Submitted:
21 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
With the aim to produce new heterocycle molecules, the previously reported 2-(aminomethyl)-2-(4-chlorophenyl)-2,3-dihydroquinazolin-4(1H)-one was converted efficiently by reacting with N,N’-dithiocarbonyldiimidazole (DTCI), to produce the substituted imidazolidine-2-thione moiety inserted in a three fused ring scaffold of the title compound. The molecular composition was confirmed by a high-resolution MS experiment, and its structure was elucidated by 1H, 13CNMR, and IR analyses. The thioacetamide form of the product was supported by density functional theory (DFT)-NMR analysis where 13C chemical shifts of thioacetamide form and of its iminothiol tautomer were calculated in chloroform at the BP86 /Jgauss-TZP2 level of theory. The very good linear correlation between 13C chemical shifts by experiment and by calculation for the NHC=S form confirmed the structure.
Keywords:
substituted imidalidine-2-thione
; heterocycles synthesis
; NMR analysis
; density functional theory (DFT)
; tautomerism.
1. Introduction
Thiourea is a common framework of various approved drugs and bioactive compounds with a broad range of therapeutic and pharmacological properties. The thiourea moiety is often fixed in a ring system able to provide restricted conformational flexibility, high selectivity and, generally, an increased oral bioavailability [1]. Of note, thioidantoin derivatives are used as chemotherapeutic agents for treating schistosomiasis a parasitic widespread tropical disease caused by worms of the genus Schistosoma [2]. Regarding a heterocyclic thiourea scaffold, imidazolidine-2-thione has a remarkable pharmaceutical interest because it is present in a series of synthetic molecules displaying antimicrobial, antifungal, and anti-HIV activities [3].
Due to N, S heteroatoms, imidazoline-2-thiones can act as polydentate ligands in coordination compounds. As ligands of metal complexes, they were structurally characterized as copper and silver complexes [4]. Furthermore, these metal complexes show broad biological properties. Representative examples are copper derivatives of substituted imidazolidine-2-thiones with activity against Staphylococcus epidermidis and Enterococcus faecalis [5], silver derivatives as antimicrobials [6,7] and as antileishmanial agents [8], and antimony(III) and bismuth(III) complexes evaluated for in vitro cytotoxicity against human breast adenocarcinoma cells [9].
From a synthetic point of view, the conversion of thione group into the corresponding oxo analog has been reported by several efficient approaches [10].
The interest in imidazolidine-2-thione structure is also related to the possibility of prototropic tautomerism, corresponding to the dynamic equilibrium thione-thiol between thioamide (NHC=S) and iminethiol (N=CSH), due to the relocation of a proton similarly to the keto-enol type. In general, tautomeric equilibria are favored by conjugation with unsaturated systems, and their position mainly depends on the phase and temperature and, in solution, are affected by the nature of the solvent and by the concentration. Moreover, tautomerism must be considered when evaluating the physico-chemical properties and designing biologically active molecules.
Various investigations on tautomerism of synthetic imidazole-2-thiones have been reported by IR, UV, NMR, and X-ray spectroscopies [11]. Detailed studies were carried out for benzoimidazole-2-thione by experimental and computational approaches [12,13].
In the present study 3a-(4-chlorophenyl)-1-thioxo-2,3,3a,4-tetrahydroimidazo[1,5-a]quinazolin-5(1H)-one was synthesized from a precursor that we previously used for producing a three cycled fused quinazolin-4(3H)-one derivative [14]. The structural characterization of the title compound is described also in terms of possible tautomerism, supported by a comparison of experimental and density functional theory (DFT) calculated 13CNMR spectra.
2. Results and Discussion
Among the various synthetic methods for the access to substituted imidazolidine-2-thione [11], we produced compound 3 by cyclization starting from 2-(aminomethyl)-2-(4-chlorophenyl)-2,3-dihydroquinazolin-4(1H)-one (1, Scheme 1). This precursor was obtained according to the efficient three-step synthesis that we reported recently, starting from the commercial 4’-chloro-2-phenacylamine hydrochloride treated with trifluoracetic anhydride, then with 2-aminobenzanamide and following cleavage of the trifluoroacetyl group [14]. In the present work, the reaction of 1 with N,N’-dithiocarbonyldiimidazole (DTCI) by refluxing in THF for 1 hour provided product 3, purified by liquid chromatography and verified by analytical RP18-HPLC analysis (Figure S1), in 78% of yield. A global yield of 60% was achieved for the four-step procedure starting from the commercial 4’-chloro-2-phenacylamine hydrochloride.
