Submitted:
16 July 2024
Posted:
17 July 2024
You are already at the latest version
Abstract
Preeclampsia is a complex pregnancy-related hypertensive disorder which poses significant risks for both maternal and fetal health. Preeclampsia affects 5-8% of the pregnancies in the United States, causing a significant public health and economic burden. Despite extensive research, the etiology and pathogenesis of preeclampsia remain elusive, but have been correlated with maternal conditions such as obesity. In the past decades, the incidence of preeclampsia increased along with the prevalence of obesity among women of reproductive age. Maternal obesity has been shown to negatively affect pregnancy in almost all aspects. However, the precise mechanisms by which obesity influences preeclampsia are unclear. Ankyrin repeat and SOCS Box Containing protein 4 (ASB4) is an E3 ubiquitin ligase that can promote the degradation of a wide range of target proteins. ASB4-null mice display a full spectrum of preeclampsia-like phenotypes during pregnancy including hypertension, proteinuria, and decreased litter size. Furthermore, maternal obesity induced by a high fat diet aggravates preeclampsia-like phenotypes in pregnant mice lacking ASB4. Variants in the ASB4 gene have been associated with obesity in humans, and a functional connection between the ASB4 gene and obesity has been established in mice. This review discusses the connections between preeclampsia, obesity, and ASB4.
Keywords:
preeclampsia
; ASB4
; Obesity
; Insulin
; Leptin
; Implantation
; Endometrium
; Trophoblast
Preeclampsia
Preeclampsia is a hypertensive disorder unique to pregnancy that occurs in approximately 5–8% of U.S. pregnancies [1]. In the United States, the incidence of preeclampsia has dramatically increased in the past three decades [2,3]. There are two major classifications of preeclampsia: early-onset preeclampsia (EOP), which develops before 34 weeks of pregnancy, and late-onset preeclampsia (LOP) which develops on or after 34 weeks of pregnancy. About 20% of preeclampsia cases are EOP, which is most commonly associated with severe clinical symptoms, while patients with LOP typically have more mild symptoms [4]. Preeclampsia is a leading cause of maternal morbidity and mortality; women who develop preeclampsia are at increased risk for pulmonary edema [5], coagulation defects, hepatic and/or renal failure, seizures, cerebral hemorrhage, blindness [6,7,8], and even death [9]. Preeclampsia is also a significant contributor to neonatal morbidity and mortality. Fetal growth restriction—likely due to chronic placental hypoperfusion—is a common complication of preeclampsia [10]. Fetal growth restriction makes women with preeclampsia 3-4 times more likely to deliver small-for-gestational age babies compared to women with normal pregnancy [11]. Placental abruption, premature separation of the placenta with disruption of blood flow to the fetus, is an uncommon but dangerous complication that occurs in up to 3% of pregnancies complicated by severe preeclampsia [12]. Preeclampsia not only affects women and their babies during pregnancy but also in the years following pregnancy. Preeclamptic mothers and their offsprings have increased risk of developing cardiovascular and cognitive problems in later of life among others [13,14,15].
At present, there are few measures to predict or prevent the development of preeclampsia. The only definitive treatment for preeclampsia is delivery of the placenta and infant as physicians did a century ago [16]. During pregnancy, treatments for preeclampsia include monitoring of maternal and fetal health, as well as administering non-teratogenic anti-hypertensive drugs such as methyldopa to lower blood pressure and an antiseizure drug magnesium sulfate to lower heart rate and prevent eclampsia, which is a rare, but life-threatening condition that is characterized by the onset of seizures or coma [17,18]. Treatment other than delivery is an unmet need for women with preeclampsia.
While the etiology and pathogenesis of preeclampsia remain unclear, defective placentation resulting from impaired trophoblast invasion is thought to be at the center of preeclampsia pathogenesis (Figure 1) [19]. Impaired trophoblast invasion during early pregnancy leads to insufficient remodeling of maternal spiral arteries which subsequently decreases blood flow to placenta [20]. The ischemic placenta releases anti-angiogenic and inflammatory factors such as sFlt-1 (soluble fms-like tyrosine kinase 1, soluble form of vascular endothelial growth factor receptor 1) into maternal circulation, causing maternal endothelial dysfunction and clinical manifestations of preeclampsia: hypertension, proteinuria and end-organ damage [21]. Pre-pregnancy health conditions including obesity, chronic hypertension, diabetes mellitus, renal disease, and autoimmune disorders increase risk for preeclampsia [22]. These pre-existing maternal conditions can impact the severity of preeclampsia by negatively affecting placentation and the maternal endothelium.
Obesity and Preeclampsia
Obesity is defined as excessive fat accumulation that presents a risk to health [23]. The incidence of obesity has continued to increase among women of reproductive age, causing approximately 50% of women worldwide to enter pregnancy overweight or obese [24,25]. Obesity is a major risk factor for preeclampsia [26]. Over the past three decades, the increase in preeclampsia reflected the increased prevalence of obesity in the United States [27,28], as the rate of preeclampsia increased with increasing body mass index (BMI) [29]. Even in non-obese women, the occurrence of preeclampsia is positively correlated with BMI [30]. Obesity increases the risk of all types of preeclampsia, namely, severe and mild preeclampsia, as well as EOP and LOP [26]. Obesity is also associated with maternal hyperinsulinemia, increased inflammation and oxidative stress, and altered adipokines and pro-/anti-angiogenic factors [31,32,33,34], which also contribute to preeclampsia pathogenesis (Figure 2). In the following sections, we will discuss the role of insulin and leptin in preeclampsia as these factors are altered in the obese mouse model of preeclampsia—i.e., feeding ASB4-null mice with a high fat diet (HFD) as describe in the last section of this review.
