Submitted:
12 June 2024
Posted:
13 June 2024
You are already at the latest version
Abstract
The dynamic equilibrium of iron homeostasis has an important role in sustaining metabolic and neurological functions during human life. Iron excretion is modest and unregulated hence this overall equilibrium is tightly regulated in normal conditions by increasing deficient or limiting excessive iron assimilation. Here we focus on neural tissue overload, dyshomeostasis that is preceded by excessive assimilation, disturbances in internal homeostasis or both. Normal steps in iron homeostasis include its uptake across the apical membrane of the enterocytes in a mildly acidic duodenal milieu by the major importer of ferrous iron, Divalent Metal Transporter 1 (DMT1) (Nramp2/DCT1/SLC11A2, solute carrier family 11 member 2) [1, 2], responsible for the uptake of Non-Transferrin Bound Iron (NTBI) and divalent metals [3], passage through enterocytes bound to a chaperone and crossing the basal membrane via the only known cellular ferrous iron exporter, ferroportin [4] with the iron bound to transferrin (Tf) as it exits. Cells in peripheral tissues largely utilize circulating iron after uptake by receptor-mediated endocytosis of Tf-bound Iron [5].
Derangement of duodenal NTBI absorption of iron and heavy metals is influenced by systemic iron metabolism associated with inflammation that leads to iron accumulation into peripheral tissues and potential development of neurodegenerative diseases, involving distribution and fine regulation of DMT1 also in the central nervous system, where inflammatory-related acidosis influences DMT1 uptake, via its action as a proton cotransporter. Pharmacological strategies to inhibit NTBI transport selectively will be illustrated in relationship to protection, particularly neuroprotection, against iron overload associated with acidosis, inflammation, neuroinflammation and subsequent neurodegeneration.
Keywords:
extracellular acidosis
; iron metabolism
; Divalent Metal Transporter-1 (DMT1)
; neuroinflammation
; neurodegeneration
1. Introduction
Iron homeostasis is an essential prerequisite for metabolic and neurological functions throughout a healthy human life, helping to maintain the dynamic equilibrium between intracellular and systemic iron metabolism. In most cells, receptor-mediated endocytosis of transferrin (Tf), the circulating iron transport protein, is the principal delivery mechanism. Uptake of transferrin-bound Iron (TBI) is tightly regulated in normal conditions to limit excessive iron assimilation and prevent iron overload.
In enterocytes, however, dietary non-heme iron crosses their apical membrane in a mildly acidic duodenal milieu via the major importer of ferrous iron, Divalent Metal Transporter 1 (DMT1) (Nramp2/DCT1/SLC11A2, solute carrier family 11 member 2) [1,2], transits the cell aided by a chaperone [6,7], exiting the basal membrane via the only known cellular ferrous iron exporter, ferroportin (FPN, MTP1, SLC40 iron transporter1) [4,8,9]. FPN expression is down-regulated by inflammation, as mediated through posttranslational degradation by the liver-derived peptide hormone, hepcidin (HAMP) that controls systemic iron homeostasis by its interaction with FPN. Iron transport to peripheral tissues and to the brain is largely as TBI [10]. DMT1, expressed in four isoforms and transporting up to eight metals, is also responsible for a substantial portion of Non-Transferrin Bound Iron transport (NTBI) [3].
Excessive duodenal iron absorption in hereditary hemochromatosis and thalassemia major leads to NTBI entering the plasma. This redox-active, potentially toxic form of iron, ordinarily rapidly cleared from plasma, leads to iron overload and dyshomeostasis in peripheral tissues, and accompanying pathologies [10].
Noteworthy, iron accumulation via entry into enterocytes is regulated by duodenal levels of DMT1 and via exit from the enterocytes into circulation by the iron exporter FPN [11]. In the central nervous system, it has been identified on the endothelial cells of the blood-brain barrier, in neurons, in glial cells, in the choroid plexus, in ependymal cells and by ultrastructural analysis, in the presynaptic vesicles of synaptosomal preparations from rat brain, along the dendrites of Purkinje cells and in the cerebellar molecular layer [12]. FPN, the only known cellular iron-exporter, plays a remarkable role in the control of systemic iron homeostasis as its expression is down-regulated by inflammation and reflects post-translational degradation driven by hepcidin, the liver-derived peptide and iron-sensing hormone [4]. Hepcidin also controls FPN levels in neurons, astrocytes and microglial cells [13,14].
2. Acid-Sensing Molecules in Acidosis and Pathology
This review focuses on the relationship between acidosis and iron uptake in specific physiological conditions: We include how the duodenal cytochrome B reductase (DCYTB) present on the enterocytic apical membrane, operating in the mildly acidic duodenal milieu, catalyses reduction of ferric iron to ferrous, the substrate for DMT1 uptake at the same surface.
DMT1 exhibits maximal uptake of iron and divalent metals in acidic conditions as observed and in HEK293 cells [3,15,16,17,18], and in Caco2 cell lines [19]. The publications just cited for apparent pH optima also cover characterization of specific DMT1 isoforms and their function, plus divalent metal affinities and competition between metals for the uptake. Although the data can look like a pH optimum, it actually reflects proton co-transport with Fe2+ [20] as originally postulated [1].
An acidic environment also develops in the endosome during the Tf cycle via the action of a vacuolar ATPase that pumps in protons [21,22 and references therein] where the conditions aid the release of Fe3+ from Tf. Here too a membrane associated reductase analogous to DCYTB – called STEAP3 (Six-Transmembrane Epithelial Antigen of the Prostate 3) [23,24] catalyses reduction to Fe2+. One suspects that both reductases perform optimally in an acidic milieu.
Nevertheless, relative to gastro-duodenal acidosis and dietary nonheme ferrous iron (soluble in acidic conditions) absorption, we also focus attention on other molecules likely involved in maintaining electrolytic equilibria, like the acid sensing iron channels (ASICs) and the Carbonic Anhydrases (CaAs), described in cancer environments and recently studied as activators of neurodegeneration. ASICs are a family of molecules predominantly expressed in both the peripheral and central nervous system, characterized as potent proton sensors to detect extracellular acidification in the periphery and brain during injury, inflammation, ischemia, stroke, and tumours [25]. The ASIC1a isoform seems involved in several neurodegeneration and neuroinflammation pathologies, including stroke, epilepsy, and inflammatory pain [26].
Normal neuronal activity requires maintenance of cytosolic pH in the physiological range, whereas deviations sustain pathological conditions with potentially serious consequences [27] In pathological conditions like ischemic strokes, seizures, and inflammation that accompany several neurodegenerative diseases, pH shifts downward, correlated with neuronal hyperexcitability [28]. ASIC1a channels, acting as proton sensors, are highly enriched in brain neurons and activated by ischemic acidosis [29]. ASIC subunits are expressed in both plasma membranes intracellular compartments of mammalian cardiomyocytes, implying a role in the cardiomyocyte physiology; the ASIC2a/3 channel seems particularly involved as a molecular sensor of cardiac ischemic pain with its pH dependent activation [30]. Tissue acidosis exacerbates ischemic injury and significantly affects cardiac function. A study of ASIC1 variants indeed shows they are associated with cardiac and cerebrovascular ischemic injuries; specific pharmacologic inhibitors of ASIC1a have cardioprotective effect and could also mediate microvascular responses to CO2 [31].
