
Submitted:
31 May 2024
Posted:
31 May 2024
You are already at the latest version
Abstract
Identification of drug targets and biochemical investigations on mechanisms of action is a are major issues in modern drug development. The present article reviews the classical “one drug” – “one target” paradigm. Novel methods for target deconvolution and for investigation of resistant strains based on protein mass spectrometry have shown that multiple gene products and adapta-tion mechanisms are involved in responses of pathogens to xenobiotics. This review is focused on protozoan pathogens. The conclusions can, however, be extended to chemotherapies against other pathogens or cancer. For this review we have searched Pubmed (pubmed.ncbi.nlm.nih.gov) and Web of Science (webofscience.com) using the key words listed below (status May 2024)
Keywords:
Adaptation
; mass spectrometry
; model system
; mode of action
; resistance
; target
1. Introduction
Modern therapeutic strategies require more than mere empirical evidence of documenting efficacy, namely insight into the macromolecular targets and the mechanisms of action related to these targets. In this respect, the fundamental paradigm “one compound – one target”, issuing from simpler enzymological or receptor models still prevails. This model predicts that an active compound interacts with one specific micromolar target, most likely a protein, thereby interfering with its biological function. Consequently, resistance to this compound should be linked to modifications of this target or to inactivation of the compound by protective mechanisms.
Evidence for the validity of this model seems to be legion, particularly in neurology and in infectiology in the case of viruses or bacteria. But more recently acquired evidence has shown that this model is flawed. First, there is no therapy without side effects, meaning that secondary targets must be involved. Second, there are domains where the mechanisms of pathogenicity involve several interlinked biological processes, such as in cancer and infections with other eukaryotes - such as protozoan parasites - with a complex cellular organization similar to that of their hosts. Exposition to drugs may thus be countered by various strategies, including modifications of the cellular envelope, expression of transporters and detoxification mechanisms, deregulation of energy and intermediate metabolism, shift to a dormant state and others. Prolonged exposition to drugs may result in drug adaptation or resistance, “fixing” successful strategies in an inheritable manner.
The rapid improvement of the methodology used to unravel the mechanisms of ac-tion of drugs has also caused doubts on the “one compound – one target” paradigm. During the last two decades, the analysis of genomes, transcriptomes and proteomes has not only opened new doors to drug target discovery as anticipated in a seminal review article [1], but also made several quantum leaps with respect to sensitivity and the amount of data that can be handled within a decent time frame [2]. As we see it now, “the target” is part of a complex dataset rather than a defined “enzyme x” or “receptor y”.
Comparing proteomic investigations of antiprotozoal chemotherapies, we present two main strategies of target deconvolution, namely (i) the investigation of proteins binding to compounds of interest or “chemoproteomics”[3] and (ii) the determination of whole-cell proteomes of drug-resistant strains by shotgun mass spectrometry. We discuss the potential and the limitations of these strategies concluding that they constitute only one element within a more holistic approach including physiological and morphological investigations. Based on our personal experience, this review will be focused on the diplomonad Giardia lamblia, a causative agent of persistent diarrhea [4] and on the apicomplexan parasite Toxoplasma gondii, responsible for abortions and neuronal disorders in humans and many animal species [5]. Both are zoonotic pathogens transmitted to humans mainly by ingestion of water or food contaminated by dormant stages, cysts in the case of G. lamblia [4,6], oocysts or bradyzoites in the case of T. gondii [7]. Parallels to other species, in particular to Neospora caninum, closely related to T. gondii and responsible for abortions in cattle [8,9], and to Plasmodium sp. causative agents of malaria, the most prominent disease caused by protozoans [10], will be drawn whenever helpful.
2. Proteomic Tools
2.1. General Remarks
The results of any empirical investigation are a function of the methodology employed for this investigation. With respect to investigations of biostructures, we all know that the term “higher resolution” indicates that more details can be visualized, thus electron microscopy provides more structural information than light microscopy. In terms of proteins, “high resolution” means: high sensitivity with respect to the correct identification of primary polypeptide sequences in solutions containing an increasing number of such polypeptides (i.e. low type II error) combined with high specificity (i.e. low type I error). As for all empirical tests, both errors cannot be reduced to zero. Consequently, false or non-reproducible results cannot be excluded [11].
2.2. From Protein Sequencing to Proteomics
In the case of enzymes, proteins can be identified and quantified by functional assays such as e.g. phosphatase, dehydrogenase, glucoside dehydrogenase and others. Yet, “phosphatase activity” may occur due to a single protein species or to hundreds of different proteins. Consequently, the identification of proteins of interest is needed. The first method allowing the investigations of proteomes was thus the sequence determination of single, purified polypeptides (primary structure) by stepwise degradation from C- or N-terminal ends [12], facilitated by the development of liquid chromatography and one- and two-dimensional polyacrylamide gel electrophoresis post 1970 as highlighted elsewhere [13]. A large booster in terms of sensitivity and amount of generated data was the development of mass-spectrometry (MS) based methods. MS is the separation of charged particles in an electric field as a function of their mass-to-charge ratio (m/z). Developed in the early 1920s for the separation of atoms and small molecules in gaseous phase after chemical ionisation [14] and applied to the identification of amino acids from purified proteins in the 1950s [15], the first methodology applicable to a mixture of proteins was developed in the 1980s. This methodology is based on the indirect ionization of proteins embedded in a solid matrix via laser, the so-called matrix assisted laser desorption ionization (MALDI). The energy transferred to the matrix dissociates the proteins into peptides, which are ionized. The resulting ions are then separated according to their time of flight (TOF), lighter ions being faster than heavier ions, and their m/z is determined [16]. MALDI-TOF-MS is still used today, e.g. for the analysis of immunoprecipitated proteins [17], for protein profiling of food products [18], causative agents of diseases such as bacteria [19], fungi [20], viruses [21,22], or for in-situ-proteomics in embedded tissues [23].
Peptides in liquid phase obtained after proteolytic digestion of a protein mixture, e. g. for whole-cell-shotgun analysis are separated by liquid chromatography (“peptide fingerprinting” [24]), ionized by a convenient method, e.g. electrospray ionization (ESI) and injected into the MS. Within currently used MS, fragmented peptides undergo a second m/z determination. The procedure is therefore referred to as MS/MS. MS are constantly optimized with respect to resolution, sensitivity, and amount of data to be handled [25,26,27]. Ion mobility TOF, where lower mobility ions are analyzed first [28], commercialized as timsTOF (Bruker, Billerica, MA, USA) is one of the leading technologies to date (mid 2024). Recent achievements comprise proteomic analysis of single cells [29] and nanopore-based sequencing of single protein molecules [30,31]. More detailed information can be found in specific reviews, such as e.g. [32,33]. A simplified typical workflow is depicted in Figure 1.
2.3. From Mass Spectra to Protein Data
As mentioned, the “raw” data of any MS based analysis are the signal intensities as a function of m/z ratios of the ionized peptides and/or their fragments. The identification and quantification of mass spectrometry data are routinely handled by complete software suites which return validated lists of quantified peptides and inferred proteins filtered to a false discovery rate (FDR) of 1%. Widely used examples of such software suites are Spectronaut (Biognosis, Schlieren, Switzerland), Fragpipe [34] and MaxQuant [35]. A typical workflow consists in searching the MS2 spectra against theoretical spectra generated by digesting in silico protein sequences from a relevant protein database (e.g. Uniprot; www.uniprot.org). Candidate peptides of the correct mass are then scored by matching theoretical and experimental spectrum peaks, each software suite using different algorithms for the task. The top scorer of each experimental spectrum is retained as a potential match. Since even good matches may happen entirely by chance, especially if the search space is large (large database, large number of post-translational modification etc.), a number of these top-scorers will be false identifications. Presently, the most widely used method to estimate the score distribution of false identifications is to concatenate the original, target database with a set of decoy sequences, typically the reverse sequences of the original [36]. The decoys, which are always false identifications, are then used to model the false identification distributions of a number of measures such as scores, mass differences, retention time, spectral or chromatographic features etc., which can be combined together to form a so-called discriminant score. An FDR can then be estimated per discriminant score threshold, and a selection of MS2 identifications filtered at 1% FDR returned. Note that due to the multiplicity of MS2 spectra mapping to the same peptide, and peptide degeneracy (multiple proteins claiming the same peptide), the FDR must be re-calculated and controlled at all the levels (MS2 identification, peptide, protein) required by the experiment. Software suites will each use different procedures to return FDR-controlled lists of peptides and protein groups. While the latter will always be inferred from peptides based on Occam’s parsimony principle, its interpretation in practice can lead to potentially different protein group reporting [33].
Efforts are continuously made to boost the number of validated peptides and proteins by including further features in the estimation of FDR [37] and rescoring in specific application contexts [38]. Since correct proteins are often identified by multiple peptides and incorrect proteins by one random match only, a good practice is to require at least two unique peptides to consider protein X as “identified”[39].
Note that adding or removing samples from an experiment may alter the list of accepted peptides and proteins from one particular sample, since most tools will use the ion features and identifications of other samples to reinforce or weaken probability of presence in a sample. Protein inference will also be evaluated differently based on the evidence from different sets of samples.
Actual quantification of the abundance of ions is performed in different manners depending on software and crucially on instrument acquisition mode and can be based on integrating MS1 peaks over time, counting corresponding MS2 spectra, or integrating corresponding fragment peaks. How to reconstitute protein intensities from constituent ions is usually considered at the same time as normalization, the latter being an essential condition of label-free quantification. A selection of some of the most frequently used measures of protein abundance are directly compared elsewhere [40]. For instance, the iBAQ (Intensity-based absolute quantification) values are the sum of the peptide intensities divided by the number of theoretically observable tryptic peptides; iBAQ values have been shown to correlate with molar content and are normalized so that the intensity of one protein can be compared to that of another in the same sample. Normalization can also be performed so that the abundance of one protein can be compared across different samples. It is usually done at the peptide level, under the assumption that most peptides have an unchanged abundance across samples. A popular measure of protein abundance, the Top3 value, is obtained by summing such normalized intensities of the three most abundant peptides of each protein. Considering for each protein only a limited number of peptides mitigates the problem that long proteins inevitably produce more peptides than short ones, which could bias the total intensity calculation. A widely used measure of protein intensity that is normalized for comparisons across samples is the LFQ (label-free quantification) value [34,35]. LFQ combines multiple peptide ratios and aims at stabilizing protein ratios between pairs of samples.
While normalization in label-free quantification (LF) aims at reducing various biases such as protein size, stability, varying total amounts between samples and so on, more accurate quantification may be needed, in which case labeling approaches may be preferred. One common method established in the 1990s consists in feeding amino acids containing 13C instead of 12C or 15N instead of 14N to cell cultures (stable isotope labeling by amino acids in cell cultures, SILAC). The labeled proteins are extracted and mixed with unlabeled proteins of another culture. By analyzing the m/z shifts of labeled vs. unlabeled peptides, the amounts of the corresponding proteins in both cultures can be compared [41]. Another methodology is isobaric chemical labelling of soluble proteins or peptides using tags with defined m/z ratios upon ionization [42]. A direct comparison of LF with SILAC and TMT based quantification reveals the best coverage for LF and the lowest performance for TMT [43].
The quantitative data may then be statistically analyzed using a suitable platform such as e.g. Perseus [44]. To facilitate statistical analysis of protein quantities between samples, e.g. for differential expression studies, missing peptide values may be imputed. This can be done at peptide level, prior to summation to protein intensities, or at protein level (see e.g. [44]). Imputation procedures are based either on the assumption that a missing value is missing “not at random” (i.e., it has missed a detection threshold and can be assumed to be of small intensity), of “completely at random” (i.e., its original intensity can be low or high and its level of intensity not known a priori). In the former case, the missing value is usually replaced by drawing a random number located near the lower left of the sample intensity distribution. In the latter case, the missing value could be replaced by calculating the maximum likelihood value from all other values. Imputing has the advantage of simulating natural variance and delivering log fold changes with a statistical measure even when a peptide or protein is absent from one condition. This approach may however be source of errors, e.g. when missing peptides are imputed in knockout compared to wildtype strains, or when strains expressing or not a transgene are compared (see. e.g. [45]). A good practice for setting significance criteria imposes a minimum log2 fold change of ≥ 1, a low value of the type I error after adjustment for the multiplicity, in general lower than 0.05. Repeating the imputation several times and recording significance is a means to mitigate the stochasticity introduced by it, as detailed elsewhere [46]. Where two different normalized parameters are used for quantification of differentially expressed proteins (e.g. LFQ and Top3), belief in the data can be enhanced by considering only proteins with significantly different levels by both parameters. In any case, both the identification and the relative quantification of proteins by LC-MS/MS are probabilistic.
