 75 ions will be detected, so this is a reliable way to eliminate these interferences. The limitation of the HPLC-ICP-MS technique may also be the analysis of liquid samples in solutions with extreme pH. Such samples may cause shifts in the retention times of analytes or contribute to the precipitation of phosphates from the mobile phase of the HPLC column. However, this risk can be avoided by using an appropriate sample preparation method. Thus, as discussed previously, the potential limitations of the technique can be eliminated, and the advantages of using ICP-MS are enormous. Analyzes of organoarsenic compounds carried out using the ICP-MS technique allow achieving a sensitivity of the ppt range and excellent stability of results. An additional benefit is the shortening of the analysis time because the sample preparation procedure is quick and straightforward, and the analysis, even of multi-component mixtures, takes a few to several minutes. Some examples of this technique's use for analyzing organoarsenic compounds are given below.
In 2003, a group of scientists led by Ishizaki made the first study of drinking water from the Kizaki area of Kamisu, Ibaraki prefecture, in Japan. [57] The research was carried out due to specific symptoms impairing the central nervous system in humans and the high mortality rate among animals living in these areas. Researchers were surprised that the patient’s health improved after leaving the area where they lived for an extended period and deteriorated a few days after returning to the city. Samples of water coming from the taps of a multi-family house were analyzed. Water was extracted from a depth of approx. 12 m from a well dug in the reclaimed area of a former gravel pit. The first analyzes were performed using GFAAS. The research showed that the cause of health disorders was arsenic compounds, the content of which in drinking water exceeded the permissible concentration (0.01 mg/dm3) 450 times. At the same time, it should be noted that the other parameters that drinking water should meet, such as the content of heavy metals, volatile organic halogens, or organophosphorus and chlorine pesticides in the collected samples, were within the norm.
Subsequent studies showed that the concentration of DPAA in drinking water samples was a maximum of 15 ppm, which meant that the inhabitants of this city could consume up to 30 mg of DPAA per day. GFAAS analysis showed that the content of inorganic arsenic is 1.5 ppm, and the total concentration of this element is 4.5 ppm. That means that the concentration of organic arsenic was 3 ppm. However, using HPLC-ICP-MS, it has been proven that arsenic contamination of drinking water comes entirely from the organic form of this element, i.e., compounds such as BDPAO, DPAA, and PAA, and contamination is the result of environmental degradation of organoarsenic warfare agents: diphenylchloroarsine and diphenylcyanoarsine (DPCA) and DPCNA).
The content of inorganic arsenic in previous analyzes (conducted with the GFAAS technique) was caused by the inaccurate process of sample preparation for analysis and the ingress of phenolic arsenic compounds into the aqueous phase during extraction. A thorough analysis using the ICP-MS technique enabled the error to be detected and the correct results to be obtained. Due to the severity of the threat, more studies of drinking water from the region have been conducted. Kinoshita et al.[58] developed a method for the simultaneous analysis of DPAA and PAA in drinking water using HPLC-ICP-MS to detect contamination quickly. Water samples were taken from six wells from about 5-6 m depths n the same area - the city of Kizaki in Japan.
The samples were passed through a membrane filter with a diameter of 0.45 μm and stored in amber glass or polypropylene or polyethylene vials to prevent contamination of the samples with trace amounts of arsenic contained in low-quality glass. The mobile phase consisting of acetonitrile and water (30 : 70) at pH=2 was used for HPLC analyses, and each injection had a volume of 5 µL. Thanks to the developed method, arsenic compounds were separated within 4 minutes, reaching the detection limit: for DPAA 2 ng/mL and PAA 1 ng/mL.
The obtained calibration plots were characterized by excellent linearity, and the method was characterized by good reproducibility (RSD = 1.7÷6%). In addition, long-term analyzes were also tested, and arsenic measurements showed stability even after 7 hours (RDS = 1.3÷5.8%). However, it is not entirely clear whether the PAA contained in the samples came from the environmental decomposition of DPAA, i.e., whether it was a product of diphenylchloroarsine and diphenylcyanoarsine (DPCA and DPCNA) decomposition, or whether it was a product of phenyldichloroarsine degradation. The optimized method, yielding excellent results, allowed the team to adapt its capabilities to analyze phenylarsenic compounds on a large scale. To extend the research and refine the method, the team of Kinoshita et al.[61] validated the HPLC-ICP-MS method for analyzing organoarsenic compounds, including Lewisite 1 degradation products. Urine samples were analyzed.
Because chlorine is often present in biological samples, the signal measurements were made considering the AsO+ ion (m/z 91). Thanks to this, the signal from the ArCl+ ion, which is the most intense source of interference, did not cross the baseline. A dynamic reaction cell (DRC) module and oxygen were used as the collision gas. Some urine samples were collected from mice orally exposed to the CVAA standard solution - the main decomposition product of Lewisite 1, which is also a toxic compound. Samples were taken after 24 hours. The analytes were CVAA and the main degradation metabolite of this compound, i.e., CVAOA. For greater accuracy of the tests, several sample HPLC columns were verified: Inertsil C4, C8, Ph (2.1×150 mm, 5μm), and CHEMCOSORB 3-Dph (2.1×150 mm, 3μm).
These columns are typically used for the analysis of phenyl compounds. The best separation of compounds and the correct shape of the peaks gave the C8 column, which is why it was used for further research. The injection volume was also verified. A series of studies led scientists to conclude that as the injection volume increased, the detection limit value decreased while maintaining the favorable shape of the peak. The injection volume was therefore set at 20 μL. The method developed in this way was successfully implemented to analyze human urine samples contaminated with degradation products of organoarsenic warfare agents: CVAA and CVAOA in the presence of DPAA and PAA. The analysis was characterized by high sensitivity and short time.
All analyzed compounds were well separated within 4 min. Only DPAA was identified after 12 min. The detection limit was: CVAA 0.2 ngAs/mL, CVAOA 0.1 ngAs/mL, DPAA 0.5 ngAs/mL, and PAA 0.3 ngAs/mL. Urine analysis of CVAA-treated mice showed the presence of several unknown compounds that are analyte metabolites. The method proposed in this paper enabled the speciation analysis of arsenic. Additionally, it provided valuable information that could not be obtained by analyzing the total arsenic content of the sample or by procedures involving derivatization as one of the sample preparation steps. That is confirmed by the fact that the HPLC-ICP-MS technique can be successfully implemented in research on the metabolism of organoarsenic compounds in living organisms. Due to reports of methylated degradation products of phenylarsine warfare agents detected in environmental samples in Japan, it was decided to continue research on adapting HPLC-ICP-MS analysis methods. The research development on this topic led to another work by the Kinoshita team.
Another Kinoshita’s research group [62] described methods for the analysis of organoarsenic compounds using HPLC-ICP-MS. By using this technique it became possible to separate several degradation products of organoarsenic warfare agents, such as phenylmethylarsinic acid (PMAA), phenyldimethylarsine oxide (PDMAO), diphenylmethylarsine oxide (DPMAO) in the presence of DPAA and PAA. One of the analyzed matrices was urine, which contributed to the broadening of knowledge on the metabolism of organoarsenic compounds in the body because several unknown organoarsenic compounds were found in addition to AB. In addition, research was also carried out on environmental samples: rice and groundwater collected from a depth of about 5-6 m from the city of Kamisu in Japan. The analysis methods used in the team's previous work did not give satisfactory results, so it was decided to improve the research. However, the DRC module with oxygen as a collision gas was used, and the monitored ion was AsO+ (m/z 91).
Several HPLC columns were verified: Inertsil C4 (150mm×2.1mm, 5µm), Inertsil NH2 (250mm×1.5mm, 5µm), and Inertsil AS (150mm×2.1mm,3µm) and guard columns, e.g., Inertsil C4 (10mm×1.5mm, 5µm), and Inertsil NH2 (10mm×1.5mm, 5µm). Patient urine samples were stored at -84°C. They were diluted three-fold for analysis and passed through a 0.45 μm membrane filter. The best separation of analytes was obtained using the Inertsil AS column, thanks to which eight arsenic compounds were separated, including MMA (V), DMAA, TMAO, TMA, AB, and ACh. The next series of samples consisted of environmental samples.
Groundwater samples were diluted to ten times the original volume and filtered. Preparing rice samples for analysis consisted of pulverizing them and weighing 0.2 g. Then, extraction was performed with 5 mL of methanol : water (1:1) at 80°C. The obtained extract was dried under reduced pressure, made up to 50 μl with water, and filtered. Environmental samples were analyzed by HPLC-ICP-MS. The final series of samples included urine samples taken from mice that had been given specially prepared, contaminated food. Contamination included the addition of PAA to the food, which was the main source of contaminants found in groundwater samples, and PMAA. The compound is found most abundantly in rice samples at a concentration of 1 μg/g of food. Samples were diluted two-fold and filtered, then analyzed by LC-ICP-MS. Selected parameters for individual analyzes are presented in Table 4.
