
Submitted:
20 June 2024
Posted:
21 June 2024
You are already at the latest version
Abstract
Purpose: Investigation for the co-occurrence of two unrelated genetic disorders of muscular dystrophy and Prader-Willi syndrome (PWS) (OMIM # 176270) using joint whole genome sequencing (WGS). Methods: Trio WGS joint analysis was performed to investigate the genetic etiology in a proband with PWS, prolonged muscular hypotonia associated hyperCKemia, and early-onset obesity. The parents were unaffected. Results: Results showed maternal isodisomy uniparental disomy (UPD) in chromosome 15 expanding from 15q11.2 to 15q22.2, including PWS regions at 15q11.2-15q13. Maternal heterodisomy was detected from 15q22.2 to 15q26.3. A pathogenic variant, NM_000070.3(CAPN3):c.550del (p.Thr184fs), was identified at 15q15.1 in a heterozygous state in the mother that was homozygous in the proband due to maternal isodisomy. Conclusion: This is the first study of the concurrent molecular etiology of PWS and calpainopathy (OMIM # 253600) in the same patient. This report highlights the utility of joint analysis and need for the assessment of autosomal recessive disease in regions of isodisomy in patients with complex and unexplained phenotype.
Keywords:
Joint-WGS analysis
; Uniparental Disomy
; Isodisomy
; Heterodisomy
; Prader-Willi Syndrome
; Calpainopathy
1. Introduction
Joint analysis of WGS data enables investigation of phasing and uniparental disomy (UPD) in complex genetic diseases [1,2]. Uniparental disomy (UPD) events may cause additional genetic abnormalities by unmasking deleterious recessive alleles inherited from the parent from whom UPD is originated. Indeed, co-occurrence of unrelated disorders such as Charcot-Marie-Tooth, Gaucher disease type 3, cystinuria, Crigler-Najjar syndrome type I and long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency have been reported [3,4,5].
Herein, we describe the investigation and molecular diagnosis of two unrelated genetic syndromes in a patient with the complex phenotype of PWS and calpainopathy due to maternal isodisomy. Prader-Willi syndrome (PWS) is a rare neurodevelopmental disorder with an estimated incidence of 1:10,000–1:30,000 live births. Maternal UPD is the second most common molecular etiology of PWS, resulting in paternal expression loss of imprinting in 15q11-q13 locus [6,7,8]. While chromosome 15 is well known for its association with PWS, it encompasses many other genes associated with autosomal recessive conditions. One such gene is CAPN3 known to cause calpainopathy [9], an autosomal recessive muscular dystrophy. With the prevalence of 1:100,000 [10], calpainopathy (MIM#253600) is the most common muscular dystrophy subtype, accounting for 30% of limb-girdle muscle dystrophies [11,12,13]. As a single gene disorder, phenotypical features of calpainopathy consist of symmetric and progressive weakness of proximal limb-girdle muscles, tiptoe walk, difficulty in running, scapular winging, waddling gait, laxity of the abdominal muscles, Achilles tendon shortening, and scoliosis. Generally, cardiac muscles are not affected [10].
This is the first report of PWS and calpainopathy co-occurrence in the same individual due to isodisomy. Given the frequency of pathogenic variants in CAPN3, this report highlights the significance of extended joint genome analysis in PWS patients with complex phenotype.
2. Participants, Materials, and Methods
2.1. Participants
The index patient, a two-year-old female, with unaffected mother and father were consented at the Department of Pediatrics, University Hospital Centre Zagreb and enrolled in the CROseq genome program. CROseq Genome Program is a collaborative research program between Brigham and Women’s Hospital (BWH) (Boston, USA), and the Department of Pediatrics, University Hospital Centre Zagreb (Zagreb, Croatia), supported by the Mila Za Sve Foundation (Rijeka, Croatia). Participants consented to the study at the University Hospital Center Zagreb.
2.2. Karyotyping
Conventional karyotyping was performed to detect chromosomal abnormalities. The lymphocytes were derived from the peripheral blood. The cultured lymphocytes were treated with 10 μl/ml colcemid when they reached the logarithmic phase. Then, the fixative was added to preserve the structure of the metaphase cells. The harvested chromosomes were applied G-banding by using Trypsin and Giemsa. The chromosomes’ morphology and number were evaluated through 20 metaphases. The karyotype result was described per The International System for Human Cytogenomic Nomenclature (ISCN) 2020.
2.3. Methylation Specific-Multiplex Ligation-dependent Probe Amplification
Genomic DNA was purified from whole peripheral blood by FlexiGene DNA Kit according to the manufacturer’s protocol (Qiagen, Hilden, Germany). ME028-C1 kit was applied for MS-MLPA (MRC-Holland, Amsterdam, The Netherlands). The probemix of ME028-C1 contains probes specific for the PWS region and probes targeting sequences restricted by methylation-sensitive HhaI endonuclease. The latter probes were responsible for determining the methylation profiling. In addition, there were reference probes for genes located outside the PWS region. Internal control probe normalization was used to normalize peak intensities, and the intensity ratios of identical probes from the sample were compared with the reference. The amplified products were quantified by ABI 3130xl capillary sequencer (Applied Biosystems, Foster City, USA). The final data was analyzed with Coffalyser software (MRC-Holland, Amsterdam, The Netherlands). If there were copy number variation (CNV), the methylation pattern would be used to determine the CNV’s parental origin. However, in the absence of CNV, the methylation profile differentiated biparental inheritance from UPD.
2.4. Whole Genome Sequencing
In total, 2ml of whole blood samples per participant were collected in an EDTA tube at the University Hospital Center Zagreb, Croatia. Genomic DNA purification and WGS were carried out at the Medical College of Wisconsin (Milwaukee, USA). Genomic DNA with a purity ratio of 1.75-2.0 was obtained from the whole peripheral blood sample. Following the robotic library preparation, the Illumina NovaSeq 6000 platform, with an average depth of coverage of 30X, was used for sequencing.
2.5. Zygosity and UPD Landscape Analysis by Trio WGS
To investigate maternal UPD in chromosome 15, the maternal and paternal contribution was assessed by joint trio WGS. All protein-coding genes over chromosome 15 were queried. The zygosity of the variants over the clinically relevant genes, as defined in OMIM, was further analyzed. To exclude the common variants that can potentially present in the father due to high population allelic frequency, the 1% allele frequency filter was applied. UPD landscape analysis by joint trio WGS was conducted using single nucleotide polymorphism analysis of both the parents and the patient. The variants homozygous in the proband, heterozygous or homozygous in the relevant parent, and wild type in the other parent are labeled as UPD supportive variants. Supportive variants were clustered. Clusters of at least 4Mbps with a minimum of 20 supporting variants were used to locate potential UPD regions. The parent of origin was assigned by the inheritance patterns located within each UPD region. UPD regions were attributed as maternal if the only contributor in the proband was maternal allele.
2.6. UPD and Region of Homozygosity (ROH) Mapping
The regions of homozygosity (ROH) were used to further analyze the UPD region. Bcftools ROH module was used to assess the distribution and size of ROH. GVCF files of proband, mother, and father were merged with GATK CombineGVFs. Then this file was processed using GATKGenotypeGVCFs. The resulting multi-sample VCF file was evaluated through UPDio (https://github.com/findingdan/UPDio). Regions of homozygosity (ROH) were graphically visualized by the variant caller that detects and outputs the runs of homozygosity from whole genome calls on autosomal human chromosomes.
2.7. Focused Trio WGS Analysis on Muscular Dystrophy
A trio-based whole genome analysis was performed due to the proband’s elevated creatinine kinase levels, muscular dystrophy and early-onset obesity without hyperphagia. A focused interrogation was performed on the variants across the genes in chromosome 15. Technical assessment of variants was conducted to include only high and medium-confidence variants. Using Human Phenotype Ontology (HPO) terms for the probands phenotypes, variants were prioritized according to the gene’s association with these findings and their potential consequence (variant type and classification).