The new compound 3 has the tricyclic 2,3,3a,4-tetrahydroimidazo[1,5-a]quinazolin-5(1H)-one not containing the C=S unit as the only most similar structure cited in the Chemical Abstracts database through SciFinder platform.
The structural characterization was supported by mass spectrometric analysis, with the molecular cluster pointing out the presence of 35Cl/37Cl atom, and by high-resolution experiment confirming the composition C16H12ClN3OS.
Vibrational analysis shows absorption IR bands at 1647 cm-1 and 1415 cm-1 attributable to C=O and C=S stretching modes, respectively, besides bands at 1214 cm-1 and 568 cm-1 due to the contribution of thione group in line with data obtained for imidazolidine-2-thione [15]. However, it is known that unlike C=O groups, C=S groups do not give rise to characteristic IR stretching bands [16] .
1H and 13C NMR spectra indicate the presence of compound 3 as a single form (Figure S2). In particular, the values 182.31 ppm and 163.44 ppm attributable to C-10 and C-4 respectively, suggest the thione tautomer, if referred to what is reported in the literature according to which the C=S nucleus resonates ca. 20-30 ppm downfield relative to the corresponding C=O [16,17].
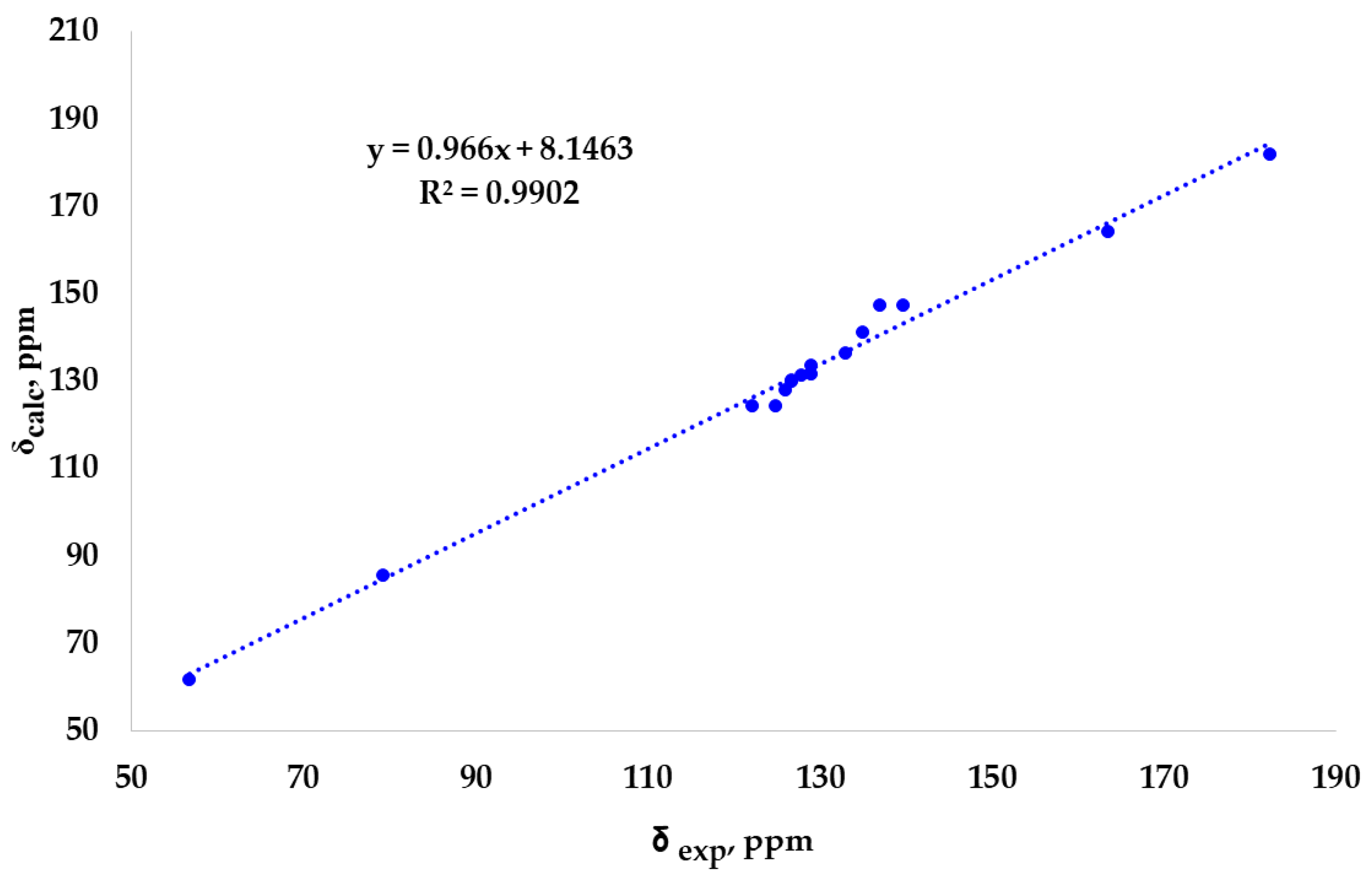
Knowing NMR spectroscopy is one of the most effective tools to study tautomerism and the predictive power of NMR DFT calculation, we decided to compare the experimental 13CNMR data with the calculated values for both thioamide and iminethiol forms (Figure 1). BP86 /Jgauss-TZP2 level of theory was selected by combining a series of functionals and basis sets, validating them for some reference compounds.
The calculation carried out in chloroform has provided a much better correlation of experimental 13C chemical shifts with the calculated values for the thioamide form (NHC=S) than the iminothiol (N=CSH) tautomer. In detail, the experimental value 182.26 ppm is in line with the calculated 181.84 ppm assigned to C-10 of the thioamide form, whereas the value 162.72 ppm was obtained for the iminethiol form. Similarly, the experimental value 56.74 ppm is better correlated with the calculated 61.74 ppm of C-9 for the thioamide form than with 78.29 ppm obtained for the tautomer (Table S1). Figure 2 shows the very good linear fit of all experimental 13C chemical shifts with the corresponding calculated values for the tioamide form of compound 3.