3. Affinity Based Target Deconvolution
3.1. Functional and Binding Assays Using Isolated Proteins
Certainly the most straigtforward strategy to investigate the interactions between anti-infective compounds and their targets is based on functional or binding assays in the presence of isolated - in most cases recombinant–enzymes or receptors. In vitro functional assays allow identifying and optimizing ligands. This strategy has some success in human vascular diseases [47], and high throughput screening systems are available [48]. Another popular example of this strategy is the fight against HIV where high-troughput screenings for inhibitors of essential steps of viral netry and proliferation are ongoing [49,50]. Well-known examples are the mandatory screening of drug candidates for interactions with the human ether-a-go-go K+ channel 1 (hERG1) in order to reduce the risk of potential side-effects [51,52] or the screening for compounds binding to acetylcholine receptors [53].
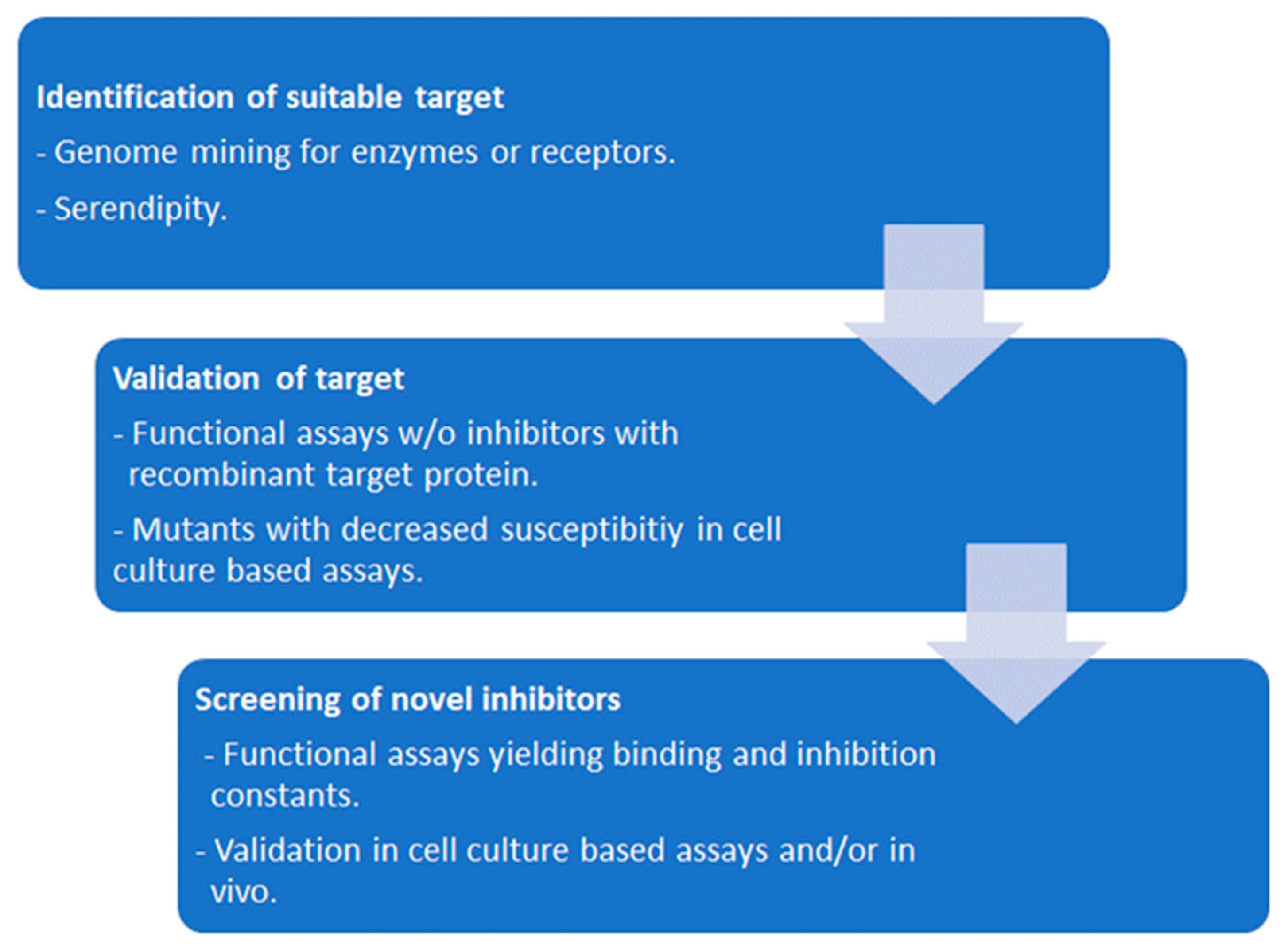
In the case of antiptotozoal compounds, the proteins of interest should be essential for the pathogen and irrelevant for the host. The first antiprotozoal compounds have been discovered by trial-and error testing of the available pharmacopeia, or just by serendipity. A good example is the discovery of artemisin antimalarials [54,55]. Modern target-based antiprotozoal drug development is based on the identification of a suitable target essential for the pathogen, but absent from the host by genome mining, often combined to in-silico modelling. Recombinant target proteins are produced in a suitable system, and functional assays are performed in oder to determine and optimize binding and inhibition constants of drug candidates. Of course, this method can be applied also to targets of existing drugs empirically identified, such as for instance anti-folates in the case of toxoplasmosis [56]. Ideally, the target is validated by overexpression and /or knock-out studies in a suitable model. Figure x represents this workflow.
Figure 2.
Workflow of antiprotozoal compound screening based on functional assays with recombinant proteins.
Figure 2.
Workflow of antiprotozoal compound screening based on functional assays with recombinant proteins.
If functional assays cannot be performed, ligand binding may be investigated using temperature shift assays. In fact, proteins with bound ligands are more resistant to chemical or thermic denaturation. Consequently, they remain in solution/soluble under conditions where proteins without ligands aggregate and can be precipitated by centrifugation [57]. Receptor and transporter antagonists are screened in whole-cell-systems (e.g. human embryonic kidney cells 293T) expressing the respective recombinant target proteins [58,59]. Table 1 gives an overview of a selected recent studies of inhibitor screenings in protozoal parasites.
As illustrated by this table, enzymes are elegant tools to identify suitable inhibitors via medium to high throughput screenings. This strategy has, however its pitfalls, in particular if the target has been identified by mere genome mining. An example for such a pitfall is a phosphodiesterase (PDE) homolog identified in the genome of Giardia lamblia [74]. The recombinant catalytic domain of this homolog expressed in yeast is inhibited by a series of inhibitors some of which also inhibit proliferation of Giardia trophozoites in culture suggesting that this enzyme may be a potential drug target against giardiasis [75]. The detail overlooked by the authors is that the protein encoded by the corresponding open reading frame is not present in trophozoites cultured under the same conditions as in the drug screenings (see the supplementary data e.g. in [76]). The fact that only PDE inhibitors with nitro groups inhibit proliferation [77] further reduces belief in PDE as a potential anti-giardial drug target. In fact, organic compounds containing nitro groups such as metronidazole or nitazoxanide commonly inhibit Giardia and other anaerobic organisms [78].
3.2. Affinity Chromatography
The main disadvantage of the strategy described above is that it is biased by the hypothesis concerning the mode of action and thereby the target of a class of compounds of interest. Therefore, in order to identify unkonwn targets, unbiased hypothesis-free strategies are required. These chemo-proteomic strategies exploit the binding affinities of proteins within the entire proteome of the the pathogen as well as of the host, in cases where side-effects are investigated [79]. The oldest and best known method is certainly affinity chromatography on a low or medium pressure liquid chromatography system. For this approach, the compound of interest is coupled onto an agarose or sepharose (trade mark by Pharmacia, now Cytiva, Marlborough, Ma, USA) column matrix via active groups such as epoxy, cyanogen bromide [80], carbomethyl, N-hydroxy-succinimide [81] or others attached via a spacer [82] thus allowing free access by binding proteins. Cell-free crude extracts are then passed through the column. After abundant washes, binding proteins are eluted by ligands in solution, via pH shift or increasing ionic strength. The eluted proteins are polished, e.g. by gel electrophoresis followed by subsequent sequencing of the most prominent bands, or can be directly identified by LC-MS/MS. A classical example of drug binding proteins of pathogens identified by affinity chromatography and confirmed via binding and mutant studies are penicillin binding proteins in Escherichia coli [83,84,85]. To enhance belief in the identified binding proteins as potential targets, controls minimizing false positive results due to unspecific binding, are paramount. This is valid for each methodology presented here.
Given the socio-economic importance of malaria, it is not surprising that a large number of chemo-proteomic studies haven been performed with mind to identify targets of either well-established or novel antimalarials, as reviewed elsewhere [79]. A selection of studies based on affinity chromatography is listed in Table 2.
During the last two decades, we have performed several target deconvolution studies in protozoal pathogens. In order to minimize the number of unspecific binding proteins identified due to the increased sensitivity of proteomic techniques, we perform differential affinity chromatography (DAC), where identical cell-free extracts are passed through a mock-coated column followed by a column coated either with the effective compound or with a ligand with high structural similarity, but ineffective against the pathogen, both columns mounted in tandem [56]. The workflow of DAC is illustrated in Figure 3.
The datasets of the studies obtained with entire eluates are much larger than those based on bands or spots excised from polyacrylamide gels (see also Table 3). This suggests that the concept “one drug – one target” may be too simplistic or at least very optimistic. The fact that – where analyzed – a good number of host cell proteins is identified within the affinoproteomes of anti-parasitic drugs throws a shadow on the claims of “specificity” of the respective compounds. A list of these studies is given in Table 3.
3.3. In-Situ-Binding
A main objection to affinity chromatography as a suitable method of target identification is that the interaction of compounds and binding proteins occurs in cell-free-extracts on columns and not under physiological conditions. Consequently, chemo-proteomic strategies have been developed, which circumvent this inconvenience via intracellular, thus in-situ or “bio-orthogonal” interactions of compounds with binding proteins. In one approach, the compounds of interest are conjugated to a suitable linker such as trans-cyclooctene [100] or biotin [101] and incubated with the cells of interest. After convenient time periods, the cells are lyzed, and the cell lysates are incubated with magnetic beads coated with a suitable linker (tetrazin-streptavidin [100] or streptavidin [101]) followed by pull-down and identification of the binding proteins by LC-MS/MS. Another approach uses ferric gold nanoparticles coated with the compound of interest [102]. In a different strategy, compounds of interest are conjugated to fluorophores and in-situ UV-cross-linked to their binding protein partners. The cell lysates are then separated via SDS-PAGE. The fluorescent bands are isolated and the proteins identified by LC-MS/MS [103]. Bio-orthogonal studies using photoactive derivatives have identified binding proteins of various antimalarials, such as albitiazolium [104], artemisinin [105], diaminoquinazoline [106], or, more recently, probes for tagging plasmepsins [107].
3.4. Thermal Proteome Profiling
The methodologies described above have the disadvantage that modified compounds are used to identify binding proteins and not the orginial compound of interest. Rather, proteins binding to the original compound in situ, thus under native conditions, should be identified. This is possible by taking advantage of the biophysical property of proteins to have a higher resistance to thermal denaturation when bound to ligands. In other words, proteins with ligands tend to denature and thus precipitate at higher temperatures than the same proteins without ligands, as illustrated by interaction with a simple fluorescent probe binding to hydrophobic residues of proteins [108]. This thermal proteome profiling (TPP) is performed using various experimental set-ups. In cellular thermal shift assays (CETSA) [109], cells are incubated with the compounds to be tested, lysed and the lysates are incubated for a given time period in a thermal cycler at various temperatures starting at 37°C [110]. Precipitated proteins are removed by centrifugation, and the supernatants are subjected to suitable functional assays [110], to one or two-dimensional gel electrophoresis [111] or to LC-MS/MS [112,113]. Classical CETSA-MS is performed by incubating cells or lysates with the compounds to be tested or with a solvent control at ten different temperatures followed by centrifugation of the precipitates, trypsinization and labelling of the soluble proteins with commercially available TMTs [114] or alternative labelling agents [115] and LC-MS analysis of the ten combined compound and control samples [116]. Given the large number of samples to be analyzed, this protocol is, however, limited by manpower needed for running the system and subsequent data analysis. Consequently, simplified protocols have been developed minimizing the amounts of samples and time. One of these protocols is the isothermal shift assay (iTSA) performing thermal denaturation at a single temperature preselected for the proteome of interest [117]. Another protocol is the single tube-TPP with uniform progression (STTPP-UP). Here, incrementally heating of a single sample is applied thereby saving time and material [118]. Generally, TPP is well suited for soluble proteins. After biotinylation-based enrichment of cell surface proteins, this method can, however, be applied to surface proteins such as receptors, as well [119]. Resistance to chemical denaturation, e.g. by solvents constitues an alternative to thermal denaturation [120]. In each experimental set-up, binding proteins are identified based on their significantly increased amounts in samples with ligand vs. samples without ligand under conditions denaturing the protein in the absence of ligands.