Based on the study's results, it was shown that PAA, after oral administration to mice, practically does not undergo metabolic changes (approx. 91% of arsenic compounds in the analyzed samples were PAA, besides PMAA and trace amounts of PDMAO). PMAA undergoes metabolic processes in approx. 12%. In contrast, when mice were contaminated with DPAA, no DPMAO was detected in the urine samples, but DPAA was present in the urine. It was shown that this compound must come from a different source, as it was also present in the blank sample.
It has also been proven that the unknown compounds seen in the chromatogram after analyzing human urine samples are not the same as the peaks from the unknown arsenates in mouse urine samples. Therefore, it is likely that the arsenic compounds in the patient's urine did not come from metabolites but from contaminated water, meaning that the types of arsenic compounds found in the patient's urine depended on the exposure conditions. Unidentified compounds may also prove the complexity of metabolic processes in living organisms. It also follows that the metabolic processes of phenylarsenic compounds differ from those of inorganic arsenic compounds, and arsenic compounds containing the same hydroxyl groups do not necessarily follow the same metabolic processes. The difference may be due to the water solubility of the phenylarsenic compounds. PAA and DPAA are slightly soluble, while PMAA is very soluble. Thanks to Kinoshita et al.,[61,62] it can be seen how important it is to assess the risk of exposure to arsenic under different conditions to obtain accurate results from analyzing biological samples. Rapid determination and speciation analysis are particularly important for monitoring the content of organic arsenic in blood and urine, which allows to verify the presence of arsenic CWA residues.
GC-MS analyses suffer from the disadvantage that it is impossible to analyze organic and inorganic arsenic compounds simultaneously. The work of Daus et al.[63] aimed to develop the HPLC-ICP-MS method for analyzing organic (CWA derivatives) and inorganic arsenic compounds in groundwater samples during a single chromatographic analysis. In addition, the authored attention to a significant relationship, namely the high content of iron (II) in groundwater, which, after contact with atmospheric air, will result in sediment precipitation in the sample and lead to the loss of some of the analytes. To preserve the sample, the researchers suggest adding phosphoric acid to a final concentration of 10 mM as soon as possible after collection. The validity of such a process has been confirmed by research. Additional protection was keeping the samples at 6°C for no longer than a week. The proposed method, based on the Shodex RSpack NN-614 column, was used to separate arsenic (III), arsenic (V), phenylarsenic acid, phenylarsinic oxide, and diphenylarsenic acid (PAA, PAO, and DPAA). The analysis time was 20 min. It has been shown that the highest DPAA contamination of groundwater is found at a depth of 2-3 m: 3.505 mg/l and the lowest at a depth of 14-15 m: 0.019 mg/l. Inorganic arsenic compounds with a total content of up to 240 μg/l were also detected. Thanks to the possibilities offered by the HPLC-ICP-MS analysis, simultaneous analysis of organic and inorganic arsenic compounds was carried out, with an uncomplicated and quick sample preparation process, obtaining satisfactory results thanks to the excellent sensitivity of the method.
The team of Stetson et al.[64] published the results of research on similar topics. It describes a method for separating four arsenic compounds: As (III), As (V), DMA, and MMA using HPLC-ICP-QQQ. The matrix was surface and groundwater. The implemented method resulted in a good separation of compounds within 12 minutes. To preserve the samples, EDTA was added to a concentration of 2.5 mM in each sample. EDTA helps reduce the redox reactions of arsenic compounds in the presence of iron, which is present in relatively significant amounts in groundwater samples. In addition, the samples were stored in a dark place at 4±2°C. Thanks to the proposed method, unprecedented detection limits were achieved: LODAs(III) = 0.03 ng/mL, LODAs(V) = 0.05 ng/mL, LODDMA = 0.03 ng/mL, LODMMA = 0.04 ng/mL.
The team of Hisatomi[65] published an interesting paper on the possible metabolic pathways of arsenic compounds. The study aimed to investigate the metabolic pathway and determine the main DPAA metabolite in anaerobic sulfate-reducing soil conditions. To study the metabolic pathway of arsenic compounds under such conditions, a soil culture was created where 30 mL of deionized water and 10.7 μg of As in the form of DPAA, 3.5 mg C/g of dry soil in the form of rice straw, and 425 μg S/g dry soil as K2SO4. The samples thus obtained were kept sealed in a dark place at 30°C for 3 weeks. After this time, the samples were analyzed. The chromatograms showed a peak of a compound of unknown origin, with a concentration higher than the DPAA concentration calculated based on the peak intensity. Thanks to the use of HPLC-ICP-MS, it was discovered that the so far unidentified arsenic compound, formed in the reaction of DPAA with hydrogen sulfide, has the same retention time as the unknown compound being a product of metabolic changes in soil culture. Due to the intensity of the peak of the unknown compound and its absence in the analyzes of soil culture samples without source C and S, as well as inconsistency with the retention times of the peaks from As(V), PAA, PMAA, and DPMAO, it was concluded that this compound is the main metabolite of DPAA degradation in anaerobic and sulfur-reducing soil conditions. The unknown compound is diphenylthioarsinic acid (DPTA). The possible dimerization of this compound has also been proven.
The metabolic pathway by which most of the arsenic that enters the body is excreted in the urine as DMAA still needs to be fully understood. Suzuki's’ team [66] attempted to determine the ongoing transformations of organoarsenic compounds.
Thanks to the analysis of rat liver using HPLC-ICP-MS, several different organoarsenic compounds containing sulfur in their structure were discovered, including two compounds unidentified at the time: dimethyldithioarsinic acid (dimethylarsinodithioic acid) (DMTA (V)) and dimethylthioarsinous acid (DMTA (III)). The rats used for the study were injected with arsenic compounds (DMAA, DMA (III), MMA (III), MMA (V)) in a single dose of 0.5 mg As/kg body weight via the portal vein under anesthesia. After 5 minutes, the rats were sacrificed, and their livers were excised for examination after whole-body perfusion. Arsenic solutions were adjusted to 1.0 mL/kg body weight.
For HPLC-ICP MS analysis, 20 µL of the sample solution was used, which was applied to a polymer-based gel filtration column (Shodex Asahipak GS-220 HQ, 300 mm 7.6 mm I.D., exclusion limit >3000) or anion exchange column (Shodex Asahipak ES-502N 7C, 100 mm, 7.6 mm I.D.). The column was eluted with 50 mM ammonium acetate buffer (pH 6.5 at 25°C) or 15 mM citric acid buffer (pH 2.0 at 19°C) gel filtration or anion exchange column at a flow rate of 0.6 or 1.0 mL/min. The outlet of the HPLC system was connected directly (with a PEEK tube 300 mm long, 0.25 mm I.D.) to the inlet of the ICP nebulizer. The m/z 75 (As+) and m/z 77 signals were monitored to compensate for ArCl+ molecular interference. Online ICP-MS data was processed using in-house developed software. Liver samples were prepared by homogenization in 4 volumes of extraction buffer (50 mM ammonium acetate buffer, pH=7.4) using a glass-teflon homogenizer under nitrogen. The homogenates were centrifuged for 60 min at 4°C to obtain the supernatant fraction. Low molecular weight arsenic compounds were well separated during the 30-minute analysis. Research has established that DMA (III) (and not DMAA) is converted to DMTA (V) and DMTA (III) in the liver supernatant. DMTA (V) may be present when the amount of DMA (III) relative to the amount of liver supernatant is high.
Good knowledge of the metabolism of organoarsenic compounds may contribute to the development of science in the context of a new decontamination approach.
In the study of Hempel et al.,[56] the authors verified for the first time the influence of natural conditions on the degradation of persistent phenylarsenic compounds in groundwater under anaerobic conditions. The research aimed was to investigate the potential of colonies of bacteria naturally occurring at the site of contamination with organoarsenic compounds to degrade PAA and DPAA under anaerobic conditions in situ.
Groundwater samples collected from an abandoned ammunition depot and CWA munition filling station in central Germany were used for the study. Samples containing groundwater mixed with fine-grained aquifer material were taken from a depth of 4.4-5.4 m below the surface of a potential source of land contamination. The samples were stored in tightly closed glass bottles at 12°C without access to light. A groundwater microcosm was created by treating samples in an anaerobic glove box under nitrogen (95%) and hydrogen (5%) to replicate environmental conditions and avoid oxygen contamination. 100 mL of the groundwater/sediment mixture (approximately 1.5 g of solids) was used for the tests.
Variants of the experiments were as follows: abiotic environment, environment without additional electron donors, and environment with lactate as an additional electron donor. All media were statically grown in the dark at 20°C for at least 13 weeks. The Desulfovibrio gigas DSM 1382 strain was used for the study. Cells were cultured in a mineral salt medium supplemented with 20 mM sodium sulfate and 10 mM sodium lactate. The first environment (microcosm) contained only mineral salt substrate with changes. Three other bottles were injected with 5 mL of freshly grown D. gigas cells. One of these bottles served as a living microcosm. All media (bottles) were sampled for sulfur and arsenic analysis within one hour after the first addition of PAA (final PAA concentration 1.5 mg/L). The analyzed samples were acidified with phosphoric (V) acid to a final concentration of 10 mM.
