
Submitted:
04 July 2024
Posted:
05 July 2024
You are already at the latest version
Abstract
Dengue (DENV) and Zika (ZIKV) virus continue to pose significant challenges globally due to their widespread prevalence and severe health implications. Given the absence of effective vaccines and specific therapeutics, targeting of the highly conserved NS5 RNA-dependent RNA polymerase (RdRp) domain has emerged as a promising strategy. However, limited efforts have been made to develop inhibitors for this crucial target. In this study, we employed an integrated in silico approach utilizing combinatorial chemistry, docking, molecular dynamics simulations, MM/GBSA, and ADMET studies to target the allosteric N-pocket of DENV3-RdRp and ZIKV-RdRp. Using this methodology, we designed lycorine analogs with natural S-enantiomers (LYCS) and R-enantiomers (LYCR) as potential inhibitors of non-structural protein 5 (NS5) in DENV3 and ZIKV. Notably, 12 lycorine analogs displayed a robust binding free energy (<-9.00 kcal/mol), surpassing that of RdRp-Ribavirin (<-7.00 kcal/mol) along with promising ADMET score predictions (<4.00), of which (LYCR728-210, LYCS728-210, LYCR728-212, LYCS505-214) displayed binding properties to both DENV3 and ZIKV targets. This study highlights the potential of non-nucleoside lycorine-based analogs with different enantiomers that may present different or even completely opposite metabolic, toxicological, and pharmacological profiles as promising candidates for inhibiting NS5-RdRp in ZIKV and DENV3, paving the way for further exploration for the development of effective antiviral agents.
Keywords:
DENV3
; ZIKV
; molecular docking
; MD simulations
; MM/GBSA
; compound library
; NS5
; lycorine
1. Introduction
Dengue and Zika fever represent two neglected infectious diseases (NIDs) of significant global concern [1]. These diseases are particularly prevalent in tropical and subtropical regions across Africa, the Americas, Asia, and the Pacific [2,3], and are caused by dengue (DENV) and Zika (ZIKV) viruses, both belonging to the Flaviviridae family, characterized as positive-sense single-stranded RNA (+ssRNA) viruses [4]. Their transmission occurs primarily through the bite of infected Aedes mosquitoes [5,6,7] with shared urban transmission cycles and initial clinical symptoms. However, the severity of complications and sequelae differed significantly between the two diseases. DENV infection can lead to severe bleeding, organ impairment, and plasma leakage [8,9], whereas ZIKV is associated with complications such as microcephaly, Guillain-Barré syndrome (GBS), and other congenital neurological disorders [10,11]. Despite ongoing efforts in disease surveillance, diagnosis, and prevention, specific treatment options for these diseases remain elusive [8,9]. Consequently, it is imperative to develop effective drug discovery strategies for targeted infection therapies. Urgency is heightened by the co-circulation of emerging DENV/ZIKV cases, particularly in Latin American countries, such as Brazil, Venezuela, and Colombia [6,12]. Therefore, there is an urgent need to identify chemical compounds capable of effectively blocking DENV and ZIKV replication, with high efficacy, selectivity, and a favorable safety profile.
The RNA-dependent RNA polymerase domain (RdRp), positioned within the C-terminal segment of the NS5 (nonstructural 5) protein, is a key target for the development of novel small-molecule drugs to combat DENV and ZIKV. This zinc metalloenzyme, found consistently among flaviviruses and notably absent in humans, plays a central role in viral genome replication [4,17]. Comprised of three subdomains—thumb (priming loop), fingers (motifs G, B, F), and palm (active site) (see Figure 1 and Figure S4)— NS5-RdRp governs RNA and NTP entry and binding, as well as the exit of the newly synthesized double-stranded RNA (dsR-NA) product. Although a handful of broad-spectrum inhibitors capable of binding and inhibiting DENV and ZIKV NS5-RdRp have been identified over the past decade [18,19], none have progressed as viable drug candidates because of concerns related to low efficacy and potential toxicity. However, recent attention has turned toward the promising potential of non-nucleoside inhibitors (NNIs), which can bind to allosteric druggable sites and induce conformational changes that affect protein function. Notably, lycorine (depicted in Figure 1, left side), a natural plant alkaloid, has shown the ability to impede RNA replication in DENV and ZIKV through weak binding to NS5-RdRp; however, a comprehensive understanding of the molecular mechanism remains elusive [16]. Given the compact size of lycorine, it serves as an optimal foundational core for the structural design and exploration of drug-sized analogs, thus enhancing its inhibitory potential against NS5-RdRp for both flaviviruses.
In this study, we aimed to model novel non-nucleoside lycorine-based compounds with the potential to serve as dual inhibitors of DENV3 and ZIKV NS5-RdRp. We conducted a comprehensive in silico protocol integrating structure-based virtual screening and chemoinformatic analysis through combinatorial chemistry, docking, molecular dynamics simulations, MM/GBSA, and ADMET studies (Figure 2). Initially, we performed virtual enumeration of a chemical library using lycorine as the core scaffold, generating a series of analogs encompassing both the naturally occurring enantiomeric isoform, S (LYCS), and the non-classical isoform, R (LYCR) at the ring D of the structure (Figure 1, left side). Subsequently, these analogs were subjected to rigorous evaluation through docking screening, molecular dynamics (MD) simulations, and MM/GBSA calculations. Additionally, the molecular descriptors and ADMET properties were estimated and compared for the complete set of designed compounds. Our analysis identified several analogs with inhibitory potential against either DENV3 or ZIKV NS5-RdRp. More significantly, certain analogs exhibited dual inhibitory potential, effectively targeting this crucial protein domain in both viral agents. Notably, analogs featuring the non-classical isoform RRRR were predicted to display more favorable interaction profiles with the target enzyme. We propose that the lycorine-based compounds assessed in this study represent a significant breakthrough in the pursuit of broad-spectrum small-molecule drugs based on natural products for combating DENV3/ZIKV and potentially other closely related flaviviruses.
2. Materials and Methods
2.1. Preparation of DENV3 and ZIKV RdRp NS5 Structures
The crystal structures of the DENV serotype 3 RdRp (PDB: 5F3Z) and ZIKV RdRp (PDB: 6LD5) were selected as targets for screening compounds with potential inhibitory effects. The protein structures for docking and MD simulations were prepared using Protein Preparation Wizard at a pH of 7.4, similar to cytosolic pH [20]. Crystallized water molecules, and other HETATM records were removed. Polar hydrogen atoms were added using the Epik module (Schrödinger, LCC). Finally, restricted minimization of protein structures was performed using the OPLS3 force field with an RMSD of 0.18 Å.
2.2. Generation of Lycorine Analogs Library
In this study, lycorine analogs with both S and R enantiomers were synthesized to evaluate their inhibitory activity against the NS5 RNA-dependent RNA polymerase (RdRp) of Zika virus (ZIKV) and Dengue virus serotype 3 (DENV3). Lycorine-derived molecules were constructed to incorporate central carbon enantiomers by structure enumeration involving specific functional groups anchored at various points of the base structure (Figure S1) and filtered by ADMET properties analysis (Figure 2), resulting in a library containing 556,276 lycorine analogs. The use of lycorine R enantiomers against the NS5 RdRp of ZIKV and DENV3 targets provides new in silico insights into the potential inhibitory activity differences between the two enantiomers and reveals aspects of their antiviral properties. A subsequent search for structural similarity of lycorine analogs was conducted in the PubChem database using the Jaccard-Tanimoto coefficient [21], with the resulting index score ranging from 0 to 1, representing the degree of intersection between pairs of elements in the compared molecules. The newly generated library, constrained within a threshold value of 0.90, formed the central structure for the anchoring points of various R groups (Figure S1), which were filtered using the Maestro program suite Schrödinger v2020-1 [11]. Moreover, a comprehensive review of the effects of prior lycorine manipulations at anchor points 1, 2, and 3 (Figure S1), as well as the nitrogen atom properties at the C-ring [22], and the stereochemistry of the lycorine structure [22,23] was undertaken. Subsequently, filtered structures were selected to assess their ability to form stable complexes with the RdRp subunit of the NS5 protein of DENV3 and ZIKV. The LigPrep module (Schröding-er, LCC) facilitated the preparation of the ligand's three-dimensional coordinates for subsequent molecular docking simulations, while the generation of ionization/tautomeric states was accomplished using the Epik module (Schrödinger, LCC) at a pH 7.4, specified chirality was retained and no tautomer were generated. ligands were energy-minimized by applying Optimized Potential for Liquid Simulations with an OPLS-2005 force field. This comprehensive approach enhances the understanding of the molecular interactions and mechanisms, also informs the design of more effective lycorine analogs, for future drug development process, improving the efficacy and safety as potential antiviral agents.