Insulin
Insulin and Implantation: Insulin is a polypeptide hormone secreted by the pancreas that functions in glucose, lipid, and protein metabolism [35]. In the uterus, insulin signaling is essential for endometrial epithelial and stromal cell proliferation and differentiation before implantation [36]. Implantation is a delicate interaction between the embryo and endometrium, and it is crucial for the establishment of successful pregnancy. Altered endometrial receptivity and/or trophoblast cell biology lead to impaired implantation and insufficient placentation [37]. Insufficient placentation subsequently causes the release of anti-angiogenic and proinflammatory factors into maternal circulation which contribute to preeclampsia development, especially in EOP [38].
Excess Insulin in Maternal Endometrial Receptivity: Hyperinsulinemia is an established characteristic of obesity [39,40]. Insulin may impair implantation by interfering with endometrial receptivity: insulin resistance does not affect early embryo development but decreases implantation rate in the in vitro maturation-in vitro fertilization-embryo transfer cycle of women with polycystic ovarian syndrome (PCOS)—a syndrome associated with insulin resistance [41]. Women with PCOS have endometrial receptivity problems such as impaired decidualization and placentation, as well as an increased risk for preeclampsia [42]. Animal studies demonstrate that insulin signaling is essential for normal endometrial structure and function [36], however, excess insulin has adverse effects on endometrial receptivity. For example, Li R. et al. reported that exogenous administration of insulin prior to mating induced hyperinsulinemia in wild type (WT) female mice [43]. The expression of markers of endometrial receptivity (i.e., estrogen receptor, progesterone receptor and homeobox A10) were altered in these mice, leading to the conclusion that endometrial receptivity in these mice was impaired by maternal hyperinsulinemia [43]. However, the authors did not observe a decreased number of implantation sites as Li M. et al. observed in mice with poor endometrial receptivity caused by haploinsufficiency for adrenomedullin, which has been established as a mice model that exhibits reduced maternal fertility with endometrial receptivity [44]. Furthermore, Li R. et al. did not identify any obvious morphological changes in the endometrium that are examined by light microscopy in pregnant WT mice with exogenous administration of insulin. Whether there is ultra-structural change is unclear [44].
High doses of insulin (5-500 nM) increased prokineticin expression during decidualization of human endometrial stromal cells, inhibiting the migratory and invasive capacity of trophoblast cells [45]. Supporting the hypothesis that insulin adversely affects endometrial receptivity, our unpublished data showed that Ishikawa cells, an epithelial cell line derived from an endometrial adenocarcinoma, had decreased normal trophoblast spheroid attachment to the Ishikawa monolayer after treatment with a high dose insulin (500 nM). Overall, insulin could cause dysfunction of endometrial epithelial and stromal cells, leading to impaired endometrial receptivity.
Excess Insulin in Trophoblast Cells: Embryo-derived trophoblasts also play an important role in implantation, but it is almost impossible to study trophoblast biology at such an early stage of human pregnancy due to ethical issues. Cell culture models and experimental animal studies provide insights on the effect of excess insulin in trophoblast cells. Vega et al. reported that human primary first trimester trophoblast cells exposed to 1 nM of insulin (the human physiological concentration of insulin is 0.1 nM) in culture had decreased cell survival due to increased DNA damage and apoptosis—as evidence by increased γ-H2AX (a DNA damage marker) and cleaved caspase-3 (a marker of apoptosis) [46]. Furthermore, pre-treatment with metformin (a drug improves insulin sensitivity by increasing peripheral glucose uptake and utilization) prevented the detrimental effects of insulin [46].
In the immortalized human first trimester trophoblast cell line HTR8/SVneo, Silva et al. found that insulin (1-10 nM for 48 hours) decreased cell proliferation in a dose and time-dependent manner, possibly in association with the activation of down-stream mediators of insulin including phosphoinositide 3 kinase (PI3K), mammalian target of rapamycin (mTOR), and p38 mitogen-activated protein kinase (p38 MAPK) [47]. Quercetin (a flavonoid supplement) and simvastatin (for lowering blood cholesterol and triglycerides levels) inhibited insulin's antiproliferative effect [47]. In contrast, exposing trophoblast cells to insulin (10 nM for 48 hours) showed no significant effect on cell viability, apoptosis, and migration capacity [47].
Later, a publication reported that by treating HTR-8/SVneo cells with higher dose of insulin (1 µM) for 48 hours, the proliferation, migration, and invasion of cells were suppressed [48]. Taken together, these in vitro data suggest that higher-than-normal insulin negatively affects trophoblast cells and could lead to impaired implantation. More study is needed to provide strong evidence to support the theory that insulin directly impairs trophoblast cells and subsequent implantation.
Insulin in Maternal Endothelium: Hyperinsulinemia may also aggravate the effects of placenta-derived anti-angiogenic/inflammatory factors on pre-existing endothelial problems. Under normal physiological conditions, insulin maintains the endothelial health by balancing the production of vasodilator [endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO)] and vasoconstrictor (endothelin-1, ET-1) using two separate pathways. Insulin increases eNOS activity and NO production through the insulin receptor (IR)/insulin receptor substrate 1 (IRS-1)/PI3K/phosphoinositide-dependent kinase-1 (PDK-1)/Ak strain transforming (AKT) pathway [49], while insulin stimulates ET-1 secretion through IR/growth factor receptor-bound protein 1(GRB-1)/rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/MAPK pathway [49]. Under hyperinsulinemia condition, the PI3K pathway in vascular tissues is seriously impaired, whereas the MAPK pathway remains unaffected, leading to increased ET-1/decreased NO production [50,51]. This imbalance caused by hyperinsulinemia could be exaggerated by detrimental factors derived from placentas in preeclamptic pregnancies.
Leptin
Leptin and Implantation: Leptin—a 146-amino acid (16 kDa) peptide hormone mainly secreted by adipose tissue—regulates food intake, body mass, and reproductive function [52]. While leptin is required before and during implantation, it is not essential for late pregnancy and parturition (giving birth) [53]. Obesity causes hyperleptinemia (elevated plasma leptin levels) in humans and mice [54,55,56], which is associated with preeclampsia [57,58]. Low plasma leptin during the first trimester has been associated with women who suffer spontaneous first trimester loss, implying the direct role of leptin in implantation [59]. Animal studies also provide clear evidence of the indispensable role of leptin during implantation. Female mice lacking leptin are infertile, however, administration of leptin (5 mg/kg twice daily) only during implantation—i.e., the first 6 days of pregnancy—leads to normal pregnancy and parturition [53].