Various organic and inorganic forms of arsenic have been detected and quantified by the speciation method described by Daus et al. [63] by HPLC-ICP-MS with an RSpak NN-14 column (150 x 6 mm, Shodex Japan).
The dominant forms/compounds of arsenic in the original sample were As (V), PAA, and DPAA. The total concentration of arsenic was 1755 μg/l. In living microcosms, after 13 weeks of incubation, more than 95% of the PAA had been removed from the solution. Total arsenic showed a similar trend, with an overall elimination of arsenic of about 77%. In contrast to the concentration of PAA, the concentration of DPAA was stable throughout the cultivation period both in the live and lactated environment. During the first and fourth weeks of culture, an unknown organoarsenic compound was detected as an intermediate. In the living microcosms enriched with lactate, PAA concentration decreased by about 98% within five weeks. PAO formed after 14 days and was removed after 7 weeks. Like unknown arsenic compounds in living environments, PAO was a metabolite in lactate-enriched culture during the PAA transformation process. Total arsenic was reduced to 81% of the initial concentration with the addition of lactate.
Since arsenic compounds were chemically stable under sterile conditions, the observed transformations of arsenic compounds in living microcosms are believed to result from microbial activity and the resulting reducing conditions. The faster reaction rate in the presence of lactate can be explained by the lower redox potential in these samples and/or the higher concentration of sulfides in the water. Studies have shown that the concentration of organic arsenic compounds in anaerobic groundwater samples can be significantly reduced through a process mediated by bacteria. The paper also presents probable structural formulas of unknown metabolites of organoarsenic compounds.
5. Concluding Remarks
As demonstrated, the HPLC-ICP-MS technique allows the quick detection of even trace amounts (<ppt) of chemical warfare agents and their degradation products. Table 5 summarizes the selected analytes and the corresponding limit of detection obtained in published studies. Each HPLC-ICP-MS analysis should consider several possible interferences related to the same mass number of different ions.
The modernization of the ICP-MS technology in the form of a collision reaction cell, or rather one of its types, the ORS, allowed for eliminating these disturbances.
A fast analysis that does not require complicated sample preparation procedures may be crucial in the face of the growing threat of using chemical warfare agents and other highly toxic substances during war, terrorist attacks, sabotage, and/or chemical accidents.
HPLC-ICP-MS can be used as a complementary to currently commonly used GC-MS and HPLC-MS techniques for analysis of CWA. In special cases, it may be the leading analytical technique.
A relatively small number of research papers describing the use of HPLC/GC-ICP-MS show the still not fully discovered possibilities of this analytical tool, and the documented results of analyzes of CWA indicate its great potential. HPLC-ICP-MS can also be successfully used to verify the Chemical Weapons Convention (CWC)[67] provisions. We believe that the HPLC-ICP-MS technique is currently underestimated in studies of the presence of CWA and their metabolic products in living organisms and the environment. The increasing availability and popularity of the HPLC-ICP-MS technique allow us to conclude that it will be more often used in CWC and Homeland Security analytical laboratories in the future.
Equipping laboratory stands with HPLC-ICP-MS is justified and will increase the possibilities, credibility, and reliability of laboratory analyses of CWA, their derivatives, and other toxic and hazardous substances.
The rapid detection of traces (<ppt) of CW agents and their degradation products by HPLC-ICP-MS has been demonstrated. Table 5 summarises the selected analytes and the corresponding detection limits obtained in published studies. Any HPLC-ICP-MS analysis should take into account several possible interferences. These are related to the same mass number of different ions.
The modernisation of ICP-MS technology in the form of a collision reaction cell, or rather one of its types, the ORS, has made it possible to eliminate these interferences.
Rapid analysis, without the need for complicated sample preparation, can be crucial in view of the growing threat of the use of chemical warfare agents and other highly toxic substances in war, terrorist attacks, sabotage and/or chemical accidents.
For the analysis of CWA, HPLC-ICP-MS can be used in addition to the GC-MS and HPLC-MS techniques currently in use. In special cases, it may even be the leading technique for the analysis.
A relatively small number of research papers describing the use of HPLC/GC-ICP-MS show the not yet fully discovered possibilities of this analytical tool. The documented results of analyses of CWA indicate its great potential. HPLC-ICP-MS can also be successfully used for the verification of the provisions of the Chemical Weapons Convention (CWC) [55]. As a complementary method, ICP-MS can be combined with gas and liquid chromatography techniques. In our opinion, ICP-MS is currently underutilised in the investigation of the presence of CWA and its metabolites in living organisms and the environment. The increasing availability and popularity of the HPLC-ICP-MS technique leads us to the conclusion that it will be more widely used in CWC and homeland security analytical laboratories in the future.
The equipment of laboratories with HPLC-ICP-MS is justified and will increase the capabilities, credibility and reliability of laboratory analysis of CWA, their derivatives and other toxic and hazardous substances.
Author Contributions
S.N. and M.K. conceptualization the topic and methodology; literature resources analysis and synthesis. SN and M.K. writing original draft of the article and editing. M.K. visualization. SN. formal analysis and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by The Polish Ministry of Education and Science for the Faculty of Advanced Technologies and Chemistry at the Military University of Technology in 2024 (project number UGB 22-722/WAT).
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Abbreviations and Acronyms
| Acronym and abbreviation | Full name | No CAS |
| AB | Arsenobetaine | 64436-13-1 |
| ACh | Acetylcholine | 51-84-3 |
| AChE | Acetylcholinesterase | 9000-81-1 |
| APCI | Atmospheric Pressure Chemical Ionization | - |
| BCVAA | Bis(2-chlorovinyl)arsinous acid | NF |
| BDPAO | Bis(diphenylarsine)oxide | 2215-16-9 |
| BHETBu | 1,4-bis(2-hydroxyethylthio)butane | 7425-93-6 |
| BHETE | 1,2-bis(2-hydroxyethylthio)ethane | 5244-34-8 |
| BHETM | Bis(2-hydroxyethylthio)methane | 44860-68-6 |
| BHETPr | 1,3-bis(2-hydroxyethylthio)propane | 16260-48-3 |
| CCT | Collision-reaction Cell Technology | - |
| CMPA | Cyclohexyl methylphosphonic acid | 1932-60-1 |
| CVAA | 2-chlorovinylarsenous acid | 85090-33-1 |
| CVAO | 2-chlorovinylarsine oxide | 3088-37-7 |
| CVAOA | 2-chlorovinylarsonic acid | 64038-44-4 |
| CWA | Chemical Warfare Agents | - |
| CWC | Chemical Weapons Convention | - |
| DEHP | Diethylhydrogen phosphate | 598-02-7 |
| DMA (III) | Dimethylarsinous acid | 55094-22-9 |
| DMAA | Dimethylarsinic acid | 75-60-5 |
| DMHP | Dimethylhydrogen phosphate | 813-78-5 |
| DMTA (III) | Dimethylthioarsinous acid | NF |
| DMTA (V) | Dimethyldithioarsinic acid (dimethylarsinodithioic acid) | NF |
| DPAA | Diphenylarsinic acid | 6217-24-9 |
| DPCA | Diphenylchloroarsine | 712-48-1 |
| DPCNA | Diphenylcyanoarsine | 23525-22-6 |
| DPMAO | Diphenylmethylarsine oxide | NF |
| DPTA | Diphenylthioarsinic acid | NF |
| DRC | Dynamic reaction cell | - |
| EDTA | Ethylenediaminetetraacetic acid | 60-00-4 |
| EHDAP | Ethyl hydrogen dimethylamidophosphate sodium salt | NF |
| EMPA | Ethyl methylphosphonic acid | 1832-53-7 |
| EMPTA | Ethyl methylphosphonothioic acid | 18005-40-8 |
| EPA | Ethylphosphonic acid | 6779-09-5 |
| EPCN | Ethylphosphorylcyanidate | NF |
| ESI | Electrospray ionization | - |
| GA | O-Ethyl N,N-dimethyl phosphoramidocyanidate (Tabun) | 77-81-6 |
| GB | Propan-2-yl methylphosphonofluoridate (Sarin) | 107-44-8 |
| GC | Gas Chromatography | - |
| GD | 3,3-Dimethylbutan-2-yl methylphosphonofluoridate (Soman) | 96-64-0 |
| GF | Cyclohexyl methylphosphonofluoridate (Cyclosarin) | 329-99-7 |
| GFAAS | Graphite Furnace Atomic Absorption Spectroscopy | - |
| HD | bis(2-chloroethyl)sulfide (Sulfur mustard) | 505-60-2 |
| HPLC | High-Performance Liquid Chromatography | - |
| IBHMP | Isobutyl hydrogen methylphosphonate | 1604-38-2 |
| IC50 | Median inhibitory concentration | - |
| ICP-MS | Inductively Coupled Plasma Mass Spectrometry | - |
| IMPA | Isopropyl methylphosphonic acid | 1832-54-8 |
| IPHEP | Isopropyl hydrogen ethylphosphonate | 170135-50-9 |
| LC | Liquid Chromatography | - |
| MMA (III) | Monomethylarsonous acid | 25400-23-1 |
| MMA (V) | Monomethylarsonic acid | 124-58-3 |
| MPA | Methylphosphonic acid | 993-13-5 |
| MS | Mass spectrometry | - |
| OES | Optical emission spectroscopy | - |
| OPNA | Organophosphorus Nerve Agents | - |
| OPNA DP | Organophosphorus Nerve Agents Degradation Products | - |
| ORS | Octopole Reaction System | - |
| PAA | Phenylarsonic acid | 98-05-5 |
| PAO | Phenylarsine oxide | 637-03-6 |
| PDCA | Phenyldichloroarsine | 696-28-6 |
| PDMAO | Phenyldimethylarsine oxide | NF |
| PMAA | Phenylmethylarsinic acid | NF |
| PMPA | Pinacolyl methylphosphonic acid | 616-52-4 |
| PPA | Propylphosphonic acid | 4672-38-2 |
| ppb | Parts per billion | - |
| ppm | Parts per million | - |
| ppt | Parts per trillion | - |
| PTE | Phosphotriesterase | - |
| QQQ | Triple quadrupole (mass spectrometer) | - |
| TBDMS | Tert-Butyldimethylsilyl ether | 124150-87-4 |
| TDG | Thiodiglycol | 111-48-8 |
| TMA | Tetramethylarsonium | NF |
| TMAO | Trimethylarsine oxide | 4964-14-1 |
| UV | Ultraviolet | - |
References
- R.S. Houk, V.A. Fassel, G.D. Flesch, H.J. Svec, A.L. Gray, C.E. Taylor, Inductively Coupled Argon Plasma as an Ion Source for Mass Spectrometric Determination of Trace Elements, Anal. Chem. 952 (1980) 2283-2289. https://masspec.scripps.edu/learn/ms/pdf/1980_HoukRS.pdf.