2.3. Molecular Docking Screening
All docking calculations were conducted using the XP scoring function in Glide (Schrödinger, LLC). Emphasis was placed on investigating the allosteric N-pocket site located proximal to the active site of the RNA-dependent RNA polymerase (RdRp) (Figure 1 and Figure S4A,C). The selection of this docking site was justified by the mechanism of action of RNA polymerases, which involves two aspartic acids that bind and position two metal ions, catalyzing the nucleotide transfer [13,24]. In the Zika (ZIKV) NS5 RdRp, these correspond to aspartates 535 and 665 [25] while in DENV3 are aspartates 663 and 664 [26] (Figure 1 and Figure S4D). The active site of ZIKV and DENV3 NS5 RdRp, as with other flavivirus RdRps, is located at the intersection of two tunnels. One tunnel, formed by the interfaces of the fingers and thumb domain, coordinates the single-strand RNA, while the second tunnel coordinates the nascent double-strand RNA (Figure 1 and Figure S4A). The priming loop, present in both DENV3 and ZIKV NS5 RdRp, is responsible for the allosteric positioning of the 3′ terminus of the RNA template at the active site [14,26].
For docking, the grid box dimensions were set to 28 × 28 × 28 Å for both DENV3 and ZIKV RdRp. The box for ZIKV-RdRp was centered around the co-crystallized ligand G80 [25] (coordinates: x = 76.1 Å, y = -2.3 Å, z = 15.4 Å), and the grid center for DENV-RdRp was positioned in the PC-79-SH52 [13] binding region (coordinates: x = 18 Å, y = 61 Å, z = 10.2 Å). A total of 556,276 compounds, including lycorine-(R) and lycorine-(S) analogs, were subjected to docking within the specified binding regions of ZIKV-RdRp and DENV3-RdRp, respectively. Compounds were ranked based on their calculated XP Glide scores (ΔG XP Glide scores). Promising candidates from the virtual screening that exhibited robust binding affinities (XP Glide score lower than -9.0 kcal/mol) for both DENV and ZIKV RdRps were selected for further analysis. To ensure the reliability of the molecular docking procedure, a validation process was conducted. This involved re-docking co-crystallized ligands from the literature (PC-79-SH52 and G8O_A_903) into their respective binding sites (PDB IDs 5F3Z and 6LD5). The validation was confirmed by comparing the Root Mean Square Deviation (RMSD) values between the co-crystallized ligand poses and the predicted poses, ensuring reproducibility and accuracy of the docking protocol (refer to Supplementary Figure S6).
2.4. Molecular Dynamics Simulations
Selected ligand poses from the molecular docking process were extracted by binding affinity scoring and underwent Quantum Mechanics (QM) electronic energy minimization utilizing the RHF/6-31G(d) method. The minimized geometries were further employed for QM-restrained electrostatic potential calculations (RESP) utilizing the Merz-Singh-Kollman scheme (MK). QM calculations were executed in the gas phase by employing singlet multiplicity and Gaussian 16. The resulting RESP charges were then incorporated for ligand parameterization using the General Amber Force Field (GAFF) through Amber-Tools20. Given the presence of zinc atoms, the force field parameters for these metal ions and coordinating residues were established using the Metal Center Parameter Builder (MCPB) tool within AmberTools20. The bond and angle force constants were determined using the Seminario and Chg-ModB methods. QM calculations were performed using Gaussian 16. Parametrization of the protein atoms was carried out using the ff14SB force field, while the ligand atoms were parameterized using the GAFF force field. Additionally, the acpype.py script was utilized to convert files into Gromacs format [27].
The protein-ligand complexes were solvated within a cubic box (1.5 nm) with water molecules parameterized using the TIP3P model. To neutralize the system charge, sodium and chlorine ions were introduced at concentration of 0.15 M NaCl. The systems underwent minimization utilizing the steepest descent algorithm with a 0.01 nm step. The subsequent heating was conducted using a Berendsen thermostat, maintaining a temperature of 310 K over a simulation time of 100 ps. The initial random atomic velocities were assigned according to the Maxwell-Boltzmann distribution at 310 K. A system equilibration phase was then implemented, spanning a simulation time of 100 ps, using a Berendsen thermostat and barostat. During heating and equilibration, a harmonic potential with constant k = 1000 kJ/ (mol nm2) was applied to restrain the backbone and ligand atoms. The Particle Mesh Ewald (PME) method was employed to address long-range electrostatic interactions, while all bonds involving hydrogen atoms were constrained using a Fourier spacing of 0.125 and a 1 nm cutoff. The entire simulation process, including heating, equilibration, and production, was conducted with a time step of 2.0 fs. The production run of the systems entailed a 500 ns simulation time, utilizing the Berendsen thermostat and Parrinello-Rahman barostat. All molecular dynamics (MD) simulations were replicated, with snapshots saved every 0.1 ns and extracted from the MD trajectory. All simulations were executed using the resources provided by Virginia Tech's Advanced Research Computing (ARC) Cluster with Nvidia T4 GPUs and MAGNUS HPC from the Universidad de Los Andes with Nvidia Tesla K40c GPUs.
2.5. MM/GBSA Free Energy Calculations
Prime software was used to calculate the energy of the optimized complex, free receptors, and ligands. Binding free energy change calculations were performed using molecular mechanics generalized Born surface area (MM/GBSA) calculations using the formula ΔG(bind) = ΔG(solv) + ΔE(MM) + ΔG(SA), where ΔGsolv is the difference in the GBSA solvation energy of the RdRp/lycorine complex and the sum of the solvation energies for the unliganded complex, ΔEMM is the difference in the minimized energies between the RdRp/lycorine complex and the sum of the energies of the unliganded complex, and ΔGSA is the difference in the surface area energies of the complex and the sum of the surface area energies of the unliganded RdRp/lycorine complex. The RdRp-ligand complexes were minimized using local optimization features in Prime Wizard of the Maestro program suite Schrödinger v2020-1 [11]. The OPLS-2005 force field was used to determine the binding energy (ΔG-bind) of each ligand, and the ligand strain energy was calculated by placing the ligand in a solution autogenerated using the VSGB 2.0 suit.
2.6. Chemoinformatic Analysis
Physicochemical properties of the ligand structures were explored to assess their developmental potential. The canonical Simplified Molecular Input Line Entry System (SMILES) of selected lycorine analogues, including lycorine and rivabirin, were subjected to a comprehensive screening for absorption, distribution, metabolism, excretion, and toxicity using the ADMET predictor (version 10.0.0.11, Simulations Plus, Lancaster, CA, USA) and replicated with SwissADME [28]. ADMET predictor program produced an ADMET Risk score, considering absorption risk, CYP risk, and TOX Risk, representing the chance of general toxicity, hepatotoxicity, mutagenicity, and adherence to Lipinski's rules of five [29]. The cumulative effect of these aspects was measured on a scale from 0 (minimal risk) to 8 (substantial risk), an ADMET Risk value of ≤ 4 indicates an acceptable risk level for the drug. The synthetic accessibility parameter (Synth Diff) of lycorine analogues were evaluated by the ADMET predictor, scoring the synthesis complexity of a compound in a rating from 1 (straightforward synthesis) to 10 (challenging synthesis). The Synth Diff evaluation is based on the molecular fragments, their frequency, and an assessment of the fragment's complexity, considering factors like heavy atoms, macrocycles, stereocenters, spirocenters, and bridges.
2.7. Activity Cliff Detection and Matched Molecular Pair Analysis
Using the ADMET predictor, we employed an automated Matched Molecular Pair Analysis (MMPA) method to identify minor structural transformations or matched molecular pairs (MMPs) [30] across each lycorine analog in the dataset. For this analysis, we set parameters to allow a maximum of 40 unmatched atoms and bonds, Tanimoto similarity thresholds of 0.50, and a maximum of one site for structural changes. The resulting "Molecular Pairs" data were used to detect subtle structural modifications that led to significant impacts on molecular properties, known as "Activity Cliffs" (ACs) [31]. To establish a framework scaffold, we utilized the Maximum Common Substructure (MCS) method, configuring rules to support a maximum atom count of 64, a maximum ring count of 32, and the exclusion of the smallest ring systems.
Distribution plots were generated to visualize the changes in values across all properties. Additionally, keys were generated using Extended Connectivity Fingerprints (ECFPs) with a minimum path length of 1 and a maximum path length of 3 [32]. This comprehensive approach allowed for the identification of critical structural changes that influence the activity of lycorine analogs, providing valuable insights for further optimization and development of these compounds as potential antiviral agents.
