
Submitted:
10 July 2024
Posted:
10 July 2024
You are already at the latest version
Abstract
Diabetic Kidney Disease (DKD) is a common complication of Type 2 Diabetes Mellitus and a major risk factor for end-stage kidney disease. The immune system plays a significant role in the pathogenesis of DKD, involving infiltration of immune cells in the kidney, upregulation of in-flammatory mediators, downregulation of anti-inflammatory mediators, and disturbance in ad-hesion molecules and growth factors. Dendritic cells are professional antigen-presenting cells acting as a bridge connecting innate and adaptive immune responses. DC is also capable of modulating inflammation. Autologous dendritic cells can be made by in vitro differentiation of peripheral blood monocytes and then utilized as a cell-based therapy. Cell-based therapy for DKD is currently focused on utilizing the anti-inflammatory properties of stem cells. However, research suggests unsatisfactory results due to several limitations. This article highlights the possibility of using DC as a cell-based therapy for DKD through its role in controlling inflammation.
Keywords:
Dendritic cells
; Diabetic Kidney Disease
; Cell Immunotherapy
; Proteinuria
; Diabetes Mellitus
1. Introduction
Twenty to fifty percent of diabetic people develop diabetic kidney disease (DKD), which is a significant risk factor for end-stage kidney disease. [1] Over the course of the year, more than two million newly diagnosed DKD are found globally, and keep increasing. [2] The management of DKD cases represents a significant financial burden. This disease also significantly decreases the quality of life. Therefore, developing effective DKD therapies that can minimize side effects is necessary.
DKD is characterized by persistent proteinuria, gradual decline in renal function, histologically appears as glomerular disease. [3] In DKD, structural and functional changes occur in the kidneys [4]. Mesangial enlargement in the glomerulus, basement membrane thickening, loss of podocytes, nodular glo-merulosclerosis, and damage to endothelial cells are among the physically pathological characteristics of DKD. [5] Tubular hypertrophy in the early stages of DKD might progresses to fibrosis in the interstitial with atrophy of tubules. [6]
In DKD, there is also an increase in albumin excretion and disruption of glomerular filtration. [7] However, there is also a non-classical presentation where DKD occurs without albuminuria. Non-albuminuric DKD (NA-DKD) is hypothesized to be caused by macroangiopathy leading to interstitial fibrosis and vascular lesions. In NA-DKD, interstitial fibrosis due to this macroangiopathy is not associated (independent) with albuminuria. [8,9] Some studies suggest that renal impairment in NA-DKD is not caused by hyperglycemia or microangiopathy but rather by genetic susceptibility, aging, and arteriosclerosis. [10] This article will focus on the classical presentation of DKD with albuminuria due to these differences in pathogenic mechanisms.
Dendritic Cells (DCs) are innate immune cells that function as antigen-presenting cells. They are considered the "master regulator" of immunity. DCs connect innate and adaptive immunity. DCs express MHC class I and MHC class II molecules, allowing the presentation of antigens to both subsets of CD4+ T cells and CD8+ T cells (cross-presentation). The immune response triggered by DCs is diverse. Some antigens presented by DCs evoke an immunogenic response, activating the immune system to fight against the antigen. However, DCs also play a role in stimulating tolerogenic (anti-inflammatory) responses, thereby limiting excessive inflammation [11].
Type 2 Diabetes Mellitus (T2DM) involves metabolic abnormalities that lead to chronic low-grade inflammation. This chronic inflammation ultimately results in various complications, such as DKD. Administering agents that can control inflammation in T2DM patients has the potential to serve as therapy as well as prevention for DKD. Therefore, this article aims to elucidate the biomechanisms that underlie the development of DC-based DKD therapy.
2. Diabetic Kidney Disease Immunopathology
The immunopathological aspects of Type 2 Diabetes Mellitus (T2DM) constitute a complex mechanism. Immune dysregulation in T2DM is caused by the interaction of genetic, immunological, metabolic, and clinical elements [12]. Although Type 2 Diabetes Mellitus (T2DM) is generally classified as a non-autoimmune disease, recent findings suggest the involvement of autoimmune processes in this condition. [13,14] However, inflammation, insulin resistance, and a decrease in pancreatic beta cells are the leading causes of Type 2 Diabetes Mellitus (T2DM). [14] Understanding the immunological processes and conditions affected by the immune system is important in developing an effective therapy for DKD.
Chronic inflammation is a primary immune system-related pathology that plays a vital role in developing T2DM disease. [15,16,17,18] Inflammation causes various microvascular and macrovascular complications in diabetics. [17] T2DM is associated with activated inflammatory signaling and abnormal cytokine production. [19] Prolonged mild inflammation and elevated pro-inflammatory markers are closely associated with the development and progression of T2DM. [17] Inflammation also causes insulin resistance, beta cell dysfunction, and complications of diabetes [20,21]
Studies have shown that inflammation, characterized by increased C-reactive protein (CRP), is closely related to insulin resistance. [22] In addition, obesity provokes chronic inflammation, contributing to the formation of insulin resistance. [23,24] Subsequent research has revealed a connection between the resistance to insulin and cardiovascular diseases in T2DM patients with coronary heart disease which is bysystemic inflammation mediated by high-sensitivity CRP (hsCRP). [25] Inflammation not only plays a role in the pathogenesis of T2DM but also its complications, including nonalcoholic fatty liver, retinopathy, DKD, and cardiovascular problems. [23] Chronic inflammation is strongly associated with insulin resistance in individuals with T2DM, so addressing inflammation is critical in the management and prevention of T2DM [22,23,24,25]
This chronic inflammatory condition in T2DM causes various immune disorders that contribute to the pathogenesis of DKD. Molecularly involved factors start from transcription factors such as Nuclear Factor Kappa B (NF-kB) [26,27,28], Janus kinase/signal transducers and activators of transcription (JAK/STAT) [29,30], and Adenosine Monophosphate Activated Protein Kinase (AMPK) [31,32,33]. In addition, infiltration of immune cells in the kidneys also affects the course of DKD disease. Cytokines, chemokines, and related adhesion molecules determine the fate and reactivity of immune and non-immune cells in the kidneys. [34,35,36] The accumulation of all these factors causes abnormalities in the renal glomerulus, resulting in DKD.
In DM, glucose and metabolites activate macrophages present in the kidneys. [37] Macrophages release cytokines, recruit peripheral monocytes/macrophages, and increase kidney cell injury, ultimately resulting in inflammation and fibrosis. [38] Macrophages can be activated into pro-inflammatory phenotypes by pathogen-associated molecules released from injured kidney cells, stimulating the recruitment of other inflammatory cells and activating renal fibroblasts. [39] In chronic kidney disease, persistent activation of pro-inflammatory monocytes and persistence of reparative macrophages contribute to glomerulosclerosis and tubulointerstitial fibrosis. [38] Macrophage accumulation in the kidneys is associated with the occurrence of glomerulonephritis. [40] Autophagy and lysosome degradation pathways also contribute to macrophage polarization, chronic inflammation, and organ fibrosis. [41] Regulation of autophagy also plays a role in developing glomerulus-related diseases. [42]. This event ultimately triggers the emergence of proteinuria in DM patients.
Several studies have shown T cells' pathogenic role in the induction of proteinuria in DKD [43,44,45]. Activation of innate cells from the immune response and release of inflammatory cytokines like interferon-y (IFN-̯y) and tumor necrosis factor-α (TNF-α) are linked to an increase in T cells in the circulation and renal cortex [46]. Once circulating, T cells are recruited into renal tissue or amplified, differentiated, and activated in the renal. These T cells then mediate various pathogenic mechanisms, such as influencing insulin resistance, mediating podocyte damage, inducing fibrosis, and regulating proteinuria [47].