- A.L. Gray, Mass-spectrometric Analysis of Solutions Using an Atmospheric Pressure Ion Source, Analyst 100 (1975) 1190, 289-299. [CrossRef]
- P. Vanninen (Ed.) P. Vanninen, ed., Recommended Operating Procedures for Analysis in the Verification of Chemical Disarmament, The Blue Book, University of Helsinki, Helsinki (2017) ISBN 978-951-51-9958-4.
- J. Nawała, K. Czupryński, S. Popiel, D. Dziedzic, J. Bełdowski, Development of the HS-SPME-GC-MS/MS method for analysis of chemical warfare agent and their degradation products in environmental samples, Anal. Chim. Acta 933 (2016) 103–116. [CrossRef]
- C.A. Valdez, R.N. Leif, S. Hok, B.R. Hart, Analysis of chemical warfare agents by gas chromatography-mass spectrometry: Methods for their direct detection and derivatization approaches for the analysis of their degradation products, Rev. Anal. Chem. 37 (2018) 1, 20170007. [CrossRef]
- T. Wada, E. Nagasawa, S. Hanaoka, Simultaneous determination of degradation products related to chemical warfare agents by high-performance liquid chromatography/mass spectrometry, Appl. Organomet. Chem. 20 (2006) 9, 573–579. [CrossRef]
- B. Muir, B.J. Slater, D.B. Cooper, C.M. Timperley, Analysis of chemical warfare agents: I. Use of aliphatic thiols in the trace level determination of Lewisite compounds in complex matrices, J. Chromatogr. A 1028 (2004) 2, 313–320. [CrossRef]
- S. Popiel, M. Sankowska, Determination of chemical warfare agents and related compounds in environmental samples by solid-phase microextraction with gas chromatography, J. Chromatogr. A 1218 (2011) 47, 8457–8479. [CrossRef]
- Terzic, I. Swahn, G. Cretu, M. Palit, G. Mallard, Gas chromatography-full scan mass spectrometry determination of traces of chemical warfare agents and their impurities in air samples by inlet based thermal desorption of sorbent tubes, J. Chromatogr. A 1225 (2012) 182–192. [CrossRef]
- J.A. Tørnes, A.M. Opstad, B.A. Johnsen, Determination of organoarsenic warfare agents in sediment samples from Skagerrak by gas chromatography-mass spectrometry, Sci. Total Environ. |356 (2006) 1-3, 235–246. [CrossRef]
- M.Y. Cheh, H.C. M.Y. Cheh, H.C. Chua, F.B. Hopkins, J.R. Riches, C.M. Timperley, H.S.N. Lee, Determination of lewisite constituents in aqueous samples using hollow-fibre liquid-phase microextraction followed by gas chromatography-mass spectrometry, Anal. Bioanal. Chem. 406 (2014) 21, 5103–5110. [CrossRef]
- T. Capoun, J. Krykorkova, Internal Standards for Quantitative Analysis of Chemical Warfare Agents by the GC/MS Method: Nerve Agents, J. Anal. Methods Chem. 2020. [CrossRef]
- S. Wilbur, Applications of ICP-MS in Homeland Security, Agilent Technologies (2004) https://www.agilent.com/Library/applications/5989-0741EN.pdf.
- STANAG 4701(1), NATO Standard AEP-66, NATO Handbook for Sampling and Identification of Biological, Chemical and Radiological Agents (SIBCRA), Edition A Version 1, NATO Standardization Agency (2015) http://nso.nato.int/nso/.
- M.-M. Blum, R.V.S.M. Mamidanna, Analytical chemistry and the Chemical Weapons Convention, Anal. Bioanal. Chem. 406 (2014) 5067–5069. [CrossRef]
- S. Mukherjee, R.D. S. Mukherjee, R.D. Gupta, Organophosphorus Nerve Agents: Types, Toxicity, and Treatments, J. Toxicol. 2020 (2020) 1–16, 3007984. [CrossRef]
- M.A. DeLuca, P.R. M.A. DeLuca, P.R. Chai, E. Goralnick, T.B. Erickson, Five Decades of Global Chemical Terror Attacks: Data Analysis to Inform Training and Preparedness, Disaster Med. Public Health Prep. 15 (2021) 6, 750–761. [CrossRef]
- P.R. Chai, B.D. Hayes, T.B. Erickson, E.W. Boyer, Novichok agents: a historical, current, and toxicological perspective, Toxicol. Commun. 2 (2018) 1, 45–48. [CrossRef]
- D.D. Palkki, L. D.D. Palkki, L. Rubin, Saddam Hussein’s role in the gassing of Halabja, The Nonproliferation Review 28 (2021) 115–129. [CrossRef]
- J.R. Hiltermann, A Poisonous Affair: America, Iraq, and the Gassing of Halabja, Cambridge University Press (2014) ISBN: 9780521876865, 0521876869.
- Sarin attacks in Japan: acute and delayed health effects in survivors, Handbook of Toxicology of Chemical Warfare Agents (Third Edition), R. C. Gupta, Academic Press, Elsevier (2020) 37–53, ISBN 9780128190906. [CrossRef]
- T. Okumura, T. Nomura, T. Suzuki, M. Sugita, Y. Takeuchi, T. Naito, S. Okumura, H. Maekawa, N. Takasu, K. Miura, K. Suzuki, The Dark Morning: The Experiences and Lessons Learned from the Tokyo Subway Sarin Attack, Chemical Warfare Agents, John Wiley & Sons (2007) 277–285, ISBN: 9780470013595. [CrossRef]
- OPCW, Note by the Technical Secretariat: Report of the OPCW Fact-Finding Mission in Syria Regarding Alleged Incidents in Ltamenah, the Syrian Arab Republic on 24 and 25 March 2018 (S/1636/2018), 2018. https://www.opcw.org/sites/default/files/documents/S_series/2018/en/s-1636-2018_e_.pdf (accessed June 6, 2024). 25 March.