In conclusion, a substituted imidazolidine-2-thione moiety was inserted by a reaction of the previously reported precursor with N,N'-dithiocarbonyldiimidazole to form a three-fused ring scaffold. The product was obtained in thioacetamide form, as supported by the comparison of the experimental 13C chemical shifts with the DFT-calculated values for the thioacetamide and iminothiol tautomers. The synthesis of the title compound can be exploited to access analogs with different aryl fragments and metal complexes to be evaluated for their biological activities.
3. Materials and Methods
3.1. Chemistry
3.1.1. General
The reactions were monitored by thin layer chromatography (TLC): silica gel F254 or reversed phase RP-18 F254 (Merck, WVR, Milan, Italy), with visualisation using UV light. Flash chromatography (FC): RP-18 Lichroprep 40-63µm (Merck, Darmstadt, Germany). HPLC analysis of 3 using a Lichroprep RP-18, (Merck, Darmstadt, Germany) in isocratic condition, flow 1mL/min, UV detection at λ=300 nm. Melting points were determined on Reichert Thermovapor microscope, and the data are uncorrected. Infrared spectra were recorded using a FT-IR Tensor 27 Bruker spectrometer (Attenuated Transmitter Reflection, ATR configuration) at 1 cm−1 resolution in the absorption region 4000–600 cm–1. A thin solid layer is obtained by evaporation of the sample’s dichloromethane solution. The instrument was purged with a constant dry air flux, and a clean ATR crystal was used as background. Spectra processing was made using Opus software package. NMR spectra were recorded on Bruker-Avance 400, 1H-NMR at 400 MHz and 13C-NMR at 100 MHz, calibrated using residual non-deuterated solvent CDCl3 with values relative to TMS (δH 7.25 ppm, and δC 77.0 ppm, respectively) with chemical shift values in ppm and J values in Hz. 13C multiplicity from attached proton test (APT) experiment and assignments confirmed by comparison with DFT simulated spectrum (Table S1). Electron impact (EI)–MS and high-resolution HR-EI-MS spectra (m/z; rel.%) were recorded using a Kratos MS80 mass spectrometer equipped with home-built computerized acquisition software.