Interestingly, multiple groups ([32] and references therein) have extensively evaluated crosstalk between acidosis and iron metabolism in terms of iron absorption, requiring an acidic environment, induced by the activity of the proton pump H(+)/K(+)ATPase (ATP4) on the gastric parietal cells. ATP4 is upregulated by hepcidin, the iron regulatory peptide, with consequent increase of gastric acidity and illustrating a connection in vivo between iron metabolism and acidosis. During acidosis, serum hepcidin was induced and its mRNA level increased in the liver but not in the stomach, another tissue where hepcidin is expressed. These changes accompany physiological downregulation of both mRNA and protein levels of intestinal DMT1 and FPN, unchanged serum iron level and Tf saturation, but significantly increased serum ferritin. An iron-rich diet results in increased ATP4A protein expression plus induction of serum, hepatic and gastric hepcidin. Hepcidin knockout (KO) mice show significant lowering of ATP4A expression, defining a reciprocal link between acidic pH and hepcidin-regulated iron balance.
Studies on CaA isoforms [33] and their catalysis of the hydration of CO2 to produce bicarbonate and a proton illustrate an important reaction for pH homeostasis that contributes to its regulation in extracellular acidification, particularly in tumour environments. Because CaAs can also contribute to the development of cerebrovascular pathologies noted in a study of FDA-approved CaA modulators [34], CaA management is a potential therapeutic approach for the treatment of Alzheimer’s disease [35].
The pathophysiology of acidosis, with an influence on inflammation, could be considered as linked to regulation of the Bcl-2 family members [36]. In fact, acidosis arises in solid and lymphoid malignancies secondary to altered nutrient supply and utilization. In lymphoma cell lines grown in acidic conditions (pH 6.5), induced apoptosis increases Bcl-2, suggesting that acidosis causes a Bcl-2 dependence, partially mediated by the acid-sensing G protein-coupled receptor, GPR65, via a MEK/ERK pathway. In this context, we mention a related mechanism involved during neuronal ischemia where the Nuclear Factor kappa B (NF-κB)/p50/RelA dimer acts as transcriptional inducer of the Bcl-2 proapoptotic family members, while the c-Rel dimers specifically promote transcription of the anti-apoptotic Bcl-xL gene with changes in the nuclear content of c-Rel dimers that strongly affect the threshold of neuron vulnerability to ischemic insult [37], a mechanism underpinning the associated DMT1 contribution to cell death, through epigenetic regulation by NF-κB/ RelA acetylation at Lysine 310 during neuronal and brain ischemia [38].
Moreover, extracellular acidification consequent to several types of stress on cells, such as hypoxia, nutrient deprivation, and cytokine inflammation, may affect the function of lysosomes with a long term dysregulation of lysosomal function not only in cancer cells [39], but also in a subclass of neurodegeneration with brain iron accumulation (NBIA) due to de novo mutations in the WDR45 gene, coding for a beta-propeller protein, involved in the initial step of the autophagic flow, so called Beta-Propeller protein-associated neurodegeneration (BPAN) with iron accumulation prevalent in the basal ganglia [40]. A cross-correlation was also found between iron metabolism and lysosomal impairment, consequent to autophagic derangement. In fact, concomitant up-regulation of DMT1 and ferrous iron was described also at the peripheral level, in the primary fibroblasts of BPAN affected patients, where ferrous iron increase was found by Turnbull’s staining during starvation, a known acidosis-dependent condition [41]. Furthermore, pathophysiology of ischemic stroke [42] now appears to include ferroptosis associated with the acidosis-dependent cardio- and cerebrovascular ischemic damage. In ischemic stroke, one sees changes in the expression of ferroptosis related genes such as for Glutathione Peroxidase 4 (GPX4), acyl-CoA long-chain fatty acids Co 4 (ACSL4) and Solute Carrier Family 7 Member 11 (SLC7A11), suggesting that ferroptosis plays a key role in ischemic stroke, raising new options for the clinical treatment of ischemic stroke.
Oxidative stress, inflammation, and mitochondrial dysfunction were among alterations associated with iron dyshomeostasis that join ferroptosis to accompany cerebral ischemic injury [43]. This paper also notes contribution from Nuclear factor erythroid 2-related factor 2 (Nrf2), a critical antioxidant transcription factor, able to coordinate various cytoprotective agents to minimize oxidative stress. Hence, targeting Nrf2 and its signalling pathway can also be a strategy to prevent damage from stroke.
Thus, we argue that the crosstalk between inflammation plus antioxidant pathways related to acidosis and iron metabolism, extensively evaluated in terms of iron absorption and facilitated by an acidic environment, influences cellular iron homeostasis, with a downstream reflection to peripheral tissues.
3. Acidosis and Iron Uptake by DMT1
Before identification of DMT1 as an importer of ferrous iron, earlier characterization of the Belgrade (b/b) rat [44,45] and the microcytic (mk/mk) mouse models [46] had led to the conclusion that their mutations severely disrupted iron homeostasis. Then two groups [1,2] published data that indicted a transporter ultimately labelled DMT1 as the intestinal importer of iron. Shortly thereafter, the latter study having identified the mk mutation as G185R, an identical mutation emerged [5] to account for the b phenotype that clearly had difficulty in cellular iron metabolism as well as GI uptake. The b mutation clearly affected ferrous iron uptake. This paper also raised the issue of isoforms of DMT1 by noting that one mRNA form had a 3’ iron responsive element (IRE) in it (+IRE) and one lacked an IRE (-IRE). Furthermore, diminished expression of DMT1 was found at the apical membrane of enterocytes in mk/mk mice [47].
Fine dissection of the transport of nonTf ferric and ferrous iron into cultured cells, however, done with both K562 human erythroleukemia cells and a human embryonic kidney cell line (HEK293T) demonstrated that ferrous iron uptake was mediated by DMT1 via extensive studies of competitive inhibition [48]. Although Conrad and collaborators postulated a separate pathway to manage NTBI as ferric iron, most investigators have relied on the role of DCYTB in reducing it to Fe2+ [49] as a means of assuring import of Fe3+ then assumed that TBI accounts for any metabolic Fe3+.
After recognition that transcription of DMT1 mRNA could start at alternative promoters [50] designated 1A and 1B with the concomitant definition of DMT1 mRNA isoforms as 1A+IRE, 1A-IRE, 1A+IRE and 1-IRE depending on 5’ and 3’ ends, it became common to refer to them as alternative splicing isoforms with recognition that their protein products also differed at the N- and C-terminal regions. Studies of ectopic over expression of different cDNA DMT1 isoforms [3,18] not only characterized some of their properties but pointed out they were due to alternative promoters and poly-adenylation sites, with the 1A isoform, responsive to hypoxia, downregulated by hepcidin in enterocytes [9] and the 1B isoform upregulated by NF-κB and inflammation [51].
Initial evidence that iron transport activity of DMT1 involved a H+/Fe2+ cotransport mechanism came in one of the earliest reports [1]. Although confirmation of H+/Fe2+ cotransport is extensive [11,52], it is critical to note that an Israeli group [20] has reviewed proton slippage, a potentially protective mechanism. They argue that normal cotransport should have a 1:1 stoichiometry, but excess protons can lead to many protons entering with one ferrous ion. Here one of our main points is that this mechanism could exist but is insufficient in many acidotic conditions and/or that if the cotransport is also a protective mechanism that seeks to get dangerous extracellular ferrous ions internalized and associated with protective chaperones [6,7], it is unable to avoid pathology when acidosis is outside of physiological limits for the tissue involved.
The plasma membrane is not the only sub-cellular location or organelle where acidosis can interact with redox-active iron. The mitochondrion, meriting particular attention in neurodegenerative diseases, can have any of all four DMT1 isoforms on the outer mitochondrial membrane (OMM) [53]. There is now evidence [54] that supports endosomal/mitochondria contact, thus also “kiss and run” [55] for the two organelles as part of iron uptake in the mitochondria. These results join evidence for DMT1 participating in acquisition across the OMM of both Fe2+ and Mn2+ [56]. Given that endosomal acidification is a physiological part of the Tf cycle too [57], the situation reminds us that acidification can be physiological or pathological for iron metabolism.