In DM, there is a disruption in the balance of inflammatory mediators (IL-6, TNF-α, TGF-β) and anti-inflammatory such as IL-10. Hyperglycemia conditions trigger resident kidney cells (mesangial and podocyte cells), interstitial tissue, and tubules to produce IL-6 [48]. JAK/STAT which mainly triggers cell proliferation are continuously activated resulting in hypertrophy of podocyte [49,50]. Furthermore, through activation of the rapamycin complex, podocyte hypertrophy continuously occurs and ultimately leads to the release of podocytes from the Basal Glomerular Membrane (MBG) (due to changes in the structure of podocytes) [51] dan apoptosis podosit [52]. IL-6 has also been shown to play a role in mesangial expansion, as evidenced by the discovery of IL-6 in the mesangium, interstitium, and renal tubules [53]. This IL-6 display also triggers the release of MCP-1 by mesangial cells, thus increasing monocyte recruits [54]. Podocyte hypertrophy, podocyte cell loss, and mesangial expansion Impaired glomerular function characterized by proteinuria and decreased glomerular filtration rate (GFR) [55].
TNF-α is produced by macrophages/monocytes that infiltrate the kidneys. Still, mesangial cells, podocytes, and tubular epithelium can also release TNF-α after being stimulated by hyperglycemia and Advanced Glycation End Products (AGEs). [56] TNF-α works by activating various secondary proteins that cause activation of gene transcription and production of reactive oxygen species or nitrogen radicals (NO). TNF-α can activate G-proteins, transcription factors (e.g., NF-kB, AP-1), protein kinases (e.g., CK-II, erk-1, erk-2, and MAP2), phospholipases, mitochondrial proteins, and proteases [57]. In DKD pathogenesis, TNF-α also increases the expression of adhesion and chemokine molecules, thus aggravating renal microinflammatory conditions. TNF-α also increases renal cell cytotoxicity/necrosis by inducing ROS and NO. In podocytes, TNF-α induces cytoskeleton reorganization and decreases cell viability. In addition, TNF-α also causes changes in intraglomerular blood flow and GFR and increases endothelial permeability [56].
Hyperglycemia leads to increased glucose transport-1 (GLUT-1) regulation, resulting in excess TGF-β expression by renal mesangial and tubular cells. Moreover, increased intraglomerular pressure, stretching of mesangial cells, activation of the renin-angiotensin system, ROS, and advanced glycation end products (AGEs) induce TGF-β production in renal mesangial and tubular cells [58]. TGF-β1 functions as a pro-fibrotic mediator in various kidney diseases [59]. TGF-β1 acts on renal mesangial cells and fibroblasts by inducing cell proliferation, cell migration, and transcription of profibrosis molecules (collagen, fibronectin, and plasminogen activator inhibitor-1 [PAI-1]). The mechanism of indirect fibrosis by TGF-β1 is still not widely studied, but Das et al. show that TGF-β1 initiates an apoptotic cascade in podocytes, leading to podocyte loss [60].
Interleukin 10 (IL-10) was initially known as an inhibitory factor of cytokine synthesis [61]. Activation of immunity and chronic inflammation contributes to the pathogenesis of T2DM as well as its progression to DKD [62]. Patients with T2DM have lower IL-10 levels [63]. This is exacerbated by the resistance of immune cells to the anti-inflammatory effects of IL-10 [64]. IL-10 has the potential to slow progression and improve the prognosis of DKD. In DKD, the accumulation of inflammatory cells (leukocytes, monocytes, and macrophages) in the kidneys synthesizes more pro-inflammatory and fibrogenic cytokines that damage kidney structure directly [65]. IL-10 can suppress this through its anti-inflammatory properties by reducing inflammatory cell infiltration, reducing fibrosis of renal interstitial, and preventing mesangial cell expansion 58 [66]. In vitro, it was found that IL-10 can reduce reactive oxygen species (ROS) level and, in embryonic mouse fibroblast cells, can reduce collagen synthesis [67]. Directly, IL-10 reduces glomerular macrophages' recruitment, activation, and proliferation in vivo. In addition, IL-10 also significantly reduces macrophage-mediated glomerular injury and improves proteinuria conditions [68].
In the kidneys, this adhesion molecule also plays a role in the attachment of leukocytes to the endothelium, promotes macrophage accumulation, increases TGF protein synthesis by tubular cells, and increases adhesion and activation of T cells [69]. Molecular adhesion can be induced by conditions commonly found in type 2 DM patients: hyperglycemia, Advanced Glycation Enda Product (AGE), oxidative stress, and hyperinsulinemia [69]. Research on experimental animal models found a causative relationship between VCAM ICAM and DKD. Okada et al. found that mice in ICAM-1 deficient diabetes models had higher rates of macrophage infiltration in the kidneys than controls. Nephropathy can be reduced by inhibiting ICAM-1 expression [70]. Chow et al. also found that ICAM-1 deficiency can reduce macrophage accumulation in the glomerulus to decrease glomerular hypertrophy and interstitial fibrosis [71]. Thus, therapeutic modalities targeting decreased VCAM-1 and ICAM-1 expression in kidney could be DKD therapy [69].
Podocytes and tubule cells will produce VEGF-A continuously, unlike other tissues that stop making it when blood vessel development is complete. In the kidneys, VEGF plays a vital role in angiogenesis, endothelial cell proliferation and survival, and regeneration of damaged tissues. Hyperglycemia conditions will trigger a chain reaction to cause accumulation of VEGF-A and ultimately cause microvascular complications of DM. The renin-angiotensin system, ROS, and AGEs64 influence this chain reaction. Angiotensin II will increase the production of VEGF-A and vascular inflammation [72]. VEGF-A expression increases as ROS production increases [73]. Meanwhile, AGEs can cause mesangial cells to apoptosis and increase VEGF expression, causing increased vascular permeability [74]. VEGF in the kidneys is mainly sourced from podocytes and tubular cells. Therefore, in the early phases of kidney damage, VEGF increases. However, severe kidney damage found a decrease in VEGF as a result of damage so extensive that cells in the kidneys were no longer able to express VEGF [75].
Matrix metalloproteinase (MMP) also plays an important role in developing DKD disease. MMP-9, or gelatinase B, is an endopeptidase that causes the degradation of extracellular matrix proteins such as collagen, fibronectin, and laminin. The main substrate of MMP-9 is type 4 collagen; in the kidney, type 4 collagen is the main structure that makes up the glomerular filtration barrier, especially the glomerular basement membrane [76]. Neutrophils, macrophages, and fibroblasts70 mostly express MMP-9 [77]. However, in diabetic kidney injury, there is also an increase in MMP-9 expression in proximal renal tubule epithelial cells [78]. An increase in MMP-9 in the kidneys leads to an increase in various activities of chemokine chemokines (CCXCL5, CXCL8), cytokines (TNF-α, IL1β, TGF-β), receptors, growth factors, and other cytokine adhesion molecules. On the other hand, MMP-9 can also inactivate CXCL1, CXCL4, CXCL5, CXCL7, CXCL12 and IL1 β [77]. MMP-9 activity causes endothelial-mesenchymal transition, tubulointerstitial, and inflammatory fibrosis [79].