3. Results
Generation of Lycorine Analogs Library
The library of lycorine analogs was generated by modifying specific anchor points on the lycorine scaffold. The enumeration of scaffold anchor points resulted in 798 modifications at R1, 797 at R2, and 200 at R3 (Figure S1). The ADMET Risk scores of these analogs averaged 3.2 ± 1.9, indicating an acceptable risk level. The average molecular weight of the analogs was 471.58 ± 51.69 g/mol, compared to the natural lycorine's molecular weight of 287.31 g/mol (Figure S2). These modifications at the anchor points led to improved affinity while maintaining a low ADMET risk [4]. Library generation demonstrated enhanced activity through molecular docking studies, with acceptable ADMET properties and other endpoints achieved through minor structural changes. The resulting library provided valuable insights into structure-activity relationships (SAR), revealing structural determinants critical for biological properties, such as target-specific activity [5,6,7,8,9].
Molecular Docking and MMGBSA
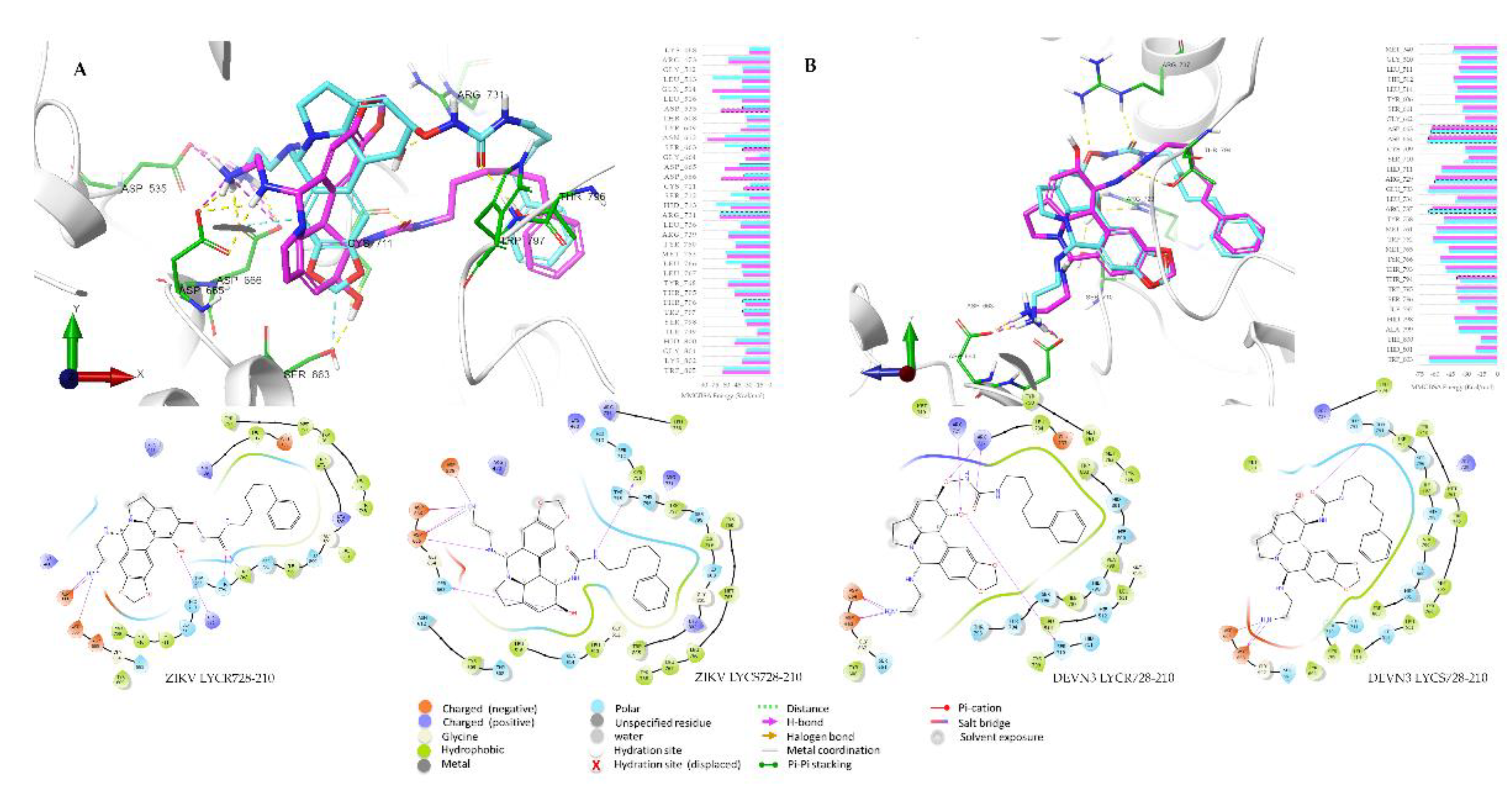
We conducted molecular docking and MM/GBSA analysis of the filtered lycorine analogs to evaluate their potential as inhibitors against DENV3 and ZIKV. Twelve analogs were selected based on their docking scores, binding free energies, and affinities for the NS5 RdRp enzyme (supp. Figure 6). The docking studies revealed that the selected lycorine analogs bind effectively to the allosteric N-pocket of the NS5 RdRp enzyme. Specifically, the R3 anchor point formed hydrogen-bonding interactions with key residues at the active site such as ASP664, ASP663 and priming loop R794 in DENV3. Furthermore, the oxydiazole ring on the B ring of lycorine displayed π-π stacking interactions with HID711 in DENV3, and π-cation interactions with ARG731 in ZIKV, with binding affinities of Δ53.90 kcal/mol and Δ75.75 kcal/mol, respectively (Figure 2). Dual and single compounds exhibited two or three hydrogen bonds with active site residues and one π-π stacking interaction with HID711, showing a binding affinity of 40.12 kcal/mol (Figure 3). Docking score, binding free energy, and binding affinities of the analogues with the RNA polymerase on docking are also tabulated in Table 1. Glide XP energy delta was used to find the binding affinities of the docked hit compounds with the viral NS5 RdRp enzyme [34]. The LYCS analogs in general demonstrated lower binding affinities compared to LYCR analogs, but all compounds were found to bind within the same pocket of the protein structure, exhibiting better theoretical binding affinities than natural lycorine and with top-ranked poses. These results highlight the potential of these lycorine R enantiomer analogs as promising candidates for further studies.
Further validation of the docking results was achieved through molecular dynamics (MD) simulations, including a replica (Supplementary data: MD replica), performed on the selected protein-ligand complexes. The MM/GBSA analysis provided a detailed thermodynamic description of residue contributions to the binding free energy, decomposing the enthalpy value (ΔG_total,GB) at a per-residue level (Figure 3). This comprehensive analysis enhances our understanding of the interaction dynamics and stability of the lycorine analogs within the NS5 RdRp binding site.
Chemoinformatic Analysis
The molecular weights (MW) ranged from 244.20 (Rivabirin) to 603.77 (LYCR727-112), and the number of heavy atoms ranged from 17 (Rivabirin) to 44 (LYCR727-112), with higher counts associated with a logical increment in the complexity of lycorine analogs. The number of hydrogen bond acceptors ranged from 5 (Lycorine) to 8 (LYCS214-510), while the number of hydrogen bond donors ranged from 1 (LYCR66-506) to 8 (LYCS214-510). Rotatable bonds varied significantly, with simpler structures like lycorine having none and more flexible molecules like LYCR728-210 having up to 14. Log P values (iLOGP [33], XLOGP3 [34], WLOGP) denoted lipophilicity, with LYCR294-114 showing the highest consensus Log P (6.00), suggesting significant lipophilicity. In contrast, Rivabirin had much lower values, reflecting its hydrophilic nature. Solubility predictions (ESOL Log S, Ali Log S, Silicos-IT LogSw) indicated that lycorine analog like LYCR66-506 and LYCS505-214 were very soluble, whereas LYCR294-114 and LYCR727-112 were poorly soluble. ADMET Risk scores were generated for absorption, distribution, metabolism, excretion, and toxicity. LYCS505-214 and LYCR66-506 had ADMET Risk values (≤ 4), indicating their potential suitability for drug development. Specific risks included absorption, CYP enzyme inhibition, and general toxicity. LYCS505-214 and LYCR66-506 demonstrated favorable profiles with minimal predicted risks. The synthetic accessibility scores ranged from 4.20 (Lycorine) to 6.57 (LYCR727-112), indicating varying levels of complexity in synthesis. Lower scores suggest simpler and more feasible synthetic routes. Lipinski, Ghose, Veber, Egan, and Muegge rule violations [28] were minimal (Table 1), indicating good drug-likeness for most analogs. LYCR294-114 and LYCR727-112 are exceptions with higher synthetic difficulties (Table 2). Bioavailability scores of 0.55 for all compounds indicate moderate likelihood of oral bioavailability. Most exhibited high GI absorption, critical for oral drug administration. None were predicted to cross the blood–brain barrier (BBB), reducing the likelihood of central nervous system (CNS) side effects. Inhibition of cytochrome P450 enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) was minimal across the analogs (Table 2), suggesting low potential for drug-drug interactions. Several were identified as Pgp substrates, which could affect drug distribution and clearance. PAINS (Pan-Assay Interference Compounds) [35] and Brenk alerts [36] were minimal, suggesting low risk of false positives in biological assays. The comprehensive chemoinformatic analysis revealed that several lycorine analogues and co-crystallized ligands demonstrated favorable physicochemical properties, ADMET profiles, and synthetic accessibility, making them promising candidates for further in vitro investigation as antiviral agents. Notably, analogs like LYCR728-210 and LYCS505-214 stood out due to their balanced profiles of solubility, lipophilicity, and synthetic feasibility and their potential capacity to target ZIKV and DENV3 NS5 RdRp. Further experimental validation and optimization were warranted to fully assess their therapeutic potential.