Excess Leptin in Maternal Endometrial Receptivity: PCOS patients with elevated serum leptin levels had increased very early pregnancy loss and decreased expression of the γ epithelial Na+ channel (γ-ENaC, important in embryo implantation) in the secretory phase of endometrium [60]. In a cell culture model, treatment with high doses of leptin (50-200 ng/mL) down-regulated the expression of γ-ENaC, which was accompanied with decreased trophoblast spheroid attachment to cultured endometrial epithelial cells (Ishikawa cells) [60]. Knockdown of STAT3 (signal transducer and activator of transcription 3), a mediator of leptin action, blocked the effects of leptin [60]. In vitro studies reported that leptin inhibits normal human endometrial stromal cell differentiation, which is critical for proper endometrial receptivity [61]. Taken together, these data suggest that excess leptin/STAT3 signaling may impair both endometrial epithelial and stromal cell function, leading to defective endometrial receptivity.
Excess Leptin in Trophoblast Cells: Though there are several studies that provide information of the role of leptin in trophoblasts, the direct effects of excess leptin on trophoblasts are not clear [62]. Using the human first trimester placenta-derived extravillous trophoblast cell line (TEV-1) [63], Liu et al. found that 5-500 ng/mL of leptin (non-obese and non-pregnant adult women has serum leptin levels around 8 ng/mL) decreased cell proliferation after 24 hours of treatment. The highest dose of leptin treatment promoting trophoblast invasion and migration [64,65], indicating that excess leptin could inhibit trophoblast proliferation but promote differentiation. Two other groups have similarly reported that leptin stimulated the invasion of another trophoblast cell line (HTR8/SVneo) [66,67]. Mechanistically, it was proposed that the crosstalk between metastasis associated 1 (MTA1)/wingless-related integration site (WNT) and PI3K/AKT pathways possibly increases matrix metallopeptidase 14 (MMP-14) [66]. On the other hand, Wang et al. postulate that the crosstalk between the neurogenic locus notch homolog protein 1 (NOTCH1) and PI3K/AKT pathways activates β-catenin, thereby driving trophoblast invasion [67]. Trophoblast progenitors must sufficiently proliferate before differentiating into distinct trophoblast cell lineages. If leptin disrupts the proliferative process, trophoblasts could differentiate prematurely and negatively affect embryo implantation.
ASB4
Ankyrin Repeat and SOCS Box-Containing protein 4 (ASB4) was initially discovered in mice as an imprinted gene that is only maternally expressed [68]. Like most proteins in the ASB4 family, ASB4 acts as an adaptor for the subunits of cullin-based ubiquitin ligases [69]. ASB4 is a substrate-recognition component of the E3 ubiquitin ligase complex [70]. Nine N-terminal ankyrin repeats of ASB4 serve as the substrate-binding site, and the C-terminal suppressor of cytokine signaling (SOCS) box interacts with elongin B/C, rendering the E3 ligase function [69,71,72,73]. Together, the E3 ligase complex mediates the ubiquitination and subsequent proteasomal degradation of the target proteins [71].
ASB4 and Obesity: Genome wide association studies have identified the link between single nucleotide polymorphisms (SNPs) in the ASB4 gene and obesity [74,75]. ASB4’s specific expression in hypothalamic arcuate nucleus (ARC) regulates energy homeostasis and food intake through insulin and leptin [74,75]. Leptin increases ASB4 expression in the ARC while insulin seems to have no effect [76]. During fasting, ASB4 expression is decreased in the hypothalamus, and it is indirectly downregulated by Agouti-related protein (AgRP), an orexigenic neuropeptide [77,78]. ASB4 is expressed in proopiomelanocortin (POMC) and neuropeptide Y (NPY) neurons in ARC [74,76]. In mice, POMC-specific ASB4 knockdown resulted in impaired glucose tolerance without obesity, while overexpressing ASB4 in POMC neurons caused an increase in food intake without obesity [77]. ASB4 is also required in calcitonin signaling. Global ASB4 deficiency in mice leads to decreased expression of calcitonin receptor (CalcR), which inhibits the anorectic effect of calcitonin and only male mice, not female mice, lacking ASB4 started becoming heavier than WT mice at 5 months of age [79].
In human subjects with type 2 diabetes, ASB4 expression is reduced in the hypothalamus, suggesting the potential role of ASB4 in satiety and glucose homeostasis [77]. Mechanistic studies have reported that ASB4 colocalizes with insulin receptor substrate 4 (IRS4) in the hypothalamus [74]. ASB4 increases IRS4 ubiquitination and decreases the phosphorylation of AKT, thereby reducing the downstream signaling of IRS4 and potentially regulating the action of insulin and leptin in the hypothalamus [74]. ASB4 also interacts with G-protein pathway suppressor 1 (GPS1), and this interaction decreases the phosphorylation of IRS1 and inhibits c-Jun NH2- terminal kinase (JNK) activity [80].
ASB4’s involvement in cancer cell lines might also provide potential links with obesity. In liver cancer, ASB4 expression is upregulated by microRNA-200a (miRNA-200a), and suppressing ASB4 mitigated the invasion and migration ability of a subset of hepatocellular carcinoma cells [81]. Although the exact mechanisms are not clear, miRNAs are identified as key factors in glucose and lipid metabolism [82]. Disruption in the ASB4-miRNA interaction could possibly play a role in the development of obesity and its related metabolic changes. ASB4 was also identified as a downstream protein of the transcription factor nuclear factor kappa b (NF-κB) in the TNF-α signaling pathway [83]. Obesity is associated with the long-term low-grade inflammation of adipocytes and macrophages [84]. NF-κB can initiate the inflammation cascade through the activation of macrophages [85], and this could be a new angle to examine the mechanistic relationships between ASB4 and obesity.