In relation to ferrous iron accumulation and neurodegeneration, we need to recall the intriguing regulatory aspect consists in selective involvement of DMT1 because of its pH-dependent uptake as a proton co-transporter. DMT1 is the elective NTBI importer in the mildly acidic duodenal compartment, as demonstrated in Caco2 cell lines, where the uptake of both ferrous iron and other divalent metals is maximal at acidic pH, selectively abrogated by DMT1 knockdown [15,16,19].
Also, the acidic pH in the duodenum is optimal for NTBI uptake because of the maximal DMT1 activity as proton-cotransporter; yet acidosis-dependent iron uptake is often pathological in tissues other than duodenum. Noteworthy in this respect, acute ferrous iron treatment in acidic conditions significantly increases iron uptake, particularly after overexpression of 1B/-IRE DMT1 in differentiated human neuroblastoma cells, with consequent increase in cell death, already after the basal DMT1 overexpression ([38] and references therein). Hence, physiological control of extracellular and likely intraorganellar acidosis is required to avoid an imbalance of ferrous iron uptake with consequent NTBI overload in peripheral tissues.
4. Acidosis-Dependent Inflammation and Iron Homeostasis
FPN, the only known cellular iron-exporter, plays a remarkable role in the control of systemic iron homeostasis as its expression is down-regulated in duodenal enterocytes by inflammation and high levels of iron, controlled by post-translational degradation driven by hepcidin, the liver-derived peptide and iron-sensing hormone [3]. Similarly, iron-recycling macrophages and Kupffer cells also rely on FPN to export iron to targets like developing erythroid cells and hepatocytes. Hepcidin also controls FPN levels in neurons, astrocytes and microglial cells [13,14]. This similar regulation in iron exit into circulation after ingestion and repeated exit from many cell types avoids having high levels of NTBI in circulation partly by minimizing unnecessary acquisition and partly by keeping iron where it is utilized or stored.
As hepcidin blocks ferroportin-mediated iron export, this peptide is downregulated during iron deficiency, allowing compensatory gastrointestinal iron uptake into circulation as well as reutilization from effete red cells. Hepcidin increases during inflammation to block iron export to the plasma, preventing iron availability to infecting bacteria or viruses. Essentially, the concentration of iron, both systemic and tissues, is maintained by hepcidin, in its role of iron-regulatory peptide hormone [9].
Interestingly, in regard to inflammation there is a report that pro-inflammatory cytokines promote cellular iron uptake by up-regulation of DMT1 expression in pancreatic β-cells inducing cell death [58], while DMT1 silencing has a protective role. The signalling pathway in this phenomenon has not been fully clarified.
Noteworthy in a similar context, an impairment in buffering ability is shown to influence those diseases characterized by glucose metabolic disorders, like diabetes mellitus, inflammation, and cancer. Thus, one sees an important relationship between acidosis and insulin metabolism or insulin receptor signalling, given that insulin induces the activation of glycolysis to counteract acidosis, and acidosis possibly enhances insulin resistance through the NF-κB inflammatory pathway [59]. Afterwards, as we discussed above, a crosstalk between iron and metabolic pathways was depicted in studies in vivo related to the requirement for an acidic environment for iron absorption from dietary sources and generated by the activity of the proton pump gastric H(+)/K(+)ATPase (ATP4), expressed in gastric parietal cells [32].
Relating what is physiological to the pathologic again, inflammation often induces extracellular acidification in the inflamed region through increased glycolysis and lactate secretion with influence of extracellular acidosis on the modulation of inflammasome signalling, albeit not clearly defined. The ensuing reduction in the intracellular pH of macrophages but not neutrophils and increased ability of macrophages to assemble the NLR family pyrin domain containing 3 (NLRP3) inflammasome and activation of the chloride intracellular channel protein 1 (CLIC1) may be thus envisaged as a pharmacological target for NLRP3 inflammasome-related diseases [60].
5. Acidosis-Dependent Neuroinflammation and Iron Overload
Iron homeostasis, important for brain function, involves the blood–brain barrier both at systemic and cellular level. Iron overload activates the Haber-Weiss and Fenton reactions leading to production of free radicals and oxidative cellular damage and triggering ferroptosis, iron-dependent programmed cell death. A growing number of publications associate neurodegenerative diseases with disrupted brain iron homeostasis. Considering also the principal molecular players of inflammation, like NF-κB, interleukins, the inflammasome activator NLRP3, one may envisage a wide perspective for a relationship between inflammation and the development of neurological diseases.
Another pointer towards iron and inflammation is evidence for increased serum hepcidin, the hepatic iron-regulatory hormone, in Alzheimer disease (AD) and patients with mild cognitive impairment (MCI) [61]. Both serum hepcidin and ferritin were found significantly higher than in controls, while Aβ40 and Aβ42 decreased. Interestingly, the authors observed an increase of iron and iron-related proteins with progression of cognitive impairment, thus highlighting a significant role for iron dyshomeostasis in AD as elevated serum biomarkers, supporting an interconnection between central nervous system and systemic homeostasis, potentially contributing to diagnosis of AD and AD progression. As recently found in a murine model of AD [62], initial deposition of Aβ aggregates in the brain is followed by a decline of the Aβ42/Aβ40 ratio in CSF and serum once the cerebral Aβ pathology becomes significant. The observation is consistent with the findings above in MCI and AD patients of an inverse relationship between hepcidin and Aβ levels in the serum.
Accordingly, as extracellular acidification consequent to cellular stress like hypoxia, nutrient deprivation, and cytokine inflammation may affect the function of lysosomes, key organelles in the regulation of autophagy, thus acting as intracellular nutrient sensing mechanism, we also mention the decrease of both autophagic ATG5 and the mitophagy-related Parkin proteins, found in the serum of patients with both AD and MCI, compared to healthy subjects, reflecting significant impairment of the autophagy and mitophagy pathways, known to be associated to acidosis, inflammation and neurodegeneration [63].
In line with the involvement of inflammation-related autophagy in AD, we remind readers autophagy involvement was implicated in Parkinson’s disease (PD) and recently described in a model with deletion of the ATG5 gene that caused PD-like symptoms in mice, with impairment in motor coordination and cognitive learning, loss of tyrosine hydroxylase (TH) neurons and reduction in dopamine levels in the striatum [64]. These results were associated to the activation of microglia and NLRP3-inflammsome upregulation of downstream IL1B/IL-1β and increased expression of macrophage migration inhibitory factor, a pro-inflammatory cytokine, found by the authors significantly increased in the serum of Parkinson patients.
Also related to PD, NFκB/c-Rel-deficient mice described as model of late-onset parkinsonism showed a significant loss of dopaminergic neurons in the substantia nigra pars compacta, assessed by loss of TH positive fibers, reduced immunoreactivity for the dopamine transporter in the caudate putamen, marked immunoreactivity for fibrillary α-synuclein in the substantia nigra pars compacta, with increased microglial reactivity in the basal ganglia and age-dependent locomotor deficit [65]. Furthermore, this NFκB/c-Rel knockout model of parkinsonism displayed the increased expression of DMT1 and iron staining in both the substantia nigra pars compacta and striatum, consistent with the PD mice model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication, where the increased DMT1 expression and concomitant iron accumulation was found in the ventral mesencephalon, with dopaminergic cell loss [66]. Interestingly, the latter report also showed increased expression of DMT1 in the SN of post-mortem slices of PD patients not to mention protection against MPTP intoxication to parkinsonism by the mk G185R DMT1 mutation in mice and vs parkinsonism-inducing 6-hydroxydopamine in the Belgrade rat model, also carrying the G185R mutation in DMT1. Remarkably, a connection between PD and degradation of 1B DMT1 (but not 1A DMT1) emerged after the discovery that Parkin was responsible [67].