3.1.2. Synthesis of 3a-(4-chlorophenyl)-1-thioxo-2,3,3a,4-tetrahydroimidazo[1,5-a] quinazolin-5(1H)-one (3)
To a solution of 1 (80 mg., 0.274 mmol) in THF (2.5 mL) thiocarbonyldiimidazole was added (70 mg, 0.360 mmol). The reaction mixture was refluxed for 1 h, the solvent was removed, and the residue was subjected to flash chromatography on silica gel (CHCl3 /CH3OH 9:1), to give pure compound 3 (70 mg, 78%).
Data: powder. M.p. 321-322°C (dec). TLC: CHCl3/MeOH 90:10 v/v, Rf: 0.3. HPLC(RP18), CH3CN/ H2O 1:1, tR 5.99 min. FT-IR(cm-1): 3220 vw, 3040 vw, 1647 s, 1481 m, 1214m, 757m, 568m. 1H-NMR (600 μL CDCl3 + 80 μl CD3OD): 8.05 (d, J = 8.1, 1H, H-5,), 7.82 (brd, J = 7.8, 1H), 7.48 (m, 1H, H-7,), 7.30 and 7.20 (two d, J = 8.2, 2H each, H-3’/H-4’ and H-2’/H-6’), 7.16 (m, 1H), 3.99 (d, J = 11.3, 1H, Ha-9,) , 3.73 (d, J = 11.3, 1H, Hb-9). 13C-NMR (600 μl CDCl3 + 80 μL CD3OD): 182.26 (s, C-10), 163.44 (s, C-4), 139.67 (s, C-1’), 137.03 (s, C-4’), 134.95 (s, C-8a), 132.96 (d, C-7), 129.01 (d, 2C, C-3’and C-5’), 127.80 (d, C-5), 126.77 (d, 2C, C-2’and C-6’), 126.02 (d, C-6), 124.88 (s, C-4a), 122.08 (d, C-8), 79.20 (s, C-2), 56.74 (t, C-9). EI-MS: m/z (%) 331 (1.3), 329 (3.6), 259 (4), 257 (13). HR-MS: m/z 329.03841± 0.001, calcd. for C16H1235ClN3O 329.03896; m/z 257.04726± 0.001, calcd. for C14H10 35ClN2O 257.04817.
3.2. DFT Calculation
DFT calculation was preoptimized in the gas phase and later performed in chloroform by using the Solvation Model Density (SMD) [18]. Calculations were carried out on a PC running at 3.4 GHz on an AMD Ryzen 9 5950X 16-core (32 threads) processor with 32 GB RAM and 1 TB hard disk with Windows 10 Home 64-bit as an operating system. The structures of compounds were built using GaussView 6.0, and the Gaussian program [19] was used in the geometry optimization at a density functional theory (DFT) level of theory. The optimized geometry was obtained by using RFO step, integral precision = superfine grid and type convergence criteria, and invoking gradient employing 6- 311G(d,p) basis set for all atoms. The electronic correlation functional B1B95, where the gradient-corrected DFT with Becke hybrid functional B1 [20] for the exchange part and the B95 for correlation function [21] was utilized. The vibrational energy calculations at the DFT levels used the optimized structural parameters to characterize all stationary points as minima. No imaginary wave number modes were obtained for the optimized structure, proving that a local minimum on the potential energy surface was actually found. NMR simulation was carried out in chloroform, employing all the combinations of three different basis sets: pcSseg-2 and x2c-TZVPPAll-s and Jgauss-TZP2 basis set [22,23,24] and three different functionals: the generalized gradient approximation (GGA) functionals with D2 version of Grimme’s dispersion B97-D and BP86 [25], the meta-generalized gradient approximation (M-GGA) TPSSTPSS functional [26,27]. Validation was performed on acetone, acetamide, thioacetamide, imidazolin-2-one, thiourea, phenylthiourea, cyanothioacetamide, ethyl dithioacetate, and N,N'-diethylthiobarbituric acid (Figure S3). Magnetic properties were calculated with GIAO [28,29,30,31,32,33] schemes. The isotropic shift constants were obtained (σ) for each nucleus, and these were converted to a chemical shift (δ) value according to the equation: δi = σTMS−σi. The reference substance was tetramethylsilane (TMS), calculated at the same level of theory. xyz Coordinates of the geometry-optimized thioacetamide and iminothiol structures of compound 3 are reported in Table S2.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: HPLC chromatogram of title compound; Figure S2: NMR spectra of tile compound; Table S1: 13CNMR chemical shifts: experimental and BP86 /Jgauss-TZP2 calculated values for the thioamide and iminethiol tautomers; Figure S3: Correlation plot of calculated vs. experimental 13CNMR chemical shifts for the model compounds used in the validation of the adopted BP86 /Jgauss-TZP2 level of theory. Table S2: xyz Coordinates of geometry- optimized structures of title compounds and its tautomer.