ASB4 and Vascular Development: During early embryogenesis, the Asb4 gene is highly expressed in the vasculature and it is spatiotemporally expressed in vasculogenic tissues such as yolk sac, dorsal aorta, and placenta [70]. In mice, embryonic day 7.5 (E7.5) embryos have low global Asb4 mRNA expression, but at E9.5, Asb4 expression was drastically increased, but begins to diminish after E10.5 and it is only expressed in tissues such as forelimb and umbilical vessels [70]. The onset of placental blood flow increases oxygen tension and triggers factors inhibiting HIF1α (FIH) to hydroxylate ASB4 at Asn 246, thereby promoting vascular development [70]. As the blood vessels mature, ASB4 mRNA expression in the highly vascularized organs (liver, lungs, kidneys) was quickly downregulated and became undetectable [70]. In addition, Asb4 gene transcription can be decreased by laminar shear stress and hypoxia insult [83], further highlighting the importance of ASB4 in the vascular system. In adults, Asb4 mRNA expression was only detected in testis, ovary, and heart [70], suggesting that ASB4 is more important for early vascular development rather than for tissue maintenance.
ASB4 and PE
ASB4, Implantation and Preeclampsia: ASB4 is known to contribute to the differentiation of trophoblast stem cells into vascular cells and giant trophoblasts, which are necessary for embryo implantation and embryogenesis in mice [86], however, its role in human pregnancy is unknown. Insufficient placentation resulting from a lack of ASB4 displays immature vascular patterning and retains the expression of placental progenitor markers, including the ID2 [86]. ID2 is a member of the anti-differentiation ID protein family, which shares significant structural similarity to the basic helix-loop-helix (bHLH) family of transcription factors but lacks the basic domain [87]. Through heterodimerization with functional factors, ID2 blocks the transcription of pro-differentiation elements by preventing bHLH dimerization and subsequent translocation into the nucleus [88]. ASB4 mediates the ubiquitination and proteasomal degradation of ID2, and these processes are essential for the differentiation of cells [86]. Thus, ASB4 is a key element in trophoblast differentiation, implantation and placentation. Asb4-/- female mice develop mild preeclampsia-like phenotypes during pregnancy, including increased systolic blood pressure (SBP) and urinary albumin excretion, as well as a decreased litter size [86]. Because a loss of ASB4 leads to impaired trophoblast cell differentiation [89], it is highly possible that Asb4-/- dams have defective implantation. However, neither blastocyst function nor endometrial receptivity—which could contribute to defective implantation—have been investigated in Asb4-/- dams.
In addition, we found that circulation VEGF levels are decreased in Asb4-/- dams, suggesting that imbalanced angiogenesis contributes to the pathogenesis of PE in these mice [90] The mechanism by which lacking ASB4 leads to decreased VEGF is not clear. We hypothesize that increased ID2 may play a role because hepatocellular carcinoma (HCC) over-expressing ID2 showed decreased VEGF secretion [91]. However, neuroblastoma cells transfected with Ad-Id2 had increased VEGF expression [92]. Therefore, future study is needed to elucidate the effects of ID2 on VEGF in placentas lacking ASB4.
ID2 is expressed by human vascular endothelial and smooth muscle cells, thus, dysregulation of this protein could cause maternal endothelial dysfunction. It would be interesting to know the levels of ID2 in vasculatures of Asb4-/- mice and if they exhibit any dysfunctions.
ASB4, Obesity and Preeclampsia: Recently, we demonstrated that high fat diet (HFD)-induced obesity worsens the preeclampsia-like phenotypes in Asb4-/- dams while the same diet treatment did not have obvious adverse effects on WT mice [93]. Therefore, we focused on comparing Asb4-/- mice on a normal chow (NC) or HFD. Asb4-/- female mice were started on a HFD (42% calories from fat) at age of 21 days for five weeks. At age of eight weeks old, Asb4-/- males were introduced for mating, and HFD was fed throughout the entire pregnancy. We have demonstrated: 1. HFD-induced obesity in Asb4-/- dams as evidenced by approximately 2x increase in visceral white fat mass in Asb4-/- mice fed HFD compared to Asb4-/- mice fed a normal diet. 2. Asb4-/- dams also had increased plasma cholesterol (1.6 x), insulin (2.9 x), and leptin (1.5 x) levels compared to NC-fed Asb4-/- dams while the plasma levels of glucose and triglyceride were not different between the two groups of mice. 3. plasma VEGF levels were decreasing in HFD-fed Asb4-/- dams along with pregnancy, although these mice had higher plasma VEGF levels than NC-fed Asb4-/- mice before pregnancy. 4. Importantly, HFD-fed Asb4-/- dams had higher blood pressure and urinary albumin excretion than NC-fed Asb4-/- dams while HFD-fed Asb4-/- dams had decreased number of surviving fetuses than NC-fed Asb4-/- dams at the term (e.g. 18.5 day post coitus), suggesting that all the preeclampsia-like phenotypes presented in Asb4-/- dams were aggravated by HFD-induced obesity.
Following this observation, we investigated the role of altered factors in increased severity of preeclampsia. We have demonstrated that placental ID2 was higher in Asb4-/- dams than NC-fed Asb4-/- dams and investigated the role of insulin and leptin in increased placental ID2. In cultured HTR8/SVneo cell, high insulin (10 nM, comparable to the concentration observed in patients with insulin resistance [94,95]) increased ID2 protein levels while leptin (5-500 ng/ml) did not in these cells. These data suggest that hyperinsulinemia could play a role in increased ID2, leading to placental problems and decreased VEGF levels. Of note, we did not measure free fatty acid levels in plasma, we cannot exclude the effects of free fatty acid on ID2 and placentation. The decreased placental VEGF could be due to increased ID2; however, this hypothesis needs to be tested.