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is an inhibitory receptor of the immunoglobulin CD28 subfamily. CTLA-4 is an essential molecule for helper T cells and podocytes. In hyperglycemia, there is an increase in CD28, which is a marker of podocyte damage. This increase in CD28 mediates T-cell infiltration, thus aggravating podocyte damage. By acting as a negative regulator of T cell activation, CTLA-4 guards against harm to podocytes. Research shows that CD28/B7/CTLA-4 polymorphisms increase susceptibility to DKD in Type 2 Diabetes patients in China [80]. Inhibition of CTLA-4 has become one of the focuses of DKD therapy development [81,82,83].
3. The Role of DC in Inducing an Anti-Inflammatory Response
DC is the primary regulator of the immune system. The role of dendritic cells in the immune system has been DC functions as a response controller to pathogens and is actively involved in inflammatory resolution [97]. Various studies have successfully revealed the anti-inflammatory properties of DC, which further adds to the complexity of DC's role in immunity [98,99,100,101]. Generally, DC plays a role in antigen presentation and T-cell activation. However, DC has also been known to play a central role in modulating inflammatory responses. DC relieves inflammation, prevents excessive tissue damage, and promotes immune homeostasis [101].
One of the critical anti-inflammatory mechanisms of DC is the secretion of anti-inflammatory cytokines. DC can produce cytokines such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which are immunosuppressive. These cytokines act on other immune cells, such as T cells and macrophages, to dampen the pro-inflammatory response and increase inflammatory resolution [98,102]. Dendritic cells are essential in promoting differentiation and activation of regulatory T cells (Tregs), a specialized subset of T cells with immunosuppressive solid functions. DC creates an environment conducive to Treg development by the presentation of antigens through tolerogenic and regulatory cytokine secretion. The Treg then directly suppresses the exaggerated immune response and contributes to the resolution of inflammation [100].
Under certain conditions, dendritic cells undergo phenotypic changes, becoming tolerogenic DCs characterized by reduced expression of co-stimulating molecules and increased production of anti-inflammatory cytokines. Tolerogenic DC actively provokes the formation of an anti-inflammatory microenvironment, influences T cell development into anti-inflammatory phenotypes, and contributes to the resolution of inflammation [103].
DC can actively suppress effector T-cell responses, thereby preventing inflammatory reactions. Through the induction of T-cell anergy or by promoting T-cell apoptosis, DC contributes to the downregulation of immune responses and inflammatory resolution [101]. Dendritic cells also affect macrophage polarization by directing macrophages into the M2 phenotype, which is anti-inflammatory. This modulation of macrophage function contributes to tissue repair and inflammatory resolution since M2 macrophages are involved in tissue remodeling and clearance of cellular debris [104]. DC engages in negative feedback regulation to control the intensity and duration of the immune response. By expressing inhibitory receptors (e.g., CTLA-4, LAG-3, PD-1) and interacting with regulatory molecules, DCs modulate their activation and prevent excessive inflammation, restoring immune homeostasis [103].
Understanding DC-mediated anti-inflammatory mechanisms opens up exciting possibilities for development as therapeutic interventions in inflammatory disorders and autoimmune diseases. Utilization of Treg cells immunoregulator properties induced by DC may be one of the therapeutic interventions. Strategies to increase Treg activity or trigger its expansion may also be a way to elicit an anti-inflammatory response. Innovations in precision medicine enable the development of personalized therapies based on individual immune profiles. Thus, interventions modulating DC function in patients can optimize therapeutic outcomes in inflammatory disorders.
4. Ex Vivo Production of Autologous Dendritic Cells
Two approaches can be used to make DC ex vivo: MoDC differentiation from peripheral blood mononuclear cells (PBMC) with a mixture of maturation cytokines and DC differentiation from CD34+ bone marrow precursors, which are hematopoietic stem cells with multipotent capabilities. PBMC stimulated with a mixture of maturation cytokines will produce a subset of MoDC. In contrast, differentiation of the CD34+ bone marrow precursor produces a mixture of a subset of MoDC, DC, which phenotypically resembles Langerhans cells (DC present in the epidermis), and myeloid cells at various levels of maturation [105,106]. Although much evidence suggests that cDC is better at cross-presenting exogenous antigens to MHC-1, cDC production is still quite difficult [107]. MoDC is the most widely available and versatile subtype of DC104 in humans [108]. MoDC was created ex vivo from PBMC, which is still the most frequently used method in developing DC-based therapies, both cancer and infection therapies. MoDC is more widely used not because of its superior clinical efficacy but because it is more widely available than CD34+ precursors in blood taken by apheresis [106]. The isolation of CD34+ from the bone marrow through bone marrow punctuation is an invasive procedure with a high risk. Thus, MoDC made from PBMC is the best choice today for manufacturing DC for therapeutic purposes.
PMBC is differentiated into DC immature by culturing cells for 5-7 days along with GM-CSF (Granulocyte-Macrophage Colony-Stimulating Factor) and Interleukin-4 (IL-4) [109]. GM-CSF is a cytokine that stimulates the differentiation of hematopoietic stem cells into various types of cells, including monocytes in PBMC [110]. GM-CSF is a cytokine that stimulates the differentiation of hematopoietic stem cells in multiple kinds of cells, including monocytes present in PBMC [111]. Thus, the mixture of the two cytokines synergistically promotes the differentiation of monocytes into MoDC and their maturation into APC.
DC can be modified to control inflammation by culturing MoDC and specific agents such as Vitamin D3 and Dexamethasone. DC with anti-inflammatory abilities is also called Tolerogenic DC (tolDC). Maturation stimuli such as LPS are given so that semi-mature tolDC is obtained [112]. MoDCs cultured with probiotic bacteria such as L. delbruekii and L. rhamnosus also exhibited tolerogenic phenotypes [113]. Another method that can be done is to induce a tolerogenic phenotype in DC through transfection [114]. Interestingly, DC made from bone marrow cultures and GM-CSF and IL-4 without being given an antigen stimulus (DC immature) also showed the ability to control inflammation [115]. Thus, DC transfer without antigen stimulus can be considered for administration as therapy in diseases based on inflammatory disorders.
5. Current State of Cell-Based Therapy for DKD
Autologous DC transfer is commonly used as a therapy for cancer, chronic infections, autoimmune diseases, and infection-preventing vaccines [116]. The use of autologous DC transfer in metabolic and degenerative disease therapy has yet to be carried out. However, there are other cell-based therapies to improve conditions in metabolic diseases that have been studied. This section will discuss some significant findings related to cell therapy performed in patients with DKD.
A study by Dubsky et al. found that systemic and local administration of autologous mononuclear cells in the form of hematologic stem cells in DM subjects with Chronic Kidney Disease (CKD) can protect against amputation due to critical limb ischemia, including patients with End Stage Renal Disease [117]. Critical limb ischemia is caused by severe occlusion of the arteries of the lower extremities (Peripheral Artery Disease) and is one of the vascular complications of DM. Hyperglycemia in DM triggers the formation of Advanced Glycation End Products (AGEs), which then increase the uptake of oxidized low-density lipoproteins by macrophages and then develop into foam cells. Foam cells accumulate in the subendothelial area of the artery wall, forming atherosclerosis lesions [118]. Atherosclerosis in this artery then blocks blood flow to the distal region, resulting in Critical Limb Ischemia.
Peripheral Artery Disease (PAD) generally occurs in the large arteries, so it is classified as macroangiopathy. However, the fact is that PAD is also accompanied by local and systemic microangiopathy [119]. PAD is closely related to DKD. This is due to endothelial dysfunction in various blood vessels, causing thickening of the basement membrane of capillary arteries, endothelial hyperplasia, decreased oxygen pressure, and hypoxia, causing disorders of multiple organs, including the kidneys. In addition, reduced kidney function is also one factor supporting the severity of PAD—AGEs are factors that trigger the formation of physiological foam cells that are eliminated by the kidneys. Thus, along with the decline in kidney function, the number of AGEs will increase, ultimately increasing the possibility of Critical Limb Ischemia.