Author Contributions
Conceptualization, A.D. and I.M.; investigation, A.D., I.M. and N.I.; writing—original draft preparation, I.M. and A.D.; writing—review and editing, I.M., N.I. and A.D.; supervision, I.M.; project administration, A.D.; funding acquisition, I.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Adriano Sterni, University of Trento for the technical contributions with mass spectra.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Riccardo Ronchetti, R.; Moroni, G.; Carotti, A.; Gioiello, A.; Camaioni, E. Recent advances in urea- and thiourea-containing compounds: focus on innovative approaches in medicinal chemistry and organic synthesis. RSC Med. Chem. 2021, 12, 1046–1064. [Google Scholar] [CrossRef] [PubMed]
- Pitta, M.G.R.; Pitta, M.G.R.; Barreto De Melo Rego, J.B.; Galdino, L.S. The Evolution of Drugs on Schistosoma Treatment: Looking to the Past to Improve the Future. Mini-Rev. Med. Chem. 2013, 13, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Savjani, J.K.; Gajjar, A.K. Pharmaceutical Importance and Synthetic Strategies for Imidazolidine-2-thione and Imidazole-2-thione Derivatives. Pakistan J Biol. Sci. 2011, 14, 1076–1089. [Google Scholar] [CrossRef]
- García-Vázquez, J.A.; Sousa-Pedrares, A.; Carabel, M.; Romero, J.; Sousa, A. Electrochemical Synthesis and Structural Characterization of Copper and Silver Complexes of Imidazolidine-2-Thione Ligands. Polyhedron 2005, 24, 2043–2054. [Google Scholar] [CrossRef]
- Lobana, T.S.; Aulakh, J.K.; Sood, H.; Arora, D.S.; Garcia-Santos, I.; Kaur, M.; Duff, C.E.; Jasinski, J.P. Synthesis, Structures and Antimicrobial Activity of Copper Derivatives of N -Substituted Imidazolidine-2-Thiones: Unusual Bio-Activity against Staphylococcus epidermidis and Enterococcus faecalis. New J. Chem. 2018, 42, 9886–9900. [Google Scholar] [CrossRef]
- Aulakh, J.K.; Lobana, T.S.; Sood, H.; Arora, D.S.; Smolinski, V.A.; Duff, C.E.; Jasinski, J.P. Synthesis, Structures, and ESI-Mass Studies of Silver(I) Derivatives of Imidazolidine-2-Thiones: Antimicrobial Potential and Biosafety Evaluation. J. Inorg. Biochem. 2018, 178, 18–31. [Google Scholar] [CrossRef]
- Aulakh, J.K.; Lobana, T.S.; Sood, H.; Arora, D.S.; Garcia-Santos, I.; Kaur, M.; Jasinski, J.P. Synthesis, Structures, and Novel Antimicrobial Activity of Silver(I) Halide Complexes of Imidazolidine-2-Thiones. Polyhedron 2020, 175, 114235. [Google Scholar] [CrossRef]
- Espuri, P.F.; Dos Reis, L.L.; De Figueiredo Peloso, E.; Gontijo, V.S.; Colombo, F.A.; Nunes, J.B.; De Oliveira, C.E.; De Almeida, E.T.; Silva, D.E.S.; Bortoletto, J.; Segura, D.F.; Netto, A.V.G.; Marques, M.J. Synthesis and Evaluation of the Antileishmanial Activity of Silver Compounds Containing Imidazolidine-2-Thione. J Biol Inorg Chem 2019, 24, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Ucar, O.; Grześkiewicz, A.M.; Banti, C.; Hadjikakou, S.K.; Ozturk, I.I. Structural Characterization and Biological Evaluation of Antimony(III) and Bismuth(III) Complexes with Imidazolidine-2-Thione. J. Mol. Struct. 2021, 1235, 130270. [Google Scholar] [CrossRef]