In the studies quoted above, TPP has been applied on mammalian cells. During the last decade, a couple of studies using TPP have been performed with mind to target identification in protozoal parasites. These studies are summarized in Table 4.
When evaluating thermal shift based methodologies with respect to drug target deconvolution, one must bear in mind that thermal stability of some proteins is also affected by binding to natural ligands such as nucleic acids and is subjected to intrinsic variation, e.g. during the cell cycle [126]. Consequently, it is imaginable that proteins binding to nucleic acids such as ribosomal proteins for instance may loose their natural target upon interactions with xenobiotics and therefore show up in the precipitated rather than the soluble fractions of TPP assays. Moreover, compounds such as transition metal ions may stabilize proteins in solution thereby generating false positive results (see e.g. [123]). A summary of affinity based methods is given in Table 5.
4. Analysis of Resistant Strains
4.1. General Considerations
A complementary strategy the mode of action of antiparasitic compounds is the analysis of resistant strains. Before going into detail, we have to define what we mean by “resistance” to a given compound. The operational definition of resistance depends on the determination of drug efficacy. One way to determine drug efficacy is to expose the organism of interest to increasing concentrations of the compounds of interest and to measure the proliferation using appropriate tools such as reporter strains (see e.g. [127] for a detailed review on T. gondii drug screening). Based on the proliferation data, the concentration corresponding to half of the proliferation obtained in absence of the compound, “the inhibitory concentration 50 %” or IC50 or other ICs e.g. IC90) is then calculated by appropriate algorithms [127]. These values are compared to corresponding data obtained with suitable host cell lines (often referred to as “effective concentration” or EC50). The higher the EC50/IC50 ratio, the more “promising” the compound seems to be and the more efforts will be spent on subsequent investigations such as in vivo studies including this compound. These IC50s are, however, only one part of the story. Another parameter of similar importance is the “minimal inhibitory” concentration (MIC), i.e. the concentration at which proliferation does not occur, anymore (in other words, the IC100). Unlike other ICs, this IC100 or the MIC cannot be calculated by extrapolation from growth assay data by suitable algorithms [128]. It has to be determined by visual investigation of individual in vitro cultures exposed to increasing concentrations of the compound of interest. Another, perhaps more elegant, way is to remove the drug pressure after a convenient time period and to screen for regrowth of the pathogen. Obviously, in the case of intracellular parasites, long term exposure of the compound of interest must not harm the host cells. There is no other way to determine the MIC. Resistance to a given compound can now be defined by higher IC50s and higher MICs in resistant as compared to susceptible strains [129]. If only the IC50 is increased, but not the MIC, the corresponding strains tolerate the compound or have adapted to it, but are not resistant (see e.g. [130] or [131] for examples). Moreover, to allow robust investigation of the resistant strains including the preparation of single clones, the resistance must not be lost in the absence of drug pressure. Due to their metabolic plasticity, eukaryotic cells (including protozoal parasites) may simply escape to drug pressure by switching to a dormant stage, as observed in the case of Plasmodium sp. exposed to artemisinin [132]. In this case, the terms “resilience” or “tolerance” are more adequate than “resistance”[133]. Another example for resilience is given by intracellular T. gondii or N. caninum exposed to calcium-dependent kinase inhibitors. Whereas infection of cell cultures is inhibited by these compounds at sub-micromolar concentrations, treatment of infected cells even by one magnitude higher concentrations does not lead to parasite death, but to inhibition of egress. Subsequently, the intracellular parasites generate multinucleated complexes (see e.g. [134]) characterized by a downregulation of more than 50% of the identified unique proteins and upregulation of a few proteins typical for dormant stages [135]. This observation cannot be generalized to all apicomplexans. C. parvum, for instance, is killed very efficiently by calcium-dependent kinase inhibitor treatments, even when applied post-infection [99]. This illustrates that (ultra)structural investigation of organisms treated with a given compound is paramount for correctly appreciating its effects.
4.2. Resistance of Transgenic Strains
The most straightforward strategy to validate potential drug targets (see Figure 3) is to perform knock-out or knock-in studies using appropriate reverse genetic tools. Overexpressing genes of interest using integrative or episomal plasmids is certainly the oldest of these methods and well established for G. lamblia [136], Plasmodium sp. [137,138], T. gondii [139] and related protozoans [140]. Knock-down or silencing of genes of interest is less straightforward. RNA interference by degradation of double stranded RNA occurs in the excavata G. lamblia, Leishmania sp., and Trypanosoma brucei, as well as in the apicomplexa Plasmodium sp. and T. gondii [141], but has been supplanted as a major tool by gene editing using the CRISPR/Cas9 system in T. gondii [142,143], where a genome-wide screening using this method has allowed to distinguish between essential and non-essential genes [144].
If the target catabolizes the compound of interest, overexpressors are more resistant than wildtype strains. A textbook example for such “resistance markers” are enzymes inactivating antibiotics such as beta-lactamases or neomycin phosphotransferases. Encoded by resistance plasmids (R plasmids or R factors), they spread through microbial communities and are considered as a major health problem [145].
If the target protein activates the compound, overexpressors are more susceptible than wildtype strains. A good example are nitroreductases from microaerophilic or anaerobic pathogens such as e.g. G. lamblia [146]. The Giardia genome contains open reading frames (ORFs) encoding at least four multifunctional quinone reductases with the ability to transfer electrons not only to quinones, but also to nitro compounds thereby recycling NAD or NADP cofactors ([90,91,147]). The nitroreductases encoded by ORF 22677 and ORF 15307 increase the susceptibility to the nitro compound nitazoxanide when overexpressed in G. lamblia trophozoites and to metronidazole when overexpressed in E. coli [91]. Into the same direction goes the overexpression of gain-of function genes. The widely used overexpression of dihydrofolate reductase (DHFR) as a resistance marker in plasmids or in CRISPR/Cas9 gene editing constructs is such an example. The antimalarial pyrimethamine inhibits the bifunctional wildtype DHFR-thymidylate synthases of Plasmodium sp. and other protozoans such as T. gondii [63]. Overexpressors of mutated enzymes are more resistant to pyrimethamine and can be selected for on pyrimethamine containing media.
Moreover, overexpression of potential drug targets may elicit “compensatory reactions” conferring drug adaptation or resistance, as shown for several, but, of course, not all antibiotics [148]. When analyzing data obtained with transgenic strains, one should keep in mind that these strains underwent a more or less challenging selection for resistance against drugs effective against this parasite. Therefore, it is not surprising to observe an alteration of proteome patterns that may affect metabolism when comparing transgenic strains with wildtypes. For instance, a Giardia line overexpressing E. coli glucuronidase A differs by nearly 10 % of the detected unique proteins from the corresponding wildtype with a major focus on altered patterns of surface proteins, referred to as “antigenic variation” [45]. Moreover, T. gondii knock-out clones of the surface antigen SAG1 created by gene editing show distinct strain-specific proteome patterns besides the intended knock-down [149].
4.3. Differential Analysis of the Proteomes of Susceptible vs Resistant Strains
An alternative to the analysis of engineered stains overexpressing or silencing defined target proteins is the comparison of susceptible strains to strains resistant to a compound of interest. This strategy does not hold only for resistant pathogens, but also for the analysis of chemotherapy resistant cancer cell lines [150]. This type of study can be performed on clinical isolates [151] of a given pathogen or on resistant laboratory strains created by increasing drug concentrations in the culture medium or by chemical mutagenesis. Clearly, the analysis of clinical isolates corresponds more to “reality”, but the creation of resistant strains under laboratory conditions offers the following advantages: i.) Resistant clinical isolates can only be obtained for established drugs, not for experimental compounds. ii.) Resistance can be induced using a reference strain with well-established molecular genetic tools. iii.) The process of resistance formation can be monitored. iv.) The corresponding wildtype strain can be maintained and cultivated under the same standardized conditions as the resistant strains (except the absence of drug in the culture medium).
The fundamental paradigm of this strategy is the investigation of Plasmodium strains resistant to antimalarials widely applied for prophylaxis and treatment. Fostered by genome sequencing efforts more than two decades ago [152], genomic, transcriptomic and genomic investigations have led to the identification of several gene products associated with resistance. The take home message from these studies is that – concerning resistance to artemisinin in particular - there is no single gene or marker associated with resistance. Rather, several genes show differences with respect to point mutations or differential expression between susceptible and resistant strains as highlighted by seminal review articles [153,154,155,156].
This view is fostered by analyses of resistant G. lamblia strains induced under laboratory conditions. Prima vista, the analysis of resistant patient isolates would be closer to reality, but depending on the genotypes, the isolates are difficult to maintain in culture. Moreover, resistance may be lost after en- and excystation [157] thereby precluding isolation of resistant strains from cyst containing feces. Resistance to nitro drugs such as metronidazole and nitazoxanide can easily be induced by increasing drug concentrations in trophozoite culture medium followed by cloning and characterization of the resistant clones [158,159]. Our knowledge about anaerobic metabolism of Giardia and other anaerobic organisms suggests three strategies for nitro drug resistance, namely restriction of electrons available for nitro reduction by downregulation of glycolysis, by downregulation of enzymes reducing nitrocompounds (nitroreductases) or by upregulation of enzymes scavenging radicals issuing from nitro reduction [146,160]. When comparing the proteome patterns of nitro drug resistant clones from different origins, it is striking that all three strategies are found depending on the resistant strains and the nitro compounds used for selection [161]. The results only partially match previously published results on differential transcriptomics [162]. A summary of selected studies illustrating this strategy is given in Table 6.
As in the case of the target deconvolution studies, the overview of studies of differentially expressed proteins in resistant vs. susceptible strains presented in Table 6 reveals a large number of differentials thus of candidate proteins involved in the mode of action of the respective drugs.
5. Combining Evidence from Chemoproteomics and Whole-Cell Proteomics
So far, research designs for drug target identification that combine proteomic, in silico and genetic approaches are gaining popularity. Is there an added value of merging outcomes of more than one proteomic approach? In other words: Is it possible to combine the two strategies discussed through this review, namely target deconvolution studies and whole-cell proteomes of resistant or resilient strains by mass spectrometry? A combination of both approaches within one model system could enhance belief in a mode of action or in the targets identified. Such studies are, however, rare. One approach is to compare the affinoproteomes of drug susceptible and resistant strains by a convenient method such as TPP. A first step is the analysis of thermal shifts of a favored target in susceptible and resistant strains, as exemplified for T. gondii susceptible or resistant to bumped kinase inhibitors and their kinase target [167]. This approach is, however, confirmative rather than explorative since the kinase target – shown by previous kinetic studies – is confirmed by another method and other more systemic effects of the compound are neglected. The study comparing the affinoproteomes of L. infantum susceptible or resistant to various antileishmanials [123] is an example for an explorative study. As mentioned in Table 4, the analysis of the affinoproteomes by TPP has revealed differentials in a good number of various proteins, previously not suspected as potential targets. How could the output of a study comparing drug binding proteins and differentially expressed proteins in the same model system look like? If drug metabolizers are the only binding proteins, one would expect to find them amongst the differentials, as well. However, if components of the cellular machinery essential for survival and therefore tolerating only minor changes in expression are the main binding proteins, one would not expect them amongst the differentials. Rather, drug metabolizers or transporters [168] would be expected here.