Dubsky et al. conducted experiments by giving mononuclear cell transfer CD34+ (Bone marrow stem cell) to patients with end-stage renal disease, which proved to protect against amputation. The therapeutic effects of CD34+ cell transfer are due to the immunomodulating impacts through direct contact with innate and adaptive immune cells and paracrine effects caused by the production of cytokines, chemokines, and growth factors [120]. Although the study did not see any impact on the kidneys, given that PAD in DM patients is closely related to kidney function, the protective effect of critical limb ischemia may also be influenced by the renoprotective effect through modulating the immune system systemically after CD34+ mononuclear cell transfer.
Some researchers have also conducted autologous cell transfer experiments as DKD therapy. A systematic evaluation and meta-analysis revealed that kidney function can be enhanced in experimental animals with DKD with the use of stem cell treatment. Reductions in proinflammatory markers (TNF-α, IFN-γ, IL-6, IL-8, MCP-1), reductions in fibrosis indicators (TGF-β, Collagen I and IV, fibronectin), and increases in anti-inflammatory markers (IL-10) were linked to improvements in kidney function [121]. However, human testing has not shown satisfactory results. A randomized placebo-controlled clinical trial on Allogeneic Mesenchymal Precursor Cells (MPC) administration in DKD patients found that there was no significant difference in the ratio of urinary creatinine-albumin, serum creatinine, and HbA1c in the test group compared to placebo [122].
Stem cells are hypothesized to improve pathology in metabolic diseases because stem cells' multipotential ability is expected to regenerate damaged cells. However, in the context of DKD therapy, the mechanism of action of cell-based therapy is mainly based on the ability to modulate immunity and anti-inflammatory effects of stem cells because there is not enough evidence to suggest the engraftment of systemically administered stem cells [122]. The use of stem cells for DKD has several disadvantages, namely the presence of metabolic memory, which causes stem cells obtained from DKD patients to experience decreased multipotency and immunomodulatory abilities, are more susceptible to apoptosis, and experience increased senescence. In addition, there is a risk of teratoma formation from stem cells [123].
Overall, the current research shows that cell-based therapy, such as DKD, can be applied as one of the approaches to metabolic disease therapy. Current research focuses on developing stem cell-based therapies. Still, current evidence suggests that the mechanism of action of stem cells as DKD therapy emphasizes paracrine immunomodulating and anti-inflammatory abilities. Thus, other cell-based products with similar capabilities also have the potential to be developed.
6. Autologous DC Mechanism of Action in DKD
DC immunotherapy is created by modifying monocytes into DC by stimulating differentiated cytokines. The product is a cell with a late differentiation stage, so teratoma formation is not risky. However, as previously explained, DC has immunomodulatory abilities through direct interaction with other immune cells and through the paracrine effect of cytokines and growth factors released. In addition, DC is a component of the immune system whose primary physiological function is to regulate the immune response. Thus, the immunomodulation function of DC is likely better than that of stem cells. Therefore, the effectiveness and mechanism of action of DC transfer as DKD therapy need further investigation.
Based on existing research, it was found that DC can induce an anti-inflammatory response [124]. Through the production of anti-inflammatory cytokines, DC can systemically inhibit low-grade chronic inflammation. In addition, DCs that express fewer co-stimulating molecules on the surface also directly inhibit the activation and anergy of kidney-infiltrating T cells. DCs with tolerogenic phenotypes also express inhibitory molecules such as CTLA-4 on their surfaces to directly inhibit T-cell function [125]. DC is also known to support the proliferation of macrophages into the anti-inflammatory M2 type. In addition, DC can stimulate the proliferation of peripheral Treg cells that firmly control the immune response [56].
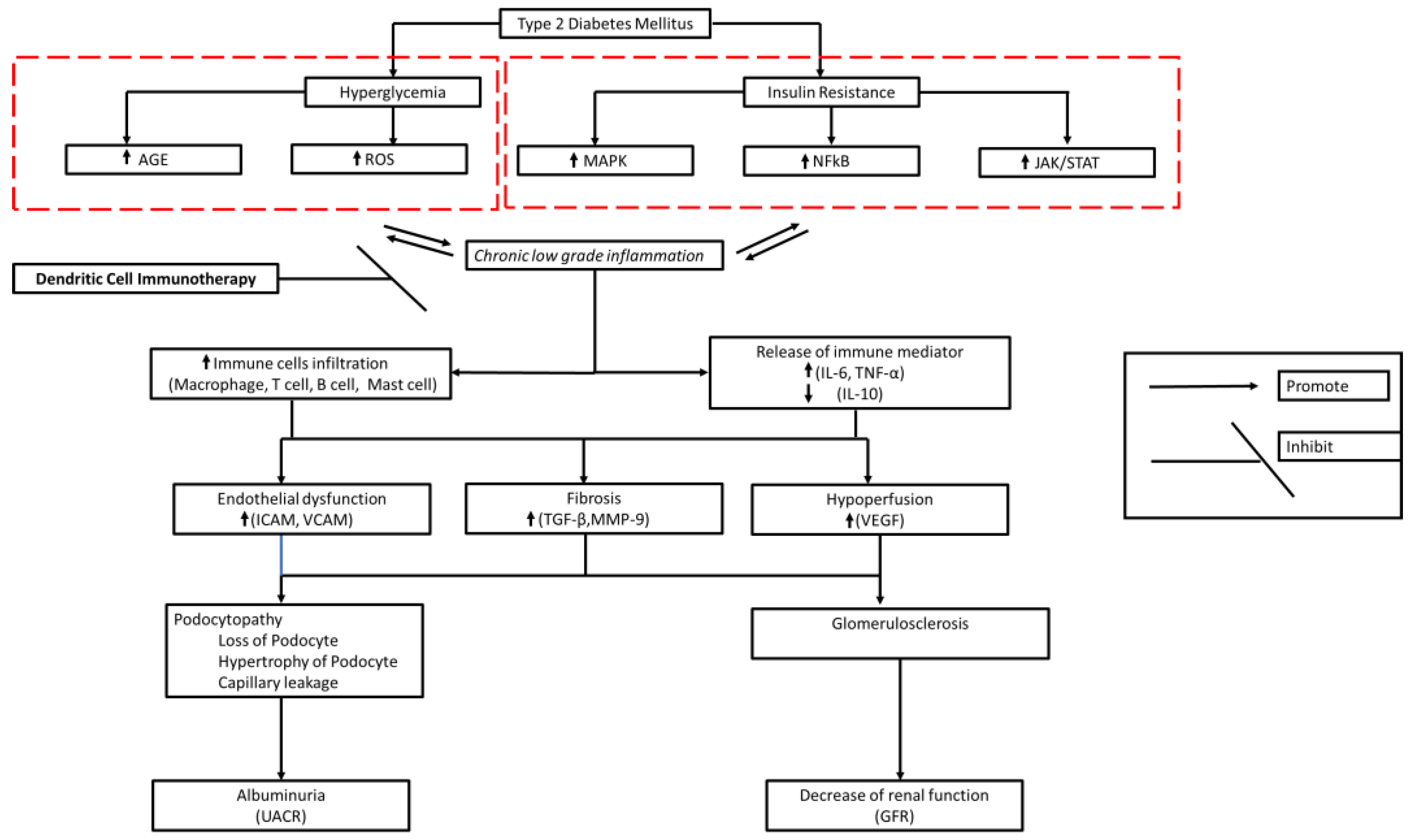
Chronic low-grade chronic inflammation in DM contributes to fibrosis, endothelial dysfunction, and hypoperfusion underlying DKD pathology [85,126]. Thus, DC therapy should be given systemically so that systemic immune immunomodulation occurs. This is expected to stop further immune cell-mediated damage to the kidneys and maintain kidney function (Figure 1). However, DKD is a progressive chronic disease. Products that can induce long-term anti-inflammatories are needed to provide clinically significant effects. Therefore, it is necessary to know the dose (number of cells), frequency, and appropriate period of administration so that a satisfactory clinical effect can be achieved.