2.8. Genome Variant Classification
The variant interpretation was performed based on the ACMG-AMP guideline [14]. The population allele frequency of the variants was evaluated with gnomAD (v2.1.1) aggregated allele frequency. Aggregated scores from the in silico prediction tools, including Polyphen, SIFT, MutationTaster, Mutation Assessor, FATHMM, FITCONS, GENOCANYON, dbscSNV ADA, dbscSNV RF, were used to predict the effects of missense variants. A score of 0.7 was assigned for the deleterious impact, and scores less than 0.15 were used as a benign threshold. For the splice variants, SpliceAI with the upper and lower thresholds of 0.7 and 0.2 respectively was used. Previous submissions in ClinVar were considered in the variant assessment. Additionally, online databases such as PubMed, OMIM, Orphanet, and GeneReviews were utilized to evaluate gene-phenotype association.
3. Results
3.1. Clinical Findings
A two-year-old female patient has been admitted to the Department of Pediatrics, University Hospital Centre Zagreb, due to neurodevelopmental delay, congenital muscular dystrophy, severe central early-onset obesity without hyperphagia, distinctive facial features and a prenatal history of intrauterine growth restriction (IUGR), oligohydramnios, and decreased fetal movements (Figure1). The postnatal physical findings of birth weight (2970 g), length (48 cm), and head circumference (32.5 cm), all below the 3rd percentile, were compliant with intrauterine growth restriction. In the physical examination, metatarsus varus and equinovarus were inspected on the right and left foot, respectively. Characteristic dysmorphic and clinical features consistent with PWS were detected.
The neuromuscular abnormalities were present shortly after birth through weak cry and poor suck, severe central generalized muscular hypotonia, and absence of deep tendon reflexes. Over the years, the motor milestones were delayed (unsupported sitting at 10 months, standing with support at 22 months, and walking at 30 months). The lack of improvement in hypotonia and deep tendon reflexes was unusual for PWS. Serum creatinine kinase (CK) level was elevated to 5X, a finding not consistent with PWS. Generalized tonic-clonic epileptic seizures with loss of consciousness presented at 3 years of age. CK level was continuously increasing and was elevated to 10X. Magnetic resonance imaging (MRI) of skeletal proximal and distal muscle groups of the lower extremities showed signs of muscle wasting in the proximal extremities and relative sparing of muscles in the distal muscle groups (Figure 2).
Additionally, unlike typical PWS, severe central obesity was prominent at 19 months beyond the PWS’s hyperphagic nutritional phase 2b onset (BW 13 kg, BMI 20.8 kg/m2) (Figure 1). Both, body weight for height and BMI had early-onset and rapid gain, which was unusual for PWS, particularly with the absence of hyperphagia in the proband.
3.2. Genomic Findings
Karyotyping and Methylation Analysis
G-banded karyotype analysis was unremarkable, showing a 46,XX finding. MS-MLPA analysis showed an aberrant methylation profile in the PWS region. It determined a hypermethylation pattern in SNRPN and MAGEL genes. The methylation level was 100% in these genes. No copy number variations (CNVs) were identified (Supplementary Figure S1). These results were consistent with PWS diagnosis. MS-MLPA could not delineate the causative molecular events for PWS including possible imprinting defects by epimutation or UPD. Additionally, the absence of deep tendon reflexes, prolonged hypotonia and elevated CK could not be explained by this finding. A comprehensive joint WGS was performed.
3.3. Ascertainment of UPD by Trio-WGS
Joint analysis of the PWS critical region in 15q11.2-q13 (MKRN3, MAGEL2, NDN, PWRN1, NPAP1, SNURF, SNRNP, SNORD107, SNORD64, SNORD108, SNORD109A, SNORD116, IPW, SNORD115, SNORD109B, ATP10A, GABRB3, GABRA5, GABRG3, OCA2, HERC2, APBA2, CHRNA7) showed maternal-only contribution in the proband. Variants in these regions were heterozygous in the mother and homozygous in the proband.
We set to investigate the extent of maternal contribution in 15q by performing a global UPD- ROH mapping in 15q to delineate the boundaries of isodisomy. In the proband, a continuous ROH was identified on chromosome 15 from 15q11.2 to 15q22.2 (Figure 3, Figure 4). The isodisomy consisting of 38.6 Mb in this region was shown by the complete mapping of ROH with UPD with only maternal contribution (Table 1, Figure 4). Telomeric to 15q22.2, the ROH were detected (Figure 5). The UPD-ROH mapping of this 41.55 Mb region, was consistent with heterodisomy, and the inheritance pattern showed this region was also maternal only (Table 1). Variant level zygosity is shown for 25,091 variants of a selected 270 OMIM genes within 15q that included the PWS region of 15q11.2-q13 (Supplementary Table 1). An allele frequency filter of 1% to exclude common variants.
3.4. Muscular Dystrophy Assessment Using Trio WGS Analysis
A separate joint analysis for congenital muscular dystrophy (HP:0003741) was performed. This analysis revealed a pathogenic variant in CAPN3 (HGNC:1480) gene that is associated with calpainopathy, a rare limb-girdle muscular dystrophy (MIM#253600). The NM_000070.3(CAPN3):c.550del (p.Thr184fs) variant is located in the 15q15.1 isodisomy region (Figure 6), and was heterozygous in the mother and homozygous in the proband. CAPN3:c.550del(p.Thr184fs) variant is a pathogenic frameshift variant (ClinVar ID: 17621) and has been reported in autosomal recessive calpainopathy patients [15]. The CAPN3 gene constraint score of o/e and pLI of CAPN3 are 0.965 (0.56 - 0.96) and 0, respectively. The population allele frequency for this variant in the gnomAD database has been reported as 0.00023. CAPN3:c.550del is the most frequent pathogenic CAPN3 variant in countries such as Croatia, Russia, Turkey, and Germany [16]. The isodisomy in the 15q15.1 region resulted in the isozygosity of the CAPN3:c.550del(p.Thr184fs) variant in the proband, which in turn led to the diagnosis of calpainopathy (Figure 7).
3.5. Assessment of the breakpoint within UPD region
The breakpoint between isodisomy and heterodisomy in 15q was located at chr15:g.60984740 (Table 1, Figure 6, Supplementary Figure S2) in a noncoding region between exon 1 and 2 of RORA (HGNC:10258) gene (NM_134261.3). RORA is associated with intellectual developmental disorder with or without epilepsy or cerebellar ataxia (MIM#618060). There was no deleterious variant identified in RORA, but genomic breakpoints can often affect a genes’ function. Both PWS and RORA phenotypes are associated with seizures, and it was not clear if any potential abnormality of RORA could be contributing to the phenotype in the proband. To account for the possibility of breakpoint affecting the RORA’s function, brain magnetic resonance imaging (MRI) was performed. In individuals with heterozygous RORA loss of function, cerebellar hypoplasia and pontocerebellar atrophy are typically indicated. The evaluation of MRI in the proband was unremarkable (Figure 8). Sagittal T1-weighted image showed normal formation of the corpus callosum and shape and volume of cerebellar vermis and brainstem. The Axial T2-weighted image displays normal white matter myelinization and basal ganglia volumes.
4. Discussion
The prevalence of UPD conditions is reportedly 1 in 3,500 [17]. Since the first description of UPD in the etiology of cystic fibrosis in 1988, there have been many studies of autosomal recessive diseases associated with UPD [18,19,20]. Co-occurrence of PWS with Tay-Sacs disease, Bloom syndrome, congenital ichthyosis, and hereditary spastic paraplegia type 11 has been reported [21,22,23,24]. These conditions were associated with UPD, therefore can also potentially be present in association with Angelman Syndrome (AS), depending on the parental contributing of the isodisomy.
To our knowledge, this is the first report of PWS co-occurrence with calpainopathy. Herein patient’s signature characteristics of PWS comprising of facial dysmorphology, muscular hypotonia, and early poor weight gain followed by severe central early-onset obesity [25,26,27] were molecularly confirmed by the MS-MLPA. However, severe obesity with an earlier onset than expected, persistent muscular hypotonia with elevated CK, and absence of deep tendon reflexes were atypical for PWS. Trio WGS analysis identified a pathogenic CAPN3:c.550del homozygous variant within the isodisomy region.