Conclusion and Future Direction
Both environmental and genetic factors contribute to the development of obesity. Environmental factors, such as a high-calorie diet, promote obesity, while genetics factors can increase susceptibility to this condition. The manifestation of obesity is influenced by multiple genetic factors and their complex interactions with each other. Better understanding of the natural relationship between obesity and preeclampsia is needed, as these conditions not only adversely affect the mother and fetus during pregnancy but also have long-term health impacts.
Feeding Asb4-/- mice with HFD (that is adjusted to diets consumed in Western societies) effectively replicates this human condition (obese combining with preeclampsia). This excellent mouse model provides a platform to better understand the molecular and genetic pathways involved in the pathogenesis of preeclampsia, including impaired uterine receptivity, trophoblast biology, and implantation process, which subsequently leads to insufficient placentation and maternal endothelial dysfunction. This mouse model could also be used to test the efficacy of drugs target the signaling pathways altered by obesity, including insulin, leptin, and free fatty acid. Future research could aim to elucidate the precise mechanisms linking obesity-related metabolic dysregulations with preeclampsia pathogenesis, as well as to explore novel targets such as ASB4 that could potentially mitigate the impact of maternal obesity on pregnancy outcomes. By addressing these gaps, there is potential to improve maternal and fetal health and reduce the burden preeclampsia brings to the global healthcare systems.
Funding
This publication was supported by the National Institutes of Health (award nos. R01HL049277 (N.M.-S.) and R01HD101485 (F.L.)).
Conflicts of Interest
None
References
- Saftlas, A.F.; et al. Epidemiology of preeclampsia and eclampsia in the United States, 1979-1986. Am J Obstet Gynecol 1990, 163, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the United States, 1980-2010: age-period-cohort analysis. BMJ 2013, 347, f6564. [Google Scholar] [CrossRef] [PubMed]
- Wallis, A.B.; et al. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987-2004. Am J Hypertens 2008, 21, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.; Peterson, E. A critical review of early-onset and late-onset preeclampsia. Obstet Gynecol Surv 2011, 66, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, T.J.; Kates, R.; Williams, V. Hemodynamic observations in severe preeclampsia complicated by pulmonary edema. Am J Obstet Gynecol 1985, 152, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Drislane, F.W.; Wang, A.M. Multifocal cerebral hemorrhage in eclampsia and severe pre-eclampsia. J Neurol 1997, 244, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Morriss, M.C.; et al. Cerebral blood flow and cranial magnetic resonance imaging in eclampsia and severe preeclampsia. Obstet Gynecol 1997, 89, 561–568. [Google Scholar] [PubMed]
- Cunningham, F.G.; Fernandez, C.O.; Hernandez, C. Blindness associated with preeclampsia and eclampsia. Am J Obstet Gynecol 1995, 172, 1291–1298. [Google Scholar] [CrossRef]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: risk factors and outcomes associated with early- versus late-onset disease. Am J Obstet Gynecol 2013, 209, 544 e1–544 e12. [Google Scholar] [CrossRef]
- Odegard, R.A.; et al. Preeclampsia and fetal growth. Obstet Gynecol 2000, 96, 950–955. [Google Scholar]
- Xiao, R.; et al. Influence of pre-eclampsia on fetal growth. J Matern Fetal Neonatal Med 2003, 13, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.M.; et al. Aggressive versus expectant management of severe preeclampsia at 28 to 32 weeks' gestation: a randomized controlled trial. Am J Obstet Gynecol 1994, 171, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Gammill, H. Pre-eclampsia and cardiovascular disease in later life. The Lancet 2005, 366, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; et al. Preeclampsia and cognitive impairment later in life. American Journal of Obstetrics and Gynecology 2017, 217, e1–e74. [Google Scholar] [CrossRef] [PubMed]
- Wojczakowski, W.; et al. Preeclampsia and Cardiovascular Risk for Offspring. Journal of Clinical Medicine 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, E.; et al. Pre-eclampsia. Nature Reviews Disease Primers 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-J.; Seow, K.-M.; Chen, K.-H. Preeclampsia: Recent Advances in Predicting, Preventing, and Managing the Maternal and Fetal Life-Threatening Condition. International Journal of Environmental Research and Public Health 2023, 20. [Google Scholar] [CrossRef]
- Odigboegwu, O.; Pan, L.J.; Chatterjee, P. Use of Antihypertensive Drugs During Preeclampsia. Frontiers in Cardiovascular Medicine 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. Journal of Reproductive Immunology 2019, 134-135, 1–10. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biology of Reproduction 2003, 69, 1–7. [Google Scholar] [CrossRef]
- Maynard, S.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. Journal of Clinical Investigation 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Catov, J.M.; et al. Risk of early or severe pre-eclampsia related to pre-existing conditions. Int J Epidemiol 2007, 36, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, M.; le Roux, C.W.; Docherty, N.G. Morbidity and mortality associated with obesity. Annals of Translational Medicine 2017, 5, 161–161. [Google Scholar] [CrossRef]
- Hill, B.; et al. Health in Preconception, Pregnancy and Postpartum Global Alliance: International Network Preconception Research Priorities for the Prevention of Maternal Obesity and Related Pregnancy and Long-Term Complications. Journal of Clinical Medicine 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ogunwole, S.M.; Zera, C.A.; Stanford, F.C. Obesity Management in Women of Reproductive Age. Jama 2021, 325. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; et al. The Role of Obesity in Preeclampsia. Pregnancy Hypertens 2011, 1, 6–16. [Google Scholar] [CrossRef]