8. Conclusions
DKD caused by kidney damage as a result of chronic low-grade inflammation in type 2 diabetes patients. Inflammation causes damage to kidney structure and function, characterized by proteinuria and progressive decline in kidney function. DC can induce a robust anti-inflammatory response through the secretion of anti-inflammatory cytokines, inhibit activation, give rise to T cell anergy, and support M2 proliferation and Treg cell stimulation. Thus, DC transfer in DKD patients has the potential to be a new therapeutic approach.
Author Contributions
J. and E.C.S. contributed equally to the conception, formal analysis, writing, and editing of the manuscript, thus sharing the first authorship of this article. L.C. provided valuable feedback and discussion in the writing of the manuscript. I.E.L. and TAP supervised and provided valuable feedback and discussion in the writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The APC for this manuscript was funded by Indonesia Army Cellcure Center.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data is available upon request to authors.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Selby, N.M.; Taal, M.W. An updated overview of diabetic nephropathy: Diagnosis, prognosis, treatment goals and latest guidelines. Diabetes, Obes. Metab. 2020, 22, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, N.; Wu, Y.; Wang, M.; Yang, S.; Zheng, Y.; Deng, X.; Xiang, D.; Zhu, Y.; Xu, P.; et al. Global, Regional, and National Burden of Diabetes-Related Chronic Kidney Disease From 1990 to 2019. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V. Chronic kidney disease. The Lancet. 2021 Aug;398(10302):786–802. [CrossRef]
- Wada, J.; Makino, H. Inflammation and the pathogenesis of diabetic nephropathy. Clin. Sci. 2012, 124, 139–152. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Jaisser, F. Pathophysiologic mechanisms in diabetic kidney disease: A focus on current and future therapeutic targets. Diabetes, Obes. Metab. 2020, 22, 16–31. [Google Scholar] [CrossRef]
- Kikkawa R, Koya D, Haneda M. Progression of diabetic nephropathy. American Journal of Kidney Diseases. 2003 Mar;41(3):S19–21. [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- D'Marco, L.; Guerra-Torres, X.; Viejo, I.; Lopez-Romero, L.; Yugueros, A.; Bermúdez, V.; de Valencia, V.H.G.U.; de Asturias, M.H.U.P.; Bolívar, F.d.C.d.l.S.U.S. Non-albuminuric Diabetic Kidney Disease Phenotype: Beyond Albuminuria. Eur. Endocrinol. 2022, 18, 102–105. [Google Scholar] [CrossRef]
- Ekinci, E.I.; Jerums, G.; Skene, A.; Crammer, P.; Power, D.; Cheong, K.Y.; Panagiotopoulos, S.; McNeil, K.; Baker, S.T.; Fioretto, P.; et al. Renal Structure in Normoalbuminuric and Albuminuric Patients With Type 2 Diabetes and Impaired Renal Function. Diabetes Care 2013, 36, 3620–3626. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, V.; Zhao, B.; Azar, K.M.; Wang, E.J.; Choi, S.; Wong, E.C.; Fortmann, S.P.; Palaniappan, L.P. Racial/Ethnic Differences in the Prevalence of Proteinuric and Nonproteinuric Diabetic Kidney Disease. Diabetes Care 2013, 36, 1215–1221. [Google Scholar] [CrossRef]
- Azeem, W.; Bakke, R.M.; Appel, S.; Øyan, A.M.; Kalland, K.-H. Dual Pro- and Anti-Inflammatory Features of Monocyte-Derived Dendritic Cells. Front. Immunol. 2020, 11, 438. [Google Scholar] [CrossRef]
- Luo S, Zhou Z. The clinical heterogeneity of diabetes challenges the accuracy of typing diagnosis | 糖尿病的临床异质性对精准分型诊断的挑战. Journal of Chinese Physician. 2022;24(2):179–83.
- Petrelli, A.; Giovenzana, A.; Insalaco, V.; Phillips, B.E.; Pietropaolo, M.; Giannoukakis, N. Autoimmune Inflammation and Insulin Resistance: Hallmarks So Far and Yet So Close to Explain Diabetes Endotypes. Curr. Diabetes Rep. 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Asfandiyarova, NS. TYPE 2 DIABETES MELLITUS – AN AUTOIMMUNE DISEASE? Russian Journal of Immunology. 2020;23(1):9–18.
- Girard, D.; Vandiedonck, C. How dysregulation of the immune system promotes diabetes mellitus and cardiovascular risk complications. Front. Cardiovasc. Med. 2022, 9, 991716. [Google Scholar] [CrossRef] [PubMed]
- Lempesis, I.G.; E Georgakopoulou, V. Physiopathological mechanisms related to inflammation in obesity and type 2 diabetes mellitus. World J. Exp. Med. 2023, 13, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Fontanella, A.M.; Merscher, S.; Fornoni, A. Lipid deposition and metaflammation in diabetic kidney disease. Curr. Opin. Pharmacol. 2020, 55, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Wu, D.; Cai, Z.; Xu, Y.; Ding, X.; Wu, W.; Lan, S.; Chen, L.; Guo, Z.; Balmer, L.; et al. Supra-additive effect of chronic inflammation and atherogenic dyslipidemia on developing type 2 diabetes among young adults: a prospective cohort study. Cardiovasc. Diabetol. 2023, 22, 1–12. [Google Scholar] [CrossRef]
- Thimmappa, P.Y.; Vasishta, S.; Ganesh, K.; Nair, A.S.; Joshi, M.B. Neutrophil (dys)function due to altered immuno-metabolic axis in type 2 diabetes: implications in combating infections. Hum. Cell 2023, 36, 1265–1282. [Google Scholar] [CrossRef] [PubMed]
- Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55(1):31–55.
- Lytrivi M, Igoillo-Esteve M, Cnop M. Inflammatory stress in islet β-cells: therapeutic implications for type 2 diabetes? Curr Opin Pharmacol. 2018;43:40–5.
- Shahid, R.; Chu, L.M.; Arnason, T.; Pahwa, P. Association Between Insulin Resistance and the Inflammatory Marker C-reactive Protein in a Representative Healthy Adult Canadian Population: Results From the Canadian Health Measures Survey. Can. J. Diabetes 2023, 47, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55(1):31–55.
- Bandawane D, Pujari R, Upaganlawar A. High-fat diets and insulin resistance. Everything You Need to Know About High-Fat Diets. 2023. 213–230 p.
- Li, T.; Wang, P.; Wang, X.; Liu, Z.; Zhang, Z.; Zhang, Y.; Wang, Z.; Feng, Y.; Wang, Q.; Guo, X.; et al. Inflammation and Insulin Resistance in Diabetic Chronic Coronary Syndrome Patients. Nutrients 2023, 15, 2808. [Google Scholar] [CrossRef] [PubMed]
- Hoorzad P, Mousavinasab F, Tofigh P, Kalahroud EM, Aghaei-Zarch SM, Salehi A, et al. Understanding the lncRNA/miRNA-NFκB regulatory network in diabetes mellitus: From function to clinical translation. Diabetes Res Clin Pract. 2023;202.