6. Conclusions
Nowadays, there is a steadily increasing number of scientific articles claiming drug targets merely based on computer simulations of interactions between a single target and a single molecule. The articles reviewed above show that the identification of drug targets and therefore the development of reason-based chemotherapies is more complicated. It is not surprising to find multiple proteins as drug interaction partners in protozoan parasites suggesting that the drug kills the parasite by “…disrupting its biological landscape…” as stated in a review on artemisinin binding proteins in P. falciparum [169]. Indeed, the examples of studies mentioned above suggest that multiple targets interact with antiprotozoal compounds, not only the usual suspects such as specific enzymes investigated via functional assays, but also components of general cellular maintenance such as cytoskeletal and ribosomal proteins, or proteins involved in replication and transcription. This suggests that multigenic adaptation or resistance mechanisms get going upon exposure to these compounds and may even be part of developmental programs established during evolution as countermeasures to exposure to xenobiotics affecting fitness. The formation of multinucleated complexes in intracellular apicomplexans upon exposure to bumped kinase inhibitors may follow such a program [134].
During evolution, protozoan pathogens have learned to cope with hostile environments generated by innate or acquired host immunity, as reviewed elsewhere [170]. The onset of antigenic variation in Giardia [171] as a response to both drug [161] and immune [172] pressure is a good example and is worth being further exploited as a model system.
Although chemoproteomics, using affinity chromatography has the potential to display a large set of drug binding proteins, the question how to distinguish “true” targets from drug-binding proteins remains. Certainly, experimental validation based on knockdown, knockout or overexpression of the target gene is feasible. Now, taken into consideration in one hand the off target molecular effects (see e.g. [149]) and the untrustworthy concept of one-drug for one-target for one-disease approach in other hand, experimental validation of drug target using genetic tools may ultimately leave more questions than answers.
Consequently, state-of-the-art proteomics play an essential, but not unique role in unraveling these mechanisms. More complex approaches involving e.g. ultrastructural and metabolomic investigations of drug treated vs. untreated and of resistant vs. susceptible strains are important to draw a more complete picture. Metabolomic investigations may even be performed on intact cells by 1H-NMR, as shown for Giardia, which again reveals its suitability as a model system to study resistance formation in protozoans [173]. This brings us to the final statement that the key for progress in studying drug target interactions and resistance formation in protozoans in particular and in any empirical science in general is the choice of standardized model systems, well-suited for the application of state-of-the-art laboratory scale methodology.
Author Contributions
“Conceptualization, J.M. and A.H.; methodology, A.C.U., S.B.-L., M.H..; writing—original draft preparation, J.M.; writing—review and editing, J.M., G. B, A.C.U., S.B.-L., M.H.. N.M., A.H.; supervision, A.H.; project administration, A.H.; funding acquisition, A.H. “All authors have read and agreed to the published version of the manuscript.”.
Funding
“This research and the APC were financed by Swiss National Science Foundation, grant number 310030_214897.”.
Institutional Review Board Statement
“Not applicable”.
Informed Consent Statement
“Not applicable.”.
Data Availability Statement
New data were not created.
Conflicts of Interest
“The authors declare no conflicts of interest.”.
References
- Lindsay, M.A. Target discovery. Nat Rev Drug Discov 2003, 2, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Paananen, J.; Fortino, V. An omics perspective on drug target discovery platforms. Brief Bioinform 2020, 21, 1937–1953. [Google Scholar] [CrossRef] [PubMed]
- Tsukidate, T.; Li, Q.; Hang, H.C. Targeted and proteome-wide analysis of metabolite-protein interactions. Curr Opin Chem Biol 2020, 54, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Dixon, B.R. Giardia duodenalis in humans and animals - Transmission and disease. Res Vet Sci 2021, 135, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.K.; Rinkenberger, N.; Dunay, I.R.; Sibley, L.D. Toxoplasma gondii infection and its implications within the central nervous system. Nat Rev Microbiol 2021, 19, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Ryan, U.M.; Feng, Y.; Fayer, R.; Xiao, L. Taxonomy and molecular epidemiology of Cryptosporidium and Giardia - a 50 year perspective (1971-2021). Int J Parasitol 2021, 51, 1099–1119. [Google Scholar] [CrossRef] [PubMed]
- Marin-Garcia, P.J.; Planas, N.; Llobat, L. Toxoplasma gondii in foods: prevalence, control, and safety. Foods 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Nayeri, T.; Moosazadeh, M.; Sarvi, S.; Daryani, A. Neospora caninum infection in aborting bovines and lost fetuses: A systematic review and meta-analysis. PLoS One 2022, 17, e0268903. [Google Scholar] [CrossRef] [PubMed]
- Reichel, M.P.; Wahl, L.C.; Ellis, J.T. Research into Neospora caninum-What Have We Learnt in the Last Thirty Years? Pathogens 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, B.M.; Bojang, K.; Whitty, C.J.; Targett, G.A. Malaria. Lancet 2005, 365, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Understanding the replication crisis as a base rate fallacy. British Journal for the Philosophy of Science 2021, 72, 965–993. [Google Scholar] [CrossRef]
- Edman, P. A method for the determination of amino acid sequence in peptides. Arch Biochem 1949, 22, 475. [Google Scholar]
- Rabilloud, T. Paleoproteomics explained to youngsters: how did the wedding of two-dimensional electrophoresis and protein sequencing spark proteomics on: let there be light. J Proteomics 2014, 107, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Downard, K.M. Historical account: Francis William Aston: the man behind the mass spectrograph. Eur J Mass Spectrom (Chichester) 2007, 13, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Biemann, K.; Seibl, J.; Gapp, F. Mass spectrometric identification of amino acids. Biochemical and Biophysical Research Communications 1959, 1, 307–311. [Google Scholar] [CrossRef]
- Karas, M.; Bachmann, D.; Hillenkamp, F. Influence of the wavelength in high-irradiance ultraviolet-laser desorption mass-spectrometry of organic-molecules. Analytical Chemistry 1985, 57, 2935–2939. [Google Scholar] [CrossRef]
- Shimada, T.; Toyama, A.; Aoki, C.; Aoki, Y.; Tanaka, K.; Sato, T.A. Direct antigen detection from immunoprecipitated beads using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; a new method for immunobeads-mass spectrometry (iMS). Rapid Commun Mass Spectrom 2011, 25, 3521–3526. [Google Scholar] [CrossRef] [PubMed]
- Sedo, O.; Roblickova, A.; Jezek, F.; Gintar, P.; Kamenik, J.; Zdrahal, Z. Discriminatory power of MALDI-TOF MS protein profiling analysis of pork meat and meat products. Food Chem 2024, 449, 139155. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wu, P.; Zhao, C.; Zheng, F.; Hu, C.; Lu, X.; Xu, G. Protein profiling analysis based on matrix-assisted laser desorption/ionization-Fourier transform ion cyclotron resonance mass spectrometry and its application in typing Streptomyces isolates. Talanta 2020, 208, 120439. [Google Scholar] [CrossRef] [PubMed]
- Horisawa, S.; Iwamoto, K. Identification and typing of strains of wood-rotting basidiomycetes by protein profiling using MALDI-TOF MS. BioTech (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Chivte, P.; LaCasse, Z.; Seethi, V.D.R.; Bharti, P.; Bland, J.; Kadkol, S.S.; Gaillard, E.R. MALDI-ToF protein profiling as a potential rapid diagnostic platform for COVID-19. J Mass Spectrom Adv Clin Lab 2021, 21, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tan, C.; Zenobi, R. Rapid profiling of the glycosylation effects on the binding of SARS-CoV-2 spike protein to angiotensin-converting enzyme 2 using MALDI-MS with high mass detection. Anal Chem 2024, 96, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.J.; Spraggins, J.M.; Caprioli, R.M. Protein identification strategies in MALDI imaging mass spectrometry: a brief review. Curr Opin Chem Biol 2019, 48, 64–72. [Google Scholar] [CrossRef] [PubMed]
- James, P.; Quadroni, M.; Carafoli, E.; Gonnet, G. Protein identification by mass profile fingerprinting. Biochem Biophys Res Commun 1993, 195, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Cox, J.; Mann, M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res 2011, 10, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Damoc, E.; Lange, O.; Denisov, E.; Nolting, D.; Muller, M.; Viner, R.; Schwartz, J.; Remes, P.; Belford, M.; et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics 2012, 11, O111–013698. [Google Scholar] [CrossRef] [PubMed]
- Shishkova, E.; Hebert, A.S.; Coon, J.J. Now, more than ever, proteomics needs better chromatography. Cell Syst 2016, 3, 321–324. [Google Scholar] [CrossRef]
- May, J.C.; Goodwin, C.R.; McLean, J.A. Ion mobility-mass spectrometry strategies for untargeted systems, synthetic, and chemical biology. Curr Opin Biotechnol 2015, 31, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Orsburn, B.C. Single cell proteomics by mass spectrometry reveals deep epigenetic insight and new targets of a class specific histone deacetylase inhibitor. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.W.F.; Reiner, J.E. The utility of nanopore technology for protein and peptide sensing. Proteomics 2018, 18, e1800026. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Penkauskas, T.; Reiner, J.E.; Kennard, C.; Uline, M.J.; Wang, Q.; Li, S.; Aksimentiev, A.; Robertson, J.W.F.; Liu, C. Engineering biological nanopore approaches toward protein sequencing. ACS Nano 2023, 17, 16369–16395. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Aebersold, R.; Chen, R.; Rush, J.; Goodlett, D.R.; McIntosh, M.W.; Zhang, J.; Brentnall, T.A. Mass spectrometry based targeted protein quantification: methods and applications. J Proteome Res 2009, 8, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Shuken, S.R. An Introduction to mass spectrometry-based proteomics. J Proteome Res 2023, 22, 2151–2171. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Haynes, S.E.; Teo, G.C.; Avtonomov, D.M.; Polasky, D.A.; Nesvizhskii, A.I. Fast quantitative analysis of timsTOF PASEF data with MSFragger and IonQuant. Molecular and Cellular Proteomics 2020, 19, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Wilhelm, M.; Hahne, H.; Kuster, B.; Bantscheff, M. A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol Cell Proteomics 2015, 14, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.L.; Yu, F.; Teo, G.C.; Li, K.; Demichev, V.; Ralser, M.; Nesvizhskii, A.I. MSBooster: improving peptide identification rates using deep learning-based features. Nat Commun 2023, 14, 4539. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.; Gabriel, W.; Laukens, K.; Picciani, M.; Wilhelm, M.; Bittremieux, W.; Boonen, K. Fragment ion intensity prediction improves the identification rate of non-tryptic peptides in timsTOF. Nat Commun 2024, 15, 3956. [Google Scholar] [CrossRef] [PubMed]
- Braga-Lagache, S.; Buchs, N.; Iacovache, M.I.; Zuber, B.; Jackson, C.B.; Heller, M. Robust label-free, quantitative profiling of circulating plasma microparticle (MP) associated proteins. Mol Cell Proteomics 2016, 15, 3640–3652. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Bouyssié, D.; Menneteau, T.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Comparison of label-free quantification methods for the determination of protein complexes subunits stoichiometry. EuPA Open Proteomics 2014, 4, 82–86. [Google Scholar] [CrossRef]
- Macleod, A.K.; Zang, T.; Riches, Z.; Henderson, C.J.; Wolf, C.R.; Huang, J.T. A targeted in vivo SILAC approach for quantification of drug metabolism enzymes: regulation by the constitutive androstane receptor. J Proteome Res 2014, 13, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Sanchez, J.C. Relative protein quantification by MS/MS using the tandem mass tag technology. Methods Mol Biol 2012, 893, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Stepath, M.; Zulch, B.; Maghnouj, A.; Schork, K.; Turewicz, M.; Eisenacher, M.; Hahn, S.; Sitek, B.; Bracht, T. Systematic Comparison of Label-Free, SILAC, and TMT Techniques to Study Early Adaption toward Inhibition of EGFR Signaling in the Colorectal Cancer Cell Line DiFi. J Proteome Res 2020, 19, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.; Braga, S.; Müller, N.; Müller, J. Transfection with plasmid causing stable expression of a foreign gene affects general proteome pattern in Giardia lamblia trophozoites. Front Cell Infect Microbiol 2020, 10, 602756. [Google Scholar] [CrossRef] [PubMed]
- Uldry, A.C.; Maciel-Dominguez, A.; Jornod, M.; Buchs, N.; Braga-Lagache, S.; Brodard, J.; Jankovic, J.; Bonadies, N.; Heller, M. Effect of sample transportation on the proteome of human circulating blood extracellular vesicles. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Waduge, P.; Tian, H.; Webster, K.A.; Li, W. Profiling disease-selective drug targets: From proteomics to ligandomics. Drug Discov Today 2023, 28, 103430. [Google Scholar] [CrossRef] [PubMed]