Similarly, activation of NFκB/RelA acetylation at Lysine 310 was also shown to be the epigenetic mechanism responsible of post-ischemic cell death in cellular and animal models of brain ischemia with concomitant iron overload mediated by the 1B/-IRE isoform of DMT1 [38]; The role of iron and DMT1 clearly contributes to a complex panel of interrelationships among aging, neurodegeneration and neuroinflammation [68]. Also, elevated neuroinflammation, where an increase in iron content exacerbates oxidative stress, was shown in a model of activated-astrocytes with excess iron that correlated with DMT1 function [69]. An effect of iron overload was shown on brain function in aging with higher striatal iron accompanying diminished fronto-striatal activity in older adults [70], but that activity was enhanced by hhigher striatal iron in younger subjects. Indeed, inflammation can cause iron accumulation into the central nervous system [13], with DMT1 upregulation in the rat hippocampus after excitotoxic injury induced by kainate [71]. Development of Parkinsonism / PD is associated with altered intracellular transport of iron and heavy metals, principally mediated by DMT1 [65,66,68,72].
Furthermore, the principal hallmark of NBIA is NTBI accumulation in basal ganglia [40] shown by MRI with progressive iron deposition in the substantia nigra and globus pallidus, initially associated to cerebral atrophy, with later insurgence of bilateral hypointensity of the globus pallidus and substantia nigra on T2-weighted sequences with reduced uptake of tracer in DAT-Scan [73,74]. Confirmatory evidence in support of the influence of extracellular acidosis in respect to iron overload comes from the finding of ischemic penumbra in post-mortem pallidum slices of an NBIA/PKAN brain [75]. In this regard, iron and DMT1 accumulation were found in the post-ischemic neurodegeneration in brain ischemia [38]. Afterwards, impairment of intracellular iron homeostasis was found in patients affected by a different subclass of NBIA with de novo mutations in the WDR45 gene, coding for a beta-propeller binding protein, designated NBIA/BPAN, where up-regulation of endogenous DMT1 and ferrous iron was found in the primary fibroblasts of affected patients [41].
Hence, acidosis frequently accompanies iron accumulation in tissues other than the duodenum, usually under inflammatory or stressful circumstances. There needs to be more experimental work to ascertain the extent to which the pairing is a metabolic effort to cope or if it is itself pathological. We next consider ways to manage the iron and how they can begin to tell us what the roles of the observed responses are.
Figure 1.
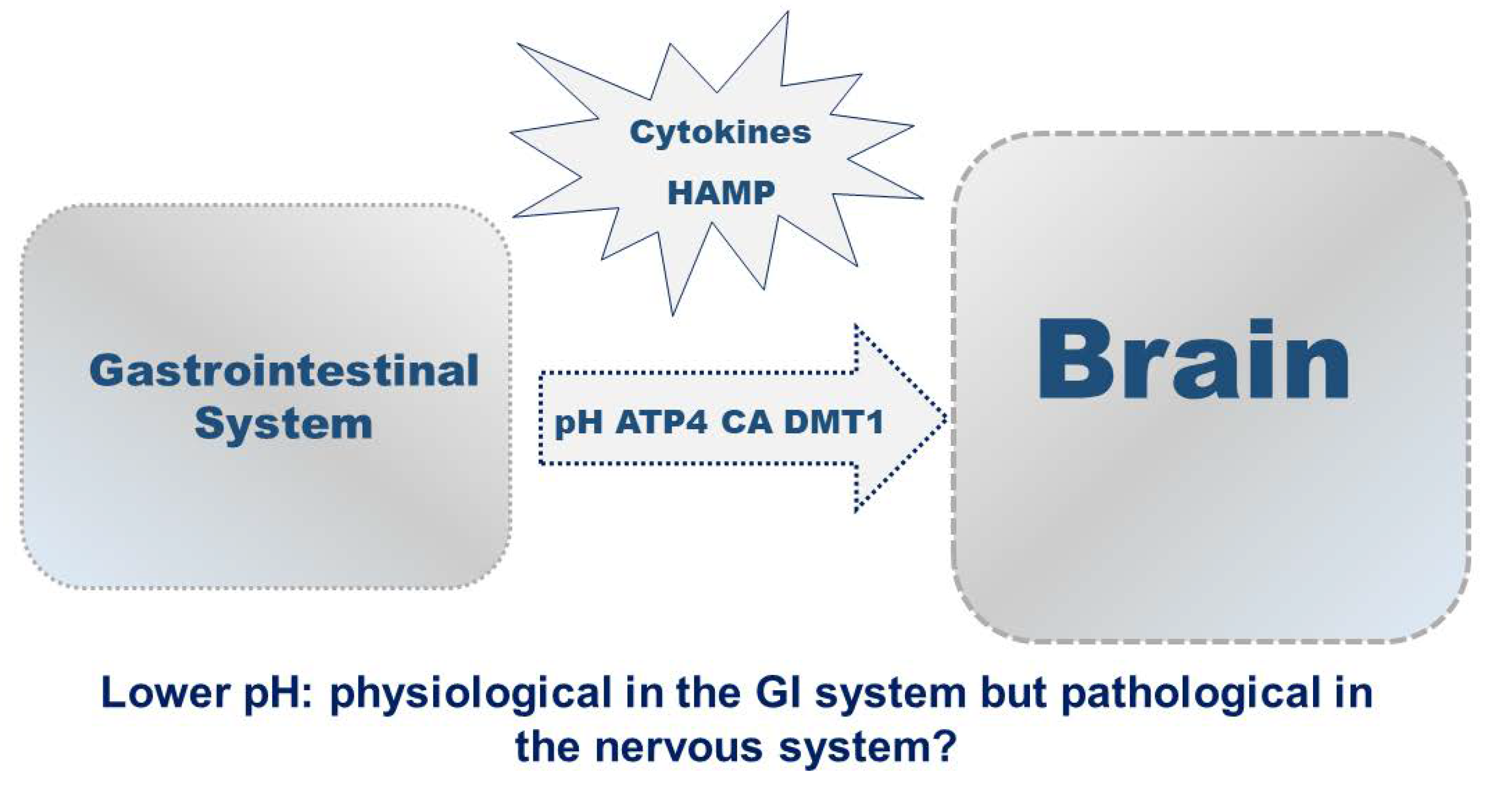
6. Iron Chelation and Transport Strategies
Disappointingly, a pharmaceutical company abandoned their synthesis strategy that afforded development of a gut-specific DMT1 inhibitor, to inhibit selectively dietary duodenal NTBI transport, without a significant impact in the physiological homeostasis of other divalent metals than iron [76]. This inhibitor and likely its relatives need to remain available to the research community given their properties [77].
MD Burke and collaborators ingeniously proposed circumstances where small molecules can replace mutated gene products like transporters or enzymes [78] then showed that hinokitiol, a natural product, would substitute for DMT1 in Belgrade rats as well as for other defective iron transporters in specific animal models. Subsequently, hinokitiol was compared to different isoforms of DMT1 in HEK293 cell lines capable of overexpressing them and shown to transport both Fe2+ and Fe3+ with Michaelis-Menten kinetics [79]. The Burke group has already extended application of this framework to other genetic disorders [80] using a different, but appropriate drug. The framework with hinokitiol (and likely modifications of the product) clearly applies to iron dyshomeostasis including perhaps neuroprotection where a conventional inhibitor recently failed in PD [81], discussed more below.