CAPN3 associated calpainopathy is a rare limb-girdle muscular dystrophy with a prevalence of 1:100000 [9,10]. As a member of the calpain family, CAPN3 has a key role in muscle cell survival, motility, and skeletal plasticity [28,29,30]. Pathogenic variants and loss of function of CAPN3 result in impairment of muscle adaptation and regeneration, and an increase in myonuclear apoptosis [31]. The cumulative muscle atrophy associated with fibrous and adipose tissue hyperplasia leads to calpainopathy. The range for age of symptoms onset is highly variable [10]. The elevated CK level in young patients is consistent with the early stage of calpainopathy in our patient.
The CAPN3:c.550del variant is reportedly a founder variant in European populations [32,33]. Among known CAPN3 variants, the c.550del is the most prevalent variant in Croatia [33], and has been frequently reported in patients with calpainopathy from countries across Europe [16,32,33,34]. Haplotype analyses have suggested that this variant is rooted in ancestral chromosomes from the ancient Mediterranean population [16,35,36]. The CAPN3:550del variant has been reported by several studies [33,36] in patients in the eastern Mediterranean region with suspected calpainopathy. Based on the finding of this study herein, and the prevalence of CAPN3:550del, testing of this variant is strongly advised in patients with PWS or AS.
Our genome analysis revealed a maternal segmental isodisomy in the PWS region along with heterodisomy in more telomeric region of chromosome 15. The presence of heterodisomy and isodisomy have been reported in 15q, often due to recombination event prior to the chromosome non-disjunction (NDJ) event [37]. After the fertilization of these gametes, postzygotic trisomy rescue preserves the disomy state [38,39]. In segmental UPD cases, homozygosity is often observed in the centromeric, and heterodisomy in telomeric regions [40].
The breakpoint between maternal isodisomy and heterodisomy was located in the noncoding region of the RORA gene. RORA is associated with autosomal dominant intellectual developmental disorder with or without epilepsy or cerebellar ataxia (MIM#618060) [41]. The clinical findings of RORA associated disorder have variable phenotype and include intellectual impairment at varying degrees from normal to severe, speech delay, seizures at multiple types, strabismus, delayed motor development and walking, ataxia, hypotonia, poor coordination and/or mild tremor, hypoplastic cerebellum, and pontocerebellar atrophy [41]. In the proband herein, the phenotypical features expected from RORA associated disorder, namely seizures, developmental delay, speech delay, hypotonia, and broad-based gait overlapped with PWS or calpainopathy phenotypes. However, abnormal RORA is reportedly associated with a distinctive brain MRI pattern of cerebellar hypoplasia and pontocerebellar atrophy in some patients. To date, MRI was unremarkable in our patient as shown Figure 4. Age related penetrance or phenotypic variability may present confounding factors. Future studies are required to examine the potential functional impact of the breakpoint in the RORA gene.
In conclusion, we report the first co-occurrence of PWS and calpainopathy in a patient with complex phenotype. Joint WGS analysis effectively identified the underlying genetic causes of these unrelated genomic conditions. Despite the rarity of CAPN3 calpainopathy, but given the high prevalence of CAPN3:c.550del variant, homodisomy of CAPN3 recessive alleles should be evaluated in patients with unexpected muscle abnormalities with PWS or AS.
Conflicts of Interests
The authors declare that they have no competing interests.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) of the proband with PWS. The upper panel shows copy number, and the bottom panel shows the methylation profile. In both panels, each black circle and blue rectangle represents the final probe ratio of the proband and reference sample, respectively. No copy number change was observed in the proband in comparison to the reference sample; Figure S2: The integrative genomics viewer (IGV) of the isodisomy and heterodisomy breakpoint at chr15:g.60984740 in the WGS data. The upper panel shows the mother`s zygosity at chr15:g.60984740 and chr15:g.60984741. The bottom pane shows proband`s zygosity at the same coordinate.
Author Contributions
Conceptualization: A.A.G., M.C.; Funding acquisition: A.A.G., M.C.; Investigation: A.A.G., M.C., B.U., A.B., C.P.H., M.W., F.A., R.B., K.C.G., D.O., Q.N., H.V.R., M.P.; Methodology: A.A.G., M.C., B.U., H.V.R., Q.N., R.B.; Supervision: A.A.G.; Visualization: A.A.G., M.C., B.U., A.B., K.C.G., D.O., Q.N., H.V.R., M.P.; Writing - original draft: A.A.G., M.C., B.U., A.B.; Review & editing: A.A.G., M.C., B.U., A.B., C.P.H., M.W., F.A., R.B., K.C.G., D.O., Q.N., H.V.R., M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This investigation was done in collaboration with Brigham and Women’s Hospital, Inc / University Hospital Centre Zagreb, with funding support provided by Mila za Sve Foundation.
Institutional Review Board Statement
Institutional Review Board at BWH and University Hospital Centre Zagreb approved this study (Class: 8.1-21/6-2; Reg. No.: 02/21 AG). Written informed consent was obtained from all the participants at the University Hospital Centre Zagreb, Croatia. This study is compliant with the General Data Protection Regulation (GDPR), approved by BWH, Mila Za Sve Foundation, and the University Hospital Centre Zagreb, Croatia.
Data Availability Statement
Data for this manuscript are subject to BWH institutional and GDPR privacy policies and restricted from inclusion in repositories. They may be available upon request.
Acknowledgments
We thank our patient and their family for their participation in the CROseq Genome Program and for permitting us to anonymously share the clinical details.
References
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Akdemir, Z.H.C.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. New Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Rosenfeld, J.A.; James, R.A.; Bainbridge, M.; Niu, Z.; Wang, X.; Dhar, S.; Wiszniewski, W.; Akdemir, Z.H.C.; Gambin, T.; et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Anesthesia Analg. 2015, 18, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Knapp A, Jagła M, Madetko-Talowska A, et al. Paternal uniparental disomy of chromosome 2 resulting in a concurrent presentation of Crigler-Najjar syndrome type I and long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Am J Med Genet A. 2022;188(6):1848-1852.
- Ohtsuka, Y.; Higashimoto, K.; Sasaki, K.; Jozaki, K.; Yoshinaga, H.; Okamoto, N.; Takama, Y.; Kubota, A.; Nakayama, M.; Yatsuki, H.; et al. Autosomal recessive cystinuria caused by genome-wide paternal uniparental isodisomy in a patient with Beckwith–Wiedemann syndrome. Clin. Genet. 2014, 88, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Benko, W.S.; Hruska, K.S.; Nagan, N.; Goker-Alpan, O.; Hart, P.S.; Schiffmann, R.; Sidransky, E. UNIPARENTAL DISOMY OF CHROMOSOME 1 CAUSING CONCURRENT CHARCOT-MARIE-TOOTH AND GAUCHER DISEASE TYPE 3. Neurology 2008, 70, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Cassidy SB, Forsythe M, Heeger S, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet. 1997;68(4):433-440.
- Gillessen-Kaesbach, G.; Robinson, W.; Lohmann, D.; Kaya-Westerloh, S.; Passarge, E.; Horsthemke, B. Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum. Genet. 1995, 96, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; Knoll, J.H.M.; Butler, M.G.; Karam, S.; Lalande, M. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader-Willi syndrome. Nature 1989, 342, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Hata, S.; Ono, Y. Expanding Members and Roles of the Calpain Superfamily and Their Genetically Modified Animals. Exp. Anim. 2010, 59, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Adam MP, Mirzaa GM, Pagon RA, et al. GeneReviews. In:1993.