- Kuptsova, A.O.; Vinogradova, E.E.; Kravchenko, A.N.; Gazieva, G.A. Methods for Substitution of the Thioxo Group with the Oxo Group in Imidazolidine-2-Thione Derivatives. Russ. Chem. Bul.l 2022, 71, 885–904. [Google Scholar] [CrossRef]
- Trzhtsinskaya, B.V.; Abramova, N.D. Imidazole-2-Thiones: Synthesis, Structure, Properties. Sulfur Rep. 1991, 10, 389–421. [Google Scholar] [CrossRef]
- Öğretir, C.; Öztürk, İ.İ.; Tay, N.F. Quantum Chemical Studies on Tautomerism, Isomerism and Deprotonation of Some 5(6)-Substituted Benzimidazole-2-Thiones. Arkivoc 2007, 75–99. [Google Scholar] [CrossRef]
- Altun, A.; Azeez, N. Structural Elucidation of Sulfur Derivatives of Benzimidazoles by Density Functional Calculations, and Vibrational and NMR Spectral Analyses. Pharm. Anal. Acta 2015, 7, 459–462. [Google Scholar] [CrossRef]
- Defant, A.; Innocenti, N.; Mancini, I. 3a-(4-Chlorophenyl)-1-Methyl-3a,4-Dihydroimidazo[1,5-a]Quinazolin-5(3H)-One: Synthesis and In Silico Evaluation as a Ligand in the µ-Opioid Receptor. Molbank 2023, 2023, M1622. [Google Scholar] [CrossRef]
- Dwarakanath, K.; Sathyanarayana, D.N. Vibrational Spectra and Assignments for the Fundamental Vibrations of Imidazolidine-2-thione and -2-selenone Bull. Chem. Soc. Jpn. 1979, 52, 2699–2704. [Google Scholar] [CrossRef]
- Sifferlen, T.; Rueping, M.; Gademann, K.; Jaun, B.; Seebach, D. β-Thiopeptides: Synthesis, NMR Solution Structure, CD Spectra, and Photochemistry. Helv. Chim. Acta 1999, 82, 2067–2093. [Google Scholar] [CrossRef]
- Kozyra, P.; Kaczor, A.; Karczmarzyk, Z.; Wysocki, W.; Pitucha, M. Experimental and Computational Studies of Tautomerism Pyridine Carbonyl Thiosemicarbazide Derivatives. Struct. Chem. 2023, 34, 1973–1984. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-Functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. 4. A new dynamical correlation functional and implications for exact exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Gauss, J. Effects of electron correlation in the calculation of nuclear magnetic resonance chemical shifts. J. Chem. Phys. 1993, 99, 3629–3643. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to K. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp.Chem., 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 194101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- London, F. The quantic theory of inter-atomic currents in aromatic combinations. J.Phys.Radium 1937, 8, 397–409. [Google Scholar] [CrossRef]
- McWeeny, R. Perturbation Theory for Fock-Dirac Density Matrix. Phys.Rev. 1962, 126, 1028–1034. [Google Scholar] [CrossRef]
- Ditchfield, R. Molecular Orbital Theory of Magnetic Shielding and Magnetic Susceptibility. J. Chem. Phys. 1972, 56, 5688–5691. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys, 1974; 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of the target compound 3 from 1, with an indication of the previously reported production of 1 [14]. Arbitrary numbering is for convenience, used for NMR assignments.