Also, with respect to protection, particularly, neuroprotection, against ferrous iron uptake exacerbated by conditions of acidosis, a small-molecule inhibitor of iron uptake through DMT1, ebselen, a seleno-compound, has been used in clinical trials as a protective agent against ischemic stroke. Ebselen inhibited Fe(II) uptake, with IC(50) ~0.22 μM, and did not influence Fe(III) transport or DMT1-mediated manganese uptake [82,83,84]. Ebselen was initially described as an organo-selenium compound with glutathione peroxidase (GPx)-like, thiol-dependent, hydroperoxide reducing activity [85,86]. A number of neuroprotective treatments were investigated in clinical trials such as hypothermia and ebselen [87] and earlier neuroprotection by ebselen was shown in a rabbit model of embolic stroke [88]. Ebselen was also effective against peripheral oxidative stress in a mouse model of sporadic AD induced by intracerebroventricular streptozotocin administration [89]. More recently, ebselen was found to protect against iron-overload-associated cardiomyopathy in thalassemic patients with iron burden [90]. In a neuronal model of senescence induced by ferric ammonium citrate treatment in human neuroblastoma SH-SY5Y cells, ebselen decreased levels of DMT1, ferritin light chains and the cytosolic labile iron pool, while increasing FPN expression [91].
Interestingly, ferrous iron uptake was selectively abrogated by DMT1 knockdown in Caco2 cell lines [3,19]. A key to the knockdown approach could be targeting the interference; in a mouse model of hemochromatosis, oral gavage of ginger nanoparticles targeted to the duodenum, achieved appropriate delivery of DMT1-specific small interfering RNA (siRNA) to lower intestinal expression levels, thus mitigating iron overload without apparent toxicity [92,93]. Another siRNA usage, where -IRE DMT1 was the target, induced neuroprotection from cell death during oxygen glucose deprivation (OGD) in differentiated human SK-NS-H neuroblastoma cells [38]. The authors observed inhibition of iron overload, by total X-ray fluorescence analysis (TXRF).
In the same model, neuroprotection was also obtained by iron chelation with desferal during OGD and post ischemic reoxygenation [38]. As mentioned above, animal models with the G186R mutation in DMT1 proved resistant to induced Parkinsonism [66]. Such data (and more covered in [81]) led this large group to trials utilizing deferiprone chelation in PD patients shortly after diagnosis. Although neuroimaging evaluation detected a greater decrease in nigrostriatal iron content in the deferiprone group than in the placebo group [81], diminution of iron not only did not slow the progression of PD disease in these participants who had never received levodopa, but treatment produced worsening of motor and nonmotor symptoms over a period of 36 weeks. Reviewing this well-designed study, Levi and Volontè underlined that a multifactorial neurodegenerative disorder like PD might require a combination of treatments to simultaneously target different mechanisms [94] and commented that the trials may have overshot on deferiprone chelation. While we hesitate to encourage trial of another iron binding compound, hinokitiol (and perhaps a modified version thereof) may be easier to ‘tune’ than deferiprone, allowing optimal levels of nigrostriatal iron.
Moreover, while considering different approaches of neuroprotection, we already mentioned the role of CaA, as modulator of acidosis. With several of its isoforms expressed in the brain and studies already performed on several classes of CaA inhibitor, they represent possible pharmacological compounds for neuroprotection during brain ischemia [95,96]. Another approach to managing neurodegeneration interventions might be to take advantage of the role of degradation by Parkin and other E3 ligases, suggested to affect ferroptosis [97], partly inspired by [67] where the Authors reported that Parkin overexpression in human neuroblastoma decreases 1B-DMT1, the associated divalent metal transport and toxicity, effects counteracted by the proteasomal inhibitor, MG-132. Thus, highlighting again the role of DMT1 expression and iron homeostasis in the neurodegeneration [68].
7. Conclusions
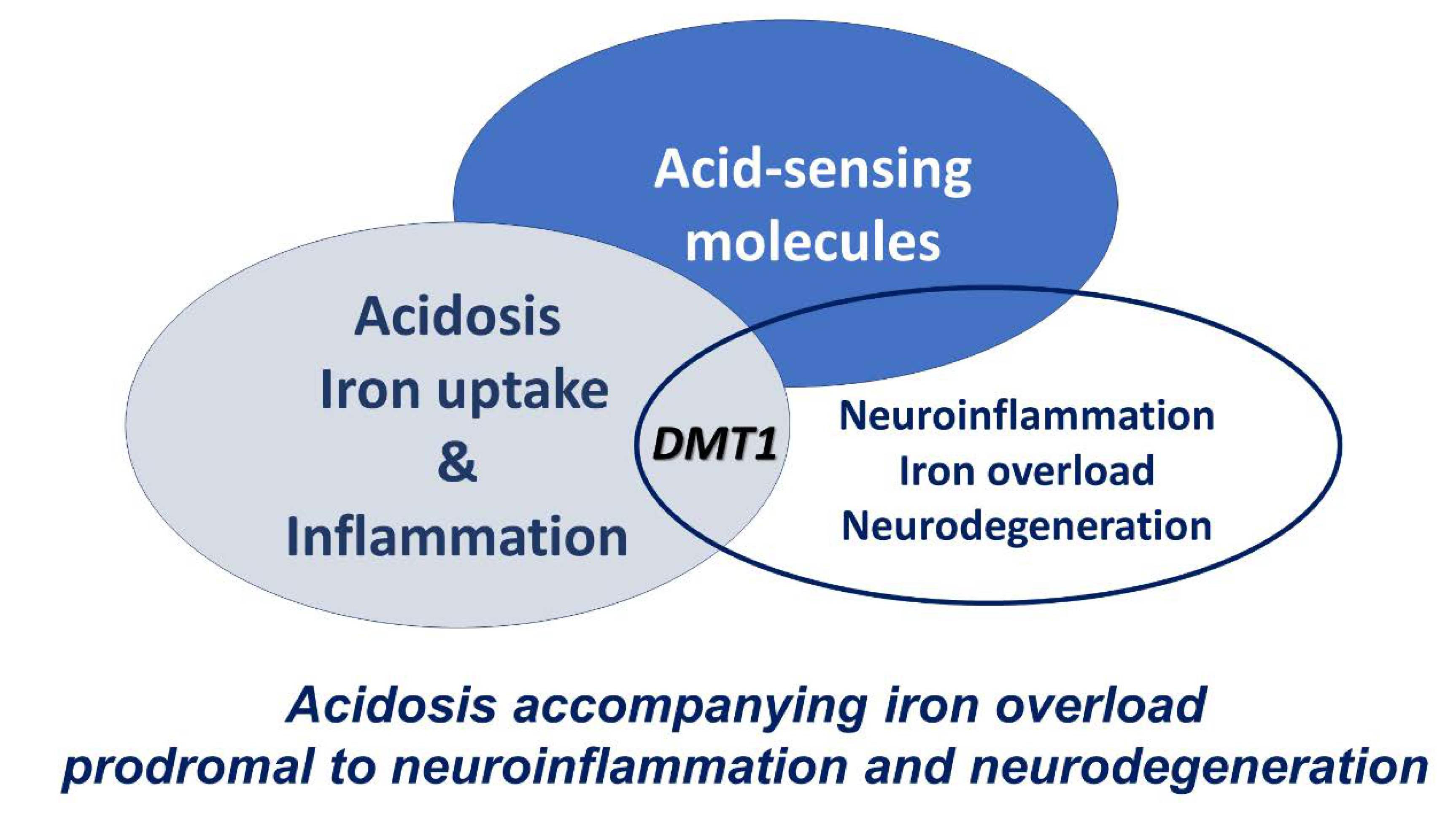
When the cellular environment is acidotic, DMT1 has maximal uptake activity - potentially much higher than an otherwise expected expression level. Inflammation is often associated with such an acidosis, frequently evoked through NFκB and interleukins, as discussed earlier. Thus, conditions can conspire to increase intracellular iron. Such a response can be protective or overwhelmed to cause ROS dependent damage to cells.