- Meldrum, D.R. ; M. A. Morris, and J.C. Gambone, Obesity pandemic: causes, consequences, and solutions-but do we have the will? Fertil Steril 2017, 107, 833–839. [Google Scholar]
- Rahman, M.M.; et al. Maternal body mass index and risk of birth and maternal health outcomes in low- and middle-income countries: a systematic review and meta-analysis. Obes Rev 2015, 16, 758–770. [Google Scholar] [CrossRef]
- Mbah, A.K.; et al. Super-obesity and risk for early and late pre-eclampsia. BJOG 2010, 117, 997–1004. [Google Scholar] [CrossRef]
- Bodnar, L.M.; et al. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann Epidemiol 2005, 15, 475–482. [Google Scholar] [CrossRef]
- Corvera, S.; Solivan-Rivera, J.; Loureiro, Z.Y. Angiogenesis in adipose tissue and obesity. Angiogenesis 2022, 25, 439–453. [Google Scholar] [CrossRef]
- Savini, I.; et al. Obesity-Associated Oxidative Stress: Strategies Finalized to Improve Redox State. International Journal of Molecular Sciences 2013, 14, 10497–10538. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; et al. Normalization of adiponectin concentrations by leptin replacement in ob/ob mice is accompanied by reductions in systemic oxidative stress and inflammation. Scientific Reports 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Fryk, E.; et al. Hyperinsulinemia and insulin resistance in the obese may develop as part of a homeostatic response to elevated free fatty acids: A mechanistic case-control and a population-based cohort study. EBioMedicine 2021, 65. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Wang, X.; Mittendorfer, B. Metabolic actions of insulin in men and women. Nutrition 2010, 26, 686–693. [Google Scholar] [CrossRef]
- Sekulovski, N.; et al. Insulin signaling is an essential regulator of endometrial proliferation and implantation in mice. The FASEB Journal 2021, 35. [Google Scholar] [CrossRef]
- Singh, M.; Chaudhry, P.; Asselin, E. Bridging endometrial receptivity and implantation: network of hormones, cytokines, and growth factors. Journal of Endocrinology 2011, 210, 5–14. [Google Scholar] [CrossRef]
- Rabaglino, M.B.; et al. Bioinformatics approach reveals evidence for impaired endometrial maturation before and during early pregnancy in women who developed preeclampsia. Hypertension 2015, 65, 421–429. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J Clin Invest 2000, 106, 473–481. [Google Scholar] [CrossRef]
- Kasumov, T.; et al. Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity (Silver Spring) 2015, 23, 1414–1421. [Google Scholar] [CrossRef]
- Chang, E.M.; et al. Insulin resistance does not affect early embryo development but lowers implantation rate in in vitro maturation–in vitro fertilization–embryo transfer cycle. Clinical Endocrinology 2013, 79, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Piltonen, T.T. Polycystic ovary syndrome: Endometrial markers. Best Pract Res Clin Obstet Gynaecol 2016, 37, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; et al. Mice endometrium receptivity in early pregnancy is impaired by maternal hyperinsulinemia. Molecular Medicine Reports 2017, 15, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, Y.; Caron, K.M. Haploinsufficiency for Adrenomedullin Reduces Pinopodes and Diminishes Uterine Receptivity in Mice1. Biology of Reproduction 2008, 79, 1169–1175. [Google Scholar] [CrossRef]
- Ujvari, D.; et al. Prokineticin 1 is up-regulated by insulin in decidualizing human endometrial stromal cells. Journal of Cellular and Molecular Medicine 2017, 22, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertility and Sterility 2019, 111, 489–496e5. [Google Scholar] [CrossRef]
- Silva, C.; et al. Insulin Exhibits an Antiproliferative and Hypertrophic Effect in First Trimester Human Extravillous Trophoblasts. Reproductive Sciences 2017, 24, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; et al. Calcium/calmodulin dependent protein kinase IV in trophoblast cells under insulin resistance: functional and metabolomic analyses. Mol Med 2023, 29, 82. [Google Scholar] [CrossRef]
- Kim, J.-a.; et al. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Cusi, K.; et al. Insulin resistance differentially affects the PI 3-kinase– and MAP kinase–mediated signaling in human muscle. Journal of Clinical Investigation 2000, 105, 311–320. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Bakris, G.L. Insulin and Endothelin: An Interplay Contributing to Hypertension Development? The Journal of Clinical Endocrinology & Metabolism 2007, 92, 379–385. [Google Scholar]
- Cervero, A.; et al. Leptin system in embryo development and implantation: a protein in search of a function. Reprod Biomed Online 2005, 10, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.M.; et al. Leptin Requirement for Conception, Implantation, and Gestation in the Mouse. Endocrinology 2001, 142, 5198–5202. [Google Scholar] [CrossRef] [PubMed]
- Frederich, R.C.; et al. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1995, 1, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Mendieta Zeron, H.; et al. Hyperleptinemia as a prognostic factor for preeclampsia: a cohort study. Acta Medica (Hradec Kralove) 2012, 55, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, F.A.; et al. Adiposity and hyperleptinemia during the first trimester among pregnant women with preeclampsia. Int J Womens Health 2017, 9, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Laird, S.M.; et al. Leptin and leptin-binding activity in women with recurrent miscarriage: correlation with pregnancy outcome. Human Reproduction 2001, 16, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-H.; et al. Leptin down-regulates γ-ENaC expression: a novel mechanism involved in low endometrial receptivity. Fertility and Sterility 2015, 103, 228–235e3. [Google Scholar] [CrossRef]