- Nascimbeni, A.C.; Fanin, M.; Tasca, E.; Angelini, C. Transcriptional and translational effects of intronic CAPN3 gene mutations. Hum. Mutat. 2010, 31, E1658–E1669. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.-L.; Angelini, C.; Daentl, D.; Garcia, C.; Greco, C.; Hausmanowa–Petrusewicz, I.; Fidzianska, A.; Wessel, H.; Hoffman, E. Calpain III mutation analysis of a heterogeneous limb–girdle muscular dystrophy population. Neurology 1999, 52, 1015–1015. [Google Scholar] [CrossRef] [PubMed]
- Zatz, M.; Vainzof, M.; Passos-Bueno, M.R. Limb-girdle muscular dystrophy: one gene with different phenotypes, one phenotype with different genes. Curr. Opin. Neurol. 2000, 13, 511–517. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Brenguier, L.; Dinçer, P.; Roudaut, C.; Bady, B.; Burgunder, J.M.; Chemaly, R.; A Garcia, C.; Halaby, G.; E Jackson, C.; et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. . 1997, 60, 1128–38. [Google Scholar] [PubMed]
- Nakka, P.; Smith, S.P.; O’donnell-Luria, A.H.; McManus, K.F.; Mountain, J.L.; Ramachandran, S.; Sathirapongsasuti, J.F.; Agee, M.; Auton, A.; Bell, R.K.; et al. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 2019, 105, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Polonis, K.; Lopes, J.L.; Cabral, H.; Babcock, H.E.; Kline, L.; Ruiz, K.M.; Schwartz, S.; Hasadsri, L.; Rowsey, R.A.; Hoppman, N.L. Uniparental disomy of multiple chromosomes in two cases with a complex phenotype. Am. J. Med Genet. Part A 2023, 191, 1978–1983. [Google Scholar] [CrossRef]
- Molloy, B.; Jones, E.R.; Linhares, N.D.; Buckley, P.G.; Leahy, T.R.; Lynch, B.; Knerr, I.; King, M.D.; Gorman, K.M. Uniparental disomy screen of Irish rare disorder cohort unmasks homozygous variants of clinical significance in the TMCO1 and PRKRA genes. Front. Genet. 2022, 13, 945296. [Google Scholar] [CrossRef]
- Chien, S.-C.; Chen, C.-P.; Liou, J.-D. Prenatal diagnosis and genetic counseling of uniparental disomy. Taiwan. J. Obstet. Gynecol. 2022, 61, 210–215. [Google Scholar] [CrossRef]
- Kunta, A.R.; Jueng, J.; Jordan, C.; Kojic, J.; Mo, A.; Ebrahimi-Fakhari, D. Blended Phenotype of Prader-Willi Syndrome and HSP- SPG11 Caused by Maternal Uniparental Isodisomy. Neurol. Genet. 2022, 8, e200041. [Google Scholar] [CrossRef]
- Muthusamy, K.; Macke, E.L.; Klee, E.W.; Tebben, P.J.; Hand, J.L.; Hasadsri, L.; Marcou, C.A.; Schimmenti, L.A. Congenital ichthyosis in Prader-Willi syndrome associated with maternal chromosome 15 uniparental disomy: Case report and review of autosomal recessive conditions unmasked byUPD. Am. J. Med Genet. Part A 2020, 182, 2442–2449. [Google Scholar] [CrossRef]
- Woodage, T.; Prasad, M.; Dixon, J.W.; E Selby, R.; Romain, D.R.; Columbano-Green, L.M.; Graham, D.; Rogan, P.K.; Seip, J.R.; Smith, A. Bloom syndrome and maternal uniparental disomy for chromosome 15. . 1994, 55, 74–80. [Google Scholar]
- Zeesman, S.; McCready, E.; Sadikovic, B.; Nowaczyk, M.J. Prader–Willi syndrome and Tay–Sachs disease in association with mixed maternal uniparental isodisomy and heterodisomy 15 in a girl who also had isochromosome Xq. Am. J. Med Genet. Part A 2014, 167, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J. Nutritional phases in Prader–Willi syndrome. Am. J. Med Genet. Part A 2011, 155, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Butler MG, Sturich J, Myers SE, Gold JA, Kimonis V, Driscoll DJ. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J Assist Reprod Genet. 2009;26(8):461-466.
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Baghdiguian S, Martin M, Richard I, et al. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat Med. 1999;5(5):503-511.
- Sorimachi, H.; Kinbara, K.; Kimura, S.; Takahashi, M.; Ishiura, S.; Sasagawa, N.; Sorimachi, N.; Shimada, H.; Tagawa, K.; Maruyama, K.; et al. Muscle-specific Calpain, p94, Responsible for Limb Girdle Muscular Dystrophy Type 2A, Associates with Connectin through IS2, a p94-specific Sequence. J. Biol. Chem. 1995, 270, 31158–31162. [Google Scholar] [CrossRef] [PubMed]
- Taveau, M.; Bourg, N.; Sillon, G.; Roudaut, C.; Bartoli, M.; Richard, I. Calpain 3 Is Activated through Autolysis within the Active Site and Lyses Sarcomeric and Sarcolemmal Components. Mol. Cell. Biol. 2003, 23, 9127–9135. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tang, F.; Gao, H.; Zhang, X.; Li, X.; Xiao, D. CAPN3: A muscle-specific calpain with an important role in the pathogenesis of diseases (Review). Int. J. Mol. Med. 2021, 48, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Zheng, Y.; Zhao, Z.; Lin, P.; Xi, J.; Zhu, W.; Lin, J.; Lu, J.; Yu, M.; Zhang, W.; et al. Molecular landscape of CAPN3 mutations in limb-girdle muscular dystrophy type R1: from a Chinese multicentre analysis to a worldwide perspective. J. Med Genet. 2020, 58, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Canki-Klain, N.; Milic, A.; Kovac, B.; Trlaja, A.; Grgicevic, D.; Zurak, N.; Fardeau, M.; Leturcq, F.; Kaplan, J.; Urtizberea, J.A.; et al. Prevalence of the 550delA mutation in calpainopathy (LGMD 2A) in Croatia. Am. J. Med Genet. Part A 2003, 125A, 152–156. [Google Scholar] [CrossRef]
- Topaloǧlu, H.; Dinçer, P.; Richard, I.; Akçören, Z.; Alehan, D.; Özme. ; Çaǧlar, M.; Karaduman, A.; Urtizberea, J.A.; Beckmann, J.S. Calpain-3 Deficiency Causes a Mild Muscular Dystrophy in Childhood. Neuropediatrics 1997, 28, 212–216. [Google Scholar] [CrossRef]
- Dincer, P.; Leturcq, F.; Richard, I.; Piccolo, F.; Yalnizoàlu, D.; De Toma, C.; Akçören, Z.; Broux, O.; Deburgrave, N.; Ba, L.B.; et al. A biochemical, genetic, and clinical survey of autosomal recessive limb girdle muscular dystrophies in Turkey. Ann. Neurol. 1997, 42, 222–229. [Google Scholar] [CrossRef]
- Pogoda TV, Krakhmaleva IN, Lipatova NA, Shakhovskaya NI, Shishkin SS, Limborska SA. High incidence of 550delA mutation of CAPN3 in LGMD2 patients from Russia. Hum Mutat. 2000;15(3):295.
- Yamazawa, K.; Ogata, T.; Ferguson-Smith, A.C. Uniparental disomy and human disease: An overview. Am. J. Med Genet. Part C: Semin. Med Genet. 2010, 154C, 329–334. [Google Scholar] [CrossRef] [PubMed]
- del Gaudio, D.; Shinawi, M.; Astbury, C.; Tayeh, M.K.; Deak, K.L.; Raca, G. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2020, 22, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Benn, P. Uniparental disomy: Origin, frequency, and clinical significance. Prenat. Diagn. 2020, 41, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K.E.; Hawley, R.S.; Sherman, S.; Hassold, T. Recombination and nondisjunction in humans and flies. Hum. Mol. Genet. 1996, 5, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Guissart, C.; Latypova, X.; Rollier, P.; Khan, T.N.; Stamberger, H.; McWalter, K.; Cho, M.T.; Kjaergaard, S.; Weckhuysen, S.; Lesca, G.; et al. Dual Molecular Effects of Dominant RORA Mutations Cause Two Variants of Syndromic Intellectual Disability with Either Autism or Cerebellar Ataxia. Am. J. Hum. Genet. 2018, 102, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLOS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Yin, T.; Cook, D.; Lawrence, M. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 2012, 13, R77–R77. [Google Scholar] [CrossRef]
Figure 1.
Initial clinical presentation of the patient and transforming effect of therapy from A-G. Collectively, photographs are shown according to age of the patient, from age 19 to 35 months and corresponding nutritional phases 1b and 2a. A-C) Early-onset, non-hyperphagic obesity in the proband on at age of 19 months (Body weight 13 kg, body height 79 cm, BMI 20.8 kg/m2). D) At age 21 months, growth hormone, diet and physical habilitation therapy was introduced. After introducing growth hormone, diet and physical habilitation therapy, BMI is progressively decreased at age of 21 months (D), 24 months (E), and 30 months (F) and 35 months of age (G) and nutritional phase 2a (14 months after therapy introduction).