Figure 2.
References
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, and Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Fleming MD, Trenor CC, Su MA, Foernzler D, Beier DR, Dietrich WF, and Andrews NC Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 1997, 16, 383–386. [CrossRef] [PubMed]
- Garrick MD, Dolan KG, Horbinski C, Ghio AJ, Higgins D, Porubcin M, Moore EG, Hainsworth LN, Umbreit JN, Conrad ME, Feng L, Lis A, Roth JA, Singleton S, Garrick LM. DMT1: a mammalian transporter for multiple metals. Biometals. 2003, 16, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998, 95, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Philpott CC, Ryu MS. Special delivery: distributing iron in the cytosol of mammalian cells. Front Pharmacol. 2014, 5, 173. [Google Scholar]
- Yanatori I, Kishi F. DMT1 and iron transport. Free Radical Biology and Medicine. 2019, 133, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, McVey Ward D, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Nemeth E, Ganz T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis Int J Mol Sci 2021, 22, 6493.
- Knutson, MD. J Iron transport proteins: Gateways of cellular and systemic iron homeostasis. J Biol Chem. 2017, 292, 12735–12743. [Google Scholar] [CrossRef]
- Mackenzie B and Garrick MD Iron imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol 2005, 289, G981–G986. [Google Scholar] [CrossRef] [PubMed]
- Wu LJ, Leenders AG, Cooperman S, Meyron-Holtz E, Smith S, Land W, Tsai RY, Berger UV, Sheng ZH, Rouault TA. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res 2004, 1001, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P. , Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- You, L. H. , Yan, C. Z., Zheng, B. J., Ci, Y. Z., Chang, S. Y., Yu, P., et al. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 2017, 8, e2676. [Google Scholar] [CrossRef] [PubMed]
- Garrick MD, Dolan KG. An expression system for a transporter of iron and other metals. In Ultrastructure and Molecular Biology stet for oxidants and Antioxidants, Methods in Molecular Biology; D. Armstrong ed; Humana press: Totawa NJ, 2002. [Google Scholar]
- Garrick MD, Kuo HC, Vargas F, Singleton S, Zhao L, Smith JJ, Paradkar P, Roth JA, Garrick LM. Comparison of mammalian cell lines expressing distinct isoforms of divalent metal transporter 1 in a tetracycline-regulated fashion. Biochem J. 2006, 398, 539–546.
- Arredondo M, Mendiburo MJ, Flores S, Singleton ST, Garrick MD. Mouse divalent metal transporter 1 is a copper transporter in HEK293 cells. Biometals 2014, 27, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Garrick MD, Singleton ST, Vargas F, Kuo HC, Zhao L, Knöpfel M, Davidson T, Costa M, Paradkar P, Roth JA, Garrick LM. DMT1: which metals does it transport? Biol Res 2006, 39, 79–85. [Google Scholar]
- et al. Effect of DMT1 knockdown on iron, cadmium, and lead uptake in Caco-2 cells. Am J Physiol Cell Physiol 2003, 284, C44–C50. [Google Scholar] [CrossRef]
- Nevo Y, Nelson N. The NRAMP family of metal-ion transporters. Biochim Biophys Acta. 2006, 1763, 609–620. [Google Scholar] [CrossRef]
- Nuñez MT, Gaete V, Watkins JA, Glass J. Mobilization of iron from endocytic vesicles. The effects of acidification and reduction. J Biol Chem 1990, 265, 6688–6692. [Google Scholar] [CrossRef]
- Watkins JA, Nunez MT, Gaete V, Alvarez O, Glass J. Kinetics of iron passage through subcellular compartments of rabbit reticulocytes. J Membr Biol 1991, 119, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Sendamarai AK, Ohgami RS, Fleming MD, Lawrence CM. Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc Natl Acad Sci U S A. 2008, 105, 7410–7415. [Google Scholar] [CrossRef] [PubMed]
- Cheng YR, Jiang BY, Chen CC. Acid-sensing ion channels: dual function proteins for chemo-sensing and mechano-sensing J Biomed Sci 2018, 25, 46.
- Mazzocchi N, De Ceglia R, Mazza D, Forti L, Muzio L, Menegon A. Fluorescence-Based Automated Screening Assay for the Study of the pH-Sensitive Channel ASIC1a. J Biomol Screen 2016, 21, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Mango D, Nisticò R. Neurodegenerative Disease: What Potential Therapeutic Role of Acid-Sensing Ion Channels? Front Cell Neurosci 2021, 15, 730641. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiaretta, M. , Latifi, S., Bramini, M., Fadda, M., Fassio, A., Benfenati, F., et al. Neuronal hyperactivity causes Na +/H + exchanger-induced extracellular acidification at active synapses. J. Cell Sci, 1435. [Google Scholar]
- Xiong ZG, Xu TL. The role of ASICS in cerebral ischemia. Wiley Interdiscip Rev Membr Transp Signal 2012, 1, 655–662. [Google Scholar] [CrossRef] [PubMed]
- López-Ramírez O, González-Garrido A. The role of acid sensing ion channels in the cardiovascular function. Front Physiol. 2023, 14, 1194948. [Google Scholar] [CrossRef] [PubMed]
- Redd MA, Scheuer SE, Saez NJ, Yoshikawa Y, Chiu HS, Gao L, Hicks M, Villanueva JE, Joshi Y, Chow CY, Cuellar-Partida G, Peart JN, See Hoe LE, Chen X, Sun Y, Suen JY, Hatch RJ, Rollo B, Xia D, Alzubaidi MAH, Maljevic S, Quaife-Ryan GA, Hudson JE, Porrello ER, White MY, Cordwell SJ, Fraser JF, Petrou S, Reichelt ME, Thomas WG, King GF, Macdonald PS, Palpant NJ. Therapeutic Inhibition of Acid-Sensing Ion Channel 1a Recovers Heart Function After Ischemia-Reperfusion Injury. Circulation. 2021, 144, 947–960. [Google Scholar] [CrossRef]
- Daher R, Ducrot N, Lefebvre T, Zineeddine S, Ausseil J, Puy H, Karim Z. Crosstalk between Acidosis and Iron Metabolism: Data from In Vivo Studies. Metabolites 2022, 12, 89. [Google Scholar] [CrossRef]
- Becker HM, Deitmer JW. Proton Transport in Cancer Cells: the role of Carbonic Anhydrases. Int J Mol Sci. 2021, 22, 3171. [Google Scholar] [CrossRef] [PubMed]
- Lemon N, Canepa E, Ilies MA, Fossati S. Carbonic Anhydrases as Potential Targets Against Neurovascular Unit Dysfunction in Alzheimer's Disease and Stroke. Front Aging Neurosci 2021, 13, 772278. [Google Scholar] [CrossRef] [PubMed]
- Poggetti V, Salerno S, Baglini E, Barresi E, Da Settimo F, Taliani S. Carbonic Anhydrase Activators for Neurodegeneration: An Overview. Molecules 2022, 27, 2544. [Google Scholar] [CrossRef] [PubMed]
- Ryder C, McColl K, Zhong F, Distelhorst CW. Acidosis promotes Bcl-2 family-mediated evasion of apoptosis: involvement of acid-sensing G protein-coupled receptor Gpr65 signaling to Mek/Erk. J Biol Chem 2012, 287, 27863–27875. [Google Scholar] [CrossRef] [PubMed]
- Sarnico I, Lanzillotta A, Benarese M, Alghis M, Baiguera C, Battistin L, Spano PF, Pizzi M. NF-kappaB dimers in the regulation of neuronal survival. Int Rev Neurobiol 2009, 85, 351–362. [Google Scholar]
- et al. 1B/(-)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-κB/RelA acetylation at Lys310. PLoS One 2012, 7, e38019. [Google Scholar]
- Ordway B, Gillies RJ, Damaghi M. Extracellular Acidification Induces Lysosomal Dysregulation Cells 2021, 10, 1188.