- Tanaka, T.; et al. Leptin inhibits decidualization and enhances cell viability of normal human endometrial stromal cells. Int J Mol Med 2003, 12, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; et al. Role of leptin in the pathophysiology of preeclampsia. Placenta 2023, 142, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.C.; et al. Establishment and characterization of a human first-trimester extravillous trophoblast cell line (TEV-1). J Soc Gynecol Investig 2005, 12, e21–e32. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; et al. Effect of leptin on cytotrophoblast proliferation and invasion. J Huazhong Univ Sci Technolog Med Sci 2009, 29, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Al-Sultan, A.I.; Al-Elq, A.H. Leptin levels in normal weight and obese saudi adults. J Family Community Med 2006, 13, 97–102. [Google Scholar] [PubMed]
- Fan, M.; et al. Leptin Promotes HTR-8/SVneo Cell Invasion via the Crosstalk between MTA1/WNT and PI3K/AKT Pathways. Disease Markers 2022, 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; et al. Leptin-Promoted Human Extravillous Trophoblast Invasion Is MMP14 Dependent and Requires the Cross Talk Between Notch1 and PI3K/Akt Signaling1. Biology of Reproduction 2014, 90. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; et al. Asb4, Ata3, and Dcn Are Novel Imprinted Genes Identified by High-Throughput Screening Using RIKEN cDNA Microarray. Biochemical and Biophysical Research Communications 2002, 290, 1499–1505. [Google Scholar] [CrossRef]
- Kohroki, J.; et al. ASB proteins interact with Cullin5 and Rbx2 to form E3 ubiquitin ligase complexes. FEBS Letters 2005, 579, 6796–6802. [Google Scholar] [CrossRef]
- Ferguson, J.E.; et al. ASB4 Is a Hydroxylation Substrate of FIH and Promotes Vascular Differentiation via an Oxygen-Dependent Mechanism. Molecular and Cellular Biology 2023, 27, 6407–6419. [Google Scholar] [CrossRef]
- Linossi, E.M. ; S. E. Nicholson, The SOCS box—Adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 2012, 64, 316–323. [Google Scholar]
- Wilcox, A.; et al. Asb6, an Adipocyte-specific Ankyrin and SOCS Box Protein, Interacts with APS to Enable Recruitment of Elongins B and C to the Insulin Receptor Signaling Complex. Journal of Biological Chemistry 2004, 279, 38881–38888. [Google Scholar] [CrossRef] [PubMed]
- Kile, B.T.; et al. Cloning and characterization of the genes encoding the ankyrin repeat and SOCS box-containing proteins Asb-1, Asb-2, Asb-3 and Asb-4. Gene 2000, 258, 31–41. [Google Scholar] [CrossRef]
- Li, J.-Y.; et al. Ankyrin repeat and SOCS box containing protein 4 (Asb-4) colocalizes with insulin receptor substrate 4 (IRS4) in the hypothalamic neurons and mediates IRS4 degradation. BMC Neuroscience 2011, 12. [Google Scholar] [CrossRef]
- Locke, A.E.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, M.W.; et al. Expression of Ankyrin Repeat and Suppressor of Cytokine Signaling Box Protein 4 (Asb-4) in Proopiomelanocortin Neurons of the Arcuate Nucleus of Mice Produces a Hyperphagic, Lean Phenotype. Endocrinology 2010, 151, 134–142. [Google Scholar]
- Vagena, E.; et al. ASB4 modulates central melanocortinergic neurons and calcitonin signaling to control satiety and glucose homeostasis. Science Signaling 2022, 15. [Google Scholar] [CrossRef]
- Li, J.Y.; et al. Arcuate Nucleus Transcriptome Profiling Identifies Ankyrin Repeat and Suppressor of Cytokine Signalling Box-Containing Protein 4 as a Gene Regulated by Fasting in Central Nervous System Feeding Circuits. Journal of Neuroendocrinology 2005, 17, 394–404. [Google Scholar] [CrossRef]
- Vagena, E.; et al. ASB4 modulates central melanocortinergic neurons and calcitonin signaling to control satiety and glucose homeostasis. Sci Signal 2022, 15, eabj8204. [Google Scholar] [CrossRef]
- Li, J.-Y.; et al. Akyrin repeat and SOCS box containing protein 4 (Asb-4) interacts with GPS1 (CSN1) and inhibits c-Jun NH2-terminal kinase activity. Cellular Signalling 2007, 19, 1185–1192. [Google Scholar] [CrossRef]
- Au, V.; et al. Expression of ankyrin repeat and SOCS box containing 4 (ASB4) confers migration and invasion properties of hepatocellular carcinoma cells. BioScience Trends 2014, 8, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Siani, A. Role of microRNAs in obesity and obesity-related diseases. Genes & Nutrition 2017, 12. [Google Scholar]
- Bode, M.; et al. Regulation of ankyrin repeat and suppressor of cytokine signalling box protein 4 expression in the immortalized murine endothelial cell lines MS1 and SVR: a role for tumour necrosis factor alpha and oxygen. Cell Biochemistry and Function 2011, 29, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; et al. Obesity: A Chronic Low-Grade Inflammation and Its Markers. Cureus, 2022.
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, Inflammation, and Metabolic Disease. Cell Metabolism 2011, 13, 11–22. [Google Scholar] [CrossRef]
- Townley-Tilson, W.H.D.; et al. The Ubiquitin Ligase ASB4 Promotes Trophoblast Differentiation through the Degradation of ID2. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Ling, F.; Kang, B.; Sun, X.H. Id proteins: small molecules, mighty regulators. Curr Top Dev Biol 2014, 110, 189–216. [Google Scholar] [PubMed]
- Liu, Y.P.; et al. Id2 is a primary partner for the E2-2 basic helix-loop-helix transcription factor in the human placenta. Mol Cell Endocrinol 2004, 222, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Townley-Tilson, W.H.; et al. The ubiquitin ligase ASB4 promotes trophoblast differentiation through the degradation of ID2. PLoS One 2014, 9, e89451. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc Natl Acad Sci U S A 2016, 113, 13450–13455. [Google Scholar] [CrossRef]
- Tsunedomi, R.; et al. Decreased ID2 promotes metastatic potentials of hepatocellular carcinoma by altering secretion of vascular endothelial growth factor. Clin Cancer Res 2008, 14, 1025–1031. [Google Scholar] [CrossRef]
- Lasorella, A.; et al. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol Cell Biol 2005, 25, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Kayashima, Y.; et al. Insulin Elevates ID2 Expression in Trophoblasts and Aggravates Preeclampsia in Obese ASB4-Null Mice. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertil Steril 2019, 111, 489–496 e5. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; et al. Insulin Exhibits an Antiproliferative and Hypertrophic Effect in First Trimester Human Extravillous Trophoblasts. Reprod Sci 2017, 24, 582–594. [Google Scholar] [CrossRef]
Figure 1.