- Neun, S.; Zurek, P.J.; Kaminski, T.S.; Hollfelder, F. Ultrahigh throughput screening for enzyme function in droplets. Methods Enzymol 2020, 643, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zankharia, U.; Cassel, J.A.; Lu, F.; Salvino, J.M.; Lieberman, P.M.; Collman, R.G. A high-throughput screening assay for silencing established HIV-1 macrophage infection identifies nucleoside analogs that perturb H3K9me3 on proviral genomes. J Virol 2023, 97, e0065323. [Google Scholar] [CrossRef] [PubMed]
- Taoda, Y.; Sugiyama, S.; Seki, T. New designs for HIV-1 integrase inhibitors: a patent review (2018-present). Expert Opin Ther Pat 2023, 33, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, A.; Sala, C.; Altadonna, G.C.; Becchetti, A.; Arcangeli, A. High throughput clone screening on overexpressed hERG1 and Kv1. 3 potassium channels using ion channel reader (ICR) label free technology. Heliyon 2023, 9, e20112. [Google Scholar] [CrossRef] [PubMed]
- Diaz, G.J.; Daniell, K.; Leitza, S.T.; Martin, R.L.; Su, Z.; McDermott, J.S.; Cox, B.F.; Gintant, G.A. The [3H]dofetilide binding assay is a predictive screening tool for hERG blockade and proarrhythmia: Comparison of intact cell and membrane preparations and effects of altering [K+]o. J Pharmacol Toxicol Methods 2004, 50, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Sichler, S.; Hofner, G.; Nitsche, V.; Niessen, K.V.; Seeger, T.; Worek, F.; Paintner, F.F.; Wanner, K.T. Screening for new ligands of the MB327-PAM-1 binding site of the nicotinic acetylcholine receptor. Toxicol Lett 2024. [Google Scholar] [CrossRef] [PubMed]
- Loo, C.S.; Lam, N.S.; Yu, D.; Su, X.Z.; Lu, F. Artemisinin and its derivatives in treating protozoan infections beyond malaria. Pharmacol Res 2017, 117, 192–217. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat Med 2011, 17, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Hemphill, A. Toxoplasma gondii infection: novel emerging therapeutic targets. Expert Opin Ther Targets 2023, 27, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Kurganov, B.I. Kinetics of protein aggregation. Quantitative estimation of the chaperone-like activity in test-systems based on suppression of protein aggregation. Biochemistry (Mosc) 2002, 67, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Kassack, M.U. Quantitative comparison of functional screening by measuring intracellular Ca2+ with radioligand binding at recombinant human dopamine receptors. AAPS PharmSci 2002, 4, E31. [Google Scholar] [CrossRef] [PubMed]
- Oostendorp, J.; Meurs, H.; Adriaan Nelemans, S.; Zaagsma, J.; Kauffman, H.F.; Postma, D.S.; Boddeke, H.W.; Biber, K. Cloning, pharmacological characterization, and polymorphism screening of the guinea pig beta(2)-adrenoceptor. Eur J Pharmacol 2002, 457, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, F.; Fisher, F.; Milne, R.; Teran, F.S.; Wiedemar, N.; Wrobel, K.; Edwards, D.; Baumann, H.; Gilbert, I.H.; Baragana, B.; et al. High-throughput screening platform to identify inhibitors of protein synthesis with potential for the treatment of malaria. Antimicrob Agents Chemother 2022, 66, e0023722. [Google Scholar] [CrossRef] [PubMed]
- Ojo, K.K.; Larson, E.T.; Keyloun, K.R.; Castaneda, L.J.; Derocher, A.E.; Inampudi, K.K.; Kim, J.E.; Arakaki, T.L.; Murphy, R.C.; Zhang, L.; et al. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nature Structural & Molecular Biology 2010, 17, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Murphy, R.C.; Geiger, J.A.; DeRocher, A.E.; Zhang, Z.; Ojo, K.K.; Larson, E.T.; Perera, B.G.; Dale, E.J.; He, P.; et al. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. J Med Chem 2012, 55, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Vanichtanankul, J.; Yoomuang, A.; Taweechai, S.; Saeyang, T.; Pengon, J.; Yuvaniyama, J.; Tarnchompoo, B.; Yuthavong, Y.; Kamchonwongpaisan, S. Structural insight into effective inhibitors binding to Toxoplasma gondii dihydrofolate reductase thymidylate synthase. ACS Chem Biol 2022, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Djapa, L.Y.; Basco, L.K.; Zelikson, R.; Rosowsky, A.; Djaman, J.A.; Yonkeu, J.N.; Bolotin-Fukuhara, M.; Mazabraud, A. Antifolate screening using yeast expressing Plasmodium vivax dihydrofolate reductase and in vitro drug susceptibility assay for Plasmodium falciparum. Mol Biochem Parasitol 2007, 156, 89–92. [Google Scholar] [CrossRef]
- Jelenska, J.; Sirikhachornkit, A.; Haselkorn, R.; Gornicki, P. The carboxyltransferase activity of the apicoplast acetyl-CoA carboxylase of Toxoplasma gondii is the target of aryloxyphenoxypropionate inhibitors. J Biol Chem 2002, 277, 23208–23215. [Google Scholar] [CrossRef] [PubMed]
- Goo, Y.K.; Yamagishi, J.; Ueno, A.; Terkawi, M.A.; Aboge, G.O.; Kwak, D.; Hong, Y.; Chung, D.I.; Igarashi, M.; Nishikawa, Y.; et al. Characterization of Toxoplasma gondii glyoxalase 1 and evaluation of inhibitory effects of curcumin on the enzyme and parasite cultures. Parasit Vectors 2015, 8, 654. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Kerscher, S.; Saleh, A.; Brandt, U.; Gross, U.; Bohne, W. The Toxoplasma gondii type-II NADH dehydrogenase TgNDH2-I is inhibited by 1-hydroxy-2-alkyl-4(1H)quinolones. Biochim Biophys Acta 2008, 1777, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Nagai, J.; Kurata, R.; Shimizu, K.; Cui, X.; Isagawa, T.; Semba, H.; Ishihara, J.; Yoshida, Y.; Takeda, N.; et al. Establishment of novel high-standard chemiluminescent assay for NTPase in two protozoans and its high-throughput screening. Mar Drugs 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Razakantoanina, V.; Florent, I.; Jaureguiberry, G. Plasmodium falciparum: functional mitochondrial ADP/ATP transporter in Escherichia coli plasmic membrane as a tool for selective drug screening. Exp Parasitol 2008, 118, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Walunj, S.B.; Dias, M.M.; Kaur, C.; Wagstaff, K.M.; Dey, V.; Hick, C.; Patankar, S.; Jans, D.A. High-throughput screening to identify inhibitors of Plasmodium falciparum importin alpha. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.B.; Melillo, B.; Mittal, P.; Sharma, M.; Sharma, A.; Fu, Y.; Uddin, T.; Gonse, A.; Comer, E.; Schreiber, S.L.; et al. Bicyclic azetidines target acute and chronic stages of Toxoplasma gondii by inhibiting parasite phenylalanyl t-RNA synthetase. Nat Commun 2022, 13, 459. [Google Scholar] [CrossRef]
- Bosch, S.S.; Lunev, S.; Batista, F.A.; Linzke, M.; Kronenberger, T.; Domling, A.S.S.; Groves, M.R.; Wrenger, C. Molecular target validation of aspartate transcarbamoylase from Plasmodium falciparum by Torin 2. ACS Infect Dis 2020, 6, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.A.; Gyau, B.; Vilacha, J.F.; Bosch, S.S.; Lunev, S.; Wrenger, C.; Groves, M.R. New directions in antimalarial target validation. Expert Opin Drug Discov 2020, 15, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Jex, A.; Svard, S.G. A chromosome-scale reference genome for Giardia intestinalis WB. Sci Data 2020, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Balmer, V.; Sterk, G.J.; Pollastri, M.P.; Leurs, R.; Müller, N.; Hemphill, A.; Spycher, C. The single cyclic nucleotide-specific phosphodiesterase of the intestinal parasite Giardia lamblia represents a potential drug target. PLoS Negl Trop Dis 2017, 11, e0005891. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Braga, S.; Uldry, A.C.; Heller, M.; Müller, N. Comparative proteomics of three Giardia lamblia strains: investigation of antigenic variation in the post-genomic era. Parasitology 2020, 147, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Müller, J.; Kunz, S.; Siderius, M.; Maes, L.; Caljon, G.; Müller, N.; Hemphill, A.; Sterk, G.J.; Leurs, R. 3-nitroimidazo[1,2-b]pyridazine as a novel scaffold for antiparasitics with sub-nanomolar anti-Giardia lamblia activity. Int J Parasitol Drugs Drug Resist 2022, 19, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D. A review on metronidazole: an old warhorse in antimicrobial chemotherapy. Parasitology 2017, 146, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Bailey, B.L.; Nguyen, W.; Cowman, A.F.; Sleebs, B.E. Chemo-proteomics in antimalarial target identification and engagement. Med Res Rev 2023, 43, 2303–2351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Li, M.; Qiao, Y.; Wang, W.; Ma, L.; Liu, K. Target discovery of bioactive natural products with native-compound-coupled CNBr-activated Sepharose 4B beads (NCCB): Applications, mechanisms and outlooks. Bioorg Med Chem 2023, 96, 117483. [Google Scholar] [CrossRef] [PubMed]
- Lechner, S.; Malgapo, M.I.P.; Gratz, C.; Steimbach, R.R.; Baron, A.; Ruther, P.; Nadal, S.; Stumpf, C.; Loos, C.; Ku, X.; et al. Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common off-target. Nat Chem Biol 2022, 18, 812–820. [Google Scholar] [CrossRef] [PubMed]
- O’Carra, P.; Barry, S.; Griffin, T. Spacer arms in affinity chromatography: use of hydrophilic arms to control or eliminate nonbiospecific adsorption effects. FEBS Lett 1974, 43, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Spratt, B.G.; Pardee, A.B. Penicillin-binding proteins and cell shape in E. coli. Nature 1975, 254, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Spratt, B.G. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A 1975, 72, 2999–3003. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.J.; Strominger, J.L. Purification of penicillin-binding protein 2 of Escherichia coli. J Bacteriol 1981, 145, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, M.; Gray, N.; Damiens, E.; Chang, Y.T.; Grellier, P.; Grant, K.; Fergusson, D.; Mottram, J.; Soete, M.; Dubremetz, J.F.; et al. Intracellular targets of cyclin-dependent kinase inhibitors: identification by affinity chromatography using immobilised inhibitors. Chem Biol 2000, 7, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Kwiek, J.J.; Fadden, P.; Ray, R.; Hardeman, K.; Coley, A.M.; Foley, M.; Haystead, T.A. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol 2002, 62, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Sanai, H.; Hiramoto, A.; Sato, A.; Hiraoka, O.; Sakura, T.; Kaneko, O.; Masuyama, A.; Nojima, M.; Wataya, Y.; et al. Plasmodium falciparum endoplasmic reticulum-resident calcium binding protein is a possible target of synthetic antimalarial endoperoxides, N-89 and N-251. J Proteome Res 2012, 11, 5704–5711. [Google Scholar] [CrossRef]
- Müller, J.; Wastling, J.; Sanderson, S.; Müller, N.; Hemphill, A. A novel Giardia lamblia nitroreductase, GlNR1, interacts with nitazoxanide and other thiazolides. Antimicrob Agents Chemother 2007, 51, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Rout, S.; Leitsch, D.; Vaithilingam, J.; Hehl, A.; Müller, N. Comparative characterisation of two nitroreductases from Giardia lamblia as potential activators of nitro compounds. Int J Parasitol Drugs Drug Resist 2015, 5, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Müller, N. Nitroreductases of bacterial origin in Giardia lamblia: Potential role in detoxification of xenobiotics. Microbiologyopen 2019, e904. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Sidler, D.; Nachbur, U.; Wastling, J.; Brunner, T.; Hemphill, A. Thiazolides inhibit growth and induce glutathione-S-transferase Pi (GSTP1)-dependent cell death in human colon cancer cells. Int J Cancer 2008, 123, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Sidler, D.; Brockmann, A.; Müller, J.; Nachbur, U.; Corazza, N.; Renzulli, P.; Hemphill, A.; Brunner, T. Thiazolide-induced apoptosis in colorectal cancer cells is mediated via the Jun kinase-Bim axis and reveals glutathione-S-transferase P1 as Achilles’ heel. Oncogene 2012, 31, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Hemphill, A. Identification of a host cell target for the thiazolide class of broad-spectrum anti-parasitic drugs. Exp Parasitol 2011, 128, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Basto, A.P.; Müller, J.; Rubbiani, R.; Stibal, D.; Giannini, F.; Suss-Fink, G.; Balmer, V.; Hemphill, A.; Gasser, G.; Furrer, J. Characterization of the activities of dinuclear thiolato-bridged arene ruthenium complexes against Toxoplasma gondii. Antimicrob Agents Chemother 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Anghel, N.; Müller, J.; Serricchio, M.; Jelk, J.; Butikofer, P.; Boubaker, G.; Imhof, D.; Ramseier, J.; Desiatkina, O.; Paunescu, E.; et al. Cellular and molecular targets of nucleotide-tagged trithiolato-bridged arene ruthenium complexes in the protozoan parasites Toxoplasma gondii and Trypanosoma brucei. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Boubaker, G.; Imhof, D.; Hanggeli, K.; Haudenschild, N.; Uldry, A.C.; Braga-Lagache, S.; Heller, M.; Ortega-Mora, L.M.; Hemphill, A. Differential affinity chromatography coupled to mass spectrometry: A suitable tool to identify common binding proteins of a broad-range antimicrobial peptide derived from leucinostatin. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Anghel, N.; Imhof, D.; Hänggeli, K.; Uldry, A.C.; Braga-Lagache, S.; Heller, M.; Ojo, K.K.; Ortega-Mora, L.M.; Van Voorhis, W.C.; et al. Common molecular targets of a quinoline based bumped kinase inhibitor in Neospora caninum and Danio rerio. Int J Mol Sci 2022, 23, 2381–2399. [Google Scholar] [CrossRef] [PubMed]
- Ajiboye, J.; Uldry, A.C.; Heller, M.; Naguleswaran, A.; Fan, E.; Van Voorhis, W.C.; Hemphill, A.; Müller, J. Molecular targets of the 5-amido-carboxamide bumped kinase inhibitor BKI-1748 in Cryptosporidium parvum and HCT-8 Host Cells. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Yang, K.S.; Budin, G.; Tassa, C.; Kister, O.; Weissleder, R. Bioorthogonal approach to identify unsuspected drug targets in live cells. Angew Chem Int Ed Engl 2013, 52, 10593–10597. [Google Scholar] [CrossRef] [PubMed]