Activity cliff analysis highlighted the differences between the various pairs of lycorine analogs, focusing on their structural mismatches, similarity scores, and changes in key properties (Scheme 1). The number of mismatches and distinct molecular sites where compounds differ significantly across pairs ranges from four mismatches (for highly similar pairs) to 22 mismatches (for more distinct pairs). Lycorine analogs LYCS728-210 and LYCR728-212 displayed high similarity scores of 0.955 and minimal changes in XP Gscore (-0.071), while LYCR66-506 and LYCS510-214 displayed an average similarity of 0.763 but the highest change in XP GScore in favor of the second analog (0,728). These changes in the XP GScore reflect the differences in binding affinity between the pairs related to the effect of the R groups added. Changes in the rule of five violations were minimal for most pairs, indicating that most analogs maintained drug-like properties. Pairs with high similarity scores and minimal changes in key properties (LYC728-210 and LYC728-212) may offer insight into minor structural modifications that can enhance activity or reduce toxicity.
Binding Poses Analysis of lycorine
Ligand–protein interactions of DENV3-RdRp and ZIKV-RdRp are situated within the thumb domain in an analogous position (Figure 11-14). Interestingly, LYCR728-210, LYCR728-212, LYCS505-214, and LYCS728-210 were anchored by the N-pocket and primming loop in DENV3 RdRps and ZIKV RdRps, despite residue differences, suggesting a structural tendency of the positive charge of the lycorine scaffold to be cavity guided by the superficial polarity of the palm domain. The active site and Motif E superficies at the palm domain are essential for the initiation of RNA replication. In the DENV3-RdRp/ LYCR294-114 complex, the priming loop residues Glu 733, Arg 737, Gln 802, Trp 803 and with ZIKV-RdRp the residues Arg 731, Glu 735 made the main contribution to the total binding energy of interaction between /LYCR66-506 at N-pocket. This interaction has the potential of block RdRp activity by a potential retraction of the priming loop (aa785-807) from the active site during enzyme elongation changing the N-pocket conformation, reducing binding affinities of the RdRp for the ligands [17,19]. This estructural change also has the potential of obstruct the substrates access by affecting the priming loop and keep RdRp in a “closed” state that may prevent the polymer from shifting from initiation to elongation stage of replication [19,37,38]. Similarly, LYCR66-506 and LYCS505-212 also interacted with highly conserved residues (GDD) in the N-pocket of ZIKV RdRp (Figure 11-14). Our study indicates that, despite the sequence differences between DENV3 and ZIKV RdRps, their common allosteric sites share similar geometric and surface properties. Lycorine derivatives may be effective against other viral polymerases with similar allosteric sites althoug. Natural products from plants and in silico derivations are promising strategies for the development of novel antiviral therapeutic agents. This development will be greatly improved in terms of time and cost. To identify DENV3 and ZIKV RdRp inhibitors using in silico derivation, we derived lycorine structures and screened them in silico. Interestingly, data from our study demonstrated for the first time that lycorine analogs may have broad-spectrum antiviral potential. It inhibits DENV3 and ZIKV replication by targeting viral RdRps.
The design by derivatization of lycorine produced numerous molecules, most of which were not synthetically feasible as is usually the case in this type of approach [39]. The reduction of the synthesis feasibility space using synthetic accessibility (SA) parameters [40] allowed the final determination of a set of hit molecules. It was considered that lycorine in its natural conformation fits within the cases presented in the literature in which the compounds to be validated initially exist, and it was taken into account that the SA scoring methods by the complexity method are susceptible to very optimistic evaluations, especially when starting from chemical structures made de novo or derived, which reduced the guarantees that these would be feasible even if the SA score was positive. In our case, the compounds that were screened for validation from natural lycorine offered acceptable synthesis probability (SA), meaning that chemical synthesis should, in principle, be feasible despite possible complications [39]. The joint efforts in this phase on molecules derived from lycorine have not been fruitful to date, as suggested in the synthesis feasibility evaluations with the team, among the possible factors discussed are the difficulty of some reactions, steric hindrances, experimentally risky reactions, among others that have been discussed in other studies with similar approaches [39].
Molecular Dynamic Simulation
The RMSD was calculated for all the complexes for 500 ns trajectory. From the RMSD (Figure S9-10). The RMSD value of the NS5 RdRp/LYCR728-210 complex showed a steady pattern for ZIKV and DENV3. The RMSF plot (Figure 4) was calculated for a 500 ns simulation for all the complexes showing a ~0,5 Å deviation between residues corresponding to palm and finger subdomains, which took participation in the ligand binding in particular the priming loop and the active site (Figure S5-8).
Validation of the Docking Performance and Accuracy
The docking protocol was validated by removing the inhibitors from their respective complexes, re-docking them, and calculating the Root Mean Square Deviation (RMSD). Specifically, the inhibitors PC-79-SH52 (PDB ID 5F3Z) [13] and G8O903 (PDB ID 6LD5) [19] were chosen as templates due to their suitable ligand-binding pockets for virtual screening. Lycorine analogs were similarly docked to the N-POCKET of DENV-RdRp, interacting with active site residues.
Superimposition of the DENV-RdRp/PC-79-SH52 and ZIKV-RdRp/G8O903 complex structures showed that both crystal structures bound to a common allosteric site. The docking parameters for GLIDE were optimized by re-docking the co-crystallized ligands PC-79-SH52 [29] and G8O903 [15] into their inhibitor-binding sites. The calculated binding energies for PC-79-SH52 and G8O903 in their respective binding sites were −5.6 and −6.2 kcal/mol, respectively, with RMSD values of 1.26 and 0.44 Å (see Supplementary Figure 6). To further assess accuracy, the interactions reproduced upon re-docking were compared to those in the native conformations of PC-79-SH52 and G8O903. For G8O903, the re-docked conformation replicated the same hydrogen bonding interactions with Arg 731 and Thr 796 of the priming loop. Similarly, the re-docked PC-79-SH52 ligand's carboxyl group formed an ionic bond with Arg 737, consistent with its native conformation. The acceptable RMSD values, both under 2.00 Å, and the reproducibility of key interactions indicated that the docking protocol was reliable and could be used effectively for further studies [41].
4. Discussion
Binding Poses Analysis of Lycorine
To understand the interaction of lycorine derivates with DENV and ZIKV RdRp, we use a theoretical approach to predict the binding mode. Published crystal structures of DENV and ZIKV RdRps were extracted from Protein Data Bank and docked to lycorine derivates library in the allosteric ligand binding region (N-Pocket) of polymerase using GLIDE XP. Structures of RdRp in complex were selected by XP Glide score (≤ 9.00), ADMET predictor score (≤ 4.00) and relaxed using molecule dynamic simulation for 10 ns. After that, MM/GBSA computational method was used to calculate the contribution of amino acids to binding free energy of lycorine hits. As shown in Figure 2, the RMSD (LYCR 728-210, 728-212) and figure S10 (LYCS 505-214, 728-210) indicated stability of lycorine hits in both DENV and ZIKV RdRp binding sites, confirmation of lycorine hits as RdRp inhibitor. Superimposition of previous reported structures show lycorine bound to a common site at the N-pocket of DENV and ZIKV RdRp. As ligand interaction analysis of the binding site display (Supplementary figures 5-8), the residues that make main contribution to lycorine derivative LYCS728-210 binding in DENV3 RdRp are (TRP762, ASP663, ASP664, ARG737, TRP803) while (ASP665, ASP66, ARG731, ASP535) in ZIKV.