- A.T. Tu, The use of VX as a terrorist agent: action by Aum Shinrikyo of Japan and the death of Kim Jong-Nam in Malaysia: four case studies, Global Security: Health, Science and Policy 5 (2020) 1, 48–56. [CrossRef]
- T. Nakagawa, A.T. Tu, Murders with VX: Aum Shinrikyo in Japan and the assassination of Kim Jong-Nam in Malaysia, Forensic Toxicol. 36 (2018) 542–544. [CrossRef]
- D. Butler, Attacks in UK and Syria highlight growing need for chemical-forensics expertise, Nature 556 (2018) 7701, 285–286. [CrossRef]
- H.K. Kim, State Terrorism as a Mechanism for Acts of Violence against Individuals: Case Studies of Kim Jong-Nam, Skripal and Khashoggi Assassinations, Journal of East Asia and International Law (JEAIL) 14 (2021) 55–78. [CrossRef]
- V. Aroniadou-Anderjaska, J.P. V. Aroniadou-Anderjaska, J.P. Apland, T.H. Figueiredo, M. De Araujo Furtado, M.F. Braga, Acetylcholinesterase inhibitors (nerve agents) as weapons of mass destruction: History, mechanisms of action, and medical countermeasures, Neuropharmacology 181 (2020) 108298. [CrossRef]
- R.M. Black, B. Muir, Derivatisation reactions in the chromatographic analysis of chemical warfare agents and their degradation products, J. Chromatogr. A 1000 (2003) 1-2, 253–281. [CrossRef]
- I.S. Che Sulaiman, B.W. I.S. Che Sulaiman, B.W. Chieng, F.E. Pojol, K.K. Ong, J.I. Abdul Rashid, W.M.Z. Wan Yunus, N.A. Mohd Kasim, N. Abdul Halim, S.A. Mohd Noor, V.F. Knight, A review on analysis methods for nerve agent hydrolysis products, Forensic Toxicol. 38 (2020) 297–313. [CrossRef]
- D.D. Richardson, J.A. D.D. Richardson, J.A. Caruso, Derivatization of organophosphorus nerve agent degradation products for gas chromatography with ICPMS and TOF-MS detection, Anal. Bioanal. Chem. 388 (2007) 4, 809–823. [CrossRef]
- C.A. Valdez, R.N. Leif, Analysis of Organophosphorus-Based Nerve Agent Degradation Products by Gas Chromatography-Mass Spectrometry (GC-MS): Current Derivatization Reactions in the Analytical Chemist’s Toolbox, Molecules 26 (2021) 4631. [CrossRef]
- D.D. Richardson, B.B.M. D.D. Richardson, B.B.M. Sadi, J.A. Caruso, Reversed phase ion-pairing HPLC-ICP-MS for analysis of organophosphorus chemical warfare agent degradation products, J. Anal. At. Spectrom. 21 (2006) 396–403. [CrossRef]
- K.M. Kubachka, D.D. K.M. Kubachka, D.D. Richardson, D.T. Heitkemper, J.A. Caruso, Detection of chemical warfare agent degradation products in foods using liquid chromatography coupled to inductively coupled plasma mass spectrometry and electrospray ionization mass spectrometry, J. Chromatogr. A 1202 (2008) 2, 124–131. [CrossRef]
- E. Carletti, L. Jacquamet, M. Loiodice, D. Rochu, P. Masson, F. Nachon, Update on biochemical properties of recombinant Pseudomonas diminuta phosphotriesterase., J. Enzyme Inhib. Med. Chem.24 (2009) 4, 1045–1055. [CrossRef]
- R.J. Duchovic, J.A. R.J. Duchovic, J.A. Vilensky, Mustard Gas: Its Pre-World War I History, J. Chem. Educ. 84 (2007) 6, 944. [CrossRef]
- UN. Secretary-General, Report of the Mission Dispatched by the Secretary-General to Investigate Allegations of the Use of Chemical Weapons in the Conflict between the Islamic Republic of Iran and Iraq, S/20060, New York (1988). https://digitallibrary.un.org/record/48547/files/S_20060-EN.
- M.H. Rabiee, M. Ghanei, H. Amini, A. Akhlaghi, Mortality rate of people exposed to Mustard Gas during Iran-Iraq war in Sardasht, Iran: a 32 years retrospective cohort study, BMC Public Health 22 (2022) 1152. [CrossRef]
- H. Abolghasemi, M.H. H. Abolghasemi, M.H. Radfar, M. Rambod, P. Salehi, H. Ghofrani, M.R. Soroush, F. Falahaty, Y. Tavakolifar, A. Sadaghianifar, S.M. Khademolhosseini, Z. Kavehmanesh, M. Joffres, F.M. Burkle, E.J. Mills, Childhood physical abnormalities following paternal exposure to sulfur mustard gas in Iran: a case-control study, Confl. Health. 4 (2010) 13. [CrossRef]
- T. Knobloch, J. Bełdowski, C. Böttcher, M. Söderström, N.P. Rühl, J. Sternheim, Baltic Marine Environment Protection Commission Chemical Munitions Dumped in the Baltic Sea Report of the ad hoc Expert Group to Update and Review the Existing Information on Dumped Chemical Munitions in the Baltic Sea (HELCOM MUNI), Balt. Sea Environ. Proc., Helsinki (2013) ISSN 0357-2994. https://helcom.fi/wp-content/uploads/2019/10/Chemical-Munitions-Dumped-in-the-Baltic-Sea-Report-of-the-ad-hoc-Expert-Group.pdf (access: 10. June 2024.).
- T. Missiaen, M. Söderström, I. Popescu, P. Vanninen, Evaluation of a chemical munition dumpsite in the Baltic Sea based on geophysical and chemical investigations, Sci. Total Environ.408 (2010) 17, 3536–3553. [CrossRef]
- J. Bełdowski, Z. Klusek, M. Szubska, R. Turja, A.I. Bulczak, D. Rak, M. Brenner, T. Lang, L. Kotwicki, K. Grzelak, J. Jakacki, N. Fricke, A. Östin, U. Olsson, J. Fabisiak, G. Garnaga, J.R. Nyholm, P. Majewski, K. Broeg, M. Söderström, P. Vanninen, S. Popiel, J. Nawała, K. Lehtonen, R. Berglind, B. Schmidt, Chemical Munitions Search & Assessment-An evaluation of the dumped munitions problem in the Baltic Sea, Deep Sea Res., Part II 128 (2016) 85–95. [CrossRef]
- N. Schlager, J. Weisblatt, D.E. Newton, Chemical Compounds, Thomson Gale, ISBN 1 4144 0467 0 (2006). https://rushim.ru/books/spravochniki/chemical-compounds.pdf (access: 10. June 2024.).
- S.L. Hoenig, Compendium of Chemical Warfare Agents, Springer Science & Business Media, LLC (2007), ISBN: 978-0-387-34626-7. [CrossRef]
- B.T. Røen, Trace determination of sulphur mustard and related compounds in environmental samples by headspace-trap GC-MS, Norwegian Defence Research Establishment (FFI), FFI-rapport 2008/02247 (2008) ISBN 978-82-464-1469-0. https://ffi-publikasjoner.archive.knowledgearc.net/bitstream/handle/20.500.12242/2250/08-02247.pdf (access: 10. June 2024.).
- N.B. Munro, S.S. N.B. Munro, S.S. Talmage, G.D. Griffin, L.C. Waters, A.P. Watson, J.F. King, V. Hauschild, The Sources, Fate, and Toxicity of Chemical Warfare Agent Degradation Products, Environ. Health Perspect. 107 (1999) 12, 933–974. [CrossRef]
- R.M. Black, R.J. Clarke, D.B. Cooper, R.W. Read, D. Utley, Application of headspace analysis, solvent extraction, thermal desorption and gas chromatography-mass spectrometry to the analysis of chemical warfare samples containing sulphur mustard and related compounds, J. Chromatogr. A 637 (1993) 1, 71–80. [CrossRef]