- Meyerovich K, Ortis F, Cardozo AK. The non-canonical NF-κB pathway and its contribution to β-cell failure in diabetes. J Mol Endocrinol. 2018;61(2):F1–6.
- Chen X, Liu Z, Liu W, Wang S, Jiang R, Hu K, et al. NF-κB-Inducing Kinase Provokes Insulin Resistance in Skeletal Muscle of Obese Mice. Inflammation. 2023;46(4):1445–57.
- Singh L, Bhatti R. Cellular and molecular mechanisms involved in metabolic disorders. Drug Delivery Systems for Metabolic Disorders. 2022. 21–29 p.
- Lourido, F.; Quenti, D.; Salgado-Canales, D.; Tobar, N. Domeless receptor loss in fat body tissue reverts insulin resistance induced by a high-sugar diet in Drosophila melanogaster. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Singh L, Bhatti R. Cellular and molecular mechanisms involved in metabolic disorders. Drug Delivery Systems for Metabolic Disorders. 2022. 21–29 p.
- Li, T.; Yang, X.; Zhu, J.; Liu, Y.; Jin, X.; Chen, G.; Ye, L. Current application status and structure–activity relationship of selective and non-selective JAK inhibitors in diseases. Int. Immunopharmacol. 2023, 122, 110660. [Google Scholar] [CrossRef] [PubMed]
- Lourido, F.; Quenti, D.; Salgado-Canales, D.; Tobar, N. Domeless receptor loss in fat body tissue reverts insulin resistance induced by a high-sugar diet in Drosophila melanogaster. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; He, J.; Li, Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 2022, 13, 958790. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liu, Y.; Hu, Q.; Zhou, J.; Lin, H. The landscape of immune cell infiltration in the glomerulus of diabetic nephropathy: evidence based on bioinformatics. BMC Nephrol. 2022, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lv, Y.; Zhang, T.; Huang, T.; Lang, Y.; Sheng, Q.; Liu, Y.; Kong, Z.; Gao, Y.; Lu, S.; et al. T cells and their products in diabetic kidney disease. Front. Immunol. 2023, 14, 1084448. [Google Scholar] [CrossRef] [PubMed]
- Rico Fontalvo J, Aroca-Martínez G, Daza-Arnedo R, Raad-Sarabia M, Luis Torres J, Pajaro-Galvis N, et al. Non-proteinuric diabetic kidney disease: State of art | Enfermedad renal diabética no proteinúrica: Estado del arte. Revista de Nefrologia, Dialisis y Trasplante. 2022;42(4):330–9.
- Lis-López L, Bauset C, Seco-Cervera M, Cosín-Roger J. Is the macrophage phenotype determinant for fibrosis development? Biomedicines. 2021;9(12).
- Bell RMB, Conway BR. Macrophages in the kidney in health, injury and repair. Vol. 367, International Review of Cell and Molecular Biology. 2022. 101–147 p.
- Chen, A.; Lee, K.; He, J.C. Treating crescentic glomerulonephritis by targeting macrophages. Kidney Int. 2022, 102, 1212–1214. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.-H.; Li, D.-Y.; Liang, S.; Yang, C.; Tang, J.-X.; Liu, H.-F. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front. Immunol. 2022, 13, 946832. [Google Scholar] [CrossRef] [PubMed]
- Cui J, Bai X, Chen X. Autophagy and glomerular diseases. Vol. 1207, Advances in Experimental Medicine and Biology. 2020. 481–486 p.
- Moon, J.-Y.; Jeong, K.-H.; Lee, T.-W.; Ihm, C.-G.; Lim, S.J.; Lee, S.-H. Aberrant Recruitment and Activation of T Cells in Diabetic Nephropathy. Am. J. Nephrol. 2012, 35, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Bending, J.J.; Lobo-Yeo, A.; Vergani, D.; Viberti, G.C. Proteinuria and activated T-lymphocytes in diabetic nephropathy. Diabetes 1988, 37, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Moriya, R.; Manivel, J.C.; Mauer, M. Juxtaglomerular apparatus T-cell infiltration affects glomerular structure in Type 1 diabetic patients. Diabetologia 2004, 47, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Andrikopoulos, S.; MacIsaac, R.J.; Mackay, L.K.; Nikolic-Paterson, D.J.; Torkamani, N.; Zafari, N.; Marin, E.C.S.; I Ekinci, E. Role of the adaptive immune system in diabetic kidney disease. J. Diabetes Investig. 2021, 13, 213–226. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, Y.; Zhang, T.; Huang, T.; Lang, Y.; Sheng, Q.; Liu, Y.; Kong, Z.; Gao, Y.; Lu, S.; et al. T cells and their products in diabetic kidney disease. Front. Immunol. 2023, 14, 1084448. [Google Scholar] [CrossRef]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O'Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Jo HA, Kim JY, Yang SH, Han SS, Joo KW, Kim YS, et al. The role of local IL6/JAK2/STAT3 signaling in high glucose–induced podocyte hypertrophy. Kidney Res Clin Pract. 2016 Dec;35(4):212–8.
- Yin L, Yu L, He JC, Chen A. Controversies in Podocyte Loss: Death or Detachment? Front Cell Dev Biol. 2021 Nov 22;9.
- Cha, D.R. Interleukin-6 signaling in podocyte hypertrophy. Kidney Res. Clin. Pr. 2016, 35, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lv, Y.; Zhang, T.; Huang, T.; Lang, Y.; Sheng, Q.; Liu, Y.; Kong, Z.; Gao, Y.; Lu, S.; et al. T cells and their products in diabetic kidney disease. Front. Immunol. 2023, 14, 1084448. [Google Scholar] [CrossRef] [PubMed]
- Coletta I, Soldo L, Polentarutti N, Mancini F, Guglielmotti A, Pinza M, et al. Selective Induction of MCP-1 in Human Mesangial Cells by the IL-6/sIL-6R Complex. Nephron Exp Nephrol. 2000 Jan 14;8(1):37–43.
- Thomas, H.Y.; Versypt, A.N.F. Pathophysiology of mesangial expansion in diabetic nephropathy: mesangial structure, glomerular biomechanics, and biochemical signaling and regulation. J. Biol. Eng. 2022, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Barutta, F.; Bruno, G.; Grimaldi, S.; Gruden, G. Inflammation in diabetic nephropathy: moving toward clinical biomarkers and targets for treatment. Endocrine 2014, 48, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Idriss HT, Naismith JH. TNF Alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc Res Tech. 2000 Aug 1;50(3):184–95.
- Braga Gomes K, Fontana Rodrigues K, Fernandes AP. The Role of Transforming Growth Factor-Beta in Diabetic Nephropathy. International Journal of Medical Genetics. 2014 Jan 28;2014(2):1–6.