The main interactions are hydrogen bonds between the ligand and R group of ASP residues of the active site (Figure 3). Another important interaction is π-cation and π-π stacking to the R groups with free hydroxyl groups and the benzodioxole group at B-ring (Figure 1), like ARG729 of DENV3 versus the in LYCS728-210 and ARG731 of ZIKV NS5 RdRp in complex with LYCR211-507. Studies about structure-activity of natural lycorine revealed that the free hydroxyl groups at C-1, C-2 and intact benzodioxole group at B-ring, the basic nitrogen, and the C3-C4 double bond are crucial for the antivirus activity of lycorine [22].
Lycorine analogues LYCS and R anchor at the N-pocket to the priming loop from DENV3 and ZIKV (~aa790--807) and interacted with the polar residues (THR793, THR794, THR796, and Ser 796) and hydrophobic residues (TRP793, TRP797, and Ile 799). The priming loop is one of the special characteristics of flavivirus RdRp and plays a key role in de novo RNA initiation. The interaction between lycorine analogues and key priming loop residues has the potential of keeps the polymerase in the closed conformation and, therefore, halts viral RNA synthesis. As shown in Figure 3, Lycorine analogues binds to the equivalent allosteric site N-Pocket in DENV and ZIKV RdRp.
The primary contributing amino acids in the DENV3 N-Pocket binding cavity were ASP663, ASP664, ARG731, and ASN612. These residues form the key interactions between the ligand and the receptor for both LYCS and LYCR analogues. Notably, there is a cationic π-interaction between ring B (Figure 1) of LYCR211-507 and the side chain of ARG731, similar to the interaction reported with RAI-13. [42].
5. Conclusions
This study was carried using chemioinformatics analysis, delineate substantial results by showing the strong affinity of the selected non-nucleoside inhibitor compounds with non-natural LYCR and natural LYCS enantiomer conformations, against Zika and DENV virus NS5 (RdRp) polymerase. The binding energies calculated through simulation were supportive of its values to the selected docking compound binding affinity values. Furthermore, molecular dynamics simulation analyses supported the docking study with stable RMSD values. The ADMET properties of all the compounds revealed drugs like characteristics.
A series of novel lycorine analogues were synthesized and there in silico inhibitor activities were evaluated. Diverse R groups were introduced into the R1, R2, and R3 positions of lycorine and for the first time a l R enantiomer was tested, these analogues exhibited good binding affinity, molecular dynamics and ADMET properties. This provides new opportunities for the investigation of novel lycorine analogues as inhibitors agents against DENV3 and ZIKV. Interestingly, a dual affinity towards DENV3 and ZIKV RdRp targets with this analogue of lycorine was observed. Compounds with dual selectivity profiles could potentially be used as treatment in patients with multiple infection of flaviviruses like ZIKV and DENV.
Authors Contributions
Adrián-Camilo Rodríguez-Ararat, Yasser Hayek-Orduz, Andrés-Felipe Vásquez, Felipe Sierra-Hurtado: Conceptualization, Methodology, Data curation, Visualization, Investigation. María-Francisca Villegas-Torres, Paola A. Caicedo-Burbano, Andrés-Fernando Gonzalez-Barrios: Supervision, writing- review and editing, project administration, funding acquisition. Luke-E.K. Achenie: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: 2D structure of the natural product lycorine; Figure S2: Lycorine enumeration ADMET properties analysis; Table S1: The docking results (Extra precision glide scores – XP GScore); Table S2: GLIDE/ADMET analysis of natural lycorine; Table S1: Analysis of the activity cliffs for lycorine analogs; Figure S2: Redocking of crystallographic ligands; Figure S3. (A) RdRp complex ZIKV, (B) electrostatic potential colored surface, (C) DENV3 and ZIKV RdRp sequence alignment; Figure S4-8. XP glide docking pose and 2D diagram for the interaction results for lycorine enantiomers; Figure S5: Molecular dynamics simulation (MDS) results of DENV3 and ZIKV RdRp-ligand complex.
Funding
This work was funded by the Sistema General de Regalías of Colombia grant number BPIN 2020000100092. PACB was also funded by Universidad Icesi - Convocatoria Interna, grant number CA0413119; and MFVT by the Assistant Professorship Funds from Universidad de los Andes.
Data Availability Statement
Supplementary Data can be found here: https://github.com/AnBio2023/Lycorine-analogs. Additional raw data will be available upon request.
Conflicts of Interest
The authors declare no competing interests.
References
- Messina, J.P.; Brady, O.J.; Golding, N.; Kraemer, M.U.G.; Wint, G.R.W.; Ray, S.E.; Pigott, D.M.; Shearer, F.M.; Johnson, K.; Earl, L.; et al. The Current and Future Global Distribution and Population at Risk of Dengue. Nat. Microbiol. 2019, 4, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The Continued Threat of Emerging Flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Young, P.R. Arboviruses: A Family on the Move. Adv. Exp. Med. Biol. 2018, 1062, 1–10. [Google Scholar] [CrossRef]
- Wu, J.; Liu, W.; Gong, P.; Gong, P. A Structural Overview of RNA-Dependent RNA Polymerases from the Flaviviridae Family. Int. J. Mol. Sci. 2015, 16, 12943–12957. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.R. Historical Perspectives on Flavivirus Research. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Hernández, M.Y.; Ruiz-Saenz, J.; Villamizar, L.J.; Gómez-Rangel, S.Y.; Martínez-Gutierrez, M. Co-Circulation and Simultaneous Co-Infection of Dengue, Chikungunya, and Zika Viruses in Patients with Febrile Syndrome at the Colombian-Venezuelan Border. BMC Infect. Dis. 2018, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paixão, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, Chikungunya and Dengue: The Causes and Threats of New and Reemerging Arboviral Diseases. BMJ Glob. Heal. 2018, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Obi, J.O.; Gutiérrez-Barbosa, H.; Chua, J. V.; Deredge, D.J. Current Trends and Limitations in Dengue Antiviral Research. Trop. Med. Infect. Dis. 2021, 6, 180. [Google Scholar] [CrossRef]
- Nasar, S.; Rashid, N.; Iftikhar, S. Dengue Proteins with Their Role in Pathogenesis, and Strategies for Developing an Effective Anti-Dengue Treatment: A Review. J. Med. Virol. 2020, 92, 941–955. [Google Scholar] [CrossRef] [PubMed]
- McEntire, C.R.S.; Song, K.W.; McInnis, R.P.; Rhee, J.Y.; Young, M.; Williams, E.; Wibecan, L.L.; Nolan, N.; Nagy, A.M.; Gluckstein, J.; et al. Neurologic Manifestations of the World Health Organization’s List of Pandemic and Epidemic Diseases. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Koppolu, V.; Shantha Raju, T. Zika Virus Outbreak: A Review of Neurological Complications, Diagnosis, and Treatment Options. J. Neurovirol. 2018, 24, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.J.R. da; Magalhães, J.J.F. de; Pena, L. Simultaneous Circulation of DENV, CHIKV, ZIKV and SARS-CoV-2 in Brazil: An Inconvenient Truth. One Heal. 2021, 12, 100205. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.G.; Lim, S.P.; Arora, R.; Yokokawa, F.; Nilar, S.; Seh, C.C.; Wright, S.K.; Benson, T.E.; Smith, P.W.; Shi, P.Y. A Conserved Pocket in the Dengue Virus Polymerase Identified through Fragment-Based Screening. J. Biol. Chem. 2016, 291, 8541–8548. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Liew, C.W.; Soh, T.S.; Otoo, D.A.; Seh, C.C.; Yue, K.; Nilar, S.; Wang, G.; Yokokawa, F.; Noble, C.G.; et al. Two RNA Tunnel Inhibitors Bind in Highly Conserved Sites in Dengue Virus NS5 Polymerase: Structural and Functional Studies. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Liu, J.N.; Pan, X.D. Synthesis and Antiviral Activity of Lycorine Derivatives. J. Asian Nat. Prod. Res. 2020, 22, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lao, Z.; Xu, J.; Li, Z.; Long, H.; Li, D.; Lin, L.; Liu, X.; Yu, L.; Liu, W.; et al. Antiviral Activity of Lycorine against Zika Virus in Vivo and in Vitro. Virology 2020, 546, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.P.; Noble, C.G.; Seh, C.C.; Soh, T.S.; El Sahili, A.; Chan, G.K.Y.; Lescar, J.; Arora, R.; Benson, T.; Nilar, S.; et al. Potent Allosteric Dengue Virus NS5 Polymerase Inhibitors: Mechanism of Action and Resistance Profiling. PLoS Pathog. 2016, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Rehman, A.U.; Badshah, S.L.; Ullah, A.; Mohammad, A.; Khan, K. Molecular Dynamics Simulation of Zika Virus NS5 RNA Dependent RNA Polymerase with Selected Novel Non-Nucleoside Inhibitors. J. Mol. Struct. 2020, 1203, 127428. [Google Scholar] [CrossRef]
- Gharbi-Ayachi, A.; Santhanakrishnan, S.; Wong, Y.H.; Chan, K.W.K.; Tan, S.T.; Bates, R.W.; Vasudevan, S.G.; El Sahili, A.; Lescar, J. Non-Nucleoside Inhibitors of Zika Virus RNA-Dependent RNA Polymerase. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular PH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Hu, X.; Hu, Y.; Vogt, M.; Stumpfe, D.; Bajorath, J. MMP-Cliffs: Systematic Identification of Activity Cliffs on the Basis of Matched Molecular Pairs. J. Chem. Inf. Model. 2012, 52, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cai, J.; Cheng, J.; Jing, C.; Yin, J.; Jiang, J.; Peng, Z.; Hao, X. Design, Synthesis and Structure-Activity Relationship Optimization of Lycorine Derivatives for HCV Inhibition. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Liang, L.; Xiao, X.; Feng, P.; Ye, M.; Liu, J. Lycorine: A Prospective Natural Lead for Anticancer Drug Discovery. Biomed. Pharmacother. 2018, 107, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.S.; Lima, G.M.A.; Oliveira, K.I.Z.; Torres, N.U.; Maluf, F. V; Guido, R.V.C.; Oliva, G. Crystal Structure of Zika Virus NS5 RNA-Dependent RNA Polymerase. Nat. Commun. 2017, 8, 14764. [Google Scholar] [CrossRef] [PubMed]
- El Sahili, A. RCSB PDB - 6LD5: Zika NS5 Polymerase Domain.