- A.E. Boyer, D. Ash, D.B. Barr, C.L. Young, W.J. Driskell, R.D. Whitehead, M. Ospina, K.E. Preston, A.R. Woolfitt, R.A. Martinez, L.A. Silks, J.R. Barr, Quantitation of the sulfur mustard metabolites 1,1′-sulfonylbis[2-(methylthio)ethane] and thiodiglycol in urine using isotope-dilution gas chromatography-tandem mass spectrometry, J. Anal. Toxicol. 28 (2004) 5, 327–332. [CrossRef]
- J. Riches, R.W. J. Riches, R.W. Read, R.M. Black, Analysis of the sulphur mustard metabolites thiodiglycol and thiodiglycol sulphoxide in urine using isotope-dilution gas chromatography-ion trap tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 845 (2007) 1, 114–120. [CrossRef]
- S. Popiel, J. Nawała, D. Dziedzic, M. Söderström, P. Vanninen, Determination of mustard gas hydrolysis products thiodiglycol and thiodiglycol sulfoxide by gas chromatography-tandem mass spectrometry after trifluoroacetylation, Anal. Chem. 86 (2014) 12, 5865–5872. [CrossRef]
- K.K. Kroening, D.D. Richardson, S. Afton, J.A. Caruso, Screening hydrolysis products of sulfur mustard agents by high-performance liquid chromatography with inductively coupled plasma mass spectrometry detection, Anal. Bioanal. Chem. 393 (2009) 8, 1949–1956. [CrossRef]
- G. Garnaga, E. Wyse, S. Azemard, A. Stankevičius, S. de Mora, Arsenic in sediments from the southeastern Baltic Sea, Environ. Pollut. 144 (2006) 3, 855–861. [CrossRef]
- M. Kuligowska, S. Neffe, Collection and identification of samples contaminated with chemical warfare agents and their degradation products with the use of military equipment for detecting chemical contamination at the site of their occurrence, Bulletin of MUT 71 (2022) 4, 73–110. [CrossRef]
- T. Bausinger, J. Preuß, Environmental remnants of the first World War: Soil contamination of a burning ground for arsenical ammunition, Bull. Environ. Contam. Toxicol. 74 (2005) 6, 1045–1052. [CrossRef]
- M. Hempel, B. Daus, C. Vogt, H. Weiss, Natural attenuation potential of phenylarsenicals in anoxic groundwaters. Environ. Sci. Technol. 2009, 18, 6989–6995. [CrossRef]
- M. Ishizaki, T. Yanaoka, M. Nakamura, T. Hakuta, S. Ueno, M. Komuro, M. Shibata, T. Kitamura, A. Honda, M. Doy, K. Ishii, A. Tamaoka, N. Shimojo, T. Ogata, E. Nagasawa, S. Hanaoka, Detection of Bis(diphenyl wrsine)oxide, Diphenylarsinic Acid and Phenylarsonic Acid, Compounds Probably Derived from Chemical Warfare Agents, in Drinking Well Water, J. Health Sci. 52 (2005) 2, 130–137. [CrossRef]
- K. Kinoshita, Y. K. Kinoshita, Y. Shida, C. Sakuma, M. Ishizaki, K. Kiso, O. Shikino, H. Ito, M. Morita, T. Ochi, T. Kaise, Determination of diphenylarsinic acid and phenylarsonic acid, the degradation products of organoarsenic chemical warfare agents, in well water by HPLC-ICP-MS, Appl. Organomet. Chem. 19 (2005) 2, 287–293. [CrossRef]
- J. Nawała, D. Gordon, D. Dziedzic, P. Rodziewicz, S. Popiel, Why does Clark I remain in the marine environment for a long time?, Sci. Total Environ. 774 (2021) 145675. [CrossRef]
- T. Brzeziński, M. Czub, J. Nawała, D. Gordon, D. Dziedzic, B. Dawidziuk, S. Popiel, P. Maszczyk, The effects of chemical warfare agent Clark I on the life histories and stable isotopes composition of Daphnia magna, Environ. Pollut. 266 (2020) 115142. [CrossRef]
- K. Kinoshita, O. Shikino, Y. Seto, T. Kaise, Determination of degradation compounds derived from Lewisite by high performance liquid chromatography/inductively coupled plasma-mass spectrometry, Appl. Organomet. Chem. 20 (2006) 9, 591–596. [CrossRef]
- K. Kinoshita, A. Noguchi, K. Ishii, A. Tamaoka, T. Ochi, T. Kaise, Urine analysis of patients exposed to phenylarsenic compounds via accidental pollution, J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci. 867 (2008) 2, 179–188. [CrossRef]
- B. Daus, J. Mattusch, R. Wennrich, H. Weiss, Analytical investigations of phenyl arsenicals in groundwater, Talanta 75 (2008) 2, 376–379. [CrossRef]
- S.J. Stetson, M.L. Erickson, J. Brenner, E.C. Berquist, C. Kanagy, S. Whitcomb, C. Lawrence, Stability of inorganic and methylated arsenic species in laboratory standards, surface water and groundwater under three different preservation regimes, J. Appl. Geochem. 125 (2021) 104814, 13. [CrossRef]
- S. Hisatomi, L. Guan, M. Nakajima, K. Fujii, M. Nonaka, N. Harada, Formation of diphenylthioarsinic acid from diphenylarsinic acid under anaerobic sulfate-reducing soil conditions, J. Hazard. Mater. 262 (2013) 25–30. [CrossRef]
- K.T. Suzuki, B.K. Mandal, A. Katagiri, Y. Sakuma, A. Kawakami, Y. Ogra, K. Yamaguchi, Y. Sei, K. Yamanaka, K. Anzai, M. Ohmichi, H. Takayama, N. Aimi, Dimethylthioarsenicals as arsenic metabolites and their chemical preparations, Chem. Res. Toxicol. 17 (2004) 7, 914–921. [CrossRef]
- OPCW, Chemical Weapon Convention on the Prohibition of the Development, Production, Stockpiling and Use of Chemical Weapons and on their Destruction, Geneva (1992). https://www.opcw.org/sites/default/files/documents/CWC/CWC_en.pdf (access: 10. June 2024.).
- T.M. Baygildiev, I.A. Rodin, A.N. Stavrianidi, A. v. Braun, D.I. Akhmerova, O.A. Shpigun, I. v. Rybalchenko, Time-efficient LC/MS/MS determination of low concentrations of methylphosphonic acid, Inorg. Mater. 53 (2017) 1382–1385. [CrossRef]
- J.Y. Lee, Y.H. Lee, Rapid screening and determination of nerve agent metabolites in human urine by LC-MS/MS, J. Anal. Chem. 69 (2014) 909–916. [CrossRef]
- T. Baygildiev, M. Vokuev, A. Braun, I. Rybalchenko, I. Rodin, Monitoring of hydrolysis products of mustard gas, some sesqui- and oxy-mustards and other chemical warfare agents in a plant material by HPLC-MS/MS, J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci. 1162 (2021) 122452. [CrossRef]
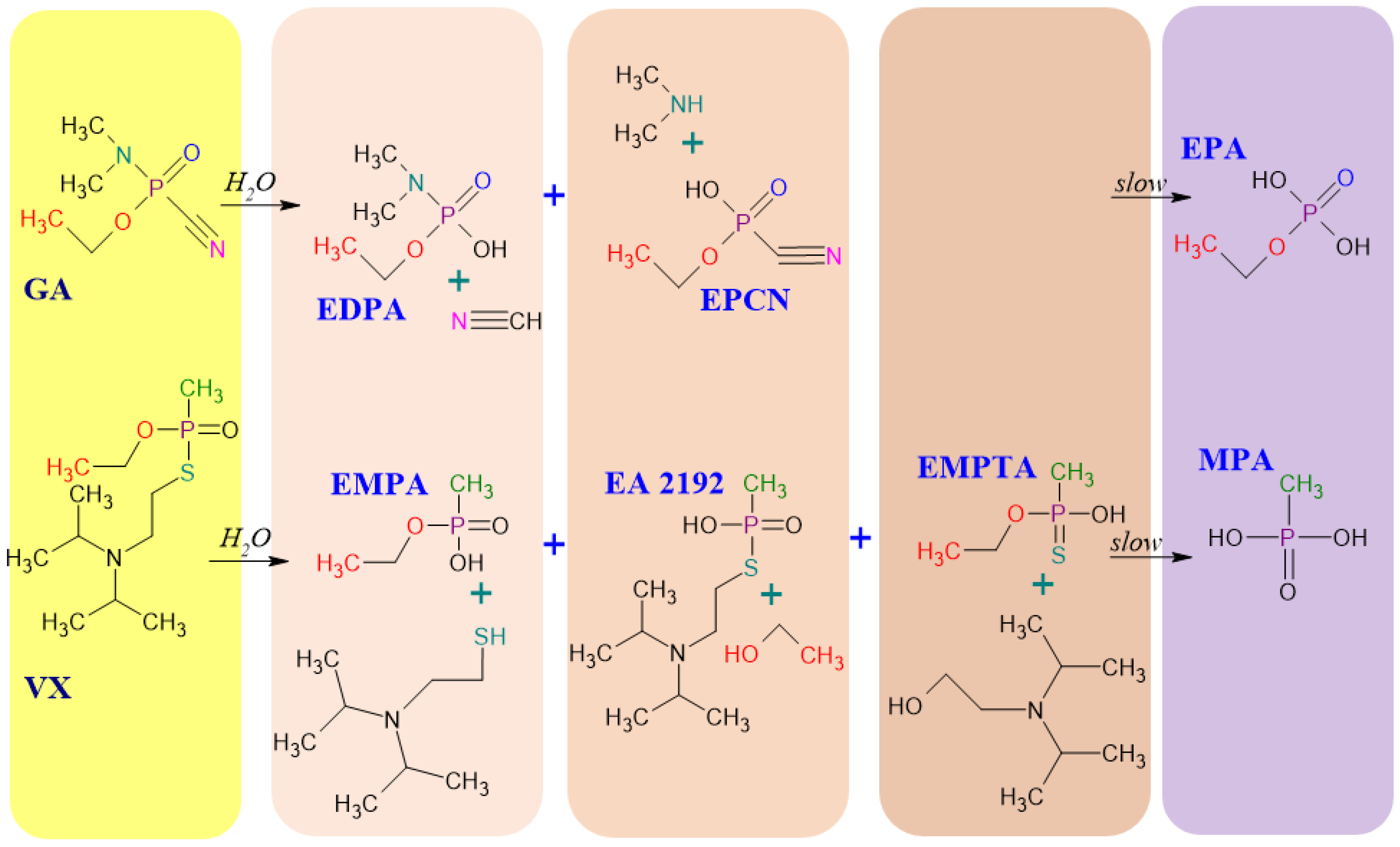
Figure 2.
Simplified scheme of the hydrolysis of RVX, sarin (GB), soman (GD), and cyclosarin (GF). Starting substances (CWA) are marked on a yellow background, intermediates are marked on a beige background, and the product of the hydrolysis of these compounds (MPA) is marked on a purple background. [5,31,32].
Figure 2.