- Wilson JL, Allison Gregory A, Kurian MA, Bushlin I, Mochel F, Emrick L, Adang L. BPAN Guideline Contributing Author roup. Hogarth P, Hayflick SJ. Consensus clinical management guideline for beta-propeller protein-associated neurodegeneration. Dev Med Child Neurol. 2021, 63, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia R, Memo M, Garavaglia B. Ferrous Iron Up-regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN). Front Genet. 2017, 8, 18. [Google Scholar]
- Hu X, Bao Y, Li M, Zhang W, Chen C. The role of ferroptosis and its mechanism in ischemic stroke. Exp Neurol. 2024, 372, 114630. [Google Scholar] [CrossRef]
- Wang L, Zhang X, Xiong X, Zhu H, Chen R, Zhang S, Chen G, Jian Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants (Basel). 2022, 11, 2377. [Google Scholar] [CrossRef] [PubMed]
- 44. Oates PS, Morgan EH. 1996 Defective iron uptake by the duodenum of Belgrade rats fed diets of different iron contents. Am J Physiol Gastrointest Liver Physiol.
- Garrick M, Scott D, Walpole S, Finkelstein E, Whitbred J, Chopra S, Trivikram L, Mayes D, Rhodes D, Cabbagestalk K, Oklu R, Sadiq A, Mascia B, Hoke J, Garrick L. Iron supplementation moderates but does not cure the Belgrade anemia. BioMetals 1997, 10, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Edwards JA, Hoke JE. Defect of intestinal mucosal iron uptake in mice with hereditary microcytic anemia. Proc Soc Exp Biol Med 1972, 141, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Canonne-Hergaux F, Fleming MD, Levy JE, Gauthier S, Ralph T, Picard V, Andrews NC, Gros P. The Nramp2/DMT1 iron transporter is induced in the duodenum of microcytic anemia mk mice but is not properly targeted to the intestinal brush border. Blood. 2000, 96, 3964–3970. [Google Scholar]
- Conrad ME, Umbreit JN, Moore EG, Hainsworth LN, Porubcin M, Simovich MJ, Nakada MT, Dolan K, Garrick MD. Separate pathways for cellular uptake of ferric and ferrous iron. Am J Physiol Gastrointest Liver Physiol. 2000, 279, G767–G774. [Google Scholar] [CrossRef] [PubMed]
- McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- 50. Hubert N, Hentze MW..Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A. 1235.
- Paradkar PN, Roth JA. Nitric oxide transcriptionally down-regulates specific isoforms of divalent metal transporter (DMT1) via NF-kappaB. J Neurochem. 2006, 96, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Shawki, A. , Anthony, S. R., Nose, Y., Engevik, M. A., Niespodzany, E. J., Barrientos, T., Öhrvik, H., Worrell, R. T., Thiele, D. J., and Mackenzie, B. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G635–G647. [Google Scholar] [PubMed]
- Wolff NA, Ghio AJ, Garrick LM, Garrick MD, Zhao L, Fenton RA, Thévenod F. Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J 2014, 28, 2134–2145. [Google Scholar] [CrossRef]
- Das A, Nag S, Mason AB, Barroso MM. Endosome-mitochondria interactions are modulated by iron release from transferrin. J Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef]
- Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood. 2007, 110, 125–132. [Google Scholar] [CrossRef]
- Wolff NA, Garrick MD, Zhao L, Garrick LM, Ghio AJ, Thévenod F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Garrick MD, Gniecko K, Liu Y, Cohan DS, Garrick LM. Transferrin and the transferrin cycle in Belgrade rat reticulocytes. J Biol Chem. 1993, 268, 14867–14874. [Google Scholar] [CrossRef]
- Kang T, Huang H, Mandrup-Poulsen T, Larsen MR. Divalent Metal Transporter 1 Knock-Down Modulates IL-1β Mediated Pancreatic Beta-Cell Pro-Apoptotic Signaling Pathways through the Autophagic Machinery. Int J Mol Sci. 2021, 22, 8013. [Google Scholar] [CrossRef] [PubMed]
- Baldini N, Avnet S. The Effects of Systemic and Local Acidosis on Insulin Resistance and Signaling. Int J Mol Sci. 2018, 20, 126. [Google Scholar] [CrossRef]
- Chae BJ, Lee KS, Hwang I, Yu JW. Extracellular Acidification Augments NLRP3-Mediated Inflammasome Signaling in Macrophages. Immune Netw 2023, 23, e23. [Google Scholar] [CrossRef]
- Sternberg Z, Hu Z, Sternberg D, Waseh S, Quinn JF, Wild K, Jeffrey K, Zhao L, Garrick M. Serum Hepcidin Levels, Iron Dyshomeostasis and Cognitive Loss in Alzheimer's Disease. Aging Dis. 2017, 8, 215–227. [Google Scholar] [CrossRef]
- Andersson E, Schultz N, Saito T, Saido TC, Blennow K, Gouras GK, Zetterberg H, Hansson O. Cerebral Abeta deposition precedes reduced cerebrospinal fluid and serum Abeta42/Abeta40 ratios in the App(NL-F/NL-F) knock-in mouse model of Alzheimer's disease. Alzheimers Res Ther. 2023, 15, 64. [Google Scholar] [CrossRef]
- Castellazzi M, Patergnani S, Donadio M, Giorgi C, Bonora M, Bosi C, Brombo G, Pugliatti M, Seripa D, Zuliani G, Pinton P. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer's disease and mild cognitive impairment. Sci Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X, Li S, Li X, Guo J, Qin L, Yu J, Ma C, Li J, Li M, Tang B, Yuan Z. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 2020, 16, 2193–2205. [Google Scholar] [CrossRef]
- Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L, Morelli M, Ingrassia R, Benarese M, Porrini V, Pellitteri M, Bertini G, Fabene PF, Sigala S, Spillantini MG, Liou HC, Spano PF, Pizzi M. Late-onset Parkinsonism in NFκB/c-Rel-deficient mice. Brain 2012, 135, 2750–2765. [Google Scholar] [CrossRef] [PubMed]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nuñez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc Natl Acad Sci U S A. 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [PubMed]
- Roth JA, Singleton S, Feng J, Garrick M, Paradkar PN. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J Neurochem. 2010, 113, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia R, Garavaglia B, Memo M. DMT1 Expression and Iron Levels at the Crossroads Between Aging and Neurodegeneration. Front Neurosci. 2019, 13, 575.
- Pelizzoni I, Zacchetti D, Campanella A, Grohovaz F, Codazzi F. Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim Biophys Acta 2013. 1832, 1326–1333.
- Salami A, Papenberg G, Sitnikov R, Laukka EJ, Persson J, Kalpouzos G. Elevated neuroinflammation contributes to the deleterious impact of iron overload on brain function in aging. Neuroimage 2021, 230, 117792. [Google Scholar] [CrossRef] [PubMed]
- Huang E, Ong WY, Go ML, Connor JR. Upregulation of iron regulatory proteins and divalent metal transporter-1 isoforms in the rat hippocampus after kainate induced neuronal injury. Exp Brain Res. 2006, 170, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, EC. Iron transport in Parkinson's disease. Parkinsonism Relat Disord. 2009, 2009 15 Suppl 3, S209–11. [Google Scholar] [CrossRef]
- Russo C, Ardissone A, Freri E, Gasperini S, Moscatelli M, Zorzi G, Panteghini C, Castellotti B, Garavaglia B, Nardocci N, Chiapparini L. Substantia Nigra Swelling and Dentate Nucleus T2 Hyperintensity May Be Early Magnetic Resonance Imaging Signs of β-Propeller Protein-Associated Neurodegeneration Mov Disord Clin Pract 2018, 6, 51–56.