Scheme 1.
Synthesis of the target compound 3 from 1, with an indication of the previously reported production of 1 [14]. Arbitrary numbering is for convenience, used for NMR assignments.
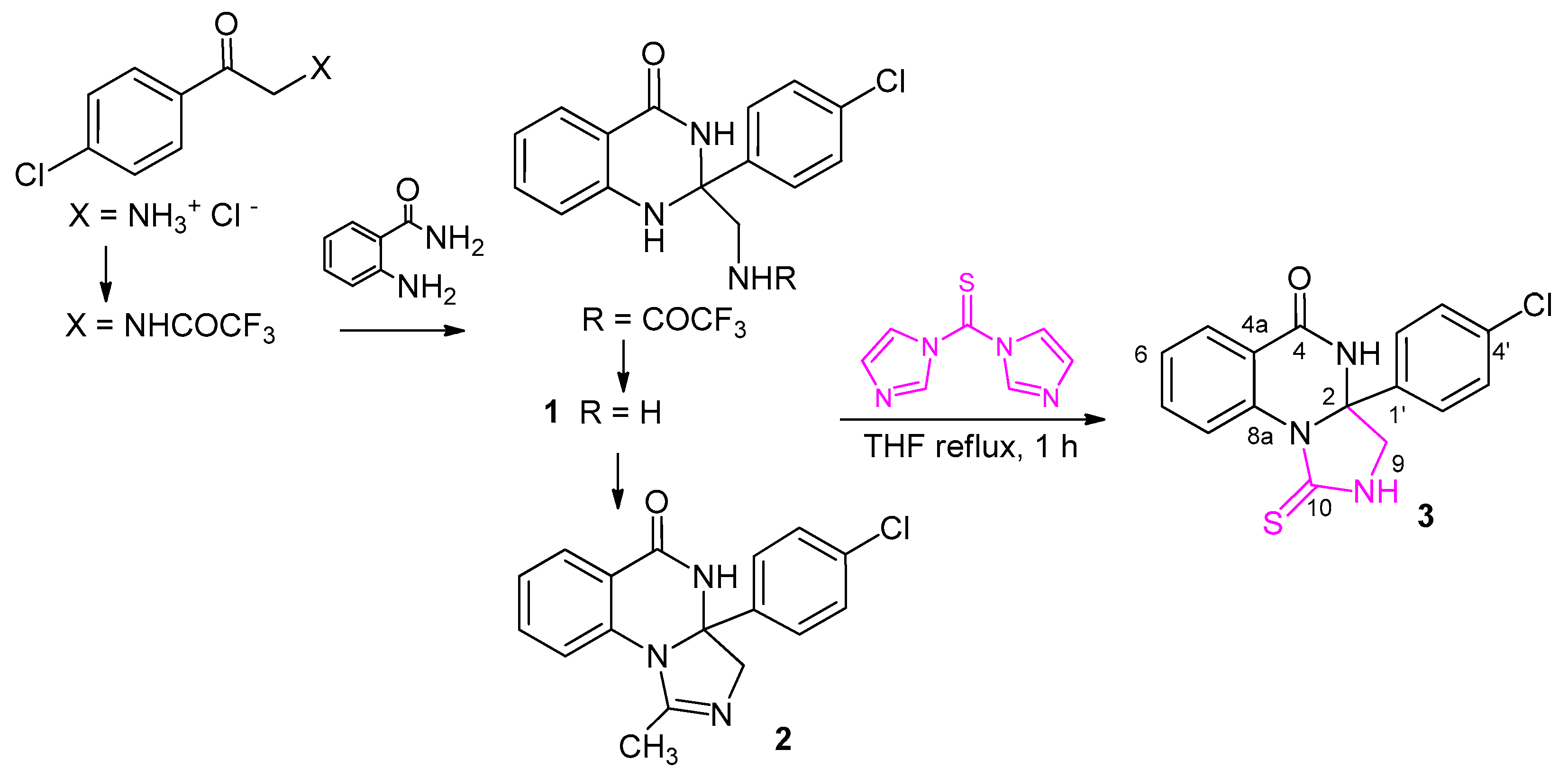
Figure 1.