Simplified scheme of the hydrolysis of RVX, sarin (GB), soman (GD), and cyclosarin (GF). Starting substances (CWA) are marked on a yellow background, intermediates are marked on a beige background, and the product of the hydrolysis of these compounds (MPA) is marked on a purple background. [5,31,32].
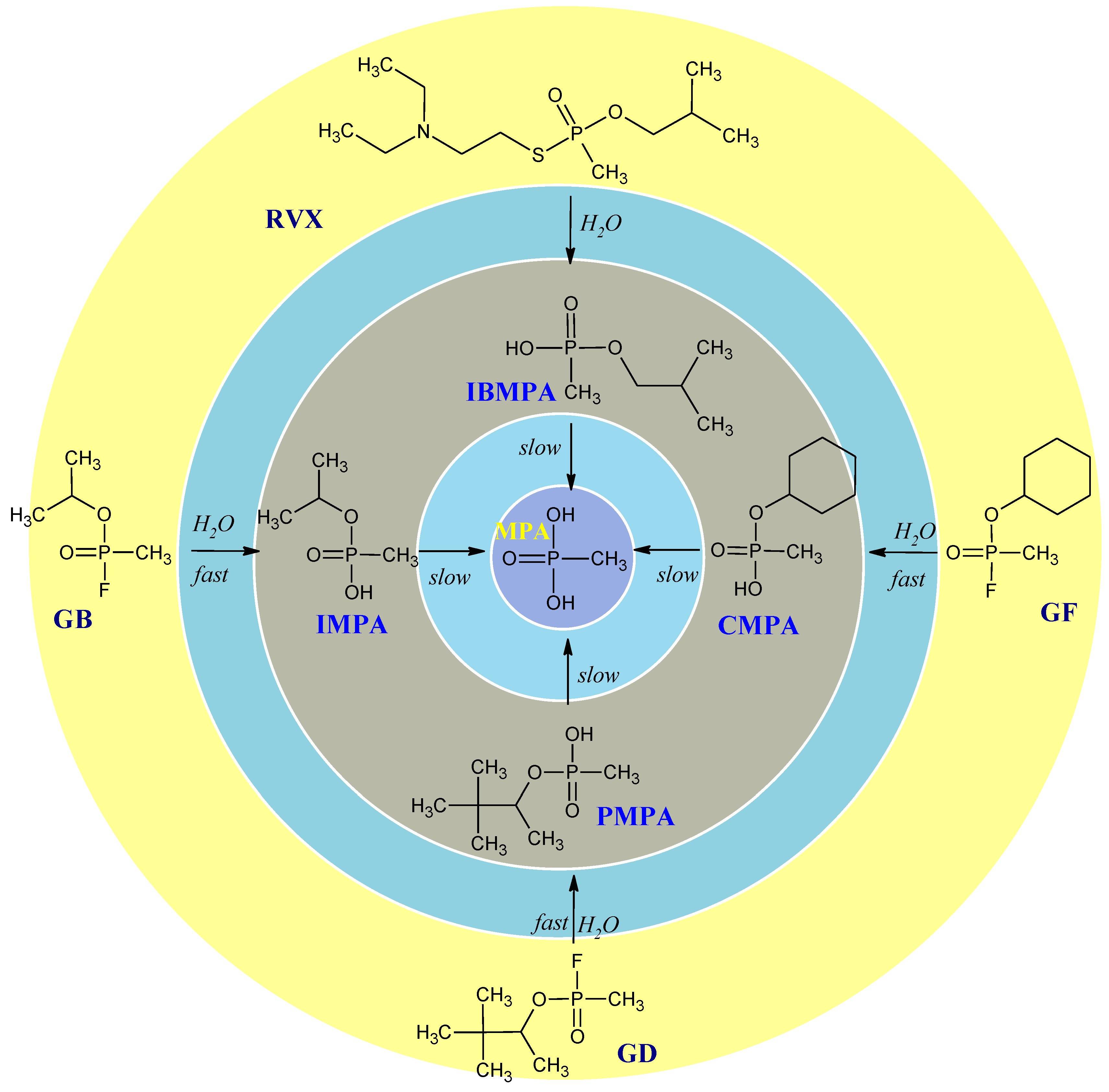
Figure 3.
The most common bis(2-chloroethyl)sulfide degradation products. Based on[47].
Figure 3.
The most common bis(2-chloroethyl)sulfide degradation products. Based on[47].
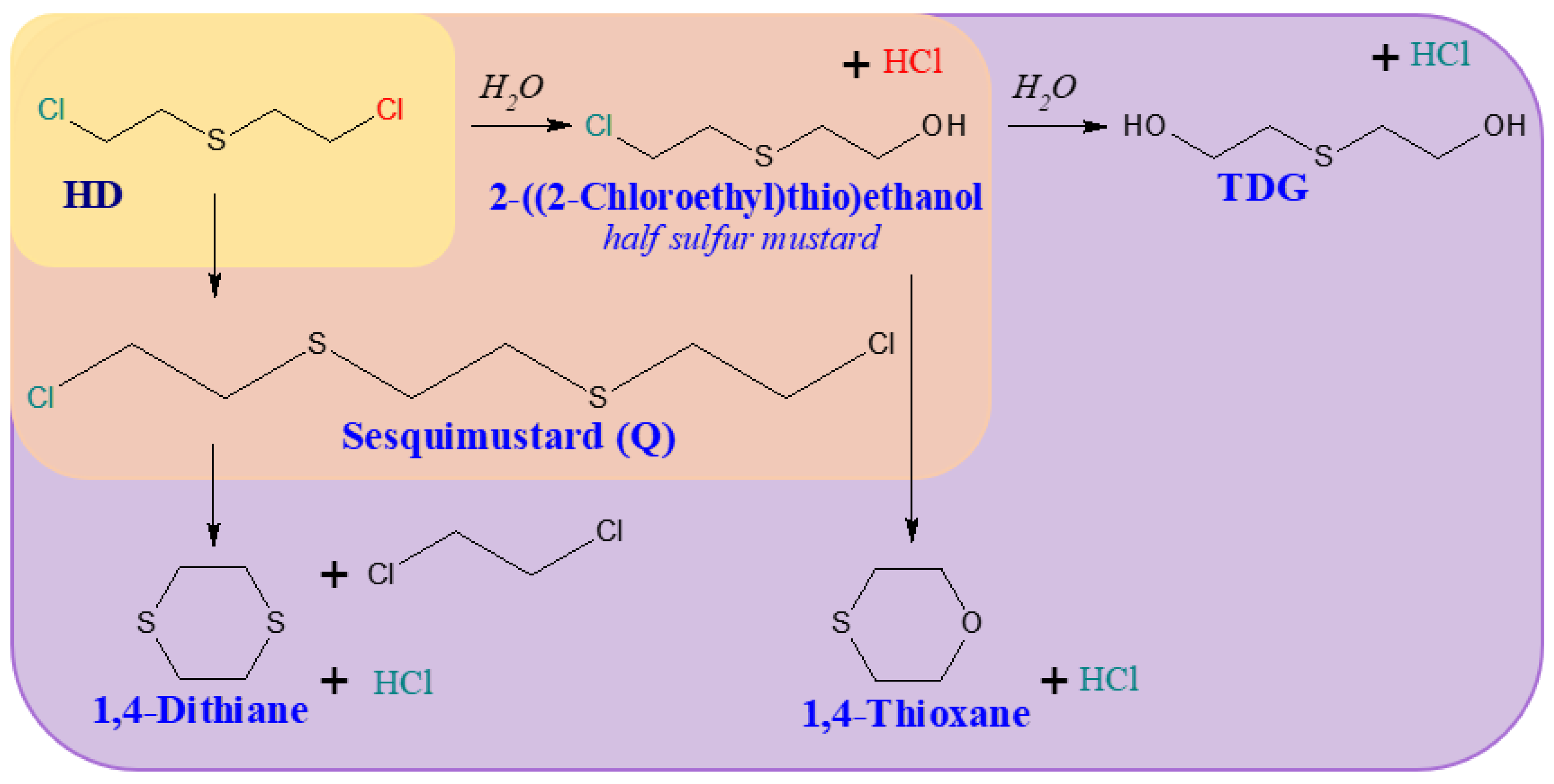
Figure 4.