- Selisko, B.; Papageorgiou, N.; Ferron, F.; Canard, B. Structural and Functional Basis of the Fidelity of Nucleotide Selection by Flavivirus RNA-Dependent RNA Polymerases. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE - AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012 51 2012, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Reports 2017 71 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Tyrchan, C.; Evertsson, E. Matched Molecular Pair Analysis in Short: Algorithms, Applications and Limitations. Comput. Struct. Biotechnol. J. 2017, 15, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Jasial, S.; Miyao, T.; Funatsu, K. Interpretation of Ligand-Based Activity Cliff Prediction Models Using the Matched Molecular Pair Kernel. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J Chem Inf Model 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Hoffmann, R.D. Methods and Principles in Medicinal Chemistry. Pharmacophores and Pharmacophore Searches.; 2006; ISBN 3527609164r9783527609161r9783527608720r3527608729.
- Tarantino, D.; Cannalire, R.; Mastrangelo, E.; Croci, R.; Querat, G.; Barreca, M.L.; Bolognesi, M.; Manfroni, G.; Cecchetti, V.; Milani, M. Targeting Flavivirus RNA Dependent RNA Polymerase through a Pyridobenzothiazole Inhibitor. Antiviral Res. 2016, 134, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Netzler, N.E.; Enosi Tuipulotu, D.; Eltahla, A.A.; Lun, J.H.; Ferla, S.; Brancale, A.; Urakova, N.; Frese, M.; Strive, T.; Mackenzie, J.M.; et al. Broad-Spectrum Non-Nucleoside Inhibitors for Caliciviruses. Antiviral Res. 2017, 146, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Liu, Z. Multi-Objective de Novo Drug Design with Conditional Graph Generative Model. J. Cheminform. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger Structure-Based Virtual Screening Using Glide. 2020.
- Yi, D.; Li, Q.; Pang, L.; Wang, Y.; Zhang, Y.; Duan, Z.; Liang, C.; Cen, S. Identification of a Broad-Spectrum Viral Inhibitor Targeting a Novel Allosteric Site in the RNA-Dependent RNA Polymerases of Dengue Virus and Norovirus. Front. Microbiol. 2020, 11, 1–14. [Google Scholar] [CrossRef]
Figure 1.
“Front view” of the overall structure of the DENV-3 RdRp (PDB code 5f3z) with lycorine analog LYCS505-214 as substrate. A ribbon diagram of the RdRp based on the report by Noble et al. (pdb entry 5F3Z) [13,14]. Interaction between LYCS505-214 (grey molecule) and the active-site residues (Asp663 and Asp664 from the GDD motif, represented as sticks), palm domain (green). The fingers and thumb domains are colored blue and yellow, respectively. The priming loop is colored red, and zinc atoms (grey). Left: Lycorine-(S) and lycorine-(R) enantiomers base structure, benzodioxol group (A), points essential for activity (B, C) [15,16]. Anchor points (R1, R2, R3), and enantiomers points (D, E) for this investigation.
Figure 1.
“Front view” of the overall structure of the DENV-3 RdRp (PDB code 5f3z) with lycorine analog LYCS505-214 as substrate. A ribbon diagram of the RdRp based on the report by Noble et al. (pdb entry 5F3Z) [13,14]. Interaction between LYCS505-214 (grey molecule) and the active-site residues (Asp663 and Asp664 from the GDD motif, represented as sticks), palm domain (green). The fingers and thumb domains are colored blue and yellow, respectively. The priming loop is colored red, and zinc atoms (grey). Left: Lycorine-(S) and lycorine-(R) enantiomers base structure, benzodioxol group (A), points essential for activity (B, C) [15,16]. Anchor points (R1, R2, R3), and enantiomers points (D, E) for this investigation.
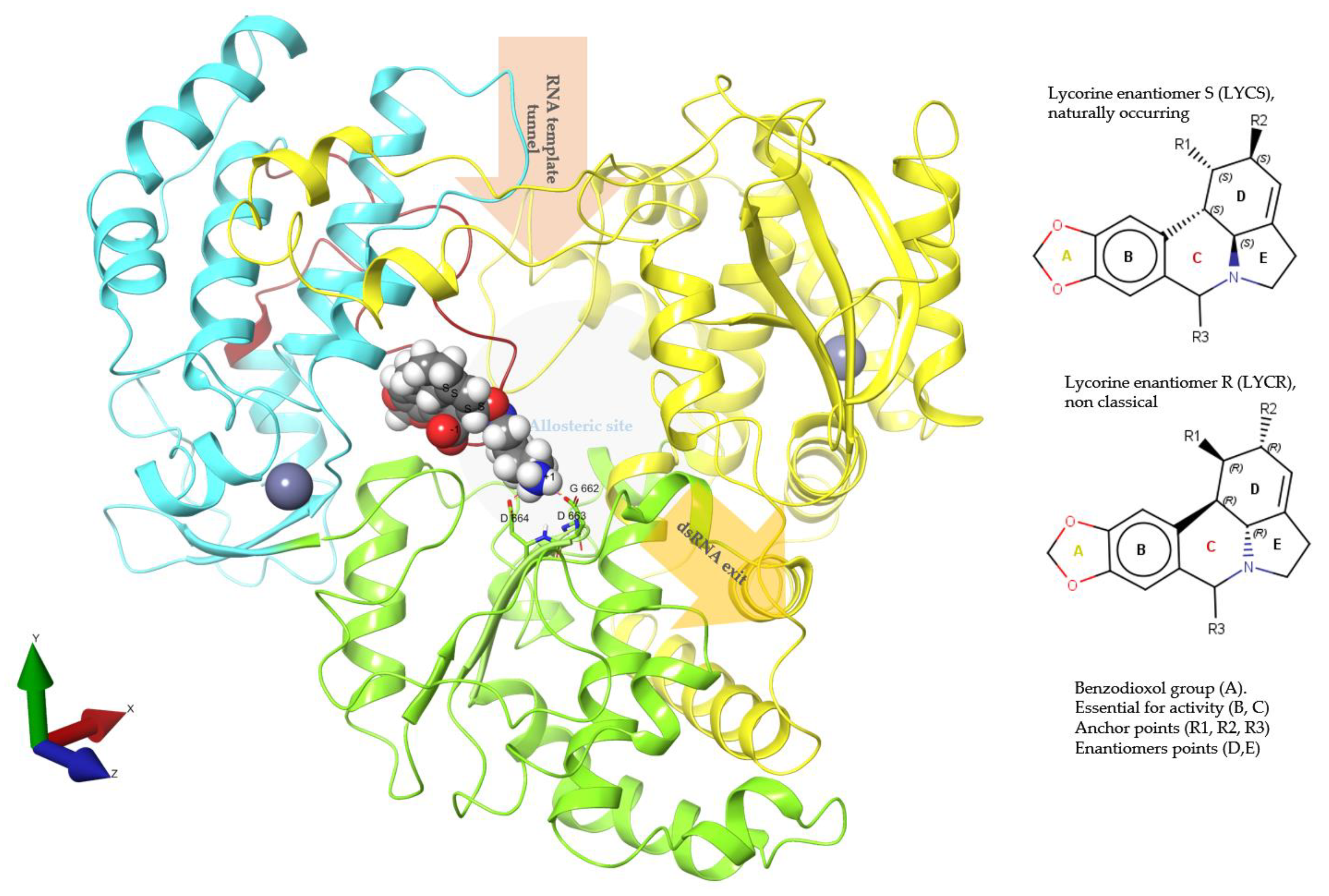
Figure 2.