Figure 1.
Initial clinical presentation of the patient and transforming effect of therapy from A-G. Collectively, photographs are shown according to age of the patient, from age 19 to 35 months and corresponding nutritional phases 1b and 2a. A-C) Early-onset, non-hyperphagic obesity in the proband on at age of 19 months (Body weight 13 kg, body height 79 cm, BMI 20.8 kg/m2). D) At age 21 months, growth hormone, diet and physical habilitation therapy was introduced. After introducing growth hormone, diet and physical habilitation therapy, BMI is progressively decreased at age of 21 months (D), 24 months (E), and 30 months (F) and 35 months of age (G) and nutritional phase 2a (14 months after therapy introduction).
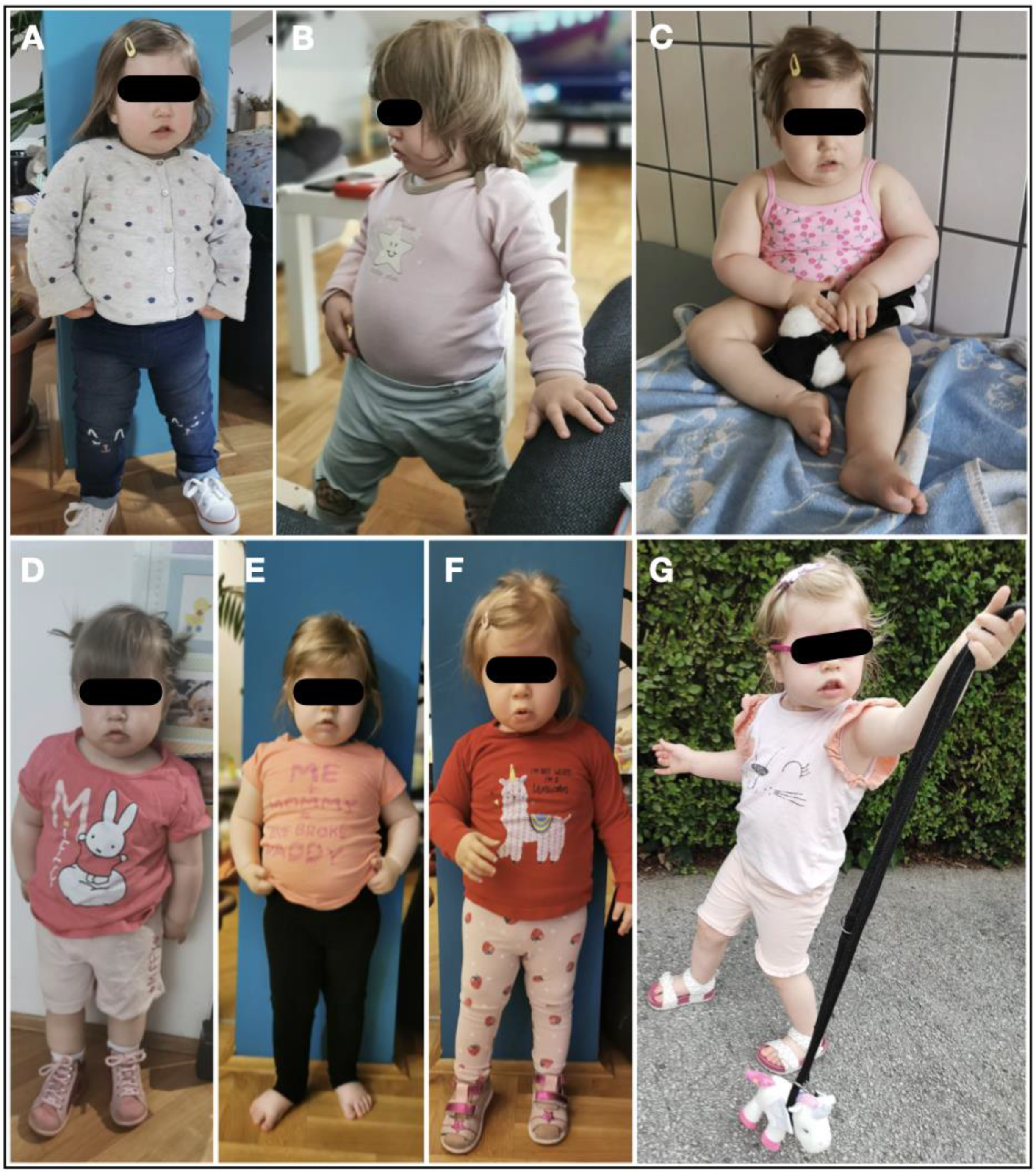
Figure 2.
Magnetic resonance imaging of skeletal muscles (MRI). Axial T1-weighted images of proximal right and left (a) and distal right and left (b) muscle groups of the lower extremities showed minimal signs of fatty replacement of quadriceps muscles and relative sparing of muscles belonging to the distal muscle groups.
Figure 2.
Magnetic resonance imaging of skeletal muscles (MRI). Axial T1-weighted images of proximal right and left (a) and distal right and left (b) muscle groups of the lower extremities showed minimal signs of fatty replacement of quadriceps muscles and relative sparing of muscles belonging to the distal muscle groups.
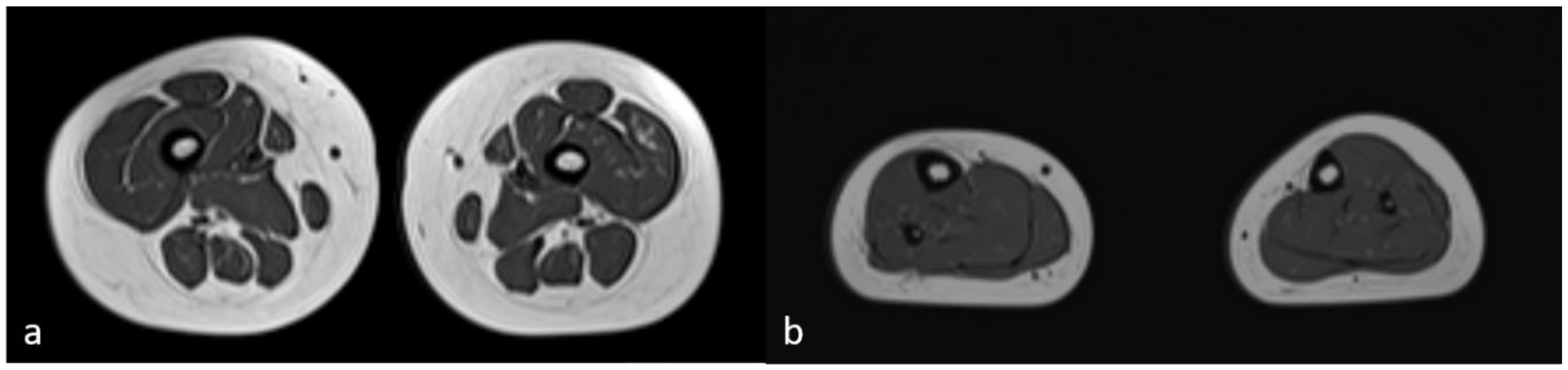
Figure 3.
Regions of homozygosity (ROH) in chromosome 15q using joint whole genome sequencing (WGS) data. The red region in the ideogram corresponds to chr15:g.22373341-g.102531392. The reference sequence (RefSeq) and variant call format (VCF) tracks are depicted below. The proband and mother’s zygosity tracks show the region of homozygosity (ROH) in the proband. The proband`s ROH is continuous within the maternal isodisomy contributed segment.
Figure 3.
Regions of homozygosity (ROH) in chromosome 15q using joint whole genome sequencing (WGS) data. The red region in the ideogram corresponds to chr15:g.22373341-g.102531392. The reference sequence (RefSeq) and variant call format (VCF) tracks are depicted below. The proband and mother’s zygosity tracks show the region of homozygosity (ROH) in the proband. The proband`s ROH is continuous within the maternal isodisomy contributed segment.