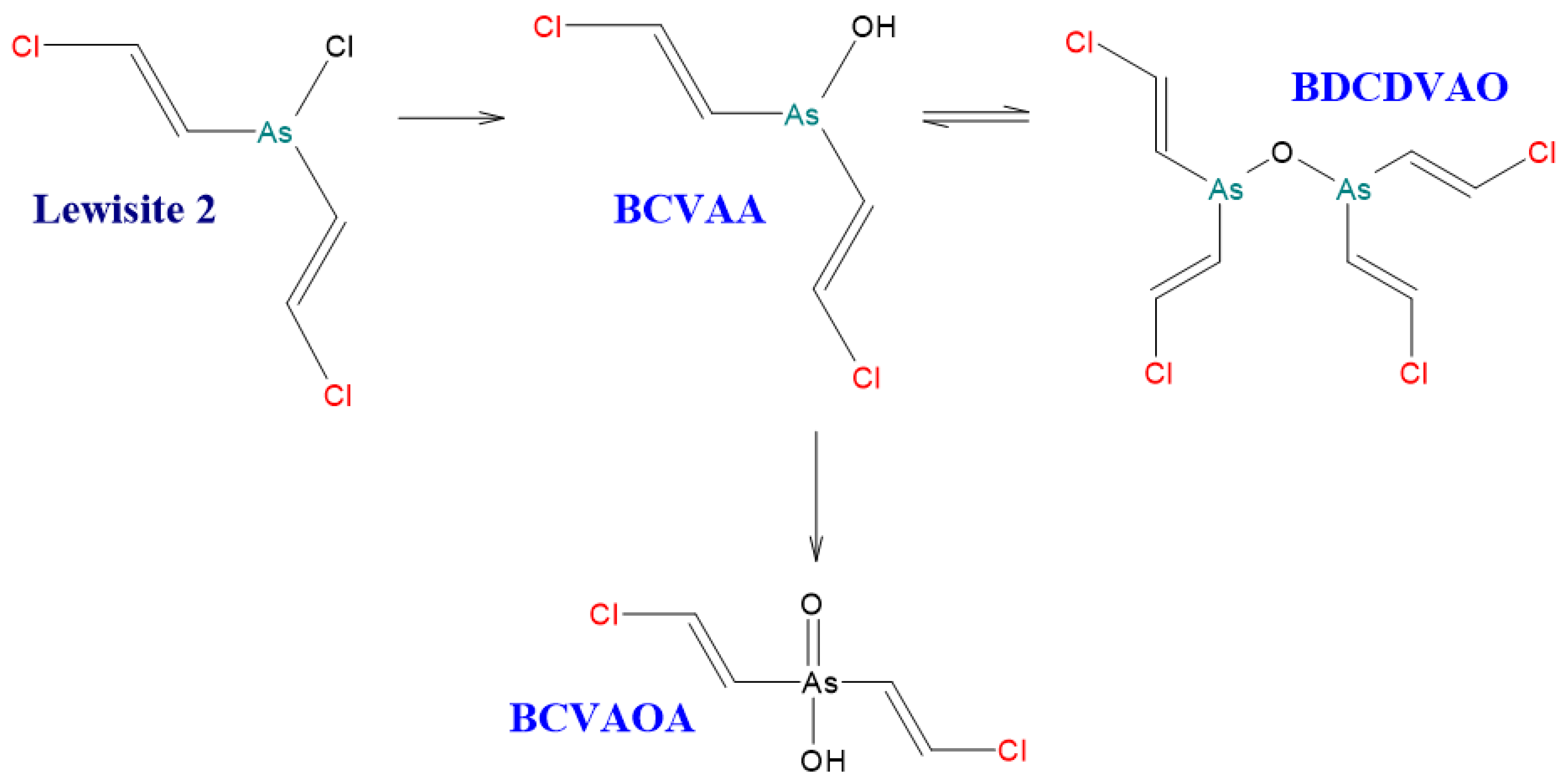
Hydrolysis of [(E)-2-Chloroethen-1-yl]arsonous dichloride (Lewisite 1). [5].
Figure 4.
Hydrolysis of [(E)-2-Chloroethen-1-yl]arsonous dichloride (Lewisite 1). [5].
Figure 5.
Hydrolysis of Bis[(E)-2-chloroethen-1-yl]arsinous chloride (Lewisite 2). [5].
Figure 5.
Hydrolysis of Bis[(E)-2-chloroethen-1-yl]arsinous chloride (Lewisite 2). [5].

| Analyte. | LOD for HPLC-ICP-MS [pg/mL] | LOD for GC-ICP-MS [pg/mL] |
|---|---|---|
| EMPA | 263 | 34,2 |
| IMPA | 183 | 20,9 |
| MPA | 139 | 49,6 |
Table 2.
Some parameters for OPNA DP separation [35].
Table 2.
Some parameters for OPNA DP separation [35].
| No. | Type of column | Mobile phase | Flow [mL/min] | V injection [μL] | Analytes | LOD [ng/mL] | Separation time [min] |
|---|---|---|---|---|---|---|---|
| 1. | Dionex IonPac AS7 4.1 mm × 250 mm, guard column | 0.4 mM acetic acid/sodium acetate, 5 mM HNO3 (pH ∼2.4) in DDW | 1.0 | 25 | EMPA IMPA MPA |
NA NA NA |
5 |
| 2. | Hamilton PRP-X100 4.6 mm × 100 mm 5μm | A= 0.5% H2CO2 (pH ∼2.3), 5% MeOH in DDW B = 0.3M NH4HCO2 (pH ∼2.3), 22% MeOH in DDW |
1.0 | 100 | EDHAP MPA EPA DMHP PPA EMPA IMPA DEHP IPHEP IBHMP |
21.7 18.3 19.9 10.0 22.9 20.4 19.5 26.6 61.5 81.1 |
25 |
| 3. | Hamilton PRP-X100 2.1 mm× 150 mm 5 μm | A= 10 mM (NH4)2CO3 (pH ∼8.5) in DDW B=50 mM (NH4)2CO3 (pH ∼8.5) in DDW |
0.5 | 100 | EDHAP MPA EPA DMHP PPA EMPA IMPA |
88.1 7.4 7.4 8.9 14.5 11.2 28.6 |
30 |
NA – not appear.
Table 3.
Selected parameters of analyzes carried out in the work of Kroening et al.[52].
Table 3.
Selected parameters of analyzes carried out in the work of Kroening et al.[52].
| STRUCTURE | 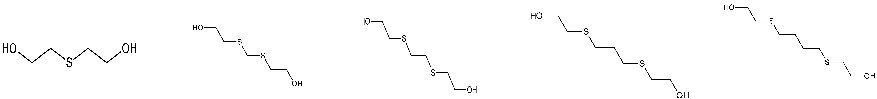 |
||||
| ACRONYM | TDG | BHETM | BHETE | BHETPr | BHETBu |
| COMPOUND NAME | Thiodiglycol | Bis(2-hydroxyethylthio)methane | 1,2-bis(2-hydroxyethylthio)ethane | 1,3-bis(2-hydroxyethylthio)propane | 1,4-bis(2-hydroxyethylthio)butane |
| LOD [NG/ML] STUDY | 4,60 | 35,50 | 79,30 | 98,50 | 73,20 |
| LOQ [NG/ML] ENVIRONMENTAL SAMPLES | 750,00 | 690,00 | 1,00 | 1,16 | 3,30 |
Table 4.
Summary of analysis parameters and the obtained limit of detection in the work of Kinoshita et al.[61,62].
| Publication[62] | Publication[61] | ||||
|---|---|---|---|---|---|
| Mechanism | Hydrophobicity | Dipole-dipole interaction | Separation of functional groups | ||
| Column | C4 | CN | NH2 | C8 | |
| Organic solvents | H2O (HNO3) / C2H5OH /ACN = 80:15:5, | citrate buffer / CH3OH /ACN = 70:20:10, | phosphate buffer/ACN = 50:50 | 0.1% HCOOH–CH3CN = 80:20 | |
| pH | 1,5 | 5,5 | 2,5 | 2,0 | |
| T column [°C] | 40 | 40 | 40 | 40 | |
| Flow [mL/min] | 0,3 | 0,3 | 0,1 | 0,2 | |
| V injection[μl] | 20 | 10 | no satisfactory separation, the NH2 column was not used for further research |
20 | |
| LOD [ng/mL As] | PAA | 0,25 | 1,0 | 0,3 | |
| PMAA | 0,25 | 0,5 | - | ||
| DPAA | 0,5 | DPAA + AB | 0,5 | ||
| PDMAO | 0,1 | 0,5 | - | ||
| DMPAO | 0,3 | 1,0 | - | ||
Table 5.
Comparison of the sensitivity of HPLC-MS/MS and HPLC-ICP-MS based on selected works in which CWA degradation products were analytes.
Table 5.
Comparison of the sensitivity of HPLC-MS/MS and HPLC-ICP-MS based on selected works in which CWA degradation products were analytes.
| No. | Analyte | LOD [ppb = ng/mL] | |||
| HPLC-MS/MS | Ref. | HPLC-ICP-MS | Ref. | ||
| 1. | MPA | 10 | [68] | 0.14a) | [34] |
| 2. | EMPA | 5* 1** |
[69] | 0.03 a) | [34] |
| 3. | IMPA | 5* 1** |
[69] | 0.02 a) | [34] |
| 4. | TDG | 40 | [70] | 4.6 | [52] |
| 5. | CVAOA | 500 | [6] | 0.1 | [61] |
| 6. | PAA | 50 | [6] | 0.25 | [62] |
| 7. | PMAA | 1 | [6] | 0.25 | [62] |
*(SIM mode), ** (SRM mode), a) approximate value to the second decimal place.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