- Kusza, D.A.; Hunter, R.; Schafer, G.; Smith, M.; Katz, A.A.; Kaschula, C.H. Activity-based proteomic identification of the S-thiolation targets of ajoene in MDA-MB-231 breast cancer cells. J Agric Food Chem 2022, 70, 14679–14692. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Sun, Q.; Xie, Y.; Zheng, Q.; Ding, Y. Virus-like iron-gold heterogeneous nanoparticles for drug target screening. Anal Chem 2023, 95, 17187–17192. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.Y.; Corson, T.W. Small molecule target identification using photo-affinity chromatography. Methods Enzymol 2019, 622, 347–374. [Google Scholar] [CrossRef] [PubMed]
- Penarete-Vargas, D.M.; Boisson, A.; Urbach, S.; Chantelauze, H.; Peyrottes, S.; Fraisse, L.; Vial, H.J. A chemical proteomics approach for the search of pharmacological targets of the antimalarial clinical candidate albitiazolium in Plasmodium falciparum using photocrosslinking and click chemistry. PLoS One 2014, 9, e113918. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Barton, V.; Phanchana, M.; Charoensutthivarakul, S.; Wong, M.H.; Hemingway, J.; Biagini, G.A.; O’Neill, P.M.; Ward, S.A. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc Natl Acad Sci U S A 2016, 113, 2080–2085. [Google Scholar] [CrossRef]
- Lubin, A.S.; Rueda-Zubiaurre, A.; Matthews, H.; Baumann, H.; Fisher, F.R.; Morales-Sanfrutos, J.; Hadavizadeh, K.S.; Nardella, F.; Tate, E.W.; Baum, J.; et al. Development of a photo-cross-linkable diaminoquinazoline inhibitor for target identification in Plasmodium falciparum. ACS Infect Dis 2018, 4, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Lisauskaite, M.; Nixon, G.L.; Woodley, C.M.; Berry, N.G.; Coninckx, A.; Qie, L.C.; Leung, S.C.; Taramelli, D.; Basilico, N.; Parapini, S.; et al. Design, synthesis and modelling of photoreactive chemical probes for investigating target engagement of plasmepsin IX and X in Plasmodium falciparum. RSC Chem Biol 2024, 5, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.H.; Light, S.H.; Dunne, S.F.; Anderson, W.F. Ligand screening using fluorescence thermal shift analysis (FTS). Methods Mol Biol 2014, 1140, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014, 9, 2100–2122. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.H.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Muroi, M.; Osada, H. Two-dimensional electrophoresis-cellular thermal shift assay (2DE-CETSA) for target identification of bioactive compounds. Methods Enzymol 2022, 675, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Friman, T. Mass spectrometry-based cellular thermal shift assay (CETSA(R)) for target deconvolution in phenotypic drug discovery. Bioorg Med Chem 2020, 28, 115174. [Google Scholar] [CrossRef] [PubMed]
- Sauer, P.; Bantscheff, M. Thermal proteome profiling for drug target identification and probing of protein states. Methods Mol Biol 2023, 2718, 73–98. [Google Scholar] [CrossRef] [PubMed]
- George, A.L.; Sidgwick, F.R.; Watt, J.E.; Martin, M.P.; Trost, M.; Marin-Rubio, J.L.; Duenas, M.E. Comparison of quantitative mass spectrometric methods for drug target identification by thermal proteome profiling. J Proteome Res 2023, 22, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Ren, Y.; Li, S.; Hao, P. Identifying drug targets with thermal proteome profiling using IBT-16plex. Rapid Commun Mass Spectrom 2024, 38, e9673. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 55-+, ARTN 1255784. [Google Scholar] [CrossRef]
- Ball, K.A.; Webb, K.J.; Coleman, S.J.; Cozzolino, K.A.; Jacobsen, J.; Jones, K.R.; Stowell, M.H.B.; Old, W.M. An isothermal shift assay for proteome scale drug-target identification. Commun Biol 2020, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Zijlmans, D.W.; Hernandez-Quiles, M.; Jansen, P.; Becher, I.; Stein, F.; Savitski, M.M.; Vermeulen, M. STPP-UP: An alternative method for drug target identification using protein thermal stability. J Biol Chem 2023, 299, 105279. [Google Scholar] [CrossRef] [PubMed]
- Kalxdorf, M.; Gunthner, I.; Becher, I.; Kurzawa, N.; Knecht, S.; Savitski, M.M.; Eberl, H.C.; Bantscheff, M. Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane. Nat Methods 2021, 18, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, L.; Ye, M. Solvent-induced protein precipitation for drug target discovery. Methods Mol Biol 2023, 2554, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Paradela, L.S.; Wall, R.J.; Carvalho, S.; Chemi, G.; Corpas-Lopez, V.; Moynihan, E.; Bello, D.; Patterson, S.; Guther, M.L.S.; Fairlamb, A.H.; et al. Multiple unbiased approaches identify oxidosqualene cyclase as the molecular target of a promising anti-leishmanial. Cell Chem Biol 2021, 28, 711–721 e718. [Google Scholar] [CrossRef] [PubMed]
- Corpas-Lopez, V.; Wyllie, S. Utilizing thermal proteome profiling to identify the molecular targets of anti-leishmanial compounds. STAR Protoc 2021, 2, 100704. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Meneses, A.V.; Corbeil, A.; Wagner, V.; Beaudry, F.; do Monte-Neto, R.L.; Fernandez-Prada, C. Exploring direct and indirect targets of current antileishmanial drugs using a novel thermal proteomics profiling approach. Front Cell Infect Microbiol 2022, 12, 954144. [Google Scholar] [CrossRef] [PubMed]
- Dziekan, J.M.; Wirjanata, G.; Dai, L.; Go, K.D.; Yu, H.; Lim, Y.T.; Chen, L.; Wang, L.C.; Puspita, B.; Prabhu, N.; et al. Cellular thermal shift assay for the identification of drug-target interactions in the Plasmodium falciparum proteome. Nat Protoc 2020, 15, 1881–1921. [Google Scholar] [CrossRef] [PubMed]
- Herneisen, A.L.; Sidik, S.M.; Markus, B.M.; Drewry, D.H.; Zuercher, W.J.; Lourido, S. Identifying the target of an antiparasitic compound in Toxoplasma using thermal proteome profiling. ACS Chem Biol 2020, 15, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Becher, I.; Andres-Pons, A.; Romanov, N.; Stein, F.; Schramm, M.; Baudin, F.; Helm, D.; Kurzawa, N.; Mateus, A.; Mackmull, M.T.; et al. Pervasive protein thermal stability variation during the cell cycle. Cell 2018, 173, 1495–1507 e1418. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Hemphill, A. In vitro screening technologies for the discovery and development of novel drugs against Toxoplasma gondii. Expert Opin Drug Discov 2024, 19, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Tjorve, K.M.C.; Tjorve, E. The use of Gompertz models in growth analyses, and new Gompertz-model approach: An addition to the Unified-Richards family. PLoS One 2017, 12, e0178691. [Google Scholar] [CrossRef] [PubMed]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol 2016, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Hemphill, A.; Müller, N. Physiological aspects of nitro drug resistance in Giardia lamblia. Int J Parasitol Drugs Drug Resist 2018, 8, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Schlange, C.; Heller, M.; Uldry, A.C.; Braga-Lagache, S.; Haynes, R.K.; Hemphill, A. Proteomic characterization of Toxoplasma gondii ME49 derived strains resistant to the artemisinin derivatives artemiside and artemisone implies potential mode of action independent of ROS formation. Int J Parasitol Drugs Drug Resist 2022, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wellems, T.E.; Sá, J.M.; Su, X.Z.; Connelly, S.V.; Ellis, A.C. ’Artemisinin Resistance’: Something New or Old? Something of a Misnomer? Trends in Parasitology 2020, 36, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, C.; Zhao, Y.; Tang, J.; Zhu, M.; Platon, L.; Culleton, R.; Zhu, G.; Menard, D.; Zhang, Q.; et al. Ring-stage growth arrest: Metabolic basis of artemisinin tolerance in Plasmodium falciparum. iScience 2023, 26, 105725. [Google Scholar] [CrossRef] [PubMed]
- Winzer, P.; Anghel, N.; Imhof, D.; Balmer, V.; Ortega-Mora, L.M.; Ojo, K.K.; Van Voorhis, W.C.; Müller, J.; Hemphill, A. Neospora caninum: Structure and Fate of Multinucleated Complexes Induced by the Bumped Kinase Inhibitor BKI-1294. Pathogens 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Winzer, P.; Müller, J.; Imhof, D.; Ritler, D.; Uldry, A.C.; Braga-Lagache, S.; Heller, M.; Ojo, K.K.; Van Voorhis, W.C.; Ortega-Mora, L.M.; et al. Neospora caninum: Differential Proteome of Multinucleated Complexes Induced by the Bumped Kinase Inhibitor BKI-1294. Microorganisms 2020, 8. [Google Scholar] [CrossRef]
- Jerlström-Hultqvist, J.; Stadelmann, B.; Birkestedt, S.; Hellman, U.; Svärd, S.G. Plasmid vectors for proteomic analyses in Giardia: purification of virulence factors and analysis of the proteasome. Eukaryot Cell 2012, 11, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Breinich, M.S.; Gilson, P.R.; Crabb, B.S. Molecular genetic tools in Toxoplasma and Plasmodium: achievements and future needs. Curr Opin Microbiol 2007, 10, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Carucci, D.J. Technologies for the study of gene and protein expression in Plasmodium. Philos Trans R Soc Lond B Biol Sci 2002, 357, 13–16. [Google Scholar] [CrossRef]
- Donald, R.G.; Roos, D.S. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc Natl Acad Sci U S A 1993, 90, 11703–11707. [Google Scholar] [CrossRef] [PubMed]
- Howe, D.K.; Sibley, L.D. Development of molecular genetics for Neospora caninum: A complementary system to Toxoplasma gondii. Methods 1997, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ullu, E.; Tschudi, C.; Chakraborty, T. RNA interference in protozoan parasites. Cell Microbiol 2004, 6, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Sidik, S.M.; Hackett, C.G.; Tran, F.; Westwood, N.J.; Lourido, S. Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 2014, 9, e100450. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Brown, K.M.; Lee, T.D.; Sibley, L.D. Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio 2014, 5, e01114–01114. [Google Scholar] [CrossRef] [PubMed]
- Sidik, S.M.; Huet, D.; Ganesan, S.M.; Huynh, M.H.; Wang, T.; Nasamu, A.S.; Thiru, P.; Saeij, J.P.; Carruthers, V.B.; Niles, J.C.; et al. A Genome-wide CRISPR screen in Toxoplasma Identifies essential apicomplexan genes. Cell 2016, 166, 1423–1435 e1412. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Barba, S.; Top, E.M.; Stalder, T. Plasmids, a molecular cornerstone of antimicrobial resistance in the One Health era. Nat Rev Microbiol 2024, 22, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D.; Williams, C.F.; Hrdy, I. Redox pathways as drug targets in microaerophilic parasites. Trends Parasitol 2018, 34, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Heller, M.; Uldry, A.C.; Braga, S.; Müller, N. Nitroreductase activites in Giardia lamblia: ORF 17150 encodes a quinone reductase with nitroreductase activity. Pathogens 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Kishony, R. Opposing effects of target overexpression reveal drug mechanisms. Nat Commun 2014, 5, 4296. [Google Scholar] [CrossRef] [PubMed]
- Hanggeli, K.P.A.; Hemphill, A.; Müller, N.; Heller, M.; Uldry, A.C.; Braga-Lagache, S.; Müller, J.; Boubaker, G. Comparative proteomic analysis of Toxoplasma gondii RH wild-type and four SRS29B (SAG1) knock-out clones reveals significant differences between individual strains. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Freisleben, F.; Behrmann, L.; Thaden, V.; Muschhammer, J.; Bokemeyer, C.; Fiedler, W.; Wellbrock, J. Downregulation of GLI3 expression mediates chemotherapy resistance in acute myeloid leukemia. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- de Koning, H.P. Drug resistance in protozoan parasites. Emerg Top Life Sci 2017, 1, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.A.; Carucci, D.J. Proteomic approaches to studying drug targets and resistance in Plasmodium. Curr Drug Targets Infect Disord 2004, 4, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, F.; Yan, H.; Yin, J.; Xia, Z. A review of malaria molecular markers for drug resistance in Plasmodium falciparum and Plasmodium vivax in China. Front Cell Infect Microbiol 2023, 13, 1167220. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.; Surolia, N.; Bhattacharjee, S.; Karmodiya, K. The many paths to artemisinin resistance in Plasmodium falciparum. Trends Parasitol 2023, 39, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Platon, L.; Menard, D. Plasmodium falciparum ring-stage plasticity and drug resistance. Trends Parasitol 2024, 40, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Ley, S.; Felger, I.; Hemphill, A.; Müller, N. Identification of differentially expressed genes in a Giardia lamblia WB C6 clone resistant to nitazoxanide and metronidazole. J Antimicrob Chemother 2008, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Townson, S.M.; Laqua, H.; Upcroft, P.; Boreham, P.F.; Upcroft, J.A. Induction of metronidazole and furazolidone resistance in Giardia. Trans R Soc Trop Med Hyg 1992, 86, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Sterk, M.; Hemphill, A.; Müller, N. Characterization of Giardia lamblia WB C6 clones resistant to nitazoxanide and to metronidazole. J Antimicrob Chemother 2007, 60, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Upcroft, J.A.; Edwards, M.R.; Upcroft, P. Anaerobic bacterial metabolism in the ancient eukaryote Giardia duodenalis. Int J Parasitol 1998, 28, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Braga, S.; Heller, M.; Müller, N. Resistance formation to nitro drugs in <italic>Giardia lamblia</italic>: No common markers identified by comparative proteomics Int J Parasitol: Drugs Drug Resist 2019, 9, 112-119.