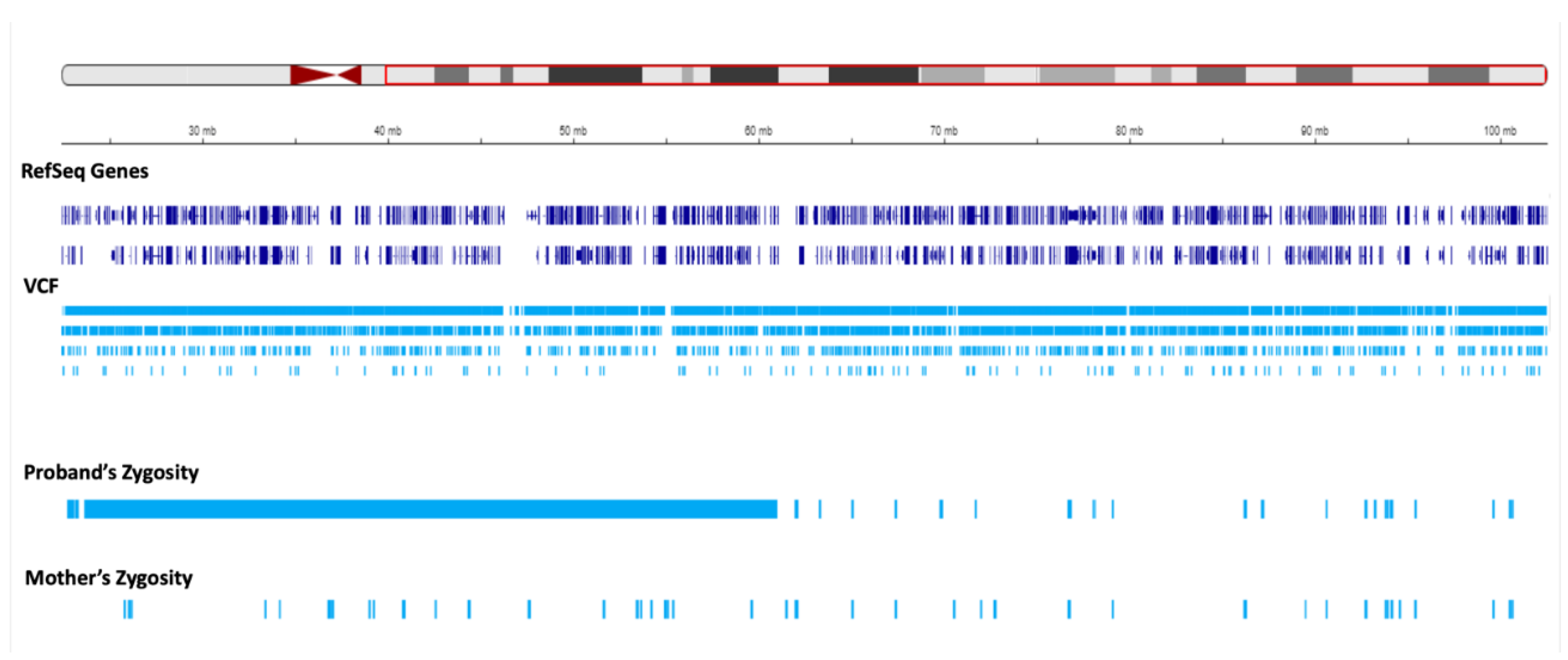
Figure 4.
Status of zygosity in the chr15:22373341-60984740 region. The red region in the ideogram shows the analysis region. The reference sequence (RefSeq) and variant call format (VCF) tracks are depicted below. The proband and mother’s zygosity tracks show homozygosity and heterozygosity status, respectively, resulting in segmental isodisomy in the proband.
Figure 4.
Status of zygosity in the chr15:22373341-60984740 region. The red region in the ideogram shows the analysis region. The reference sequence (RefSeq) and variant call format (VCF) tracks are depicted below. The proband and mother’s zygosity tracks show homozygosity and heterozygosity status, respectively, resulting in segmental isodisomy in the proband.
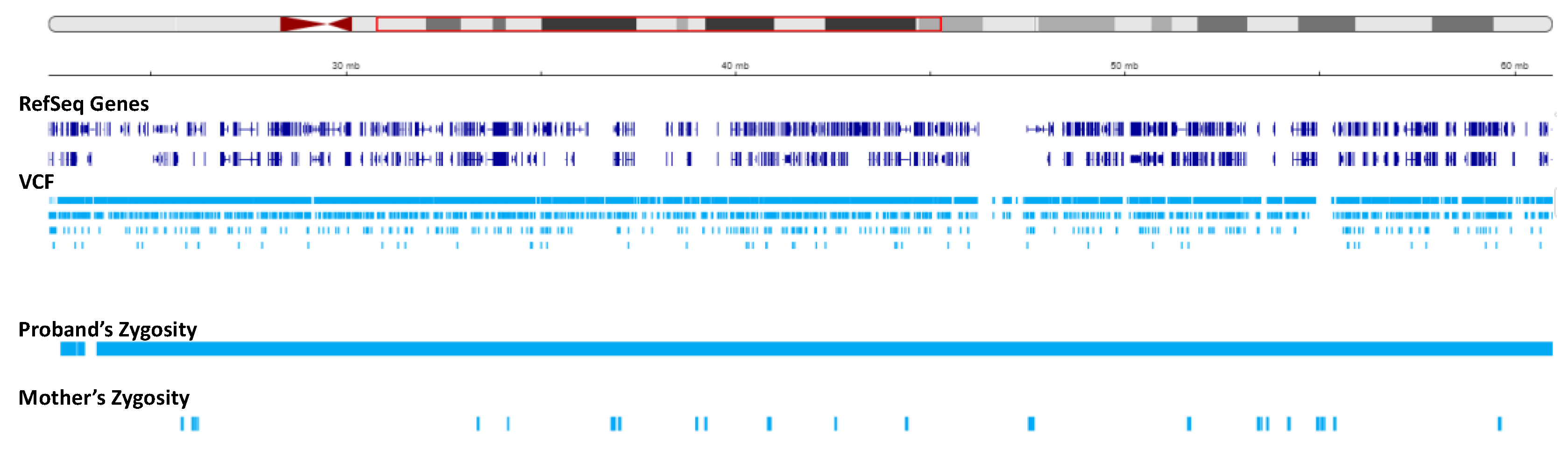
Figure 5.
Status of zygosity in the chr15:60984741-102531392 region. The red region in the ideogram shows the analysis region. The reference sequence (RefSeq) and variant call format (VCF) tracks are depicted below. The proband and mother’s zygosity tracks both show heterozygosity status in this region, corresponding to the segmental heterodisomy in the proband.
Figure 5.
Status of zygosity in the chr15:60984741-102531392 region. The red region in the ideogram shows the analysis region. The reference sequence (RefSeq) and variant call format (VCF) tracks are depicted below. The proband and mother’s zygosity tracks both show heterozygosity status in this region, corresponding to the segmental heterodisomy in the proband.