General workflow of molecular docking calculations. Target DENV3 and ZIKV 3D structures preparation and structure comparison. Lycorine analogs library ligands preparation: protonation states and partial charges. Screening round by molecular docking, ADMET score and molecular dynamics.
Figure 2.
General workflow of molecular docking calculations. Target DENV3 and ZIKV 3D structures preparation and structure comparison. Lycorine analogs library ligands preparation: protonation states and partial charges. Screening round by molecular docking, ADMET score and molecular dynamics.
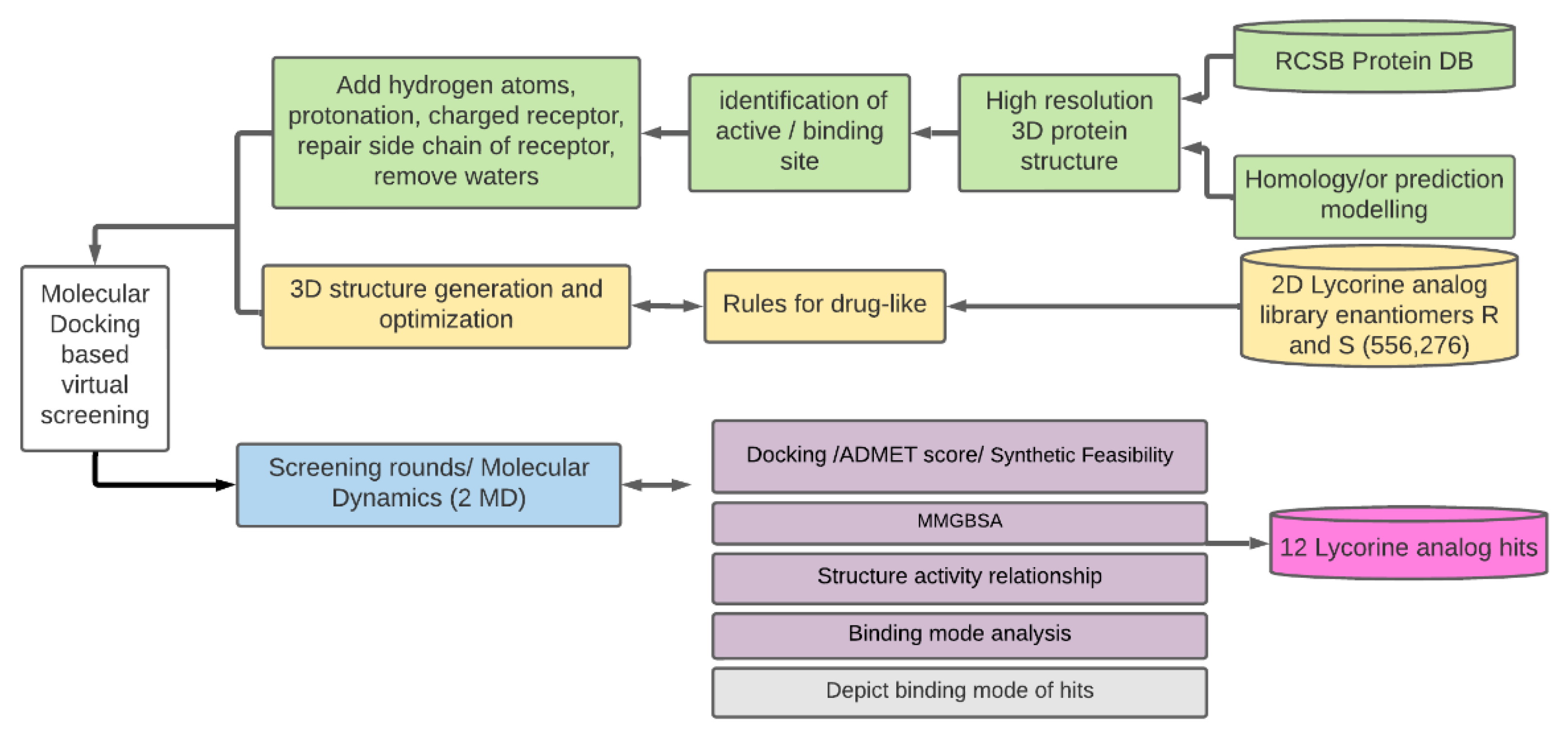
Figure 3.
Molecular docking results: Close-up view of dual lycorine analogue 728-210 enantiomers R (cyan) and S (pink) at RdRp the binding site N-pocket, interacting residues shown in green sticks, hydrogen bonds (yellow), π-π stackin (cyan), and salt bridges (pink), and MMGBSA binding free energy profiles, highlighting the interacting amino acid residues (dashes). For DENV3 (A) and ZIKV (B) RdRp.
Figure 3.
Molecular docking results: Close-up view of dual lycorine analogue 728-210 enantiomers R (cyan) and S (pink) at RdRp the binding site N-pocket, interacting residues shown in green sticks, hydrogen bonds (yellow), π-π stackin (cyan), and salt bridges (pink), and MMGBSA binding free energy profiles, highlighting the interacting amino acid residues (dashes). For DENV3 (A) and ZIKV (B) RdRp.
Scheme 1.
Activity cliff of dual lycorine derivates analogs LYCR728-210 and LYCR728-212 containing modifications at the anchor points R1, R2, R3 and their affinity, ADMET properties and scaffold. XP Gscore Glide binding energy values and Binding site interactions from molecular docking. SynthDiff: synthetic differential ADMET simulation plus, XP Gscore: Extra precision glide docking score.
Scheme 1.
Activity cliff of dual lycorine derivates analogs LYCR728-210 and LYCR728-212 containing modifications at the anchor points R1, R2, R3 and their affinity, ADMET properties and scaffold. XP Gscore Glide binding energy values and Binding site interactions from molecular docking. SynthDiff: synthetic differential ADMET simulation plus, XP Gscore: Extra precision glide docking score.
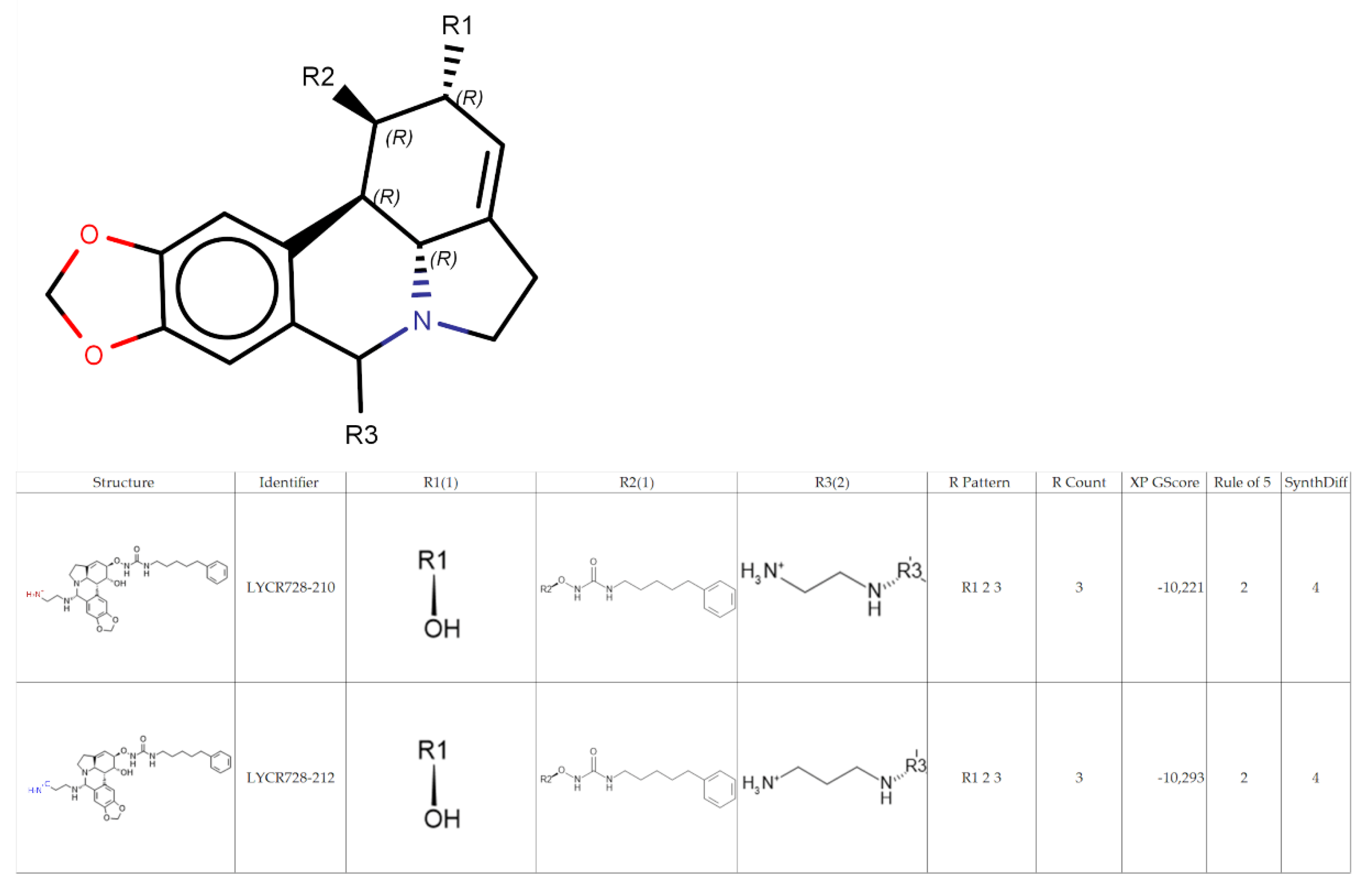
Figure 4.