- Ansell, B.R.; Baker, L.; Emery, S.J.; McConville, M.J.; Svard, S.G.; Gasser, R.B.; Jex, A.R. Transcriptomics indicates active and passive metronidazole resistance mechanisms in three seminal Giardia lines. Front Microbiol 2017, 8, 398. [Google Scholar] [CrossRef] [PubMed]
- Emery, S.J.; Baker, L.; Ansell, B.R.E.; Mirzaei, M.; Haynes, P.A.; McConville, M.J.; Svard, S.G.; Jex, A.R. Differential protein expression and post-translational modifications in metronidazole-resistant Giardia duodenalis. Gigascience 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Koncarevic, S.; Bogumil, R.; Becker, K. SELDI-TOF-MS analysis of chloroquine resistant and sensitive Plasmodium falciparum strains. Proteomics 2007, 7, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Doliwa, C.; Xia, D.; Escotte-Binet, S.; Newsham, E.L.; Sanya, J.S.; Aubert, D.; Randle, N.; Wastling, J.M.; Villena, I. Identification of differentially expressed proteins in sulfadiazine resistant and sensitive strains of Toxoplasma gondii using difference-gel electrophoresis (DIGE). Int J Parasitol Drugs Drug Resist 2013, 3, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Schlange, C.; Heller, M.; Uldry, A.C.; Braga-Lagache, S.; Haynes, R.K.; Hemphill, A. Proteomic characterization of Toxoplasma gondii ME49 derived strains resistant to the artemisinin derivatives artemiside and artemisone implies potential mode of action independent of ROS formation. Int J Parasitol Drugs Drug Resist 2023, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Scheele, S.; Geiger, J.A.; DeRocher, A.E.; Choi, R.; Smith, T.R.; Hulverson, M.A.; Vidadala, R.S.R.; Barrett, L.K.; Maly, D.J.; Merritt, E.A.; et al. Toxoplasma calcium-dependent protein kinase 1 inhibitors: Probing activity and resistance using cellular thermal shift assays. Antimicrob Agents Chemother 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Burata, O.E.; Yeh, T.J.; Macdonald, C.B.; Stockbridge, R.B. Still rocking in the structural era: A molecular overview of the small multidrug resistance (SMR) transporter family. J Biol Chem 2022, 298, 102482. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Q. Chemical proteomics approach reveals the direct targets and the heme-dependent activation mechanism of artemisinin in Plasmodium falciparum using an artemisinin-based activity probe. Microb Cell 2016, 3, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Rios-Barros, L.V.; Silva-Moreira, A.L.; Horta, M.F.; Gontijo, N.F.; Castro-Gomes, T. How to get away with murder: The multiple strategies employed by pathogenic protozoa to avoid complement killing. Mol Immunol 2022, 149, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Prucca, C.G.; Lujan, H.D. Antigenic variation in Giardia lamblia. Cell Microbiol 2009, 11, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Gottstein, B. Antigenic variation and the murine immune response to Giardia lamblia. Int J Parasitol 1998, 28, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Vermathen, M.; Leitsch, D.; Vermathen, P.; Müller, N. Metabolomic profiling of wildtype and transgenic Giardia lamblia Strains by (1)H HR-MAS NMR Spectroscopy. Metabolites 2020, 10. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Simplified workflow of protein LC-MS/MS.
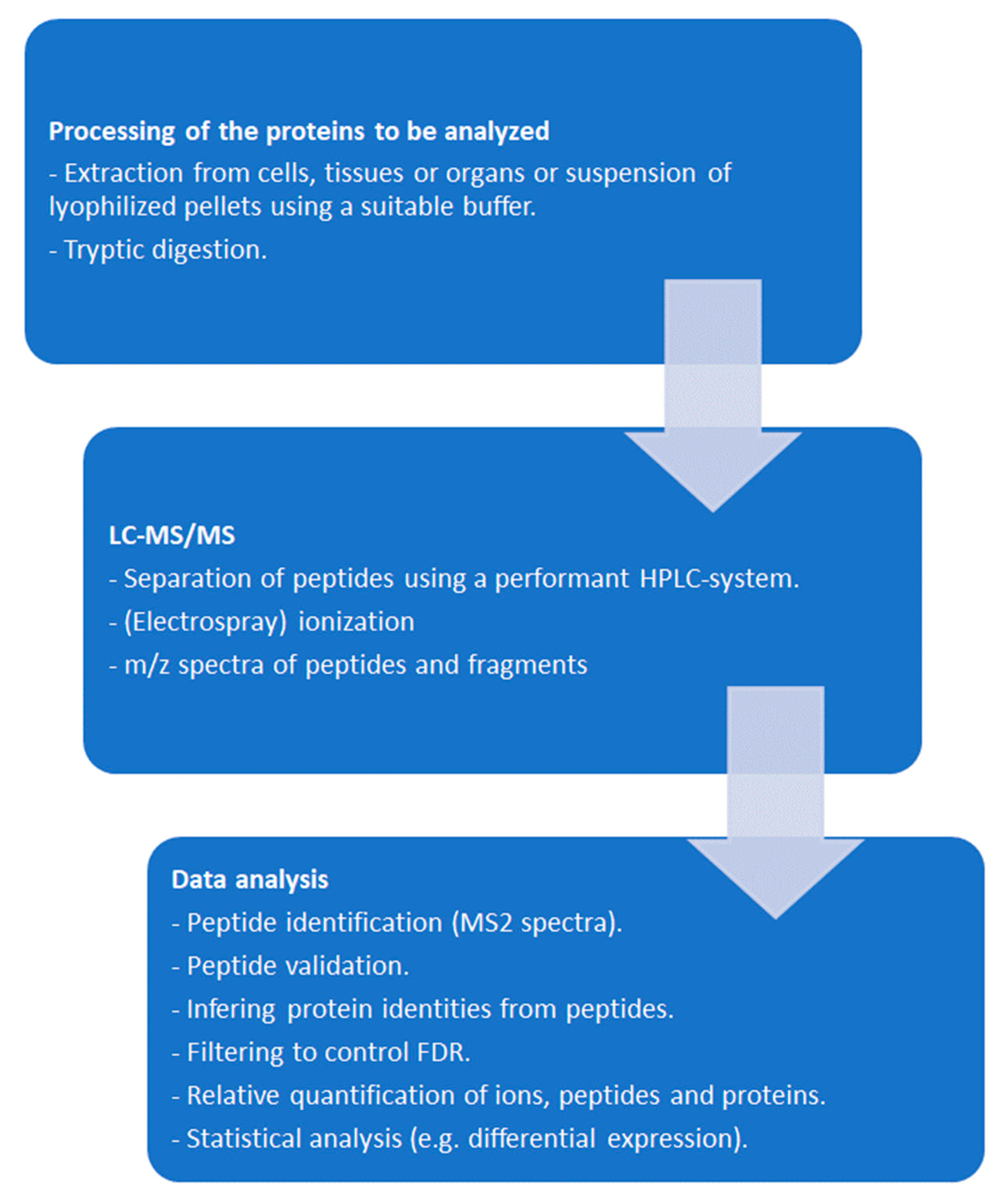
Figure 3.
Workflow of differential affinity chromatography (DAC).