- Gu, Y.-Y.; Liu, X.-S.; Huang, X.-R.; Yu, X.-Q.; Lan, H.-Y. Diverse Role of TGF-β in Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.-K.; Park, K.-S. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10andRelatedCytokines andReceptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef]
- Duran-Salgado, M.B. Diabetic nephropathy and inflammation. World J. Diabetes 2014, 5, 393–8. [Google Scholar] [CrossRef]
- Naz, S.; Shafique, N.; Sharif, S.; Manzoor, F.; Saifi, S.Z.; Firasat, S.; Kaul, H. Association of Interleukin 10 (IL-10) Gene with Type 2 Diabetes Mellitus by Single Nucleotide Polymorphism of Its Promotor Region G/A 1082. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.C.; Shakibakho, S.; Durrer, C.; Simtchouk, S.; Jawanda, K.K.; Cheung, S.T.; Mui, A.L.; Little, J.P. Hyporesponsiveness to the anti-inflammatory action of interleukin-10 in type 2 diabetes. Sci. Rep. 2016, 6, 21244. [Google Scholar] [CrossRef] [PubMed]
- Samsu, N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Res. Int. 2021, 2021, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, X.; Liu, L.C.; Kim, A.Y.; Curley, S.P.; Chen, X.; Dworkin, L.D.; Cooper, C.J.; Gupta, R. Interleukin-10 attenuates renal injury after myocardial infarction in diabetes. J. Investig. Med. 2022, 70, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhao, Y.; Zhang, Y.; Jin, H.; Shou, S. The role of IL-10 in kidney disease. Int. Immunopharmacol. 2022, 108, 108917. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhao, Y.; Zhang, Y.; Jin, H.; Shou, S. The role of IL-10 in kidney disease. Int. Immunopharmacol. 2022, 108, 108917. [Google Scholar] [CrossRef] [PubMed]
- Lenz, O.; Fornoni, A.; Ijaz, A.; Tejada, T. Role of Inflammation in Diabetic Nephropathy. Curr. Diabetes Rev. 2008, 4, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Shikata, K.; Matsuda, M.; Ogawa, D.; Usui, H.; Kido, Y.; Nagase, R.; Wada, J.; Shikata, Y.; Makino, H. Intercellular Adhesion Molecule-1–Deficient Mice Are Resistant Against Renal Injury After Induction of Diabetes. Diabetes 2003, 52, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Tesch, G.H. Intercellular Adhesion Molecule-1 Deficiency Is Protective against Nephropathy in Type 2 Diabetic db/db Mice. J. Am. Soc. Nephrol. 2005, 16, 1711–1722. [Google Scholar] [CrossRef]
- Zhao, Q.; Ishibashi, M.; Hiasa, K.-I.; Tan, C.; Takeshita, A.; Egashira, K.; S, F.; K, E.; T, K.; T, O.; et al. Essential Role of Vascular Endothelial Growth Factor in Angiotensin II–Induced Vascular Inflammation and Remodeling. Hypertension 2004, 44, 264–270. [Google Scholar] [CrossRef]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-Induced Reactive Oxygen Species: Mechanism of Their Generation and Role in Renal Injury. J. Diabetes Res. 2017, 2017, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.; Rheault, M.N. Genetic Basis of Type IV Collagen Disorders of the Kidney. Clin. J. Am. Soc. Nephrol. 2021, 16, 1101–1109. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix Metalloproteinase-9: Many Shades of Function in Cardiovascular Disease. Physiology 2013, 28, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, H.; Liu, F.; Ma, Q. Up-regulation of matrix metalloproteinases-9 in the kidneys of diabetic rats and the association with neutrophil gelatinase-associated lipocalin. BMC Nephrol. 2021, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Arcos-Sacramento VG, Sampieri CL, Sandoval-Lozano VH, Orozco-Ortega RA, Acuña-Hernández MA, Morales-Romero J, et al. Urinary MMP-9/UCr association with albumin concentration and albumin-creatinine-ratio in Mexican patients with type 2 diabetes mellitus. PeerJ. 2020 Dec 16;8:e10474.
- Li, Y.; Jin, L.; Yan, J.; Zhang, H.; Hu, C. CD28 Genetic Variants Increase Susceptibility to Diabetic Kidney Disease in Chinese Patients with Type 2 Diabetes: A Cross-Sectional Case Control Study. Mediat. Inflamm. 2021, 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, P.; Li, X.; Dong, Y.; Wu, S.; Xu, M.; Chen, X.; Wang, S.; Zheng, C.; Zou, C. Combination CTLA-4 immunoglobulin treatment and ultrasound microbubble-mediated exposure improve renal function in a rat model of diabetic nephropathy. Aging 2021, 13, 8524–8540. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Söderberg, M.; Sabirsh, A.; Valastro, B.; Mölne, J.; Santamaria, B.; Valverde, A.M.; Guionaud, S.; Heasman, S.; Bigley, A.; et al. Inhibition of T-cell activation by the CTLA4-Fc Abatacept is sufficient to ameliorate proteinuric kidney disease. Am. J. Physiol. Physiol. 2017, 312, F748–F759. [Google Scholar] [CrossRef]
- Zou CP, Li XY, Wei KN, Yang Y, Zhao YZ, Wang P, et al. Therapeutic effects of CTLA-4-Ig on diabetic nephropathy in type 2 diabetes mellitus rats ascribed to protection of CTLA-4-Ig on podocytes. Int J Clin Exp Med. 2018;11(10):10516–25.
- Chen, J.; Liu, Q.; He, J.; Li, Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 2022, 13, 958790. [Google Scholar] [CrossRef]
- Hickey, F.B.; Martin, F. Role of the Immune System in Diabetic Kidney Disease. Curr. Diabetes Rep. 2018, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Balan S, Saxena M, Bhardwaj N. Dendritic cell subsets and locations. 1st ed. Vol. 348, International Review of Cell and Molecular Biology. Elsevier Inc.; 2019. 1–68 p.
- Saadeh, D.; Kurban, M.; Abbas, O. Update on the role of plasmacytoid dendritic cells in inflammatory/autoimmune skin diseases. Exp. Dermatol. 2016, 25, 415–421. [Google Scholar] [CrossRef]
- Takagi, H.; Arimura, K.; Uto, T.; Fukaya, T.; Nakamura, T.; Choijookhuu, N.; Hishikawa, Y.; Sato, K. Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation. Sci. Rep. 2016, 6, 24477–24477. [Google Scholar] [CrossRef] [PubMed]
- Kisielow, P. How does the immune system learn to distinguish between good and evil? The first definitive studies of T cell central tolerance and positive selection. Immunogenetics 2019, 71, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Macri C, Pang ES, Patton T, O’Keeffe M. Dendritic cell subsets. Semin Cell Dev Biol. 2018;84:11–21.
- Kim, M.K.; Kim, J. Properties of immature and mature dendritic cells: phenotype, morphology, phagocytosis, and migration. RSC Adv. 2019, 9, 11230–11238. [Google Scholar] [CrossRef] [PubMed]
- Ugur, M.; Mueller, S.N. T cell and dendritic cell interactions in lymphoid organs: More than just being in the right place at the right time. Immunol. Rev. 2019, 289, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Cresswell, P. Antigen Processing and Presentation Mechanisms in Myeloid Cells. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel). 2016 Mar 15;8(3):36.
- Park C, Choi YS. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology. 2005 Jan 16;114(1):2–10.
- Passeri, L.; Marta, F.; Bassi, V.; Gregori, S. Tolerogenic Dendritic Cell-Based Approaches in Autoimmunity. Int. J. Mol. Sci. 2021, 22, 8415. [Google Scholar] [CrossRef]
- Comi, M.; Avancini, D.; de Sio, F.S.; Villa, M.; Uyeda, M.J.; Floris, M.; Tomasoni, D.; Bulfone, A.; Roncarolo, M.G.; Gregori, S. Coexpression of CD163 and CD141 identifies human circulating IL-10-producing dendritic cells (DC-10). Cell. Mol. Immunol. 2019, 17, 95–107. [Google Scholar] [CrossRef]
- Boks MA, Kager-Groenland JR, Haasjes MSP, Zwaginga JJ, van Ham SM, ten Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction — A comparative study of human clinical-applicable DC. Clinical Immunology. 2012 Mar;142(3):332–42.