MD simulation results at 500 ns timescale for LYCR728-210 and LYCR728-212 complexes with RdRp NS5 (A), RMSD ligand (B), RMSF(C).
Figure 4.
MD simulation results at 500 ns timescale for LYCR728-210 and LYCR728-212 complexes with RdRp NS5 (A), RMSD ligand (B), RMSF(C).
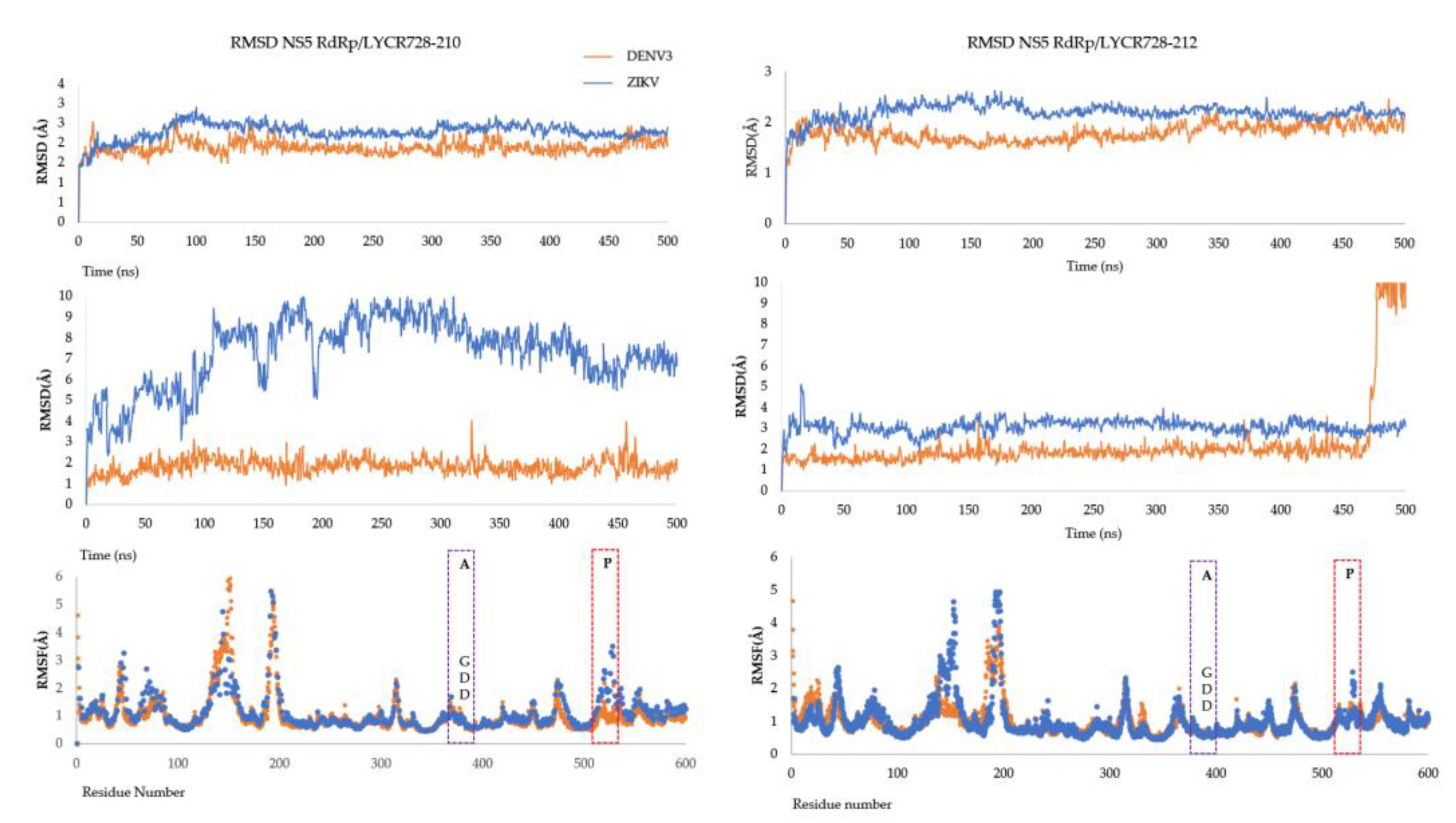

Table 1.
Predicted drug-likeness based on the Lipinski, Ghose,Veber, Egan, and Muegge rules and synthetic accessibility of the selected compounds.
Table 1.
Predicted drug-likeness based on the Lipinski, Ghose,Veber, Egan, and Muegge rules and synthetic accessibility of the selected compounds.
| Molecule | Lipinski | Ghose | Veber | Egan | Muegge | Synthetic Accessibility |
| Rivabirin | 0 | 1 | 1 | 1 | 0 | 3,89 |
| Lycorine | 0 | 0 | 0 | 0 | 0 | 4,2 |
| LYCS214-507 | 0 | 1 | 0 | 0 | 0 | 4,94 |
| LYCS505-214 | 0 | 1 | 0 | 0 | 1 | 5,04 |
| LYCR66-506 | 0 | 0 | 0 | 0 | 0 | 5,09 |
| LYCS510-212 | 0 | 1 | 0 | 0 | 0 | 5,18 |
| LYCR211-507 | 0 | 1 | 0 | 0 | 0 | 5,27 |
| LYCS510-214 | 0 | 2 | 1 | 0 | 0 | 5,31 |
| LYCS214-510 | 0 | 3 | 1 | 0 | 0 | 5,76 |
| LYCS728-210 | 1 | 3 | 1 | 0 | 0 | 5,87 |
| LYCR728-210 | 1 | 3 | 1 | 1 | 0 | 6,11 |
| LYCR728-212 | 1 | 3 | 1 | 1 | 0 | 6,24 |
| LYCR294-114 | 1 | 3 | 1 | 0 | 0 | 6,41 |
| LYCR727-112 | 1 | 3 | 1 | 0 | 1 | 6,57 |
Table 2.
Predicted ADMET properties of the selected compounds.
| Molecule | GI absorption | BBB permeant | Bioavailability Score | CYP-Substrate/inhibitor | ||||
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | ||||
| Rivabirin | Low | No | 0,55 | No | No | No | No | No |
| Lycorine | High | No | 0,55 | No | No | No | Yes | No |
| LYCS214-507 | High | No | 0,55 | No | No | No | No | No |
| LYCS505-214 | High | No | 0,55 | No | No | No | No | No |
| LYCR66-506 | High | No | 0,55 | No | No | No | No | No |
| LYCS510-212 | High | No | 0,55 | No | No | No | No | No |
| LYCR211-507 | High | No | 0,55 | No | No | No | No | No |
| LYCS510-214 | High | No | 0,55 | No | No | No | No | No |
| LYCS214-510 | High | No | 0,55 | No | No | No | No | Yes |
| LYCS728-210 | High | No | 0,55 | No | No | No | No | Yes |
| LYCR728-210 | High | No | 0,55 | No | No | No | No | Yes |
| LYCR728-212 | High | No | 0,55 | No | No | No | No | Yes |
| LYCR294-114 | High | No | 0,55 | No | No | No | No | No |
| LYCR727-112 | High | No | 0,55 | No | No | No | No | Yes |
GI gastrointestinal system, BBB blood–brain barrier, CYP cytochrome P.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