- Manti F, Panteghini C, Garavaglia B, Leuzzi V. Neurodevelopmental Disorder and Late-Onset Degenerative Parkinsonism in a Patient with a WDR45 Defect. Mov Disord Clin Pract 2021, 9, 110–112. [Google Scholar]
- et al. Pallidal neuronal apolipoprotein E in pantothenate kinase-associated neurodegeneration recapitulates ischemic injury to the globus pallidus. Mol Genet Metab 2015, 116, 289–97. [Google Scholar] [CrossRef]
- Cutts A, Chowdhury S, Ratkay LG, Eyers M, Young C, Namdari R, Cadieux JA, Chahal N, Grimwood M, Zhang Z, Lin S, Tietjen I, Xie Z, Robinette L, Sojo L, Waldbrook M, Hayden M, Mansour T, Pimstone S, Goldberg YP, Webb M, Cohen CJ. Potent, Gut-Restricted Inhibitors of Divalent Metal Transporter 1: Preclinical Efficacy against Iron Overload and Safety Evaluation. J Pharmacol Exp Ther. 2023, 386, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Garrick, MD. Managing Iron Overload: A Gut Check. J Pharmacol Exp Ther 2023, 386, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Grillo AS, SantaMaria AM, Kafina MD, Cioffi AG, Huston NC, Han M, Seo YA, Yien YY, Nardone C, Menon AV, Fan J, Svoboda DC, Anderson JB, Hong JD, Nicolau BG, Subedi K, Gewirth AA, Wessling-Resnick M, Kim J, Paw BH, Burke MD. Restored iron transport by a small molecule promotes absorption and hemoglobinization in animals. Science 2017, 356, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Garrick MD, Garrick LM, Zhao L, Collins JF, Soukup J, Ghio AJ. A direct comparison of divalent metal-ion transporter (DMT1) and hinokitiol, a potential small molecule replacement. Biometals 2019, 32, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Muraglia KA, Chorghade RS, Kim BR, Tang XX, Shah VS, Grillo AS, Daniels PN, Cioffi AG, Karp PH, Zhu L, Welsh MJ, Burke MD. Small-molecule ion channels increase host defences in cystic fibrosis airway epithelia. Nature 2019, 567, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Devos D, Labreuche J, Rascol O, Corvol JC, Duhamel A, Guyon Delannoy P, Poewe W, Compta Y, Pavese N, Růžička E, Dušek P, Post B, Bloem BR, Berg D, Maetzler W, Otto M, Habert MO, Lehericy S, Ferreira J, Dodel R, Tranchant C, Eusebio A, Thobois S, Marques AR, Meissner WG, Ory-Magne F, Walter U, de Bie RMA, Gago M, Vilas D, Kulisevsky J, Januario C, Coelho MVS, Behnke S, Worth P, Seppi K, Ouk T, Potey C, Leclercq C, Viard R, Kuchcinski G, Lopes R, Pruvo JP, Pigny P, Garçon G, Simonin O, Carpentier J, Rolland AS, Nyholm D, Scherfler C, Mangin JF, Chupin M, Bordet R, Dexter DT, Fradette C, Spino M, Tricta F, Ayton S, Bush AI, Devedjian JC, Duce JA, Cabantchik I, Defebvre L, Deplanque D, Moreau C; FAIRPARK-II Study Group. Trial of Deferiprone in Parkinson's Disease. N Engl J Med. 2022, 387, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Wetli HA, Buckett PD, Wessling-Resnick M. Small-molecule screening identifies the selanazal drug ebselen as a potent inhibitor of DMT1-mediated iron uptake. Chem Biol. 2006, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A. , Yoshimoto, T., Kikuchi, H., Sano, K., Saito, I., Yamaguchi, T., and Yasuhara, H. Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial. Cerebrovasc. Dis.
- Yamaguchi, T. , Sano, K., Takakura, K., Saito, I., Shinohara,Y, Asano, T., and Yasuhara, H. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Müller A, Cadenas E, Graf P, Sies H. A novel biologically active seleno-organic compound--I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem Pharmacol 1984, 33, 3235–3239. [Google Scholar]
- Paernham MJ, Sies H. The early research and development of ebselen. Biochem Pharmacol. 2013, 86, 1248–1253. [Google Scholar] [CrossRef]
- Minnerup J, Sutherland BA, M Buchan AM, Kleinschnitz C. Neuroprotection for stroke: current status and future perspectives. Int J Mol Sci 2012, 13, 11753–11772.
- Lapchak PA, Zivin JA. Ebselen, a seleno-organic antioxidant, is neuroprotective after embolic strokes in rabbits: synergism with low-dose tissue plasminogen activator. Stroke 2003, 34, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Klann IP, Martini F, Gonçalves Rosa S, Nogueira CW. Ebselen reversed peripheral oxidative stress induced by a mouse model of sporadic Alzheimer's disease. Mol Biol Rep 2020, 47, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Ghazaiean M, Aliasgharian A, Karami H, Darvishi-Khezri H. Ebselen: A promising therapy protecting cardiomyocytes from excess iron in iron-overloaded thalassemia patients. Open Med (Wars) 2023, 18, 20230733. [Google Scholar] [CrossRef] [PubMed]
- Mukem S, Sayoh I, Maungchanburi S, Thongbuakaew T. Ebselen, Iron Uptake Inhibitor, Alleviates Iron Overload-Induced Senescence-Like Neuronal Cells SH-SY5Y via Suppressing the mTORC1 Signaling Pathway. Adv Pharmacol Pharm Sci 2023, 2023, 6641347. [Google Scholar]
- Wang X, Zhang M, Flores SRL, Woloshun RR, Yang C, Yin L, Xiang P, Xu X, Garrick MD, Vidyasagar S, Merlin D and Collins JF Oral gavage of ginger nanoparticle-derived lipid vectors carrying Dmt1 siRNA blunts iron loading in murine hereditary hemochromatosis. Mol Ther 2019, 27, 493–506. [CrossRef] [PubMed]
- Wang X, Zhang M, Woloshun RR, Yu Y, Lee JK, Flores SRL, Merlin D, and Collins JF. Oral administration of ginger-derived lipid nanoparticles and Dmt1 siRNA potentiates the effect of dietary iron restriction and mitigates pre-existing iron overload in Hamp KO mice. Nutrients 2021, 13, 1686. [Google Scholar] [CrossRef] [PubMed]
- Levi S, Volonté MA. Iron chelation in early Parkinson's disease. Lancet Neurol 2023, 22, 290–291. [Google Scholar] [CrossRef]
- Bulli I, Dettori I, Coppi E, Cherchi F, Venturini M, Di Cesare Mannelli L, Ghelardini C, Nocentini A, Supuran CT, Pugliese AM, Pedata F. Role of Carbonic Anhydrase in Cerebral Ischemia and Carbonic Anhydrase Inhibitors as Putative Protective Agents. Int J Mol Sci 2021, 22, 5029. [Google Scholar] [CrossRef]
- Supuran, CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat. 2018, 28, 709–712. [Google Scholar] [CrossRef]
- Lu L, Jifu C, Xia J, Wang J. E3 ligases and DUBs target ferroptosis: A potential therapeutic strategy for neurodegenerative diseases. Biomed Pharmacother. 2024, 175, 116753. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