- Bakdash, G.; Vogelpoel, L.T.; van Capel, T.M.; Kapsenberg, M.; de Jong, E. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol. 2015, 8, 265–278. [Google Scholar] [CrossRef]
- Morante-Palacios, O.; Fondelli, F.; Ballestar, E.; Martínez-Cáceres, E.M. Tolerogenic Dendritic Cells in Autoimmunity and Inflammatory Diseases. Trends Immunol. 2020, 42, 59–75. [Google Scholar] [CrossRef]
- Ochando, J.; Ordikhani, F.; Jordan, S.; Boros, P.; Thomson, A.W. Tolerogenic dendritic cells in organ transplantation. Transpl. Int. 2019, 33, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Pulendran, B. Dendritic cell control of tolerogenic responses. Immunol. Rev. 2011, 241, 206–227. [Google Scholar] [CrossRef]
- Choo, E.H.; Lee, J.-H.; Park, E.-H.; Park, H.E.; Jung, N.-C.; Kim, T.-H.; Koh, Y.-S.; Kim, E.; Seung, K.-B.; Park, C.; et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circ. 2017, 135, 1444–1457. [Google Scholar] [CrossRef]
- Chung DJ, Sharma S, Rangesa M, DeWolf S, Elhanati Y, Perica K, et al. Langerhans dendritic cell vaccine bearing mRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv. 2022 Mar 8;6(5):1547–58.
- Constantino, J.; Gomes, C.; Falcão, A.; Neves, B.M.; Cruz, M.T. Dendritic cell-based immunotherapy: a basic review and recent advances. Immunol. Res. 2017, 65, 798–810. [Google Scholar] [CrossRef]
- Makino K, Long MD, Kajihara R, Matsueda S, Oba T, Kanehira K, et al. Generation of cDC-like cells from human induced pluripotent stem cells via Notch signaling. J Immunother Cancer. 2022 Jan 31;10(1):e003827.
- Sung SSJ. Monocyte-Derived Dendritic Cells as Antigen-Presenting Cells in T-Cell Proliferation and Cytokine Production. In 2019. p. 131–41. Available from: http://link.springer.com/10. 1007.
- Sung SSJ. Monocyte-Derived Dendritic Cells as Antigen-Presenting Cells in T-Cell Proliferation and Cytokine Production. In 2019. p. 131–41.
- Bhattacharya, P.; Thiruppathi, M.; Elshabrawy, H.A.; Alharshawi, K.; Kumar, P.; Prabhakar, B.S. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 2015, 75, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Cechim, G.; Chies, J.A. In vitro generation of human monocyte-derived dendritic cells methodological aspects in a comprehensive review. An. da Acad. Bras. de Cienc. 2019, 91. [Google Scholar] [CrossRef]
- Wu, H.J.; Lo, Y.; Luk, D.; Lau, C.S.; Lu, L.; Mok, M.Y. Alternatively activated dendritic cells derived from systemic lupus erythematosus patients have tolerogenic phenotype and function. Clin. Immunol. 2015, 156, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, S.; Mahmoudi, M.; Rezaieyazdi, Z.; Sahebari, M.; Tabasi, N.; Sahebkar, A.; Rastin, M. Generation of tolerogenic dendritic cells using Lactobacillus rhamnosus and Lactobacillus delbrueckii as tolerogenic probiotics. J. Cell. Biochem. 2018, 119, 7865–7872. [Google Scholar] [CrossRef]
- Li CH, Zhang J, Baylink DJ, Wang X, Goparaju NB, Xu Y, et al. Dendritic cells, engineered to overexpress 25-hydroxyvitamin D 1α-hydroxylase and pulsed with a myelin antigen, provide myelin-specific suppression of ongoing experimental allergic encephalomyelitis. FASEB Journal. 2017;31(7):2996–3006.
- Lo, J.; Xia, C.-Q.; Peng, R.; Clare-Salzler, M.J. Immature Dendritic Cell Therapy Confers Durable Immune Modulation in an Antigen-Dependent and Antigen-Independent Manner in Nonobese Diabetic Mice. J. Immunol. Res. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Jonny, J.; Putranto, T.A.; Sitepu, E.C.; Irfon, R. Dendritic cell vaccine as a potential strategy to end the COVID-19 pandemic. Why should it be Ex Vivo? Expert Rev. Vaccines 2022, 21, 1111–1120. [Google Scholar] [CrossRef]
- Dubský, M.; Jirkovská, A.; Bem, R.; Němcová, A.; Fejfarová, V.; Hazdrová, J.; Sutoris, K.; Chlupáč, J.; Skibová, J.; Jude, E.B. Impact of severe diabetic kidney disease on the clinical outcome of autologous cell therapy in people with diabetes and critical limb ischaemia. Diabet. Med. 2019, 36, 1133–1140. [Google Scholar] [CrossRef]
- Ying, A.F.; Tang, T.Y.; Jin, A.; Chong, T.T.; Hausenloy, D.J.; Koh, W.-P. Diabetes and other vascular risk factors in association with the risk of lower extremity amputation in chronic limb-threatening ischemia: a prospective cohort study. Cardiovasc. Diabetol. 2022, 21, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: molecular mechanisms and therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 1–29. [Google Scholar] [CrossRef]
- Song, N.; Scholtemeijer, M.; Shah, K. Mesenchymal Stem Cell Immunomodulation: Mechanisms and Therapeutic Potential. Trends Pharmacol. Sci. 2020, 41, 653–664. [Google Scholar] [CrossRef]
- Hickson, L.J.; Abedalqader, T.; Ben-Bernard, G.; Mondy, J.M.; Bian, X.; Conley, S.M.; Zhu, X.; Herrmann, S.M.; Kukla, A.; Lorenz, E.C.; et al. A Systematic Review and Meta-Analysis of Cell-Based Interventions in Experimental Diabetic Kidney Disease. STEM CELLS Transl. Med. 2021, 10, 1304–1319. [Google Scholar] [CrossRef]
- Packham, D.K.; Fraser, I.R.; Kerr, P.G.; Segal, K.R. Allogeneic Mesenchymal Precursor Cells (MPC) in Diabetic Nephropathy: A Randomized, Placebo-controlled, Dose Escalation Study. EBioMedicine 2016, 12, 263–269. [Google Scholar] [CrossRef]
- Sávio-Silva, C.; Beyerstedt, S.; Soinski-Sousa, P.E.; Casaro, E.B.; Balby-Rocha, M.T.A.; Simplício-Filho, A.; Alves-Silva, J.; Rangel. B. Mesenchymal Stem Cell Therapy for Diabetic Kidney Disease: A Review of the Studies Using Syngeneic, Autologous, Allogeneic, and Xenogeneic Cells. Stem Cells Int. 2020, 2020, 1–28. [Google Scholar] [CrossRef]
- Waisman, A.; Lukas, D.; Clausen, B.E.; Yogev, N. Dendritic cells as gatekeepers of tolerance. Semin. Immunopathol. 2016, 39, 153–163. [Google Scholar] [CrossRef]
- Van Coillie S, Wiernicki B, Xu J. Molecular and Cellular Functions of CTLA-4. In 2020. p. 7–32.
- Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KAM, Zoungas S. Diabetic kidney disease. Nat Rev Dis Primers. 30;1(1):15018.
Figure 1.
Proposed Action Mechanism of DC Immunotherapy for DKD. Immunomodulation activity of DC can inhibit chronic low-grade-inflammation which is expected to stop renal damage and maintain kidney function.
Figure 1.
Proposed Action Mechanism of DC Immunotherapy for DKD. Immunomodulation activity of DC can inhibit chronic low-grade-inflammation which is expected to stop renal damage and maintain kidney function.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