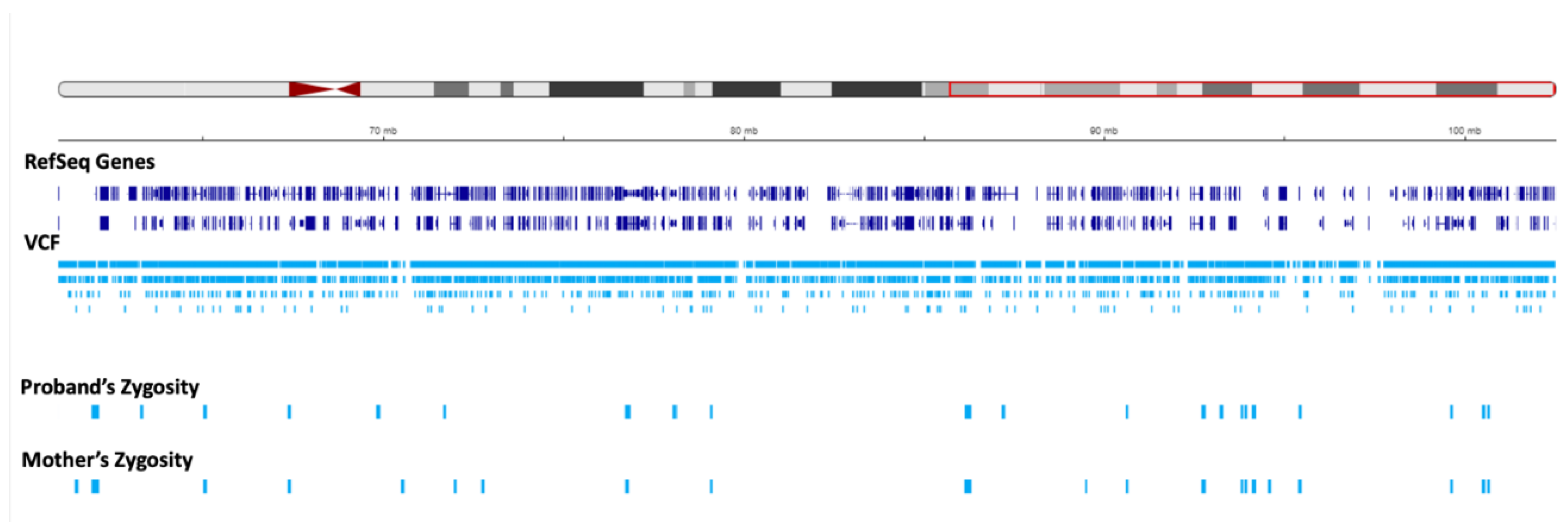
Figure 6.
Ideogram of chromosome 15 and the schematic map of maternal uniparental disomy. The maternal isodisomy and heterodisomy contributions are coded with blue and green colors, respectively. The orange rectangular box highlights Angelman Syndrome (AS) and Prader-Willi Syndrome (PWS) critical regions. The imprinting center (IC) is located in this region. In the isodisomy contribution, CAPN3 is marked in red. The breakpoint between isodisomy and heterodisomy occurs at RORA. The blue box illustrates the part of RORA within the maternal isodisomy region, and the green one shows the region with maternal heterodisomy. The template ideogram has been generated in R version 4.2.2 with GenomicRanges and ggbio packages [42,43] based on UCSC hg 19.
Figure 6.
Ideogram of chromosome 15 and the schematic map of maternal uniparental disomy. The maternal isodisomy and heterodisomy contributions are coded with blue and green colors, respectively. The orange rectangular box highlights Angelman Syndrome (AS) and Prader-Willi Syndrome (PWS) critical regions. The imprinting center (IC) is located in this region. In the isodisomy contribution, CAPN3 is marked in red. The breakpoint between isodisomy and heterodisomy occurs at RORA. The blue box illustrates the part of RORA within the maternal isodisomy region, and the green one shows the region with maternal heterodisomy. The template ideogram has been generated in R version 4.2.2 with GenomicRanges and ggbio packages [42,43] based on UCSC hg 19.
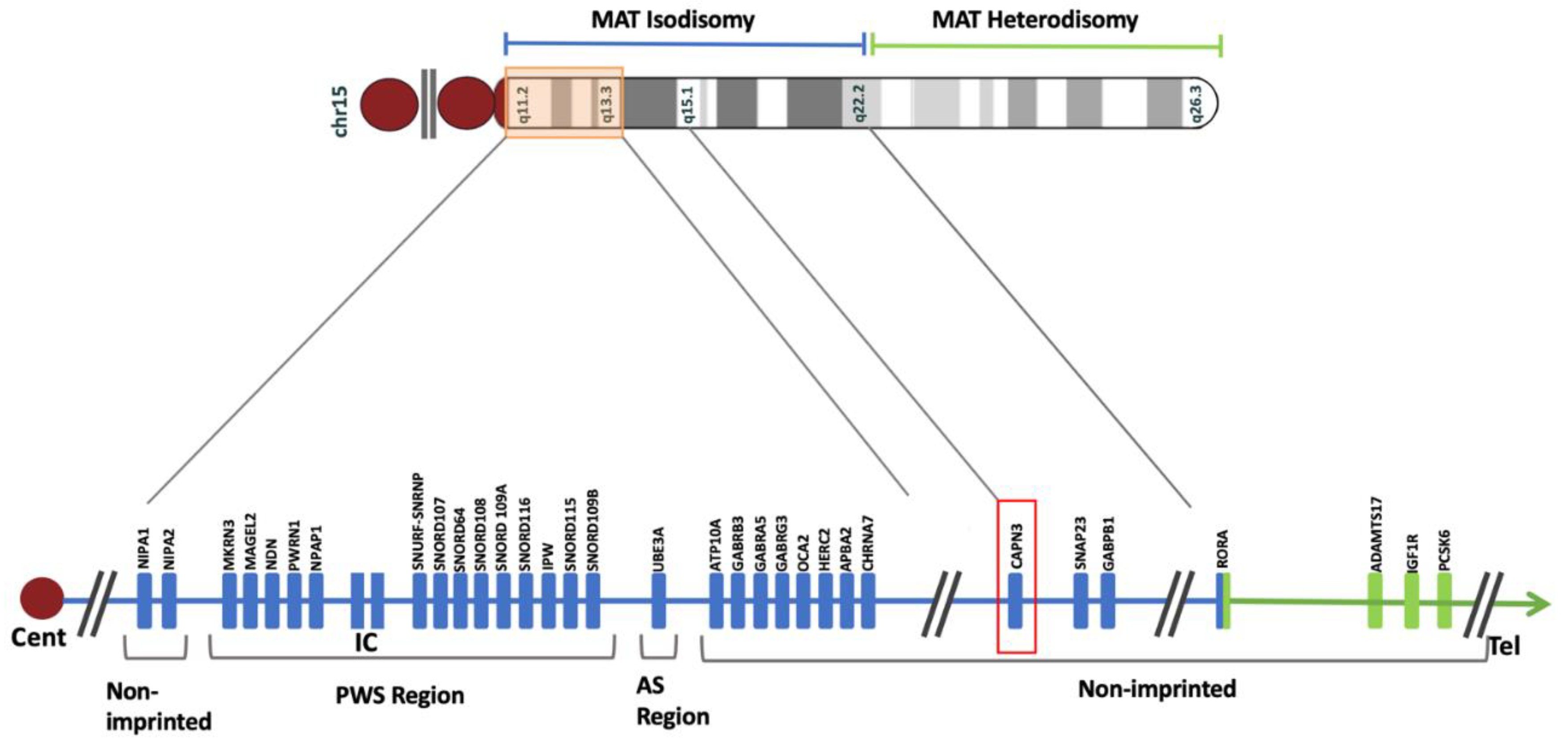
Figure 7.
The pedigree of the family schematically demonstrating the homodisomy of the maternal NM_000070.3(CAPN3):c.550del (p.Thr184fs) variant in the proband. Circles and squares represent female and male family members, respectively. The arrow indicates the proband diagnosed with Prader-Willi syndrome and Calpainopathy. The blue and green ideograms present the parent of origin (maternal in blue and paternal in green). The red mark highlights the location of the CAPN3:c.550del variant.
Figure 7.
The pedigree of the family schematically demonstrating the homodisomy of the maternal NM_000070.3(CAPN3):c.550del (p.Thr184fs) variant in the proband. Circles and squares represent female and male family members, respectively. The arrow indicates the proband diagnosed with Prader-Willi syndrome and Calpainopathy. The blue and green ideograms present the parent of origin (maternal in blue and paternal in green). The red mark highlights the location of the CAPN3:c.550del variant.