Energy minimized structures of (a)thioamide and (b) iminethiol forms of compound 3.
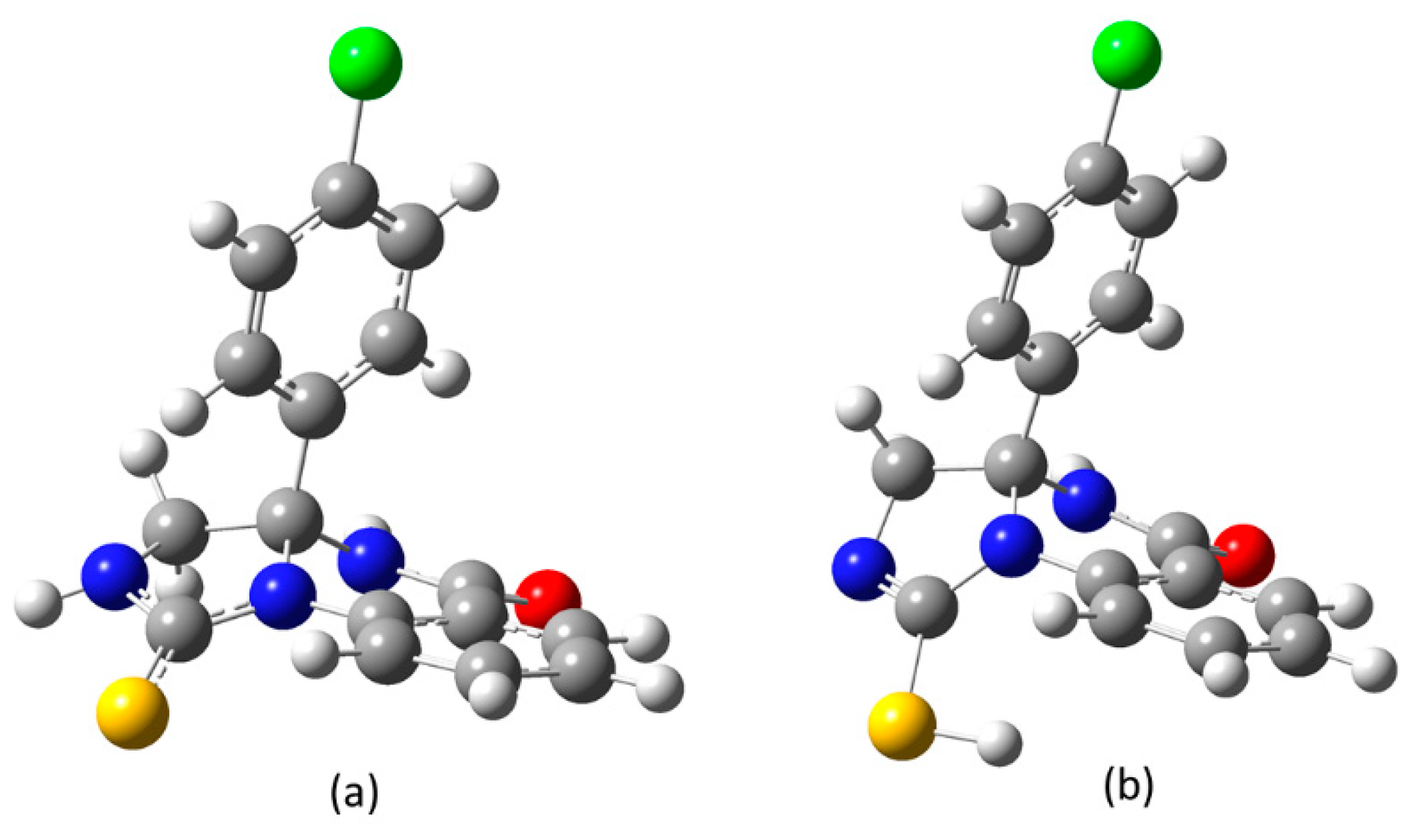
Figure 2.
Correlation plot of calculated vs. experimental 13CNMR chemical shifts for the thioamide form of compound 3.
Figure 2.
Correlation plot of calculated vs. experimental 13CNMR chemical shifts for the thioamide form of compound 3.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