Summary of the widely accepted mechanism of preeclampsia pathogenesis. Defects during trophoblast invasion cause shallow remodeling of uteroplacental spiral arteries. Abnormal vascular remodeling contributes to poor placentation due to the reduction in placental perfusion, making the placenta hypoxic/ischemic. The abnormal placenta releases inflammatory cytokines, anti-angiogenic factors, and reactive oxygen species into systemic circulation. These factors subsequently cause systemic endothelial dysfunction, leading to the clinical manifestations of preeclampsia (end organ damage, proteinuria, and hypertension). Downward arrows denote causation. All images are created with BioRender.com.
Figure 1.
Summary of the widely accepted mechanism of preeclampsia pathogenesis. Defects during trophoblast invasion cause shallow remodeling of uteroplacental spiral arteries. Abnormal vascular remodeling contributes to poor placentation due to the reduction in placental perfusion, making the placenta hypoxic/ischemic. The abnormal placenta releases inflammatory cytokines, anti-angiogenic factors, and reactive oxygen species into systemic circulation. These factors subsequently cause systemic endothelial dysfunction, leading to the clinical manifestations of preeclampsia (end organ damage, proteinuria, and hypertension). Downward arrows denote causation. All images are created with BioRender.com.
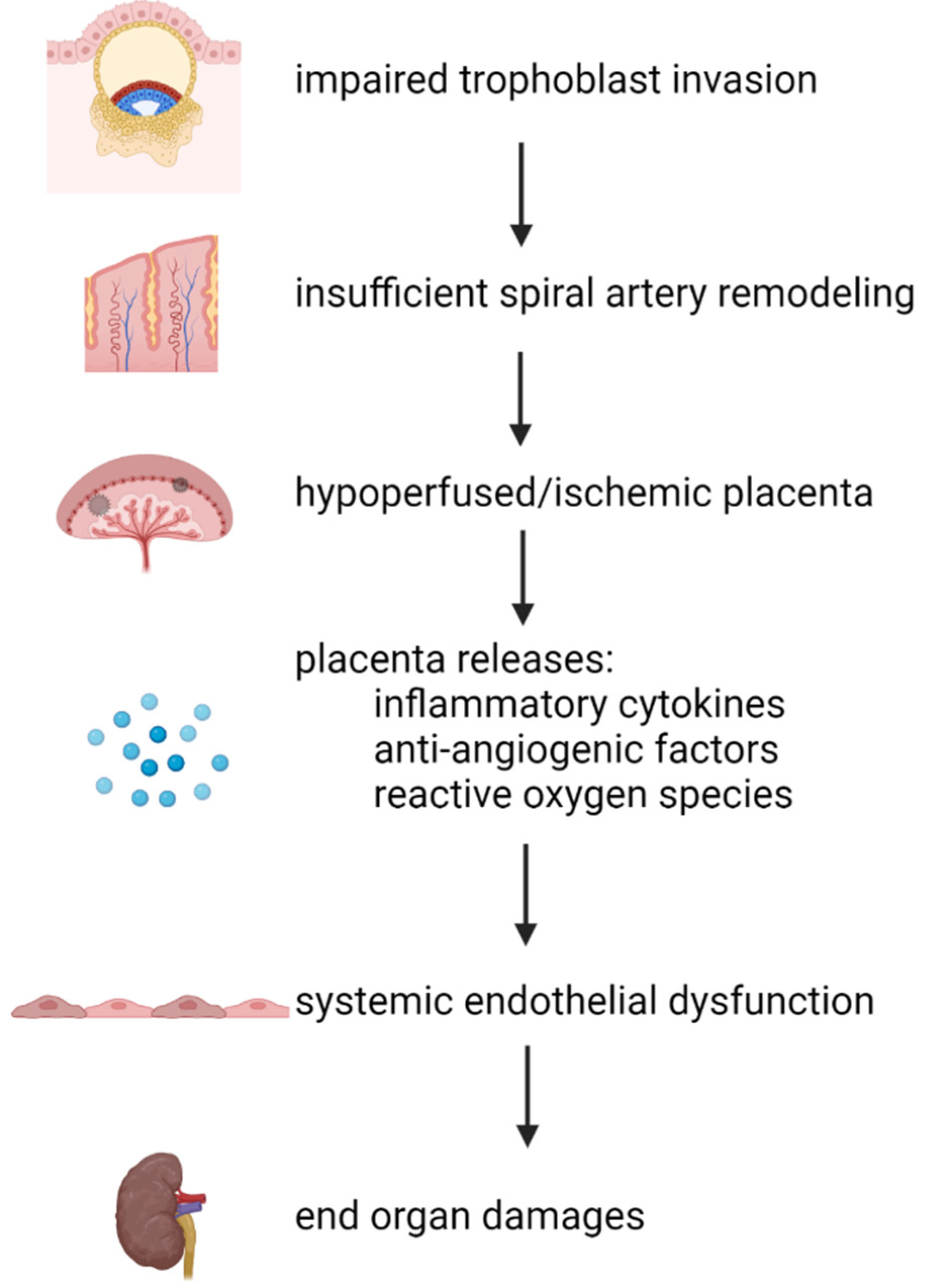
Figure 2.
A graphical overview of how maternal obesity caused by excessive adipose tissue (denoted as yellowish spheres around the abdominal area of the pregnant mother) contributes to the pathogenesis of preeclampsia. All images are created with BioRender.com.
Figure 2.
A graphical overview of how maternal obesity caused by excessive adipose tissue (denoted as yellowish spheres around the abdominal area of the pregnant mother) contributes to the pathogenesis of preeclampsia. All images are created with BioRender.com.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