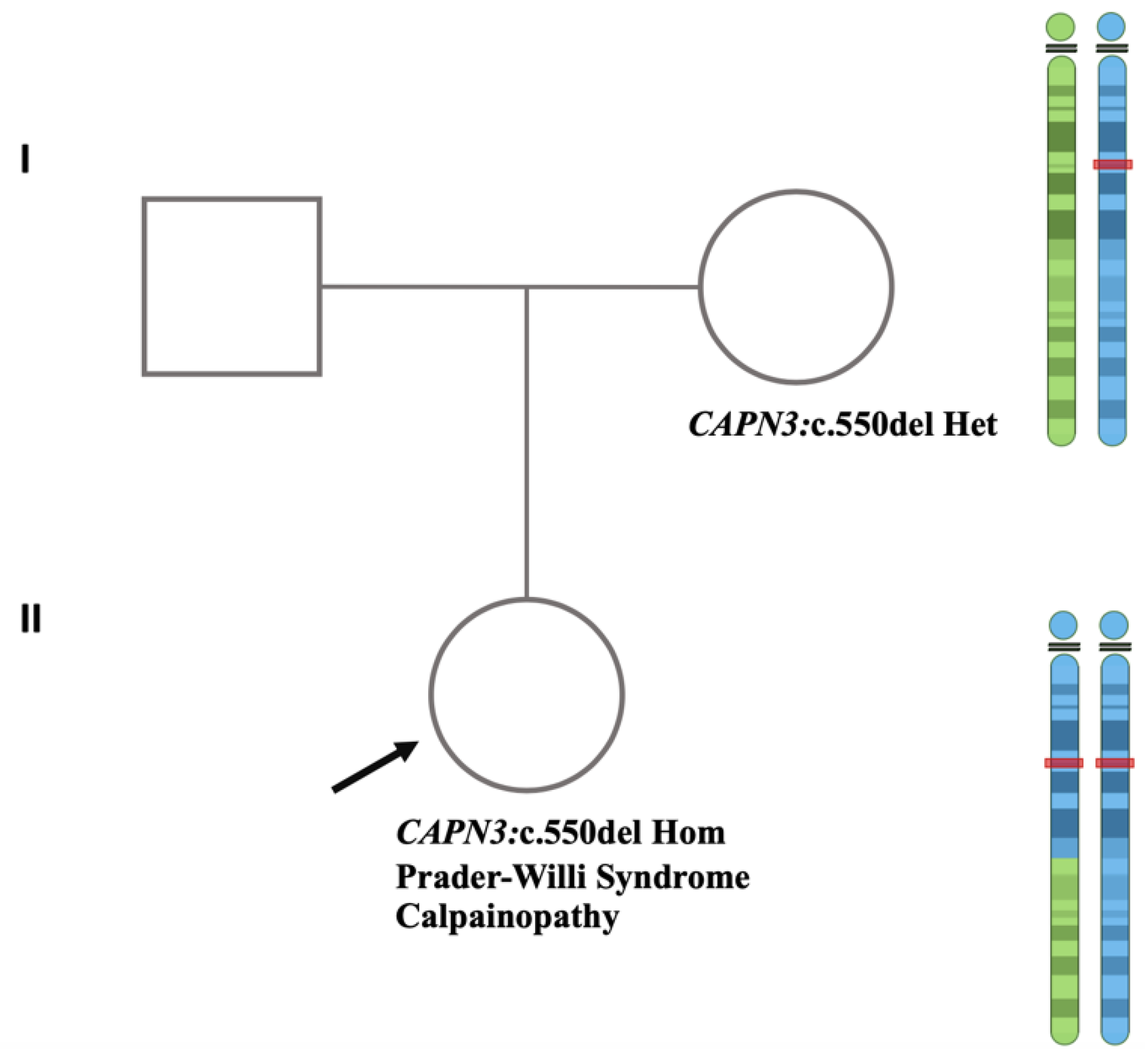
Figure 8.
Brain MRI. a) Sagittal T1-weighted image showing complete formation of the corpus callosum and normal shape and volume of cerebellar vermis and brainstem. b) Axial T2-weighted image displays adequate white matter myelinization and normal basal ganglia volumes.
Figure 8.
Brain MRI. a) Sagittal T1-weighted image showing complete formation of the corpus callosum and normal shape and volume of cerebellar vermis and brainstem. b) Axial T2-weighted image displays adequate white matter myelinization and normal basal ganglia volumes.
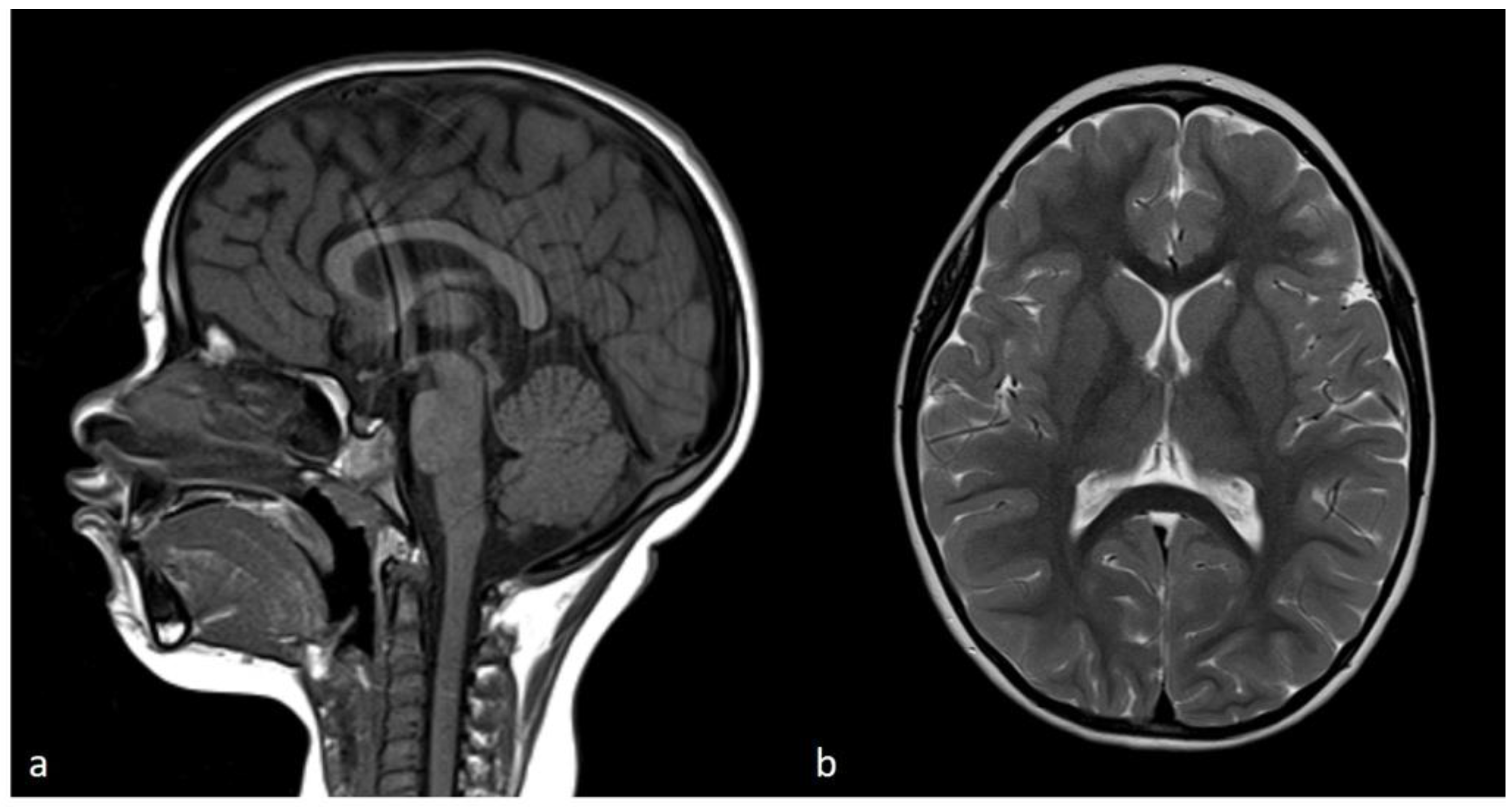

Table 1.
Disomy distribution shows that UPD identified in the proband is segmental isodisomy.
| Cytoband | Size | Location | UPD Type |
|---|---|---|---|
| 15q11.2-15q22.2 | 38.61 Mbp | Chr15: 22373341-60984740 | Mat Isodisomy |
| 15q22.2-15q26.3 | 41.55 Mbp | Chr15: 60984741-102531392 | Mat Heterodisomy |
Chr:Chromosome, UPD:Uniparental Disomy, Mat:Maternal.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