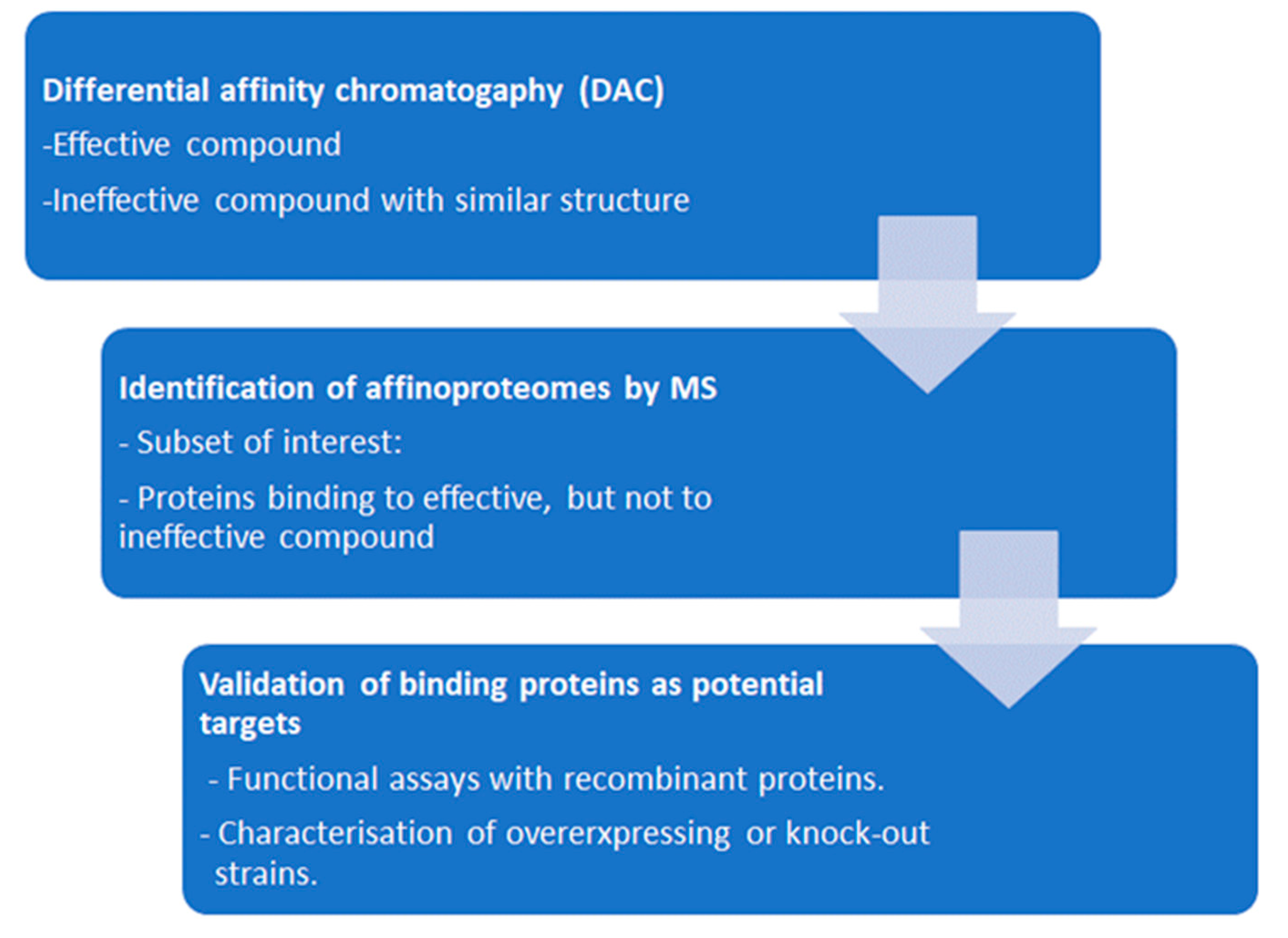

Table 1.
Overview of studies presenting in vitro assays with selected target proteins.
| Protein Target for Inhibitor Screenings | Pathogen | Methodology | Reference |
|---|---|---|---|
| Protein biosynthesis | P. falciparum | Luciferase assay | [60] |
| Calcium-dependent protein kinase 1 | T. gondii | Kinase assay Cocrystallization |
[61,62] |
| Dihydrofolate reductase thymidylate synthase | T. gondii | Functional assay | [63] |
| Dihydrofolate reductase | P. vivax | Heterologous expression in yeast Growth assay |
[64] |
| Acetyl-CoA carboxylase | T. gondii | Functional assay | [65] |
| Glyoxalase 1 | T. gondii | Functional assay | [66] |
| Type-II NADH dehydrogenase | T. gondii | Functional assay | [67] |
| Nucleoside triphosphate hydrolase | N. caninum, T. gondii | Chemoluminescence assay | [68] |
| Mitochondrial ADP/ATP | P. falciparum | Heterologous expression in E. coli Radioactive uptake assay |
[69] |
| Importin alpha binding to nuclear localization signal | P. falciparum | Alpha screen binding assay | [70] |
| Phenylalanyl t-RNA synthetase | T. gondii | Functional assay | [71] |
| Aspartate transcarbamoylase | P. falciparum | Functional assay, protein interference assay | [72,73] |
Table 2.
Overview of affinity chromatography studies identifying antimalarial binding proteins. AC, affinity chromatography; BP, binding protein; DAC, differential affinity chromatography; FA, functional assay.
Table 2.
Overview of affinity chromatography studies identifying antimalarial binding proteins. AC, affinity chromatography; BP, binding protein; DAC, differential affinity chromatography; FA, functional assay.
| Antimalarial | Methodology | Remarks | Reference |
|---|---|---|---|
| Kinase inhibitors | Cell-free extracts from various cell types and organisms. DAC with active and inactive purines. SDS-PAGE followed by digestion of binding proteins and microsequencing of the peptides. | Detection of known kinases by Western Blotting. Some of the peptide sequences match to other kinases and other proteins. | [86] |
| Quinolines | Cell-free extracts of infected human erythrocytes. DAC with ATP as a control, elution with various quinoline antimalarials, SDS-PAGE followed by Edman mixed peptide sequencing. | Human aldehyde dehydrogenase 1 and quinone reductase 2 major BP. Validated as potential target by FA. | [87] |
| Endoperoxides | P. falciparum trophozoite lysates. AC with an artemisinin analog, followed by 2-D SDS-PAGE and MALDI-TOF MS. | Identification of 9 P. falciparum BPs. Major BP is a calcium binding protein. | [88] |
Table 3.
Overview of affinity chromatography (AC) and differential affinity chromatography (DAC) studies performed in protozoal pathogens and host cells. All studies were performed using epoxy-activated sepharose. BP, binding protein; FA, functional assay; GST, glutathione-S-transferase.
Table 3.
Overview of affinity chromatography (AC) and differential affinity chromatography (DAC) studies performed in protozoal pathogens and host cells. All studies were performed using epoxy-activated sepharose. BP, binding protein; FA, functional assay; GST, glutathione-S-transferase.
| Organism | Ligand | Methodology | Remarks | Reference |
|---|---|---|---|---|
| G. lamblia | Thiazolide | AC, elution with ligand, SDS-PAGE followed by LC-MS/MS. | Nitroreductase NR1 major BP. Validated as potential target by FA and in subsequent studies. | [89,90,91] |
|
H. sapiens Caco2 |
Thiazolide | AC, elution with ligand, SDS-PAGE followed by LC-MS/MS. | Human GSTP1 major BP. Validated as potential target by FA and in subsequent studies. | [92,93] |
|
H. sapiens Fibroblasts |
Thiazolide | AC, elution with ligand, SDS-PAGE followed by LC-MS/MS. | Human quinone reductase 1 major BP in N. caninum infected cells. Validation by FA. | [94] |
| T. gondii | Ruthenium complex | DAC with mock column only; elution by pH shift; SDS-PAGE followed by LC-MS/MS. | Translation elongation factor 1 alpha and two ribosomal proteins identified as binding proteins. |
[95] |
|
T. gondii T. brucei |
Ruthenium complex | Comparative DAC with two ineffective complexes in two pathogens, elution with pH shift, LC-MS/MS on entire eluates. | 128 specific T. gondii BPs and 46 specific T. brucei BPs. Major T. brucei BP mitochondrial ATP synthase subunit validated by FA. | [96] |
|
T. gondii M. musculus splenocytes |
Antimicrobial peptide | Comparative DAC with ineffective peptide, elution with pH shift, LC-MS/MS on entire eluates. | Several hundred BPs in eluates from both organisms suggesting common modes of action. | [97] |
|
N. caninum D. rerio |
Bumped kinase inhibitor with quinoline core | Comparative DAC with quinine, elution with pH shift, LC-MS/MS on entire eluates. | 12 specific N. caninum BPs and 13 specific D. rerio BPs. Many BPs in both organism in quinine eluates, as well. Majority involved in RNAbinding or modification. | [98] |
|
C. parvum H. sapiens HCT-8 cells |
Bumped kinase in-hibitor with quino-line core | Comparative DAC with quinine, elution with pH shift, LC-MS/MS on entire eluates. | No specific binding proteins in C. parvum, 25 specific BPs in host cells. 29 C. parvum and 224 host cell BPs also in quinine eluates. Common targets in RNA binding or modification. | [99] |
Table 4.
Overview of thermal proteome profiling based target deconvolution studies performed in protozoal pathogens. CETSA, cellular thermal shift assay; MS, mass spectrometry.
Table 4.
Overview of thermal proteome profiling based target deconvolution studies performed in protozoal pathogens. CETSA, cellular thermal shift assay; MS, mass spectrometry.
| Organism | Methodology | Remarks | Reference |
|---|---|---|---|
| Leishmania donovani | Classical CETSA-MS on promastigotes with an inhibitor of sterol biosynthesis. | Oxidosqualene cyclase identified as a target of this inhibitor. | [121,122] |
| L. infantum | Classical CETSA-MS on cell-free extracts of amphotericine B, antimony or miltefosine susceptible and resistant lines incubated with the respective drugs. | Up to several hundred proteins with altered melting profiles depending on the compound. Sb tend to stabilize ribosomal proteins. | [123] |
| P. falciparum | Comparison of classical and isothermal CETSA on intraerythrocytic stages using pyrimethamine as a proof of concept. | Conceptual study. No data on novel binding proteins directly available. | [124] |
| T. gondii | Classical CETSA-MS with calcium egress inhibitor ENH1 as ligand. | 82 proteins with enhanced thermal stability identified including Calcium dependent protein kinase 1. | [125] |
Table 5.
Summary of affinity-based methods identifying drug interacting proteomes. AC, affinity chromatography; DAC, differential affinity chromatography; TPP, thermal protein profiling.
Table 5.
Summary of affinity-based methods identifying drug interacting proteomes. AC, affinity chromatography; DAC, differential affinity chromatography; TPP, thermal protein profiling.
| Methodology | Advantages | Inconvenients |
|---|---|---|
| AC–elution with ligand | Well established. Does not need sophisticated equipment. Fast. | Modification of original ligand necessary to create column matrix. Identification of major binding proteins after PAGE, resulting in low yields, bias. Cell-free extracts. |
| DAC–unspecific elution | See above. LC-MS/MS if elution with compatible solvent. | Needs ineffective control compound with similar structure. Cell-free extracts. |
| Affinity labelling | Interaction occurs intracellularly, therefore under physiological conditions. Fast. | Modification of original ligand necessary to create compound for affinity labelling. Polishing of labelled proteins by PAGE, therefore low yields and bias. Label may interfere with subsequent MS. |
| TPP | Flexible, since interaction of proteomes and ligands are investigated under physiological conditions or in cell free extracts. Unmodified compounds may be used. | Time and cost intensive. Use of isobaric labels. Large data volumes needing appropriate bioinformatic tools. |
Table 6.
Overview of selected studies comparing whole-cell proteomes of resistant vs. susceptible strains. DIGE, differential gel electrophoresis; LC, liquid chromatography; MS, mass spectrometry, SELDI, surface-enhanced laser desorption/ ionization; TOF, time of flight.
Table 6.
Overview of selected studies comparing whole-cell proteomes of resistant vs. susceptible strains. DIGE, differential gel electrophoresis; LC, liquid chromatography; MS, mass spectrometry, SELDI, surface-enhanced laser desorption/ ionization; TOF, time of flight.
| Organism | Drug | Methodology | Remarks | Reference |
|---|---|---|---|---|
| G. lamblia | Metronidazole | Comparison of three resistant cell lines created by increasing drug concentrations plus UV irradiation with susceptible parental strains. Analysis of proteomes and post-translational modifications by broad panel of proteome analytical methods. | 265, 171, and 76 differentially expressed proteins depending on the strains. High isolate dependent variability of adaptation mechanisms. | [163] |
| G. lamblia | Nitazoxanide Metronidazole |
Comparison of a strain generated by increasing nitazoxanide concentrations and two metronidazole resistant strains from study quoted above with their corresponding wildtypes. All resistant strains were resistant to both drugs and were grown int the presence of either drug prior to analysis by shotgun LC/MS-MS. | 225, 248, and 304 differentially expressed proteins in the presence of nitazoxanide, 510, 287, and 216 in the presence of metronidazole. No common markers for nitro resistance. Common pattern of antigenic variation in all metronidazole resistant vs. susceptible strains. Strategies of coping with nitro reduction strain and drug dependent. | [161] |
| P. falciparum | Chloroquine | Comparison of two clinical isolates resistant to chloroquine with two susceptible isolates using SELDI-TOF. | Study focused on the methodology. One of the susceptible strains and both resistant strains are resistant to pyrimethamine, one resistant strain is resistant to quinine and sulfadoxine, as well. 10 “marker proteins” identified. | [164] |
|
T. gondii |
Sulfadizine | Resistant clinical isolates, susceptible reference strains. Comparison of proteomes by DIGE-MS. | 31 unique differential proteins were identified. |
[165] |
| T. gondii | Artemisone Artemiside |
Generation of resistant strains by treating the reference strain ME49 with increasing concentrations. Whole-cell-shotgun LC/MS-MS. | 215 proteins downregulated in the artemisone resistant strain, 8 proteins in the artemiside resistant strain. | [166] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



